User login
The electronic medical record’s role in ObGyn burnout and patient care

Physician burnout has been labeled a public health crisis by the Harvard School of Public Health and other institutions.1 A 2018 Physician’s Foundation survey found that 78% of physicians had symptoms of burnout,2 which result from chronic workplace stress and include feeling depleted of energy or exhausted, mentally distanced from or cynical about one’s job, and problems getting one’s job done successfully.3 Among ObGyns, almost half (46%) report burnout.4 One-third of ObGyns responded on a recent Medscape Burnout Report that the computerization of practice is contributing to their burnout, and 54% said too many bureaucratic tasks, including charting, were adding to their burnout.5
Inefficient electronic medical records (EMRs) have been implicated as one reason for burnout, with improvements in efficiency cited as one of several potential resolutions to the problem. About 96% of hospitals have adopted EMRs today, compared with only 9% in 2008,6 and many physicians report recognizing value in the technology. For instance, 60% of participants in Stanford Medicine’s 2018 National Physician Poll said EMRs had led to improved patient care. At the same time, however, about as many (59%) said EMRs needed a “complete overhaul” and that the systems had detracted from their professional satisfaction (54%) as well as from their clinical effectiveness (49%).7
With this roundtable, we explore the concerns with hours spent on the EMR with several experts, and whether it is a problem that has been contributing to burnout among staff at their institutions. In addition, are there solutions that their institutions have implemented that they can share to help to cope with the problem?
John J. Dougherty, MD, MBA: Yes, absolutely. There is not a day that goes by that I don’t hear about or experience “Epic Fails.” (We use Epic’s EMR product at our institution.) Too many clicks are needed to navigate even the simplest tasks—finding notes or results, documenting visits, and billing for services are all unnecessarily complex. In addition, we are being held accountable for achieving a long and growing list of “metrics” measures, education projects (HealthStream), and productivity goals. When do we have time to treat patients? And it is not just practicing physicians and clinicians. Our resident physicians spend an inordinate amount of time in front of the computer documenting, placing orders, and transferring patients using a system with a very inefficient user interface, to say the least.
Megan L. Evans, MD, MPH: I absolutely agree. Over the years, my institution has created a conglomerate of EMRs, requiring physicians across the hospital to be fluent in a multitude of systems. For example, you finish your clinic notes in one system, sign off on discharge summaries in another, and complete your operative notes in an entirely different system. As busy attendings, it is hard to keep ahead of all of these tasks, especially when the systems do not talk to one another. Fortunately, my hospital is changing our EMR to a single system within the next year. Until then, however, we will work in this piecemeal system.
Mark Woodland, MS, MD: EMR and computerization of medicine is the number 1 issue relating to dissatisfaction by ObGyn providers in our institution. Providers are earnest in their attempt to be compliant with EMR requirements, but the reality is that they are dealing with an automated system that does not have realistic expectations for management of results, follow-up tasks, and patient communications for a human provider. The actual charting, ordering of tests and consults, and communication between providers has been enhanced. However, the “in-basket” of tasks to be accomplished are extraordinary and much of it relies on the provider, which requires an inordinate amount of time. Additionally, while other members of the medical staff are stationary at a desk, physicians and other providers are not. They are mobile between inpatient units, labor and delivery, operating rooms, and emergency rooms. Time management does not always allow for providers to access computers from all of these areas to facilitate their managing the expectations of the EMR. This requires providers to access the EMR at off hours, extending their workload. Finally, the EMR is neither personal nor friendly. It is not designed with the clinician in mind, and it is not fun or engaging for a provider.
EMRs are not just inefficient and contributing to physician burnout, according to a joint report from Kaiser Health News (KHN) and Fortune magazine, they are inadequate and contributing to patient safety concerns.1 This was not the intended goal of the HITECH Act, signed into law in 2009 as part of the stimulus bill. HITECH was intended to promote the adoption of meaningful use of health information technology by providing financial incentives to clinicians to adopt electronic medical records (EMRs). It also intended to increase security for health care data--achieved through larger penalties for HIPAA violations.2
Ten years later, however, "America has little to show" for its $36 billion investment, according to KHN and Fortune. Yes, 96% of hospitals have one of the currently available EMRs, among thousands, but they are disconnected. And they are "glitchy." At least 2 EMR vendors have reached settlements with the federal government over egregious patient errors. At least 7 deaths have resulted from errors related to the EMR, according to the firm Quantros, reports KHN and Fortune, and the number of EMR-related safety events tops 18,000. The problem is that information, critical to a patient's well-being, may get buried in the EMR. Clinicians may not have been aware of, because they did not see, a critical medication allergy or piece of patient history.1
The problems with health information technology usability do have solutions, however, asserts Raj M. Ratwani, MD, and colleagues. In a recent article published in the Journal of the American Medical Association, the researchers propose 5 priorities for achieving progress3:
- Establishment of a national database of usability and safety issues. This database should allow sharing of safety information among EMR vendors, hospitals, and clinicians, and make the public aware of any technology risks.
- Establishment of basic design standards, which should promote innovation and be regulated by a board composed of all stakeholders: EMR vendors, researchers, clinicians, and health care organizations.
- Addressing unintended harms. Causes of harm could include "vendor design and development, vendor and health care organization implementation, and customization by the health care organization." Along with shared responsibility and collaboration comes shared liability for harms caused by inadequate usability.
- Simplification of mandated documentation requirements that affect usability. Reducing clinician's "busy work" would go a long way toward simplifying documentation requirements.
- Development of standard usability and safety measures so that progress can be tracked and the market can react. EMR vendors cannot be directly compared currently, since no standards for usability are in place.
Ratwani and colleagues cite shared responsibility and commitment among all of the parties invested in EMR usability success as keys to solving the current challenges affecting health information technology, with policy makers at the helm.3 The federal government is attempting to respond: As part of the 2016 21st Century Cures Act and with an aim toward alleviating physician time spent on the EMR, the Department of Health and Human Services is required to recommend reductions to current EMR burdens required under the HITECH Act. It plans to revise E&M codes, lessening documentation. And the Centers for Medicare and Medicaid Services aims to make meaningful use requirements more flexible, require information exchange between providers and patients, and provide incentive to clinicians to allow patient access to EMRs.4,5
References
- Fry E, Schulte F. Death by a thousand clicks. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- Burde H. The HITECH Act: an overview. AMA J Ethics. March 2011. https://journalofethics.ama-assn.org/article/hitech-act-overview/2011-03. Accessed September 9, 2019.
- Ratwani R, Reider J, Singh H. A decade of health information technology usability challenges and the path forward. JAMA. 2019;321:743-744.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Morris G, Anthony ES. 21st Century Cures Act overview for states. Office of the National Coordinator for Health Information Technology. https://www.healthit.gov/sites/default/files/curesactlearningsession_1_v6_10818.pdf. Accessed September 11, 2019.
Continue to:
Dr. Dougherty: When our institution compared EMR offerings, EMR companies put their best collective marketing feet forward. The general notion, at least with the Epic EMR, was that “you can customize Epic to your liking.” It did not take long for a bunch of motivated Epic users to create “smart” stuff (lists, phrases, and texts) in an effort to customize workflows and create fancy-looking electronic notes. Shortly thereafter, it was obvious that, as an institution, our reporting efforts kept coming up short—our reports lacked accuracy and meaning. Everyone was documenting in different ways and in different areas. Considering that reports are currently generated using (mostly) discrete data entries (data placed in specific fields within the EMR), it became obvious that our data entry paradigm needed to change. Therefore, standardization became the leading buzzword. Our institution recently initiated a project aimed at standardizing our workflows and documentation habits. In addition, we have incorporated a third-party information exchange product into our health system data aggregation and analysis workflow. Much more needs to be done, but it is a start.
Dr. Evans: At my institution, as a group, we have created templates for routine procedures and visits that also auto populate billing codes. I know that some departments have used scribes. From the hospital side, there has been improved access to the EMR from home. Some of my colleagues like this feature; however, others, like myself, believe this contributes to some of our burnout. I like to leave work at work. Having the ability to continue working at home is not a solution in my mind.
Dr. Woodland: At our institution, we have engaged our chaperones and medical assistants to help facilitate completion of the medical records during the office visit. Providers work with their assistants to accommodate documentation of history and physical findings while also listening to the provider as they are speaking in order to document patient care plans and orders. This saves the clinicians time in reviewing and editing the record as well as making sure the appropriate care plan is instituted. Our EMR provider recently has begun experimenting with personalization of color themes as well as pictures as part of the interface. Having said this, I still ask, “Why have medical professionals allowed non–clinical agencies and information technology groups to run this show?” It is also inconceivable to me that this unfunded mandate—that has increased cost, decreased clinical efficiency, and decreased clinician satisfaction—has not been addressed by national and international medical communities.
Dr. Woodland: I feel that we need to appropriately manage expectations of the EMR and the institution with relation to EMR and providers. By this I mean that we need to make the EMR more user-friendly and appropriate for different clinicians as well as patients. We also need to manage expectations of our patients. In a digital age where immediate contact is the norm, we need to address the issue that the EMR is not social media but rather a communication tool for routine contact and information transmission. Emergencies are not typically addressed well through the EMR platform; they are better handled with a more appropriate communication interface.
Dr. Dougherty: I feel that the biggest change needed is a competent, simple, and standard user-interface. Our old charting methods were great on a number of levels. For instance, if I wanted to add an order, I flipped to the ”Orders” tab and entered an order. If I needed to document a note, I flipped to the “Notes” tab and started writing, etc. Obviously, manual charting had its downsides—like trying to decipher handwriting art! EMRs could easily adopt the stuff that worked from our old methods of documentation, while leveraging the advantages that computerized workflows can bring to practitioners, including efficient transfer of records, meaningful reporting, simple electronic ordering, and interprofessional communication portals.
Dr. Evans: Our systems need to better communicate with one another. I am in an academic practice, and I should be able to see labs, consultant notes, imaging, all in one spot to improve efficiency and ease with patient visits. Minimizing clicks would be helpful as well. I try to write as much as I can while in the room with a patient to avoid after-hours note writing, but it takes away from my interaction with each patient.
Continue to:
Dr. Evans: When I first started as a new attending, it would take me hours to finish my notes, partly because of the level of detail I would write in my history of present illness (HPI) and assessment and plan. One great piece of advice I received was to be satisfied with good notes, not perfect notes. I worked to consolidate my thoughts and use preconstructed phrases/paragraphs on common problems I saw. This saved time to focus on other aspects of my academic job.
Dr. Dougherty: We need to refocus on the patient first, and mold our systems to meet that priority. Much too often, we have our backs to the patients or spend too much time interfacing with our EMR systems, and our patients are not happy about it (as many surveys have demonstrated). More importantly, a renewed focus on patient care, not EMR care, would allow our practitioners to do what they signed up for—treating patients. In the meantime, I would suggest that practitioners stay away from EMR gimmicks and go back to old-style documentation practices (like those established by the Centers for Medicare and Medicaid Services in 1997 and 1998), and ask the IT folks to help with molding the EMR systems to meet your own standards, not the standards established by EMR companies. I am also very hopeful that the consumer will drive most of the health care-related data collection in the near future, thereby marginalizing the current generation of EMR systems.
Dr. Woodland: I would add that providers need to manage the EMR and not let the EMR manage them. Set up task reminders at point times to handle results and communications from the EMR and set up time in your schedule where you can facilitate meeting these tasks. When providers are out on vacation, make sure to have an out-of-office reminder built into their EMR so that patients and others know timing of potential responses. Try to make the EMR as enjoyable as possible and focus on the good points of the EMR, such as legibility, order verification, safety, and documentation.
1. Engage the computer in your patient encounter, says Rey Wuerth and colleagues. Share the screen, and any test results you are highlighting, with your patient by turning it toward her during your discussion. This can increase patient satisfaction.1
2. Go mobile at the point of care, suggests Tom Giannulli, MD, MS, Chief Medical Information Officer at Kareo. By using a tablet or mobile device, you can enter data while facing a patient or on the go.2
3. Use templates when documenting data, advises Wuerth and colleagues, as pre-filled templates, that are provided through the EMR or that you create within the EMR, can reduce the time required to enter patient visits, findings, and referrals.1
4. Delegate responsibility for routing documents, says Brian Anderson, MD. Hand off to staff administrative duties, such as patient forms and routine negative test results.3
5. Involve medical assistants (MAs) in the process. Make the MA feel part of the team, says R. Scott Eden, and assign them history-taking responsibilities, utilizing your EMR's templates. Assign them other tasks as well, including medication reconciliation, referrals, refills, routine screening, and patient education.4
6. Employ physical or virtual scribes who are specifically assigned to EMR duty. Although drawbacks can include patient privacy concerns and reduced practice income due to salary requirements, employing a scribe (often a pre-medical or graduate student), who trails you on patient visits, or who is connected virtually, can leave the clinician free to interact with patients.5,6
References
- Wuerth R, Campbell C, Peng MD, et al. Top 10 tips for effective use of electronic health records. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959973/. Paediatr Child Health. 2014;19:138.
- Giannulli T. 7 time-saving EHR use tips to boost physician productivity. April 28, 2016. https://ehrintelligence.com/news/7-time-saving-emr-use-tips-to-boost-physician-productivity. Accessed September 9, 2019.
- Anderson B. 5 ways to increase your EMR efficiency. October 28, 2014. https://www.kevinmd.com/blog/2014/10/5-ways-increase-emr-efficiency.html. Accessed September 9, 2019.
- Eden RS. Maximizing your medical assistant's role. Fam Pract Manag. 2016;23:5-7. https://www.aafp.org/fpm/2016/0500/p5.html.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Caliri A. The case for virtual scribes. January 2, 2019. Becker's Hospital Review. https://www.beckershospitalreview.com/hospital-physician-relationships/the-case-for-virtual-scribes.html. Accessed September 20, 2019.
Dr. Evans: Yes and no. Yes, in that it can be much easier to follow a patient’s health care history from other provider notes or prior surgeries. Information is searchable and legible. If an EMR is built correctly, it can save time for providers, through smart phrases and templates, and it can help providers with proper billing codes and documentation requirements. No, in that it can take away from important patient interaction. We are required to see more patients in less time all while using, at times, a cumbersome EMR system.
Dr. Woodland: This is a tricky question because the EMR has both positive and negative attributes. Certainly, the legibility and order verification has improved, but the ease of accessing information in the EMR has changed. Additionally, there has been a drastic increase in provider dissatisfaction that has not been addressed. Provider dissatisfaction can lead to problems in patient care. If there was a clear-cut increased value for the cost, I do not think the EMR would be such a huge focus of negative attention. Providers need to take back control of their EMR and their profession so that they can utilize the EMR as the tool it was supposed to be and not the dissatisfier that it has become.
Dr. Dougherty: I do not believe patient care has been improved by EMR systems, for all of the reasons we have discussed, and then some. But there is an enormous amount of potential, if we get the interface between humans and EMR systems right!
- A crisis in health care: a call to action on physician burnout. Massachusetts Health and Hospital Association. Massachusetts Medical Society. Harvard T.H. Chan School of Public Health. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/21/2019/01/PhysicianBurnoutReport2018FINAL.pdf. Accessed September 9, 2019.
- Physician’s Foundation. 2018 survey of America’s physicians practice patterns and perspectives. https://physiciansfoundation.org/wp-content/uploads/2018/09/physicians-survey-results-final-2018.pdf. Accessed September 9, 2019.
- Burn-out. ICD-11 for Mortality and Morbidity Statistics. Version 04/2019. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/129180281. Accessed September 11, 2019.
- Peckham C. Medscape National Physician Burnout & Depression Report 2018. January 17, 2018. https://www.medscape.com/slideshow/2018-lifestyle-burnout-depression-6009235#3. Accessed September 9, 2019.
- Kane L. Medscape National Physician Burnout, Depression & Suicide Report 2019. January 16, 2019. https://www.medscape.com/slideshow/2019-lifestyle-burnout-depression-6011056#5. Accessed September 9, 2019.
- Fry E, Schulte F. Death by a thousand clicks: where electronic health records went wrong. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- How doctors feel about electronic health records: National Physician Poll by The Harris Poll. https://med.stanford.edu/content/dam/sm/ehr/documents/EHR-Poll-Presentation.pdf. Accessed September 9, 2019.

Megan L. Evans, MD, MPH
Dr. Evans is Assistant Professor, Tufts University School of Medicine, and Associate Resident Program Director, Department of Obstetrics and Gynecology, Tufts Medical Center, Boston, Massachusetts.
John J. Dougherty, MD, MBA
Dr. Dougherty is Medical Director, Women’s Health Center, and Associate Residency Program Director, Reading Hospital, Tower Health, Reading, Pennsylvania.
Mark B. Woodland, MS, MD
Dr. Woodland is Chair, Obstetrics and Gynecology, Reading Health System, and Clinical Professor, Obstetrics and Gynecology, Drexel University College of Medicine, Philadelphia, Pennsylvania.
The authors report no financial relationships relevant to this article.
Megan L. Evans, MD, MPH
Dr. Evans is Assistant Professor, Tufts University School of Medicine, and Associate Resident Program Director, Department of Obstetrics and Gynecology, Tufts Medical Center, Boston, Massachusetts.
John J. Dougherty, MD, MBA
Dr. Dougherty is Medical Director, Women’s Health Center, and Associate Residency Program Director, Reading Hospital, Tower Health, Reading, Pennsylvania.
Mark B. Woodland, MS, MD
Dr. Woodland is Chair, Obstetrics and Gynecology, Reading Health System, and Clinical Professor, Obstetrics and Gynecology, Drexel University College of Medicine, Philadelphia, Pennsylvania.
The authors report no financial relationships relevant to this article.
Megan L. Evans, MD, MPH
Dr. Evans is Assistant Professor, Tufts University School of Medicine, and Associate Resident Program Director, Department of Obstetrics and Gynecology, Tufts Medical Center, Boston, Massachusetts.
John J. Dougherty, MD, MBA
Dr. Dougherty is Medical Director, Women’s Health Center, and Associate Residency Program Director, Reading Hospital, Tower Health, Reading, Pennsylvania.
Mark B. Woodland, MS, MD
Dr. Woodland is Chair, Obstetrics and Gynecology, Reading Health System, and Clinical Professor, Obstetrics and Gynecology, Drexel University College of Medicine, Philadelphia, Pennsylvania.
The authors report no financial relationships relevant to this article.
Physician burnout has been labeled a public health crisis by the Harvard School of Public Health and other institutions.1 A 2018 Physician’s Foundation survey found that 78% of physicians had symptoms of burnout,2 which result from chronic workplace stress and include feeling depleted of energy or exhausted, mentally distanced from or cynical about one’s job, and problems getting one’s job done successfully.3 Among ObGyns, almost half (46%) report burnout.4 One-third of ObGyns responded on a recent Medscape Burnout Report that the computerization of practice is contributing to their burnout, and 54% said too many bureaucratic tasks, including charting, were adding to their burnout.5
Inefficient electronic medical records (EMRs) have been implicated as one reason for burnout, with improvements in efficiency cited as one of several potential resolutions to the problem. About 96% of hospitals have adopted EMRs today, compared with only 9% in 2008,6 and many physicians report recognizing value in the technology. For instance, 60% of participants in Stanford Medicine’s 2018 National Physician Poll said EMRs had led to improved patient care. At the same time, however, about as many (59%) said EMRs needed a “complete overhaul” and that the systems had detracted from their professional satisfaction (54%) as well as from their clinical effectiveness (49%).7
With this roundtable, we explore the concerns with hours spent on the EMR with several experts, and whether it is a problem that has been contributing to burnout among staff at their institutions. In addition, are there solutions that their institutions have implemented that they can share to help to cope with the problem?
John J. Dougherty, MD, MBA: Yes, absolutely. There is not a day that goes by that I don’t hear about or experience “Epic Fails.” (We use Epic’s EMR product at our institution.) Too many clicks are needed to navigate even the simplest tasks—finding notes or results, documenting visits, and billing for services are all unnecessarily complex. In addition, we are being held accountable for achieving a long and growing list of “metrics” measures, education projects (HealthStream), and productivity goals. When do we have time to treat patients? And it is not just practicing physicians and clinicians. Our resident physicians spend an inordinate amount of time in front of the computer documenting, placing orders, and transferring patients using a system with a very inefficient user interface, to say the least.
Megan L. Evans, MD, MPH: I absolutely agree. Over the years, my institution has created a conglomerate of EMRs, requiring physicians across the hospital to be fluent in a multitude of systems. For example, you finish your clinic notes in one system, sign off on discharge summaries in another, and complete your operative notes in an entirely different system. As busy attendings, it is hard to keep ahead of all of these tasks, especially when the systems do not talk to one another. Fortunately, my hospital is changing our EMR to a single system within the next year. Until then, however, we will work in this piecemeal system.
Mark Woodland, MS, MD: EMR and computerization of medicine is the number 1 issue relating to dissatisfaction by ObGyn providers in our institution. Providers are earnest in their attempt to be compliant with EMR requirements, but the reality is that they are dealing with an automated system that does not have realistic expectations for management of results, follow-up tasks, and patient communications for a human provider. The actual charting, ordering of tests and consults, and communication between providers has been enhanced. However, the “in-basket” of tasks to be accomplished are extraordinary and much of it relies on the provider, which requires an inordinate amount of time. Additionally, while other members of the medical staff are stationary at a desk, physicians and other providers are not. They are mobile between inpatient units, labor and delivery, operating rooms, and emergency rooms. Time management does not always allow for providers to access computers from all of these areas to facilitate their managing the expectations of the EMR. This requires providers to access the EMR at off hours, extending their workload. Finally, the EMR is neither personal nor friendly. It is not designed with the clinician in mind, and it is not fun or engaging for a provider.
EMRs are not just inefficient and contributing to physician burnout, according to a joint report from Kaiser Health News (KHN) and Fortune magazine, they are inadequate and contributing to patient safety concerns.1 This was not the intended goal of the HITECH Act, signed into law in 2009 as part of the stimulus bill. HITECH was intended to promote the adoption of meaningful use of health information technology by providing financial incentives to clinicians to adopt electronic medical records (EMRs). It also intended to increase security for health care data--achieved through larger penalties for HIPAA violations.2
Ten years later, however, "America has little to show" for its $36 billion investment, according to KHN and Fortune. Yes, 96% of hospitals have one of the currently available EMRs, among thousands, but they are disconnected. And they are "glitchy." At least 2 EMR vendors have reached settlements with the federal government over egregious patient errors. At least 7 deaths have resulted from errors related to the EMR, according to the firm Quantros, reports KHN and Fortune, and the number of EMR-related safety events tops 18,000. The problem is that information, critical to a patient's well-being, may get buried in the EMR. Clinicians may not have been aware of, because they did not see, a critical medication allergy or piece of patient history.1
The problems with health information technology usability do have solutions, however, asserts Raj M. Ratwani, MD, and colleagues. In a recent article published in the Journal of the American Medical Association, the researchers propose 5 priorities for achieving progress3:
- Establishment of a national database of usability and safety issues. This database should allow sharing of safety information among EMR vendors, hospitals, and clinicians, and make the public aware of any technology risks.
- Establishment of basic design standards, which should promote innovation and be regulated by a board composed of all stakeholders: EMR vendors, researchers, clinicians, and health care organizations.
- Addressing unintended harms. Causes of harm could include "vendor design and development, vendor and health care organization implementation, and customization by the health care organization." Along with shared responsibility and collaboration comes shared liability for harms caused by inadequate usability.
- Simplification of mandated documentation requirements that affect usability. Reducing clinician's "busy work" would go a long way toward simplifying documentation requirements.
- Development of standard usability and safety measures so that progress can be tracked and the market can react. EMR vendors cannot be directly compared currently, since no standards for usability are in place.
Ratwani and colleagues cite shared responsibility and commitment among all of the parties invested in EMR usability success as keys to solving the current challenges affecting health information technology, with policy makers at the helm.3 The federal government is attempting to respond: As part of the 2016 21st Century Cures Act and with an aim toward alleviating physician time spent on the EMR, the Department of Health and Human Services is required to recommend reductions to current EMR burdens required under the HITECH Act. It plans to revise E&M codes, lessening documentation. And the Centers for Medicare and Medicaid Services aims to make meaningful use requirements more flexible, require information exchange between providers and patients, and provide incentive to clinicians to allow patient access to EMRs.4,5
References
- Fry E, Schulte F. Death by a thousand clicks. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- Burde H. The HITECH Act: an overview. AMA J Ethics. March 2011. https://journalofethics.ama-assn.org/article/hitech-act-overview/2011-03. Accessed September 9, 2019.
- Ratwani R, Reider J, Singh H. A decade of health information technology usability challenges and the path forward. JAMA. 2019;321:743-744.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Morris G, Anthony ES. 21st Century Cures Act overview for states. Office of the National Coordinator for Health Information Technology. https://www.healthit.gov/sites/default/files/curesactlearningsession_1_v6_10818.pdf. Accessed September 11, 2019.
Continue to:
Dr. Dougherty: When our institution compared EMR offerings, EMR companies put their best collective marketing feet forward. The general notion, at least with the Epic EMR, was that “you can customize Epic to your liking.” It did not take long for a bunch of motivated Epic users to create “smart” stuff (lists, phrases, and texts) in an effort to customize workflows and create fancy-looking electronic notes. Shortly thereafter, it was obvious that, as an institution, our reporting efforts kept coming up short—our reports lacked accuracy and meaning. Everyone was documenting in different ways and in different areas. Considering that reports are currently generated using (mostly) discrete data entries (data placed in specific fields within the EMR), it became obvious that our data entry paradigm needed to change. Therefore, standardization became the leading buzzword. Our institution recently initiated a project aimed at standardizing our workflows and documentation habits. In addition, we have incorporated a third-party information exchange product into our health system data aggregation and analysis workflow. Much more needs to be done, but it is a start.
Dr. Evans: At my institution, as a group, we have created templates for routine procedures and visits that also auto populate billing codes. I know that some departments have used scribes. From the hospital side, there has been improved access to the EMR from home. Some of my colleagues like this feature; however, others, like myself, believe this contributes to some of our burnout. I like to leave work at work. Having the ability to continue working at home is not a solution in my mind.
Dr. Woodland: At our institution, we have engaged our chaperones and medical assistants to help facilitate completion of the medical records during the office visit. Providers work with their assistants to accommodate documentation of history and physical findings while also listening to the provider as they are speaking in order to document patient care plans and orders. This saves the clinicians time in reviewing and editing the record as well as making sure the appropriate care plan is instituted. Our EMR provider recently has begun experimenting with personalization of color themes as well as pictures as part of the interface. Having said this, I still ask, “Why have medical professionals allowed non–clinical agencies and information technology groups to run this show?” It is also inconceivable to me that this unfunded mandate—that has increased cost, decreased clinical efficiency, and decreased clinician satisfaction—has not been addressed by national and international medical communities.
Dr. Woodland: I feel that we need to appropriately manage expectations of the EMR and the institution with relation to EMR and providers. By this I mean that we need to make the EMR more user-friendly and appropriate for different clinicians as well as patients. We also need to manage expectations of our patients. In a digital age where immediate contact is the norm, we need to address the issue that the EMR is not social media but rather a communication tool for routine contact and information transmission. Emergencies are not typically addressed well through the EMR platform; they are better handled with a more appropriate communication interface.
Dr. Dougherty: I feel that the biggest change needed is a competent, simple, and standard user-interface. Our old charting methods were great on a number of levels. For instance, if I wanted to add an order, I flipped to the ”Orders” tab and entered an order. If I needed to document a note, I flipped to the “Notes” tab and started writing, etc. Obviously, manual charting had its downsides—like trying to decipher handwriting art! EMRs could easily adopt the stuff that worked from our old methods of documentation, while leveraging the advantages that computerized workflows can bring to practitioners, including efficient transfer of records, meaningful reporting, simple electronic ordering, and interprofessional communication portals.
Dr. Evans: Our systems need to better communicate with one another. I am in an academic practice, and I should be able to see labs, consultant notes, imaging, all in one spot to improve efficiency and ease with patient visits. Minimizing clicks would be helpful as well. I try to write as much as I can while in the room with a patient to avoid after-hours note writing, but it takes away from my interaction with each patient.
Continue to:
Dr. Evans: When I first started as a new attending, it would take me hours to finish my notes, partly because of the level of detail I would write in my history of present illness (HPI) and assessment and plan. One great piece of advice I received was to be satisfied with good notes, not perfect notes. I worked to consolidate my thoughts and use preconstructed phrases/paragraphs on common problems I saw. This saved time to focus on other aspects of my academic job.
Dr. Dougherty: We need to refocus on the patient first, and mold our systems to meet that priority. Much too often, we have our backs to the patients or spend too much time interfacing with our EMR systems, and our patients are not happy about it (as many surveys have demonstrated). More importantly, a renewed focus on patient care, not EMR care, would allow our practitioners to do what they signed up for—treating patients. In the meantime, I would suggest that practitioners stay away from EMR gimmicks and go back to old-style documentation practices (like those established by the Centers for Medicare and Medicaid Services in 1997 and 1998), and ask the IT folks to help with molding the EMR systems to meet your own standards, not the standards established by EMR companies. I am also very hopeful that the consumer will drive most of the health care-related data collection in the near future, thereby marginalizing the current generation of EMR systems.
Dr. Woodland: I would add that providers need to manage the EMR and not let the EMR manage them. Set up task reminders at point times to handle results and communications from the EMR and set up time in your schedule where you can facilitate meeting these tasks. When providers are out on vacation, make sure to have an out-of-office reminder built into their EMR so that patients and others know timing of potential responses. Try to make the EMR as enjoyable as possible and focus on the good points of the EMR, such as legibility, order verification, safety, and documentation.
1. Engage the computer in your patient encounter, says Rey Wuerth and colleagues. Share the screen, and any test results you are highlighting, with your patient by turning it toward her during your discussion. This can increase patient satisfaction.1
2. Go mobile at the point of care, suggests Tom Giannulli, MD, MS, Chief Medical Information Officer at Kareo. By using a tablet or mobile device, you can enter data while facing a patient or on the go.2
3. Use templates when documenting data, advises Wuerth and colleagues, as pre-filled templates, that are provided through the EMR or that you create within the EMR, can reduce the time required to enter patient visits, findings, and referrals.1
4. Delegate responsibility for routing documents, says Brian Anderson, MD. Hand off to staff administrative duties, such as patient forms and routine negative test results.3
5. Involve medical assistants (MAs) in the process. Make the MA feel part of the team, says R. Scott Eden, and assign them history-taking responsibilities, utilizing your EMR's templates. Assign them other tasks as well, including medication reconciliation, referrals, refills, routine screening, and patient education.4
6. Employ physical or virtual scribes who are specifically assigned to EMR duty. Although drawbacks can include patient privacy concerns and reduced practice income due to salary requirements, employing a scribe (often a pre-medical or graduate student), who trails you on patient visits, or who is connected virtually, can leave the clinician free to interact with patients.5,6
References
- Wuerth R, Campbell C, Peng MD, et al. Top 10 tips for effective use of electronic health records. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959973/. Paediatr Child Health. 2014;19:138.
- Giannulli T. 7 time-saving EHR use tips to boost physician productivity. April 28, 2016. https://ehrintelligence.com/news/7-time-saving-emr-use-tips-to-boost-physician-productivity. Accessed September 9, 2019.
- Anderson B. 5 ways to increase your EMR efficiency. October 28, 2014. https://www.kevinmd.com/blog/2014/10/5-ways-increase-emr-efficiency.html. Accessed September 9, 2019.
- Eden RS. Maximizing your medical assistant's role. Fam Pract Manag. 2016;23:5-7. https://www.aafp.org/fpm/2016/0500/p5.html.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Caliri A. The case for virtual scribes. January 2, 2019. Becker's Hospital Review. https://www.beckershospitalreview.com/hospital-physician-relationships/the-case-for-virtual-scribes.html. Accessed September 20, 2019.
Dr. Evans: Yes and no. Yes, in that it can be much easier to follow a patient’s health care history from other provider notes or prior surgeries. Information is searchable and legible. If an EMR is built correctly, it can save time for providers, through smart phrases and templates, and it can help providers with proper billing codes and documentation requirements. No, in that it can take away from important patient interaction. We are required to see more patients in less time all while using, at times, a cumbersome EMR system.
Dr. Woodland: This is a tricky question because the EMR has both positive and negative attributes. Certainly, the legibility and order verification has improved, but the ease of accessing information in the EMR has changed. Additionally, there has been a drastic increase in provider dissatisfaction that has not been addressed. Provider dissatisfaction can lead to problems in patient care. If there was a clear-cut increased value for the cost, I do not think the EMR would be such a huge focus of negative attention. Providers need to take back control of their EMR and their profession so that they can utilize the EMR as the tool it was supposed to be and not the dissatisfier that it has become.
Dr. Dougherty: I do not believe patient care has been improved by EMR systems, for all of the reasons we have discussed, and then some. But there is an enormous amount of potential, if we get the interface between humans and EMR systems right!
Physician burnout has been labeled a public health crisis by the Harvard School of Public Health and other institutions.1 A 2018 Physician’s Foundation survey found that 78% of physicians had symptoms of burnout,2 which result from chronic workplace stress and include feeling depleted of energy or exhausted, mentally distanced from or cynical about one’s job, and problems getting one’s job done successfully.3 Among ObGyns, almost half (46%) report burnout.4 One-third of ObGyns responded on a recent Medscape Burnout Report that the computerization of practice is contributing to their burnout, and 54% said too many bureaucratic tasks, including charting, were adding to their burnout.5
Inefficient electronic medical records (EMRs) have been implicated as one reason for burnout, with improvements in efficiency cited as one of several potential resolutions to the problem. About 96% of hospitals have adopted EMRs today, compared with only 9% in 2008,6 and many physicians report recognizing value in the technology. For instance, 60% of participants in Stanford Medicine’s 2018 National Physician Poll said EMRs had led to improved patient care. At the same time, however, about as many (59%) said EMRs needed a “complete overhaul” and that the systems had detracted from their professional satisfaction (54%) as well as from their clinical effectiveness (49%).7
With this roundtable, we explore the concerns with hours spent on the EMR with several experts, and whether it is a problem that has been contributing to burnout among staff at their institutions. In addition, are there solutions that their institutions have implemented that they can share to help to cope with the problem?
John J. Dougherty, MD, MBA: Yes, absolutely. There is not a day that goes by that I don’t hear about or experience “Epic Fails.” (We use Epic’s EMR product at our institution.) Too many clicks are needed to navigate even the simplest tasks—finding notes or results, documenting visits, and billing for services are all unnecessarily complex. In addition, we are being held accountable for achieving a long and growing list of “metrics” measures, education projects (HealthStream), and productivity goals. When do we have time to treat patients? And it is not just practicing physicians and clinicians. Our resident physicians spend an inordinate amount of time in front of the computer documenting, placing orders, and transferring patients using a system with a very inefficient user interface, to say the least.
Megan L. Evans, MD, MPH: I absolutely agree. Over the years, my institution has created a conglomerate of EMRs, requiring physicians across the hospital to be fluent in a multitude of systems. For example, you finish your clinic notes in one system, sign off on discharge summaries in another, and complete your operative notes in an entirely different system. As busy attendings, it is hard to keep ahead of all of these tasks, especially when the systems do not talk to one another. Fortunately, my hospital is changing our EMR to a single system within the next year. Until then, however, we will work in this piecemeal system.
Mark Woodland, MS, MD: EMR and computerization of medicine is the number 1 issue relating to dissatisfaction by ObGyn providers in our institution. Providers are earnest in their attempt to be compliant with EMR requirements, but the reality is that they are dealing with an automated system that does not have realistic expectations for management of results, follow-up tasks, and patient communications for a human provider. The actual charting, ordering of tests and consults, and communication between providers has been enhanced. However, the “in-basket” of tasks to be accomplished are extraordinary and much of it relies on the provider, which requires an inordinate amount of time. Additionally, while other members of the medical staff are stationary at a desk, physicians and other providers are not. They are mobile between inpatient units, labor and delivery, operating rooms, and emergency rooms. Time management does not always allow for providers to access computers from all of these areas to facilitate their managing the expectations of the EMR. This requires providers to access the EMR at off hours, extending their workload. Finally, the EMR is neither personal nor friendly. It is not designed with the clinician in mind, and it is not fun or engaging for a provider.
EMRs are not just inefficient and contributing to physician burnout, according to a joint report from Kaiser Health News (KHN) and Fortune magazine, they are inadequate and contributing to patient safety concerns.1 This was not the intended goal of the HITECH Act, signed into law in 2009 as part of the stimulus bill. HITECH was intended to promote the adoption of meaningful use of health information technology by providing financial incentives to clinicians to adopt electronic medical records (EMRs). It also intended to increase security for health care data--achieved through larger penalties for HIPAA violations.2
Ten years later, however, "America has little to show" for its $36 billion investment, according to KHN and Fortune. Yes, 96% of hospitals have one of the currently available EMRs, among thousands, but they are disconnected. And they are "glitchy." At least 2 EMR vendors have reached settlements with the federal government over egregious patient errors. At least 7 deaths have resulted from errors related to the EMR, according to the firm Quantros, reports KHN and Fortune, and the number of EMR-related safety events tops 18,000. The problem is that information, critical to a patient's well-being, may get buried in the EMR. Clinicians may not have been aware of, because they did not see, a critical medication allergy or piece of patient history.1
The problems with health information technology usability do have solutions, however, asserts Raj M. Ratwani, MD, and colleagues. In a recent article published in the Journal of the American Medical Association, the researchers propose 5 priorities for achieving progress3:
- Establishment of a national database of usability and safety issues. This database should allow sharing of safety information among EMR vendors, hospitals, and clinicians, and make the public aware of any technology risks.
- Establishment of basic design standards, which should promote innovation and be regulated by a board composed of all stakeholders: EMR vendors, researchers, clinicians, and health care organizations.
- Addressing unintended harms. Causes of harm could include "vendor design and development, vendor and health care organization implementation, and customization by the health care organization." Along with shared responsibility and collaboration comes shared liability for harms caused by inadequate usability.
- Simplification of mandated documentation requirements that affect usability. Reducing clinician's "busy work" would go a long way toward simplifying documentation requirements.
- Development of standard usability and safety measures so that progress can be tracked and the market can react. EMR vendors cannot be directly compared currently, since no standards for usability are in place.
Ratwani and colleagues cite shared responsibility and commitment among all of the parties invested in EMR usability success as keys to solving the current challenges affecting health information technology, with policy makers at the helm.3 The federal government is attempting to respond: As part of the 2016 21st Century Cures Act and with an aim toward alleviating physician time spent on the EMR, the Department of Health and Human Services is required to recommend reductions to current EMR burdens required under the HITECH Act. It plans to revise E&M codes, lessening documentation. And the Centers for Medicare and Medicaid Services aims to make meaningful use requirements more flexible, require information exchange between providers and patients, and provide incentive to clinicians to allow patient access to EMRs.4,5
References
- Fry E, Schulte F. Death by a thousand clicks. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- Burde H. The HITECH Act: an overview. AMA J Ethics. March 2011. https://journalofethics.ama-assn.org/article/hitech-act-overview/2011-03. Accessed September 9, 2019.
- Ratwani R, Reider J, Singh H. A decade of health information technology usability challenges and the path forward. JAMA. 2019;321:743-744.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Morris G, Anthony ES. 21st Century Cures Act overview for states. Office of the National Coordinator for Health Information Technology. https://www.healthit.gov/sites/default/files/curesactlearningsession_1_v6_10818.pdf. Accessed September 11, 2019.
Continue to:
Dr. Dougherty: When our institution compared EMR offerings, EMR companies put their best collective marketing feet forward. The general notion, at least with the Epic EMR, was that “you can customize Epic to your liking.” It did not take long for a bunch of motivated Epic users to create “smart” stuff (lists, phrases, and texts) in an effort to customize workflows and create fancy-looking electronic notes. Shortly thereafter, it was obvious that, as an institution, our reporting efforts kept coming up short—our reports lacked accuracy and meaning. Everyone was documenting in different ways and in different areas. Considering that reports are currently generated using (mostly) discrete data entries (data placed in specific fields within the EMR), it became obvious that our data entry paradigm needed to change. Therefore, standardization became the leading buzzword. Our institution recently initiated a project aimed at standardizing our workflows and documentation habits. In addition, we have incorporated a third-party information exchange product into our health system data aggregation and analysis workflow. Much more needs to be done, but it is a start.
Dr. Evans: At my institution, as a group, we have created templates for routine procedures and visits that also auto populate billing codes. I know that some departments have used scribes. From the hospital side, there has been improved access to the EMR from home. Some of my colleagues like this feature; however, others, like myself, believe this contributes to some of our burnout. I like to leave work at work. Having the ability to continue working at home is not a solution in my mind.
Dr. Woodland: At our institution, we have engaged our chaperones and medical assistants to help facilitate completion of the medical records during the office visit. Providers work with their assistants to accommodate documentation of history and physical findings while also listening to the provider as they are speaking in order to document patient care plans and orders. This saves the clinicians time in reviewing and editing the record as well as making sure the appropriate care plan is instituted. Our EMR provider recently has begun experimenting with personalization of color themes as well as pictures as part of the interface. Having said this, I still ask, “Why have medical professionals allowed non–clinical agencies and information technology groups to run this show?” It is also inconceivable to me that this unfunded mandate—that has increased cost, decreased clinical efficiency, and decreased clinician satisfaction—has not been addressed by national and international medical communities.
Dr. Woodland: I feel that we need to appropriately manage expectations of the EMR and the institution with relation to EMR and providers. By this I mean that we need to make the EMR more user-friendly and appropriate for different clinicians as well as patients. We also need to manage expectations of our patients. In a digital age where immediate contact is the norm, we need to address the issue that the EMR is not social media but rather a communication tool for routine contact and information transmission. Emergencies are not typically addressed well through the EMR platform; they are better handled with a more appropriate communication interface.
Dr. Dougherty: I feel that the biggest change needed is a competent, simple, and standard user-interface. Our old charting methods were great on a number of levels. For instance, if I wanted to add an order, I flipped to the ”Orders” tab and entered an order. If I needed to document a note, I flipped to the “Notes” tab and started writing, etc. Obviously, manual charting had its downsides—like trying to decipher handwriting art! EMRs could easily adopt the stuff that worked from our old methods of documentation, while leveraging the advantages that computerized workflows can bring to practitioners, including efficient transfer of records, meaningful reporting, simple electronic ordering, and interprofessional communication portals.
Dr. Evans: Our systems need to better communicate with one another. I am in an academic practice, and I should be able to see labs, consultant notes, imaging, all in one spot to improve efficiency and ease with patient visits. Minimizing clicks would be helpful as well. I try to write as much as I can while in the room with a patient to avoid after-hours note writing, but it takes away from my interaction with each patient.
Continue to:
Dr. Evans: When I first started as a new attending, it would take me hours to finish my notes, partly because of the level of detail I would write in my history of present illness (HPI) and assessment and plan. One great piece of advice I received was to be satisfied with good notes, not perfect notes. I worked to consolidate my thoughts and use preconstructed phrases/paragraphs on common problems I saw. This saved time to focus on other aspects of my academic job.
Dr. Dougherty: We need to refocus on the patient first, and mold our systems to meet that priority. Much too often, we have our backs to the patients or spend too much time interfacing with our EMR systems, and our patients are not happy about it (as many surveys have demonstrated). More importantly, a renewed focus on patient care, not EMR care, would allow our practitioners to do what they signed up for—treating patients. In the meantime, I would suggest that practitioners stay away from EMR gimmicks and go back to old-style documentation practices (like those established by the Centers for Medicare and Medicaid Services in 1997 and 1998), and ask the IT folks to help with molding the EMR systems to meet your own standards, not the standards established by EMR companies. I am also very hopeful that the consumer will drive most of the health care-related data collection in the near future, thereby marginalizing the current generation of EMR systems.
Dr. Woodland: I would add that providers need to manage the EMR and not let the EMR manage them. Set up task reminders at point times to handle results and communications from the EMR and set up time in your schedule where you can facilitate meeting these tasks. When providers are out on vacation, make sure to have an out-of-office reminder built into their EMR so that patients and others know timing of potential responses. Try to make the EMR as enjoyable as possible and focus on the good points of the EMR, such as legibility, order verification, safety, and documentation.
1. Engage the computer in your patient encounter, says Rey Wuerth and colleagues. Share the screen, and any test results you are highlighting, with your patient by turning it toward her during your discussion. This can increase patient satisfaction.1
2. Go mobile at the point of care, suggests Tom Giannulli, MD, MS, Chief Medical Information Officer at Kareo. By using a tablet or mobile device, you can enter data while facing a patient or on the go.2
3. Use templates when documenting data, advises Wuerth and colleagues, as pre-filled templates, that are provided through the EMR or that you create within the EMR, can reduce the time required to enter patient visits, findings, and referrals.1
4. Delegate responsibility for routing documents, says Brian Anderson, MD. Hand off to staff administrative duties, such as patient forms and routine negative test results.3
5. Involve medical assistants (MAs) in the process. Make the MA feel part of the team, says R. Scott Eden, and assign them history-taking responsibilities, utilizing your EMR's templates. Assign them other tasks as well, including medication reconciliation, referrals, refills, routine screening, and patient education.4
6. Employ physical or virtual scribes who are specifically assigned to EMR duty. Although drawbacks can include patient privacy concerns and reduced practice income due to salary requirements, employing a scribe (often a pre-medical or graduate student), who trails you on patient visits, or who is connected virtually, can leave the clinician free to interact with patients.5,6
References
- Wuerth R, Campbell C, Peng MD, et al. Top 10 tips for effective use of electronic health records. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959973/. Paediatr Child Health. 2014;19:138.
- Giannulli T. 7 time-saving EHR use tips to boost physician productivity. April 28, 2016. https://ehrintelligence.com/news/7-time-saving-emr-use-tips-to-boost-physician-productivity. Accessed September 9, 2019.
- Anderson B. 5 ways to increase your EMR efficiency. October 28, 2014. https://www.kevinmd.com/blog/2014/10/5-ways-increase-emr-efficiency.html. Accessed September 9, 2019.
- Eden RS. Maximizing your medical assistant's role. Fam Pract Manag. 2016;23:5-7. https://www.aafp.org/fpm/2016/0500/p5.html.
- Hoffman S. Healing the healers: legal remedies for physician burnout. Case Western Reserve University School of Law. September 2018.
- Caliri A. The case for virtual scribes. January 2, 2019. Becker's Hospital Review. https://www.beckershospitalreview.com/hospital-physician-relationships/the-case-for-virtual-scribes.html. Accessed September 20, 2019.
Dr. Evans: Yes and no. Yes, in that it can be much easier to follow a patient’s health care history from other provider notes or prior surgeries. Information is searchable and legible. If an EMR is built correctly, it can save time for providers, through smart phrases and templates, and it can help providers with proper billing codes and documentation requirements. No, in that it can take away from important patient interaction. We are required to see more patients in less time all while using, at times, a cumbersome EMR system.
Dr. Woodland: This is a tricky question because the EMR has both positive and negative attributes. Certainly, the legibility and order verification has improved, but the ease of accessing information in the EMR has changed. Additionally, there has been a drastic increase in provider dissatisfaction that has not been addressed. Provider dissatisfaction can lead to problems in patient care. If there was a clear-cut increased value for the cost, I do not think the EMR would be such a huge focus of negative attention. Providers need to take back control of their EMR and their profession so that they can utilize the EMR as the tool it was supposed to be and not the dissatisfier that it has become.
Dr. Dougherty: I do not believe patient care has been improved by EMR systems, for all of the reasons we have discussed, and then some. But there is an enormous amount of potential, if we get the interface between humans and EMR systems right!
- A crisis in health care: a call to action on physician burnout. Massachusetts Health and Hospital Association. Massachusetts Medical Society. Harvard T.H. Chan School of Public Health. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/21/2019/01/PhysicianBurnoutReport2018FINAL.pdf. Accessed September 9, 2019.
- Physician’s Foundation. 2018 survey of America’s physicians practice patterns and perspectives. https://physiciansfoundation.org/wp-content/uploads/2018/09/physicians-survey-results-final-2018.pdf. Accessed September 9, 2019.
- Burn-out. ICD-11 for Mortality and Morbidity Statistics. Version 04/2019. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/129180281. Accessed September 11, 2019.
- Peckham C. Medscape National Physician Burnout & Depression Report 2018. January 17, 2018. https://www.medscape.com/slideshow/2018-lifestyle-burnout-depression-6009235#3. Accessed September 9, 2019.
- Kane L. Medscape National Physician Burnout, Depression & Suicide Report 2019. January 16, 2019. https://www.medscape.com/slideshow/2019-lifestyle-burnout-depression-6011056#5. Accessed September 9, 2019.
- Fry E, Schulte F. Death by a thousand clicks: where electronic health records went wrong. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- How doctors feel about electronic health records: National Physician Poll by The Harris Poll. https://med.stanford.edu/content/dam/sm/ehr/documents/EHR-Poll-Presentation.pdf. Accessed September 9, 2019.
- A crisis in health care: a call to action on physician burnout. Massachusetts Health and Hospital Association. Massachusetts Medical Society. Harvard T.H. Chan School of Public Health. https://cdn1.sph.harvard.edu/wp-content/uploads/sites/21/2019/01/PhysicianBurnoutReport2018FINAL.pdf. Accessed September 9, 2019.
- Physician’s Foundation. 2018 survey of America’s physicians practice patterns and perspectives. https://physiciansfoundation.org/wp-content/uploads/2018/09/physicians-survey-results-final-2018.pdf. Accessed September 9, 2019.
- Burn-out. ICD-11 for Mortality and Morbidity Statistics. Version 04/2019. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/129180281. Accessed September 11, 2019.
- Peckham C. Medscape National Physician Burnout & Depression Report 2018. January 17, 2018. https://www.medscape.com/slideshow/2018-lifestyle-burnout-depression-6009235#3. Accessed September 9, 2019.
- Kane L. Medscape National Physician Burnout, Depression & Suicide Report 2019. January 16, 2019. https://www.medscape.com/slideshow/2019-lifestyle-burnout-depression-6011056#5. Accessed September 9, 2019.
- Fry E, Schulte F. Death by a thousand clicks: where electronic health records went wrong. Fortune. March 18, 2019. http://fortune.com/longform/medical-records/. Accessed September 9, 2019.
- How doctors feel about electronic health records: National Physician Poll by The Harris Poll. https://med.stanford.edu/content/dam/sm/ehr/documents/EHR-Poll-Presentation.pdf. Accessed September 9, 2019.

Product Update: Menstrual pain relief, Exparel, STI assay, new ART option
MENSTRUAL PAIN RELIEF THROUGH MICRO-PULSES

Livia, by iPulse Medical Ltd, is a US Food and Drug Administration (FDA) approved, drug-free option to treat menstrual pain through the transmission of electrical pulses. Electrodes are placed on the body at the source of menstrual pain and send a frequency to the nerves to reduce pain. Livia was designed based on the principles of the “gate control” theory of pain, says iPulse Medical. When the nerves are stimulated by the device’s electrodes, the nerve gate is closed, preventing pain signals from being received or felt in the brain.
The device can be worn in public or at home and allows the user to adjust the frequency of the electrical signal to correspond with her pain intensity. According to iPulse Medical, there are no adverse effects and the user will not build up a tolerance; however, the device should not be worn if the user has a pacemaker or is undergoing fertility treatment.
FOR MORE INFORMATION, VISIT: https://mylivia.com/
EXPAREL FOR CESAREAN DELIVERY
Pacira BioSciences an-nounced completion of their Phase 4 study of Exparel (bupivacaine lipsome injectable suspension), a local analgesic given to patients undergoing planned cesarean delivery (CD), aimed at reducing postsurgical pain and total opioid consumption through the first 72 hours postsurgery. Exparel is administered through transversus abdominis plane field block.
Pacira’s multicenter, randomized, double-blind study of 186 patients showed that those receiving Exparel plus bupivacaine HCl had a 52% reduction in total opioid consumption and significantly lower pain scores through the first 72 hours after CD, compared with those receiving only bupivacaine HCl. The most common adverse effects are itching and nausea. Exparel should not be used for patients under the age of 18 and should be used cautiously in patients with hepatic disease.
FOR MORE INFORMATION, VISIT: https://www.exparel.com/
M GENITALIUM ASSAY DETECTS THE STI

Hologic’s Aptima® Mycoplasma genitalium assay is the first FDA-cleared diagnostic test for this sexually transmitted infection (STI), which has been identified by the Centers for Disease Control and Prevention as an emerging public health threat. The assay is an in vitro nucleic acid amplification test that can be used to verify swab or urine samples from women and men. In published studies, the ribosomal RNA-based assay displayed greater sensitivity than lab-developed or CE-marked DNA-based tests. Early detection is important, Hologic asserts, because M genitalium is increasing in prevalence among higher-risk populations; however, it is not well known and often misdiagnosed, leading to incorrect treatment as well as risk for transmission and recurrence.
Hologic cites several studies that have shown M genitalium can be asymptomatic; however, it also can be associated with nongonococcal urethritis in men and cervicitis in women, as well as increased risk for pelvic inflammatory disease, preterm birth, spontaneous abortion, and infertility. A high percentage of infected people have an antibiotic-resistant strain, demonstrating a need for early detection and screening.
FOR MORE INFORMATION, VISIT: https://www.hologic.com
NEW ART OPTION

FOR MORE INFORMATION, VISIT: https://www.ferringusa.com
MENSTRUAL PAIN RELIEF THROUGH MICRO-PULSES
Livia, by iPulse Medical Ltd, is a US Food and Drug Administration (FDA) approved, drug-free option to treat menstrual pain through the transmission of electrical pulses. Electrodes are placed on the body at the source of menstrual pain and send a frequency to the nerves to reduce pain. Livia was designed based on the principles of the “gate control” theory of pain, says iPulse Medical. When the nerves are stimulated by the device’s electrodes, the nerve gate is closed, preventing pain signals from being received or felt in the brain.
The device can be worn in public or at home and allows the user to adjust the frequency of the electrical signal to correspond with her pain intensity. According to iPulse Medical, there are no adverse effects and the user will not build up a tolerance; however, the device should not be worn if the user has a pacemaker or is undergoing fertility treatment.
FOR MORE INFORMATION, VISIT: https://mylivia.com/
EXPAREL FOR CESAREAN DELIVERY
Pacira BioSciences an-nounced completion of their Phase 4 study of Exparel (bupivacaine lipsome injectable suspension), a local analgesic given to patients undergoing planned cesarean delivery (CD), aimed at reducing postsurgical pain and total opioid consumption through the first 72 hours postsurgery. Exparel is administered through transversus abdominis plane field block.
Pacira’s multicenter, randomized, double-blind study of 186 patients showed that those receiving Exparel plus bupivacaine HCl had a 52% reduction in total opioid consumption and significantly lower pain scores through the first 72 hours after CD, compared with those receiving only bupivacaine HCl. The most common adverse effects are itching and nausea. Exparel should not be used for patients under the age of 18 and should be used cautiously in patients with hepatic disease.
FOR MORE INFORMATION, VISIT: https://www.exparel.com/
M GENITALIUM ASSAY DETECTS THE STI
Hologic’s Aptima® Mycoplasma genitalium assay is the first FDA-cleared diagnostic test for this sexually transmitted infection (STI), which has been identified by the Centers for Disease Control and Prevention as an emerging public health threat. The assay is an in vitro nucleic acid amplification test that can be used to verify swab or urine samples from women and men. In published studies, the ribosomal RNA-based assay displayed greater sensitivity than lab-developed or CE-marked DNA-based tests. Early detection is important, Hologic asserts, because M genitalium is increasing in prevalence among higher-risk populations; however, it is not well known and often misdiagnosed, leading to incorrect treatment as well as risk for transmission and recurrence.
Hologic cites several studies that have shown M genitalium can be asymptomatic; however, it also can be associated with nongonococcal urethritis in men and cervicitis in women, as well as increased risk for pelvic inflammatory disease, preterm birth, spontaneous abortion, and infertility. A high percentage of infected people have an antibiotic-resistant strain, demonstrating a need for early detection and screening.
FOR MORE INFORMATION, VISIT: https://www.hologic.com
NEW ART OPTION
FOR MORE INFORMATION, VISIT: https://www.ferringusa.com
MENSTRUAL PAIN RELIEF THROUGH MICRO-PULSES
Livia, by iPulse Medical Ltd, is a US Food and Drug Administration (FDA) approved, drug-free option to treat menstrual pain through the transmission of electrical pulses. Electrodes are placed on the body at the source of menstrual pain and send a frequency to the nerves to reduce pain. Livia was designed based on the principles of the “gate control” theory of pain, says iPulse Medical. When the nerves are stimulated by the device’s electrodes, the nerve gate is closed, preventing pain signals from being received or felt in the brain.
The device can be worn in public or at home and allows the user to adjust the frequency of the electrical signal to correspond with her pain intensity. According to iPulse Medical, there are no adverse effects and the user will not build up a tolerance; however, the device should not be worn if the user has a pacemaker or is undergoing fertility treatment.
FOR MORE INFORMATION, VISIT: https://mylivia.com/
EXPAREL FOR CESAREAN DELIVERY
Pacira BioSciences an-nounced completion of their Phase 4 study of Exparel (bupivacaine lipsome injectable suspension), a local analgesic given to patients undergoing planned cesarean delivery (CD), aimed at reducing postsurgical pain and total opioid consumption through the first 72 hours postsurgery. Exparel is administered through transversus abdominis plane field block.
Pacira’s multicenter, randomized, double-blind study of 186 patients showed that those receiving Exparel plus bupivacaine HCl had a 52% reduction in total opioid consumption and significantly lower pain scores through the first 72 hours after CD, compared with those receiving only bupivacaine HCl. The most common adverse effects are itching and nausea. Exparel should not be used for patients under the age of 18 and should be used cautiously in patients with hepatic disease.
FOR MORE INFORMATION, VISIT: https://www.exparel.com/
M GENITALIUM ASSAY DETECTS THE STI
Hologic’s Aptima® Mycoplasma genitalium assay is the first FDA-cleared diagnostic test for this sexually transmitted infection (STI), which has been identified by the Centers for Disease Control and Prevention as an emerging public health threat. The assay is an in vitro nucleic acid amplification test that can be used to verify swab or urine samples from women and men. In published studies, the ribosomal RNA-based assay displayed greater sensitivity than lab-developed or CE-marked DNA-based tests. Early detection is important, Hologic asserts, because M genitalium is increasing in prevalence among higher-risk populations; however, it is not well known and often misdiagnosed, leading to incorrect treatment as well as risk for transmission and recurrence.
Hologic cites several studies that have shown M genitalium can be asymptomatic; however, it also can be associated with nongonococcal urethritis in men and cervicitis in women, as well as increased risk for pelvic inflammatory disease, preterm birth, spontaneous abortion, and infertility. A high percentage of infected people have an antibiotic-resistant strain, demonstrating a need for early detection and screening.
FOR MORE INFORMATION, VISIT: https://www.hologic.com
NEW ART OPTION
FOR MORE INFORMATION, VISIT: https://www.ferringusa.com
Systemic sclerosis raises risk of breast cancer, lung cancer, melanoma
in a population-linked cohort study published in Arthritis Care & Research.
Kathleen Morrisroe, MBBS, PhD, of St. Vincent’s Hospital Melbourne and colleagues matched deidentified patient data in the Australian Scleroderma Cohort Study (ASCS) with patients’ respective state cancer registry data between January 2008 and December 2015. The researchers also used the Australian Medical Benefit Schedule (MBS) to track health care costs for hospital admissions, presentations to the ED, other health visits, pathology, and imaging, as well as other associated costs for care, in each state. Based on this information, Dr. Morrisroe and colleagues calculated standardized incidence ratios (SIR) and standardized mortality ratios (SMR) for these patients by comparing them with the general population in Australia.
The results included 1,727 patients with systemic sclerosis (SSc) and cancer in the cohort, which consisted of mostly white (92.1%) women (85.9%) who had limited cutaneous SSc (73.9%). They were a mean of 46.6 years old when they were diagnosed with SSc and had a mean disease duration of 10.9 years. The incidence of cancer was 1.3% per year, and the overall prevalence for the cohort was 14.2%, which was higher than the general Australian population (SIR, 2.15; 95% confidence interval, 1.84-2.49). Breast cancer, melanoma, hematologic cancer, and lung cancer were the most common types of cancers found in the cohort, with early breast cancer (SIR, 3.07; 95% CI, 1.47-5.64), lung cancer (SIR, 3.07; 95% CI, 1.21-3.44), and early melanoma (SIR, 3.40; 95% CI, 1.10-7.93) having a higher incidence than the general population.
Patients with RNA polymerase III (RNAP) autoantibody had a higher incidence of early onset cancer (odds ratio, 2.9; P = .044), defined as a cancer diagnosis within 5 years of SSc diagnosis. Interstitial lung disease was also linked to an increased risk of lung cancer (OR, 2.83; P = .031), which persisted after the researchers performed a multivariate analysis.
Another factor that increased the overall risk of cancer was calcium channel blockers (OR, 1.47; P = .016), which also increased the risk of breast (OR, 1.61; P = .051) and melanoma-specific cancers (OR, 2.01; P = .042), a finding the researchers said was “unexpected, but has been reported in the literature with conflicting results.”
“This association is hypothesized to be related to the role of calcium in cell apoptosis, such as activation of the caspase pathway, induction of endonuclease activity and mitochondrial permeation,” Dr. Morrisroe and colleagues wrote.
SSc patients had more than a doubling of risk of mortality with incident cancer in comparison with SSc patients who did not have cancer (hazard ratio, 2.85; 95% CI, 1.51-5.37; P = .001). The average cost of health care annually for an SSc patient with cancer was AUD $1,496 (P less than .001), the researchers said.
This study was funded in part by Scleroderma Australia, Arthritis Australia, Actelion Australia, Bayer, CSL Biotherapies, GlaxoSmithKline Australia, and Pfizer. Dr. Morrisroe reported receiving support from Arthritis Australia and Royal Australasian College of Physicians Research Establishment Fellowships. Another author reported receiving a fellowship from the National Health and Medical Research Council of Australia. The other authors reported no relevant conflicts of interest.
SOURCE: Morrisroe K et al. Arthritis Care Res. 2019 Sep 20. doi: 10.1002/acr.24076
in a population-linked cohort study published in Arthritis Care & Research.
Kathleen Morrisroe, MBBS, PhD, of St. Vincent’s Hospital Melbourne and colleagues matched deidentified patient data in the Australian Scleroderma Cohort Study (ASCS) with patients’ respective state cancer registry data between January 2008 and December 2015. The researchers also used the Australian Medical Benefit Schedule (MBS) to track health care costs for hospital admissions, presentations to the ED, other health visits, pathology, and imaging, as well as other associated costs for care, in each state. Based on this information, Dr. Morrisroe and colleagues calculated standardized incidence ratios (SIR) and standardized mortality ratios (SMR) for these patients by comparing them with the general population in Australia.
The results included 1,727 patients with systemic sclerosis (SSc) and cancer in the cohort, which consisted of mostly white (92.1%) women (85.9%) who had limited cutaneous SSc (73.9%). They were a mean of 46.6 years old when they were diagnosed with SSc and had a mean disease duration of 10.9 years. The incidence of cancer was 1.3% per year, and the overall prevalence for the cohort was 14.2%, which was higher than the general Australian population (SIR, 2.15; 95% confidence interval, 1.84-2.49). Breast cancer, melanoma, hematologic cancer, and lung cancer were the most common types of cancers found in the cohort, with early breast cancer (SIR, 3.07; 95% CI, 1.47-5.64), lung cancer (SIR, 3.07; 95% CI, 1.21-3.44), and early melanoma (SIR, 3.40; 95% CI, 1.10-7.93) having a higher incidence than the general population.
Patients with RNA polymerase III (RNAP) autoantibody had a higher incidence of early onset cancer (odds ratio, 2.9; P = .044), defined as a cancer diagnosis within 5 years of SSc diagnosis. Interstitial lung disease was also linked to an increased risk of lung cancer (OR, 2.83; P = .031), which persisted after the researchers performed a multivariate analysis.
Another factor that increased the overall risk of cancer was calcium channel blockers (OR, 1.47; P = .016), which also increased the risk of breast (OR, 1.61; P = .051) and melanoma-specific cancers (OR, 2.01; P = .042), a finding the researchers said was “unexpected, but has been reported in the literature with conflicting results.”
“This association is hypothesized to be related to the role of calcium in cell apoptosis, such as activation of the caspase pathway, induction of endonuclease activity and mitochondrial permeation,” Dr. Morrisroe and colleagues wrote.
SSc patients had more than a doubling of risk of mortality with incident cancer in comparison with SSc patients who did not have cancer (hazard ratio, 2.85; 95% CI, 1.51-5.37; P = .001). The average cost of health care annually for an SSc patient with cancer was AUD $1,496 (P less than .001), the researchers said.
This study was funded in part by Scleroderma Australia, Arthritis Australia, Actelion Australia, Bayer, CSL Biotherapies, GlaxoSmithKline Australia, and Pfizer. Dr. Morrisroe reported receiving support from Arthritis Australia and Royal Australasian College of Physicians Research Establishment Fellowships. Another author reported receiving a fellowship from the National Health and Medical Research Council of Australia. The other authors reported no relevant conflicts of interest.
SOURCE: Morrisroe K et al. Arthritis Care Res. 2019 Sep 20. doi: 10.1002/acr.24076
in a population-linked cohort study published in Arthritis Care & Research.
Kathleen Morrisroe, MBBS, PhD, of St. Vincent’s Hospital Melbourne and colleagues matched deidentified patient data in the Australian Scleroderma Cohort Study (ASCS) with patients’ respective state cancer registry data between January 2008 and December 2015. The researchers also used the Australian Medical Benefit Schedule (MBS) to track health care costs for hospital admissions, presentations to the ED, other health visits, pathology, and imaging, as well as other associated costs for care, in each state. Based on this information, Dr. Morrisroe and colleagues calculated standardized incidence ratios (SIR) and standardized mortality ratios (SMR) for these patients by comparing them with the general population in Australia.
The results included 1,727 patients with systemic sclerosis (SSc) and cancer in the cohort, which consisted of mostly white (92.1%) women (85.9%) who had limited cutaneous SSc (73.9%). They were a mean of 46.6 years old when they were diagnosed with SSc and had a mean disease duration of 10.9 years. The incidence of cancer was 1.3% per year, and the overall prevalence for the cohort was 14.2%, which was higher than the general Australian population (SIR, 2.15; 95% confidence interval, 1.84-2.49). Breast cancer, melanoma, hematologic cancer, and lung cancer were the most common types of cancers found in the cohort, with early breast cancer (SIR, 3.07; 95% CI, 1.47-5.64), lung cancer (SIR, 3.07; 95% CI, 1.21-3.44), and early melanoma (SIR, 3.40; 95% CI, 1.10-7.93) having a higher incidence than the general population.
Patients with RNA polymerase III (RNAP) autoantibody had a higher incidence of early onset cancer (odds ratio, 2.9; P = .044), defined as a cancer diagnosis within 5 years of SSc diagnosis. Interstitial lung disease was also linked to an increased risk of lung cancer (OR, 2.83; P = .031), which persisted after the researchers performed a multivariate analysis.
Another factor that increased the overall risk of cancer was calcium channel blockers (OR, 1.47; P = .016), which also increased the risk of breast (OR, 1.61; P = .051) and melanoma-specific cancers (OR, 2.01; P = .042), a finding the researchers said was “unexpected, but has been reported in the literature with conflicting results.”
“This association is hypothesized to be related to the role of calcium in cell apoptosis, such as activation of the caspase pathway, induction of endonuclease activity and mitochondrial permeation,” Dr. Morrisroe and colleagues wrote.
SSc patients had more than a doubling of risk of mortality with incident cancer in comparison with SSc patients who did not have cancer (hazard ratio, 2.85; 95% CI, 1.51-5.37; P = .001). The average cost of health care annually for an SSc patient with cancer was AUD $1,496 (P less than .001), the researchers said.
This study was funded in part by Scleroderma Australia, Arthritis Australia, Actelion Australia, Bayer, CSL Biotherapies, GlaxoSmithKline Australia, and Pfizer. Dr. Morrisroe reported receiving support from Arthritis Australia and Royal Australasian College of Physicians Research Establishment Fellowships. Another author reported receiving a fellowship from the National Health and Medical Research Council of Australia. The other authors reported no relevant conflicts of interest.
SOURCE: Morrisroe K et al. Arthritis Care Res. 2019 Sep 20. doi: 10.1002/acr.24076
FROM ARTHRITIS CARE & RESEARCH
2019 Update on contraception
Long-acting reversible contraception (LARC) use continues to increase in the United States. According to the most recent estimates from 2014, 14% of women use either an intrauterine device (IUD) or the etonogestrel implant.1 Forms of LARC currently available in the United States include:
- 4 hormone-releasing IUDs
- 1 nonhormonal copper IUD, and
- 1 hormonal subdermal implant.
The hormone-releasing IUDs all contain levonorgestrel (LNG). These include two 52-mg LNG products and a 19.5-mg LNG IUD, which are currently approved by the US Food and Drug Administration (FDA) for contraception for 5 continuous years of use. In addition, a 13.5-mg LNG IUD is FDA-approved for 3 years of use. The hormonal subdermal implant, which contains etonogestrel, is FDA-approved for 3 years of use. Although major complications with IUDs (perforation, expulsion, intrauterine infection)and implants (subfascial implantation, distant migration) are rare, adverse effects that can affect continuation—such as irregular bleeding—are more common.2,3
Contraceptive discontinuation due to bleeding concerns occurs more frequently with the etonogestrel implant than with LNG IUDs (TABLE 1). In a large prospective study in the United States, 13% of women discontinued the implant during 3 years of follow-up due to bleeding pattern changes.
Notably, it is important to use standardized definitions to understand and compare bleeding concerns with LARC use. The Belsey criteria of the World Health Organization (WHO), a standard used for decades, describe bleeding patterns using 90-day reference periods or intervals (TABLE 2).9 Bleeding patterns that decrease flow (amenorrhea, infrequent bleeding) often are considered favorable, and those that increase bleeding or irregularity often are considered unfavorable. These criteria are commonly used in package labeling to describe bleeding patterns with extended use.
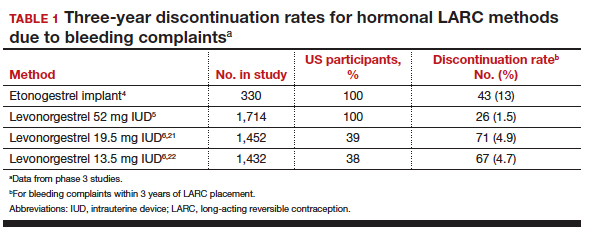
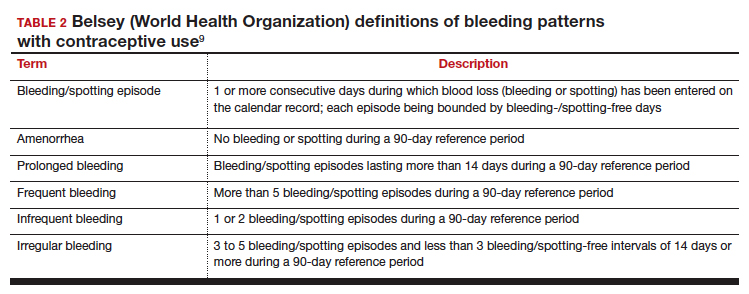
In this Update, we examine recent data evaluating differences in bleeding patterns with the 3 doses of the LNG IUD, predictors of abnormal bleeding with the etonogestrel implant, and the impact of timing on postpartum etonogestrel implant placement.
Continue to: Bleeding patterns with progestin-containing IUDs vary according to the LNG dose...
Bleeding patterns with progestin-containing IUDs vary according to the LNG dose
Goldthwaite LM, Creinin MD. Comparing bleeding patterns for the levonorgestrel 52 mg, 19.5 mg, and 13.5 mg intrauterine systems. Contraception. 2019;100:128-131.
Counseling on IUDs' different hormonal doses requires an understanding of patients' desires for contraceptive efficacy and bleeding expectations. A recent study provides guidance on what patients typically can expect for their bleeding patterns over the first few years with the 3 different doses of LNG IUDs.
Goldthwaite and Creinin used existing published or publicly available data to analyze differences in bleeding patterns associated with the 52-mg, 19.5-mg, and 13.5-mg LNG IUDs. Although two 52-mg LNG IUDs are available, published data using the WHO Belsey criteria are available only for one (Liletta; Allergan, Medicines360). The 2 products have been shown previously to have similar drug-release rates and LNG levels over 5 years.8
Comparing favorable bleeding patterns: Amenorrhea and infrequent bleeding
Among favorable bleeding patterns, amenorrhea was uncommon in the first 90 days and increased over time for all 3 IUDs. However, starting as soon as the second 90-day reference period, amenorrhea rates were significantly higher with the 52-mg LNG IUD compared with both of the lower-LNG dose IUDs, and this difference increased through 3 years of use (FIGURE 1).
Similarly, the 19.5-mg LNG IUD users had significantly higher rates of amenorrhea than the 13.5-mg LNG IUD users for all periods starting with the second 90-day reference period. At 3 years, 36% of women using the 52-mg LNG IUD had amenorrhea compared with 20% of those using the 19.5-mg LNG IUD (P<.0001) and 12% of those using the 13.5-mg LNG IUD (P<.0001).
Infrequent bleeding was similar for all 3 LNG IUDs in the first 90-day period, and it then increased most rapidly in the 52-mg LNG IUD users. At the end of year 1, 30% of the 52-mg LNG IUD users had infrequent bleeding compared with 26% of the 19.5-mg users (P = .01) and 20% of the 13.5-mg users (P<.0001). Although there was no difference in infrequent bleeding rates between the 52-mg and the 19.5-mg LNG IUD users at the end of year 1, those using a 52-mg LNG IUD had significantly higher rates of infrequent bleeding compared with the 13.5-mg LNG IUD at all time points.
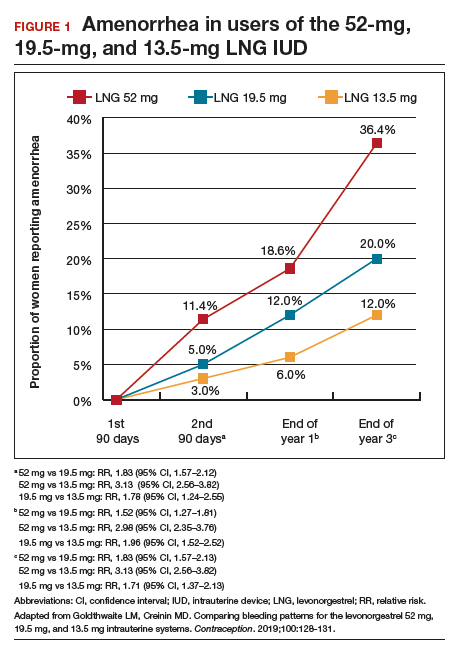
Comparing unfavorable bleeding patterns: Frequent, prolonged, and irregular bleeding
Frequent and prolonged bleeding were uncommon with all LNG doses. Irregular bleeding rates declined for users of the 3 IUDs over time. However, significantly fewer users of the 52-mg LNG IUD reported irregular bleeding at 1 year (6%) compared with users of the 19.5-mg (16.5%, P<.0001) and 13.5-mg (23%, P<.0001) LNG IUD (FIGURE 2).
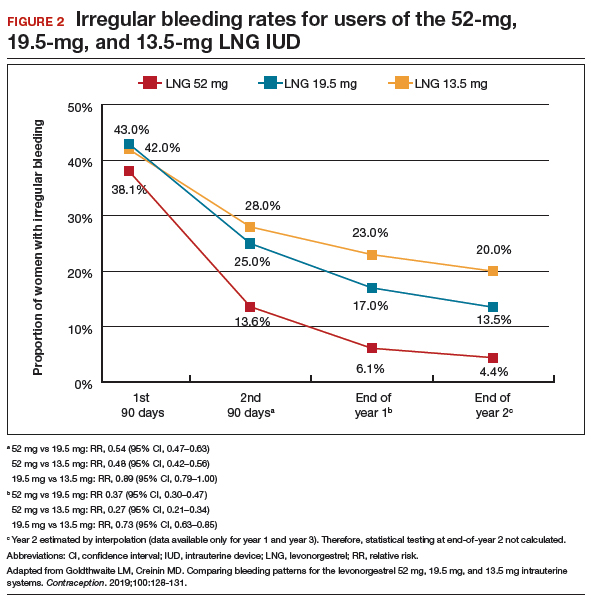
Study limitations
Comparing the data from different studies has limitations. For example, the data were collected from different populations, with the lower-dose LNG products tested in women who had a lower body mass index (BMI) and higher parity. However, prior analysis of the data on the 52-mg LNG IUD demonstrated that bleeding pattern changes did not vary based on these factors.10
When considering the different progestin-based IUD options, it is important to counsel patients according to their preferences for potential adverse effects. A randomized trial during product development found no difference in systemic adverse effects with the 3 doses of LNG IUD, likely because the systemic hormone levels are incredibly low for all 3 products.11 The summary data in this report helps explain why women using the lower-dose LNG products have slightly higher discontinuation rates for bleeding complaints, a fact we can explain to our patients during counseling.
Overall, the 52-mg LNG IUD is associated with a higher likelihood of favorable bleeding patterns over the first few years of use, with higher rates of amenorrhea and infrequent bleeding and lower rates of irregular bleeding. For women who prefer to not have periods or to have infrequent periods, the 52-mg LNG IUD is most likely to provide that outcome. For a patient who prefers to have periods, there is no evidence that the lower-dose IUDs result in “regular” or “normal” menstrual bleeding, even though they do result in more bleeding/spotting days overall. To the contrary, the available data show that these women have a significantly higher likelihood of experiencing prolonged, frequent, and irregular bleeding. In fact, no studies have reported rates of “normal” bleeding with the progestin IUDs, likely because women uncommonly have “normal” bleeding with these contraception methods. If a patient does not desire amenorrhea or strongly prefers to have “regular bleeding,” alternative methods such as a copper IUD should be considered rather than counseling her toward a lower-dose progestin IUD.
Continue to: Predicting long-term bleeding patterns after etonogestrel implant insertion...
Predicting long-term bleeding patterns after etonogestrel implant insertion
Mansour D, Fraser IS, Edelman A, et al. Can initial vaginal bleeding patterns in etonogestrel implant users predict subsequent bleeding in the first two years of use? Contraception. 2019. doi: 10.1016/j.contraception.2019.05.017.
Data from 2014 indicate that the etonogestrel implant was used by nearly 1 million women in the United States and by 3% of women using contraception.1 The primary reason women discontinue implant use is because of changes in bleeding patterns. Given the high prevalence of bleeding concerns with the etonogestrel implant, we need more data to help counsel our patients on how they can expect their bleeding to change with implant use.
Etonogestral implant and bleeding pattern trends
Mansour and colleagues completed a secondary analysis of 12 phase 3 studies to evaluate the correlation between bleeding patterns early after placement of the etonogestrel implant (days 29-118) compared with bleeding patterns through 90-day intervals during the rest of the first year of use. To account for differences in timing of etonogestrel implant placement relative to the menstrual cycle and discontinuation of other methods like oral contraceptives, bleeding outcomes on days 0-28 were excluded. They also sought to investigate the correlation between bleeding patterns in year 1 compared with those in year 2.
Overall, these studies included 923 individuals across 11 countries; however, for the current analysis, the researchers excluded women from Asian countries who comprised more than 28% of the study population. These women report significantly fewer bleeding/spotting days with the etonogestrel implant and have a lower average body weight compared with European and American women.12
A prior analysis of the same data set looked at the number of bleeding/spotting days in groups of users rather than trends in individual patients, and, as mentioned, it also included Asian women, which diluted the overall number of bleeding days.12 In this new analysis, Mansour and colleagues used the Belsey criteria to analyze individual bleeding patterns as favorable (amenorrhea, infrequent bleeding, normal bleeding) or unfavorable (prolonged and/or frequent bleeding) from a patient perspective. In this way, we can understand trends in bleeding patterns for each patient over time, rather than seeing a static (cross-sectional) report of bleeding patterns at one point in time. Data were analyzed from 537 women in year 1 and 428 women in year 2. During the first 90-day reference period (days 29-118 after implant insertion), 61% of women reported favorable bleeding, and 39% reported unfavorable bleeding.
Favorable bleeding correlates with favorable patterns later
A favorable bleeding pattern in this first reference period correlated with favorable bleeding patterns through year 1, with 85%, 80%, and 80% of these women having a favorable pattern in reference periods 2, 3, and 4, respectively. Overall, 61% of women with a favorable pattern in reference period 1 had favorable bleeding throughout the entire first year of use. Only 3.7% of women with favorable bleeding in the first reference period discontinued the implant for bleeding in year 1. Further, women with favorable bleeding at year 1 commonly continued to have favorable bleeding in year 2, with a low discontinuation rate (2.5%) in year 2.
Individual patients who have a favorable bleeding pattern initially with etonogestrel implant placement are highly likely to continue having favorable bleeding at year 1 and year 2. Notably, of women with a favorable bleeding pattern in any 90-day reference period, about 80% will continue to have a favorable bleeding pattern in the next reference period. These women can be counseled that, even if they have a 90-day period with unfavorable bleeding, about two-thirds will have a favorable pattern in the next reference period. For those with initial unfavorable patterns, about one-third to one-half change to a favorable pattern in subsequent 90-day reference periods. For women who require intervention for unfavorable bleeding but wish to keep their etonogestrel implant, prior data support use of combined oral contraceptive pills, although bleeding resolution seems to be temporary, with 86% of women having bleeding recurrence within 10 days after treatment.13
Initial unfavorable bleeding portends less favorable patterns later
Women who had an unfavorable bleeding pattern initially, however, had a less predictable course over the first year. For those with an initial unfavorable pattern, only 37%, 47%, and 51% reported a favorable pattern in reference periods 2, 3, and 4. Despite these relatively low rates of favorable bleeding, only 13% of the women with an initial unfavorable bleeding pattern discontinued implant use for a bleeding complaint by the end of year 1; this rate was significantly higher than that for women with a favorable initial bleeding pattern (P<.0001). The discontinuation rate for bleeding complaints also remained higher in year 2, at 16.5%.
Limitations and strengths to consider
Although the etonogestrel implant is FDA-approved for 3 years of use, the bleeding data from the combined trials included information for only up to 2 years after placement. The studies included also did not uniformly assess BMI, which makes it difficult to find correlations between bleeding patterns and BMI. Importantly, the studies did not include women who were more than 30% above their ideal body weight, so these assessments do not apply to obese users.12 Exclusion of women from Southeast Asia in this analysis makes this study's findings more generalizable to populations in the United States and Europe.
Continue to: Early versus delayed postpartum etonogestrel implant insertion...
Early versus delayed postpartum etonogestrel implant insertion: Similar impacts on 12-month bleeding patterns
Vieira CS, de Nadai MN, de Melo Pereira do Carmo LS, et al. Timing of postpartum etonogestrel-releasing implant insertion and bleeding patterns, weight change, 12-month continuation and satisfaction rates: a randomized controlled trial. Contraception. 2019. doi:10.1016/j.contraception.2019.05.007.
Initiation of a desired LARC method shortly after delivery is associated with significant reductions in short interpregnancy intervals.14 With that goal in mind, Vieira and colleagues compared bleeding patterns in women who received an etonogestrel implant within 48 hours of delivery with those who received an implant at 6 weeks postdelivery.
The study was a secondary analysis of data from a randomized controlled trial of early versus delayed postpartum insertion of the etonogestrel implant conducted in Sao Paulo, Brazil. That primary trial's goal was to examine the impact of early versus delayed implant insertion on infant growth (100 women were randomly assigned to the 2 implant groups); no difference in infant growth at 12 months was seen in the 2 groups.15 In the secondary analysis, bleeding patterns and BMI were evaluated every 90 days for 12 months. The mean BMI at enrollment postpartum was 29.4 kg/m2 in the early-insertion group and 30.2 kg/m2 for the delayed-insertion group.
Bleeding patterns with early or delayed implant insertion were similar
Vieira and colleagues found similar bleeding patterns between the groups over 12 months of follow-up. Amenorrhea was reported by 56% of the early-insertion group in the first 90 days and by 62% in the delayed-insertion group. During the last 90 days of the year, 52% of the early-insertion and 46% of the delayed-insertion group reported amenorrhea. Amenorrhea rates did not differ between women who were exclusively breastfeeding and those nonexclusively breastfeeding.
Continuation rates were high at 1 year
Prolonged bleeding episodes were uncommon in both groups, with only 2% of women reporting prolonged bleeding in any given reference period. Twelve-month implant continuation rates were high in both groups: 98% in the early- and 100% in the delayed-insertion group. Additionally, the investigators found that both groups experienced a BMI decrease, with no difference between groups (10.3% and 11% in the early- and delayed-insertion groups, respectively).
Study limitations and strengths
This study included a larger number of participants than prior randomized, controlled trials that evaluated bleeding patterns with postpartum etonogestrel implant insertion, and it had very low rates of loss to follow-up. The study's low rate of 12-month implant discontinuation (2%) is lower than that of other studies that reported rates of 6% to 14%.16,17 Although the authors stated that this low rate may be due to thorough anticipatory counseling prior to placement, it is also possible that this study population does not reflect all populations. Regardless, the data clearly show that placing an etonogestrel implant prior to hospital discharge, compared with waiting for later placement, does not impact bleeding patterns over the ensuing year.
For patients who desire an etonogestrel implant for contraception postpartum, we now have additional information to counsel about the impact of implant placement on postpartum bleeding patterns. Overall, bleeding patterns are highly favorable and do not vary whether the implant is placed in the hospital or later. Additionally, the timing of placement does not impact implant continuation rates or BMI changes over 1 year. Further, the primary study assessed infant growth in the early- versus delayed-placement groups and found no differences in infant growth. Although the data are limited, immediate postpartum etonogestrel implant placement does not seem to affect the rate of breastfeeding or the volume of breast milk.18,19 Timing of implant placement, assuming adequate resources, should be based primarily on patient preference. And, given the correlation of immediate postpartum LARC placement to increased interpregnancy interval, particular efforts should be made to provide the implant in the immediate postpartum period, if the patient desires.20
- Kavanaugh ML, Jerman J. Contraceptive method use in the United States: trends and characteristics between 2008, 2012 and 2014. Contraception. 2018;97:14-21.
- Trussell J. Contraceptive failure in the United States. Contraception. 2011;83:397-404.
- Odom EB, Eisenberg DL, Fox IK. Difficult removal of subdermal contraceptive implants: a multidisciplinary approach involving a peripheral nerve expert. Contraception. 2017;96: 89-95.
- Funk S, Miller MM, Mishell DR Jr, et al; Implanon US Study Group. Safety and efficacy of Implanon, a single-rod implantable contraceptive containing etonogestrel. Contraception. 2005;71:319-326.
- Eisenberg DL, Schreiber CA, Turok DK, et al; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92:10-16.
- Nelson A, Apter D, Hauck B, et al. Two low-dose levonorgestrel intrauterine contraceptive systems: a randomized controlled trial. Obstet Gynecol. 2013;122:1205-1213.
- Beckert V, Ahlers C, Frenz AK, et al. Bleeding patterns with the 19.5mg LNG-IUS, with special focus on the first year of use: implications for counselling. Eur J Contracept Reprod Health Care. 2019;24:251-259.
- Teal SB, Turok DK, Chen BA, et al. Five-year contraceptive efficacy and safety of a levonorgestrel 52-mg intrauterine system. Obstet Gynecol. 2019;133:63-70.
- Belsey EM, Machines D, d’Arcangues C. The analysis of vaginal bleeding patterns induced by fertility regulating methods. Contraception. 1986;34:253-260.
- Schreiber CA, Teal SB, Blumenthal PD, et al. Bleeding patterns for the Liletta® levonorgestrel 52mg intrauterine system. Eur J Contracept Reprod Health Care. 2018;23:116–120.
- Gemzell-Danielsson K, Schellschmidt I, Apter D. A randomized, phase II study describing the efficacy, bleeding profile, and safety of two low-dose levonorgestrel-releasing intrauterine contraceptive systems and Mirena. Fertil Steril. 2012;97:616-22.e1-3.
- Mansour D, Korver T, Marintcheva-Petrova M, et al. The effects of Implanon on menstrual bleeding patterns. Eur J Contracept Reprod Health Care. 2008;13(suppl 1):13-28.
- Guiahi M, McBride M, Sheeder J, et al. Short-term treatment of bothersome bleeding for etonogestrel implant users using a 14-day oral contraceptive pill regimen: a randomized controlled trial. Obstet Gynecol. 2015;126:508-513.
- Brunson MR, Klein DA, Olsen CH, et al. Postpartum contraception: initiation and effectiveness in a large universal healthcare system. Am J Obstet Gynecol. 2017;217:55.e1-55.e9
- de Melo Pereira Carmo LS, Braga GC, Ferriani RA, et al. Timing of etonogestrel-releasing implants and growth of breastfed infants: a randomized controlled trial. Obstet Gynecol. 2017;130:100-107.
- Crockett AH, Pickell LB, Heberlein EC, et al. Six- and twelve-month documented removal rates among women electing postpartum inpatient compared to delayed or interval contraceptive implant insertions after Medicaid payment reform. Contraception. 2017;95:71-76.
- Wilson S, Tennant C, Sammel MD, et al. Immediate postpartum etonogestrel implant: a contraception option with long-term continuation. Contraception. 2014;90:259-264.
- Sothornwit J, Werawatakul Y, Kaewrudee S, et al. Immediate versus delayed postpartum insertion of contraceptive implant for contraception. Cochrane Database Syst Rev. 2017;4:CD011913.
- Braga GC, Ferriolli E, Quintana SM, et al. Immediate postpartum initiation of etonogestrel-releasing implant: a randomized controlled trial on breastfeeding impact. Contraception. 2015;92:536-542.
- Thiel de Bocanegra H, Chang R, Howell M, et al. Interpregnancy intervals: impact of postpartum contraceptive effectiveness and coverage. Am J Obstet Gynecol. 2014;210:311.e1-8.
- Kyleena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc;2016.
- Skyla [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
Long-acting reversible contraception (LARC) use continues to increase in the United States. According to the most recent estimates from 2014, 14% of women use either an intrauterine device (IUD) or the etonogestrel implant.1 Forms of LARC currently available in the United States include:
- 4 hormone-releasing IUDs
- 1 nonhormonal copper IUD, and
- 1 hormonal subdermal implant.
The hormone-releasing IUDs all contain levonorgestrel (LNG). These include two 52-mg LNG products and a 19.5-mg LNG IUD, which are currently approved by the US Food and Drug Administration (FDA) for contraception for 5 continuous years of use. In addition, a 13.5-mg LNG IUD is FDA-approved for 3 years of use. The hormonal subdermal implant, which contains etonogestrel, is FDA-approved for 3 years of use. Although major complications with IUDs (perforation, expulsion, intrauterine infection)and implants (subfascial implantation, distant migration) are rare, adverse effects that can affect continuation—such as irregular bleeding—are more common.2,3
Contraceptive discontinuation due to bleeding concerns occurs more frequently with the etonogestrel implant than with LNG IUDs (TABLE 1). In a large prospective study in the United States, 13% of women discontinued the implant during 3 years of follow-up due to bleeding pattern changes.
Notably, it is important to use standardized definitions to understand and compare bleeding concerns with LARC use. The Belsey criteria of the World Health Organization (WHO), a standard used for decades, describe bleeding patterns using 90-day reference periods or intervals (TABLE 2).9 Bleeding patterns that decrease flow (amenorrhea, infrequent bleeding) often are considered favorable, and those that increase bleeding or irregularity often are considered unfavorable. These criteria are commonly used in package labeling to describe bleeding patterns with extended use.
In this Update, we examine recent data evaluating differences in bleeding patterns with the 3 doses of the LNG IUD, predictors of abnormal bleeding with the etonogestrel implant, and the impact of timing on postpartum etonogestrel implant placement.
Continue to: Bleeding patterns with progestin-containing IUDs vary according to the LNG dose...
Bleeding patterns with progestin-containing IUDs vary according to the LNG dose
Goldthwaite LM, Creinin MD. Comparing bleeding patterns for the levonorgestrel 52 mg, 19.5 mg, and 13.5 mg intrauterine systems. Contraception. 2019;100:128-131.
Counseling on IUDs' different hormonal doses requires an understanding of patients' desires for contraceptive efficacy and bleeding expectations. A recent study provides guidance on what patients typically can expect for their bleeding patterns over the first few years with the 3 different doses of LNG IUDs.
Goldthwaite and Creinin used existing published or publicly available data to analyze differences in bleeding patterns associated with the 52-mg, 19.5-mg, and 13.5-mg LNG IUDs. Although two 52-mg LNG IUDs are available, published data using the WHO Belsey criteria are available only for one (Liletta; Allergan, Medicines360). The 2 products have been shown previously to have similar drug-release rates and LNG levels over 5 years.8
Comparing favorable bleeding patterns: Amenorrhea and infrequent bleeding
Among favorable bleeding patterns, amenorrhea was uncommon in the first 90 days and increased over time for all 3 IUDs. However, starting as soon as the second 90-day reference period, amenorrhea rates were significantly higher with the 52-mg LNG IUD compared with both of the lower-LNG dose IUDs, and this difference increased through 3 years of use (FIGURE 1).
Similarly, the 19.5-mg LNG IUD users had significantly higher rates of amenorrhea than the 13.5-mg LNG IUD users for all periods starting with the second 90-day reference period. At 3 years, 36% of women using the 52-mg LNG IUD had amenorrhea compared with 20% of those using the 19.5-mg LNG IUD (P<.0001) and 12% of those using the 13.5-mg LNG IUD (P<.0001).
Infrequent bleeding was similar for all 3 LNG IUDs in the first 90-day period, and it then increased most rapidly in the 52-mg LNG IUD users. At the end of year 1, 30% of the 52-mg LNG IUD users had infrequent bleeding compared with 26% of the 19.5-mg users (P = .01) and 20% of the 13.5-mg users (P<.0001). Although there was no difference in infrequent bleeding rates between the 52-mg and the 19.5-mg LNG IUD users at the end of year 1, those using a 52-mg LNG IUD had significantly higher rates of infrequent bleeding compared with the 13.5-mg LNG IUD at all time points.
Comparing unfavorable bleeding patterns: Frequent, prolonged, and irregular bleeding
Frequent and prolonged bleeding were uncommon with all LNG doses. Irregular bleeding rates declined for users of the 3 IUDs over time. However, significantly fewer users of the 52-mg LNG IUD reported irregular bleeding at 1 year (6%) compared with users of the 19.5-mg (16.5%, P<.0001) and 13.5-mg (23%, P<.0001) LNG IUD (FIGURE 2).
Study limitations
Comparing the data from different studies has limitations. For example, the data were collected from different populations, with the lower-dose LNG products tested in women who had a lower body mass index (BMI) and higher parity. However, prior analysis of the data on the 52-mg LNG IUD demonstrated that bleeding pattern changes did not vary based on these factors.10
When considering the different progestin-based IUD options, it is important to counsel patients according to their preferences for potential adverse effects. A randomized trial during product development found no difference in systemic adverse effects with the 3 doses of LNG IUD, likely because the systemic hormone levels are incredibly low for all 3 products.11 The summary data in this report helps explain why women using the lower-dose LNG products have slightly higher discontinuation rates for bleeding complaints, a fact we can explain to our patients during counseling.
Overall, the 52-mg LNG IUD is associated with a higher likelihood of favorable bleeding patterns over the first few years of use, with higher rates of amenorrhea and infrequent bleeding and lower rates of irregular bleeding. For women who prefer to not have periods or to have infrequent periods, the 52-mg LNG IUD is most likely to provide that outcome. For a patient who prefers to have periods, there is no evidence that the lower-dose IUDs result in “regular” or “normal” menstrual bleeding, even though they do result in more bleeding/spotting days overall. To the contrary, the available data show that these women have a significantly higher likelihood of experiencing prolonged, frequent, and irregular bleeding. In fact, no studies have reported rates of “normal” bleeding with the progestin IUDs, likely because women uncommonly have “normal” bleeding with these contraception methods. If a patient does not desire amenorrhea or strongly prefers to have “regular bleeding,” alternative methods such as a copper IUD should be considered rather than counseling her toward a lower-dose progestin IUD.
Continue to: Predicting long-term bleeding patterns after etonogestrel implant insertion...
Predicting long-term bleeding patterns after etonogestrel implant insertion
Mansour D, Fraser IS, Edelman A, et al. Can initial vaginal bleeding patterns in etonogestrel implant users predict subsequent bleeding in the first two years of use? Contraception. 2019. doi: 10.1016/j.contraception.2019.05.017.
Data from 2014 indicate that the etonogestrel implant was used by nearly 1 million women in the United States and by 3% of women using contraception.1 The primary reason women discontinue implant use is because of changes in bleeding patterns. Given the high prevalence of bleeding concerns with the etonogestrel implant, we need more data to help counsel our patients on how they can expect their bleeding to change with implant use.
Etonogestral implant and bleeding pattern trends
Mansour and colleagues completed a secondary analysis of 12 phase 3 studies to evaluate the correlation between bleeding patterns early after placement of the etonogestrel implant (days 29-118) compared with bleeding patterns through 90-day intervals during the rest of the first year of use. To account for differences in timing of etonogestrel implant placement relative to the menstrual cycle and discontinuation of other methods like oral contraceptives, bleeding outcomes on days 0-28 were excluded. They also sought to investigate the correlation between bleeding patterns in year 1 compared with those in year 2.
Overall, these studies included 923 individuals across 11 countries; however, for the current analysis, the researchers excluded women from Asian countries who comprised more than 28% of the study population. These women report significantly fewer bleeding/spotting days with the etonogestrel implant and have a lower average body weight compared with European and American women.12
A prior analysis of the same data set looked at the number of bleeding/spotting days in groups of users rather than trends in individual patients, and, as mentioned, it also included Asian women, which diluted the overall number of bleeding days.12 In this new analysis, Mansour and colleagues used the Belsey criteria to analyze individual bleeding patterns as favorable (amenorrhea, infrequent bleeding, normal bleeding) or unfavorable (prolonged and/or frequent bleeding) from a patient perspective. In this way, we can understand trends in bleeding patterns for each patient over time, rather than seeing a static (cross-sectional) report of bleeding patterns at one point in time. Data were analyzed from 537 women in year 1 and 428 women in year 2. During the first 90-day reference period (days 29-118 after implant insertion), 61% of women reported favorable bleeding, and 39% reported unfavorable bleeding.
Favorable bleeding correlates with favorable patterns later
A favorable bleeding pattern in this first reference period correlated with favorable bleeding patterns through year 1, with 85%, 80%, and 80% of these women having a favorable pattern in reference periods 2, 3, and 4, respectively. Overall, 61% of women with a favorable pattern in reference period 1 had favorable bleeding throughout the entire first year of use. Only 3.7% of women with favorable bleeding in the first reference period discontinued the implant for bleeding in year 1. Further, women with favorable bleeding at year 1 commonly continued to have favorable bleeding in year 2, with a low discontinuation rate (2.5%) in year 2.
Individual patients who have a favorable bleeding pattern initially with etonogestrel implant placement are highly likely to continue having favorable bleeding at year 1 and year 2. Notably, of women with a favorable bleeding pattern in any 90-day reference period, about 80% will continue to have a favorable bleeding pattern in the next reference period. These women can be counseled that, even if they have a 90-day period with unfavorable bleeding, about two-thirds will have a favorable pattern in the next reference period. For those with initial unfavorable patterns, about one-third to one-half change to a favorable pattern in subsequent 90-day reference periods. For women who require intervention for unfavorable bleeding but wish to keep their etonogestrel implant, prior data support use of combined oral contraceptive pills, although bleeding resolution seems to be temporary, with 86% of women having bleeding recurrence within 10 days after treatment.13
Initial unfavorable bleeding portends less favorable patterns later
Women who had an unfavorable bleeding pattern initially, however, had a less predictable course over the first year. For those with an initial unfavorable pattern, only 37%, 47%, and 51% reported a favorable pattern in reference periods 2, 3, and 4. Despite these relatively low rates of favorable bleeding, only 13% of the women with an initial unfavorable bleeding pattern discontinued implant use for a bleeding complaint by the end of year 1; this rate was significantly higher than that for women with a favorable initial bleeding pattern (P<.0001). The discontinuation rate for bleeding complaints also remained higher in year 2, at 16.5%.
Limitations and strengths to consider
Although the etonogestrel implant is FDA-approved for 3 years of use, the bleeding data from the combined trials included information for only up to 2 years after placement. The studies included also did not uniformly assess BMI, which makes it difficult to find correlations between bleeding patterns and BMI. Importantly, the studies did not include women who were more than 30% above their ideal body weight, so these assessments do not apply to obese users.12 Exclusion of women from Southeast Asia in this analysis makes this study's findings more generalizable to populations in the United States and Europe.
Continue to: Early versus delayed postpartum etonogestrel implant insertion...
Early versus delayed postpartum etonogestrel implant insertion: Similar impacts on 12-month bleeding patterns
Vieira CS, de Nadai MN, de Melo Pereira do Carmo LS, et al. Timing of postpartum etonogestrel-releasing implant insertion and bleeding patterns, weight change, 12-month continuation and satisfaction rates: a randomized controlled trial. Contraception. 2019. doi:10.1016/j.contraception.2019.05.007.
Initiation of a desired LARC method shortly after delivery is associated with significant reductions in short interpregnancy intervals.14 With that goal in mind, Vieira and colleagues compared bleeding patterns in women who received an etonogestrel implant within 48 hours of delivery with those who received an implant at 6 weeks postdelivery.
The study was a secondary analysis of data from a randomized controlled trial of early versus delayed postpartum insertion of the etonogestrel implant conducted in Sao Paulo, Brazil. That primary trial's goal was to examine the impact of early versus delayed implant insertion on infant growth (100 women were randomly assigned to the 2 implant groups); no difference in infant growth at 12 months was seen in the 2 groups.15 In the secondary analysis, bleeding patterns and BMI were evaluated every 90 days for 12 months. The mean BMI at enrollment postpartum was 29.4 kg/m2 in the early-insertion group and 30.2 kg/m2 for the delayed-insertion group.
Bleeding patterns with early or delayed implant insertion were similar
Vieira and colleagues found similar bleeding patterns between the groups over 12 months of follow-up. Amenorrhea was reported by 56% of the early-insertion group in the first 90 days and by 62% in the delayed-insertion group. During the last 90 days of the year, 52% of the early-insertion and 46% of the delayed-insertion group reported amenorrhea. Amenorrhea rates did not differ between women who were exclusively breastfeeding and those nonexclusively breastfeeding.
Continuation rates were high at 1 year
Prolonged bleeding episodes were uncommon in both groups, with only 2% of women reporting prolonged bleeding in any given reference period. Twelve-month implant continuation rates were high in both groups: 98% in the early- and 100% in the delayed-insertion group. Additionally, the investigators found that both groups experienced a BMI decrease, with no difference between groups (10.3% and 11% in the early- and delayed-insertion groups, respectively).
Study limitations and strengths
This study included a larger number of participants than prior randomized, controlled trials that evaluated bleeding patterns with postpartum etonogestrel implant insertion, and it had very low rates of loss to follow-up. The study's low rate of 12-month implant discontinuation (2%) is lower than that of other studies that reported rates of 6% to 14%.16,17 Although the authors stated that this low rate may be due to thorough anticipatory counseling prior to placement, it is also possible that this study population does not reflect all populations. Regardless, the data clearly show that placing an etonogestrel implant prior to hospital discharge, compared with waiting for later placement, does not impact bleeding patterns over the ensuing year.
For patients who desire an etonogestrel implant for contraception postpartum, we now have additional information to counsel about the impact of implant placement on postpartum bleeding patterns. Overall, bleeding patterns are highly favorable and do not vary whether the implant is placed in the hospital or later. Additionally, the timing of placement does not impact implant continuation rates or BMI changes over 1 year. Further, the primary study assessed infant growth in the early- versus delayed-placement groups and found no differences in infant growth. Although the data are limited, immediate postpartum etonogestrel implant placement does not seem to affect the rate of breastfeeding or the volume of breast milk.18,19 Timing of implant placement, assuming adequate resources, should be based primarily on patient preference. And, given the correlation of immediate postpartum LARC placement to increased interpregnancy interval, particular efforts should be made to provide the implant in the immediate postpartum period, if the patient desires.20
Long-acting reversible contraception (LARC) use continues to increase in the United States. According to the most recent estimates from 2014, 14% of women use either an intrauterine device (IUD) or the etonogestrel implant.1 Forms of LARC currently available in the United States include:
- 4 hormone-releasing IUDs
- 1 nonhormonal copper IUD, and
- 1 hormonal subdermal implant.
The hormone-releasing IUDs all contain levonorgestrel (LNG). These include two 52-mg LNG products and a 19.5-mg LNG IUD, which are currently approved by the US Food and Drug Administration (FDA) for contraception for 5 continuous years of use. In addition, a 13.5-mg LNG IUD is FDA-approved for 3 years of use. The hormonal subdermal implant, which contains etonogestrel, is FDA-approved for 3 years of use. Although major complications with IUDs (perforation, expulsion, intrauterine infection)and implants (subfascial implantation, distant migration) are rare, adverse effects that can affect continuation—such as irregular bleeding—are more common.2,3
Contraceptive discontinuation due to bleeding concerns occurs more frequently with the etonogestrel implant than with LNG IUDs (TABLE 1). In a large prospective study in the United States, 13% of women discontinued the implant during 3 years of follow-up due to bleeding pattern changes.
Notably, it is important to use standardized definitions to understand and compare bleeding concerns with LARC use. The Belsey criteria of the World Health Organization (WHO), a standard used for decades, describe bleeding patterns using 90-day reference periods or intervals (TABLE 2).9 Bleeding patterns that decrease flow (amenorrhea, infrequent bleeding) often are considered favorable, and those that increase bleeding or irregularity often are considered unfavorable. These criteria are commonly used in package labeling to describe bleeding patterns with extended use.
In this Update, we examine recent data evaluating differences in bleeding patterns with the 3 doses of the LNG IUD, predictors of abnormal bleeding with the etonogestrel implant, and the impact of timing on postpartum etonogestrel implant placement.
Continue to: Bleeding patterns with progestin-containing IUDs vary according to the LNG dose...
Bleeding patterns with progestin-containing IUDs vary according to the LNG dose
Goldthwaite LM, Creinin MD. Comparing bleeding patterns for the levonorgestrel 52 mg, 19.5 mg, and 13.5 mg intrauterine systems. Contraception. 2019;100:128-131.
Counseling on IUDs' different hormonal doses requires an understanding of patients' desires for contraceptive efficacy and bleeding expectations. A recent study provides guidance on what patients typically can expect for their bleeding patterns over the first few years with the 3 different doses of LNG IUDs.
Goldthwaite and Creinin used existing published or publicly available data to analyze differences in bleeding patterns associated with the 52-mg, 19.5-mg, and 13.5-mg LNG IUDs. Although two 52-mg LNG IUDs are available, published data using the WHO Belsey criteria are available only for one (Liletta; Allergan, Medicines360). The 2 products have been shown previously to have similar drug-release rates and LNG levels over 5 years.8
Comparing favorable bleeding patterns: Amenorrhea and infrequent bleeding
Among favorable bleeding patterns, amenorrhea was uncommon in the first 90 days and increased over time for all 3 IUDs. However, starting as soon as the second 90-day reference period, amenorrhea rates were significantly higher with the 52-mg LNG IUD compared with both of the lower-LNG dose IUDs, and this difference increased through 3 years of use (FIGURE 1).
Similarly, the 19.5-mg LNG IUD users had significantly higher rates of amenorrhea than the 13.5-mg LNG IUD users for all periods starting with the second 90-day reference period. At 3 years, 36% of women using the 52-mg LNG IUD had amenorrhea compared with 20% of those using the 19.5-mg LNG IUD (P<.0001) and 12% of those using the 13.5-mg LNG IUD (P<.0001).
Infrequent bleeding was similar for all 3 LNG IUDs in the first 90-day period, and it then increased most rapidly in the 52-mg LNG IUD users. At the end of year 1, 30% of the 52-mg LNG IUD users had infrequent bleeding compared with 26% of the 19.5-mg users (P = .01) and 20% of the 13.5-mg users (P<.0001). Although there was no difference in infrequent bleeding rates between the 52-mg and the 19.5-mg LNG IUD users at the end of year 1, those using a 52-mg LNG IUD had significantly higher rates of infrequent bleeding compared with the 13.5-mg LNG IUD at all time points.
Comparing unfavorable bleeding patterns: Frequent, prolonged, and irregular bleeding
Frequent and prolonged bleeding were uncommon with all LNG doses. Irregular bleeding rates declined for users of the 3 IUDs over time. However, significantly fewer users of the 52-mg LNG IUD reported irregular bleeding at 1 year (6%) compared with users of the 19.5-mg (16.5%, P<.0001) and 13.5-mg (23%, P<.0001) LNG IUD (FIGURE 2).
Study limitations
Comparing the data from different studies has limitations. For example, the data were collected from different populations, with the lower-dose LNG products tested in women who had a lower body mass index (BMI) and higher parity. However, prior analysis of the data on the 52-mg LNG IUD demonstrated that bleeding pattern changes did not vary based on these factors.10
When considering the different progestin-based IUD options, it is important to counsel patients according to their preferences for potential adverse effects. A randomized trial during product development found no difference in systemic adverse effects with the 3 doses of LNG IUD, likely because the systemic hormone levels are incredibly low for all 3 products.11 The summary data in this report helps explain why women using the lower-dose LNG products have slightly higher discontinuation rates for bleeding complaints, a fact we can explain to our patients during counseling.
Overall, the 52-mg LNG IUD is associated with a higher likelihood of favorable bleeding patterns over the first few years of use, with higher rates of amenorrhea and infrequent bleeding and lower rates of irregular bleeding. For women who prefer to not have periods or to have infrequent periods, the 52-mg LNG IUD is most likely to provide that outcome. For a patient who prefers to have periods, there is no evidence that the lower-dose IUDs result in “regular” or “normal” menstrual bleeding, even though they do result in more bleeding/spotting days overall. To the contrary, the available data show that these women have a significantly higher likelihood of experiencing prolonged, frequent, and irregular bleeding. In fact, no studies have reported rates of “normal” bleeding with the progestin IUDs, likely because women uncommonly have “normal” bleeding with these contraception methods. If a patient does not desire amenorrhea or strongly prefers to have “regular bleeding,” alternative methods such as a copper IUD should be considered rather than counseling her toward a lower-dose progestin IUD.
Continue to: Predicting long-term bleeding patterns after etonogestrel implant insertion...
Predicting long-term bleeding patterns after etonogestrel implant insertion
Mansour D, Fraser IS, Edelman A, et al. Can initial vaginal bleeding patterns in etonogestrel implant users predict subsequent bleeding in the first two years of use? Contraception. 2019. doi: 10.1016/j.contraception.2019.05.017.
Data from 2014 indicate that the etonogestrel implant was used by nearly 1 million women in the United States and by 3% of women using contraception.1 The primary reason women discontinue implant use is because of changes in bleeding patterns. Given the high prevalence of bleeding concerns with the etonogestrel implant, we need more data to help counsel our patients on how they can expect their bleeding to change with implant use.
Etonogestral implant and bleeding pattern trends
Mansour and colleagues completed a secondary analysis of 12 phase 3 studies to evaluate the correlation between bleeding patterns early after placement of the etonogestrel implant (days 29-118) compared with bleeding patterns through 90-day intervals during the rest of the first year of use. To account for differences in timing of etonogestrel implant placement relative to the menstrual cycle and discontinuation of other methods like oral contraceptives, bleeding outcomes on days 0-28 were excluded. They also sought to investigate the correlation between bleeding patterns in year 1 compared with those in year 2.
Overall, these studies included 923 individuals across 11 countries; however, for the current analysis, the researchers excluded women from Asian countries who comprised more than 28% of the study population. These women report significantly fewer bleeding/spotting days with the etonogestrel implant and have a lower average body weight compared with European and American women.12
A prior analysis of the same data set looked at the number of bleeding/spotting days in groups of users rather than trends in individual patients, and, as mentioned, it also included Asian women, which diluted the overall number of bleeding days.12 In this new analysis, Mansour and colleagues used the Belsey criteria to analyze individual bleeding patterns as favorable (amenorrhea, infrequent bleeding, normal bleeding) or unfavorable (prolonged and/or frequent bleeding) from a patient perspective. In this way, we can understand trends in bleeding patterns for each patient over time, rather than seeing a static (cross-sectional) report of bleeding patterns at one point in time. Data were analyzed from 537 women in year 1 and 428 women in year 2. During the first 90-day reference period (days 29-118 after implant insertion), 61% of women reported favorable bleeding, and 39% reported unfavorable bleeding.
Favorable bleeding correlates with favorable patterns later
A favorable bleeding pattern in this first reference period correlated with favorable bleeding patterns through year 1, with 85%, 80%, and 80% of these women having a favorable pattern in reference periods 2, 3, and 4, respectively. Overall, 61% of women with a favorable pattern in reference period 1 had favorable bleeding throughout the entire first year of use. Only 3.7% of women with favorable bleeding in the first reference period discontinued the implant for bleeding in year 1. Further, women with favorable bleeding at year 1 commonly continued to have favorable bleeding in year 2, with a low discontinuation rate (2.5%) in year 2.
Individual patients who have a favorable bleeding pattern initially with etonogestrel implant placement are highly likely to continue having favorable bleeding at year 1 and year 2. Notably, of women with a favorable bleeding pattern in any 90-day reference period, about 80% will continue to have a favorable bleeding pattern in the next reference period. These women can be counseled that, even if they have a 90-day period with unfavorable bleeding, about two-thirds will have a favorable pattern in the next reference period. For those with initial unfavorable patterns, about one-third to one-half change to a favorable pattern in subsequent 90-day reference periods. For women who require intervention for unfavorable bleeding but wish to keep their etonogestrel implant, prior data support use of combined oral contraceptive pills, although bleeding resolution seems to be temporary, with 86% of women having bleeding recurrence within 10 days after treatment.13
Initial unfavorable bleeding portends less favorable patterns later
Women who had an unfavorable bleeding pattern initially, however, had a less predictable course over the first year. For those with an initial unfavorable pattern, only 37%, 47%, and 51% reported a favorable pattern in reference periods 2, 3, and 4. Despite these relatively low rates of favorable bleeding, only 13% of the women with an initial unfavorable bleeding pattern discontinued implant use for a bleeding complaint by the end of year 1; this rate was significantly higher than that for women with a favorable initial bleeding pattern (P<.0001). The discontinuation rate for bleeding complaints also remained higher in year 2, at 16.5%.
Limitations and strengths to consider
Although the etonogestrel implant is FDA-approved for 3 years of use, the bleeding data from the combined trials included information for only up to 2 years after placement. The studies included also did not uniformly assess BMI, which makes it difficult to find correlations between bleeding patterns and BMI. Importantly, the studies did not include women who were more than 30% above their ideal body weight, so these assessments do not apply to obese users.12 Exclusion of women from Southeast Asia in this analysis makes this study's findings more generalizable to populations in the United States and Europe.
Continue to: Early versus delayed postpartum etonogestrel implant insertion...
Early versus delayed postpartum etonogestrel implant insertion: Similar impacts on 12-month bleeding patterns
Vieira CS, de Nadai MN, de Melo Pereira do Carmo LS, et al. Timing of postpartum etonogestrel-releasing implant insertion and bleeding patterns, weight change, 12-month continuation and satisfaction rates: a randomized controlled trial. Contraception. 2019. doi:10.1016/j.contraception.2019.05.007.
Initiation of a desired LARC method shortly after delivery is associated with significant reductions in short interpregnancy intervals.14 With that goal in mind, Vieira and colleagues compared bleeding patterns in women who received an etonogestrel implant within 48 hours of delivery with those who received an implant at 6 weeks postdelivery.
The study was a secondary analysis of data from a randomized controlled trial of early versus delayed postpartum insertion of the etonogestrel implant conducted in Sao Paulo, Brazil. That primary trial's goal was to examine the impact of early versus delayed implant insertion on infant growth (100 women were randomly assigned to the 2 implant groups); no difference in infant growth at 12 months was seen in the 2 groups.15 In the secondary analysis, bleeding patterns and BMI were evaluated every 90 days for 12 months. The mean BMI at enrollment postpartum was 29.4 kg/m2 in the early-insertion group and 30.2 kg/m2 for the delayed-insertion group.
Bleeding patterns with early or delayed implant insertion were similar
Vieira and colleagues found similar bleeding patterns between the groups over 12 months of follow-up. Amenorrhea was reported by 56% of the early-insertion group in the first 90 days and by 62% in the delayed-insertion group. During the last 90 days of the year, 52% of the early-insertion and 46% of the delayed-insertion group reported amenorrhea. Amenorrhea rates did not differ between women who were exclusively breastfeeding and those nonexclusively breastfeeding.
Continuation rates were high at 1 year
Prolonged bleeding episodes were uncommon in both groups, with only 2% of women reporting prolonged bleeding in any given reference period. Twelve-month implant continuation rates were high in both groups: 98% in the early- and 100% in the delayed-insertion group. Additionally, the investigators found that both groups experienced a BMI decrease, with no difference between groups (10.3% and 11% in the early- and delayed-insertion groups, respectively).
Study limitations and strengths
This study included a larger number of participants than prior randomized, controlled trials that evaluated bleeding patterns with postpartum etonogestrel implant insertion, and it had very low rates of loss to follow-up. The study's low rate of 12-month implant discontinuation (2%) is lower than that of other studies that reported rates of 6% to 14%.16,17 Although the authors stated that this low rate may be due to thorough anticipatory counseling prior to placement, it is also possible that this study population does not reflect all populations. Regardless, the data clearly show that placing an etonogestrel implant prior to hospital discharge, compared with waiting for later placement, does not impact bleeding patterns over the ensuing year.
For patients who desire an etonogestrel implant for contraception postpartum, we now have additional information to counsel about the impact of implant placement on postpartum bleeding patterns. Overall, bleeding patterns are highly favorable and do not vary whether the implant is placed in the hospital or later. Additionally, the timing of placement does not impact implant continuation rates or BMI changes over 1 year. Further, the primary study assessed infant growth in the early- versus delayed-placement groups and found no differences in infant growth. Although the data are limited, immediate postpartum etonogestrel implant placement does not seem to affect the rate of breastfeeding or the volume of breast milk.18,19 Timing of implant placement, assuming adequate resources, should be based primarily on patient preference. And, given the correlation of immediate postpartum LARC placement to increased interpregnancy interval, particular efforts should be made to provide the implant in the immediate postpartum period, if the patient desires.20
- Kavanaugh ML, Jerman J. Contraceptive method use in the United States: trends and characteristics between 2008, 2012 and 2014. Contraception. 2018;97:14-21.
- Trussell J. Contraceptive failure in the United States. Contraception. 2011;83:397-404.
- Odom EB, Eisenberg DL, Fox IK. Difficult removal of subdermal contraceptive implants: a multidisciplinary approach involving a peripheral nerve expert. Contraception. 2017;96: 89-95.
- Funk S, Miller MM, Mishell DR Jr, et al; Implanon US Study Group. Safety and efficacy of Implanon, a single-rod implantable contraceptive containing etonogestrel. Contraception. 2005;71:319-326.
- Eisenberg DL, Schreiber CA, Turok DK, et al; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92:10-16.
- Nelson A, Apter D, Hauck B, et al. Two low-dose levonorgestrel intrauterine contraceptive systems: a randomized controlled trial. Obstet Gynecol. 2013;122:1205-1213.
- Beckert V, Ahlers C, Frenz AK, et al. Bleeding patterns with the 19.5mg LNG-IUS, with special focus on the first year of use: implications for counselling. Eur J Contracept Reprod Health Care. 2019;24:251-259.
- Teal SB, Turok DK, Chen BA, et al. Five-year contraceptive efficacy and safety of a levonorgestrel 52-mg intrauterine system. Obstet Gynecol. 2019;133:63-70.
- Belsey EM, Machines D, d’Arcangues C. The analysis of vaginal bleeding patterns induced by fertility regulating methods. Contraception. 1986;34:253-260.
- Schreiber CA, Teal SB, Blumenthal PD, et al. Bleeding patterns for the Liletta® levonorgestrel 52mg intrauterine system. Eur J Contracept Reprod Health Care. 2018;23:116–120.
- Gemzell-Danielsson K, Schellschmidt I, Apter D. A randomized, phase II study describing the efficacy, bleeding profile, and safety of two low-dose levonorgestrel-releasing intrauterine contraceptive systems and Mirena. Fertil Steril. 2012;97:616-22.e1-3.
- Mansour D, Korver T, Marintcheva-Petrova M, et al. The effects of Implanon on menstrual bleeding patterns. Eur J Contracept Reprod Health Care. 2008;13(suppl 1):13-28.
- Guiahi M, McBride M, Sheeder J, et al. Short-term treatment of bothersome bleeding for etonogestrel implant users using a 14-day oral contraceptive pill regimen: a randomized controlled trial. Obstet Gynecol. 2015;126:508-513.
- Brunson MR, Klein DA, Olsen CH, et al. Postpartum contraception: initiation and effectiveness in a large universal healthcare system. Am J Obstet Gynecol. 2017;217:55.e1-55.e9
- de Melo Pereira Carmo LS, Braga GC, Ferriani RA, et al. Timing of etonogestrel-releasing implants and growth of breastfed infants: a randomized controlled trial. Obstet Gynecol. 2017;130:100-107.
- Crockett AH, Pickell LB, Heberlein EC, et al. Six- and twelve-month documented removal rates among women electing postpartum inpatient compared to delayed or interval contraceptive implant insertions after Medicaid payment reform. Contraception. 2017;95:71-76.
- Wilson S, Tennant C, Sammel MD, et al. Immediate postpartum etonogestrel implant: a contraception option with long-term continuation. Contraception. 2014;90:259-264.
- Sothornwit J, Werawatakul Y, Kaewrudee S, et al. Immediate versus delayed postpartum insertion of contraceptive implant for contraception. Cochrane Database Syst Rev. 2017;4:CD011913.
- Braga GC, Ferriolli E, Quintana SM, et al. Immediate postpartum initiation of etonogestrel-releasing implant: a randomized controlled trial on breastfeeding impact. Contraception. 2015;92:536-542.
- Thiel de Bocanegra H, Chang R, Howell M, et al. Interpregnancy intervals: impact of postpartum contraceptive effectiveness and coverage. Am J Obstet Gynecol. 2014;210:311.e1-8.
- Kyleena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc;2016.
- Skyla [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Kavanaugh ML, Jerman J. Contraceptive method use in the United States: trends and characteristics between 2008, 2012 and 2014. Contraception. 2018;97:14-21.
- Trussell J. Contraceptive failure in the United States. Contraception. 2011;83:397-404.
- Odom EB, Eisenberg DL, Fox IK. Difficult removal of subdermal contraceptive implants: a multidisciplinary approach involving a peripheral nerve expert. Contraception. 2017;96: 89-95.
- Funk S, Miller MM, Mishell DR Jr, et al; Implanon US Study Group. Safety and efficacy of Implanon, a single-rod implantable contraceptive containing etonogestrel. Contraception. 2005;71:319-326.
- Eisenberg DL, Schreiber CA, Turok DK, et al; ACCESS IUS Investigators. Three-year efficacy and safety of a new 52-mg levonorgestrel-releasing intrauterine system. Contraception. 2015;92:10-16.
- Nelson A, Apter D, Hauck B, et al. Two low-dose levonorgestrel intrauterine contraceptive systems: a randomized controlled trial. Obstet Gynecol. 2013;122:1205-1213.
- Beckert V, Ahlers C, Frenz AK, et al. Bleeding patterns with the 19.5mg LNG-IUS, with special focus on the first year of use: implications for counselling. Eur J Contracept Reprod Health Care. 2019;24:251-259.
- Teal SB, Turok DK, Chen BA, et al. Five-year contraceptive efficacy and safety of a levonorgestrel 52-mg intrauterine system. Obstet Gynecol. 2019;133:63-70.
- Belsey EM, Machines D, d’Arcangues C. The analysis of vaginal bleeding patterns induced by fertility regulating methods. Contraception. 1986;34:253-260.
- Schreiber CA, Teal SB, Blumenthal PD, et al. Bleeding patterns for the Liletta® levonorgestrel 52mg intrauterine system. Eur J Contracept Reprod Health Care. 2018;23:116–120.
- Gemzell-Danielsson K, Schellschmidt I, Apter D. A randomized, phase II study describing the efficacy, bleeding profile, and safety of two low-dose levonorgestrel-releasing intrauterine contraceptive systems and Mirena. Fertil Steril. 2012;97:616-22.e1-3.
- Mansour D, Korver T, Marintcheva-Petrova M, et al. The effects of Implanon on menstrual bleeding patterns. Eur J Contracept Reprod Health Care. 2008;13(suppl 1):13-28.
- Guiahi M, McBride M, Sheeder J, et al. Short-term treatment of bothersome bleeding for etonogestrel implant users using a 14-day oral contraceptive pill regimen: a randomized controlled trial. Obstet Gynecol. 2015;126:508-513.
- Brunson MR, Klein DA, Olsen CH, et al. Postpartum contraception: initiation and effectiveness in a large universal healthcare system. Am J Obstet Gynecol. 2017;217:55.e1-55.e9
- de Melo Pereira Carmo LS, Braga GC, Ferriani RA, et al. Timing of etonogestrel-releasing implants and growth of breastfed infants: a randomized controlled trial. Obstet Gynecol. 2017;130:100-107.
- Crockett AH, Pickell LB, Heberlein EC, et al. Six- and twelve-month documented removal rates among women electing postpartum inpatient compared to delayed or interval contraceptive implant insertions after Medicaid payment reform. Contraception. 2017;95:71-76.
- Wilson S, Tennant C, Sammel MD, et al. Immediate postpartum etonogestrel implant: a contraception option with long-term continuation. Contraception. 2014;90:259-264.
- Sothornwit J, Werawatakul Y, Kaewrudee S, et al. Immediate versus delayed postpartum insertion of contraceptive implant for contraception. Cochrane Database Syst Rev. 2017;4:CD011913.
- Braga GC, Ferriolli E, Quintana SM, et al. Immediate postpartum initiation of etonogestrel-releasing implant: a randomized controlled trial on breastfeeding impact. Contraception. 2015;92:536-542.
- Thiel de Bocanegra H, Chang R, Howell M, et al. Interpregnancy intervals: impact of postpartum contraceptive effectiveness and coverage. Am J Obstet Gynecol. 2014;210:311.e1-8.
- Kyleena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc;2016.
- Skyla [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
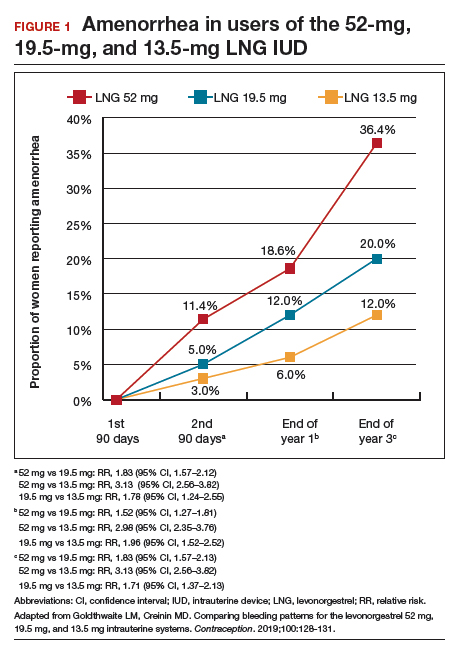
Liraglutide ‘option’ for treating pediatric type 2 diabetes
BARCELONA – The glucagon-like peptide-1 receptor agonist (GLP-1 RA) liraglutide added onto metformin with or without basal insulin effectively reduced hemoglobin A1c and fasting plasma glucose levels in children with type 2 diabetes in the 52-week ELLIPSE study.

The primary endpoint of the trial, which was the mean change in HbA1c from baseline to 26 weeks, was met, with a greater percentage point decrease with liraglutide (Victoza) than placebo (–0.64 vs. +0.42), with an estimated treatment difference of –1.06 percentage points (P less than .001). At the end of the study, the percentage point changes were –0.50 and +0.80, with a between-group difference of –1.30 in favor of liraglutide.
“Those of us working in pediatric practice are seeing an increasing demand for our clinical services in children with type 2 diabetes,” study investigator Timothy Barrett, PhD, MBBS, observed at the annual meeting of the European Association for the Study of Diabetes. This reflects the increasing prevalence of type 2 diabetes in this age group and is most likely linked to the rising rates of obesity and overweight that have been reported widely in young people in recent years, he added.
“Unfortunately, we look with envy upon our adult physician colleagues, and the range of treatments they have available to treat type 2 diabetes in adults.” In pediatrics, the only licensed treatments that have been available until recently were metformin and insulin, with the latter being an “illogical treatment to treat those with obesity-related diabetes.” The study’s findings, however, support liraglutide as another option to consider, said Dr. Barrett, a pediatric endocrinologist and professor of pediatrics and child health based at the University of Birmingham, England.
“Liraglutide at doses of up to 1.8 mg/day when added to metformin, and basal insulin if required, does seem to offer an additional treatment option for children and young people with type 2 diabetes who require improved glycemic control after they’ve reached a maximum dose of metformin,” he said.
ELLIPSE (Evaluation of Liraglutide in Pediatrics with Diabetes) was a multicenter, randomized, parallel group, placebo-controlled trial to assess the efficacy and safety of liraglutide as an add-on treatment to metformin, with or without basal insulin, in 134 overweight or obese children and adolescents (aged 10-17) with type 2 diabetes.
For inclusion, patients had to be able to complete the trial before their 18th birthday, and have an HbA1c of at least 7% if being treated with diet and exercise, or 6.5% or higher if already being treated with metformin, with or without insulin. Body mass index had to be above the 85the percentile for their age and sex.
Of 307 children and adolescents screened at 84 centers in 25 countries, 135 were randomized and 134 were treated between 2012 and 2018. Screening took place over a period of 2 weeks, after which time those eligible for the trial underwent a 3- to 4-week period where their dose of metformin was titrated if needed followed by an 8-week maintenance period. Only after that was randomization to liraglutide or placebo done, with the GLP-1R started at a subcutaneous dose of 0.6 mg and titrated up to 1.2 or 1.8 mg over 3 days to achieve a fasting plasma glucose (FPG) of less than 6.1 mmol/L (110 mg/dL). However, not all patients were escalated to the top dose, Dr. Barrett noted.
The mean age of patients in the trial was 14.5 years; about 60% of patients were female. The duration of diabetes was about 1.9 years and the average body weight and BMI a respective 91 kg and 33 kg/m2.
Over the course of the study, FPG fell by 1.06 mmol/L at week 26 and 1.03 mmol/L at week 52 in the liraglutide group but rose in the placebo group by 0.80 and 0.78 mmol/L, respectively. The estimated treatment difference was –1.88 (P = .002) and –1.81 at 26 an 52 weeks, respectively.
What was “a really gratifying to see,” said Dr. Barrett, was that the proportion of children and young people achieving a glycemic target of an HbA1c of less than 7% by the end of the double-blind treatment period was significantly higher in the liraglutide than placebo group, at 63.7% and 36.5%, respectively.
Most of the adverse effects seen in the study were gastrointestinal symptoms, including nausea, vomiting, and diarrhea, in about 20% of liraglutide-treated patients, compared with roughly 10% of placebo-treated patients. “This is really reflected in the adult studies as well, and many of these were thankfully transient.”
As for hypoglycemia, Dr. Barrett reported that there was a higher rate in liraglutide- than placebo-treated patients (45.5% vs. 25% for any event), although there were no severe episodes in the liraglutide group and one in the placebo group. Almost a third (31%) of hypoglycemic episodes were asymptomatic, versus 17.6% for the placebo group.
“This is the first successfully completed phase 3 trial showing efficacy of a noninsulin agent, in this case, for children who do not get managed solely on metformin monotherapy,” Dr. Barrett said.
The Food and Drug Administration has approved liraglutide for use in pediatric patients 10 years or older with type 2 diabetes, based in part on results of the ELLIPSE results, Novo Nordisk announced in June. The trial results were published prior to the EASD meeting (Tamborlane WV et al. N Engl J Med. 2019 Aug 15;381:637-46).
Novo Nordisk initiated and funded the trial, and most of the investigators reported receiving funds from the company outside the submitted work. Dr Barrett disclosed being a consultant to and/or receiving honoraria from AstraZeneca, Novo Nordisk and Servier.
SOURCE: Barrett T et al. EASD 2019. Abstract 84.
BARCELONA – The glucagon-like peptide-1 receptor agonist (GLP-1 RA) liraglutide added onto metformin with or without basal insulin effectively reduced hemoglobin A1c and fasting plasma glucose levels in children with type 2 diabetes in the 52-week ELLIPSE study.
The primary endpoint of the trial, which was the mean change in HbA1c from baseline to 26 weeks, was met, with a greater percentage point decrease with liraglutide (Victoza) than placebo (–0.64 vs. +0.42), with an estimated treatment difference of –1.06 percentage points (P less than .001). At the end of the study, the percentage point changes were –0.50 and +0.80, with a between-group difference of –1.30 in favor of liraglutide.
“Those of us working in pediatric practice are seeing an increasing demand for our clinical services in children with type 2 diabetes,” study investigator Timothy Barrett, PhD, MBBS, observed at the annual meeting of the European Association for the Study of Diabetes. This reflects the increasing prevalence of type 2 diabetes in this age group and is most likely linked to the rising rates of obesity and overweight that have been reported widely in young people in recent years, he added.
“Unfortunately, we look with envy upon our adult physician colleagues, and the range of treatments they have available to treat type 2 diabetes in adults.” In pediatrics, the only licensed treatments that have been available until recently were metformin and insulin, with the latter being an “illogical treatment to treat those with obesity-related diabetes.” The study’s findings, however, support liraglutide as another option to consider, said Dr. Barrett, a pediatric endocrinologist and professor of pediatrics and child health based at the University of Birmingham, England.
“Liraglutide at doses of up to 1.8 mg/day when added to metformin, and basal insulin if required, does seem to offer an additional treatment option for children and young people with type 2 diabetes who require improved glycemic control after they’ve reached a maximum dose of metformin,” he said.
ELLIPSE (Evaluation of Liraglutide in Pediatrics with Diabetes) was a multicenter, randomized, parallel group, placebo-controlled trial to assess the efficacy and safety of liraglutide as an add-on treatment to metformin, with or without basal insulin, in 134 overweight or obese children and adolescents (aged 10-17) with type 2 diabetes.
For inclusion, patients had to be able to complete the trial before their 18th birthday, and have an HbA1c of at least 7% if being treated with diet and exercise, or 6.5% or higher if already being treated with metformin, with or without insulin. Body mass index had to be above the 85the percentile for their age and sex.
Of 307 children and adolescents screened at 84 centers in 25 countries, 135 were randomized and 134 were treated between 2012 and 2018. Screening took place over a period of 2 weeks, after which time those eligible for the trial underwent a 3- to 4-week period where their dose of metformin was titrated if needed followed by an 8-week maintenance period. Only after that was randomization to liraglutide or placebo done, with the GLP-1R started at a subcutaneous dose of 0.6 mg and titrated up to 1.2 or 1.8 mg over 3 days to achieve a fasting plasma glucose (FPG) of less than 6.1 mmol/L (110 mg/dL). However, not all patients were escalated to the top dose, Dr. Barrett noted.
The mean age of patients in the trial was 14.5 years; about 60% of patients were female. The duration of diabetes was about 1.9 years and the average body weight and BMI a respective 91 kg and 33 kg/m2.
Over the course of the study, FPG fell by 1.06 mmol/L at week 26 and 1.03 mmol/L at week 52 in the liraglutide group but rose in the placebo group by 0.80 and 0.78 mmol/L, respectively. The estimated treatment difference was –1.88 (P = .002) and –1.81 at 26 an 52 weeks, respectively.
What was “a really gratifying to see,” said Dr. Barrett, was that the proportion of children and young people achieving a glycemic target of an HbA1c of less than 7% by the end of the double-blind treatment period was significantly higher in the liraglutide than placebo group, at 63.7% and 36.5%, respectively.
Most of the adverse effects seen in the study were gastrointestinal symptoms, including nausea, vomiting, and diarrhea, in about 20% of liraglutide-treated patients, compared with roughly 10% of placebo-treated patients. “This is really reflected in the adult studies as well, and many of these were thankfully transient.”
As for hypoglycemia, Dr. Barrett reported that there was a higher rate in liraglutide- than placebo-treated patients (45.5% vs. 25% for any event), although there were no severe episodes in the liraglutide group and one in the placebo group. Almost a third (31%) of hypoglycemic episodes were asymptomatic, versus 17.6% for the placebo group.
“This is the first successfully completed phase 3 trial showing efficacy of a noninsulin agent, in this case, for children who do not get managed solely on metformin monotherapy,” Dr. Barrett said.
The Food and Drug Administration has approved liraglutide for use in pediatric patients 10 years or older with type 2 diabetes, based in part on results of the ELLIPSE results, Novo Nordisk announced in June. The trial results were published prior to the EASD meeting (Tamborlane WV et al. N Engl J Med. 2019 Aug 15;381:637-46).
Novo Nordisk initiated and funded the trial, and most of the investigators reported receiving funds from the company outside the submitted work. Dr Barrett disclosed being a consultant to and/or receiving honoraria from AstraZeneca, Novo Nordisk and Servier.
SOURCE: Barrett T et al. EASD 2019. Abstract 84.
BARCELONA – The glucagon-like peptide-1 receptor agonist (GLP-1 RA) liraglutide added onto metformin with or without basal insulin effectively reduced hemoglobin A1c and fasting plasma glucose levels in children with type 2 diabetes in the 52-week ELLIPSE study.
The primary endpoint of the trial, which was the mean change in HbA1c from baseline to 26 weeks, was met, with a greater percentage point decrease with liraglutide (Victoza) than placebo (–0.64 vs. +0.42), with an estimated treatment difference of –1.06 percentage points (P less than .001). At the end of the study, the percentage point changes were –0.50 and +0.80, with a between-group difference of –1.30 in favor of liraglutide.
“Those of us working in pediatric practice are seeing an increasing demand for our clinical services in children with type 2 diabetes,” study investigator Timothy Barrett, PhD, MBBS, observed at the annual meeting of the European Association for the Study of Diabetes. This reflects the increasing prevalence of type 2 diabetes in this age group and is most likely linked to the rising rates of obesity and overweight that have been reported widely in young people in recent years, he added.
“Unfortunately, we look with envy upon our adult physician colleagues, and the range of treatments they have available to treat type 2 diabetes in adults.” In pediatrics, the only licensed treatments that have been available until recently were metformin and insulin, with the latter being an “illogical treatment to treat those with obesity-related diabetes.” The study’s findings, however, support liraglutide as another option to consider, said Dr. Barrett, a pediatric endocrinologist and professor of pediatrics and child health based at the University of Birmingham, England.
“Liraglutide at doses of up to 1.8 mg/day when added to metformin, and basal insulin if required, does seem to offer an additional treatment option for children and young people with type 2 diabetes who require improved glycemic control after they’ve reached a maximum dose of metformin,” he said.
ELLIPSE (Evaluation of Liraglutide in Pediatrics with Diabetes) was a multicenter, randomized, parallel group, placebo-controlled trial to assess the efficacy and safety of liraglutide as an add-on treatment to metformin, with or without basal insulin, in 134 overweight or obese children and adolescents (aged 10-17) with type 2 diabetes.
For inclusion, patients had to be able to complete the trial before their 18th birthday, and have an HbA1c of at least 7% if being treated with diet and exercise, or 6.5% or higher if already being treated with metformin, with or without insulin. Body mass index had to be above the 85the percentile for their age and sex.
Of 307 children and adolescents screened at 84 centers in 25 countries, 135 were randomized and 134 were treated between 2012 and 2018. Screening took place over a period of 2 weeks, after which time those eligible for the trial underwent a 3- to 4-week period where their dose of metformin was titrated if needed followed by an 8-week maintenance period. Only after that was randomization to liraglutide or placebo done, with the GLP-1R started at a subcutaneous dose of 0.6 mg and titrated up to 1.2 or 1.8 mg over 3 days to achieve a fasting plasma glucose (FPG) of less than 6.1 mmol/L (110 mg/dL). However, not all patients were escalated to the top dose, Dr. Barrett noted.
The mean age of patients in the trial was 14.5 years; about 60% of patients were female. The duration of diabetes was about 1.9 years and the average body weight and BMI a respective 91 kg and 33 kg/m2.
Over the course of the study, FPG fell by 1.06 mmol/L at week 26 and 1.03 mmol/L at week 52 in the liraglutide group but rose in the placebo group by 0.80 and 0.78 mmol/L, respectively. The estimated treatment difference was –1.88 (P = .002) and –1.81 at 26 an 52 weeks, respectively.
What was “a really gratifying to see,” said Dr. Barrett, was that the proportion of children and young people achieving a glycemic target of an HbA1c of less than 7% by the end of the double-blind treatment period was significantly higher in the liraglutide than placebo group, at 63.7% and 36.5%, respectively.
Most of the adverse effects seen in the study were gastrointestinal symptoms, including nausea, vomiting, and diarrhea, in about 20% of liraglutide-treated patients, compared with roughly 10% of placebo-treated patients. “This is really reflected in the adult studies as well, and many of these were thankfully transient.”
As for hypoglycemia, Dr. Barrett reported that there was a higher rate in liraglutide- than placebo-treated patients (45.5% vs. 25% for any event), although there were no severe episodes in the liraglutide group and one in the placebo group. Almost a third (31%) of hypoglycemic episodes were asymptomatic, versus 17.6% for the placebo group.
“This is the first successfully completed phase 3 trial showing efficacy of a noninsulin agent, in this case, for children who do not get managed solely on metformin monotherapy,” Dr. Barrett said.
The Food and Drug Administration has approved liraglutide for use in pediatric patients 10 years or older with type 2 diabetes, based in part on results of the ELLIPSE results, Novo Nordisk announced in June. The trial results were published prior to the EASD meeting (Tamborlane WV et al. N Engl J Med. 2019 Aug 15;381:637-46).
Novo Nordisk initiated and funded the trial, and most of the investigators reported receiving funds from the company outside the submitted work. Dr Barrett disclosed being a consultant to and/or receiving honoraria from AstraZeneca, Novo Nordisk and Servier.
SOURCE: Barrett T et al. EASD 2019. Abstract 84.
REPORTING FROM EASD 2019
Using slings for the surgical management of urinary incontinence: A safe, effective, evidence-based approach
Urinary incontinence affects approximately 50% of women, with up to 80% of these women experiencing stress urinary incontinence (SUI) at some point in their lives.1-3 While conservative measures can offer some improvement in symptoms, the mainstay of treatment for SUI is surgical intervention.4,5 The lifetime risk of undergoing surgery for SUI is 13.6%, and surgery leads to a major improvement in quality of life and productivity.1,6
Types of slings used for SUI
Sling procedures are the most commonly used surgical approach for the treatment of SUI. Two types of urethral slings are used: the midurethral sling and the autologous fascial (pubovaginal) sling. The midurethral sling, which is the most frequently used sling today, can be further characterized as the retropubic sling, the transobturator sling, and the mini sling (FIGURE 1).
Retropubic sling
A retropubic sling is a midurethral mesh sling that is placed beneath the urethra at the midpoint between the urethral meatus and the bladder neck. The arms of the sling extend behind the pubic symphysis, providing a hammock-like support that helps prevent leakage with increased abdominal pressures. The retropubic sling is the most commonly used type of sling. For women presenting with uncomplicated SUI who desire surgical correction, it often is the best choice for providing long-term treatment success.7
Transobturator sling
A transobturator sling is a midurethral mesh sling that is placed beneath the urethra as described above, but the arms of the sling extend outward through the obturator foramen and into the groin. This enables support of the midurethra, but this sling is less likely to result in such complications as bladder perforation or postoperative urinary retention. Transobturator slings also are associated with lower rates of voiding dysfunction and urinary urgency than retropubic slings.7-9 However, transobturator slings have higher rates of groin pain, and they are less effective in maintaining long-term cure of SUI.7
First introduced in 1996, the midurethral sling quickly grew in popularity for the treatment of SUI because of its high success rates and its minimally invasive approach.10 Both retropubic and transobturator slings are safe, extensively researched surgical approaches for the management of SUI.3 Midurethral slings have a very high rate of incontinence cure (80%–90%) and extremely high patient satisfaction rates (85%–90%), as even patients without complete cure report meaningful symptomatic improvement.7,8,11
Single-incision (mini) sling
A single-incision sling is a midurethral mesh sling that is designed to be shorter in length than standard midurethral slings. The placed sling lies under the midurethra and extends toward the superior edge of the obturator foramen but does not penetrate it. The sling is held in place by small pledgets on either side of the mesh hammock that anchor it in place to the obturator internus muscular fascia. Because this “mini” sling was introduced in 2006, fewer long-term data are available for this sling than for standard midurethral slings.
Continue to: Autologous (fascial) sling...
Autologous (fascial) sling
An autologous sling is a retropubic sling made from the patient’s own fascia; it is harvested from either the fascia lata of the lateral thigh or the rectus fascia of the abdomen. The sling is placed beneath the urethra in the bladder neck region, and sutures affixed to the sling edges pass behind the pubic symphysis and through the abdominal fascia to anchor it in place.
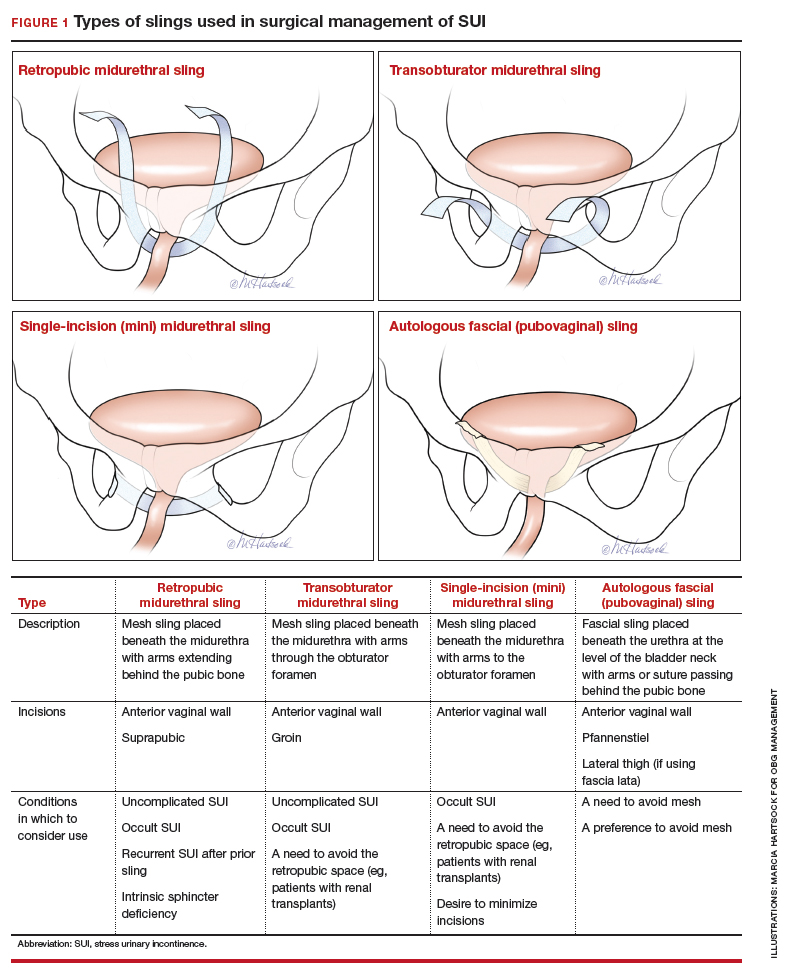
Choose a sling based on the clinical situation and patient goals
Consider the unique features of each sling when selecting the proper sling; this should be a shared decision with the patient after thorough counseling. Below, we present 4 clinical cases to exemplify scenarios in which different slings are appropriate, and we review the rationale for each selection.
CASE 1 SUI that interferes with exercise routine
Ms. P. is a 46-year-old (G3P3) active mother. She loves to exercise, but she has been working out less frequently because of embarrassing urinary leakage that occurs with activity. She has tried pelvic floor exercises and changing her fluid intake habits, but improvements have been minimal with these interventions. On evaluation, she has a positive cough stress test with a recently emptied bladder and a normal postvoid residual volume.
What type of sling would be best?
Because this patient is young, active, and has significant leakage with an empty bladder, a sling with good long-term treatment success is likely to provide her with the best results (Figure 1). We therefore offered her a retropubic midurethral sling. The retropubic approach is preferred here as it is less likely than the transobturator sling to cause groin/thigh pain, which is an important consideration in this young, active patient.
Further testing is not needed
For women with uncomplicated SUI who demonstrate leakage with stress (coughing, Valsalva stress test) and who have a normal postvoid residual volume, additional testing, such as urodynamic evaluation, is not necessary.12 These patients can be counseled on the range of conservative management options and as well as surgical inventions.
CASE 2 Return of SUI symptoms after transobturator sling placement
Ms. E. is a 70-year-old woman who had a transobturator sling placed 5 years ago. Initially, her SUI symptoms improved after surgery. Recently, however, she noticed a return of her SUI, which she finds bothersome and limiting to her quality of life.
How would you manage this patient?
While midurethral slings are highly effective, there are instances in which patients will have symptom recurrence. For women who already have a midurethral sling, consider the following important questions.
Is this truly recurrent SUI, or is it a new process?
Like any reconstructive procedure, midurethral sling success rates decline over time and recurrent SUI can develop.7 However, it also is possible for urge urinary incontinence to develop as a new process, and it is important to distinguish which type of urinary incontinence your patient has prior to counseling about treatment options.
To further evaluate patients with recurrent incontinence and a prior sling, we recommend urodynamic studies with cystoscopy (in addition to a detailed history and physical exam). This not only helps rule out other forms of incontinence, such as overactive bladder, but also evaluates for possible mesh erosion into the urethra or bladder, which can cause irritative voiding symptoms and incontinence.
Continue to: What type of sling did the patient have initially...
What type of sling did the patient have initially, and how does this impact a repeat procedure?
Regardless of the initial sling type used, repeat midurethral sling procedures have a significantly lower cure rate than primary midurethral sling procedures.13 Retropubic slings are more effective than transobturator slings for patients with recurrent SUI who have failed a prior sling. When a patient presents with recurrent SUI after a prior transobturator sling, the best option for a repeat procedure is usually a retropubic sling, as it achieves higher objective and subjective cure rates.13,14 (See FIGURE 2 for a comparison of retropubic and transobturator slings.)
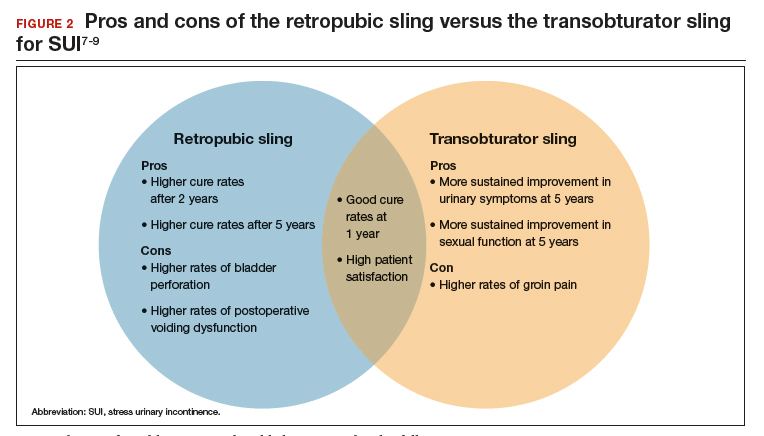
Should I remove the old sling prior to placing a new one?
While it is recommended to remove the vaginal portion of the sling if the patient has a mesh exposure or is experiencing other symptoms, such as pain or bleeding, removal of the old sling is not necessarily indicated prior to (or during) a repeat incontinence procedure.15,16 Removing the sling, removing a portion of the sling, or leaving the sling in situ are all reasonable options.
CASE 3 Treated SUI has mesh exposure
Ms. R. is a 60-year-old woman with a history of SUI that was previously managed with a retropubic midurethral sling placed at an outside hospital. She is a smoker and has developed a vaginal mesh exposure. Although she would like the mesh removed, she does not want her incontinence to come back. She tells you that she does not think she would be able to quit smoking.
What would be a reasonable next option for Ms. R.?
While complications from a midurethral sling are rare, mesh exposures occur in approximately 2% of patients, and urinary retention requiring release of the sling occurs in about 1% of patients.3,6 It often helps to clarify for patients that the US Food and Drug Administration public health advisories on the use of transvaginal mesh have been directed specifically toward the use of transvaginal mesh for the treatment of pelvic organ prolapse (POP), not the use of mesh for midurethral slings for SUI or transabdominal mesh for POP.10,17
When considering use of a mesh sling, a thorough discussion of the potential risks, as well as the benefits and alternatives, is imperative. Patients must personally balance the probability of benefit with the potential risk of complications, and while physicians can help outline the benefits and risks through shared decision-making, ultimately it is the patient who should make this decision.
Certain patient populations may be at higher risk for mesh complications18 (See "Risk factors for mesh-related complications," below). These complications are managed in various ways (FIGURE 3). Patients who have experienced mesh complications previously are typically not good candidates for a repeat mesh sling, particularly when the risk factor for complications cannot be modified.
• Smoking
• Poorly controlled diabetes
• Decreased estrogen status
• Chronic steroid use
• Prior urethral surgery (urethral diverticulum, urethroplasty)
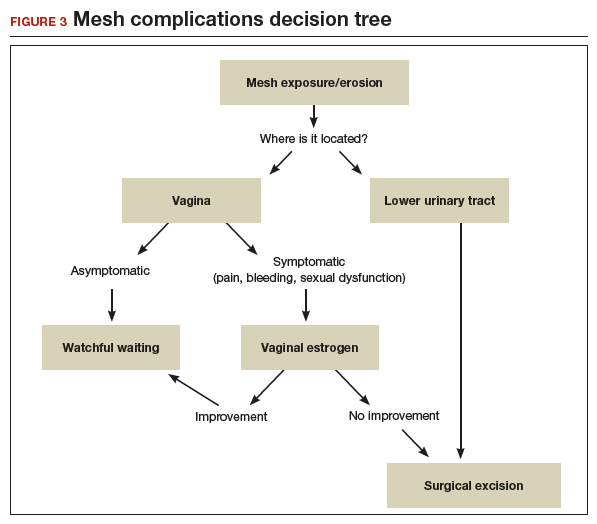
A mesh sling alternative
The most effective way to manage SUI in patients who are not good candidates for a mesh sling is to consider employing a sling that uses the patient’s own tissue.19-21 Common approaches include harvesting a graft of rectus fascia through a Pfannenstiel skin incision or using fascia lata from the patient’s iliotibial band in the lateral thigh. Autologous slings are safe and effective, and even after a mesh sling has failed, autologous slings have an almost 70% cure rate for SUI.20,21
Continue to: Timing of mesh removal and placement of an autologous fascial sling...
Timing of mesh removal and placement of an autologous fascial sling
Either concomitant or delayed placement of a pubovaginal sling is acceptable when removing mesh, though this should be a joint decision with the patient after counseling. If the risk for surgical complications is modifiable (for example, poorly controlled diabetes that could be improved with blood glucose control), it may be advisable to delay the fascial sling until the risk factors have been addressed. Similarly, if the reason for mesh removal is pain, it may be advisable to remove the mesh prior to placing a new sling to ensure that the pain resolves completely. Otherwise, if pain persists, it can be unclear whether the new sling is contributing to the pain, and this may lead to difficulties treating pain or incontinence in the future.
In this patient, who was an active smoker, we excised the exposed mesh and concomitantly placed an autologous fascial sling utilizing rectus fascia. This maintained continence without introducing mesh in a high-risk patient.
CASE 4 POP and occult SUI
Ms. B. is a 79-year-old woman with stage 3 POP planned for surgical repair. While she does not report urinary leakage, preoperative urodynamic testing revealed occult SUI with reduction of her prolapse. Her priorities are to avoid needing another surgery and to limit the chances of postoperative leakage, but she is nervous about her postoperative recovery and wants to avoid pain.
What approach would be appropriate?
Consider a mini sling for this patient
The single-incision (mini) sling is an option to consider for patients with mild incontinence or for those without evidence of intrinsic sphincter deficiency. It is also a good option for those who want to avoid the additional incisions required for full-length slings.
While currently there is not sufficient evidence to clearly state if single-incision slings are equivalent to other slings, recent studies show that single-incision slings appear to be safe and effective in the short term, with possibly fewer complications than traditional transobturator slings.22-24 As patients are often concerned about the potential for groin pain with a transobturator sling, a single-incision sling is an acceptable alternative that avoids groin incisions and also avoids the retropubic space.
Patient counseling is crucial
Regardless of the route, sling procedures are highly effective and safe for treating women with SUI.3 Understanding the characteristics of each type of sling and the distinct surgical approaches enables informed counseling for patients who are navigating the treatment options for SUI.
- Wu JM, Matthews CA, Conover MM, et al. Lifetime risk of stress urinary incontinence or pelvic organ prolapse surgery. Obstet Gynecol. 2014;123:1201-1206.
- Jonsson Funk M, Levin PJ, Wu JM. Trends in the surgical management of stress urinary incontinence. Obstet Gynecol. 2012;119:845-851.
- Ford AA, Rogerson L, Cody JD, et al. Mid-urethral sling operations for stress urinary incontinence in women. Cochrane Database Syst Rev. 2017;7:CD006375.
- Dumoulin C, Hay-Smith J, Habee-Seguin GM, et al. Pelvic floor muscle training versus no treatment, or inactive control treatments, for urinary incontinence in women: a short version Cochrane systematic review with meta-analysis. Neurourol Urodyn. 2015;34:300-308.
- Cox A, Herschorn S, Lee L. Surgical management of female SUI: is there a gold standard? Nat Rev Urol. 2013;10:78-89.
- Schimpf MO, Rahn DD, Wheeler TL, et al; Society of Gynecologic Surgeons Systematic Review Group. Sling surgery for stress urinary incontinence in women: a systematic review and metaanalysis. Am J Obstet Gynecol. 2014;211:71.e1-71.e27.
- Kenton K, Stoddard AM, Zyczynski H, et al. 5-year longitudinal followup after retropubic and transobturator mid urethral slings. J Urol. 2015;193:203-210.
- Richter HE, Albo ME, Zyczynski HM, et al; Urinary Incontinence Treatment Network. Retropubic versus transobturator midurethral slings for stress incontinence. N Engl J Med. 2010;362:2066-2076.
- Albo ME, Litman HJ, Richter HE, et al; Urinary Incontinence Treatment Network. Treatment success of retropubic and transobturator midurethral slings at 24-months. J Urol. 2012;188:2281-2287.
- US Food and Drug Administration. Urogynecologic surgical mesh: update on the safety and effectiveness of transvaginal placement for pelvic organ prolapse. July 2011;1-15. https://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM262760.pdf. Accessed September 16, 2019.
- Nilsson CG, Palva K, Aarnio R, et al. Seventeen years’ follow up of the tension-free vaginal tape procedure for female stress urinary incontinence. Int Urogynecol J. 2013;24:1265-1269.
- Nager CW, Brubaker L, Litman HJ, et al; Urinary Incontinence Treatment Network. A randomized trial of urodynamic testing before stress-incontinence surgery. N Engl J Med. 2012;366:1987-1997.
- Stav K, Dwyer PL, Rosamilia A, et al. Repeat synthetic mid urethral sling procedure for women with recurrent stress urinary incontinence. J Urol. 2010;183:241-246.
- Kim A, Kim MS, Park YJ, et al. Retropubic versus transobturator mid urethral slings in patients at high risk for recurrent stress incontinence: a systematic review and meta-analysis. J Urol. 2019;202:132-142.
- Kavanagh A, Sanaee M, Carison KV, et al. Management of patients with stress urinary incontinence after failed midurethral sling. Can Urol Assoc J. 2017;11(6 suppl 2):S143-S146.
- Steele SE, Hill AJ, Unger CA. Concurrent midurethral sling excision or lysis at the time of repeat sling for treatment of recurrent or persistent stress urinary incontinence. Int Urogynecol J. 2018;29:285-290.
- US Food and Drug Administration. Urogynecologic surgical mesh implants. https://www.fda.gov/medicaldevices/productsandmedicalprocedures/implantsandprosthetics/urogynsurgicalmesh/. Content current as of July 10, 2019. Accessed September 16, 2019.
- Kokanali MK, Doganay M, Aksakal O, et al. Risk factors for mesh erosion after vaginal sling procedures for urinary incontinence. Eur J Obstet Gynecol Reprod Biol. 2014;177:146-150.
- Nikolopoulos KI, Betschart C, Doumouchtsis SK. The surgical management of recurrent stress urinary incontinence: a systematic review. Acta Obstet Gynecol Scand. 2015;94:568-576.
- Milose JC, Sharp KM, He C, et al. Success of autologous pubovaginal sling after failed synthetic mid urethral sling. J Urol. 2015;193:916-920.
- Albo ME, Richter HE, Brubaker L, et al; Urinary Incontinence Treatment Network. Burch colposuspension versus fascial sling to reduce urinary stress incontinence. N Engl J Med. 2007;356:2143-2155.
- Imamura M, Hudson J, Wallace SA, et al. Surgical interventions for women with stress urinary incontinence: systematic review and network meta-analysis of randomised controlled trials. BMJ. 2019;365:I1842.
- Jiao B, Lai S, Xu X, et al. A systematic review and meta-analysis of single-incision mini-slings (MiniArc) versus transobturator mid-urethral slings in surgical management of female stress urinary incontinence. Medicine (Baltimore). 2018;97:e0283.
- Sun Z, Wang X, Lang J, et al. Comparison of outcomes between single-incision sling and transobturator sling for treating stress urinary incontinence: a 10-year prospective study. Neurourol Urodyn. 2019;38:1852-1858.

Dr. Ringel is Clinical Fellow, Section of Female Pelvic Medicine and Reconstructive Surgery, MedStar Georgetown/Washington Hospital Center, Washington, DC.

Dr. Richter is Assistant Professor of Urology and Obstetrics and Gynecology, Georgetown University School of Medicine, Section of Female Pelvic Medicine and Reconstructive Surgery, MedStar Washington Hospital Center, Washington, DC.
The authors report no financial relationships relevant to this article.
Dr. Ringel is Clinical Fellow, Section of Female Pelvic Medicine and Reconstructive Surgery, MedStar Georgetown/Washington Hospital Center, Washington, DC.
Dr. Richter is Assistant Professor of Urology and Obstetrics and Gynecology, Georgetown University School of Medicine, Section of Female Pelvic Medicine and Reconstructive Surgery, MedStar Washington Hospital Center, Washington, DC.
The authors report no financial relationships relevant to this article.
Dr. Ringel is Clinical Fellow, Section of Female Pelvic Medicine and Reconstructive Surgery, MedStar Georgetown/Washington Hospital Center, Washington, DC.
Dr. Richter is Assistant Professor of Urology and Obstetrics and Gynecology, Georgetown University School of Medicine, Section of Female Pelvic Medicine and Reconstructive Surgery, MedStar Washington Hospital Center, Washington, DC.
The authors report no financial relationships relevant to this article.
Urinary incontinence affects approximately 50% of women, with up to 80% of these women experiencing stress urinary incontinence (SUI) at some point in their lives.1-3 While conservative measures can offer some improvement in symptoms, the mainstay of treatment for SUI is surgical intervention.4,5 The lifetime risk of undergoing surgery for SUI is 13.6%, and surgery leads to a major improvement in quality of life and productivity.1,6
Types of slings used for SUI
Sling procedures are the most commonly used surgical approach for the treatment of SUI. Two types of urethral slings are used: the midurethral sling and the autologous fascial (pubovaginal) sling. The midurethral sling, which is the most frequently used sling today, can be further characterized as the retropubic sling, the transobturator sling, and the mini sling (FIGURE 1).
Retropubic sling
A retropubic sling is a midurethral mesh sling that is placed beneath the urethra at the midpoint between the urethral meatus and the bladder neck. The arms of the sling extend behind the pubic symphysis, providing a hammock-like support that helps prevent leakage with increased abdominal pressures. The retropubic sling is the most commonly used type of sling. For women presenting with uncomplicated SUI who desire surgical correction, it often is the best choice for providing long-term treatment success.7
Transobturator sling
A transobturator sling is a midurethral mesh sling that is placed beneath the urethra as described above, but the arms of the sling extend outward through the obturator foramen and into the groin. This enables support of the midurethra, but this sling is less likely to result in such complications as bladder perforation or postoperative urinary retention. Transobturator slings also are associated with lower rates of voiding dysfunction and urinary urgency than retropubic slings.7-9 However, transobturator slings have higher rates of groin pain, and they are less effective in maintaining long-term cure of SUI.7
First introduced in 1996, the midurethral sling quickly grew in popularity for the treatment of SUI because of its high success rates and its minimally invasive approach.10 Both retropubic and transobturator slings are safe, extensively researched surgical approaches for the management of SUI.3 Midurethral slings have a very high rate of incontinence cure (80%–90%) and extremely high patient satisfaction rates (85%–90%), as even patients without complete cure report meaningful symptomatic improvement.7,8,11
Single-incision (mini) sling
A single-incision sling is a midurethral mesh sling that is designed to be shorter in length than standard midurethral slings. The placed sling lies under the midurethra and extends toward the superior edge of the obturator foramen but does not penetrate it. The sling is held in place by small pledgets on either side of the mesh hammock that anchor it in place to the obturator internus muscular fascia. Because this “mini” sling was introduced in 2006, fewer long-term data are available for this sling than for standard midurethral slings.
Continue to: Autologous (fascial) sling...
Autologous (fascial) sling
An autologous sling is a retropubic sling made from the patient’s own fascia; it is harvested from either the fascia lata of the lateral thigh or the rectus fascia of the abdomen. The sling is placed beneath the urethra in the bladder neck region, and sutures affixed to the sling edges pass behind the pubic symphysis and through the abdominal fascia to anchor it in place.
Choose a sling based on the clinical situation and patient goals
Consider the unique features of each sling when selecting the proper sling; this should be a shared decision with the patient after thorough counseling. Below, we present 4 clinical cases to exemplify scenarios in which different slings are appropriate, and we review the rationale for each selection.
CASE 1 SUI that interferes with exercise routine
Ms. P. is a 46-year-old (G3P3) active mother. She loves to exercise, but she has been working out less frequently because of embarrassing urinary leakage that occurs with activity. She has tried pelvic floor exercises and changing her fluid intake habits, but improvements have been minimal with these interventions. On evaluation, she has a positive cough stress test with a recently emptied bladder and a normal postvoid residual volume.
What type of sling would be best?
Because this patient is young, active, and has significant leakage with an empty bladder, a sling with good long-term treatment success is likely to provide her with the best results (Figure 1). We therefore offered her a retropubic midurethral sling. The retropubic approach is preferred here as it is less likely than the transobturator sling to cause groin/thigh pain, which is an important consideration in this young, active patient.
Further testing is not needed
For women with uncomplicated SUI who demonstrate leakage with stress (coughing, Valsalva stress test) and who have a normal postvoid residual volume, additional testing, such as urodynamic evaluation, is not necessary.12 These patients can be counseled on the range of conservative management options and as well as surgical inventions.
CASE 2 Return of SUI symptoms after transobturator sling placement
Ms. E. is a 70-year-old woman who had a transobturator sling placed 5 years ago. Initially, her SUI symptoms improved after surgery. Recently, however, she noticed a return of her SUI, which she finds bothersome and limiting to her quality of life.
How would you manage this patient?
While midurethral slings are highly effective, there are instances in which patients will have symptom recurrence. For women who already have a midurethral sling, consider the following important questions.
Is this truly recurrent SUI, or is it a new process?
Like any reconstructive procedure, midurethral sling success rates decline over time and recurrent SUI can develop.7 However, it also is possible for urge urinary incontinence to develop as a new process, and it is important to distinguish which type of urinary incontinence your patient has prior to counseling about treatment options.
To further evaluate patients with recurrent incontinence and a prior sling, we recommend urodynamic studies with cystoscopy (in addition to a detailed history and physical exam). This not only helps rule out other forms of incontinence, such as overactive bladder, but also evaluates for possible mesh erosion into the urethra or bladder, which can cause irritative voiding symptoms and incontinence.
Continue to: What type of sling did the patient have initially...
What type of sling did the patient have initially, and how does this impact a repeat procedure?
Regardless of the initial sling type used, repeat midurethral sling procedures have a significantly lower cure rate than primary midurethral sling procedures.13 Retropubic slings are more effective than transobturator slings for patients with recurrent SUI who have failed a prior sling. When a patient presents with recurrent SUI after a prior transobturator sling, the best option for a repeat procedure is usually a retropubic sling, as it achieves higher objective and subjective cure rates.13,14 (See FIGURE 2 for a comparison of retropubic and transobturator slings.)
Should I remove the old sling prior to placing a new one?
While it is recommended to remove the vaginal portion of the sling if the patient has a mesh exposure or is experiencing other symptoms, such as pain or bleeding, removal of the old sling is not necessarily indicated prior to (or during) a repeat incontinence procedure.15,16 Removing the sling, removing a portion of the sling, or leaving the sling in situ are all reasonable options.
CASE 3 Treated SUI has mesh exposure
Ms. R. is a 60-year-old woman with a history of SUI that was previously managed with a retropubic midurethral sling placed at an outside hospital. She is a smoker and has developed a vaginal mesh exposure. Although she would like the mesh removed, she does not want her incontinence to come back. She tells you that she does not think she would be able to quit smoking.
What would be a reasonable next option for Ms. R.?
While complications from a midurethral sling are rare, mesh exposures occur in approximately 2% of patients, and urinary retention requiring release of the sling occurs in about 1% of patients.3,6 It often helps to clarify for patients that the US Food and Drug Administration public health advisories on the use of transvaginal mesh have been directed specifically toward the use of transvaginal mesh for the treatment of pelvic organ prolapse (POP), not the use of mesh for midurethral slings for SUI or transabdominal mesh for POP.10,17
When considering use of a mesh sling, a thorough discussion of the potential risks, as well as the benefits and alternatives, is imperative. Patients must personally balance the probability of benefit with the potential risk of complications, and while physicians can help outline the benefits and risks through shared decision-making, ultimately it is the patient who should make this decision.
Certain patient populations may be at higher risk for mesh complications18 (See "Risk factors for mesh-related complications," below). These complications are managed in various ways (FIGURE 3). Patients who have experienced mesh complications previously are typically not good candidates for a repeat mesh sling, particularly when the risk factor for complications cannot be modified.
• Smoking
• Poorly controlled diabetes
• Decreased estrogen status
• Chronic steroid use
• Prior urethral surgery (urethral diverticulum, urethroplasty)
A mesh sling alternative
The most effective way to manage SUI in patients who are not good candidates for a mesh sling is to consider employing a sling that uses the patient’s own tissue.19-21 Common approaches include harvesting a graft of rectus fascia through a Pfannenstiel skin incision or using fascia lata from the patient’s iliotibial band in the lateral thigh. Autologous slings are safe and effective, and even after a mesh sling has failed, autologous slings have an almost 70% cure rate for SUI.20,21
Continue to: Timing of mesh removal and placement of an autologous fascial sling...
Timing of mesh removal and placement of an autologous fascial sling
Either concomitant or delayed placement of a pubovaginal sling is acceptable when removing mesh, though this should be a joint decision with the patient after counseling. If the risk for surgical complications is modifiable (for example, poorly controlled diabetes that could be improved with blood glucose control), it may be advisable to delay the fascial sling until the risk factors have been addressed. Similarly, if the reason for mesh removal is pain, it may be advisable to remove the mesh prior to placing a new sling to ensure that the pain resolves completely. Otherwise, if pain persists, it can be unclear whether the new sling is contributing to the pain, and this may lead to difficulties treating pain or incontinence in the future.
In this patient, who was an active smoker, we excised the exposed mesh and concomitantly placed an autologous fascial sling utilizing rectus fascia. This maintained continence without introducing mesh in a high-risk patient.
CASE 4 POP and occult SUI
Ms. B. is a 79-year-old woman with stage 3 POP planned for surgical repair. While she does not report urinary leakage, preoperative urodynamic testing revealed occult SUI with reduction of her prolapse. Her priorities are to avoid needing another surgery and to limit the chances of postoperative leakage, but she is nervous about her postoperative recovery and wants to avoid pain.
What approach would be appropriate?
Consider a mini sling for this patient
The single-incision (mini) sling is an option to consider for patients with mild incontinence or for those without evidence of intrinsic sphincter deficiency. It is also a good option for those who want to avoid the additional incisions required for full-length slings.
While currently there is not sufficient evidence to clearly state if single-incision slings are equivalent to other slings, recent studies show that single-incision slings appear to be safe and effective in the short term, with possibly fewer complications than traditional transobturator slings.22-24 As patients are often concerned about the potential for groin pain with a transobturator sling, a single-incision sling is an acceptable alternative that avoids groin incisions and also avoids the retropubic space.
Patient counseling is crucial
Regardless of the route, sling procedures are highly effective and safe for treating women with SUI.3 Understanding the characteristics of each type of sling and the distinct surgical approaches enables informed counseling for patients who are navigating the treatment options for SUI.
Urinary incontinence affects approximately 50% of women, with up to 80% of these women experiencing stress urinary incontinence (SUI) at some point in their lives.1-3 While conservative measures can offer some improvement in symptoms, the mainstay of treatment for SUI is surgical intervention.4,5 The lifetime risk of undergoing surgery for SUI is 13.6%, and surgery leads to a major improvement in quality of life and productivity.1,6
Types of slings used for SUI
Sling procedures are the most commonly used surgical approach for the treatment of SUI. Two types of urethral slings are used: the midurethral sling and the autologous fascial (pubovaginal) sling. The midurethral sling, which is the most frequently used sling today, can be further characterized as the retropubic sling, the transobturator sling, and the mini sling (FIGURE 1).
Retropubic sling
A retropubic sling is a midurethral mesh sling that is placed beneath the urethra at the midpoint between the urethral meatus and the bladder neck. The arms of the sling extend behind the pubic symphysis, providing a hammock-like support that helps prevent leakage with increased abdominal pressures. The retropubic sling is the most commonly used type of sling. For women presenting with uncomplicated SUI who desire surgical correction, it often is the best choice for providing long-term treatment success.7
Transobturator sling
A transobturator sling is a midurethral mesh sling that is placed beneath the urethra as described above, but the arms of the sling extend outward through the obturator foramen and into the groin. This enables support of the midurethra, but this sling is less likely to result in such complications as bladder perforation or postoperative urinary retention. Transobturator slings also are associated with lower rates of voiding dysfunction and urinary urgency than retropubic slings.7-9 However, transobturator slings have higher rates of groin pain, and they are less effective in maintaining long-term cure of SUI.7
First introduced in 1996, the midurethral sling quickly grew in popularity for the treatment of SUI because of its high success rates and its minimally invasive approach.10 Both retropubic and transobturator slings are safe, extensively researched surgical approaches for the management of SUI.3 Midurethral slings have a very high rate of incontinence cure (80%–90%) and extremely high patient satisfaction rates (85%–90%), as even patients without complete cure report meaningful symptomatic improvement.7,8,11
Single-incision (mini) sling
A single-incision sling is a midurethral mesh sling that is designed to be shorter in length than standard midurethral slings. The placed sling lies under the midurethra and extends toward the superior edge of the obturator foramen but does not penetrate it. The sling is held in place by small pledgets on either side of the mesh hammock that anchor it in place to the obturator internus muscular fascia. Because this “mini” sling was introduced in 2006, fewer long-term data are available for this sling than for standard midurethral slings.
Continue to: Autologous (fascial) sling...
Autologous (fascial) sling
An autologous sling is a retropubic sling made from the patient’s own fascia; it is harvested from either the fascia lata of the lateral thigh or the rectus fascia of the abdomen. The sling is placed beneath the urethra in the bladder neck region, and sutures affixed to the sling edges pass behind the pubic symphysis and through the abdominal fascia to anchor it in place.
Choose a sling based on the clinical situation and patient goals
Consider the unique features of each sling when selecting the proper sling; this should be a shared decision with the patient after thorough counseling. Below, we present 4 clinical cases to exemplify scenarios in which different slings are appropriate, and we review the rationale for each selection.
CASE 1 SUI that interferes with exercise routine
Ms. P. is a 46-year-old (G3P3) active mother. She loves to exercise, but she has been working out less frequently because of embarrassing urinary leakage that occurs with activity. She has tried pelvic floor exercises and changing her fluid intake habits, but improvements have been minimal with these interventions. On evaluation, she has a positive cough stress test with a recently emptied bladder and a normal postvoid residual volume.
What type of sling would be best?
Because this patient is young, active, and has significant leakage with an empty bladder, a sling with good long-term treatment success is likely to provide her with the best results (Figure 1). We therefore offered her a retropubic midurethral sling. The retropubic approach is preferred here as it is less likely than the transobturator sling to cause groin/thigh pain, which is an important consideration in this young, active patient.
Further testing is not needed
For women with uncomplicated SUI who demonstrate leakage with stress (coughing, Valsalva stress test) and who have a normal postvoid residual volume, additional testing, such as urodynamic evaluation, is not necessary.12 These patients can be counseled on the range of conservative management options and as well as surgical inventions.
CASE 2 Return of SUI symptoms after transobturator sling placement
Ms. E. is a 70-year-old woman who had a transobturator sling placed 5 years ago. Initially, her SUI symptoms improved after surgery. Recently, however, she noticed a return of her SUI, which she finds bothersome and limiting to her quality of life.
How would you manage this patient?
While midurethral slings are highly effective, there are instances in which patients will have symptom recurrence. For women who already have a midurethral sling, consider the following important questions.
Is this truly recurrent SUI, or is it a new process?
Like any reconstructive procedure, midurethral sling success rates decline over time and recurrent SUI can develop.7 However, it also is possible for urge urinary incontinence to develop as a new process, and it is important to distinguish which type of urinary incontinence your patient has prior to counseling about treatment options.
To further evaluate patients with recurrent incontinence and a prior sling, we recommend urodynamic studies with cystoscopy (in addition to a detailed history and physical exam). This not only helps rule out other forms of incontinence, such as overactive bladder, but also evaluates for possible mesh erosion into the urethra or bladder, which can cause irritative voiding symptoms and incontinence.
Continue to: What type of sling did the patient have initially...
What type of sling did the patient have initially, and how does this impact a repeat procedure?
Regardless of the initial sling type used, repeat midurethral sling procedures have a significantly lower cure rate than primary midurethral sling procedures.13 Retropubic slings are more effective than transobturator slings for patients with recurrent SUI who have failed a prior sling. When a patient presents with recurrent SUI after a prior transobturator sling, the best option for a repeat procedure is usually a retropubic sling, as it achieves higher objective and subjective cure rates.13,14 (See FIGURE 2 for a comparison of retropubic and transobturator slings.)
Should I remove the old sling prior to placing a new one?
While it is recommended to remove the vaginal portion of the sling if the patient has a mesh exposure or is experiencing other symptoms, such as pain or bleeding, removal of the old sling is not necessarily indicated prior to (or during) a repeat incontinence procedure.15,16 Removing the sling, removing a portion of the sling, or leaving the sling in situ are all reasonable options.
CASE 3 Treated SUI has mesh exposure
Ms. R. is a 60-year-old woman with a history of SUI that was previously managed with a retropubic midurethral sling placed at an outside hospital. She is a smoker and has developed a vaginal mesh exposure. Although she would like the mesh removed, she does not want her incontinence to come back. She tells you that she does not think she would be able to quit smoking.
What would be a reasonable next option for Ms. R.?
While complications from a midurethral sling are rare, mesh exposures occur in approximately 2% of patients, and urinary retention requiring release of the sling occurs in about 1% of patients.3,6 It often helps to clarify for patients that the US Food and Drug Administration public health advisories on the use of transvaginal mesh have been directed specifically toward the use of transvaginal mesh for the treatment of pelvic organ prolapse (POP), not the use of mesh for midurethral slings for SUI or transabdominal mesh for POP.10,17
When considering use of a mesh sling, a thorough discussion of the potential risks, as well as the benefits and alternatives, is imperative. Patients must personally balance the probability of benefit with the potential risk of complications, and while physicians can help outline the benefits and risks through shared decision-making, ultimately it is the patient who should make this decision.
Certain patient populations may be at higher risk for mesh complications18 (See "Risk factors for mesh-related complications," below). These complications are managed in various ways (FIGURE 3). Patients who have experienced mesh complications previously are typically not good candidates for a repeat mesh sling, particularly when the risk factor for complications cannot be modified.
• Smoking
• Poorly controlled diabetes
• Decreased estrogen status
• Chronic steroid use
• Prior urethral surgery (urethral diverticulum, urethroplasty)
A mesh sling alternative
The most effective way to manage SUI in patients who are not good candidates for a mesh sling is to consider employing a sling that uses the patient’s own tissue.19-21 Common approaches include harvesting a graft of rectus fascia through a Pfannenstiel skin incision or using fascia lata from the patient’s iliotibial band in the lateral thigh. Autologous slings are safe and effective, and even after a mesh sling has failed, autologous slings have an almost 70% cure rate for SUI.20,21
Continue to: Timing of mesh removal and placement of an autologous fascial sling...
Timing of mesh removal and placement of an autologous fascial sling
Either concomitant or delayed placement of a pubovaginal sling is acceptable when removing mesh, though this should be a joint decision with the patient after counseling. If the risk for surgical complications is modifiable (for example, poorly controlled diabetes that could be improved with blood glucose control), it may be advisable to delay the fascial sling until the risk factors have been addressed. Similarly, if the reason for mesh removal is pain, it may be advisable to remove the mesh prior to placing a new sling to ensure that the pain resolves completely. Otherwise, if pain persists, it can be unclear whether the new sling is contributing to the pain, and this may lead to difficulties treating pain or incontinence in the future.
In this patient, who was an active smoker, we excised the exposed mesh and concomitantly placed an autologous fascial sling utilizing rectus fascia. This maintained continence without introducing mesh in a high-risk patient.
CASE 4 POP and occult SUI
Ms. B. is a 79-year-old woman with stage 3 POP planned for surgical repair. While she does not report urinary leakage, preoperative urodynamic testing revealed occult SUI with reduction of her prolapse. Her priorities are to avoid needing another surgery and to limit the chances of postoperative leakage, but she is nervous about her postoperative recovery and wants to avoid pain.
What approach would be appropriate?
Consider a mini sling for this patient
The single-incision (mini) sling is an option to consider for patients with mild incontinence or for those without evidence of intrinsic sphincter deficiency. It is also a good option for those who want to avoid the additional incisions required for full-length slings.
While currently there is not sufficient evidence to clearly state if single-incision slings are equivalent to other slings, recent studies show that single-incision slings appear to be safe and effective in the short term, with possibly fewer complications than traditional transobturator slings.22-24 As patients are often concerned about the potential for groin pain with a transobturator sling, a single-incision sling is an acceptable alternative that avoids groin incisions and also avoids the retropubic space.
Patient counseling is crucial
Regardless of the route, sling procedures are highly effective and safe for treating women with SUI.3 Understanding the characteristics of each type of sling and the distinct surgical approaches enables informed counseling for patients who are navigating the treatment options for SUI.
- Wu JM, Matthews CA, Conover MM, et al. Lifetime risk of stress urinary incontinence or pelvic organ prolapse surgery. Obstet Gynecol. 2014;123:1201-1206.
- Jonsson Funk M, Levin PJ, Wu JM. Trends in the surgical management of stress urinary incontinence. Obstet Gynecol. 2012;119:845-851.
- Ford AA, Rogerson L, Cody JD, et al. Mid-urethral sling operations for stress urinary incontinence in women. Cochrane Database Syst Rev. 2017;7:CD006375.
- Dumoulin C, Hay-Smith J, Habee-Seguin GM, et al. Pelvic floor muscle training versus no treatment, or inactive control treatments, for urinary incontinence in women: a short version Cochrane systematic review with meta-analysis. Neurourol Urodyn. 2015;34:300-308.
- Cox A, Herschorn S, Lee L. Surgical management of female SUI: is there a gold standard? Nat Rev Urol. 2013;10:78-89.
- Schimpf MO, Rahn DD, Wheeler TL, et al; Society of Gynecologic Surgeons Systematic Review Group. Sling surgery for stress urinary incontinence in women: a systematic review and metaanalysis. Am J Obstet Gynecol. 2014;211:71.e1-71.e27.
- Kenton K, Stoddard AM, Zyczynski H, et al. 5-year longitudinal followup after retropubic and transobturator mid urethral slings. J Urol. 2015;193:203-210.
- Richter HE, Albo ME, Zyczynski HM, et al; Urinary Incontinence Treatment Network. Retropubic versus transobturator midurethral slings for stress incontinence. N Engl J Med. 2010;362:2066-2076.
- Albo ME, Litman HJ, Richter HE, et al; Urinary Incontinence Treatment Network. Treatment success of retropubic and transobturator midurethral slings at 24-months. J Urol. 2012;188:2281-2287.
- US Food and Drug Administration. Urogynecologic surgical mesh: update on the safety and effectiveness of transvaginal placement for pelvic organ prolapse. July 2011;1-15. https://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM262760.pdf. Accessed September 16, 2019.
- Nilsson CG, Palva K, Aarnio R, et al. Seventeen years’ follow up of the tension-free vaginal tape procedure for female stress urinary incontinence. Int Urogynecol J. 2013;24:1265-1269.
- Nager CW, Brubaker L, Litman HJ, et al; Urinary Incontinence Treatment Network. A randomized trial of urodynamic testing before stress-incontinence surgery. N Engl J Med. 2012;366:1987-1997.
- Stav K, Dwyer PL, Rosamilia A, et al. Repeat synthetic mid urethral sling procedure for women with recurrent stress urinary incontinence. J Urol. 2010;183:241-246.
- Kim A, Kim MS, Park YJ, et al. Retropubic versus transobturator mid urethral slings in patients at high risk for recurrent stress incontinence: a systematic review and meta-analysis. J Urol. 2019;202:132-142.
- Kavanagh A, Sanaee M, Carison KV, et al. Management of patients with stress urinary incontinence after failed midurethral sling. Can Urol Assoc J. 2017;11(6 suppl 2):S143-S146.
- Steele SE, Hill AJ, Unger CA. Concurrent midurethral sling excision or lysis at the time of repeat sling for treatment of recurrent or persistent stress urinary incontinence. Int Urogynecol J. 2018;29:285-290.
- US Food and Drug Administration. Urogynecologic surgical mesh implants. https://www.fda.gov/medicaldevices/productsandmedicalprocedures/implantsandprosthetics/urogynsurgicalmesh/. Content current as of July 10, 2019. Accessed September 16, 2019.
- Kokanali MK, Doganay M, Aksakal O, et al. Risk factors for mesh erosion after vaginal sling procedures for urinary incontinence. Eur J Obstet Gynecol Reprod Biol. 2014;177:146-150.
- Nikolopoulos KI, Betschart C, Doumouchtsis SK. The surgical management of recurrent stress urinary incontinence: a systematic review. Acta Obstet Gynecol Scand. 2015;94:568-576.
- Milose JC, Sharp KM, He C, et al. Success of autologous pubovaginal sling after failed synthetic mid urethral sling. J Urol. 2015;193:916-920.
- Albo ME, Richter HE, Brubaker L, et al; Urinary Incontinence Treatment Network. Burch colposuspension versus fascial sling to reduce urinary stress incontinence. N Engl J Med. 2007;356:2143-2155.
- Imamura M, Hudson J, Wallace SA, et al. Surgical interventions for women with stress urinary incontinence: systematic review and network meta-analysis of randomised controlled trials. BMJ. 2019;365:I1842.
- Jiao B, Lai S, Xu X, et al. A systematic review and meta-analysis of single-incision mini-slings (MiniArc) versus transobturator mid-urethral slings in surgical management of female stress urinary incontinence. Medicine (Baltimore). 2018;97:e0283.
- Sun Z, Wang X, Lang J, et al. Comparison of outcomes between single-incision sling and transobturator sling for treating stress urinary incontinence: a 10-year prospective study. Neurourol Urodyn. 2019;38:1852-1858.
- Wu JM, Matthews CA, Conover MM, et al. Lifetime risk of stress urinary incontinence or pelvic organ prolapse surgery. Obstet Gynecol. 2014;123:1201-1206.
- Jonsson Funk M, Levin PJ, Wu JM. Trends in the surgical management of stress urinary incontinence. Obstet Gynecol. 2012;119:845-851.
- Ford AA, Rogerson L, Cody JD, et al. Mid-urethral sling operations for stress urinary incontinence in women. Cochrane Database Syst Rev. 2017;7:CD006375.
- Dumoulin C, Hay-Smith J, Habee-Seguin GM, et al. Pelvic floor muscle training versus no treatment, or inactive control treatments, for urinary incontinence in women: a short version Cochrane systematic review with meta-analysis. Neurourol Urodyn. 2015;34:300-308.
- Cox A, Herschorn S, Lee L. Surgical management of female SUI: is there a gold standard? Nat Rev Urol. 2013;10:78-89.
- Schimpf MO, Rahn DD, Wheeler TL, et al; Society of Gynecologic Surgeons Systematic Review Group. Sling surgery for stress urinary incontinence in women: a systematic review and metaanalysis. Am J Obstet Gynecol. 2014;211:71.e1-71.e27.
- Kenton K, Stoddard AM, Zyczynski H, et al. 5-year longitudinal followup after retropubic and transobturator mid urethral slings. J Urol. 2015;193:203-210.
- Richter HE, Albo ME, Zyczynski HM, et al; Urinary Incontinence Treatment Network. Retropubic versus transobturator midurethral slings for stress incontinence. N Engl J Med. 2010;362:2066-2076.
- Albo ME, Litman HJ, Richter HE, et al; Urinary Incontinence Treatment Network. Treatment success of retropubic and transobturator midurethral slings at 24-months. J Urol. 2012;188:2281-2287.
- US Food and Drug Administration. Urogynecologic surgical mesh: update on the safety and effectiveness of transvaginal placement for pelvic organ prolapse. July 2011;1-15. https://www.fda.gov/downloads/MedicalDevices/Safety/AlertsandNotices/UCM262760.pdf. Accessed September 16, 2019.
- Nilsson CG, Palva K, Aarnio R, et al. Seventeen years’ follow up of the tension-free vaginal tape procedure for female stress urinary incontinence. Int Urogynecol J. 2013;24:1265-1269.
- Nager CW, Brubaker L, Litman HJ, et al; Urinary Incontinence Treatment Network. A randomized trial of urodynamic testing before stress-incontinence surgery. N Engl J Med. 2012;366:1987-1997.
- Stav K, Dwyer PL, Rosamilia A, et al. Repeat synthetic mid urethral sling procedure for women with recurrent stress urinary incontinence. J Urol. 2010;183:241-246.
- Kim A, Kim MS, Park YJ, et al. Retropubic versus transobturator mid urethral slings in patients at high risk for recurrent stress incontinence: a systematic review and meta-analysis. J Urol. 2019;202:132-142.
- Kavanagh A, Sanaee M, Carison KV, et al. Management of patients with stress urinary incontinence after failed midurethral sling. Can Urol Assoc J. 2017;11(6 suppl 2):S143-S146.
- Steele SE, Hill AJ, Unger CA. Concurrent midurethral sling excision or lysis at the time of repeat sling for treatment of recurrent or persistent stress urinary incontinence. Int Urogynecol J. 2018;29:285-290.
- US Food and Drug Administration. Urogynecologic surgical mesh implants. https://www.fda.gov/medicaldevices/productsandmedicalprocedures/implantsandprosthetics/urogynsurgicalmesh/. Content current as of July 10, 2019. Accessed September 16, 2019.
- Kokanali MK, Doganay M, Aksakal O, et al. Risk factors for mesh erosion after vaginal sling procedures for urinary incontinence. Eur J Obstet Gynecol Reprod Biol. 2014;177:146-150.
- Nikolopoulos KI, Betschart C, Doumouchtsis SK. The surgical management of recurrent stress urinary incontinence: a systematic review. Acta Obstet Gynecol Scand. 2015;94:568-576.
- Milose JC, Sharp KM, He C, et al. Success of autologous pubovaginal sling after failed synthetic mid urethral sling. J Urol. 2015;193:916-920.
- Albo ME, Richter HE, Brubaker L, et al; Urinary Incontinence Treatment Network. Burch colposuspension versus fascial sling to reduce urinary stress incontinence. N Engl J Med. 2007;356:2143-2155.
- Imamura M, Hudson J, Wallace SA, et al. Surgical interventions for women with stress urinary incontinence: systematic review and network meta-analysis of randomised controlled trials. BMJ. 2019;365:I1842.
- Jiao B, Lai S, Xu X, et al. A systematic review and meta-analysis of single-incision mini-slings (MiniArc) versus transobturator mid-urethral slings in surgical management of female stress urinary incontinence. Medicine (Baltimore). 2018;97:e0283.
- Sun Z, Wang X, Lang J, et al. Comparison of outcomes between single-incision sling and transobturator sling for treating stress urinary incontinence: a 10-year prospective study. Neurourol Urodyn. 2019;38:1852-1858.
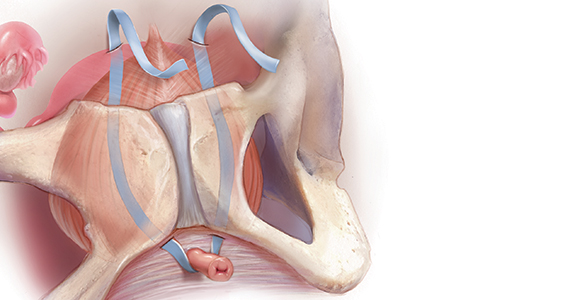
Cosmetic surgery and the secret world of Instagram dolls
They use names and hashtags that connect the work to their provider. So, for example, KathySmithDoll would be a woman who underwent surgery with a Dr. Kathy Smith.
In an era of patient empowerment, these pages – they’re called “Sx pages,” with Sx mimicking the prescriptive “Rx” – form a just-out-of-sight Instagram community. They serve as a cosmetic surgery shopping guide, a best-practices education system, and can also sound the alarm about bad experiences with practitioners. Some presurgery doll pages are more like inspiration pages or mood boards, collecting images of desired shapes.
That way, “other girls doing research can find someone with a similar build to theirs and follow their journey for a glimpse at what they might look like if they got similar procedures,” said Tai Hall, a massage therapist in Maryland. On her Instagram page, she showcases before-and-after body-contouring results; in her Facebook group, she teaches postoperative self-massage and how people can best take care of themselves while healing.
These Instagram pages, she said, “are really big deals.”
The Sx Instagram pages are private and anonymous, to some extent, and follow strict rules to stay that way, particularly since many feature nudity. (As a social media practice, Sx pages are fairly similar to teenager’s private “finsta” friends-only accounts. They are similarly unverified and what they report is unverifiable.) Many of the bios on these pages indicate they won’t allow access to men.
Each Instagram page bio often unveils elaborate details, often including height and weight. The patient – the doll – will list surgery dates and tag her surgeon, recovery house, any postoperative care specialists or private nurses, and her postoperative massage therapist.
Recovery houses, surgery providers, and massage therapists also use the hashtags to promote their services. Some of these are flooded with ads or spam. Some are used by practitioners for education about surgery.
The surgery age
According to the American Society of Plastic Surgeons, more than 1.8 million cosmetic surgeries were performed in the United States in 2018. Breast augmentation and liposuction accounted for about a third of those.
And the number of “cosmetic minimally invasive procedures” – Botox, laser hair removal, soft-tissue fillers, and more – has grown rapidly in the United States. There were fewer than 5 million procedures in 2000. In 2018, there were nearly 16 million. (Almost half of those procedures are Botox treatments.)
Cosmetic procedures are also becoming more popular among people of color. The American Society for Aesthetic Plastic Surgery reports that cosmetic augmentation, like liposuctions and buttocks lifts, increased 56% among African Americans from 2005 to 2013, and is still rising.
As the number of savvy customers grows, doll pages provide a useful glimpse into the less-glamorous side of before and after – the details that people like to overlook, like bruising, drainage, and the often painfully long process of healing after significant surgeries.
Patients become online advertisements for their surgeons. Surgeons develop a reputation on social media for being the best at certain procedures, for delivering a desired look, or for working with certain ethnic groups and body types.
“They’ll cry and upload videos of pain and success and their struggles, or whatever they’re going through, and their surgery sisters help uplift them,” Ms. Hall said.
And there is a lot to talk about, from surgeons to procedures to recovery houses to advice on how to travel with the least hassle from airport security or airline staff when patients are clad in fajas – a kind of postoperative girdle – or other foam paddings.
How we shop for surgeons now
Sx pages can be an effective patient empowerment tool if done honestly and fairly, said Alan Matarasso, MD, a plastic surgeon in New York and the president of the American Society of Plastic Surgeons.
“It makes sense because this is a small group of people,” he said. “Not a lot of doctors do Brazilian butt lifts, but patients need to realize that they are not rating a restaurant.”
Dr. Matarasso encourages prospective patients who rely on Sx pages to research and prepare in other ways as well.
“The standards have to be even greater than if you had a sick gallbladder, because you don’t have to do this,” he said. “This is not like vetting a hotel room. You have to be careful.”
Dr. Matarasso recommends that prospective patients ask to see the surgeon’s best and worst results, or a random case – say, the 37th case they did that year. He suggests that prospective clients visit the American Board of Plastic Surgery websites to do research and that patients query the licensing state and find out what, if any, violations a surgeon may have had. Patients can ask board-certified surgeons their specialty and whether they are certified in it.
Ms. Hall, the massage therapist, warned that patients may see women who heal faster or achieve different results than they might. As is often the case on Instagram, people tend to post fewer of their struggles and more of their highlight reels.
Patients taking care of patients
Sx pages might be even more valuable for patients who plan to travel internationally for their surgery. Many people in the United States do this to save money. Doll pages serve to warn prospective patients about problems that surgeons and hospitals don’t disclose.
After surgery, especially if extensive travel is needed, patients may recuperate at recovery houses for a few days. Procedures like fat transfer to the buttocks leave patients unable to move around or sit; doctors may install drains to help remove fluid after surgery.
In a recovery house, a caretaker can tend to their incisions; help with bathing, food, and pain medications; and even perform regular postoperative massages.
In May, the mother of an Instagram model named Yatnaa Rivera died during a procedure in the Dominican Republic. Ms. Rivera took to Instagram to ask for help and to warn others. The doctor who performed the operation, Hector Cabral, MD, had been fined for operating in the United States without a license. He is linked to several deaths and is still practicing. (Dr. Cabral did not respond to inquiries via social media; his office answered calls but said he was on vacation.)
Instagram accounts tagged into his doll hashtag (#CabralDoll) to spread the message.
Every day women are bombarded with images of beauty. With filters and editing apps, and the army of social media influencers who receive money or free cosmetic services in exchange for their Instagram posts, it’s often hard to know what’s real. Authentic depictions of what cosmetic surgery entails can be a reality check on what is attainable with cosmetic surgery.
In May 2019, the American College of Surgeons released voluntary ethical guidelines for social media by surgeons. Many of them address patient privacy, but they also advise practitioners to provide trustworthy medical advice and to be cautious around these “powerful educational tools.” Even so, now a real-time, crowdsourced system allows patients to cut through the surgeons’ marketing and advertising efforts.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
They use names and hashtags that connect the work to their provider. So, for example, KathySmithDoll would be a woman who underwent surgery with a Dr. Kathy Smith.
In an era of patient empowerment, these pages – they’re called “Sx pages,” with Sx mimicking the prescriptive “Rx” – form a just-out-of-sight Instagram community. They serve as a cosmetic surgery shopping guide, a best-practices education system, and can also sound the alarm about bad experiences with practitioners. Some presurgery doll pages are more like inspiration pages or mood boards, collecting images of desired shapes.
That way, “other girls doing research can find someone with a similar build to theirs and follow their journey for a glimpse at what they might look like if they got similar procedures,” said Tai Hall, a massage therapist in Maryland. On her Instagram page, she showcases before-and-after body-contouring results; in her Facebook group, she teaches postoperative self-massage and how people can best take care of themselves while healing.
These Instagram pages, she said, “are really big deals.”
The Sx Instagram pages are private and anonymous, to some extent, and follow strict rules to stay that way, particularly since many feature nudity. (As a social media practice, Sx pages are fairly similar to teenager’s private “finsta” friends-only accounts. They are similarly unverified and what they report is unverifiable.) Many of the bios on these pages indicate they won’t allow access to men.
Each Instagram page bio often unveils elaborate details, often including height and weight. The patient – the doll – will list surgery dates and tag her surgeon, recovery house, any postoperative care specialists or private nurses, and her postoperative massage therapist.
Recovery houses, surgery providers, and massage therapists also use the hashtags to promote their services. Some of these are flooded with ads or spam. Some are used by practitioners for education about surgery.
The surgery age
According to the American Society of Plastic Surgeons, more than 1.8 million cosmetic surgeries were performed in the United States in 2018. Breast augmentation and liposuction accounted for about a third of those.
And the number of “cosmetic minimally invasive procedures” – Botox, laser hair removal, soft-tissue fillers, and more – has grown rapidly in the United States. There were fewer than 5 million procedures in 2000. In 2018, there were nearly 16 million. (Almost half of those procedures are Botox treatments.)
Cosmetic procedures are also becoming more popular among people of color. The American Society for Aesthetic Plastic Surgery reports that cosmetic augmentation, like liposuctions and buttocks lifts, increased 56% among African Americans from 2005 to 2013, and is still rising.
As the number of savvy customers grows, doll pages provide a useful glimpse into the less-glamorous side of before and after – the details that people like to overlook, like bruising, drainage, and the often painfully long process of healing after significant surgeries.
Patients become online advertisements for their surgeons. Surgeons develop a reputation on social media for being the best at certain procedures, for delivering a desired look, or for working with certain ethnic groups and body types.
“They’ll cry and upload videos of pain and success and their struggles, or whatever they’re going through, and their surgery sisters help uplift them,” Ms. Hall said.
And there is a lot to talk about, from surgeons to procedures to recovery houses to advice on how to travel with the least hassle from airport security or airline staff when patients are clad in fajas – a kind of postoperative girdle – or other foam paddings.
How we shop for surgeons now
Sx pages can be an effective patient empowerment tool if done honestly and fairly, said Alan Matarasso, MD, a plastic surgeon in New York and the president of the American Society of Plastic Surgeons.
“It makes sense because this is a small group of people,” he said. “Not a lot of doctors do Brazilian butt lifts, but patients need to realize that they are not rating a restaurant.”
Dr. Matarasso encourages prospective patients who rely on Sx pages to research and prepare in other ways as well.
“The standards have to be even greater than if you had a sick gallbladder, because you don’t have to do this,” he said. “This is not like vetting a hotel room. You have to be careful.”
Dr. Matarasso recommends that prospective patients ask to see the surgeon’s best and worst results, or a random case – say, the 37th case they did that year. He suggests that prospective clients visit the American Board of Plastic Surgery websites to do research and that patients query the licensing state and find out what, if any, violations a surgeon may have had. Patients can ask board-certified surgeons their specialty and whether they are certified in it.
Ms. Hall, the massage therapist, warned that patients may see women who heal faster or achieve different results than they might. As is often the case on Instagram, people tend to post fewer of their struggles and more of their highlight reels.
Patients taking care of patients
Sx pages might be even more valuable for patients who plan to travel internationally for their surgery. Many people in the United States do this to save money. Doll pages serve to warn prospective patients about problems that surgeons and hospitals don’t disclose.
After surgery, especially if extensive travel is needed, patients may recuperate at recovery houses for a few days. Procedures like fat transfer to the buttocks leave patients unable to move around or sit; doctors may install drains to help remove fluid after surgery.
In a recovery house, a caretaker can tend to their incisions; help with bathing, food, and pain medications; and even perform regular postoperative massages.
In May, the mother of an Instagram model named Yatnaa Rivera died during a procedure in the Dominican Republic. Ms. Rivera took to Instagram to ask for help and to warn others. The doctor who performed the operation, Hector Cabral, MD, had been fined for operating in the United States without a license. He is linked to several deaths and is still practicing. (Dr. Cabral did not respond to inquiries via social media; his office answered calls but said he was on vacation.)
Instagram accounts tagged into his doll hashtag (#CabralDoll) to spread the message.
Every day women are bombarded with images of beauty. With filters and editing apps, and the army of social media influencers who receive money or free cosmetic services in exchange for their Instagram posts, it’s often hard to know what’s real. Authentic depictions of what cosmetic surgery entails can be a reality check on what is attainable with cosmetic surgery.
In May 2019, the American College of Surgeons released voluntary ethical guidelines for social media by surgeons. Many of them address patient privacy, but they also advise practitioners to provide trustworthy medical advice and to be cautious around these “powerful educational tools.” Even so, now a real-time, crowdsourced system allows patients to cut through the surgeons’ marketing and advertising efforts.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
They use names and hashtags that connect the work to their provider. So, for example, KathySmithDoll would be a woman who underwent surgery with a Dr. Kathy Smith.
In an era of patient empowerment, these pages – they’re called “Sx pages,” with Sx mimicking the prescriptive “Rx” – form a just-out-of-sight Instagram community. They serve as a cosmetic surgery shopping guide, a best-practices education system, and can also sound the alarm about bad experiences with practitioners. Some presurgery doll pages are more like inspiration pages or mood boards, collecting images of desired shapes.
That way, “other girls doing research can find someone with a similar build to theirs and follow their journey for a glimpse at what they might look like if they got similar procedures,” said Tai Hall, a massage therapist in Maryland. On her Instagram page, she showcases before-and-after body-contouring results; in her Facebook group, she teaches postoperative self-massage and how people can best take care of themselves while healing.
These Instagram pages, she said, “are really big deals.”
The Sx Instagram pages are private and anonymous, to some extent, and follow strict rules to stay that way, particularly since many feature nudity. (As a social media practice, Sx pages are fairly similar to teenager’s private “finsta” friends-only accounts. They are similarly unverified and what they report is unverifiable.) Many of the bios on these pages indicate they won’t allow access to men.
Each Instagram page bio often unveils elaborate details, often including height and weight. The patient – the doll – will list surgery dates and tag her surgeon, recovery house, any postoperative care specialists or private nurses, and her postoperative massage therapist.
Recovery houses, surgery providers, and massage therapists also use the hashtags to promote their services. Some of these are flooded with ads or spam. Some are used by practitioners for education about surgery.
The surgery age
According to the American Society of Plastic Surgeons, more than 1.8 million cosmetic surgeries were performed in the United States in 2018. Breast augmentation and liposuction accounted for about a third of those.
And the number of “cosmetic minimally invasive procedures” – Botox, laser hair removal, soft-tissue fillers, and more – has grown rapidly in the United States. There were fewer than 5 million procedures in 2000. In 2018, there were nearly 16 million. (Almost half of those procedures are Botox treatments.)
Cosmetic procedures are also becoming more popular among people of color. The American Society for Aesthetic Plastic Surgery reports that cosmetic augmentation, like liposuctions and buttocks lifts, increased 56% among African Americans from 2005 to 2013, and is still rising.
As the number of savvy customers grows, doll pages provide a useful glimpse into the less-glamorous side of before and after – the details that people like to overlook, like bruising, drainage, and the often painfully long process of healing after significant surgeries.
Patients become online advertisements for their surgeons. Surgeons develop a reputation on social media for being the best at certain procedures, for delivering a desired look, or for working with certain ethnic groups and body types.
“They’ll cry and upload videos of pain and success and their struggles, or whatever they’re going through, and their surgery sisters help uplift them,” Ms. Hall said.
And there is a lot to talk about, from surgeons to procedures to recovery houses to advice on how to travel with the least hassle from airport security or airline staff when patients are clad in fajas – a kind of postoperative girdle – or other foam paddings.
How we shop for surgeons now
Sx pages can be an effective patient empowerment tool if done honestly and fairly, said Alan Matarasso, MD, a plastic surgeon in New York and the president of the American Society of Plastic Surgeons.
“It makes sense because this is a small group of people,” he said. “Not a lot of doctors do Brazilian butt lifts, but patients need to realize that they are not rating a restaurant.”
Dr. Matarasso encourages prospective patients who rely on Sx pages to research and prepare in other ways as well.
“The standards have to be even greater than if you had a sick gallbladder, because you don’t have to do this,” he said. “This is not like vetting a hotel room. You have to be careful.”
Dr. Matarasso recommends that prospective patients ask to see the surgeon’s best and worst results, or a random case – say, the 37th case they did that year. He suggests that prospective clients visit the American Board of Plastic Surgery websites to do research and that patients query the licensing state and find out what, if any, violations a surgeon may have had. Patients can ask board-certified surgeons their specialty and whether they are certified in it.
Ms. Hall, the massage therapist, warned that patients may see women who heal faster or achieve different results than they might. As is often the case on Instagram, people tend to post fewer of their struggles and more of their highlight reels.
Patients taking care of patients
Sx pages might be even more valuable for patients who plan to travel internationally for their surgery. Many people in the United States do this to save money. Doll pages serve to warn prospective patients about problems that surgeons and hospitals don’t disclose.
After surgery, especially if extensive travel is needed, patients may recuperate at recovery houses for a few days. Procedures like fat transfer to the buttocks leave patients unable to move around or sit; doctors may install drains to help remove fluid after surgery.
In a recovery house, a caretaker can tend to their incisions; help with bathing, food, and pain medications; and even perform regular postoperative massages.
In May, the mother of an Instagram model named Yatnaa Rivera died during a procedure in the Dominican Republic. Ms. Rivera took to Instagram to ask for help and to warn others. The doctor who performed the operation, Hector Cabral, MD, had been fined for operating in the United States without a license. He is linked to several deaths and is still practicing. (Dr. Cabral did not respond to inquiries via social media; his office answered calls but said he was on vacation.)
Instagram accounts tagged into his doll hashtag (#CabralDoll) to spread the message.
Every day women are bombarded with images of beauty. With filters and editing apps, and the army of social media influencers who receive money or free cosmetic services in exchange for their Instagram posts, it’s often hard to know what’s real. Authentic depictions of what cosmetic surgery entails can be a reality check on what is attainable with cosmetic surgery.
In May 2019, the American College of Surgeons released voluntary ethical guidelines for social media by surgeons. Many of them address patient privacy, but they also advise practitioners to provide trustworthy medical advice and to be cautious around these “powerful educational tools.” Even so, now a real-time, crowdsourced system allows patients to cut through the surgeons’ marketing and advertising efforts.
Kaiser Health News is a nonprofit national health policy news service. It is an editorially independent program of the Henry J. Kaiser Family Foundation that is not affiliated with Kaiser Permanente.

Effects of Diet and Probiotics on Migraine + IBS
IgG elimination diet combined with probiotics may be beneficial to migraine plus irritable bowel syndrome (IBS), a new study found. Researchers investigated the therapeutic potential of diet based on IgG elimination combined with probiotics on 60 patients with migraine plus IBS. IgG antibodies against 266 food varieties were detected by ELISA. Participants were randomized into 3 groups for treatment of IgG elimination diet or probiotics, or diet combined with probiotics. Among the findings:
- Improvement of migraine and gut symptom was achieved at a certain time point.
- Reduced over the counter (OTC) analgesics was seen in all groups.
- Use of triptans did not show significant difference.
- An increased serum serotonin level was seen in participants treated with elimination diet and elimination diet combined with probiotics.
Xie Y, et al. Effects of diet based on IgG elimination combined with probiotics on migraine plus irritable bowel syndrome. [Published online ahead of print August 21, 2019]. Pain Res Manag. doi: 10.1155/2019/7890461.
IgG elimination diet combined with probiotics may be beneficial to migraine plus irritable bowel syndrome (IBS), a new study found. Researchers investigated the therapeutic potential of diet based on IgG elimination combined with probiotics on 60 patients with migraine plus IBS. IgG antibodies against 266 food varieties were detected by ELISA. Participants were randomized into 3 groups for treatment of IgG elimination diet or probiotics, or diet combined with probiotics. Among the findings:
- Improvement of migraine and gut symptom was achieved at a certain time point.
- Reduced over the counter (OTC) analgesics was seen in all groups.
- Use of triptans did not show significant difference.
- An increased serum serotonin level was seen in participants treated with elimination diet and elimination diet combined with probiotics.
Xie Y, et al. Effects of diet based on IgG elimination combined with probiotics on migraine plus irritable bowel syndrome. [Published online ahead of print August 21, 2019]. Pain Res Manag. doi: 10.1155/2019/7890461.
IgG elimination diet combined with probiotics may be beneficial to migraine plus irritable bowel syndrome (IBS), a new study found. Researchers investigated the therapeutic potential of diet based on IgG elimination combined with probiotics on 60 patients with migraine plus IBS. IgG antibodies against 266 food varieties were detected by ELISA. Participants were randomized into 3 groups for treatment of IgG elimination diet or probiotics, or diet combined with probiotics. Among the findings:
- Improvement of migraine and gut symptom was achieved at a certain time point.
- Reduced over the counter (OTC) analgesics was seen in all groups.
- Use of triptans did not show significant difference.
- An increased serum serotonin level was seen in participants treated with elimination diet and elimination diet combined with probiotics.
Xie Y, et al. Effects of diet based on IgG elimination combined with probiotics on migraine plus irritable bowel syndrome. [Published online ahead of print August 21, 2019]. Pain Res Manag. doi: 10.1155/2019/7890461.
Non-Invasive Brain Stimulation in Migraine
Excitatory non-invasive brain stimulation (NIBS) of the excitatory primary motor cortex (M1) is likely to reduce headache intensity and the frequency of headache attacks in patients with migraine, a new study found. Researchers quantitatively reviewed the efficacy of repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS) in randomized controlled trials (RTCs) in modifying headache intensity and frequency of headache attacks in patients with migraine. A random meta-analysis was performed to pool effect sizes of outcomes. Among the findings:
- Nine RCTs with 276 participants were included.
- Meta-analysis of excitatory M1 stimulation demonstrated significant effects on reducing headache intensity in patients with migraine.
- Meta-analysis of excitatory M1 stimulation showed significant effects on reducing frequency of headache attacks in patients with migraine.
Feng Y, et al. Effects of non-invasive brain stimulation on headache intensity and frequency of headache attacks in patients with migraine: A systematic review and meta-analysis. [Published online ahead of print September 18, 2019]. Headache. doi: 10.1111/head.13645.
Excitatory non-invasive brain stimulation (NIBS) of the excitatory primary motor cortex (M1) is likely to reduce headache intensity and the frequency of headache attacks in patients with migraine, a new study found. Researchers quantitatively reviewed the efficacy of repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS) in randomized controlled trials (RTCs) in modifying headache intensity and frequency of headache attacks in patients with migraine. A random meta-analysis was performed to pool effect sizes of outcomes. Among the findings:
- Nine RCTs with 276 participants were included.
- Meta-analysis of excitatory M1 stimulation demonstrated significant effects on reducing headache intensity in patients with migraine.
- Meta-analysis of excitatory M1 stimulation showed significant effects on reducing frequency of headache attacks in patients with migraine.
Feng Y, et al. Effects of non-invasive brain stimulation on headache intensity and frequency of headache attacks in patients with migraine: A systematic review and meta-analysis. [Published online ahead of print September 18, 2019]. Headache. doi: 10.1111/head.13645.
Excitatory non-invasive brain stimulation (NIBS) of the excitatory primary motor cortex (M1) is likely to reduce headache intensity and the frequency of headache attacks in patients with migraine, a new study found. Researchers quantitatively reviewed the efficacy of repetitive transcranial magnetic stimulation (rTMS) and transcranial direct current stimulation (tDCS) in randomized controlled trials (RTCs) in modifying headache intensity and frequency of headache attacks in patients with migraine. A random meta-analysis was performed to pool effect sizes of outcomes. Among the findings:
- Nine RCTs with 276 participants were included.
- Meta-analysis of excitatory M1 stimulation demonstrated significant effects on reducing headache intensity in patients with migraine.
- Meta-analysis of excitatory M1 stimulation showed significant effects on reducing frequency of headache attacks in patients with migraine.
Feng Y, et al. Effects of non-invasive brain stimulation on headache intensity and frequency of headache attacks in patients with migraine: A systematic review and meta-analysis. [Published online ahead of print September 18, 2019]. Headache. doi: 10.1111/head.13645.
Osmophobia is a Clinical Marker of Migraine
Osmophobia is a specific clinical marker of migraine, but not tension-type headache, a new study found. Researchers conducted a prospective study on 193 patients suffering from migraine without aura, migraine with aura, episodic tension-type headache, or a combination of these. Each patient was asked to describe in detail osmophobia, when present, in the 4 headache attacks. Among the findings:
- 45.7% of migraine with aura attacks were associated with osmophobia.
- 67.2% of patients with migraine reported osmophobia in at least a quarter of the attacks.
- No tension-type headache attack was associated with osmophobia.
Terrin A, et al. A prospective study on osmophobia in migraine versus tension-type headache in a large series of attacks. [Published online ahead of print September 19, 2019]. Cephalalgia. doi: 10.1177/0333102419877661.
Osmophobia is a specific clinical marker of migraine, but not tension-type headache, a new study found. Researchers conducted a prospective study on 193 patients suffering from migraine without aura, migraine with aura, episodic tension-type headache, or a combination of these. Each patient was asked to describe in detail osmophobia, when present, in the 4 headache attacks. Among the findings:
- 45.7% of migraine with aura attacks were associated with osmophobia.
- 67.2% of patients with migraine reported osmophobia in at least a quarter of the attacks.
- No tension-type headache attack was associated with osmophobia.
Terrin A, et al. A prospective study on osmophobia in migraine versus tension-type headache in a large series of attacks. [Published online ahead of print September 19, 2019]. Cephalalgia. doi: 10.1177/0333102419877661.
Osmophobia is a specific clinical marker of migraine, but not tension-type headache, a new study found. Researchers conducted a prospective study on 193 patients suffering from migraine without aura, migraine with aura, episodic tension-type headache, or a combination of these. Each patient was asked to describe in detail osmophobia, when present, in the 4 headache attacks. Among the findings:
- 45.7% of migraine with aura attacks were associated with osmophobia.
- 67.2% of patients with migraine reported osmophobia in at least a quarter of the attacks.
- No tension-type headache attack was associated with osmophobia.
Terrin A, et al. A prospective study on osmophobia in migraine versus tension-type headache in a large series of attacks. [Published online ahead of print September 19, 2019]. Cephalalgia. doi: 10.1177/0333102419877661.