User login
Hospitalists as Triagists: Description of the Triagist Role across Academic Medical Centers
Hospital medicine has grown dramatically over the past 20 years.1,2 A recent survey regarding hospitalists’ clinical roles showed an expansion to triaging emergency department (ED) medical admissions and transfers from outside hospitals.3 From the hospitalist perspective, triaging involves the evaluation of patients for potential admission.4 With scrutiny on ED metrics, such as wait times (https://www.medicare.gov/hospitalcompare/search.html), health system administrators have heightened expectations for efficient patient flow, which increasingly falls to hospitalists.5-7
Despite the growth in hospitalists’ triagist activities, there has been little formal assessment of their role. We hypothesized that this role differs from inpatient care in significant ways.6-8 We sought to describe the triagist role in adult academic inpatient medicine settings to understand the responsibilities and skill set required.
METHODS
Ten academic medical center (AMC) sites were recruited from Research Committee session attendees at the 2014 Society of Hospital Medicine national meeting and the 2014 Society of General Internal Medicine southern regional meeting. The AMCs were geographically diverse: three Western, two Midwestern, two Southern, one Northeastern, and two Southeastern. Site representatives were identified and completed a web-based questionnaire about their AMC (see Appendix 1 for the information collected). Clarifications regarding survey responses were performed via conference calls between the authors (STV, ESW) and site representatives.
Hospitalist Survey
In January 2018, surveys were sent to 583 physicians who worked as triagists. Participants received an anonymous 28-item RedCap survey by e-mail and were sent up to five reminder e-mails over six weeks (see Appendix 2 for the questions analyzed in this paper). Respondents were given the option to be entered in a gift card drawing.
Demographic information and individual workflow/practices were obtained. A 5-point Likert scale (strongly disagree – strongly agree) was used to assess hospitalists’ concurrence with current providers (eg, ED, clinic providers) regarding the management and whether patients must meet the utilization management (UM) criteria for admission. Time estimates used 5% increments and were categorized into four frequency categories based on the local modes provided in responses: Seldom (0%-10%), Occasional (15%-35%), Half-the-Time (40%-60%), and Frequently (65%-100%). Free text responses on effective/ineffective triagist qualities were elicited. Responses were included for analysis if at least 70% of questions were completed.
Data Analysis
Quantitative
Descriptive statistics were calculated for each variable. The Kruskal-Wallis test was used to evaluate differences across AMCs in the time spent on in-person evaluation and communication. Weighting, based on the ratio of hospitalists to survey respondents at each AMC, was used to calculate the average institutional percentages across the study sample.
Qualitative
Responses to open-ended questions were analyzed using thematic analysis.9 Three independent reviewers (STV, JC, ESW) read, analyzed, and grouped the responses by codes. Codes were then assessed for overlap and grouped into themes by one reviewer (STV). A table of themes with supporting quotes and the number of mentions was subsequently developed by all three reviewers. Similar themes were combined to create domains. The domains were reviewed by the steering committee members to create a consensus description (Appendix 3).
The University of Texas Health San Antonio’s Institutional Review Board and participating institutions approved the study as exempt.
RESULTS
Site Characteristics
Representatives from 10 AMCs reported data on a range of one to four hospitals for a total of 22 hospitals. The median reported that the number of medical patients admitted in a 24-hour period was 31-40 (range, 11-20 to >50). The median group size of hospitalists was 41-50 (range, 0-10 to >70).
The survey response rate was 40% (n = 235), ranging from 9%-70% between institutions. Self-identified female hospitalists accounted for 52% of respondents. Four percent were 25-29 years old, 66% were 30-39 years old, 24% were 40-49 years old, and 6% were ≥50 years old. The average clinical time spent as a triagist was 16%.
Description of Triagist Activities
The activities identified by the majority of respondents across all sites included transferring patients within the hospital (73%), and assessing/approving patient transfers from outside hospitals and clinics (82%). Internal transfer activities reported by >50% of respondents included allocating patients within the hospital or bed capacity coordination, assessing intensive care unit transfers, assigning ED admissions, and consulting other services. The ED accounted for an average of 55% of calls received. Respondents also reported being involved with the documentation related to these activities.
Similarities and Differences across AMCs
Two AMCs did not have a dedicated triagist; instead, physicians supervised residents and advanced practice providers. Among the eight sites with triagists, triaging was predominantly done by faculty physicians contacted via pagers. At seven of these sites, 100% of hospitalists worked as triagists. The triage service was covered by faculty physicians from 8-24 hours per day.
Bed boards and transfer centers staffed by registered nurses, nurse coordinators, house supervisors, or physicians were common support systems, though this infrastructure was organized differently across institutions. A UM review before admission was performed at three institutions 24 hours/day. The remaining institutions reviewed patients retrospectively.
Twenty-eight percent of hospitalists across all sites “Disagreed” or “Strongly disagreed” that a patient must meet UM criteria for admission. Forty-two percent had “Frequent” different opinions regarding patient management than the consulting provider.
Triagist and current provider communication practices varied widely across AMCs (Figure). There was significant variability in verbal communication (P = .02), with >70% of respondents at two AMCs reporting verbal communication at least half the time, but <30% reporting this frequency at two other AMCs. Respondents reported variable use of electronic communication (ie, notes/orders in the electronic health record) across AMCs (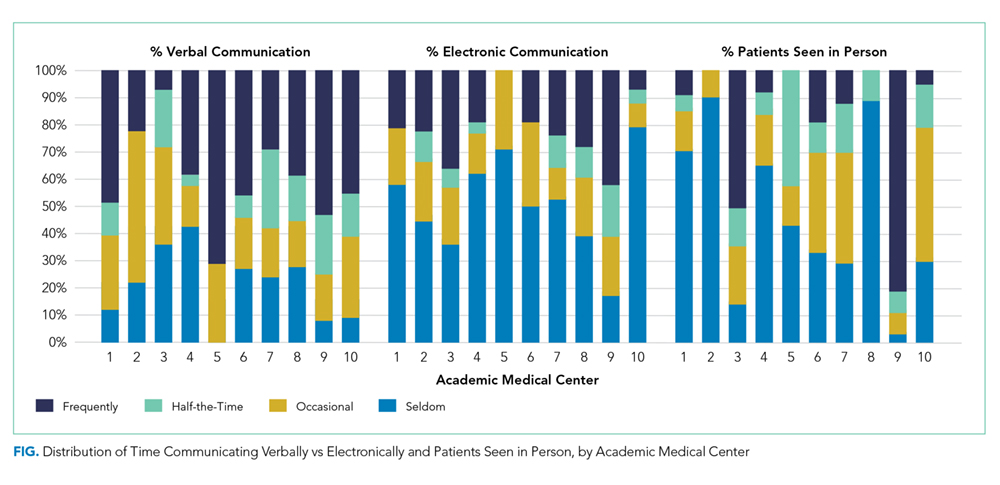
The practice of evaluating patients in person also varied significantly across AMCs (P < .0001, Figure). Across hospitalists, only 28% see patients in person about “Half-the-Time” or more.
Differences within AMCs
Variability within AMCs was greatest for the rate of verbal communication practices, with a typical interquartile range (IQR) of 20% to 90% among the hospitalists within a given AMC and for the rate of electronic communication with a typical IQR of 0% to 50%. For other survey questions, the IQR was typically 15 to 20 percentage points.
Thematic Analysis
We received 207 and 203 responses (88% and 86%, respectively) to the open-ended questions “What qualities does an effective triagist have?’ and ‘What qualities make a triagist ineffective?” We identified 22 themes for effective and ineffective qualities, which were grouped into seven domains (Table). All themes had at least three mentions by respondents. The three most frequently mentioned themes, communication skills, efficiency, and systems knowledge, had greater than 60 mentions.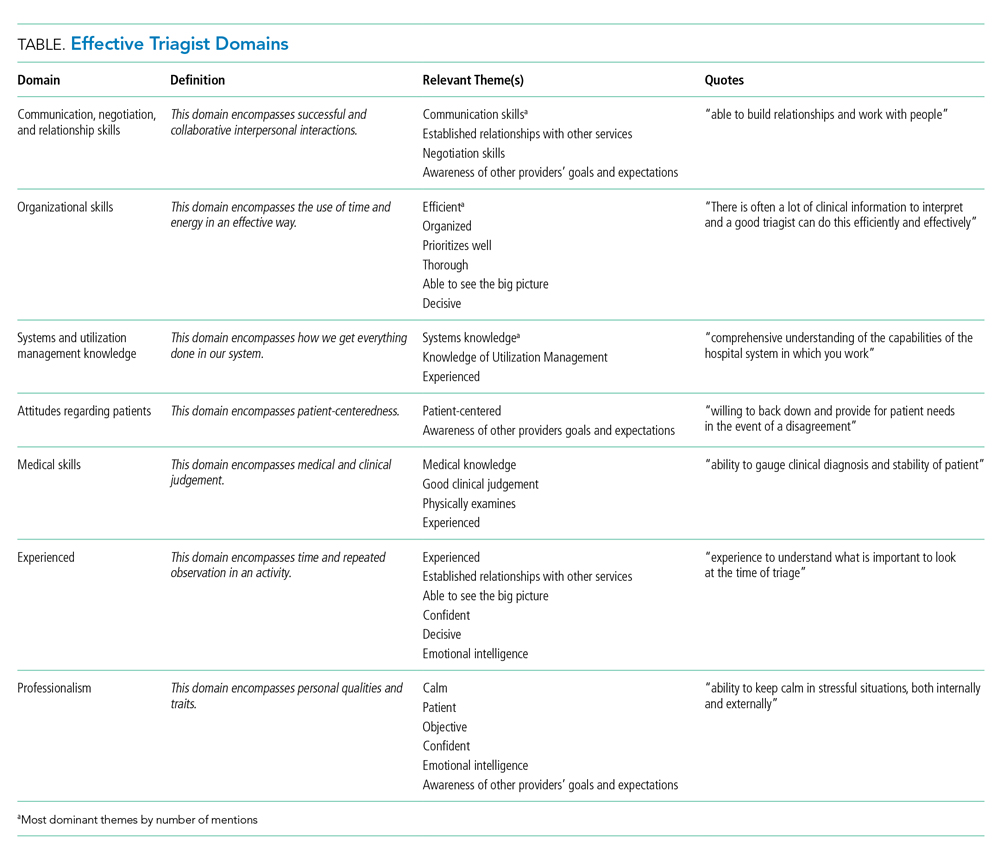
DISCUSSION
Our study of the triagist role at 10 AMCs describes critical triagist functions and identifies key findings across and within AMCs. Twenty-eight percent of hospitalists reported admitting patients even when the patient did not meet the admission criteria, consistent with previous research demonstrating the influence of factors other than clinical disease severity on triage decisions.10 However, preventable admissions remain a hospital-level quality metric.11,12 Triagists must often balance each patient’s circumstances with the complexities of the system. Juggling the competing demands of the system while providing patient-centered care can be challenging and may explain why attending physicians are more frequently filling this role.13
Local context/culture is likely to play a role in the variation across sites; however, compensation for the time spent may also be a factor. If triage activities are not reimbursable, this could lead to less documentation and a lower likelihood that patients are evaluated in person.14 This reason may also explain why all hospitalists were required to serve as a triagist at most sites.
Currently, no consensus definition of the triagist role has been developed. Our results demonstrate that this role is heterogeneous and grounded in the local healthcare system practices. We propose the following working definition of the triagist: a physician who assesses patients for admission, actively supporting the transition of the patient from the outpatient to the inpatient setting. A triagist should be equipped with a skill set that includes not only clinical knowledge but also emphasizes systems knowledge, awareness of others’ goals, efficiency, an ability to communicate effectively, and the knowledge of UM. We recommend that medical directors of hospitalist programs focus their attention on locally specific, systems-based skills development when orienting new hospitalists. The financial aspects of cost should be considered and delineated as well.
Our analysis is limited in several respects. Participant AMCs were not randomly chosen, but do represent a broad array of facility types, group size, and geographic regions. The low response rates at some AMCs may result in an inaccurate representation of those sites. Data was not obtained on hospitalists that did not respond to the survey; therefore, nonresponse bias may affect outcomes. This research used self-report rather than direct observation, which could be subject to recall and social desirability bias. Finally, our results may not be generalizable to nonacademic institutions.
CONCLUSION
The hospitalist role as triagist at AMCs emphasizes communication, organizational skills, efficiency, systems-based practice, and UM knowledge. Although we found significant variation across and within AMCs, internal transfer activities were common across programs. Hospitalist programs should focus on systems-based skills development to prepare hospitalists for the role. The skill set necessary for triagist responsibilities also has implications for internal medicine resident education.4 With increasing emphasis on value and system effectiveness in care delivery, further studies of the triagist role should be undertaken.
Acknowledgments
The TRIAGIST Collaborative Group consists of: Maralyssa Bann, MD, Andrew White, MD (University of Washington); Jagriti Chadha, MD (University of Kentucky); Joel Boggan, MD (Duke University); Sherwin Hsu, MD (UCLA); Jeff Liao, MD (Harvard Medical School); Tabatha Matthias, DO (University of Nebraska Medical Center); Tresa McNeal, MD (Scott and White Texas A&M); Roxana Naderi, MD, Khooshbu Shah, MD (University of Colorado); David Schmit, MD (University of Texas Health San Antonio); Manivannan Veerasamy, MD (Michigan State University).
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the po
1. Kisuule F, Howell EE. Hospitalists and their impact on quality, patient safety, and satisfaction. Obstet Gynecol Clin North Am. 2015; 42(3):433-446. https://doi.org/10.1016/j.ogc.2015.05.003.
2. Wachter, RM, Goldman, L. Zero to 50,000-The 20th anniversary of the hospitalist. N Engl J Med. 2016;375(11): 1009-1011. https://doi.org/10.1056/NEJMp1607958.
3. Vasilevskis EE, Knebel RJ, Wachter RM, Auerbach AD. California hospital leaders’ views of hospitalists: meeting needs of the present and future. J Hosp Med. 2009;4:528-534. https://doi.org/10.1002/jhm.529.
4. Wang ES, Velásquez ST, Smith CJ, et al. Triaging inpatient admissions: an opportunity for resident education. J Gen Intern Med. 2019; 34(5):754-757. https://doi.org/10.1007/s11606-019-04882-2.
5. Briones A, Markoff B, Kathuria N, et al. A model of a hospitalist role in the care of admitted patients in the emergency department. J Hosp Med. 2010;5(6):360-364. https://doi.org/10.1002/jhm.636.
6. Howell EE, Bessman ES, Rubin HR. Hospitalists and an innovative emergency department admission process. J Gen Intern Med. 2004;19:266-268. https://doi.org/10.1111/j.1525-1497.2004.30431.x.
7. Howell E, Bessman E, Marshall R, Wright S. Hospitalist bed management effecting throughput from the emergency department to the intensive care unit. J Crit Care. 2010;25:184-189. https://doi.org/10.1016/j.jcrc.2009.08.004.
8. Chadaga SR, Shockley L, Keniston A, et al. Hospitalist-led medicine emergency department team: associations with throughput, timeliness of patient care, and satisfaction. J Hosp Med. 2012;7:562-566. https://doi.org/10.1002/jhm.1957.
9. Braun, V. Clarke, V. Using thematic analysis in psychology. Qualitative Research in Psychology. 2006;77-101. https://doi.org/10.1191/1478088706qp063oa.
10. Lewis Hunter AE, Spatz ES, Bernstein SL, Rosenthal MS. Factors influencing hospital admission of non-critically ill patients presenting to the emergency department: a cross-sectional study. J Gen Intern Med. 2016;31(1):37-44. https://doi.org/10.1007/s11606-015-3438-8.
11. Patel KK, Vakharia N, Pile J, Howell EH, Rothberg MB. Preventable admissions on a general medicine service: prevalence, causes and comparison with AHRQ prevention quality indicators-a cross-sectional analysis. J Gen Intern Med. 2016;31(6):597-601. https://doi.org/10.1007/s11606-016-3615-4.
12. Daniels LM1, Sorita A2, Kashiwagi DT, et al. Characterizing potentially preventable admissions: a mixed methods study of rates, associated factors, outcomes, and physician decision-making. J Gen Intern Med. 2018;33(5):737-744. https://doi.org/10.1007/s11606-017-4285-6.
13. Howard-Anderson J, Lonowski S, Vangala S, Tseng CH, Busuttil A, Afsar-Manesh N. Readmissions in the era of patient engagement. JAMA Intern Med. 2014;174(11):1870-1872. https://doi.org/10.1001/jamainternmed.2014.4782.
14. Hinami K, Whelan CT, Miller JA, Wolosin RJ, Wetterneck TB, Society of Hospital Medicine Career Satisfaction Task Force. Job characteristics, satisfaction, and burnout across hospitalist practice models. J Hosp Med. 2012;7(5):402-410. https://doi.org/10.1002/jhm.1907
Hospital medicine has grown dramatically over the past 20 years.1,2 A recent survey regarding hospitalists’ clinical roles showed an expansion to triaging emergency department (ED) medical admissions and transfers from outside hospitals.3 From the hospitalist perspective, triaging involves the evaluation of patients for potential admission.4 With scrutiny on ED metrics, such as wait times (https://www.medicare.gov/hospitalcompare/search.html), health system administrators have heightened expectations for efficient patient flow, which increasingly falls to hospitalists.5-7
Despite the growth in hospitalists’ triagist activities, there has been little formal assessment of their role. We hypothesized that this role differs from inpatient care in significant ways.6-8 We sought to describe the triagist role in adult academic inpatient medicine settings to understand the responsibilities and skill set required.
METHODS
Ten academic medical center (AMC) sites were recruited from Research Committee session attendees at the 2014 Society of Hospital Medicine national meeting and the 2014 Society of General Internal Medicine southern regional meeting. The AMCs were geographically diverse: three Western, two Midwestern, two Southern, one Northeastern, and two Southeastern. Site representatives were identified and completed a web-based questionnaire about their AMC (see Appendix 1 for the information collected). Clarifications regarding survey responses were performed via conference calls between the authors (STV, ESW) and site representatives.
Hospitalist Survey
In January 2018, surveys were sent to 583 physicians who worked as triagists. Participants received an anonymous 28-item RedCap survey by e-mail and were sent up to five reminder e-mails over six weeks (see Appendix 2 for the questions analyzed in this paper). Respondents were given the option to be entered in a gift card drawing.
Demographic information and individual workflow/practices were obtained. A 5-point Likert scale (strongly disagree – strongly agree) was used to assess hospitalists’ concurrence with current providers (eg, ED, clinic providers) regarding the management and whether patients must meet the utilization management (UM) criteria for admission. Time estimates used 5% increments and were categorized into four frequency categories based on the local modes provided in responses: Seldom (0%-10%), Occasional (15%-35%), Half-the-Time (40%-60%), and Frequently (65%-100%). Free text responses on effective/ineffective triagist qualities were elicited. Responses were included for analysis if at least 70% of questions were completed.
Data Analysis
Quantitative
Descriptive statistics were calculated for each variable. The Kruskal-Wallis test was used to evaluate differences across AMCs in the time spent on in-person evaluation and communication. Weighting, based on the ratio of hospitalists to survey respondents at each AMC, was used to calculate the average institutional percentages across the study sample.
Qualitative
Responses to open-ended questions were analyzed using thematic analysis.9 Three independent reviewers (STV, JC, ESW) read, analyzed, and grouped the responses by codes. Codes were then assessed for overlap and grouped into themes by one reviewer (STV). A table of themes with supporting quotes and the number of mentions was subsequently developed by all three reviewers. Similar themes were combined to create domains. The domains were reviewed by the steering committee members to create a consensus description (Appendix 3).
The University of Texas Health San Antonio’s Institutional Review Board and participating institutions approved the study as exempt.
RESULTS
Site Characteristics
Representatives from 10 AMCs reported data on a range of one to four hospitals for a total of 22 hospitals. The median reported that the number of medical patients admitted in a 24-hour period was 31-40 (range, 11-20 to >50). The median group size of hospitalists was 41-50 (range, 0-10 to >70).
The survey response rate was 40% (n = 235), ranging from 9%-70% between institutions. Self-identified female hospitalists accounted for 52% of respondents. Four percent were 25-29 years old, 66% were 30-39 years old, 24% were 40-49 years old, and 6% were ≥50 years old. The average clinical time spent as a triagist was 16%.
Description of Triagist Activities
The activities identified by the majority of respondents across all sites included transferring patients within the hospital (73%), and assessing/approving patient transfers from outside hospitals and clinics (82%). Internal transfer activities reported by >50% of respondents included allocating patients within the hospital or bed capacity coordination, assessing intensive care unit transfers, assigning ED admissions, and consulting other services. The ED accounted for an average of 55% of calls received. Respondents also reported being involved with the documentation related to these activities.
Similarities and Differences across AMCs
Two AMCs did not have a dedicated triagist; instead, physicians supervised residents and advanced practice providers. Among the eight sites with triagists, triaging was predominantly done by faculty physicians contacted via pagers. At seven of these sites, 100% of hospitalists worked as triagists. The triage service was covered by faculty physicians from 8-24 hours per day.
Bed boards and transfer centers staffed by registered nurses, nurse coordinators, house supervisors, or physicians were common support systems, though this infrastructure was organized differently across institutions. A UM review before admission was performed at three institutions 24 hours/day. The remaining institutions reviewed patients retrospectively.
Twenty-eight percent of hospitalists across all sites “Disagreed” or “Strongly disagreed” that a patient must meet UM criteria for admission. Forty-two percent had “Frequent” different opinions regarding patient management than the consulting provider.
Triagist and current provider communication practices varied widely across AMCs (Figure). There was significant variability in verbal communication (P = .02), with >70% of respondents at two AMCs reporting verbal communication at least half the time, but <30% reporting this frequency at two other AMCs. Respondents reported variable use of electronic communication (ie, notes/orders in the electronic health record) across AMCs (
The practice of evaluating patients in person also varied significantly across AMCs (P < .0001, Figure). Across hospitalists, only 28% see patients in person about “Half-the-Time” or more.
Differences within AMCs
Variability within AMCs was greatest for the rate of verbal communication practices, with a typical interquartile range (IQR) of 20% to 90% among the hospitalists within a given AMC and for the rate of electronic communication with a typical IQR of 0% to 50%. For other survey questions, the IQR was typically 15 to 20 percentage points.
Thematic Analysis
We received 207 and 203 responses (88% and 86%, respectively) to the open-ended questions “What qualities does an effective triagist have?’ and ‘What qualities make a triagist ineffective?” We identified 22 themes for effective and ineffective qualities, which were grouped into seven domains (Table). All themes had at least three mentions by respondents. The three most frequently mentioned themes, communication skills, efficiency, and systems knowledge, had greater than 60 mentions.
DISCUSSION
Our study of the triagist role at 10 AMCs describes critical triagist functions and identifies key findings across and within AMCs. Twenty-eight percent of hospitalists reported admitting patients even when the patient did not meet the admission criteria, consistent with previous research demonstrating the influence of factors other than clinical disease severity on triage decisions.10 However, preventable admissions remain a hospital-level quality metric.11,12 Triagists must often balance each patient’s circumstances with the complexities of the system. Juggling the competing demands of the system while providing patient-centered care can be challenging and may explain why attending physicians are more frequently filling this role.13
Local context/culture is likely to play a role in the variation across sites; however, compensation for the time spent may also be a factor. If triage activities are not reimbursable, this could lead to less documentation and a lower likelihood that patients are evaluated in person.14 This reason may also explain why all hospitalists were required to serve as a triagist at most sites.
Currently, no consensus definition of the triagist role has been developed. Our results demonstrate that this role is heterogeneous and grounded in the local healthcare system practices. We propose the following working definition of the triagist: a physician who assesses patients for admission, actively supporting the transition of the patient from the outpatient to the inpatient setting. A triagist should be equipped with a skill set that includes not only clinical knowledge but also emphasizes systems knowledge, awareness of others’ goals, efficiency, an ability to communicate effectively, and the knowledge of UM. We recommend that medical directors of hospitalist programs focus their attention on locally specific, systems-based skills development when orienting new hospitalists. The financial aspects of cost should be considered and delineated as well.
Our analysis is limited in several respects. Participant AMCs were not randomly chosen, but do represent a broad array of facility types, group size, and geographic regions. The low response rates at some AMCs may result in an inaccurate representation of those sites. Data was not obtained on hospitalists that did not respond to the survey; therefore, nonresponse bias may affect outcomes. This research used self-report rather than direct observation, which could be subject to recall and social desirability bias. Finally, our results may not be generalizable to nonacademic institutions.
CONCLUSION
The hospitalist role as triagist at AMCs emphasizes communication, organizational skills, efficiency, systems-based practice, and UM knowledge. Although we found significant variation across and within AMCs, internal transfer activities were common across programs. Hospitalist programs should focus on systems-based skills development to prepare hospitalists for the role. The skill set necessary for triagist responsibilities also has implications for internal medicine resident education.4 With increasing emphasis on value and system effectiveness in care delivery, further studies of the triagist role should be undertaken.
Acknowledgments
The TRIAGIST Collaborative Group consists of: Maralyssa Bann, MD, Andrew White, MD (University of Washington); Jagriti Chadha, MD (University of Kentucky); Joel Boggan, MD (Duke University); Sherwin Hsu, MD (UCLA); Jeff Liao, MD (Harvard Medical School); Tabatha Matthias, DO (University of Nebraska Medical Center); Tresa McNeal, MD (Scott and White Texas A&M); Roxana Naderi, MD, Khooshbu Shah, MD (University of Colorado); David Schmit, MD (University of Texas Health San Antonio); Manivannan Veerasamy, MD (Michigan State University).
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the po
Hospital medicine has grown dramatically over the past 20 years.1,2 A recent survey regarding hospitalists’ clinical roles showed an expansion to triaging emergency department (ED) medical admissions and transfers from outside hospitals.3 From the hospitalist perspective, triaging involves the evaluation of patients for potential admission.4 With scrutiny on ED metrics, such as wait times (https://www.medicare.gov/hospitalcompare/search.html), health system administrators have heightened expectations for efficient patient flow, which increasingly falls to hospitalists.5-7
Despite the growth in hospitalists’ triagist activities, there has been little formal assessment of their role. We hypothesized that this role differs from inpatient care in significant ways.6-8 We sought to describe the triagist role in adult academic inpatient medicine settings to understand the responsibilities and skill set required.
METHODS
Ten academic medical center (AMC) sites were recruited from Research Committee session attendees at the 2014 Society of Hospital Medicine national meeting and the 2014 Society of General Internal Medicine southern regional meeting. The AMCs were geographically diverse: three Western, two Midwestern, two Southern, one Northeastern, and two Southeastern. Site representatives were identified and completed a web-based questionnaire about their AMC (see Appendix 1 for the information collected). Clarifications regarding survey responses were performed via conference calls between the authors (STV, ESW) and site representatives.
Hospitalist Survey
In January 2018, surveys were sent to 583 physicians who worked as triagists. Participants received an anonymous 28-item RedCap survey by e-mail and were sent up to five reminder e-mails over six weeks (see Appendix 2 for the questions analyzed in this paper). Respondents were given the option to be entered in a gift card drawing.
Demographic information and individual workflow/practices were obtained. A 5-point Likert scale (strongly disagree – strongly agree) was used to assess hospitalists’ concurrence with current providers (eg, ED, clinic providers) regarding the management and whether patients must meet the utilization management (UM) criteria for admission. Time estimates used 5% increments and were categorized into four frequency categories based on the local modes provided in responses: Seldom (0%-10%), Occasional (15%-35%), Half-the-Time (40%-60%), and Frequently (65%-100%). Free text responses on effective/ineffective triagist qualities were elicited. Responses were included for analysis if at least 70% of questions were completed.
Data Analysis
Quantitative
Descriptive statistics were calculated for each variable. The Kruskal-Wallis test was used to evaluate differences across AMCs in the time spent on in-person evaluation and communication. Weighting, based on the ratio of hospitalists to survey respondents at each AMC, was used to calculate the average institutional percentages across the study sample.
Qualitative
Responses to open-ended questions were analyzed using thematic analysis.9 Three independent reviewers (STV, JC, ESW) read, analyzed, and grouped the responses by codes. Codes were then assessed for overlap and grouped into themes by one reviewer (STV). A table of themes with supporting quotes and the number of mentions was subsequently developed by all three reviewers. Similar themes were combined to create domains. The domains were reviewed by the steering committee members to create a consensus description (Appendix 3).
The University of Texas Health San Antonio’s Institutional Review Board and participating institutions approved the study as exempt.
RESULTS
Site Characteristics
Representatives from 10 AMCs reported data on a range of one to four hospitals for a total of 22 hospitals. The median reported that the number of medical patients admitted in a 24-hour period was 31-40 (range, 11-20 to >50). The median group size of hospitalists was 41-50 (range, 0-10 to >70).
The survey response rate was 40% (n = 235), ranging from 9%-70% between institutions. Self-identified female hospitalists accounted for 52% of respondents. Four percent were 25-29 years old, 66% were 30-39 years old, 24% were 40-49 years old, and 6% were ≥50 years old. The average clinical time spent as a triagist was 16%.
Description of Triagist Activities
The activities identified by the majority of respondents across all sites included transferring patients within the hospital (73%), and assessing/approving patient transfers from outside hospitals and clinics (82%). Internal transfer activities reported by >50% of respondents included allocating patients within the hospital or bed capacity coordination, assessing intensive care unit transfers, assigning ED admissions, and consulting other services. The ED accounted for an average of 55% of calls received. Respondents also reported being involved with the documentation related to these activities.
Similarities and Differences across AMCs
Two AMCs did not have a dedicated triagist; instead, physicians supervised residents and advanced practice providers. Among the eight sites with triagists, triaging was predominantly done by faculty physicians contacted via pagers. At seven of these sites, 100% of hospitalists worked as triagists. The triage service was covered by faculty physicians from 8-24 hours per day.
Bed boards and transfer centers staffed by registered nurses, nurse coordinators, house supervisors, or physicians were common support systems, though this infrastructure was organized differently across institutions. A UM review before admission was performed at three institutions 24 hours/day. The remaining institutions reviewed patients retrospectively.
Twenty-eight percent of hospitalists across all sites “Disagreed” or “Strongly disagreed” that a patient must meet UM criteria for admission. Forty-two percent had “Frequent” different opinions regarding patient management than the consulting provider.
Triagist and current provider communication practices varied widely across AMCs (Figure). There was significant variability in verbal communication (P = .02), with >70% of respondents at two AMCs reporting verbal communication at least half the time, but <30% reporting this frequency at two other AMCs. Respondents reported variable use of electronic communication (ie, notes/orders in the electronic health record) across AMCs (
The practice of evaluating patients in person also varied significantly across AMCs (P < .0001, Figure). Across hospitalists, only 28% see patients in person about “Half-the-Time” or more.
Differences within AMCs
Variability within AMCs was greatest for the rate of verbal communication practices, with a typical interquartile range (IQR) of 20% to 90% among the hospitalists within a given AMC and for the rate of electronic communication with a typical IQR of 0% to 50%. For other survey questions, the IQR was typically 15 to 20 percentage points.
Thematic Analysis
We received 207 and 203 responses (88% and 86%, respectively) to the open-ended questions “What qualities does an effective triagist have?’ and ‘What qualities make a triagist ineffective?” We identified 22 themes for effective and ineffective qualities, which were grouped into seven domains (Table). All themes had at least three mentions by respondents. The three most frequently mentioned themes, communication skills, efficiency, and systems knowledge, had greater than 60 mentions.
DISCUSSION
Our study of the triagist role at 10 AMCs describes critical triagist functions and identifies key findings across and within AMCs. Twenty-eight percent of hospitalists reported admitting patients even when the patient did not meet the admission criteria, consistent with previous research demonstrating the influence of factors other than clinical disease severity on triage decisions.10 However, preventable admissions remain a hospital-level quality metric.11,12 Triagists must often balance each patient’s circumstances with the complexities of the system. Juggling the competing demands of the system while providing patient-centered care can be challenging and may explain why attending physicians are more frequently filling this role.13
Local context/culture is likely to play a role in the variation across sites; however, compensation for the time spent may also be a factor. If triage activities are not reimbursable, this could lead to less documentation and a lower likelihood that patients are evaluated in person.14 This reason may also explain why all hospitalists were required to serve as a triagist at most sites.
Currently, no consensus definition of the triagist role has been developed. Our results demonstrate that this role is heterogeneous and grounded in the local healthcare system practices. We propose the following working definition of the triagist: a physician who assesses patients for admission, actively supporting the transition of the patient from the outpatient to the inpatient setting. A triagist should be equipped with a skill set that includes not only clinical knowledge but also emphasizes systems knowledge, awareness of others’ goals, efficiency, an ability to communicate effectively, and the knowledge of UM. We recommend that medical directors of hospitalist programs focus their attention on locally specific, systems-based skills development when orienting new hospitalists. The financial aspects of cost should be considered and delineated as well.
Our analysis is limited in several respects. Participant AMCs were not randomly chosen, but do represent a broad array of facility types, group size, and geographic regions. The low response rates at some AMCs may result in an inaccurate representation of those sites. Data was not obtained on hospitalists that did not respond to the survey; therefore, nonresponse bias may affect outcomes. This research used self-report rather than direct observation, which could be subject to recall and social desirability bias. Finally, our results may not be generalizable to nonacademic institutions.
CONCLUSION
The hospitalist role as triagist at AMCs emphasizes communication, organizational skills, efficiency, systems-based practice, and UM knowledge. Although we found significant variation across and within AMCs, internal transfer activities were common across programs. Hospitalist programs should focus on systems-based skills development to prepare hospitalists for the role. The skill set necessary for triagist responsibilities also has implications for internal medicine resident education.4 With increasing emphasis on value and system effectiveness in care delivery, further studies of the triagist role should be undertaken.
Acknowledgments
The TRIAGIST Collaborative Group consists of: Maralyssa Bann, MD, Andrew White, MD (University of Washington); Jagriti Chadha, MD (University of Kentucky); Joel Boggan, MD (Duke University); Sherwin Hsu, MD (UCLA); Jeff Liao, MD (Harvard Medical School); Tabatha Matthias, DO (University of Nebraska Medical Center); Tresa McNeal, MD (Scott and White Texas A&M); Roxana Naderi, MD, Khooshbu Shah, MD (University of Colorado); David Schmit, MD (University of Texas Health San Antonio); Manivannan Veerasamy, MD (Michigan State University).
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the po
1. Kisuule F, Howell EE. Hospitalists and their impact on quality, patient safety, and satisfaction. Obstet Gynecol Clin North Am. 2015; 42(3):433-446. https://doi.org/10.1016/j.ogc.2015.05.003.
2. Wachter, RM, Goldman, L. Zero to 50,000-The 20th anniversary of the hospitalist. N Engl J Med. 2016;375(11): 1009-1011. https://doi.org/10.1056/NEJMp1607958.
3. Vasilevskis EE, Knebel RJ, Wachter RM, Auerbach AD. California hospital leaders’ views of hospitalists: meeting needs of the present and future. J Hosp Med. 2009;4:528-534. https://doi.org/10.1002/jhm.529.
4. Wang ES, Velásquez ST, Smith CJ, et al. Triaging inpatient admissions: an opportunity for resident education. J Gen Intern Med. 2019; 34(5):754-757. https://doi.org/10.1007/s11606-019-04882-2.
5. Briones A, Markoff B, Kathuria N, et al. A model of a hospitalist role in the care of admitted patients in the emergency department. J Hosp Med. 2010;5(6):360-364. https://doi.org/10.1002/jhm.636.
6. Howell EE, Bessman ES, Rubin HR. Hospitalists and an innovative emergency department admission process. J Gen Intern Med. 2004;19:266-268. https://doi.org/10.1111/j.1525-1497.2004.30431.x.
7. Howell E, Bessman E, Marshall R, Wright S. Hospitalist bed management effecting throughput from the emergency department to the intensive care unit. J Crit Care. 2010;25:184-189. https://doi.org/10.1016/j.jcrc.2009.08.004.
8. Chadaga SR, Shockley L, Keniston A, et al. Hospitalist-led medicine emergency department team: associations with throughput, timeliness of patient care, and satisfaction. J Hosp Med. 2012;7:562-566. https://doi.org/10.1002/jhm.1957.
9. Braun, V. Clarke, V. Using thematic analysis in psychology. Qualitative Research in Psychology. 2006;77-101. https://doi.org/10.1191/1478088706qp063oa.
10. Lewis Hunter AE, Spatz ES, Bernstein SL, Rosenthal MS. Factors influencing hospital admission of non-critically ill patients presenting to the emergency department: a cross-sectional study. J Gen Intern Med. 2016;31(1):37-44. https://doi.org/10.1007/s11606-015-3438-8.
11. Patel KK, Vakharia N, Pile J, Howell EH, Rothberg MB. Preventable admissions on a general medicine service: prevalence, causes and comparison with AHRQ prevention quality indicators-a cross-sectional analysis. J Gen Intern Med. 2016;31(6):597-601. https://doi.org/10.1007/s11606-016-3615-4.
12. Daniels LM1, Sorita A2, Kashiwagi DT, et al. Characterizing potentially preventable admissions: a mixed methods study of rates, associated factors, outcomes, and physician decision-making. J Gen Intern Med. 2018;33(5):737-744. https://doi.org/10.1007/s11606-017-4285-6.
13. Howard-Anderson J, Lonowski S, Vangala S, Tseng CH, Busuttil A, Afsar-Manesh N. Readmissions in the era of patient engagement. JAMA Intern Med. 2014;174(11):1870-1872. https://doi.org/10.1001/jamainternmed.2014.4782.
14. Hinami K, Whelan CT, Miller JA, Wolosin RJ, Wetterneck TB, Society of Hospital Medicine Career Satisfaction Task Force. Job characteristics, satisfaction, and burnout across hospitalist practice models. J Hosp Med. 2012;7(5):402-410. https://doi.org/10.1002/jhm.1907
1. Kisuule F, Howell EE. Hospitalists and their impact on quality, patient safety, and satisfaction. Obstet Gynecol Clin North Am. 2015; 42(3):433-446. https://doi.org/10.1016/j.ogc.2015.05.003.
2. Wachter, RM, Goldman, L. Zero to 50,000-The 20th anniversary of the hospitalist. N Engl J Med. 2016;375(11): 1009-1011. https://doi.org/10.1056/NEJMp1607958.
3. Vasilevskis EE, Knebel RJ, Wachter RM, Auerbach AD. California hospital leaders’ views of hospitalists: meeting needs of the present and future. J Hosp Med. 2009;4:528-534. https://doi.org/10.1002/jhm.529.
4. Wang ES, Velásquez ST, Smith CJ, et al. Triaging inpatient admissions: an opportunity for resident education. J Gen Intern Med. 2019; 34(5):754-757. https://doi.org/10.1007/s11606-019-04882-2.
5. Briones A, Markoff B, Kathuria N, et al. A model of a hospitalist role in the care of admitted patients in the emergency department. J Hosp Med. 2010;5(6):360-364. https://doi.org/10.1002/jhm.636.
6. Howell EE, Bessman ES, Rubin HR. Hospitalists and an innovative emergency department admission process. J Gen Intern Med. 2004;19:266-268. https://doi.org/10.1111/j.1525-1497.2004.30431.x.
7. Howell E, Bessman E, Marshall R, Wright S. Hospitalist bed management effecting throughput from the emergency department to the intensive care unit. J Crit Care. 2010;25:184-189. https://doi.org/10.1016/j.jcrc.2009.08.004.
8. Chadaga SR, Shockley L, Keniston A, et al. Hospitalist-led medicine emergency department team: associations with throughput, timeliness of patient care, and satisfaction. J Hosp Med. 2012;7:562-566. https://doi.org/10.1002/jhm.1957.
9. Braun, V. Clarke, V. Using thematic analysis in psychology. Qualitative Research in Psychology. 2006;77-101. https://doi.org/10.1191/1478088706qp063oa.
10. Lewis Hunter AE, Spatz ES, Bernstein SL, Rosenthal MS. Factors influencing hospital admission of non-critically ill patients presenting to the emergency department: a cross-sectional study. J Gen Intern Med. 2016;31(1):37-44. https://doi.org/10.1007/s11606-015-3438-8.
11. Patel KK, Vakharia N, Pile J, Howell EH, Rothberg MB. Preventable admissions on a general medicine service: prevalence, causes and comparison with AHRQ prevention quality indicators-a cross-sectional analysis. J Gen Intern Med. 2016;31(6):597-601. https://doi.org/10.1007/s11606-016-3615-4.
12. Daniels LM1, Sorita A2, Kashiwagi DT, et al. Characterizing potentially preventable admissions: a mixed methods study of rates, associated factors, outcomes, and physician decision-making. J Gen Intern Med. 2018;33(5):737-744. https://doi.org/10.1007/s11606-017-4285-6.
13. Howard-Anderson J, Lonowski S, Vangala S, Tseng CH, Busuttil A, Afsar-Manesh N. Readmissions in the era of patient engagement. JAMA Intern Med. 2014;174(11):1870-1872. https://doi.org/10.1001/jamainternmed.2014.4782.
14. Hinami K, Whelan CT, Miller JA, Wolosin RJ, Wetterneck TB, Society of Hospital Medicine Career Satisfaction Task Force. Job characteristics, satisfaction, and burnout across hospitalist practice models. J Hosp Med. 2012;7(5):402-410. https://doi.org/10.1002/jhm.1907
© 2019 Society of Hospital Medicine
Clinical Guideline Highlights for the Hospitalist: Initial Management of Acute Pancreatitis in the Hospitalized Adult
Acute pancreatitis (AP) is the most common gastrointestinal discharge diagnosis in the United States, with a mortality rate of 1%-5%.1 Recent data demonstrate increasing AP-related admissions, making AP management of utmost importance to hospitalists.1 The American Gastroenterological Association (AGA) guideline specifically addresses AP management in the initial 48-72 hours of admission, during which management decisions can alter disease course and length of stay. AP requires two of the following three criteria for diagnosis: characteristic abdominal pain, elevation of lipase or amylase ≥3 times the upper limit of normal, and/or radiographic evidence of pancreatitis on cross-sectional imaging. The guideline provides eight recommendations, which we consolidated to highlight practice changing recommendations: fluids, nutrition, management of the most common causes, and prophylactic antibiotics.2,3
KEY RECOMMENDATIONS FOR THE HOSPITALIST
Fluids
Recommendation 1. In patients with AP, use goal-directed isotonic crystalloids for fluid management (conditional recommendation, very low-quality evidence).
The guideline emphasizes goal-directed fluid management despite low-quality, heterogeneous evidence and does not recommend Ringer’s lactate over normal saline. “Goal-directed” fluid management involves the use of crystalloid infusions titrated to improve physiologic and biochemical markers, but no target volume is specified by the guideline. Frequent reassessments should look for signs of volume overload, the primary risk of harm with fluid therapy. Despite failure to reduce mortality or morbidities such as pancreatic necrosis or persistent multi-organ failure, the AGA cites the mortality benefit of goal-directed therapy in sepsis as justification for this approach in AP, given the similar physiologic abnormalities.
Nutrition
Recommendation 2. Begin feeding early in patients with AP regardless of predicted severity. If oral nutrition is not tolerated, enteral feeding with either a nasogastric or nasojejunal tube is preferred to parenteral nutrition (strong recommendation, moderate-quality evidence).
Early feeding (ie, within 24 hours) is recommended regardless of AP severity. This represents a change from prior practices of bowel rest, theorized to prevent continued stimulation of an inflamed pancreas. Although early feeding has not been linked to improved mortality, it has demonstrated lower rates of multi-organ failure and infected pancreatic necrosis, possibly due to maintenance of the gut mucosal barrier and reduced bacterial translocation. When oral feeding is not tolerated, enteral nutrition is preferred over parenteral nutrition due to less risks. The preferred dietary composition guidance for patients with persistent pain or ileus is not addressed.
Management of the Most Common Causes of AP in Adults
Recommendation 3. Patients with mild acute biliary pancreatitis should have cholecystectomy during the initial admission (strong recommendation, moderate-quality evidence).
All patients with suspected biliary pancreatitis should receive a surgical consultation for cholecystectomy during the index admission. At the time of the guideline release, only one trial was available to support the recommendation of early cholecystectomy; however, newer studies similarly support cholecystectomy during index admission by demonstrating reductions in composite outcomes of mortality and gallstone-related complications, readmission for pancreatitis, and other pancreatobiliary complications.4 A Cochrane review included in the guideline found no differences in complication rates even in patients with severe biliary pancreatitis. In the absence of cholangitis, urgent endoscopic retrograde cholangiography (ERCP) is not indicated as most stones causing biliary pancreatitis pass spontaneously.
Recommendation 4. In patients with acute alcoholic pancreatitis, brief alcohol intervention should occur during admission (strong recommendation, moderate-quality evidence).
Ongoing alcohol consumption is a risk factor for recurrent acute and chronic pancreatitis. Only one trial assessed the impact of inpatient alcohol cessation counseling on recurrent AP, noting a trend toward reduced readmissions.5 However, indirect evidence from similar interventions in ambulatory settings demonstrates reductions in alcohol intake, leading to the AGA recommendation for inpatients with alcohol-induced AP.3
Antibiotics
Recommendation 5. Avoid empiric antibiotics in patients with AP who otherwise lack an indication, regardless of predicted severity (conditional recommendation, low-quality evidence).
Since 2002, well performed trials have consistently failed to demonstrate improvement in outcomes such as multi-organ failure or length of stay with use of prophylactic antibiotics for AP, even severe AP and pancreatic necrosis. Therefore, the AGA recommends against prophylactic antibiotics in initial management of AP regardless of disease severity. Lack of blinding in the majority of trial designs conducted before 2002 contributed to the overall assessment of low-quality evidence. The guideline does not address acute biliary pancreatitis with cholangitis, for which antibiotics and ERCP for decompression are critical.
CRITIQUE
The AGA Institute supported this guideline development and employed the rigorous and standardized GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology. This approach allowed the guideline panel members to account not only for evidence quality, but also the benefits and harms of an intervention and resource utilization. None of the authors had any stated conflicts of interest.
The guideline heavily weighted results from randomized control trials, most of which excluded key populations cared for by hospitalists (eg, patients older than 75 years, with end-stage renal disease). Particular areas where this creates challenges for clinicians and patients alike include goal-directed fluid therapy and when to consider more invasive interventions such as ERCP and early cholecystectomy. For example, patients considered to be poor surgical candidates may benefit from ERCP with biliary sphincterotomy to reduce the risk of recurrent biliary pancreatitis.
Lack of specificity in the guidelines for goal-directed fluid management and enteral feeding regimens makes it challenging to standardize hospitalists’ approach to the early care of patients with AP. Interestingly, the 2013 American College of Gastroenterology (ACG) Guideline for the Management of AP included strong recommendations for the use of Ringer’s lactate and volume targets in the initial management of AP.6 Evidence supporting the use of Ringer’s lactate versus normal saline is based largely upon improved inflammatory markers, theoretical potentiation of pancreatic enzyme activation with hypercholemic metabolic acidosis, and small studies demonstrating trends toward improved mortality.7 The ACG guideline was released prior to mounting evidence suggesting that goal-directed fluid therapy in sepsis does not improve mortality versus usual care.8 The growing uncertainty regarding the efficacy of goal-directed fluids for septic shock, as well limitations of studies on AP, may contribute to the differences between the AGA and ACG recommendations.
Finally, as the guideline covers the initial therapeutic management of AP, no recommendations are made for diagnostic studies such as right upper quadrant ultrasound. This noninvasive and readily available test plays a critical role in evaluating for presence of gallstones and other potential etiologies of abdominal pain.
AREAS IN NEED OF FUTURE STUDY
Additional research is needed to better understand goal-directed fluid therapy with respect to the fluid type, amount, and target outcomes. Similarly, determining the optimal enteral feeding regimens for patients failing oral intake would help clinicians meet the recommendation for early nutrition. Finally, clarification on the roles and timing of endoscopic and surgical procedures for patients with severe biliary pancreatitis, as well as geriatric and medically complex populations, would help hospitalists advocate for a multidisciplinary approach to this common and often serious disease.
Disclosures
The authors have nothing to disclose.
1. Krishna SG, Kamboj AK, Hart PA, Hinton A, Conwell DL. The changing epidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas. 2017;46(4):482-488. https://doi.org/10.1097/MPA.0000000000000783.
2. Crockett SD, Wani S, Gardner TB, et al. American Gastroenterological Association Institute Guideline on initial management of acute pancreatitis. Gastroenterology. 2018;154(4):1096-1101. https://doi.org/10.1053/j.gastro.2018.01.032.
3. Vege SS, DiMagno MJ, Forsmark CE, Martel M, Barkun AN. Initial medical treatment of acute pancreatitis: American Gastroenterological Association Institute technical review. Gastroenterology. 2018;154(4):1103-1139. https://doi.org/10.1053/j.gastro.2018.01.031.
4 Noel R, Arnelo U, Lundell L, et al. Index versus delayed cholecystectomy in mild gallstone pancreatitis: results of a randomized controlled trial. HPB (Oxford). 2018;20(10):932-938. https://doi.org/10.1016/j.hpb.2018.03.016.
5. Kaner EF, Beyer F, Dickinson HO, et al. Effectiveness of brief alcohol interventions in primary care populations. Cochrane Database Syst Rev. 2007:CD004148. https://doi.org/10.1002/14651858.CD004148.pub3.
6. Tenner S, Baillie J, DeWitt J, Vege SS. American College of Gastroenterology guideline: Management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415. https://doi.org/10.1038/ajg.2013.218.
7. de-Madaria E, Herrera-Marante I, González-Camacho V, et al. Fluid resuscitation with lactated Ringer’s solution vs normal saline in acute pancreatitis: a triple-blind, randomized, controlled trial. United European Gastroenterol J. 2018;6(1):63-72. https://doi.org/10.1177/2050640617707864
8. The PRISM Investigators. Early, goal-directed therapy for septic shock — a patient-level meta-analysis. New Engl J Med. 2017;376(23):2223-2234. https://doi.org/10.1056/NEJMoa1701380.
Acute pancreatitis (AP) is the most common gastrointestinal discharge diagnosis in the United States, with a mortality rate of 1%-5%.1 Recent data demonstrate increasing AP-related admissions, making AP management of utmost importance to hospitalists.1 The American Gastroenterological Association (AGA) guideline specifically addresses AP management in the initial 48-72 hours of admission, during which management decisions can alter disease course and length of stay. AP requires two of the following three criteria for diagnosis: characteristic abdominal pain, elevation of lipase or amylase ≥3 times the upper limit of normal, and/or radiographic evidence of pancreatitis on cross-sectional imaging. The guideline provides eight recommendations, which we consolidated to highlight practice changing recommendations: fluids, nutrition, management of the most common causes, and prophylactic antibiotics.2,3
KEY RECOMMENDATIONS FOR THE HOSPITALIST
Fluids
Recommendation 1. In patients with AP, use goal-directed isotonic crystalloids for fluid management (conditional recommendation, very low-quality evidence).
The guideline emphasizes goal-directed fluid management despite low-quality, heterogeneous evidence and does not recommend Ringer’s lactate over normal saline. “Goal-directed” fluid management involves the use of crystalloid infusions titrated to improve physiologic and biochemical markers, but no target volume is specified by the guideline. Frequent reassessments should look for signs of volume overload, the primary risk of harm with fluid therapy. Despite failure to reduce mortality or morbidities such as pancreatic necrosis or persistent multi-organ failure, the AGA cites the mortality benefit of goal-directed therapy in sepsis as justification for this approach in AP, given the similar physiologic abnormalities.
Nutrition
Recommendation 2. Begin feeding early in patients with AP regardless of predicted severity. If oral nutrition is not tolerated, enteral feeding with either a nasogastric or nasojejunal tube is preferred to parenteral nutrition (strong recommendation, moderate-quality evidence).
Early feeding (ie, within 24 hours) is recommended regardless of AP severity. This represents a change from prior practices of bowel rest, theorized to prevent continued stimulation of an inflamed pancreas. Although early feeding has not been linked to improved mortality, it has demonstrated lower rates of multi-organ failure and infected pancreatic necrosis, possibly due to maintenance of the gut mucosal barrier and reduced bacterial translocation. When oral feeding is not tolerated, enteral nutrition is preferred over parenteral nutrition due to less risks. The preferred dietary composition guidance for patients with persistent pain or ileus is not addressed.
Management of the Most Common Causes of AP in Adults
Recommendation 3. Patients with mild acute biliary pancreatitis should have cholecystectomy during the initial admission (strong recommendation, moderate-quality evidence).
All patients with suspected biliary pancreatitis should receive a surgical consultation for cholecystectomy during the index admission. At the time of the guideline release, only one trial was available to support the recommendation of early cholecystectomy; however, newer studies similarly support cholecystectomy during index admission by demonstrating reductions in composite outcomes of mortality and gallstone-related complications, readmission for pancreatitis, and other pancreatobiliary complications.4 A Cochrane review included in the guideline found no differences in complication rates even in patients with severe biliary pancreatitis. In the absence of cholangitis, urgent endoscopic retrograde cholangiography (ERCP) is not indicated as most stones causing biliary pancreatitis pass spontaneously.
Recommendation 4. In patients with acute alcoholic pancreatitis, brief alcohol intervention should occur during admission (strong recommendation, moderate-quality evidence).
Ongoing alcohol consumption is a risk factor for recurrent acute and chronic pancreatitis. Only one trial assessed the impact of inpatient alcohol cessation counseling on recurrent AP, noting a trend toward reduced readmissions.5 However, indirect evidence from similar interventions in ambulatory settings demonstrates reductions in alcohol intake, leading to the AGA recommendation for inpatients with alcohol-induced AP.3
Antibiotics
Recommendation 5. Avoid empiric antibiotics in patients with AP who otherwise lack an indication, regardless of predicted severity (conditional recommendation, low-quality evidence).
Since 2002, well performed trials have consistently failed to demonstrate improvement in outcomes such as multi-organ failure or length of stay with use of prophylactic antibiotics for AP, even severe AP and pancreatic necrosis. Therefore, the AGA recommends against prophylactic antibiotics in initial management of AP regardless of disease severity. Lack of blinding in the majority of trial designs conducted before 2002 contributed to the overall assessment of low-quality evidence. The guideline does not address acute biliary pancreatitis with cholangitis, for which antibiotics and ERCP for decompression are critical.
CRITIQUE
The AGA Institute supported this guideline development and employed the rigorous and standardized GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology. This approach allowed the guideline panel members to account not only for evidence quality, but also the benefits and harms of an intervention and resource utilization. None of the authors had any stated conflicts of interest.
The guideline heavily weighted results from randomized control trials, most of which excluded key populations cared for by hospitalists (eg, patients older than 75 years, with end-stage renal disease). Particular areas where this creates challenges for clinicians and patients alike include goal-directed fluid therapy and when to consider more invasive interventions such as ERCP and early cholecystectomy. For example, patients considered to be poor surgical candidates may benefit from ERCP with biliary sphincterotomy to reduce the risk of recurrent biliary pancreatitis.
Lack of specificity in the guidelines for goal-directed fluid management and enteral feeding regimens makes it challenging to standardize hospitalists’ approach to the early care of patients with AP. Interestingly, the 2013 American College of Gastroenterology (ACG) Guideline for the Management of AP included strong recommendations for the use of Ringer’s lactate and volume targets in the initial management of AP.6 Evidence supporting the use of Ringer’s lactate versus normal saline is based largely upon improved inflammatory markers, theoretical potentiation of pancreatic enzyme activation with hypercholemic metabolic acidosis, and small studies demonstrating trends toward improved mortality.7 The ACG guideline was released prior to mounting evidence suggesting that goal-directed fluid therapy in sepsis does not improve mortality versus usual care.8 The growing uncertainty regarding the efficacy of goal-directed fluids for septic shock, as well limitations of studies on AP, may contribute to the differences between the AGA and ACG recommendations.
Finally, as the guideline covers the initial therapeutic management of AP, no recommendations are made for diagnostic studies such as right upper quadrant ultrasound. This noninvasive and readily available test plays a critical role in evaluating for presence of gallstones and other potential etiologies of abdominal pain.
AREAS IN NEED OF FUTURE STUDY
Additional research is needed to better understand goal-directed fluid therapy with respect to the fluid type, amount, and target outcomes. Similarly, determining the optimal enteral feeding regimens for patients failing oral intake would help clinicians meet the recommendation for early nutrition. Finally, clarification on the roles and timing of endoscopic and surgical procedures for patients with severe biliary pancreatitis, as well as geriatric and medically complex populations, would help hospitalists advocate for a multidisciplinary approach to this common and often serious disease.
Disclosures
The authors have nothing to disclose.
Acute pancreatitis (AP) is the most common gastrointestinal discharge diagnosis in the United States, with a mortality rate of 1%-5%.1 Recent data demonstrate increasing AP-related admissions, making AP management of utmost importance to hospitalists.1 The American Gastroenterological Association (AGA) guideline specifically addresses AP management in the initial 48-72 hours of admission, during which management decisions can alter disease course and length of stay. AP requires two of the following three criteria for diagnosis: characteristic abdominal pain, elevation of lipase or amylase ≥3 times the upper limit of normal, and/or radiographic evidence of pancreatitis on cross-sectional imaging. The guideline provides eight recommendations, which we consolidated to highlight practice changing recommendations: fluids, nutrition, management of the most common causes, and prophylactic antibiotics.2,3
KEY RECOMMENDATIONS FOR THE HOSPITALIST
Fluids
Recommendation 1. In patients with AP, use goal-directed isotonic crystalloids for fluid management (conditional recommendation, very low-quality evidence).
The guideline emphasizes goal-directed fluid management despite low-quality, heterogeneous evidence and does not recommend Ringer’s lactate over normal saline. “Goal-directed” fluid management involves the use of crystalloid infusions titrated to improve physiologic and biochemical markers, but no target volume is specified by the guideline. Frequent reassessments should look for signs of volume overload, the primary risk of harm with fluid therapy. Despite failure to reduce mortality or morbidities such as pancreatic necrosis or persistent multi-organ failure, the AGA cites the mortality benefit of goal-directed therapy in sepsis as justification for this approach in AP, given the similar physiologic abnormalities.
Nutrition
Recommendation 2. Begin feeding early in patients with AP regardless of predicted severity. If oral nutrition is not tolerated, enteral feeding with either a nasogastric or nasojejunal tube is preferred to parenteral nutrition (strong recommendation, moderate-quality evidence).
Early feeding (ie, within 24 hours) is recommended regardless of AP severity. This represents a change from prior practices of bowel rest, theorized to prevent continued stimulation of an inflamed pancreas. Although early feeding has not been linked to improved mortality, it has demonstrated lower rates of multi-organ failure and infected pancreatic necrosis, possibly due to maintenance of the gut mucosal barrier and reduced bacterial translocation. When oral feeding is not tolerated, enteral nutrition is preferred over parenteral nutrition due to less risks. The preferred dietary composition guidance for patients with persistent pain or ileus is not addressed.
Management of the Most Common Causes of AP in Adults
Recommendation 3. Patients with mild acute biliary pancreatitis should have cholecystectomy during the initial admission (strong recommendation, moderate-quality evidence).
All patients with suspected biliary pancreatitis should receive a surgical consultation for cholecystectomy during the index admission. At the time of the guideline release, only one trial was available to support the recommendation of early cholecystectomy; however, newer studies similarly support cholecystectomy during index admission by demonstrating reductions in composite outcomes of mortality and gallstone-related complications, readmission for pancreatitis, and other pancreatobiliary complications.4 A Cochrane review included in the guideline found no differences in complication rates even in patients with severe biliary pancreatitis. In the absence of cholangitis, urgent endoscopic retrograde cholangiography (ERCP) is not indicated as most stones causing biliary pancreatitis pass spontaneously.
Recommendation 4. In patients with acute alcoholic pancreatitis, brief alcohol intervention should occur during admission (strong recommendation, moderate-quality evidence).
Ongoing alcohol consumption is a risk factor for recurrent acute and chronic pancreatitis. Only one trial assessed the impact of inpatient alcohol cessation counseling on recurrent AP, noting a trend toward reduced readmissions.5 However, indirect evidence from similar interventions in ambulatory settings demonstrates reductions in alcohol intake, leading to the AGA recommendation for inpatients with alcohol-induced AP.3
Antibiotics
Recommendation 5. Avoid empiric antibiotics in patients with AP who otherwise lack an indication, regardless of predicted severity (conditional recommendation, low-quality evidence).
Since 2002, well performed trials have consistently failed to demonstrate improvement in outcomes such as multi-organ failure or length of stay with use of prophylactic antibiotics for AP, even severe AP and pancreatic necrosis. Therefore, the AGA recommends against prophylactic antibiotics in initial management of AP regardless of disease severity. Lack of blinding in the majority of trial designs conducted before 2002 contributed to the overall assessment of low-quality evidence. The guideline does not address acute biliary pancreatitis with cholangitis, for which antibiotics and ERCP for decompression are critical.
CRITIQUE
The AGA Institute supported this guideline development and employed the rigorous and standardized GRADE (Grading of Recommendations Assessment, Development and Evaluation) methodology. This approach allowed the guideline panel members to account not only for evidence quality, but also the benefits and harms of an intervention and resource utilization. None of the authors had any stated conflicts of interest.
The guideline heavily weighted results from randomized control trials, most of which excluded key populations cared for by hospitalists (eg, patients older than 75 years, with end-stage renal disease). Particular areas where this creates challenges for clinicians and patients alike include goal-directed fluid therapy and when to consider more invasive interventions such as ERCP and early cholecystectomy. For example, patients considered to be poor surgical candidates may benefit from ERCP with biliary sphincterotomy to reduce the risk of recurrent biliary pancreatitis.
Lack of specificity in the guidelines for goal-directed fluid management and enteral feeding regimens makes it challenging to standardize hospitalists’ approach to the early care of patients with AP. Interestingly, the 2013 American College of Gastroenterology (ACG) Guideline for the Management of AP included strong recommendations for the use of Ringer’s lactate and volume targets in the initial management of AP.6 Evidence supporting the use of Ringer’s lactate versus normal saline is based largely upon improved inflammatory markers, theoretical potentiation of pancreatic enzyme activation with hypercholemic metabolic acidosis, and small studies demonstrating trends toward improved mortality.7 The ACG guideline was released prior to mounting evidence suggesting that goal-directed fluid therapy in sepsis does not improve mortality versus usual care.8 The growing uncertainty regarding the efficacy of goal-directed fluids for septic shock, as well limitations of studies on AP, may contribute to the differences between the AGA and ACG recommendations.
Finally, as the guideline covers the initial therapeutic management of AP, no recommendations are made for diagnostic studies such as right upper quadrant ultrasound. This noninvasive and readily available test plays a critical role in evaluating for presence of gallstones and other potential etiologies of abdominal pain.
AREAS IN NEED OF FUTURE STUDY
Additional research is needed to better understand goal-directed fluid therapy with respect to the fluid type, amount, and target outcomes. Similarly, determining the optimal enteral feeding regimens for patients failing oral intake would help clinicians meet the recommendation for early nutrition. Finally, clarification on the roles and timing of endoscopic and surgical procedures for patients with severe biliary pancreatitis, as well as geriatric and medically complex populations, would help hospitalists advocate for a multidisciplinary approach to this common and often serious disease.
Disclosures
The authors have nothing to disclose.
1. Krishna SG, Kamboj AK, Hart PA, Hinton A, Conwell DL. The changing epidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas. 2017;46(4):482-488. https://doi.org/10.1097/MPA.0000000000000783.
2. Crockett SD, Wani S, Gardner TB, et al. American Gastroenterological Association Institute Guideline on initial management of acute pancreatitis. Gastroenterology. 2018;154(4):1096-1101. https://doi.org/10.1053/j.gastro.2018.01.032.
3. Vege SS, DiMagno MJ, Forsmark CE, Martel M, Barkun AN. Initial medical treatment of acute pancreatitis: American Gastroenterological Association Institute technical review. Gastroenterology. 2018;154(4):1103-1139. https://doi.org/10.1053/j.gastro.2018.01.031.
4 Noel R, Arnelo U, Lundell L, et al. Index versus delayed cholecystectomy in mild gallstone pancreatitis: results of a randomized controlled trial. HPB (Oxford). 2018;20(10):932-938. https://doi.org/10.1016/j.hpb.2018.03.016.
5. Kaner EF, Beyer F, Dickinson HO, et al. Effectiveness of brief alcohol interventions in primary care populations. Cochrane Database Syst Rev. 2007:CD004148. https://doi.org/10.1002/14651858.CD004148.pub3.
6. Tenner S, Baillie J, DeWitt J, Vege SS. American College of Gastroenterology guideline: Management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415. https://doi.org/10.1038/ajg.2013.218.
7. de-Madaria E, Herrera-Marante I, González-Camacho V, et al. Fluid resuscitation with lactated Ringer’s solution vs normal saline in acute pancreatitis: a triple-blind, randomized, controlled trial. United European Gastroenterol J. 2018;6(1):63-72. https://doi.org/10.1177/2050640617707864
8. The PRISM Investigators. Early, goal-directed therapy for septic shock — a patient-level meta-analysis. New Engl J Med. 2017;376(23):2223-2234. https://doi.org/10.1056/NEJMoa1701380.
1. Krishna SG, Kamboj AK, Hart PA, Hinton A, Conwell DL. The changing epidemiology of acute pancreatitis hospitalizations: a decade of trends and the impact of chronic pancreatitis. Pancreas. 2017;46(4):482-488. https://doi.org/10.1097/MPA.0000000000000783.
2. Crockett SD, Wani S, Gardner TB, et al. American Gastroenterological Association Institute Guideline on initial management of acute pancreatitis. Gastroenterology. 2018;154(4):1096-1101. https://doi.org/10.1053/j.gastro.2018.01.032.
3. Vege SS, DiMagno MJ, Forsmark CE, Martel M, Barkun AN. Initial medical treatment of acute pancreatitis: American Gastroenterological Association Institute technical review. Gastroenterology. 2018;154(4):1103-1139. https://doi.org/10.1053/j.gastro.2018.01.031.
4 Noel R, Arnelo U, Lundell L, et al. Index versus delayed cholecystectomy in mild gallstone pancreatitis: results of a randomized controlled trial. HPB (Oxford). 2018;20(10):932-938. https://doi.org/10.1016/j.hpb.2018.03.016.
5. Kaner EF, Beyer F, Dickinson HO, et al. Effectiveness of brief alcohol interventions in primary care populations. Cochrane Database Syst Rev. 2007:CD004148. https://doi.org/10.1002/14651858.CD004148.pub3.
6. Tenner S, Baillie J, DeWitt J, Vege SS. American College of Gastroenterology guideline: Management of acute pancreatitis. Am J Gastroenterol. 2013;108(9):1400-1415. https://doi.org/10.1038/ajg.2013.218.
7. de-Madaria E, Herrera-Marante I, González-Camacho V, et al. Fluid resuscitation with lactated Ringer’s solution vs normal saline in acute pancreatitis: a triple-blind, randomized, controlled trial. United European Gastroenterol J. 2018;6(1):63-72. https://doi.org/10.1177/2050640617707864
8. The PRISM Investigators. Early, goal-directed therapy for septic shock — a patient-level meta-analysis. New Engl J Med. 2017;376(23):2223-2234. https://doi.org/10.1056/NEJMoa1701380.
© 2019 Society of Hospital Medicine
Hospital Medicine Update: High-Impact Literature from March 2018 to April 2019
Given the breadth and depth of patients cared for by hospital medicine providers, it is challenging to remain current with the literature. The authors critically appraised the literature from March 2018 to April 2019 for high-quality studies relevant to hospital medicine. Articles were selected based on methodologic rigor and likelihood to impact clinical practice. Thirty articles were selected by the presenting authors for the Hospital Medicine Updates at the 2019 Society of Hospital Medicine (CH, CM) and Society of General Internal Medicine Annual Meetings (BS, AB). After two sequential rounds of voting and group discussion to adjudicate voting discrepancies, the authors selected the 10 most impactful articles for this review. Each article is described below with the key points summarized in the Table.
ESSENTIAL PUBLICATIONS
Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). McDonald LC, et al. Clin Infect Dis. 2018;66(7):e1–e48.1
Background. In the United States, approximately 500,000 Clostridioides difficile infections (CDI) occur annually with 15,000-30,000 deaths. CDI has become a marker of hospital quality and has been placed under numerous “pay for performance” metrics. The Infectious Diseases Society of America/Society of Healthcare Epidemiology of America updated their guidelines from 2010 regarding hospital surveillance, diagnostic testing, treatment, and infection precautions and control.
Findings. The panel included 14 multidisciplinary experts in epidemiology, diagnosis, infection control, and clinical management of adult and pediatric CDI. They used problem intervention comparison-outcome (PICO)-formatted, evidence-based questions. The selection of data and final recommendations were made in accordance with the GRADE criteria. A total of 35 recommendations were made.
Key clinical recommendations for hospitalists caring for adults: (1) Prescribe vancomycin or fidaxomicin over metronidazole for the initial treatment of CDI (strong recommendation, high quality of evidence); (2) Limit testing to the patients with unexplained new onset diarrhea, which is defined as greater than or equal to 3 unformed stools in 24 hours (weak recommendation, very low-quality evidence); (3) Avoid routine repeat testing within seven days, and only test asymptomatic patients for epidemiologic reasons (strong recommendation, moderate-quality evidence); (4) Minimize the frequency and duration of high-risk antibiotic therapy and the number of antibiotic agents prescribed (strong recommendation, moderate quality of evidence); (5) Discontinue therapy with the inciting antibiotic agent as soon as possible (strong recommendation, moderate quality of evidence).
Caveats. As with the clinical application of any guidelines, individual case adjustments may be required.
Implications. Vancomycin or fidaxomicin should be used for the initial episode of CDI instead of metronidazole.
Mortality and Morbidity in Acutely Ill Adults Treated with Liberal versus Conservative Oxygen Therapy (IOTA): a Systematic Review and Meta-analysis. Chu DK, et al. Lancet. 2018;391(10131):1693-1705.2
Background. Supplemental oxygen is often given to acutely ill hospitalized adults, even when they are not hypoxic or dyspneic. The safety and efficacy of this practice is unknown.
Findings. This systematic review and meta-analysis evaluated 25 randomized controlled trials enrolling 16,037 patients. Patients presented with several conditions, including sepsis, critical illness, stroke, myocardial infarction, and emergency surgery. The fraction of inspired oxygen in the liberal arms varied from 30% to 100%. Most patients randomized to the conservative arm received no supplemental oxygen. Delivery of liberal oxygen to acutely ill adults was associated with increased in-hospital mortality (relative risk [RR]: 1.21; 95% CI: 1.03-1.43), 30-day mortality (RR: 1.14; 95% CI: 1.01-1.29), and 90-day mortality (RR: 1.10; 95% CI: 1.00-1.20). The results were believed to be of high quality and were robust across multiple sensitivity analyses. It seemed that the mortality began to increase when supplemental oxygen raised the peripheral oxygen saturation (Sp02) above a range of 94%-96%.
Caveats. Heterogeneity was observed in the study settings and oxygen delivery. In addition, the cause for increased mortality could not be determined.
Implications. In hospitalized acutely ill adults, “liberal” supplemental oxygen was associated with increased in-hospital and longer-term mortality. The study authors postulated that this finding resulted from the direct toxic effects of oxygen or that oxygen delivery may “mask” illness and lead to delays in diagnosis and treatment. A subsequent clinical practice guideline recommends (1) a target SpO2 of less than 96% for patients receiving oxygen therapy; (2) a target SpO2 range of 90%-94% seems appropriate for most hospitalized adults.3
Do Words Matter? Stigmatizing Language and the Transmission of Bias in the Medical Record. P Goddu A, et al. J Gen Intern Med. 2018;33(5):68-91.4
Background. Previous work has shown that clinician bias affects health outcomes, often worsening health disparities. It is unknown whether clinicians’ language in medical records biases other clinicians and whether this affects patients.
Findings. The investigators randomized medical students and residents in internal and emergency medicine at one academic medical center to review one of two vignettes in the format of notes on the same hypothetical patient with sickle cell disease (SCD) admitted with a pain crisis. One vignette contained stigmatizing language, and the other contained neutral language. The trainees exposed to the vignettes with stigmatizing language showed a more negative attitude toward the patient, as measured by a previously validated scale of attitudes toward patients with SCD (20.6 stigmatizing vs 25.6 neutral, with a total score range of 7-35 for the instrument; higher scores indicate more positive attitudes; P < .001). Furthermore, the intensity of pain treatment was assessed in the resident group and was less aggressive when residents were exposed to stigmatizing language (5.56 stigmatizing vs 6.22 neutral on a scale of 2-7, with higher scores indicating more aggressive pain treatment; P = .003).
Cautions. This research was a single-center study of residents and medical students in two departments. Additionally, the study used vignettes on a hypothetical patient so trainees in the study group might have witnessed stronger stigmatizing language than what is typically observed in an actual patients’ notes.
Implications. Stigmatizing language used in medical records possibly contributed to health disparities by negatively impacting other physicians’ biases and prescribing practices toward patients with SCD at an academic medical center. Clinicians should avoid stigmatizing language in medical records.
Catheter Ablation for Atrial Fibrillation with Heart Failure. Marrouche, NF et al. New Engl J Med. 2018;378:417-427.5
Background. Atrial fibrillation (AF) in patients with heart failure is associated with increased mortality and morbidity. Small-scale studies have suggested that ablation of AF may benefit patients with heart failure.
Findings. This multicenter trial included 398 patients with heart failure and symptomatic AF. Patients had New York Heart Association Class II-IV heart failure, an ejection fraction (EF) of 35% or less, and an internal cardiac defibrillator (ICD). Patients were randomized to either ablation or medical therapy. All enrolled patients either refused, failed, or showed poor tolerance to antiarrhythmic therapy for AF. The primary outcome was death from any cause or hospitalization for heart failure.
The composite endpoint occurred in 28.5% of the ablation group versus 44.6% of patients in the medical therapy group (hazard ratio [HR]: 0.62; 95% CI: 0.43-0.87). Fewer patients in the ablation group died (13% vs 25%; HR: 0.53; 95% CI: 0.32-0.86) or were hospitalized for heart failure (21% vs 36%; HR: 0.56; 95% CI: 0.37-0.83). The patients in the ablation group had higher EF increases above baseline and a greater proportion were in sinus rhythm at the 60-month follow-up visit.
Cautions. The trial was terminated early due to slow recruitment and lower than expected events. Over twice as many patients were lost to follow-up in the ablation group versus the medical therapy group, and by 60 months, AF recurred in 50% of patients who underwent ablation. The sample size was small, and the trial was unblinded.
Implications. Ablation should be considered for AF in patients with heart failure. Additional studies to evaluate ablation versus medical therapy for patients with heart failure and AF are underway.
Medication for Opioid Use Disorder after Nonfatal Opioid Overdose and Association with Mortality. Larochelle MR, et al. Ann Intern Med. 2018;169(3):137-145.6
Background. More than 70,000 Americans died of drug overdose in 2017; this number is higher than the deaths resulting from human immunodeficiency virus, car crash, or gun violence at their peaks.7 Methadone, buprenorphine, and naltrexone are approved by the Federal Drug Administration for the treatment of opioid use disorder (OUD). These medications increase treatment retention; methadone and buprenorphine have been associated with significant decreases in all-cause and overdose mortality.8 However, whether receipt of these medications following a nonfatal opioid overdose reduces mortality is unknown.
Findings. This retrospective cohort study included 17,568 opioid overdose survivors from the Massachusetts’s Public Health Dataset between 2012 and 2014. Only three in 10 of these patients received any medications for OUD over 12 months following overdose. All-cause mortality was 4.7 deaths (95% CI: 4.4-5.0 deaths) per 100 person-years. The relative risk for all-cause mortality was 53% lower with methadone (adjusted hazard ratio [aHR]: 0.47; 95% CI: 0.32-0.71) and 37% lower with buprenorphine (aHR: 0.63; 95% CI: 0.46-0.87).
Caveats. This cohort study may have missed confounders explaining why certain patients received medications for OUD. As a result, association cannot be interpreted as causation.
Implications. Methadone and buprenorphine are associated with a reduction in preventable deaths in patients with OUD who have survived an overdose. All patients with OUD should be considered for therapy.
Outcomes Associated with Apixaban Use in Patients with End-Stage Kidney Disease and Atrial Fibrillation in the United States. Siontis, KC, et al. Circulation. 2018;138:1519–1529.9
Background. Patients with end-stage kidney disease (ESKD) have poor outcomes when treated with warfarin for AF. These patients were excluded from clinical trials of direct oral anticoagulants. The goal of this study was to determine the outcomes of the use of apixaban in patients with ESKD and AF.
Findings. This retrospective cohort study included 25,523 Medicare patients with ESKD and AF on anticoagulants. A 3:1 propensity score match was performed between patients on warfarin and apixaban. Time without stroke/systemic embolism, bleeding (major, gastrointestinal, and intracranial), and death were assessed. A total of 2,351 patients were on apixaban, and 23,172 patients were on warfarin. No difference was observed in the risk of stroke/systemic embolism between apixaban and warfarin (HR 0.88; 95% CI: 0.69-1.12). Apixaban was associated with a lower risk of major bleeding (HR: 0.72; 95% CI: 0.59-0.87). Standard-dose apixaban (5 mg twice a day) was associated with lower risks of stroke/systemic embolism and death compared with reduced-dose apixaban (2.5 mg twice a day; n = 1,317; HR: 0.61; 95% CI: 0.37-0.98; P = .04 for stroke/systemic embolism; HR: 0.64; 95% CI: 0.45-0.92; P = .01 for death) or warfarin (HR: 0.64; 95% CI: 0.42-0.97; P = .04 for stroke/systemic embolism; HR: 0.63; 95% CI: 0.46-0.85; P = .003 for death).
Cautions. There may be unique patient factors that led providers to prescribe apixaban to patients with ESKD.
Implications. The use of standard-dose apixaban appears safe and potentially preferable in patients with ESKD and AF due to reductions in major bleeding, thromboembolism, and mortality risk compared with warfarin. Several additional studies are pending to evaluate the use and dose of apixaban in patients with ESKD and AF.
Outcomes Associated with De-escalating Therapy for Methicillin-Resistant Staphylococcus aureus in Culture-Negative Nosocomial Pneumonia. Cowley MC, et al. Chest. 2019;155(1):53-59.10
Background. Patients diagnosed with hospital-acquired pneumonia (HAP) are often treated empirically with broad-spectrum antibiotics. In many patients with HAP, cultures remain negative, and providers must decide if antibiotics can safely be narrowed. Specifically, the safety of deciding to “de-escalate” and discontinue the coverage for methicillin-resistant Staphylococcus aureus (MRSA) if cultures remain negative is unclear.
Findings. In this single-center retrospective cohort study, 279 patients who were (1) diagnosed with HAP and (2) had negative sputum cultures were enrolled. The patients in whom MRSA coverage was de-escalated by day four were compared with those with continued anti-MRSA coverage. No difference was observed between the two groups in terms of degree of illness or comorbidities. The patients who were de-escalated received five fewer days of anti-MRSA coverage than patients who were not. No difference was noted in the 28-day mortality between the two groups (de-escalation: 23% vs no de-escalation: 28%; 95% CI: −16.1%-6.5%). The incidence of acute kidney injury (AKI) was significantly lower in the de-escalation group (36% vs 50%; 95% CI: −26.9- 0.04), and the overall length of stay was five days shorter in the de-escalation group (95% CI: 0.1-6.4 days).
Caveats. Given the retrospective nature, unmeasured confounders may have impacted the decision to de-escalate anti-MRSA coverage. The observed lower risk of AKI in the de-escalation group may be due to the simultaneous de-escalation of anti-Pseudomonas antibiotic agents in addition to the de-escalation of anti-MRSA coverage, as opposed to de-escalation of the anti-MRSA coverage alone.
Implications. De-escalation of anti-MRSA coverage in patients with HAP with negative cultures is associated with fewer antibiotic days, less AKI, and possibly shorter length of stay.
Partial Oral versus Intravenous Antibiotic Treatment for Endocarditis (POET). Iversen K et al. New Engl J Med. 2019;380(5):415-424.11
Background. Patients with left-sided infective endocarditis are typically treated with up to six weeks of intravenous (IV) antibiotics. The investigators studied the effectiveness and safety of switching to oral antibiotics after at least 10 days of IV therapy.
Findings. This randomized, multicenter, noninferiority trial at cardiac centers across Denmark included 400 adults with left-sided endocarditis who were clinically stable after at least 10 days of IV antibiotics. Half of the patients were randomized to continue IV therapy, whereas the other half was switched to oral antibiotics to complete the treatment course. Six months after therapy, no significant difference was observed between the two groups in terms of the primary composite outcomes, including all-cause mortality, unplanned cardiac surgery, embolic events, or relapse of bacteremia with the primary pathogen (IV-treated group: 12.1%; orally treated group: 9.0% [between-group difference: 3.1%; P = .40]).
Caveats. A total of 20% of the screened population (1,954 adults) was randomized, and about 1% (5/400) of patients used injection drugs. None of the patients had MRSA. Patients in the oral group were assessed two to three times per week as outpatients, which may not be feasible in most settings.
Implications. Switching to oral antibiotics after at least 10 days of IV therapy appears to be safe and effective in selected patients with left-sided endocarditis. However, this study largely excluded patients with injection drug use and/or MRSA infections.
Oral versus Intravenous Antibiotics for Bone and Joint Infection (OVIVA). Li HK, et al. New Engl J Med. 2019;380(5):425-436.12
Background. Most complex orthopedic infections are treated with several weeks of IV antibiotics. This study sought to determine whether oral antibiotics are noninferior to IV antibiotics for bone and joint infections.
Findings. This randomized, multicenter, noninferiority, open-label trial of 1,054 adults with bone and joint infections in the United Kingdom included patients with prosthetic joints, other indwelling joint hardware, and native joint infections. Within seven days of antibiotic medication or within seven days of surgery (if performed), the patients received either IV or oral antibiotics for six weeks with a primary endpoint of treatment failure one year after the study randomization. The choice and duration of antibiotic treatment were determined by the involved infectious disease physician. A majority (77%) of patients received greater than six weeks of therapy. Treatment failure was defined by clinical, microbiologic, or histologic criteria. Most enrolled patients were infected with Staphylococcus aureus, with 10% having methicillin-resistant S. aureus. Treatment failure was more frequent in the IV group than the oral group (14.6% vs 13.2%), and these findings were consistent across all subgroups. More patients discontinued treatment in the IV group than the oral group.
Cautions. This study included a heterogenous population of patients with bone and joint infections, with or without hardware, and with different species of bacteria. Patients with bacteremia, endocarditis, or another indication for IV therapy were excluded. Limited injection drug use history was available for the enrolled patients. Most patients had lower limb infections. Thus, these findings are less applicable to vertebral osteomyelitis. Additionally, the study offered no comparison of specific antibiotics.
Implications. With appropriate oversight from infectious disease specialists, targeted oral therapy may be appropriate for the treatment of osteomyelitis. This shift in practice likely requires more study before broad implementation.
Prognostic Accuracy of the HEART Score for Prediction of Major Adverse Cardiac Events in Patients Presenting with Chest Pain: A Systematic Review and Meta‐analysis. Fernando S, et al. Acad Emerg Med. 2019;26(2):140-151.13
Background. Chest pain accounts for over eight million emergency department (ED) visits yearly in the United States. Of those presenting with chest pain, 10%-20% will experience acute coronary syndrome (ACS) requiring further medical treatment. Given the fear of missing ACS, many low-risk patients are hospitalized. The American Heart Association has advocated using validated predictive scoring models to identify patients with chest pain who are at low risk for short-term major cardiovascular adverse event (MACE) for potential discharge without further testing. The authors evaluated the prognostic accuracy of higher risk scores to predict MACE in adult ED patients presenting with chest pain.
Findings. This study was a systematic review and meta-analysis of 30 prospective and retrospective studies evaluating the history–electrocardiogram–age–risk factors–troponin (HEART) score through May 1, 2018. Meta-analysis compared the sensitivity, specificity, positive likelihood ratios, negative likelihood ratios, and diagnostic odds ratios of the HEART score and the Thrombolysis in Myocardial Infarction (TIMI) score when reported. An intermediate HEART score of 4-6 had a sensitivity of 95.9% and a specificity of 44.6%. A high HEART score of greater than or equal to 7 had a sensitivity of 39.5% and a specificity of 95.0%. Similarly, a high TIMI score of great than or equal to 6 had a sensitivity of only 2.8% and a specificity of 99.6%. The authors concluded that a HEART score of greater than or equal to 4 best identifies patients at risk of MACE who need greater consideration for additional testing.
Caveats. This meta-analysis failed to assess the potential adverse effects of false positive downstream testing. Additionally, no study compared the HEART score with the experienced clinician gestalt, which has often been equivalent to decision rules.
Implication. A HEART score greater than or equal to 4 risk stratifies ED patients with chest pain requiring further consideration for evaluation versus those that can be discharged with low risk for short-term MACE.
1. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66(7):e1-e48. https://doi.org/10.1093/cid/cix1085.
2. Chu DK, Kim LH, Young PJ, et al. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet. 2018;391(10131):1693-1705. https://doi.org/10.1016/S0140-6736(18)30479-3.
3. Siemieniuk RAC, Chu DK, Kim LH, et al. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ. 2018;363:k4169. https://doi.org/https://doi.org/10.1136/bmj.k4169
4. A PG, O’Conor KJ, Lanzkron S, et al. Do words matter? Stigmatizing language and the transmission of bias in the medical record. J Gen Intern Med. 2018;33(5):685-691. https://doi.org/10.1007/s11606-017-4289-2.
5. Marrouche NF, Kheirkhahan M, Brachmann J. Catheter ablation for atrial fibrillation with heart failure. N Engl J Med. 2018;379(5):492. https://doi.org/10.1056/NEJMoa1707855.
6. Larochelle MR, Bernson D, Land T, et al. Medication for opioid use disorder after nonfatal opioid overdose and association with mortality: a cohort study. Ann Intern Med. 2018;169(3):137-145. https://doi.org/10.7326/M17-3107.
7. Hedegaard HM, A; Warner, M. Drug Overdose Deaths in the United States, 1999-2017. 2018; https://www.cdc.gov/nchs/products/databriefs/db329.htm. Accessed March 07, 2019.
8. Medications for Opioid Use Disorder Save Lives. 2019; http://www.nationalacademies.org/hmd/Reports/2019/medications-for-opioid-use-disorder-save-lives.aspx. Accessed March 07, 2019.
9. Siontis KC, Zhang X, Eckard A, et al. Outcomes associated with apixaban use in patients with end-stage kidney disease and atrial fibrillation in the United States. Circulation. 2018;138(15):1519-1529. https://doi.org/10.1161/CIRCULATIONAHA.118.035418.
10. Cowley MC, Ritchie DJ, Hampton N, Kollef MH, Micek ST. Outcomes Associated With De-escalating Therapy for Methicillin-Resistant Staphylococcus aureus in Culture-Negative Nosocomial Pneumonia. Chest. 2019;155(1):53-59. https://doi.org/10.1016/j.chest.2018.10.014
11. Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Engl J Med. 2019;380(5):415-424. https://doi.org/10.1056/NEJMoa1808312
12. Li HK, Rombach I, Zambellas R, et al. Oral versus Intravenous Antibiotics for Bone and Joint Infection. N Engl J Med. 2019;380(5):425-436. https://doi.org/10.1056/NEJMoa1710926
13. Fernando SM, Tran A, Cheng W, et al. Prognostic accuracy of the HEART score for prediction of major adverse cardiac events in patients presenting with chest pain: a systematic review and meta-analysis. Acad Emerg Med. 2019;26(2):140-151. https://doi.org/10.1111/acem.13649.
Given the breadth and depth of patients cared for by hospital medicine providers, it is challenging to remain current with the literature. The authors critically appraised the literature from March 2018 to April 2019 for high-quality studies relevant to hospital medicine. Articles were selected based on methodologic rigor and likelihood to impact clinical practice. Thirty articles were selected by the presenting authors for the Hospital Medicine Updates at the 2019 Society of Hospital Medicine (CH, CM) and Society of General Internal Medicine Annual Meetings (BS, AB). After two sequential rounds of voting and group discussion to adjudicate voting discrepancies, the authors selected the 10 most impactful articles for this review. Each article is described below with the key points summarized in the Table.
ESSENTIAL PUBLICATIONS
Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). McDonald LC, et al. Clin Infect Dis. 2018;66(7):e1–e48.1
Background. In the United States, approximately 500,000 Clostridioides difficile infections (CDI) occur annually with 15,000-30,000 deaths. CDI has become a marker of hospital quality and has been placed under numerous “pay for performance” metrics. The Infectious Diseases Society of America/Society of Healthcare Epidemiology of America updated their guidelines from 2010 regarding hospital surveillance, diagnostic testing, treatment, and infection precautions and control.
Findings. The panel included 14 multidisciplinary experts in epidemiology, diagnosis, infection control, and clinical management of adult and pediatric CDI. They used problem intervention comparison-outcome (PICO)-formatted, evidence-based questions. The selection of data and final recommendations were made in accordance with the GRADE criteria. A total of 35 recommendations were made.
Key clinical recommendations for hospitalists caring for adults: (1) Prescribe vancomycin or fidaxomicin over metronidazole for the initial treatment of CDI (strong recommendation, high quality of evidence); (2) Limit testing to the patients with unexplained new onset diarrhea, which is defined as greater than or equal to 3 unformed stools in 24 hours (weak recommendation, very low-quality evidence); (3) Avoid routine repeat testing within seven days, and only test asymptomatic patients for epidemiologic reasons (strong recommendation, moderate-quality evidence); (4) Minimize the frequency and duration of high-risk antibiotic therapy and the number of antibiotic agents prescribed (strong recommendation, moderate quality of evidence); (5) Discontinue therapy with the inciting antibiotic agent as soon as possible (strong recommendation, moderate quality of evidence).
Caveats. As with the clinical application of any guidelines, individual case adjustments may be required.
Implications. Vancomycin or fidaxomicin should be used for the initial episode of CDI instead of metronidazole.
Mortality and Morbidity in Acutely Ill Adults Treated with Liberal versus Conservative Oxygen Therapy (IOTA): a Systematic Review and Meta-analysis. Chu DK, et al. Lancet. 2018;391(10131):1693-1705.2
Background. Supplemental oxygen is often given to acutely ill hospitalized adults, even when they are not hypoxic or dyspneic. The safety and efficacy of this practice is unknown.
Findings. This systematic review and meta-analysis evaluated 25 randomized controlled trials enrolling 16,037 patients. Patients presented with several conditions, including sepsis, critical illness, stroke, myocardial infarction, and emergency surgery. The fraction of inspired oxygen in the liberal arms varied from 30% to 100%. Most patients randomized to the conservative arm received no supplemental oxygen. Delivery of liberal oxygen to acutely ill adults was associated with increased in-hospital mortality (relative risk [RR]: 1.21; 95% CI: 1.03-1.43), 30-day mortality (RR: 1.14; 95% CI: 1.01-1.29), and 90-day mortality (RR: 1.10; 95% CI: 1.00-1.20). The results were believed to be of high quality and were robust across multiple sensitivity analyses. It seemed that the mortality began to increase when supplemental oxygen raised the peripheral oxygen saturation (Sp02) above a range of 94%-96%.
Caveats. Heterogeneity was observed in the study settings and oxygen delivery. In addition, the cause for increased mortality could not be determined.
Implications. In hospitalized acutely ill adults, “liberal” supplemental oxygen was associated with increased in-hospital and longer-term mortality. The study authors postulated that this finding resulted from the direct toxic effects of oxygen or that oxygen delivery may “mask” illness and lead to delays in diagnosis and treatment. A subsequent clinical practice guideline recommends (1) a target SpO2 of less than 96% for patients receiving oxygen therapy; (2) a target SpO2 range of 90%-94% seems appropriate for most hospitalized adults.3
Do Words Matter? Stigmatizing Language and the Transmission of Bias in the Medical Record. P Goddu A, et al. J Gen Intern Med. 2018;33(5):68-91.4
Background. Previous work has shown that clinician bias affects health outcomes, often worsening health disparities. It is unknown whether clinicians’ language in medical records biases other clinicians and whether this affects patients.
Findings. The investigators randomized medical students and residents in internal and emergency medicine at one academic medical center to review one of two vignettes in the format of notes on the same hypothetical patient with sickle cell disease (SCD) admitted with a pain crisis. One vignette contained stigmatizing language, and the other contained neutral language. The trainees exposed to the vignettes with stigmatizing language showed a more negative attitude toward the patient, as measured by a previously validated scale of attitudes toward patients with SCD (20.6 stigmatizing vs 25.6 neutral, with a total score range of 7-35 for the instrument; higher scores indicate more positive attitudes; P < .001). Furthermore, the intensity of pain treatment was assessed in the resident group and was less aggressive when residents were exposed to stigmatizing language (5.56 stigmatizing vs 6.22 neutral on a scale of 2-7, with higher scores indicating more aggressive pain treatment; P = .003).
Cautions. This research was a single-center study of residents and medical students in two departments. Additionally, the study used vignettes on a hypothetical patient so trainees in the study group might have witnessed stronger stigmatizing language than what is typically observed in an actual patients’ notes.
Implications. Stigmatizing language used in medical records possibly contributed to health disparities by negatively impacting other physicians’ biases and prescribing practices toward patients with SCD at an academic medical center. Clinicians should avoid stigmatizing language in medical records.
Catheter Ablation for Atrial Fibrillation with Heart Failure. Marrouche, NF et al. New Engl J Med. 2018;378:417-427.5
Background. Atrial fibrillation (AF) in patients with heart failure is associated with increased mortality and morbidity. Small-scale studies have suggested that ablation of AF may benefit patients with heart failure.
Findings. This multicenter trial included 398 patients with heart failure and symptomatic AF. Patients had New York Heart Association Class II-IV heart failure, an ejection fraction (EF) of 35% or less, and an internal cardiac defibrillator (ICD). Patients were randomized to either ablation or medical therapy. All enrolled patients either refused, failed, or showed poor tolerance to antiarrhythmic therapy for AF. The primary outcome was death from any cause or hospitalization for heart failure.
The composite endpoint occurred in 28.5% of the ablation group versus 44.6% of patients in the medical therapy group (hazard ratio [HR]: 0.62; 95% CI: 0.43-0.87). Fewer patients in the ablation group died (13% vs 25%; HR: 0.53; 95% CI: 0.32-0.86) or were hospitalized for heart failure (21% vs 36%; HR: 0.56; 95% CI: 0.37-0.83). The patients in the ablation group had higher EF increases above baseline and a greater proportion were in sinus rhythm at the 60-month follow-up visit.
Cautions. The trial was terminated early due to slow recruitment and lower than expected events. Over twice as many patients were lost to follow-up in the ablation group versus the medical therapy group, and by 60 months, AF recurred in 50% of patients who underwent ablation. The sample size was small, and the trial was unblinded.
Implications. Ablation should be considered for AF in patients with heart failure. Additional studies to evaluate ablation versus medical therapy for patients with heart failure and AF are underway.
Medication for Opioid Use Disorder after Nonfatal Opioid Overdose and Association with Mortality. Larochelle MR, et al. Ann Intern Med. 2018;169(3):137-145.6
Background. More than 70,000 Americans died of drug overdose in 2017; this number is higher than the deaths resulting from human immunodeficiency virus, car crash, or gun violence at their peaks.7 Methadone, buprenorphine, and naltrexone are approved by the Federal Drug Administration for the treatment of opioid use disorder (OUD). These medications increase treatment retention; methadone and buprenorphine have been associated with significant decreases in all-cause and overdose mortality.8 However, whether receipt of these medications following a nonfatal opioid overdose reduces mortality is unknown.
Findings. This retrospective cohort study included 17,568 opioid overdose survivors from the Massachusetts’s Public Health Dataset between 2012 and 2014. Only three in 10 of these patients received any medications for OUD over 12 months following overdose. All-cause mortality was 4.7 deaths (95% CI: 4.4-5.0 deaths) per 100 person-years. The relative risk for all-cause mortality was 53% lower with methadone (adjusted hazard ratio [aHR]: 0.47; 95% CI: 0.32-0.71) and 37% lower with buprenorphine (aHR: 0.63; 95% CI: 0.46-0.87).
Caveats. This cohort study may have missed confounders explaining why certain patients received medications for OUD. As a result, association cannot be interpreted as causation.
Implications. Methadone and buprenorphine are associated with a reduction in preventable deaths in patients with OUD who have survived an overdose. All patients with OUD should be considered for therapy.
Outcomes Associated with Apixaban Use in Patients with End-Stage Kidney Disease and Atrial Fibrillation in the United States. Siontis, KC, et al. Circulation. 2018;138:1519–1529.9
Background. Patients with end-stage kidney disease (ESKD) have poor outcomes when treated with warfarin for AF. These patients were excluded from clinical trials of direct oral anticoagulants. The goal of this study was to determine the outcomes of the use of apixaban in patients with ESKD and AF.
Findings. This retrospective cohort study included 25,523 Medicare patients with ESKD and AF on anticoagulants. A 3:1 propensity score match was performed between patients on warfarin and apixaban. Time without stroke/systemic embolism, bleeding (major, gastrointestinal, and intracranial), and death were assessed. A total of 2,351 patients were on apixaban, and 23,172 patients were on warfarin. No difference was observed in the risk of stroke/systemic embolism between apixaban and warfarin (HR 0.88; 95% CI: 0.69-1.12). Apixaban was associated with a lower risk of major bleeding (HR: 0.72; 95% CI: 0.59-0.87). Standard-dose apixaban (5 mg twice a day) was associated with lower risks of stroke/systemic embolism and death compared with reduced-dose apixaban (2.5 mg twice a day; n = 1,317; HR: 0.61; 95% CI: 0.37-0.98; P = .04 for stroke/systemic embolism; HR: 0.64; 95% CI: 0.45-0.92; P = .01 for death) or warfarin (HR: 0.64; 95% CI: 0.42-0.97; P = .04 for stroke/systemic embolism; HR: 0.63; 95% CI: 0.46-0.85; P = .003 for death).
Cautions. There may be unique patient factors that led providers to prescribe apixaban to patients with ESKD.
Implications. The use of standard-dose apixaban appears safe and potentially preferable in patients with ESKD and AF due to reductions in major bleeding, thromboembolism, and mortality risk compared with warfarin. Several additional studies are pending to evaluate the use and dose of apixaban in patients with ESKD and AF.
Outcomes Associated with De-escalating Therapy for Methicillin-Resistant Staphylococcus aureus in Culture-Negative Nosocomial Pneumonia. Cowley MC, et al. Chest. 2019;155(1):53-59.10
Background. Patients diagnosed with hospital-acquired pneumonia (HAP) are often treated empirically with broad-spectrum antibiotics. In many patients with HAP, cultures remain negative, and providers must decide if antibiotics can safely be narrowed. Specifically, the safety of deciding to “de-escalate” and discontinue the coverage for methicillin-resistant Staphylococcus aureus (MRSA) if cultures remain negative is unclear.
Findings. In this single-center retrospective cohort study, 279 patients who were (1) diagnosed with HAP and (2) had negative sputum cultures were enrolled. The patients in whom MRSA coverage was de-escalated by day four were compared with those with continued anti-MRSA coverage. No difference was observed between the two groups in terms of degree of illness or comorbidities. The patients who were de-escalated received five fewer days of anti-MRSA coverage than patients who were not. No difference was noted in the 28-day mortality between the two groups (de-escalation: 23% vs no de-escalation: 28%; 95% CI: −16.1%-6.5%). The incidence of acute kidney injury (AKI) was significantly lower in the de-escalation group (36% vs 50%; 95% CI: −26.9- 0.04), and the overall length of stay was five days shorter in the de-escalation group (95% CI: 0.1-6.4 days).
Caveats. Given the retrospective nature, unmeasured confounders may have impacted the decision to de-escalate anti-MRSA coverage. The observed lower risk of AKI in the de-escalation group may be due to the simultaneous de-escalation of anti-Pseudomonas antibiotic agents in addition to the de-escalation of anti-MRSA coverage, as opposed to de-escalation of the anti-MRSA coverage alone.
Implications. De-escalation of anti-MRSA coverage in patients with HAP with negative cultures is associated with fewer antibiotic days, less AKI, and possibly shorter length of stay.
Partial Oral versus Intravenous Antibiotic Treatment for Endocarditis (POET). Iversen K et al. New Engl J Med. 2019;380(5):415-424.11
Background. Patients with left-sided infective endocarditis are typically treated with up to six weeks of intravenous (IV) antibiotics. The investigators studied the effectiveness and safety of switching to oral antibiotics after at least 10 days of IV therapy.
Findings. This randomized, multicenter, noninferiority trial at cardiac centers across Denmark included 400 adults with left-sided endocarditis who were clinically stable after at least 10 days of IV antibiotics. Half of the patients were randomized to continue IV therapy, whereas the other half was switched to oral antibiotics to complete the treatment course. Six months after therapy, no significant difference was observed between the two groups in terms of the primary composite outcomes, including all-cause mortality, unplanned cardiac surgery, embolic events, or relapse of bacteremia with the primary pathogen (IV-treated group: 12.1%; orally treated group: 9.0% [between-group difference: 3.1%; P = .40]).
Caveats. A total of 20% of the screened population (1,954 adults) was randomized, and about 1% (5/400) of patients used injection drugs. None of the patients had MRSA. Patients in the oral group were assessed two to three times per week as outpatients, which may not be feasible in most settings.
Implications. Switching to oral antibiotics after at least 10 days of IV therapy appears to be safe and effective in selected patients with left-sided endocarditis. However, this study largely excluded patients with injection drug use and/or MRSA infections.
Oral versus Intravenous Antibiotics for Bone and Joint Infection (OVIVA). Li HK, et al. New Engl J Med. 2019;380(5):425-436.12
Background. Most complex orthopedic infections are treated with several weeks of IV antibiotics. This study sought to determine whether oral antibiotics are noninferior to IV antibiotics for bone and joint infections.
Findings. This randomized, multicenter, noninferiority, open-label trial of 1,054 adults with bone and joint infections in the United Kingdom included patients with prosthetic joints, other indwelling joint hardware, and native joint infections. Within seven days of antibiotic medication or within seven days of surgery (if performed), the patients received either IV or oral antibiotics for six weeks with a primary endpoint of treatment failure one year after the study randomization. The choice and duration of antibiotic treatment were determined by the involved infectious disease physician. A majority (77%) of patients received greater than six weeks of therapy. Treatment failure was defined by clinical, microbiologic, or histologic criteria. Most enrolled patients were infected with Staphylococcus aureus, with 10% having methicillin-resistant S. aureus. Treatment failure was more frequent in the IV group than the oral group (14.6% vs 13.2%), and these findings were consistent across all subgroups. More patients discontinued treatment in the IV group than the oral group.
Cautions. This study included a heterogenous population of patients with bone and joint infections, with or without hardware, and with different species of bacteria. Patients with bacteremia, endocarditis, or another indication for IV therapy were excluded. Limited injection drug use history was available for the enrolled patients. Most patients had lower limb infections. Thus, these findings are less applicable to vertebral osteomyelitis. Additionally, the study offered no comparison of specific antibiotics.
Implications. With appropriate oversight from infectious disease specialists, targeted oral therapy may be appropriate for the treatment of osteomyelitis. This shift in practice likely requires more study before broad implementation.
Prognostic Accuracy of the HEART Score for Prediction of Major Adverse Cardiac Events in Patients Presenting with Chest Pain: A Systematic Review and Meta‐analysis. Fernando S, et al. Acad Emerg Med. 2019;26(2):140-151.13
Background. Chest pain accounts for over eight million emergency department (ED) visits yearly in the United States. Of those presenting with chest pain, 10%-20% will experience acute coronary syndrome (ACS) requiring further medical treatment. Given the fear of missing ACS, many low-risk patients are hospitalized. The American Heart Association has advocated using validated predictive scoring models to identify patients with chest pain who are at low risk for short-term major cardiovascular adverse event (MACE) for potential discharge without further testing. The authors evaluated the prognostic accuracy of higher risk scores to predict MACE in adult ED patients presenting with chest pain.
Findings. This study was a systematic review and meta-analysis of 30 prospective and retrospective studies evaluating the history–electrocardiogram–age–risk factors–troponin (HEART) score through May 1, 2018. Meta-analysis compared the sensitivity, specificity, positive likelihood ratios, negative likelihood ratios, and diagnostic odds ratios of the HEART score and the Thrombolysis in Myocardial Infarction (TIMI) score when reported. An intermediate HEART score of 4-6 had a sensitivity of 95.9% and a specificity of 44.6%. A high HEART score of greater than or equal to 7 had a sensitivity of 39.5% and a specificity of 95.0%. Similarly, a high TIMI score of great than or equal to 6 had a sensitivity of only 2.8% and a specificity of 99.6%. The authors concluded that a HEART score of greater than or equal to 4 best identifies patients at risk of MACE who need greater consideration for additional testing.
Caveats. This meta-analysis failed to assess the potential adverse effects of false positive downstream testing. Additionally, no study compared the HEART score with the experienced clinician gestalt, which has often been equivalent to decision rules.
Implication. A HEART score greater than or equal to 4 risk stratifies ED patients with chest pain requiring further consideration for evaluation versus those that can be discharged with low risk for short-term MACE.
Given the breadth and depth of patients cared for by hospital medicine providers, it is challenging to remain current with the literature. The authors critically appraised the literature from March 2018 to April 2019 for high-quality studies relevant to hospital medicine. Articles were selected based on methodologic rigor and likelihood to impact clinical practice. Thirty articles were selected by the presenting authors for the Hospital Medicine Updates at the 2019 Society of Hospital Medicine (CH, CM) and Society of General Internal Medicine Annual Meetings (BS, AB). After two sequential rounds of voting and group discussion to adjudicate voting discrepancies, the authors selected the 10 most impactful articles for this review. Each article is described below with the key points summarized in the Table.
ESSENTIAL PUBLICATIONS
Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). McDonald LC, et al. Clin Infect Dis. 2018;66(7):e1–e48.1
Background. In the United States, approximately 500,000 Clostridioides difficile infections (CDI) occur annually with 15,000-30,000 deaths. CDI has become a marker of hospital quality and has been placed under numerous “pay for performance” metrics. The Infectious Diseases Society of America/Society of Healthcare Epidemiology of America updated their guidelines from 2010 regarding hospital surveillance, diagnostic testing, treatment, and infection precautions and control.
Findings. The panel included 14 multidisciplinary experts in epidemiology, diagnosis, infection control, and clinical management of adult and pediatric CDI. They used problem intervention comparison-outcome (PICO)-formatted, evidence-based questions. The selection of data and final recommendations were made in accordance with the GRADE criteria. A total of 35 recommendations were made.
Key clinical recommendations for hospitalists caring for adults: (1) Prescribe vancomycin or fidaxomicin over metronidazole for the initial treatment of CDI (strong recommendation, high quality of evidence); (2) Limit testing to the patients with unexplained new onset diarrhea, which is defined as greater than or equal to 3 unformed stools in 24 hours (weak recommendation, very low-quality evidence); (3) Avoid routine repeat testing within seven days, and only test asymptomatic patients for epidemiologic reasons (strong recommendation, moderate-quality evidence); (4) Minimize the frequency and duration of high-risk antibiotic therapy and the number of antibiotic agents prescribed (strong recommendation, moderate quality of evidence); (5) Discontinue therapy with the inciting antibiotic agent as soon as possible (strong recommendation, moderate quality of evidence).
Caveats. As with the clinical application of any guidelines, individual case adjustments may be required.
Implications. Vancomycin or fidaxomicin should be used for the initial episode of CDI instead of metronidazole.
Mortality and Morbidity in Acutely Ill Adults Treated with Liberal versus Conservative Oxygen Therapy (IOTA): a Systematic Review and Meta-analysis. Chu DK, et al. Lancet. 2018;391(10131):1693-1705.2
Background. Supplemental oxygen is often given to acutely ill hospitalized adults, even when they are not hypoxic or dyspneic. The safety and efficacy of this practice is unknown.
Findings. This systematic review and meta-analysis evaluated 25 randomized controlled trials enrolling 16,037 patients. Patients presented with several conditions, including sepsis, critical illness, stroke, myocardial infarction, and emergency surgery. The fraction of inspired oxygen in the liberal arms varied from 30% to 100%. Most patients randomized to the conservative arm received no supplemental oxygen. Delivery of liberal oxygen to acutely ill adults was associated with increased in-hospital mortality (relative risk [RR]: 1.21; 95% CI: 1.03-1.43), 30-day mortality (RR: 1.14; 95% CI: 1.01-1.29), and 90-day mortality (RR: 1.10; 95% CI: 1.00-1.20). The results were believed to be of high quality and were robust across multiple sensitivity analyses. It seemed that the mortality began to increase when supplemental oxygen raised the peripheral oxygen saturation (Sp02) above a range of 94%-96%.
Caveats. Heterogeneity was observed in the study settings and oxygen delivery. In addition, the cause for increased mortality could not be determined.
Implications. In hospitalized acutely ill adults, “liberal” supplemental oxygen was associated with increased in-hospital and longer-term mortality. The study authors postulated that this finding resulted from the direct toxic effects of oxygen or that oxygen delivery may “mask” illness and lead to delays in diagnosis and treatment. A subsequent clinical practice guideline recommends (1) a target SpO2 of less than 96% for patients receiving oxygen therapy; (2) a target SpO2 range of 90%-94% seems appropriate for most hospitalized adults.3
Do Words Matter? Stigmatizing Language and the Transmission of Bias in the Medical Record. P Goddu A, et al. J Gen Intern Med. 2018;33(5):68-91.4
Background. Previous work has shown that clinician bias affects health outcomes, often worsening health disparities. It is unknown whether clinicians’ language in medical records biases other clinicians and whether this affects patients.
Findings. The investigators randomized medical students and residents in internal and emergency medicine at one academic medical center to review one of two vignettes in the format of notes on the same hypothetical patient with sickle cell disease (SCD) admitted with a pain crisis. One vignette contained stigmatizing language, and the other contained neutral language. The trainees exposed to the vignettes with stigmatizing language showed a more negative attitude toward the patient, as measured by a previously validated scale of attitudes toward patients with SCD (20.6 stigmatizing vs 25.6 neutral, with a total score range of 7-35 for the instrument; higher scores indicate more positive attitudes; P < .001). Furthermore, the intensity of pain treatment was assessed in the resident group and was less aggressive when residents were exposed to stigmatizing language (5.56 stigmatizing vs 6.22 neutral on a scale of 2-7, with higher scores indicating more aggressive pain treatment; P = .003).
Cautions. This research was a single-center study of residents and medical students in two departments. Additionally, the study used vignettes on a hypothetical patient so trainees in the study group might have witnessed stronger stigmatizing language than what is typically observed in an actual patients’ notes.
Implications. Stigmatizing language used in medical records possibly contributed to health disparities by negatively impacting other physicians’ biases and prescribing practices toward patients with SCD at an academic medical center. Clinicians should avoid stigmatizing language in medical records.
Catheter Ablation for Atrial Fibrillation with Heart Failure. Marrouche, NF et al. New Engl J Med. 2018;378:417-427.5
Background. Atrial fibrillation (AF) in patients with heart failure is associated with increased mortality and morbidity. Small-scale studies have suggested that ablation of AF may benefit patients with heart failure.
Findings. This multicenter trial included 398 patients with heart failure and symptomatic AF. Patients had New York Heart Association Class II-IV heart failure, an ejection fraction (EF) of 35% or less, and an internal cardiac defibrillator (ICD). Patients were randomized to either ablation or medical therapy. All enrolled patients either refused, failed, or showed poor tolerance to antiarrhythmic therapy for AF. The primary outcome was death from any cause or hospitalization for heart failure.
The composite endpoint occurred in 28.5% of the ablation group versus 44.6% of patients in the medical therapy group (hazard ratio [HR]: 0.62; 95% CI: 0.43-0.87). Fewer patients in the ablation group died (13% vs 25%; HR: 0.53; 95% CI: 0.32-0.86) or were hospitalized for heart failure (21% vs 36%; HR: 0.56; 95% CI: 0.37-0.83). The patients in the ablation group had higher EF increases above baseline and a greater proportion were in sinus rhythm at the 60-month follow-up visit.
Cautions. The trial was terminated early due to slow recruitment and lower than expected events. Over twice as many patients were lost to follow-up in the ablation group versus the medical therapy group, and by 60 months, AF recurred in 50% of patients who underwent ablation. The sample size was small, and the trial was unblinded.
Implications. Ablation should be considered for AF in patients with heart failure. Additional studies to evaluate ablation versus medical therapy for patients with heart failure and AF are underway.
Medication for Opioid Use Disorder after Nonfatal Opioid Overdose and Association with Mortality. Larochelle MR, et al. Ann Intern Med. 2018;169(3):137-145.6
Background. More than 70,000 Americans died of drug overdose in 2017; this number is higher than the deaths resulting from human immunodeficiency virus, car crash, or gun violence at their peaks.7 Methadone, buprenorphine, and naltrexone are approved by the Federal Drug Administration for the treatment of opioid use disorder (OUD). These medications increase treatment retention; methadone and buprenorphine have been associated with significant decreases in all-cause and overdose mortality.8 However, whether receipt of these medications following a nonfatal opioid overdose reduces mortality is unknown.
Findings. This retrospective cohort study included 17,568 opioid overdose survivors from the Massachusetts’s Public Health Dataset between 2012 and 2014. Only three in 10 of these patients received any medications for OUD over 12 months following overdose. All-cause mortality was 4.7 deaths (95% CI: 4.4-5.0 deaths) per 100 person-years. The relative risk for all-cause mortality was 53% lower with methadone (adjusted hazard ratio [aHR]: 0.47; 95% CI: 0.32-0.71) and 37% lower with buprenorphine (aHR: 0.63; 95% CI: 0.46-0.87).
Caveats. This cohort study may have missed confounders explaining why certain patients received medications for OUD. As a result, association cannot be interpreted as causation.
Implications. Methadone and buprenorphine are associated with a reduction in preventable deaths in patients with OUD who have survived an overdose. All patients with OUD should be considered for therapy.
Outcomes Associated with Apixaban Use in Patients with End-Stage Kidney Disease and Atrial Fibrillation in the United States. Siontis, KC, et al. Circulation. 2018;138:1519–1529.9
Background. Patients with end-stage kidney disease (ESKD) have poor outcomes when treated with warfarin for AF. These patients were excluded from clinical trials of direct oral anticoagulants. The goal of this study was to determine the outcomes of the use of apixaban in patients with ESKD and AF.
Findings. This retrospective cohort study included 25,523 Medicare patients with ESKD and AF on anticoagulants. A 3:1 propensity score match was performed between patients on warfarin and apixaban. Time without stroke/systemic embolism, bleeding (major, gastrointestinal, and intracranial), and death were assessed. A total of 2,351 patients were on apixaban, and 23,172 patients were on warfarin. No difference was observed in the risk of stroke/systemic embolism between apixaban and warfarin (HR 0.88; 95% CI: 0.69-1.12). Apixaban was associated with a lower risk of major bleeding (HR: 0.72; 95% CI: 0.59-0.87). Standard-dose apixaban (5 mg twice a day) was associated with lower risks of stroke/systemic embolism and death compared with reduced-dose apixaban (2.5 mg twice a day; n = 1,317; HR: 0.61; 95% CI: 0.37-0.98; P = .04 for stroke/systemic embolism; HR: 0.64; 95% CI: 0.45-0.92; P = .01 for death) or warfarin (HR: 0.64; 95% CI: 0.42-0.97; P = .04 for stroke/systemic embolism; HR: 0.63; 95% CI: 0.46-0.85; P = .003 for death).
Cautions. There may be unique patient factors that led providers to prescribe apixaban to patients with ESKD.
Implications. The use of standard-dose apixaban appears safe and potentially preferable in patients with ESKD and AF due to reductions in major bleeding, thromboembolism, and mortality risk compared with warfarin. Several additional studies are pending to evaluate the use and dose of apixaban in patients with ESKD and AF.
Outcomes Associated with De-escalating Therapy for Methicillin-Resistant Staphylococcus aureus in Culture-Negative Nosocomial Pneumonia. Cowley MC, et al. Chest. 2019;155(1):53-59.10
Background. Patients diagnosed with hospital-acquired pneumonia (HAP) are often treated empirically with broad-spectrum antibiotics. In many patients with HAP, cultures remain negative, and providers must decide if antibiotics can safely be narrowed. Specifically, the safety of deciding to “de-escalate” and discontinue the coverage for methicillin-resistant Staphylococcus aureus (MRSA) if cultures remain negative is unclear.
Findings. In this single-center retrospective cohort study, 279 patients who were (1) diagnosed with HAP and (2) had negative sputum cultures were enrolled. The patients in whom MRSA coverage was de-escalated by day four were compared with those with continued anti-MRSA coverage. No difference was observed between the two groups in terms of degree of illness or comorbidities. The patients who were de-escalated received five fewer days of anti-MRSA coverage than patients who were not. No difference was noted in the 28-day mortality between the two groups (de-escalation: 23% vs no de-escalation: 28%; 95% CI: −16.1%-6.5%). The incidence of acute kidney injury (AKI) was significantly lower in the de-escalation group (36% vs 50%; 95% CI: −26.9- 0.04), and the overall length of stay was five days shorter in the de-escalation group (95% CI: 0.1-6.4 days).
Caveats. Given the retrospective nature, unmeasured confounders may have impacted the decision to de-escalate anti-MRSA coverage. The observed lower risk of AKI in the de-escalation group may be due to the simultaneous de-escalation of anti-Pseudomonas antibiotic agents in addition to the de-escalation of anti-MRSA coverage, as opposed to de-escalation of the anti-MRSA coverage alone.
Implications. De-escalation of anti-MRSA coverage in patients with HAP with negative cultures is associated with fewer antibiotic days, less AKI, and possibly shorter length of stay.
Partial Oral versus Intravenous Antibiotic Treatment for Endocarditis (POET). Iversen K et al. New Engl J Med. 2019;380(5):415-424.11
Background. Patients with left-sided infective endocarditis are typically treated with up to six weeks of intravenous (IV) antibiotics. The investigators studied the effectiveness and safety of switching to oral antibiotics after at least 10 days of IV therapy.
Findings. This randomized, multicenter, noninferiority trial at cardiac centers across Denmark included 400 adults with left-sided endocarditis who were clinically stable after at least 10 days of IV antibiotics. Half of the patients were randomized to continue IV therapy, whereas the other half was switched to oral antibiotics to complete the treatment course. Six months after therapy, no significant difference was observed between the two groups in terms of the primary composite outcomes, including all-cause mortality, unplanned cardiac surgery, embolic events, or relapse of bacteremia with the primary pathogen (IV-treated group: 12.1%; orally treated group: 9.0% [between-group difference: 3.1%; P = .40]).
Caveats. A total of 20% of the screened population (1,954 adults) was randomized, and about 1% (5/400) of patients used injection drugs. None of the patients had MRSA. Patients in the oral group were assessed two to three times per week as outpatients, which may not be feasible in most settings.
Implications. Switching to oral antibiotics after at least 10 days of IV therapy appears to be safe and effective in selected patients with left-sided endocarditis. However, this study largely excluded patients with injection drug use and/or MRSA infections.
Oral versus Intravenous Antibiotics for Bone and Joint Infection (OVIVA). Li HK, et al. New Engl J Med. 2019;380(5):425-436.12
Background. Most complex orthopedic infections are treated with several weeks of IV antibiotics. This study sought to determine whether oral antibiotics are noninferior to IV antibiotics for bone and joint infections.
Findings. This randomized, multicenter, noninferiority, open-label trial of 1,054 adults with bone and joint infections in the United Kingdom included patients with prosthetic joints, other indwelling joint hardware, and native joint infections. Within seven days of antibiotic medication or within seven days of surgery (if performed), the patients received either IV or oral antibiotics for six weeks with a primary endpoint of treatment failure one year after the study randomization. The choice and duration of antibiotic treatment were determined by the involved infectious disease physician. A majority (77%) of patients received greater than six weeks of therapy. Treatment failure was defined by clinical, microbiologic, or histologic criteria. Most enrolled patients were infected with Staphylococcus aureus, with 10% having methicillin-resistant S. aureus. Treatment failure was more frequent in the IV group than the oral group (14.6% vs 13.2%), and these findings were consistent across all subgroups. More patients discontinued treatment in the IV group than the oral group.
Cautions. This study included a heterogenous population of patients with bone and joint infections, with or without hardware, and with different species of bacteria. Patients with bacteremia, endocarditis, or another indication for IV therapy were excluded. Limited injection drug use history was available for the enrolled patients. Most patients had lower limb infections. Thus, these findings are less applicable to vertebral osteomyelitis. Additionally, the study offered no comparison of specific antibiotics.
Implications. With appropriate oversight from infectious disease specialists, targeted oral therapy may be appropriate for the treatment of osteomyelitis. This shift in practice likely requires more study before broad implementation.
Prognostic Accuracy of the HEART Score for Prediction of Major Adverse Cardiac Events in Patients Presenting with Chest Pain: A Systematic Review and Meta‐analysis. Fernando S, et al. Acad Emerg Med. 2019;26(2):140-151.13
Background. Chest pain accounts for over eight million emergency department (ED) visits yearly in the United States. Of those presenting with chest pain, 10%-20% will experience acute coronary syndrome (ACS) requiring further medical treatment. Given the fear of missing ACS, many low-risk patients are hospitalized. The American Heart Association has advocated using validated predictive scoring models to identify patients with chest pain who are at low risk for short-term major cardiovascular adverse event (MACE) for potential discharge without further testing. The authors evaluated the prognostic accuracy of higher risk scores to predict MACE in adult ED patients presenting with chest pain.
Findings. This study was a systematic review and meta-analysis of 30 prospective and retrospective studies evaluating the history–electrocardiogram–age–risk factors–troponin (HEART) score through May 1, 2018. Meta-analysis compared the sensitivity, specificity, positive likelihood ratios, negative likelihood ratios, and diagnostic odds ratios of the HEART score and the Thrombolysis in Myocardial Infarction (TIMI) score when reported. An intermediate HEART score of 4-6 had a sensitivity of 95.9% and a specificity of 44.6%. A high HEART score of greater than or equal to 7 had a sensitivity of 39.5% and a specificity of 95.0%. Similarly, a high TIMI score of great than or equal to 6 had a sensitivity of only 2.8% and a specificity of 99.6%. The authors concluded that a HEART score of greater than or equal to 4 best identifies patients at risk of MACE who need greater consideration for additional testing.
Caveats. This meta-analysis failed to assess the potential adverse effects of false positive downstream testing. Additionally, no study compared the HEART score with the experienced clinician gestalt, which has often been equivalent to decision rules.
Implication. A HEART score greater than or equal to 4 risk stratifies ED patients with chest pain requiring further consideration for evaluation versus those that can be discharged with low risk for short-term MACE.
1. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66(7):e1-e48. https://doi.org/10.1093/cid/cix1085.
2. Chu DK, Kim LH, Young PJ, et al. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet. 2018;391(10131):1693-1705. https://doi.org/10.1016/S0140-6736(18)30479-3.
3. Siemieniuk RAC, Chu DK, Kim LH, et al. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ. 2018;363:k4169. https://doi.org/https://doi.org/10.1136/bmj.k4169
4. A PG, O’Conor KJ, Lanzkron S, et al. Do words matter? Stigmatizing language and the transmission of bias in the medical record. J Gen Intern Med. 2018;33(5):685-691. https://doi.org/10.1007/s11606-017-4289-2.
5. Marrouche NF, Kheirkhahan M, Brachmann J. Catheter ablation for atrial fibrillation with heart failure. N Engl J Med. 2018;379(5):492. https://doi.org/10.1056/NEJMoa1707855.
6. Larochelle MR, Bernson D, Land T, et al. Medication for opioid use disorder after nonfatal opioid overdose and association with mortality: a cohort study. Ann Intern Med. 2018;169(3):137-145. https://doi.org/10.7326/M17-3107.
7. Hedegaard HM, A; Warner, M. Drug Overdose Deaths in the United States, 1999-2017. 2018; https://www.cdc.gov/nchs/products/databriefs/db329.htm. Accessed March 07, 2019.
8. Medications for Opioid Use Disorder Save Lives. 2019; http://www.nationalacademies.org/hmd/Reports/2019/medications-for-opioid-use-disorder-save-lives.aspx. Accessed March 07, 2019.
9. Siontis KC, Zhang X, Eckard A, et al. Outcomes associated with apixaban use in patients with end-stage kidney disease and atrial fibrillation in the United States. Circulation. 2018;138(15):1519-1529. https://doi.org/10.1161/CIRCULATIONAHA.118.035418.
10. Cowley MC, Ritchie DJ, Hampton N, Kollef MH, Micek ST. Outcomes Associated With De-escalating Therapy for Methicillin-Resistant Staphylococcus aureus in Culture-Negative Nosocomial Pneumonia. Chest. 2019;155(1):53-59. https://doi.org/10.1016/j.chest.2018.10.014
11. Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Engl J Med. 2019;380(5):415-424. https://doi.org/10.1056/NEJMoa1808312
12. Li HK, Rombach I, Zambellas R, et al. Oral versus Intravenous Antibiotics for Bone and Joint Infection. N Engl J Med. 2019;380(5):425-436. https://doi.org/10.1056/NEJMoa1710926
13. Fernando SM, Tran A, Cheng W, et al. Prognostic accuracy of the HEART score for prediction of major adverse cardiac events in patients presenting with chest pain: a systematic review and meta-analysis. Acad Emerg Med. 2019;26(2):140-151. https://doi.org/10.1111/acem.13649.
1. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis. 2018;66(7):e1-e48. https://doi.org/10.1093/cid/cix1085.
2. Chu DK, Kim LH, Young PJ, et al. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet. 2018;391(10131):1693-1705. https://doi.org/10.1016/S0140-6736(18)30479-3.
3. Siemieniuk RAC, Chu DK, Kim LH, et al. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ. 2018;363:k4169. https://doi.org/https://doi.org/10.1136/bmj.k4169
4. A PG, O’Conor KJ, Lanzkron S, et al. Do words matter? Stigmatizing language and the transmission of bias in the medical record. J Gen Intern Med. 2018;33(5):685-691. https://doi.org/10.1007/s11606-017-4289-2.
5. Marrouche NF, Kheirkhahan M, Brachmann J. Catheter ablation for atrial fibrillation with heart failure. N Engl J Med. 2018;379(5):492. https://doi.org/10.1056/NEJMoa1707855.
6. Larochelle MR, Bernson D, Land T, et al. Medication for opioid use disorder after nonfatal opioid overdose and association with mortality: a cohort study. Ann Intern Med. 2018;169(3):137-145. https://doi.org/10.7326/M17-3107.
7. Hedegaard HM, A; Warner, M. Drug Overdose Deaths in the United States, 1999-2017. 2018; https://www.cdc.gov/nchs/products/databriefs/db329.htm. Accessed March 07, 2019.
8. Medications for Opioid Use Disorder Save Lives. 2019; http://www.nationalacademies.org/hmd/Reports/2019/medications-for-opioid-use-disorder-save-lives.aspx. Accessed March 07, 2019.
9. Siontis KC, Zhang X, Eckard A, et al. Outcomes associated with apixaban use in patients with end-stage kidney disease and atrial fibrillation in the United States. Circulation. 2018;138(15):1519-1529. https://doi.org/10.1161/CIRCULATIONAHA.118.035418.
10. Cowley MC, Ritchie DJ, Hampton N, Kollef MH, Micek ST. Outcomes Associated With De-escalating Therapy for Methicillin-Resistant Staphylococcus aureus in Culture-Negative Nosocomial Pneumonia. Chest. 2019;155(1):53-59. https://doi.org/10.1016/j.chest.2018.10.014
11. Iversen K, Ihlemann N, Gill SU, et al. Partial oral versus intravenous antibiotic treatment of endocarditis. N Engl J Med. 2019;380(5):415-424. https://doi.org/10.1056/NEJMoa1808312
12. Li HK, Rombach I, Zambellas R, et al. Oral versus Intravenous Antibiotics for Bone and Joint Infection. N Engl J Med. 2019;380(5):425-436. https://doi.org/10.1056/NEJMoa1710926
13. Fernando SM, Tran A, Cheng W, et al. Prognostic accuracy of the HEART score for prediction of major adverse cardiac events in patients presenting with chest pain: a systematic review and meta-analysis. Acad Emerg Med. 2019;26(2):140-151. https://doi.org/10.1111/acem.13649.
© 2019 Society of Hospital Medicine
Perspectives of Clinicians, Staff, and Veterans in Transitioning Veterans from non-VA Hospitals to Primary Care in a Single VA Healthcare System

As the VA moves toward increased utilization of non-VA care, it is crucial to understand and address the challenges of transitional care faced by dual-use veterans to provide high-quality care that improves healthcare outcomes.7,11,12 The VA implemented a shift in policy from the Veterans Access, Choice, and Accountability Act of 2014 (Public Law 113-146; “Choice Act”) to the VA Maintaining Internal Systems and Strengthening Integrated Outside Networks (MISSION) Act beginning June 6, 2019.13,14 Under the MISSION Act, veterans have more ways to access healthcare within the VA’s network and through approved non-VA medical providers in the community known as “community care providers.”15 This shift expanded the existing VA Choice Act of 2014, where the program allowed those veterans who are unable to schedule an appointment within 30 days of their preferred date or the clinically appropriate date, or on the basis of their place of residence, to elect to receive care from eligible non-VA healthcare entities or providers.14,15 These efforts to better serve veterans by increasing non-VA care might present added care coordination challenges for patients and their providers when they seek care in the VA.
High-quality transitional care prevents poor outcomes such as hospital readmissions.16-18 When communication and coordination across healthcare delivery systems are lacking, patients and their families often find themselves at risk for adverse events.19,20 Past research shows that patients have fewer adverse events when they receive comprehensive postdischarge care, including instructions on medications and self-care, symptom recognition and management, and reminders to attend follow-up appointments.17,21,22 Although researchers have identified the components of effective transitional care,23 barriers persist. The communication and collaboration needed to provide coordinated care across healthcare delivery systems are difficult due to the lack of standardized approaches between systems.24 Consequently, follow-up care may be delayed or missed altogether. To our knowledge, there is no published research identifying transitional care challenges for clinicians, staff, and veterans in transitioning from non-VA hospitals to a VA primary care setting.
The objective of this quality assessment was to understand VA and non-VA hospital clinicians’ and staff as well as veterans’ perspectives of the barriers and facilitators to providing high-quality transitional care.
METHODS
Study Design
We conducted a qualitative assessment within the VA Eastern Colorado Health Care System, an urban tertiary medical center, as well as urban and rural non-VA hospitals used by veterans. Semi-structured interview guides informed by the practical robust implementation and sustainability (PRISM) model, the Lean approach, and the Ideal Transitions of Care Bridge were used.25-27 We explored the PRISM domains such as recipient’s characteristics, the interaction with the external environment, and the implementation and sustainability infrastructure to inform the design and implementation of the intervention.25 The Lean approach included methods to optimize processes by maximizing efficiency and minimizing waste.26 The Ideal Transitions of Care Bridge was used to identify the domains in transitions of care such as discharge planning, communication of information, and care coordination.27
Setting and Participants
We identified the top 10 non-VA hospitals serving the most urban and rural veterans in 2015 using VA administrative data. Purposive sampling was used to ensure that urban and rural non-VA hospitals and different roles within these hospitals were represented. VA clinicians and staff were selected from the Denver VA Medical Center, a tertiary hospital within the Eastern Colorado Health Care System and one VA Community-Based Outpatient Clinic (CBOC) that primarily serves rural veterans. The Denver VA Medical Center has three clinics staffed by Patient Aligned Care Teams (PACTs), a model built on the concept of Patient-Centered Medical Home.28 Hospital leadership were initially approached for permission to recruit their staff and to be involved as key informants, and all agreed. To ensure representativeness, diversity of roles was recruited, including PACT primary care physicians, nurses, and other staff members such as medical assistants and administrators. Veterans were approached for sampling if they were discharged from a non-VA hospital during June–September 2015 and used the VA for primary care. This was to ensure that they remembered the process they went through postdischarge at the time of the interview.
Data Collection and Analysis
The evaluation team members (RA, EL, and MM) conducted the interviews from November 2015 to July 2016. Clinicians, staff, and veterans were asked semi-structured questions about their experiences and their role in transitioning VA patients across systems (see Appendix for interview guides). Veterans were asked to describe their experience and satisfaction with the current postdischarge transition process. We stopped the interviews when we reached data saturation.29
Interviews were audio-recorded, transcribed verbatim, and validated (transcribed interviews were double-checked against recording) to ensure data quality and accuracy. Coding was guided by a conventional content analysis technique30, 31 using a deductive and inductive coding approach.31 The deductive coding approach was drawn from the Ideal Transitions of Care Bridge and PRISM domains. 32,33 Two evaluation team members (RA and EL) defined the initial code book by independently coding the first three interviews, worked to clarify the meanings of emergent codes, and came to a consensus when disagreements occurred. Next, a priori codes were added by team members to include the PRISM domains. These PRISM domains included the implementation and sustainability infrastructure, the external environment, the characteristics of intervention recipients, and the organizational and patient perspectives of an intervention.
Additional emergent codes were added to the code book and agreed upon by team members (RA, EL, and MM). Consistent with previously used methods, consensus building was achieved by identifying and resolving differences by discussing with team members (RA, EL, MM, CB, and RB).29 Codes were examined and organized into themes by team members.29,34-36 This process was continued until no new themes were identified. Results were reviewed by all evaluation team members to assess thoroughness and comprehensiveness.34,35 In addition, team members triangulated the findings with VA and non-VA participants to ensure validity and reduce researcher bias.29,37
RESULTS
We conducted a total of 70 interviews with 23 VA and 29 non-VA hospital clinicians and staff and 18 veterans (Table 1). Overall, we found that there was no standardized process for transitioning veterans across healthcare delivery systems. Participants reported that transitions were often inefficient when non-VA hospitals could not (1) identify patients as veterans and notify VA primary care of discharge; (2) transfer non-VA hospital medical records to VA primary care; (3) obtain follow-up care appointments with VA primary care; and (4) write VA formulary medications for veterans to fill at VA pharmacies. In addition, participants discussed about facilitators and suggestions to overcome these inefficiencies and improve transitional care (Table2). We mapped the identified barriers as well as the suggestions for improvement to the PRISM and the Ideal Transitions of Care Bridge domains (Table 3).
Unable to Identify Patients as Veterans and Notify VA Primary Care of Discharge
VA and non-VA participants reported difficulty in communicating about veterans’ hospitalizations and discharge follow-up needs across systems. Non-VA clinicians referenced difficulty in identifying patients as veterans to communicate with VA, except in instances where the VA is a payor, while VA providers described feeling largely uninformed of the veterans non-VA hospitalization. For non-VA clinicians, the lack of a systematic method for veteran identification often left them to inadvertently identify veteran status by asking about their primary care clinicians and insurance and even through an offhanded comment made by the veteran. If a veteran was identified, non-VA clinicians described being uncertain about the best way to notify VA primary care of the patient’s impending discharge. Veterans described instances of the non-VA hospital knowing their veteran status upon admission, but accounts varied on whether the non-VA hospital notified the VA primary care of their hospitalization (Table 2, Theme 1).
Unable to Transfer Non-VA Hospital Medical Records to VA Primary Care
VA clinicians discussed about the challenges associated with obtaining the veteran’s medical record from the non-VA hospitals, and when it was received, it was often incomplete information and significantly delayed. They described relying on the veteran’s description of the care received, which was not complete or accurate information needed to make clinical judgment or coordinate follow-up care. Non-VA clinicians mentioned about trying several methods for transferring the medical record to VA primary care, including discharge summary via electronic system and sometimes solely relying on patients to deliver discharge paperwork to their primary care clinicians. In instances where non-VA hospitals sent discharge paperwork to VA, there was no way for non-VA hospitals to verify whether the faxed electronic medical record was received by the VA hospital. Most of the veterans discussed receiving written postdischarge instructions to take to their VA primary care clinicians; however, they were unsure whether the VA primary care received their medical record or any other information from the non-VA hospital (Table 2, Theme 2).
Unable to Obtain Follow-Up Care Appointments with VA Primary Care
All participants described how difficult it was to obtain a follow-up appointment for veterans with VA primary care. This often resulted in delayed follow-up care. VA clinicians also shared that a non-VA hospitalization can be the impetus for a veteran to seek care at the VA for the very first time. Once eligibility is determined, the veteran is assigned a VA primary care clinician. This process may take up to six weeks, and in the meantime, the veteran is scheduled in VA urgent care for immediate postdischarge care. This lag in primary care assignment creates delayed and fragmented care (Table 2, Theme 3).
Non-VA clinicians, administrators, and staff also discussed the difficulties in scheduling follow-up care with VA primary care. Although discharge paperwork instructed patients to see their VA clinicians, there was no process in place for non-VA clinicians to confirm whether the follow-up care was received due to lack of bilateral communication. In addition, veterans discussed the inefficiencies in scheduling follow-up appointments with VA clinicians where attempts to follow-up with primary care clinicians took eight weeks or more. Several veterans described walking into the clinic without an appointment asking to be seen postdischarge or utilizing the VA emergency department for follow-up care after discharge from a non-VA hospital. Veterans admitted utilizing the VA emergency department for nonemergent reasons such as filling their prescriptions because they are unable to see a VA PCP in a timely manner (Table 2, Theme 3).
Unable to Write VA Formulary Medications for Veterans to Fill at VA Pharmacies
All participants described the difficulties in obtaining medications at VA pharmacies when prescribed by the non-VA hospital clinicians. VA clinicians often had to reassess, and rewrite prescriptions written by clinicians, causing delays. Moreover, rural VA clinicians described lack of VA pharmacies in their locations, where veterans had to mail order medications, causing further delays in needed medications. Non-VA clinicians echoed these frustrations. They noted that veterans were confused about their VA pharmacy benefits as well as the need for the non-VA clinicians to follow VA formulary guidelines. Veterans expressed that it was especially challenging to physically go to the VA pharmacy to pick up medications after discharge due to lack of transportation, limited VA pharmacy hours, and long wait times. Several veterans discussed paying for their prescriptions out of pocket even though they had VA pharmacy benefits because it was more convenient to use the non-VA pharmacy. In other instances, veterans discussed going to a VA emergency department and waiting for hours to have their non-VA clinician prescription rewritten by a VA clinician (Table 2, Theme 4).
Facilitators of the Current Transition Process
Several participants provided examples of when transitional care communication between systems occurred seamlessly. VA staff and veterans noted that the VA increased the availability of urgent care appointments, which allowed for timelier postacute care follow-up appointments. Non-VA hospital clinicians also noted the availability of additional appointment slots but stated that they did not learn about these additional appointments directly from the VA. Instead, they learned of these through medical residents caring for patients at both VA and non-VA hospitals. One VA CBOC designated two nurses to care for walk-in veterans for their postdischarge follow-up needs. Some VA participants also noted that the VA Call Center Nurses occasionally called veterans upon discharge to schedule a follow-up appointment and facilitated timely care.
Participants from a VA CBOC discussed being part of a Community Transitions Consortium aimed at identifying high-utilizing patients (veteran and nonveteran) and improving communication across systems. The consortium members discussed each facility’s transition-of-care process, described having access to local non-VA hospital medical records and a backline phone number at the non-VA hospitals to coordinate transitional care. This allowed the VA clinicians to learn about non-VA hospital processes and veteran needs.
Suggestions for Improving the Transitional Care Process
VA and non-VA clinicians suggested hiring a VA liaison, preferably with a clinical background to facilitate care coordination across healthcare systems. They recommended that this person work closely with VA primary care, strengthen the relationship with non-VA hospitals, and help veterans learn more about the transition-of-care processes. Topics discussed for veteran education included how to (1) access their primary care tea
Veterans agreed that improvements to the current process should include an efficient system for obtaining medications and the ability to schedule timely follow-up appointments. Furthermore, veterans wanted education about the VA transition-of-care process following a non-VA hospitalization, including payment and VA notification processes (Table 2, Theme 5).
DISCUSSION
Participants described the current transitional care process as inefficient with specific barriers that have negative consequences on patient care and clinician and staff work processes. They described difficulties in obtaining medications prescribed by non-VA clinicians from VA pharmacies, delays in follow-up appointments at the VA, and lack of bilateral communication between systems and medical record transfer. Participants also provided concrete suggestions to improving the current process, including a care coordinator with clinical background. These findings are important in the context of VA increasing veteran access to care in the community.
Despite an increasing emphasis on veteran access to non-VA care as a result of the VA strategic goals and several new programs,7,12,13 there has not been a close examination of the current transition-of-care process from non-VA hospitals to VA primary care. Several studies have shown that the period following a hospitalization is especially vulnerable and associated with adverse events such as readmission, high cost, and death.12,31,32 Our findings agree with previous research that identified medical record transfer across systems as one of the most challenging issues contributing to deficits in communication between care teams.33 In addition, our study brought into focus the significant challenges faced by veterans in obtaining medications post non-VA hospital discharge. Addressing these key barriers in transitional care will improve the quality, safety, and value of healthcare in the current transition process.38,39
Based on our findings, our participants’ concern in transitional care can be addressed in various ways. First, as veterans are increasingly receiving care in the community, identifying their veteran status early on in the non-VA hospital setting could help in improved, real time communication with the VA. This could be done by updating patient intake forms to ask patients whether they are veterans or not. Second, VA policy-level changes should work to provide veterans access to non-VA pharmacy benefits equivalent to the access patients are receiving for hospital, specialty, and outpatient care. Third, patient and provider satisfaction for dual-use veterans should be examined closely. Although participants expressed frustration with the overall transitions of care from non-VA hospitals to VA primary care setting, influence of this on the Quadruple Aim-improving patient outcomes, experience, and reducing clinician and staff burnout should be examined closely.40 Fourth, evidence-based interventions such as nurse-led transitional care programs that have proven helpful in reducing adverse outcomes in both VA and non-VA settings will be useful to implement.41-45 Such programs could be located in the VA, and a care coordinator role could help facilitate transitional care needs for veterans by working with multiple non-VA hospitals.
The limitations of this study are that the perspectives shared by these participants may not represent all VA and non-VA hospitals as well as veterans’ experiences with transition of care. In addition, the study was conducted in one state and the findings may not be applicable to other healthcare systems. However, our study highlighted the consistent challenges of receiving care across VA and other hospital systems. Two strengths of this study are that it was conducted by multidisciplinary research team members with expertise in qualitative research, clinical care, and implementation science and that we obtained convergent information from VA, non-VA, and veteran participants.
Our current transition-of-care process has several shortcomings. There was a clear agreement on barriers, facilitators, and suggestions for improving the current transitions-of-care process among VA and non-VA hospital participants, as well as from veterans who experienced transitions across different delivery systems. Transitioning veterans to VA primary care following a non-VA hospitalization is a crucial first step for improving care for veterans and reducing adverse outcomes such as avoidable hospital readmissions and death.
These results describe the inefficiencies experienced by patients, clinicians, and staff and their suggestions to alleviate these barriers for optimal continuum of care. To avoid frustration and inefficiencies, the increased emphasis of providing non-VA care for veterans should consider the challenges experienced in transitional care and the opportunities for increased coordination of care.
1. Borowsky SJ, Cowper DC. Dual use of VA and non-VA primary care. J Gen Intern Med. 1999;14(5):274-280. https://doi.org/10.1046/j.1525-1497.1999.00335.x.
2. Charlton ME, Mengeling MA, Schlichting JA, et al. Veteran use of health care systems in rural states. Comparing VA and Non-VA health care use among privately insured veterans under age 65. J Rural Health. 2016;32(4):407-417. https://doi.org/10.1111/jrh.12206.
3. Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138(3):161. https://doi.org/10.7326/0003-4819-138-3-200302040-00007.
4. Nguyen KA, Haggstrom DA, Ofner S, et al. Medication use among veterans across health care systems. Appl Clin Inform. 2017;26(1):235-249. https://doi.org/10.4338/ACI-2016-10-RA-0184.
5. Nayar P, Apenteng B, Yu F, Woodbridge P, Fetrick A. Rural veterans’ perspectives of dual care. J Commun Health. 2013;38(1):70-77. https://doi.org/10.1007/s10900-012-9583-7.
6. West AN, Charlton ME. Insured veterans’ use of VA and Non-VA health care in a rural state. J Rural Health. 2016;32(4):387-396. https://doi.org/10.1111/jrh.12196.
7. Gellad WF. The veterans choice act and dual health system use. J Gen Intern Med. 2016;31(2):153-154. https://doi.org/10.1007/s11606-015-3492-2.
8. Axon RN, Gebregziabher M, Everett CJ, Heidenreich P, Hunt KJ. Dual health care system use is associated with higher rates of hospitalization and hospital readmission among veterans with heart failure. Am Heart J. 2016;174:157-163. https://doi.org/10.1016/j.ahj.2015.09.023.
9. Humensky J, Carretta H, de Groot K, et al. Service utilization of veterans dually eligible for VA and medicare fee-for-service: 1999–2004. Medicare Medicaid Res Rev. 2012;2(3). https://doi.org/10.5600/mmrr.002.03.A06.
10. West AN, Charlton ME, Vaughan-Sarrazin M. Dual use of VA and non-VA hospitals by veterans with multiple hospitalizations. BMC Health Serv Res. 2015;15(1):431. https://doi.org/10.1186/s12913-015-1069-8.
11. Gaglioti A, Cozad A, Wittrock S, et al. Non-VA primary care providers’ perspectives on comanagement for rural veterans. Mil Med. 2014;179(11):1236-1243. https://doi.org/10.7205/MILMED-D-13-00342.
12. Department of Veterans Affairs. Expanded access to non-VA care through the veterans choice program. Final rule. Fed Regist. 2018;83(92):21893-21897.
13. Shuster B. Text-H.R.3236-114th Congress. Surface Transportation and Veterans Health Care Choice Improvement Act of 2015.. https://www.congress.gov/bill/114th-congress/house-bill/3236/text/pl. Accessed April 16, 2017; 2015-2016.
14. Veterans Affairs Mission Act. MISSIONAct.va.gov Available at. https://missionact.va.gov/. Accessed August 9, 2019.
15. Veterans Choice Program (VCP). Community care. https://www.va.gov/COMMUNITYCARE/programs/veterans/VCP/index.asp. Accessed August 9, 2019.
16. A Decade of Transitional Care Research with Vulnerable Elder… : journal of cardiovascular nursing. LWW. http://journals.lww.com/jcnjournal/Fulltext/2000/04000/A_Decade_of_Transitional_Care_Research_with.4.aspx. Accessed April 16, 2017.
17. Coleman EA, Boult C. Improving the quality of transitional care for persons with complex care needs. J Am Geriatr Soc. 2003;51(4):556-557. https://doi.org/10.1046/j.1532-5415.2003.51186.x.
18. Krichbaum K. GAPN postacute care coordination improves hip fracture outcomes. West J Nurs Res. 2007;29(5):523-544. https://doi.org/10.1177/0193945906293817.
19. Kripalani S, Jackson AT, Schnipper JL, Coleman EA. Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314-323. https://doi.org/10.1002/jhm.228.
20. Coleman EA, Mahoney E, Parry C. Assessing the quality of preparation for posthospital care from the patient’s perspective: the care transitions measure. Med Care. 2005;43(3):246-255. https://doi.org/10.1097/00005650-200503000-00007.
21. Naylor MD, Aiken LH, Kurtzman ET, Olds DM, Hirschman KB. The importance of transitional care in achieving health reform. Health Aff (Millwood). 2011;30(4):746-754. https://doi.org/10.1377/hlthaff.2011.0041.
22. Naylor MD, Brooten DA, Campbell RL, et al. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc. 2004;52(5):675-684. https://doi.org/10.1111/j.1532-5415.2004.52202.x.
23. Snow V, Beck D, Budnitz T, et al. Transitions of care consensus policy statement: American College of Physicians, Society of General Internal Medicine, society of hospital medicine, American Geriatrics Society, American College of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364-370. https://doi.org/10.1002/jhm.510.
24. Coleman EA. Falling through the cracks: challenges and opportunities for improving transitional care for persons with continuous complex care needs. J Am Geriatr Soc. 2003;51(4):549-555. https://doi.org/10.1046/j.1532-5415.2003.51185.x.
25. Feldstein AC, Glasgow RE. A practical, robust implementation and sustainability model (PRISM) for integrating research findings into practice. Jt Comm J Qual Patient Saf. 2008;34(4):228-243. https://doi.org/10.1016/S1553-7250(08)34030-6.
26. Schweikhart SA, Dembe AE. The applicability of lean and six sigma techniques to clinical and translational research. J Investig Med. 2009;57(7):748-755. https://doi.org/10.2310/JIM.0b013e3181b91b3a.
27. Burke RE, Kripalani S, Vasilevskis EE, Schnipper JL. Moving beyond readmission penalties: creating an ideal process to improve transitional care. J Hosp Med. 2013;8(2):102-109. https://doi.org/10.1002/jhm.1990.
28. Patient Aligned Care Team (PACT)-Patient Care. Services. https://www.patientcare.va.gov/primarycare/PACT.asp. Accessed November 20, 2017.
29. Morse JM. Critical analysis of strategies for determining rigor in qualitative inquiry. Qual Health Res. 2015;25(9):1212-1222. https://doi.org/10.1177/1049732315588501.
30. Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15(9):1277-1288. https://doi.org/10.1177/1049732305276687.
31. Fereday J, Muir-Cochrane E. Demonstrating rigor using thematic analysis: a hybrid approach of inductive and deductive coding and theme development. Int J Qual Methods. 2006;5(1):80-92. https://doi.org/10.1177/160940690600500107.
32. Ayele RA, Lawrence E, McCreight M, et al. Study protocol: improving the transition of care from a non-network hospital back to the patient’s medical home. BMC Health Serv Res. 2017;17(1):123. https://doi.org/10.1186/s12913-017-2048-z.
33. Burke RE, Kripalani S, Vasilevskis EE, Schnipper JL. Moving beyond readmission penalties: creating an ideal process to improve transitional care. J Hosp Med. 2013;8(2):102-109. https://doi.org/10.1002/jhm.1990.
34. Qualitative research & evaluation methods. https://us.sagepub.com/en-us/nam/qualitative-research-evaluation-methods/book232962. Accessed April 16, 2017. SAGE Publications Inc.
35. Curry LA, Nembhard IM, Bradley EH. Qualitative and mixed methods provide unique contributions to outcomes research. Circulation. 2009;119(10):1442-1452. https://doi.org/10.1161/CIRCULATIONAHA.107.742775.
36. Creswell JW, Hanson WE, Clark Plano VL, Morales A. Qualitative research designs: selection and implementation. Couns Psychol. 2007;35(2):236-264. https://doi.org/10.1177/0011000006287390.
37. Carter N, Bryant-Lukosius D, DiCenso A, Blythe J, Neville AJ. The use of triangulation in qualitative research. Oncol Nurs Forum. 2014;41(5):545-547. https://doi.org/10.1188/14.ONF.545-547.
38. Krumholz HM. Post-hospital syndrome—an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. https://doi.org/10.1056/NEJMp1212324.
39. Improving Care Transitions. Health affairs-health policy briefs. http://www.healthaffairs.org/healthpolicybriefs/brief.php?brief_id=76. Accessed August 13, 2016.
40. Bodenheimer T, Sinsky C. From triple to quadruple aim: care of the patient requires care of the provider. Ann Fam Med. 2014;12(6):573-576. https://doi.org/10.1370/afm.1713.
41. Burke RE, Kelley L, Gunzburger E, et al. Improving transitions of care for veterans transferred to tertiary VA medical centers. Am J Med Qual. 2018;33(2):147-153. https://doi.org/10.1177/1062860617715508.
42. Capp R, Misky GJ, Lindrooth RC, et al. Coordination program reduced acute care use and increased primary care visits among frequent emergency care users. Health Aff (Millwood). 2017;36(10):1705-1711. https://doi.org/10.1377/hlthaff.2017.0612.
43. Kind AJH, Brenny-Fitzpatrick M, Leahy-Gross K, et al. Harnessing protocolized adaptation in dissemination: successful implementation and sustainment of the veterans affairs coordinated-transitional care program in a non-veterans affairs hospital. J Am Geriatr Soc. 2016;64(2):409-416. https://doi.org/10.1111/jgs.13935.
44. Kind AJH, Jensen L, Barczi S, et al. Low-cost transitional care with nurse managers making mostly phone contact With patients cut rehospitalization at a VA Hospital. Health Aff. 2012;31(12):2659-2668. https://doi.org/10.1377/hlthaff.2012.0366.
45. Reese RL, Clement SA, Syeda S, et al. Coordinated-transitional care for veterans with heart failure and chronic lung disease. J Am Geriatr Soc. 2019;67(7):1502-1507. https://doi.org/10.1111/jgs.15978.
As the VA moves toward increased utilization of non-VA care, it is crucial to understand and address the challenges of transitional care faced by dual-use veterans to provide high-quality care that improves healthcare outcomes.7,11,12 The VA implemented a shift in policy from the Veterans Access, Choice, and Accountability Act of 2014 (Public Law 113-146; “Choice Act”) to the VA Maintaining Internal Systems and Strengthening Integrated Outside Networks (MISSION) Act beginning June 6, 2019.13,14 Under the MISSION Act, veterans have more ways to access healthcare within the VA’s network and through approved non-VA medical providers in the community known as “community care providers.”15 This shift expanded the existing VA Choice Act of 2014, where the program allowed those veterans who are unable to schedule an appointment within 30 days of their preferred date or the clinically appropriate date, or on the basis of their place of residence, to elect to receive care from eligible non-VA healthcare entities or providers.14,15 These efforts to better serve veterans by increasing non-VA care might present added care coordination challenges for patients and their providers when they seek care in the VA.
High-quality transitional care prevents poor outcomes such as hospital readmissions.16-18 When communication and coordination across healthcare delivery systems are lacking, patients and their families often find themselves at risk for adverse events.19,20 Past research shows that patients have fewer adverse events when they receive comprehensive postdischarge care, including instructions on medications and self-care, symptom recognition and management, and reminders to attend follow-up appointments.17,21,22 Although researchers have identified the components of effective transitional care,23 barriers persist. The communication and collaboration needed to provide coordinated care across healthcare delivery systems are difficult due to the lack of standardized approaches between systems.24 Consequently, follow-up care may be delayed or missed altogether. To our knowledge, there is no published research identifying transitional care challenges for clinicians, staff, and veterans in transitioning from non-VA hospitals to a VA primary care setting.
The objective of this quality assessment was to understand VA and non-VA hospital clinicians’ and staff as well as veterans’ perspectives of the barriers and facilitators to providing high-quality transitional care.
METHODS
Study Design
We conducted a qualitative assessment within the VA Eastern Colorado Health Care System, an urban tertiary medical center, as well as urban and rural non-VA hospitals used by veterans. Semi-structured interview guides informed by the practical robust implementation and sustainability (PRISM) model, the Lean approach, and the Ideal Transitions of Care Bridge were used.25-27 We explored the PRISM domains such as recipient’s characteristics, the interaction with the external environment, and the implementation and sustainability infrastructure to inform the design and implementation of the intervention.25 The Lean approach included methods to optimize processes by maximizing efficiency and minimizing waste.26 The Ideal Transitions of Care Bridge was used to identify the domains in transitions of care such as discharge planning, communication of information, and care coordination.27
Setting and Participants
We identified the top 10 non-VA hospitals serving the most urban and rural veterans in 2015 using VA administrative data. Purposive sampling was used to ensure that urban and rural non-VA hospitals and different roles within these hospitals were represented. VA clinicians and staff were selected from the Denver VA Medical Center, a tertiary hospital within the Eastern Colorado Health Care System and one VA Community-Based Outpatient Clinic (CBOC) that primarily serves rural veterans. The Denver VA Medical Center has three clinics staffed by Patient Aligned Care Teams (PACTs), a model built on the concept of Patient-Centered Medical Home.28 Hospital leadership were initially approached for permission to recruit their staff and to be involved as key informants, and all agreed. To ensure representativeness, diversity of roles was recruited, including PACT primary care physicians, nurses, and other staff members such as medical assistants and administrators. Veterans were approached for sampling if they were discharged from a non-VA hospital during June–September 2015 and used the VA for primary care. This was to ensure that they remembered the process they went through postdischarge at the time of the interview.
Data Collection and Analysis
The evaluation team members (RA, EL, and MM) conducted the interviews from November 2015 to July 2016. Clinicians, staff, and veterans were asked semi-structured questions about their experiences and their role in transitioning VA patients across systems (see Appendix for interview guides). Veterans were asked to describe their experience and satisfaction with the current postdischarge transition process. We stopped the interviews when we reached data saturation.29
Interviews were audio-recorded, transcribed verbatim, and validated (transcribed interviews were double-checked against recording) to ensure data quality and accuracy. Coding was guided by a conventional content analysis technique30, 31 using a deductive and inductive coding approach.31 The deductive coding approach was drawn from the Ideal Transitions of Care Bridge and PRISM domains. 32,33 Two evaluation team members (RA and EL) defined the initial code book by independently coding the first three interviews, worked to clarify the meanings of emergent codes, and came to a consensus when disagreements occurred. Next, a priori codes were added by team members to include the PRISM domains. These PRISM domains included the implementation and sustainability infrastructure, the external environment, the characteristics of intervention recipients, and the organizational and patient perspectives of an intervention.
Additional emergent codes were added to the code book and agreed upon by team members (RA, EL, and MM). Consistent with previously used methods, consensus building was achieved by identifying and resolving differences by discussing with team members (RA, EL, MM, CB, and RB).29 Codes were examined and organized into themes by team members.29,34-36 This process was continued until no new themes were identified. Results were reviewed by all evaluation team members to assess thoroughness and comprehensiveness.34,35 In addition, team members triangulated the findings with VA and non-VA participants to ensure validity and reduce researcher bias.29,37
RESULTS
We conducted a total of 70 interviews with 23 VA and 29 non-VA hospital clinicians and staff and 18 veterans (Table 1). Overall, we found that there was no standardized process for transitioning veterans across healthcare delivery systems. Participants reported that transitions were often inefficient when non-VA hospitals could not (1) identify patients as veterans and notify VA primary care of discharge; (2) transfer non-VA hospital medical records to VA primary care; (3) obtain follow-up care appointments with VA primary care; and (4) write VA formulary medications for veterans to fill at VA pharmacies. In addition, participants discussed about facilitators and suggestions to overcome these inefficiencies and improve transitional care (Table2). We mapped the identified barriers as well as the suggestions for improvement to the PRISM and the Ideal Transitions of Care Bridge domains (Table 3).
Unable to Identify Patients as Veterans and Notify VA Primary Care of Discharge
VA and non-VA participants reported difficulty in communicating about veterans’ hospitalizations and discharge follow-up needs across systems. Non-VA clinicians referenced difficulty in identifying patients as veterans to communicate with VA, except in instances where the VA is a payor, while VA providers described feeling largely uninformed of the veterans non-VA hospitalization. For non-VA clinicians, the lack of a systematic method for veteran identification often left them to inadvertently identify veteran status by asking about their primary care clinicians and insurance and even through an offhanded comment made by the veteran. If a veteran was identified, non-VA clinicians described being uncertain about the best way to notify VA primary care of the patient’s impending discharge. Veterans described instances of the non-VA hospital knowing their veteran status upon admission, but accounts varied on whether the non-VA hospital notified the VA primary care of their hospitalization (Table 2, Theme 1).
Unable to Transfer Non-VA Hospital Medical Records to VA Primary Care
VA clinicians discussed about the challenges associated with obtaining the veteran’s medical record from the non-VA hospitals, and when it was received, it was often incomplete information and significantly delayed. They described relying on the veteran’s description of the care received, which was not complete or accurate information needed to make clinical judgment or coordinate follow-up care. Non-VA clinicians mentioned about trying several methods for transferring the medical record to VA primary care, including discharge summary via electronic system and sometimes solely relying on patients to deliver discharge paperwork to their primary care clinicians. In instances where non-VA hospitals sent discharge paperwork to VA, there was no way for non-VA hospitals to verify whether the faxed electronic medical record was received by the VA hospital. Most of the veterans discussed receiving written postdischarge instructions to take to their VA primary care clinicians; however, they were unsure whether the VA primary care received their medical record or any other information from the non-VA hospital (Table 2, Theme 2).
Unable to Obtain Follow-Up Care Appointments with VA Primary Care
All participants described how difficult it was to obtain a follow-up appointment for veterans with VA primary care. This often resulted in delayed follow-up care. VA clinicians also shared that a non-VA hospitalization can be the impetus for a veteran to seek care at the VA for the very first time. Once eligibility is determined, the veteran is assigned a VA primary care clinician. This process may take up to six weeks, and in the meantime, the veteran is scheduled in VA urgent care for immediate postdischarge care. This lag in primary care assignment creates delayed and fragmented care (Table 2, Theme 3).
Non-VA clinicians, administrators, and staff also discussed the difficulties in scheduling follow-up care with VA primary care. Although discharge paperwork instructed patients to see their VA clinicians, there was no process in place for non-VA clinicians to confirm whether the follow-up care was received due to lack of bilateral communication. In addition, veterans discussed the inefficiencies in scheduling follow-up appointments with VA clinicians where attempts to follow-up with primary care clinicians took eight weeks or more. Several veterans described walking into the clinic without an appointment asking to be seen postdischarge or utilizing the VA emergency department for follow-up care after discharge from a non-VA hospital. Veterans admitted utilizing the VA emergency department for nonemergent reasons such as filling their prescriptions because they are unable to see a VA PCP in a timely manner (Table 2, Theme 3).
Unable to Write VA Formulary Medications for Veterans to Fill at VA Pharmacies
All participants described the difficulties in obtaining medications at VA pharmacies when prescribed by the non-VA hospital clinicians. VA clinicians often had to reassess, and rewrite prescriptions written by clinicians, causing delays. Moreover, rural VA clinicians described lack of VA pharmacies in their locations, where veterans had to mail order medications, causing further delays in needed medications. Non-VA clinicians echoed these frustrations. They noted that veterans were confused about their VA pharmacy benefits as well as the need for the non-VA clinicians to follow VA formulary guidelines. Veterans expressed that it was especially challenging to physically go to the VA pharmacy to pick up medications after discharge due to lack of transportation, limited VA pharmacy hours, and long wait times. Several veterans discussed paying for their prescriptions out of pocket even though they had VA pharmacy benefits because it was more convenient to use the non-VA pharmacy. In other instances, veterans discussed going to a VA emergency department and waiting for hours to have their non-VA clinician prescription rewritten by a VA clinician (Table 2, Theme 4).
Facilitators of the Current Transition Process
Several participants provided examples of when transitional care communication between systems occurred seamlessly. VA staff and veterans noted that the VA increased the availability of urgent care appointments, which allowed for timelier postacute care follow-up appointments. Non-VA hospital clinicians also noted the availability of additional appointment slots but stated that they did not learn about these additional appointments directly from the VA. Instead, they learned of these through medical residents caring for patients at both VA and non-VA hospitals. One VA CBOC designated two nurses to care for walk-in veterans for their postdischarge follow-up needs. Some VA participants also noted that the VA Call Center Nurses occasionally called veterans upon discharge to schedule a follow-up appointment and facilitated timely care.
Participants from a VA CBOC discussed being part of a Community Transitions Consortium aimed at identifying high-utilizing patients (veteran and nonveteran) and improving communication across systems. The consortium members discussed each facility’s transition-of-care process, described having access to local non-VA hospital medical records and a backline phone number at the non-VA hospitals to coordinate transitional care. This allowed the VA clinicians to learn about non-VA hospital processes and veteran needs.
Suggestions for Improving the Transitional Care Process
VA and non-VA clinicians suggested hiring a VA liaison, preferably with a clinical background to facilitate care coordination across healthcare systems. They recommended that this person work closely with VA primary care, strengthen the relationship with non-VA hospitals, and help veterans learn more about the transition-of-care processes. Topics discussed for veteran education included how to (1) access their primary care tea
Veterans agreed that improvements to the current process should include an efficient system for obtaining medications and the ability to schedule timely follow-up appointments. Furthermore, veterans wanted education about the VA transition-of-care process following a non-VA hospitalization, including payment and VA notification processes (Table 2, Theme 5).
DISCUSSION
Participants described the current transitional care process as inefficient with specific barriers that have negative consequences on patient care and clinician and staff work processes. They described difficulties in obtaining medications prescribed by non-VA clinicians from VA pharmacies, delays in follow-up appointments at the VA, and lack of bilateral communication between systems and medical record transfer. Participants also provided concrete suggestions to improving the current process, including a care coordinator with clinical background. These findings are important in the context of VA increasing veteran access to care in the community.
Despite an increasing emphasis on veteran access to non-VA care as a result of the VA strategic goals and several new programs,7,12,13 there has not been a close examination of the current transition-of-care process from non-VA hospitals to VA primary care. Several studies have shown that the period following a hospitalization is especially vulnerable and associated with adverse events such as readmission, high cost, and death.12,31,32 Our findings agree with previous research that identified medical record transfer across systems as one of the most challenging issues contributing to deficits in communication between care teams.33 In addition, our study brought into focus the significant challenges faced by veterans in obtaining medications post non-VA hospital discharge. Addressing these key barriers in transitional care will improve the quality, safety, and value of healthcare in the current transition process.38,39
Based on our findings, our participants’ concern in transitional care can be addressed in various ways. First, as veterans are increasingly receiving care in the community, identifying their veteran status early on in the non-VA hospital setting could help in improved, real time communication with the VA. This could be done by updating patient intake forms to ask patients whether they are veterans or not. Second, VA policy-level changes should work to provide veterans access to non-VA pharmacy benefits equivalent to the access patients are receiving for hospital, specialty, and outpatient care. Third, patient and provider satisfaction for dual-use veterans should be examined closely. Although participants expressed frustration with the overall transitions of care from non-VA hospitals to VA primary care setting, influence of this on the Quadruple Aim-improving patient outcomes, experience, and reducing clinician and staff burnout should be examined closely.40 Fourth, evidence-based interventions such as nurse-led transitional care programs that have proven helpful in reducing adverse outcomes in both VA and non-VA settings will be useful to implement.41-45 Such programs could be located in the VA, and a care coordinator role could help facilitate transitional care needs for veterans by working with multiple non-VA hospitals.
The limitations of this study are that the perspectives shared by these participants may not represent all VA and non-VA hospitals as well as veterans’ experiences with transition of care. In addition, the study was conducted in one state and the findings may not be applicable to other healthcare systems. However, our study highlighted the consistent challenges of receiving care across VA and other hospital systems. Two strengths of this study are that it was conducted by multidisciplinary research team members with expertise in qualitative research, clinical care, and implementation science and that we obtained convergent information from VA, non-VA, and veteran participants.
Our current transition-of-care process has several shortcomings. There was a clear agreement on barriers, facilitators, and suggestions for improving the current transitions-of-care process among VA and non-VA hospital participants, as well as from veterans who experienced transitions across different delivery systems. Transitioning veterans to VA primary care following a non-VA hospitalization is a crucial first step for improving care for veterans and reducing adverse outcomes such as avoidable hospital readmissions and death.
These results describe the inefficiencies experienced by patients, clinicians, and staff and their suggestions to alleviate these barriers for optimal continuum of care. To avoid frustration and inefficiencies, the increased emphasis of providing non-VA care for veterans should consider the challenges experienced in transitional care and the opportunities for increased coordination of care.
As the VA moves toward increased utilization of non-VA care, it is crucial to understand and address the challenges of transitional care faced by dual-use veterans to provide high-quality care that improves healthcare outcomes.7,11,12 The VA implemented a shift in policy from the Veterans Access, Choice, and Accountability Act of 2014 (Public Law 113-146; “Choice Act”) to the VA Maintaining Internal Systems and Strengthening Integrated Outside Networks (MISSION) Act beginning June 6, 2019.13,14 Under the MISSION Act, veterans have more ways to access healthcare within the VA’s network and through approved non-VA medical providers in the community known as “community care providers.”15 This shift expanded the existing VA Choice Act of 2014, where the program allowed those veterans who are unable to schedule an appointment within 30 days of their preferred date or the clinically appropriate date, or on the basis of their place of residence, to elect to receive care from eligible non-VA healthcare entities or providers.14,15 These efforts to better serve veterans by increasing non-VA care might present added care coordination challenges for patients and their providers when they seek care in the VA.
High-quality transitional care prevents poor outcomes such as hospital readmissions.16-18 When communication and coordination across healthcare delivery systems are lacking, patients and their families often find themselves at risk for adverse events.19,20 Past research shows that patients have fewer adverse events when they receive comprehensive postdischarge care, including instructions on medications and self-care, symptom recognition and management, and reminders to attend follow-up appointments.17,21,22 Although researchers have identified the components of effective transitional care,23 barriers persist. The communication and collaboration needed to provide coordinated care across healthcare delivery systems are difficult due to the lack of standardized approaches between systems.24 Consequently, follow-up care may be delayed or missed altogether. To our knowledge, there is no published research identifying transitional care challenges for clinicians, staff, and veterans in transitioning from non-VA hospitals to a VA primary care setting.
The objective of this quality assessment was to understand VA and non-VA hospital clinicians’ and staff as well as veterans’ perspectives of the barriers and facilitators to providing high-quality transitional care.
METHODS
Study Design
We conducted a qualitative assessment within the VA Eastern Colorado Health Care System, an urban tertiary medical center, as well as urban and rural non-VA hospitals used by veterans. Semi-structured interview guides informed by the practical robust implementation and sustainability (PRISM) model, the Lean approach, and the Ideal Transitions of Care Bridge were used.25-27 We explored the PRISM domains such as recipient’s characteristics, the interaction with the external environment, and the implementation and sustainability infrastructure to inform the design and implementation of the intervention.25 The Lean approach included methods to optimize processes by maximizing efficiency and minimizing waste.26 The Ideal Transitions of Care Bridge was used to identify the domains in transitions of care such as discharge planning, communication of information, and care coordination.27
Setting and Participants
We identified the top 10 non-VA hospitals serving the most urban and rural veterans in 2015 using VA administrative data. Purposive sampling was used to ensure that urban and rural non-VA hospitals and different roles within these hospitals were represented. VA clinicians and staff were selected from the Denver VA Medical Center, a tertiary hospital within the Eastern Colorado Health Care System and one VA Community-Based Outpatient Clinic (CBOC) that primarily serves rural veterans. The Denver VA Medical Center has three clinics staffed by Patient Aligned Care Teams (PACTs), a model built on the concept of Patient-Centered Medical Home.28 Hospital leadership were initially approached for permission to recruit their staff and to be involved as key informants, and all agreed. To ensure representativeness, diversity of roles was recruited, including PACT primary care physicians, nurses, and other staff members such as medical assistants and administrators. Veterans were approached for sampling if they were discharged from a non-VA hospital during June–September 2015 and used the VA for primary care. This was to ensure that they remembered the process they went through postdischarge at the time of the interview.
Data Collection and Analysis
The evaluation team members (RA, EL, and MM) conducted the interviews from November 2015 to July 2016. Clinicians, staff, and veterans were asked semi-structured questions about their experiences and their role in transitioning VA patients across systems (see Appendix for interview guides). Veterans were asked to describe their experience and satisfaction with the current postdischarge transition process. We stopped the interviews when we reached data saturation.29
Interviews were audio-recorded, transcribed verbatim, and validated (transcribed interviews were double-checked against recording) to ensure data quality and accuracy. Coding was guided by a conventional content analysis technique30, 31 using a deductive and inductive coding approach.31 The deductive coding approach was drawn from the Ideal Transitions of Care Bridge and PRISM domains. 32,33 Two evaluation team members (RA and EL) defined the initial code book by independently coding the first three interviews, worked to clarify the meanings of emergent codes, and came to a consensus when disagreements occurred. Next, a priori codes were added by team members to include the PRISM domains. These PRISM domains included the implementation and sustainability infrastructure, the external environment, the characteristics of intervention recipients, and the organizational and patient perspectives of an intervention.
Additional emergent codes were added to the code book and agreed upon by team members (RA, EL, and MM). Consistent with previously used methods, consensus building was achieved by identifying and resolving differences by discussing with team members (RA, EL, MM, CB, and RB).29 Codes were examined and organized into themes by team members.29,34-36 This process was continued until no new themes were identified. Results were reviewed by all evaluation team members to assess thoroughness and comprehensiveness.34,35 In addition, team members triangulated the findings with VA and non-VA participants to ensure validity and reduce researcher bias.29,37
RESULTS
We conducted a total of 70 interviews with 23 VA and 29 non-VA hospital clinicians and staff and 18 veterans (Table 1). Overall, we found that there was no standardized process for transitioning veterans across healthcare delivery systems. Participants reported that transitions were often inefficient when non-VA hospitals could not (1) identify patients as veterans and notify VA primary care of discharge; (2) transfer non-VA hospital medical records to VA primary care; (3) obtain follow-up care appointments with VA primary care; and (4) write VA formulary medications for veterans to fill at VA pharmacies. In addition, participants discussed about facilitators and suggestions to overcome these inefficiencies and improve transitional care (Table2). We mapped the identified barriers as well as the suggestions for improvement to the PRISM and the Ideal Transitions of Care Bridge domains (Table 3).
Unable to Identify Patients as Veterans and Notify VA Primary Care of Discharge
VA and non-VA participants reported difficulty in communicating about veterans’ hospitalizations and discharge follow-up needs across systems. Non-VA clinicians referenced difficulty in identifying patients as veterans to communicate with VA, except in instances where the VA is a payor, while VA providers described feeling largely uninformed of the veterans non-VA hospitalization. For non-VA clinicians, the lack of a systematic method for veteran identification often left them to inadvertently identify veteran status by asking about their primary care clinicians and insurance and even through an offhanded comment made by the veteran. If a veteran was identified, non-VA clinicians described being uncertain about the best way to notify VA primary care of the patient’s impending discharge. Veterans described instances of the non-VA hospital knowing their veteran status upon admission, but accounts varied on whether the non-VA hospital notified the VA primary care of their hospitalization (Table 2, Theme 1).
Unable to Transfer Non-VA Hospital Medical Records to VA Primary Care
VA clinicians discussed about the challenges associated with obtaining the veteran’s medical record from the non-VA hospitals, and when it was received, it was often incomplete information and significantly delayed. They described relying on the veteran’s description of the care received, which was not complete or accurate information needed to make clinical judgment or coordinate follow-up care. Non-VA clinicians mentioned about trying several methods for transferring the medical record to VA primary care, including discharge summary via electronic system and sometimes solely relying on patients to deliver discharge paperwork to their primary care clinicians. In instances where non-VA hospitals sent discharge paperwork to VA, there was no way for non-VA hospitals to verify whether the faxed electronic medical record was received by the VA hospital. Most of the veterans discussed receiving written postdischarge instructions to take to their VA primary care clinicians; however, they were unsure whether the VA primary care received their medical record or any other information from the non-VA hospital (Table 2, Theme 2).
Unable to Obtain Follow-Up Care Appointments with VA Primary Care
All participants described how difficult it was to obtain a follow-up appointment for veterans with VA primary care. This often resulted in delayed follow-up care. VA clinicians also shared that a non-VA hospitalization can be the impetus for a veteran to seek care at the VA for the very first time. Once eligibility is determined, the veteran is assigned a VA primary care clinician. This process may take up to six weeks, and in the meantime, the veteran is scheduled in VA urgent care for immediate postdischarge care. This lag in primary care assignment creates delayed and fragmented care (Table 2, Theme 3).
Non-VA clinicians, administrators, and staff also discussed the difficulties in scheduling follow-up care with VA primary care. Although discharge paperwork instructed patients to see their VA clinicians, there was no process in place for non-VA clinicians to confirm whether the follow-up care was received due to lack of bilateral communication. In addition, veterans discussed the inefficiencies in scheduling follow-up appointments with VA clinicians where attempts to follow-up with primary care clinicians took eight weeks or more. Several veterans described walking into the clinic without an appointment asking to be seen postdischarge or utilizing the VA emergency department for follow-up care after discharge from a non-VA hospital. Veterans admitted utilizing the VA emergency department for nonemergent reasons such as filling their prescriptions because they are unable to see a VA PCP in a timely manner (Table 2, Theme 3).
Unable to Write VA Formulary Medications for Veterans to Fill at VA Pharmacies
All participants described the difficulties in obtaining medications at VA pharmacies when prescribed by the non-VA hospital clinicians. VA clinicians often had to reassess, and rewrite prescriptions written by clinicians, causing delays. Moreover, rural VA clinicians described lack of VA pharmacies in their locations, where veterans had to mail order medications, causing further delays in needed medications. Non-VA clinicians echoed these frustrations. They noted that veterans were confused about their VA pharmacy benefits as well as the need for the non-VA clinicians to follow VA formulary guidelines. Veterans expressed that it was especially challenging to physically go to the VA pharmacy to pick up medications after discharge due to lack of transportation, limited VA pharmacy hours, and long wait times. Several veterans discussed paying for their prescriptions out of pocket even though they had VA pharmacy benefits because it was more convenient to use the non-VA pharmacy. In other instances, veterans discussed going to a VA emergency department and waiting for hours to have their non-VA clinician prescription rewritten by a VA clinician (Table 2, Theme 4).
Facilitators of the Current Transition Process
Several participants provided examples of when transitional care communication between systems occurred seamlessly. VA staff and veterans noted that the VA increased the availability of urgent care appointments, which allowed for timelier postacute care follow-up appointments. Non-VA hospital clinicians also noted the availability of additional appointment slots but stated that they did not learn about these additional appointments directly from the VA. Instead, they learned of these through medical residents caring for patients at both VA and non-VA hospitals. One VA CBOC designated two nurses to care for walk-in veterans for their postdischarge follow-up needs. Some VA participants also noted that the VA Call Center Nurses occasionally called veterans upon discharge to schedule a follow-up appointment and facilitated timely care.
Participants from a VA CBOC discussed being part of a Community Transitions Consortium aimed at identifying high-utilizing patients (veteran and nonveteran) and improving communication across systems. The consortium members discussed each facility’s transition-of-care process, described having access to local non-VA hospital medical records and a backline phone number at the non-VA hospitals to coordinate transitional care. This allowed the VA clinicians to learn about non-VA hospital processes and veteran needs.
Suggestions for Improving the Transitional Care Process
VA and non-VA clinicians suggested hiring a VA liaison, preferably with a clinical background to facilitate care coordination across healthcare systems. They recommended that this person work closely with VA primary care, strengthen the relationship with non-VA hospitals, and help veterans learn more about the transition-of-care processes. Topics discussed for veteran education included how to (1) access their primary care tea
Veterans agreed that improvements to the current process should include an efficient system for obtaining medications and the ability to schedule timely follow-up appointments. Furthermore, veterans wanted education about the VA transition-of-care process following a non-VA hospitalization, including payment and VA notification processes (Table 2, Theme 5).
DISCUSSION
Participants described the current transitional care process as inefficient with specific barriers that have negative consequences on patient care and clinician and staff work processes. They described difficulties in obtaining medications prescribed by non-VA clinicians from VA pharmacies, delays in follow-up appointments at the VA, and lack of bilateral communication between systems and medical record transfer. Participants also provided concrete suggestions to improving the current process, including a care coordinator with clinical background. These findings are important in the context of VA increasing veteran access to care in the community.
Despite an increasing emphasis on veteran access to non-VA care as a result of the VA strategic goals and several new programs,7,12,13 there has not been a close examination of the current transition-of-care process from non-VA hospitals to VA primary care. Several studies have shown that the period following a hospitalization is especially vulnerable and associated with adverse events such as readmission, high cost, and death.12,31,32 Our findings agree with previous research that identified medical record transfer across systems as one of the most challenging issues contributing to deficits in communication between care teams.33 In addition, our study brought into focus the significant challenges faced by veterans in obtaining medications post non-VA hospital discharge. Addressing these key barriers in transitional care will improve the quality, safety, and value of healthcare in the current transition process.38,39
Based on our findings, our participants’ concern in transitional care can be addressed in various ways. First, as veterans are increasingly receiving care in the community, identifying their veteran status early on in the non-VA hospital setting could help in improved, real time communication with the VA. This could be done by updating patient intake forms to ask patients whether they are veterans or not. Second, VA policy-level changes should work to provide veterans access to non-VA pharmacy benefits equivalent to the access patients are receiving for hospital, specialty, and outpatient care. Third, patient and provider satisfaction for dual-use veterans should be examined closely. Although participants expressed frustration with the overall transitions of care from non-VA hospitals to VA primary care setting, influence of this on the Quadruple Aim-improving patient outcomes, experience, and reducing clinician and staff burnout should be examined closely.40 Fourth, evidence-based interventions such as nurse-led transitional care programs that have proven helpful in reducing adverse outcomes in both VA and non-VA settings will be useful to implement.41-45 Such programs could be located in the VA, and a care coordinator role could help facilitate transitional care needs for veterans by working with multiple non-VA hospitals.
The limitations of this study are that the perspectives shared by these participants may not represent all VA and non-VA hospitals as well as veterans’ experiences with transition of care. In addition, the study was conducted in one state and the findings may not be applicable to other healthcare systems. However, our study highlighted the consistent challenges of receiving care across VA and other hospital systems. Two strengths of this study are that it was conducted by multidisciplinary research team members with expertise in qualitative research, clinical care, and implementation science and that we obtained convergent information from VA, non-VA, and veteran participants.
Our current transition-of-care process has several shortcomings. There was a clear agreement on barriers, facilitators, and suggestions for improving the current transitions-of-care process among VA and non-VA hospital participants, as well as from veterans who experienced transitions across different delivery systems. Transitioning veterans to VA primary care following a non-VA hospitalization is a crucial first step for improving care for veterans and reducing adverse outcomes such as avoidable hospital readmissions and death.
These results describe the inefficiencies experienced by patients, clinicians, and staff and their suggestions to alleviate these barriers for optimal continuum of care. To avoid frustration and inefficiencies, the increased emphasis of providing non-VA care for veterans should consider the challenges experienced in transitional care and the opportunities for increased coordination of care.
1. Borowsky SJ, Cowper DC. Dual use of VA and non-VA primary care. J Gen Intern Med. 1999;14(5):274-280. https://doi.org/10.1046/j.1525-1497.1999.00335.x.
2. Charlton ME, Mengeling MA, Schlichting JA, et al. Veteran use of health care systems in rural states. Comparing VA and Non-VA health care use among privately insured veterans under age 65. J Rural Health. 2016;32(4):407-417. https://doi.org/10.1111/jrh.12206.
3. Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138(3):161. https://doi.org/10.7326/0003-4819-138-3-200302040-00007.
4. Nguyen KA, Haggstrom DA, Ofner S, et al. Medication use among veterans across health care systems. Appl Clin Inform. 2017;26(1):235-249. https://doi.org/10.4338/ACI-2016-10-RA-0184.
5. Nayar P, Apenteng B, Yu F, Woodbridge P, Fetrick A. Rural veterans’ perspectives of dual care. J Commun Health. 2013;38(1):70-77. https://doi.org/10.1007/s10900-012-9583-7.
6. West AN, Charlton ME. Insured veterans’ use of VA and Non-VA health care in a rural state. J Rural Health. 2016;32(4):387-396. https://doi.org/10.1111/jrh.12196.
7. Gellad WF. The veterans choice act and dual health system use. J Gen Intern Med. 2016;31(2):153-154. https://doi.org/10.1007/s11606-015-3492-2.
8. Axon RN, Gebregziabher M, Everett CJ, Heidenreich P, Hunt KJ. Dual health care system use is associated with higher rates of hospitalization and hospital readmission among veterans with heart failure. Am Heart J. 2016;174:157-163. https://doi.org/10.1016/j.ahj.2015.09.023.
9. Humensky J, Carretta H, de Groot K, et al. Service utilization of veterans dually eligible for VA and medicare fee-for-service: 1999–2004. Medicare Medicaid Res Rev. 2012;2(3). https://doi.org/10.5600/mmrr.002.03.A06.
10. West AN, Charlton ME, Vaughan-Sarrazin M. Dual use of VA and non-VA hospitals by veterans with multiple hospitalizations. BMC Health Serv Res. 2015;15(1):431. https://doi.org/10.1186/s12913-015-1069-8.
11. Gaglioti A, Cozad A, Wittrock S, et al. Non-VA primary care providers’ perspectives on comanagement for rural veterans. Mil Med. 2014;179(11):1236-1243. https://doi.org/10.7205/MILMED-D-13-00342.
12. Department of Veterans Affairs. Expanded access to non-VA care through the veterans choice program. Final rule. Fed Regist. 2018;83(92):21893-21897.
13. Shuster B. Text-H.R.3236-114th Congress. Surface Transportation and Veterans Health Care Choice Improvement Act of 2015.. https://www.congress.gov/bill/114th-congress/house-bill/3236/text/pl. Accessed April 16, 2017; 2015-2016.
14. Veterans Affairs Mission Act. MISSIONAct.va.gov Available at. https://missionact.va.gov/. Accessed August 9, 2019.
15. Veterans Choice Program (VCP). Community care. https://www.va.gov/COMMUNITYCARE/programs/veterans/VCP/index.asp. Accessed August 9, 2019.
16. A Decade of Transitional Care Research with Vulnerable Elder… : journal of cardiovascular nursing. LWW. http://journals.lww.com/jcnjournal/Fulltext/2000/04000/A_Decade_of_Transitional_Care_Research_with.4.aspx. Accessed April 16, 2017.
17. Coleman EA, Boult C. Improving the quality of transitional care for persons with complex care needs. J Am Geriatr Soc. 2003;51(4):556-557. https://doi.org/10.1046/j.1532-5415.2003.51186.x.
18. Krichbaum K. GAPN postacute care coordination improves hip fracture outcomes. West J Nurs Res. 2007;29(5):523-544. https://doi.org/10.1177/0193945906293817.
19. Kripalani S, Jackson AT, Schnipper JL, Coleman EA. Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314-323. https://doi.org/10.1002/jhm.228.
20. Coleman EA, Mahoney E, Parry C. Assessing the quality of preparation for posthospital care from the patient’s perspective: the care transitions measure. Med Care. 2005;43(3):246-255. https://doi.org/10.1097/00005650-200503000-00007.
21. Naylor MD, Aiken LH, Kurtzman ET, Olds DM, Hirschman KB. The importance of transitional care in achieving health reform. Health Aff (Millwood). 2011;30(4):746-754. https://doi.org/10.1377/hlthaff.2011.0041.
22. Naylor MD, Brooten DA, Campbell RL, et al. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc. 2004;52(5):675-684. https://doi.org/10.1111/j.1532-5415.2004.52202.x.
23. Snow V, Beck D, Budnitz T, et al. Transitions of care consensus policy statement: American College of Physicians, Society of General Internal Medicine, society of hospital medicine, American Geriatrics Society, American College of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364-370. https://doi.org/10.1002/jhm.510.
24. Coleman EA. Falling through the cracks: challenges and opportunities for improving transitional care for persons with continuous complex care needs. J Am Geriatr Soc. 2003;51(4):549-555. https://doi.org/10.1046/j.1532-5415.2003.51185.x.
25. Feldstein AC, Glasgow RE. A practical, robust implementation and sustainability model (PRISM) for integrating research findings into practice. Jt Comm J Qual Patient Saf. 2008;34(4):228-243. https://doi.org/10.1016/S1553-7250(08)34030-6.
26. Schweikhart SA, Dembe AE. The applicability of lean and six sigma techniques to clinical and translational research. J Investig Med. 2009;57(7):748-755. https://doi.org/10.2310/JIM.0b013e3181b91b3a.
27. Burke RE, Kripalani S, Vasilevskis EE, Schnipper JL. Moving beyond readmission penalties: creating an ideal process to improve transitional care. J Hosp Med. 2013;8(2):102-109. https://doi.org/10.1002/jhm.1990.
28. Patient Aligned Care Team (PACT)-Patient Care. Services. https://www.patientcare.va.gov/primarycare/PACT.asp. Accessed November 20, 2017.
29. Morse JM. Critical analysis of strategies for determining rigor in qualitative inquiry. Qual Health Res. 2015;25(9):1212-1222. https://doi.org/10.1177/1049732315588501.
30. Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15(9):1277-1288. https://doi.org/10.1177/1049732305276687.
31. Fereday J, Muir-Cochrane E. Demonstrating rigor using thematic analysis: a hybrid approach of inductive and deductive coding and theme development. Int J Qual Methods. 2006;5(1):80-92. https://doi.org/10.1177/160940690600500107.
32. Ayele RA, Lawrence E, McCreight M, et al. Study protocol: improving the transition of care from a non-network hospital back to the patient’s medical home. BMC Health Serv Res. 2017;17(1):123. https://doi.org/10.1186/s12913-017-2048-z.
33. Burke RE, Kripalani S, Vasilevskis EE, Schnipper JL. Moving beyond readmission penalties: creating an ideal process to improve transitional care. J Hosp Med. 2013;8(2):102-109. https://doi.org/10.1002/jhm.1990.
34. Qualitative research & evaluation methods. https://us.sagepub.com/en-us/nam/qualitative-research-evaluation-methods/book232962. Accessed April 16, 2017. SAGE Publications Inc.
35. Curry LA, Nembhard IM, Bradley EH. Qualitative and mixed methods provide unique contributions to outcomes research. Circulation. 2009;119(10):1442-1452. https://doi.org/10.1161/CIRCULATIONAHA.107.742775.
36. Creswell JW, Hanson WE, Clark Plano VL, Morales A. Qualitative research designs: selection and implementation. Couns Psychol. 2007;35(2):236-264. https://doi.org/10.1177/0011000006287390.
37. Carter N, Bryant-Lukosius D, DiCenso A, Blythe J, Neville AJ. The use of triangulation in qualitative research. Oncol Nurs Forum. 2014;41(5):545-547. https://doi.org/10.1188/14.ONF.545-547.
38. Krumholz HM. Post-hospital syndrome—an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. https://doi.org/10.1056/NEJMp1212324.
39. Improving Care Transitions. Health affairs-health policy briefs. http://www.healthaffairs.org/healthpolicybriefs/brief.php?brief_id=76. Accessed August 13, 2016.
40. Bodenheimer T, Sinsky C. From triple to quadruple aim: care of the patient requires care of the provider. Ann Fam Med. 2014;12(6):573-576. https://doi.org/10.1370/afm.1713.
41. Burke RE, Kelley L, Gunzburger E, et al. Improving transitions of care for veterans transferred to tertiary VA medical centers. Am J Med Qual. 2018;33(2):147-153. https://doi.org/10.1177/1062860617715508.
42. Capp R, Misky GJ, Lindrooth RC, et al. Coordination program reduced acute care use and increased primary care visits among frequent emergency care users. Health Aff (Millwood). 2017;36(10):1705-1711. https://doi.org/10.1377/hlthaff.2017.0612.
43. Kind AJH, Brenny-Fitzpatrick M, Leahy-Gross K, et al. Harnessing protocolized adaptation in dissemination: successful implementation and sustainment of the veterans affairs coordinated-transitional care program in a non-veterans affairs hospital. J Am Geriatr Soc. 2016;64(2):409-416. https://doi.org/10.1111/jgs.13935.
44. Kind AJH, Jensen L, Barczi S, et al. Low-cost transitional care with nurse managers making mostly phone contact With patients cut rehospitalization at a VA Hospital. Health Aff. 2012;31(12):2659-2668. https://doi.org/10.1377/hlthaff.2012.0366.
45. Reese RL, Clement SA, Syeda S, et al. Coordinated-transitional care for veterans with heart failure and chronic lung disease. J Am Geriatr Soc. 2019;67(7):1502-1507. https://doi.org/10.1111/jgs.15978.
1. Borowsky SJ, Cowper DC. Dual use of VA and non-VA primary care. J Gen Intern Med. 1999;14(5):274-280. https://doi.org/10.1046/j.1525-1497.1999.00335.x.
2. Charlton ME, Mengeling MA, Schlichting JA, et al. Veteran use of health care systems in rural states. Comparing VA and Non-VA health care use among privately insured veterans under age 65. J Rural Health. 2016;32(4):407-417. https://doi.org/10.1111/jrh.12206.
3. Forster AJ, Murff HJ, Peterson JF, Gandhi TK, Bates DW. The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138(3):161. https://doi.org/10.7326/0003-4819-138-3-200302040-00007.
4. Nguyen KA, Haggstrom DA, Ofner S, et al. Medication use among veterans across health care systems. Appl Clin Inform. 2017;26(1):235-249. https://doi.org/10.4338/ACI-2016-10-RA-0184.
5. Nayar P, Apenteng B, Yu F, Woodbridge P, Fetrick A. Rural veterans’ perspectives of dual care. J Commun Health. 2013;38(1):70-77. https://doi.org/10.1007/s10900-012-9583-7.
6. West AN, Charlton ME. Insured veterans’ use of VA and Non-VA health care in a rural state. J Rural Health. 2016;32(4):387-396. https://doi.org/10.1111/jrh.12196.
7. Gellad WF. The veterans choice act and dual health system use. J Gen Intern Med. 2016;31(2):153-154. https://doi.org/10.1007/s11606-015-3492-2.
8. Axon RN, Gebregziabher M, Everett CJ, Heidenreich P, Hunt KJ. Dual health care system use is associated with higher rates of hospitalization and hospital readmission among veterans with heart failure. Am Heart J. 2016;174:157-163. https://doi.org/10.1016/j.ahj.2015.09.023.
9. Humensky J, Carretta H, de Groot K, et al. Service utilization of veterans dually eligible for VA and medicare fee-for-service: 1999–2004. Medicare Medicaid Res Rev. 2012;2(3). https://doi.org/10.5600/mmrr.002.03.A06.
10. West AN, Charlton ME, Vaughan-Sarrazin M. Dual use of VA and non-VA hospitals by veterans with multiple hospitalizations. BMC Health Serv Res. 2015;15(1):431. https://doi.org/10.1186/s12913-015-1069-8.
11. Gaglioti A, Cozad A, Wittrock S, et al. Non-VA primary care providers’ perspectives on comanagement for rural veterans. Mil Med. 2014;179(11):1236-1243. https://doi.org/10.7205/MILMED-D-13-00342.
12. Department of Veterans Affairs. Expanded access to non-VA care through the veterans choice program. Final rule. Fed Regist. 2018;83(92):21893-21897.
13. Shuster B. Text-H.R.3236-114th Congress. Surface Transportation and Veterans Health Care Choice Improvement Act of 2015.. https://www.congress.gov/bill/114th-congress/house-bill/3236/text/pl. Accessed April 16, 2017; 2015-2016.
14. Veterans Affairs Mission Act. MISSIONAct.va.gov Available at. https://missionact.va.gov/. Accessed August 9, 2019.
15. Veterans Choice Program (VCP). Community care. https://www.va.gov/COMMUNITYCARE/programs/veterans/VCP/index.asp. Accessed August 9, 2019.
16. A Decade of Transitional Care Research with Vulnerable Elder… : journal of cardiovascular nursing. LWW. http://journals.lww.com/jcnjournal/Fulltext/2000/04000/A_Decade_of_Transitional_Care_Research_with.4.aspx. Accessed April 16, 2017.
17. Coleman EA, Boult C. Improving the quality of transitional care for persons with complex care needs. J Am Geriatr Soc. 2003;51(4):556-557. https://doi.org/10.1046/j.1532-5415.2003.51186.x.
18. Krichbaum K. GAPN postacute care coordination improves hip fracture outcomes. West J Nurs Res. 2007;29(5):523-544. https://doi.org/10.1177/0193945906293817.
19. Kripalani S, Jackson AT, Schnipper JL, Coleman EA. Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314-323. https://doi.org/10.1002/jhm.228.
20. Coleman EA, Mahoney E, Parry C. Assessing the quality of preparation for posthospital care from the patient’s perspective: the care transitions measure. Med Care. 2005;43(3):246-255. https://doi.org/10.1097/00005650-200503000-00007.
21. Naylor MD, Aiken LH, Kurtzman ET, Olds DM, Hirschman KB. The importance of transitional care in achieving health reform. Health Aff (Millwood). 2011;30(4):746-754. https://doi.org/10.1377/hlthaff.2011.0041.
22. Naylor MD, Brooten DA, Campbell RL, et al. Transitional care of older adults hospitalized with heart failure: a randomized, controlled trial. J Am Geriatr Soc. 2004;52(5):675-684. https://doi.org/10.1111/j.1532-5415.2004.52202.x.
23. Snow V, Beck D, Budnitz T, et al. Transitions of care consensus policy statement: American College of Physicians, Society of General Internal Medicine, society of hospital medicine, American Geriatrics Society, American College of Emergency Physicians, and Society for Academic Emergency Medicine. J Hosp Med. 2009;4(6):364-370. https://doi.org/10.1002/jhm.510.
24. Coleman EA. Falling through the cracks: challenges and opportunities for improving transitional care for persons with continuous complex care needs. J Am Geriatr Soc. 2003;51(4):549-555. https://doi.org/10.1046/j.1532-5415.2003.51185.x.
25. Feldstein AC, Glasgow RE. A practical, robust implementation and sustainability model (PRISM) for integrating research findings into practice. Jt Comm J Qual Patient Saf. 2008;34(4):228-243. https://doi.org/10.1016/S1553-7250(08)34030-6.
26. Schweikhart SA, Dembe AE. The applicability of lean and six sigma techniques to clinical and translational research. J Investig Med. 2009;57(7):748-755. https://doi.org/10.2310/JIM.0b013e3181b91b3a.
27. Burke RE, Kripalani S, Vasilevskis EE, Schnipper JL. Moving beyond readmission penalties: creating an ideal process to improve transitional care. J Hosp Med. 2013;8(2):102-109. https://doi.org/10.1002/jhm.1990.
28. Patient Aligned Care Team (PACT)-Patient Care. Services. https://www.patientcare.va.gov/primarycare/PACT.asp. Accessed November 20, 2017.
29. Morse JM. Critical analysis of strategies for determining rigor in qualitative inquiry. Qual Health Res. 2015;25(9):1212-1222. https://doi.org/10.1177/1049732315588501.
30. Hsieh H-F, Shannon SE. Three approaches to qualitative content analysis. Qual Health Res. 2005;15(9):1277-1288. https://doi.org/10.1177/1049732305276687.
31. Fereday J, Muir-Cochrane E. Demonstrating rigor using thematic analysis: a hybrid approach of inductive and deductive coding and theme development. Int J Qual Methods. 2006;5(1):80-92. https://doi.org/10.1177/160940690600500107.
32. Ayele RA, Lawrence E, McCreight M, et al. Study protocol: improving the transition of care from a non-network hospital back to the patient’s medical home. BMC Health Serv Res. 2017;17(1):123. https://doi.org/10.1186/s12913-017-2048-z.
33. Burke RE, Kripalani S, Vasilevskis EE, Schnipper JL. Moving beyond readmission penalties: creating an ideal process to improve transitional care. J Hosp Med. 2013;8(2):102-109. https://doi.org/10.1002/jhm.1990.
34. Qualitative research & evaluation methods. https://us.sagepub.com/en-us/nam/qualitative-research-evaluation-methods/book232962. Accessed April 16, 2017. SAGE Publications Inc.
35. Curry LA, Nembhard IM, Bradley EH. Qualitative and mixed methods provide unique contributions to outcomes research. Circulation. 2009;119(10):1442-1452. https://doi.org/10.1161/CIRCULATIONAHA.107.742775.
36. Creswell JW, Hanson WE, Clark Plano VL, Morales A. Qualitative research designs: selection and implementation. Couns Psychol. 2007;35(2):236-264. https://doi.org/10.1177/0011000006287390.
37. Carter N, Bryant-Lukosius D, DiCenso A, Blythe J, Neville AJ. The use of triangulation in qualitative research. Oncol Nurs Forum. 2014;41(5):545-547. https://doi.org/10.1188/14.ONF.545-547.
38. Krumholz HM. Post-hospital syndrome—an acquired, transient condition of generalized risk. N Engl J Med. 2013;368(2):100-102. https://doi.org/10.1056/NEJMp1212324.
39. Improving Care Transitions. Health affairs-health policy briefs. http://www.healthaffairs.org/healthpolicybriefs/brief.php?brief_id=76. Accessed August 13, 2016.
40. Bodenheimer T, Sinsky C. From triple to quadruple aim: care of the patient requires care of the provider. Ann Fam Med. 2014;12(6):573-576. https://doi.org/10.1370/afm.1713.
41. Burke RE, Kelley L, Gunzburger E, et al. Improving transitions of care for veterans transferred to tertiary VA medical centers. Am J Med Qual. 2018;33(2):147-153. https://doi.org/10.1177/1062860617715508.
42. Capp R, Misky GJ, Lindrooth RC, et al. Coordination program reduced acute care use and increased primary care visits among frequent emergency care users. Health Aff (Millwood). 2017;36(10):1705-1711. https://doi.org/10.1377/hlthaff.2017.0612.
43. Kind AJH, Brenny-Fitzpatrick M, Leahy-Gross K, et al. Harnessing protocolized adaptation in dissemination: successful implementation and sustainment of the veterans affairs coordinated-transitional care program in a non-veterans affairs hospital. J Am Geriatr Soc. 2016;64(2):409-416. https://doi.org/10.1111/jgs.13935.
44. Kind AJH, Jensen L, Barczi S, et al. Low-cost transitional care with nurse managers making mostly phone contact With patients cut rehospitalization at a VA Hospital. Health Aff. 2012;31(12):2659-2668. https://doi.org/10.1377/hlthaff.2012.0366.
45. Reese RL, Clement SA, Syeda S, et al. Coordinated-transitional care for veterans with heart failure and chronic lung disease. J Am Geriatr Soc. 2019;67(7):1502-1507. https://doi.org/10.1111/jgs.15978.
© 2020 Society of Hospital Medicine
Things We Do for No Reason™: Lumbar Punctures in Low-Risk Febrile Infants with Bronchiolitis

CLINICAL SCENARIO
A 22-day-old full-term previously healthy male infant was evaluated in the emergency department (ED). The patient’s mother reported a three-day history of nasal congestion, cough and labored breathing, decreased oral intake, and subjective fever.
In the ED, the patient was found to have a rectal temperature of 101.3 °F (38.3 °C), heart rate of 112 beats per minute, and a respiratory rate of 54 breaths per minute, with subcostal retractions and diffuse expiratory wheezing. His appearance was otherwise unremarkable. His evaluation in the ED included a normal complete blood count (CBC) with differential, a normal urinalysis, and a chest radiograph with diffuse peribronchial thickening. Blood and catheterized urine cultures were also collected. The patient’s provider informs the parents that a lumbar puncture (LP) would be performed to rule out bacterial meningitis. Is it necessary for this patient to receive an LP?
INTRODUCTION
Fever in an infant <90 days old is a common clinical presentation.1 Because a newborn’s immune system is still developing, there is a heightened concern for bacterial infections in this age group. These include bloodstream infections, meningitis, pneumonia, urinary tract infections (UTIs), skin/soft tissue infections, and osteoarticular infections. Bacterial infections collectively account for approximately 10% of illness in young febrile infants <90 days.2 Of these, UTIs are the most common. The most recent literature has narrowed the focus on infants <60 days old as the risk of serious infection is inversely correlated with age. Meningitis accounts for 1% of infections or less in children <60 days of age who present with a fever.3
Frequently, the evaluation of fever in young infants leads to cerebrospinal fluid (CSF) collection and hospitalization.4 Among febrile infants, current practice patterns regarding LPs vary across institutions.5 Some clinical practice guidelines recommend universal CSF testing for all febrile infants ≤56 days old.6
Bronchiolitis is also a common presentation. Up to 90% of children are infected with respiratory syncytial virus, the most common viral cause of bronchiolitis, within the first two years of life.7 Fever may be a presenting symptom in infants with bronchiolitis and one study found approximately 11% of febrile infants less than 90 days old met clinical criteria for bronchiolitis.8
WHY YOU MIGHT THINK LUMBAR PUNCTURE IN FEBRILE INFANTS WITH BRONCHIOLITIS IS HELPFUL
While clinical guidelines for bronchiolitis are well established,7 the evaluation and management of fever in an infant <90 days old remains a challenge because of concern for missing a bloodstream infection or meningitis. Meningitis can devastate an infant neurologically.9 Signs and symptoms of bacterial meningitis in infants are not specific, including the physical exam.10 Blood cultures are only concomitantly positive in 62% of cases of culture-confirmed bacterial meningitis.11
Several risk stratification algorithms exist to evaluate the likelihood of bacterial infections in febrile infants (Table). Two of the most common criteria—the Boston and Philadelphia—were validated using CSF cell count data. Other algorithms do not require an LP.12-15 All of the fever criteria algorithms have several limitations including lack of robust validation studies, under-powered methodologies (particularly for meningitis), and different inclusion criteria.2 Even with these risk stratification algorithms, some providers may continue to feel more comfortable obtaining CSF due to fear of missing meningitis in well-appearing, low-risk infants.
WHY LUMBAR PUNCTURE IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS IS NOT NECESSARY
Bacterial meningitis, even in young infants, is rare. A recent meta-analysis estimated the general prevalence of meningitis in febrile neonates (regardless of risk stratification or bronchiolitis symptoms) in their first and second months of life were 1.2% (95% CI, 0.8%-1.9%) and 0.4% (95% CI, 0.2%-1.0%), respectively.3
Febrile infant risk stratification algorithms have high negative predictive values (NPVs) in ruling out meningitis. The Rochester criteria, which does not utilize CSF, has an NPV of greater than 98%.12 A recent Pediatric Emergency Care Applied Research Network Clinical Prediction Rule has an NPV of 99.9% among febrile infants <60 days, using only absolute neutrophil count, urinalysis, and procalcitonin.15
Among the patients that are already a low risk, concomitant viral infections further decrease the pretest probability. Febrile infants with lab-confirmed respiratory viral infections are at lower risk for serious bacterial infections.16,17 Multiple retrospective and prospective observational studies have demonstrated that low-risk patients with bronchiolitis symptoms are extremely unlikely to have bacterial meningitis.8,18-22 A systematic review of 1749 febrile patients under 90 days of age with clinical bronchiolitis demonstrated no cases of meningitis.23 Many of these studies included infants aged <28 days. Though the total number of neonates (<28 days) in all studies is somewhat unclear, it suggests that the cut-off to avoid an LP may be even lower.
Recent literature has advocated outpatient observation without an LP for low-risk infants as a cost-effective management tool,24 and this is particularly true in patients with concomitant viral bronchiolitis.
Based on the latest data confirming the low prevalence of meningitis among all infants,3 the ability to identify low-risk infants based on risk stratification algorithms (Table), and the decreased prevalence of meningitis in patients with clinical bronchiolitis,23 low-risk infants with bronchiolitis seem to have minimal, if any, risk of meningitis. Therefore, low-risk infants with bronchiolitis do not warrant an LP.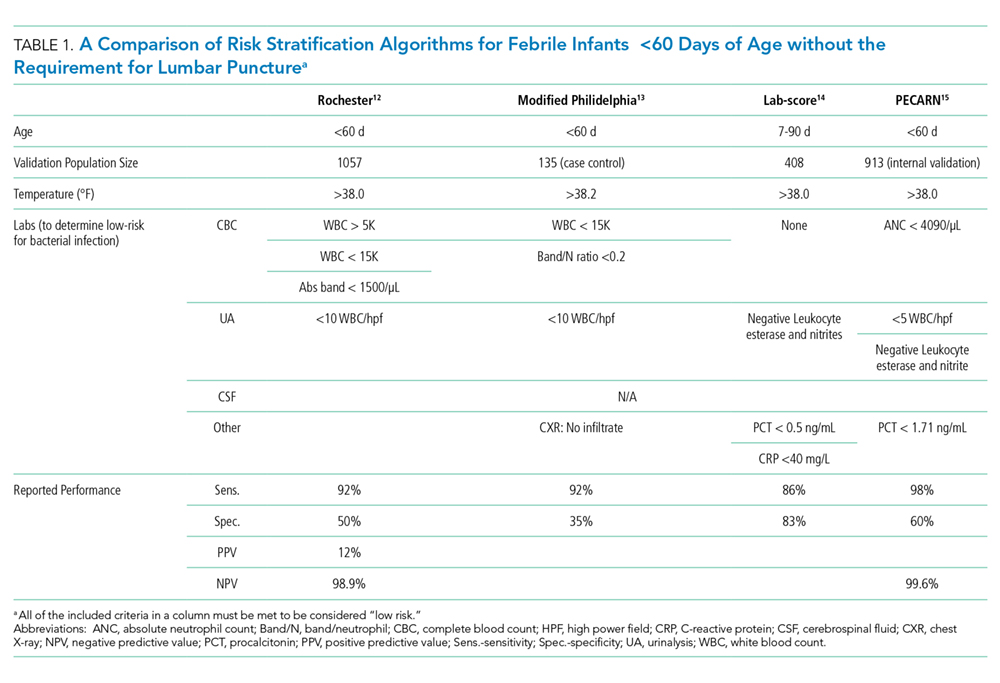
Importantly, LPs are not risk neutral. Their benefit versus harm should be weighed every time they are considered. Approximately 19% of LP attempts in infants under 90 days old are either traumatic or unsuccessful.25 Infants aged 28 to 60 days with traumatic or unsuccessful LPs are more frequently hospitalized.25 Increased hospitalizations are associated with higher costs.4 The majority of positive CSF cultures are deemed to be “contaminants” (87% in one study26), but the positive result still leads to unnecessary further evaluation, hospitalization, repeated invasive procedures, and family distress.27 These data further support refraining from pursuing an LP in low-risk infants with bronchiolitis.
WHY LUMBAR PUNCTURE MIGHT BE HELPFUL IN CERTAIN CIRCUMSTANCES
If the patient is not low risk based on criteria or does not have clinical bronchiolitis, consider performing an LP. A recent study demonstrated a 0.4% incidence of bacterial meningitis in febrile infants with viral co-infection,29 though it is not determined if the patients presented with symptoms of bronchiolitis or were risk-stratified using the algorithms discussed.
In the studies looking at viral infections in febrile infants, each has important exclusion criteria including prematurity, comorbidities, and recent antibiotic administration.23 For these patients, an LP may be warranted (though the evidence is lacking). In addition, in very young infants (less than seven-14 days old), viral infections may be less common than in older infants, resulting in a desire to rule out bacterial infections more thoroughly in this population.
WHAT YOU SHOULD DO INSTEAD: AVOID AN LP IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS
For low-risk febrile infants with signs of bronchiolitis, evaluation for bacterial meningitis is not necessary. The low prevalence of meningitis in this age range along with the even lower likelihood of meningitis when bronchiolitis is identified suggests that the procedure is unnecessary. Moreover, the risks associated with LP—including trauma, hospitalization, costs, and family stress—likely outweigh the benefits of CSF analysis.
RECOMMENDATIONS
- In febrile infants, determine the risk of serious bacterial infections using published algorithms (Table) before considering lumbar puncture.
- In low-risk febrile infants with typical bronchiolitis, evaluation for bacterial meningitis with an LP is not necessary.
CONCLUSION
Infants under 90 days of age often present to care with fever. While there is a concern for missing bacterial meningitis, the prevalence of such an infection in infants is very low. Moreover, in low-risk patients that present with typical bronchiolitis symptoms, the prevalence is effectively zero. LP practices vary by institution and can be associated with risks. In low-risk infants with typical bronchiolitis symptoms, an LP is one of the Things We Do for No Reason™.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing [email protected].
1. Cioffredi L-A, Jhaveri R. Evaluation and management of febrile children. JAMA Pediatr. 2016;170(8):794. https://doi.org/10.1001/jamapediatrics.2016.0596.
2. Huppler AR, Eickhoff JC, Wald ER. Performance of low-risk criteria in the evaluation of young infants with fever: review of the literature. Pediatrics. 2010;125(2):228-233. https://doi.org/10.1542/peds.2009-1070.
3. Biondi EA, Lee B, Ralston SL, et al. Prevalence of bacteremia and bacterial meningitis in febrile neonates and infants in the second month of life a systematic review and meta-analysis + supplemental content. JAMA Netw Open. 2019;2(3):190874. https://doi.org/10.1001/jamanetworkopen.2019.0874.
4. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
5. Aronson PL, Thurm C, Alpern ER, et al. Variation in care of the febrile young infant <90 days in us pediatric emergency departments. Pediatrics. 2014;134(4):667-677. https://doi.org/10.1542/peds.2014-1382.
6. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
7. Mendonca EA, Meissner HC, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. https://doi.org/10.1542/peds.2014-2742.
8. Melendez E, Harper MB. Utility of sepsis evaluation in infants 90 days of age or younger with fever and clinical bronchiolitis. Pediatr Infect Dis J. 2003;22(12):1053-1056. https://doi.org/10.1097/01.inf.0000101296.68993.4d.
9. Pruitt CM, Neuman MI, Shah SS, et al. Factors associated with adverse outcomes among febrile young infants with invasive bacterial infections. J. Pediatr. 2018;204:177-182. https://doi.org/10.1016/j.jpeds.2018.08.066.
10. Casper TC, Mahajan PV., Tzimenatos L, et al. The Yale Observation Scale Score and the risk of serious bacterial infections in febrile infants. Pediatrics. 2017;140(1):e20170695. https://doi.org/10.1542/peds.2017-0695.
11. Garges HP. Neonatal meningitis: what is the correlation among cerebrospinal fluid cultures, blood cultures, and cerebrospinal fluid parameters? Pediatrics. 2006;117(4):1094-1100. https://doi.org/10.1542/peds.2005-1132.
12. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection-an appraisal of the Rochester criteria and implications for management. Pediatrics. 1994;94(3):390-396. http://www.ncbi.nlm.nih.gov/pubmed/8065869. Accessed March 23, 2019.
13. Aronson P, Wang M, Shapiro E, et al. Risk stratification of febrile infants ≤60 days old without routine lumbar puncture. Pediatrics. 2018;142(6):e20181879. https://doi.org/10.1542/peds.2018-1879.
14. Galetto-Lacour A, Zamora SA, Andreola B, et al. Validation of a laboratory risk index score for the identification of severe bacterial infection in children with fever without source. Arch Dis Child. 2010;95(12):968-973. https://doi.org/10.1136/adc.2009.176800.
15. Kuppermann N, Dayan PS, Levine DA, et al. A clinical prediction rule to identify febrile infants 60 days and younger at low risk for serious bacterial infections. JAMA Pediatr. 2019;173(4):342. https://doi.org/10.1001/jamapediatrics.2018.5501.
16. Byington CL, Enriquez FR, Hoff C, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics. 2004;113(6):1662-1666. https://doi.org/10.1542/peds.113.6.1662.
17. Cioffredi LA, Jhaveri R. Evaluation and management of febrile children: a review. JAMA Pediatr. 2016;170(8):794-800. https://doi.org/10.1001/jamapediatrics.2016.0596.
18. Dayan PS, Roskind CG, Levine DA, Kuppermann N. Controversies in the management of children with bronchiolitis. Clin Pediatr Emerg Med. 2004;5(1):41-53. https://doi.org/10.1016/j.cpem.2003.11.001.
19. Oray-Schrom P, Phoenix C, St. Martin D, Amoateng-Adjepong Y. Sepsis workup in febrile infants 0-90 days of age with respiratory syncytial virus infection. Pediatr Emerg Care. 2003;19(5):314-319. https://doi.org/10.1097/01.pec.0000092576.40174.28.
20. Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156(4):322-324. https://doi.org/10.1001/archpedi.156.4.322.
21. Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23(3):267-269. https://doi.org/10.1097/01.inf.0000116759.21252.29.
22. Yarden-Bilavsky H, Ashkenazi-Hoffnung L, Livni G, Amir J, Bilavsky E. Month-by-month age analysis of the risk for serious bacterial infections in febrile infants with bronchiolitis. Clin Pediatr (Phila). 2011;50(11):1052-1056. https://doi.org/10.1177/0009922811412949.
23. Ralston S, Hill V, Waters A. Occult serious bacterial infection in infants younger than 60 to 90 days with bronchiolitis: a systematic review. Arch Pediatr Adolesc Med. 2011;165(10):951-956. https://doi.org/10.1001/archpediatrics.2011.155.
24. Lee TJ, Aronson PL. To spinal tap or not to spinal tap, that is the question. Hosp Pediatr. 2018;8(4):236-238. https://doi.org/10.1542/hpeds.2017-0207.
25. Pingree EW, Kimia AA, Nigrovic LE. The effect of traumatic lumbar puncture on hospitalization rate for febrile infants 28 to 60 days of age. Acad Emerg Med. 2015;22(2):240-243. https://doi.org/10.1111/acem.12582.
26. Leazer R, Erickson N, Paulson J, et al. epidemiology of cerebrospinal fluid cultures and time to detection in term infants. Pediatrics. 2017;139(5):e20163268. https://doi.org/10.1542/peds.2016-3268.
27. Paxton RD, Byington CL. An examination of the unintended consequences of the rule-out sepsis evaluation: a parental perspective. Clin Pediatr (Phila). 2001;40(2):71-77. https://doi.org/10.1177/000992280104000202.
28. Mahajan P, Br owne LR, Levine DA, et al. Risk of bacterial coinfections in febrile infants 60 days old and younger with documented viral infections. J Pediatr. 2018;203:86-91.e2. https://doi.org/10.1016/j.jpeds.2018.07.073.
CLINICAL SCENARIO
A 22-day-old full-term previously healthy male infant was evaluated in the emergency department (ED). The patient’s mother reported a three-day history of nasal congestion, cough and labored breathing, decreased oral intake, and subjective fever.
In the ED, the patient was found to have a rectal temperature of 101.3 °F (38.3 °C), heart rate of 112 beats per minute, and a respiratory rate of 54 breaths per minute, with subcostal retractions and diffuse expiratory wheezing. His appearance was otherwise unremarkable. His evaluation in the ED included a normal complete blood count (CBC) with differential, a normal urinalysis, and a chest radiograph with diffuse peribronchial thickening. Blood and catheterized urine cultures were also collected. The patient’s provider informs the parents that a lumbar puncture (LP) would be performed to rule out bacterial meningitis. Is it necessary for this patient to receive an LP?
INTRODUCTION
Fever in an infant <90 days old is a common clinical presentation.1 Because a newborn’s immune system is still developing, there is a heightened concern for bacterial infections in this age group. These include bloodstream infections, meningitis, pneumonia, urinary tract infections (UTIs), skin/soft tissue infections, and osteoarticular infections. Bacterial infections collectively account for approximately 10% of illness in young febrile infants <90 days.2 Of these, UTIs are the most common. The most recent literature has narrowed the focus on infants <60 days old as the risk of serious infection is inversely correlated with age. Meningitis accounts for 1% of infections or less in children <60 days of age who present with a fever.3
Frequently, the evaluation of fever in young infants leads to cerebrospinal fluid (CSF) collection and hospitalization.4 Among febrile infants, current practice patterns regarding LPs vary across institutions.5 Some clinical practice guidelines recommend universal CSF testing for all febrile infants ≤56 days old.6
Bronchiolitis is also a common presentation. Up to 90% of children are infected with respiratory syncytial virus, the most common viral cause of bronchiolitis, within the first two years of life.7 Fever may be a presenting symptom in infants with bronchiolitis and one study found approximately 11% of febrile infants less than 90 days old met clinical criteria for bronchiolitis.8
WHY YOU MIGHT THINK LUMBAR PUNCTURE IN FEBRILE INFANTS WITH BRONCHIOLITIS IS HELPFUL
While clinical guidelines for bronchiolitis are well established,7 the evaluation and management of fever in an infant <90 days old remains a challenge because of concern for missing a bloodstream infection or meningitis. Meningitis can devastate an infant neurologically.9 Signs and symptoms of bacterial meningitis in infants are not specific, including the physical exam.10 Blood cultures are only concomitantly positive in 62% of cases of culture-confirmed bacterial meningitis.11
Several risk stratification algorithms exist to evaluate the likelihood of bacterial infections in febrile infants (Table). Two of the most common criteria—the Boston and Philadelphia—were validated using CSF cell count data. Other algorithms do not require an LP.12-15 All of the fever criteria algorithms have several limitations including lack of robust validation studies, under-powered methodologies (particularly for meningitis), and different inclusion criteria.2 Even with these risk stratification algorithms, some providers may continue to feel more comfortable obtaining CSF due to fear of missing meningitis in well-appearing, low-risk infants.
WHY LUMBAR PUNCTURE IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS IS NOT NECESSARY
Bacterial meningitis, even in young infants, is rare. A recent meta-analysis estimated the general prevalence of meningitis in febrile neonates (regardless of risk stratification or bronchiolitis symptoms) in their first and second months of life were 1.2% (95% CI, 0.8%-1.9%) and 0.4% (95% CI, 0.2%-1.0%), respectively.3
Febrile infant risk stratification algorithms have high negative predictive values (NPVs) in ruling out meningitis. The Rochester criteria, which does not utilize CSF, has an NPV of greater than 98%.12 A recent Pediatric Emergency Care Applied Research Network Clinical Prediction Rule has an NPV of 99.9% among febrile infants <60 days, using only absolute neutrophil count, urinalysis, and procalcitonin.15
Among the patients that are already a low risk, concomitant viral infections further decrease the pretest probability. Febrile infants with lab-confirmed respiratory viral infections are at lower risk for serious bacterial infections.16,17 Multiple retrospective and prospective observational studies have demonstrated that low-risk patients with bronchiolitis symptoms are extremely unlikely to have bacterial meningitis.8,18-22 A systematic review of 1749 febrile patients under 90 days of age with clinical bronchiolitis demonstrated no cases of meningitis.23 Many of these studies included infants aged <28 days. Though the total number of neonates (<28 days) in all studies is somewhat unclear, it suggests that the cut-off to avoid an LP may be even lower.
Recent literature has advocated outpatient observation without an LP for low-risk infants as a cost-effective management tool,24 and this is particularly true in patients with concomitant viral bronchiolitis.
Based on the latest data confirming the low prevalence of meningitis among all infants,3 the ability to identify low-risk infants based on risk stratification algorithms (Table), and the decreased prevalence of meningitis in patients with clinical bronchiolitis,23 low-risk infants with bronchiolitis seem to have minimal, if any, risk of meningitis. Therefore, low-risk infants with bronchiolitis do not warrant an LP.
Importantly, LPs are not risk neutral. Their benefit versus harm should be weighed every time they are considered. Approximately 19% of LP attempts in infants under 90 days old are either traumatic or unsuccessful.25 Infants aged 28 to 60 days with traumatic or unsuccessful LPs are more frequently hospitalized.25 Increased hospitalizations are associated with higher costs.4 The majority of positive CSF cultures are deemed to be “contaminants” (87% in one study26), but the positive result still leads to unnecessary further evaluation, hospitalization, repeated invasive procedures, and family distress.27 These data further support refraining from pursuing an LP in low-risk infants with bronchiolitis.
WHY LUMBAR PUNCTURE MIGHT BE HELPFUL IN CERTAIN CIRCUMSTANCES
If the patient is not low risk based on criteria or does not have clinical bronchiolitis, consider performing an LP. A recent study demonstrated a 0.4% incidence of bacterial meningitis in febrile infants with viral co-infection,29 though it is not determined if the patients presented with symptoms of bronchiolitis or were risk-stratified using the algorithms discussed.
In the studies looking at viral infections in febrile infants, each has important exclusion criteria including prematurity, comorbidities, and recent antibiotic administration.23 For these patients, an LP may be warranted (though the evidence is lacking). In addition, in very young infants (less than seven-14 days old), viral infections may be less common than in older infants, resulting in a desire to rule out bacterial infections more thoroughly in this population.
WHAT YOU SHOULD DO INSTEAD: AVOID AN LP IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS
For low-risk febrile infants with signs of bronchiolitis, evaluation for bacterial meningitis is not necessary. The low prevalence of meningitis in this age range along with the even lower likelihood of meningitis when bronchiolitis is identified suggests that the procedure is unnecessary. Moreover, the risks associated with LP—including trauma, hospitalization, costs, and family stress—likely outweigh the benefits of CSF analysis.
RECOMMENDATIONS
- In febrile infants, determine the risk of serious bacterial infections using published algorithms (Table) before considering lumbar puncture.
- In low-risk febrile infants with typical bronchiolitis, evaluation for bacterial meningitis with an LP is not necessary.
CONCLUSION
Infants under 90 days of age often present to care with fever. While there is a concern for missing bacterial meningitis, the prevalence of such an infection in infants is very low. Moreover, in low-risk patients that present with typical bronchiolitis symptoms, the prevalence is effectively zero. LP practices vary by institution and can be associated with risks. In low-risk infants with typical bronchiolitis symptoms, an LP is one of the Things We Do for No Reason™.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing [email protected].
CLINICAL SCENARIO
A 22-day-old full-term previously healthy male infant was evaluated in the emergency department (ED). The patient’s mother reported a three-day history of nasal congestion, cough and labored breathing, decreased oral intake, and subjective fever.
In the ED, the patient was found to have a rectal temperature of 101.3 °F (38.3 °C), heart rate of 112 beats per minute, and a respiratory rate of 54 breaths per minute, with subcostal retractions and diffuse expiratory wheezing. His appearance was otherwise unremarkable. His evaluation in the ED included a normal complete blood count (CBC) with differential, a normal urinalysis, and a chest radiograph with diffuse peribronchial thickening. Blood and catheterized urine cultures were also collected. The patient’s provider informs the parents that a lumbar puncture (LP) would be performed to rule out bacterial meningitis. Is it necessary for this patient to receive an LP?
INTRODUCTION
Fever in an infant <90 days old is a common clinical presentation.1 Because a newborn’s immune system is still developing, there is a heightened concern for bacterial infections in this age group. These include bloodstream infections, meningitis, pneumonia, urinary tract infections (UTIs), skin/soft tissue infections, and osteoarticular infections. Bacterial infections collectively account for approximately 10% of illness in young febrile infants <90 days.2 Of these, UTIs are the most common. The most recent literature has narrowed the focus on infants <60 days old as the risk of serious infection is inversely correlated with age. Meningitis accounts for 1% of infections or less in children <60 days of age who present with a fever.3
Frequently, the evaluation of fever in young infants leads to cerebrospinal fluid (CSF) collection and hospitalization.4 Among febrile infants, current practice patterns regarding LPs vary across institutions.5 Some clinical practice guidelines recommend universal CSF testing for all febrile infants ≤56 days old.6
Bronchiolitis is also a common presentation. Up to 90% of children are infected with respiratory syncytial virus, the most common viral cause of bronchiolitis, within the first two years of life.7 Fever may be a presenting symptom in infants with bronchiolitis and one study found approximately 11% of febrile infants less than 90 days old met clinical criteria for bronchiolitis.8
WHY YOU MIGHT THINK LUMBAR PUNCTURE IN FEBRILE INFANTS WITH BRONCHIOLITIS IS HELPFUL
While clinical guidelines for bronchiolitis are well established,7 the evaluation and management of fever in an infant <90 days old remains a challenge because of concern for missing a bloodstream infection or meningitis. Meningitis can devastate an infant neurologically.9 Signs and symptoms of bacterial meningitis in infants are not specific, including the physical exam.10 Blood cultures are only concomitantly positive in 62% of cases of culture-confirmed bacterial meningitis.11
Several risk stratification algorithms exist to evaluate the likelihood of bacterial infections in febrile infants (Table). Two of the most common criteria—the Boston and Philadelphia—were validated using CSF cell count data. Other algorithms do not require an LP.12-15 All of the fever criteria algorithms have several limitations including lack of robust validation studies, under-powered methodologies (particularly for meningitis), and different inclusion criteria.2 Even with these risk stratification algorithms, some providers may continue to feel more comfortable obtaining CSF due to fear of missing meningitis in well-appearing, low-risk infants.
WHY LUMBAR PUNCTURE IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS IS NOT NECESSARY
Bacterial meningitis, even in young infants, is rare. A recent meta-analysis estimated the general prevalence of meningitis in febrile neonates (regardless of risk stratification or bronchiolitis symptoms) in their first and second months of life were 1.2% (95% CI, 0.8%-1.9%) and 0.4% (95% CI, 0.2%-1.0%), respectively.3
Febrile infant risk stratification algorithms have high negative predictive values (NPVs) in ruling out meningitis. The Rochester criteria, which does not utilize CSF, has an NPV of greater than 98%.12 A recent Pediatric Emergency Care Applied Research Network Clinical Prediction Rule has an NPV of 99.9% among febrile infants <60 days, using only absolute neutrophil count, urinalysis, and procalcitonin.15
Among the patients that are already a low risk, concomitant viral infections further decrease the pretest probability. Febrile infants with lab-confirmed respiratory viral infections are at lower risk for serious bacterial infections.16,17 Multiple retrospective and prospective observational studies have demonstrated that low-risk patients with bronchiolitis symptoms are extremely unlikely to have bacterial meningitis.8,18-22 A systematic review of 1749 febrile patients under 90 days of age with clinical bronchiolitis demonstrated no cases of meningitis.23 Many of these studies included infants aged <28 days. Though the total number of neonates (<28 days) in all studies is somewhat unclear, it suggests that the cut-off to avoid an LP may be even lower.
Recent literature has advocated outpatient observation without an LP for low-risk infants as a cost-effective management tool,24 and this is particularly true in patients with concomitant viral bronchiolitis.
Based on the latest data confirming the low prevalence of meningitis among all infants,3 the ability to identify low-risk infants based on risk stratification algorithms (Table), and the decreased prevalence of meningitis in patients with clinical bronchiolitis,23 low-risk infants with bronchiolitis seem to have minimal, if any, risk of meningitis. Therefore, low-risk infants with bronchiolitis do not warrant an LP.
Importantly, LPs are not risk neutral. Their benefit versus harm should be weighed every time they are considered. Approximately 19% of LP attempts in infants under 90 days old are either traumatic or unsuccessful.25 Infants aged 28 to 60 days with traumatic or unsuccessful LPs are more frequently hospitalized.25 Increased hospitalizations are associated with higher costs.4 The majority of positive CSF cultures are deemed to be “contaminants” (87% in one study26), but the positive result still leads to unnecessary further evaluation, hospitalization, repeated invasive procedures, and family distress.27 These data further support refraining from pursuing an LP in low-risk infants with bronchiolitis.
WHY LUMBAR PUNCTURE MIGHT BE HELPFUL IN CERTAIN CIRCUMSTANCES
If the patient is not low risk based on criteria or does not have clinical bronchiolitis, consider performing an LP. A recent study demonstrated a 0.4% incidence of bacterial meningitis in febrile infants with viral co-infection,29 though it is not determined if the patients presented with symptoms of bronchiolitis or were risk-stratified using the algorithms discussed.
In the studies looking at viral infections in febrile infants, each has important exclusion criteria including prematurity, comorbidities, and recent antibiotic administration.23 For these patients, an LP may be warranted (though the evidence is lacking). In addition, in very young infants (less than seven-14 days old), viral infections may be less common than in older infants, resulting in a desire to rule out bacterial infections more thoroughly in this population.
WHAT YOU SHOULD DO INSTEAD: AVOID AN LP IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS
For low-risk febrile infants with signs of bronchiolitis, evaluation for bacterial meningitis is not necessary. The low prevalence of meningitis in this age range along with the even lower likelihood of meningitis when bronchiolitis is identified suggests that the procedure is unnecessary. Moreover, the risks associated with LP—including trauma, hospitalization, costs, and family stress—likely outweigh the benefits of CSF analysis.
RECOMMENDATIONS
- In febrile infants, determine the risk of serious bacterial infections using published algorithms (Table) before considering lumbar puncture.
- In low-risk febrile infants with typical bronchiolitis, evaluation for bacterial meningitis with an LP is not necessary.
CONCLUSION
Infants under 90 days of age often present to care with fever. While there is a concern for missing bacterial meningitis, the prevalence of such an infection in infants is very low. Moreover, in low-risk patients that present with typical bronchiolitis symptoms, the prevalence is effectively zero. LP practices vary by institution and can be associated with risks. In low-risk infants with typical bronchiolitis symptoms, an LP is one of the Things We Do for No Reason™.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing [email protected].
1. Cioffredi L-A, Jhaveri R. Evaluation and management of febrile children. JAMA Pediatr. 2016;170(8):794. https://doi.org/10.1001/jamapediatrics.2016.0596.
2. Huppler AR, Eickhoff JC, Wald ER. Performance of low-risk criteria in the evaluation of young infants with fever: review of the literature. Pediatrics. 2010;125(2):228-233. https://doi.org/10.1542/peds.2009-1070.
3. Biondi EA, Lee B, Ralston SL, et al. Prevalence of bacteremia and bacterial meningitis in febrile neonates and infants in the second month of life a systematic review and meta-analysis + supplemental content. JAMA Netw Open. 2019;2(3):190874. https://doi.org/10.1001/jamanetworkopen.2019.0874.
4. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
5. Aronson PL, Thurm C, Alpern ER, et al. Variation in care of the febrile young infant <90 days in us pediatric emergency departments. Pediatrics. 2014;134(4):667-677. https://doi.org/10.1542/peds.2014-1382.
6. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
7. Mendonca EA, Meissner HC, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. https://doi.org/10.1542/peds.2014-2742.
8. Melendez E, Harper MB. Utility of sepsis evaluation in infants 90 days of age or younger with fever and clinical bronchiolitis. Pediatr Infect Dis J. 2003;22(12):1053-1056. https://doi.org/10.1097/01.inf.0000101296.68993.4d.
9. Pruitt CM, Neuman MI, Shah SS, et al. Factors associated with adverse outcomes among febrile young infants with invasive bacterial infections. J. Pediatr. 2018;204:177-182. https://doi.org/10.1016/j.jpeds.2018.08.066.
10. Casper TC, Mahajan PV., Tzimenatos L, et al. The Yale Observation Scale Score and the risk of serious bacterial infections in febrile infants. Pediatrics. 2017;140(1):e20170695. https://doi.org/10.1542/peds.2017-0695.
11. Garges HP. Neonatal meningitis: what is the correlation among cerebrospinal fluid cultures, blood cultures, and cerebrospinal fluid parameters? Pediatrics. 2006;117(4):1094-1100. https://doi.org/10.1542/peds.2005-1132.
12. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection-an appraisal of the Rochester criteria and implications for management. Pediatrics. 1994;94(3):390-396. http://www.ncbi.nlm.nih.gov/pubmed/8065869. Accessed March 23, 2019.
13. Aronson P, Wang M, Shapiro E, et al. Risk stratification of febrile infants ≤60 days old without routine lumbar puncture. Pediatrics. 2018;142(6):e20181879. https://doi.org/10.1542/peds.2018-1879.
14. Galetto-Lacour A, Zamora SA, Andreola B, et al. Validation of a laboratory risk index score for the identification of severe bacterial infection in children with fever without source. Arch Dis Child. 2010;95(12):968-973. https://doi.org/10.1136/adc.2009.176800.
15. Kuppermann N, Dayan PS, Levine DA, et al. A clinical prediction rule to identify febrile infants 60 days and younger at low risk for serious bacterial infections. JAMA Pediatr. 2019;173(4):342. https://doi.org/10.1001/jamapediatrics.2018.5501.
16. Byington CL, Enriquez FR, Hoff C, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics. 2004;113(6):1662-1666. https://doi.org/10.1542/peds.113.6.1662.
17. Cioffredi LA, Jhaveri R. Evaluation and management of febrile children: a review. JAMA Pediatr. 2016;170(8):794-800. https://doi.org/10.1001/jamapediatrics.2016.0596.
18. Dayan PS, Roskind CG, Levine DA, Kuppermann N. Controversies in the management of children with bronchiolitis. Clin Pediatr Emerg Med. 2004;5(1):41-53. https://doi.org/10.1016/j.cpem.2003.11.001.
19. Oray-Schrom P, Phoenix C, St. Martin D, Amoateng-Adjepong Y. Sepsis workup in febrile infants 0-90 days of age with respiratory syncytial virus infection. Pediatr Emerg Care. 2003;19(5):314-319. https://doi.org/10.1097/01.pec.0000092576.40174.28.
20. Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156(4):322-324. https://doi.org/10.1001/archpedi.156.4.322.
21. Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23(3):267-269. https://doi.org/10.1097/01.inf.0000116759.21252.29.
22. Yarden-Bilavsky H, Ashkenazi-Hoffnung L, Livni G, Amir J, Bilavsky E. Month-by-month age analysis of the risk for serious bacterial infections in febrile infants with bronchiolitis. Clin Pediatr (Phila). 2011;50(11):1052-1056. https://doi.org/10.1177/0009922811412949.
23. Ralston S, Hill V, Waters A. Occult serious bacterial infection in infants younger than 60 to 90 days with bronchiolitis: a systematic review. Arch Pediatr Adolesc Med. 2011;165(10):951-956. https://doi.org/10.1001/archpediatrics.2011.155.
24. Lee TJ, Aronson PL. To spinal tap or not to spinal tap, that is the question. Hosp Pediatr. 2018;8(4):236-238. https://doi.org/10.1542/hpeds.2017-0207.
25. Pingree EW, Kimia AA, Nigrovic LE. The effect of traumatic lumbar puncture on hospitalization rate for febrile infants 28 to 60 days of age. Acad Emerg Med. 2015;22(2):240-243. https://doi.org/10.1111/acem.12582.
26. Leazer R, Erickson N, Paulson J, et al. epidemiology of cerebrospinal fluid cultures and time to detection in term infants. Pediatrics. 2017;139(5):e20163268. https://doi.org/10.1542/peds.2016-3268.
27. Paxton RD, Byington CL. An examination of the unintended consequences of the rule-out sepsis evaluation: a parental perspective. Clin Pediatr (Phila). 2001;40(2):71-77. https://doi.org/10.1177/000992280104000202.
28. Mahajan P, Br owne LR, Levine DA, et al. Risk of bacterial coinfections in febrile infants 60 days old and younger with documented viral infections. J Pediatr. 2018;203:86-91.e2. https://doi.org/10.1016/j.jpeds.2018.07.073.
1. Cioffredi L-A, Jhaveri R. Evaluation and management of febrile children. JAMA Pediatr. 2016;170(8):794. https://doi.org/10.1001/jamapediatrics.2016.0596.
2. Huppler AR, Eickhoff JC, Wald ER. Performance of low-risk criteria in the evaluation of young infants with fever: review of the literature. Pediatrics. 2010;125(2):228-233. https://doi.org/10.1542/peds.2009-1070.
3. Biondi EA, Lee B, Ralston SL, et al. Prevalence of bacteremia and bacterial meningitis in febrile neonates and infants in the second month of life a systematic review and meta-analysis + supplemental content. JAMA Netw Open. 2019;2(3):190874. https://doi.org/10.1001/jamanetworkopen.2019.0874.
4. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
5. Aronson PL, Thurm C, Alpern ER, et al. Variation in care of the febrile young infant <90 days in us pediatric emergency departments. Pediatrics. 2014;134(4):667-677. https://doi.org/10.1542/peds.2014-1382.
6. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
7. Mendonca EA, Meissner HC, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. https://doi.org/10.1542/peds.2014-2742.
8. Melendez E, Harper MB. Utility of sepsis evaluation in infants 90 days of age or younger with fever and clinical bronchiolitis. Pediatr Infect Dis J. 2003;22(12):1053-1056. https://doi.org/10.1097/01.inf.0000101296.68993.4d.
9. Pruitt CM, Neuman MI, Shah SS, et al. Factors associated with adverse outcomes among febrile young infants with invasive bacterial infections. J. Pediatr. 2018;204:177-182. https://doi.org/10.1016/j.jpeds.2018.08.066.
10. Casper TC, Mahajan PV., Tzimenatos L, et al. The Yale Observation Scale Score and the risk of serious bacterial infections in febrile infants. Pediatrics. 2017;140(1):e20170695. https://doi.org/10.1542/peds.2017-0695.
11. Garges HP. Neonatal meningitis: what is the correlation among cerebrospinal fluid cultures, blood cultures, and cerebrospinal fluid parameters? Pediatrics. 2006;117(4):1094-1100. https://doi.org/10.1542/peds.2005-1132.
12. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection-an appraisal of the Rochester criteria and implications for management. Pediatrics. 1994;94(3):390-396. http://www.ncbi.nlm.nih.gov/pubmed/8065869. Accessed March 23, 2019.
13. Aronson P, Wang M, Shapiro E, et al. Risk stratification of febrile infants ≤60 days old without routine lumbar puncture. Pediatrics. 2018;142(6):e20181879. https://doi.org/10.1542/peds.2018-1879.
14. Galetto-Lacour A, Zamora SA, Andreola B, et al. Validation of a laboratory risk index score for the identification of severe bacterial infection in children with fever without source. Arch Dis Child. 2010;95(12):968-973. https://doi.org/10.1136/adc.2009.176800.
15. Kuppermann N, Dayan PS, Levine DA, et al. A clinical prediction rule to identify febrile infants 60 days and younger at low risk for serious bacterial infections. JAMA Pediatr. 2019;173(4):342. https://doi.org/10.1001/jamapediatrics.2018.5501.
16. Byington CL, Enriquez FR, Hoff C, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics. 2004;113(6):1662-1666. https://doi.org/10.1542/peds.113.6.1662.
17. Cioffredi LA, Jhaveri R. Evaluation and management of febrile children: a review. JAMA Pediatr. 2016;170(8):794-800. https://doi.org/10.1001/jamapediatrics.2016.0596.
18. Dayan PS, Roskind CG, Levine DA, Kuppermann N. Controversies in the management of children with bronchiolitis. Clin Pediatr Emerg Med. 2004;5(1):41-53. https://doi.org/10.1016/j.cpem.2003.11.001.
19. Oray-Schrom P, Phoenix C, St. Martin D, Amoateng-Adjepong Y. Sepsis workup in febrile infants 0-90 days of age with respiratory syncytial virus infection. Pediatr Emerg Care. 2003;19(5):314-319. https://doi.org/10.1097/01.pec.0000092576.40174.28.
20. Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156(4):322-324. https://doi.org/10.1001/archpedi.156.4.322.
21. Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23(3):267-269. https://doi.org/10.1097/01.inf.0000116759.21252.29.
22. Yarden-Bilavsky H, Ashkenazi-Hoffnung L, Livni G, Amir J, Bilavsky E. Month-by-month age analysis of the risk for serious bacterial infections in febrile infants with bronchiolitis. Clin Pediatr (Phila). 2011;50(11):1052-1056. https://doi.org/10.1177/0009922811412949.
23. Ralston S, Hill V, Waters A. Occult serious bacterial infection in infants younger than 60 to 90 days with bronchiolitis: a systematic review. Arch Pediatr Adolesc Med. 2011;165(10):951-956. https://doi.org/10.1001/archpediatrics.2011.155.
24. Lee TJ, Aronson PL. To spinal tap or not to spinal tap, that is the question. Hosp Pediatr. 2018;8(4):236-238. https://doi.org/10.1542/hpeds.2017-0207.
25. Pingree EW, Kimia AA, Nigrovic LE. The effect of traumatic lumbar puncture on hospitalization rate for febrile infants 28 to 60 days of age. Acad Emerg Med. 2015;22(2):240-243. https://doi.org/10.1111/acem.12582.
26. Leazer R, Erickson N, Paulson J, et al. epidemiology of cerebrospinal fluid cultures and time to detection in term infants. Pediatrics. 2017;139(5):e20163268. https://doi.org/10.1542/peds.2016-3268.
27. Paxton RD, Byington CL. An examination of the unintended consequences of the rule-out sepsis evaluation: a parental perspective. Clin Pediatr (Phila). 2001;40(2):71-77. https://doi.org/10.1177/000992280104000202.
28. Mahajan P, Br owne LR, Levine DA, et al. Risk of bacterial coinfections in febrile infants 60 days old and younger with documented viral infections. J Pediatr. 2018;203:86-91.e2. https://doi.org/10.1016/j.jpeds.2018.07.073.
© 2019 Society of Hospital Medicine
A Branching Algorithm
A 21-year-old man with a history of hypertension presented to the emergency department with four days of generalized abdominal pain, nausea, and vomiting as well as one month of loose stools. He also had a headache (not further specified) for one day. Due to his nausea, he had been unable to take his medications for two days. Home blood pressure measurements over the preceding two days revealed systolic pressures exceeding 200 mm Hg. He did not experience fever, dyspnea, chest pain, vision changes, numbness, weakness, diaphoresis, or palpitations.
Abdominal pain with vomiting and diarrhea is often caused by a self-limited gastroenteritis. However, the priority initially is to exclude serious intraabdominal processes including arterial insufficiency, bowel obstruction, organ perforation, or organ-based infection or inflammation (eg, appendicitis, cholecystitis, pancreatitis). Essential hypertension accounts for 95% of cases of hypertension in the United States, but given this patient’s young age, secondary causes should be evaluated. These include primary aldosteronism (the most common endocrine cause for hypertension in young patients), chronic kidney disease, fibromuscular dysplasia, illicit drug use, hypercortisolism, pheochromocytoma, and coarctation of the aorta. Thyrotoxicosis can elevate blood pressure (although usually not to this extent) and cause hyperdefecation. While the etiology of the chronic hypertension is uncertain, the proximate cause of the acute rise in blood pressure is likely the stress of his acute illness and the inability to take his prescribed antihypertensive medications. In the setting of severe hypertension, his headache may reflect an intracranial hemorrhage and his abdominal pain could signal an aortic dissection.
His medical history included hypertension diagnosed at age 16 as well as anxiety diagnosed following a panic attack at age 19. Over the past year, he had also developed persistent nausea, which was attributed to gastroesophageal reflux disease. His medications included metoprolol 50 mg daily, amlodipine 5 mg daily, hydrochlorothiazide 12.5 mg daily, escitalopram 20 mg daily, and omeprazole 20 mg daily. His father and 15-year-old brother also had hypertension. He was a part-time student while working at a car dealership. He did not smoke or use drugs and he rarely drank alcohol.
The need for three antihypertensive medications (albeit at submaximal doses) reflects the severity of his hypertension (provided challenges with medication adherence have been excluded). His family history, especially that of his brother who was diagnosed with hypertension at an early age, and the patient’s own early onset hypertension point toward an inherited form of hypertension. Autosomal dominant polycystic kidney disease often results in hypertension before chronic kidney disease develops. Rare inherited forms of hypertension include familial hyperaldosteronism, apparent mineralocorticoid excess, Liddle syndrome, or a hereditary endocrine tumor syndrome predisposing to pheochromocytoma. Even among patients who report classic pheochromocytoma symptoms, such as headache and anxiety, the diagnosis remains unlikely as these symptoms are nonspecific and highly prevalent in the general population. However, once secondary hypertension is plausible or suspected, testing for hyperadrenergic states, which can also cause nausea and vomiting during times of catecholamine excess, should be pursued.
His temperature was 97.5°F, heart rate 95 beats per minute and regular, respiratory rate 18 breaths per minute, blood pressure 181/118 mm Hg (systolic and diastolic pressures in each arm were within 10 mm Hg), and oxygen saturation 100% on room air. Systolic and diastolic pressures did not decrease by more than 20 mm Hg and 10 mm Hg, respectively, after he stood for two minutes. His body mass index was 24 kg/m2. He was alert and appeared slightly anxious. There was a bounding point of maximal impulse in the fifth intercostal space at the midclavicular line and a 3/6 systolic murmur at the left upper sternal border with radiation to the carotid arteries. His abdomen was soft with generalized tenderness to palpation and without rebound tenderness, masses, organomegaly, or bruits. There was no costovertebral angle tenderness. No lymphadenopathy was present. His fundoscopic, pulmonary, skin and neurologic examinations were normal.
Laboratory studies revealed a white blood cell count of 13.3 × 103/uL with a normal differential, hemoglobin 13.9 g/dL, platelet count 373 × 103/uL, sodium 142 mmol/L, potassium 3.8 mmol/L, chloride 103 mmol/L,bicarbonate 25 mmol/L, blood urea nitrogen 12 mg/dL, creatinine 1.3 mg/dL (a baseline creatinine level was not available), glucose 88 mg/dL, calcium 10.6 mg/dL, albumin 4.9 g/dL, aspartate aminotransferase 27 IU/L, alanine aminotransferase 37 IU/L, and lipase 40 IU/L. Urinalysis revealed 5-10 white blood cells per high power field without casts and 10 mg/dL protein. Urine toxicology was not performed. Electrocardiogram (ECG) showed left ventricular hypertrophy (LVH). Chest radiography was normal.
The abdominal examination does not suggest peritonitis. The laboratory tests do not suggest inflammation of the liver, pancreas, or biliary tree as the cause of his abdominal pain or diarrhea. The murmur may indicate hypertrophic cardiomyopathy or a congenital anomaly such as bicuspid aortic valve; but neither would explain hypertension unless they were associated with another developmental abnormality, such as coarctation of the aorta. Tricuspid regurgitation is conceivable and if confirmed, might raise concern for carcinoid syndrome, which can cause diarrhea. The normal neurologic examination, including the absence of papilledema, lowers suspicion of intracranial hemorrhage as a cause of his headache.
The albumin of 4.9 g/dL likely reflects hypovolemia resulting from vomiting and diarrhea. Vasoconstriction associated with pheochromocytoma can cause pressure diuresis and resultant hypovolemia. Hyperaldosteronism arising from bilateral adrenal hyperplasia or adrenal adenoma commonly causes hypokalemia, although this is not a universal feature.
The duration of his mildly decreased glomerular filtration rate is uncertain. He may have chronic kidney disease from sustained hypertension, or acute kidney injury from hypovolemia. The mild pyuria could indicate infection or renal calculi, either of which could account for generalized abdominal pain or could reflect an acute renal injury from acute interstitial nephritis from his proton pump inhibitor or hydrochlorothiazide.
LVH on the ECG indicates longstanding hypertension. The chest radiograph does not reveal clues to the etiology of or sequelae from hypertension. In particular, there is no widened aorta to suggest aortic dissection, no pulmonary edema to indicate heart failure, and no rib notching that points toward aortic coarctation. A transthoracic echocardiogram to assess for valvular and other structural abnormalities is warranted.
Tests for secondary hypertension should be sent, including serum aldosterone and renin levels to assess for primary aldosteronism and plasma or 24-hour urine normetanephrine and metanephrine levels to assess for pheochromocytoma. Biochemical evaluation is the mainstay for endocrine hypertension evaluation and should be followed by imaging if abnormal results are found.
Intact parathyroid hormone (PTH) was 78 pg/mL (normal, 10-65 pg/mL), thyroid stimulating hormone 3.6 mIU/L (normal, 0.30-5.50 mIU/L), and morning cortisol 4.1 ug/dL (normal, >7.0 ug/dL). Plasma aldosterone was 14.6 ng/dL (normal, 1-16 ng/dL), plasma renin activity 3.6 ng/mL/hr (normal, 0.5-3.5 ng/mL/hr), and aldosterone-renin ratio 4.1 (normal, <20). Transthoracic echocardiogram showed LVH with normal valves, wall motion, and proximal aorta; the left ventricular ejection fraction was 70%. Magnetic resonance angiography of the renal vessels demonstrated no abnormalities.
Computed tomography (CT) of the abdomen and pelvis with oral and intravenous contrast revealed a 5 cm heterogeneous enhancing mass associated with the prostate gland extending into the base of the bladder. The mass obstructed the right renal collecting system and ureter causing severe right-sided ureterectasis and hydronephrosis. There was also 2.8 cm right-sided paracaval lymph node enlargement and 2.1 cm right-sided and 1.5 cm left-sided external iliac lymph node enlargement (Figure 1). There were no adrenal masses.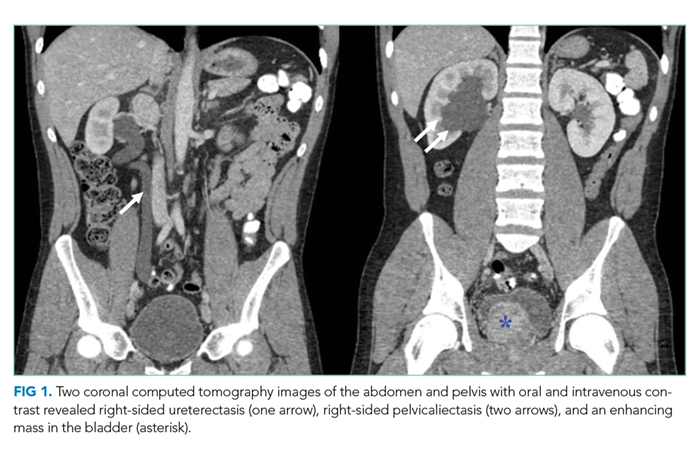
He is young for prostate, bladder, or colorectal cancer, but early onset variations of these tumors, along with metastatic testicular cancer, must be considered for the pelvic mass and associated lymphadenopathy. Prostatic masses can be infectious (eg, abscess) or malignant (eg, adenocarcinoma, small cell carcinoma). Additional considerations for abdominopelvic cancer are sarcomas, germ cell tumors, or lymphoma. A low aldosterone-renin ratio coupled with a normal potassium level makes primary aldosteronism unlikely. The normal angiography excludes renovascular hypertension.
His abdominal pain and gastrointestinal symptoms could arise from irritation of the bowel, distension of the right-sided urinary collecting system, or products secreted from the mass (eg, catecholamines). The hyperdynamic precordium, elevated ejection fraction, and murmur may reflect augmented blood flow from a hyperadrenergic state. A unifying diagnosis would be a pheochromocytoma. However, given the normal appearance of the adrenal glands on CT imaging, catecholamines arising from a paraganglioma, a tumor of the autonomic nervous system, is more likely. These tumors often secrete catecholamines and can be metastatic (suggested here by the lymphadenopathy). Functional imaging or biopsy of either the mass or an adjacent lymph node is indicated. However, because of the possibility of a catecholamine-secreting tumor, he should be treated with an alpha-adrenergic receptor antagonist before undergoing a biopsy to prevent unopposed vasoconstriction from catecholamine leakage.
Scrotal ultrasound revealed no evidence of a testicular tumor. Lactate dehydrogenase (LDH) was 179 IU/L (normal, 120-240 IU/L) and prostate specific antigen (PSA) was 0.7 ng/mL (normal, <2.5 ng/mL). The patient was given amlodipine and labetalol with improvement of blood pressures to 160s/100s. His creatinine decreased to 1.1 mg/dL. He underwent CT-guided biopsy of a pelvic lymph node. CT of the head without intravenous contrast demonstrated no intracranial abnormalities. His headache resolved with improvement in blood pressure, and he had minimal gastrointestinal symptoms during his hospitalization. No stool studies were sent. A right-sided percutaneous nephrostomy was placed which yielded >15 L of urine from the tube over the next four days.
Upon the first episode of micturition through the urethra four days after percutaneous nephrostomy placement, he experienced severe lightheadedness, diaphoresis, and palpitations. These symptoms prompted him to recall similar episodes following micturition for several months prior to his hospitalization.
It is likely that contraction of the bladder during episodes of urination caused irritation of the pelvic mass, leading to catecholamine secretion. Another explanation for his recurrent lightheadedness would be a neurocardiogenic reflex with micturition (which when it culminates with loss of consciousness is called micturition syncope), but this would not explain his hypertension or bladder mass.
Biochemical tests that were ordered on admission but sent to a reference lab then returned. Plasma metanephrine was 0.2 nmol/L (normal, <0.5 nmol/L) and plasma normetanephrine 34.6 nmol/L (normal, <0.9 nmol/L). His 24-hour urine metanephrine was 72 ug/24 hr (normal, 0-300 ug/24 hr) and normetanephrine 8,511 ug/24 hr (normal, 50-800 ug/24 hr).
The markedly elevated plasma and urine normetanephrine levels confirm a diagnosis of a catecholamine-secreting tumor (paraganglioma). The tissue obtained from the CT-guided lymph node biopsy should be sent for markers of neuroendocrine tumors including chromogranin.
Lymph node biopsy revealed metastatic paraganglioma that was chromogranin A and synaptophysin positive (Figure 2). A fluorodeoxyglucose positron emission tomography (FDG-PET) scan disclosed skull metastases. He was treated with phenoxybenzamine, amlodipine, and labetalol. Surgical resection of the pelvic mass was discussed, but the patient elected to defer surgery as the location of the primary tumor made it challenging to resect and would have required an ileal conduit.
After the diagnosis was made, the patient’s family recalled that a maternal uncle had been diagnosed with a paraganglioma of the carotid body. Genetic testing of the patient identified a succinate dehydrogenase complex subunit B (SDHB) pathogenic variant and confirmed hereditary paraganglioma syndrome (HPGL). One year after the diagnosis, liver and lung metastases developed. He was treated with lanreotide (somatostatin analogue), capecitabine, and temozolomide, as well as a craniotomy and radiotherapy for palliation of bony metastases. The patient died less than two years after diagnosis.
DISCUSSION
Most patients with hypertension (defined as blood pressure >130/80 mm Hg1) do not have an identifiable etiology (primary hypertension). Many components of this patient’s history, however, including his young age of onset, a teenage sibling with hypertension, lack of obesity, hypertension refractory to multiple medications, and LVH suggested secondary hypertension. Hypertension onset at an age less than 30 years, resistance to three or more medications,1,2 and/or acute onset hypertension at any age should prompt an evaluation for secondary causes.1 The prevalence of secondary hypertension is approximately 30% in hypertensive patients ages 18 to 40 years compared with 5%-10% in the overall adult population with hypertension.3 Among children and adolescents ages 0 to 19 years with hypertension, the prevalence of secondary hypertension may be as high as 57%.4
The most common etiology of secondary hypertension is primary aldosteronism.5,6 However, in young adults (ages 19 to 39 years), common etiologies also include renovascular disease and renal parenchymal disease.7 Other causes include obstructive sleep apnea, medications, stimulants (cocaine and amphetamines),8 and endocrinopathies such as thyrotoxicosis, Cushing syndrome, and catecholamine-secreting tumors.7 Less than 1% of secondary hypertension in all adults is due to catecholamine-secreting tumors, and the minority of those catecholamine-secreting tumors are paragangliomas.9
Paragangliomas are tumors of the peripheral autonomic nervous system. These neoplasms arise in the sympathetic and parasympathetic chains along the paravertebral and paraaortic axes. They are closely related to pheochromocytomas, which arise in the adrenal medulla.9 Most head and neck paragangliomas are biochemically silent and are generally discovered due to mass effect.10 The subset of paragangliomas that secrete catecholamines most often arise in the abdomen and pelvis, and their clinical presentation mimics that of pheochromocytomas, including episodic hypertension, palpitations, pallor, and diaphoresis.
This patient had persistent, nonepisodic hypertension, while palpitations and diaphoresis only manifested following micturition. Other cases of urinary bladder paragangliomas have described micturition-associated symptoms and hypertensive crises. Three-fold increases of catecholamine secretion after micturition have been observed in these patients, likely due to muscle contraction and pressure changes in the bladder leading to the systemic release of catecholamines.11
Epinephrine and norepinephrine are monoamine neurotransmitters that activate alpha-adrenergic and beta-adrenergic receptors. Adrenergic receptors are present in all tissues of the body but have prominent effects on the smooth muscle in the vasculature, gastrointestinal tract, urinary tract, and airways.12 Alpha-adrenergic vasoconstriction causes hypertension, which is commonly observed in patients with catecholamine-secreting tumors.10 Catecholamine excess due to secretion from these tumors causes headache in 60%-80% of patients, tachycardia/palpitations in 50%-70%, anxiety in 20%-40%, and nausea in 20%-25%.10 Other symptoms include sweating, pallor, dyspnea, and vertigo.9,10 This patient’s chronic nausea, which was attributed to gastroesophageal reflux, and his anxiety, attributed to generalized anxiety disorder, were likely symptoms of catecholamine excess.13
The best test for the diagnosis of paragangliomas and pheochromocytomas is the measurement of plasma free or 24-hour urinary fractionated metanephrines (test sensitivity of >90% and >90%, respectively).14 Screening for pheochromocytoma should be considered in hypertensive patients who have symptoms of catecholamine excess, refractory or paroxysmal hypertension, and/or familial pheochromocytoma/paraganglioma syndromes.15 Screening for pheochromocytoma should also be performed in children and adolescents with systolic or diastolic blood pressure that is greater than the 95th percentile for their age plus 5 mm Hg.16
While a typical tumor location and elevated metanephrine levels are sufficient to make the diagnosis of a pheochromocytoma or catecholamine-secreting paraganglioma, functional imaging with FDG-PET, Ga-DOTATATE-PET, or 123I-meta-iodobenzylguanidine (123I-MIBG) can further confirm the diagnosis and detect distant metastases. However, imaging has low sensitivity for these tumors and thus should only be considered for patients in whom metastatic disease is suspected.14 Biopsy is rarely needed and should be reserved for unusual metastatic locations. Treatment with an alpha-adrenergic receptor antagonist often reduces symptoms and lowers blood pressure. Definitive management typically involves surgical resection for benign disease. Surgery, radionuclide therapy, or chemotherapy is used for malignant disease.
While most pheochromocytomas are sporadic, up to 40% of paragangliomas are due to germline pathogenic variants.17 Mutations in the succinate dehydrogenase (SDH) group of genes are the most common germline pathogenic variants in the autosomal dominant hereditary paraganglioma syndrome (HPGL). Most paragangliomas and pheochromocytomas are localized and benign, but 10%-15% are metastatic.18 SDHB mutations are associated with a high risk of metastasis.19 Thus, genetic testing for patients and subsequent cascade testing to identify at-risk family members is advised in all patients with pheochromocytomas or paragangliomas.20 This patient’s younger brother and mother were both found to carry the same pathogenic SDHB variant, but neither was found to have paragangliomas. Annual metanephrine levels (urine or plasma) and every other year whole-body magnetic resonance imaging (MRI) scans were recommended for tumor surveillance.
The clinician team followed a logical branching algorithm for the diagnosis of severe hypertension with biochemical testing, advanced imaging, histology, and genetic testing to arrive at the final diagnosis of hereditary paraganglioma syndrome. Although this patient presented for urgent care because of the acute effects of catecholamine excess, he suffered from chronic effects (nausea, anxiety, and hypertension) for years. Each symptom had been diagnosed and treated in isolation, but the combination and severity in a young patient suggested a unifying diagnosis. The family history of hypertension (brother and father) suggested an inherited diagnosis from the father’s family, but the final answer rested on the other branch (maternal uncle) of the family tree.
KEY TEACHING POINTS
- Hypertension in a young adult is due to a secondary cause in up to 30% of patients.
- Pathologic catecholamine excess leads to hypertension, tachycardia, pallor, sweating, anxiety, and nausea. A sustained and unexplained combination of these symptoms should prompt a biochemical evaluation for pheochromocytoma or paraganglioma.
- Paragangliomas are tumors of the autonomic nervous system. The frequency of catecholamine secretion depends on their location in the body, and they are commonly caused by germline pathogenic variants.
Acknowledgments
This conundrum was presented during a live Grand Rounds with the expert clinician’s responses recorded and edited for space and clarity.
Disclosures
Dr. Dhaliwal reports speaking honoraria from ISMIE Mutual Insurance Company and GE Healthcare. All other authors have nothing to disclose.
Funding
No sources of funding.
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13-e115. https://doi.org/10.1161/HYP.0000000000000065.
2. Acelajado MC, Calhoun DA. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin. 2010;28(4):639-654. https://doi.org/10.1016/j.ccl.2010.07.002.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206-1252. https://doi.org/10.1161/01.HYP.0000107251.49515.c2.
4. Gupta-Malhotra M, Banker A, Shete S, et al. Essential hypertension vs. secondary hypertension among children. Am J Hypertens. 2015;28(1):73-80. https://doi.org/10.1093/ajh/hpu083.
5. Mosso L, Carvajal C, Gonzalez A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42(2):161-165. https://doi.org/10.1161/01.HYP.0000079505.25750.11.
6. Kayser SC, Dekkers T, Groenewoud HJ, et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab. 2016;101(7):2826-2835. https://doi.org/10.1210/jc.2016-1472.
7. Charles L, Triscott J, Dobbs B. Secondary hypertension: discovering the underlying cause. Am Fam Physician. 2017;96(7):453-461.
8. Aronow WS. Drug-induced causes of secondary hypertension. Ann Transl Med. 2017;5(17):349. https://doi.org/10.21037/atm.2017.06.16.
9. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675. https://doi.org/10.1016/S0140-6736(05)67139-5.
10. Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26(4):507-515. https://doi.org/10.1016/j.beem.2011.10.008.
11. Kappers MH, van den Meiracker AH, Alwani RA, Kats E, Baggen MG. Paraganglioma of the urinary bladder. Neth J Med. 2008;66(4):163-165.
12. Paravati S, Warrington SJ. Physiology, Catecholamines. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC; 2019.
13. King KS, Darmani NA, Hughes MS, Adams KT, Pacak K. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37(3):403-407. https://doi.org/10.1007/s12020-010-9319-3.
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. https://doi.org/10.1210/jc.2014-1498.
15. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). 2017;32(2):152-161. https://doi.org/10.3803/EnM.2017.32.2.152.
16. National High Blood Pressure Education Program Working Group. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555-576.
17. Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 1993.
18. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756. https://doi.org/10.1097/00000658-199906000-00001.
19. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822-3828. https://doi.org/10.1210/jc.2007-0709.
20. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111. https://doi.org/10.1038/nrendo.2014.188.
A 21-year-old man with a history of hypertension presented to the emergency department with four days of generalized abdominal pain, nausea, and vomiting as well as one month of loose stools. He also had a headache (not further specified) for one day. Due to his nausea, he had been unable to take his medications for two days. Home blood pressure measurements over the preceding two days revealed systolic pressures exceeding 200 mm Hg. He did not experience fever, dyspnea, chest pain, vision changes, numbness, weakness, diaphoresis, or palpitations.
Abdominal pain with vomiting and diarrhea is often caused by a self-limited gastroenteritis. However, the priority initially is to exclude serious intraabdominal processes including arterial insufficiency, bowel obstruction, organ perforation, or organ-based infection or inflammation (eg, appendicitis, cholecystitis, pancreatitis). Essential hypertension accounts for 95% of cases of hypertension in the United States, but given this patient’s young age, secondary causes should be evaluated. These include primary aldosteronism (the most common endocrine cause for hypertension in young patients), chronic kidney disease, fibromuscular dysplasia, illicit drug use, hypercortisolism, pheochromocytoma, and coarctation of the aorta. Thyrotoxicosis can elevate blood pressure (although usually not to this extent) and cause hyperdefecation. While the etiology of the chronic hypertension is uncertain, the proximate cause of the acute rise in blood pressure is likely the stress of his acute illness and the inability to take his prescribed antihypertensive medications. In the setting of severe hypertension, his headache may reflect an intracranial hemorrhage and his abdominal pain could signal an aortic dissection.
His medical history included hypertension diagnosed at age 16 as well as anxiety diagnosed following a panic attack at age 19. Over the past year, he had also developed persistent nausea, which was attributed to gastroesophageal reflux disease. His medications included metoprolol 50 mg daily, amlodipine 5 mg daily, hydrochlorothiazide 12.5 mg daily, escitalopram 20 mg daily, and omeprazole 20 mg daily. His father and 15-year-old brother also had hypertension. He was a part-time student while working at a car dealership. He did not smoke or use drugs and he rarely drank alcohol.
The need for three antihypertensive medications (albeit at submaximal doses) reflects the severity of his hypertension (provided challenges with medication adherence have been excluded). His family history, especially that of his brother who was diagnosed with hypertension at an early age, and the patient’s own early onset hypertension point toward an inherited form of hypertension. Autosomal dominant polycystic kidney disease often results in hypertension before chronic kidney disease develops. Rare inherited forms of hypertension include familial hyperaldosteronism, apparent mineralocorticoid excess, Liddle syndrome, or a hereditary endocrine tumor syndrome predisposing to pheochromocytoma. Even among patients who report classic pheochromocytoma symptoms, such as headache and anxiety, the diagnosis remains unlikely as these symptoms are nonspecific and highly prevalent in the general population. However, once secondary hypertension is plausible or suspected, testing for hyperadrenergic states, which can also cause nausea and vomiting during times of catecholamine excess, should be pursued.
His temperature was 97.5°F, heart rate 95 beats per minute and regular, respiratory rate 18 breaths per minute, blood pressure 181/118 mm Hg (systolic and diastolic pressures in each arm were within 10 mm Hg), and oxygen saturation 100% on room air. Systolic and diastolic pressures did not decrease by more than 20 mm Hg and 10 mm Hg, respectively, after he stood for two minutes. His body mass index was 24 kg/m2. He was alert and appeared slightly anxious. There was a bounding point of maximal impulse in the fifth intercostal space at the midclavicular line and a 3/6 systolic murmur at the left upper sternal border with radiation to the carotid arteries. His abdomen was soft with generalized tenderness to palpation and without rebound tenderness, masses, organomegaly, or bruits. There was no costovertebral angle tenderness. No lymphadenopathy was present. His fundoscopic, pulmonary, skin and neurologic examinations were normal.
Laboratory studies revealed a white blood cell count of 13.3 × 103/uL with a normal differential, hemoglobin 13.9 g/dL, platelet count 373 × 103/uL, sodium 142 mmol/L, potassium 3.8 mmol/L, chloride 103 mmol/L,bicarbonate 25 mmol/L, blood urea nitrogen 12 mg/dL, creatinine 1.3 mg/dL (a baseline creatinine level was not available), glucose 88 mg/dL, calcium 10.6 mg/dL, albumin 4.9 g/dL, aspartate aminotransferase 27 IU/L, alanine aminotransferase 37 IU/L, and lipase 40 IU/L. Urinalysis revealed 5-10 white blood cells per high power field without casts and 10 mg/dL protein. Urine toxicology was not performed. Electrocardiogram (ECG) showed left ventricular hypertrophy (LVH). Chest radiography was normal.
The abdominal examination does not suggest peritonitis. The laboratory tests do not suggest inflammation of the liver, pancreas, or biliary tree as the cause of his abdominal pain or diarrhea. The murmur may indicate hypertrophic cardiomyopathy or a congenital anomaly such as bicuspid aortic valve; but neither would explain hypertension unless they were associated with another developmental abnormality, such as coarctation of the aorta. Tricuspid regurgitation is conceivable and if confirmed, might raise concern for carcinoid syndrome, which can cause diarrhea. The normal neurologic examination, including the absence of papilledema, lowers suspicion of intracranial hemorrhage as a cause of his headache.
The albumin of 4.9 g/dL likely reflects hypovolemia resulting from vomiting and diarrhea. Vasoconstriction associated with pheochromocytoma can cause pressure diuresis and resultant hypovolemia. Hyperaldosteronism arising from bilateral adrenal hyperplasia or adrenal adenoma commonly causes hypokalemia, although this is not a universal feature.
The duration of his mildly decreased glomerular filtration rate is uncertain. He may have chronic kidney disease from sustained hypertension, or acute kidney injury from hypovolemia. The mild pyuria could indicate infection or renal calculi, either of which could account for generalized abdominal pain or could reflect an acute renal injury from acute interstitial nephritis from his proton pump inhibitor or hydrochlorothiazide.
LVH on the ECG indicates longstanding hypertension. The chest radiograph does not reveal clues to the etiology of or sequelae from hypertension. In particular, there is no widened aorta to suggest aortic dissection, no pulmonary edema to indicate heart failure, and no rib notching that points toward aortic coarctation. A transthoracic echocardiogram to assess for valvular and other structural abnormalities is warranted.
Tests for secondary hypertension should be sent, including serum aldosterone and renin levels to assess for primary aldosteronism and plasma or 24-hour urine normetanephrine and metanephrine levels to assess for pheochromocytoma. Biochemical evaluation is the mainstay for endocrine hypertension evaluation and should be followed by imaging if abnormal results are found.
Intact parathyroid hormone (PTH) was 78 pg/mL (normal, 10-65 pg/mL), thyroid stimulating hormone 3.6 mIU/L (normal, 0.30-5.50 mIU/L), and morning cortisol 4.1 ug/dL (normal, >7.0 ug/dL). Plasma aldosterone was 14.6 ng/dL (normal, 1-16 ng/dL), plasma renin activity 3.6 ng/mL/hr (normal, 0.5-3.5 ng/mL/hr), and aldosterone-renin ratio 4.1 (normal, <20). Transthoracic echocardiogram showed LVH with normal valves, wall motion, and proximal aorta; the left ventricular ejection fraction was 70%. Magnetic resonance angiography of the renal vessels demonstrated no abnormalities.
Computed tomography (CT) of the abdomen and pelvis with oral and intravenous contrast revealed a 5 cm heterogeneous enhancing mass associated with the prostate gland extending into the base of the bladder. The mass obstructed the right renal collecting system and ureter causing severe right-sided ureterectasis and hydronephrosis. There was also 2.8 cm right-sided paracaval lymph node enlargement and 2.1 cm right-sided and 1.5 cm left-sided external iliac lymph node enlargement (Figure 1). There were no adrenal masses.
He is young for prostate, bladder, or colorectal cancer, but early onset variations of these tumors, along with metastatic testicular cancer, must be considered for the pelvic mass and associated lymphadenopathy. Prostatic masses can be infectious (eg, abscess) or malignant (eg, adenocarcinoma, small cell carcinoma). Additional considerations for abdominopelvic cancer are sarcomas, germ cell tumors, or lymphoma. A low aldosterone-renin ratio coupled with a normal potassium level makes primary aldosteronism unlikely. The normal angiography excludes renovascular hypertension.
His abdominal pain and gastrointestinal symptoms could arise from irritation of the bowel, distension of the right-sided urinary collecting system, or products secreted from the mass (eg, catecholamines). The hyperdynamic precordium, elevated ejection fraction, and murmur may reflect augmented blood flow from a hyperadrenergic state. A unifying diagnosis would be a pheochromocytoma. However, given the normal appearance of the adrenal glands on CT imaging, catecholamines arising from a paraganglioma, a tumor of the autonomic nervous system, is more likely. These tumors often secrete catecholamines and can be metastatic (suggested here by the lymphadenopathy). Functional imaging or biopsy of either the mass or an adjacent lymph node is indicated. However, because of the possibility of a catecholamine-secreting tumor, he should be treated with an alpha-adrenergic receptor antagonist before undergoing a biopsy to prevent unopposed vasoconstriction from catecholamine leakage.
Scrotal ultrasound revealed no evidence of a testicular tumor. Lactate dehydrogenase (LDH) was 179 IU/L (normal, 120-240 IU/L) and prostate specific antigen (PSA) was 0.7 ng/mL (normal, <2.5 ng/mL). The patient was given amlodipine and labetalol with improvement of blood pressures to 160s/100s. His creatinine decreased to 1.1 mg/dL. He underwent CT-guided biopsy of a pelvic lymph node. CT of the head without intravenous contrast demonstrated no intracranial abnormalities. His headache resolved with improvement in blood pressure, and he had minimal gastrointestinal symptoms during his hospitalization. No stool studies were sent. A right-sided percutaneous nephrostomy was placed which yielded >15 L of urine from the tube over the next four days.
Upon the first episode of micturition through the urethra four days after percutaneous nephrostomy placement, he experienced severe lightheadedness, diaphoresis, and palpitations. These symptoms prompted him to recall similar episodes following micturition for several months prior to his hospitalization.
It is likely that contraction of the bladder during episodes of urination caused irritation of the pelvic mass, leading to catecholamine secretion. Another explanation for his recurrent lightheadedness would be a neurocardiogenic reflex with micturition (which when it culminates with loss of consciousness is called micturition syncope), but this would not explain his hypertension or bladder mass.
Biochemical tests that were ordered on admission but sent to a reference lab then returned. Plasma metanephrine was 0.2 nmol/L (normal, <0.5 nmol/L) and plasma normetanephrine 34.6 nmol/L (normal, <0.9 nmol/L). His 24-hour urine metanephrine was 72 ug/24 hr (normal, 0-300 ug/24 hr) and normetanephrine 8,511 ug/24 hr (normal, 50-800 ug/24 hr).
The markedly elevated plasma and urine normetanephrine levels confirm a diagnosis of a catecholamine-secreting tumor (paraganglioma). The tissue obtained from the CT-guided lymph node biopsy should be sent for markers of neuroendocrine tumors including chromogranin.
Lymph node biopsy revealed metastatic paraganglioma that was chromogranin A and synaptophysin positive (Figure 2). A fluorodeoxyglucose positron emission tomography (FDG-PET) scan disclosed skull metastases. He was treated with phenoxybenzamine, amlodipine, and labetalol. Surgical resection of the pelvic mass was discussed, but the patient elected to defer surgery as the location of the primary tumor made it challenging to resect and would have required an ileal conduit.
After the diagnosis was made, the patient’s family recalled that a maternal uncle had been diagnosed with a paraganglioma of the carotid body. Genetic testing of the patient identified a succinate dehydrogenase complex subunit B (SDHB) pathogenic variant and confirmed hereditary paraganglioma syndrome (HPGL). One year after the diagnosis, liver and lung metastases developed. He was treated with lanreotide (somatostatin analogue), capecitabine, and temozolomide, as well as a craniotomy and radiotherapy for palliation of bony metastases. The patient died less than two years after diagnosis.
DISCUSSION
Most patients with hypertension (defined as blood pressure >130/80 mm Hg1) do not have an identifiable etiology (primary hypertension). Many components of this patient’s history, however, including his young age of onset, a teenage sibling with hypertension, lack of obesity, hypertension refractory to multiple medications, and LVH suggested secondary hypertension. Hypertension onset at an age less than 30 years, resistance to three or more medications,1,2 and/or acute onset hypertension at any age should prompt an evaluation for secondary causes.1 The prevalence of secondary hypertension is approximately 30% in hypertensive patients ages 18 to 40 years compared with 5%-10% in the overall adult population with hypertension.3 Among children and adolescents ages 0 to 19 years with hypertension, the prevalence of secondary hypertension may be as high as 57%.4
The most common etiology of secondary hypertension is primary aldosteronism.5,6 However, in young adults (ages 19 to 39 years), common etiologies also include renovascular disease and renal parenchymal disease.7 Other causes include obstructive sleep apnea, medications, stimulants (cocaine and amphetamines),8 and endocrinopathies such as thyrotoxicosis, Cushing syndrome, and catecholamine-secreting tumors.7 Less than 1% of secondary hypertension in all adults is due to catecholamine-secreting tumors, and the minority of those catecholamine-secreting tumors are paragangliomas.9
Paragangliomas are tumors of the peripheral autonomic nervous system. These neoplasms arise in the sympathetic and parasympathetic chains along the paravertebral and paraaortic axes. They are closely related to pheochromocytomas, which arise in the adrenal medulla.9 Most head and neck paragangliomas are biochemically silent and are generally discovered due to mass effect.10 The subset of paragangliomas that secrete catecholamines most often arise in the abdomen and pelvis, and their clinical presentation mimics that of pheochromocytomas, including episodic hypertension, palpitations, pallor, and diaphoresis.
This patient had persistent, nonepisodic hypertension, while palpitations and diaphoresis only manifested following micturition. Other cases of urinary bladder paragangliomas have described micturition-associated symptoms and hypertensive crises. Three-fold increases of catecholamine secretion after micturition have been observed in these patients, likely due to muscle contraction and pressure changes in the bladder leading to the systemic release of catecholamines.11
Epinephrine and norepinephrine are monoamine neurotransmitters that activate alpha-adrenergic and beta-adrenergic receptors. Adrenergic receptors are present in all tissues of the body but have prominent effects on the smooth muscle in the vasculature, gastrointestinal tract, urinary tract, and airways.12 Alpha-adrenergic vasoconstriction causes hypertension, which is commonly observed in patients with catecholamine-secreting tumors.10 Catecholamine excess due to secretion from these tumors causes headache in 60%-80% of patients, tachycardia/palpitations in 50%-70%, anxiety in 20%-40%, and nausea in 20%-25%.10 Other symptoms include sweating, pallor, dyspnea, and vertigo.9,10 This patient’s chronic nausea, which was attributed to gastroesophageal reflux, and his anxiety, attributed to generalized anxiety disorder, were likely symptoms of catecholamine excess.13
The best test for the diagnosis of paragangliomas and pheochromocytomas is the measurement of plasma free or 24-hour urinary fractionated metanephrines (test sensitivity of >90% and >90%, respectively).14 Screening for pheochromocytoma should be considered in hypertensive patients who have symptoms of catecholamine excess, refractory or paroxysmal hypertension, and/or familial pheochromocytoma/paraganglioma syndromes.15 Screening for pheochromocytoma should also be performed in children and adolescents with systolic or diastolic blood pressure that is greater than the 95th percentile for their age plus 5 mm Hg.16
While a typical tumor location and elevated metanephrine levels are sufficient to make the diagnosis of a pheochromocytoma or catecholamine-secreting paraganglioma, functional imaging with FDG-PET, Ga-DOTATATE-PET, or 123I-meta-iodobenzylguanidine (123I-MIBG) can further confirm the diagnosis and detect distant metastases. However, imaging has low sensitivity for these tumors and thus should only be considered for patients in whom metastatic disease is suspected.14 Biopsy is rarely needed and should be reserved for unusual metastatic locations. Treatment with an alpha-adrenergic receptor antagonist often reduces symptoms and lowers blood pressure. Definitive management typically involves surgical resection for benign disease. Surgery, radionuclide therapy, or chemotherapy is used for malignant disease.
While most pheochromocytomas are sporadic, up to 40% of paragangliomas are due to germline pathogenic variants.17 Mutations in the succinate dehydrogenase (SDH) group of genes are the most common germline pathogenic variants in the autosomal dominant hereditary paraganglioma syndrome (HPGL). Most paragangliomas and pheochromocytomas are localized and benign, but 10%-15% are metastatic.18 SDHB mutations are associated with a high risk of metastasis.19 Thus, genetic testing for patients and subsequent cascade testing to identify at-risk family members is advised in all patients with pheochromocytomas or paragangliomas.20 This patient’s younger brother and mother were both found to carry the same pathogenic SDHB variant, but neither was found to have paragangliomas. Annual metanephrine levels (urine or plasma) and every other year whole-body magnetic resonance imaging (MRI) scans were recommended for tumor surveillance.
The clinician team followed a logical branching algorithm for the diagnosis of severe hypertension with biochemical testing, advanced imaging, histology, and genetic testing to arrive at the final diagnosis of hereditary paraganglioma syndrome. Although this patient presented for urgent care because of the acute effects of catecholamine excess, he suffered from chronic effects (nausea, anxiety, and hypertension) for years. Each symptom had been diagnosed and treated in isolation, but the combination and severity in a young patient suggested a unifying diagnosis. The family history of hypertension (brother and father) suggested an inherited diagnosis from the father’s family, but the final answer rested on the other branch (maternal uncle) of the family tree.
KEY TEACHING POINTS
- Hypertension in a young adult is due to a secondary cause in up to 30% of patients.
- Pathologic catecholamine excess leads to hypertension, tachycardia, pallor, sweating, anxiety, and nausea. A sustained and unexplained combination of these symptoms should prompt a biochemical evaluation for pheochromocytoma or paraganglioma.
- Paragangliomas are tumors of the autonomic nervous system. The frequency of catecholamine secretion depends on their location in the body, and they are commonly caused by germline pathogenic variants.
Acknowledgments
This conundrum was presented during a live Grand Rounds with the expert clinician’s responses recorded and edited for space and clarity.
Disclosures
Dr. Dhaliwal reports speaking honoraria from ISMIE Mutual Insurance Company and GE Healthcare. All other authors have nothing to disclose.
Funding
No sources of funding.
A 21-year-old man with a history of hypertension presented to the emergency department with four days of generalized abdominal pain, nausea, and vomiting as well as one month of loose stools. He also had a headache (not further specified) for one day. Due to his nausea, he had been unable to take his medications for two days. Home blood pressure measurements over the preceding two days revealed systolic pressures exceeding 200 mm Hg. He did not experience fever, dyspnea, chest pain, vision changes, numbness, weakness, diaphoresis, or palpitations.
Abdominal pain with vomiting and diarrhea is often caused by a self-limited gastroenteritis. However, the priority initially is to exclude serious intraabdominal processes including arterial insufficiency, bowel obstruction, organ perforation, or organ-based infection or inflammation (eg, appendicitis, cholecystitis, pancreatitis). Essential hypertension accounts for 95% of cases of hypertension in the United States, but given this patient’s young age, secondary causes should be evaluated. These include primary aldosteronism (the most common endocrine cause for hypertension in young patients), chronic kidney disease, fibromuscular dysplasia, illicit drug use, hypercortisolism, pheochromocytoma, and coarctation of the aorta. Thyrotoxicosis can elevate blood pressure (although usually not to this extent) and cause hyperdefecation. While the etiology of the chronic hypertension is uncertain, the proximate cause of the acute rise in blood pressure is likely the stress of his acute illness and the inability to take his prescribed antihypertensive medications. In the setting of severe hypertension, his headache may reflect an intracranial hemorrhage and his abdominal pain could signal an aortic dissection.
His medical history included hypertension diagnosed at age 16 as well as anxiety diagnosed following a panic attack at age 19. Over the past year, he had also developed persistent nausea, which was attributed to gastroesophageal reflux disease. His medications included metoprolol 50 mg daily, amlodipine 5 mg daily, hydrochlorothiazide 12.5 mg daily, escitalopram 20 mg daily, and omeprazole 20 mg daily. His father and 15-year-old brother also had hypertension. He was a part-time student while working at a car dealership. He did not smoke or use drugs and he rarely drank alcohol.
The need for three antihypertensive medications (albeit at submaximal doses) reflects the severity of his hypertension (provided challenges with medication adherence have been excluded). His family history, especially that of his brother who was diagnosed with hypertension at an early age, and the patient’s own early onset hypertension point toward an inherited form of hypertension. Autosomal dominant polycystic kidney disease often results in hypertension before chronic kidney disease develops. Rare inherited forms of hypertension include familial hyperaldosteronism, apparent mineralocorticoid excess, Liddle syndrome, or a hereditary endocrine tumor syndrome predisposing to pheochromocytoma. Even among patients who report classic pheochromocytoma symptoms, such as headache and anxiety, the diagnosis remains unlikely as these symptoms are nonspecific and highly prevalent in the general population. However, once secondary hypertension is plausible or suspected, testing for hyperadrenergic states, which can also cause nausea and vomiting during times of catecholamine excess, should be pursued.
His temperature was 97.5°F, heart rate 95 beats per minute and regular, respiratory rate 18 breaths per minute, blood pressure 181/118 mm Hg (systolic and diastolic pressures in each arm were within 10 mm Hg), and oxygen saturation 100% on room air. Systolic and diastolic pressures did not decrease by more than 20 mm Hg and 10 mm Hg, respectively, after he stood for two minutes. His body mass index was 24 kg/m2. He was alert and appeared slightly anxious. There was a bounding point of maximal impulse in the fifth intercostal space at the midclavicular line and a 3/6 systolic murmur at the left upper sternal border with radiation to the carotid arteries. His abdomen was soft with generalized tenderness to palpation and without rebound tenderness, masses, organomegaly, or bruits. There was no costovertebral angle tenderness. No lymphadenopathy was present. His fundoscopic, pulmonary, skin and neurologic examinations were normal.
Laboratory studies revealed a white blood cell count of 13.3 × 103/uL with a normal differential, hemoglobin 13.9 g/dL, platelet count 373 × 103/uL, sodium 142 mmol/L, potassium 3.8 mmol/L, chloride 103 mmol/L,bicarbonate 25 mmol/L, blood urea nitrogen 12 mg/dL, creatinine 1.3 mg/dL (a baseline creatinine level was not available), glucose 88 mg/dL, calcium 10.6 mg/dL, albumin 4.9 g/dL, aspartate aminotransferase 27 IU/L, alanine aminotransferase 37 IU/L, and lipase 40 IU/L. Urinalysis revealed 5-10 white blood cells per high power field without casts and 10 mg/dL protein. Urine toxicology was not performed. Electrocardiogram (ECG) showed left ventricular hypertrophy (LVH). Chest radiography was normal.
The abdominal examination does not suggest peritonitis. The laboratory tests do not suggest inflammation of the liver, pancreas, or biliary tree as the cause of his abdominal pain or diarrhea. The murmur may indicate hypertrophic cardiomyopathy or a congenital anomaly such as bicuspid aortic valve; but neither would explain hypertension unless they were associated with another developmental abnormality, such as coarctation of the aorta. Tricuspid regurgitation is conceivable and if confirmed, might raise concern for carcinoid syndrome, which can cause diarrhea. The normal neurologic examination, including the absence of papilledema, lowers suspicion of intracranial hemorrhage as a cause of his headache.
The albumin of 4.9 g/dL likely reflects hypovolemia resulting from vomiting and diarrhea. Vasoconstriction associated with pheochromocytoma can cause pressure diuresis and resultant hypovolemia. Hyperaldosteronism arising from bilateral adrenal hyperplasia or adrenal adenoma commonly causes hypokalemia, although this is not a universal feature.
The duration of his mildly decreased glomerular filtration rate is uncertain. He may have chronic kidney disease from sustained hypertension, or acute kidney injury from hypovolemia. The mild pyuria could indicate infection or renal calculi, either of which could account for generalized abdominal pain or could reflect an acute renal injury from acute interstitial nephritis from his proton pump inhibitor or hydrochlorothiazide.
LVH on the ECG indicates longstanding hypertension. The chest radiograph does not reveal clues to the etiology of or sequelae from hypertension. In particular, there is no widened aorta to suggest aortic dissection, no pulmonary edema to indicate heart failure, and no rib notching that points toward aortic coarctation. A transthoracic echocardiogram to assess for valvular and other structural abnormalities is warranted.
Tests for secondary hypertension should be sent, including serum aldosterone and renin levels to assess for primary aldosteronism and plasma or 24-hour urine normetanephrine and metanephrine levels to assess for pheochromocytoma. Biochemical evaluation is the mainstay for endocrine hypertension evaluation and should be followed by imaging if abnormal results are found.
Intact parathyroid hormone (PTH) was 78 pg/mL (normal, 10-65 pg/mL), thyroid stimulating hormone 3.6 mIU/L (normal, 0.30-5.50 mIU/L), and morning cortisol 4.1 ug/dL (normal, >7.0 ug/dL). Plasma aldosterone was 14.6 ng/dL (normal, 1-16 ng/dL), plasma renin activity 3.6 ng/mL/hr (normal, 0.5-3.5 ng/mL/hr), and aldosterone-renin ratio 4.1 (normal, <20). Transthoracic echocardiogram showed LVH with normal valves, wall motion, and proximal aorta; the left ventricular ejection fraction was 70%. Magnetic resonance angiography of the renal vessels demonstrated no abnormalities.
Computed tomography (CT) of the abdomen and pelvis with oral and intravenous contrast revealed a 5 cm heterogeneous enhancing mass associated with the prostate gland extending into the base of the bladder. The mass obstructed the right renal collecting system and ureter causing severe right-sided ureterectasis and hydronephrosis. There was also 2.8 cm right-sided paracaval lymph node enlargement and 2.1 cm right-sided and 1.5 cm left-sided external iliac lymph node enlargement (Figure 1). There were no adrenal masses.
He is young for prostate, bladder, or colorectal cancer, but early onset variations of these tumors, along with metastatic testicular cancer, must be considered for the pelvic mass and associated lymphadenopathy. Prostatic masses can be infectious (eg, abscess) or malignant (eg, adenocarcinoma, small cell carcinoma). Additional considerations for abdominopelvic cancer are sarcomas, germ cell tumors, or lymphoma. A low aldosterone-renin ratio coupled with a normal potassium level makes primary aldosteronism unlikely. The normal angiography excludes renovascular hypertension.
His abdominal pain and gastrointestinal symptoms could arise from irritation of the bowel, distension of the right-sided urinary collecting system, or products secreted from the mass (eg, catecholamines). The hyperdynamic precordium, elevated ejection fraction, and murmur may reflect augmented blood flow from a hyperadrenergic state. A unifying diagnosis would be a pheochromocytoma. However, given the normal appearance of the adrenal glands on CT imaging, catecholamines arising from a paraganglioma, a tumor of the autonomic nervous system, is more likely. These tumors often secrete catecholamines and can be metastatic (suggested here by the lymphadenopathy). Functional imaging or biopsy of either the mass or an adjacent lymph node is indicated. However, because of the possibility of a catecholamine-secreting tumor, he should be treated with an alpha-adrenergic receptor antagonist before undergoing a biopsy to prevent unopposed vasoconstriction from catecholamine leakage.
Scrotal ultrasound revealed no evidence of a testicular tumor. Lactate dehydrogenase (LDH) was 179 IU/L (normal, 120-240 IU/L) and prostate specific antigen (PSA) was 0.7 ng/mL (normal, <2.5 ng/mL). The patient was given amlodipine and labetalol with improvement of blood pressures to 160s/100s. His creatinine decreased to 1.1 mg/dL. He underwent CT-guided biopsy of a pelvic lymph node. CT of the head without intravenous contrast demonstrated no intracranial abnormalities. His headache resolved with improvement in blood pressure, and he had minimal gastrointestinal symptoms during his hospitalization. No stool studies were sent. A right-sided percutaneous nephrostomy was placed which yielded >15 L of urine from the tube over the next four days.
Upon the first episode of micturition through the urethra four days after percutaneous nephrostomy placement, he experienced severe lightheadedness, diaphoresis, and palpitations. These symptoms prompted him to recall similar episodes following micturition for several months prior to his hospitalization.
It is likely that contraction of the bladder during episodes of urination caused irritation of the pelvic mass, leading to catecholamine secretion. Another explanation for his recurrent lightheadedness would be a neurocardiogenic reflex with micturition (which when it culminates with loss of consciousness is called micturition syncope), but this would not explain his hypertension or bladder mass.
Biochemical tests that were ordered on admission but sent to a reference lab then returned. Plasma metanephrine was 0.2 nmol/L (normal, <0.5 nmol/L) and plasma normetanephrine 34.6 nmol/L (normal, <0.9 nmol/L). His 24-hour urine metanephrine was 72 ug/24 hr (normal, 0-300 ug/24 hr) and normetanephrine 8,511 ug/24 hr (normal, 50-800 ug/24 hr).
The markedly elevated plasma and urine normetanephrine levels confirm a diagnosis of a catecholamine-secreting tumor (paraganglioma). The tissue obtained from the CT-guided lymph node biopsy should be sent for markers of neuroendocrine tumors including chromogranin.
Lymph node biopsy revealed metastatic paraganglioma that was chromogranin A and synaptophysin positive (Figure 2). A fluorodeoxyglucose positron emission tomography (FDG-PET) scan disclosed skull metastases. He was treated with phenoxybenzamine, amlodipine, and labetalol. Surgical resection of the pelvic mass was discussed, but the patient elected to defer surgery as the location of the primary tumor made it challenging to resect and would have required an ileal conduit.
After the diagnosis was made, the patient’s family recalled that a maternal uncle had been diagnosed with a paraganglioma of the carotid body. Genetic testing of the patient identified a succinate dehydrogenase complex subunit B (SDHB) pathogenic variant and confirmed hereditary paraganglioma syndrome (HPGL). One year after the diagnosis, liver and lung metastases developed. He was treated with lanreotide (somatostatin analogue), capecitabine, and temozolomide, as well as a craniotomy and radiotherapy for palliation of bony metastases. The patient died less than two years after diagnosis.
DISCUSSION
Most patients with hypertension (defined as blood pressure >130/80 mm Hg1) do not have an identifiable etiology (primary hypertension). Many components of this patient’s history, however, including his young age of onset, a teenage sibling with hypertension, lack of obesity, hypertension refractory to multiple medications, and LVH suggested secondary hypertension. Hypertension onset at an age less than 30 years, resistance to three or more medications,1,2 and/or acute onset hypertension at any age should prompt an evaluation for secondary causes.1 The prevalence of secondary hypertension is approximately 30% in hypertensive patients ages 18 to 40 years compared with 5%-10% in the overall adult population with hypertension.3 Among children and adolescents ages 0 to 19 years with hypertension, the prevalence of secondary hypertension may be as high as 57%.4
The most common etiology of secondary hypertension is primary aldosteronism.5,6 However, in young adults (ages 19 to 39 years), common etiologies also include renovascular disease and renal parenchymal disease.7 Other causes include obstructive sleep apnea, medications, stimulants (cocaine and amphetamines),8 and endocrinopathies such as thyrotoxicosis, Cushing syndrome, and catecholamine-secreting tumors.7 Less than 1% of secondary hypertension in all adults is due to catecholamine-secreting tumors, and the minority of those catecholamine-secreting tumors are paragangliomas.9
Paragangliomas are tumors of the peripheral autonomic nervous system. These neoplasms arise in the sympathetic and parasympathetic chains along the paravertebral and paraaortic axes. They are closely related to pheochromocytomas, which arise in the adrenal medulla.9 Most head and neck paragangliomas are biochemically silent and are generally discovered due to mass effect.10 The subset of paragangliomas that secrete catecholamines most often arise in the abdomen and pelvis, and their clinical presentation mimics that of pheochromocytomas, including episodic hypertension, palpitations, pallor, and diaphoresis.
This patient had persistent, nonepisodic hypertension, while palpitations and diaphoresis only manifested following micturition. Other cases of urinary bladder paragangliomas have described micturition-associated symptoms and hypertensive crises. Three-fold increases of catecholamine secretion after micturition have been observed in these patients, likely due to muscle contraction and pressure changes in the bladder leading to the systemic release of catecholamines.11
Epinephrine and norepinephrine are monoamine neurotransmitters that activate alpha-adrenergic and beta-adrenergic receptors. Adrenergic receptors are present in all tissues of the body but have prominent effects on the smooth muscle in the vasculature, gastrointestinal tract, urinary tract, and airways.12 Alpha-adrenergic vasoconstriction causes hypertension, which is commonly observed in patients with catecholamine-secreting tumors.10 Catecholamine excess due to secretion from these tumors causes headache in 60%-80% of patients, tachycardia/palpitations in 50%-70%, anxiety in 20%-40%, and nausea in 20%-25%.10 Other symptoms include sweating, pallor, dyspnea, and vertigo.9,10 This patient’s chronic nausea, which was attributed to gastroesophageal reflux, and his anxiety, attributed to generalized anxiety disorder, were likely symptoms of catecholamine excess.13
The best test for the diagnosis of paragangliomas and pheochromocytomas is the measurement of plasma free or 24-hour urinary fractionated metanephrines (test sensitivity of >90% and >90%, respectively).14 Screening for pheochromocytoma should be considered in hypertensive patients who have symptoms of catecholamine excess, refractory or paroxysmal hypertension, and/or familial pheochromocytoma/paraganglioma syndromes.15 Screening for pheochromocytoma should also be performed in children and adolescents with systolic or diastolic blood pressure that is greater than the 95th percentile for their age plus 5 mm Hg.16
While a typical tumor location and elevated metanephrine levels are sufficient to make the diagnosis of a pheochromocytoma or catecholamine-secreting paraganglioma, functional imaging with FDG-PET, Ga-DOTATATE-PET, or 123I-meta-iodobenzylguanidine (123I-MIBG) can further confirm the diagnosis and detect distant metastases. However, imaging has low sensitivity for these tumors and thus should only be considered for patients in whom metastatic disease is suspected.14 Biopsy is rarely needed and should be reserved for unusual metastatic locations. Treatment with an alpha-adrenergic receptor antagonist often reduces symptoms and lowers blood pressure. Definitive management typically involves surgical resection for benign disease. Surgery, radionuclide therapy, or chemotherapy is used for malignant disease.
While most pheochromocytomas are sporadic, up to 40% of paragangliomas are due to germline pathogenic variants.17 Mutations in the succinate dehydrogenase (SDH) group of genes are the most common germline pathogenic variants in the autosomal dominant hereditary paraganglioma syndrome (HPGL). Most paragangliomas and pheochromocytomas are localized and benign, but 10%-15% are metastatic.18 SDHB mutations are associated with a high risk of metastasis.19 Thus, genetic testing for patients and subsequent cascade testing to identify at-risk family members is advised in all patients with pheochromocytomas or paragangliomas.20 This patient’s younger brother and mother were both found to carry the same pathogenic SDHB variant, but neither was found to have paragangliomas. Annual metanephrine levels (urine or plasma) and every other year whole-body magnetic resonance imaging (MRI) scans were recommended for tumor surveillance.
The clinician team followed a logical branching algorithm for the diagnosis of severe hypertension with biochemical testing, advanced imaging, histology, and genetic testing to arrive at the final diagnosis of hereditary paraganglioma syndrome. Although this patient presented for urgent care because of the acute effects of catecholamine excess, he suffered from chronic effects (nausea, anxiety, and hypertension) for years. Each symptom had been diagnosed and treated in isolation, but the combination and severity in a young patient suggested a unifying diagnosis. The family history of hypertension (brother and father) suggested an inherited diagnosis from the father’s family, but the final answer rested on the other branch (maternal uncle) of the family tree.
KEY TEACHING POINTS
- Hypertension in a young adult is due to a secondary cause in up to 30% of patients.
- Pathologic catecholamine excess leads to hypertension, tachycardia, pallor, sweating, anxiety, and nausea. A sustained and unexplained combination of these symptoms should prompt a biochemical evaluation for pheochromocytoma or paraganglioma.
- Paragangliomas are tumors of the autonomic nervous system. The frequency of catecholamine secretion depends on their location in the body, and they are commonly caused by germline pathogenic variants.
Acknowledgments
This conundrum was presented during a live Grand Rounds with the expert clinician’s responses recorded and edited for space and clarity.
Disclosures
Dr. Dhaliwal reports speaking honoraria from ISMIE Mutual Insurance Company and GE Healthcare. All other authors have nothing to disclose.
Funding
No sources of funding.
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13-e115. https://doi.org/10.1161/HYP.0000000000000065.
2. Acelajado MC, Calhoun DA. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin. 2010;28(4):639-654. https://doi.org/10.1016/j.ccl.2010.07.002.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206-1252. https://doi.org/10.1161/01.HYP.0000107251.49515.c2.
4. Gupta-Malhotra M, Banker A, Shete S, et al. Essential hypertension vs. secondary hypertension among children. Am J Hypertens. 2015;28(1):73-80. https://doi.org/10.1093/ajh/hpu083.
5. Mosso L, Carvajal C, Gonzalez A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42(2):161-165. https://doi.org/10.1161/01.HYP.0000079505.25750.11.
6. Kayser SC, Dekkers T, Groenewoud HJ, et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab. 2016;101(7):2826-2835. https://doi.org/10.1210/jc.2016-1472.
7. Charles L, Triscott J, Dobbs B. Secondary hypertension: discovering the underlying cause. Am Fam Physician. 2017;96(7):453-461.
8. Aronow WS. Drug-induced causes of secondary hypertension. Ann Transl Med. 2017;5(17):349. https://doi.org/10.21037/atm.2017.06.16.
9. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675. https://doi.org/10.1016/S0140-6736(05)67139-5.
10. Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26(4):507-515. https://doi.org/10.1016/j.beem.2011.10.008.
11. Kappers MH, van den Meiracker AH, Alwani RA, Kats E, Baggen MG. Paraganglioma of the urinary bladder. Neth J Med. 2008;66(4):163-165.
12. Paravati S, Warrington SJ. Physiology, Catecholamines. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC; 2019.
13. King KS, Darmani NA, Hughes MS, Adams KT, Pacak K. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37(3):403-407. https://doi.org/10.1007/s12020-010-9319-3.
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. https://doi.org/10.1210/jc.2014-1498.
15. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). 2017;32(2):152-161. https://doi.org/10.3803/EnM.2017.32.2.152.
16. National High Blood Pressure Education Program Working Group. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555-576.
17. Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 1993.
18. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756. https://doi.org/10.1097/00000658-199906000-00001.
19. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822-3828. https://doi.org/10.1210/jc.2007-0709.
20. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111. https://doi.org/10.1038/nrendo.2014.188.
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13-e115. https://doi.org/10.1161/HYP.0000000000000065.
2. Acelajado MC, Calhoun DA. Resistant hypertension, secondary hypertension, and hypertensive crises: diagnostic evaluation and treatment. Cardiol Clin. 2010;28(4):639-654. https://doi.org/10.1016/j.ccl.2010.07.002.
3. Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42(6):1206-1252. https://doi.org/10.1161/01.HYP.0000107251.49515.c2.
4. Gupta-Malhotra M, Banker A, Shete S, et al. Essential hypertension vs. secondary hypertension among children. Am J Hypertens. 2015;28(1):73-80. https://doi.org/10.1093/ajh/hpu083.
5. Mosso L, Carvajal C, Gonzalez A, et al. Primary aldosteronism and hypertensive disease. Hypertension. 2003;42(2):161-165. https://doi.org/10.1161/01.HYP.0000079505.25750.11.
6. Kayser SC, Dekkers T, Groenewoud HJ, et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab. 2016;101(7):2826-2835. https://doi.org/10.1210/jc.2016-1472.
7. Charles L, Triscott J, Dobbs B. Secondary hypertension: discovering the underlying cause. Am Fam Physician. 2017;96(7):453-461.
8. Aronow WS. Drug-induced causes of secondary hypertension. Ann Transl Med. 2017;5(17):349. https://doi.org/10.21037/atm.2017.06.16.
9. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665-675. https://doi.org/10.1016/S0140-6736(05)67139-5.
10. Mannelli M, Lenders JW, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26(4):507-515. https://doi.org/10.1016/j.beem.2011.10.008.
11. Kappers MH, van den Meiracker AH, Alwani RA, Kats E, Baggen MG. Paraganglioma of the urinary bladder. Neth J Med. 2008;66(4):163-165.
12. Paravati S, Warrington SJ. Physiology, Catecholamines. In: StatPearls. Treasure Island, FL: StatPearls Publishing LLC; 2019.
13. King KS, Darmani NA, Hughes MS, Adams KT, Pacak K. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37(3):403-407. https://doi.org/10.1007/s12020-010-9319-3.
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. https://doi.org/10.1210/jc.2014-1498.
15. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). 2017;32(2):152-161. https://doi.org/10.3803/EnM.2017.32.2.152.
16. National High Blood Pressure Education Program Working Group. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2):555-576.
17. Else T, Greenberg S, Fishbein L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 1993.
18. Goldstein RE, O’Neill JA, Jr., Holcomb GW, 3rd, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755-764; discussion 764-756. https://doi.org/10.1097/00000658-199906000-00001.
19. Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822-3828. https://doi.org/10.1210/jc.2007-0709.
20. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101-111. https://doi.org/10.1038/nrendo.2014.188.
© 2019 Society of Hospital Medicine
Clinical Progress Note: Addressing Prognosis in Advanced Dementia
Advanced dementia (AD) is a serious terminal illness. Some features of AD include significant memory deficits (inability to recognize family members), inability to ambulate, very limited verbal communication, and needing assistance with all activities of daily living.1 AD carries a six-month mortality of 25% and a median survival of 1.3 years.1
Despite a limited life expectancy, patients with AD face increasingly significant symptom burden and use of burdensome interventions2 near the end of life. Among the most common interventions hospitalists routinely navigate during a hospitalization is tube feeding for enteral artificial nutrition, which has not been shown to prolong survival, improve quality of life, decrease risk of aspiration pneumonia, or decrease the risk of pressure ulcers.3,4 Recent data show that rates of hospitalizations in the last 90 days of life, especially in the last three days of life, are increasing.5 These late transitions can have significant negative impact on family perceptions of quality of care, including not being treated with respect, receiving care inconsistent with goals, receiving inadequate communication about care decisions, and not being fully informed of the medical conditions.6
Therefore, hospitalization in AD, especially a readmission, indicates a critical change in a patient’s illness, marking an opportune time to have discussions on prognosis and improve care at the end of life. While determining and sharing prognosis can be challenging in the setting of many chronic diseases, resources exist to help clinicians share prognosis in AD and understand the goals of care for each patient.7 The aim of this paper is to assist hospitalists in addressing prognosis in the setting of AD. We identify and present key knowledge and recommendations from relevant articles identified from a hand-search of articles, published in 2018, from leading palliative care journals, as well as a MEDLINE search from 2003 through December 2018 using the key words “dementia” and “prognosis.” Final presented articles and recommendations were determined based on scientific rigor and relevance to hospital-based care of patients with AD.
IMPORTANCE OF PROGNOSIS DISCUSSIONS IN ADVANCED DEMENTIA
For a myriad of reasons, most AD caregivers do not receive adequate information on the complications of dementia or prognosis.2 Conversations that provide prognostic estimates and aim to understand the goals, preferences, and values of AD patients and their surrogates can help in providing goal-concordant care. A prospective study of nursing-home patients with AD showed that having goals of care discussions was strongly associated with surrogates’ likelihood of estimating a life expectancy of less than six months in AD patients.8 Having this perception was associated with a lower likelihood of patients with AD undergoing burdensome interventions such as hospitalizations, parenteral therapy, venipuncture, feeding tube, or urinary catheterization.8 To help improve goal-concordant care, it is important that hospitalists be prepared to have prognostic conversations with patients and their caregivers.
“FORESEEING” PROGNOSIS IN ADVANCED DEMENTIA
Offering a clinical prognosis involves components of foreseeing (estimating prognosis) and foretelling (sharing prognosis).9 Foreseeing prognosis in AD can be complex due to the highly variable but slow, dwindling clinical course of AD. As a practical matter, determining if a patient has a six-month prognosis is most helpful as eligibility for hospice services may allow for a discharge to a supportive home setting instead of a transfer to an institution.10 An evidence-based clinical prediction rule, the Advanced Dementia Prognostic Tool (ADEPT, Appendix Table),11 can be used to estimate prognosis by a composite of 12 risk factors in nursing-home patients. Although the consensus-based National Hospice and Palliative Care Organization (NHPCO) guidelines for Medicare hospice eligibility12 (Table) do not perform well in predicting individual mortality, they are used as criteria for hospice enrollment. Given the variability of course of AD, evaluating the mortality risk for acute illnesses leading to hospitalization, like pneumonia or hip fracture, can further help estimate prognosis. The website, www.eprognosis.com, combines various prediction tools to help estimate prognosis. Although using these tools can often help clinicians satisfy the entry requirements to offer hospice, the ADEPT tool and the NHPCO criteria both perform poorly in discriminating those who will or will not actually die in six months. ADEPT, as a prognostic tool, has not yet been validated for community-dwelling patients. Clinicians should exercise caution in making a highly specific estimate of survival in AD, but can and should communicate the expected decline in function over time.
“FORETELLING” PROGNOSIS IN ADVANCED DEMENTIA
Having goals of care conversations and sharing prognosis has many benefits. A large multistate cohort study showed that having goals of care conversations among patients with terminal cancer was associated with less use of intensive care units, mechanical ventilation, and cardio-pulmonary resuscitation.13 Caregivers may also benefit from prognosis discussions through identifying resources to care for the patient at home and by potentially limiting their risk of major depressive order and regret, common among those witnessing patients undergoing aggressive treatment at the end of life.13 How to share prognosis can be challenging; however, tools such as the Serious Illness Conversation Guide14 can provide step-by-step guidance for providers. The key aspects of the guide are asking permission, assessing illness understanding, and exploring goals, fears, worries, and tradeoffs.
Before exploring goals, it is helpful to explain the serious illness by using “I wish,” “I worry,” and “I wonder” statements such as “I wish I had better news for you; your mom’s dementia has progressed given the recent complication of aspiration,” “I worry that she will not be able to eat on her own and will develop another serious infection very soon,” or “I wonder whether it is a good time to talk about what your mom would want if she cannot eat on her own.”
An example of an effective conversation about artificial nutrition and hydration with a surrogate of a patient with AD with recurrent aspirations may include the following elements:14,15
- Obtain the caregiver’s and/or patient’s perception of illness: “Is it OK if we have a conversation about what may lie ahead with your mother? Is there anyone else that should be present? What is your understanding of your mother’s illness?”
- Give relevant data: “Based on her current level of decline with complications and repetitive hospitalizations, I am worried that her life expectancy is likely measured in months rather than years.”
- Address emotions: “This must be very hard to hear. I cannot imagine how difficult it must be to see her in the hospital so often.”
- Elicit concerns and goals based on understanding key values: “Tell me what you are hoping for regarding your mother’s future care and what worries you have. Tell me what your mother would say if she could fully understand her current situation.”
- Present goals based on patient and caregiver values: “Based on what you have told me about your mother, she valued her interactions with family and her independence, and she would not want measures that would cause distress, especially when facing a terminal illness.”
- Be mindful of prognostic uncertainty: “While we cannot know for certain what will happen next, I am very worried that your mother will continue to aspirate even with a feeding tube.”
- Make a recommendation with permission: “From our conversation, I have an idea of what treatment might make sense to your mom. May I share my recommendation with you?” If they are willing, you might say: “As evidence shows that feeding tubes do not improve the level of family interaction or independence in patients with dementia and as your mother would not want any distressing procedures, I recommend that we do not place a feeding-tube.”
- Balance realism and hope: “Instead, we can focus on other ways to maintain dignity and quality of life for her even without a feeding tube.”
RESPONDING TO CHALLENGES
Conversations about goals of care and prognosis can be challenging and time consuming. At times, the conversations can be strained. The following tips are based on authors’ shared experiences to help in those challenging situations:
- Caregivers may show signs of emotional and/or cognitive strain: Recognize and name the emotional response and consider asking the family if they need a break to avoid overtaxing them.
“I can see that this is very difficult for you. Do you want to take a break and meet again?”
- Caregivers may have unrealistic hopes: Confirm the caregivers’ understanding of the situation, before assuming their hope is unrealistic. Try to reframe what they/we can hope for by validating their goals while avoiding unnecessary burdens or discomfort.
“I want to be sure that I have explained your mom’s situation clearly. Can you tell me in your words, what I have told you?” as this gives you an opportunity to clarify misunderstandings that may manifest as “false hope”.
“Together we can hope for the best and see if your mother can tolerate hand-feeding safely without causing any harm or distress.”
- Avoid assumptions about cultural and religious beliefs: Be curious and demonstrate cultural humility to all patients.
“Are there any cultural or spiritual beliefs that are important to you or your mother?”
- Avoid spending too much time on clinical details: Give families time to share stories about the patient in better days as this gives you an opportunity to get to know the patient.
“Tell me more about what your mother was like when she was healthy.”
- Listen first, recommend second: Refrain from making recommendations about the patient’s care before you understand his/her values and preferences.
“What would your mother say is most important to her as her health worsens?”
- Use active listening techniques: Using reflection statements can confirm your understanding of the caregiver’s view point.
“So, I hear that your mother valued being at home and being comfortable. Is that correct?”
These conversations are often an iterative process of helping the patient and family traverse the course of AD. Therefore, starting the process even during a hospitalization earlier in the course of AD can help engage in preparedness planning to provide goal-concordant care and help optimize the patient’s quality of life.
CONCLUSION
Hospitalization among patients with AD can signal a significant change in prognosis and represents an important opportunity for further dialogue. A patient- and caregiver-centered conversation, sharing prognosis and learning about values important to the patient and family, has the potential to lead to less burdensome interventions. Doing so can minimize harm, promote quality of life, and reduce unnecessary care transitions near the end of life.
Disclosures
The authors have nothing to disclose.
Funding
Dr. Havyer was supported, in part, by the Mayo Clinic Department of Medicine Catalyst for Advancing in Academics grant. Dr. Abedini was supported by the National Clinician Scholars Program at the Institute for Healthcare Policy and Innovation, University of Michigan, Ann Arbor, MI.
1. Mitchell SL. Advanced dementia. N Engl J Med. 2015;373(13):1276-1277. https://doi.org/10.1056/NEJMcp1412652.
2. Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009;361(16):1529-1538. https://doi.org/10.1056/NEJMoa0902234.
3. Teno JM, Gozalo PL, Mitchell SL, et al. Does feeding tube insertion and its timing improve survival? J Am Geriatr Soc. 2012;60(10):1918-1921. https://doi.org/10.1111/j.1532-5415.2012.04148.x.
4. Teno JM, Gozalo P, Mitchell SL, Kuo S, Fulton AT, Mor V. Feeding tubes and the prevention or healing of pressure ulcers. Arch Intern Med. 2012;172(9):697-701. https://doi.org/10.1001/archinternmed.2012.1200.
5. Teno JM, Gozalo PL, Bynum JP, et al. Change in end-of-life care for medicare beneficiaries: site of death, place of care, and health care transitions in 2000, 2005, and 2009. JAMA. 2013;309(5):470-477. https://doi.org/10.1001/jama.2012.207624.
6. Makaroun LK, Teno JM, Freedman VA, Kasper JD, Gozalo P, Mor V. Late transitions and bereaved family member perceptions of quality of end-of-life care. J Am Geriatr Soc. 2018;66(9):1730-1736. https://doi.org/10.1111/jgs.15455.
7. Ansari AA, Pomerantz DH, Jayes RL, Aguirre EA, Havyer RD. Promoting primary palliative care in severe chronic obstructive pulmonary disease: symptom management and preparedness planning. J Palliat Care. 2019;34(2):85-91. https://doi.org/10.1177/0825859718819437.
8. Loizeau AJ, Shaffer ML, Habtemariam DA, Hanson LC, Volandes AE, Mitchell SL. Association of prognostic estimates with burdensome interventions in nursing home residents with advanced dementia. JAMA Intern Med. 2018;178(7):922-929. https://doi.org/10.1001/jamainternmed.2018.1413.
9. Glare PA, Sinclair CT. Palliative medicine review: Prognostication. J Palliat Med. 2008;11(1):84-103. https://doi.org/10.1089/jpm.2008.9992.
10. Jayes RL, Arnold RM, Fromme EK. Does this dementia patient meet the prognosis eligibility requirements for hospice enrollment? J Pain Symptom Manage. 2012;44(5):750-756. https://doi.org/10.1016/j.jpainsymman.2012.08.004.
11. Mitchell SL, Miller SC, Teno JM, Kiely DK, Davis RB, Shaffer ML. Prediction of 6-month survival of nursing home residents with advanced dementia using ADEPT vs hospice eligibility guidelines. JAMA. 2010;304(17):1929-1935. https://doi.org/10.1001/jama.2010.1572.
12. Schonwetter RS, Han B, Small BJ, et al. Predictors of six-month survival among patients with dementia: an evaluation of hospice Medicare guidelines. Amer J Hospice & Pall Care. 2003;20(2):105-113. https://doi.org/10.1177/104990910302000208
13. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment. JAMA. 2008;300(14):1665-1673. https://doi.org/10.1001/jama.300.14.1665.
14. Bernacki R, Hutchings M, Vick J, et al. Development of the serious illness care program: a randomised controlled trial of a palliative care communication intervention. BMJ Open. 2015;5(10):e009032-2015-009032. https://doi.org/10.1136/bmjopen-2015-009032
15. Ansari A, Pomerantz D, Smith K. Being mindful: difficult decisions in advanced dementia and end stage renal disease. SGIM Forum. 2017;40(3):4, 13.
Advanced dementia (AD) is a serious terminal illness. Some features of AD include significant memory deficits (inability to recognize family members), inability to ambulate, very limited verbal communication, and needing assistance with all activities of daily living.1 AD carries a six-month mortality of 25% and a median survival of 1.3 years.1
Despite a limited life expectancy, patients with AD face increasingly significant symptom burden and use of burdensome interventions2 near the end of life. Among the most common interventions hospitalists routinely navigate during a hospitalization is tube feeding for enteral artificial nutrition, which has not been shown to prolong survival, improve quality of life, decrease risk of aspiration pneumonia, or decrease the risk of pressure ulcers.3,4 Recent data show that rates of hospitalizations in the last 90 days of life, especially in the last three days of life, are increasing.5 These late transitions can have significant negative impact on family perceptions of quality of care, including not being treated with respect, receiving care inconsistent with goals, receiving inadequate communication about care decisions, and not being fully informed of the medical conditions.6
Therefore, hospitalization in AD, especially a readmission, indicates a critical change in a patient’s illness, marking an opportune time to have discussions on prognosis and improve care at the end of life. While determining and sharing prognosis can be challenging in the setting of many chronic diseases, resources exist to help clinicians share prognosis in AD and understand the goals of care for each patient.7 The aim of this paper is to assist hospitalists in addressing prognosis in the setting of AD. We identify and present key knowledge and recommendations from relevant articles identified from a hand-search of articles, published in 2018, from leading palliative care journals, as well as a MEDLINE search from 2003 through December 2018 using the key words “dementia” and “prognosis.” Final presented articles and recommendations were determined based on scientific rigor and relevance to hospital-based care of patients with AD.
IMPORTANCE OF PROGNOSIS DISCUSSIONS IN ADVANCED DEMENTIA
For a myriad of reasons, most AD caregivers do not receive adequate information on the complications of dementia or prognosis.2 Conversations that provide prognostic estimates and aim to understand the goals, preferences, and values of AD patients and their surrogates can help in providing goal-concordant care. A prospective study of nursing-home patients with AD showed that having goals of care discussions was strongly associated with surrogates’ likelihood of estimating a life expectancy of less than six months in AD patients.8 Having this perception was associated with a lower likelihood of patients with AD undergoing burdensome interventions such as hospitalizations, parenteral therapy, venipuncture, feeding tube, or urinary catheterization.8 To help improve goal-concordant care, it is important that hospitalists be prepared to have prognostic conversations with patients and their caregivers.
“FORESEEING” PROGNOSIS IN ADVANCED DEMENTIA
Offering a clinical prognosis involves components of foreseeing (estimating prognosis) and foretelling (sharing prognosis).9 Foreseeing prognosis in AD can be complex due to the highly variable but slow, dwindling clinical course of AD. As a practical matter, determining if a patient has a six-month prognosis is most helpful as eligibility for hospice services may allow for a discharge to a supportive home setting instead of a transfer to an institution.10 An evidence-based clinical prediction rule, the Advanced Dementia Prognostic Tool (ADEPT, Appendix Table),11 can be used to estimate prognosis by a composite of 12 risk factors in nursing-home patients. Although the consensus-based National Hospice and Palliative Care Organization (NHPCO) guidelines for Medicare hospice eligibility12 (Table) do not perform well in predicting individual mortality, they are used as criteria for hospice enrollment. Given the variability of course of AD, evaluating the mortality risk for acute illnesses leading to hospitalization, like pneumonia or hip fracture, can further help estimate prognosis. The website, www.eprognosis.com, combines various prediction tools to help estimate prognosis. Although using these tools can often help clinicians satisfy the entry requirements to offer hospice, the ADEPT tool and the NHPCO criteria both perform poorly in discriminating those who will or will not actually die in six months. ADEPT, as a prognostic tool, has not yet been validated for community-dwelling patients. Clinicians should exercise caution in making a highly specific estimate of survival in AD, but can and should communicate the expected decline in function over time.
“FORETELLING” PROGNOSIS IN ADVANCED DEMENTIA
Having goals of care conversations and sharing prognosis has many benefits. A large multistate cohort study showed that having goals of care conversations among patients with terminal cancer was associated with less use of intensive care units, mechanical ventilation, and cardio-pulmonary resuscitation.13 Caregivers may also benefit from prognosis discussions through identifying resources to care for the patient at home and by potentially limiting their risk of major depressive order and regret, common among those witnessing patients undergoing aggressive treatment at the end of life.13 How to share prognosis can be challenging; however, tools such as the Serious Illness Conversation Guide14 can provide step-by-step guidance for providers. The key aspects of the guide are asking permission, assessing illness understanding, and exploring goals, fears, worries, and tradeoffs.
Before exploring goals, it is helpful to explain the serious illness by using “I wish,” “I worry,” and “I wonder” statements such as “I wish I had better news for you; your mom’s dementia has progressed given the recent complication of aspiration,” “I worry that she will not be able to eat on her own and will develop another serious infection very soon,” or “I wonder whether it is a good time to talk about what your mom would want if she cannot eat on her own.”
An example of an effective conversation about artificial nutrition and hydration with a surrogate of a patient with AD with recurrent aspirations may include the following elements:14,15
- Obtain the caregiver’s and/or patient’s perception of illness: “Is it OK if we have a conversation about what may lie ahead with your mother? Is there anyone else that should be present? What is your understanding of your mother’s illness?”
- Give relevant data: “Based on her current level of decline with complications and repetitive hospitalizations, I am worried that her life expectancy is likely measured in months rather than years.”
- Address emotions: “This must be very hard to hear. I cannot imagine how difficult it must be to see her in the hospital so often.”
- Elicit concerns and goals based on understanding key values: “Tell me what you are hoping for regarding your mother’s future care and what worries you have. Tell me what your mother would say if she could fully understand her current situation.”
- Present goals based on patient and caregiver values: “Based on what you have told me about your mother, she valued her interactions with family and her independence, and she would not want measures that would cause distress, especially when facing a terminal illness.”
- Be mindful of prognostic uncertainty: “While we cannot know for certain what will happen next, I am very worried that your mother will continue to aspirate even with a feeding tube.”
- Make a recommendation with permission: “From our conversation, I have an idea of what treatment might make sense to your mom. May I share my recommendation with you?” If they are willing, you might say: “As evidence shows that feeding tubes do not improve the level of family interaction or independence in patients with dementia and as your mother would not want any distressing procedures, I recommend that we do not place a feeding-tube.”
- Balance realism and hope: “Instead, we can focus on other ways to maintain dignity and quality of life for her even without a feeding tube.”
RESPONDING TO CHALLENGES
Conversations about goals of care and prognosis can be challenging and time consuming. At times, the conversations can be strained. The following tips are based on authors’ shared experiences to help in those challenging situations:
- Caregivers may show signs of emotional and/or cognitive strain: Recognize and name the emotional response and consider asking the family if they need a break to avoid overtaxing them.
“I can see that this is very difficult for you. Do you want to take a break and meet again?”
- Caregivers may have unrealistic hopes: Confirm the caregivers’ understanding of the situation, before assuming their hope is unrealistic. Try to reframe what they/we can hope for by validating their goals while avoiding unnecessary burdens or discomfort.
“I want to be sure that I have explained your mom’s situation clearly. Can you tell me in your words, what I have told you?” as this gives you an opportunity to clarify misunderstandings that may manifest as “false hope”.
“Together we can hope for the best and see if your mother can tolerate hand-feeding safely without causing any harm or distress.”
- Avoid assumptions about cultural and religious beliefs: Be curious and demonstrate cultural humility to all patients.
“Are there any cultural or spiritual beliefs that are important to you or your mother?”
- Avoid spending too much time on clinical details: Give families time to share stories about the patient in better days as this gives you an opportunity to get to know the patient.
“Tell me more about what your mother was like when she was healthy.”
- Listen first, recommend second: Refrain from making recommendations about the patient’s care before you understand his/her values and preferences.
“What would your mother say is most important to her as her health worsens?”
- Use active listening techniques: Using reflection statements can confirm your understanding of the caregiver’s view point.
“So, I hear that your mother valued being at home and being comfortable. Is that correct?”
These conversations are often an iterative process of helping the patient and family traverse the course of AD. Therefore, starting the process even during a hospitalization earlier in the course of AD can help engage in preparedness planning to provide goal-concordant care and help optimize the patient’s quality of life.
CONCLUSION
Hospitalization among patients with AD can signal a significant change in prognosis and represents an important opportunity for further dialogue. A patient- and caregiver-centered conversation, sharing prognosis and learning about values important to the patient and family, has the potential to lead to less burdensome interventions. Doing so can minimize harm, promote quality of life, and reduce unnecessary care transitions near the end of life.
Disclosures
The authors have nothing to disclose.
Funding
Dr. Havyer was supported, in part, by the Mayo Clinic Department of Medicine Catalyst for Advancing in Academics grant. Dr. Abedini was supported by the National Clinician Scholars Program at the Institute for Healthcare Policy and Innovation, University of Michigan, Ann Arbor, MI.
Advanced dementia (AD) is a serious terminal illness. Some features of AD include significant memory deficits (inability to recognize family members), inability to ambulate, very limited verbal communication, and needing assistance with all activities of daily living.1 AD carries a six-month mortality of 25% and a median survival of 1.3 years.1
Despite a limited life expectancy, patients with AD face increasingly significant symptom burden and use of burdensome interventions2 near the end of life. Among the most common interventions hospitalists routinely navigate during a hospitalization is tube feeding for enteral artificial nutrition, which has not been shown to prolong survival, improve quality of life, decrease risk of aspiration pneumonia, or decrease the risk of pressure ulcers.3,4 Recent data show that rates of hospitalizations in the last 90 days of life, especially in the last three days of life, are increasing.5 These late transitions can have significant negative impact on family perceptions of quality of care, including not being treated with respect, receiving care inconsistent with goals, receiving inadequate communication about care decisions, and not being fully informed of the medical conditions.6
Therefore, hospitalization in AD, especially a readmission, indicates a critical change in a patient’s illness, marking an opportune time to have discussions on prognosis and improve care at the end of life. While determining and sharing prognosis can be challenging in the setting of many chronic diseases, resources exist to help clinicians share prognosis in AD and understand the goals of care for each patient.7 The aim of this paper is to assist hospitalists in addressing prognosis in the setting of AD. We identify and present key knowledge and recommendations from relevant articles identified from a hand-search of articles, published in 2018, from leading palliative care journals, as well as a MEDLINE search from 2003 through December 2018 using the key words “dementia” and “prognosis.” Final presented articles and recommendations were determined based on scientific rigor and relevance to hospital-based care of patients with AD.
IMPORTANCE OF PROGNOSIS DISCUSSIONS IN ADVANCED DEMENTIA
For a myriad of reasons, most AD caregivers do not receive adequate information on the complications of dementia or prognosis.2 Conversations that provide prognostic estimates and aim to understand the goals, preferences, and values of AD patients and their surrogates can help in providing goal-concordant care. A prospective study of nursing-home patients with AD showed that having goals of care discussions was strongly associated with surrogates’ likelihood of estimating a life expectancy of less than six months in AD patients.8 Having this perception was associated with a lower likelihood of patients with AD undergoing burdensome interventions such as hospitalizations, parenteral therapy, venipuncture, feeding tube, or urinary catheterization.8 To help improve goal-concordant care, it is important that hospitalists be prepared to have prognostic conversations with patients and their caregivers.
“FORESEEING” PROGNOSIS IN ADVANCED DEMENTIA
Offering a clinical prognosis involves components of foreseeing (estimating prognosis) and foretelling (sharing prognosis).9 Foreseeing prognosis in AD can be complex due to the highly variable but slow, dwindling clinical course of AD. As a practical matter, determining if a patient has a six-month prognosis is most helpful as eligibility for hospice services may allow for a discharge to a supportive home setting instead of a transfer to an institution.10 An evidence-based clinical prediction rule, the Advanced Dementia Prognostic Tool (ADEPT, Appendix Table),11 can be used to estimate prognosis by a composite of 12 risk factors in nursing-home patients. Although the consensus-based National Hospice and Palliative Care Organization (NHPCO) guidelines for Medicare hospice eligibility12 (Table) do not perform well in predicting individual mortality, they are used as criteria for hospice enrollment. Given the variability of course of AD, evaluating the mortality risk for acute illnesses leading to hospitalization, like pneumonia or hip fracture, can further help estimate prognosis. The website, www.eprognosis.com, combines various prediction tools to help estimate prognosis. Although using these tools can often help clinicians satisfy the entry requirements to offer hospice, the ADEPT tool and the NHPCO criteria both perform poorly in discriminating those who will or will not actually die in six months. ADEPT, as a prognostic tool, has not yet been validated for community-dwelling patients. Clinicians should exercise caution in making a highly specific estimate of survival in AD, but can and should communicate the expected decline in function over time.
“FORETELLING” PROGNOSIS IN ADVANCED DEMENTIA
Having goals of care conversations and sharing prognosis has many benefits. A large multistate cohort study showed that having goals of care conversations among patients with terminal cancer was associated with less use of intensive care units, mechanical ventilation, and cardio-pulmonary resuscitation.13 Caregivers may also benefit from prognosis discussions through identifying resources to care for the patient at home and by potentially limiting their risk of major depressive order and regret, common among those witnessing patients undergoing aggressive treatment at the end of life.13 How to share prognosis can be challenging; however, tools such as the Serious Illness Conversation Guide14 can provide step-by-step guidance for providers. The key aspects of the guide are asking permission, assessing illness understanding, and exploring goals, fears, worries, and tradeoffs.
Before exploring goals, it is helpful to explain the serious illness by using “I wish,” “I worry,” and “I wonder” statements such as “I wish I had better news for you; your mom’s dementia has progressed given the recent complication of aspiration,” “I worry that she will not be able to eat on her own and will develop another serious infection very soon,” or “I wonder whether it is a good time to talk about what your mom would want if she cannot eat on her own.”
An example of an effective conversation about artificial nutrition and hydration with a surrogate of a patient with AD with recurrent aspirations may include the following elements:14,15
- Obtain the caregiver’s and/or patient’s perception of illness: “Is it OK if we have a conversation about what may lie ahead with your mother? Is there anyone else that should be present? What is your understanding of your mother’s illness?”
- Give relevant data: “Based on her current level of decline with complications and repetitive hospitalizations, I am worried that her life expectancy is likely measured in months rather than years.”
- Address emotions: “This must be very hard to hear. I cannot imagine how difficult it must be to see her in the hospital so often.”
- Elicit concerns and goals based on understanding key values: “Tell me what you are hoping for regarding your mother’s future care and what worries you have. Tell me what your mother would say if she could fully understand her current situation.”
- Present goals based on patient and caregiver values: “Based on what you have told me about your mother, she valued her interactions with family and her independence, and she would not want measures that would cause distress, especially when facing a terminal illness.”
- Be mindful of prognostic uncertainty: “While we cannot know for certain what will happen next, I am very worried that your mother will continue to aspirate even with a feeding tube.”
- Make a recommendation with permission: “From our conversation, I have an idea of what treatment might make sense to your mom. May I share my recommendation with you?” If they are willing, you might say: “As evidence shows that feeding tubes do not improve the level of family interaction or independence in patients with dementia and as your mother would not want any distressing procedures, I recommend that we do not place a feeding-tube.”
- Balance realism and hope: “Instead, we can focus on other ways to maintain dignity and quality of life for her even without a feeding tube.”
RESPONDING TO CHALLENGES
Conversations about goals of care and prognosis can be challenging and time consuming. At times, the conversations can be strained. The following tips are based on authors’ shared experiences to help in those challenging situations:
- Caregivers may show signs of emotional and/or cognitive strain: Recognize and name the emotional response and consider asking the family if they need a break to avoid overtaxing them.
“I can see that this is very difficult for you. Do you want to take a break and meet again?”
- Caregivers may have unrealistic hopes: Confirm the caregivers’ understanding of the situation, before assuming their hope is unrealistic. Try to reframe what they/we can hope for by validating their goals while avoiding unnecessary burdens or discomfort.
“I want to be sure that I have explained your mom’s situation clearly. Can you tell me in your words, what I have told you?” as this gives you an opportunity to clarify misunderstandings that may manifest as “false hope”.
“Together we can hope for the best and see if your mother can tolerate hand-feeding safely without causing any harm or distress.”
- Avoid assumptions about cultural and religious beliefs: Be curious and demonstrate cultural humility to all patients.
“Are there any cultural or spiritual beliefs that are important to you or your mother?”
- Avoid spending too much time on clinical details: Give families time to share stories about the patient in better days as this gives you an opportunity to get to know the patient.
“Tell me more about what your mother was like when she was healthy.”
- Listen first, recommend second: Refrain from making recommendations about the patient’s care before you understand his/her values and preferences.
“What would your mother say is most important to her as her health worsens?”
- Use active listening techniques: Using reflection statements can confirm your understanding of the caregiver’s view point.
“So, I hear that your mother valued being at home and being comfortable. Is that correct?”
These conversations are often an iterative process of helping the patient and family traverse the course of AD. Therefore, starting the process even during a hospitalization earlier in the course of AD can help engage in preparedness planning to provide goal-concordant care and help optimize the patient’s quality of life.
CONCLUSION
Hospitalization among patients with AD can signal a significant change in prognosis and represents an important opportunity for further dialogue. A patient- and caregiver-centered conversation, sharing prognosis and learning about values important to the patient and family, has the potential to lead to less burdensome interventions. Doing so can minimize harm, promote quality of life, and reduce unnecessary care transitions near the end of life.
Disclosures
The authors have nothing to disclose.
Funding
Dr. Havyer was supported, in part, by the Mayo Clinic Department of Medicine Catalyst for Advancing in Academics grant. Dr. Abedini was supported by the National Clinician Scholars Program at the Institute for Healthcare Policy and Innovation, University of Michigan, Ann Arbor, MI.
1. Mitchell SL. Advanced dementia. N Engl J Med. 2015;373(13):1276-1277. https://doi.org/10.1056/NEJMcp1412652.
2. Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009;361(16):1529-1538. https://doi.org/10.1056/NEJMoa0902234.
3. Teno JM, Gozalo PL, Mitchell SL, et al. Does feeding tube insertion and its timing improve survival? J Am Geriatr Soc. 2012;60(10):1918-1921. https://doi.org/10.1111/j.1532-5415.2012.04148.x.
4. Teno JM, Gozalo P, Mitchell SL, Kuo S, Fulton AT, Mor V. Feeding tubes and the prevention or healing of pressure ulcers. Arch Intern Med. 2012;172(9):697-701. https://doi.org/10.1001/archinternmed.2012.1200.
5. Teno JM, Gozalo PL, Bynum JP, et al. Change in end-of-life care for medicare beneficiaries: site of death, place of care, and health care transitions in 2000, 2005, and 2009. JAMA. 2013;309(5):470-477. https://doi.org/10.1001/jama.2012.207624.
6. Makaroun LK, Teno JM, Freedman VA, Kasper JD, Gozalo P, Mor V. Late transitions and bereaved family member perceptions of quality of end-of-life care. J Am Geriatr Soc. 2018;66(9):1730-1736. https://doi.org/10.1111/jgs.15455.
7. Ansari AA, Pomerantz DH, Jayes RL, Aguirre EA, Havyer RD. Promoting primary palliative care in severe chronic obstructive pulmonary disease: symptom management and preparedness planning. J Palliat Care. 2019;34(2):85-91. https://doi.org/10.1177/0825859718819437.
8. Loizeau AJ, Shaffer ML, Habtemariam DA, Hanson LC, Volandes AE, Mitchell SL. Association of prognostic estimates with burdensome interventions in nursing home residents with advanced dementia. JAMA Intern Med. 2018;178(7):922-929. https://doi.org/10.1001/jamainternmed.2018.1413.
9. Glare PA, Sinclair CT. Palliative medicine review: Prognostication. J Palliat Med. 2008;11(1):84-103. https://doi.org/10.1089/jpm.2008.9992.
10. Jayes RL, Arnold RM, Fromme EK. Does this dementia patient meet the prognosis eligibility requirements for hospice enrollment? J Pain Symptom Manage. 2012;44(5):750-756. https://doi.org/10.1016/j.jpainsymman.2012.08.004.
11. Mitchell SL, Miller SC, Teno JM, Kiely DK, Davis RB, Shaffer ML. Prediction of 6-month survival of nursing home residents with advanced dementia using ADEPT vs hospice eligibility guidelines. JAMA. 2010;304(17):1929-1935. https://doi.org/10.1001/jama.2010.1572.
12. Schonwetter RS, Han B, Small BJ, et al. Predictors of six-month survival among patients with dementia: an evaluation of hospice Medicare guidelines. Amer J Hospice & Pall Care. 2003;20(2):105-113. https://doi.org/10.1177/104990910302000208
13. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment. JAMA. 2008;300(14):1665-1673. https://doi.org/10.1001/jama.300.14.1665.
14. Bernacki R, Hutchings M, Vick J, et al. Development of the serious illness care program: a randomised controlled trial of a palliative care communication intervention. BMJ Open. 2015;5(10):e009032-2015-009032. https://doi.org/10.1136/bmjopen-2015-009032
15. Ansari A, Pomerantz D, Smith K. Being mindful: difficult decisions in advanced dementia and end stage renal disease. SGIM Forum. 2017;40(3):4, 13.
1. Mitchell SL. Advanced dementia. N Engl J Med. 2015;373(13):1276-1277. https://doi.org/10.1056/NEJMcp1412652.
2. Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009;361(16):1529-1538. https://doi.org/10.1056/NEJMoa0902234.
3. Teno JM, Gozalo PL, Mitchell SL, et al. Does feeding tube insertion and its timing improve survival? J Am Geriatr Soc. 2012;60(10):1918-1921. https://doi.org/10.1111/j.1532-5415.2012.04148.x.
4. Teno JM, Gozalo P, Mitchell SL, Kuo S, Fulton AT, Mor V. Feeding tubes and the prevention or healing of pressure ulcers. Arch Intern Med. 2012;172(9):697-701. https://doi.org/10.1001/archinternmed.2012.1200.
5. Teno JM, Gozalo PL, Bynum JP, et al. Change in end-of-life care for medicare beneficiaries: site of death, place of care, and health care transitions in 2000, 2005, and 2009. JAMA. 2013;309(5):470-477. https://doi.org/10.1001/jama.2012.207624.
6. Makaroun LK, Teno JM, Freedman VA, Kasper JD, Gozalo P, Mor V. Late transitions and bereaved family member perceptions of quality of end-of-life care. J Am Geriatr Soc. 2018;66(9):1730-1736. https://doi.org/10.1111/jgs.15455.
7. Ansari AA, Pomerantz DH, Jayes RL, Aguirre EA, Havyer RD. Promoting primary palliative care in severe chronic obstructive pulmonary disease: symptom management and preparedness planning. J Palliat Care. 2019;34(2):85-91. https://doi.org/10.1177/0825859718819437.
8. Loizeau AJ, Shaffer ML, Habtemariam DA, Hanson LC, Volandes AE, Mitchell SL. Association of prognostic estimates with burdensome interventions in nursing home residents with advanced dementia. JAMA Intern Med. 2018;178(7):922-929. https://doi.org/10.1001/jamainternmed.2018.1413.
9. Glare PA, Sinclair CT. Palliative medicine review: Prognostication. J Palliat Med. 2008;11(1):84-103. https://doi.org/10.1089/jpm.2008.9992.
10. Jayes RL, Arnold RM, Fromme EK. Does this dementia patient meet the prognosis eligibility requirements for hospice enrollment? J Pain Symptom Manage. 2012;44(5):750-756. https://doi.org/10.1016/j.jpainsymman.2012.08.004.
11. Mitchell SL, Miller SC, Teno JM, Kiely DK, Davis RB, Shaffer ML. Prediction of 6-month survival of nursing home residents with advanced dementia using ADEPT vs hospice eligibility guidelines. JAMA. 2010;304(17):1929-1935. https://doi.org/10.1001/jama.2010.1572.
12. Schonwetter RS, Han B, Small BJ, et al. Predictors of six-month survival among patients with dementia: an evaluation of hospice Medicare guidelines. Amer J Hospice & Pall Care. 2003;20(2):105-113. https://doi.org/10.1177/104990910302000208
13. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment. JAMA. 2008;300(14):1665-1673. https://doi.org/10.1001/jama.300.14.1665.
14. Bernacki R, Hutchings M, Vick J, et al. Development of the serious illness care program: a randomised controlled trial of a palliative care communication intervention. BMJ Open. 2015;5(10):e009032-2015-009032. https://doi.org/10.1136/bmjopen-2015-009032
15. Ansari A, Pomerantz D, Smith K. Being mindful: difficult decisions in advanced dementia and end stage renal disease. SGIM Forum. 2017;40(3):4, 13.
© 2019 Society of Hospital Medicine
Things We Do for No Reason™: Supplemental Oxygen for Patients without Hypoxemia

Inspired by the ABIM Foundation’s Choosing Wisely® campaign, the “Things We Do for No Reason™” (TWDFNR) series reviews practices that have become common parts of hospital care but may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent clear-cut conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion.
CLINICAL SCENARIO
A 65-year-old woman with hypertension presents to the emergency department with three days of dyspnea, malaise, and pleuritic chest pain. Her temperature is 100.1°F, heart rate 110 beats per minute, and blood pressure 110/60 mm Hg. She is breathing 24 times per minute and has an oxygen saturation (SpO2) of 94% on room air. Her exam is remarkable for dry mucous membranes and right lower lung crackles. Her nurse places her on 3 L of oxygen per minute via nasal cannula, and her SpO2 rises to 99%.
WHY YOU MIGHT THINK SUPPLEMENTAL OXYGEN FOR NORMOXEMIC PATIENTS IS HELPFUL
Shortly after the discovery of oxygen in the late 18th century, physicians began using it to treat a variety of conditions including tuberculosis, pneumonia, respiratory failure, and angina. By the 1970s, most medical texts recommended oxygen use in suspected myocardial infarction (MI) because of the theoretical appeal of increasing delivery of oxygen to the heart and other vital organs.1 Additionally, there is a tendency to believe that supplemental oxygen alleviates dyspnea regardless of etiology or oxygen saturation. Recent studies have shown widespread use of oxygen in scenarios without clear indications and without oxygen saturation goals. A 2010 survey of clinicians managing acute MI found that 98% “always or usually” used oxygen and 55% believed that oxygen “definitely or probably reduces the risk of death.”2 In a Danish prehospital study, supplemental oxygen was used in 34% of ambulance patients even though only 17% of these patients had an SpO2 less than 94%.3 A study of critically ill patients found that most of the time, SpO2 exceeded 98%. Even when the fraction of inspired oxygen (FiO2) was between 0.3 and 0.4, no one adjusted the oxygen dose.4
WHY IT IS NOT HELPFUL TO PROVIDE SUPPLEMENTAL OXYGEN TO NORMOXEMIC PATIENTS
The reflexive use of oxygen in patients with acute respiratory or cardiovascular illness is problematic for several reasons. First, when oxygen saturation is near-normal, the potential benefit from supplemental oxygen lacks physiologic plausibility. More compellingly, evidence exists that hyperoxemia may cause significant harm. Finally, the unnecessary use of supplemental oxygen incurs practical inconveniences and expenses.
To understand why the physiologic basis for reflexive oxygen use is weak, it is important to distinguish hypoxemia (low arterial oxygen tension and hemoglobin oxygen saturation), tissue hypoxia (which can occur from hypoxemia or focal abnormalities in perfusion), and dyspnea (a subjective experience of breathing discomfort). A variety of mechanisms cause dyspnea, most of which do not involve hypoxemia. A patient with acute heart failure may experience severe dyspnea caused by activation of pressure-sensitive J-receptors in the lung, even if oxygen saturation and tissue perfusion are intact. This process will be relieved by reducing pulmonary capillary pressures, but it is unaffected by supplemental oxygen. Coronary occlusion causes hypoxia of the heart muscle, but restoring perfusion is the most effective treatment. The instinct to maximize the oxygen-carrying capacity of the remaining blood flow is understandable. However, in a normoxemic patient, increasing the inspired fraction of oxygen has a marginal effect on oxygen-carrying capacity, since hemoglobin saturation and concentration rather than arterial oxygen tension (PaO2) predominantly determine oxygen-carrying capacity. On the other hand, supraphysiologic levels of dissolved oxygen may lead to toxicity.5
For over a century, we have known the potential harms of hyperoxia. Original studies in animal models showed that hyperoxia led to lung injury, altered hemodynamics, endothelial cell dysfunction, and inflammatory activation.5 Many of these detrimental effects involve the generation of reactive oxygen species and oxidative stress.5 High levels of inspired oxygen can also cause increased pulmonary shunting through inhibition of physiologic hypoxic vasoconstriction and due to absorption atelectasis.6 Oxygen negatively affects cardiovascular function by reducing coronary blood flow, increasing systemic vascular resistance, and reducing cardiac output.1
Chronic obstructive pulmonary disease (COPD) is the clinical setting in which risks of supplemental oxygen are most well-recognized historically. In patients with COPD at risk for hypercarbia, oxygen titrated to a goal SpO2 outside 88%-92% is associated with a two-fold risk of mortality.7 Worsening ventilation-perfusion matching and the Haldane effect (decreased affinity of hemoglobin for carbon dioxide as the PaO2 rises), rather than the previously theorized decrease in hypoxic drive, are now believed to contribute most to hyperoxia-induced hypercarbia. These unintended consequences may also occur in patients with other forms of acute and chronic lung disease.
The British Medical Journal published the first randomized controlled trial of oxygen use in suspected MI in 1976.1 Patients who received oxygen at 6 L per minute for 24 hours had more episodes of sinus tachycardia without any improvement in mortality, analgesic use, or infarct size.1 More recent and robust trials comparing outcomes in normoxemic patients randomized to supplemental oxygen versus room air have had similar findings: no difference in mortality, infarct size, or pain ratings.8,9 One found a significantly increased rate of MI recurrence with the use of oxygen.8 These data have led the latest guidelines for the management of ST-elevation MI from the European Society of Cardiology to discourage the use of supplemental oxygen unless SpO2 is <90%.10
Two recent trials investigated the effects of hyperoxia in critically ill patients.11,12 Girardis and colleagues randomized 480 critically ill patients in an Italian medical-surgical intensive care unit to conservative (SpO2 between 94% and 98% or PaO2 between 70 and 100 mm Hg) versus conventional oxygenation targets (SpO2 between 97% and 100% and PaO2 up to 150 mm Hg). Compared with conventional oxygen targets, conservative oxygen use was associated with an absolute risk reduction in mortality of 8.6% (11.6% vs 20.2%; P =.01).11 Another trial from 22 centers in France compared outcomes in mechanically ventilated patients with septic shock who received FiO2 at 1.0 compared with those with oxygen titration to SpO2 between 88% and 95%. The trial was stopped early for safety concerns. Those in the hyperoxemia group had a higher incidence of serious adverse events (85% vs 76%; P =.02), including pneumothorax, clinically relevant bleeding, myocardial infarction, and arrhythmias, as well as a trend toward increased mortality.12
Trials of liberal oxygen use in other settings of acute illness,13 including ischemic stroke,14 traumatic brain injury,15 and postcardiac arrest,16 have also linked liberal oxygen use with increased risk of mortality and other adverse events. “Liberal” use in these trials ranged from an FiO2 of 0.28 (equivalent to 2 L of nasal cannula) to 1.0. Significant secondary outcomes included fewer hospital-free and ventilator-free days in patients with liberal oxygen use. Furthermore, a meta-analysis of 25 trials including over 16,000 patients found dose-dependent toxicity: for every 1% increase in SpO2 above 94%-96% (the median SpO2 in the liberal oxygen groups), there was a 25% relative increase in in-hospital mortality.13
In addition to the data above, there are practical reasons to avoid unnecessary use of supplemental oxygen. Providing supplemental oxygen to a patient who is not hypoxemic may delay the recognition of cardiopulmonary decompensation by delaying detection of hypoxemia.6 Beyond the effects of oxygen itself, oxygen delivery methods carry their own potential adverse effects. These include epistaxis (with nasal cannula), claustrophobia (with face masks), decreased mobility, falls, and delirium.17 Finally, oxygen administration has direct and indirect financial costs, including those of supplies, care coordination, and monitoring.
WHEN SUPPLEMENTAL OXYGEN MIGHT BE HELPFUL
Importantly, the above discussion pertains to normoxemic patients receiving supplemental oxygen. There is no dispute that significantly hypoxemic patients should receive supplemental oxygen. There are also instances where the use of supplemental oxygen in normoxemic patients may be beneficial, such as in carbon monoxide poisoning, decompression injury, gas embolism, cluster headaches, sickle cell crisis, and pneumothorax.17
WHAT YOU SHOULD DO INSTEAD
Like any other drug, oxygen should be administered after assessment of its indications, intended benefits, and possible harms. Both significant hypoxemia and hyperoxemia should be avoided. In patients with neither hypoxemia nor the indications above, clinicians should not administer supplemental oxygen. Recent society guidelines can be applied in various clinical contexts. In patients with suspected MI, oxygen should be administered if SpO2 is <90%.10 For most other acutely ill patients, clinicians should administer supplemental oxygen if SpO2 <90%-92% and target an SpO2 of no higher than 94%-96%,18-19 as meta-analyses found evidence of harm above this level.13 Results of randomized trials currently underway should add supporting evidence for more specific oxygenation targets in different patient populations. With respect to implementation, it must be noted that factors beyond physician decision influence the use of supplemental oxygen. Appropriate institutional policies, standards of care, and educational efforts to all hospital providers must be enacted in order to reduce the unnecessary use of supplemental oxygen.
RECOMMENDATIONS
- For most acutely ill patients, do not administer supplemental oxygen when SpO2 >92%. If supplemental oxygen is used, the SpO2 should not exceed 94%-96%.
- For patients with suspected MI, only start supplemental oxygen for SpO2 <90%.
- For patients at risk for hypercapnic respiratory failure (eg, COPD patients), target SpO2 of 88%-92%.
- Provide supplemental oxygen to normoxemic patients with carbon monoxide poisoning, decompression injury, gas embolism, cluster headache, sickle cell crisis, and pneumothorax.
- Review and revise institutional practices and policies that contribute to unnecessary use of supplemental oxygen.
CONCLUSIONS
In the opening case, the patient is acutely ill and requires further workup. Her current SpO2 of 99% puts her at risk for adverse events and death, and supplemental oxygen should be titrated down or stopped to avoid SpO2 greater than 94%-96%. For years, clinicians have erred on the side of using supplemental oxygen, without recognizing its dangers. However, over a century of evidence from pathophysiologic experiments and randomized trials across multiple clinical settings have associated hyperoxemia with adverse outcomes and increased mortality. Professional societies are adopting this evidence into their guideline recommendations, and clinicians should use supplemental oxygen judiciously in their daily practice.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing [email protected].
1. Rawles JM, Kenmure AC. Controlled trial of oxygen in uncomplicated myocardial infarction. Br Med J. 1976;1(6018):1121-1123. https://doi.org/10.1136/bmj.1.6018.1121.
2. Burls A, Emparanza JI, Quinn T, Cabello J. Oxygen use in acute myocardial infarction: an online survey of health professionals’ practice and beliefs. Emerg Med J. 2010;27(4):283-286. https://doi.org/10.1136/emj.2009.077370.
3. Hale KE, Gavin C, O’Driscoll BR. Audit of oxygen use in emergency ambulances and in a hospital emergency department. Emerg Med J. 2008;25(11):773-776. https://doi.org/10.1136/emj.2008.059287.
4. Suzuki S, Eastwood G, Peck L, Glassford N, Bellomo R. Oxygen management in mechanically ventilated patients: a prospective observational cohort study. Aust Crit Care. 2014;27(1):50-51. https://doi.org/10.1016/j.aucc.2013.10.025.
5. Helmerhorst HJ, Schultz MJ, van der Voort PH, de Jonge E, van Wasterloo DJ. Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care. 2015;19(1):284. https://doi.org/10.1186/s13054-015-0996-4.
6. Downs JB. Has oxygen administration delayed appropriate respiratory care? Fallacies regarding oxygen therapy. Respir Care. 2003;48(6):611-620.
7. Austin MA, Willis KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ. 2010;341:c5462. https://doi.org/10.2307/20800296.
8. Stub D, Smith K, Bernard S, et al. Air versus oxygen in ST-segment-elevation myocardial infarction. Circulation. 2015;131(24):2143-2150. https://doi.org/10.1161/CIRCULATIONAHA.114.014494.
9. Hofman R. Witt N, Lagergvist B, et al. Oxygen therapy in ST-elevation myocardial infarction. Eur Heart J. 2018;39(29):2730-2739. https://doi.org/10.1093/eurheartj/ehy326.
10. Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018:39(2):119-177. https://doi.org/10.1093/eurheartj/ehx393.
11. Girardis M, Busani S, Damiani E, et al. Effect of conservative vs conventional oxygen therapy on mortality among patients in an intensive care unit. JAMA. 2016;316(15):1583-1589. https://doi.org/10.1001/jama.2016.11993.
12. Asfar P, Schortgen F, Boisramé-Helms J, et al. Hyperoxia and hypertonic saline in patients with septic shock (HYPERS2S): a two-by-two factorial, multicentre, randomised, clinical trial. Lancet Respir Med. 2017:5(3):180-190. https://doi.org/10.1016/S2213-2600(17)30046-2.
13. Chu DK, Kim LH, Young PJ, et al. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet. 2018;391(10131):1693-1705. https://doi.org/10.1016/S0140-6736(18)30479-3.
14. Rincon F, Kang J, Maltenfort M, et al. Association between hyperoxia and mortality after stroke: a multicenter cohort study. Crit Care Med. 2014;42(2):387-396. https://doi.org/10.1097/CCM.0b013e3182a27732.
15. Brenner M, Stein D, Hu P, Kufera J, Woodford M, Scalea T. Association between early hyperoxia and worse outcomes after traumatic brain injury. Arch Surg. 2012;147(11):1042-1046. https://doi.org/10.1001/archsurg.2012.1560.
16. Kilgannon JH, Jones AE, Shapiro NI, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303(21):2165-2171. https://doi.org/10.1
17. Siemieniuk RA, Chu DK, Kim L, et al. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ. 2018;363:k4169. https://doi.org/10.1136/bmj.k4169.
18. O’Driscoll BR, Howard LS, Earis J, et al. BTS guideline for oxygen use in adults in healthcare and emergency settings. Thorax. 2017;72(1):ii1-ii90. https://doi.org/10.1136/thoraxjnl-2016-209729.
19. Beasley R, Chien J, Douglas J, et al. Thoracic Society of Australia and New Zealand oxygen guidelines for acute oxygen use in adults: ‘Swimming between the flags’. Respirology. 2015;20(8):1182-1191. https://doi.org/10.1111/resp.12620.
Inspired by the ABIM Foundation’s Choosing Wisely® campaign, the “Things We Do for No Reason™” (TWDFNR) series reviews practices that have become common parts of hospital care but may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent clear-cut conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion.
CLINICAL SCENARIO
A 65-year-old woman with hypertension presents to the emergency department with three days of dyspnea, malaise, and pleuritic chest pain. Her temperature is 100.1°F, heart rate 110 beats per minute, and blood pressure 110/60 mm Hg. She is breathing 24 times per minute and has an oxygen saturation (SpO2) of 94% on room air. Her exam is remarkable for dry mucous membranes and right lower lung crackles. Her nurse places her on 3 L of oxygen per minute via nasal cannula, and her SpO2 rises to 99%.
WHY YOU MIGHT THINK SUPPLEMENTAL OXYGEN FOR NORMOXEMIC PATIENTS IS HELPFUL
Shortly after the discovery of oxygen in the late 18th century, physicians began using it to treat a variety of conditions including tuberculosis, pneumonia, respiratory failure, and angina. By the 1970s, most medical texts recommended oxygen use in suspected myocardial infarction (MI) because of the theoretical appeal of increasing delivery of oxygen to the heart and other vital organs.1 Additionally, there is a tendency to believe that supplemental oxygen alleviates dyspnea regardless of etiology or oxygen saturation. Recent studies have shown widespread use of oxygen in scenarios without clear indications and without oxygen saturation goals. A 2010 survey of clinicians managing acute MI found that 98% “always or usually” used oxygen and 55% believed that oxygen “definitely or probably reduces the risk of death.”2 In a Danish prehospital study, supplemental oxygen was used in 34% of ambulance patients even though only 17% of these patients had an SpO2 less than 94%.3 A study of critically ill patients found that most of the time, SpO2 exceeded 98%. Even when the fraction of inspired oxygen (FiO2) was between 0.3 and 0.4, no one adjusted the oxygen dose.4
WHY IT IS NOT HELPFUL TO PROVIDE SUPPLEMENTAL OXYGEN TO NORMOXEMIC PATIENTS
The reflexive use of oxygen in patients with acute respiratory or cardiovascular illness is problematic for several reasons. First, when oxygen saturation is near-normal, the potential benefit from supplemental oxygen lacks physiologic plausibility. More compellingly, evidence exists that hyperoxemia may cause significant harm. Finally, the unnecessary use of supplemental oxygen incurs practical inconveniences and expenses.
To understand why the physiologic basis for reflexive oxygen use is weak, it is important to distinguish hypoxemia (low arterial oxygen tension and hemoglobin oxygen saturation), tissue hypoxia (which can occur from hypoxemia or focal abnormalities in perfusion), and dyspnea (a subjective experience of breathing discomfort). A variety of mechanisms cause dyspnea, most of which do not involve hypoxemia. A patient with acute heart failure may experience severe dyspnea caused by activation of pressure-sensitive J-receptors in the lung, even if oxygen saturation and tissue perfusion are intact. This process will be relieved by reducing pulmonary capillary pressures, but it is unaffected by supplemental oxygen. Coronary occlusion causes hypoxia of the heart muscle, but restoring perfusion is the most effective treatment. The instinct to maximize the oxygen-carrying capacity of the remaining blood flow is understandable. However, in a normoxemic patient, increasing the inspired fraction of oxygen has a marginal effect on oxygen-carrying capacity, since hemoglobin saturation and concentration rather than arterial oxygen tension (PaO2) predominantly determine oxygen-carrying capacity. On the other hand, supraphysiologic levels of dissolved oxygen may lead to toxicity.5
For over a century, we have known the potential harms of hyperoxia. Original studies in animal models showed that hyperoxia led to lung injury, altered hemodynamics, endothelial cell dysfunction, and inflammatory activation.5 Many of these detrimental effects involve the generation of reactive oxygen species and oxidative stress.5 High levels of inspired oxygen can also cause increased pulmonary shunting through inhibition of physiologic hypoxic vasoconstriction and due to absorption atelectasis.6 Oxygen negatively affects cardiovascular function by reducing coronary blood flow, increasing systemic vascular resistance, and reducing cardiac output.1
Chronic obstructive pulmonary disease (COPD) is the clinical setting in which risks of supplemental oxygen are most well-recognized historically. In patients with COPD at risk for hypercarbia, oxygen titrated to a goal SpO2 outside 88%-92% is associated with a two-fold risk of mortality.7 Worsening ventilation-perfusion matching and the Haldane effect (decreased affinity of hemoglobin for carbon dioxide as the PaO2 rises), rather than the previously theorized decrease in hypoxic drive, are now believed to contribute most to hyperoxia-induced hypercarbia. These unintended consequences may also occur in patients with other forms of acute and chronic lung disease.
The British Medical Journal published the first randomized controlled trial of oxygen use in suspected MI in 1976.1 Patients who received oxygen at 6 L per minute for 24 hours had more episodes of sinus tachycardia without any improvement in mortality, analgesic use, or infarct size.1 More recent and robust trials comparing outcomes in normoxemic patients randomized to supplemental oxygen versus room air have had similar findings: no difference in mortality, infarct size, or pain ratings.8,9 One found a significantly increased rate of MI recurrence with the use of oxygen.8 These data have led the latest guidelines for the management of ST-elevation MI from the European Society of Cardiology to discourage the use of supplemental oxygen unless SpO2 is <90%.10
Two recent trials investigated the effects of hyperoxia in critically ill patients.11,12 Girardis and colleagues randomized 480 critically ill patients in an Italian medical-surgical intensive care unit to conservative (SpO2 between 94% and 98% or PaO2 between 70 and 100 mm Hg) versus conventional oxygenation targets (SpO2 between 97% and 100% and PaO2 up to 150 mm Hg). Compared with conventional oxygen targets, conservative oxygen use was associated with an absolute risk reduction in mortality of 8.6% (11.6% vs 20.2%; P =.01).11 Another trial from 22 centers in France compared outcomes in mechanically ventilated patients with septic shock who received FiO2 at 1.0 compared with those with oxygen titration to SpO2 between 88% and 95%. The trial was stopped early for safety concerns. Those in the hyperoxemia group had a higher incidence of serious adverse events (85% vs 76%; P =.02), including pneumothorax, clinically relevant bleeding, myocardial infarction, and arrhythmias, as well as a trend toward increased mortality.12
Trials of liberal oxygen use in other settings of acute illness,13 including ischemic stroke,14 traumatic brain injury,15 and postcardiac arrest,16 have also linked liberal oxygen use with increased risk of mortality and other adverse events. “Liberal” use in these trials ranged from an FiO2 of 0.28 (equivalent to 2 L of nasal cannula) to 1.0. Significant secondary outcomes included fewer hospital-free and ventilator-free days in patients with liberal oxygen use. Furthermore, a meta-analysis of 25 trials including over 16,000 patients found dose-dependent toxicity: for every 1% increase in SpO2 above 94%-96% (the median SpO2 in the liberal oxygen groups), there was a 25% relative increase in in-hospital mortality.13
In addition to the data above, there are practical reasons to avoid unnecessary use of supplemental oxygen. Providing supplemental oxygen to a patient who is not hypoxemic may delay the recognition of cardiopulmonary decompensation by delaying detection of hypoxemia.6 Beyond the effects of oxygen itself, oxygen delivery methods carry their own potential adverse effects. These include epistaxis (with nasal cannula), claustrophobia (with face masks), decreased mobility, falls, and delirium.17 Finally, oxygen administration has direct and indirect financial costs, including those of supplies, care coordination, and monitoring.
WHEN SUPPLEMENTAL OXYGEN MIGHT BE HELPFUL
Importantly, the above discussion pertains to normoxemic patients receiving supplemental oxygen. There is no dispute that significantly hypoxemic patients should receive supplemental oxygen. There are also instances where the use of supplemental oxygen in normoxemic patients may be beneficial, such as in carbon monoxide poisoning, decompression injury, gas embolism, cluster headaches, sickle cell crisis, and pneumothorax.17
WHAT YOU SHOULD DO INSTEAD
Like any other drug, oxygen should be administered after assessment of its indications, intended benefits, and possible harms. Both significant hypoxemia and hyperoxemia should be avoided. In patients with neither hypoxemia nor the indications above, clinicians should not administer supplemental oxygen. Recent society guidelines can be applied in various clinical contexts. In patients with suspected MI, oxygen should be administered if SpO2 is <90%.10 For most other acutely ill patients, clinicians should administer supplemental oxygen if SpO2 <90%-92% and target an SpO2 of no higher than 94%-96%,18-19 as meta-analyses found evidence of harm above this level.13 Results of randomized trials currently underway should add supporting evidence for more specific oxygenation targets in different patient populations. With respect to implementation, it must be noted that factors beyond physician decision influence the use of supplemental oxygen. Appropriate institutional policies, standards of care, and educational efforts to all hospital providers must be enacted in order to reduce the unnecessary use of supplemental oxygen.
RECOMMENDATIONS
- For most acutely ill patients, do not administer supplemental oxygen when SpO2 >92%. If supplemental oxygen is used, the SpO2 should not exceed 94%-96%.
- For patients with suspected MI, only start supplemental oxygen for SpO2 <90%.
- For patients at risk for hypercapnic respiratory failure (eg, COPD patients), target SpO2 of 88%-92%.
- Provide supplemental oxygen to normoxemic patients with carbon monoxide poisoning, decompression injury, gas embolism, cluster headache, sickle cell crisis, and pneumothorax.
- Review and revise institutional practices and policies that contribute to unnecessary use of supplemental oxygen.
CONCLUSIONS
In the opening case, the patient is acutely ill and requires further workup. Her current SpO2 of 99% puts her at risk for adverse events and death, and supplemental oxygen should be titrated down or stopped to avoid SpO2 greater than 94%-96%. For years, clinicians have erred on the side of using supplemental oxygen, without recognizing its dangers. However, over a century of evidence from pathophysiologic experiments and randomized trials across multiple clinical settings have associated hyperoxemia with adverse outcomes and increased mortality. Professional societies are adopting this evidence into their guideline recommendations, and clinicians should use supplemental oxygen judiciously in their daily practice.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing [email protected].
Inspired by the ABIM Foundation’s Choosing Wisely® campaign, the “Things We Do for No Reason™” (TWDFNR) series reviews practices that have become common parts of hospital care but may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent clear-cut conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion.
CLINICAL SCENARIO
A 65-year-old woman with hypertension presents to the emergency department with three days of dyspnea, malaise, and pleuritic chest pain. Her temperature is 100.1°F, heart rate 110 beats per minute, and blood pressure 110/60 mm Hg. She is breathing 24 times per minute and has an oxygen saturation (SpO2) of 94% on room air. Her exam is remarkable for dry mucous membranes and right lower lung crackles. Her nurse places her on 3 L of oxygen per minute via nasal cannula, and her SpO2 rises to 99%.
WHY YOU MIGHT THINK SUPPLEMENTAL OXYGEN FOR NORMOXEMIC PATIENTS IS HELPFUL
Shortly after the discovery of oxygen in the late 18th century, physicians began using it to treat a variety of conditions including tuberculosis, pneumonia, respiratory failure, and angina. By the 1970s, most medical texts recommended oxygen use in suspected myocardial infarction (MI) because of the theoretical appeal of increasing delivery of oxygen to the heart and other vital organs.1 Additionally, there is a tendency to believe that supplemental oxygen alleviates dyspnea regardless of etiology or oxygen saturation. Recent studies have shown widespread use of oxygen in scenarios without clear indications and without oxygen saturation goals. A 2010 survey of clinicians managing acute MI found that 98% “always or usually” used oxygen and 55% believed that oxygen “definitely or probably reduces the risk of death.”2 In a Danish prehospital study, supplemental oxygen was used in 34% of ambulance patients even though only 17% of these patients had an SpO2 less than 94%.3 A study of critically ill patients found that most of the time, SpO2 exceeded 98%. Even when the fraction of inspired oxygen (FiO2) was between 0.3 and 0.4, no one adjusted the oxygen dose.4
WHY IT IS NOT HELPFUL TO PROVIDE SUPPLEMENTAL OXYGEN TO NORMOXEMIC PATIENTS
The reflexive use of oxygen in patients with acute respiratory or cardiovascular illness is problematic for several reasons. First, when oxygen saturation is near-normal, the potential benefit from supplemental oxygen lacks physiologic plausibility. More compellingly, evidence exists that hyperoxemia may cause significant harm. Finally, the unnecessary use of supplemental oxygen incurs practical inconveniences and expenses.
To understand why the physiologic basis for reflexive oxygen use is weak, it is important to distinguish hypoxemia (low arterial oxygen tension and hemoglobin oxygen saturation), tissue hypoxia (which can occur from hypoxemia or focal abnormalities in perfusion), and dyspnea (a subjective experience of breathing discomfort). A variety of mechanisms cause dyspnea, most of which do not involve hypoxemia. A patient with acute heart failure may experience severe dyspnea caused by activation of pressure-sensitive J-receptors in the lung, even if oxygen saturation and tissue perfusion are intact. This process will be relieved by reducing pulmonary capillary pressures, but it is unaffected by supplemental oxygen. Coronary occlusion causes hypoxia of the heart muscle, but restoring perfusion is the most effective treatment. The instinct to maximize the oxygen-carrying capacity of the remaining blood flow is understandable. However, in a normoxemic patient, increasing the inspired fraction of oxygen has a marginal effect on oxygen-carrying capacity, since hemoglobin saturation and concentration rather than arterial oxygen tension (PaO2) predominantly determine oxygen-carrying capacity. On the other hand, supraphysiologic levels of dissolved oxygen may lead to toxicity.5
For over a century, we have known the potential harms of hyperoxia. Original studies in animal models showed that hyperoxia led to lung injury, altered hemodynamics, endothelial cell dysfunction, and inflammatory activation.5 Many of these detrimental effects involve the generation of reactive oxygen species and oxidative stress.5 High levels of inspired oxygen can also cause increased pulmonary shunting through inhibition of physiologic hypoxic vasoconstriction and due to absorption atelectasis.6 Oxygen negatively affects cardiovascular function by reducing coronary blood flow, increasing systemic vascular resistance, and reducing cardiac output.1
Chronic obstructive pulmonary disease (COPD) is the clinical setting in which risks of supplemental oxygen are most well-recognized historically. In patients with COPD at risk for hypercarbia, oxygen titrated to a goal SpO2 outside 88%-92% is associated with a two-fold risk of mortality.7 Worsening ventilation-perfusion matching and the Haldane effect (decreased affinity of hemoglobin for carbon dioxide as the PaO2 rises), rather than the previously theorized decrease in hypoxic drive, are now believed to contribute most to hyperoxia-induced hypercarbia. These unintended consequences may also occur in patients with other forms of acute and chronic lung disease.
The British Medical Journal published the first randomized controlled trial of oxygen use in suspected MI in 1976.1 Patients who received oxygen at 6 L per minute for 24 hours had more episodes of sinus tachycardia without any improvement in mortality, analgesic use, or infarct size.1 More recent and robust trials comparing outcomes in normoxemic patients randomized to supplemental oxygen versus room air have had similar findings: no difference in mortality, infarct size, or pain ratings.8,9 One found a significantly increased rate of MI recurrence with the use of oxygen.8 These data have led the latest guidelines for the management of ST-elevation MI from the European Society of Cardiology to discourage the use of supplemental oxygen unless SpO2 is <90%.10
Two recent trials investigated the effects of hyperoxia in critically ill patients.11,12 Girardis and colleagues randomized 480 critically ill patients in an Italian medical-surgical intensive care unit to conservative (SpO2 between 94% and 98% or PaO2 between 70 and 100 mm Hg) versus conventional oxygenation targets (SpO2 between 97% and 100% and PaO2 up to 150 mm Hg). Compared with conventional oxygen targets, conservative oxygen use was associated with an absolute risk reduction in mortality of 8.6% (11.6% vs 20.2%; P =.01).11 Another trial from 22 centers in France compared outcomes in mechanically ventilated patients with septic shock who received FiO2 at 1.0 compared with those with oxygen titration to SpO2 between 88% and 95%. The trial was stopped early for safety concerns. Those in the hyperoxemia group had a higher incidence of serious adverse events (85% vs 76%; P =.02), including pneumothorax, clinically relevant bleeding, myocardial infarction, and arrhythmias, as well as a trend toward increased mortality.12
Trials of liberal oxygen use in other settings of acute illness,13 including ischemic stroke,14 traumatic brain injury,15 and postcardiac arrest,16 have also linked liberal oxygen use with increased risk of mortality and other adverse events. “Liberal” use in these trials ranged from an FiO2 of 0.28 (equivalent to 2 L of nasal cannula) to 1.0. Significant secondary outcomes included fewer hospital-free and ventilator-free days in patients with liberal oxygen use. Furthermore, a meta-analysis of 25 trials including over 16,000 patients found dose-dependent toxicity: for every 1% increase in SpO2 above 94%-96% (the median SpO2 in the liberal oxygen groups), there was a 25% relative increase in in-hospital mortality.13
In addition to the data above, there are practical reasons to avoid unnecessary use of supplemental oxygen. Providing supplemental oxygen to a patient who is not hypoxemic may delay the recognition of cardiopulmonary decompensation by delaying detection of hypoxemia.6 Beyond the effects of oxygen itself, oxygen delivery methods carry their own potential adverse effects. These include epistaxis (with nasal cannula), claustrophobia (with face masks), decreased mobility, falls, and delirium.17 Finally, oxygen administration has direct and indirect financial costs, including those of supplies, care coordination, and monitoring.
WHEN SUPPLEMENTAL OXYGEN MIGHT BE HELPFUL
Importantly, the above discussion pertains to normoxemic patients receiving supplemental oxygen. There is no dispute that significantly hypoxemic patients should receive supplemental oxygen. There are also instances where the use of supplemental oxygen in normoxemic patients may be beneficial, such as in carbon monoxide poisoning, decompression injury, gas embolism, cluster headaches, sickle cell crisis, and pneumothorax.17
WHAT YOU SHOULD DO INSTEAD
Like any other drug, oxygen should be administered after assessment of its indications, intended benefits, and possible harms. Both significant hypoxemia and hyperoxemia should be avoided. In patients with neither hypoxemia nor the indications above, clinicians should not administer supplemental oxygen. Recent society guidelines can be applied in various clinical contexts. In patients with suspected MI, oxygen should be administered if SpO2 is <90%.10 For most other acutely ill patients, clinicians should administer supplemental oxygen if SpO2 <90%-92% and target an SpO2 of no higher than 94%-96%,18-19 as meta-analyses found evidence of harm above this level.13 Results of randomized trials currently underway should add supporting evidence for more specific oxygenation targets in different patient populations. With respect to implementation, it must be noted that factors beyond physician decision influence the use of supplemental oxygen. Appropriate institutional policies, standards of care, and educational efforts to all hospital providers must be enacted in order to reduce the unnecessary use of supplemental oxygen.
RECOMMENDATIONS
- For most acutely ill patients, do not administer supplemental oxygen when SpO2 >92%. If supplemental oxygen is used, the SpO2 should not exceed 94%-96%.
- For patients with suspected MI, only start supplemental oxygen for SpO2 <90%.
- For patients at risk for hypercapnic respiratory failure (eg, COPD patients), target SpO2 of 88%-92%.
- Provide supplemental oxygen to normoxemic patients with carbon monoxide poisoning, decompression injury, gas embolism, cluster headache, sickle cell crisis, and pneumothorax.
- Review and revise institutional practices and policies that contribute to unnecessary use of supplemental oxygen.
CONCLUSIONS
In the opening case, the patient is acutely ill and requires further workup. Her current SpO2 of 99% puts her at risk for adverse events and death, and supplemental oxygen should be titrated down or stopped to avoid SpO2 greater than 94%-96%. For years, clinicians have erred on the side of using supplemental oxygen, without recognizing its dangers. However, over a century of evidence from pathophysiologic experiments and randomized trials across multiple clinical settings have associated hyperoxemia with adverse outcomes and increased mortality. Professional societies are adopting this evidence into their guideline recommendations, and clinicians should use supplemental oxygen judiciously in their daily practice.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing [email protected].
1. Rawles JM, Kenmure AC. Controlled trial of oxygen in uncomplicated myocardial infarction. Br Med J. 1976;1(6018):1121-1123. https://doi.org/10.1136/bmj.1.6018.1121.
2. Burls A, Emparanza JI, Quinn T, Cabello J. Oxygen use in acute myocardial infarction: an online survey of health professionals’ practice and beliefs. Emerg Med J. 2010;27(4):283-286. https://doi.org/10.1136/emj.2009.077370.
3. Hale KE, Gavin C, O’Driscoll BR. Audit of oxygen use in emergency ambulances and in a hospital emergency department. Emerg Med J. 2008;25(11):773-776. https://doi.org/10.1136/emj.2008.059287.
4. Suzuki S, Eastwood G, Peck L, Glassford N, Bellomo R. Oxygen management in mechanically ventilated patients: a prospective observational cohort study. Aust Crit Care. 2014;27(1):50-51. https://doi.org/10.1016/j.aucc.2013.10.025.
5. Helmerhorst HJ, Schultz MJ, van der Voort PH, de Jonge E, van Wasterloo DJ. Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care. 2015;19(1):284. https://doi.org/10.1186/s13054-015-0996-4.
6. Downs JB. Has oxygen administration delayed appropriate respiratory care? Fallacies regarding oxygen therapy. Respir Care. 2003;48(6):611-620.
7. Austin MA, Willis KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ. 2010;341:c5462. https://doi.org/10.2307/20800296.
8. Stub D, Smith K, Bernard S, et al. Air versus oxygen in ST-segment-elevation myocardial infarction. Circulation. 2015;131(24):2143-2150. https://doi.org/10.1161/CIRCULATIONAHA.114.014494.
9. Hofman R. Witt N, Lagergvist B, et al. Oxygen therapy in ST-elevation myocardial infarction. Eur Heart J. 2018;39(29):2730-2739. https://doi.org/10.1093/eurheartj/ehy326.
10. Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018:39(2):119-177. https://doi.org/10.1093/eurheartj/ehx393.
11. Girardis M, Busani S, Damiani E, et al. Effect of conservative vs conventional oxygen therapy on mortality among patients in an intensive care unit. JAMA. 2016;316(15):1583-1589. https://doi.org/10.1001/jama.2016.11993.
12. Asfar P, Schortgen F, Boisramé-Helms J, et al. Hyperoxia and hypertonic saline in patients with septic shock (HYPERS2S): a two-by-two factorial, multicentre, randomised, clinical trial. Lancet Respir Med. 2017:5(3):180-190. https://doi.org/10.1016/S2213-2600(17)30046-2.
13. Chu DK, Kim LH, Young PJ, et al. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet. 2018;391(10131):1693-1705. https://doi.org/10.1016/S0140-6736(18)30479-3.
14. Rincon F, Kang J, Maltenfort M, et al. Association between hyperoxia and mortality after stroke: a multicenter cohort study. Crit Care Med. 2014;42(2):387-396. https://doi.org/10.1097/CCM.0b013e3182a27732.
15. Brenner M, Stein D, Hu P, Kufera J, Woodford M, Scalea T. Association between early hyperoxia and worse outcomes after traumatic brain injury. Arch Surg. 2012;147(11):1042-1046. https://doi.org/10.1001/archsurg.2012.1560.
16. Kilgannon JH, Jones AE, Shapiro NI, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303(21):2165-2171. https://doi.org/10.1
17. Siemieniuk RA, Chu DK, Kim L, et al. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ. 2018;363:k4169. https://doi.org/10.1136/bmj.k4169.
18. O’Driscoll BR, Howard LS, Earis J, et al. BTS guideline for oxygen use in adults in healthcare and emergency settings. Thorax. 2017;72(1):ii1-ii90. https://doi.org/10.1136/thoraxjnl-2016-209729.
19. Beasley R, Chien J, Douglas J, et al. Thoracic Society of Australia and New Zealand oxygen guidelines for acute oxygen use in adults: ‘Swimming between the flags’. Respirology. 2015;20(8):1182-1191. https://doi.org/10.1111/resp.12620.
1. Rawles JM, Kenmure AC. Controlled trial of oxygen in uncomplicated myocardial infarction. Br Med J. 1976;1(6018):1121-1123. https://doi.org/10.1136/bmj.1.6018.1121.
2. Burls A, Emparanza JI, Quinn T, Cabello J. Oxygen use in acute myocardial infarction: an online survey of health professionals’ practice and beliefs. Emerg Med J. 2010;27(4):283-286. https://doi.org/10.1136/emj.2009.077370.
3. Hale KE, Gavin C, O’Driscoll BR. Audit of oxygen use in emergency ambulances and in a hospital emergency department. Emerg Med J. 2008;25(11):773-776. https://doi.org/10.1136/emj.2008.059287.
4. Suzuki S, Eastwood G, Peck L, Glassford N, Bellomo R. Oxygen management in mechanically ventilated patients: a prospective observational cohort study. Aust Crit Care. 2014;27(1):50-51. https://doi.org/10.1016/j.aucc.2013.10.025.
5. Helmerhorst HJ, Schultz MJ, van der Voort PH, de Jonge E, van Wasterloo DJ. Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care. 2015;19(1):284. https://doi.org/10.1186/s13054-015-0996-4.
6. Downs JB. Has oxygen administration delayed appropriate respiratory care? Fallacies regarding oxygen therapy. Respir Care. 2003;48(6):611-620.
7. Austin MA, Willis KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ. 2010;341:c5462. https://doi.org/10.2307/20800296.
8. Stub D, Smith K, Bernard S, et al. Air versus oxygen in ST-segment-elevation myocardial infarction. Circulation. 2015;131(24):2143-2150. https://doi.org/10.1161/CIRCULATIONAHA.114.014494.
9. Hofman R. Witt N, Lagergvist B, et al. Oxygen therapy in ST-elevation myocardial infarction. Eur Heart J. 2018;39(29):2730-2739. https://doi.org/10.1093/eurheartj/ehy326.
10. Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018:39(2):119-177. https://doi.org/10.1093/eurheartj/ehx393.
11. Girardis M, Busani S, Damiani E, et al. Effect of conservative vs conventional oxygen therapy on mortality among patients in an intensive care unit. JAMA. 2016;316(15):1583-1589. https://doi.org/10.1001/jama.2016.11993.
12. Asfar P, Schortgen F, Boisramé-Helms J, et al. Hyperoxia and hypertonic saline in patients with septic shock (HYPERS2S): a two-by-two factorial, multicentre, randomised, clinical trial. Lancet Respir Med. 2017:5(3):180-190. https://doi.org/10.1016/S2213-2600(17)30046-2.
13. Chu DK, Kim LH, Young PJ, et al. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta-analysis. Lancet. 2018;391(10131):1693-1705. https://doi.org/10.1016/S0140-6736(18)30479-3.
14. Rincon F, Kang J, Maltenfort M, et al. Association between hyperoxia and mortality after stroke: a multicenter cohort study. Crit Care Med. 2014;42(2):387-396. https://doi.org/10.1097/CCM.0b013e3182a27732.
15. Brenner M, Stein D, Hu P, Kufera J, Woodford M, Scalea T. Association between early hyperoxia and worse outcomes after traumatic brain injury. Arch Surg. 2012;147(11):1042-1046. https://doi.org/10.1001/archsurg.2012.1560.
16. Kilgannon JH, Jones AE, Shapiro NI, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303(21):2165-2171. https://doi.org/10.1
17. Siemieniuk RA, Chu DK, Kim L, et al. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ. 2018;363:k4169. https://doi.org/10.1136/bmj.k4169.
18. O’Driscoll BR, Howard LS, Earis J, et al. BTS guideline for oxygen use in adults in healthcare and emergency settings. Thorax. 2017;72(1):ii1-ii90. https://doi.org/10.1136/thoraxjnl-2016-209729.
19. Beasley R, Chien J, Douglas J, et al. Thoracic Society of Australia and New Zealand oxygen guidelines for acute oxygen use in adults: ‘Swimming between the flags’. Respirology. 2015;20(8):1182-1191. https://doi.org/10.1111/resp.12620.
© 2019 Society of Hospital Medicine
Long Peripheral Catheters: A Retrospective Review of Major Complications
Introduced in the 1950s, midline catheters have become a popular option for intravenous (IV) access.1,2 Ranging from 8 to 25 cm in length, they are inserted in the veins of the upper arm. Unlike peripherally inserted central catheters (PICCs), the tip of midline catheters terminates proximal to the axillary vein; thus, midlines are peripheral, not central venous access devices.1-3 One popular variation of a midline catheter, though nebulously defined, is the long peripheral catheter (LPC), a device ranging from 6 to 15 cm in length.4,5
Concerns regarding inappropriate use and complications such as thrombosis and central line-associated bloodstream infection (CLABSI) have spurred growth in the use of LPCs.6 However, data regarding complication rates with these devices are limited. Whether LPCs are a safe and viable option for IV access is unclear. We conducted a retrospective study to examine indications, patterns of use, and complications following LPC insertion in hospitalized patients.
METHODS
Device Selection
Our institution is a 470-bed tertiary care, safety-net hospital in Chicago, Illinois. Our vascular access team (VAT) performs a patient assessment and selects IV devices based upon published standards for device appropriateness. 7 We retrospectively collated electronic requests for LPC insertion on adult inpatients between October 2015 and June 2017. Cases where (1) duplicate orders, (2) patient refusal, (3) peripheral intravenous catheter of any length, or (4) PICCs were placed were excluded from this analysis.
VAT and Device Characteristics
We used Bard PowerGlide® (Bard Access Systems, Inc., Salt Lake City, Utah), an 18-gauge, 8-10 cm long, power-injectable, polyurethane LPC. Bundled kits (ie, device, gown, dressing, etc.) were utilized, and VAT providers underwent two weeks of training prior to the study period. All LPCs were inserted in the upper extremities under sterile technique using ultrasound guidance (accelerated Seldinger technique). Placement confirmation was verified by aspiration, flush, and ultrasound visualization of the catheter tip within the vein. An antimicrobial dressing was applied to the catheter insertion site, and daily saline flushes and weekly dressing changes by bedside nurses were used for device maintenance. LPC placement was available on all nonholiday weekdays from 8
Data Selection
For each LPC recipient, demographic and comorbidity data were collected to calculate the Charlson Comorbidity Index (Table 1). Every LPC recipient’s history of deep vein thrombosis (DVT) and catheter-related infection (CRI) was recorded. Procedural information (eg, inserter, vein, and number of attempts) was obtained from insertion notes. All data were extracted from the electronic medical record via chart review. Two reviewers verified outcomes to ensure concordance with stated definitions (ie, DVT, CRI). Device parameters, including dwell time, indication, and time to complication(s) were also collected.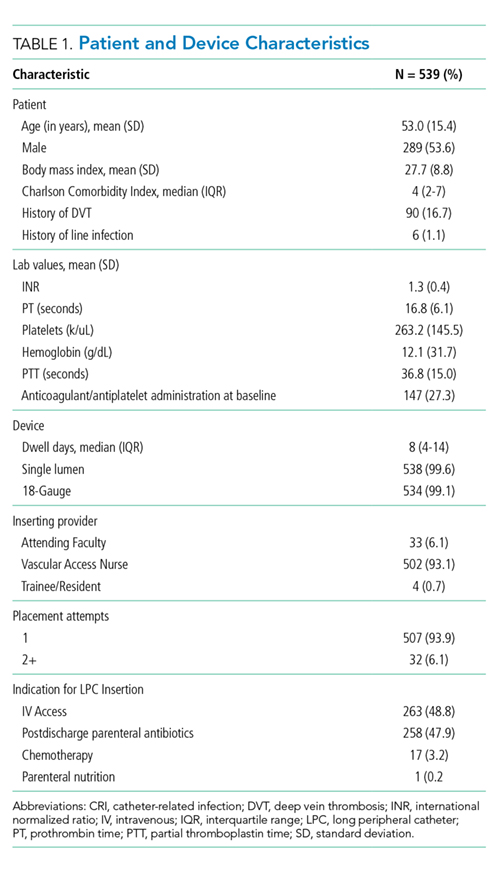
Primary Outcomes
The primary outcome was the incidence of DVT and CRI (Table 2). DVT was defined as radiographically confirmed (eg, ultrasound, computed tomography) thrombosis in the presence of patient signs or symptoms. CRI was defined in accordance with Timsit et al.8 as follows: catheter-related clinical sepsis without bloodstream infection defined as (1) combination of fever (body temperature >38.5°C) or hypothermia (body temperature <36.5°C), (2) catheter-tip culture yielding ≥103 CFUs/mL, (3) pus at the insertion site or resolution of clinical sepsis after catheter removal, and (4) absence of any other infectious focus or catheter-related bloodstream infection (CRBSI). CRBSI was defined as a combination of (1) one or more positive peripheral blood cultures sampled immediately before or within 48 hours after catheter removal, (2) a quantitative catheter-tip culture testing positive for the same microorganisms (same species and susceptibility pattern) or a differential time to positivity of blood cultures ≥2 hours, and (3) no other infectious focus explaining the positive blood culture result.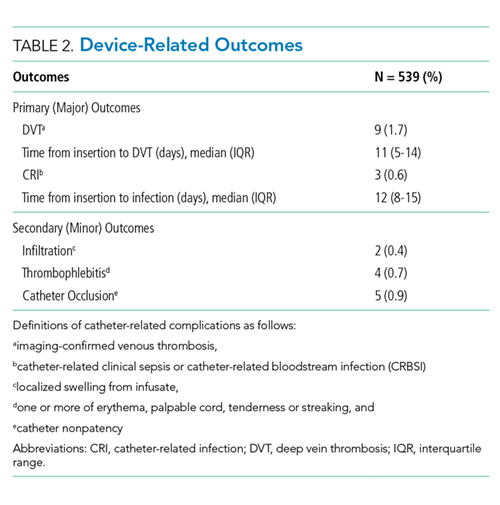
Secondary Outcomes
Secondary outcomes, defined as minor complications, included infiltration, thrombophlebitis, and catheter occlusion. Infiltration was defined as localized swelling due to infusate or site leakage. Thrombophlebitis was defined as one or more of the following: localized erythema, palpable cord, tenderness, or streaking. Occlusion was defined as nonpatency of the catheter due to the inability to flush or aspirate. Definitions for secondary outcomes are consistent with those used in prior studies.9
Statistical Analysis
Patient and LPC characteristics were analyzed using descriptive statistics. Results were reported as percentages, means, medians (interquartile range [IQR]), and rates per 1,000 catheter days. All analyses were conducted in Stata v.15 (StataCorp, College Station, Texas).
RESULTS
Within the 20-month study period, a total of 539 LPCs representing 5,543 catheter days were available for analysis. The mean patient age was 53 years. A total of 90 patients (16.7%) had a history of DVT, while 6 (1.1%) had a history of CRI. We calculated a median Charlson index of 4 (interquartile range [IQR], 2-7), suggesting an estimated one-year postdischarge survival of 53% (Table 1).
The majority of LPCs (99.6% [537/539]) were single lumen catheters. No patient had more than one concurrent LPC. The cannulation success rate on the first attempt was 93.9% (507/539). The brachial or basilic veins were primarily targeted (98.7%, [532/539]). Difficult intravenous access represented 48.8% (263/539) of indications, and postdischarge parenteral antibiotics constituted 47.9% (258/539). The median catheter dwell time was eight days (IQR, 4-14 days).
Nine DVTs (1.7% [9/539]) occurred in patients with LPCs. The incidence of DVT was higher in patients with a history of DVT (5.7%, 5/90). The median time from insertion to DVT was 11 (IQR, 5-14) days. DVTs were managed with LPC removal and systemic anticoagulation in accordance with catheter-related DVT guidelines. The rate of CRI was 0.6% (3/539), or 0.54 per 1,000 catheter days. Two CRIs had positive blood cultures, while one had negative cultures. Infections occurred after a median of 12 (IQR, 8-15) days of catheter dwell. Each was treated with LPC removal and IV antibiotics, with two patients receiving two weeks and one receiving six weeks of antibiotic therapy (Table 2).
With respect to secondary outcomes, the incidence of infiltration was 0.4% (2/539), thrombophlebitis 0.7% (4/539), and catheter occlusion 0.9% (5/539). The time to event was 8.5, 3.75, and 5.4 days, respectively. Collectively, 2.0% of devices experienced a minor complication.
DISCUSSION
In our single-center study, LPCs were primarily inserted for difficult venous access or parenteral antibiotics. Despite a clinically complex population with a high number of comorbidities, rates of major and minor complications associated with LPCs were low. These data suggest that LPCs are a safe alternative to PICCs and other central access devices for short-term use.
Our incidence of CRI of 0.6% (0.54 per 1,000 catheter days) is similar to or lower than other studies.2,10,11 An incidence of 0%-1.5% was observed in two recent publications about midline catheters, with rates across individual studies and hospital sites varying widely.12,13 A systematic review of intravascular devices reported CRI rates of 0.4% (0.2 per 1,000 catheter days) for midlines and 0.1% (0.5 per 1,000 catheter days for peripheral IVs), in contrast to PICCs at 3.1% (1.1 per 1,000 catheter days).14 However, catheters of varying lengths and diameters were used in studies within the review, potentially leading to heterogeneous outcomes. In accordance with existing data, CRI incidence in our study increased with catheter dwell time.10
The 1.7% rate of DVT observed in our study is on the lower end of existing data (1.4%-5.9%).12-15 Compared with PICCs (2%-15%), the incidence of venous thrombosis appears to be lower with midlines/LPCs—justifying their use as an alternative device for IV access.7,9,12,14 There was an overall low rate of minor complications, similar to recently published results.10 As rates were greater in patients with a history of DVT (5.7%), caution is warranted when using these devices in this population.
Our experience with LPCs suggests financial and patient benefits. The cost of LPCs is lower than central access devices.4 As rates of CRI were low, costs related to CLABSIs from PICC use may be reduced by appropriate LPC use. LPCs may allow the ability to draw blood routinely, which could improve the patient experience—albeit with its own risks. Current recommendations support the use of PICCs or LPCs, somewhat interchangeably, for patients with appropriate indications needing IV therapy for more than five to six days.2,7 However, LPCs now account for 57% of vascular access procedures in our center and have led to a decrease in reliance on PICCs and attendant complications.
Our study has several limitations. First, LPCs and midlines are often used interchangeably in the literature.4,5 Therefore, reported complication rates may not reflect those of LPCs alone and may limit comparisons. Second, ours was a single-center study with experts assessing device appropriateness and performing ultrasound-guided insertions; our findings may not be generalizable to dissimilar settings. Third, we did not track LPC complications such as nonpatency and leakage. As prior studies reported high rates of complications such as these events, caution is advised when interpreting our findings.15 Finally, we retrospectively extracted data from our medical records; limitations in documentation may influence our findings.
CONCLUSION
In patients requiring short-term IV therapy, these data suggest LPCs have low complication rates and may be safely used as an alternative option for venous access.
Acknowledgments
The authors thank Drs. Laura Hernandez, Andres Mendez Hernandez, and Victor Prado for their assistance in data collection. The authors also thank Mr. Onofre Donceras and Dr. Sharon Welbel from the John H. Stroger, Jr. Hospital of Cook County Department of Infection Control & Epidemiology for their assistance in reviewing local line infection data.
Drs. Patel and Chopra developed the study design. Drs. Patel, Araujo, Parra Rodriguez, Ramirez Sanchez, and Chopra contributed to manuscript writing. Ms. Snyder provided statistical analysis. All authors have seen and approved the final manuscript for submission.
Disclosures
The authors have nothing to disclose.
1. Anderson NR. Midline catheters: the middle ground of intravenous therapy administration. J Infus Nurs. 2004;27(5):313-321.
2. Adams DZ, Little A, Vinsant C, et al. The midline catheter: a clinical review. J Emerg Med. 2016;51(3):252-258. https://doi.org/10.1016/j.jemermed.2016.05.029.
3. Scoppettuolo G, Pittiruti M, Pitoni S, et al. Ultrasound-guided “short” midline catheters for difficult venous access in the emergency department: a retrospective analysis. Int J Emerg Med. 2016;9(1):3. https://doi.org/10.1186/s12245-016-0100-0.
4. Qin KR, Nataraja RM, Pacilli M. Long peripheral catheters: is it time to address the confusion? J Vasc Access. 2018;20(5). https://doi.org/10.1177/1129729818819730.
5. Pittiruti M, Scoppettuolo G. The GAVeCeLT Manual of PICC and Midlines. Milano: EDRA; 2016.
6. Dawson RB, Moureau NL. Midline catheters: an essential tool in CLABSI reduction. Infection Control Today. https://www.infectioncontroltoday.com/clabsi/midline-catheters-essential-tool-clabsi-reduction. Accessed February 19, 2018
7. Chopra V, Flanders SA, Saint S, et al. The Michigan Appropriateness Guide for Intravenous Catheters (MAGIC): results from a multispecialty panel using the RAND/UCLA appropriateness method. Ann Intern Med. 2015;163(6):S1-S40. https://doi.org/10.7326/M15-0744.
8. Timsit JF, Schwebel C, Bouadma L, et al. Chlorhexidine-impregnated sponges and less frequent dressing changes for prevention of catheter-related infections in critically ill adults: a randomized controlled trial. JAMA. 2009;301(12):1231-1241. https://doi.org/10.1001/jama.2009.376.
9. Bahl A, Karabon P, Chu D. Comparison of venous thrombosis complications in midlines versus peripherally inserted central catheters: are midlines the safer option? Clin Appl Thromb Hemost. 2019;25. https://doi.org/10.1177/1076029619839150.
10. Goetz AM, Miller J, Wagener MM, et al. Complications related to intravenous midline catheter usage. A 2-year study. J Intraven Nurs. 1998;21(2):76-80.
11. Xu T, Kingsley L, DiNucci S, et al. Safety and utilization of peripherally inserted central catheters versus midline catheters at a large academic medical center. Am J Infect Control. 2016;44(12):1458-1461. https://doi.org/10.1016/j.ajic.2016.09.010.
12. Chopra V, Kaatz S, Swaminathan L, et al. Variation in use and outcomes related to midline catheters: results from a multicentre pilot study. BMJ Qual Saf. 2019;28(9):714-720. https://doi.org/10.1136/bmjqs-2018-008554.
13. Badger J. Long peripheral catheters for deep arm vein venous access: A systematic review of complications. Heart Lung. 2019;48(3):222-225. https://doi.org/10.1016/j.hrtlng.2019.01.002.
14. Maki DG, Kluger DM, Crnich CJ. The risk of bloodstream infection in adults with different intravascular devices: a systematic review of 200 published prospective studies. Mayo Clin Proc. 2006;81(9):1159-1171. https://doi.org/10.4065/81.9.1159.
15. Zerla PA, Caravella G, De Luca G, et al. Open- vs closed-tip valved peripherally inserted central catheters and midlines: Findings from a vascular access database. J Assoc Vasc Access. 2015;20(3):169-176. https://doi.org/10.1016/j.java.2015.06.001.
Introduced in the 1950s, midline catheters have become a popular option for intravenous (IV) access.1,2 Ranging from 8 to 25 cm in length, they are inserted in the veins of the upper arm. Unlike peripherally inserted central catheters (PICCs), the tip of midline catheters terminates proximal to the axillary vein; thus, midlines are peripheral, not central venous access devices.1-3 One popular variation of a midline catheter, though nebulously defined, is the long peripheral catheter (LPC), a device ranging from 6 to 15 cm in length.4,5
Concerns regarding inappropriate use and complications such as thrombosis and central line-associated bloodstream infection (CLABSI) have spurred growth in the use of LPCs.6 However, data regarding complication rates with these devices are limited. Whether LPCs are a safe and viable option for IV access is unclear. We conducted a retrospective study to examine indications, patterns of use, and complications following LPC insertion in hospitalized patients.
METHODS
Device Selection
Our institution is a 470-bed tertiary care, safety-net hospital in Chicago, Illinois. Our vascular access team (VAT) performs a patient assessment and selects IV devices based upon published standards for device appropriateness. 7 We retrospectively collated electronic requests for LPC insertion on adult inpatients between October 2015 and June 2017. Cases where (1) duplicate orders, (2) patient refusal, (3) peripheral intravenous catheter of any length, or (4) PICCs were placed were excluded from this analysis.
VAT and Device Characteristics
We used Bard PowerGlide® (Bard Access Systems, Inc., Salt Lake City, Utah), an 18-gauge, 8-10 cm long, power-injectable, polyurethane LPC. Bundled kits (ie, device, gown, dressing, etc.) were utilized, and VAT providers underwent two weeks of training prior to the study period. All LPCs were inserted in the upper extremities under sterile technique using ultrasound guidance (accelerated Seldinger technique). Placement confirmation was verified by aspiration, flush, and ultrasound visualization of the catheter tip within the vein. An antimicrobial dressing was applied to the catheter insertion site, and daily saline flushes and weekly dressing changes by bedside nurses were used for device maintenance. LPC placement was available on all nonholiday weekdays from 8
Data Selection
For each LPC recipient, demographic and comorbidity data were collected to calculate the Charlson Comorbidity Index (Table 1). Every LPC recipient’s history of deep vein thrombosis (DVT) and catheter-related infection (CRI) was recorded. Procedural information (eg, inserter, vein, and number of attempts) was obtained from insertion notes. All data were extracted from the electronic medical record via chart review. Two reviewers verified outcomes to ensure concordance with stated definitions (ie, DVT, CRI). Device parameters, including dwell time, indication, and time to complication(s) were also collected.
Primary Outcomes
The primary outcome was the incidence of DVT and CRI (Table 2). DVT was defined as radiographically confirmed (eg, ultrasound, computed tomography) thrombosis in the presence of patient signs or symptoms. CRI was defined in accordance with Timsit et al.8 as follows: catheter-related clinical sepsis without bloodstream infection defined as (1) combination of fever (body temperature >38.5°C) or hypothermia (body temperature <36.5°C), (2) catheter-tip culture yielding ≥103 CFUs/mL, (3) pus at the insertion site or resolution of clinical sepsis after catheter removal, and (4) absence of any other infectious focus or catheter-related bloodstream infection (CRBSI). CRBSI was defined as a combination of (1) one or more positive peripheral blood cultures sampled immediately before or within 48 hours after catheter removal, (2) a quantitative catheter-tip culture testing positive for the same microorganisms (same species and susceptibility pattern) or a differential time to positivity of blood cultures ≥2 hours, and (3) no other infectious focus explaining the positive blood culture result.
Secondary Outcomes
Secondary outcomes, defined as minor complications, included infiltration, thrombophlebitis, and catheter occlusion. Infiltration was defined as localized swelling due to infusate or site leakage. Thrombophlebitis was defined as one or more of the following: localized erythema, palpable cord, tenderness, or streaking. Occlusion was defined as nonpatency of the catheter due to the inability to flush or aspirate. Definitions for secondary outcomes are consistent with those used in prior studies.9
Statistical Analysis
Patient and LPC characteristics were analyzed using descriptive statistics. Results were reported as percentages, means, medians (interquartile range [IQR]), and rates per 1,000 catheter days. All analyses were conducted in Stata v.15 (StataCorp, College Station, Texas).
RESULTS
Within the 20-month study period, a total of 539 LPCs representing 5,543 catheter days were available for analysis. The mean patient age was 53 years. A total of 90 patients (16.7%) had a history of DVT, while 6 (1.1%) had a history of CRI. We calculated a median Charlson index of 4 (interquartile range [IQR], 2-7), suggesting an estimated one-year postdischarge survival of 53% (Table 1).
The majority of LPCs (99.6% [537/539]) were single lumen catheters. No patient had more than one concurrent LPC. The cannulation success rate on the first attempt was 93.9% (507/539). The brachial or basilic veins were primarily targeted (98.7%, [532/539]). Difficult intravenous access represented 48.8% (263/539) of indications, and postdischarge parenteral antibiotics constituted 47.9% (258/539). The median catheter dwell time was eight days (IQR, 4-14 days).
Nine DVTs (1.7% [9/539]) occurred in patients with LPCs. The incidence of DVT was higher in patients with a history of DVT (5.7%, 5/90). The median time from insertion to DVT was 11 (IQR, 5-14) days. DVTs were managed with LPC removal and systemic anticoagulation in accordance with catheter-related DVT guidelines. The rate of CRI was 0.6% (3/539), or 0.54 per 1,000 catheter days. Two CRIs had positive blood cultures, while one had negative cultures. Infections occurred after a median of 12 (IQR, 8-15) days of catheter dwell. Each was treated with LPC removal and IV antibiotics, with two patients receiving two weeks and one receiving six weeks of antibiotic therapy (Table 2).
With respect to secondary outcomes, the incidence of infiltration was 0.4% (2/539), thrombophlebitis 0.7% (4/539), and catheter occlusion 0.9% (5/539). The time to event was 8.5, 3.75, and 5.4 days, respectively. Collectively, 2.0% of devices experienced a minor complication.
DISCUSSION
In our single-center study, LPCs were primarily inserted for difficult venous access or parenteral antibiotics. Despite a clinically complex population with a high number of comorbidities, rates of major and minor complications associated with LPCs were low. These data suggest that LPCs are a safe alternative to PICCs and other central access devices for short-term use.
Our incidence of CRI of 0.6% (0.54 per 1,000 catheter days) is similar to or lower than other studies.2,10,11 An incidence of 0%-1.5% was observed in two recent publications about midline catheters, with rates across individual studies and hospital sites varying widely.12,13 A systematic review of intravascular devices reported CRI rates of 0.4% (0.2 per 1,000 catheter days) for midlines and 0.1% (0.5 per 1,000 catheter days for peripheral IVs), in contrast to PICCs at 3.1% (1.1 per 1,000 catheter days).14 However, catheters of varying lengths and diameters were used in studies within the review, potentially leading to heterogeneous outcomes. In accordance with existing data, CRI incidence in our study increased with catheter dwell time.10
The 1.7% rate of DVT observed in our study is on the lower end of existing data (1.4%-5.9%).12-15 Compared with PICCs (2%-15%), the incidence of venous thrombosis appears to be lower with midlines/LPCs—justifying their use as an alternative device for IV access.7,9,12,14 There was an overall low rate of minor complications, similar to recently published results.10 As rates were greater in patients with a history of DVT (5.7%), caution is warranted when using these devices in this population.
Our experience with LPCs suggests financial and patient benefits. The cost of LPCs is lower than central access devices.4 As rates of CRI were low, costs related to CLABSIs from PICC use may be reduced by appropriate LPC use. LPCs may allow the ability to draw blood routinely, which could improve the patient experience—albeit with its own risks. Current recommendations support the use of PICCs or LPCs, somewhat interchangeably, for patients with appropriate indications needing IV therapy for more than five to six days.2,7 However, LPCs now account for 57% of vascular access procedures in our center and have led to a decrease in reliance on PICCs and attendant complications.
Our study has several limitations. First, LPCs and midlines are often used interchangeably in the literature.4,5 Therefore, reported complication rates may not reflect those of LPCs alone and may limit comparisons. Second, ours was a single-center study with experts assessing device appropriateness and performing ultrasound-guided insertions; our findings may not be generalizable to dissimilar settings. Third, we did not track LPC complications such as nonpatency and leakage. As prior studies reported high rates of complications such as these events, caution is advised when interpreting our findings.15 Finally, we retrospectively extracted data from our medical records; limitations in documentation may influence our findings.
CONCLUSION
In patients requiring short-term IV therapy, these data suggest LPCs have low complication rates and may be safely used as an alternative option for venous access.
Acknowledgments
The authors thank Drs. Laura Hernandez, Andres Mendez Hernandez, and Victor Prado for their assistance in data collection. The authors also thank Mr. Onofre Donceras and Dr. Sharon Welbel from the John H. Stroger, Jr. Hospital of Cook County Department of Infection Control & Epidemiology for their assistance in reviewing local line infection data.
Drs. Patel and Chopra developed the study design. Drs. Patel, Araujo, Parra Rodriguez, Ramirez Sanchez, and Chopra contributed to manuscript writing. Ms. Snyder provided statistical analysis. All authors have seen and approved the final manuscript for submission.
Disclosures
The authors have nothing to disclose.
Introduced in the 1950s, midline catheters have become a popular option for intravenous (IV) access.1,2 Ranging from 8 to 25 cm in length, they are inserted in the veins of the upper arm. Unlike peripherally inserted central catheters (PICCs), the tip of midline catheters terminates proximal to the axillary vein; thus, midlines are peripheral, not central venous access devices.1-3 One popular variation of a midline catheter, though nebulously defined, is the long peripheral catheter (LPC), a device ranging from 6 to 15 cm in length.4,5
Concerns regarding inappropriate use and complications such as thrombosis and central line-associated bloodstream infection (CLABSI) have spurred growth in the use of LPCs.6 However, data regarding complication rates with these devices are limited. Whether LPCs are a safe and viable option for IV access is unclear. We conducted a retrospective study to examine indications, patterns of use, and complications following LPC insertion in hospitalized patients.
METHODS
Device Selection
Our institution is a 470-bed tertiary care, safety-net hospital in Chicago, Illinois. Our vascular access team (VAT) performs a patient assessment and selects IV devices based upon published standards for device appropriateness. 7 We retrospectively collated electronic requests for LPC insertion on adult inpatients between October 2015 and June 2017. Cases where (1) duplicate orders, (2) patient refusal, (3) peripheral intravenous catheter of any length, or (4) PICCs were placed were excluded from this analysis.
VAT and Device Characteristics
We used Bard PowerGlide® (Bard Access Systems, Inc., Salt Lake City, Utah), an 18-gauge, 8-10 cm long, power-injectable, polyurethane LPC. Bundled kits (ie, device, gown, dressing, etc.) were utilized, and VAT providers underwent two weeks of training prior to the study period. All LPCs were inserted in the upper extremities under sterile technique using ultrasound guidance (accelerated Seldinger technique). Placement confirmation was verified by aspiration, flush, and ultrasound visualization of the catheter tip within the vein. An antimicrobial dressing was applied to the catheter insertion site, and daily saline flushes and weekly dressing changes by bedside nurses were used for device maintenance. LPC placement was available on all nonholiday weekdays from 8
Data Selection
For each LPC recipient, demographic and comorbidity data were collected to calculate the Charlson Comorbidity Index (Table 1). Every LPC recipient’s history of deep vein thrombosis (DVT) and catheter-related infection (CRI) was recorded. Procedural information (eg, inserter, vein, and number of attempts) was obtained from insertion notes. All data were extracted from the electronic medical record via chart review. Two reviewers verified outcomes to ensure concordance with stated definitions (ie, DVT, CRI). Device parameters, including dwell time, indication, and time to complication(s) were also collected.
Primary Outcomes
The primary outcome was the incidence of DVT and CRI (Table 2). DVT was defined as radiographically confirmed (eg, ultrasound, computed tomography) thrombosis in the presence of patient signs or symptoms. CRI was defined in accordance with Timsit et al.8 as follows: catheter-related clinical sepsis without bloodstream infection defined as (1) combination of fever (body temperature >38.5°C) or hypothermia (body temperature <36.5°C), (2) catheter-tip culture yielding ≥103 CFUs/mL, (3) pus at the insertion site or resolution of clinical sepsis after catheter removal, and (4) absence of any other infectious focus or catheter-related bloodstream infection (CRBSI). CRBSI was defined as a combination of (1) one or more positive peripheral blood cultures sampled immediately before or within 48 hours after catheter removal, (2) a quantitative catheter-tip culture testing positive for the same microorganisms (same species and susceptibility pattern) or a differential time to positivity of blood cultures ≥2 hours, and (3) no other infectious focus explaining the positive blood culture result.
Secondary Outcomes
Secondary outcomes, defined as minor complications, included infiltration, thrombophlebitis, and catheter occlusion. Infiltration was defined as localized swelling due to infusate or site leakage. Thrombophlebitis was defined as one or more of the following: localized erythema, palpable cord, tenderness, or streaking. Occlusion was defined as nonpatency of the catheter due to the inability to flush or aspirate. Definitions for secondary outcomes are consistent with those used in prior studies.9
Statistical Analysis
Patient and LPC characteristics were analyzed using descriptive statistics. Results were reported as percentages, means, medians (interquartile range [IQR]), and rates per 1,000 catheter days. All analyses were conducted in Stata v.15 (StataCorp, College Station, Texas).
RESULTS
Within the 20-month study period, a total of 539 LPCs representing 5,543 catheter days were available for analysis. The mean patient age was 53 years. A total of 90 patients (16.7%) had a history of DVT, while 6 (1.1%) had a history of CRI. We calculated a median Charlson index of 4 (interquartile range [IQR], 2-7), suggesting an estimated one-year postdischarge survival of 53% (Table 1).
The majority of LPCs (99.6% [537/539]) were single lumen catheters. No patient had more than one concurrent LPC. The cannulation success rate on the first attempt was 93.9% (507/539). The brachial or basilic veins were primarily targeted (98.7%, [532/539]). Difficult intravenous access represented 48.8% (263/539) of indications, and postdischarge parenteral antibiotics constituted 47.9% (258/539). The median catheter dwell time was eight days (IQR, 4-14 days).
Nine DVTs (1.7% [9/539]) occurred in patients with LPCs. The incidence of DVT was higher in patients with a history of DVT (5.7%, 5/90). The median time from insertion to DVT was 11 (IQR, 5-14) days. DVTs were managed with LPC removal and systemic anticoagulation in accordance with catheter-related DVT guidelines. The rate of CRI was 0.6% (3/539), or 0.54 per 1,000 catheter days. Two CRIs had positive blood cultures, while one had negative cultures. Infections occurred after a median of 12 (IQR, 8-15) days of catheter dwell. Each was treated with LPC removal and IV antibiotics, with two patients receiving two weeks and one receiving six weeks of antibiotic therapy (Table 2).
With respect to secondary outcomes, the incidence of infiltration was 0.4% (2/539), thrombophlebitis 0.7% (4/539), and catheter occlusion 0.9% (5/539). The time to event was 8.5, 3.75, and 5.4 days, respectively. Collectively, 2.0% of devices experienced a minor complication.
DISCUSSION
In our single-center study, LPCs were primarily inserted for difficult venous access or parenteral antibiotics. Despite a clinically complex population with a high number of comorbidities, rates of major and minor complications associated with LPCs were low. These data suggest that LPCs are a safe alternative to PICCs and other central access devices for short-term use.
Our incidence of CRI of 0.6% (0.54 per 1,000 catheter days) is similar to or lower than other studies.2,10,11 An incidence of 0%-1.5% was observed in two recent publications about midline catheters, with rates across individual studies and hospital sites varying widely.12,13 A systematic review of intravascular devices reported CRI rates of 0.4% (0.2 per 1,000 catheter days) for midlines and 0.1% (0.5 per 1,000 catheter days for peripheral IVs), in contrast to PICCs at 3.1% (1.1 per 1,000 catheter days).14 However, catheters of varying lengths and diameters were used in studies within the review, potentially leading to heterogeneous outcomes. In accordance with existing data, CRI incidence in our study increased with catheter dwell time.10
The 1.7% rate of DVT observed in our study is on the lower end of existing data (1.4%-5.9%).12-15 Compared with PICCs (2%-15%), the incidence of venous thrombosis appears to be lower with midlines/LPCs—justifying their use as an alternative device for IV access.7,9,12,14 There was an overall low rate of minor complications, similar to recently published results.10 As rates were greater in patients with a history of DVT (5.7%), caution is warranted when using these devices in this population.
Our experience with LPCs suggests financial and patient benefits. The cost of LPCs is lower than central access devices.4 As rates of CRI were low, costs related to CLABSIs from PICC use may be reduced by appropriate LPC use. LPCs may allow the ability to draw blood routinely, which could improve the patient experience—albeit with its own risks. Current recommendations support the use of PICCs or LPCs, somewhat interchangeably, for patients with appropriate indications needing IV therapy for more than five to six days.2,7 However, LPCs now account for 57% of vascular access procedures in our center and have led to a decrease in reliance on PICCs and attendant complications.
Our study has several limitations. First, LPCs and midlines are often used interchangeably in the literature.4,5 Therefore, reported complication rates may not reflect those of LPCs alone and may limit comparisons. Second, ours was a single-center study with experts assessing device appropriateness and performing ultrasound-guided insertions; our findings may not be generalizable to dissimilar settings. Third, we did not track LPC complications such as nonpatency and leakage. As prior studies reported high rates of complications such as these events, caution is advised when interpreting our findings.15 Finally, we retrospectively extracted data from our medical records; limitations in documentation may influence our findings.
CONCLUSION
In patients requiring short-term IV therapy, these data suggest LPCs have low complication rates and may be safely used as an alternative option for venous access.
Acknowledgments
The authors thank Drs. Laura Hernandez, Andres Mendez Hernandez, and Victor Prado for their assistance in data collection. The authors also thank Mr. Onofre Donceras and Dr. Sharon Welbel from the John H. Stroger, Jr. Hospital of Cook County Department of Infection Control & Epidemiology for their assistance in reviewing local line infection data.
Drs. Patel and Chopra developed the study design. Drs. Patel, Araujo, Parra Rodriguez, Ramirez Sanchez, and Chopra contributed to manuscript writing. Ms. Snyder provided statistical analysis. All authors have seen and approved the final manuscript for submission.
Disclosures
The authors have nothing to disclose.
1. Anderson NR. Midline catheters: the middle ground of intravenous therapy administration. J Infus Nurs. 2004;27(5):313-321.
2. Adams DZ, Little A, Vinsant C, et al. The midline catheter: a clinical review. J Emerg Med. 2016;51(3):252-258. https://doi.org/10.1016/j.jemermed.2016.05.029.
3. Scoppettuolo G, Pittiruti M, Pitoni S, et al. Ultrasound-guided “short” midline catheters for difficult venous access in the emergency department: a retrospective analysis. Int J Emerg Med. 2016;9(1):3. https://doi.org/10.1186/s12245-016-0100-0.
4. Qin KR, Nataraja RM, Pacilli M. Long peripheral catheters: is it time to address the confusion? J Vasc Access. 2018;20(5). https://doi.org/10.1177/1129729818819730.
5. Pittiruti M, Scoppettuolo G. The GAVeCeLT Manual of PICC and Midlines. Milano: EDRA; 2016.
6. Dawson RB, Moureau NL. Midline catheters: an essential tool in CLABSI reduction. Infection Control Today. https://www.infectioncontroltoday.com/clabsi/midline-catheters-essential-tool-clabsi-reduction. Accessed February 19, 2018
7. Chopra V, Flanders SA, Saint S, et al. The Michigan Appropriateness Guide for Intravenous Catheters (MAGIC): results from a multispecialty panel using the RAND/UCLA appropriateness method. Ann Intern Med. 2015;163(6):S1-S40. https://doi.org/10.7326/M15-0744.
8. Timsit JF, Schwebel C, Bouadma L, et al. Chlorhexidine-impregnated sponges and less frequent dressing changes for prevention of catheter-related infections in critically ill adults: a randomized controlled trial. JAMA. 2009;301(12):1231-1241. https://doi.org/10.1001/jama.2009.376.
9. Bahl A, Karabon P, Chu D. Comparison of venous thrombosis complications in midlines versus peripherally inserted central catheters: are midlines the safer option? Clin Appl Thromb Hemost. 2019;25. https://doi.org/10.1177/1076029619839150.
10. Goetz AM, Miller J, Wagener MM, et al. Complications related to intravenous midline catheter usage. A 2-year study. J Intraven Nurs. 1998;21(2):76-80.
11. Xu T, Kingsley L, DiNucci S, et al. Safety and utilization of peripherally inserted central catheters versus midline catheters at a large academic medical center. Am J Infect Control. 2016;44(12):1458-1461. https://doi.org/10.1016/j.ajic.2016.09.010.
12. Chopra V, Kaatz S, Swaminathan L, et al. Variation in use and outcomes related to midline catheters: results from a multicentre pilot study. BMJ Qual Saf. 2019;28(9):714-720. https://doi.org/10.1136/bmjqs-2018-008554.
13. Badger J. Long peripheral catheters for deep arm vein venous access: A systematic review of complications. Heart Lung. 2019;48(3):222-225. https://doi.org/10.1016/j.hrtlng.2019.01.002.
14. Maki DG, Kluger DM, Crnich CJ. The risk of bloodstream infection in adults with different intravascular devices: a systematic review of 200 published prospective studies. Mayo Clin Proc. 2006;81(9):1159-1171. https://doi.org/10.4065/81.9.1159.
15. Zerla PA, Caravella G, De Luca G, et al. Open- vs closed-tip valved peripherally inserted central catheters and midlines: Findings from a vascular access database. J Assoc Vasc Access. 2015;20(3):169-176. https://doi.org/10.1016/j.java.2015.06.001.
1. Anderson NR. Midline catheters: the middle ground of intravenous therapy administration. J Infus Nurs. 2004;27(5):313-321.
2. Adams DZ, Little A, Vinsant C, et al. The midline catheter: a clinical review. J Emerg Med. 2016;51(3):252-258. https://doi.org/10.1016/j.jemermed.2016.05.029.
3. Scoppettuolo G, Pittiruti M, Pitoni S, et al. Ultrasound-guided “short” midline catheters for difficult venous access in the emergency department: a retrospective analysis. Int J Emerg Med. 2016;9(1):3. https://doi.org/10.1186/s12245-016-0100-0.
4. Qin KR, Nataraja RM, Pacilli M. Long peripheral catheters: is it time to address the confusion? J Vasc Access. 2018;20(5). https://doi.org/10.1177/1129729818819730.
5. Pittiruti M, Scoppettuolo G. The GAVeCeLT Manual of PICC and Midlines. Milano: EDRA; 2016.
6. Dawson RB, Moureau NL. Midline catheters: an essential tool in CLABSI reduction. Infection Control Today. https://www.infectioncontroltoday.com/clabsi/midline-catheters-essential-tool-clabsi-reduction. Accessed February 19, 2018
7. Chopra V, Flanders SA, Saint S, et al. The Michigan Appropriateness Guide for Intravenous Catheters (MAGIC): results from a multispecialty panel using the RAND/UCLA appropriateness method. Ann Intern Med. 2015;163(6):S1-S40. https://doi.org/10.7326/M15-0744.
8. Timsit JF, Schwebel C, Bouadma L, et al. Chlorhexidine-impregnated sponges and less frequent dressing changes for prevention of catheter-related infections in critically ill adults: a randomized controlled trial. JAMA. 2009;301(12):1231-1241. https://doi.org/10.1001/jama.2009.376.
9. Bahl A, Karabon P, Chu D. Comparison of venous thrombosis complications in midlines versus peripherally inserted central catheters: are midlines the safer option? Clin Appl Thromb Hemost. 2019;25. https://doi.org/10.1177/1076029619839150.
10. Goetz AM, Miller J, Wagener MM, et al. Complications related to intravenous midline catheter usage. A 2-year study. J Intraven Nurs. 1998;21(2):76-80.
11. Xu T, Kingsley L, DiNucci S, et al. Safety and utilization of peripherally inserted central catheters versus midline catheters at a large academic medical center. Am J Infect Control. 2016;44(12):1458-1461. https://doi.org/10.1016/j.ajic.2016.09.010.
12. Chopra V, Kaatz S, Swaminathan L, et al. Variation in use and outcomes related to midline catheters: results from a multicentre pilot study. BMJ Qual Saf. 2019;28(9):714-720. https://doi.org/10.1136/bmjqs-2018-008554.
13. Badger J. Long peripheral catheters for deep arm vein venous access: A systematic review of complications. Heart Lung. 2019;48(3):222-225. https://doi.org/10.1016/j.hrtlng.2019.01.002.
14. Maki DG, Kluger DM, Crnich CJ. The risk of bloodstream infection in adults with different intravascular devices: a systematic review of 200 published prospective studies. Mayo Clin Proc. 2006;81(9):1159-1171. https://doi.org/10.4065/81.9.1159.
15. Zerla PA, Caravella G, De Luca G, et al. Open- vs closed-tip valved peripherally inserted central catheters and midlines: Findings from a vascular access database. J Assoc Vasc Access. 2015;20(3):169-176. https://doi.org/10.1016/j.java.2015.06.001.
© 2019 Society of Hospital Medicine
Clinical Guideline Highlights for the Hospitalist: Clostridium difficile Infections in Children
Clostridium difficile (name changed to Clostridioides difficile [CDI]) are a major public health problem, with 500,000 infections annually in the United States, 15,000-30,000 associated deaths, and acute care costs exceeding $4.8 billion. The recent clinical practice guideline for CDI provides recommendations about the epidemiology, diagnosis, treatment, prevention, and environmental management. A total of 52 recommendations are included, and we will review 11 with pertinence to pediatrics in this highlight.
KEY RECOMMENDATIONS FOR THE HOSPITALIST
Recommendation 1. In infants ≤12 months of age, testing for CDI should never be routinely recommended because of the high prevalence of asymptomatic carriage of toxigenic C. difficile in infants (strong recommendation, moderate quality of evidence).
Recommendation 2. In children 1-2 years of age, testing should not be routinely performed unless other causes have been excluded (weak recommendation, low quality of evidence).Recommendation 3. In children ≥2 years of age, testing is recommended for patients with prolonged or worsening diarrhea and risk factors (eg, underlying inflammatory bowel disease) or immunocompromising conditions) or relevant exposures (eg, contact with the healthcare system or recent antibiotics) (weak recommendation, moderate quality of evidence).
The rate of C. difficile colonization among asymptomatic infants can exceed 40%. This rate declines over the first year but remains 15% at 12 months of age.1 Therefore, the guideline recommends against routinely testing infants ≤12 months of age as a positive test probably reflects colonization rather than disease. Testing in infants is recommended only when other causes have been excluded and a concern for pseudomembranous colitis, toxic megacolon, or clinically significant diarrhea exists.
The rate of asymptomatic colonization remains elevated in the second year of life. By 2-3 years, the rate is 1%-3% which is similar to that in healthy adults. However, the role of C. difficile in community-onset diarrhea in otherwise healthy children is controversial. In a study of 100 hospitalized children aged <2 years with CDI and diarrhea, all had resolution of diarrhea regardless of whether therapy was administered.2 Another study found an alternative pathogen in >50% of hospitalized children with CDI.3 Therefore, the guideline recommends against testing in children aged 1-2 years unless other causes have been excluded and in children aged >2 years only when they have prolonged or worsening diarrhea along with risk factors or exposures.
Recommendation 4. In institutions without specific required criteria for stool submissions, use a stool toxin test as part of a multistep algorithm (ie, glutamate dehydrogenase [GDH] plus toxin, GDH plus toxin arbitrated by nucleic-acid amplification tests [NAAT], or NAAT plus toxin) rather than a NAAT alone (weak recommendation, low quality of evidence).
Recommendation 5. In institutions with specific required criteria for stool submissions, use a NAAT alone or a multistep algorithm for testing (ie, GDH plus toxin, GDH plus toxin arbitrated by NAAT, or NAAT plus toxin) rather than a toxin test alone (weak recommendation, low quality of evidence).
There are a variety of testing approaches for CDI and recommendations vary based on local practice. If laboratories accept all stools, a more specific approach is recommended, including a toxin test as part of a multistep algorithm to limit false positives. If laboratories first screen for symptoms and antibiotic exposure before accepting stool samples, a more sensitive approach is recommended including NAAT alone or a multistep algorithm rather than toxin alone.
Infection Prevention and Control
Recommendation 6. There is insufficient evidence for discontinuation of PPIs (proton pump inhibitors) as a measure for preventing CDI (no recommendation).
The guideline acknowledges data suggesting an association between PPI use and CDI, but not a causal relationship. Due to the lack of high-quality evidence, it does not recommend stopping PPIs to prevent CDI.
Recommendation 7. There are insufficient data to recommend probiotics for primary prevention of CDI outside of clinical trials (no recommendation).
The guideline notes that although several meta-analyses indicate that probiotics may prevent CDI; however there were limitations, including a high incidence of CDI in placebo arms and differences in probiotic formulations and duration of use, leading to insufficient data to recommend probiotic use to prevent CDI.
Treatment
Recommendation 8. Either per os (PO) metronidazole or PO vancomycin is recommended for an initial episode or first recurrence of nonsevere pediatric CDI (weak recommendation, low quality of evidence).
Data assessing the optimal treatment for nonsevere pediatric CDI are limited. Emerging data support the use of vancomycin,4 which is now recommended for initial episodes of CDI in adults. However, there are insufficient data to recommend vancomycin over metronidazole for nonsevere pediatric CDI; therefore, either option is recommended.
Recommendation 9. For children with an initial episode of severe CDI, oral vancomycin with or without IV metronidazole is recommended over metronidazole alone (strong recommendation, moderate quality of evidence).
Recommendation 10. For children with a second or greater episode of recurrent CDI, oral vancomycin is recommended over metronidazole (weak recommendation, low quality of evidence).
There is no well-designed trial comparing metronidazole and vancomycin for severe or recurrent pediatric CDI. For children previously treated with metronidazole, vancomycin is recommended based on adult literature.4 For children previously treated with metronidazole and vancomycin, an extended course of tapered or pulse regimen vancomycin or vancomycin followed by rifaximin is recommended.
Recommendations must weigh potential harms. Metronidazole has been associated with neuropathies,5 cramping, and nausea. PO vancomycin has poor enteral absorption, minimizing systemic effects. Both vancomycin and metronidazole may promote carriage of resistant enterococci.
Recommendation 11. Fecal microbiota transplantation (FMT) should be considered for pediatric patients with multiple recurrences of CDI following standard treatments (weak recommendation, very low quality of evidence).
There are no robust data examining the effectiveness of pediatric FMT. Recommendations are guided by adult studies. Limited evidence suggests that FMT can be effective in children with multiple recurrent CDI.6 Concerns include procedure-related risks, transmission of resistant organisms and blood-borne pathogens, and induced metabolic or immunologic disorders.
CRITIQUE
Methods in Preparing a Guideline
The strength of a guideline includes representation from a diverse panel, including the Infectious Diseases Society of America (IDSA), the Society for Healthcare Epidemiology of America, the American Society of Health-Systems Pharmacists, the Society of Infectious Diseases Pharmacists, and the Pediatric Infectious Diseases Society.
The panel utilized the Grading of Recommendations Assessment, Development, and Evaluation system to weigh the strength and quality of evidence.
From a pediatric perspective, the current guideline added pediatric-specific recommendations based on a comprehensive review of the literature from 1977 to 2016. The strength of these recommendations is somewhat limited by the lack of well-designed pediatric studies. An additional limitation is that treatment recommendations are based on illness severity, although the definitions used to classify severity are not pediatric-specific and are based on unvalidated expert opinion.
Sources of Potential Conflicts or Interest or Bias
The panel complied with the IDSA policy on conflicts of interest and disclosed any interest that might be construed as a conflict, regardless of relevancy. These were evaluated by the IDSA Standards and Practice Guidelines Committee.
Generalizability
Guideline generalizability may be impacted by testing availabilities within a particular setting. Cost factors and local formularies may also limit treatment options within a given setting.
Areas in Need of Future Study
Research gaps exist regarding at what age C. difficile is pathogenic given the prevalence of asymptomatic carriage. Future studies can also focus on a newly available molecular polymerase chain reaction test platform that detects C. difficile.7
There is limited pediatric evidence to recommend metronidazole versus vancomycin in children, particularly in nonsevere cases. There is also an opportunity to further explore alternative therapies, including fidaxomicin (not currently approved for children) and bezlotoxumab, a new agent approved as adult adjunctive therapy.8
1. Donta ST, Myers MG. Clostridium difficile toxin in asymptomatic neonates. J Pediatr. 1982;100(3):431-434. https://doi.org/10.1016/s0022-3476(82)80454-x.
2. González-Del Vecchio M, Álvarez-Uria A, Marin M, et al. Clinical significance of Clostridium difficile in children less than 2 years old: a case-control study. Pediatr Infect Dis J. 2016;35(3):281-285. https://doi.org/10.1097/INF.0000000000001008.
3. Valentini D, Vittucci AC, Grandin A, et al. Coinfection in acute gastroenteritis predicts a more severe clinical course in children. Eur J Clin Microbiol Infect Dis. 2013;32(7):909-915. https://doi.org/10.1007/s10096-013-1825-9.
4. Johnson S, Louie TJ, Gerding DN, et al. Vancomycin, metronidazole, or tolevamer for Clostridium difficile infection: results from two multinational, randomized, controlled trials. Clin Infect Dis. 2014;59(3):345-354. https://doi.org/10.1093/cid/ciu313.
5. Yamamoto T, Abe K, Anjiki H, Ishii T, Kuyama Y. Metronidazole-induced neurotoxicity developed in liver cirrhosis. J Clin Med Res. 2012;4(4):295-298. https://doi.org/10.4021/jocmr893w.
6. Russell G, Kaplan J, Ferraro M, Michelow IC. Fecal bacteriotherapy for relapsing Clostridium difficile infection in a child: a proposed treatment protocol. Pediatrics. 2010;126(1):e239-e242. https://doi.org/10.1542/peds.2009-3363.
7. Zhang H, Morrison S, Tang YW. Multiplex polymerase chain reaction tests for detection of pathogens associated with gastroenteritis. Clin Lab Med. 2015;35(2):461-486. https://doi.org/10.1016/j.cll.2015.02.006.
8. Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med. 2017;376(4):305-317. https://doi.org/10.1056/NEJMoa1602615.
Clostridium difficile (name changed to Clostridioides difficile [CDI]) are a major public health problem, with 500,000 infections annually in the United States, 15,000-30,000 associated deaths, and acute care costs exceeding $4.8 billion. The recent clinical practice guideline for CDI provides recommendations about the epidemiology, diagnosis, treatment, prevention, and environmental management. A total of 52 recommendations are included, and we will review 11 with pertinence to pediatrics in this highlight.
KEY RECOMMENDATIONS FOR THE HOSPITALIST
Recommendation 1. In infants ≤12 months of age, testing for CDI should never be routinely recommended because of the high prevalence of asymptomatic carriage of toxigenic C. difficile in infants (strong recommendation, moderate quality of evidence).
Recommendation 2. In children 1-2 years of age, testing should not be routinely performed unless other causes have been excluded (weak recommendation, low quality of evidence).Recommendation 3. In children ≥2 years of age, testing is recommended for patients with prolonged or worsening diarrhea and risk factors (eg, underlying inflammatory bowel disease) or immunocompromising conditions) or relevant exposures (eg, contact with the healthcare system or recent antibiotics) (weak recommendation, moderate quality of evidence).
The rate of C. difficile colonization among asymptomatic infants can exceed 40%. This rate declines over the first year but remains 15% at 12 months of age.1 Therefore, the guideline recommends against routinely testing infants ≤12 months of age as a positive test probably reflects colonization rather than disease. Testing in infants is recommended only when other causes have been excluded and a concern for pseudomembranous colitis, toxic megacolon, or clinically significant diarrhea exists.
The rate of asymptomatic colonization remains elevated in the second year of life. By 2-3 years, the rate is 1%-3% which is similar to that in healthy adults. However, the role of C. difficile in community-onset diarrhea in otherwise healthy children is controversial. In a study of 100 hospitalized children aged <2 years with CDI and diarrhea, all had resolution of diarrhea regardless of whether therapy was administered.2 Another study found an alternative pathogen in >50% of hospitalized children with CDI.3 Therefore, the guideline recommends against testing in children aged 1-2 years unless other causes have been excluded and in children aged >2 years only when they have prolonged or worsening diarrhea along with risk factors or exposures.
Recommendation 4. In institutions without specific required criteria for stool submissions, use a stool toxin test as part of a multistep algorithm (ie, glutamate dehydrogenase [GDH] plus toxin, GDH plus toxin arbitrated by nucleic-acid amplification tests [NAAT], or NAAT plus toxin) rather than a NAAT alone (weak recommendation, low quality of evidence).
Recommendation 5. In institutions with specific required criteria for stool submissions, use a NAAT alone or a multistep algorithm for testing (ie, GDH plus toxin, GDH plus toxin arbitrated by NAAT, or NAAT plus toxin) rather than a toxin test alone (weak recommendation, low quality of evidence).
There are a variety of testing approaches for CDI and recommendations vary based on local practice. If laboratories accept all stools, a more specific approach is recommended, including a toxin test as part of a multistep algorithm to limit false positives. If laboratories first screen for symptoms and antibiotic exposure before accepting stool samples, a more sensitive approach is recommended including NAAT alone or a multistep algorithm rather than toxin alone.
Infection Prevention and Control
Recommendation 6. There is insufficient evidence for discontinuation of PPIs (proton pump inhibitors) as a measure for preventing CDI (no recommendation).
The guideline acknowledges data suggesting an association between PPI use and CDI, but not a causal relationship. Due to the lack of high-quality evidence, it does not recommend stopping PPIs to prevent CDI.
Recommendation 7. There are insufficient data to recommend probiotics for primary prevention of CDI outside of clinical trials (no recommendation).
The guideline notes that although several meta-analyses indicate that probiotics may prevent CDI; however there were limitations, including a high incidence of CDI in placebo arms and differences in probiotic formulations and duration of use, leading to insufficient data to recommend probiotic use to prevent CDI.
Treatment
Recommendation 8. Either per os (PO) metronidazole or PO vancomycin is recommended for an initial episode or first recurrence of nonsevere pediatric CDI (weak recommendation, low quality of evidence).
Data assessing the optimal treatment for nonsevere pediatric CDI are limited. Emerging data support the use of vancomycin,4 which is now recommended for initial episodes of CDI in adults. However, there are insufficient data to recommend vancomycin over metronidazole for nonsevere pediatric CDI; therefore, either option is recommended.
Recommendation 9. For children with an initial episode of severe CDI, oral vancomycin with or without IV metronidazole is recommended over metronidazole alone (strong recommendation, moderate quality of evidence).
Recommendation 10. For children with a second or greater episode of recurrent CDI, oral vancomycin is recommended over metronidazole (weak recommendation, low quality of evidence).
There is no well-designed trial comparing metronidazole and vancomycin for severe or recurrent pediatric CDI. For children previously treated with metronidazole, vancomycin is recommended based on adult literature.4 For children previously treated with metronidazole and vancomycin, an extended course of tapered or pulse regimen vancomycin or vancomycin followed by rifaximin is recommended.
Recommendations must weigh potential harms. Metronidazole has been associated with neuropathies,5 cramping, and nausea. PO vancomycin has poor enteral absorption, minimizing systemic effects. Both vancomycin and metronidazole may promote carriage of resistant enterococci.
Recommendation 11. Fecal microbiota transplantation (FMT) should be considered for pediatric patients with multiple recurrences of CDI following standard treatments (weak recommendation, very low quality of evidence).
There are no robust data examining the effectiveness of pediatric FMT. Recommendations are guided by adult studies. Limited evidence suggests that FMT can be effective in children with multiple recurrent CDI.6 Concerns include procedure-related risks, transmission of resistant organisms and blood-borne pathogens, and induced metabolic or immunologic disorders.
CRITIQUE
Methods in Preparing a Guideline
The strength of a guideline includes representation from a diverse panel, including the Infectious Diseases Society of America (IDSA), the Society for Healthcare Epidemiology of America, the American Society of Health-Systems Pharmacists, the Society of Infectious Diseases Pharmacists, and the Pediatric Infectious Diseases Society.
The panel utilized the Grading of Recommendations Assessment, Development, and Evaluation system to weigh the strength and quality of evidence.
From a pediatric perspective, the current guideline added pediatric-specific recommendations based on a comprehensive review of the literature from 1977 to 2016. The strength of these recommendations is somewhat limited by the lack of well-designed pediatric studies. An additional limitation is that treatment recommendations are based on illness severity, although the definitions used to classify severity are not pediatric-specific and are based on unvalidated expert opinion.
Sources of Potential Conflicts or Interest or Bias
The panel complied with the IDSA policy on conflicts of interest and disclosed any interest that might be construed as a conflict, regardless of relevancy. These were evaluated by the IDSA Standards and Practice Guidelines Committee.
Generalizability
Guideline generalizability may be impacted by testing availabilities within a particular setting. Cost factors and local formularies may also limit treatment options within a given setting.
Areas in Need of Future Study
Research gaps exist regarding at what age C. difficile is pathogenic given the prevalence of asymptomatic carriage. Future studies can also focus on a newly available molecular polymerase chain reaction test platform that detects C. difficile.7
There is limited pediatric evidence to recommend metronidazole versus vancomycin in children, particularly in nonsevere cases. There is also an opportunity to further explore alternative therapies, including fidaxomicin (not currently approved for children) and bezlotoxumab, a new agent approved as adult adjunctive therapy.8
Clostridium difficile (name changed to Clostridioides difficile [CDI]) are a major public health problem, with 500,000 infections annually in the United States, 15,000-30,000 associated deaths, and acute care costs exceeding $4.8 billion. The recent clinical practice guideline for CDI provides recommendations about the epidemiology, diagnosis, treatment, prevention, and environmental management. A total of 52 recommendations are included, and we will review 11 with pertinence to pediatrics in this highlight.
KEY RECOMMENDATIONS FOR THE HOSPITALIST
Recommendation 1. In infants ≤12 months of age, testing for CDI should never be routinely recommended because of the high prevalence of asymptomatic carriage of toxigenic C. difficile in infants (strong recommendation, moderate quality of evidence).
Recommendation 2. In children 1-2 years of age, testing should not be routinely performed unless other causes have been excluded (weak recommendation, low quality of evidence).Recommendation 3. In children ≥2 years of age, testing is recommended for patients with prolonged or worsening diarrhea and risk factors (eg, underlying inflammatory bowel disease) or immunocompromising conditions) or relevant exposures (eg, contact with the healthcare system or recent antibiotics) (weak recommendation, moderate quality of evidence).
The rate of C. difficile colonization among asymptomatic infants can exceed 40%. This rate declines over the first year but remains 15% at 12 months of age.1 Therefore, the guideline recommends against routinely testing infants ≤12 months of age as a positive test probably reflects colonization rather than disease. Testing in infants is recommended only when other causes have been excluded and a concern for pseudomembranous colitis, toxic megacolon, or clinically significant diarrhea exists.
The rate of asymptomatic colonization remains elevated in the second year of life. By 2-3 years, the rate is 1%-3% which is similar to that in healthy adults. However, the role of C. difficile in community-onset diarrhea in otherwise healthy children is controversial. In a study of 100 hospitalized children aged <2 years with CDI and diarrhea, all had resolution of diarrhea regardless of whether therapy was administered.2 Another study found an alternative pathogen in >50% of hospitalized children with CDI.3 Therefore, the guideline recommends against testing in children aged 1-2 years unless other causes have been excluded and in children aged >2 years only when they have prolonged or worsening diarrhea along with risk factors or exposures.
Recommendation 4. In institutions without specific required criteria for stool submissions, use a stool toxin test as part of a multistep algorithm (ie, glutamate dehydrogenase [GDH] plus toxin, GDH plus toxin arbitrated by nucleic-acid amplification tests [NAAT], or NAAT plus toxin) rather than a NAAT alone (weak recommendation, low quality of evidence).
Recommendation 5. In institutions with specific required criteria for stool submissions, use a NAAT alone or a multistep algorithm for testing (ie, GDH plus toxin, GDH plus toxin arbitrated by NAAT, or NAAT plus toxin) rather than a toxin test alone (weak recommendation, low quality of evidence).
There are a variety of testing approaches for CDI and recommendations vary based on local practice. If laboratories accept all stools, a more specific approach is recommended, including a toxin test as part of a multistep algorithm to limit false positives. If laboratories first screen for symptoms and antibiotic exposure before accepting stool samples, a more sensitive approach is recommended including NAAT alone or a multistep algorithm rather than toxin alone.
Infection Prevention and Control
Recommendation 6. There is insufficient evidence for discontinuation of PPIs (proton pump inhibitors) as a measure for preventing CDI (no recommendation).
The guideline acknowledges data suggesting an association between PPI use and CDI, but not a causal relationship. Due to the lack of high-quality evidence, it does not recommend stopping PPIs to prevent CDI.
Recommendation 7. There are insufficient data to recommend probiotics for primary prevention of CDI outside of clinical trials (no recommendation).
The guideline notes that although several meta-analyses indicate that probiotics may prevent CDI; however there were limitations, including a high incidence of CDI in placebo arms and differences in probiotic formulations and duration of use, leading to insufficient data to recommend probiotic use to prevent CDI.
Treatment
Recommendation 8. Either per os (PO) metronidazole or PO vancomycin is recommended for an initial episode or first recurrence of nonsevere pediatric CDI (weak recommendation, low quality of evidence).
Data assessing the optimal treatment for nonsevere pediatric CDI are limited. Emerging data support the use of vancomycin,4 which is now recommended for initial episodes of CDI in adults. However, there are insufficient data to recommend vancomycin over metronidazole for nonsevere pediatric CDI; therefore, either option is recommended.
Recommendation 9. For children with an initial episode of severe CDI, oral vancomycin with or without IV metronidazole is recommended over metronidazole alone (strong recommendation, moderate quality of evidence).
Recommendation 10. For children with a second or greater episode of recurrent CDI, oral vancomycin is recommended over metronidazole (weak recommendation, low quality of evidence).
There is no well-designed trial comparing metronidazole and vancomycin for severe or recurrent pediatric CDI. For children previously treated with metronidazole, vancomycin is recommended based on adult literature.4 For children previously treated with metronidazole and vancomycin, an extended course of tapered or pulse regimen vancomycin or vancomycin followed by rifaximin is recommended.
Recommendations must weigh potential harms. Metronidazole has been associated with neuropathies,5 cramping, and nausea. PO vancomycin has poor enteral absorption, minimizing systemic effects. Both vancomycin and metronidazole may promote carriage of resistant enterococci.
Recommendation 11. Fecal microbiota transplantation (FMT) should be considered for pediatric patients with multiple recurrences of CDI following standard treatments (weak recommendation, very low quality of evidence).
There are no robust data examining the effectiveness of pediatric FMT. Recommendations are guided by adult studies. Limited evidence suggests that FMT can be effective in children with multiple recurrent CDI.6 Concerns include procedure-related risks, transmission of resistant organisms and blood-borne pathogens, and induced metabolic or immunologic disorders.
CRITIQUE
Methods in Preparing a Guideline
The strength of a guideline includes representation from a diverse panel, including the Infectious Diseases Society of America (IDSA), the Society for Healthcare Epidemiology of America, the American Society of Health-Systems Pharmacists, the Society of Infectious Diseases Pharmacists, and the Pediatric Infectious Diseases Society.
The panel utilized the Grading of Recommendations Assessment, Development, and Evaluation system to weigh the strength and quality of evidence.
From a pediatric perspective, the current guideline added pediatric-specific recommendations based on a comprehensive review of the literature from 1977 to 2016. The strength of these recommendations is somewhat limited by the lack of well-designed pediatric studies. An additional limitation is that treatment recommendations are based on illness severity, although the definitions used to classify severity are not pediatric-specific and are based on unvalidated expert opinion.
Sources of Potential Conflicts or Interest or Bias
The panel complied with the IDSA policy on conflicts of interest and disclosed any interest that might be construed as a conflict, regardless of relevancy. These were evaluated by the IDSA Standards and Practice Guidelines Committee.
Generalizability
Guideline generalizability may be impacted by testing availabilities within a particular setting. Cost factors and local formularies may also limit treatment options within a given setting.
Areas in Need of Future Study
Research gaps exist regarding at what age C. difficile is pathogenic given the prevalence of asymptomatic carriage. Future studies can also focus on a newly available molecular polymerase chain reaction test platform that detects C. difficile.7
There is limited pediatric evidence to recommend metronidazole versus vancomycin in children, particularly in nonsevere cases. There is also an opportunity to further explore alternative therapies, including fidaxomicin (not currently approved for children) and bezlotoxumab, a new agent approved as adult adjunctive therapy.8
1. Donta ST, Myers MG. Clostridium difficile toxin in asymptomatic neonates. J Pediatr. 1982;100(3):431-434. https://doi.org/10.1016/s0022-3476(82)80454-x.
2. González-Del Vecchio M, Álvarez-Uria A, Marin M, et al. Clinical significance of Clostridium difficile in children less than 2 years old: a case-control study. Pediatr Infect Dis J. 2016;35(3):281-285. https://doi.org/10.1097/INF.0000000000001008.
3. Valentini D, Vittucci AC, Grandin A, et al. Coinfection in acute gastroenteritis predicts a more severe clinical course in children. Eur J Clin Microbiol Infect Dis. 2013;32(7):909-915. https://doi.org/10.1007/s10096-013-1825-9.
4. Johnson S, Louie TJ, Gerding DN, et al. Vancomycin, metronidazole, or tolevamer for Clostridium difficile infection: results from two multinational, randomized, controlled trials. Clin Infect Dis. 2014;59(3):345-354. https://doi.org/10.1093/cid/ciu313.
5. Yamamoto T, Abe K, Anjiki H, Ishii T, Kuyama Y. Metronidazole-induced neurotoxicity developed in liver cirrhosis. J Clin Med Res. 2012;4(4):295-298. https://doi.org/10.4021/jocmr893w.
6. Russell G, Kaplan J, Ferraro M, Michelow IC. Fecal bacteriotherapy for relapsing Clostridium difficile infection in a child: a proposed treatment protocol. Pediatrics. 2010;126(1):e239-e242. https://doi.org/10.1542/peds.2009-3363.
7. Zhang H, Morrison S, Tang YW. Multiplex polymerase chain reaction tests for detection of pathogens associated with gastroenteritis. Clin Lab Med. 2015;35(2):461-486. https://doi.org/10.1016/j.cll.2015.02.006.
8. Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med. 2017;376(4):305-317. https://doi.org/10.1056/NEJMoa1602615.
1. Donta ST, Myers MG. Clostridium difficile toxin in asymptomatic neonates. J Pediatr. 1982;100(3):431-434. https://doi.org/10.1016/s0022-3476(82)80454-x.
2. González-Del Vecchio M, Álvarez-Uria A, Marin M, et al. Clinical significance of Clostridium difficile in children less than 2 years old: a case-control study. Pediatr Infect Dis J. 2016;35(3):281-285. https://doi.org/10.1097/INF.0000000000001008.
3. Valentini D, Vittucci AC, Grandin A, et al. Coinfection in acute gastroenteritis predicts a more severe clinical course in children. Eur J Clin Microbiol Infect Dis. 2013;32(7):909-915. https://doi.org/10.1007/s10096-013-1825-9.
4. Johnson S, Louie TJ, Gerding DN, et al. Vancomycin, metronidazole, or tolevamer for Clostridium difficile infection: results from two multinational, randomized, controlled trials. Clin Infect Dis. 2014;59(3):345-354. https://doi.org/10.1093/cid/ciu313.
5. Yamamoto T, Abe K, Anjiki H, Ishii T, Kuyama Y. Metronidazole-induced neurotoxicity developed in liver cirrhosis. J Clin Med Res. 2012;4(4):295-298. https://doi.org/10.4021/jocmr893w.
6. Russell G, Kaplan J, Ferraro M, Michelow IC. Fecal bacteriotherapy for relapsing Clostridium difficile infection in a child: a proposed treatment protocol. Pediatrics. 2010;126(1):e239-e242. https://doi.org/10.1542/peds.2009-3363.
7. Zhang H, Morrison S, Tang YW. Multiplex polymerase chain reaction tests for detection of pathogens associated with gastroenteritis. Clin Lab Med. 2015;35(2):461-486. https://doi.org/10.1016/j.cll.2015.02.006.
8. Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med. 2017;376(4):305-317. https://doi.org/10.1056/NEJMoa1602615.
© 2019 Society of Hospital Medicine