User login
Daniel Claassen, MD, on working with patients experiencing levodopa-induced dyskinesia

How do we recognize and explain the necessity for a peak dose to patients with levodopa-induced dyskinesia (LID)?
The way I usually do it with my patients is I draw a graph. On the X axis I have time; on the Y axis I have levodopa levels. Then I show them on the graph a sinusoidal wave, displaying how, if they take their medication at 8 in the morning, the medication will grow in terms of concentration and then it will wear off.
With the sinusoidal wave I'm able to illustrate that at the peak a patient may have what we call "peak-dose dyskinesia." Typically for patients that's anywhere from 30 minutes to 45 minutes after they take their carbidopa/levodopa medication.
In terms of recognizing those symptoms, I usually spend some time with a patient describing what dyskinesia looks or feels like. There are certain cases where patients are unaware of their dyskinetic movements, when instead their caregiver, spouse, or partner are the ones actually recognizing the movements.
Typically, we talk about facial movements or lip/jaw movements and we can talk about upper extremity truncal writhing or hyperkinetic movements.
Sometimes, I'll have the patient come to the clinic having not taken their medication and then we evaluate them and have them take their medicine. About 30-45 minutes later we reevaluate the patient, which is when we can see the movements and have the patient look at themselves in the mirror to see what we're talking about, or at least explain movements to the caregiver.
Ultimately what we are trying to do is link the timing of medication and the timing of these side effects to help the patient and their family member understand the nature of what these symptoms are and when they're happening in relation to the medication that they're taking.
How can practitioners assess patients as their LID might become unpredictable?
I think the first step is to recognize that there are certain patients with Parkinson disease who are more likely to have adverse responses to levodopa. They're typically younger and they usually have other symptoms they are concerned about.
For example, it is very common for the patient to describe feeling stressed. I have had a number of patients tell me that when they're having anxiety or stress-related issues, such as at work or in their interpersonal relationships, that their dyskinesia might come on and progress a little bit more suddenly or unpredictably outside of that window when we typically expect it to peak.
Other triggers could be food. For instance, if a patient has changed their diet or changed the timing of their food intake, there may be issues related to gastric emptying or gastrointestinal symptoms that may influence the onset of these symptoms.
What we try to do is recognize the movements and then associate them with the environment or the timing behind the medication. When these things happen outside of the regular time when they're accustomed to getting them, we talk about these as unpredictable movements.
The other side effect is that patients can often have dystonia, or a forced muscle contraction. We are not only focused on the dyskinesia movement; dystonic movements are important as well.
Overall, I think practitioners can explain to patients the difference between these predictable and unpredictable movements. Additionally, we must help patients better recognize their symptoms and maybe the triggers for them, so that they may better manage them over time.
What are some aspects of LID that are most important to discuss with patients before a treatment?
The most important thing to talk about is the rationale for why a person would want to initiate levodopa or another medication.
Usually when a clinician is talking with a patient about pharmacotherapy they explain symptoms and how the individuals quality of life would improve if we treat them with levodopa.
We spend time explaining that the dose that's going to be required to get optimal control of their symptoms may differ in one person from another. And part of that dose selection is the fact that we're going to have to balance between not enough medication to too much medication.
So, I think the most important thing to discuss with patients is developing a strategy to come up with an individualized plan for medication management and explain to the patients the idea of on and off, explaining the idea of things that could interfere with medication, such as food or timing of medication use because of sleeping or changes to their day.
Basically, we are trying to give patients the framework for why we're dosing at certain times of the day regularly, why we're starting at a certain dose, and why we gradually increase the dose until we find resolution of their symptoms. We might explain why we may gradually reduce the dose if they are having symptoms like LID, and then why we may add other medications if they are experiencing LID. Also, we explain how and why we might not reduce the dose if a dose reduction would likely trigger worse symptoms.
Our aim is to give patients an outline of on/off factors that can affect drug availability and explain the long-term treatment goal, which is to optimize their motor symptoms to give them the best quality of life.
How do we recognize and explain the necessity for a peak dose to patients with levodopa-induced dyskinesia (LID)?
The way I usually do it with my patients is I draw a graph. On the X axis I have time; on the Y axis I have levodopa levels. Then I show them on the graph a sinusoidal wave, displaying how, if they take their medication at 8 in the morning, the medication will grow in terms of concentration and then it will wear off.
With the sinusoidal wave I'm able to illustrate that at the peak a patient may have what we call "peak-dose dyskinesia." Typically for patients that's anywhere from 30 minutes to 45 minutes after they take their carbidopa/levodopa medication.
In terms of recognizing those symptoms, I usually spend some time with a patient describing what dyskinesia looks or feels like. There are certain cases where patients are unaware of their dyskinetic movements, when instead their caregiver, spouse, or partner are the ones actually recognizing the movements.
Typically, we talk about facial movements or lip/jaw movements and we can talk about upper extremity truncal writhing or hyperkinetic movements.
Sometimes, I'll have the patient come to the clinic having not taken their medication and then we evaluate them and have them take their medicine. About 30-45 minutes later we reevaluate the patient, which is when we can see the movements and have the patient look at themselves in the mirror to see what we're talking about, or at least explain movements to the caregiver.
Ultimately what we are trying to do is link the timing of medication and the timing of these side effects to help the patient and their family member understand the nature of what these symptoms are and when they're happening in relation to the medication that they're taking.
How can practitioners assess patients as their LID might become unpredictable?
I think the first step is to recognize that there are certain patients with Parkinson disease who are more likely to have adverse responses to levodopa. They're typically younger and they usually have other symptoms they are concerned about.
For example, it is very common for the patient to describe feeling stressed. I have had a number of patients tell me that when they're having anxiety or stress-related issues, such as at work or in their interpersonal relationships, that their dyskinesia might come on and progress a little bit more suddenly or unpredictably outside of that window when we typically expect it to peak.
Other triggers could be food. For instance, if a patient has changed their diet or changed the timing of their food intake, there may be issues related to gastric emptying or gastrointestinal symptoms that may influence the onset of these symptoms.
What we try to do is recognize the movements and then associate them with the environment or the timing behind the medication. When these things happen outside of the regular time when they're accustomed to getting them, we talk about these as unpredictable movements.
The other side effect is that patients can often have dystonia, or a forced muscle contraction. We are not only focused on the dyskinesia movement; dystonic movements are important as well.
Overall, I think practitioners can explain to patients the difference between these predictable and unpredictable movements. Additionally, we must help patients better recognize their symptoms and maybe the triggers for them, so that they may better manage them over time.
What are some aspects of LID that are most important to discuss with patients before a treatment?
The most important thing to talk about is the rationale for why a person would want to initiate levodopa or another medication.
Usually when a clinician is talking with a patient about pharmacotherapy they explain symptoms and how the individuals quality of life would improve if we treat them with levodopa.
We spend time explaining that the dose that's going to be required to get optimal control of their symptoms may differ in one person from another. And part of that dose selection is the fact that we're going to have to balance between not enough medication to too much medication.
So, I think the most important thing to discuss with patients is developing a strategy to come up with an individualized plan for medication management and explain to the patients the idea of on and off, explaining the idea of things that could interfere with medication, such as food or timing of medication use because of sleeping or changes to their day.
Basically, we are trying to give patients the framework for why we're dosing at certain times of the day regularly, why we're starting at a certain dose, and why we gradually increase the dose until we find resolution of their symptoms. We might explain why we may gradually reduce the dose if they are having symptoms like LID, and then why we may add other medications if they are experiencing LID. Also, we explain how and why we might not reduce the dose if a dose reduction would likely trigger worse symptoms.
Our aim is to give patients an outline of on/off factors that can affect drug availability and explain the long-term treatment goal, which is to optimize their motor symptoms to give them the best quality of life.
How do we recognize and explain the necessity for a peak dose to patients with levodopa-induced dyskinesia (LID)?
The way I usually do it with my patients is I draw a graph. On the X axis I have time; on the Y axis I have levodopa levels. Then I show them on the graph a sinusoidal wave, displaying how, if they take their medication at 8 in the morning, the medication will grow in terms of concentration and then it will wear off.
With the sinusoidal wave I'm able to illustrate that at the peak a patient may have what we call "peak-dose dyskinesia." Typically for patients that's anywhere from 30 minutes to 45 minutes after they take their carbidopa/levodopa medication.
In terms of recognizing those symptoms, I usually spend some time with a patient describing what dyskinesia looks or feels like. There are certain cases where patients are unaware of their dyskinetic movements, when instead their caregiver, spouse, or partner are the ones actually recognizing the movements.
Typically, we talk about facial movements or lip/jaw movements and we can talk about upper extremity truncal writhing or hyperkinetic movements.
Sometimes, I'll have the patient come to the clinic having not taken their medication and then we evaluate them and have them take their medicine. About 30-45 minutes later we reevaluate the patient, which is when we can see the movements and have the patient look at themselves in the mirror to see what we're talking about, or at least explain movements to the caregiver.
Ultimately what we are trying to do is link the timing of medication and the timing of these side effects to help the patient and their family member understand the nature of what these symptoms are and when they're happening in relation to the medication that they're taking.
How can practitioners assess patients as their LID might become unpredictable?
I think the first step is to recognize that there are certain patients with Parkinson disease who are more likely to have adverse responses to levodopa. They're typically younger and they usually have other symptoms they are concerned about.
For example, it is very common for the patient to describe feeling stressed. I have had a number of patients tell me that when they're having anxiety or stress-related issues, such as at work or in their interpersonal relationships, that their dyskinesia might come on and progress a little bit more suddenly or unpredictably outside of that window when we typically expect it to peak.
Other triggers could be food. For instance, if a patient has changed their diet or changed the timing of their food intake, there may be issues related to gastric emptying or gastrointestinal symptoms that may influence the onset of these symptoms.
What we try to do is recognize the movements and then associate them with the environment or the timing behind the medication. When these things happen outside of the regular time when they're accustomed to getting them, we talk about these as unpredictable movements.
The other side effect is that patients can often have dystonia, or a forced muscle contraction. We are not only focused on the dyskinesia movement; dystonic movements are important as well.
Overall, I think practitioners can explain to patients the difference between these predictable and unpredictable movements. Additionally, we must help patients better recognize their symptoms and maybe the triggers for them, so that they may better manage them over time.
What are some aspects of LID that are most important to discuss with patients before a treatment?
The most important thing to talk about is the rationale for why a person would want to initiate levodopa or another medication.
Usually when a clinician is talking with a patient about pharmacotherapy they explain symptoms and how the individuals quality of life would improve if we treat them with levodopa.
We spend time explaining that the dose that's going to be required to get optimal control of their symptoms may differ in one person from another. And part of that dose selection is the fact that we're going to have to balance between not enough medication to too much medication.
So, I think the most important thing to discuss with patients is developing a strategy to come up with an individualized plan for medication management and explain to the patients the idea of on and off, explaining the idea of things that could interfere with medication, such as food or timing of medication use because of sleeping or changes to their day.
Basically, we are trying to give patients the framework for why we're dosing at certain times of the day regularly, why we're starting at a certain dose, and why we gradually increase the dose until we find resolution of their symptoms. We might explain why we may gradually reduce the dose if they are having symptoms like LID, and then why we may add other medications if they are experiencing LID. Also, we explain how and why we might not reduce the dose if a dose reduction would likely trigger worse symptoms.
Our aim is to give patients an outline of on/off factors that can affect drug availability and explain the long-term treatment goal, which is to optimize their motor symptoms to give them the best quality of life.
David Charles, MD, and Thomas Davis, MD, on updates on levodopa-induced dyskinesia treatment and research
David Charles, MD, and Thomas Davis, MD, of the Vanderbilt University Department of Neurology, recently spoke with Neurology Reviews about the treatment pipeline and latest research in levodopa-induced dyskinesia in Parkinson's disease.
How is the treatment pipeline advancing for different types of levodopa-induced dyskinesia (LID)?
Dr. Thomas Davis: Dyskinesia has traditionally been hard to quantify, and we have been lacking any US Food and Drug Administration (FDA)-approved anti-dyskinesia drugs. The pipeline has historically been strongest for wearing-off because it is easier to measure on time than to quantify involuntary movements.
The Unified Dyskinesia Rating Scale (UDysRS), released in 2008 by the Movement Disorder Society, provided a standardized scale that allowed dyskinesia clinical trials to move forward. The UDysRS was used as the primary outcome for the extended release amantadine capsule study. This was important because it demonstrated the possibility of a successful clinical trial design to get a drug approved for dyskinesia, which will encourage others to test more potential new treatments.
What is the status of research on deep brain stimulation (DBS) for Parkinson's disease, and when might it be considered?
Dr. David Charles: This is one of the areas of research that we're focused on here at Vanderbilt. All 3 of the FDA-approved device manufacturers have been conducting research in technology refinement and improvements. These advances include not only patient programmers and physician programmers, but also new sensing capability and lead designs. Some of the manufacturers now have leads that allow the physician to steer the current in one direction or another, where traditionally the current has been delivered in a circumferential contact that's shaped like a cylinder, where the energy is transmitted 360 degrees from the lead. The new designs allow you to steer the current hopefully toward areas that provide more efficacy and away from areas that cause side effects. Even more exciting is the emerging sensing capability that may allow the development of stimulating technology that is responsive to fluctuating symptoms. There is keen research interest in understanding whether a device could detect a specific neuronal firing pattern and then respond with an individually tailored stimulation to improve symptoms as needed. Will the next generation of deep brain stimulating devices detect the pattern and deliver energy in a more targeted and precise way, responsive to what it's sensing from the patient's brain? I think that's an area of research that's really exciting.
In regard to when it might be considered: The ability to steer the current is already available in 2 of the 3 systems that are on the market today. Having current that is steerable in all 3 will be coming in the not too distant future. The available devices already have improved programming platforms for health care providers as well.
Our research at Vanderbilt is focused on DBS in early-stage Parkinson's disease. There is a paper published in Neurology that reports Class II evidencet hat DBS applied in early-stage Parkinson's disease slows the progression of tremor. This is exciting because none of the available treatments change the progression of disease—they're currently accepted as symptomatic therapies only. In this publication, we report that participants receiving DBS in the very earliest stages of Parkinson's disease it may slow the progression of rest tremor. We now have approval from the FDA to conduct a large-scale phase 3, multicenter, clinical trial of DBS in early-stage Parkinson's disease, with the primary endpoint focused on slowing progression of tremor, a cardinal feature of the disease. This upcoming trial is approved by the FDA as a pivotal trial, meaning that the findings could potentially be used to change the labeling of DBS devices. Our goal is to obtain Class I evidence of slowing the progression of tremor or other elements of the disease.
Dr. Thomas Davis: If you are a device manufacturer for DBS it is natural for you to aim to make better devices, better batteries, better programming, and better electrodes than your competitor. That's really where the industry-based research is right now. Clinicians are still determining which device is the best candidate, where the best target in the brain is, and when to use DBS.
When would a health care practitioner decide to try frequent smaller dosages or immediate-release formulations of dopaminergic drugs to control levodopa-induced dyskinesia (LID), compared with non-dopaminergic treatments that are available? What are some pros and cons of each approach?
Dr. David Charles: If you have a patient who's already on levodopa, it's not uncommon that—separate from the way we prescribe the medicine—the patients experiment with their medicine to some degree. At the very least, people occasionally forget to take a dose and they feel the effect of a missed dose. They may take an extra dose or an extra half dose, particularly if they feel that the last dose isn't working as well, or in the event they have some special occasion coming up.
Over time, patients and physicians learn that sometimes smaller, more frequent dosing of levodopa can be a helpful strategy for certain individuals. One advantage is that it's the medication that the patient is already taking, and they're just simply breaking tablets. Many pharmacies will break tablets for patients so they can take some smaller doses more frequently. Obviously, there can be downsides to that, such as it becoming harder to remember to take more frequent doses.
Dr. Thomas Davis: I would agree that the biggest advantage of taking more frequent, smaller doses is that it's cheaper than adding an adjunct or moving to a more invasive therapy. More frequent smaller doses of levodopa also generally has no side effects because if you're taking 2 carbidopa/levodopa 3 times a day and you're tolerating it, but you're having peak dose problems, you can then switch to 1.5 tablets 4 times a day. It involves more work and planning, but it's no more total medication than the patient is taking already, so this strategy usually does not have any unexpected side effects. It really boils down to how much work the patient wants to put in, how adherent they are to medication dosing, and whether they want to add another medication.
Most of the adjunctive medications to treat motor fluctuations are approved to improve on-time in Parkinson's disease patients with wearing off. These include the monoamine oxidase inhibitors, COMT inhibitors, and adenosine A2A antagonists. For treatment of dyskinesia, only the extended release capsule formulation of amantadine has FDA approval, although all formulations are approved for Parkinson's disease and are used clinically to dampen dyskinesia. How long to try these strategies before moving to one of the more advanced therapies, like DBS or jejunal infusion of levodopa, is not clear. It's great to have options, but it makes the decisions a lot harder.
Dr. David Charles: Dr. Davis raised the question of what medicine to choose, and what's your next choice in a patient who's having wearing-off dyskinesia or LID and so forth. There is an increasing number of options for those mid-stage patients. The pitfall is feeling that you have to try every available medication and combination before moving to a more advanced therapy. The physician risks churning through the various combinations for so long that the benefit of an advanced therapy becomes shortened or lost altogether.
Take epilepsy, for example. In operative candidates, surgery is often more beneficial when applied earlier. There's solid data to support that adding on multiple anti-epileptics medications is not always helpful. Continuing to add or change medications can actually diminish returns, particularly in a person who could receive benefit from surgery for epilepsy.
I get the sense that the same may be true for dyskinesia. In clinical practice, we often receive DBS referrals when a patient's community-based physician has tried various medications and combination therapies until the point that the patient and the physician have become totally frustrated. By the time they are referred, the patient may benefit from DBS, but not nearly as well and as for long as they could have if they had received it earlier. We as physicians have to be mindful that while we have these increasing number of options—which is a good thing for both patients and physicians—that we don't continue to use them to the point that it takes away the option of more advanced therapies for appropriate candidates.
Metabotropic glutamate (mGlu) receptors have been receiving attention as potential therapeutic targets for LID. How do these compare with other receptors such as N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxyl-5-
Dr. Thomas Davis: NMDA and AMPA are traditional ionotropic receptors, meaning that they are ligand gated, that they are almost exclusively excitatory, and that they generally have to do with the flow of potassium.
mGlu receptors are protein coupled receptors. They have more elaborate action and may be either excitatory or inhibitory. Though mGlu, NMDA, and AMPA are completely different, they are all activated by glutamate. Pharmacologically utilizing the mGlu receptors is a relatively new and novel idea. Specifically, mGlu-5 receptors have received the most attention as potentially having an anti-parkinsonian effect and possibly dampening dyskinesia. The mGlu-5 receptors are an attractive target because they are concentrated in the striatum, as opposed to other glutamatergic receptors that are more diffusely located. It was felt that mGlu-5 modulators would be more specific and have less of the potential adverse effects of other glutamates. Most drugs that we think of affecting glutamate, like amantadine and memantine (used for Alzheimer's disease), have some NMDA antagonist effect, but this is relatively mild.
David Charles, MD, and Thomas Davis, MD, of the Vanderbilt University Department of Neurology, recently spoke with Neurology Reviews about the treatment pipeline and latest research in levodopa-induced dyskinesia in Parkinson's disease.
How is the treatment pipeline advancing for different types of levodopa-induced dyskinesia (LID)?
Dr. Thomas Davis: Dyskinesia has traditionally been hard to quantify, and we have been lacking any US Food and Drug Administration (FDA)-approved anti-dyskinesia drugs. The pipeline has historically been strongest for wearing-off because it is easier to measure on time than to quantify involuntary movements.
The Unified Dyskinesia Rating Scale (UDysRS), released in 2008 by the Movement Disorder Society, provided a standardized scale that allowed dyskinesia clinical trials to move forward. The UDysRS was used as the primary outcome for the extended release amantadine capsule study. This was important because it demonstrated the possibility of a successful clinical trial design to get a drug approved for dyskinesia, which will encourage others to test more potential new treatments.
What is the status of research on deep brain stimulation (DBS) for Parkinson's disease, and when might it be considered?
Dr. David Charles: This is one of the areas of research that we're focused on here at Vanderbilt. All 3 of the FDA-approved device manufacturers have been conducting research in technology refinement and improvements. These advances include not only patient programmers and physician programmers, but also new sensing capability and lead designs. Some of the manufacturers now have leads that allow the physician to steer the current in one direction or another, where traditionally the current has been delivered in a circumferential contact that's shaped like a cylinder, where the energy is transmitted 360 degrees from the lead. The new designs allow you to steer the current hopefully toward areas that provide more efficacy and away from areas that cause side effects. Even more exciting is the emerging sensing capability that may allow the development of stimulating technology that is responsive to fluctuating symptoms. There is keen research interest in understanding whether a device could detect a specific neuronal firing pattern and then respond with an individually tailored stimulation to improve symptoms as needed. Will the next generation of deep brain stimulating devices detect the pattern and deliver energy in a more targeted and precise way, responsive to what it's sensing from the patient's brain? I think that's an area of research that's really exciting.
In regard to when it might be considered: The ability to steer the current is already available in 2 of the 3 systems that are on the market today. Having current that is steerable in all 3 will be coming in the not too distant future. The available devices already have improved programming platforms for health care providers as well.
Our research at Vanderbilt is focused on DBS in early-stage Parkinson's disease. There is a paper published in Neurology that reports Class II evidencet hat DBS applied in early-stage Parkinson's disease slows the progression of tremor. This is exciting because none of the available treatments change the progression of disease—they're currently accepted as symptomatic therapies only. In this publication, we report that participants receiving DBS in the very earliest stages of Parkinson's disease it may slow the progression of rest tremor. We now have approval from the FDA to conduct a large-scale phase 3, multicenter, clinical trial of DBS in early-stage Parkinson's disease, with the primary endpoint focused on slowing progression of tremor, a cardinal feature of the disease. This upcoming trial is approved by the FDA as a pivotal trial, meaning that the findings could potentially be used to change the labeling of DBS devices. Our goal is to obtain Class I evidence of slowing the progression of tremor or other elements of the disease.
Dr. Thomas Davis: If you are a device manufacturer for DBS it is natural for you to aim to make better devices, better batteries, better programming, and better electrodes than your competitor. That's really where the industry-based research is right now. Clinicians are still determining which device is the best candidate, where the best target in the brain is, and when to use DBS.
When would a health care practitioner decide to try frequent smaller dosages or immediate-release formulations of dopaminergic drugs to control levodopa-induced dyskinesia (LID), compared with non-dopaminergic treatments that are available? What are some pros and cons of each approach?
Dr. David Charles: If you have a patient who's already on levodopa, it's not uncommon that—separate from the way we prescribe the medicine—the patients experiment with their medicine to some degree. At the very least, people occasionally forget to take a dose and they feel the effect of a missed dose. They may take an extra dose or an extra half dose, particularly if they feel that the last dose isn't working as well, or in the event they have some special occasion coming up.
Over time, patients and physicians learn that sometimes smaller, more frequent dosing of levodopa can be a helpful strategy for certain individuals. One advantage is that it's the medication that the patient is already taking, and they're just simply breaking tablets. Many pharmacies will break tablets for patients so they can take some smaller doses more frequently. Obviously, there can be downsides to that, such as it becoming harder to remember to take more frequent doses.
Dr. Thomas Davis: I would agree that the biggest advantage of taking more frequent, smaller doses is that it's cheaper than adding an adjunct or moving to a more invasive therapy. More frequent smaller doses of levodopa also generally has no side effects because if you're taking 2 carbidopa/levodopa 3 times a day and you're tolerating it, but you're having peak dose problems, you can then switch to 1.5 tablets 4 times a day. It involves more work and planning, but it's no more total medication than the patient is taking already, so this strategy usually does not have any unexpected side effects. It really boils down to how much work the patient wants to put in, how adherent they are to medication dosing, and whether they want to add another medication.
Most of the adjunctive medications to treat motor fluctuations are approved to improve on-time in Parkinson's disease patients with wearing off. These include the monoamine oxidase inhibitors, COMT inhibitors, and adenosine A2A antagonists. For treatment of dyskinesia, only the extended release capsule formulation of amantadine has FDA approval, although all formulations are approved for Parkinson's disease and are used clinically to dampen dyskinesia. How long to try these strategies before moving to one of the more advanced therapies, like DBS or jejunal infusion of levodopa, is not clear. It's great to have options, but it makes the decisions a lot harder.
Dr. David Charles: Dr. Davis raised the question of what medicine to choose, and what's your next choice in a patient who's having wearing-off dyskinesia or LID and so forth. There is an increasing number of options for those mid-stage patients. The pitfall is feeling that you have to try every available medication and combination before moving to a more advanced therapy. The physician risks churning through the various combinations for so long that the benefit of an advanced therapy becomes shortened or lost altogether.
Take epilepsy, for example. In operative candidates, surgery is often more beneficial when applied earlier. There's solid data to support that adding on multiple anti-epileptics medications is not always helpful. Continuing to add or change medications can actually diminish returns, particularly in a person who could receive benefit from surgery for epilepsy.
I get the sense that the same may be true for dyskinesia. In clinical practice, we often receive DBS referrals when a patient's community-based physician has tried various medications and combination therapies until the point that the patient and the physician have become totally frustrated. By the time they are referred, the patient may benefit from DBS, but not nearly as well and as for long as they could have if they had received it earlier. We as physicians have to be mindful that while we have these increasing number of options—which is a good thing for both patients and physicians—that we don't continue to use them to the point that it takes away the option of more advanced therapies for appropriate candidates.
Metabotropic glutamate (mGlu) receptors have been receiving attention as potential therapeutic targets for LID. How do these compare with other receptors such as N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxyl-5-
Dr. Thomas Davis: NMDA and AMPA are traditional ionotropic receptors, meaning that they are ligand gated, that they are almost exclusively excitatory, and that they generally have to do with the flow of potassium.
mGlu receptors are protein coupled receptors. They have more elaborate action and may be either excitatory or inhibitory. Though mGlu, NMDA, and AMPA are completely different, they are all activated by glutamate. Pharmacologically utilizing the mGlu receptors is a relatively new and novel idea. Specifically, mGlu-5 receptors have received the most attention as potentially having an anti-parkinsonian effect and possibly dampening dyskinesia. The mGlu-5 receptors are an attractive target because they are concentrated in the striatum, as opposed to other glutamatergic receptors that are more diffusely located. It was felt that mGlu-5 modulators would be more specific and have less of the potential adverse effects of other glutamates. Most drugs that we think of affecting glutamate, like amantadine and memantine (used for Alzheimer's disease), have some NMDA antagonist effect, but this is relatively mild.
David Charles, MD, and Thomas Davis, MD, of the Vanderbilt University Department of Neurology, recently spoke with Neurology Reviews about the treatment pipeline and latest research in levodopa-induced dyskinesia in Parkinson's disease.
How is the treatment pipeline advancing for different types of levodopa-induced dyskinesia (LID)?
Dr. Thomas Davis: Dyskinesia has traditionally been hard to quantify, and we have been lacking any US Food and Drug Administration (FDA)-approved anti-dyskinesia drugs. The pipeline has historically been strongest for wearing-off because it is easier to measure on time than to quantify involuntary movements.
The Unified Dyskinesia Rating Scale (UDysRS), released in 2008 by the Movement Disorder Society, provided a standardized scale that allowed dyskinesia clinical trials to move forward. The UDysRS was used as the primary outcome for the extended release amantadine capsule study. This was important because it demonstrated the possibility of a successful clinical trial design to get a drug approved for dyskinesia, which will encourage others to test more potential new treatments.
What is the status of research on deep brain stimulation (DBS) for Parkinson's disease, and when might it be considered?
Dr. David Charles: This is one of the areas of research that we're focused on here at Vanderbilt. All 3 of the FDA-approved device manufacturers have been conducting research in technology refinement and improvements. These advances include not only patient programmers and physician programmers, but also new sensing capability and lead designs. Some of the manufacturers now have leads that allow the physician to steer the current in one direction or another, where traditionally the current has been delivered in a circumferential contact that's shaped like a cylinder, where the energy is transmitted 360 degrees from the lead. The new designs allow you to steer the current hopefully toward areas that provide more efficacy and away from areas that cause side effects. Even more exciting is the emerging sensing capability that may allow the development of stimulating technology that is responsive to fluctuating symptoms. There is keen research interest in understanding whether a device could detect a specific neuronal firing pattern and then respond with an individually tailored stimulation to improve symptoms as needed. Will the next generation of deep brain stimulating devices detect the pattern and deliver energy in a more targeted and precise way, responsive to what it's sensing from the patient's brain? I think that's an area of research that's really exciting.
In regard to when it might be considered: The ability to steer the current is already available in 2 of the 3 systems that are on the market today. Having current that is steerable in all 3 will be coming in the not too distant future. The available devices already have improved programming platforms for health care providers as well.
Our research at Vanderbilt is focused on DBS in early-stage Parkinson's disease. There is a paper published in Neurology that reports Class II evidencet hat DBS applied in early-stage Parkinson's disease slows the progression of tremor. This is exciting because none of the available treatments change the progression of disease—they're currently accepted as symptomatic therapies only. In this publication, we report that participants receiving DBS in the very earliest stages of Parkinson's disease it may slow the progression of rest tremor. We now have approval from the FDA to conduct a large-scale phase 3, multicenter, clinical trial of DBS in early-stage Parkinson's disease, with the primary endpoint focused on slowing progression of tremor, a cardinal feature of the disease. This upcoming trial is approved by the FDA as a pivotal trial, meaning that the findings could potentially be used to change the labeling of DBS devices. Our goal is to obtain Class I evidence of slowing the progression of tremor or other elements of the disease.
Dr. Thomas Davis: If you are a device manufacturer for DBS it is natural for you to aim to make better devices, better batteries, better programming, and better electrodes than your competitor. That's really where the industry-based research is right now. Clinicians are still determining which device is the best candidate, where the best target in the brain is, and when to use DBS.
When would a health care practitioner decide to try frequent smaller dosages or immediate-release formulations of dopaminergic drugs to control levodopa-induced dyskinesia (LID), compared with non-dopaminergic treatments that are available? What are some pros and cons of each approach?
Dr. David Charles: If you have a patient who's already on levodopa, it's not uncommon that—separate from the way we prescribe the medicine—the patients experiment with their medicine to some degree. At the very least, people occasionally forget to take a dose and they feel the effect of a missed dose. They may take an extra dose or an extra half dose, particularly if they feel that the last dose isn't working as well, or in the event they have some special occasion coming up.
Over time, patients and physicians learn that sometimes smaller, more frequent dosing of levodopa can be a helpful strategy for certain individuals. One advantage is that it's the medication that the patient is already taking, and they're just simply breaking tablets. Many pharmacies will break tablets for patients so they can take some smaller doses more frequently. Obviously, there can be downsides to that, such as it becoming harder to remember to take more frequent doses.
Dr. Thomas Davis: I would agree that the biggest advantage of taking more frequent, smaller doses is that it's cheaper than adding an adjunct or moving to a more invasive therapy. More frequent smaller doses of levodopa also generally has no side effects because if you're taking 2 carbidopa/levodopa 3 times a day and you're tolerating it, but you're having peak dose problems, you can then switch to 1.5 tablets 4 times a day. It involves more work and planning, but it's no more total medication than the patient is taking already, so this strategy usually does not have any unexpected side effects. It really boils down to how much work the patient wants to put in, how adherent they are to medication dosing, and whether they want to add another medication.
Most of the adjunctive medications to treat motor fluctuations are approved to improve on-time in Parkinson's disease patients with wearing off. These include the monoamine oxidase inhibitors, COMT inhibitors, and adenosine A2A antagonists. For treatment of dyskinesia, only the extended release capsule formulation of amantadine has FDA approval, although all formulations are approved for Parkinson's disease and are used clinically to dampen dyskinesia. How long to try these strategies before moving to one of the more advanced therapies, like DBS or jejunal infusion of levodopa, is not clear. It's great to have options, but it makes the decisions a lot harder.
Dr. David Charles: Dr. Davis raised the question of what medicine to choose, and what's your next choice in a patient who's having wearing-off dyskinesia or LID and so forth. There is an increasing number of options for those mid-stage patients. The pitfall is feeling that you have to try every available medication and combination before moving to a more advanced therapy. The physician risks churning through the various combinations for so long that the benefit of an advanced therapy becomes shortened or lost altogether.
Take epilepsy, for example. In operative candidates, surgery is often more beneficial when applied earlier. There's solid data to support that adding on multiple anti-epileptics medications is not always helpful. Continuing to add or change medications can actually diminish returns, particularly in a person who could receive benefit from surgery for epilepsy.
I get the sense that the same may be true for dyskinesia. In clinical practice, we often receive DBS referrals when a patient's community-based physician has tried various medications and combination therapies until the point that the patient and the physician have become totally frustrated. By the time they are referred, the patient may benefit from DBS, but not nearly as well and as for long as they could have if they had received it earlier. We as physicians have to be mindful that while we have these increasing number of options—which is a good thing for both patients and physicians—that we don't continue to use them to the point that it takes away the option of more advanced therapies for appropriate candidates.
Metabotropic glutamate (mGlu) receptors have been receiving attention as potential therapeutic targets for LID. How do these compare with other receptors such as N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxyl-5-
Dr. Thomas Davis: NMDA and AMPA are traditional ionotropic receptors, meaning that they are ligand gated, that they are almost exclusively excitatory, and that they generally have to do with the flow of potassium.
mGlu receptors are protein coupled receptors. They have more elaborate action and may be either excitatory or inhibitory. Though mGlu, NMDA, and AMPA are completely different, they are all activated by glutamate. Pharmacologically utilizing the mGlu receptors is a relatively new and novel idea. Specifically, mGlu-5 receptors have received the most attention as potentially having an anti-parkinsonian effect and possibly dampening dyskinesia. The mGlu-5 receptors are an attractive target because they are concentrated in the striatum, as opposed to other glutamatergic receptors that are more diffusely located. It was felt that mGlu-5 modulators would be more specific and have less of the potential adverse effects of other glutamates. Most drugs that we think of affecting glutamate, like amantadine and memantine (used for Alzheimer's disease), have some NMDA antagonist effect, but this is relatively mild.
COVID-19: Adjusting practice in acute leukemia care
The SARS-CoV-2 pandemic poses significant risks to leukemia patients and their providers, impacting every aspect of care from diagnosis through therapy, according to an editorial letter published online in Leukemia Research.
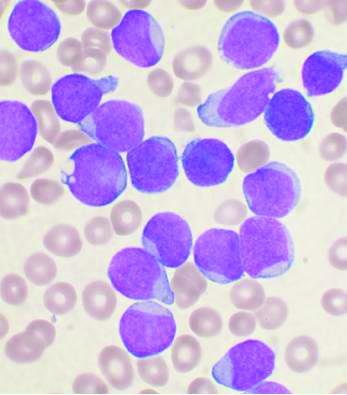
One key concern to be considered is the risk of missed or delayed diagnosis due to the pandemic conditions. An estimated 50%-75% of patients with acute leukemia are febrile at diagnosis and this puts them at high risk of a misdiagnosis of COVID-19 upon initial evaluation. As with other oncological conditions (primary mediastinal lymphoma or lung cancer, for example), which often present with a cough with or without fever, their symptoms “are likely to be considered trivial after a negative SARS-CoV-2 test,” with patients then being sent home without further assessment. In a rapidly progressing disease such as acute leukemia, this could lead to critical delays in therapeutic intervention.
The authors, from the Service and Central Laboratory of Hematology, Lausanne (Switzerland) University Hospital, also discussed the problems that might occur with regard to most standard forms of therapy. In particular, they addressed potential impacts of the pandemic on chemotherapy, bone marrow transplantation, maintenance treatments, supportive measures, and targeted therapies.
Of particular concern, “most patients may suffer from postponed chemotherapy, due to a shortage of isolation beds and blood products or the wish to avoid immunosuppressive treatments,” the authors noted, warning that “delay in chemotherapy initiation may negatively affect prognosis, [particularly in patients under age 60] with favorable- or intermediate-risk disease.”
With regard to stem cell transplantation, the authors detail the many potential difficulties with regard to procedures involving both donors and recipients, and warn that in some cases, delay in transplant could result in the reappearance of a significant minimal residual disease, which has a well-established negative impact on survival.
The authors also noted that blood product shortages have already begun in most affected countries, and how, in response, transfusion societies have called for conservative transfusion policies in strict adherence to evidence-based guidelines for patient’s blood management.
“COVID-19 will result in numerous casualties. Acute leukemia patients are at a higher risk of severe complications,” the authors stated. In particular, physicians should especially be aware of how treatment for acute leukemia may have “interactions with other drugs used to treat SARS-CoV-2–related infections/complications such as antibiotics, antiviral drugs, and various other drugs that prolong QTc or impact targeted-therapy pharmacokinetics,” they concluded.
The authors reported that they received no government or private funding for this research, and that they had no conflicts of interest.
SOURCE: Gavillet M et al. Leuk. Res. 2020. doi.org/10.1016/j.leukres.2020.106353.
The SARS-CoV-2 pandemic poses significant risks to leukemia patients and their providers, impacting every aspect of care from diagnosis through therapy, according to an editorial letter published online in Leukemia Research.
One key concern to be considered is the risk of missed or delayed diagnosis due to the pandemic conditions. An estimated 50%-75% of patients with acute leukemia are febrile at diagnosis and this puts them at high risk of a misdiagnosis of COVID-19 upon initial evaluation. As with other oncological conditions (primary mediastinal lymphoma or lung cancer, for example), which often present with a cough with or without fever, their symptoms “are likely to be considered trivial after a negative SARS-CoV-2 test,” with patients then being sent home without further assessment. In a rapidly progressing disease such as acute leukemia, this could lead to critical delays in therapeutic intervention.
The authors, from the Service and Central Laboratory of Hematology, Lausanne (Switzerland) University Hospital, also discussed the problems that might occur with regard to most standard forms of therapy. In particular, they addressed potential impacts of the pandemic on chemotherapy, bone marrow transplantation, maintenance treatments, supportive measures, and targeted therapies.
Of particular concern, “most patients may suffer from postponed chemotherapy, due to a shortage of isolation beds and blood products or the wish to avoid immunosuppressive treatments,” the authors noted, warning that “delay in chemotherapy initiation may negatively affect prognosis, [particularly in patients under age 60] with favorable- or intermediate-risk disease.”
With regard to stem cell transplantation, the authors detail the many potential difficulties with regard to procedures involving both donors and recipients, and warn that in some cases, delay in transplant could result in the reappearance of a significant minimal residual disease, which has a well-established negative impact on survival.
The authors also noted that blood product shortages have already begun in most affected countries, and how, in response, transfusion societies have called for conservative transfusion policies in strict adherence to evidence-based guidelines for patient’s blood management.
“COVID-19 will result in numerous casualties. Acute leukemia patients are at a higher risk of severe complications,” the authors stated. In particular, physicians should especially be aware of how treatment for acute leukemia may have “interactions with other drugs used to treat SARS-CoV-2–related infections/complications such as antibiotics, antiviral drugs, and various other drugs that prolong QTc or impact targeted-therapy pharmacokinetics,” they concluded.
The authors reported that they received no government or private funding for this research, and that they had no conflicts of interest.
SOURCE: Gavillet M et al. Leuk. Res. 2020. doi.org/10.1016/j.leukres.2020.106353.
The SARS-CoV-2 pandemic poses significant risks to leukemia patients and their providers, impacting every aspect of care from diagnosis through therapy, according to an editorial letter published online in Leukemia Research.
One key concern to be considered is the risk of missed or delayed diagnosis due to the pandemic conditions. An estimated 50%-75% of patients with acute leukemia are febrile at diagnosis and this puts them at high risk of a misdiagnosis of COVID-19 upon initial evaluation. As with other oncological conditions (primary mediastinal lymphoma or lung cancer, for example), which often present with a cough with or without fever, their symptoms “are likely to be considered trivial after a negative SARS-CoV-2 test,” with patients then being sent home without further assessment. In a rapidly progressing disease such as acute leukemia, this could lead to critical delays in therapeutic intervention.
The authors, from the Service and Central Laboratory of Hematology, Lausanne (Switzerland) University Hospital, also discussed the problems that might occur with regard to most standard forms of therapy. In particular, they addressed potential impacts of the pandemic on chemotherapy, bone marrow transplantation, maintenance treatments, supportive measures, and targeted therapies.
Of particular concern, “most patients may suffer from postponed chemotherapy, due to a shortage of isolation beds and blood products or the wish to avoid immunosuppressive treatments,” the authors noted, warning that “delay in chemotherapy initiation may negatively affect prognosis, [particularly in patients under age 60] with favorable- or intermediate-risk disease.”
With regard to stem cell transplantation, the authors detail the many potential difficulties with regard to procedures involving both donors and recipients, and warn that in some cases, delay in transplant could result in the reappearance of a significant minimal residual disease, which has a well-established negative impact on survival.
The authors also noted that blood product shortages have already begun in most affected countries, and how, in response, transfusion societies have called for conservative transfusion policies in strict adherence to evidence-based guidelines for patient’s blood management.
“COVID-19 will result in numerous casualties. Acute leukemia patients are at a higher risk of severe complications,” the authors stated. In particular, physicians should especially be aware of how treatment for acute leukemia may have “interactions with other drugs used to treat SARS-CoV-2–related infections/complications such as antibiotics, antiviral drugs, and various other drugs that prolong QTc or impact targeted-therapy pharmacokinetics,” they concluded.
The authors reported that they received no government or private funding for this research, and that they had no conflicts of interest.
SOURCE: Gavillet M et al. Leuk. Res. 2020. doi.org/10.1016/j.leukres.2020.106353.
FROM LEUKEMIA RESEARCH
Noninvasive fibrosis scores not sensitive in people with fatty liver disease and T2D
Noninvasive fibrosis scores, which are widely used to predict advanced fibrosis in people with nonalcoholic fatty liver disease (NAFLD), do not do a good job of picking up advanced fibrosis in patients with underlying diabetes, according to a new study.
Advanced fibrosis is associated with an increased risk of cirrhosis, end-stage liver disease, and liver failure. Underlying diabetes is a risk factor for both advanced fibrosis and death in patients with NAFLD.
While liver biopsy remains the gold standard for detecting advanced fibrosis, high costs and risks limit its use. Noninvasive scores such as the AST/ALT ratio; AST to platelet ratio index (APRI); fibrosis-4 (FIB-4) index; and NAFLD fibrosis score (NFS) have gained popularity in recent years, as they offer the compelling advantage of using easily and cheaply attained clinical and laboratory measures to assess likelihood of disease.
But their accuracy has come into question, particularly for people with diabetes.
In research published in the Journal of Clinical Gastroenterology, Amandeep Singh, MD, and colleagues at the Cleveland Clinic looked at their center’s records for 1,157 patients with type 2 diabetes (65% women, 88% white, 85% with obesity) who had undergone a liver biopsy for suspected advanced fibrosis between 2000 and 2015. Biopsy results revealed that a third of the cohort (32%) was positive for advanced fibrosis.
The investigators then pulled patients’ laboratory results for AST, ALT, cholesterol, triglycerides, fasting glucose, hemoglobin A1c, bilirubin, albumin, platelet count, alkaline phosphatase, albumin, and lipid levels, all collected within a year of biopsy. After plugging these into the algorithms of four different scoring systems for advanced fibrosis, they compared results with results from the biopsies.
The scores of AST/ALT greater than 1.4, APRI of at least 1.5, NFS greater than 0.676, and FIB-4 index greater than 2.67 had high specificities of 84%, 97%, 70%, and 93%, respectively, but sensitivities of only 27%, 17%, 64%, and 44%. Even when the cutoff measures were tightened, the scoring systems still missed a lot of disease. This suggests, Dr. Singh and colleagues wrote, that “the presence of diabetes could decrease the predictive value of these scores to detect advanced disease in NAFLD patients.” Reliable noninvasive biomarkers are “urgently needed” for this patient population.
In an interview, Dr. Singh advised that clinicians continue to use current noninvasive scores in patients with diabetes – preferably the NFS – “until we have a better scoring system.” If clinicians suspect advanced fibrosis based on lab tests and clinical data, then “liver biopsy should be considered,” he said.
The investigators described among the limitations of their study its retrospective, single-center design, with patients who were mostly white and from one geographic region.
Dr. Singh and colleagues reported no conflicts of interest or outside funding for their study.
SOURCE: Singh A et al. J Clin Gastroenterol. 2020 Mar 11. doi: 10.1097/MCG.0000000000001339.
Noninvasive fibrosis scores, which are widely used to predict advanced fibrosis in people with nonalcoholic fatty liver disease (NAFLD), do not do a good job of picking up advanced fibrosis in patients with underlying diabetes, according to a new study.
Advanced fibrosis is associated with an increased risk of cirrhosis, end-stage liver disease, and liver failure. Underlying diabetes is a risk factor for both advanced fibrosis and death in patients with NAFLD.
While liver biopsy remains the gold standard for detecting advanced fibrosis, high costs and risks limit its use. Noninvasive scores such as the AST/ALT ratio; AST to platelet ratio index (APRI); fibrosis-4 (FIB-4) index; and NAFLD fibrosis score (NFS) have gained popularity in recent years, as they offer the compelling advantage of using easily and cheaply attained clinical and laboratory measures to assess likelihood of disease.
But their accuracy has come into question, particularly for people with diabetes.
In research published in the Journal of Clinical Gastroenterology, Amandeep Singh, MD, and colleagues at the Cleveland Clinic looked at their center’s records for 1,157 patients with type 2 diabetes (65% women, 88% white, 85% with obesity) who had undergone a liver biopsy for suspected advanced fibrosis between 2000 and 2015. Biopsy results revealed that a third of the cohort (32%) was positive for advanced fibrosis.
The investigators then pulled patients’ laboratory results for AST, ALT, cholesterol, triglycerides, fasting glucose, hemoglobin A1c, bilirubin, albumin, platelet count, alkaline phosphatase, albumin, and lipid levels, all collected within a year of biopsy. After plugging these into the algorithms of four different scoring systems for advanced fibrosis, they compared results with results from the biopsies.
The scores of AST/ALT greater than 1.4, APRI of at least 1.5, NFS greater than 0.676, and FIB-4 index greater than 2.67 had high specificities of 84%, 97%, 70%, and 93%, respectively, but sensitivities of only 27%, 17%, 64%, and 44%. Even when the cutoff measures were tightened, the scoring systems still missed a lot of disease. This suggests, Dr. Singh and colleagues wrote, that “the presence of diabetes could decrease the predictive value of these scores to detect advanced disease in NAFLD patients.” Reliable noninvasive biomarkers are “urgently needed” for this patient population.
In an interview, Dr. Singh advised that clinicians continue to use current noninvasive scores in patients with diabetes – preferably the NFS – “until we have a better scoring system.” If clinicians suspect advanced fibrosis based on lab tests and clinical data, then “liver biopsy should be considered,” he said.
The investigators described among the limitations of their study its retrospective, single-center design, with patients who were mostly white and from one geographic region.
Dr. Singh and colleagues reported no conflicts of interest or outside funding for their study.
SOURCE: Singh A et al. J Clin Gastroenterol. 2020 Mar 11. doi: 10.1097/MCG.0000000000001339.
Noninvasive fibrosis scores, which are widely used to predict advanced fibrosis in people with nonalcoholic fatty liver disease (NAFLD), do not do a good job of picking up advanced fibrosis in patients with underlying diabetes, according to a new study.
Advanced fibrosis is associated with an increased risk of cirrhosis, end-stage liver disease, and liver failure. Underlying diabetes is a risk factor for both advanced fibrosis and death in patients with NAFLD.
While liver biopsy remains the gold standard for detecting advanced fibrosis, high costs and risks limit its use. Noninvasive scores such as the AST/ALT ratio; AST to platelet ratio index (APRI); fibrosis-4 (FIB-4) index; and NAFLD fibrosis score (NFS) have gained popularity in recent years, as they offer the compelling advantage of using easily and cheaply attained clinical and laboratory measures to assess likelihood of disease.
But their accuracy has come into question, particularly for people with diabetes.
In research published in the Journal of Clinical Gastroenterology, Amandeep Singh, MD, and colleagues at the Cleveland Clinic looked at their center’s records for 1,157 patients with type 2 diabetes (65% women, 88% white, 85% with obesity) who had undergone a liver biopsy for suspected advanced fibrosis between 2000 and 2015. Biopsy results revealed that a third of the cohort (32%) was positive for advanced fibrosis.
The investigators then pulled patients’ laboratory results for AST, ALT, cholesterol, triglycerides, fasting glucose, hemoglobin A1c, bilirubin, albumin, platelet count, alkaline phosphatase, albumin, and lipid levels, all collected within a year of biopsy. After plugging these into the algorithms of four different scoring systems for advanced fibrosis, they compared results with results from the biopsies.
The scores of AST/ALT greater than 1.4, APRI of at least 1.5, NFS greater than 0.676, and FIB-4 index greater than 2.67 had high specificities of 84%, 97%, 70%, and 93%, respectively, but sensitivities of only 27%, 17%, 64%, and 44%. Even when the cutoff measures were tightened, the scoring systems still missed a lot of disease. This suggests, Dr. Singh and colleagues wrote, that “the presence of diabetes could decrease the predictive value of these scores to detect advanced disease in NAFLD patients.” Reliable noninvasive biomarkers are “urgently needed” for this patient population.
In an interview, Dr. Singh advised that clinicians continue to use current noninvasive scores in patients with diabetes – preferably the NFS – “until we have a better scoring system.” If clinicians suspect advanced fibrosis based on lab tests and clinical data, then “liver biopsy should be considered,” he said.
The investigators described among the limitations of their study its retrospective, single-center design, with patients who were mostly white and from one geographic region.
Dr. Singh and colleagues reported no conflicts of interest or outside funding for their study.
SOURCE: Singh A et al. J Clin Gastroenterol. 2020 Mar 11. doi: 10.1097/MCG.0000000000001339.
FROM THE JOURNAL OF CLINICAL GASTROENTEROLOGY
AASLD: Liver transplants should proceed despite COVID-19
In liver transplant recipients or patients with autoimmune hepatitis on immunosuppressive therapy, acute cellular rejection or disease flare should not be presumed in the face of active coronavirus disease 2019 (COVID-19), according to the American Association for the Study of Liver Diseases (AASLD).
Signs that would normally be interpreted as flare or rejection need to be considered more cautiously now because the virus attacks the liver, and elevated aspartate aminotransferase, alanine aminotransferase, and slightly elevated bilirubin are common, ranging from a prevalence of 14% to 53% in COVID-19 patients. Acute liver injury is possible, especially in more severe cases, the group said.
The advice comes from a recently released document from AASLD, called “Clinical Insights for Hepatology and Liver Transplant Providers During the Covid-19 Pandemic,” to help hepatologists and liver transplant providers negotiate the pandemic, according to the latest data. It’s a far-ranging work that contains a lot of now familiar steps for providers to take to protect themselves and patients from the virus, but also much advice specific to liver medicine.
For instance, the group said it’s important to keep in mind that experimental treatments for the infection, including statins, remdesivir, and tocilizumab, can be hepatotoxic. Abnormal liver biochemistries are not a contraindication, but liver biochemistries need to be followed regularly in COVID-19 patients, especially those treated with remdesivir or tocilizumab, regardless of baseline values.
Also, lopinavir/ritonavir is a potent inhibitor of cytochrome P450 enzymes involved with calcineurin inhibitor metabolism, so if it’s used, AASLD said to reduce tacrolimus dosages to 1/20–1/50 of baseline.
The group cautioned against anticipatory adjustments to immunosuppressive drugs or dosages in patients without COVID-19, but if immunosuppressed liver disease patients do get the infection, prednisone doses should be reduced but kept above 10 mg/day to avoid adrenal insufficiency. In the setting of lymphopenia, fever, or worsening COVID-19 pneumonia, it advised reduction of azathioprine and mycophenolate dosages and reduction of, but not stopping, calcineurin inhibitors.
Liver transplants should not be postponed. However, to minimize exposure to the hospital environment, AASLD advised to “consider evaluating only patients with HCC [hepatocellular carcinoma] or those patients with severe disease and high MELD [model for end-stage liver disease] scores who are likely to benefit from immediate liver transplant.”
“An argument that has been put forward to justify deferring some transplants is concern about immunosuppressing patients during the COVID-19 pandemic,” the group said, but “data suggest the innate immune response may be the main driver for pulmonary injury due to COVID-19 and [that] immunosuppression may be protective. ... Posttransplant immunosuppression was not a risk factor for mortality associated with” the severe acute respiratory syndrome pandemic in 2003-2004 or the ongoing Middle East respiratory syndrome pandemic, both also caused by coronaviruses.
AASLD advised against reducing immunosuppression or stopping mycophenolate for asymptomatic patients after transplant, but COVID-19 prevention measures should be emphasized, including frequent hand washing and staying away from large crowds.
People who test positive for COVID-19 are ineligible for organ donation. Bronchoalveolar lavage is the most sensitive test (93%), followed by nasal swabs (63%) and pharyngeal swabs (32%).
In general, the group said elective procedures should be postponed, but urgent ones, such as biliary surgery and transjugular intrahepatic portosystemic shunts for bleeding varices, in addition to liver transplants, should not.
Also, HCC patients “should not wait until the pandemic abates to undergo [surveillance] imaging because the prospective duration of the pandemic is unknown. ... An arbitrary delay of 2 months is reasonable” for imaging based on patient and facility circumstances, but otherwise, “proceed with HCC treatments rather than delaying them due to the pandemic,” the group said.
As for who to bring into the office for an initial consult, “consider seeing in person only new adult and pediatric patients with urgent issues and clinically significant liver disease (e.g., jaundice, elevated ALT or AST above 500 U/L, recent onset of hepatic decompensation),” AASLD said.
In liver transplant recipients or patients with autoimmune hepatitis on immunosuppressive therapy, acute cellular rejection or disease flare should not be presumed in the face of active coronavirus disease 2019 (COVID-19), according to the American Association for the Study of Liver Diseases (AASLD).
Signs that would normally be interpreted as flare or rejection need to be considered more cautiously now because the virus attacks the liver, and elevated aspartate aminotransferase, alanine aminotransferase, and slightly elevated bilirubin are common, ranging from a prevalence of 14% to 53% in COVID-19 patients. Acute liver injury is possible, especially in more severe cases, the group said.
The advice comes from a recently released document from AASLD, called “Clinical Insights for Hepatology and Liver Transplant Providers During the Covid-19 Pandemic,” to help hepatologists and liver transplant providers negotiate the pandemic, according to the latest data. It’s a far-ranging work that contains a lot of now familiar steps for providers to take to protect themselves and patients from the virus, but also much advice specific to liver medicine.
For instance, the group said it’s important to keep in mind that experimental treatments for the infection, including statins, remdesivir, and tocilizumab, can be hepatotoxic. Abnormal liver biochemistries are not a contraindication, but liver biochemistries need to be followed regularly in COVID-19 patients, especially those treated with remdesivir or tocilizumab, regardless of baseline values.
Also, lopinavir/ritonavir is a potent inhibitor of cytochrome P450 enzymes involved with calcineurin inhibitor metabolism, so if it’s used, AASLD said to reduce tacrolimus dosages to 1/20–1/50 of baseline.
The group cautioned against anticipatory adjustments to immunosuppressive drugs or dosages in patients without COVID-19, but if immunosuppressed liver disease patients do get the infection, prednisone doses should be reduced but kept above 10 mg/day to avoid adrenal insufficiency. In the setting of lymphopenia, fever, or worsening COVID-19 pneumonia, it advised reduction of azathioprine and mycophenolate dosages and reduction of, but not stopping, calcineurin inhibitors.
Liver transplants should not be postponed. However, to minimize exposure to the hospital environment, AASLD advised to “consider evaluating only patients with HCC [hepatocellular carcinoma] or those patients with severe disease and high MELD [model for end-stage liver disease] scores who are likely to benefit from immediate liver transplant.”
“An argument that has been put forward to justify deferring some transplants is concern about immunosuppressing patients during the COVID-19 pandemic,” the group said, but “data suggest the innate immune response may be the main driver for pulmonary injury due to COVID-19 and [that] immunosuppression may be protective. ... Posttransplant immunosuppression was not a risk factor for mortality associated with” the severe acute respiratory syndrome pandemic in 2003-2004 or the ongoing Middle East respiratory syndrome pandemic, both also caused by coronaviruses.
AASLD advised against reducing immunosuppression or stopping mycophenolate for asymptomatic patients after transplant, but COVID-19 prevention measures should be emphasized, including frequent hand washing and staying away from large crowds.
People who test positive for COVID-19 are ineligible for organ donation. Bronchoalveolar lavage is the most sensitive test (93%), followed by nasal swabs (63%) and pharyngeal swabs (32%).
In general, the group said elective procedures should be postponed, but urgent ones, such as biliary surgery and transjugular intrahepatic portosystemic shunts for bleeding varices, in addition to liver transplants, should not.
Also, HCC patients “should not wait until the pandemic abates to undergo [surveillance] imaging because the prospective duration of the pandemic is unknown. ... An arbitrary delay of 2 months is reasonable” for imaging based on patient and facility circumstances, but otherwise, “proceed with HCC treatments rather than delaying them due to the pandemic,” the group said.
As for who to bring into the office for an initial consult, “consider seeing in person only new adult and pediatric patients with urgent issues and clinically significant liver disease (e.g., jaundice, elevated ALT or AST above 500 U/L, recent onset of hepatic decompensation),” AASLD said.
In liver transplant recipients or patients with autoimmune hepatitis on immunosuppressive therapy, acute cellular rejection or disease flare should not be presumed in the face of active coronavirus disease 2019 (COVID-19), according to the American Association for the Study of Liver Diseases (AASLD).
Signs that would normally be interpreted as flare or rejection need to be considered more cautiously now because the virus attacks the liver, and elevated aspartate aminotransferase, alanine aminotransferase, and slightly elevated bilirubin are common, ranging from a prevalence of 14% to 53% in COVID-19 patients. Acute liver injury is possible, especially in more severe cases, the group said.
The advice comes from a recently released document from AASLD, called “Clinical Insights for Hepatology and Liver Transplant Providers During the Covid-19 Pandemic,” to help hepatologists and liver transplant providers negotiate the pandemic, according to the latest data. It’s a far-ranging work that contains a lot of now familiar steps for providers to take to protect themselves and patients from the virus, but also much advice specific to liver medicine.
For instance, the group said it’s important to keep in mind that experimental treatments for the infection, including statins, remdesivir, and tocilizumab, can be hepatotoxic. Abnormal liver biochemistries are not a contraindication, but liver biochemistries need to be followed regularly in COVID-19 patients, especially those treated with remdesivir or tocilizumab, regardless of baseline values.
Also, lopinavir/ritonavir is a potent inhibitor of cytochrome P450 enzymes involved with calcineurin inhibitor metabolism, so if it’s used, AASLD said to reduce tacrolimus dosages to 1/20–1/50 of baseline.
The group cautioned against anticipatory adjustments to immunosuppressive drugs or dosages in patients without COVID-19, but if immunosuppressed liver disease patients do get the infection, prednisone doses should be reduced but kept above 10 mg/day to avoid adrenal insufficiency. In the setting of lymphopenia, fever, or worsening COVID-19 pneumonia, it advised reduction of azathioprine and mycophenolate dosages and reduction of, but not stopping, calcineurin inhibitors.
Liver transplants should not be postponed. However, to minimize exposure to the hospital environment, AASLD advised to “consider evaluating only patients with HCC [hepatocellular carcinoma] or those patients with severe disease and high MELD [model for end-stage liver disease] scores who are likely to benefit from immediate liver transplant.”
“An argument that has been put forward to justify deferring some transplants is concern about immunosuppressing patients during the COVID-19 pandemic,” the group said, but “data suggest the innate immune response may be the main driver for pulmonary injury due to COVID-19 and [that] immunosuppression may be protective. ... Posttransplant immunosuppression was not a risk factor for mortality associated with” the severe acute respiratory syndrome pandemic in 2003-2004 or the ongoing Middle East respiratory syndrome pandemic, both also caused by coronaviruses.
AASLD advised against reducing immunosuppression or stopping mycophenolate for asymptomatic patients after transplant, but COVID-19 prevention measures should be emphasized, including frequent hand washing and staying away from large crowds.
People who test positive for COVID-19 are ineligible for organ donation. Bronchoalveolar lavage is the most sensitive test (93%), followed by nasal swabs (63%) and pharyngeal swabs (32%).
In general, the group said elective procedures should be postponed, but urgent ones, such as biliary surgery and transjugular intrahepatic portosystemic shunts for bleeding varices, in addition to liver transplants, should not.
Also, HCC patients “should not wait until the pandemic abates to undergo [surveillance] imaging because the prospective duration of the pandemic is unknown. ... An arbitrary delay of 2 months is reasonable” for imaging based on patient and facility circumstances, but otherwise, “proceed with HCC treatments rather than delaying them due to the pandemic,” the group said.
As for who to bring into the office for an initial consult, “consider seeing in person only new adult and pediatric patients with urgent issues and clinically significant liver disease (e.g., jaundice, elevated ALT or AST above 500 U/L, recent onset of hepatic decompensation),” AASLD said.
COVID-19 experiences from the ob.gyn. front line
As the COVID-19 pandemic continues to spread across the United States, several members of the Ob.Gyn. News Editorial Advisory Board shared their experiences.

Catherine Cansino, MD, MPH, who is an associate clinical professor in the department of obstetrics and gynecology at the University of California, Davis, discussed the changes COVID-19 has had on local and regional practice in Sacramento and northern California.
There has been a dramatic increase in telehealth, using video, phone, and apps such as Zoom. Although ob.gyns. at the university are limiting outpatient appointments to essential visits only, we are continuing to offer telehealth to a few nonessential visits. This will be readdressed when the COVID-19 cases peak, Dr. Cansino said.
All patients admitted to labor & delivery undergo COVID-19 testing regardless of symptoms. For patients in the clinic who are expected to be induced or scheduled for cesarean delivery, we are screening them within 72 hours before admission.
In gynecology, only essential or urgent surgeries at UC Davis are being performed and include indications such as cancer, serious benign conditions unresponsive to conservative treatment (e.g., tubo-ovarian abscess, large symptomatic adnexal mass), and pregnancy termination. We are preserving access to abortion and reproductive health services since these are essential services.
We limit the number of providers involved in direct contact with inpatients to one or two, including a physician, nurse, and/or resident, Dr. Cansino said in an interview. Based on recent Liaison Committee on Medical Education policies related to concerns about educational experience during the pandemic, no medical students are allowed at the hospital at present. We also severely restrict the number of visitors in the inpatient and outpatient settings, including only two attendants (partner, doula, and such) during labor and delivery, and consider the impact on patients’ well-being when we restrict their visitors.
We are following University of California guidelines regarding face mask use, which have been in evolution over the last month. Face masks are used for patients and the health care providers primarily when patients either have known COVID-19 infection or are considered as patients under investigation or if the employee had a high-risk exposure. The use of face masks is becoming more permissive, rather than mandatory, to conserve personal protective equipment (PPE) for when the surge arrives.
Education is ongoing about caring for our families and ourselves if we get infected and need to isolate within our own homes. The department and health system is trying to balance the challenges of urgent patient care needs against the wellness concerns for the faculty, staff, and residents. Many physicians are also struggling with childcare problems, which add to our personal stress. There is anxiety among many physicians about exposure to asymptomatic carriers, including themselves, patients, and their families, Dr. Cansino said.

David Forstein, DO, dean and professor of obstetrics and gynecology at Touro College of Osteopathic Medicine, New York, said in an interview that the COVID-19 pandemic has “totally disrupted medical education. At almost all medical schools, didactics have moved completely online – ZOOM sessions abound, but labs become demonstrations, if at all, during the preclinical years. The clinical years have been put on hold, as well as student rotations suspended, out of caution for the students because hospitals needed to conserve PPE for the essential personnel and because administrators knew there would be less time for teaching. After initially requesting a pause, many hospitals now are asking students to come back because so many physicians, nurses, and residents have become ill with COVID-19 and either are quarantined or are patients in the hospital themselves.
“There has been a state-by-state call to consider graduating health professions students early, and press them into service, before their residencies actually begin. Some locations are looking for these new graduates to volunteer; some are willing to pay them a resident’s salary level. Medical schools are auditing their student records now to see which students would qualify to graduate early,” Dr. Forstein noted.

David M. Jaspan, DO, chairman of the department of obstetrics and gynecology at the Einstein Health Care Network in Philadelphia, described in an email interview how COVID-19 has changed practice.
To minimize the number of providers on the front line, we have developed a Monday to Friday rotating schedule of three teams of five members, he explained. There will be a hospital-based team, an office-based team, and a telehealth-based team who will provide their services from home. On-call responsibilities remain the same.
The hospital team, working 7 a.m. to 5 p.m., will rotate through assignments each day:
- One person will cover labor and delivery.
- One person will cover triage and help on labor and delivery.
- One person will be assigned to the resident office.
- One person will be assigned to cover the team of the post call attending (Sunday through Thursday call).
- One person will be assigned to gynecology coverage, consults, and postpartum rounds.
To further minimize the patient interactions, when possible, each patient should be seen by the attending physician with the resident. This is a change from usual practice, where the patient is first seen by the resident, who reports back to the attending, and then both physicians see the patient together.
The network’s offices now open from 9 a.m. (many offices had been offering early-morning hours starting at 7 a.m.), and the physicians and advanced practice providers will work through the last scheduled patient appointment, Dr. Jaspan explained. “The office-based team will preferentially see in-person visits.”
Several offices have been closed so that ob.gyns. and staff can be reassigned to telehealth. The remaining five offices generally have one attending physician and one advanced practice provider.
The remaining team of ob.gyns. provides telehealth with the help of staff members. This involves an initial call to the patient by staff letting them know the doctor will be calling, checking them in, verifying insurance, and collecting payment, followed by the actual telehealth visit. If follow-up is needed, the staff member schedules the follow-up.
Dr. Jaspan called the new approach to prenatal care because of COVID-19 a “cataclysmic change in how we care for our patients. We have decided to further limit our obstetrical in-person visits. It is our feeling that these changes will enable patients to remain outside of the office and in the safety of their homes, provide appropriate social distancing, and diminish potential exposures to the office staff providers and patients.”
In-person visits will occur at: the initial visit, between 24 and 28 weeks, at 32 weeks, and at 36 or 37 weeks; if the patient at 36/37 has a blood pressure cuff, they will not have additional scheduled in-patient visits. We have partnered with the insurance companies to provide more than 88% of obstetrical patients with home blood pressure cuffs.
Obstetrical visits via telehealth will continue at our standard intervals: monthly until 26 weeks; twice monthly during 26-36 weeks; and weekly from 37 weeks to delivery. These visits should use a video component such as Zoom, Doxy.me, or FaceTime.
“If the patient has concerns or problems, we will see them at any time. However, the new standard will be telehealth visits and the exception will be the in-person visit,” Dr. Jaspan said.
In addition, we have worked our division of maternal-fetal medicine to adjust the antenatal testing schedules, and we have curtailed the frequency of ultrasound, he noted.
He emphasized the importance of documenting telehealth interactions with obstetrical patients, in addition to “providing adequate teaching and education for patients regarding kick counts to ensure fetal well-being.” It also is key to “properly document conversations with patients regarding bleeding, rupture of membranes, fetal movement, headache, visual changes, fevers, cough, nausea and vomiting, diarrhea, fatigue, muscle aches, etc.”
The residents’ schedule also has been modified to diminish their exposure. Within our new paradigm, we have scheduled video conferences to enable our program to maintain our commitment to academics.
It is imperative that we keep our patients safe, and it is critical to protect our staff members. Those who provide women’s health cannot be replaced by other nurses or physicians.

Mark P. Trolice, MD, is director of Fertility CARE: the IVF Center in Winter Park, Fla., and professor of obstetrics and gynecology at the University of Central Florida, Orlando. He related in an email interview that, on March 17, 2020, the American Society for Reproductive Medicine (ASRM) released “Patient Management and Clinical Recommendations During the Coronavirus (COVID-19) Pandemic.” This document serves as guidance on fertility care during the current crisis. Specifically, the recommendations include the following:
- Suspend initiation of new treatment cycles, including ovulation induction, intrauterine inseminations, in vitro fertilization including retrievals and frozen embryo transfers, and nonurgent gamete cryopreservation.
- Strongly consider cancellation of all embryo transfers, whether fresh or frozen.
- Continue to care for patients who are currently “in cycle” or who require urgent stimulation and cryopreservation.
- Suspend elective surgeries and nonurgent diagnostic procedures.
- Minimize in-person interactions and increase utilization of telehealth.
As a member of ASRM for more than 2 decades and a participant of several of their committees, my practice immediately ceased treatment cycles to comply with this guidance.
Then on March 20, 2020, the Florida governor’s executive order 20-72 was released, stating, “All hospitals, ambulatory surgical centers, office surgery centers, dental, orthodontic and endodontic offices, and other health care practitioners’ offices in the State of Florida are prohibited from providing any medically unnecessary, nonurgent or nonemergency procedure or surgery which, if delayed, does not place a patient’s immediate health, safety, or well-being at risk, or will, if delayed, not contribute to the worsening of a serious or life-threatening medical condition.”
As a result, my practice has been limited to telemedicine consultations. While the ASRM guidance and the gubernatorial executive order pose a significant financial hardship on my center and all applicable medical clinics in my state, resulting in expected layoffs, salary reductions, and requests for government stimulus loans, the greater good takes priority and we pray for all the victims of this devastating pandemic.
The governor’s current executive order is set to expire on May 9, 2020, unless it is extended.
ASRM released an update of their guidance on March 30, 2020, offering no change from their prior recommendations. The organization plans to reevaluate the guidance at 2-week intervals.
Sangeeta Sinha, MD, an ob.gyn. in private practice at Stone Springs Hospital Center, Dulles, Va. said in an interview, “COVID 19 has put fear in all aspects of our daily activities which we are attempting to cope with.”
She related several changes made to her office and hospital environments. “In our office, we are now wearing a mask at all times, gloves to examine every patient. We have staggered physicians in the office to take televisits and in-office patients. We are screening all new patients on the phone to determine if they are sick, have traveled to high-risk, hot spot areas of the country, or have had contact with someone who tested positive for COVID-19. We are only seeing our pregnant women and have also pushed out their return appointments to 4 weeks if possible. There are several staff who are not working due to fear or are in self quarantine so we have shortage of staff in the office. At the hospital as well we are wearing a mask at all times, using personal protective equipment for deliveries and C-sections.
“We have had several scares, including a new transfer of an 18-year-old pregnant patient at 30 weeks with cough and sore throat, who later reported that her roommate is very sick and he works with someone who has tested positive for COVID-19. Thankfully she is healthy and well. We learned several lessons from this one.”
As the COVID-19 pandemic continues to spread across the United States, several members of the Ob.Gyn. News Editorial Advisory Board shared their experiences.
Catherine Cansino, MD, MPH, who is an associate clinical professor in the department of obstetrics and gynecology at the University of California, Davis, discussed the changes COVID-19 has had on local and regional practice in Sacramento and northern California.
There has been a dramatic increase in telehealth, using video, phone, and apps such as Zoom. Although ob.gyns. at the university are limiting outpatient appointments to essential visits only, we are continuing to offer telehealth to a few nonessential visits. This will be readdressed when the COVID-19 cases peak, Dr. Cansino said.
All patients admitted to labor & delivery undergo COVID-19 testing regardless of symptoms. For patients in the clinic who are expected to be induced or scheduled for cesarean delivery, we are screening them within 72 hours before admission.
In gynecology, only essential or urgent surgeries at UC Davis are being performed and include indications such as cancer, serious benign conditions unresponsive to conservative treatment (e.g., tubo-ovarian abscess, large symptomatic adnexal mass), and pregnancy termination. We are preserving access to abortion and reproductive health services since these are essential services.
We limit the number of providers involved in direct contact with inpatients to one or two, including a physician, nurse, and/or resident, Dr. Cansino said in an interview. Based on recent Liaison Committee on Medical Education policies related to concerns about educational experience during the pandemic, no medical students are allowed at the hospital at present. We also severely restrict the number of visitors in the inpatient and outpatient settings, including only two attendants (partner, doula, and such) during labor and delivery, and consider the impact on patients’ well-being when we restrict their visitors.
We are following University of California guidelines regarding face mask use, which have been in evolution over the last month. Face masks are used for patients and the health care providers primarily when patients either have known COVID-19 infection or are considered as patients under investigation or if the employee had a high-risk exposure. The use of face masks is becoming more permissive, rather than mandatory, to conserve personal protective equipment (PPE) for when the surge arrives.
Education is ongoing about caring for our families and ourselves if we get infected and need to isolate within our own homes. The department and health system is trying to balance the challenges of urgent patient care needs against the wellness concerns for the faculty, staff, and residents. Many physicians are also struggling with childcare problems, which add to our personal stress. There is anxiety among many physicians about exposure to asymptomatic carriers, including themselves, patients, and their families, Dr. Cansino said.
David Forstein, DO, dean and professor of obstetrics and gynecology at Touro College of Osteopathic Medicine, New York, said in an interview that the COVID-19 pandemic has “totally disrupted medical education. At almost all medical schools, didactics have moved completely online – ZOOM sessions abound, but labs become demonstrations, if at all, during the preclinical years. The clinical years have been put on hold, as well as student rotations suspended, out of caution for the students because hospitals needed to conserve PPE for the essential personnel and because administrators knew there would be less time for teaching. After initially requesting a pause, many hospitals now are asking students to come back because so many physicians, nurses, and residents have become ill with COVID-19 and either are quarantined or are patients in the hospital themselves.
“There has been a state-by-state call to consider graduating health professions students early, and press them into service, before their residencies actually begin. Some locations are looking for these new graduates to volunteer; some are willing to pay them a resident’s salary level. Medical schools are auditing their student records now to see which students would qualify to graduate early,” Dr. Forstein noted.
David M. Jaspan, DO, chairman of the department of obstetrics and gynecology at the Einstein Health Care Network in Philadelphia, described in an email interview how COVID-19 has changed practice.
To minimize the number of providers on the front line, we have developed a Monday to Friday rotating schedule of three teams of five members, he explained. There will be a hospital-based team, an office-based team, and a telehealth-based team who will provide their services from home. On-call responsibilities remain the same.
The hospital team, working 7 a.m. to 5 p.m., will rotate through assignments each day:
- One person will cover labor and delivery.
- One person will cover triage and help on labor and delivery.
- One person will be assigned to the resident office.
- One person will be assigned to cover the team of the post call attending (Sunday through Thursday call).
- One person will be assigned to gynecology coverage, consults, and postpartum rounds.
To further minimize the patient interactions, when possible, each patient should be seen by the attending physician with the resident. This is a change from usual practice, where the patient is first seen by the resident, who reports back to the attending, and then both physicians see the patient together.
The network’s offices now open from 9 a.m. (many offices had been offering early-morning hours starting at 7 a.m.), and the physicians and advanced practice providers will work through the last scheduled patient appointment, Dr. Jaspan explained. “The office-based team will preferentially see in-person visits.”
Several offices have been closed so that ob.gyns. and staff can be reassigned to telehealth. The remaining five offices generally have one attending physician and one advanced practice provider.
The remaining team of ob.gyns. provides telehealth with the help of staff members. This involves an initial call to the patient by staff letting them know the doctor will be calling, checking them in, verifying insurance, and collecting payment, followed by the actual telehealth visit. If follow-up is needed, the staff member schedules the follow-up.
Dr. Jaspan called the new approach to prenatal care because of COVID-19 a “cataclysmic change in how we care for our patients. We have decided to further limit our obstetrical in-person visits. It is our feeling that these changes will enable patients to remain outside of the office and in the safety of their homes, provide appropriate social distancing, and diminish potential exposures to the office staff providers and patients.”
In-person visits will occur at: the initial visit, between 24 and 28 weeks, at 32 weeks, and at 36 or 37 weeks; if the patient at 36/37 has a blood pressure cuff, they will not have additional scheduled in-patient visits. We have partnered with the insurance companies to provide more than 88% of obstetrical patients with home blood pressure cuffs.
Obstetrical visits via telehealth will continue at our standard intervals: monthly until 26 weeks; twice monthly during 26-36 weeks; and weekly from 37 weeks to delivery. These visits should use a video component such as Zoom, Doxy.me, or FaceTime.
“If the patient has concerns or problems, we will see them at any time. However, the new standard will be telehealth visits and the exception will be the in-person visit,” Dr. Jaspan said.
In addition, we have worked our division of maternal-fetal medicine to adjust the antenatal testing schedules, and we have curtailed the frequency of ultrasound, he noted.
He emphasized the importance of documenting telehealth interactions with obstetrical patients, in addition to “providing adequate teaching and education for patients regarding kick counts to ensure fetal well-being.” It also is key to “properly document conversations with patients regarding bleeding, rupture of membranes, fetal movement, headache, visual changes, fevers, cough, nausea and vomiting, diarrhea, fatigue, muscle aches, etc.”
The residents’ schedule also has been modified to diminish their exposure. Within our new paradigm, we have scheduled video conferences to enable our program to maintain our commitment to academics.
It is imperative that we keep our patients safe, and it is critical to protect our staff members. Those who provide women’s health cannot be replaced by other nurses or physicians.
Mark P. Trolice, MD, is director of Fertility CARE: the IVF Center in Winter Park, Fla., and professor of obstetrics and gynecology at the University of Central Florida, Orlando. He related in an email interview that, on March 17, 2020, the American Society for Reproductive Medicine (ASRM) released “Patient Management and Clinical Recommendations During the Coronavirus (COVID-19) Pandemic.” This document serves as guidance on fertility care during the current crisis. Specifically, the recommendations include the following:
- Suspend initiation of new treatment cycles, including ovulation induction, intrauterine inseminations, in vitro fertilization including retrievals and frozen embryo transfers, and nonurgent gamete cryopreservation.
- Strongly consider cancellation of all embryo transfers, whether fresh or frozen.
- Continue to care for patients who are currently “in cycle” or who require urgent stimulation and cryopreservation.
- Suspend elective surgeries and nonurgent diagnostic procedures.
- Minimize in-person interactions and increase utilization of telehealth.
As a member of ASRM for more than 2 decades and a participant of several of their committees, my practice immediately ceased treatment cycles to comply with this guidance.
Then on March 20, 2020, the Florida governor’s executive order 20-72 was released, stating, “All hospitals, ambulatory surgical centers, office surgery centers, dental, orthodontic and endodontic offices, and other health care practitioners’ offices in the State of Florida are prohibited from providing any medically unnecessary, nonurgent or nonemergency procedure or surgery which, if delayed, does not place a patient’s immediate health, safety, or well-being at risk, or will, if delayed, not contribute to the worsening of a serious or life-threatening medical condition.”
As a result, my practice has been limited to telemedicine consultations. While the ASRM guidance and the gubernatorial executive order pose a significant financial hardship on my center and all applicable medical clinics in my state, resulting in expected layoffs, salary reductions, and requests for government stimulus loans, the greater good takes priority and we pray for all the victims of this devastating pandemic.
The governor’s current executive order is set to expire on May 9, 2020, unless it is extended.
ASRM released an update of their guidance on March 30, 2020, offering no change from their prior recommendations. The organization plans to reevaluate the guidance at 2-week intervals.
Sangeeta Sinha, MD, an ob.gyn. in private practice at Stone Springs Hospital Center, Dulles, Va. said in an interview, “COVID 19 has put fear in all aspects of our daily activities which we are attempting to cope with.”
She related several changes made to her office and hospital environments. “In our office, we are now wearing a mask at all times, gloves to examine every patient. We have staggered physicians in the office to take televisits and in-office patients. We are screening all new patients on the phone to determine if they are sick, have traveled to high-risk, hot spot areas of the country, or have had contact with someone who tested positive for COVID-19. We are only seeing our pregnant women and have also pushed out their return appointments to 4 weeks if possible. There are several staff who are not working due to fear or are in self quarantine so we have shortage of staff in the office. At the hospital as well we are wearing a mask at all times, using personal protective equipment for deliveries and C-sections.
“We have had several scares, including a new transfer of an 18-year-old pregnant patient at 30 weeks with cough and sore throat, who later reported that her roommate is very sick and he works with someone who has tested positive for COVID-19. Thankfully she is healthy and well. We learned several lessons from this one.”
As the COVID-19 pandemic continues to spread across the United States, several members of the Ob.Gyn. News Editorial Advisory Board shared their experiences.
Catherine Cansino, MD, MPH, who is an associate clinical professor in the department of obstetrics and gynecology at the University of California, Davis, discussed the changes COVID-19 has had on local and regional practice in Sacramento and northern California.
There has been a dramatic increase in telehealth, using video, phone, and apps such as Zoom. Although ob.gyns. at the university are limiting outpatient appointments to essential visits only, we are continuing to offer telehealth to a few nonessential visits. This will be readdressed when the COVID-19 cases peak, Dr. Cansino said.
All patients admitted to labor & delivery undergo COVID-19 testing regardless of symptoms. For patients in the clinic who are expected to be induced or scheduled for cesarean delivery, we are screening them within 72 hours before admission.
In gynecology, only essential or urgent surgeries at UC Davis are being performed and include indications such as cancer, serious benign conditions unresponsive to conservative treatment (e.g., tubo-ovarian abscess, large symptomatic adnexal mass), and pregnancy termination. We are preserving access to abortion and reproductive health services since these are essential services.
We limit the number of providers involved in direct contact with inpatients to one or two, including a physician, nurse, and/or resident, Dr. Cansino said in an interview. Based on recent Liaison Committee on Medical Education policies related to concerns about educational experience during the pandemic, no medical students are allowed at the hospital at present. We also severely restrict the number of visitors in the inpatient and outpatient settings, including only two attendants (partner, doula, and such) during labor and delivery, and consider the impact on patients’ well-being when we restrict their visitors.
We are following University of California guidelines regarding face mask use, which have been in evolution over the last month. Face masks are used for patients and the health care providers primarily when patients either have known COVID-19 infection or are considered as patients under investigation or if the employee had a high-risk exposure. The use of face masks is becoming more permissive, rather than mandatory, to conserve personal protective equipment (PPE) for when the surge arrives.
Education is ongoing about caring for our families and ourselves if we get infected and need to isolate within our own homes. The department and health system is trying to balance the challenges of urgent patient care needs against the wellness concerns for the faculty, staff, and residents. Many physicians are also struggling with childcare problems, which add to our personal stress. There is anxiety among many physicians about exposure to asymptomatic carriers, including themselves, patients, and their families, Dr. Cansino said.
David Forstein, DO, dean and professor of obstetrics and gynecology at Touro College of Osteopathic Medicine, New York, said in an interview that the COVID-19 pandemic has “totally disrupted medical education. At almost all medical schools, didactics have moved completely online – ZOOM sessions abound, but labs become demonstrations, if at all, during the preclinical years. The clinical years have been put on hold, as well as student rotations suspended, out of caution for the students because hospitals needed to conserve PPE for the essential personnel and because administrators knew there would be less time for teaching. After initially requesting a pause, many hospitals now are asking students to come back because so many physicians, nurses, and residents have become ill with COVID-19 and either are quarantined or are patients in the hospital themselves.
“There has been a state-by-state call to consider graduating health professions students early, and press them into service, before their residencies actually begin. Some locations are looking for these new graduates to volunteer; some are willing to pay them a resident’s salary level. Medical schools are auditing their student records now to see which students would qualify to graduate early,” Dr. Forstein noted.
David M. Jaspan, DO, chairman of the department of obstetrics and gynecology at the Einstein Health Care Network in Philadelphia, described in an email interview how COVID-19 has changed practice.
To minimize the number of providers on the front line, we have developed a Monday to Friday rotating schedule of three teams of five members, he explained. There will be a hospital-based team, an office-based team, and a telehealth-based team who will provide their services from home. On-call responsibilities remain the same.
The hospital team, working 7 a.m. to 5 p.m., will rotate through assignments each day:
- One person will cover labor and delivery.
- One person will cover triage and help on labor and delivery.
- One person will be assigned to the resident office.
- One person will be assigned to cover the team of the post call attending (Sunday through Thursday call).
- One person will be assigned to gynecology coverage, consults, and postpartum rounds.
To further minimize the patient interactions, when possible, each patient should be seen by the attending physician with the resident. This is a change from usual practice, where the patient is first seen by the resident, who reports back to the attending, and then both physicians see the patient together.
The network’s offices now open from 9 a.m. (many offices had been offering early-morning hours starting at 7 a.m.), and the physicians and advanced practice providers will work through the last scheduled patient appointment, Dr. Jaspan explained. “The office-based team will preferentially see in-person visits.”
Several offices have been closed so that ob.gyns. and staff can be reassigned to telehealth. The remaining five offices generally have one attending physician and one advanced practice provider.
The remaining team of ob.gyns. provides telehealth with the help of staff members. This involves an initial call to the patient by staff letting them know the doctor will be calling, checking them in, verifying insurance, and collecting payment, followed by the actual telehealth visit. If follow-up is needed, the staff member schedules the follow-up.
Dr. Jaspan called the new approach to prenatal care because of COVID-19 a “cataclysmic change in how we care for our patients. We have decided to further limit our obstetrical in-person visits. It is our feeling that these changes will enable patients to remain outside of the office and in the safety of their homes, provide appropriate social distancing, and diminish potential exposures to the office staff providers and patients.”
In-person visits will occur at: the initial visit, between 24 and 28 weeks, at 32 weeks, and at 36 or 37 weeks; if the patient at 36/37 has a blood pressure cuff, they will not have additional scheduled in-patient visits. We have partnered with the insurance companies to provide more than 88% of obstetrical patients with home blood pressure cuffs.
Obstetrical visits via telehealth will continue at our standard intervals: monthly until 26 weeks; twice monthly during 26-36 weeks; and weekly from 37 weeks to delivery. These visits should use a video component such as Zoom, Doxy.me, or FaceTime.
“If the patient has concerns or problems, we will see them at any time. However, the new standard will be telehealth visits and the exception will be the in-person visit,” Dr. Jaspan said.
In addition, we have worked our division of maternal-fetal medicine to adjust the antenatal testing schedules, and we have curtailed the frequency of ultrasound, he noted.
He emphasized the importance of documenting telehealth interactions with obstetrical patients, in addition to “providing adequate teaching and education for patients regarding kick counts to ensure fetal well-being.” It also is key to “properly document conversations with patients regarding bleeding, rupture of membranes, fetal movement, headache, visual changes, fevers, cough, nausea and vomiting, diarrhea, fatigue, muscle aches, etc.”
The residents’ schedule also has been modified to diminish their exposure. Within our new paradigm, we have scheduled video conferences to enable our program to maintain our commitment to academics.
It is imperative that we keep our patients safe, and it is critical to protect our staff members. Those who provide women’s health cannot be replaced by other nurses or physicians.
Mark P. Trolice, MD, is director of Fertility CARE: the IVF Center in Winter Park, Fla., and professor of obstetrics and gynecology at the University of Central Florida, Orlando. He related in an email interview that, on March 17, 2020, the American Society for Reproductive Medicine (ASRM) released “Patient Management and Clinical Recommendations During the Coronavirus (COVID-19) Pandemic.” This document serves as guidance on fertility care during the current crisis. Specifically, the recommendations include the following:
- Suspend initiation of new treatment cycles, including ovulation induction, intrauterine inseminations, in vitro fertilization including retrievals and frozen embryo transfers, and nonurgent gamete cryopreservation.
- Strongly consider cancellation of all embryo transfers, whether fresh or frozen.
- Continue to care for patients who are currently “in cycle” or who require urgent stimulation and cryopreservation.
- Suspend elective surgeries and nonurgent diagnostic procedures.
- Minimize in-person interactions and increase utilization of telehealth.
As a member of ASRM for more than 2 decades and a participant of several of their committees, my practice immediately ceased treatment cycles to comply with this guidance.
Then on March 20, 2020, the Florida governor’s executive order 20-72 was released, stating, “All hospitals, ambulatory surgical centers, office surgery centers, dental, orthodontic and endodontic offices, and other health care practitioners’ offices in the State of Florida are prohibited from providing any medically unnecessary, nonurgent or nonemergency procedure or surgery which, if delayed, does not place a patient’s immediate health, safety, or well-being at risk, or will, if delayed, not contribute to the worsening of a serious or life-threatening medical condition.”
As a result, my practice has been limited to telemedicine consultations. While the ASRM guidance and the gubernatorial executive order pose a significant financial hardship on my center and all applicable medical clinics in my state, resulting in expected layoffs, salary reductions, and requests for government stimulus loans, the greater good takes priority and we pray for all the victims of this devastating pandemic.
The governor’s current executive order is set to expire on May 9, 2020, unless it is extended.
ASRM released an update of their guidance on March 30, 2020, offering no change from their prior recommendations. The organization plans to reevaluate the guidance at 2-week intervals.
Sangeeta Sinha, MD, an ob.gyn. in private practice at Stone Springs Hospital Center, Dulles, Va. said in an interview, “COVID 19 has put fear in all aspects of our daily activities which we are attempting to cope with.”
She related several changes made to her office and hospital environments. “In our office, we are now wearing a mask at all times, gloves to examine every patient. We have staggered physicians in the office to take televisits and in-office patients. We are screening all new patients on the phone to determine if they are sick, have traveled to high-risk, hot spot areas of the country, or have had contact with someone who tested positive for COVID-19. We are only seeing our pregnant women and have also pushed out their return appointments to 4 weeks if possible. There are several staff who are not working due to fear or are in self quarantine so we have shortage of staff in the office. At the hospital as well we are wearing a mask at all times, using personal protective equipment for deliveries and C-sections.
“We have had several scares, including a new transfer of an 18-year-old pregnant patient at 30 weeks with cough and sore throat, who later reported that her roommate is very sick and he works with someone who has tested positive for COVID-19. Thankfully she is healthy and well. We learned several lessons from this one.”
Patients with preexisting diabetes benefit less from bariatric surgery
according to a retrospective review of patients receiving both sleeve gastrectomy and gastric bypass.
The difference was particularly pronounced and persistent for patients who had gastric bypass, Yingying Luo, MD, said during a virtual news conference held by the Endocrine Society. The study was slated for presentation during ENDO 2020, the society’s annual meeting, which was canceled because of the COVID-19 pandemic.
“Our findings demonstrated that having bariatric surgery before developing diabetes may result in greater weight loss from the surgery, especially within the first 3 years after surgery and in patients undergoing gastric bypass surgery,” said Dr. Luo.
More than a third of U.S. adults have obesity, and more than half the population is overweight or has obesity, said Dr. Luo, citing data from the Centers for Disease Control and Prevention.
Bariatric surgery not only reduces body weight, but also “can lead to remission of many metabolic disorders, including diabetes, hypertension, and dyslipidemia,” said Dr. Luo, a visiting scholar at the University of Michigan’s division of metabolism, endocrinology, and diabetes. However, until now, it has not been known how diabetes interacts with bariatric surgery to affect weight loss outcomes.
To address that question, Dr. Luo and her colleagues looked at patients in the Michigan Bariatric Surgery Cohort who were at least 18 years old and had a body mass index (BMI) of more than 40 kg/m2, or of more than 35 kg/m2 with comorbidities.
The researchers followed 380 patients who received gastric bypass and 334 who received sleeve gastrectomy for at least 5 years. Over time, sleeve gastrectomy became the predominant type of surgery conducted, noted Dr. Luo.
At baseline, and yearly for 5 years thereafter, the researchers recorded participants’ BMI as well as their lipid levels and other laboratory values. Medication use was also tracked. Patients with a diagnosis of diabetes also had their hemoglobin A1c levels recorded at each visit.
Overall, patients in the sleeve gastrectomy group were more overweight, and those in the gastric bypass group had higher HbA1c and total cholesterol levels. The mean baseline weight for the sleeve gastrectomy recipients was 141.5 kg, compared with 133.5 kg for those receiving gastric bypass (BMI, 49.9 vs. 47.3 kg/m2, respectively; P < .01 for both measures). Mean HbA1c was 6.5% for the gastric bypass group, compared with 6.3% for the sleeve gastrectomy group (P = .03).
At baseline, 149 (39.2%) of the gastric bypass patients had diabetes, compared with 108 (32.3%) of the sleeve gastrectomy patients, but the difference was not statistically significant.
About two-thirds of the full cohort were tracked for at least 5 years, which is still considered “a good follow-up rate in a real-world study,” said Dr. Luo.
Total weight loss was defined as the difference between initial weight and postoperative weight at a given point in time. Excess weight was the difference between initial weight and an individual’s ideal weight, that is, what their weight would have been if they had a BMI of 25 kg/m2.
“The probability of achieving a BMI of less than 30 kg/m2 or excess weight loss of 50% or more was higher in patients who did not have diabetes diagnosis at baseline. We found that the presence of diabetes at baseline substantially impacted the probability of achieving both indicators,” said Dr. Luo. “Individuals without diabetes had a 1.5 times higher chance of achieving a BMI of under 30 kg/m2, and … [they also] had a 1.6 times higher chance of achieving excess body weight loss of 50%, or more.” Both of those differences were statistically significant on univariate analysis (P = .0249 and .0021, respectively).
The researchers conducted further statistical analysis – adjusted for age, gender, surgery type, and baseline weight – to examine whether diabetes still predicted future weight loss after bariatric surgery. After those adjustments, they still found that “the presence of diabetes before surgery is an indicator of future weight loss outcomes,” said Dr. Luo.
The differences in outcomes for those with and without diabetes tended to diminish over time in looking at the cohort as a whole. However, greater BMI reduction for those without diabetes persisted for the full 5 years of follow-up for the gastric bypass recipients. Those trends held when the researchers looked at the proportion of patients whose BMI dropped to below 30 kg/m2, and those who achieved excess weight loss of more than 50%.
Dr. Luo acknowledged that an ideal study would track patients for longer than 5 years and that studies involving more patients would also be useful. Still, she said, “our study opens the door for further research to understand why diabetes diminishes the weight loss effect of bariatric surgery.”
The research will be published in a special supplemental issue of the Journal of the Endocrine Society. In addition to a series of news conferences on March 30-31, the society will host ENDO Online 2020 during June 8-22, which will present programming for clinicians and researchers.
Dr. Luo reported no outside sources of funding and no conflicts of interest.
SOURCE: Luo Y et al. ENDO 2020, Abstract 590.
according to a retrospective review of patients receiving both sleeve gastrectomy and gastric bypass.
The difference was particularly pronounced and persistent for patients who had gastric bypass, Yingying Luo, MD, said during a virtual news conference held by the Endocrine Society. The study was slated for presentation during ENDO 2020, the society’s annual meeting, which was canceled because of the COVID-19 pandemic.
“Our findings demonstrated that having bariatric surgery before developing diabetes may result in greater weight loss from the surgery, especially within the first 3 years after surgery and in patients undergoing gastric bypass surgery,” said Dr. Luo.
More than a third of U.S. adults have obesity, and more than half the population is overweight or has obesity, said Dr. Luo, citing data from the Centers for Disease Control and Prevention.
Bariatric surgery not only reduces body weight, but also “can lead to remission of many metabolic disorders, including diabetes, hypertension, and dyslipidemia,” said Dr. Luo, a visiting scholar at the University of Michigan’s division of metabolism, endocrinology, and diabetes. However, until now, it has not been known how diabetes interacts with bariatric surgery to affect weight loss outcomes.
To address that question, Dr. Luo and her colleagues looked at patients in the Michigan Bariatric Surgery Cohort who were at least 18 years old and had a body mass index (BMI) of more than 40 kg/m2, or of more than 35 kg/m2 with comorbidities.
The researchers followed 380 patients who received gastric bypass and 334 who received sleeve gastrectomy for at least 5 years. Over time, sleeve gastrectomy became the predominant type of surgery conducted, noted Dr. Luo.
At baseline, and yearly for 5 years thereafter, the researchers recorded participants’ BMI as well as their lipid levels and other laboratory values. Medication use was also tracked. Patients with a diagnosis of diabetes also had their hemoglobin A1c levels recorded at each visit.
Overall, patients in the sleeve gastrectomy group were more overweight, and those in the gastric bypass group had higher HbA1c and total cholesterol levels. The mean baseline weight for the sleeve gastrectomy recipients was 141.5 kg, compared with 133.5 kg for those receiving gastric bypass (BMI, 49.9 vs. 47.3 kg/m2, respectively; P < .01 for both measures). Mean HbA1c was 6.5% for the gastric bypass group, compared with 6.3% for the sleeve gastrectomy group (P = .03).
At baseline, 149 (39.2%) of the gastric bypass patients had diabetes, compared with 108 (32.3%) of the sleeve gastrectomy patients, but the difference was not statistically significant.
About two-thirds of the full cohort were tracked for at least 5 years, which is still considered “a good follow-up rate in a real-world study,” said Dr. Luo.
Total weight loss was defined as the difference between initial weight and postoperative weight at a given point in time. Excess weight was the difference between initial weight and an individual’s ideal weight, that is, what their weight would have been if they had a BMI of 25 kg/m2.
“The probability of achieving a BMI of less than 30 kg/m2 or excess weight loss of 50% or more was higher in patients who did not have diabetes diagnosis at baseline. We found that the presence of diabetes at baseline substantially impacted the probability of achieving both indicators,” said Dr. Luo. “Individuals without diabetes had a 1.5 times higher chance of achieving a BMI of under 30 kg/m2, and … [they also] had a 1.6 times higher chance of achieving excess body weight loss of 50%, or more.” Both of those differences were statistically significant on univariate analysis (P = .0249 and .0021, respectively).
The researchers conducted further statistical analysis – adjusted for age, gender, surgery type, and baseline weight – to examine whether diabetes still predicted future weight loss after bariatric surgery. After those adjustments, they still found that “the presence of diabetes before surgery is an indicator of future weight loss outcomes,” said Dr. Luo.
The differences in outcomes for those with and without diabetes tended to diminish over time in looking at the cohort as a whole. However, greater BMI reduction for those without diabetes persisted for the full 5 years of follow-up for the gastric bypass recipients. Those trends held when the researchers looked at the proportion of patients whose BMI dropped to below 30 kg/m2, and those who achieved excess weight loss of more than 50%.
Dr. Luo acknowledged that an ideal study would track patients for longer than 5 years and that studies involving more patients would also be useful. Still, she said, “our study opens the door for further research to understand why diabetes diminishes the weight loss effect of bariatric surgery.”
The research will be published in a special supplemental issue of the Journal of the Endocrine Society. In addition to a series of news conferences on March 30-31, the society will host ENDO Online 2020 during June 8-22, which will present programming for clinicians and researchers.
Dr. Luo reported no outside sources of funding and no conflicts of interest.
SOURCE: Luo Y et al. ENDO 2020, Abstract 590.
according to a retrospective review of patients receiving both sleeve gastrectomy and gastric bypass.
The difference was particularly pronounced and persistent for patients who had gastric bypass, Yingying Luo, MD, said during a virtual news conference held by the Endocrine Society. The study was slated for presentation during ENDO 2020, the society’s annual meeting, which was canceled because of the COVID-19 pandemic.
“Our findings demonstrated that having bariatric surgery before developing diabetes may result in greater weight loss from the surgery, especially within the first 3 years after surgery and in patients undergoing gastric bypass surgery,” said Dr. Luo.
More than a third of U.S. adults have obesity, and more than half the population is overweight or has obesity, said Dr. Luo, citing data from the Centers for Disease Control and Prevention.
Bariatric surgery not only reduces body weight, but also “can lead to remission of many metabolic disorders, including diabetes, hypertension, and dyslipidemia,” said Dr. Luo, a visiting scholar at the University of Michigan’s division of metabolism, endocrinology, and diabetes. However, until now, it has not been known how diabetes interacts with bariatric surgery to affect weight loss outcomes.
To address that question, Dr. Luo and her colleagues looked at patients in the Michigan Bariatric Surgery Cohort who were at least 18 years old and had a body mass index (BMI) of more than 40 kg/m2, or of more than 35 kg/m2 with comorbidities.
The researchers followed 380 patients who received gastric bypass and 334 who received sleeve gastrectomy for at least 5 years. Over time, sleeve gastrectomy became the predominant type of surgery conducted, noted Dr. Luo.
At baseline, and yearly for 5 years thereafter, the researchers recorded participants’ BMI as well as their lipid levels and other laboratory values. Medication use was also tracked. Patients with a diagnosis of diabetes also had their hemoglobin A1c levels recorded at each visit.
Overall, patients in the sleeve gastrectomy group were more overweight, and those in the gastric bypass group had higher HbA1c and total cholesterol levels. The mean baseline weight for the sleeve gastrectomy recipients was 141.5 kg, compared with 133.5 kg for those receiving gastric bypass (BMI, 49.9 vs. 47.3 kg/m2, respectively; P < .01 for both measures). Mean HbA1c was 6.5% for the gastric bypass group, compared with 6.3% for the sleeve gastrectomy group (P = .03).
At baseline, 149 (39.2%) of the gastric bypass patients had diabetes, compared with 108 (32.3%) of the sleeve gastrectomy patients, but the difference was not statistically significant.
About two-thirds of the full cohort were tracked for at least 5 years, which is still considered “a good follow-up rate in a real-world study,” said Dr. Luo.
Total weight loss was defined as the difference between initial weight and postoperative weight at a given point in time. Excess weight was the difference between initial weight and an individual’s ideal weight, that is, what their weight would have been if they had a BMI of 25 kg/m2.
“The probability of achieving a BMI of less than 30 kg/m2 or excess weight loss of 50% or more was higher in patients who did not have diabetes diagnosis at baseline. We found that the presence of diabetes at baseline substantially impacted the probability of achieving both indicators,” said Dr. Luo. “Individuals without diabetes had a 1.5 times higher chance of achieving a BMI of under 30 kg/m2, and … [they also] had a 1.6 times higher chance of achieving excess body weight loss of 50%, or more.” Both of those differences were statistically significant on univariate analysis (P = .0249 and .0021, respectively).
The researchers conducted further statistical analysis – adjusted for age, gender, surgery type, and baseline weight – to examine whether diabetes still predicted future weight loss after bariatric surgery. After those adjustments, they still found that “the presence of diabetes before surgery is an indicator of future weight loss outcomes,” said Dr. Luo.
The differences in outcomes for those with and without diabetes tended to diminish over time in looking at the cohort as a whole. However, greater BMI reduction for those without diabetes persisted for the full 5 years of follow-up for the gastric bypass recipients. Those trends held when the researchers looked at the proportion of patients whose BMI dropped to below 30 kg/m2, and those who achieved excess weight loss of more than 50%.
Dr. Luo acknowledged that an ideal study would track patients for longer than 5 years and that studies involving more patients would also be useful. Still, she said, “our study opens the door for further research to understand why diabetes diminishes the weight loss effect of bariatric surgery.”
The research will be published in a special supplemental issue of the Journal of the Endocrine Society. In addition to a series of news conferences on March 30-31, the society will host ENDO Online 2020 during June 8-22, which will present programming for clinicians and researchers.
Dr. Luo reported no outside sources of funding and no conflicts of interest.
SOURCE: Luo Y et al. ENDO 2020, Abstract 590.
FROM ENDO 2020
Financial toxicity is a common complication of gynecologic cancers
More than one-fifth of patients being treated for gynecologic malignancies experience financial toxicity, results of a single-center study suggest.
Among 5,188 patients treated for gynecologic cancers, 1,155 (22%) experienced financial toxicity, measured by bills sent to collection, financial assistance, bankruptcy, or similar measures, reported Emeline Aviki, MD, of Memorial Sloan Kettering Cancer Center (MSKCC) in New York, and colleagues.
“In any clinical study reporting that over 20% of patients develop a serious complication as a result of treatment, financial toxicity in this case, future efforts to address the complication are critically important,” Dr. Aviki said in an interview.
Her group’s study is detailed in an abstract that had been slated for presentation at the Society of Gynecologic Oncology’s Annual Meeting on Women’s Cancer. The meeting was canceled because of the COVID-19 pandemic.
Study details
To address financial problems patients with gynecologic cancer face, MSKCC assembled a multidisciplinary team that included the strategy and innovation department, the patient financial services department, medical oncologists, radiation oncologists, and surgical oncologists.
The team’s first priority was to measure the prevalence of financial burden among the center’s patients using readily available institutional data. Financial toxicity was defined as one or more of the following:
- Two or more bills sent for collection.
- Application for and granting of a time-payment plan.
- Bill settlement.
- Bankruptcy.
- Enrollment in a financial assistance plan.
- Finance-related social work visit.
In a univariate analysis, factors significantly associated with financial toxicity, and the proportion of patients in each category affected, included cervical cancer (31%), stage 3 (29%) or 4 disease (27%), age younger than 30 years (32%), nonpartnered marital status (28%), black (38%) or Hispanic (33%) race/ethnicity, self-pay (42%) or commercial insurance (26%), clinical trial participation (27%), nine or more imaging studies (33%), one or more emergency department visits (31%), inpatient stays of 20 days or longer (35%), and 20 or more outpatient clinic visits (35%).
In a multivariate analysis controlling for disease and demographics, factors that remained significantly associated with financial toxicity (P < .05) included younger age, nonpartnered marital status, black and Hispanic race/ethnicity, commercial insurance, more imaging studies, and more outpatient physician visits.
Implications for patients and providers
“We were really surprised to see the significant increase in financial toxicity associated with patients undergoing more frequent imaging studies,” Dr. Aviki said. “There are randomized controlled studies showing that patients with ovarian cancer do not benefit from more frequent surveillance imaging. However, many providers across the country still order scans every 3 or 4 months. With this new data showing increased financial toxicity in patients who undergo more frequent scans, I think many will pause before ordering their next surveillance scan or at least have the conversation with patients to make sure no financial harm is being done.”
Dr. Aviki and colleagues used the data from this study to create a risk-stratification tool that can be employed to identify patients with gynecologic cancers who are at increased risk for financial toxicity, who can then be offered help through patient financial services.
In addition, the investigators are working to improve provider knowledge about the costs and financial implications surveillance imaging can have for patients.
“When considering interventions that might reduce patient financial burden, we questioned what role providers should play in patient affordability issues,” Dr. Aviki said. “Many providers may believe it is unethical to be informed of their patient’s risk of financial toxicity as it may affect their treatment recommendations. Others may believe it is important for them to be fully aware of any and all treatment-related risks their patients face.”
To get a better sense of how providers see their role in patient finances and care affordability, Dr. Aviki and colleagues surveyed more than 350 attending physicians at MSKCC. The investigators plan to use the results to develop provider-focused interventions.
The study was internally funded. Dr. Aviki reported no conflicts of interest.
SOURCE: Aviki EM et al. SGO 2020, Abstract 144.
More than one-fifth of patients being treated for gynecologic malignancies experience financial toxicity, results of a single-center study suggest.
Among 5,188 patients treated for gynecologic cancers, 1,155 (22%) experienced financial toxicity, measured by bills sent to collection, financial assistance, bankruptcy, or similar measures, reported Emeline Aviki, MD, of Memorial Sloan Kettering Cancer Center (MSKCC) in New York, and colleagues.
“In any clinical study reporting that over 20% of patients develop a serious complication as a result of treatment, financial toxicity in this case, future efforts to address the complication are critically important,” Dr. Aviki said in an interview.
Her group’s study is detailed in an abstract that had been slated for presentation at the Society of Gynecologic Oncology’s Annual Meeting on Women’s Cancer. The meeting was canceled because of the COVID-19 pandemic.
Study details
To address financial problems patients with gynecologic cancer face, MSKCC assembled a multidisciplinary team that included the strategy and innovation department, the patient financial services department, medical oncologists, radiation oncologists, and surgical oncologists.
The team’s first priority was to measure the prevalence of financial burden among the center’s patients using readily available institutional data. Financial toxicity was defined as one or more of the following:
- Two or more bills sent for collection.
- Application for and granting of a time-payment plan.
- Bill settlement.
- Bankruptcy.
- Enrollment in a financial assistance plan.
- Finance-related social work visit.
In a univariate analysis, factors significantly associated with financial toxicity, and the proportion of patients in each category affected, included cervical cancer (31%), stage 3 (29%) or 4 disease (27%), age younger than 30 years (32%), nonpartnered marital status (28%), black (38%) or Hispanic (33%) race/ethnicity, self-pay (42%) or commercial insurance (26%), clinical trial participation (27%), nine or more imaging studies (33%), one or more emergency department visits (31%), inpatient stays of 20 days or longer (35%), and 20 or more outpatient clinic visits (35%).
In a multivariate analysis controlling for disease and demographics, factors that remained significantly associated with financial toxicity (P < .05) included younger age, nonpartnered marital status, black and Hispanic race/ethnicity, commercial insurance, more imaging studies, and more outpatient physician visits.
Implications for patients and providers
“We were really surprised to see the significant increase in financial toxicity associated with patients undergoing more frequent imaging studies,” Dr. Aviki said. “There are randomized controlled studies showing that patients with ovarian cancer do not benefit from more frequent surveillance imaging. However, many providers across the country still order scans every 3 or 4 months. With this new data showing increased financial toxicity in patients who undergo more frequent scans, I think many will pause before ordering their next surveillance scan or at least have the conversation with patients to make sure no financial harm is being done.”
Dr. Aviki and colleagues used the data from this study to create a risk-stratification tool that can be employed to identify patients with gynecologic cancers who are at increased risk for financial toxicity, who can then be offered help through patient financial services.
In addition, the investigators are working to improve provider knowledge about the costs and financial implications surveillance imaging can have for patients.
“When considering interventions that might reduce patient financial burden, we questioned what role providers should play in patient affordability issues,” Dr. Aviki said. “Many providers may believe it is unethical to be informed of their patient’s risk of financial toxicity as it may affect their treatment recommendations. Others may believe it is important for them to be fully aware of any and all treatment-related risks their patients face.”
To get a better sense of how providers see their role in patient finances and care affordability, Dr. Aviki and colleagues surveyed more than 350 attending physicians at MSKCC. The investigators plan to use the results to develop provider-focused interventions.
The study was internally funded. Dr. Aviki reported no conflicts of interest.
SOURCE: Aviki EM et al. SGO 2020, Abstract 144.
More than one-fifth of patients being treated for gynecologic malignancies experience financial toxicity, results of a single-center study suggest.
Among 5,188 patients treated for gynecologic cancers, 1,155 (22%) experienced financial toxicity, measured by bills sent to collection, financial assistance, bankruptcy, or similar measures, reported Emeline Aviki, MD, of Memorial Sloan Kettering Cancer Center (MSKCC) in New York, and colleagues.
“In any clinical study reporting that over 20% of patients develop a serious complication as a result of treatment, financial toxicity in this case, future efforts to address the complication are critically important,” Dr. Aviki said in an interview.
Her group’s study is detailed in an abstract that had been slated for presentation at the Society of Gynecologic Oncology’s Annual Meeting on Women’s Cancer. The meeting was canceled because of the COVID-19 pandemic.
Study details
To address financial problems patients with gynecologic cancer face, MSKCC assembled a multidisciplinary team that included the strategy and innovation department, the patient financial services department, medical oncologists, radiation oncologists, and surgical oncologists.
The team’s first priority was to measure the prevalence of financial burden among the center’s patients using readily available institutional data. Financial toxicity was defined as one or more of the following:
- Two or more bills sent for collection.
- Application for and granting of a time-payment plan.
- Bill settlement.
- Bankruptcy.
- Enrollment in a financial assistance plan.
- Finance-related social work visit.
In a univariate analysis, factors significantly associated with financial toxicity, and the proportion of patients in each category affected, included cervical cancer (31%), stage 3 (29%) or 4 disease (27%), age younger than 30 years (32%), nonpartnered marital status (28%), black (38%) or Hispanic (33%) race/ethnicity, self-pay (42%) or commercial insurance (26%), clinical trial participation (27%), nine or more imaging studies (33%), one or more emergency department visits (31%), inpatient stays of 20 days or longer (35%), and 20 or more outpatient clinic visits (35%).
In a multivariate analysis controlling for disease and demographics, factors that remained significantly associated with financial toxicity (P < .05) included younger age, nonpartnered marital status, black and Hispanic race/ethnicity, commercial insurance, more imaging studies, and more outpatient physician visits.
Implications for patients and providers
“We were really surprised to see the significant increase in financial toxicity associated with patients undergoing more frequent imaging studies,” Dr. Aviki said. “There are randomized controlled studies showing that patients with ovarian cancer do not benefit from more frequent surveillance imaging. However, many providers across the country still order scans every 3 or 4 months. With this new data showing increased financial toxicity in patients who undergo more frequent scans, I think many will pause before ordering their next surveillance scan or at least have the conversation with patients to make sure no financial harm is being done.”
Dr. Aviki and colleagues used the data from this study to create a risk-stratification tool that can be employed to identify patients with gynecologic cancers who are at increased risk for financial toxicity, who can then be offered help through patient financial services.
In addition, the investigators are working to improve provider knowledge about the costs and financial implications surveillance imaging can have for patients.
“When considering interventions that might reduce patient financial burden, we questioned what role providers should play in patient affordability issues,” Dr. Aviki said. “Many providers may believe it is unethical to be informed of their patient’s risk of financial toxicity as it may affect their treatment recommendations. Others may believe it is important for them to be fully aware of any and all treatment-related risks their patients face.”
To get a better sense of how providers see their role in patient finances and care affordability, Dr. Aviki and colleagues surveyed more than 350 attending physicians at MSKCC. The investigators plan to use the results to develop provider-focused interventions.
The study was internally funded. Dr. Aviki reported no conflicts of interest.
SOURCE: Aviki EM et al. SGO 2020, Abstract 144.
FROM SGO 2020
Maintaining cancer care in the face of COVID-19
Medical oncologist Anne Chiang, MD, PhD, is scrambling to maintain cancer care in New Haven, Connecticut, while COVID-19 advances unrelentingly. As deputy chief medical officer of the Smilow Cancer Network, the largest cancer care delivery system in Connecticut and Rhode Island, she has no illusions about dodging what’s unfolding just 2 hours down the road in New York City.
“They’re trying their best to continue active cancer treatment but it’s getting harder,” she says of her colleagues in the thick of the pandemic. “We have to be prepared for it here.”
In anticipation of what’s coming, her team has just emptied the top three floors of the Smilow Cancer Hospital, moving 60 patients by ambulance and other medical transport to a different hospital nearby.
The move frees the Smilow Cancer hospital’s negative-pressure wards for the anticipated wave of COVID-19 patients. It will keep the virus sealed off from the rest of the hospital. But in other locations it’s harder to shield patients with cancer from the infection.
Around the state, Smilow Cancer Network’s affiliated hospitals are already treating a growing number of COVID-19 patients, especially at Greenwich Hospital, right on the border with New York state.
To protect patients with cancer, who are among the most vulnerable to the virus, oncologists are embracing telemedicine to allow most patients to stay home.
“We’re really concentrating on decreasing the risk to these patients, with a widespread massive-scale conversion to telehealth,” said Chiang. “This is something that, in the space of about a week, has transformed the care of our patients — it’s a really amazing transformation.”
If anything good comes out of the COVID-19 pandemic, it will be this global adoption of virtual healthcare.
Across the US border in Canada, the medical director of Toronto’s Princess Margaret Cancer Centre is directing a similar transformation.
“We have converted probably about 70% to 80% of our clinic visits to virtual visits,” says radiation oncologist Mary Gospodarowicz, MD.
“We have three priorities: number one, to keep our patients safe; number two, to keep our staff safe, because if staff are sick we won’t be treating anybody; and number three, to treat as many patients with cancer as possible.”
Gospodarowicz woke up last week to a local headline about a woman whose mastectomy had been canceled “because of the coronavirus.” The story exposed the many layers of the COVID-19 crisis. “A lot of hospitals have canceled elective surgeries,” she acknowledged. “For patients who have treatment or surgery deferred, we have a database and we’ll make sure we look after those patients eventually. We have a priority system, so low-risk prostate cancer, very low-risk breast cancer patients are waiting. All the urgent head and neck, breast, and other higher priority surgeries are still being done, but it just depends how it goes. The situation changes every day.”
It’s similar in Los Angeles, at the University of Southern California, says Elizabeth David, MD, a cardiothoracic surgeon with Keck Medicine.
“For thoracic, we just had a conference call with about 30 surgeons around the country going through really nitty-gritty specifics to help with our decision making about what could wait without detriment to the patient – hopefully – and what should be done now,” she told Medscape Medical News.
“There are some hospitals where they are not doing anything but life and death emergency operations, whereas we are still doing our emergent cancer operations in our institution, but we all know – and patients know – that could change from one day to the next. They may think they’re having surgery tomorrow but may get a call saying we can’t do it,” David said.
Many of David’s patients have non–small cell lung cancer, putting them at particular risk with a pulmonary infection like COVID-19. For now, she says delivery of postsurgical chemotherapy and radiotherapy has not been impacted in her area, but her videoconference discussions with patients are much longer – and harder – these days.
“I’ve been in practice a while now and I’ve had numerous conversations with patients this week that I never trained for, and I’ve never known anyone else who has. It’s really hard as a provider to know what to say,” she said.
In cardiothoracic surgery, David said guidance on clinical decision making is coming from the American College of Surgeons, Society of Thoracic Surgery, and American Association of Thoracic Surgeons. Yet, she says each patient is being assessed – and reassessed – individually.
“You have to balance the risk of delaying the intervention with supply issues, hospital exposure issues, the danger to the patient of being in the hospital environment – there’s just so many factors. We’re spending so much time talking through cases, and a lot of times we’re talking about cases we already talked about, but we’re just making sure that based on today’s numbers we should still be moving forward,” she commented.
In Connecticut, Chiang said treatment decisions are also mostly on a case-by-case basis at the moment, although more standardized guidelines are being worked out.
“Our disease teams have been really proactive in terms of offering alternative solutions to patients, creative ways to basically keep them out of the hospital and also reduce the immunosuppressive regimens that we give them,” she said.
Examples include offering endocrine therapy to patients who can’t get breast cancer surgery, or offering alternative drug regimens and dosing schedules. “At this point we haven’t needed to ration actual treatment – patients are continuing to get active therapy if that’s appropriate – it’s more about how can we protect them,” she said. “It’s a complex puzzle of moving pieces.”
In Toronto, Gospodarowicz says newly published medical and radiation oncology guidelines from France are the backbone of her hospital’s policy discussions about treating cancer and protecting patients from COVID-19.
While patients’ concerns are understandable, she says even in the current hot spots of infection, it’s encouraging to know that cancer patients are not being forgotten.
“I recently had email communication with a radiation oncologist in Brescia, one of the worst-affected areas in Italy, and he told me the radiotherapy department has been 60% to 70% capacity, so they still treat 70% these patients, just taking precautions and separating the COVID-positive and negative ones. When we read the stats it looks horrible, but life still goes on and people are still being treated,” she said.
Although telemedicine offers meaningful solutions to the COVID-19 crisis in North America, it may not be possible in other parts of the world.
Web consultations were only just approved in Brazil this week. “We are still discussing how to make it official and reimbursed,” says Rachel Riechelmann, MD, head of clinical oncology at AC Camargo Cancer Center in São Paulo.
To minimize infection risk for patients, Riechelmann says her hospital is doing the following: postponing surgeries in cases where there is good evidence of neoadjuvant treatment, such as total neoadjuvant therapy for rectal cancer; avoiding adjuvant chemo for stage 2 colon cancer; moving to hypofractionated radiotherapy if possible; adopting watchful waiting in grade 1 nonfunctional neuroendocrine tumors; and postponing follow-up visits.
“We do our best,” she wrote in an email. “We keep treating cancer if treatment cannot wait.”
Riechelmann’s center has just launched a trial of hydroxychloroquine and tocilizumab therapy in patients with cancer who have severe COVID-19 and acute respiratory distress syndrome (ARDS).
Meanwhile in New Haven, Chiang says for patients with cancer who are infected with COVID-19, her team is also prognosticating about the fair allocation of limited resources such as ventilators.
“If it ever gets to the point where somebody has to choose between a cancer patient and a noncancer patient in providing life support, it’s really important that people understand that cancer patients are doing very well nowadays and even with a diagnosis of cancer they can potentially live for many years, so that shouldn’t necessarily be a decision-point,” she emphasized.
This article first appeared on Medscape.com.
Medical oncologist Anne Chiang, MD, PhD, is scrambling to maintain cancer care in New Haven, Connecticut, while COVID-19 advances unrelentingly. As deputy chief medical officer of the Smilow Cancer Network, the largest cancer care delivery system in Connecticut and Rhode Island, she has no illusions about dodging what’s unfolding just 2 hours down the road in New York City.
“They’re trying their best to continue active cancer treatment but it’s getting harder,” she says of her colleagues in the thick of the pandemic. “We have to be prepared for it here.”
In anticipation of what’s coming, her team has just emptied the top three floors of the Smilow Cancer Hospital, moving 60 patients by ambulance and other medical transport to a different hospital nearby.
The move frees the Smilow Cancer hospital’s negative-pressure wards for the anticipated wave of COVID-19 patients. It will keep the virus sealed off from the rest of the hospital. But in other locations it’s harder to shield patients with cancer from the infection.
Around the state, Smilow Cancer Network’s affiliated hospitals are already treating a growing number of COVID-19 patients, especially at Greenwich Hospital, right on the border with New York state.
To protect patients with cancer, who are among the most vulnerable to the virus, oncologists are embracing telemedicine to allow most patients to stay home.
“We’re really concentrating on decreasing the risk to these patients, with a widespread massive-scale conversion to telehealth,” said Chiang. “This is something that, in the space of about a week, has transformed the care of our patients — it’s a really amazing transformation.”
If anything good comes out of the COVID-19 pandemic, it will be this global adoption of virtual healthcare.
Across the US border in Canada, the medical director of Toronto’s Princess Margaret Cancer Centre is directing a similar transformation.
“We have converted probably about 70% to 80% of our clinic visits to virtual visits,” says radiation oncologist Mary Gospodarowicz, MD.
“We have three priorities: number one, to keep our patients safe; number two, to keep our staff safe, because if staff are sick we won’t be treating anybody; and number three, to treat as many patients with cancer as possible.”
Gospodarowicz woke up last week to a local headline about a woman whose mastectomy had been canceled “because of the coronavirus.” The story exposed the many layers of the COVID-19 crisis. “A lot of hospitals have canceled elective surgeries,” she acknowledged. “For patients who have treatment or surgery deferred, we have a database and we’ll make sure we look after those patients eventually. We have a priority system, so low-risk prostate cancer, very low-risk breast cancer patients are waiting. All the urgent head and neck, breast, and other higher priority surgeries are still being done, but it just depends how it goes. The situation changes every day.”
It’s similar in Los Angeles, at the University of Southern California, says Elizabeth David, MD, a cardiothoracic surgeon with Keck Medicine.
“For thoracic, we just had a conference call with about 30 surgeons around the country going through really nitty-gritty specifics to help with our decision making about what could wait without detriment to the patient – hopefully – and what should be done now,” she told Medscape Medical News.
“There are some hospitals where they are not doing anything but life and death emergency operations, whereas we are still doing our emergent cancer operations in our institution, but we all know – and patients know – that could change from one day to the next. They may think they’re having surgery tomorrow but may get a call saying we can’t do it,” David said.
Many of David’s patients have non–small cell lung cancer, putting them at particular risk with a pulmonary infection like COVID-19. For now, she says delivery of postsurgical chemotherapy and radiotherapy has not been impacted in her area, but her videoconference discussions with patients are much longer – and harder – these days.
“I’ve been in practice a while now and I’ve had numerous conversations with patients this week that I never trained for, and I’ve never known anyone else who has. It’s really hard as a provider to know what to say,” she said.
In cardiothoracic surgery, David said guidance on clinical decision making is coming from the American College of Surgeons, Society of Thoracic Surgery, and American Association of Thoracic Surgeons. Yet, she says each patient is being assessed – and reassessed – individually.
“You have to balance the risk of delaying the intervention with supply issues, hospital exposure issues, the danger to the patient of being in the hospital environment – there’s just so many factors. We’re spending so much time talking through cases, and a lot of times we’re talking about cases we already talked about, but we’re just making sure that based on today’s numbers we should still be moving forward,” she commented.
In Connecticut, Chiang said treatment decisions are also mostly on a case-by-case basis at the moment, although more standardized guidelines are being worked out.
“Our disease teams have been really proactive in terms of offering alternative solutions to patients, creative ways to basically keep them out of the hospital and also reduce the immunosuppressive regimens that we give them,” she said.
Examples include offering endocrine therapy to patients who can’t get breast cancer surgery, or offering alternative drug regimens and dosing schedules. “At this point we haven’t needed to ration actual treatment – patients are continuing to get active therapy if that’s appropriate – it’s more about how can we protect them,” she said. “It’s a complex puzzle of moving pieces.”
In Toronto, Gospodarowicz says newly published medical and radiation oncology guidelines from France are the backbone of her hospital’s policy discussions about treating cancer and protecting patients from COVID-19.
While patients’ concerns are understandable, she says even in the current hot spots of infection, it’s encouraging to know that cancer patients are not being forgotten.
“I recently had email communication with a radiation oncologist in Brescia, one of the worst-affected areas in Italy, and he told me the radiotherapy department has been 60% to 70% capacity, so they still treat 70% these patients, just taking precautions and separating the COVID-positive and negative ones. When we read the stats it looks horrible, but life still goes on and people are still being treated,” she said.
Although telemedicine offers meaningful solutions to the COVID-19 crisis in North America, it may not be possible in other parts of the world.
Web consultations were only just approved in Brazil this week. “We are still discussing how to make it official and reimbursed,” says Rachel Riechelmann, MD, head of clinical oncology at AC Camargo Cancer Center in São Paulo.
To minimize infection risk for patients, Riechelmann says her hospital is doing the following: postponing surgeries in cases where there is good evidence of neoadjuvant treatment, such as total neoadjuvant therapy for rectal cancer; avoiding adjuvant chemo for stage 2 colon cancer; moving to hypofractionated radiotherapy if possible; adopting watchful waiting in grade 1 nonfunctional neuroendocrine tumors; and postponing follow-up visits.
“We do our best,” she wrote in an email. “We keep treating cancer if treatment cannot wait.”
Riechelmann’s center has just launched a trial of hydroxychloroquine and tocilizumab therapy in patients with cancer who have severe COVID-19 and acute respiratory distress syndrome (ARDS).
Meanwhile in New Haven, Chiang says for patients with cancer who are infected with COVID-19, her team is also prognosticating about the fair allocation of limited resources such as ventilators.
“If it ever gets to the point where somebody has to choose between a cancer patient and a noncancer patient in providing life support, it’s really important that people understand that cancer patients are doing very well nowadays and even with a diagnosis of cancer they can potentially live for many years, so that shouldn’t necessarily be a decision-point,” she emphasized.
This article first appeared on Medscape.com.
Medical oncologist Anne Chiang, MD, PhD, is scrambling to maintain cancer care in New Haven, Connecticut, while COVID-19 advances unrelentingly. As deputy chief medical officer of the Smilow Cancer Network, the largest cancer care delivery system in Connecticut and Rhode Island, she has no illusions about dodging what’s unfolding just 2 hours down the road in New York City.
“They’re trying their best to continue active cancer treatment but it’s getting harder,” she says of her colleagues in the thick of the pandemic. “We have to be prepared for it here.”
In anticipation of what’s coming, her team has just emptied the top three floors of the Smilow Cancer Hospital, moving 60 patients by ambulance and other medical transport to a different hospital nearby.
The move frees the Smilow Cancer hospital’s negative-pressure wards for the anticipated wave of COVID-19 patients. It will keep the virus sealed off from the rest of the hospital. But in other locations it’s harder to shield patients with cancer from the infection.
Around the state, Smilow Cancer Network’s affiliated hospitals are already treating a growing number of COVID-19 patients, especially at Greenwich Hospital, right on the border with New York state.
To protect patients with cancer, who are among the most vulnerable to the virus, oncologists are embracing telemedicine to allow most patients to stay home.
“We’re really concentrating on decreasing the risk to these patients, with a widespread massive-scale conversion to telehealth,” said Chiang. “This is something that, in the space of about a week, has transformed the care of our patients — it’s a really amazing transformation.”
If anything good comes out of the COVID-19 pandemic, it will be this global adoption of virtual healthcare.
Across the US border in Canada, the medical director of Toronto’s Princess Margaret Cancer Centre is directing a similar transformation.
“We have converted probably about 70% to 80% of our clinic visits to virtual visits,” says radiation oncologist Mary Gospodarowicz, MD.
“We have three priorities: number one, to keep our patients safe; number two, to keep our staff safe, because if staff are sick we won’t be treating anybody; and number three, to treat as many patients with cancer as possible.”
Gospodarowicz woke up last week to a local headline about a woman whose mastectomy had been canceled “because of the coronavirus.” The story exposed the many layers of the COVID-19 crisis. “A lot of hospitals have canceled elective surgeries,” she acknowledged. “For patients who have treatment or surgery deferred, we have a database and we’ll make sure we look after those patients eventually. We have a priority system, so low-risk prostate cancer, very low-risk breast cancer patients are waiting. All the urgent head and neck, breast, and other higher priority surgeries are still being done, but it just depends how it goes. The situation changes every day.”
It’s similar in Los Angeles, at the University of Southern California, says Elizabeth David, MD, a cardiothoracic surgeon with Keck Medicine.
“For thoracic, we just had a conference call with about 30 surgeons around the country going through really nitty-gritty specifics to help with our decision making about what could wait without detriment to the patient – hopefully – and what should be done now,” she told Medscape Medical News.
“There are some hospitals where they are not doing anything but life and death emergency operations, whereas we are still doing our emergent cancer operations in our institution, but we all know – and patients know – that could change from one day to the next. They may think they’re having surgery tomorrow but may get a call saying we can’t do it,” David said.
Many of David’s patients have non–small cell lung cancer, putting them at particular risk with a pulmonary infection like COVID-19. For now, she says delivery of postsurgical chemotherapy and radiotherapy has not been impacted in her area, but her videoconference discussions with patients are much longer – and harder – these days.
“I’ve been in practice a while now and I’ve had numerous conversations with patients this week that I never trained for, and I’ve never known anyone else who has. It’s really hard as a provider to know what to say,” she said.
In cardiothoracic surgery, David said guidance on clinical decision making is coming from the American College of Surgeons, Society of Thoracic Surgery, and American Association of Thoracic Surgeons. Yet, she says each patient is being assessed – and reassessed – individually.
“You have to balance the risk of delaying the intervention with supply issues, hospital exposure issues, the danger to the patient of being in the hospital environment – there’s just so many factors. We’re spending so much time talking through cases, and a lot of times we’re talking about cases we already talked about, but we’re just making sure that based on today’s numbers we should still be moving forward,” she commented.
In Connecticut, Chiang said treatment decisions are also mostly on a case-by-case basis at the moment, although more standardized guidelines are being worked out.
“Our disease teams have been really proactive in terms of offering alternative solutions to patients, creative ways to basically keep them out of the hospital and also reduce the immunosuppressive regimens that we give them,” she said.
Examples include offering endocrine therapy to patients who can’t get breast cancer surgery, or offering alternative drug regimens and dosing schedules. “At this point we haven’t needed to ration actual treatment – patients are continuing to get active therapy if that’s appropriate – it’s more about how can we protect them,” she said. “It’s a complex puzzle of moving pieces.”
In Toronto, Gospodarowicz says newly published medical and radiation oncology guidelines from France are the backbone of her hospital’s policy discussions about treating cancer and protecting patients from COVID-19.
While patients’ concerns are understandable, she says even in the current hot spots of infection, it’s encouraging to know that cancer patients are not being forgotten.
“I recently had email communication with a radiation oncologist in Brescia, one of the worst-affected areas in Italy, and he told me the radiotherapy department has been 60% to 70% capacity, so they still treat 70% these patients, just taking precautions and separating the COVID-positive and negative ones. When we read the stats it looks horrible, but life still goes on and people are still being treated,” she said.
Although telemedicine offers meaningful solutions to the COVID-19 crisis in North America, it may not be possible in other parts of the world.
Web consultations were only just approved in Brazil this week. “We are still discussing how to make it official and reimbursed,” says Rachel Riechelmann, MD, head of clinical oncology at AC Camargo Cancer Center in São Paulo.
To minimize infection risk for patients, Riechelmann says her hospital is doing the following: postponing surgeries in cases where there is good evidence of neoadjuvant treatment, such as total neoadjuvant therapy for rectal cancer; avoiding adjuvant chemo for stage 2 colon cancer; moving to hypofractionated radiotherapy if possible; adopting watchful waiting in grade 1 nonfunctional neuroendocrine tumors; and postponing follow-up visits.
“We do our best,” she wrote in an email. “We keep treating cancer if treatment cannot wait.”
Riechelmann’s center has just launched a trial of hydroxychloroquine and tocilizumab therapy in patients with cancer who have severe COVID-19 and acute respiratory distress syndrome (ARDS).
Meanwhile in New Haven, Chiang says for patients with cancer who are infected with COVID-19, her team is also prognosticating about the fair allocation of limited resources such as ventilators.
“If it ever gets to the point where somebody has to choose between a cancer patient and a noncancer patient in providing life support, it’s really important that people understand that cancer patients are doing very well nowadays and even with a diagnosis of cancer they can potentially live for many years, so that shouldn’t necessarily be a decision-point,” she emphasized.
This article first appeared on Medscape.com.