User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Aplastic Anemia and MDS International Foundation Plans Regional Patient/Family Conference
The Aplastic Anemia and MDS International Foundation presents the Regional Patient and Family Conference in Newark, NJ, on Sept. 16, 2017. Patients with aplastic anemia, myelodysplastic syndromes, or paroxysmal nocturnal hemoglobinuria (PNH), as well as family members and health care professionals, area invited.
The Aplastic Anemia and MDS International Foundation presents the Regional Patient and Family Conference in Newark, NJ, on Sept. 16, 2017. Patients with aplastic anemia, myelodysplastic syndromes, or paroxysmal nocturnal hemoglobinuria (PNH), as well as family members and health care professionals, area invited.
The Aplastic Anemia and MDS International Foundation presents the Regional Patient and Family Conference in Newark, NJ, on Sept. 16, 2017. Patients with aplastic anemia, myelodysplastic syndromes, or paroxysmal nocturnal hemoglobinuria (PNH), as well as family members and health care professionals, area invited.
Alport Syndrome Research Funding Is Awarded
Researchers at the University of Miami in Florida and University of Cyprus have each been awarded $100,000 to explore two drug repurposing projects to identify possible treatments for Alport syndrome. The funding was provided by the Alport Syndrome Foundation, Pedersen Family, and the Kidney Foundation of Canada.
Alport syndrome is a genetic condition characterized by kidney disease, hearing loss and eye abnormalities. When the Alport Syndrome Foundation was established in 2007, there was little funding or interest in researching this rare condition. In recent years, partly as a result of the foundation’s support for research and development of basic resources to assist researchers, several potential new treatments have entered the pipeline and are in development. Read more about this and the recent funding awards on the foundation’s website.
Researchers at the University of Miami in Florida and University of Cyprus have each been awarded $100,000 to explore two drug repurposing projects to identify possible treatments for Alport syndrome. The funding was provided by the Alport Syndrome Foundation, Pedersen Family, and the Kidney Foundation of Canada.
Alport syndrome is a genetic condition characterized by kidney disease, hearing loss and eye abnormalities. When the Alport Syndrome Foundation was established in 2007, there was little funding or interest in researching this rare condition. In recent years, partly as a result of the foundation’s support for research and development of basic resources to assist researchers, several potential new treatments have entered the pipeline and are in development. Read more about this and the recent funding awards on the foundation’s website.
Researchers at the University of Miami in Florida and University of Cyprus have each been awarded $100,000 to explore two drug repurposing projects to identify possible treatments for Alport syndrome. The funding was provided by the Alport Syndrome Foundation, Pedersen Family, and the Kidney Foundation of Canada.
Alport syndrome is a genetic condition characterized by kidney disease, hearing loss and eye abnormalities. When the Alport Syndrome Foundation was established in 2007, there was little funding or interest in researching this rare condition. In recent years, partly as a result of the foundation’s support for research and development of basic resources to assist researchers, several potential new treatments have entered the pipeline and are in development. Read more about this and the recent funding awards on the foundation’s website.
NORD Outlines Recommendations for Health Care Reform in Letter to Congressional Leadership
In a letter to the Senate and House leadership, NORD President and CEO Peter L. Saltonstall has outlined short-term fixes to stabilize the current health care markets and long-term reforms to improve health coverage for patients in the future. Mr. Saltonstall’s letter was written on behalf of the 30 million Americans who have rare diseases and those seeking to improve their lives.
On Feb. 28, 2017, NORD and several advocacy partners submitted to Congressional leadership a letter with “Principles for Health Coverage Reform.” The principles outline various provisions that NORD and its advocacy partners believe are necessary in any health care reform plan to assure adequate coverage for Americans with rare diseases.
NORD’s more recent letter, sent on Aug. 16, outlines specific short-term reforms to provide needed stability and viability to the current insurance marketplaces established by the Affordable Care Act as well as long-term proposals to improve health care in future health coverage reform legislation.
In a letter to the Senate and House leadership, NORD President and CEO Peter L. Saltonstall has outlined short-term fixes to stabilize the current health care markets and long-term reforms to improve health coverage for patients in the future. Mr. Saltonstall’s letter was written on behalf of the 30 million Americans who have rare diseases and those seeking to improve their lives.
On Feb. 28, 2017, NORD and several advocacy partners submitted to Congressional leadership a letter with “Principles for Health Coverage Reform.” The principles outline various provisions that NORD and its advocacy partners believe are necessary in any health care reform plan to assure adequate coverage for Americans with rare diseases.
NORD’s more recent letter, sent on Aug. 16, outlines specific short-term reforms to provide needed stability and viability to the current insurance marketplaces established by the Affordable Care Act as well as long-term proposals to improve health care in future health coverage reform legislation.
In a letter to the Senate and House leadership, NORD President and CEO Peter L. Saltonstall has outlined short-term fixes to stabilize the current health care markets and long-term reforms to improve health coverage for patients in the future. Mr. Saltonstall’s letter was written on behalf of the 30 million Americans who have rare diseases and those seeking to improve their lives.
On Feb. 28, 2017, NORD and several advocacy partners submitted to Congressional leadership a letter with “Principles for Health Coverage Reform.” The principles outline various provisions that NORD and its advocacy partners believe are necessary in any health care reform plan to assure adequate coverage for Americans with rare diseases.
NORD’s more recent letter, sent on Aug. 16, outlines specific short-term reforms to provide needed stability and viability to the current insurance marketplaces established by the Affordable Care Act as well as long-term proposals to improve health care in future health coverage reform legislation.
Rare Disease Film Festival Is Planned
Films from around the world featuring the challenges of life with a rare disease will be shown in the first-ever Rare Disease Film Festival to take place in Boston on Oct. 2-3. Tickets are on sale now and the event is open to all. Additional information is available here.
Films from around the world featuring the challenges of life with a rare disease will be shown in the first-ever Rare Disease Film Festival to take place in Boston on Oct. 2-3. Tickets are on sale now and the event is open to all. Additional information is available here.
Films from around the world featuring the challenges of life with a rare disease will be shown in the first-ever Rare Disease Film Festival to take place in Boston on Oct. 2-3. Tickets are on sale now and the event is open to all. Additional information is available here.
Join the National Organization for Rare Disorders and the University of Massachusetts for Two Upcoming CME-Accredited Events
Do you currently treat patients with rare diseases? Do you suspect that you might have a patient with an undiagnosed rare disease? The National Organization for Rare Disorders (NORD) and the University of Massachusetts Medical School have designed two events for you and your colleagues to learn more about rare diseases and the available diagnostic tools.
An interactive half-day symposium on diagnostic hurdles, tools, and resources is planned for Sunday, October 16, 2017, at the Marriott Wardman Park Hotel in Washington DC. This event, “Finding a Zebra Among Many Horses,” will focus on challenges and resources related to rare disease diagnosis.
On the following two days, Oct. 16 and 17, NORD’s Rare Diseases and Orphan Products Breakthrough Summit will take place at the same location and is also CME-accredited. Topics to be covered will include:
- Assuring Patient Access: Future Outlook for Patient Assistance Programs
- The Challenge of Health Care Costs and Treatment Prices
- An Evergreen and Sustainable Approach to Orphan Drugs
- Next-Generation Treatments and Advancing Clinical Trials
The NORD Breakthrough Summit attracts approximately 600 participants from rare disease stakeholder groups, including clinicians, researchers, patient advocates, and government partners from the NIH and FDA.
Online registration is available now for either or both events.
Do you currently treat patients with rare diseases? Do you suspect that you might have a patient with an undiagnosed rare disease? The National Organization for Rare Disorders (NORD) and the University of Massachusetts Medical School have designed two events for you and your colleagues to learn more about rare diseases and the available diagnostic tools.
An interactive half-day symposium on diagnostic hurdles, tools, and resources is planned for Sunday, October 16, 2017, at the Marriott Wardman Park Hotel in Washington DC. This event, “Finding a Zebra Among Many Horses,” will focus on challenges and resources related to rare disease diagnosis.
On the following two days, Oct. 16 and 17, NORD’s Rare Diseases and Orphan Products Breakthrough Summit will take place at the same location and is also CME-accredited. Topics to be covered will include:
- Assuring Patient Access: Future Outlook for Patient Assistance Programs
- The Challenge of Health Care Costs and Treatment Prices
- An Evergreen and Sustainable Approach to Orphan Drugs
- Next-Generation Treatments and Advancing Clinical Trials
The NORD Breakthrough Summit attracts approximately 600 participants from rare disease stakeholder groups, including clinicians, researchers, patient advocates, and government partners from the NIH and FDA.
Online registration is available now for either or both events.
Do you currently treat patients with rare diseases? Do you suspect that you might have a patient with an undiagnosed rare disease? The National Organization for Rare Disorders (NORD) and the University of Massachusetts Medical School have designed two events for you and your colleagues to learn more about rare diseases and the available diagnostic tools.
An interactive half-day symposium on diagnostic hurdles, tools, and resources is planned for Sunday, October 16, 2017, at the Marriott Wardman Park Hotel in Washington DC. This event, “Finding a Zebra Among Many Horses,” will focus on challenges and resources related to rare disease diagnosis.
On the following two days, Oct. 16 and 17, NORD’s Rare Diseases and Orphan Products Breakthrough Summit will take place at the same location and is also CME-accredited. Topics to be covered will include:
- Assuring Patient Access: Future Outlook for Patient Assistance Programs
- The Challenge of Health Care Costs and Treatment Prices
- An Evergreen and Sustainable Approach to Orphan Drugs
- Next-Generation Treatments and Advancing Clinical Trials
The NORD Breakthrough Summit attracts approximately 600 participants from rare disease stakeholder groups, including clinicians, researchers, patient advocates, and government partners from the NIH and FDA.
Online registration is available now for either or both events.
Hidradenitis Suppurativa: A New Indication for Adalimumab


Recalcitrant Ulcer on the Lower Leg
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
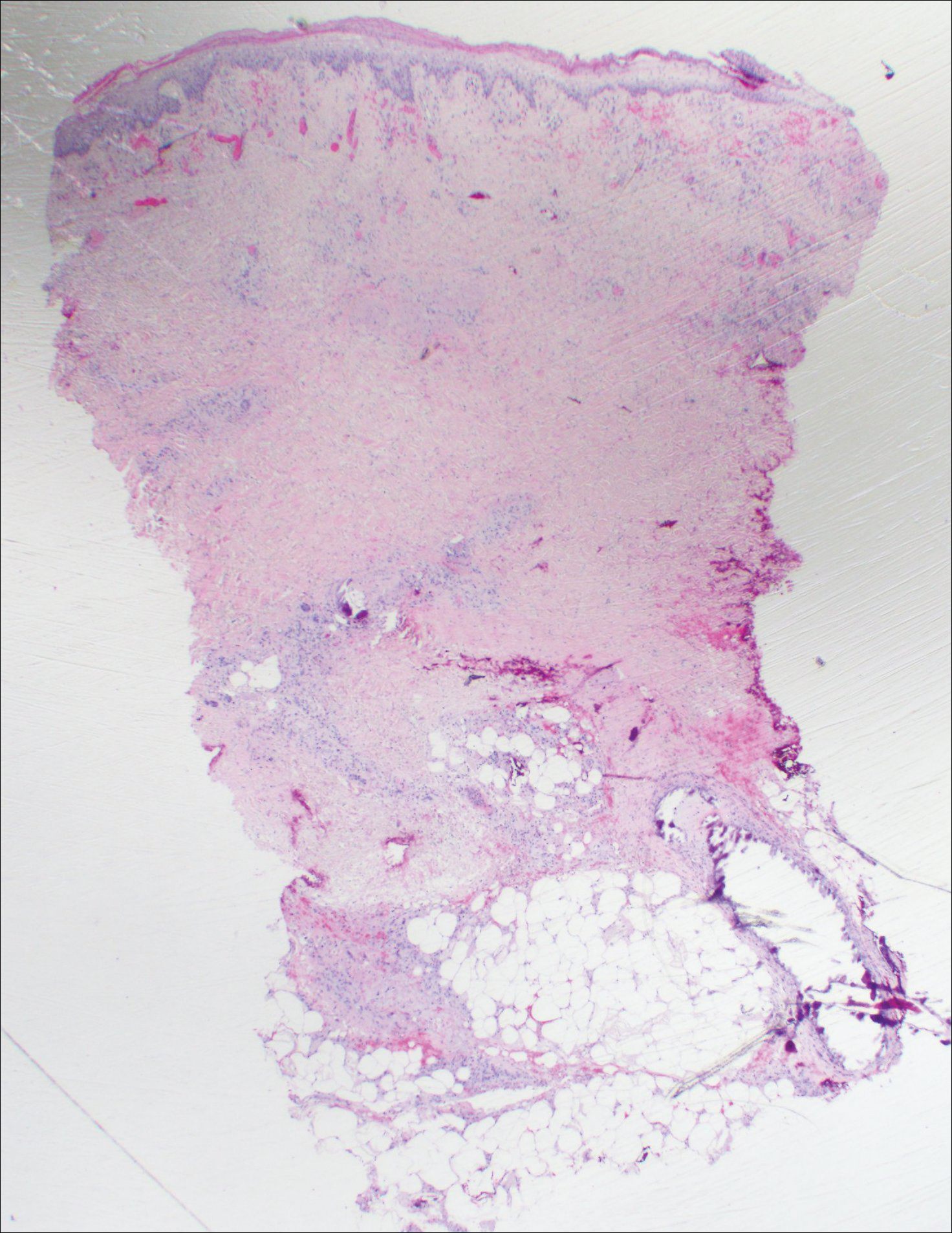
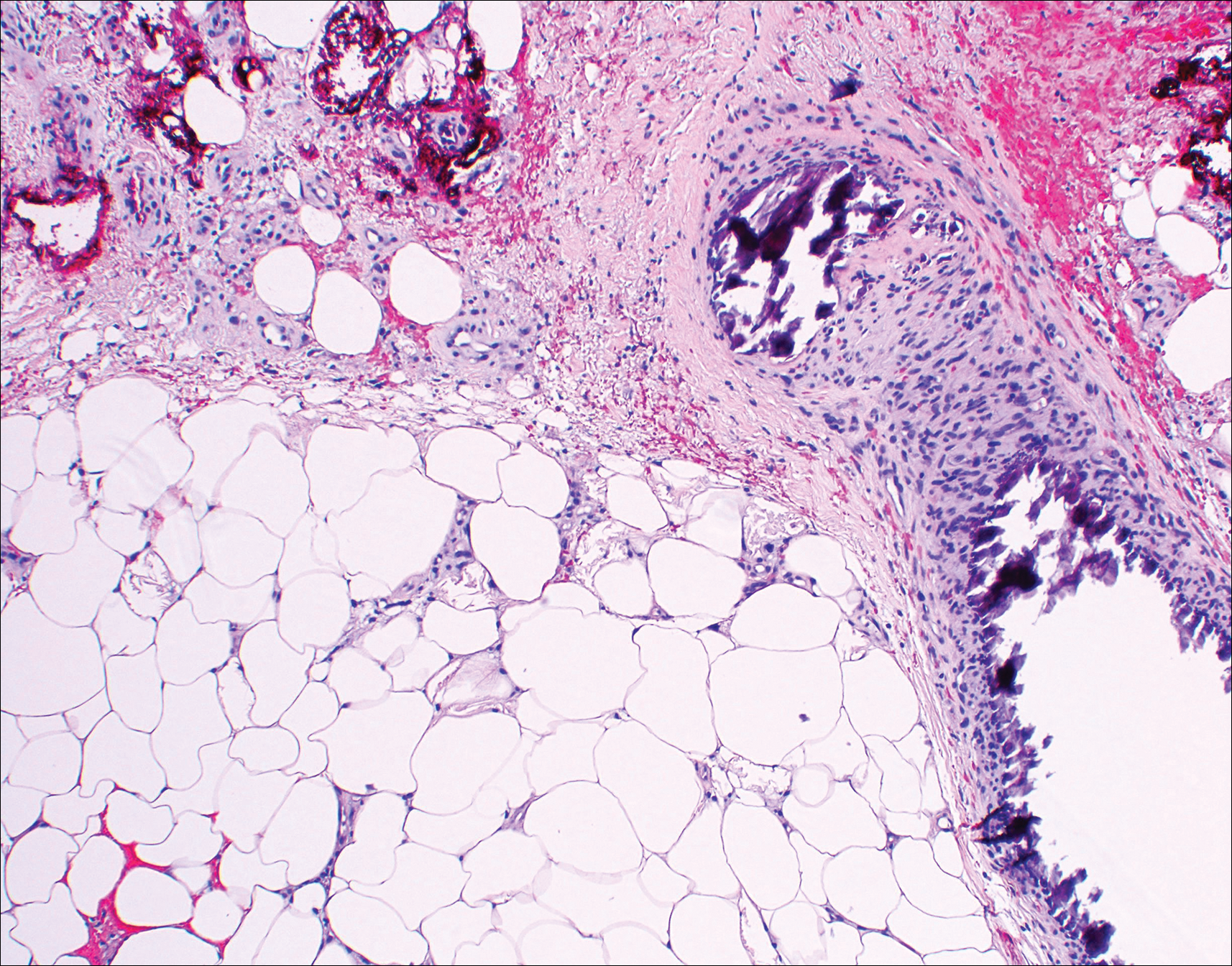
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
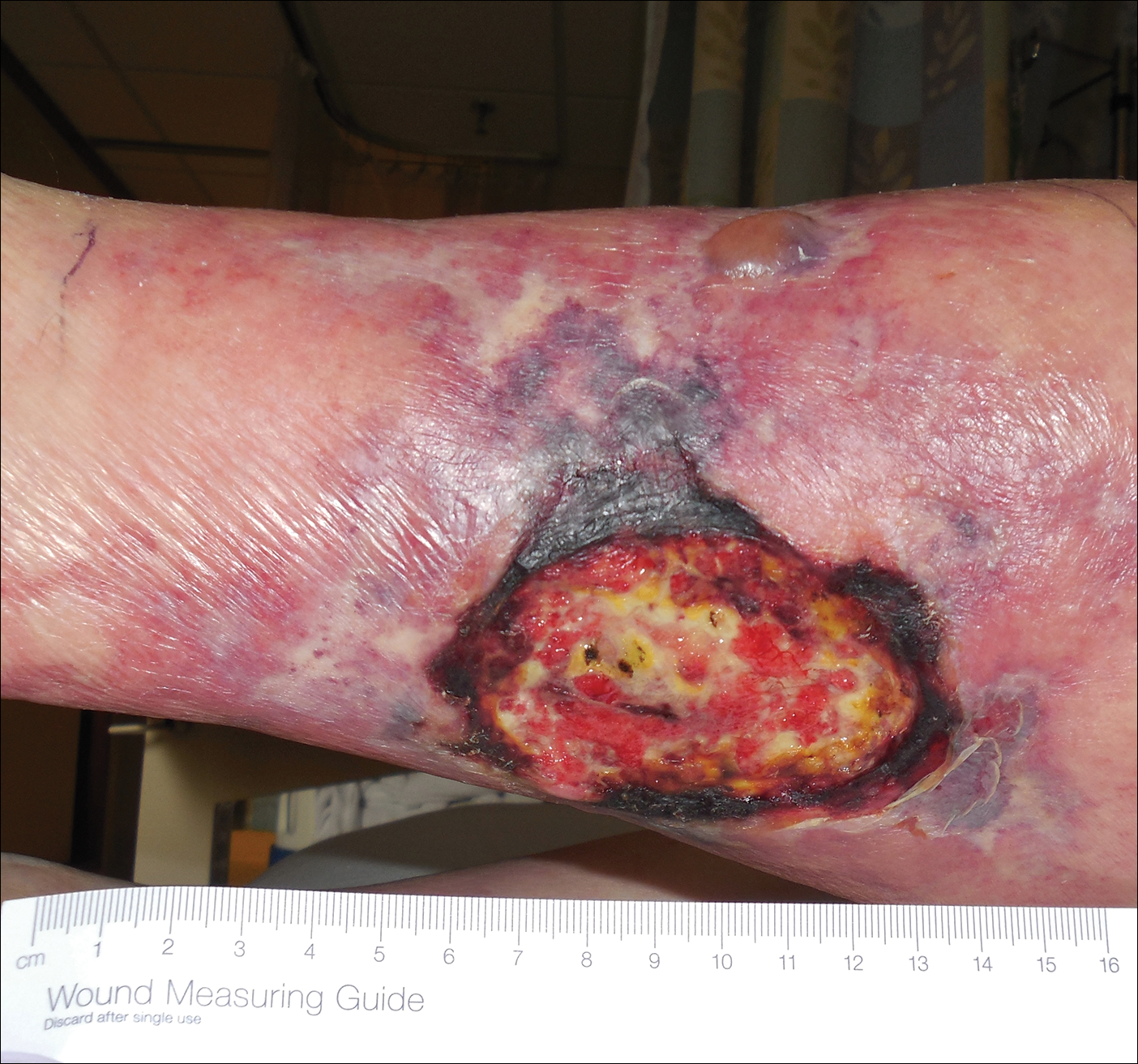
An 80-year-old woman with a medical history notable for obesity (body mass index, 31.2), type 2 diabetes mellitus, hypertension, and chronic atrial fibrillation treated with warfarin presented with a chronic painful wound on the left lower calf of 1 month's duration. A 7×7-cm ulcer on the posterior aspect of the left calf with necrotic debris was seen surrounded by skin of mottled purple discoloration. The edge of the ulcer was not undermined. There were tense nonhemorrhagic bullae on the medial aspect of the left leg and on bilateral anterior tibial areas. Two punch biopsy specimens were obtained from the anterior tibial bulla and the edge of the ulcer.
Imipramine-Induced Hyperpigmentation
Imipramine is a tricyclic medication uncommonly used to treat depression, anxiety, and other psychiatric illnesses. Although relatively rare, it has been associated with hyperpigmentation of the skin including slate gray discoloration of sun-exposed areas.
We present the case of a 63-year-old woman who had been taking imipramine for more than 20 years when she developed bluish gray discoloration on the face and neck. Histopathology of biopsy specimens showed numerous perivascular and interstitial brown globules in the dermis that were composed of melanin only, as evidenced by positive Fontana-Masson staining and negative Perls Prussian blue staining. A diagnosis of imipramine-induced hyperpigmentation was made based on histopathology and clinical history.
In addition to the case presentation, we provide a review of drugs that commonly cause hyperpigmentation as well as their associated histopathologic staining characteristics.
Case Report
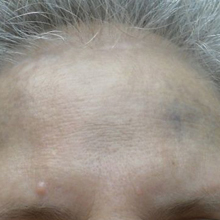
A 63-year-old woman presented with blue-gray discoloration on the face and neck. She first noted the discoloration on the left side of the forehead 3 years prior; it then spread to the right side of the forehead, cheeks, and neck. She denied pruritus, pain, redness, and scaling of the involved areas; any recent changes in medications; or the use of any topical products on the affected areas. Her medical history was remarkable for hypertension, which was inconsistently controlled with lisinopril and hydrochlorothiazide, and depression, which had been managed with oral imipramine.
Physical examination disclosed blue-gray hyperpigmented patches with irregular borders on the bilateral forehead, temples, and periorbital skin (Figure 1). Reticulated brown patches were noted on the bilateral cheeks, and the neck displayed diffuse muddy brown patches with sparing of the submental areas.
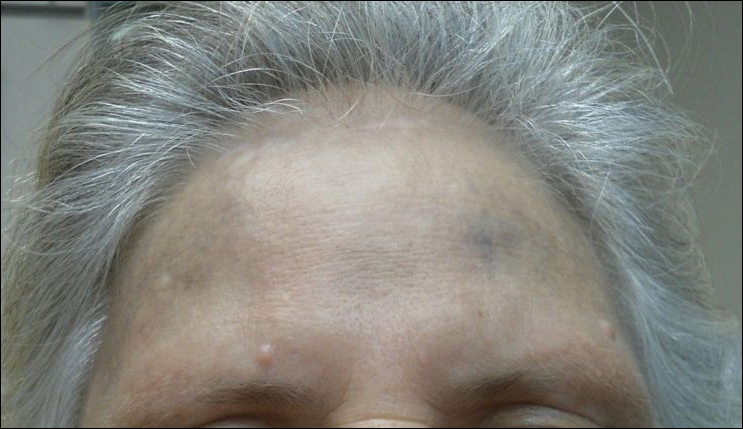
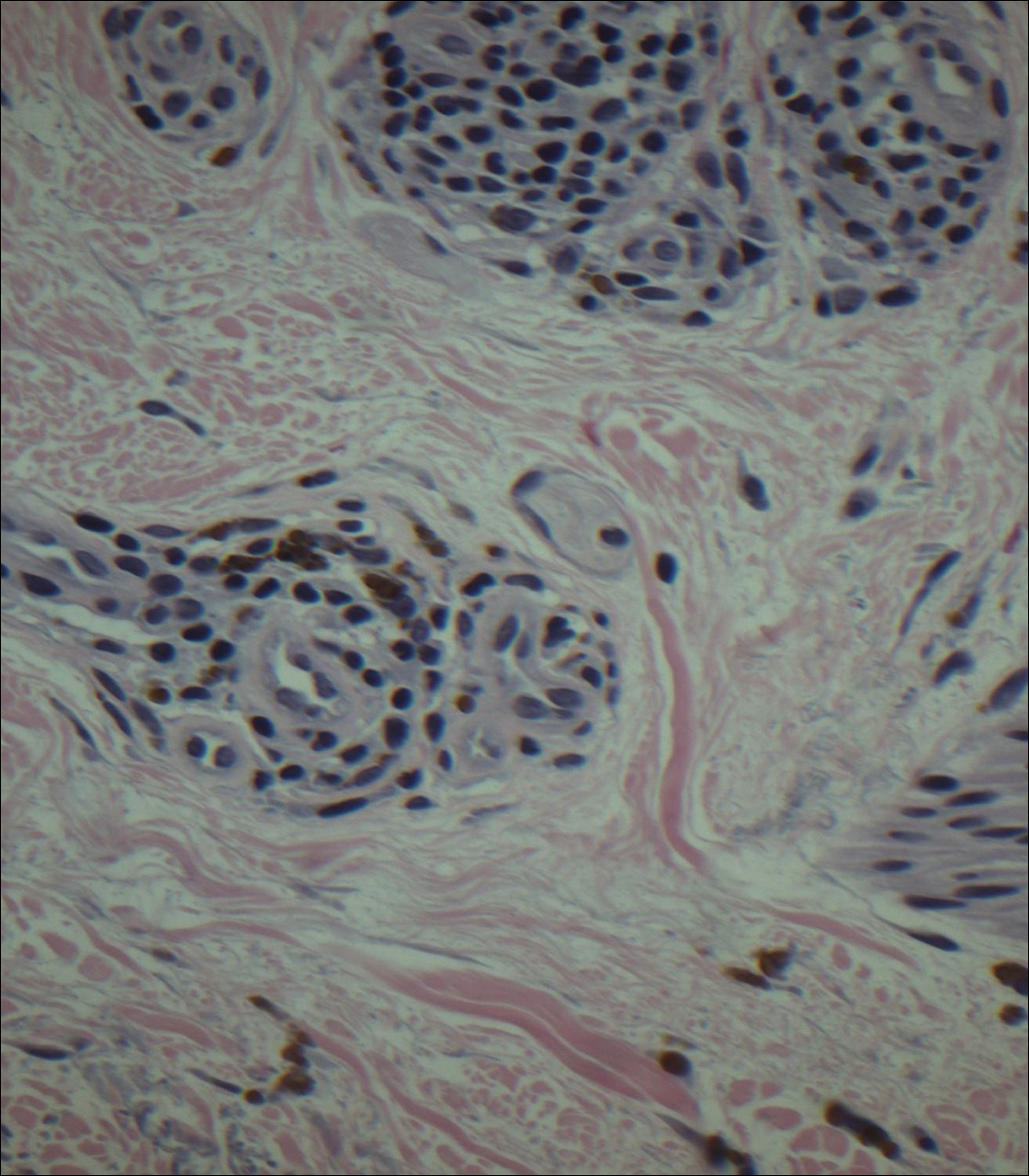
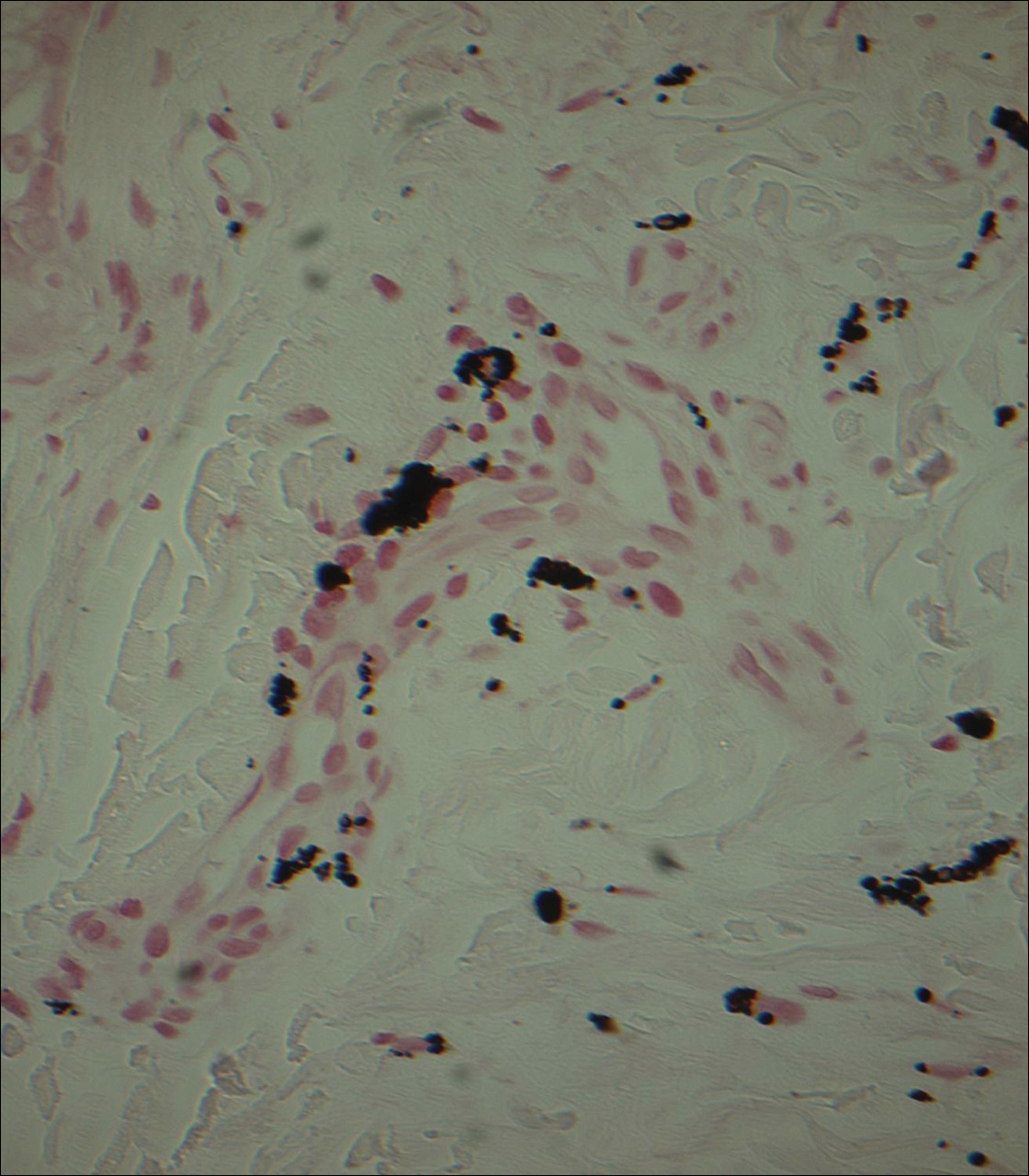
Punch biopsies obtained from the lateral forehead showed an unremarkable epidermis with deposition of numerous golden brown granules in the upper and mid dermis and in perivascular macrophages (Figure 2). The pigmented granules showed positive staining with Fontana-Masson (Figure 3), and a Perls Prussian blue stain for hemosiderin was negative. Based on the clinical history, a diagnosis of imipramine-induced hyperpigmentation was made.
The patient revealed that she had taken imipramine for more than 20 years for depression as prescribed by her mental health professional. She had tried several other antidepressants but none were as effective as imipramine. Therefore, she was not willing to discontinue it despite the likelihood that the hyperpigmentation would persist and could worsen with continued use of the medication. Diligent photoprotection was advised. Additionally, she started taking lisinopril some time after the appearance of the hyperpigmentation presented and had not taken hydrochlorothiazide consistently for several years. Although these drugs are known to cause various cutaneous reactions, it was not considered likely in this case.
Comment
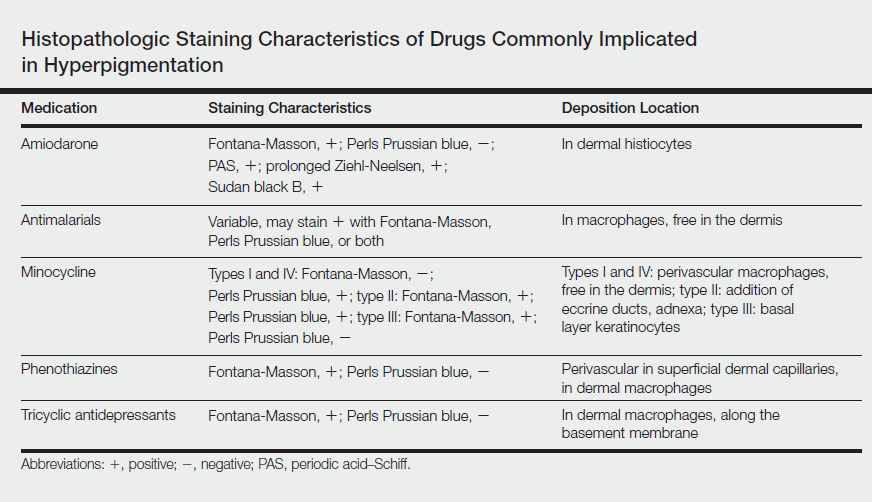
Drug-induced hyperpigmentation accounts for 10% to 20% of all cases of acquired hyperpigmentation.1 Common causative drugs include amiodarone, antimalarials, minocycline, and rarely psychotropics including phenothiazines and tricyclic antidepressants such as imipramine.1-4 Although amiodarone-induced hyperpigmentation is associated with lipofuscin in addition to melanin, most other medications, including imipramine, induce cutaneous effects through deposition of melanin and/or hemosiderin. A review of the histopathologic staining characteristics in pigment anomalies caused by these drugs is summarized in the Table.
Imipramine-induced hyperpigmentation presents as slate gray discrete macules and patches on sun-exposed skin that may appear anywhere from 2 to 22 years after initiating the medication.1-4 Affected areas include the malar cheeks, temples, periorbital areas, hands, forearms, and seldom the iris and sclera.2-4 Although the blue to slate gray coloring is classic, other colors have been described including brown, golden brown, and purple.2
Histopathology of imipramine-induced hyperpigmentation shows golden brown, round to oval granules in the superficial dermis and within dermal macrophages.1,3 Generally, Fontana-Masson staining is positive for melanin and Perls Prussian blue staining is negative for iron.1,2,4
Imipramine-induced hyperpigmentation likely results from photoexcitation of imipramine or one of its metabolites. These compounds activate tyrosinase, increasing melanogenesis and leading to formation of melanin-imipramine or melanin-metabolite complexes.1-3 Complexes are deposited in the dermis and basal layer or are engulfed by dermal macrophages and darkened on sun exposure due to their high melanin content.1 Other possible mechanisms of hyperpigmentation include nonspecific inflammation caused by the drug in the skin, hemosiderin deposition from vessel damage and subsequent erythrocyte extravasation, or deposition of newly formed pigments related to the drug.1
Most patients report satisfactory resolution of imipramine-induced discoloration within 1 year of stopping imipramine or switching to a different antidepressant.1,4 Patients who are unwilling to discontinue imipramine may achieve resolution with alexandrite or Q-switched ruby laser therapy.1,4 Strict sun protective measures are necessary, both to prevent new deposition of melanin and to prevent darkening of existing pigment.
Despite the advent of new psychotropic medications, imipramine remains the antidepressant of choice for many patients. Although rare, it is important to be able to recognize imipramine-induced hyperpigmentation and to encourage patient-psychiatrist communication to determine an antidepressant regimen that avoids unnecessary cutaneous side effects.
- D’Agostino ML, Risser J, Robinson-Bostom L. Imipramine-induced hyperpigmentation: a case report and review of the literature. J Cutan Pathol. 2009;36:799-803.
- Ming ME, Bhawan J, Stefanato CM, et al. Imipramine-induced hyperpigmentation: four cases and a review of the literature. J Am Acad Dermatol. 1999;40(2, pt 1):159-166.
- Sicari MC, Lebwohl M, Baral J, et al. Photoinduced dermal pigmentation in patients taking tricyclic antidepressants: histology, electron microscopy, and energy dispersive spectroscopy. J Am Acad Dermatol.1999;40(2, pt 2):290-293.
- Atkin DH, Fitzpatrick RE. Laser treatment of imipramine-induced hyperpigmentation. J Am Acad Dermatol. 2000;43(1, pt 1):77-80.
Imipramine is a tricyclic medication uncommonly used to treat depression, anxiety, and other psychiatric illnesses. Although relatively rare, it has been associated with hyperpigmentation of the skin including slate gray discoloration of sun-exposed areas.
We present the case of a 63-year-old woman who had been taking imipramine for more than 20 years when she developed bluish gray discoloration on the face and neck. Histopathology of biopsy specimens showed numerous perivascular and interstitial brown globules in the dermis that were composed of melanin only, as evidenced by positive Fontana-Masson staining and negative Perls Prussian blue staining. A diagnosis of imipramine-induced hyperpigmentation was made based on histopathology and clinical history.
In addition to the case presentation, we provide a review of drugs that commonly cause hyperpigmentation as well as their associated histopathologic staining characteristics.
Case Report
A 63-year-old woman presented with blue-gray discoloration on the face and neck. She first noted the discoloration on the left side of the forehead 3 years prior; it then spread to the right side of the forehead, cheeks, and neck. She denied pruritus, pain, redness, and scaling of the involved areas; any recent changes in medications; or the use of any topical products on the affected areas. Her medical history was remarkable for hypertension, which was inconsistently controlled with lisinopril and hydrochlorothiazide, and depression, which had been managed with oral imipramine.
Physical examination disclosed blue-gray hyperpigmented patches with irregular borders on the bilateral forehead, temples, and periorbital skin (Figure 1). Reticulated brown patches were noted on the bilateral cheeks, and the neck displayed diffuse muddy brown patches with sparing of the submental areas.
Punch biopsies obtained from the lateral forehead showed an unremarkable epidermis with deposition of numerous golden brown granules in the upper and mid dermis and in perivascular macrophages (Figure 2). The pigmented granules showed positive staining with Fontana-Masson (Figure 3), and a Perls Prussian blue stain for hemosiderin was negative. Based on the clinical history, a diagnosis of imipramine-induced hyperpigmentation was made.
The patient revealed that she had taken imipramine for more than 20 years for depression as prescribed by her mental health professional. She had tried several other antidepressants but none were as effective as imipramine. Therefore, she was not willing to discontinue it despite the likelihood that the hyperpigmentation would persist and could worsen with continued use of the medication. Diligent photoprotection was advised. Additionally, she started taking lisinopril some time after the appearance of the hyperpigmentation presented and had not taken hydrochlorothiazide consistently for several years. Although these drugs are known to cause various cutaneous reactions, it was not considered likely in this case.
Comment
Drug-induced hyperpigmentation accounts for 10% to 20% of all cases of acquired hyperpigmentation.1 Common causative drugs include amiodarone, antimalarials, minocycline, and rarely psychotropics including phenothiazines and tricyclic antidepressants such as imipramine.1-4 Although amiodarone-induced hyperpigmentation is associated with lipofuscin in addition to melanin, most other medications, including imipramine, induce cutaneous effects through deposition of melanin and/or hemosiderin. A review of the histopathologic staining characteristics in pigment anomalies caused by these drugs is summarized in the Table.
Imipramine-induced hyperpigmentation presents as slate gray discrete macules and patches on sun-exposed skin that may appear anywhere from 2 to 22 years after initiating the medication.1-4 Affected areas include the malar cheeks, temples, periorbital areas, hands, forearms, and seldom the iris and sclera.2-4 Although the blue to slate gray coloring is classic, other colors have been described including brown, golden brown, and purple.2
Histopathology of imipramine-induced hyperpigmentation shows golden brown, round to oval granules in the superficial dermis and within dermal macrophages.1,3 Generally, Fontana-Masson staining is positive for melanin and Perls Prussian blue staining is negative for iron.1,2,4
Imipramine-induced hyperpigmentation likely results from photoexcitation of imipramine or one of its metabolites. These compounds activate tyrosinase, increasing melanogenesis and leading to formation of melanin-imipramine or melanin-metabolite complexes.1-3 Complexes are deposited in the dermis and basal layer or are engulfed by dermal macrophages and darkened on sun exposure due to their high melanin content.1 Other possible mechanisms of hyperpigmentation include nonspecific inflammation caused by the drug in the skin, hemosiderin deposition from vessel damage and subsequent erythrocyte extravasation, or deposition of newly formed pigments related to the drug.1
Most patients report satisfactory resolution of imipramine-induced discoloration within 1 year of stopping imipramine or switching to a different antidepressant.1,4 Patients who are unwilling to discontinue imipramine may achieve resolution with alexandrite or Q-switched ruby laser therapy.1,4 Strict sun protective measures are necessary, both to prevent new deposition of melanin and to prevent darkening of existing pigment.
Despite the advent of new psychotropic medications, imipramine remains the antidepressant of choice for many patients. Although rare, it is important to be able to recognize imipramine-induced hyperpigmentation and to encourage patient-psychiatrist communication to determine an antidepressant regimen that avoids unnecessary cutaneous side effects.
Imipramine is a tricyclic medication uncommonly used to treat depression, anxiety, and other psychiatric illnesses. Although relatively rare, it has been associated with hyperpigmentation of the skin including slate gray discoloration of sun-exposed areas.
We present the case of a 63-year-old woman who had been taking imipramine for more than 20 years when she developed bluish gray discoloration on the face and neck. Histopathology of biopsy specimens showed numerous perivascular and interstitial brown globules in the dermis that were composed of melanin only, as evidenced by positive Fontana-Masson staining and negative Perls Prussian blue staining. A diagnosis of imipramine-induced hyperpigmentation was made based on histopathology and clinical history.
In addition to the case presentation, we provide a review of drugs that commonly cause hyperpigmentation as well as their associated histopathologic staining characteristics.
Case Report
A 63-year-old woman presented with blue-gray discoloration on the face and neck. She first noted the discoloration on the left side of the forehead 3 years prior; it then spread to the right side of the forehead, cheeks, and neck. She denied pruritus, pain, redness, and scaling of the involved areas; any recent changes in medications; or the use of any topical products on the affected areas. Her medical history was remarkable for hypertension, which was inconsistently controlled with lisinopril and hydrochlorothiazide, and depression, which had been managed with oral imipramine.
Physical examination disclosed blue-gray hyperpigmented patches with irregular borders on the bilateral forehead, temples, and periorbital skin (Figure 1). Reticulated brown patches were noted on the bilateral cheeks, and the neck displayed diffuse muddy brown patches with sparing of the submental areas.
Punch biopsies obtained from the lateral forehead showed an unremarkable epidermis with deposition of numerous golden brown granules in the upper and mid dermis and in perivascular macrophages (Figure 2). The pigmented granules showed positive staining with Fontana-Masson (Figure 3), and a Perls Prussian blue stain for hemosiderin was negative. Based on the clinical history, a diagnosis of imipramine-induced hyperpigmentation was made.
The patient revealed that she had taken imipramine for more than 20 years for depression as prescribed by her mental health professional. She had tried several other antidepressants but none were as effective as imipramine. Therefore, she was not willing to discontinue it despite the likelihood that the hyperpigmentation would persist and could worsen with continued use of the medication. Diligent photoprotection was advised. Additionally, she started taking lisinopril some time after the appearance of the hyperpigmentation presented and had not taken hydrochlorothiazide consistently for several years. Although these drugs are known to cause various cutaneous reactions, it was not considered likely in this case.
Comment
Drug-induced hyperpigmentation accounts for 10% to 20% of all cases of acquired hyperpigmentation.1 Common causative drugs include amiodarone, antimalarials, minocycline, and rarely psychotropics including phenothiazines and tricyclic antidepressants such as imipramine.1-4 Although amiodarone-induced hyperpigmentation is associated with lipofuscin in addition to melanin, most other medications, including imipramine, induce cutaneous effects through deposition of melanin and/or hemosiderin. A review of the histopathologic staining characteristics in pigment anomalies caused by these drugs is summarized in the Table.
Imipramine-induced hyperpigmentation presents as slate gray discrete macules and patches on sun-exposed skin that may appear anywhere from 2 to 22 years after initiating the medication.1-4 Affected areas include the malar cheeks, temples, periorbital areas, hands, forearms, and seldom the iris and sclera.2-4 Although the blue to slate gray coloring is classic, other colors have been described including brown, golden brown, and purple.2
Histopathology of imipramine-induced hyperpigmentation shows golden brown, round to oval granules in the superficial dermis and within dermal macrophages.1,3 Generally, Fontana-Masson staining is positive for melanin and Perls Prussian blue staining is negative for iron.1,2,4
Imipramine-induced hyperpigmentation likely results from photoexcitation of imipramine or one of its metabolites. These compounds activate tyrosinase, increasing melanogenesis and leading to formation of melanin-imipramine or melanin-metabolite complexes.1-3 Complexes are deposited in the dermis and basal layer or are engulfed by dermal macrophages and darkened on sun exposure due to their high melanin content.1 Other possible mechanisms of hyperpigmentation include nonspecific inflammation caused by the drug in the skin, hemosiderin deposition from vessel damage and subsequent erythrocyte extravasation, or deposition of newly formed pigments related to the drug.1
Most patients report satisfactory resolution of imipramine-induced discoloration within 1 year of stopping imipramine or switching to a different antidepressant.1,4 Patients who are unwilling to discontinue imipramine may achieve resolution with alexandrite or Q-switched ruby laser therapy.1,4 Strict sun protective measures are necessary, both to prevent new deposition of melanin and to prevent darkening of existing pigment.
Despite the advent of new psychotropic medications, imipramine remains the antidepressant of choice for many patients. Although rare, it is important to be able to recognize imipramine-induced hyperpigmentation and to encourage patient-psychiatrist communication to determine an antidepressant regimen that avoids unnecessary cutaneous side effects.
- D’Agostino ML, Risser J, Robinson-Bostom L. Imipramine-induced hyperpigmentation: a case report and review of the literature. J Cutan Pathol. 2009;36:799-803.
- Ming ME, Bhawan J, Stefanato CM, et al. Imipramine-induced hyperpigmentation: four cases and a review of the literature. J Am Acad Dermatol. 1999;40(2, pt 1):159-166.
- Sicari MC, Lebwohl M, Baral J, et al. Photoinduced dermal pigmentation in patients taking tricyclic antidepressants: histology, electron microscopy, and energy dispersive spectroscopy. J Am Acad Dermatol.1999;40(2, pt 2):290-293.
- Atkin DH, Fitzpatrick RE. Laser treatment of imipramine-induced hyperpigmentation. J Am Acad Dermatol. 2000;43(1, pt 1):77-80.
- D’Agostino ML, Risser J, Robinson-Bostom L. Imipramine-induced hyperpigmentation: a case report and review of the literature. J Cutan Pathol. 2009;36:799-803.
- Ming ME, Bhawan J, Stefanato CM, et al. Imipramine-induced hyperpigmentation: four cases and a review of the literature. J Am Acad Dermatol. 1999;40(2, pt 1):159-166.
- Sicari MC, Lebwohl M, Baral J, et al. Photoinduced dermal pigmentation in patients taking tricyclic antidepressants: histology, electron microscopy, and energy dispersive spectroscopy. J Am Acad Dermatol.1999;40(2, pt 2):290-293.
- Atkin DH, Fitzpatrick RE. Laser treatment of imipramine-induced hyperpigmentation. J Am Acad Dermatol. 2000;43(1, pt 1):77-80.
Practice Points
- Imipramine is a tricyclic medication used for the treatment of depression and mood disorders.
- A rare side effect of treatment with imipramine is a blue-gray discoloration of the skin.
- Thorough medication review is important in patients who present with skin discoloration.
Pityriasis Rubra Pilaris and Severe Hypereosinophilia
To the Editor:
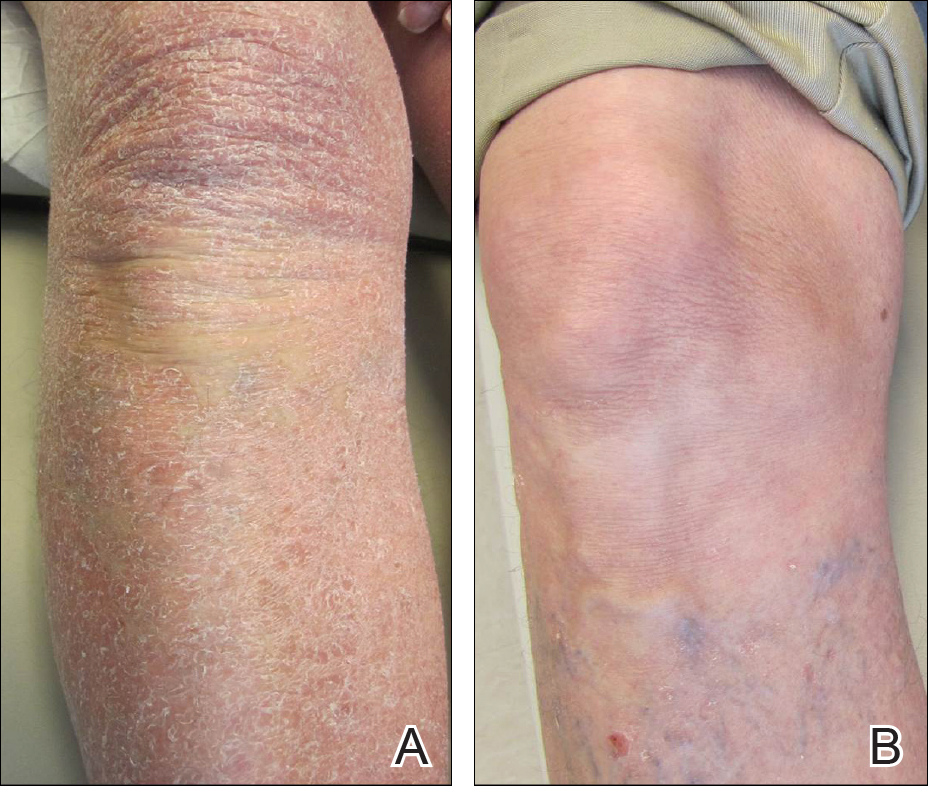
A 63-year-old man presented with a prior diagnosis of severe psoriasis affecting the extremities, neck, face, and scalp of 1 year’s duration. He reported pain, itching, and swelling in the affected areas. He felt the rash was worst on the hands and feet, and pain made performing activities of daily living difficult. His treatment regimen at presentation included triamcinolone cream 0.1% and azathioprine 150 mg daily as prescribed by an outside dermatologist without any response. Physical examination revealed diffuse erythema with lichenification and thick, white, flaking scale on the arms and legs (Figure 1A), face, neck, palms, and soles with islands of sparing. Multiple salmon-colored, follicular-based papules topped with central hyperkeratosis were scattered on these same areas. The palms and soles had severe confluent keratoderma (Figure 2A). Histologic examination of a follicular-based papule showed foci of parakeratosis and hypergranulosis consistent with the patient’s clinical picture of pityriasis rubra pilaris (PRP).
Baseline laboratory tests at the time of PRP diagnosis revealed 20.8% eosinophils (reference range, 0%–7%) and an absolute eosinophil count of 2.17×109/L (reference range, 0–0.7×109/L). Laboratory test results from an outside dermatologist conducted 10 to 12 months prior to the current presentation showed 12% eosinophils with a white blood cell count of 8.9×109/L (reference range, 4.5–11.0×109/L) around the time of rash onset and before treatment with azathioprine, making a drug reaction an unlikely cause of the eosinophilia.
After consulting with the hematology department, a hypereosinophilia workup including erythrocyte sedimentation rate, lactate dehydrogenase, serum protein electrophoresis, urine protein electrophoresis, tryptase, double-stranded DNA antibody, human T-lymphotrophic virus I/II, stool ova, and parasites, as well as a Strongyloides antibody titer, were performed; all were within reference range. His antinuclear antibody level was mildly elevated at 1:160, but the patient had no clinical manifestations of lupus. Given this negative workup, the most likely explanation for the hypereosinophilia was a reactive process secondary to the extreme inflammatory state.

The patient was started on isotretinoin 40 mg daily in addition to urea cream 40% mixed with clobetasol ointment at least once daily to the extremities. Hydrocortisone ointment 2.5% and petrolatum-based ointment were applied to the face, and hydroxyzine was used as needed for pruritus. One month after initiating isotretinoin, erythema had decreased and a repeat complete blood cell count with differential showed a decrease of eosinophils to 14.7% and an absolute eosinophil count of 1.56×109/L. After 2 months of therapy, the patient showed remarkable improvement. After 3.5 months of therapy, the keratoderma on the palms and soles was almost completely resolved, the follicular-based papules disappeared, and the patient had no areas of lichenification (Figures 1B and 2B). After 5 months of therapy, the patient experienced resolution of the PRP, except for residual facial erythema. His eosinophil count continued to trend downward during these 5 months, reaching 7.6% with an absolute eosinophil count of 0.93×109/L. Three years after the initial onset of the rash and 2 years after completing isotretinoin, his eosinophil level was normal at 5.3% with an absolute eosinophil count of 0.7×109/L.
We present a case of PRP and severe eosinophilia. We initially considered a second disease process to explain the extremely elevated eosinophil count; however, a negative eosinophilia workup and simultaneous resolution of these problems suggest that the eosinophilia was related to the severity of the PRP.
To the Editor:
A 63-year-old man presented with a prior diagnosis of severe psoriasis affecting the extremities, neck, face, and scalp of 1 year’s duration. He reported pain, itching, and swelling in the affected areas. He felt the rash was worst on the hands and feet, and pain made performing activities of daily living difficult. His treatment regimen at presentation included triamcinolone cream 0.1% and azathioprine 150 mg daily as prescribed by an outside dermatologist without any response. Physical examination revealed diffuse erythema with lichenification and thick, white, flaking scale on the arms and legs (Figure 1A), face, neck, palms, and soles with islands of sparing. Multiple salmon-colored, follicular-based papules topped with central hyperkeratosis were scattered on these same areas. The palms and soles had severe confluent keratoderma (Figure 2A). Histologic examination of a follicular-based papule showed foci of parakeratosis and hypergranulosis consistent with the patient’s clinical picture of pityriasis rubra pilaris (PRP).
Baseline laboratory tests at the time of PRP diagnosis revealed 20.8% eosinophils (reference range, 0%–7%) and an absolute eosinophil count of 2.17×109/L (reference range, 0–0.7×109/L). Laboratory test results from an outside dermatologist conducted 10 to 12 months prior to the current presentation showed 12% eosinophils with a white blood cell count of 8.9×109/L (reference range, 4.5–11.0×109/L) around the time of rash onset and before treatment with azathioprine, making a drug reaction an unlikely cause of the eosinophilia.
After consulting with the hematology department, a hypereosinophilia workup including erythrocyte sedimentation rate, lactate dehydrogenase, serum protein electrophoresis, urine protein electrophoresis, tryptase, double-stranded DNA antibody, human T-lymphotrophic virus I/II, stool ova, and parasites, as well as a Strongyloides antibody titer, were performed; all were within reference range. His antinuclear antibody level was mildly elevated at 1:160, but the patient had no clinical manifestations of lupus. Given this negative workup, the most likely explanation for the hypereosinophilia was a reactive process secondary to the extreme inflammatory state.
The patient was started on isotretinoin 40 mg daily in addition to urea cream 40% mixed with clobetasol ointment at least once daily to the extremities. Hydrocortisone ointment 2.5% and petrolatum-based ointment were applied to the face, and hydroxyzine was used as needed for pruritus. One month after initiating isotretinoin, erythema had decreased and a repeat complete blood cell count with differential showed a decrease of eosinophils to 14.7% and an absolute eosinophil count of 1.56×109/L. After 2 months of therapy, the patient showed remarkable improvement. After 3.5 months of therapy, the keratoderma on the palms and soles was almost completely resolved, the follicular-based papules disappeared, and the patient had no areas of lichenification (Figures 1B and 2B). After 5 months of therapy, the patient experienced resolution of the PRP, except for residual facial erythema. His eosinophil count continued to trend downward during these 5 months, reaching 7.6% with an absolute eosinophil count of 0.93×109/L. Three years after the initial onset of the rash and 2 years after completing isotretinoin, his eosinophil level was normal at 5.3% with an absolute eosinophil count of 0.7×109/L.
We present a case of PRP and severe eosinophilia. We initially considered a second disease process to explain the extremely elevated eosinophil count; however, a negative eosinophilia workup and simultaneous resolution of these problems suggest that the eosinophilia was related to the severity of the PRP.
To the Editor:
A 63-year-old man presented with a prior diagnosis of severe psoriasis affecting the extremities, neck, face, and scalp of 1 year’s duration. He reported pain, itching, and swelling in the affected areas. He felt the rash was worst on the hands and feet, and pain made performing activities of daily living difficult. His treatment regimen at presentation included triamcinolone cream 0.1% and azathioprine 150 mg daily as prescribed by an outside dermatologist without any response. Physical examination revealed diffuse erythema with lichenification and thick, white, flaking scale on the arms and legs (Figure 1A), face, neck, palms, and soles with islands of sparing. Multiple salmon-colored, follicular-based papules topped with central hyperkeratosis were scattered on these same areas. The palms and soles had severe confluent keratoderma (Figure 2A). Histologic examination of a follicular-based papule showed foci of parakeratosis and hypergranulosis consistent with the patient’s clinical picture of pityriasis rubra pilaris (PRP).
Baseline laboratory tests at the time of PRP diagnosis revealed 20.8% eosinophils (reference range, 0%–7%) and an absolute eosinophil count of 2.17×109/L (reference range, 0–0.7×109/L). Laboratory test results from an outside dermatologist conducted 10 to 12 months prior to the current presentation showed 12% eosinophils with a white blood cell count of 8.9×109/L (reference range, 4.5–11.0×109/L) around the time of rash onset and before treatment with azathioprine, making a drug reaction an unlikely cause of the eosinophilia.
After consulting with the hematology department, a hypereosinophilia workup including erythrocyte sedimentation rate, lactate dehydrogenase, serum protein electrophoresis, urine protein electrophoresis, tryptase, double-stranded DNA antibody, human T-lymphotrophic virus I/II, stool ova, and parasites, as well as a Strongyloides antibody titer, were performed; all were within reference range. His antinuclear antibody level was mildly elevated at 1:160, but the patient had no clinical manifestations of lupus. Given this negative workup, the most likely explanation for the hypereosinophilia was a reactive process secondary to the extreme inflammatory state.
The patient was started on isotretinoin 40 mg daily in addition to urea cream 40% mixed with clobetasol ointment at least once daily to the extremities. Hydrocortisone ointment 2.5% and petrolatum-based ointment were applied to the face, and hydroxyzine was used as needed for pruritus. One month after initiating isotretinoin, erythema had decreased and a repeat complete blood cell count with differential showed a decrease of eosinophils to 14.7% and an absolute eosinophil count of 1.56×109/L. After 2 months of therapy, the patient showed remarkable improvement. After 3.5 months of therapy, the keratoderma on the palms and soles was almost completely resolved, the follicular-based papules disappeared, and the patient had no areas of lichenification (Figures 1B and 2B). After 5 months of therapy, the patient experienced resolution of the PRP, except for residual facial erythema. His eosinophil count continued to trend downward during these 5 months, reaching 7.6% with an absolute eosinophil count of 0.93×109/L. Three years after the initial onset of the rash and 2 years after completing isotretinoin, his eosinophil level was normal at 5.3% with an absolute eosinophil count of 0.7×109/L.
We present a case of PRP and severe eosinophilia. We initially considered a second disease process to explain the extremely elevated eosinophil count; however, a negative eosinophilia workup and simultaneous resolution of these problems suggest that the eosinophilia was related to the severity of the PRP.
Practice Points
- Pityriasis rubra pilaris (PRP) can clinically mimic psoriasis. Look for islands of sparing and palmar and plantar hyperkeratosis to help diagnose PRP. A biopsy may be useful to help with this differentiation.
- Pityriasis rubra pilaris may be associated with eosinophilia, but one should rule out other causes of eosinophilia first.
