User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Vascular Nodule on the Upper Chest
Vascular Nodule on the Upper Chest
THE DIAGNOSIS: Metastatic Renal Cell Carcinoma
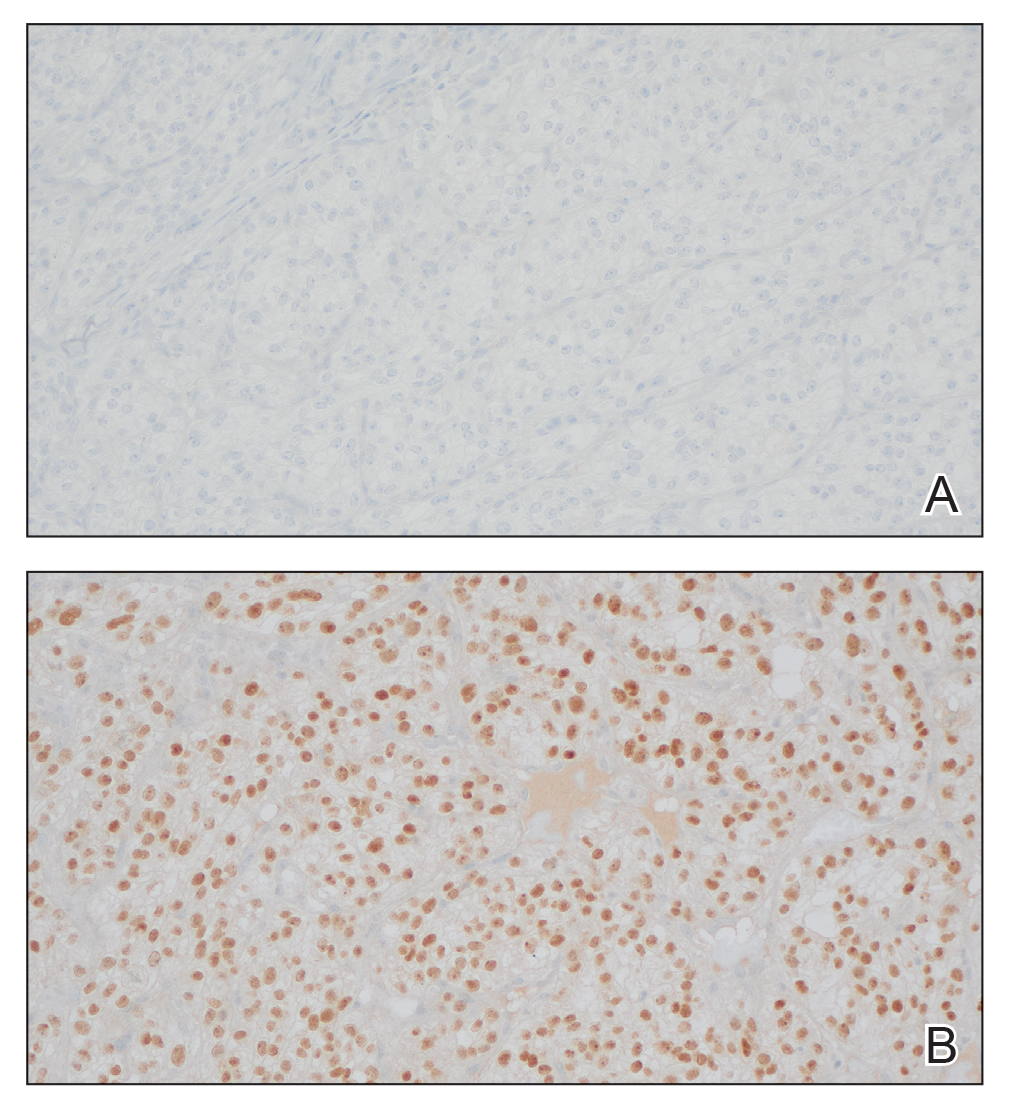
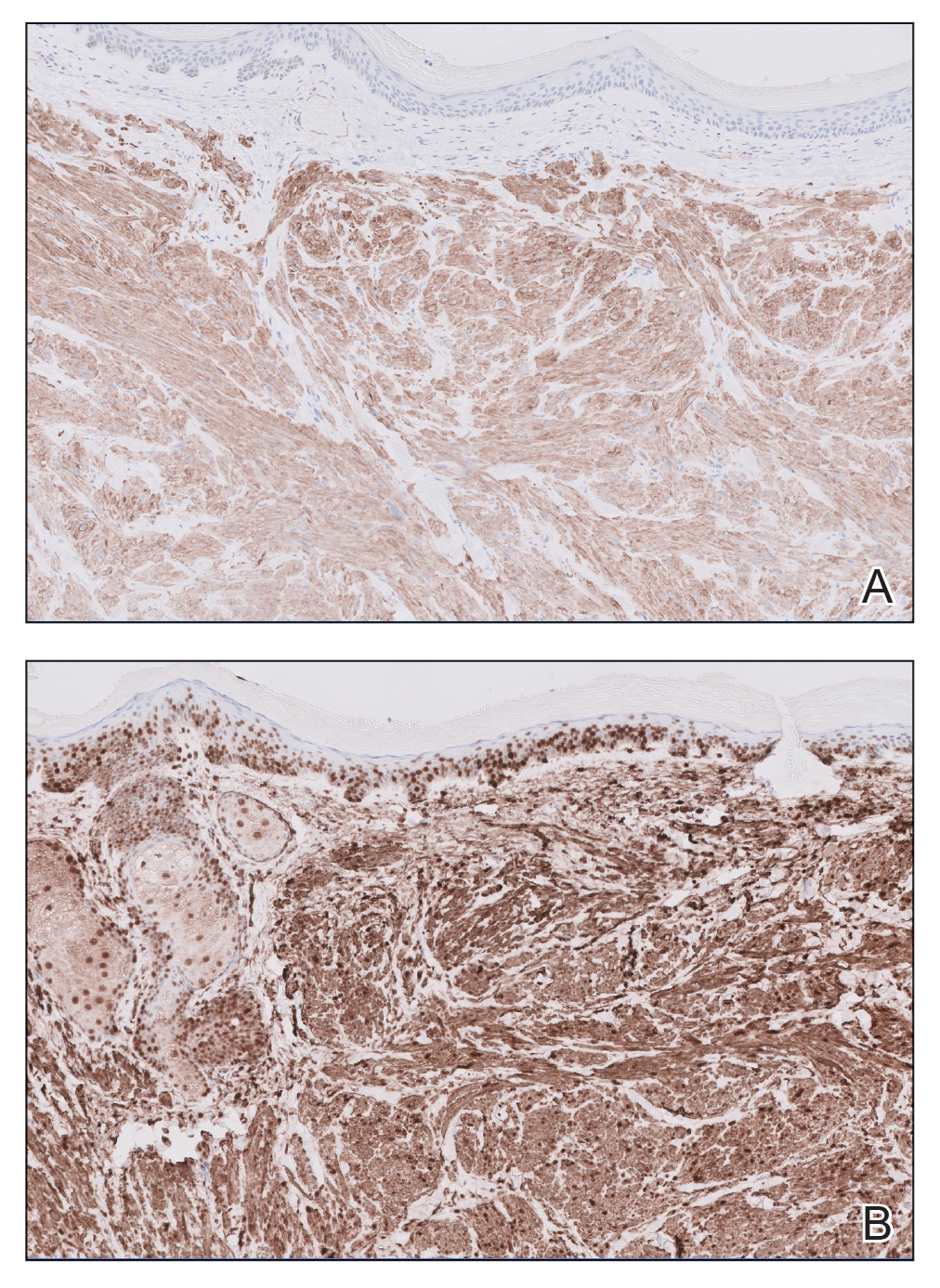
The shave biopsy revealed large cells with prominent nucleoli, clear cytoplasm, and thin cell borders in a nestlike arrangement (Figure 1). Immunohistochemical examination was negative for cytokeratin 5/6 and positive for PAX8 (Figure 2), which finalized the diagnosis of metastatic renal cell carcinoma (RCC). Later, our patient had a core biopsy-proven metastasis to the C6 spinous process, with concern for additional metastasis to the liver and lungs on positron emission tomography. Our patient’s treatment plan included pembrolizumab and axitinib to manage further cutaneous metastasis and radiation therapy for the C6 spinous process metastasis.
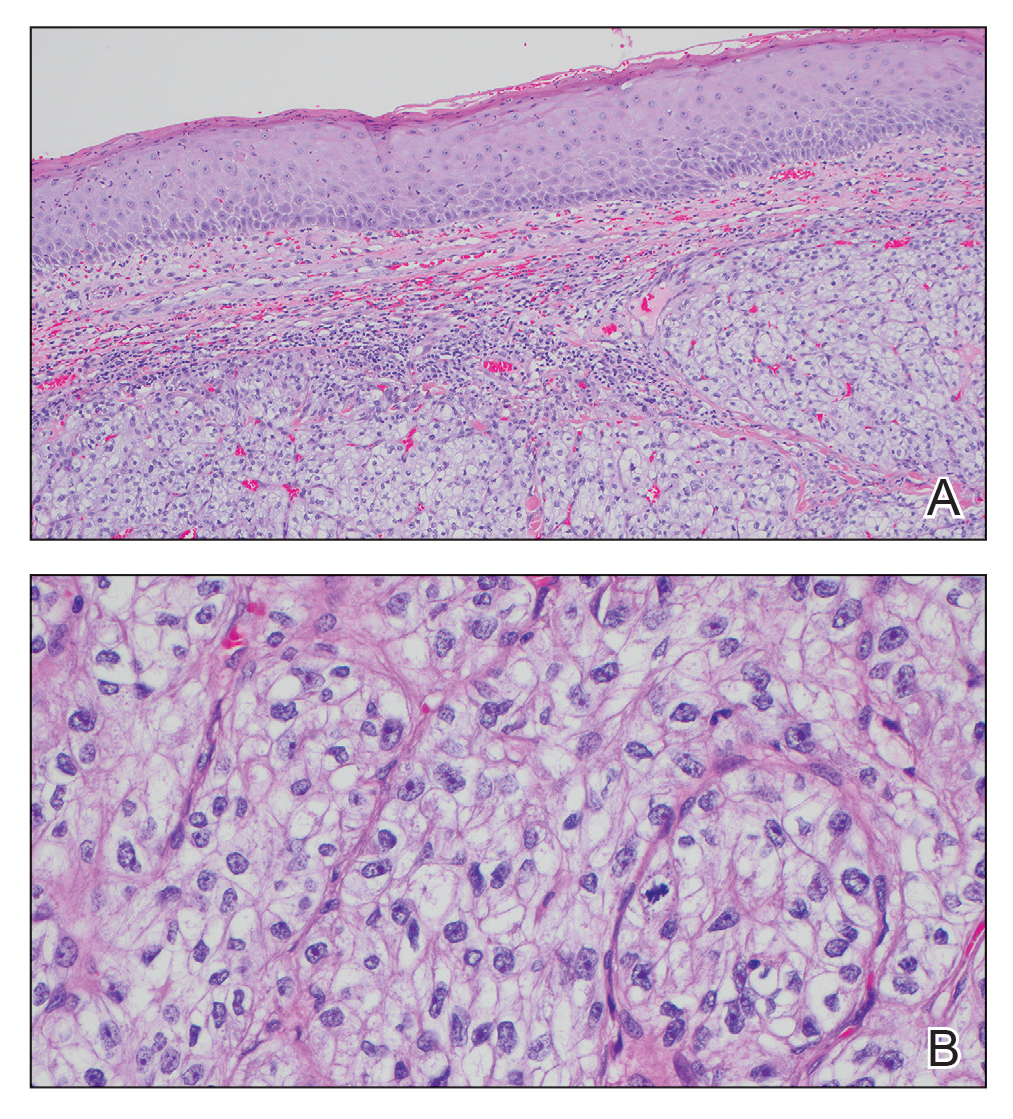
Renal cell carcinoma denotes cancer originating from the renal epithelium and is the most common kidney tumor in adults.1 Renal cell carcinoma accounts for more than 90% of kidney malignancies in the United States and has 3 main subtypes: clear cell RCC, papillary RCC, and chromophobe RCC.2 About 25% of cases metastasize, commonly to the lungs, liver, bones, lymph nodes, contralateral kidney, and adrenal glands.3
Cutaneous metastasis of RCC is rare, with an incidence of approximately 3.3%.4 Notably, 80% to 90% of patients with metastatic skin lesions had a prior diagnosis of RCC.2 Skin metastases associated with RCC predominantly are found on the face and scalp, appearing as nodular, swiftly expanding, circular, or oval-shaped growths. The robust vascular element of these lesions can lead to confusion with regard to the proper diagnosis, as they often resemble hemangiomas, pyogenic granulomas, or Kaposi sarcomas.4
Many cutaneous metastases linked to RCC exhibit a histomorphologic pattern consistent with clear cell adenocarcinoma.2 The malignant cells are large and possess transparent cytoplasm, round to oval nuclei, and prominent nucleoli. The cells can form glandular, acinar, or papillary arrangements; extravasated red blood cells frequently are found within the surrounding fibrovascular tissue.5 The presence of cytoplasmic glycogen can be revealed through periodic acid–Schiff staining. Other immunohistochemical markers commonly used to identify skin metastasis of RCC include epithelioid membrane antigen, carcinoembryonic antigen, and CD-10.1
Various mechanisms are involved in the cutaneous metastases of RCC. The most common pathway involves infiltration of the skin directly overlying the malignant renal mass; additional potential mechanisms include the introduction of abnormal cells into the skin during surgical or diagnostic interventions and their dissemination through the lymphatic system or bloodstream.1 Among urogenital malignancies other than RCC, skin metastases predominantly manifest in the abdominal region.2 Conversely, the head and neck region are more frequently impacted in RCC. The vascular composition of these tumors plays a role in facilitating the extension of cancer cells through the bloodstream, fostering the emergence of distant metastases.6
The development of cutaneous metastasis in RCC is associated with a poor prognosis, as most patients die within 6 months of detection.3 Treatment options thus are limited and palliative. Although local excision is an alternative treatment for localized cutaneous metastasis, it often provides little benefit in the presence of extensive metastasis; radiotherapy also has been shown to have a limited effect on primary RCC, though its devascularization of the lesion may be effective in metastatic cases.5 Immune checkpoint inhibitors such as nivolumab and ipilimumab have improved progression-free survival in patients with metastatic RCC, though uncertainty remains regarding their efficacy in attenuating cutaneous metastasis.5,6
- Kanwal R. Metastasis in renal cell carcinoma: biology and treatment. Adv Cancer Biol Metastasis. 2023;7:100094. doi:10.1016 /j.adcanc.2023.100094
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Bianchi M, Sun M, Jeldres C, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. 2012;23:973-980. doi:10.1093/annonc/mdr362
- Lorenzo-Rios D, Cruzval-O’Reilly E, Rabelo-Cartagena J. Facial cutaneous metastasis in renal cell carcinoma. Cureus. 2020;12:E12093. doi:10.7759/cureus.12093
- Iliescu CA, Beiu C, Racovit·a¢ A, et al. Atypical presentation of rapidly progressive cutaneous metastases of clear cell renal carcinoma: a case report. Medicina. 2024;60:1797. doi:10.3390/medicina60111797
- Joyce MJ. Management of skeletal metastases in renal cell carcinoma patients. In: Bukowski RM, Novick AC, eds. Clinical Management of Renal Tumors. Springer; 2008: 421-459.
THE DIAGNOSIS: Metastatic Renal Cell Carcinoma
The shave biopsy revealed large cells with prominent nucleoli, clear cytoplasm, and thin cell borders in a nestlike arrangement (Figure 1). Immunohistochemical examination was negative for cytokeratin 5/6 and positive for PAX8 (Figure 2), which finalized the diagnosis of metastatic renal cell carcinoma (RCC). Later, our patient had a core biopsy-proven metastasis to the C6 spinous process, with concern for additional metastasis to the liver and lungs on positron emission tomography. Our patient’s treatment plan included pembrolizumab and axitinib to manage further cutaneous metastasis and radiation therapy for the C6 spinous process metastasis.
Renal cell carcinoma denotes cancer originating from the renal epithelium and is the most common kidney tumor in adults.1 Renal cell carcinoma accounts for more than 90% of kidney malignancies in the United States and has 3 main subtypes: clear cell RCC, papillary RCC, and chromophobe RCC.2 About 25% of cases metastasize, commonly to the lungs, liver, bones, lymph nodes, contralateral kidney, and adrenal glands.3
Cutaneous metastasis of RCC is rare, with an incidence of approximately 3.3%.4 Notably, 80% to 90% of patients with metastatic skin lesions had a prior diagnosis of RCC.2 Skin metastases associated with RCC predominantly are found on the face and scalp, appearing as nodular, swiftly expanding, circular, or oval-shaped growths. The robust vascular element of these lesions can lead to confusion with regard to the proper diagnosis, as they often resemble hemangiomas, pyogenic granulomas, or Kaposi sarcomas.4
Many cutaneous metastases linked to RCC exhibit a histomorphologic pattern consistent with clear cell adenocarcinoma.2 The malignant cells are large and possess transparent cytoplasm, round to oval nuclei, and prominent nucleoli. The cells can form glandular, acinar, or papillary arrangements; extravasated red blood cells frequently are found within the surrounding fibrovascular tissue.5 The presence of cytoplasmic glycogen can be revealed through periodic acid–Schiff staining. Other immunohistochemical markers commonly used to identify skin metastasis of RCC include epithelioid membrane antigen, carcinoembryonic antigen, and CD-10.1
Various mechanisms are involved in the cutaneous metastases of RCC. The most common pathway involves infiltration of the skin directly overlying the malignant renal mass; additional potential mechanisms include the introduction of abnormal cells into the skin during surgical or diagnostic interventions and their dissemination through the lymphatic system or bloodstream.1 Among urogenital malignancies other than RCC, skin metastases predominantly manifest in the abdominal region.2 Conversely, the head and neck region are more frequently impacted in RCC. The vascular composition of these tumors plays a role in facilitating the extension of cancer cells through the bloodstream, fostering the emergence of distant metastases.6
The development of cutaneous metastasis in RCC is associated with a poor prognosis, as most patients die within 6 months of detection.3 Treatment options thus are limited and palliative. Although local excision is an alternative treatment for localized cutaneous metastasis, it often provides little benefit in the presence of extensive metastasis; radiotherapy also has been shown to have a limited effect on primary RCC, though its devascularization of the lesion may be effective in metastatic cases.5 Immune checkpoint inhibitors such as nivolumab and ipilimumab have improved progression-free survival in patients with metastatic RCC, though uncertainty remains regarding their efficacy in attenuating cutaneous metastasis.5,6
THE DIAGNOSIS: Metastatic Renal Cell Carcinoma
The shave biopsy revealed large cells with prominent nucleoli, clear cytoplasm, and thin cell borders in a nestlike arrangement (Figure 1). Immunohistochemical examination was negative for cytokeratin 5/6 and positive for PAX8 (Figure 2), which finalized the diagnosis of metastatic renal cell carcinoma (RCC). Later, our patient had a core biopsy-proven metastasis to the C6 spinous process, with concern for additional metastasis to the liver and lungs on positron emission tomography. Our patient’s treatment plan included pembrolizumab and axitinib to manage further cutaneous metastasis and radiation therapy for the C6 spinous process metastasis.
Renal cell carcinoma denotes cancer originating from the renal epithelium and is the most common kidney tumor in adults.1 Renal cell carcinoma accounts for more than 90% of kidney malignancies in the United States and has 3 main subtypes: clear cell RCC, papillary RCC, and chromophobe RCC.2 About 25% of cases metastasize, commonly to the lungs, liver, bones, lymph nodes, contralateral kidney, and adrenal glands.3
Cutaneous metastasis of RCC is rare, with an incidence of approximately 3.3%.4 Notably, 80% to 90% of patients with metastatic skin lesions had a prior diagnosis of RCC.2 Skin metastases associated with RCC predominantly are found on the face and scalp, appearing as nodular, swiftly expanding, circular, or oval-shaped growths. The robust vascular element of these lesions can lead to confusion with regard to the proper diagnosis, as they often resemble hemangiomas, pyogenic granulomas, or Kaposi sarcomas.4
Many cutaneous metastases linked to RCC exhibit a histomorphologic pattern consistent with clear cell adenocarcinoma.2 The malignant cells are large and possess transparent cytoplasm, round to oval nuclei, and prominent nucleoli. The cells can form glandular, acinar, or papillary arrangements; extravasated red blood cells frequently are found within the surrounding fibrovascular tissue.5 The presence of cytoplasmic glycogen can be revealed through periodic acid–Schiff staining. Other immunohistochemical markers commonly used to identify skin metastasis of RCC include epithelioid membrane antigen, carcinoembryonic antigen, and CD-10.1
Various mechanisms are involved in the cutaneous metastases of RCC. The most common pathway involves infiltration of the skin directly overlying the malignant renal mass; additional potential mechanisms include the introduction of abnormal cells into the skin during surgical or diagnostic interventions and their dissemination through the lymphatic system or bloodstream.1 Among urogenital malignancies other than RCC, skin metastases predominantly manifest in the abdominal region.2 Conversely, the head and neck region are more frequently impacted in RCC. The vascular composition of these tumors plays a role in facilitating the extension of cancer cells through the bloodstream, fostering the emergence of distant metastases.6
The development of cutaneous metastasis in RCC is associated with a poor prognosis, as most patients die within 6 months of detection.3 Treatment options thus are limited and palliative. Although local excision is an alternative treatment for localized cutaneous metastasis, it often provides little benefit in the presence of extensive metastasis; radiotherapy also has been shown to have a limited effect on primary RCC, though its devascularization of the lesion may be effective in metastatic cases.5 Immune checkpoint inhibitors such as nivolumab and ipilimumab have improved progression-free survival in patients with metastatic RCC, though uncertainty remains regarding their efficacy in attenuating cutaneous metastasis.5,6
- Kanwal R. Metastasis in renal cell carcinoma: biology and treatment. Adv Cancer Biol Metastasis. 2023;7:100094. doi:10.1016 /j.adcanc.2023.100094
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Bianchi M, Sun M, Jeldres C, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. 2012;23:973-980. doi:10.1093/annonc/mdr362
- Lorenzo-Rios D, Cruzval-O’Reilly E, Rabelo-Cartagena J. Facial cutaneous metastasis in renal cell carcinoma. Cureus. 2020;12:E12093. doi:10.7759/cureus.12093
- Iliescu CA, Beiu C, Racovit·a¢ A, et al. Atypical presentation of rapidly progressive cutaneous metastases of clear cell renal carcinoma: a case report. Medicina. 2024;60:1797. doi:10.3390/medicina60111797
- Joyce MJ. Management of skeletal metastases in renal cell carcinoma patients. In: Bukowski RM, Novick AC, eds. Clinical Management of Renal Tumors. Springer; 2008: 421-459.
- Kanwal R. Metastasis in renal cell carcinoma: biology and treatment. Adv Cancer Biol Metastasis. 2023;7:100094. doi:10.1016 /j.adcanc.2023.100094
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Bianchi M, Sun M, Jeldres C, et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Ann Oncol. 2012;23:973-980. doi:10.1093/annonc/mdr362
- Lorenzo-Rios D, Cruzval-O’Reilly E, Rabelo-Cartagena J. Facial cutaneous metastasis in renal cell carcinoma. Cureus. 2020;12:E12093. doi:10.7759/cureus.12093
- Iliescu CA, Beiu C, Racovit·a¢ A, et al. Atypical presentation of rapidly progressive cutaneous metastases of clear cell renal carcinoma: a case report. Medicina. 2024;60:1797. doi:10.3390/medicina60111797
- Joyce MJ. Management of skeletal metastases in renal cell carcinoma patients. In: Bukowski RM, Novick AC, eds. Clinical Management of Renal Tumors. Springer; 2008: 421-459.
Vascular Nodule on the Upper Chest
Vascular Nodule on the Upper Chest
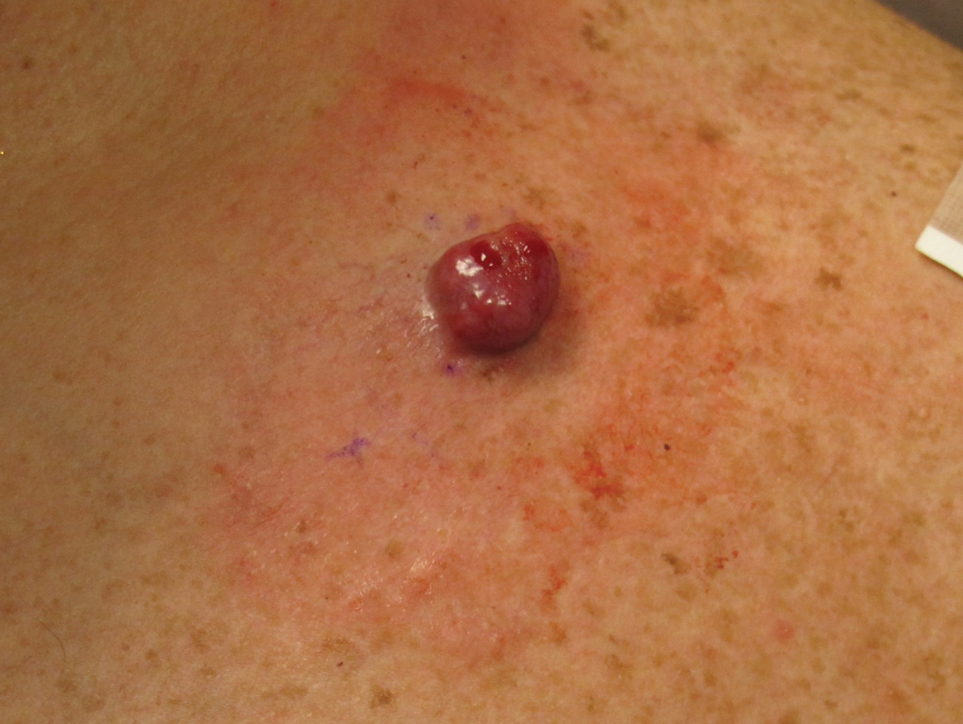
A 45-year-old man presented to the dermatology clinic with a bleeding nodule on the upper chest of 2 months’ duration. He had a history of a low-grade mucoepidermoid carcinoma of the left parotid gland that was diagnosed 14 years prior and was treated via parotidectomy with 1 positive lymph node removed. Two months prior to the current presentation, the patient presented to the emergency department with unintentional weight loss and fatigue and subsequently was diagnosed with clear cell renal cell carcinoma that was treated via radical nephrectomy.
At the current presentation, the patient denied any recent fatigue, fever, weight loss, shortness of breath, or abdominal pain but reported neck stiffness. Physical examination revealed a solitary, smooth, vascular, 1.5×1.5 cm nodule on the left upper chest with no overlying skin changes. The remainder of the skin examination was unremarkable. A shave biopsy of the nodule was performed.

A Painful Flesh-Colored Papule on the Shoulder
A Painful Flesh-Colored Papule on the Shoulder
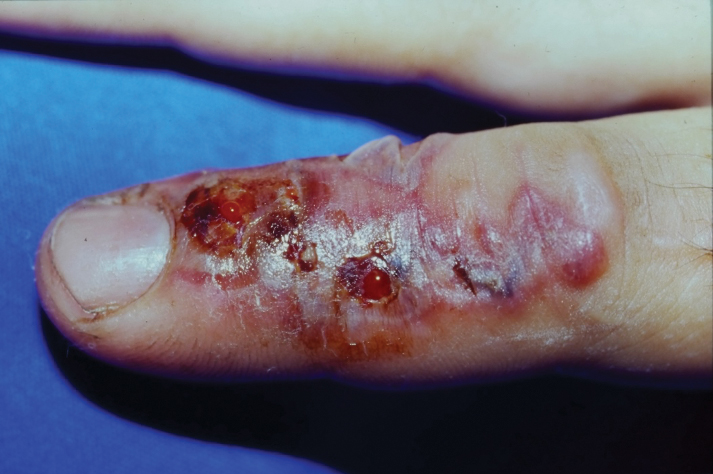
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
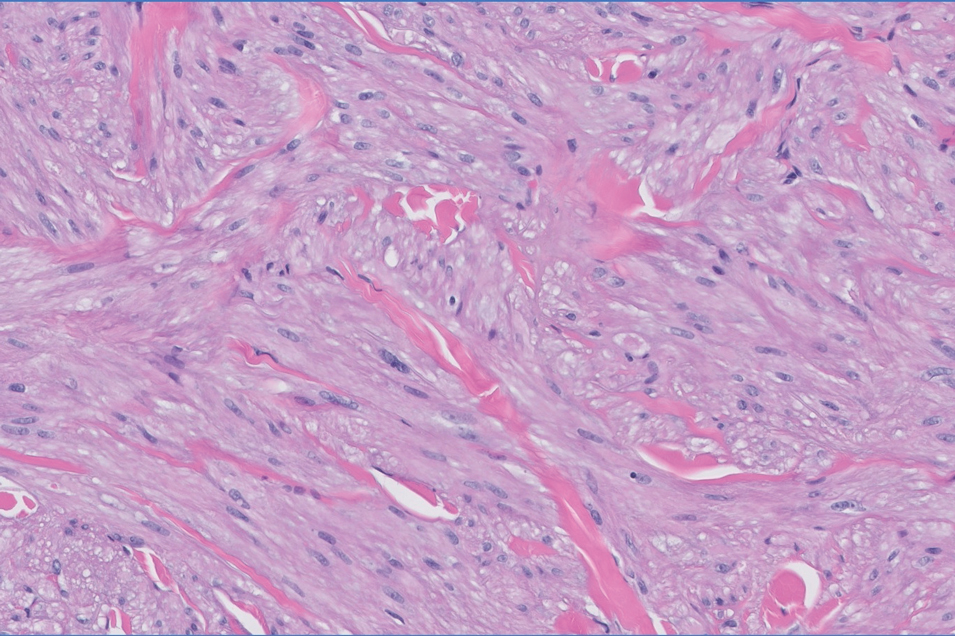

Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
The Diagnosis: Leiomyoma
Histopathology revealed a dermal mesenchymal tumor composed of fascicles of bland spindle cells with tapered nuclei, perinuclear vacuoles, eosinophilic cytoplasm, and low cellularity (Figure 1). Immunohistochemical studies of the cells stained strongly positive for smooth muscle actin and desmin, consistent with a smooth muscle neoplasm (Figure 2). Fumarate hydratase (FH) staining revealed loss of expression in tumor cells, consistent with FH deficiency (Figure 3). A diagnosis of cutaneous leiomyoma was made, and although the clinical and histologic findings suggested hereditary leiomyomatosis and renal cell cancer (HLRCC), genetic testing was negative for an FH gene mutation. This negative result indicated that HLRCC was unlikely despite the initial concerns based on the findings.
Leiomyomas are benign neoplasms that are challenging to diagnose based on the clinical picture alone. Leiomyomas most commonly are found in the genitourinary and gastrointestinal systems, with cutaneous manifestation being the second most common presentation.1 These benign smooth muscle tumors manifest as tender, firm, flesh-colored, pink or reddish-brown nodules that are subcategorized based on the derivation of the smooth muscle within the tumor.2 Angioleiomyomas, the most common type, arise from the tunica media of blood vessels, whereas piloleiomyomas and genital leiomyomas arise from the arrector pili musculature of the hair follicle and the smooth muscle found in the scrotum, labia, or nipple.2 Rare cases of cutaneous leiomyosarcomas and angioleiomyosarcomas have been reported in the literature.3,4 Solitary leiomyomas tend to develop on the lower extremities, whereas multiple lesions frequently manifest on the extensor surfaces of extremities and the trunk. Lesions often are painful, either spontaneously or in association with applied pressure, emotional stress, or exposure to cold temperatures.2
Although leiomyomas themselves are benign, patients with multiple cutaneous leiomyomas may have an underlying genetic mutation that increases their risk of developing HLRCC, an autosomal-dominant syndrome.5 Referral should be considered for individuals with a personal history of or a first-degree relative with cutaneous leiomyomas or renal cell carcinoma (RCC) with histology typical of hereditary leiomyomatosis and RCC, as recommended by the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors.6 In this case, the decision to refer the patient for genetic testing was based on her family history, specifically her paternal uncle having multiple similar lesions, which, while not a first-degree relative, still raised concerns about potential hereditary risks and warranted further evaluation. A germline mutation in the FH gene, which encodes an enzyme that converts fumarate to malate in the Krebs cycle and plays a role in tumor suppression, is the cause of HLRCC.2,7 When part of this genetic condition, cutaneous leiomyomas tend to occur around 25 years of age (range, 10-50 years).2 A diagnosis of HLRCC should be strongly considered if a patient displays multiple cutaneous leiomyomas with at least 1 histologically confirmed lesion or at least 2 of the following: solitary cutaneous leiomyoma with family history of HLRCC, onset of severely symptomatic uterine fibroids before age 40 years, type II papillary or collecting duct renal cell cancer before age 40 years, or a first-degree family member who meets 1 of these criteria.5,8
Diagnosis of cutaneous leiomyoma may be accomplished by microscopic examination of a tissue sample; however, further diagnostic workup is warranted due to the strong correlation with HLRCC.2 A definitive diagnosis of HLRCC is confirmed with a germline mutation in the FH gene, and genetic screening should be offered to patients before renal cancer surveillance to avoid unwarranted investigations.8 Timely clinical diagnosis enables early genetic testing and enhanced outcomes for patients with confirmed HLRCC who may need a multidisciplinary approach of dermatologists, gynecologists, and urologic oncologists.5,8
Cutaneous leiomyomas can be excised, and this typically is the gold standard of care for small and localized lesions, although the use of cryosurgery and carbon dioxide lasers has been reported as well.2,9,10 For more widespread lesions or for patients who are not appropriate candidates for surgery, pharmacologic therapies (α-blockers, calcium channel blockers, nitroglycerin), intralesional corticosteroids, and/or botulinum toxin injections can be utilized.2,11
The acronym BLEND AN EGG encompasses the clinical differential diagnosis for painful skin tumors: blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor. Blue rubber bleb nevi are deep blue in color, and angiolipomas sit under the skin and present as subcutaneous swellings. Dermatofibromas and neurofibromas also are included in the differential.12 Dermatofibromas are firm solitary lesions that have a pathognomonic pinch sign. Neurofibromas are soft and rubbery, have a buttonhole sign, and stain positively for S-100 protein and SOX-10 but negatively for actin and desmin.12
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Bernett CN, Mammino JJ. Cutaneous leiomyomas. In: StatPearls. StatPearls Publishing; 2023.
- Chayed Z, Kristensen LK, Ousager LB, et al. Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis. 2021;16:34. doi:10.1186/s13023-020-01653-9
- Perkins J, Scarbrough C, Sammons D, et al. Reed syndrome: an atypical presentation of a rare disease. Dermatol Online J. 2014;21: 13030/qt5k35r5pn.
- Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253-260. doi:10.2147 /IJNRD.S42097
- Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med. 2015;17:70-87. doi:10.1038/gim.2014.147
- Alam NA, Barclay E, Rowan AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol. 2005;141:199-206. doi:10.1001 /archderm.141.2.199
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644. doi:10.1007/s10689-014-9735-2
- Uyar B, Acar EM, Subas¸ıog˘lu A. Treatment of three hereditary leiomyomatosis patients with cryotherapy. Dermatol Ther. 2020;33:e13226. doi:10.1111/dth.13226
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322. doi:10.1046/j.1524-4725.2000.99250.x
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma- related pain. J Am Acad Dermatol. 2009;60:325-328. doi:10.1016 /j.jaad.2008.05.044
- Clarey DD, Lauer SR, Adams JL. Painful papules on the arms. Cutis. 2020;106:232-249. doi:10.12788/cutis.0109
A Painful Flesh-Colored Papule on the Shoulder
A Painful Flesh-Colored Papule on the Shoulder
A 65-year-old woman with a history of metabolic syndrome presented to the family medicine clinic for evaluation of a papule on the right shoulder that had started small and increased in size over the past 3 years. Physical examination revealed a 1.0×0.8×0.1-cm, smooth, flesh-colored to light brown papule on the right shoulder that was notably tender to palpation. The patient reported that her paternal uncle had multiple skin lesions of similar morphology dispersed on the bilateral upper extremities. A shave biopsy of the lesion was performed.
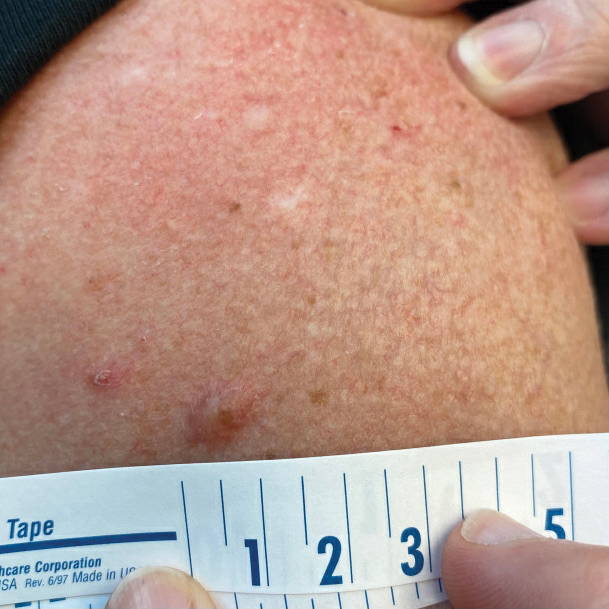
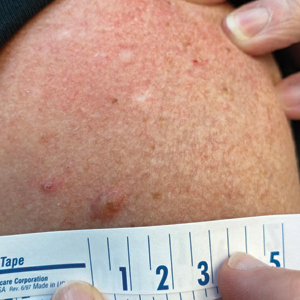
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
To the Editor:
A 22-year-old man was referred to our dermatology outpatient department for wartlike growths that gradually developed around a postoperative enteroatmospheric fistula and stoma over the past 4 months. The patient presented for an emergency exploratory laparotomy with a history of perforation peritonitis 1.5 years prior to the current presentation. He also had a small bowel obstruction 5 months prior to the current presentation that resulted in the resection of a large segment of the small bowel. He underwent a diverting loop ileostomy when the abdominal closure was not achieved because of bowel edema, following which he developed a postoperative enteroatmospheric fistula. In addition, the stoma retracted and was followed by dermal dehiscence, which led to notable leakage and resulted in heavy fecal contamination of the midline wound.
At the current presentation, physical examination revealed multiple grayish-white, dome-shaped, moist papules coalescing to form a peristomal pseudoverrucous mass on the lower side of the stoma (Figure 1). The patient experienced mild itching. The lesion showed no signs of erosion, bleeding, or purulent discharge, and there were no nearby lumps or enlarged lymph nodes. The differential diagnosis included peristomal pyoderma gangrenosum, human papillomavirus (HPV) infection, pseudoverrucous papules and nodules (PPNs), squamous cell carcinoma, and exuberant granulation tissue. A skin biopsy was performed, and histopathology revealed hyperkeratosis, moderate papillomatosis, and marked acanthotic hyperplasia seen as downgrowths into the dermis (Figure 2). No koilocytes, atypia, or mitotic figures were present. Abundant neutrophils and few eosinophils were seen in the dermal infiltrate. A final diagnosis of PPN was made based on clinicopathologic correlation. The patient was advised to use a smaller stoma bag and to change the collection pouch frequently to reduce skin contact with fecal matter.
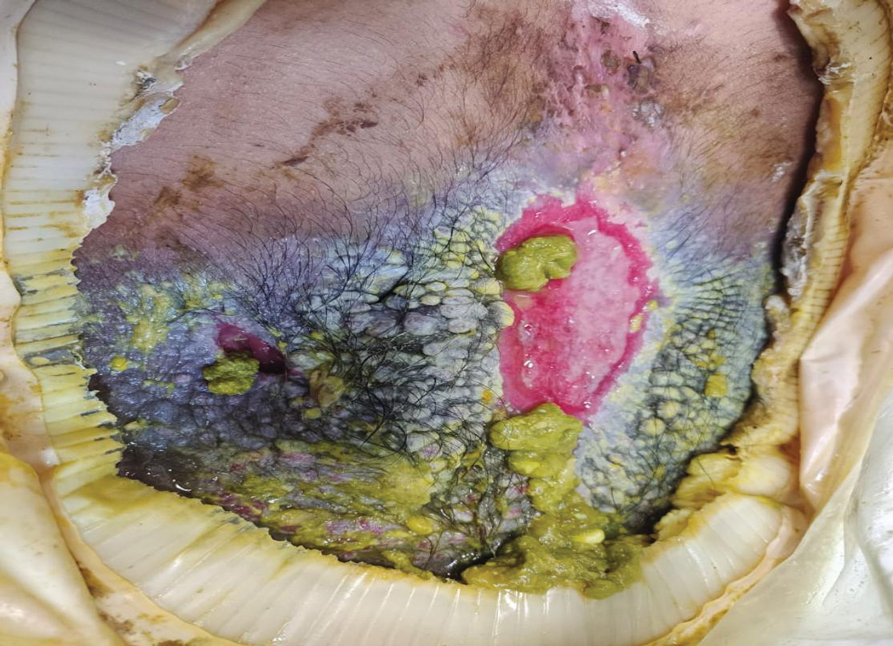
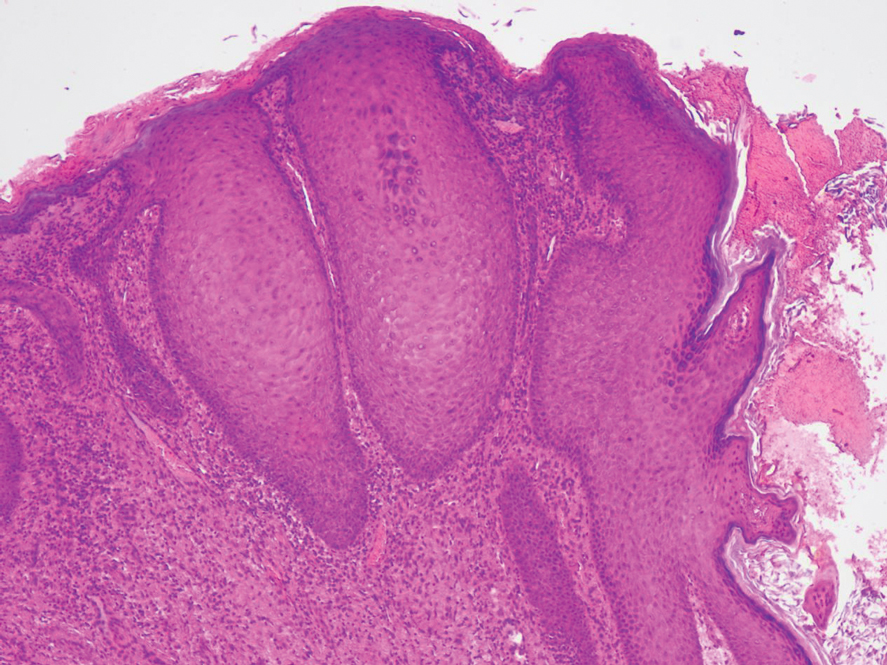
Peristomal skin conditions are reported in 18% to 55% of patients with stomas and include allergic contact dermatitis, mechanical dermatitis, infections, pyoderma gangrenosum, and irritant contact dermatitis.1,2 Pseudoverrucous papules (also called chronic papillomatous dermatitis or pseudoverrucous lesions) is a rare dermatologic complication found on the skin around stomas,3 most commonly around urostomy stomas. The presence of PPNs around colostomy stomas and the perianal region is extremely rare.2,4 This condition is the result of chronic irritant dermatitis from frequent exposure to urine or feces, leading to maceration and epidermal hyperplasia. It occurs because of improper sizing of the stoma bag or incorrect positioning or construction of the stoma.5
the overuse of topical benzocaine-resorcinol, leading to chronic irritation.6 It is clinically characterized by multiple grayish-white, wartlike, confluent papulonodules around areas chronically exposed to moisture. Differential diagnoses such as secondary neoplasms, HPV infection, exuberant granulation tissue, and candidal infections should be considered.3 Final diagnosis is based on clinicopathologic findings, similar to our case. Epidermal growth factor and transforming growth factor are thought to play a role in the pathophysiology of pseudoepitheliomatous hyperplasia. Increased expression of these mediators leads to proliferation of the epidermis into the dermis.7 The role of HPV in PPN remains unclear, as not all PPN lesions are positive for HPV and the cutaneous lesions resolve once the source of irritation is removed. Recommended treatment includes local skin care; stoma refitting; and, in severe cases, excision and revision of the stoma.2 Dermatologists must be aware of this often-underdiagnosed condition.
- Alslaim F, Al Farajat F, Alslaim HS, et al. Etiology and management of peristomal pseudoepitheliomatous hyperplasia. Cureus. 2021;13 :E20196. doi:10.7759/cureus.20196
- Rambhia PH, Conic RZ, Honda K, et al. Chronic papillomatous dermatitis in a patient with a urinary ileal diversion: a case report and review of the literature. Dermatol Arch. 2017;1:47-50. doi:10.36959/661/297
- Latour-Álvarez I, García-Peris E, Pestana-Eliche MM, et al. Nodular peristomal lesions. Actas Dermosifiliogr. 2016;108:363-364. doi:10.1016/j.ad.2016.02.018
- Dandale A, Dhurat R, Ghate S. Perianal pseudoverrucous papules and nodules. Indian J Sex Transm Dis AIDS. 2013;34:44-46. doi:10.4103/0253-7184.112939
- Brogna L. Prevention and management of pseudoverrucous lesions: a review and case scenarios. Adv Skin Wound Care. 2021;34:461-471. doi:10.1097/01.ASW.0000758620.93518.39
- Robson KJ, Maughan JA, Purcell SD, et al. Erosive papulonodular dermatosis associated with topical benzocaine: a report of two cases and evidence that granuloma gluteale, pseudoverrucous papules, and Jacquet’s erosive dermatitis are a disease spectrum. J Am Acad Dermatol. 2006;55(5 suppl):S74-S80. doi:10.1016/j .jaad.2005.12.025
- Oğuz ID, Vural S, Cinar E, et al. Peristomal pseudoverrucous lesions: a rare skin complication of colostomy. Cureus. 2023;15:E38068. doi:10.7759/cureus.38068
To the Editor:
A 22-year-old man was referred to our dermatology outpatient department for wartlike growths that gradually developed around a postoperative enteroatmospheric fistula and stoma over the past 4 months. The patient presented for an emergency exploratory laparotomy with a history of perforation peritonitis 1.5 years prior to the current presentation. He also had a small bowel obstruction 5 months prior to the current presentation that resulted in the resection of a large segment of the small bowel. He underwent a diverting loop ileostomy when the abdominal closure was not achieved because of bowel edema, following which he developed a postoperative enteroatmospheric fistula. In addition, the stoma retracted and was followed by dermal dehiscence, which led to notable leakage and resulted in heavy fecal contamination of the midline wound.
At the current presentation, physical examination revealed multiple grayish-white, dome-shaped, moist papules coalescing to form a peristomal pseudoverrucous mass on the lower side of the stoma (Figure 1). The patient experienced mild itching. The lesion showed no signs of erosion, bleeding, or purulent discharge, and there were no nearby lumps or enlarged lymph nodes. The differential diagnosis included peristomal pyoderma gangrenosum, human papillomavirus (HPV) infection, pseudoverrucous papules and nodules (PPNs), squamous cell carcinoma, and exuberant granulation tissue. A skin biopsy was performed, and histopathology revealed hyperkeratosis, moderate papillomatosis, and marked acanthotic hyperplasia seen as downgrowths into the dermis (Figure 2). No koilocytes, atypia, or mitotic figures were present. Abundant neutrophils and few eosinophils were seen in the dermal infiltrate. A final diagnosis of PPN was made based on clinicopathologic correlation. The patient was advised to use a smaller stoma bag and to change the collection pouch frequently to reduce skin contact with fecal matter.
Peristomal skin conditions are reported in 18% to 55% of patients with stomas and include allergic contact dermatitis, mechanical dermatitis, infections, pyoderma gangrenosum, and irritant contact dermatitis.1,2 Pseudoverrucous papules (also called chronic papillomatous dermatitis or pseudoverrucous lesions) is a rare dermatologic complication found on the skin around stomas,3 most commonly around urostomy stomas. The presence of PPNs around colostomy stomas and the perianal region is extremely rare.2,4 This condition is the result of chronic irritant dermatitis from frequent exposure to urine or feces, leading to maceration and epidermal hyperplasia. It occurs because of improper sizing of the stoma bag or incorrect positioning or construction of the stoma.5
the overuse of topical benzocaine-resorcinol, leading to chronic irritation.6 It is clinically characterized by multiple grayish-white, wartlike, confluent papulonodules around areas chronically exposed to moisture. Differential diagnoses such as secondary neoplasms, HPV infection, exuberant granulation tissue, and candidal infections should be considered.3 Final diagnosis is based on clinicopathologic findings, similar to our case. Epidermal growth factor and transforming growth factor are thought to play a role in the pathophysiology of pseudoepitheliomatous hyperplasia. Increased expression of these mediators leads to proliferation of the epidermis into the dermis.7 The role of HPV in PPN remains unclear, as not all PPN lesions are positive for HPV and the cutaneous lesions resolve once the source of irritation is removed. Recommended treatment includes local skin care; stoma refitting; and, in severe cases, excision and revision of the stoma.2 Dermatologists must be aware of this often-underdiagnosed condition.
To the Editor:
A 22-year-old man was referred to our dermatology outpatient department for wartlike growths that gradually developed around a postoperative enteroatmospheric fistula and stoma over the past 4 months. The patient presented for an emergency exploratory laparotomy with a history of perforation peritonitis 1.5 years prior to the current presentation. He also had a small bowel obstruction 5 months prior to the current presentation that resulted in the resection of a large segment of the small bowel. He underwent a diverting loop ileostomy when the abdominal closure was not achieved because of bowel edema, following which he developed a postoperative enteroatmospheric fistula. In addition, the stoma retracted and was followed by dermal dehiscence, which led to notable leakage and resulted in heavy fecal contamination of the midline wound.
At the current presentation, physical examination revealed multiple grayish-white, dome-shaped, moist papules coalescing to form a peristomal pseudoverrucous mass on the lower side of the stoma (Figure 1). The patient experienced mild itching. The lesion showed no signs of erosion, bleeding, or purulent discharge, and there were no nearby lumps or enlarged lymph nodes. The differential diagnosis included peristomal pyoderma gangrenosum, human papillomavirus (HPV) infection, pseudoverrucous papules and nodules (PPNs), squamous cell carcinoma, and exuberant granulation tissue. A skin biopsy was performed, and histopathology revealed hyperkeratosis, moderate papillomatosis, and marked acanthotic hyperplasia seen as downgrowths into the dermis (Figure 2). No koilocytes, atypia, or mitotic figures were present. Abundant neutrophils and few eosinophils were seen in the dermal infiltrate. A final diagnosis of PPN was made based on clinicopathologic correlation. The patient was advised to use a smaller stoma bag and to change the collection pouch frequently to reduce skin contact with fecal matter.
Peristomal skin conditions are reported in 18% to 55% of patients with stomas and include allergic contact dermatitis, mechanical dermatitis, infections, pyoderma gangrenosum, and irritant contact dermatitis.1,2 Pseudoverrucous papules (also called chronic papillomatous dermatitis or pseudoverrucous lesions) is a rare dermatologic complication found on the skin around stomas,3 most commonly around urostomy stomas. The presence of PPNs around colostomy stomas and the perianal region is extremely rare.2,4 This condition is the result of chronic irritant dermatitis from frequent exposure to urine or feces, leading to maceration and epidermal hyperplasia. It occurs because of improper sizing of the stoma bag or incorrect positioning or construction of the stoma.5
the overuse of topical benzocaine-resorcinol, leading to chronic irritation.6 It is clinically characterized by multiple grayish-white, wartlike, confluent papulonodules around areas chronically exposed to moisture. Differential diagnoses such as secondary neoplasms, HPV infection, exuberant granulation tissue, and candidal infections should be considered.3 Final diagnosis is based on clinicopathologic findings, similar to our case. Epidermal growth factor and transforming growth factor are thought to play a role in the pathophysiology of pseudoepitheliomatous hyperplasia. Increased expression of these mediators leads to proliferation of the epidermis into the dermis.7 The role of HPV in PPN remains unclear, as not all PPN lesions are positive for HPV and the cutaneous lesions resolve once the source of irritation is removed. Recommended treatment includes local skin care; stoma refitting; and, in severe cases, excision and revision of the stoma.2 Dermatologists must be aware of this often-underdiagnosed condition.
- Alslaim F, Al Farajat F, Alslaim HS, et al. Etiology and management of peristomal pseudoepitheliomatous hyperplasia. Cureus. 2021;13 :E20196. doi:10.7759/cureus.20196
- Rambhia PH, Conic RZ, Honda K, et al. Chronic papillomatous dermatitis in a patient with a urinary ileal diversion: a case report and review of the literature. Dermatol Arch. 2017;1:47-50. doi:10.36959/661/297
- Latour-Álvarez I, García-Peris E, Pestana-Eliche MM, et al. Nodular peristomal lesions. Actas Dermosifiliogr. 2016;108:363-364. doi:10.1016/j.ad.2016.02.018
- Dandale A, Dhurat R, Ghate S. Perianal pseudoverrucous papules and nodules. Indian J Sex Transm Dis AIDS. 2013;34:44-46. doi:10.4103/0253-7184.112939
- Brogna L. Prevention and management of pseudoverrucous lesions: a review and case scenarios. Adv Skin Wound Care. 2021;34:461-471. doi:10.1097/01.ASW.0000758620.93518.39
- Robson KJ, Maughan JA, Purcell SD, et al. Erosive papulonodular dermatosis associated with topical benzocaine: a report of two cases and evidence that granuloma gluteale, pseudoverrucous papules, and Jacquet’s erosive dermatitis are a disease spectrum. J Am Acad Dermatol. 2006;55(5 suppl):S74-S80. doi:10.1016/j .jaad.2005.12.025
- Oğuz ID, Vural S, Cinar E, et al. Peristomal pseudoverrucous lesions: a rare skin complication of colostomy. Cureus. 2023;15:E38068. doi:10.7759/cureus.38068
- Alslaim F, Al Farajat F, Alslaim HS, et al. Etiology and management of peristomal pseudoepitheliomatous hyperplasia. Cureus. 2021;13 :E20196. doi:10.7759/cureus.20196
- Rambhia PH, Conic RZ, Honda K, et al. Chronic papillomatous dermatitis in a patient with a urinary ileal diversion: a case report and review of the literature. Dermatol Arch. 2017;1:47-50. doi:10.36959/661/297
- Latour-Álvarez I, García-Peris E, Pestana-Eliche MM, et al. Nodular peristomal lesions. Actas Dermosifiliogr. 2016;108:363-364. doi:10.1016/j.ad.2016.02.018
- Dandale A, Dhurat R, Ghate S. Perianal pseudoverrucous papules and nodules. Indian J Sex Transm Dis AIDS. 2013;34:44-46. doi:10.4103/0253-7184.112939
- Brogna L. Prevention and management of pseudoverrucous lesions: a review and case scenarios. Adv Skin Wound Care. 2021;34:461-471. doi:10.1097/01.ASW.0000758620.93518.39
- Robson KJ, Maughan JA, Purcell SD, et al. Erosive papulonodular dermatosis associated with topical benzocaine: a report of two cases and evidence that granuloma gluteale, pseudoverrucous papules, and Jacquet’s erosive dermatitis are a disease spectrum. J Am Acad Dermatol. 2006;55(5 suppl):S74-S80. doi:10.1016/j .jaad.2005.12.025
- Oğuz ID, Vural S, Cinar E, et al. Peristomal pseudoverrucous lesions: a rare skin complication of colostomy. Cureus. 2023;15:E38068. doi:10.7759/cureus.38068
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
Pseudoverrucous Papules and Nodules Around a Surgical Stoma
PRACTICE POINTS
- Pseudoverrucous papules and nodules (PPNs) can develop around stomas due to chronic irritant dermatitis from fecal or urinary exposure.
- Proper stoma management, including the use of appropriately sized stoma bags and frequent changes, is essential to prevent skin complications such as PPN.
- When evaluating peristomal lesions, consider a broad differential diagnosis, including infections, neoplasms, and dermatitis, and ensure thorough clinicopathologic correlation for accurate diagnosis and treatment.

Key Features of North American Venomous Snake Bites
Key Features of North American Venomous Snake Bites
North American venomous snakes traditionally are classified as members of either the Viperidae (eg, rattlesnakes, copperheads, cottonmouths) or Elapidae (eg, coral snakes) families and account for roughly 5000 to 10,000 reported envenomations annually.1,2 In 2021, America’s Poison Centers reported 2287 calls related to copperheads, 71 related to coral snakes, 229 related to cottonmouths, 1184 related to rattlesnakes, and 524 related to unknown snakes.3 The majority of calls related to snake bites were for adult patients, resulting in absent to minor outcomes. Only 1 death due to a rattlesnake bite was reported.3 Death by envenomation from a North American snake species is considered rare and typically is attributed to a lapse in medical attention; however, rattlesnakes are the most common reported cause of death by snake envenomation (Figure 1).1,3 A study comparing snake bites and hospital stays in the southeast vs southwest United States found that the southeast had the highest incidence of copperhead bites (37%), while the southwest had a higher incidence of rattlesnake bites (70%); those who were bitten by a rattlesnake were reported to have more severe symptoms and greater need for medical attention and antivenin.4 Some reports have linked pediatric and elderly patients to worse outcomes.5 However, one study examining 24,388 emergency department visits for snake bites from 2006 through 2014 found that the majority of pediatric cases were handled by non– trauma centers in the southern United States,6 supporting evidence found by Campbell et al7 indicating that most snake bites in children can be managed with conservative care. Though reported complications—including weakness, paralysis, hypovolemic shock, thrombocytopenia, and death—from North American venomous snake bites are low, they are still considered a medical emergency.8 It is essential for physicians to understand the clinical manifestations and treatment of North American venomous snake bites and to educate patients on how to protect themselves against and avoid provoking snakes, particularly in rural areas.2 In this article, we review the characteristics of common North American venomous snakes and the clinical manifestations of their bites. We also discuss the appropriate measures for staging, evaluating, and treating snake envenomation to improve patient management and care.

Features of North American Venomous Snakes
Individual snakes within the Viperidae family vary in size, markings and coloration, activity, and region, and physicians should consult their local health departments regarding snakes that are common in their area.2 Cottonmouth snakes are semiaquatic and traditionally are found within the southern and central United States. With a spade-shaped head and distinct two-tone coloration, cottonmouths may be mistaken for other nonvenomous water snakes in these regions (Figure 2).2 Copperheads, true to their name, are red in color; they inhabit a large portion of the southeastern United States and eastern Texas regions and are the cause of the majority of venomous snake bites in North America (Figure 3). Both cottonmouths and copperheads are believed to bite and envenomate as a defensive mechanism when provoked.


Coral snakes, found in the eastern United States and Texas regions, are the only subspecies of the Elapidae family (Figure 4).2,9 They can be distinguished from the nonvenomous milk snake by their characteristic banding, as coral snakes are patterned in a red-yellow-black band sequence and milk snakes are patterned in a red-black-yellow or white sequence. The differences in appearance of these snakes often is remembered by the phrase “red on yellow kills a fellow.”

Anatomic differences between the Viperidae and Elapidae families, including fang size, placement, and type, as well as venom composition, are directly linked to clinical manifestations of the bites. Viperidae fangs extend from the maxillary bones and are mobile, long, and hollow, making it easy for the snake to control fang movement and envenomation.9 Viperidae snakes are uniquely capable of inflicting puncture wounds without the injection of venom, known as dry bites. In contrast, Elapidae snakes have short, hollow, and fixed fangs, and thus patients can protect themselves by wearing appropriate clothing and covered footwear.9 Currently, identifying the type of snake responsible for the bite relies on visualization of the snake and/or the identification of clinical symptoms of envenomation by a dermatologist.
Clinical Manifestations of Venomous Snake Bites
Clinical manifestations and cutaneous findings often are used to grade the severity of venomous snake bites as well as to dictate treatment procedures. Grade 0 indicates a bite has occurred without envenomation, while grades I to V describe the progression and severity of envenomation.10 Grade I describes minimal erythema and edema around the site (fang marks may or may not be present) and no systemic symptoms. Grade II describes erythema and edema extending up the extremity to the first joint (eg, hand to wrist), pain, some systemic symptoms if there is rapid progression, and potential bleeding at the site. Grade III describes erythema and edema spreading to the second joint in the extremity, pain, and systemic symptoms, including coagulation defects. Grade IV describes erythema and edema of the whole extremity, a rapid reaction and progression following the bite, and risk for compartment syndrome. Grade V includes erythema and edema beyond the extremity and increasing systemic symptoms.10
Local pain and edema, usually on easily accessible or exposed extremities, are the most common clinical symptoms reported following a Viperidae snake bite.11 Due to their capability of producing a dry bite, puncture markings alone do not indicate envenomation. Patients will need to be monitored for several hours for signs of envenomation, which may include swelling, pain, ecchymosis, and indications of systemic manifestation (eg, weakness, dizziness, nausea, severe hypotension, thrombocytopenia).11 Viperidae venom hemorrhagic metalloproteinases act on capillary blood vessels by cleaving basement membrane proteins and allowing for extravasation of fluid into local tissue.12 The inflammatory response produced at the site of envenomation likely is due to the release of tumor necrosis factor á and endogenous matrix metalloprotein.12 There is a higher risk for death associated with bites from rattlesnakes within the Viperidae family because their venom contains a unique neurotoxin that works by blocking presynaptic junctions and causing a range of paralytic symptoms from ptosis to respiratory failure.13
The severity of Elapidae bites is thought to be related to the amount of venom injected, the size of the victim, and the length of the snake. Though clothing may offer protection, envenomation occurs in 75% of coral snake bites and can produce devastating consequences due to the venom content.14 In a retrospective study between 2002 and 2004, 90% of Elapidae snake bite patients (n=82) reported local pain, redness, and paresthesia, while around 7% developed systemic symptoms.15 Elapidae venom primarily is neurotoxic and is thought to spread via lymphatics.16 Delayed reactions are common and may take up to 12 hours to develop. Patients should be monitored, as local reactions may progress to weakness, fasciculations, extremity paralysis, and lastly, respiratory paralysis. Due to the risk for progression, all patients with likely coral snake bites should be given antivenin.8,15,17
Much like the North American coral snake, the venomous snake species Gloydius blomhoffii—referred to as the salmosa or mamushi snake depending on the region of origin (ie, Korea or Japan)—is a frequent source of devastating rural snake bites due to neurotoxins (Figure 5). The species’ slender fangs are thought to directly inject the snake’s potent venom, which contains hemorrhagic toxins and α-neurotoxins and Β-neurotoxins, into the bloodstream; however, the salmosa is considered a viper like the North American cottonmouth and copperhead because of its triangular head shape and hollow fangs, which allow for the accommodation of venom-containing glands and mechanism of venom injection. Salmosa venom shares both Viperidae and Elapidae characteristics. Cutaneous findings such as progressive edema, erythema, and bleeding frequently are reported and are attributed to the proteases and hemorrhagic toxins characteristic of vipers (Figure 6). α-Neurotoxins and Β-neurotoxins, similar to the proteolytic venom of the Elapidae family, are responsible for the unique visual disturbances (binocular diplopia) caused by the salmosa.12,18,19

Treatment
Treating snake bites begins with assessing the patient’s airway, breathing, and circulation, followed by a thorough medical and encounter history (including description of how the bite occurred). Due to the range of Viperidae symptoms, it generally is recommended that patients remove any restrictive clothing or jewelry near the bite and/or over the affected limb or body part, place the affected body part at the level of the heart, and go to the nearest medical facility for prompt care. Historically, empiric antibiotics often were used to prevent wound infections; however, studies have since demonstrated that antibiotics are not necessary and lack efficacy in uncomplicated snake bites.16,20 In a study of 114 pediatric cases from 1995 to 2005, it was determined that most patients could be managed with conservative treatment directed at pain management and swelling reduction via elevation of the affected extremity.6 While conservative management may be all that is needed to care for the majority of cases, one retrospective study from Texas indicated that 70% of pediatric venomous snake bites were treated with either intravenous antibiotics and/or antivenin, highlighting the variability in management and opportunity for improvement.21
Antivenin, specifically antivenin (Crotalidae) polyvalent, is the indicated treatment for Viperidae hemorrhagic or coagulopathic envenomation.13,22 Per guidelines from the World Health Organization, physical examination will yield a grading of the snake bite based on cutaneous findings. Grades III to V are considered moderate to severe and should be given antivenin.23 Physicians should look for signs of progressive injury and coagulopathy, such as increased swelling, bruising, hypotension, or altered mental status.22 Due to the major neurotoxic risks associated with Elapidae venom, all coral snake bites should be treated with antivenin; early intubation and ventilation may be considered.13 Similarly, patients who report a salmosa snake bite require prompt treatment with antivenin and/or cepharanthine, an additive agent to reduce swelling and pain.18 Due to the nature of the neurotoxins contained in the salmosa venom (α-neurotoxin causing postsynaptic inhibition of the neuromuscular junction and Β-neurotoxin inhibiting neurotransmitter release from the presynaptic terminal), anticholinesterases, which work by blocking the enzymatic breakdown of the neurotransmitter acetylcholine, should not be used.19 While bleeding and skin and systemic changes may be reversed by antivenin, visual changes are unlikely to resolve with antivenin administration due to the presynaptic binding of Β-neurotoxin and the blockade of neuromuscular signaling.19
Antivenin should be administered intravenously for the fastest onset of action in a setting suitable for the management of anaphylaxis.24 In situations when the benefits may outweigh the risks (eg, if the patient has had a prior allergic reaction or is not in an environment where they can be watched for at least 8 hours for progression of envenomation or adverse reactions), premedication with an antihistamine or epinephrine may be considered.17 Per the World Allergy Organization and World Health Organization, adverse reactions should be treated with crystalloid solutions and antihistamines, corticosteroids, or epinephrine as indicated.25 In a qualitative analysis of emergency physicians’ attitudes toward antivenin, most expressed treatment hesitancy due to lack of knowledge and experience using the medication.26 When possible, snake bites should thus be managed in consultation with a toxicologist.2
Conclusion
Snake bites and envenomation occur commonly in the United States due to exposure to a variety of venomous snakes in the North American Viperidae and Elapidae families. Appropriate and successful management of snake bites by physicians requires general knowledge of regional snakes, the cutaneous and systemic manifestations of snake bites and envenomation, and current treatment methods.
- Greene SC, Folt J, Wyatt K, et al. Epidemiology of fatal snakebites in the United States 1981-2018. Am J Emerg Med. 2021;45:309-316.
- Wozniak EJ, Wisser J, Schwartz M. Venomous adversaries: a reference to snake identification, field safety, and bite-victim first aid for disaster-response personnel deploying into the hurricaneprone regions of North America. Wilderness Environ Med. 2006; 17:246-266.
- Gummin DD, Mowry JB, Beuhler MC, et al. 2021 annual report of National Poison Data System (NPDS) from America’s Poison Centers: 39th Annual Report. Clin Toxicol (Phila). 2022;60:1381-1643.
- Chotai PN, Watlington J, Lewis S, et al. Pediatric snakebites: comparing patients in two geographic locations in the United States. J Surg Res. 2021;265:297-302.
- Johnson PN, McGoodwin L, Banner W Jr. Utilisation of Crotalidae polyvalent immune fab (ovine) for Viperidae envenomations in children. Emerg Med J. 2008;25:793-798.
- Tadros A, Sharon M, Davis S, et al. Emergency department visits by pediatric patients for snakebites. Pediatr Emerg Care. 2022; 38:279-282.
- Campbell BT, Corsi JM, Boneti C, et al. Pediatric snake bites: lessons learned from 114 cases. J Pediatr Surg. 2008;43:1338-1341.
- Peterson ME. Snake bites: coral snakes. Clin Tech Small Anim Pract. 2006;21:183-186.
- Porter KR. Herpetology. WB Saunders Company; 1972.
- Rana A, Kheora S. Grading and envenomation of the snake bite among the emergency cases in a medical college in rural India. Hmlyn Jr Appl Med Sci Res. 2021;2:33-36.
- Peterson ME. Snake bite: pit vipers. Clin Tech Small Anim Pract. 2006;21:174-182.
- Gutierrez JM, Rucavado A. Snake venom metalloproteinases: their role in the pathogenesis of local tissue damage. Biochimie. 2000;82:841-850.
- Weinstein SA, Dart RC, Staples A, et al. Envenomations: an overview of clinical toxicology for the primary care physician. Am Fam Physician. 2009;80:793-802.
- Kitchens CS, Van Mierop LH. Envenomation by the eastern coral snake (Micrurus fulvius fulvius): a study of 39 victims. JAMA. 1987;258:1615-1618.
- Morgan DL, Borys DJ, Stanford R, et al. Texas coral snake (Micrurus tener) bites. South Med J. 2007;100:152-156.
- Clark RF, Delden BS, Furbee B. The incidence of wound infection following crotalid envenomation. J Emerg Med. 1993; 11:583-586.
- Gold BS, Dart RC, Barish RA. Bites of venomous snakes. N Engl J Med. 2002;347:347-356.
- Hifumi T, Sakai A, Kondo Y, et al. Venomous snake bites: clinical diagnosis and treatment. J Intensive Care. 2015;3:16.
- Igari R, Iseki K, Abe S, et al. Binocular diplopia and ptosis due to snake bite (Agkistrodon blomhoffi “mamushi”) case report. Brain Nerve. 2010;62:273-277.
- Kerrigan KR, Mertz BL, Nelson SJ, et al. Antibiotic prophylaxis for pit viper envenomation: prospective, controlled trial. World J Surg. 1997;21:369-372.
- Correa JA, Fallon SC, Cruz AT, et al. Management of pediatric snake bites: are we doing too much? J Pediatr Surg. 2014;49:1009-1015.
- Dart RC, McNally J. Efficacy, safety and use of snake antivenoms in the United States. Ann Emerg Med. 2001;47:181-188.
- World Health Organization Regional Office for South-East Asia. Guidelines for the Management of Snakebites. 2nd ed. World Health Organization; 2016.
- Clark RF, McKinney PE, Chase PB, et al. Immediate and delayed allergic reactions to Crotalidae polyvalent immune Fab (ovine) antivenom. Ann Emerg Med. 2002;39:671-676.
- World Health Organization. WHO Guidelines for the production, control, and regulation of snake antivenom immunoglobulins. Accessed November 25, 2024. https://extranet.who.int/prequal/vaccines/guidelines-production-control-and-regulation-snake-antivenom-immunoglobulins
- Tupetz A, Barcenas LK, Phillips AJ, et al. Bites study: a qualitive analysis among emergency medicine physicians on snake envenomation management practices. PloS One. 2022;17:E0262215.
North American venomous snakes traditionally are classified as members of either the Viperidae (eg, rattlesnakes, copperheads, cottonmouths) or Elapidae (eg, coral snakes) families and account for roughly 5000 to 10,000 reported envenomations annually.1,2 In 2021, America’s Poison Centers reported 2287 calls related to copperheads, 71 related to coral snakes, 229 related to cottonmouths, 1184 related to rattlesnakes, and 524 related to unknown snakes.3 The majority of calls related to snake bites were for adult patients, resulting in absent to minor outcomes. Only 1 death due to a rattlesnake bite was reported.3 Death by envenomation from a North American snake species is considered rare and typically is attributed to a lapse in medical attention; however, rattlesnakes are the most common reported cause of death by snake envenomation (Figure 1).1,3 A study comparing snake bites and hospital stays in the southeast vs southwest United States found that the southeast had the highest incidence of copperhead bites (37%), while the southwest had a higher incidence of rattlesnake bites (70%); those who were bitten by a rattlesnake were reported to have more severe symptoms and greater need for medical attention and antivenin.4 Some reports have linked pediatric and elderly patients to worse outcomes.5 However, one study examining 24,388 emergency department visits for snake bites from 2006 through 2014 found that the majority of pediatric cases were handled by non– trauma centers in the southern United States,6 supporting evidence found by Campbell et al7 indicating that most snake bites in children can be managed with conservative care. Though reported complications—including weakness, paralysis, hypovolemic shock, thrombocytopenia, and death—from North American venomous snake bites are low, they are still considered a medical emergency.8 It is essential for physicians to understand the clinical manifestations and treatment of North American venomous snake bites and to educate patients on how to protect themselves against and avoid provoking snakes, particularly in rural areas.2 In this article, we review the characteristics of common North American venomous snakes and the clinical manifestations of their bites. We also discuss the appropriate measures for staging, evaluating, and treating snake envenomation to improve patient management and care.
Features of North American Venomous Snakes
Individual snakes within the Viperidae family vary in size, markings and coloration, activity, and region, and physicians should consult their local health departments regarding snakes that are common in their area.2 Cottonmouth snakes are semiaquatic and traditionally are found within the southern and central United States. With a spade-shaped head and distinct two-tone coloration, cottonmouths may be mistaken for other nonvenomous water snakes in these regions (Figure 2).2 Copperheads, true to their name, are red in color; they inhabit a large portion of the southeastern United States and eastern Texas regions and are the cause of the majority of venomous snake bites in North America (Figure 3). Both cottonmouths and copperheads are believed to bite and envenomate as a defensive mechanism when provoked.
Coral snakes, found in the eastern United States and Texas regions, are the only subspecies of the Elapidae family (Figure 4).2,9 They can be distinguished from the nonvenomous milk snake by their characteristic banding, as coral snakes are patterned in a red-yellow-black band sequence and milk snakes are patterned in a red-black-yellow or white sequence. The differences in appearance of these snakes often is remembered by the phrase “red on yellow kills a fellow.”
Anatomic differences between the Viperidae and Elapidae families, including fang size, placement, and type, as well as venom composition, are directly linked to clinical manifestations of the bites. Viperidae fangs extend from the maxillary bones and are mobile, long, and hollow, making it easy for the snake to control fang movement and envenomation.9 Viperidae snakes are uniquely capable of inflicting puncture wounds without the injection of venom, known as dry bites. In contrast, Elapidae snakes have short, hollow, and fixed fangs, and thus patients can protect themselves by wearing appropriate clothing and covered footwear.9 Currently, identifying the type of snake responsible for the bite relies on visualization of the snake and/or the identification of clinical symptoms of envenomation by a dermatologist.
Clinical Manifestations of Venomous Snake Bites
Clinical manifestations and cutaneous findings often are used to grade the severity of venomous snake bites as well as to dictate treatment procedures. Grade 0 indicates a bite has occurred without envenomation, while grades I to V describe the progression and severity of envenomation.10 Grade I describes minimal erythema and edema around the site (fang marks may or may not be present) and no systemic symptoms. Grade II describes erythema and edema extending up the extremity to the first joint (eg, hand to wrist), pain, some systemic symptoms if there is rapid progression, and potential bleeding at the site. Grade III describes erythema and edema spreading to the second joint in the extremity, pain, and systemic symptoms, including coagulation defects. Grade IV describes erythema and edema of the whole extremity, a rapid reaction and progression following the bite, and risk for compartment syndrome. Grade V includes erythema and edema beyond the extremity and increasing systemic symptoms.10
Local pain and edema, usually on easily accessible or exposed extremities, are the most common clinical symptoms reported following a Viperidae snake bite.11 Due to their capability of producing a dry bite, puncture markings alone do not indicate envenomation. Patients will need to be monitored for several hours for signs of envenomation, which may include swelling, pain, ecchymosis, and indications of systemic manifestation (eg, weakness, dizziness, nausea, severe hypotension, thrombocytopenia).11 Viperidae venom hemorrhagic metalloproteinases act on capillary blood vessels by cleaving basement membrane proteins and allowing for extravasation of fluid into local tissue.12 The inflammatory response produced at the site of envenomation likely is due to the release of tumor necrosis factor á and endogenous matrix metalloprotein.12 There is a higher risk for death associated with bites from rattlesnakes within the Viperidae family because their venom contains a unique neurotoxin that works by blocking presynaptic junctions and causing a range of paralytic symptoms from ptosis to respiratory failure.13
The severity of Elapidae bites is thought to be related to the amount of venom injected, the size of the victim, and the length of the snake. Though clothing may offer protection, envenomation occurs in 75% of coral snake bites and can produce devastating consequences due to the venom content.14 In a retrospective study between 2002 and 2004, 90% of Elapidae snake bite patients (n=82) reported local pain, redness, and paresthesia, while around 7% developed systemic symptoms.15 Elapidae venom primarily is neurotoxic and is thought to spread via lymphatics.16 Delayed reactions are common and may take up to 12 hours to develop. Patients should be monitored, as local reactions may progress to weakness, fasciculations, extremity paralysis, and lastly, respiratory paralysis. Due to the risk for progression, all patients with likely coral snake bites should be given antivenin.8,15,17
Much like the North American coral snake, the venomous snake species Gloydius blomhoffii—referred to as the salmosa or mamushi snake depending on the region of origin (ie, Korea or Japan)—is a frequent source of devastating rural snake bites due to neurotoxins (Figure 5). The species’ slender fangs are thought to directly inject the snake’s potent venom, which contains hemorrhagic toxins and α-neurotoxins and Β-neurotoxins, into the bloodstream; however, the salmosa is considered a viper like the North American cottonmouth and copperhead because of its triangular head shape and hollow fangs, which allow for the accommodation of venom-containing glands and mechanism of venom injection. Salmosa venom shares both Viperidae and Elapidae characteristics. Cutaneous findings such as progressive edema, erythema, and bleeding frequently are reported and are attributed to the proteases and hemorrhagic toxins characteristic of vipers (Figure 6). α-Neurotoxins and Β-neurotoxins, similar to the proteolytic venom of the Elapidae family, are responsible for the unique visual disturbances (binocular diplopia) caused by the salmosa.12,18,19
Treatment
Treating snake bites begins with assessing the patient’s airway, breathing, and circulation, followed by a thorough medical and encounter history (including description of how the bite occurred). Due to the range of Viperidae symptoms, it generally is recommended that patients remove any restrictive clothing or jewelry near the bite and/or over the affected limb or body part, place the affected body part at the level of the heart, and go to the nearest medical facility for prompt care. Historically, empiric antibiotics often were used to prevent wound infections; however, studies have since demonstrated that antibiotics are not necessary and lack efficacy in uncomplicated snake bites.16,20 In a study of 114 pediatric cases from 1995 to 2005, it was determined that most patients could be managed with conservative treatment directed at pain management and swelling reduction via elevation of the affected extremity.6 While conservative management may be all that is needed to care for the majority of cases, one retrospective study from Texas indicated that 70% of pediatric venomous snake bites were treated with either intravenous antibiotics and/or antivenin, highlighting the variability in management and opportunity for improvement.21
Antivenin, specifically antivenin (Crotalidae) polyvalent, is the indicated treatment for Viperidae hemorrhagic or coagulopathic envenomation.13,22 Per guidelines from the World Health Organization, physical examination will yield a grading of the snake bite based on cutaneous findings. Grades III to V are considered moderate to severe and should be given antivenin.23 Physicians should look for signs of progressive injury and coagulopathy, such as increased swelling, bruising, hypotension, or altered mental status.22 Due to the major neurotoxic risks associated with Elapidae venom, all coral snake bites should be treated with antivenin; early intubation and ventilation may be considered.13 Similarly, patients who report a salmosa snake bite require prompt treatment with antivenin and/or cepharanthine, an additive agent to reduce swelling and pain.18 Due to the nature of the neurotoxins contained in the salmosa venom (α-neurotoxin causing postsynaptic inhibition of the neuromuscular junction and Β-neurotoxin inhibiting neurotransmitter release from the presynaptic terminal), anticholinesterases, which work by blocking the enzymatic breakdown of the neurotransmitter acetylcholine, should not be used.19 While bleeding and skin and systemic changes may be reversed by antivenin, visual changes are unlikely to resolve with antivenin administration due to the presynaptic binding of Β-neurotoxin and the blockade of neuromuscular signaling.19
Antivenin should be administered intravenously for the fastest onset of action in a setting suitable for the management of anaphylaxis.24 In situations when the benefits may outweigh the risks (eg, if the patient has had a prior allergic reaction or is not in an environment where they can be watched for at least 8 hours for progression of envenomation or adverse reactions), premedication with an antihistamine or epinephrine may be considered.17 Per the World Allergy Organization and World Health Organization, adverse reactions should be treated with crystalloid solutions and antihistamines, corticosteroids, or epinephrine as indicated.25 In a qualitative analysis of emergency physicians’ attitudes toward antivenin, most expressed treatment hesitancy due to lack of knowledge and experience using the medication.26 When possible, snake bites should thus be managed in consultation with a toxicologist.2
Conclusion
Snake bites and envenomation occur commonly in the United States due to exposure to a variety of venomous snakes in the North American Viperidae and Elapidae families. Appropriate and successful management of snake bites by physicians requires general knowledge of regional snakes, the cutaneous and systemic manifestations of snake bites and envenomation, and current treatment methods.
North American venomous snakes traditionally are classified as members of either the Viperidae (eg, rattlesnakes, copperheads, cottonmouths) or Elapidae (eg, coral snakes) families and account for roughly 5000 to 10,000 reported envenomations annually.1,2 In 2021, America’s Poison Centers reported 2287 calls related to copperheads, 71 related to coral snakes, 229 related to cottonmouths, 1184 related to rattlesnakes, and 524 related to unknown snakes.3 The majority of calls related to snake bites were for adult patients, resulting in absent to minor outcomes. Only 1 death due to a rattlesnake bite was reported.3 Death by envenomation from a North American snake species is considered rare and typically is attributed to a lapse in medical attention; however, rattlesnakes are the most common reported cause of death by snake envenomation (Figure 1).1,3 A study comparing snake bites and hospital stays in the southeast vs southwest United States found that the southeast had the highest incidence of copperhead bites (37%), while the southwest had a higher incidence of rattlesnake bites (70%); those who were bitten by a rattlesnake were reported to have more severe symptoms and greater need for medical attention and antivenin.4 Some reports have linked pediatric and elderly patients to worse outcomes.5 However, one study examining 24,388 emergency department visits for snake bites from 2006 through 2014 found that the majority of pediatric cases were handled by non– trauma centers in the southern United States,6 supporting evidence found by Campbell et al7 indicating that most snake bites in children can be managed with conservative care. Though reported complications—including weakness, paralysis, hypovolemic shock, thrombocytopenia, and death—from North American venomous snake bites are low, they are still considered a medical emergency.8 It is essential for physicians to understand the clinical manifestations and treatment of North American venomous snake bites and to educate patients on how to protect themselves against and avoid provoking snakes, particularly in rural areas.2 In this article, we review the characteristics of common North American venomous snakes and the clinical manifestations of their bites. We also discuss the appropriate measures for staging, evaluating, and treating snake envenomation to improve patient management and care.
Features of North American Venomous Snakes
Individual snakes within the Viperidae family vary in size, markings and coloration, activity, and region, and physicians should consult their local health departments regarding snakes that are common in their area.2 Cottonmouth snakes are semiaquatic and traditionally are found within the southern and central United States. With a spade-shaped head and distinct two-tone coloration, cottonmouths may be mistaken for other nonvenomous water snakes in these regions (Figure 2).2 Copperheads, true to their name, are red in color; they inhabit a large portion of the southeastern United States and eastern Texas regions and are the cause of the majority of venomous snake bites in North America (Figure 3). Both cottonmouths and copperheads are believed to bite and envenomate as a defensive mechanism when provoked.
Coral snakes, found in the eastern United States and Texas regions, are the only subspecies of the Elapidae family (Figure 4).2,9 They can be distinguished from the nonvenomous milk snake by their characteristic banding, as coral snakes are patterned in a red-yellow-black band sequence and milk snakes are patterned in a red-black-yellow or white sequence. The differences in appearance of these snakes often is remembered by the phrase “red on yellow kills a fellow.”
Anatomic differences between the Viperidae and Elapidae families, including fang size, placement, and type, as well as venom composition, are directly linked to clinical manifestations of the bites. Viperidae fangs extend from the maxillary bones and are mobile, long, and hollow, making it easy for the snake to control fang movement and envenomation.9 Viperidae snakes are uniquely capable of inflicting puncture wounds without the injection of venom, known as dry bites. In contrast, Elapidae snakes have short, hollow, and fixed fangs, and thus patients can protect themselves by wearing appropriate clothing and covered footwear.9 Currently, identifying the type of snake responsible for the bite relies on visualization of the snake and/or the identification of clinical symptoms of envenomation by a dermatologist.
Clinical Manifestations of Venomous Snake Bites
Clinical manifestations and cutaneous findings often are used to grade the severity of venomous snake bites as well as to dictate treatment procedures. Grade 0 indicates a bite has occurred without envenomation, while grades I to V describe the progression and severity of envenomation.10 Grade I describes minimal erythema and edema around the site (fang marks may or may not be present) and no systemic symptoms. Grade II describes erythema and edema extending up the extremity to the first joint (eg, hand to wrist), pain, some systemic symptoms if there is rapid progression, and potential bleeding at the site. Grade III describes erythema and edema spreading to the second joint in the extremity, pain, and systemic symptoms, including coagulation defects. Grade IV describes erythema and edema of the whole extremity, a rapid reaction and progression following the bite, and risk for compartment syndrome. Grade V includes erythema and edema beyond the extremity and increasing systemic symptoms.10
Local pain and edema, usually on easily accessible or exposed extremities, are the most common clinical symptoms reported following a Viperidae snake bite.11 Due to their capability of producing a dry bite, puncture markings alone do not indicate envenomation. Patients will need to be monitored for several hours for signs of envenomation, which may include swelling, pain, ecchymosis, and indications of systemic manifestation (eg, weakness, dizziness, nausea, severe hypotension, thrombocytopenia).11 Viperidae venom hemorrhagic metalloproteinases act on capillary blood vessels by cleaving basement membrane proteins and allowing for extravasation of fluid into local tissue.12 The inflammatory response produced at the site of envenomation likely is due to the release of tumor necrosis factor á and endogenous matrix metalloprotein.12 There is a higher risk for death associated with bites from rattlesnakes within the Viperidae family because their venom contains a unique neurotoxin that works by blocking presynaptic junctions and causing a range of paralytic symptoms from ptosis to respiratory failure.13
The severity of Elapidae bites is thought to be related to the amount of venom injected, the size of the victim, and the length of the snake. Though clothing may offer protection, envenomation occurs in 75% of coral snake bites and can produce devastating consequences due to the venom content.14 In a retrospective study between 2002 and 2004, 90% of Elapidae snake bite patients (n=82) reported local pain, redness, and paresthesia, while around 7% developed systemic symptoms.15 Elapidae venom primarily is neurotoxic and is thought to spread via lymphatics.16 Delayed reactions are common and may take up to 12 hours to develop. Patients should be monitored, as local reactions may progress to weakness, fasciculations, extremity paralysis, and lastly, respiratory paralysis. Due to the risk for progression, all patients with likely coral snake bites should be given antivenin.8,15,17
Much like the North American coral snake, the venomous snake species Gloydius blomhoffii—referred to as the salmosa or mamushi snake depending on the region of origin (ie, Korea or Japan)—is a frequent source of devastating rural snake bites due to neurotoxins (Figure 5). The species’ slender fangs are thought to directly inject the snake’s potent venom, which contains hemorrhagic toxins and α-neurotoxins and Β-neurotoxins, into the bloodstream; however, the salmosa is considered a viper like the North American cottonmouth and copperhead because of its triangular head shape and hollow fangs, which allow for the accommodation of venom-containing glands and mechanism of venom injection. Salmosa venom shares both Viperidae and Elapidae characteristics. Cutaneous findings such as progressive edema, erythema, and bleeding frequently are reported and are attributed to the proteases and hemorrhagic toxins characteristic of vipers (Figure 6). α-Neurotoxins and Β-neurotoxins, similar to the proteolytic venom of the Elapidae family, are responsible for the unique visual disturbances (binocular diplopia) caused by the salmosa.12,18,19
Treatment
Treating snake bites begins with assessing the patient’s airway, breathing, and circulation, followed by a thorough medical and encounter history (including description of how the bite occurred). Due to the range of Viperidae symptoms, it generally is recommended that patients remove any restrictive clothing or jewelry near the bite and/or over the affected limb or body part, place the affected body part at the level of the heart, and go to the nearest medical facility for prompt care. Historically, empiric antibiotics often were used to prevent wound infections; however, studies have since demonstrated that antibiotics are not necessary and lack efficacy in uncomplicated snake bites.16,20 In a study of 114 pediatric cases from 1995 to 2005, it was determined that most patients could be managed with conservative treatment directed at pain management and swelling reduction via elevation of the affected extremity.6 While conservative management may be all that is needed to care for the majority of cases, one retrospective study from Texas indicated that 70% of pediatric venomous snake bites were treated with either intravenous antibiotics and/or antivenin, highlighting the variability in management and opportunity for improvement.21
Antivenin, specifically antivenin (Crotalidae) polyvalent, is the indicated treatment for Viperidae hemorrhagic or coagulopathic envenomation.13,22 Per guidelines from the World Health Organization, physical examination will yield a grading of the snake bite based on cutaneous findings. Grades III to V are considered moderate to severe and should be given antivenin.23 Physicians should look for signs of progressive injury and coagulopathy, such as increased swelling, bruising, hypotension, or altered mental status.22 Due to the major neurotoxic risks associated with Elapidae venom, all coral snake bites should be treated with antivenin; early intubation and ventilation may be considered.13 Similarly, patients who report a salmosa snake bite require prompt treatment with antivenin and/or cepharanthine, an additive agent to reduce swelling and pain.18 Due to the nature of the neurotoxins contained in the salmosa venom (α-neurotoxin causing postsynaptic inhibition of the neuromuscular junction and Β-neurotoxin inhibiting neurotransmitter release from the presynaptic terminal), anticholinesterases, which work by blocking the enzymatic breakdown of the neurotransmitter acetylcholine, should not be used.19 While bleeding and skin and systemic changes may be reversed by antivenin, visual changes are unlikely to resolve with antivenin administration due to the presynaptic binding of Β-neurotoxin and the blockade of neuromuscular signaling.19
Antivenin should be administered intravenously for the fastest onset of action in a setting suitable for the management of anaphylaxis.24 In situations when the benefits may outweigh the risks (eg, if the patient has had a prior allergic reaction or is not in an environment where they can be watched for at least 8 hours for progression of envenomation or adverse reactions), premedication with an antihistamine or epinephrine may be considered.17 Per the World Allergy Organization and World Health Organization, adverse reactions should be treated with crystalloid solutions and antihistamines, corticosteroids, or epinephrine as indicated.25 In a qualitative analysis of emergency physicians’ attitudes toward antivenin, most expressed treatment hesitancy due to lack of knowledge and experience using the medication.26 When possible, snake bites should thus be managed in consultation with a toxicologist.2
Conclusion
Snake bites and envenomation occur commonly in the United States due to exposure to a variety of venomous snakes in the North American Viperidae and Elapidae families. Appropriate and successful management of snake bites by physicians requires general knowledge of regional snakes, the cutaneous and systemic manifestations of snake bites and envenomation, and current treatment methods.
- Greene SC, Folt J, Wyatt K, et al. Epidemiology of fatal snakebites in the United States 1981-2018. Am J Emerg Med. 2021;45:309-316.
- Wozniak EJ, Wisser J, Schwartz M. Venomous adversaries: a reference to snake identification, field safety, and bite-victim first aid for disaster-response personnel deploying into the hurricaneprone regions of North America. Wilderness Environ Med. 2006; 17:246-266.
- Gummin DD, Mowry JB, Beuhler MC, et al. 2021 annual report of National Poison Data System (NPDS) from America’s Poison Centers: 39th Annual Report. Clin Toxicol (Phila). 2022;60:1381-1643.
- Chotai PN, Watlington J, Lewis S, et al. Pediatric snakebites: comparing patients in two geographic locations in the United States. J Surg Res. 2021;265:297-302.
- Johnson PN, McGoodwin L, Banner W Jr. Utilisation of Crotalidae polyvalent immune fab (ovine) for Viperidae envenomations in children. Emerg Med J. 2008;25:793-798.
- Tadros A, Sharon M, Davis S, et al. Emergency department visits by pediatric patients for snakebites. Pediatr Emerg Care. 2022; 38:279-282.
- Campbell BT, Corsi JM, Boneti C, et al. Pediatric snake bites: lessons learned from 114 cases. J Pediatr Surg. 2008;43:1338-1341.
- Peterson ME. Snake bites: coral snakes. Clin Tech Small Anim Pract. 2006;21:183-186.
- Porter KR. Herpetology. WB Saunders Company; 1972.
- Rana A, Kheora S. Grading and envenomation of the snake bite among the emergency cases in a medical college in rural India. Hmlyn Jr Appl Med Sci Res. 2021;2:33-36.
- Peterson ME. Snake bite: pit vipers. Clin Tech Small Anim Pract. 2006;21:174-182.
- Gutierrez JM, Rucavado A. Snake venom metalloproteinases: their role in the pathogenesis of local tissue damage. Biochimie. 2000;82:841-850.
- Weinstein SA, Dart RC, Staples A, et al. Envenomations: an overview of clinical toxicology for the primary care physician. Am Fam Physician. 2009;80:793-802.
- Kitchens CS, Van Mierop LH. Envenomation by the eastern coral snake (Micrurus fulvius fulvius): a study of 39 victims. JAMA. 1987;258:1615-1618.
- Morgan DL, Borys DJ, Stanford R, et al. Texas coral snake (Micrurus tener) bites. South Med J. 2007;100:152-156.
- Clark RF, Delden BS, Furbee B. The incidence of wound infection following crotalid envenomation. J Emerg Med. 1993; 11:583-586.
- Gold BS, Dart RC, Barish RA. Bites of venomous snakes. N Engl J Med. 2002;347:347-356.
- Hifumi T, Sakai A, Kondo Y, et al. Venomous snake bites: clinical diagnosis and treatment. J Intensive Care. 2015;3:16.
- Igari R, Iseki K, Abe S, et al. Binocular diplopia and ptosis due to snake bite (Agkistrodon blomhoffi “mamushi”) case report. Brain Nerve. 2010;62:273-277.
- Kerrigan KR, Mertz BL, Nelson SJ, et al. Antibiotic prophylaxis for pit viper envenomation: prospective, controlled trial. World J Surg. 1997;21:369-372.
- Correa JA, Fallon SC, Cruz AT, et al. Management of pediatric snake bites: are we doing too much? J Pediatr Surg. 2014;49:1009-1015.
- Dart RC, McNally J. Efficacy, safety and use of snake antivenoms in the United States. Ann Emerg Med. 2001;47:181-188.
- World Health Organization Regional Office for South-East Asia. Guidelines for the Management of Snakebites. 2nd ed. World Health Organization; 2016.
- Clark RF, McKinney PE, Chase PB, et al. Immediate and delayed allergic reactions to Crotalidae polyvalent immune Fab (ovine) antivenom. Ann Emerg Med. 2002;39:671-676.
- World Health Organization. WHO Guidelines for the production, control, and regulation of snake antivenom immunoglobulins. Accessed November 25, 2024. https://extranet.who.int/prequal/vaccines/guidelines-production-control-and-regulation-snake-antivenom-immunoglobulins
- Tupetz A, Barcenas LK, Phillips AJ, et al. Bites study: a qualitive analysis among emergency medicine physicians on snake envenomation management practices. PloS One. 2022;17:E0262215.
- Greene SC, Folt J, Wyatt K, et al. Epidemiology of fatal snakebites in the United States 1981-2018. Am J Emerg Med. 2021;45:309-316.
- Wozniak EJ, Wisser J, Schwartz M. Venomous adversaries: a reference to snake identification, field safety, and bite-victim first aid for disaster-response personnel deploying into the hurricaneprone regions of North America. Wilderness Environ Med. 2006; 17:246-266.
- Gummin DD, Mowry JB, Beuhler MC, et al. 2021 annual report of National Poison Data System (NPDS) from America’s Poison Centers: 39th Annual Report. Clin Toxicol (Phila). 2022;60:1381-1643.
- Chotai PN, Watlington J, Lewis S, et al. Pediatric snakebites: comparing patients in two geographic locations in the United States. J Surg Res. 2021;265:297-302.
- Johnson PN, McGoodwin L, Banner W Jr. Utilisation of Crotalidae polyvalent immune fab (ovine) for Viperidae envenomations in children. Emerg Med J. 2008;25:793-798.
- Tadros A, Sharon M, Davis S, et al. Emergency department visits by pediatric patients for snakebites. Pediatr Emerg Care. 2022; 38:279-282.
- Campbell BT, Corsi JM, Boneti C, et al. Pediatric snake bites: lessons learned from 114 cases. J Pediatr Surg. 2008;43:1338-1341.
- Peterson ME. Snake bites: coral snakes. Clin Tech Small Anim Pract. 2006;21:183-186.
- Porter KR. Herpetology. WB Saunders Company; 1972.
- Rana A, Kheora S. Grading and envenomation of the snake bite among the emergency cases in a medical college in rural India. Hmlyn Jr Appl Med Sci Res. 2021;2:33-36.
- Peterson ME. Snake bite: pit vipers. Clin Tech Small Anim Pract. 2006;21:174-182.
- Gutierrez JM, Rucavado A. Snake venom metalloproteinases: their role in the pathogenesis of local tissue damage. Biochimie. 2000;82:841-850.
- Weinstein SA, Dart RC, Staples A, et al. Envenomations: an overview of clinical toxicology for the primary care physician. Am Fam Physician. 2009;80:793-802.
- Kitchens CS, Van Mierop LH. Envenomation by the eastern coral snake (Micrurus fulvius fulvius): a study of 39 victims. JAMA. 1987;258:1615-1618.
- Morgan DL, Borys DJ, Stanford R, et al. Texas coral snake (Micrurus tener) bites. South Med J. 2007;100:152-156.
- Clark RF, Delden BS, Furbee B. The incidence of wound infection following crotalid envenomation. J Emerg Med. 1993; 11:583-586.
- Gold BS, Dart RC, Barish RA. Bites of venomous snakes. N Engl J Med. 2002;347:347-356.
- Hifumi T, Sakai A, Kondo Y, et al. Venomous snake bites: clinical diagnosis and treatment. J Intensive Care. 2015;3:16.
- Igari R, Iseki K, Abe S, et al. Binocular diplopia and ptosis due to snake bite (Agkistrodon blomhoffi “mamushi”) case report. Brain Nerve. 2010;62:273-277.
- Kerrigan KR, Mertz BL, Nelson SJ, et al. Antibiotic prophylaxis for pit viper envenomation: prospective, controlled trial. World J Surg. 1997;21:369-372.
- Correa JA, Fallon SC, Cruz AT, et al. Management of pediatric snake bites: are we doing too much? J Pediatr Surg. 2014;49:1009-1015.
- Dart RC, McNally J. Efficacy, safety and use of snake antivenoms in the United States. Ann Emerg Med. 2001;47:181-188.
- World Health Organization Regional Office for South-East Asia. Guidelines for the Management of Snakebites. 2nd ed. World Health Organization; 2016.
- Clark RF, McKinney PE, Chase PB, et al. Immediate and delayed allergic reactions to Crotalidae polyvalent immune Fab (ovine) antivenom. Ann Emerg Med. 2002;39:671-676.
- World Health Organization. WHO Guidelines for the production, control, and regulation of snake antivenom immunoglobulins. Accessed November 25, 2024. https://extranet.who.int/prequal/vaccines/guidelines-production-control-and-regulation-snake-antivenom-immunoglobulins
- Tupetz A, Barcenas LK, Phillips AJ, et al. Bites study: a qualitive analysis among emergency medicine physicians on snake envenomation management practices. PloS One. 2022;17:E0262215.
Key Features of North American Venomous Snake Bites
Key Features of North American Venomous Snake Bites
PRACTICE POINTS
- Venomous snake bites require prompt medical attention and assessment of symptoms to determine the optimal course of management and need for antivenin.
- Envenomation may cause may cause discoloration and swelling of the skin as well as thrombotic or paralytic changes.

Erythematous Annular Scaly Plaques on the Upper Chest
Erythematous Annular Scaly Plaques on the Upper Chest
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.

Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.
Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
THE DIAGNOSIS: Tinea Corporis
Due to the scaly and acute nature of the rash, a potassium hydroxide (KOH) preparation was performed, and hyphal elements were floridly present. After further questioning, the patient reported finding a stray kitten a few weeks before the onset of the eruption and shared a picture of it lying on her chest in the area corresponding with the main distribution of the rash (Figure). Based on the patient’s personal history and the positive KOH preparation, a diagnosis of tinea corporis was made. She was immediately started on fluconazole 300 mg once weekly for 4 weeks and naftifine gel 1%, which she used for 6 to 8 weeks with complete resolution of the eruption.
Tinea corporis is a dermatophyte infection that typically affects exposed areas of the skin such as the chest, arms, and legs. Spread via human-to-human contact, Trichophyton rubrum is the most common cause worldwide. The second most common is Trichophyton mentagrophytes, which is spread through animal-to-human contact.1,2
Symptoms of tinea corporis usually appear 1 to 3 weeks after exposure and manifest as itchy scaly papules that spread outward, forming annular, circinate, and petaloid erythematous plaques with central clearing. The condition most commonly is diagnosed through the examination of scale from the affected area using a KOH preparation, which will reveal hyphae when positive.2-4 Cultures are the gold standard for identifying dermatophyte species,5 but results can take several weeks. Biopsy also can confirm the diagnosis by showing the presence of hyphae in the stratum corneum, which can be highlighted using periodic acid–Schiff or silver stains.3
Topical antifungals are the first-line treatment for cutaneous dermatophyte infections.3-5 The most effective topical therapies are allylamines and azoles, which work by inhibiting the growth of the fungus. Allylamines are more effective than azoles due to their fungicidal properties and ability to penetrate the skin more effectively.6,7 Topical medications should be applied at least 2 cm beyond the infected area for 2 to 4 weeks or until the infection has cleared.3 Systemic antifungals may be necessary in more complicated cases.
It is important to consider a broad differential and take into consideration the distribution of the plaques, the patient’s history, and other clinical features when differentiating tinea corporis from other conditions. Erythema annulare centrifugum more often presents as nonpruritic annular plaques with a trailing scale instead of a leading scale seen in tinea corporis. Biopsy exhibits a dense, perivascular, lymphocytic infiltrate in superficial vessels, resembling a coat sleeve.3,8 Pemphigus foliaceous can manifest with painful crusted scaly plaques and vesicles in a seborrheic distribution. Biopsy reveals subcorneal acantholytic vesicles and can be confirmed on direct immunofluorescence.3,8 Subacute cutaneous lupus erythematosus presents with annular plaques that often are symmetric and most prominent in sun-exposed areas, sparing the face.3,9,10 It can be associated with other autoimmune conditions as well as medications such as thiazides, terbinafine, and calcium channel blockers. Additionally, 76% to 90% of patients are Ro/SSA antibody positive.3 Biopsy often demonstrates follicular plugging, perivascular and periadnexal lymphocytic infiltrates, and mucin.3,10 Lastly, pityriasis rosea typically begins with a herald patch, followed by a widespread rash that often appears in a Christmas tree distribution.3
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51 (suppl 4):2-15. doi: 10.1111 /j.1439-0507.2008.01606.x
- Yee G, Al Aboud AM. Tinea corporis. 2022 Aug 8. In: StatPearls [Internet]. StatPearls Publishing; 2023
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Elsevier; 2018.
- Diseases resulting from fungal and yeast. In: James WD, Berger TG, Elston DM, et al, eds. Andrews’ Diseases of The Skin: Clinical Dermatology. 12th ed. Elsevier; 2016: 289-290.
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6 . doi:10.7573/dic.2020-5-6
- El-Gohary M, van Zuuren EJ, Fedorowicz Z, et al. Topical antifungal treatments for tinea cruris and tinea corporis. Cochrane Database Syst Rev. 2014;2014:CD009992. doi:10.1002/14651858 .CD009992.pub2
- Wolverton SE. Comprehensive Dermatologic Drug Therapy. 4th ed. Elsevier; 2018.
- Burgdorf W. Erythema annulare centrifugum and other figurate erythemas. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. McGraw-Hill; 2008: 366-368.
- Modi GM, Maender JL, Coleman N, et al. Tinea corporis masquerading as subacute cutaneous lupus erythematosus. Dermatol Online J. 2008;14:8.
- Stavropoulos PG, Goules AV, Avgerinou G, et al. Pathogenesis of subacute cutaneous lupus erythematosus. J Eur Acad Dermatol Venereol. 2008;22:1281.
Erythematous Annular Scaly Plaques on the Upper Chest
Erythematous Annular Scaly Plaques on the Upper Chest
A 60-year-old woman with a history of keratinocyte carcinomas, hypertension, diabetes mellitus, and anxiety presented to the dermatology department with a widespread rash of more than 2 weeks’ duration. The patient had tried 1 to 2 days of self-treatment with triamcinolone cream that she had previously been prescribed for an unknown dermatitis and zinc oxide cream, which caused considerable inflammation of the rash and prompted her to discontinue use. She could not recall any recent use of new personal care products or medications or eating any new foods. She also denied any recent yard work, known arthropod bites, illnesses, prolonged sun exposure, or constitutional symptoms. Her medications included metformin, hydrochlorothiazide, losartan, and sertraline. She also reported taking daily supplements of vitamins D, K, and C as well as acetaminophen and ibuprofen as needed. Physical examination revealed several 2- to 4-cm, erythematous, annular, circinate, petaloid plaques with scale mostly on photodistributed areas of the central anterior chest, neck, lower cheeks, and chin as well as a few scattered lesions with similar morphology on the arms, lower abdomen, left buttock, and back.

Historical Perspectives on Hair Care and Common Styling Practices in Black Women
Historical Perspectives on Hair Care and Common Styling Practices in Black Women
Patients often ask dermatologists how to best care for their specific hair type; however, there are no formal recommendations that apply to the many different hair care practices utilized by Black patients, as hair types in this community can range from wavy to tightly coiled.1 Understanding the the history of hair care in those of African ancestry and various styling practices in this population is necessary to adequately counsel patients and gain trust in the doctor-patient relationship. In this article, we provide an overview of hair care recommendations based on common styling practices in Black women.
A PubMed search of articles indexed for MEDLINE using the terms Black hair care, African American hair management, hair loss prevention, hair care practices, natural hair, natural-hair styles, alopecia, hairdressing, hair breakage, hair fragility, heat-stressed hair, traction alopecia, and natural hair care yielded 305 results; 107 duplicates were identified and removed, leaving 198 articles to be screened for eligibility (ie, English-language studies created in the past 15 years). Sixty-eight full-text articles were screened against the exclusion criteria, which included case reports and case series, articles not focused on Afro-textured hair, and cancer-related hair loss. Three additional fulltext articles were identified via resources from Wayne State University library (Detroit, Michigan) that were not available on PubMed. A total of 29 full-text articles were included in our review.
Background on Hair Care and Styling in African Populations
It is difficult to understand the history of hair in those of African ancestry in the United States.2 Prior to slavery, hair styling was considered a way of identification, classification, and communication as well as a medium through which to connect with the spiritual world in many parts of Africa. Hair-styling practices in Africa included elaborate cornrows, threading, and braiding with many accessories. Notable hair-styling products included natural butters, herbs, and powders to assist with moisture retention. Scarves also were used during this time for ceremonies or protection.3 During the mass enslavement of African populations and their transportation to the Americas by Europeans, slaveholders routinely cut off all the hair of both men and women in order to objectify and erase the culture of African hair styling passed down through generations.4,5 Hair texture then was weaponized to create a caste system in plantation life, in which Black slaves with straight hair textures were granted the “privilege” of domestic work, while those with kinky hair were relegated to arduous manual labor in the fields.4 Years later, during the 1800s, laws were enacted in the United States to prohibit Black women from wearing tightly coiled natural hair in public places.5 Over the next few centuries from the 1800s to the early 2000s, various hair-styling trends such as the use of hot combs, perms, afros, and Jheri curls developed as a means for Black individuals to conform to societal pressure to adopt more European features; however, as time progressed, afros, braids, locs, and natural hair would become more dominant as statements against these same societal pressures.5
The natural hair movement, which emerged in the United States in the 2000s, encouraged Black women to abandon the use of toxic chemical hair straighteners, cultivate healthier hair care practices, disrupt Eurocentric standards of wearing straightened hair, and facilitate self-definition of beauty ideals from the Civil Rights Movement of the 1960s.4,5 It is estimated that between 30% and 70% of all Black women in the United States wear natural hair, including 79% of millennial Black women younger than 30 years6; however, several new trends such as wigs and weaves have grown in popularity since the early 2000s due to mainstream pop culture and improvements in creating natural hairlines.7,8
Key Features of Afro-Textured Hair
Individuals of African descent have the most diverse hair texture phenotypes, ranging from straight to tightly coiled.9 Although hair is chemically similar across various racial groups, differences are noted mainly in the shape of the hair shaft, with elliptical and curved shapes seen in Afrotextured hair. These differences yield more tightly curled strands than in other hair types; however, these features also contribute to fragility, as it creates points of weakness and decreases the tensile strength of the hair shaft.10 This inherent fragility leads to higher rates of hair breakage as well as lower moisture content and slower growth rates, which is why Afro-textured hair requires special care.9
Afro-textured hair generally falls into 2 main categories of the Andre Walker hair typing system: 4A-4C and 3A-3C.11 In the 4A-4C category, hair is described as coily or kinky. Common concerns related to this hair type include dryness and brittleness with increased susceptibility to breakage. The 3A-3C category is described as loose to corkscrew curls, with a common concern of dryness.11,12 Additionally, Loussouarn et al13 established a method to further define natural hair curliness using curve diameter and curl meters on glass plates to measure the curvature of hair strands. This method allows for assessing diversity and range of curliness within various races without relying on ethnic origin.13
Common Hair Care Practices
A description of each hair type and recommended styling practices with their levels of evidence can be found in the eTable.
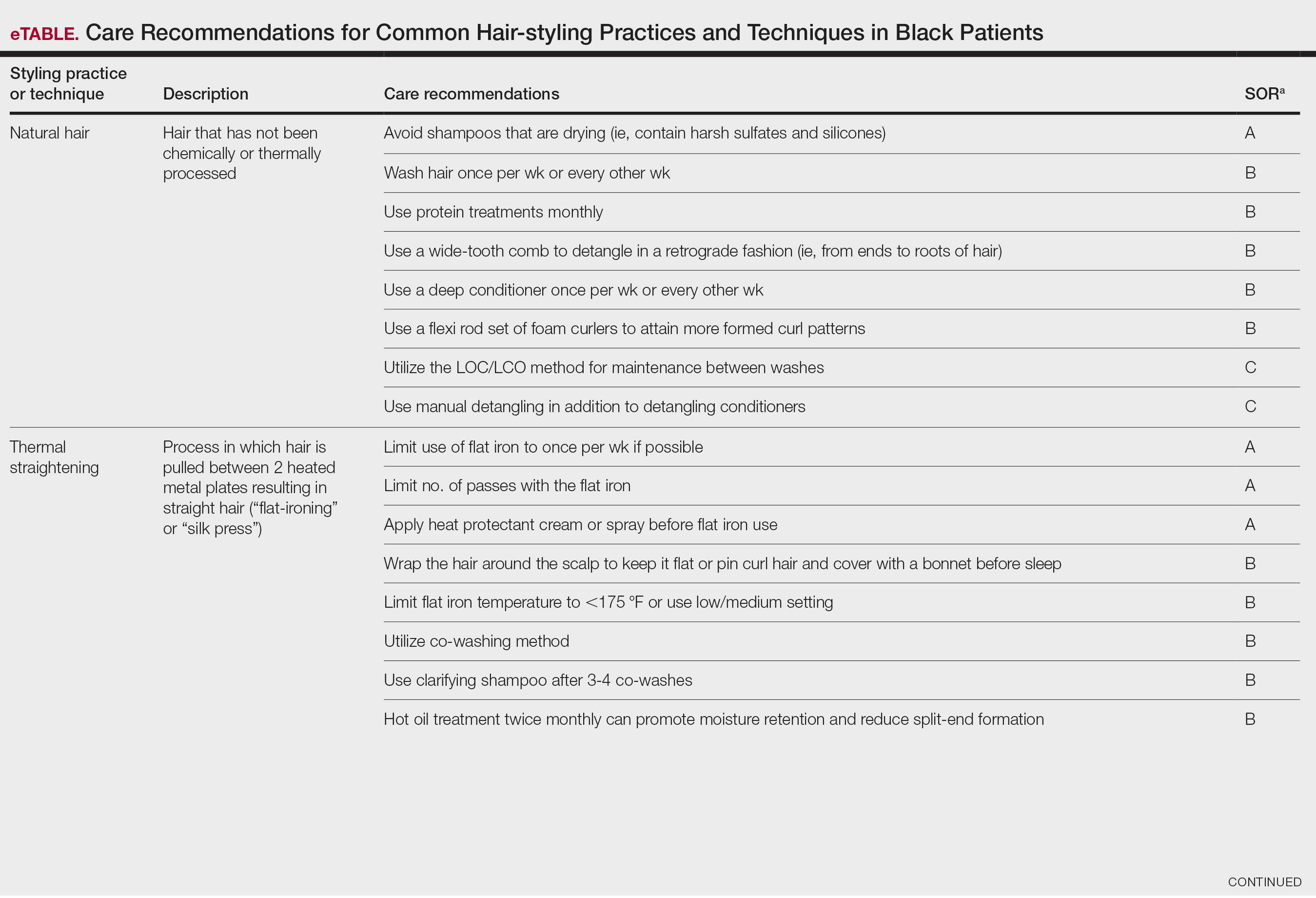
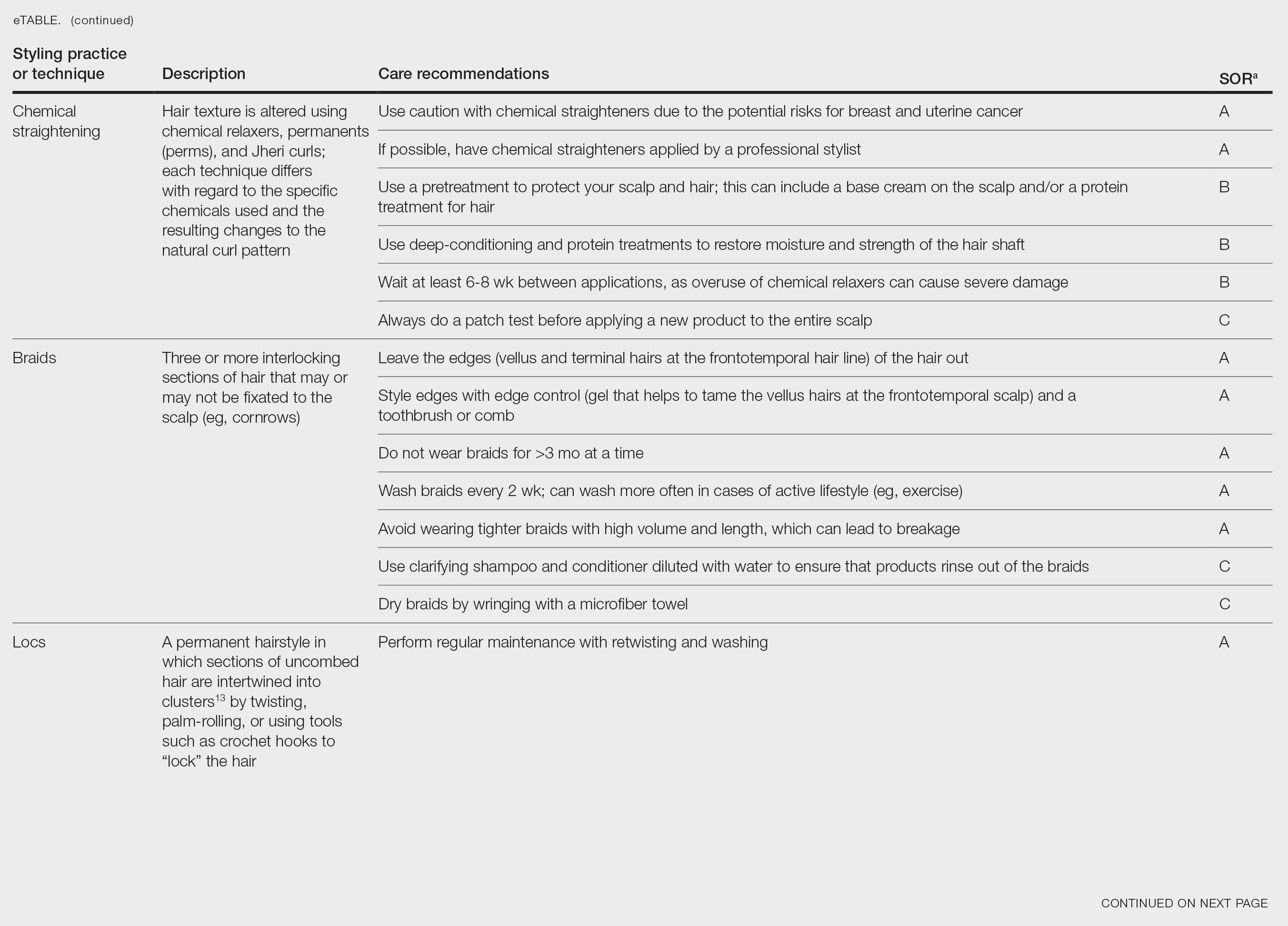
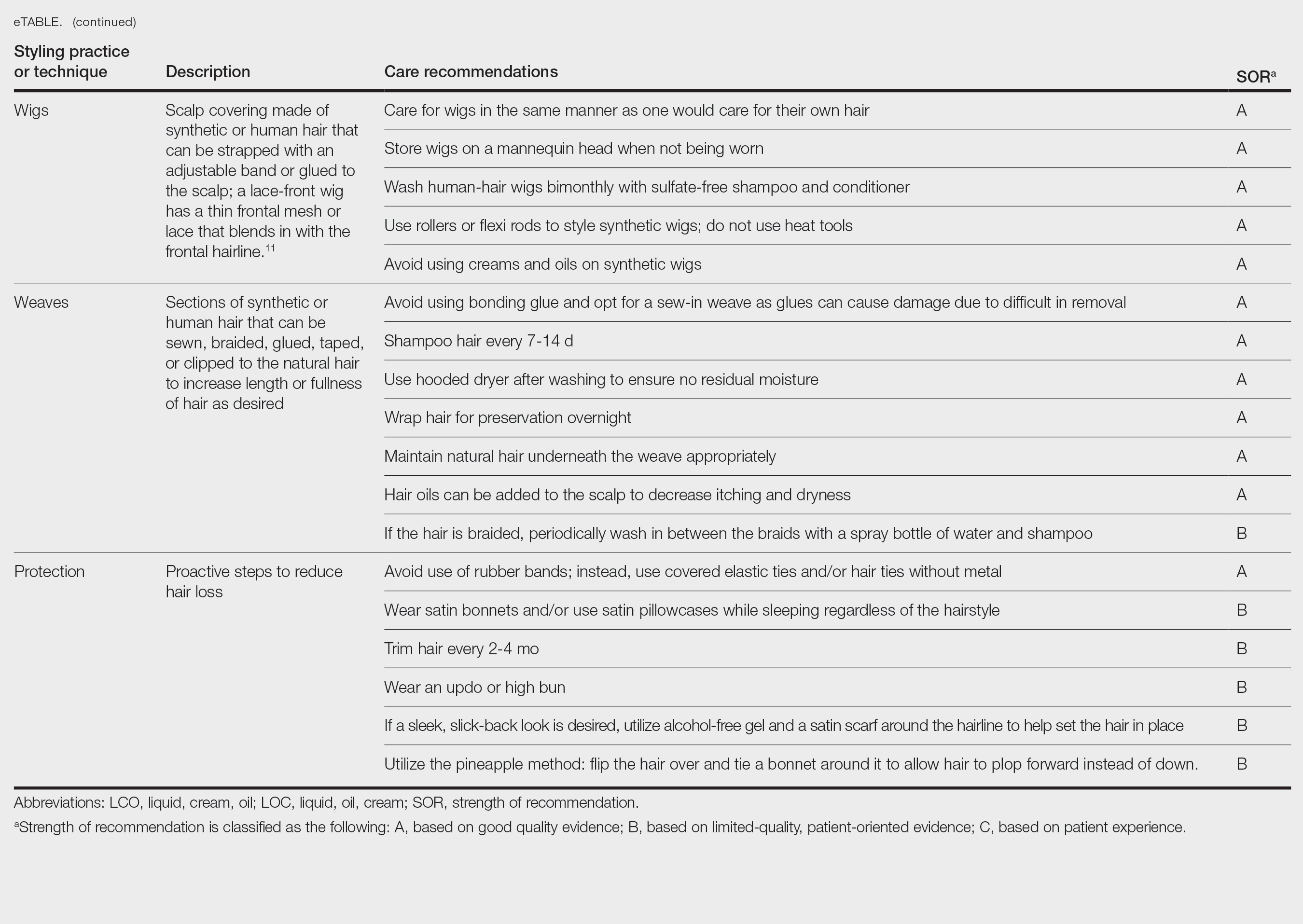
Natural Hair—Natural hair is classified as hair that has not been chemically changed by perms, heat, or other straightening treatments.12,14 For natural hair, retaining the moisture of the hair shaft should be the main focus, as moisture loss leads to considerable dryness.14 Generally, it is recommended to wash natural hair once per week or every other week; however, this can change based on hair length and oil production on the scalp. Washing daily may be ideal for shorter hair and monthly for longer hair to help prevent product build-up that can have a drying effect.15 Avoid shampoos that are drying (eg, sulfate and silicone products). The co-washing method also can be utilized, which entails washing the hair with a conditioning cleanser instead of shampoo and conditioner. However, this technique is not meant to completely replace shampoo.16 In fact, a clarifying shampoo is recommended after co-washing 3 or 4 times.16 The use of a hot oil treatment twice per month can promote moisture retention and reduce split-end formation.17 For maintenance between washes, many utilize the liquid, oil, cream (LOC) or liquid, cream, oil (LCO) methods, which describe regimens that utilize water, an oil of choice, and cream such as shea butter to lock in moisture.18 This method can be used as often as needed for dry hair.
Due to the susceptibility of Afro-textured hair to tangle and knot, using a wide-tooth comb, detangling brush, or detangling conditioners is a grade B recommendation for care (eTable). Though not widely documented in the literature, many of our patients have had anecdotal success detangling their hair simply by pulling hair strands apart by hand or “finger detangling” as well as using wide-tooth combs. Although both hair types are healthier in their natural states, kinky hair (type 4A-4C) is extremely fragile and more difficult to manage than less kinky hair (type 3A-3C).18
Special care is needed when detangling due to strands being weaker when wet.19 Detangling should be performed in a retrograde fashion. Deep conditioning can aid in moisture retention and should be performed weekly or biweekly.17-20 Depending on the health of the hair, protein treatments can be considered on a monthly basis to help preserve the cuticle. Styling with braids, twists, or other protective styles can then be completed on an individual basis.
Thermal Straightening—A blowout involves straightening the hair after a wash with the use of a hair dryer.21 This common hair-styling method does not employ the use of chemicals beyond light hair oils and heat-protectant creams or sprays, typically resulting in a less kinky afro or semi-straight hair. Thermal straightening utilizes heat to temporarily straighten hair strands. Flat irons with heated metal plates then can be used after blow-drying the hair to fully straighten and smooth the strands. These processes combined commonly are known as a silk press.21-22
For thermally straightened hair, it is recommended to either wrap the hair around the scalp to keep it flat or pin curl the hair and cover with a bonnet to sleep. Safe straightening techniques with the use of a flat iron include setting the temperature no higher than 175 °F or a low/medium setting while also limiting use to once per week if possible.23 The number of passes of the flat iron also should be limited to 1 to 2 to reduce breakage. A heat-protectant cream or spray also can be applied to the hair before flat ironing to minimize damage. Applying heat protectant to the hair prior to styling will help minimize heat damage by distributing the heat along the hair fiber surface, avoiding water boiling in the hair shaft and the development of bubble hair leading to damage.24
Chemical Straightening—Similar to how relaxers, perms, and Jheri curl treatments chemically modify hair texture using distinct chemicals yielding different curl patterns, the Brazilian blowout similarly straightens hair using a hair dryer and chemicals applied to hair strands after washing.21-24 Relaxers utilize sodium or guanidine hydroxide for straightening, perms use ammonium thioglycolate for curling, and Jheri curl treatments employ thioglycolates or mercaptans for defined curls. However, these treatments generally are cautioned against due to potential hair damage and recent associations with uterine and breast cancer in Black women. Research has suggested that endocrine disrupters in these products, especially those marketed to Black women, contribute to hormone-related disease processes.25,26 One study found higher concentrations of alkylphenols, the fragrance marker diethyl phthalate, and parabens in relaxers27; however, more research is needed to determine specific chemicals associated with these cancers.
Braids and Locs—Braiding is a technique that involves interlocking 3 or more sections of hair that may or may not be fixated to the scalp like a cornrow,11 and one can utilize extensions or natural hair depending on the desired outcome. Intended for long-term wear (ie, weeks to months), braids minimize breakage and reduce daily styling needs. Two popular styles—cornrows and individual braids—differ in preparation and weaving techniques. Cornrows are an Afro-centric style involving uniform, tightly woven braids that are close to the scalp, creating distinct patterns. Conversely, individual braids weave separate hair sections, offering diverse styling possibilities. Braiding practices should exclude hairline edges—often termed baby hairs—to prevent traction alopecia. Minimal use of edge gel, which helps to tame the vellus hairs at the frontotemporal scalp, as well as mindful weave volume, weight, and length are recommended to avert breakage. Braids that cause pain are too tight, can damage hair, and may cause traction alopecia.11 Braids should not be worn for longer than 3 months at a time and require biweekly washing with diluted shampoo and conditioner. Proper drying by wringing the hair with a microfiber towel is essential to avoid frizz and mold formation.
Locs are a low-maintenance hairstyle considered permanent until cut.28 This style involves twisting, palm rolling, or using tools such as crochet hooks to “lock” the hair. Regular maintenance with retwisting and cleaning is vital for loc health. Increased weight and tight twisting of locs can cause damage to the scalp and hair strands; however, locs are known to increase hair volume over time, often due to the accumulation of hairs that would otherwise have been shed in the telogen phase.28
Wigs and Weaves—Wigs consist of synthetic or human hair that can be strapped to the head with an adjustable band or glued to the scalp depending on the desired style.29 Wigs are removed daily, which allows for quick access to hair for cleansing and moisturizing. In contrast, weaves typically are sewn into the natural hair, which may make it difficult to reach the scalp for cleansing, leading to dryness and product build-up.29 Notably, there is evidence of a relationship between long-term use of weaves and traction alopecia.30
Wigs can have a fully synthetic hair line or lace hair line and can range from very affordable to expensive. When applied correctly, both styles offer an easy way to cover and protect the natural hair by reducing the amount of physical trauma related to daily hair styling. A lace-front wig contains a frontal thin mesh or lace that camouflages the natural frontal hairline.29,30 A risk of lace-front wigs is that they can cause friction alopecia secondary to repeated use of adhesives and repeated friction against the hairline. Generally, wigs and weaves should be cared for as one would care for one’s own hair.
Hair Care in Black Children—Children’s hair care begins with washing the hair and scalp with shampoo, applying conditioner, and detangling as needed.31 After rinsing out the conditioner, a leave-in conditioner can assist with moisture retention and further detangling. The hair is then styled, either wet or dry. Recommendations for hair care practices in Black children include loose hairstyles that do not strain hair roots and nightly removal of root-securing accessories (eg, barrettes, elastic hairbeads). Frequent cornrow styling and friction on chemically straightened hair were identified by a survey as considerable traction alopecia risk factors.32 Thus, educating caregivers on appropriate hair-grooming practices for children is important.
Hair Protection—Proactive steps to reduce hair loss include wearing satin bonnets and/or using satin pillowcases while sleeping regardless of hairstyle. Although evidence is limited, it is thought that satin and silk allow the hair to retain its moisture and natural oils, preventing breakage and friction.33,34 Frequent hair trimming every 2 to 4 months can reduce breakage when doing thermal treatments.35,36 When prolonged or repetitive styles are used, it is encouraged to give the hair a break between styles to recover from the repeated stress. Wearing an intermittent updo or high bun—a hairstyle in which the hair is pulled upward—can prevent breakage by reducing heavy strain on the hair; however, it is important to avoid the use of rubber bands due to friction and risk for tangling of hair strands. Instead, the use of covered elastic ties and/or those without metal is preferred.11 Alternatively, if a polished and neat appearance with slicked-back hair is desired, the practice of tautly pulling the hair is not recommended. Instead, use of an alcohol-free gel is suggested along with a satin scarf wrapped around the hairline to facilitate the setting of the hair in place.11
A common practice to preserve curly hairstyles while sleeping is known as the pineapple method, which protects the hair and aids in preserving the freshness and style of the curls.37 It consists of a loosely tied high ponytail at the top of the head allowing the curls to fall forward. This minimizes frizz and prevents the curls from forming knots.
Conclusion
Hair care recommendations in Black women can be complex due to a wide range of personal care preferences and styling techniques in this population. While evidence in the literature is limited, it still is important for dermatologists to be familiar with the different hair care practices utilized by Black women so they can effectively counsel patients and improve hair health. Knowledge of optimal hair care practices can aid in the prevention of common hair disorders that disproportionately affect this patient population, such as traction alopecia and trichorrhexis nodosa or breakage.
- Hall RR, Francis S, Whitt-Glover M, et al. Hair care practices as a barrier to physical activity in African American women. JAMA Dermatol. 2013;149:310-314. doi:10.1001/jamadermatol.2013.1946
- Johnson T, Bankhead T. Hair it is: examining the experiences of Black women with natural hair. Open J Soc Sci. 2014;02:86-100. doi:10.4236/jss.2014.21010
- Byrd AD, Tharps LL. Hair Story: Untangling the Roots of Black Hair in America. 2nd ed. St Martin’s Griffin; 2014.
- Mbilishaka AM, Clemons K, Hudlin M, et al. Don’t get it twisted: untangling the psychology of hair discrimination within Black communities. Am J Orthopsychiatry. 2020;90:590-599. doi:10.1037 /ort0000468
- Khumalo NP. On the history of African hair care: more treasures await discovery. J Cosmet Dermatol. 2008;7:231. doi:10.1111/j.1473- 2165.2008.00396.x
- Johnson AM, Godsil RD, MacFarlane J, et al. The “good hair” study: explicit and implicit attitudes toward Black women’s hair. Perception Institute. February 2017. Accessed February 11, 2025. https://perception.org/publications/goodhairstudy/
- Haskin A, Aguh C. All hairstyles are not created equal: what the dermatologist needs to know about black hairstyling practices and the risk of traction alopecia (TA). J Am Acad Dermatol. 2016;75:606-611. doi:10.1016/j.jaad.2016.02.1162
- Roseborough IE, McMichael AJ. Hair care practices in African- American patients. Semin Cutan Med Surg. 2009;28:103-108. doi:10.1016/j.sder.2009.04.007
- Menkart J Wolfram LJ Mao I. Caucasian hair, Negro hair and wool: similarities and differences. J Soc Cosmet Chem. 1996;17:769-787.
- Crawford K, Hernandez C. A review of hair care products for black individuals. Cutis. 2014;93:289-293.
- Mayo TT, Callender VD. The art of prevention: it’s too tight-loosen up and let your hair down. Int J Womens Dermatol. 2021;7:174-179. doi:10.1016/j.ijwd.2021.01.019
- De Sá Dias TC, Baby AR, Kaneko TM, et al. Relaxing/straightening of Afro-ethnic hair: historical overview. J Cosmet Dermatol. 2007;6:2-5. doi:10.1111/j.1473-2165.2007.00294.x
- Loussouarn G, Garcel AL, Lozano I, et al. Worldwide diversity of hair curliness: a new method of assessment. Int J Dermatol. 2007;46 (suppl 1):2-6. doi:10.1111/j.1365-4632.2007.03453.x
- Barba C, Mendez S, Marti M, et al. Water content of hair and nails. Thermochimica Acta. 2009;494:136-140. doi:10.1016/j.tca.2009.05.005
- Gray J. Hair care and hair care products. Clin Dermatol. 2001;19:227-236. doi:10.1016/s0738-081x(00)00133-4
- Gavazzoni Dias MFR. Pro and contra of cleansing conditioners. Skin Appendage Disord. 2019;5:131-134. doi:10.1159/000493588
- Gavazzoni Dias MFR. Hair cosmetics: an overview. Int J Trichology. 2015;7:2-15. doi:10.4103/0974-7753.153450
- Beal AC, Villarosa L, Abner A. The Black Parenting Book. 1999.
- Davis-Sivasothy A. The Science of Black Hair: A Comprehensive Guide to Textured Care. Saga Publishing; 2011.
- Robbins CR. The Physical Properties and Cosmetic Behavior of Hair. In: Robbins CR. Chemical and Physical Behavior of Human Hair. 3rd ed. Springer Nature; 1994:299-370. doi:10.1007/978-1-4757-3898-8_8
- Weathersby C, McMichael A. Brazilian keratin hair treatment: a review. J Cosmet Dermatol. 2013;12:144-148. doi:10.1111/jocd.12030
- Barreto T, Weffort F, Frattini S, et al. Straight to the point: what do we know so far on hair straightening? Skin Appendage Disord. 2021;7:265-271. doi:10.1159/000514367
- Dussaud A, Rana B, Lam HT. Progressive hair straightening using an automated flat iron: function of silicones. J Cosmet Sci. 2013;64:119-131.
- Zhou Y, Rigoletto R, Koelmel D, et al. The effect of various cosmetic pretreatments on protecting hair from thermal damage by hot flat ironing. J Cosmet Sci. 2011;62:265-282.
- Chang CJ, O’Brien KM, Keil AP, et al. Use of straighteners and other hair products and incident uterine cancer. J Natl Cancer Inst. 2022;114:1636-1645. doi:10.1093/jnci/djac165
- White AJ, Gregoire AM, Taylor KW, et al. Adolescent use of hair dyes, straighteners and perms in relation to breast cancer risk. Int J Cancer. 2021;148:2255-2263. doi:10.1002/ijc.33413
- Helm JS, Nishioka M, Brody JG, et al. Measurement of endocrine disrupting and asthma-associated chemicals in hair products used by Black women. Environ Res. 2018;165:448-458.
- Asbeck S, Riley-Prescott C, Glaser E, et al. Afro-ethnic hairstyling trends, risks, and recommendations. Cosmetics. 2022;9:17. doi:10.3390 /cosmetics9010017
- Saed S, Ibrahim O, Bergfeld WF. Hair camouflage: a comprehensive review. Int J Womens Dermatol. 2016;2:122-127. doi:10.1016 /j.ijwd.2016.09.002
- Billero V, Miteva M. Traction alopecia: the root of the problem. Clin Cosmet Investig Dermatol. 2018;11:149-159. doi:10.2147/CCID .S137296
- Jones NL, Heath CR. Hair at the intersection of dermatology and anthropology: a conversation on race and relationships. Pediatr Dermatol. 2021;38(suppl 2):158-160. doi:10.1111/pde.14721
- Rucker Wright D, Gathers R, Kapke A, et al. Hair care practices and their association with scalp and hair disorders in African American girls. J Am Acad Dermatol. 2011;64:253-262. doi:10.1016/j.jaad.2010.05.037
- Carefoot H. Silk pillowcases for better hair and skin: what to know. The Washington Post. April 6, 2021. Accessed February 10, 2025. https://www.washingtonpost.com/lifestyle/wellness/silk-pillowcases-hair-skin-benefits-myths/2021/04/05/a7dcad7c-866a-11eb-82bc-e58213caa38e_story.html
- Samrao A, McMichael A, Mirmirani P. Nocturnal traction: techniques used for hair style maintenance while sleeping may be a risk factor for traction alopecia. Skin Appendage Disord. 2021;7:220-223. doi:10.1159/000513088
- Callender VD, McMichael AJ, Cohen GF. Medical and surgical therapies for alopecias in black women. Dermatol Ther. 2004;17:164-176. doi:10.1111/j.1396-0296.2004.04017.x
- McMichael AJ. Hair breakage in normal and weathered hair: focus on the Black patient. J Investig Dermatol Symp Proc. 2007;12:6-9. doi:10.1038/sj.jidsymp.5650047
- Bosley RE, Daveluy S. A primer to natural hair care practices in black patients. Cutis. 2015;95:78-80,106.
Patients often ask dermatologists how to best care for their specific hair type; however, there are no formal recommendations that apply to the many different hair care practices utilized by Black patients, as hair types in this community can range from wavy to tightly coiled.1 Understanding the the history of hair care in those of African ancestry and various styling practices in this population is necessary to adequately counsel patients and gain trust in the doctor-patient relationship. In this article, we provide an overview of hair care recommendations based on common styling practices in Black women.
A PubMed search of articles indexed for MEDLINE using the terms Black hair care, African American hair management, hair loss prevention, hair care practices, natural hair, natural-hair styles, alopecia, hairdressing, hair breakage, hair fragility, heat-stressed hair, traction alopecia, and natural hair care yielded 305 results; 107 duplicates were identified and removed, leaving 198 articles to be screened for eligibility (ie, English-language studies created in the past 15 years). Sixty-eight full-text articles were screened against the exclusion criteria, which included case reports and case series, articles not focused on Afro-textured hair, and cancer-related hair loss. Three additional fulltext articles were identified via resources from Wayne State University library (Detroit, Michigan) that were not available on PubMed. A total of 29 full-text articles were included in our review.
Background on Hair Care and Styling in African Populations
It is difficult to understand the history of hair in those of African ancestry in the United States.2 Prior to slavery, hair styling was considered a way of identification, classification, and communication as well as a medium through which to connect with the spiritual world in many parts of Africa. Hair-styling practices in Africa included elaborate cornrows, threading, and braiding with many accessories. Notable hair-styling products included natural butters, herbs, and powders to assist with moisture retention. Scarves also were used during this time for ceremonies or protection.3 During the mass enslavement of African populations and their transportation to the Americas by Europeans, slaveholders routinely cut off all the hair of both men and women in order to objectify and erase the culture of African hair styling passed down through generations.4,5 Hair texture then was weaponized to create a caste system in plantation life, in which Black slaves with straight hair textures were granted the “privilege” of domestic work, while those with kinky hair were relegated to arduous manual labor in the fields.4 Years later, during the 1800s, laws were enacted in the United States to prohibit Black women from wearing tightly coiled natural hair in public places.5 Over the next few centuries from the 1800s to the early 2000s, various hair-styling trends such as the use of hot combs, perms, afros, and Jheri curls developed as a means for Black individuals to conform to societal pressure to adopt more European features; however, as time progressed, afros, braids, locs, and natural hair would become more dominant as statements against these same societal pressures.5
The natural hair movement, which emerged in the United States in the 2000s, encouraged Black women to abandon the use of toxic chemical hair straighteners, cultivate healthier hair care practices, disrupt Eurocentric standards of wearing straightened hair, and facilitate self-definition of beauty ideals from the Civil Rights Movement of the 1960s.4,5 It is estimated that between 30% and 70% of all Black women in the United States wear natural hair, including 79% of millennial Black women younger than 30 years6; however, several new trends such as wigs and weaves have grown in popularity since the early 2000s due to mainstream pop culture and improvements in creating natural hairlines.7,8
Key Features of Afro-Textured Hair
Individuals of African descent have the most diverse hair texture phenotypes, ranging from straight to tightly coiled.9 Although hair is chemically similar across various racial groups, differences are noted mainly in the shape of the hair shaft, with elliptical and curved shapes seen in Afrotextured hair. These differences yield more tightly curled strands than in other hair types; however, these features also contribute to fragility, as it creates points of weakness and decreases the tensile strength of the hair shaft.10 This inherent fragility leads to higher rates of hair breakage as well as lower moisture content and slower growth rates, which is why Afro-textured hair requires special care.9
Afro-textured hair generally falls into 2 main categories of the Andre Walker hair typing system: 4A-4C and 3A-3C.11 In the 4A-4C category, hair is described as coily or kinky. Common concerns related to this hair type include dryness and brittleness with increased susceptibility to breakage. The 3A-3C category is described as loose to corkscrew curls, with a common concern of dryness.11,12 Additionally, Loussouarn et al13 established a method to further define natural hair curliness using curve diameter and curl meters on glass plates to measure the curvature of hair strands. This method allows for assessing diversity and range of curliness within various races without relying on ethnic origin.13
Common Hair Care Practices
A description of each hair type and recommended styling practices with their levels of evidence can be found in the eTable.
Natural Hair—Natural hair is classified as hair that has not been chemically changed by perms, heat, or other straightening treatments.12,14 For natural hair, retaining the moisture of the hair shaft should be the main focus, as moisture loss leads to considerable dryness.14 Generally, it is recommended to wash natural hair once per week or every other week; however, this can change based on hair length and oil production on the scalp. Washing daily may be ideal for shorter hair and monthly for longer hair to help prevent product build-up that can have a drying effect.15 Avoid shampoos that are drying (eg, sulfate and silicone products). The co-washing method also can be utilized, which entails washing the hair with a conditioning cleanser instead of shampoo and conditioner. However, this technique is not meant to completely replace shampoo.16 In fact, a clarifying shampoo is recommended after co-washing 3 or 4 times.16 The use of a hot oil treatment twice per month can promote moisture retention and reduce split-end formation.17 For maintenance between washes, many utilize the liquid, oil, cream (LOC) or liquid, cream, oil (LCO) methods, which describe regimens that utilize water, an oil of choice, and cream such as shea butter to lock in moisture.18 This method can be used as often as needed for dry hair.
Due to the susceptibility of Afro-textured hair to tangle and knot, using a wide-tooth comb, detangling brush, or detangling conditioners is a grade B recommendation for care (eTable). Though not widely documented in the literature, many of our patients have had anecdotal success detangling their hair simply by pulling hair strands apart by hand or “finger detangling” as well as using wide-tooth combs. Although both hair types are healthier in their natural states, kinky hair (type 4A-4C) is extremely fragile and more difficult to manage than less kinky hair (type 3A-3C).18
Special care is needed when detangling due to strands being weaker when wet.19 Detangling should be performed in a retrograde fashion. Deep conditioning can aid in moisture retention and should be performed weekly or biweekly.17-20 Depending on the health of the hair, protein treatments can be considered on a monthly basis to help preserve the cuticle. Styling with braids, twists, or other protective styles can then be completed on an individual basis.
Thermal Straightening—A blowout involves straightening the hair after a wash with the use of a hair dryer.21 This common hair-styling method does not employ the use of chemicals beyond light hair oils and heat-protectant creams or sprays, typically resulting in a less kinky afro or semi-straight hair. Thermal straightening utilizes heat to temporarily straighten hair strands. Flat irons with heated metal plates then can be used after blow-drying the hair to fully straighten and smooth the strands. These processes combined commonly are known as a silk press.21-22
For thermally straightened hair, it is recommended to either wrap the hair around the scalp to keep it flat or pin curl the hair and cover with a bonnet to sleep. Safe straightening techniques with the use of a flat iron include setting the temperature no higher than 175 °F or a low/medium setting while also limiting use to once per week if possible.23 The number of passes of the flat iron also should be limited to 1 to 2 to reduce breakage. A heat-protectant cream or spray also can be applied to the hair before flat ironing to minimize damage. Applying heat protectant to the hair prior to styling will help minimize heat damage by distributing the heat along the hair fiber surface, avoiding water boiling in the hair shaft and the development of bubble hair leading to damage.24
Chemical Straightening—Similar to how relaxers, perms, and Jheri curl treatments chemically modify hair texture using distinct chemicals yielding different curl patterns, the Brazilian blowout similarly straightens hair using a hair dryer and chemicals applied to hair strands after washing.21-24 Relaxers utilize sodium or guanidine hydroxide for straightening, perms use ammonium thioglycolate for curling, and Jheri curl treatments employ thioglycolates or mercaptans for defined curls. However, these treatments generally are cautioned against due to potential hair damage and recent associations with uterine and breast cancer in Black women. Research has suggested that endocrine disrupters in these products, especially those marketed to Black women, contribute to hormone-related disease processes.25,26 One study found higher concentrations of alkylphenols, the fragrance marker diethyl phthalate, and parabens in relaxers27; however, more research is needed to determine specific chemicals associated with these cancers.
Braids and Locs—Braiding is a technique that involves interlocking 3 or more sections of hair that may or may not be fixated to the scalp like a cornrow,11 and one can utilize extensions or natural hair depending on the desired outcome. Intended for long-term wear (ie, weeks to months), braids minimize breakage and reduce daily styling needs. Two popular styles—cornrows and individual braids—differ in preparation and weaving techniques. Cornrows are an Afro-centric style involving uniform, tightly woven braids that are close to the scalp, creating distinct patterns. Conversely, individual braids weave separate hair sections, offering diverse styling possibilities. Braiding practices should exclude hairline edges—often termed baby hairs—to prevent traction alopecia. Minimal use of edge gel, which helps to tame the vellus hairs at the frontotemporal scalp, as well as mindful weave volume, weight, and length are recommended to avert breakage. Braids that cause pain are too tight, can damage hair, and may cause traction alopecia.11 Braids should not be worn for longer than 3 months at a time and require biweekly washing with diluted shampoo and conditioner. Proper drying by wringing the hair with a microfiber towel is essential to avoid frizz and mold formation.
Locs are a low-maintenance hairstyle considered permanent until cut.28 This style involves twisting, palm rolling, or using tools such as crochet hooks to “lock” the hair. Regular maintenance with retwisting and cleaning is vital for loc health. Increased weight and tight twisting of locs can cause damage to the scalp and hair strands; however, locs are known to increase hair volume over time, often due to the accumulation of hairs that would otherwise have been shed in the telogen phase.28
Wigs and Weaves—Wigs consist of synthetic or human hair that can be strapped to the head with an adjustable band or glued to the scalp depending on the desired style.29 Wigs are removed daily, which allows for quick access to hair for cleansing and moisturizing. In contrast, weaves typically are sewn into the natural hair, which may make it difficult to reach the scalp for cleansing, leading to dryness and product build-up.29 Notably, there is evidence of a relationship between long-term use of weaves and traction alopecia.30
Wigs can have a fully synthetic hair line or lace hair line and can range from very affordable to expensive. When applied correctly, both styles offer an easy way to cover and protect the natural hair by reducing the amount of physical trauma related to daily hair styling. A lace-front wig contains a frontal thin mesh or lace that camouflages the natural frontal hairline.29,30 A risk of lace-front wigs is that they can cause friction alopecia secondary to repeated use of adhesives and repeated friction against the hairline. Generally, wigs and weaves should be cared for as one would care for one’s own hair.
Hair Care in Black Children—Children’s hair care begins with washing the hair and scalp with shampoo, applying conditioner, and detangling as needed.31 After rinsing out the conditioner, a leave-in conditioner can assist with moisture retention and further detangling. The hair is then styled, either wet or dry. Recommendations for hair care practices in Black children include loose hairstyles that do not strain hair roots and nightly removal of root-securing accessories (eg, barrettes, elastic hairbeads). Frequent cornrow styling and friction on chemically straightened hair were identified by a survey as considerable traction alopecia risk factors.32 Thus, educating caregivers on appropriate hair-grooming practices for children is important.
Hair Protection—Proactive steps to reduce hair loss include wearing satin bonnets and/or using satin pillowcases while sleeping regardless of hairstyle. Although evidence is limited, it is thought that satin and silk allow the hair to retain its moisture and natural oils, preventing breakage and friction.33,34 Frequent hair trimming every 2 to 4 months can reduce breakage when doing thermal treatments.35,36 When prolonged or repetitive styles are used, it is encouraged to give the hair a break between styles to recover from the repeated stress. Wearing an intermittent updo or high bun—a hairstyle in which the hair is pulled upward—can prevent breakage by reducing heavy strain on the hair; however, it is important to avoid the use of rubber bands due to friction and risk for tangling of hair strands. Instead, the use of covered elastic ties and/or those without metal is preferred.11 Alternatively, if a polished and neat appearance with slicked-back hair is desired, the practice of tautly pulling the hair is not recommended. Instead, use of an alcohol-free gel is suggested along with a satin scarf wrapped around the hairline to facilitate the setting of the hair in place.11
A common practice to preserve curly hairstyles while sleeping is known as the pineapple method, which protects the hair and aids in preserving the freshness and style of the curls.37 It consists of a loosely tied high ponytail at the top of the head allowing the curls to fall forward. This minimizes frizz and prevents the curls from forming knots.
Conclusion
Hair care recommendations in Black women can be complex due to a wide range of personal care preferences and styling techniques in this population. While evidence in the literature is limited, it still is important for dermatologists to be familiar with the different hair care practices utilized by Black women so they can effectively counsel patients and improve hair health. Knowledge of optimal hair care practices can aid in the prevention of common hair disorders that disproportionately affect this patient population, such as traction alopecia and trichorrhexis nodosa or breakage.
Patients often ask dermatologists how to best care for their specific hair type; however, there are no formal recommendations that apply to the many different hair care practices utilized by Black patients, as hair types in this community can range from wavy to tightly coiled.1 Understanding the the history of hair care in those of African ancestry and various styling practices in this population is necessary to adequately counsel patients and gain trust in the doctor-patient relationship. In this article, we provide an overview of hair care recommendations based on common styling practices in Black women.
A PubMed search of articles indexed for MEDLINE using the terms Black hair care, African American hair management, hair loss prevention, hair care practices, natural hair, natural-hair styles, alopecia, hairdressing, hair breakage, hair fragility, heat-stressed hair, traction alopecia, and natural hair care yielded 305 results; 107 duplicates were identified and removed, leaving 198 articles to be screened for eligibility (ie, English-language studies created in the past 15 years). Sixty-eight full-text articles were screened against the exclusion criteria, which included case reports and case series, articles not focused on Afro-textured hair, and cancer-related hair loss. Three additional fulltext articles were identified via resources from Wayne State University library (Detroit, Michigan) that were not available on PubMed. A total of 29 full-text articles were included in our review.
Background on Hair Care and Styling in African Populations
It is difficult to understand the history of hair in those of African ancestry in the United States.2 Prior to slavery, hair styling was considered a way of identification, classification, and communication as well as a medium through which to connect with the spiritual world in many parts of Africa. Hair-styling practices in Africa included elaborate cornrows, threading, and braiding with many accessories. Notable hair-styling products included natural butters, herbs, and powders to assist with moisture retention. Scarves also were used during this time for ceremonies or protection.3 During the mass enslavement of African populations and their transportation to the Americas by Europeans, slaveholders routinely cut off all the hair of both men and women in order to objectify and erase the culture of African hair styling passed down through generations.4,5 Hair texture then was weaponized to create a caste system in plantation life, in which Black slaves with straight hair textures were granted the “privilege” of domestic work, while those with kinky hair were relegated to arduous manual labor in the fields.4 Years later, during the 1800s, laws were enacted in the United States to prohibit Black women from wearing tightly coiled natural hair in public places.5 Over the next few centuries from the 1800s to the early 2000s, various hair-styling trends such as the use of hot combs, perms, afros, and Jheri curls developed as a means for Black individuals to conform to societal pressure to adopt more European features; however, as time progressed, afros, braids, locs, and natural hair would become more dominant as statements against these same societal pressures.5
The natural hair movement, which emerged in the United States in the 2000s, encouraged Black women to abandon the use of toxic chemical hair straighteners, cultivate healthier hair care practices, disrupt Eurocentric standards of wearing straightened hair, and facilitate self-definition of beauty ideals from the Civil Rights Movement of the 1960s.4,5 It is estimated that between 30% and 70% of all Black women in the United States wear natural hair, including 79% of millennial Black women younger than 30 years6; however, several new trends such as wigs and weaves have grown in popularity since the early 2000s due to mainstream pop culture and improvements in creating natural hairlines.7,8
Key Features of Afro-Textured Hair
Individuals of African descent have the most diverse hair texture phenotypes, ranging from straight to tightly coiled.9 Although hair is chemically similar across various racial groups, differences are noted mainly in the shape of the hair shaft, with elliptical and curved shapes seen in Afrotextured hair. These differences yield more tightly curled strands than in other hair types; however, these features also contribute to fragility, as it creates points of weakness and decreases the tensile strength of the hair shaft.10 This inherent fragility leads to higher rates of hair breakage as well as lower moisture content and slower growth rates, which is why Afro-textured hair requires special care.9
Afro-textured hair generally falls into 2 main categories of the Andre Walker hair typing system: 4A-4C and 3A-3C.11 In the 4A-4C category, hair is described as coily or kinky. Common concerns related to this hair type include dryness and brittleness with increased susceptibility to breakage. The 3A-3C category is described as loose to corkscrew curls, with a common concern of dryness.11,12 Additionally, Loussouarn et al13 established a method to further define natural hair curliness using curve diameter and curl meters on glass plates to measure the curvature of hair strands. This method allows for assessing diversity and range of curliness within various races without relying on ethnic origin.13
Common Hair Care Practices
A description of each hair type and recommended styling practices with their levels of evidence can be found in the eTable.
Natural Hair—Natural hair is classified as hair that has not been chemically changed by perms, heat, or other straightening treatments.12,14 For natural hair, retaining the moisture of the hair shaft should be the main focus, as moisture loss leads to considerable dryness.14 Generally, it is recommended to wash natural hair once per week or every other week; however, this can change based on hair length and oil production on the scalp. Washing daily may be ideal for shorter hair and monthly for longer hair to help prevent product build-up that can have a drying effect.15 Avoid shampoos that are drying (eg, sulfate and silicone products). The co-washing method also can be utilized, which entails washing the hair with a conditioning cleanser instead of shampoo and conditioner. However, this technique is not meant to completely replace shampoo.16 In fact, a clarifying shampoo is recommended after co-washing 3 or 4 times.16 The use of a hot oil treatment twice per month can promote moisture retention and reduce split-end formation.17 For maintenance between washes, many utilize the liquid, oil, cream (LOC) or liquid, cream, oil (LCO) methods, which describe regimens that utilize water, an oil of choice, and cream such as shea butter to lock in moisture.18 This method can be used as often as needed for dry hair.
Due to the susceptibility of Afro-textured hair to tangle and knot, using a wide-tooth comb, detangling brush, or detangling conditioners is a grade B recommendation for care (eTable). Though not widely documented in the literature, many of our patients have had anecdotal success detangling their hair simply by pulling hair strands apart by hand or “finger detangling” as well as using wide-tooth combs. Although both hair types are healthier in their natural states, kinky hair (type 4A-4C) is extremely fragile and more difficult to manage than less kinky hair (type 3A-3C).18
Special care is needed when detangling due to strands being weaker when wet.19 Detangling should be performed in a retrograde fashion. Deep conditioning can aid in moisture retention and should be performed weekly or biweekly.17-20 Depending on the health of the hair, protein treatments can be considered on a monthly basis to help preserve the cuticle. Styling with braids, twists, or other protective styles can then be completed on an individual basis.
Thermal Straightening—A blowout involves straightening the hair after a wash with the use of a hair dryer.21 This common hair-styling method does not employ the use of chemicals beyond light hair oils and heat-protectant creams or sprays, typically resulting in a less kinky afro or semi-straight hair. Thermal straightening utilizes heat to temporarily straighten hair strands. Flat irons with heated metal plates then can be used after blow-drying the hair to fully straighten and smooth the strands. These processes combined commonly are known as a silk press.21-22
For thermally straightened hair, it is recommended to either wrap the hair around the scalp to keep it flat or pin curl the hair and cover with a bonnet to sleep. Safe straightening techniques with the use of a flat iron include setting the temperature no higher than 175 °F or a low/medium setting while also limiting use to once per week if possible.23 The number of passes of the flat iron also should be limited to 1 to 2 to reduce breakage. A heat-protectant cream or spray also can be applied to the hair before flat ironing to minimize damage. Applying heat protectant to the hair prior to styling will help minimize heat damage by distributing the heat along the hair fiber surface, avoiding water boiling in the hair shaft and the development of bubble hair leading to damage.24
Chemical Straightening—Similar to how relaxers, perms, and Jheri curl treatments chemically modify hair texture using distinct chemicals yielding different curl patterns, the Brazilian blowout similarly straightens hair using a hair dryer and chemicals applied to hair strands after washing.21-24 Relaxers utilize sodium or guanidine hydroxide for straightening, perms use ammonium thioglycolate for curling, and Jheri curl treatments employ thioglycolates or mercaptans for defined curls. However, these treatments generally are cautioned against due to potential hair damage and recent associations with uterine and breast cancer in Black women. Research has suggested that endocrine disrupters in these products, especially those marketed to Black women, contribute to hormone-related disease processes.25,26 One study found higher concentrations of alkylphenols, the fragrance marker diethyl phthalate, and parabens in relaxers27; however, more research is needed to determine specific chemicals associated with these cancers.
Braids and Locs—Braiding is a technique that involves interlocking 3 or more sections of hair that may or may not be fixated to the scalp like a cornrow,11 and one can utilize extensions or natural hair depending on the desired outcome. Intended for long-term wear (ie, weeks to months), braids minimize breakage and reduce daily styling needs. Two popular styles—cornrows and individual braids—differ in preparation and weaving techniques. Cornrows are an Afro-centric style involving uniform, tightly woven braids that are close to the scalp, creating distinct patterns. Conversely, individual braids weave separate hair sections, offering diverse styling possibilities. Braiding practices should exclude hairline edges—often termed baby hairs—to prevent traction alopecia. Minimal use of edge gel, which helps to tame the vellus hairs at the frontotemporal scalp, as well as mindful weave volume, weight, and length are recommended to avert breakage. Braids that cause pain are too tight, can damage hair, and may cause traction alopecia.11 Braids should not be worn for longer than 3 months at a time and require biweekly washing with diluted shampoo and conditioner. Proper drying by wringing the hair with a microfiber towel is essential to avoid frizz and mold formation.
Locs are a low-maintenance hairstyle considered permanent until cut.28 This style involves twisting, palm rolling, or using tools such as crochet hooks to “lock” the hair. Regular maintenance with retwisting and cleaning is vital for loc health. Increased weight and tight twisting of locs can cause damage to the scalp and hair strands; however, locs are known to increase hair volume over time, often due to the accumulation of hairs that would otherwise have been shed in the telogen phase.28
Wigs and Weaves—Wigs consist of synthetic or human hair that can be strapped to the head with an adjustable band or glued to the scalp depending on the desired style.29 Wigs are removed daily, which allows for quick access to hair for cleansing and moisturizing. In contrast, weaves typically are sewn into the natural hair, which may make it difficult to reach the scalp for cleansing, leading to dryness and product build-up.29 Notably, there is evidence of a relationship between long-term use of weaves and traction alopecia.30
Wigs can have a fully synthetic hair line or lace hair line and can range from very affordable to expensive. When applied correctly, both styles offer an easy way to cover and protect the natural hair by reducing the amount of physical trauma related to daily hair styling. A lace-front wig contains a frontal thin mesh or lace that camouflages the natural frontal hairline.29,30 A risk of lace-front wigs is that they can cause friction alopecia secondary to repeated use of adhesives and repeated friction against the hairline. Generally, wigs and weaves should be cared for as one would care for one’s own hair.
Hair Care in Black Children—Children’s hair care begins with washing the hair and scalp with shampoo, applying conditioner, and detangling as needed.31 After rinsing out the conditioner, a leave-in conditioner can assist with moisture retention and further detangling. The hair is then styled, either wet or dry. Recommendations for hair care practices in Black children include loose hairstyles that do not strain hair roots and nightly removal of root-securing accessories (eg, barrettes, elastic hairbeads). Frequent cornrow styling and friction on chemically straightened hair were identified by a survey as considerable traction alopecia risk factors.32 Thus, educating caregivers on appropriate hair-grooming practices for children is important.
Hair Protection—Proactive steps to reduce hair loss include wearing satin bonnets and/or using satin pillowcases while sleeping regardless of hairstyle. Although evidence is limited, it is thought that satin and silk allow the hair to retain its moisture and natural oils, preventing breakage and friction.33,34 Frequent hair trimming every 2 to 4 months can reduce breakage when doing thermal treatments.35,36 When prolonged or repetitive styles are used, it is encouraged to give the hair a break between styles to recover from the repeated stress. Wearing an intermittent updo or high bun—a hairstyle in which the hair is pulled upward—can prevent breakage by reducing heavy strain on the hair; however, it is important to avoid the use of rubber bands due to friction and risk for tangling of hair strands. Instead, the use of covered elastic ties and/or those without metal is preferred.11 Alternatively, if a polished and neat appearance with slicked-back hair is desired, the practice of tautly pulling the hair is not recommended. Instead, use of an alcohol-free gel is suggested along with a satin scarf wrapped around the hairline to facilitate the setting of the hair in place.11
A common practice to preserve curly hairstyles while sleeping is known as the pineapple method, which protects the hair and aids in preserving the freshness and style of the curls.37 It consists of a loosely tied high ponytail at the top of the head allowing the curls to fall forward. This minimizes frizz and prevents the curls from forming knots.
Conclusion
Hair care recommendations in Black women can be complex due to a wide range of personal care preferences and styling techniques in this population. While evidence in the literature is limited, it still is important for dermatologists to be familiar with the different hair care practices utilized by Black women so they can effectively counsel patients and improve hair health. Knowledge of optimal hair care practices can aid in the prevention of common hair disorders that disproportionately affect this patient population, such as traction alopecia and trichorrhexis nodosa or breakage.
- Hall RR, Francis S, Whitt-Glover M, et al. Hair care practices as a barrier to physical activity in African American women. JAMA Dermatol. 2013;149:310-314. doi:10.1001/jamadermatol.2013.1946
- Johnson T, Bankhead T. Hair it is: examining the experiences of Black women with natural hair. Open J Soc Sci. 2014;02:86-100. doi:10.4236/jss.2014.21010
- Byrd AD, Tharps LL. Hair Story: Untangling the Roots of Black Hair in America. 2nd ed. St Martin’s Griffin; 2014.
- Mbilishaka AM, Clemons K, Hudlin M, et al. Don’t get it twisted: untangling the psychology of hair discrimination within Black communities. Am J Orthopsychiatry. 2020;90:590-599. doi:10.1037 /ort0000468
- Khumalo NP. On the history of African hair care: more treasures await discovery. J Cosmet Dermatol. 2008;7:231. doi:10.1111/j.1473- 2165.2008.00396.x
- Johnson AM, Godsil RD, MacFarlane J, et al. The “good hair” study: explicit and implicit attitudes toward Black women’s hair. Perception Institute. February 2017. Accessed February 11, 2025. https://perception.org/publications/goodhairstudy/
- Haskin A, Aguh C. All hairstyles are not created equal: what the dermatologist needs to know about black hairstyling practices and the risk of traction alopecia (TA). J Am Acad Dermatol. 2016;75:606-611. doi:10.1016/j.jaad.2016.02.1162
- Roseborough IE, McMichael AJ. Hair care practices in African- American patients. Semin Cutan Med Surg. 2009;28:103-108. doi:10.1016/j.sder.2009.04.007
- Menkart J Wolfram LJ Mao I. Caucasian hair, Negro hair and wool: similarities and differences. J Soc Cosmet Chem. 1996;17:769-787.
- Crawford K, Hernandez C. A review of hair care products for black individuals. Cutis. 2014;93:289-293.
- Mayo TT, Callender VD. The art of prevention: it’s too tight-loosen up and let your hair down. Int J Womens Dermatol. 2021;7:174-179. doi:10.1016/j.ijwd.2021.01.019
- De Sá Dias TC, Baby AR, Kaneko TM, et al. Relaxing/straightening of Afro-ethnic hair: historical overview. J Cosmet Dermatol. 2007;6:2-5. doi:10.1111/j.1473-2165.2007.00294.x
- Loussouarn G, Garcel AL, Lozano I, et al. Worldwide diversity of hair curliness: a new method of assessment. Int J Dermatol. 2007;46 (suppl 1):2-6. doi:10.1111/j.1365-4632.2007.03453.x
- Barba C, Mendez S, Marti M, et al. Water content of hair and nails. Thermochimica Acta. 2009;494:136-140. doi:10.1016/j.tca.2009.05.005
- Gray J. Hair care and hair care products. Clin Dermatol. 2001;19:227-236. doi:10.1016/s0738-081x(00)00133-4
- Gavazzoni Dias MFR. Pro and contra of cleansing conditioners. Skin Appendage Disord. 2019;5:131-134. doi:10.1159/000493588
- Gavazzoni Dias MFR. Hair cosmetics: an overview. Int J Trichology. 2015;7:2-15. doi:10.4103/0974-7753.153450
- Beal AC, Villarosa L, Abner A. The Black Parenting Book. 1999.
- Davis-Sivasothy A. The Science of Black Hair: A Comprehensive Guide to Textured Care. Saga Publishing; 2011.
- Robbins CR. The Physical Properties and Cosmetic Behavior of Hair. In: Robbins CR. Chemical and Physical Behavior of Human Hair. 3rd ed. Springer Nature; 1994:299-370. doi:10.1007/978-1-4757-3898-8_8
- Weathersby C, McMichael A. Brazilian keratin hair treatment: a review. J Cosmet Dermatol. 2013;12:144-148. doi:10.1111/jocd.12030
- Barreto T, Weffort F, Frattini S, et al. Straight to the point: what do we know so far on hair straightening? Skin Appendage Disord. 2021;7:265-271. doi:10.1159/000514367
- Dussaud A, Rana B, Lam HT. Progressive hair straightening using an automated flat iron: function of silicones. J Cosmet Sci. 2013;64:119-131.
- Zhou Y, Rigoletto R, Koelmel D, et al. The effect of various cosmetic pretreatments on protecting hair from thermal damage by hot flat ironing. J Cosmet Sci. 2011;62:265-282.
- Chang CJ, O’Brien KM, Keil AP, et al. Use of straighteners and other hair products and incident uterine cancer. J Natl Cancer Inst. 2022;114:1636-1645. doi:10.1093/jnci/djac165
- White AJ, Gregoire AM, Taylor KW, et al. Adolescent use of hair dyes, straighteners and perms in relation to breast cancer risk. Int J Cancer. 2021;148:2255-2263. doi:10.1002/ijc.33413
- Helm JS, Nishioka M, Brody JG, et al. Measurement of endocrine disrupting and asthma-associated chemicals in hair products used by Black women. Environ Res. 2018;165:448-458.
- Asbeck S, Riley-Prescott C, Glaser E, et al. Afro-ethnic hairstyling trends, risks, and recommendations. Cosmetics. 2022;9:17. doi:10.3390 /cosmetics9010017
- Saed S, Ibrahim O, Bergfeld WF. Hair camouflage: a comprehensive review. Int J Womens Dermatol. 2016;2:122-127. doi:10.1016 /j.ijwd.2016.09.002
- Billero V, Miteva M. Traction alopecia: the root of the problem. Clin Cosmet Investig Dermatol. 2018;11:149-159. doi:10.2147/CCID .S137296
- Jones NL, Heath CR. Hair at the intersection of dermatology and anthropology: a conversation on race and relationships. Pediatr Dermatol. 2021;38(suppl 2):158-160. doi:10.1111/pde.14721
- Rucker Wright D, Gathers R, Kapke A, et al. Hair care practices and their association with scalp and hair disorders in African American girls. J Am Acad Dermatol. 2011;64:253-262. doi:10.1016/j.jaad.2010.05.037
- Carefoot H. Silk pillowcases for better hair and skin: what to know. The Washington Post. April 6, 2021. Accessed February 10, 2025. https://www.washingtonpost.com/lifestyle/wellness/silk-pillowcases-hair-skin-benefits-myths/2021/04/05/a7dcad7c-866a-11eb-82bc-e58213caa38e_story.html
- Samrao A, McMichael A, Mirmirani P. Nocturnal traction: techniques used for hair style maintenance while sleeping may be a risk factor for traction alopecia. Skin Appendage Disord. 2021;7:220-223. doi:10.1159/000513088
- Callender VD, McMichael AJ, Cohen GF. Medical and surgical therapies for alopecias in black women. Dermatol Ther. 2004;17:164-176. doi:10.1111/j.1396-0296.2004.04017.x
- McMichael AJ. Hair breakage in normal and weathered hair: focus on the Black patient. J Investig Dermatol Symp Proc. 2007;12:6-9. doi:10.1038/sj.jidsymp.5650047
- Bosley RE, Daveluy S. A primer to natural hair care practices in black patients. Cutis. 2015;95:78-80,106.
- Hall RR, Francis S, Whitt-Glover M, et al. Hair care practices as a barrier to physical activity in African American women. JAMA Dermatol. 2013;149:310-314. doi:10.1001/jamadermatol.2013.1946
- Johnson T, Bankhead T. Hair it is: examining the experiences of Black women with natural hair. Open J Soc Sci. 2014;02:86-100. doi:10.4236/jss.2014.21010
- Byrd AD, Tharps LL. Hair Story: Untangling the Roots of Black Hair in America. 2nd ed. St Martin’s Griffin; 2014.
- Mbilishaka AM, Clemons K, Hudlin M, et al. Don’t get it twisted: untangling the psychology of hair discrimination within Black communities. Am J Orthopsychiatry. 2020;90:590-599. doi:10.1037 /ort0000468
- Khumalo NP. On the history of African hair care: more treasures await discovery. J Cosmet Dermatol. 2008;7:231. doi:10.1111/j.1473- 2165.2008.00396.x
- Johnson AM, Godsil RD, MacFarlane J, et al. The “good hair” study: explicit and implicit attitudes toward Black women’s hair. Perception Institute. February 2017. Accessed February 11, 2025. https://perception.org/publications/goodhairstudy/
- Haskin A, Aguh C. All hairstyles are not created equal: what the dermatologist needs to know about black hairstyling practices and the risk of traction alopecia (TA). J Am Acad Dermatol. 2016;75:606-611. doi:10.1016/j.jaad.2016.02.1162
- Roseborough IE, McMichael AJ. Hair care practices in African- American patients. Semin Cutan Med Surg. 2009;28:103-108. doi:10.1016/j.sder.2009.04.007
- Menkart J Wolfram LJ Mao I. Caucasian hair, Negro hair and wool: similarities and differences. J Soc Cosmet Chem. 1996;17:769-787.
- Crawford K, Hernandez C. A review of hair care products for black individuals. Cutis. 2014;93:289-293.
- Mayo TT, Callender VD. The art of prevention: it’s too tight-loosen up and let your hair down. Int J Womens Dermatol. 2021;7:174-179. doi:10.1016/j.ijwd.2021.01.019
- De Sá Dias TC, Baby AR, Kaneko TM, et al. Relaxing/straightening of Afro-ethnic hair: historical overview. J Cosmet Dermatol. 2007;6:2-5. doi:10.1111/j.1473-2165.2007.00294.x
- Loussouarn G, Garcel AL, Lozano I, et al. Worldwide diversity of hair curliness: a new method of assessment. Int J Dermatol. 2007;46 (suppl 1):2-6. doi:10.1111/j.1365-4632.2007.03453.x
- Barba C, Mendez S, Marti M, et al. Water content of hair and nails. Thermochimica Acta. 2009;494:136-140. doi:10.1016/j.tca.2009.05.005
- Gray J. Hair care and hair care products. Clin Dermatol. 2001;19:227-236. doi:10.1016/s0738-081x(00)00133-4
- Gavazzoni Dias MFR. Pro and contra of cleansing conditioners. Skin Appendage Disord. 2019;5:131-134. doi:10.1159/000493588
- Gavazzoni Dias MFR. Hair cosmetics: an overview. Int J Trichology. 2015;7:2-15. doi:10.4103/0974-7753.153450
- Beal AC, Villarosa L, Abner A. The Black Parenting Book. 1999.
- Davis-Sivasothy A. The Science of Black Hair: A Comprehensive Guide to Textured Care. Saga Publishing; 2011.
- Robbins CR. The Physical Properties and Cosmetic Behavior of Hair. In: Robbins CR. Chemical and Physical Behavior of Human Hair. 3rd ed. Springer Nature; 1994:299-370. doi:10.1007/978-1-4757-3898-8_8
- Weathersby C, McMichael A. Brazilian keratin hair treatment: a review. J Cosmet Dermatol. 2013;12:144-148. doi:10.1111/jocd.12030
- Barreto T, Weffort F, Frattini S, et al. Straight to the point: what do we know so far on hair straightening? Skin Appendage Disord. 2021;7:265-271. doi:10.1159/000514367
- Dussaud A, Rana B, Lam HT. Progressive hair straightening using an automated flat iron: function of silicones. J Cosmet Sci. 2013;64:119-131.
- Zhou Y, Rigoletto R, Koelmel D, et al. The effect of various cosmetic pretreatments on protecting hair from thermal damage by hot flat ironing. J Cosmet Sci. 2011;62:265-282.
- Chang CJ, O’Brien KM, Keil AP, et al. Use of straighteners and other hair products and incident uterine cancer. J Natl Cancer Inst. 2022;114:1636-1645. doi:10.1093/jnci/djac165
- White AJ, Gregoire AM, Taylor KW, et al. Adolescent use of hair dyes, straighteners and perms in relation to breast cancer risk. Int J Cancer. 2021;148:2255-2263. doi:10.1002/ijc.33413
- Helm JS, Nishioka M, Brody JG, et al. Measurement of endocrine disrupting and asthma-associated chemicals in hair products used by Black women. Environ Res. 2018;165:448-458.
- Asbeck S, Riley-Prescott C, Glaser E, et al. Afro-ethnic hairstyling trends, risks, and recommendations. Cosmetics. 2022;9:17. doi:10.3390 /cosmetics9010017
- Saed S, Ibrahim O, Bergfeld WF. Hair camouflage: a comprehensive review. Int J Womens Dermatol. 2016;2:122-127. doi:10.1016 /j.ijwd.2016.09.002
- Billero V, Miteva M. Traction alopecia: the root of the problem. Clin Cosmet Investig Dermatol. 2018;11:149-159. doi:10.2147/CCID .S137296
- Jones NL, Heath CR. Hair at the intersection of dermatology and anthropology: a conversation on race and relationships. Pediatr Dermatol. 2021;38(suppl 2):158-160. doi:10.1111/pde.14721
- Rucker Wright D, Gathers R, Kapke A, et al. Hair care practices and their association with scalp and hair disorders in African American girls. J Am Acad Dermatol. 2011;64:253-262. doi:10.1016/j.jaad.2010.05.037
- Carefoot H. Silk pillowcases for better hair and skin: what to know. The Washington Post. April 6, 2021. Accessed February 10, 2025. https://www.washingtonpost.com/lifestyle/wellness/silk-pillowcases-hair-skin-benefits-myths/2021/04/05/a7dcad7c-866a-11eb-82bc-e58213caa38e_story.html
- Samrao A, McMichael A, Mirmirani P. Nocturnal traction: techniques used for hair style maintenance while sleeping may be a risk factor for traction alopecia. Skin Appendage Disord. 2021;7:220-223. doi:10.1159/000513088
- Callender VD, McMichael AJ, Cohen GF. Medical and surgical therapies for alopecias in black women. Dermatol Ther. 2004;17:164-176. doi:10.1111/j.1396-0296.2004.04017.x
- McMichael AJ. Hair breakage in normal and weathered hair: focus on the Black patient. J Investig Dermatol Symp Proc. 2007;12:6-9. doi:10.1038/sj.jidsymp.5650047
- Bosley RE, Daveluy S. A primer to natural hair care practices in black patients. Cutis. 2015;95:78-80,106.
Historical Perspectives on Hair Care and Common Styling Practices in Black Women
Historical Perspectives on Hair Care and Common Styling Practices in Black Women
PRACTICE POINTS
- There is a dearth in understanding of hair care practices in Black women among health care professionals.
- Increased knowledge and cultural understanding of past and present hair care practices in Black women enhances patient care.

A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.

Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.
Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immunologic phenomenon characterized by a systemic inflammatory response syndrome—like clinical picture with additional features, including hepatosplenomegaly, hyperferritinemia, and increased natural killer cell activity. Clinical manifestations of HLH often are nonspecific, making HLH diagnosis challenging. High persistent fever is a key feature of HLH; patients also may report gastrointestinal distress, lethargy, and/or widespread rash.1
Hemophagocytic lymphohistiocytosis is believed to stem from inherited defects in several genes, such as perforin (PRF1), as well as immune dysregulation due to infections, rheumatologic diseases, hematologic malignancies, or drug reactions.2 The primary mechanism of HLH is hypothesized to be driven by aberrant immune activation, interferon gamma released from CD8+ T cells, and uncontrolled phagocytosis by activated macrophages. The cytokine cascade results in tissue injury and multiorgan dysfunction.3,4
Although HLH historically has been categorized as primary (familial) or secondary (acquired), the most recent guidelines suggest the etiology is not always binary.3,5 That said, the concept of secondary causes is useful in understanding risk factors for developing HLH. Both forms of the disease are thought to be elicited by a trigger (eg, infection), even when inherited genetic mutations exist.6 The primary form commonly affects the pediatric population,4,6-8 whereas the secondary form is more common in adults.7
Several sets of diagnostic criteria for HLH have been developed, the most well-known being the HLH-2004 criteria.1,3 The HLH-2009 modified criteria were developed after further evidence provided a refined sense of how the HLH-2004 criteria should be stratified.9 Finally, Fardet et al10 presented the HScore as an estimation of likelihood of diagnosis of HLH. These sets of HLH criteria include clinical and laboratory features that demonstrate inflammation, natrual killer cell activity, hemophagocytosis, end-organ damage, and cell lineage effects. The HScore differs from the other sets of HLH criteria in that it is designed to estimate an individual patient’s risk of having reactive hemophagocytic syndrome, which likely is equivalent to secondary HLH, although the authors do not use this exact terminology.10
While these criteria provide a framework for diagnosing HLH, they may fail to distinguish between HLH disease and HLH disease mimics, a concept described by the North American Consortium for Histiocytosis that may impact the success of immunosuppressive treatment.3 Individuals with HLH syndrome meet the aforementioned diagnostic criteria; HLH syndrome is further divided into HLH disease and HLH disease mimics (Figure 1). The “disease” label describes the traditional concept of HLH, driven by aberrant immune overactivation, in which patients benefit from immunosuppression. In contrast, HLH mimics include a subset of patients who meet the HLH criteria but are unlikely to benefit from immunosuppression because the primary mechanism driving their condition is not owed to immune overactivation, as is the case with HLH disease. Examples of HLH mimics include certain infections, such as Epstein-Barr virus (EBV), that may demonstrate clinical findings consistent with HLH but would not benefit from immunosuppression. Ironically, infections (including EBV) also are known triggers of HLH disease, making this concept difficult to understand and adopt. In this study, we refer to HLH disease simply as HLH.
Although cutaneous manifestations of HLH are not included in the diagnostic criteria, skin findings are common and may coincide with the severity and progression of the disease.11 Despite the fact that HLH can manifest with rash,1 comprehensive reviews of reported cutaneous findings in adult HLH are lacking. Thus, the goal of this study was to provide an organized characterization of reported cutaneous findings in adults with HLH and context for how the dermatologic examination may support the diagnosis or uncover the underlying etiology of this condition.
Methods
A search of PubMed articles indexed for MEDLINE using the phrase (cutaneous OR dermatologic OR skin) findings) AND hemophagocytic lymphohistiocytosis performed on September 20, 2023, yielded 423 results (Figure 2). Filters to exclude non–English language publications and pediatric populations were applied, resulting in 161 articles. Other exclusion criteria included the absence of a description of dermatologic findings. Seventy-five articles remained after screening titles and abstracts, and full-text review yielded 55 articles that were deemed appropriate for inclusion in the study. Subsequent reference searches and use of the online resource Litmaps revealed 45 additional publications that underwent full-text screening; of these articles, 5 were included in the final review.
Results
Sixty studies were included in this systematic review.5,7,11-68 The reported prevalence of skin findings among patients with HLH from the included retrospective studies ranged from 15% to 85%.12-15 Several literature reviews reported similarly varied prevalence among adult patients with HLH.7,16 Fardet et al14 categorized cutaneous manifestations of HLH into 3 types: direct manifestations of HLH not explained by systemic features (eg, generalized maculopapular eruption), indirect manifestations of HLH that are explained by systemic features of the disease (eg, purpura due to HLH-induced coagulopathy), and findings specific to the underlying etiology of HLH (eg, malar rash seen in systemic lupus erythematosus [SLE]–associated HLH). This categorization served as the outline for the results below, providing an organized review of cutaneous findings and context for how they may support the diagnosis or uncover the underlying etiology of HLH.
Category I: Direct Manifestations of HLH
Several articles reported cutaneous findings that seemed to be the direct result of HLH and not attributed to an underlying trigger or sequalae of HLH.11,14,16-31 The most common descriptions were a generalized, morbilliform, or nonspecific eruption that encompasses large areas of the skin, commonly the trunk and extremities, sometimes extending to the face and scalp.14,16-23,25,31,32 There were variations in secondary features such as pruritus and tenderness; some studies also described violaceous discoloration in addition to erythema.16,23
Other skin findings thought to be a direct result of HLH were described in detail by Zerah and DeWitt11 in their retrospective study, including pyoderma gangrenosum, panniculitis, Stevens-Johnson syndrome, atypical targetoid lesions, and bullous eruptions. The authors also analyzed dermatopathologic data that ultimately revealed that pathologic analysis was largely inconsistent and nondescript.11 There was a single case report of purpura fulminans arising alongside signs and symptoms of HLH,26 and several case reports described Sweet syndrome developing around the same time as HLH.27-29 Lastly, Collins et al30 described a case of HLH manifesting with violaceous ulcerating papules and nodules scattered across the legs, abdomen, and arms. Biopsy of this patient’s lesions showed a diffuse dermal infiltrate of histiocytes and hemophagocytosis.
Category II: Secondary Complications and Sequelae of HLH
This was the smallest group among the 3 categories, comprising a few case reports and retrospective cohort studies primarily reporting jaundice/icterus and hemorrhagic lesions such as purpura, petechiae, and scleral hemorrhage.11,21,23,33-35 Several literature reviews described these conditions as nonspecific findings in HLH.16,20 The cause of jaundice in HLH likely can be attributed to its characteristic hepatic dysfunction, whereas hemorrhagic lesions likely are the result of both hepatic and bone marrow dysfunction resulting in coagulopathy.
Category III: Manifestations of Underlying Etiology or Triggers of HLH
Infectious—Infection is known to be one of the most common triggers of HLH, with several retrospective studies reporting infectious triggers in approximately 20% of cases.13,15 Although many pathogens have been implicated, only a few of these infection-induced HLH reports described cutaneous findings, which included a case of varicella zoster virus, Escherichia coli necrotizing fasciitis, leprosy, EBV reactivation, parvovirus B19, and both focal and disseminated herpes simplex virus 2.36-42 Most of these patients presented with classic findings of each disease. The case of varicella zoster virus exhibited pruritic erythematous papules on the face, trunk, and limbs.36 The necrotizing fasciitis case presented with tender erythematous swelling of the lower extremity.37 The patient with leprosy exhibited leonine facies and numerous erythematous nodules, plaques, and superficial ulcerating plaques over the trunk and limbs with palmoplantar involvement,39 and both cases of herpes simplex virus 2 reported small bullae either diffusely over the face, trunk, and extremities or over the genitalia.38,40 Interestingly, the cases of parvovirus B19 and EBV reactivation both exhibited polyarteritis nodosa and occurred in patients with underlying autoimmune conditions, raising the question of whether these cases of HLH had a single trigger or were the result of the overall immunologic dysregulation induced by both infection and autoimmunity.41,42
Rheumatologic—Several articles reported dermatologic findings associated with macrophage activation syndrome, a term that often is used to describe HLH associated with autoimmune conditions. Cases of HLH in adult-onset Still disease, dermatomyositis, polyarteritis nodosa, and SLE described skin findings characteristic of the underlying rheumatologic disease, sometimes with acutely worse dermatologic findings at the time of HLH presentation.35,41-48 With regard to SLE, the acute manifestation of classic findings of the disease with HLH has sometimes been described as acute lupus hemophagocytic syndrome (HPS).48 Lambotte at al48 described common findings of acute lupus hemophagocytic syndrome in their retrospective study as malar rash, weight loss, polyarthralgia, and nephritis in addition to classic HLH findings including fever, lymphadenopathy, and hepatosplenomegaly. Many other rheumatologic conditions have been associated with HLH, including rheumatoid arthritis, mixed connective tissue disease, systemic sclerosis, and Sjögren disease. All these conditions can have dermatologic manifestations; however, no descriptions of dermatologic findings in cases of HLH associated with these diseases were found.13
Malignancy—Several cases of malignancy-induced HLH described cutaneous findings, the majority being cutaneous lymphomas, namely subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Other less commonly reported malignancies in this group included Kaposi sarcoma, intravascular lymphoma, Sézary syndrome, mycosis fungoides, cutaneous diffuse large B-cell lymphoma, and several subtypes of primary cutaneous T-cell lymphoma.2,32,49-60 The most common description of SPTCL included multiple scattered plaques and subcutaneous nodules, some associated with tenderness, induration, drainage, or hemorrhagic features.32,50,52,55,57,60 Cases of mycosis fungoides and Sézary syndrome presented with variations in size and distribution of erythroderma with associated lymphadenopathy.2 A unique case of HLH developing in a patient with intravascular lymphoma described an eruption of multiple telangiectasias and petechial hemorrhages on the trunk,58 while one case associated with primary cutaneous anaplastic large cell lymphoma presented with a rapidly enlarging tumor with central ulceration and eschar.59
Drug Induced—Interestingly, most of the drug-induced cases of HLH identified in our search were secondary to biologic therapies used in the treatment of metastatic melanoma, specifically the immune checkpoint inhibitors (ICIs), which have been reported to have an association with HLH in prior literature reviews.61-65 Choi et al66 described an interesting case of ICI-induced HLH presenting with a concurrent severe lichenoid drug eruption that progressed from a pruritic truncal rash to mucocutaneous bullae, erosions, and desquamation resembling a Stevens-Johnson syndrome–type picture. This patient had treatment-refractory, HIV-negative Kaposi sarcoma, where the underlying immunologic dysregulation may explain the more severe cutaneous presentation not observed in other reported cases of ICI-induced HLH.
Yang et al’s67 review of 23 cases with concurrent diagnoses of HLH and DIHS found that 61% (14/23) of cases were diagnosed initially as DIHS before failing treatment and receiving a diagnosis of HLH several weeks later. Additionally, the authors found that several cases met criteria for one diagnosis while clinically presenting strongly for the other.67 This overlap in clinical presentation also was demonstrated in Zerah and DeWitt’s11 retrospective study regarding cutaneous findings in HLH, in which several of the morbilliform eruptions thought to be contributed to HLH ultimately were decided to be drug reactions.
Comment
Regarding direct (or primary) cutaneous findings in HLH (category I), there seem to be 2 groups of features associated with the onset of HLH that are not related to its characteristic hepatic dysfunction (category II) nor its underlying triggers (category III): a nonspecific, generalized, erythematous eruption; and dermatologic conditions separate from HLH itself (eg, Sweet syndrome, pyoderma gangrenosum). Whether the latter group truly is a direct manifestation of HLH is difficult to discern with the evidence available. Nevertheless, we can conclude that there is some type of association between these dermatologic diseases and HLH, and this association can serve as both a diagnostic tool for clinicians and a point of interest for further clinical research.
The relatively low number of articles identified through our systematic review that specifically reported secondary findings, such as jaundice or coagulopathy-associated hemorrhagic lesions, may lead one to believe that these are not common findings in HLH; however, it is possible that these are not regularly reported in the literature simply because these findings are nonspecific and can be considered expected results of the characteristic organ dysfunction in HLH.
As suspected, the skin findings in category III were the most broad given the variety of underlying etiologies that have been associated with HLH. Like the other 2 categories, these skin findings generally are nonspecific to HLH; however, the ones in category III are specific to underlying etiology of HLH and may aid in identifying and treating the underlying cause of a patient’s HLH when indicated.
Most of the rheumatologic diseases seem to have been known at the time of HLH development and diagnosis, which may highlight the importance of considering a diagnosis of HLH early on in patients with known autoimmune disease and systemic signs of illness or acutely worsening signs and symptoms of their underlying autoimmune disease.
Interestingly, several cases of malignancy-associated HLH reported signs and symptoms of HLH at initial presentation of the malignant disease.32,50,59 This situation seems to be somewhat common, as Go and Wester’s68 systematic analysis of 156 patients with SPTCL found HLH was the presenting feature in 37% of patients included in their study. This may call attention to the importance of considering cutaneous lymphomas as the cause of skin lesions in patients with signs and symptoms of HLH, where it may be easy to assume that skin findings are a result of their systemic disease.
In highlighting cases of HLH related to medication use, we found it pertinent to include and discuss the complex relationship between drug-induced hypersensitivity syndrome (DIHS [formerly known as drug rash with eosinophilia and systemic symptoms [DRESS] syndrome) and HLH. The results of this study suggest that DIHS may have considerable clinical overlap with HLH11 and may even lead to development of HLH,67 creating difficulty in distinguishing between these conditions where there may be similar findings, such as cutaneous eruptions, fever, and hepatic or other internal organ involvement. We agree with Yang et al67 that there can be large overlap in symptomology between these two conditions and that more investigation is necessary to explore the relationship between them.
Conclusion
Diagnosis of HLH in adults continues to be challenging, with several diagnostic tools but no true gold standard. In addition to the nonspecific symptomology, there is a myriad of cutaneous findings that can be present in adults with HLH (eTable), all of which are also nonspecific. Even so, awareness of which dermatologic findings have been associated with HLH may provide a cue to consider HLH in the systemically ill patient with a notable dermatologic examination. Furthermore, there are several avenues for further investigation that can be drawn, including further dermatologic analysis among nonspecific eruptions attributed to HLH, clinical and pathologic differentiation between DIHS/DRESS and HLH, and correlation between severity of skin manifestations and severity of HLH disease.
Limitations of this study included a lack of clarity in diagnosis of HLH in patients described in the included articles, as some reports use variable terminology (HLH vs hemophagocytic syndrome vs macrophage activation syndrome, etc), and it is impossible to know if all authors used the same diagnostic criteria—or any validated diagnostic criteria—unless specifically stated. Additionally, including case reports in our study limited the amount and quality of information described in each report. Despite its limitations, this systematic review outlines the cutaneous manifestations associated with HLH. These data will promote clinical awareness of this complex condition and allow for consideration of HLH in patients meeting criteria for HLH syndrome. More studies ultimately are needed to differentiate HLH from its mimics.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
- Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131. doi:10.1002/pbc.21039
- Blom A, Beylot-Barry M, D’Incan M, et al. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/ Sézary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404-410. doi:10.1016/j.jaad.2010.05.029
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019;66:e27929. doi:10.1002/pbc.27929
- Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi:10.1016/j.berh.2020.101515
- Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395-411.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681. doi:10.1182/blood-2016-01-690636
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503-1516. doi:10.1016/s0140-6736(13)61048-x
- Sieni E, Cetica V, Piccin A, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS One. 2012;7:e44649. doi:10.1371/journal.pone.0044649
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. 2009:127-131. doi:10.1182 /asheducation-2009.1.127
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66:2613-2620. doi:10.1002/art.38690
- Zerah ML, DeWitt CA. Cutaneous findings in hemophagocytic lymphohistiocytosis. Dermatology. 2015;230:234-243. doi:10.1159/000368552
- Fardet L, Galicier L, Vignon-Pennamen MD, et al. Frequency, clinical features and prognosis of cutaneous manifestations in adult patients with reactive haemophagocytic syndrome. Br J Dermatol. 2010;162:547-553. doi:10.1111/j.1365-2133.2009.09549.x
- Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49:633-639. doi:10.1002/art.11368
- Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore). 2014;93:100-105. doi:10.1097/md.0000000000000022
- Tudesq JJ, Valade S, Galicier L, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114-122. doi:10.1002 /hon.2819
- Sakai H, Otsubo S, Miura T, et al. Hemophagocytic syndrome presenting with a facial erythema in a patient with systemic lupus erythematosus. J Am Acad Dermatol. 2007;57(5 Suppl):S111-S114. doi:10.1016/j .jaad.2006.11.024
- Chung SM, Song JY, Kim W, et al. Dengue-associated hemophagocytic lymphohistiocytosis in an adult: a case report and literature review. Medicine (Baltimore). 2017;96:e6159. doi:10.1097/md.0000000000006159
- Esmaili H, Rahmani O, Fouladi RF. Hemophagocytic syndrome in patients with unexplained cytopenia: report of 15 cases. Turk Patoloji Derg. 2013;29:15-18. doi:10.5146/tjpath.2013.01142
- Jiwnani S, Karimundackal G, Kulkarni A, et al. Hemophagocytic syndrome complicating lung resection. Asian Cardiovasc Thorac Ann. 2012;20:341-343. doi:10.1177/0218492311435686
- Lee WJ, Lee DW, Kim CH, et al. Dermatopathic lymphadenitis with generalized erythroderma in a patient with Epstein-Barr virusassociated hemophagocytic lymphohistiocytosis. Am J Dermatopathol. 2010;32:357-361. doi:10.1097/DAD.0b013e3181b2a50f
- Lovisari F, Terzi V, Lippi MG, et al. Hemophagocytic lymphohistiocytosis complicated by multiorgan failure: a case report. Medicine (Baltimore). 2017;96:e9198. doi:10.1097/md.0000000000009198
- Miechowiecki J, Stainer W, Wallner G, et al. Severe complication during remission of Crohn’s disease: hemophagocytic lymphohistiocytosis due to acute cytomegalovirus infection. Z Gastroenterol. 2018;56:259-263. doi:10.1055/s-0043-123999
- Ochoa S, Cheng K, Fleury CM, et al. A 28-year-old woman with fever, rash, and pancytopenia. Allergy Asthma Proc. 2017;38:322-327. doi:10.2500/aap.2017.38.4042
- Tokoro S, Namiki T, Miura K, et al. Chronic active Epstein-Barr virus infection with cutaneous lymphoproliferation: haemophagocytosis in the skin and haemophagocytic syndrome. J Eur Acad Dermatol Venereol. 2018;32:e116-e117. doi:10.1111/jdv.14640
- Tzeng HE, Teng CL, Yang Y, et al. Occult subcutaneous panniculitislike T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106 (2 Suppl):S55-S59. doi:10.1016/s0929-6646(09)60354-5
- Honjo O, Kubo T, Sugaya F, et al. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: a case report. J Immunother Cancer. 2019;7:97. doi:10.1186/s40425- 019-0582-4
- Kao RL, Jacobsen AA, Billington CJ Jr, et al. A case of VEXAS syndrome associated with EBV-associated hemophagocytic lymphohistiocytosis. Blood Cells Mol Dis. 2022;93:102636. doi:10.1016/j .bcmd.2021.102636
- Koga T, Takano K, Horai Y, et al. Sweet’s syndrome complicated by Kikuchi’s disease and hemophagocytic syndrome which presented with retinoic acid-inducible gene-I in both the skin epidermal basal layer and the cervical lymph nodes. Intern Med. 2013;52:1839-1843. doi:10.2169 /internalmedicine.52.9542
- Lin WL, Lin WC, Chiu CS, et al. Paraneoplastic Sweet’s syndrome in a patient with hemophagocytic syndrome. Int J Dermatol. 2008;3:305-307.
- Collins MK, Ho J, Akilov OE. Case 52. A unique presentation of hemophagocytic lymphohistiocytosis with ulcerating papulonodules. In: Akilov OE, ed. Cutaneous Lymphomas: Unusual Cases 3. Springer International Publishing; 2021:126-127.
- Chakrapani A, Avery A, Warnke R. Primary cutaneous gamma delta T-cell lymphoma with brain involvement and hemophagocytic syndrome. Am J Dermatopathol. 2013;35:270-272. doi:10.1097 /DAD.0b013e3182624e98
- Sullivan C, Loghmani A, Thomas K, et al. Hemophagocytic lymphohistiocytosis as the initial presentation of subcutaneous panniculitis-like T-cell lymphoma: a rare case responding to cyclosporine A and steroids. J Investig Med High Impact Case Rep. 2020;8:2324709620981531. doi:10.1177/2324709620981531
- Darmawan G, Salido EO, Concepcion ML, et al. Hemophagocytic lymphohistiocytosis: “a dreadful mimic.” Int J Rheum Dis. 2015; 18:810-812. doi:10.1111/1756-185x.12506
- Maus MV, Leick MB, Cornejo KM, et al. Case 35-2019: a 66-year-old man with pancytopenia and rash. N Engl J Med. 2019;381:1951-1960. doi:10.1056/NEJMcpc1909627
- Chamseddin B, Marks E, Dominguez A, et al. Refractory macrophage activation syndrome in the setting of adult-onset Still disease with hemophagocytic lymphohistiocytosis detected on skin biopsy treated with canakinumab and tacrolimus. J Cutan Pathol. 2019;46:528-531. doi:10.1111/cup.13466
- Bérar A, Ardois S, Walter-Moraux P, et al. Primary varicella-zoster virus infection of the immunocompromised associated with acute pancreatitis and hemophagocytic lymphohistiocytosis: a case report. Medicine (Baltimore). 2021;100:e25351. doi:10.1097 /md.0000000000025351
- Chang CC, Hsiao PJ, Chiu CC, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168-171. doi:10.1016/j.cca.2014.10.025
- Kurosawa S, Sekiya N, Fukushima K, et al. Unusual manifestation of disseminated herpes simplex virus type 2 infection associated with pharyngotonsilitis, esophagitis, and hemophagocytic lymphohisitocytosis without genital involvement. BMC Infect Dis. 2019;19:65. doi:10.1186/s12879-019-3721-0
- Saidi W, Gammoudi R, Korbi M, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54: 1054-1059. doi:10.1111/ijd.12792
- Yamaguchi K, Yamamoto A, Hisano M, et al. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244. doi:10.1097 /01.AOG.0000157757.54948.9b
- Hayakawa I, Shirasaki F, Ikeda H, et al. Reactive hemophagocytic syndrome in a patient with polyarteritis nodosa associated with Epstein- Barr virus reactivation. Rheumatol Int. 2006;26:573-576. doi:10.1007 /s00296-005-0024-0
- Jeong JY, Park JY, Ham JY, et al. Molecular evidence of parvovirus B19 in the cutaneous polyarteritis nodosa tissue from a patient with parvovirus-associated hemophagocytic syndrome: case report. Medicine (Baltimore). 2020;99:e22079. doi:10.1097 /md.0000000000022079
- Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478. doi:10.2169 /internalmedicine.1121-18
- Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune- associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246. doi:10 .1080/03009742.2019.1653493
- Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19:449-451. doi:10.1097/rhu.0000000000000040
- Kerl K, Wolf IH, Cerroni L, et al. Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol. 2015;37:539-543. doi:10.1097 /dad.0000000000000166
- Komiya Y, Saito T, Mizoguchi F, et al. Hemophagocytic syndrome complicated with dermatomyositis controlled successfully with infliximab and conventional therapies. Intern Med. 2017;56:3237-3241. doi:10.2169 /internalmedicine.7966-16
- Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosusassociated hemophagocytic syndrome. Medicine (Baltimore). 2006;85: 169-182. doi:10.1097/01.md.0000224708.62510.d1
- Guitart J, Mangold AR, Martinez-Escala ME, et al. Clinical and pathological characteristics and outcomes among patients with subcutaneous panniculitis-like T-cell lymphoma and related adipotropic lymphoproliferative disorders. JAMA Dermatol. 2022;158:1167-1174. doi:10.1001/jamadermatol.2022.3347
- Hung GD, Chen YH, Chen DY, et al. Subcutaneous panniculitis-like T-cell lymphoma presenting with hemophagocytic lymphohistiocytosis and skin lesions with characteristic high-resolution ultrasonographic findings. Clin Rheumatol. 2007;26:775-778. doi:10.1007/s10067 -005-0193-y
- Jamil A, Nadzri N, Harun N, et al. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222-3. doi:10.1016/j.jaad.2012.04.021
- LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitis-like T-cell lymphoma: an association between subcutaneous panniculitislike T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48:572-577. doi:10.1111/cup.13863
- Lee DE, Martinez-Escala ME, Serrano LM, et al. Hemophagocytic lymphohistiocytosis in cutaneous T-cell lymphoma. JAMA Dermatol. 2018;154:828-831. doi:10.1001/jamadermatol.2018.1264
- Maejima H, Tanei R, Morioka T, et al. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339-340. doi:10.2340/00015555-0981
- Mody A, Cherry D, Georgescu G, et al. A rare case of subcutaneous panniculitis-like T cell lymphoma with hemophagocytic lymphohistiocytosis mimicking cellulitis. Am J Case Rep. 2021;22:E927142. doi:10.12659/ajcr.927142
- Park YJ, Bae HJ, Chang JY, et al. Development of Kaposi sarcoma and hemophagocytic lymphohistiocytosis associated with human herpesvirus 8 in a renal transplant recipient. Korean J Intern Med. 2017;4:750-752.
- Phatak S, Gupta L, Aggarwal A. A young woman with panniculitis and cytopenia who later developed coagulopathy. J Assoc Physicians India. 2016;64:65-67.
- Pongpairoj K, Rerknimitr P, Wititsuwannakul J, et al. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696-698. doi:10.1111/ced.12859
- Shimizu Y, Tanae K, Takahashi N, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263-266. doi:10.1016/j.leukres.2009.07.001
- Sirka CS, Pradhan S, Patra S, et al. Hemophagocytic lymphohistiocytosis: a rare, potentially fatal complication in subcutaneous panniculitis like T cell lymphoma. Indian J Dermatol Venereol Leprol. 2019;5:481-485.
- Chin CK, Hall S, Green C, et al. Secondary haemophagocytic lymphohistiocytosis due to checkpoint inhibitor therapy. Eur J Cancer. 2019;115: 84-87. doi:10.1016/j.ejca.2019.04.026
- Dudda M, Mann C, Heinz J, et al. Hemophagocytic lymphohistiocytosis of a melanoma patient under BRAF/MEK-inhibitor therapy following anti-PD1 inhibitor treatment: a case report and review to the literature. Melanoma Res. 2021;31:81-84. doi:10.1097 /cmr.0000000000000703
- Mizuta H, Nakano E, Takahashi A, et al. Hemophagocytic lymphohistiocytosis with advanced malignant melanoma accompanied by ipilimumab and nivolumab: a case report and literature review. Dermatol Ther. 2020;33:e13321. doi:10.1111/dth.13321
- Satzger I, Ivanyi P, Länger F, et al. Treatment-related hemophagocytic lymphohistiocytosis secondary to checkpoint inhibition with nivolumab plus ipilimumab. Eur J Cancer. 2018;93:150-153. doi:10.1016/j.ejca.2018.01.063
- Michot JM, Lazarovici J, Tieu A, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. 2019;122:72-90. doi:10.1016/J.EJCA.2019.07.014
- Choi S, Zhou M, Bahrani E, et al. Rare and fatal complication of immune checkpoint inhibition: a case report of haemophagocytic lymphohistiocytosis with severe lichenoid dermatitis. Br J Haematol. 2021;193:e44-e47. doi:10.1111/BJH.17442
- Yang JJ, Lei DK, Ravi V, et al. Overlap between hemophagocytic lymphohistiocytosis and drug reaction and eosinophilia with systemic symptoms: a review. Int J Dermatol. 2021;60:925-932. doi:10.1111 /ijd.15196
- Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101:1404-1413. doi:10.1002/cncr.20502
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
A Systematic Review of Dermatologic Findings in Adults With Hemophagocytic Lymphohistiocytosis
PRACTICE POINTS
- Hemophagocytic lymphohistiocytosis (HLH) is a complex, life-threatening immunologic condition that is associated with various diagnostic tools.
- Physicians who care for patients with HLH should know that skin findings are not uncommon but are largely nonspecific and can be a direct result of HLH itself, systemic complications, or the underlying etiology of the condition.
- There is a myriad of cutaneous findings that can manifest in adult patients with HLH. Awareness of HLH-associated dermatologic conditions and available diagnostic tools among multidisciplinary teams will aid in diagnosis.
Cyclically Bleeding Umbilical Papules
Cyclically Bleeding Umbilical Papules
THE DIAGNOSIS: Cutaneous Endometriosis
On histopathology, a biopsy specimen of an umbilical papule showed a dermal lymphohistiocyticrich infiltrate, hemorrhage, and ectopic endometrial glands consistent with cutaneous endometriosis (CE)(Figure). Cutaneous endometriosis is a rare condition that typically affects females of reproductive potential and is characterized by endometrial glands and stroma within the dermis and hypodermis. Cutaneous endometriosis is classified as primary or secondary. There is no surgical history of the abdomen or pelvis in primary CE. In contrast, a history of abdominopelvic surgery is the defining characteristic of secondary CE, which is more common than primary CE and typically manifests as painful red, brown, or purple papules along preexisting surgical scars of the umbilicus, lower abdomen, or pelvic region.1 Our patient may have developed secondary CE related to the laparoscopic cholecystectomy performed 10 years prior. Surgical excision is considered the definitive treatment for CE, and hormonal therapy with danazol or leuprolide may help ameliorate symptoms.1 Our patient deferred any hormonal or surgical interventions to undergo fertility treatments for pregnancy.
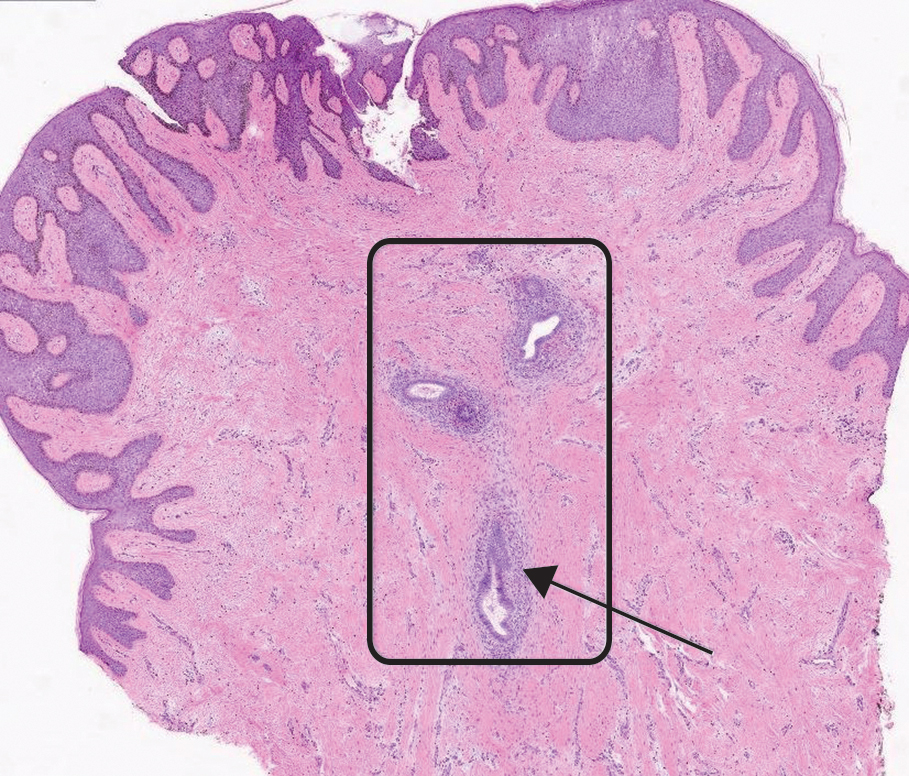
Cyclical bleeding and pain that coincides with menstruation is consistent with CE; however, cyclical symptoms are not always present, which can lead to delayed or incorrect diagnosis. Biopsy and histopathologic analysis are required for definitive diagnosis and are critical for distinguishing CE from other conditions. The differential diagnosis in our patient included pyogenic granuloma, dermatofibrosarcoma protuberans, keloid, and cutaneous metastasis of a primary malignancy. Vascular lesions such as pyogenic granuloma can manifest with bleeding but have a characteristic histopathologic lobular capillary arrangement that was not present in our patient.
Dermatofibrosarcoma protuberans is a rare, slow-growing, malignant soft-tissue sarcoma that most commonly manifests on the trunk, arms, and legs.2 It is characterized by a slow-growing, indurated plaque that often is present for years and may suddenly progress into a smooth, red-brown, multinodular mass. Histopathology typically shows spindle cells infiltrating the dermis and subcutaneous tissue in storiform or whorled pattern with variations based on the tumor stage, as well as diffuse CD34 immunoreactivity.2
Keloids are dense, raised, hyperpigmented, fibrous nodules—sometimes with accompanying telangiectasias—that typically grow secondary to trauma and project past the boundaries of the initial trauma site.1 Keloids are more commonly seen in individuals with darker skin types and tend to grow larger in this population. Histopathology reveals thickened hyalinized collagen bundles, which were not seen in our patient.1
Metastatic skin lesions of the umbilicus are rare but can arise from internal malignancies including cancers of the lung, colon, and breast.3 We considered Sister Mary Joseph nodule, which is caused most commonly by metastasis of a primary gastrointestinal cancer and signifies poor prognosis. The histopathology of metastatic lesions would reveal the presence of atypical cells with cancer-specific markers. Histopathology along with the patient’s personal and family history, a comprehensive review of symptoms, and cancer screening may help with reaching the correct diagnosis.
The average duration between abdominopelvic surgery and onset of secondary CE symptoms is 3.7 to 5.3 years.4 Our patient presented 10 years post surgery and after cessation of oral contraception, which may suggest a potential role of hormonal contraception in delayed CE onset. Diagnosis of CE can be challenging due to atypical signs or symptoms, delayed onset, and lack of awareness among health care professionals. Patients with delayed diagnosis may endure multiple procedures, prolonged physical pain, and emotional distress. Furthermore, 30% to 50% of females with endometriosis experience infertility. Delayed diagnosis of CE compounded with associated age-related increase in oocyte atresia could potentially worsen fecundity as patients age.5 It is important to consider CE in the differential diagnosis of females of reproductive age who present with cyclical bleeding and abdominal or umbilical nodules.
- James WD, Elston D, Treat JR, et al. Andrews Diseases of the Skin: Clinical Dermatology. 13th ed. Elsevier; 2019. Accessed March 19, 2024. https://search.worldcat.org/title/1084979207
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Komurcugil I, Arslan Z, Bal ZI, et al. Cutaneous metastases different clinical presentations: case series and review of the literature. Dermatol Reports. 2022;15:9553.
- Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Published online September 16, 2019. Eur J Obstet Gynecol Reprod Biol X.
- Missmer SA, Hankinson SE, Spiegelman D, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784-796.
THE DIAGNOSIS: Cutaneous Endometriosis
On histopathology, a biopsy specimen of an umbilical papule showed a dermal lymphohistiocyticrich infiltrate, hemorrhage, and ectopic endometrial glands consistent with cutaneous endometriosis (CE)(Figure). Cutaneous endometriosis is a rare condition that typically affects females of reproductive potential and is characterized by endometrial glands and stroma within the dermis and hypodermis. Cutaneous endometriosis is classified as primary or secondary. There is no surgical history of the abdomen or pelvis in primary CE. In contrast, a history of abdominopelvic surgery is the defining characteristic of secondary CE, which is more common than primary CE and typically manifests as painful red, brown, or purple papules along preexisting surgical scars of the umbilicus, lower abdomen, or pelvic region.1 Our patient may have developed secondary CE related to the laparoscopic cholecystectomy performed 10 years prior. Surgical excision is considered the definitive treatment for CE, and hormonal therapy with danazol or leuprolide may help ameliorate symptoms.1 Our patient deferred any hormonal or surgical interventions to undergo fertility treatments for pregnancy.
Cyclical bleeding and pain that coincides with menstruation is consistent with CE; however, cyclical symptoms are not always present, which can lead to delayed or incorrect diagnosis. Biopsy and histopathologic analysis are required for definitive diagnosis and are critical for distinguishing CE from other conditions. The differential diagnosis in our patient included pyogenic granuloma, dermatofibrosarcoma protuberans, keloid, and cutaneous metastasis of a primary malignancy. Vascular lesions such as pyogenic granuloma can manifest with bleeding but have a characteristic histopathologic lobular capillary arrangement that was not present in our patient.
Dermatofibrosarcoma protuberans is a rare, slow-growing, malignant soft-tissue sarcoma that most commonly manifests on the trunk, arms, and legs.2 It is characterized by a slow-growing, indurated plaque that often is present for years and may suddenly progress into a smooth, red-brown, multinodular mass. Histopathology typically shows spindle cells infiltrating the dermis and subcutaneous tissue in storiform or whorled pattern with variations based on the tumor stage, as well as diffuse CD34 immunoreactivity.2
Keloids are dense, raised, hyperpigmented, fibrous nodules—sometimes with accompanying telangiectasias—that typically grow secondary to trauma and project past the boundaries of the initial trauma site.1 Keloids are more commonly seen in individuals with darker skin types and tend to grow larger in this population. Histopathology reveals thickened hyalinized collagen bundles, which were not seen in our patient.1
Metastatic skin lesions of the umbilicus are rare but can arise from internal malignancies including cancers of the lung, colon, and breast.3 We considered Sister Mary Joseph nodule, which is caused most commonly by metastasis of a primary gastrointestinal cancer and signifies poor prognosis. The histopathology of metastatic lesions would reveal the presence of atypical cells with cancer-specific markers. Histopathology along with the patient’s personal and family history, a comprehensive review of symptoms, and cancer screening may help with reaching the correct diagnosis.
The average duration between abdominopelvic surgery and onset of secondary CE symptoms is 3.7 to 5.3 years.4 Our patient presented 10 years post surgery and after cessation of oral contraception, which may suggest a potential role of hormonal contraception in delayed CE onset. Diagnosis of CE can be challenging due to atypical signs or symptoms, delayed onset, and lack of awareness among health care professionals. Patients with delayed diagnosis may endure multiple procedures, prolonged physical pain, and emotional distress. Furthermore, 30% to 50% of females with endometriosis experience infertility. Delayed diagnosis of CE compounded with associated age-related increase in oocyte atresia could potentially worsen fecundity as patients age.5 It is important to consider CE in the differential diagnosis of females of reproductive age who present with cyclical bleeding and abdominal or umbilical nodules.
THE DIAGNOSIS: Cutaneous Endometriosis
On histopathology, a biopsy specimen of an umbilical papule showed a dermal lymphohistiocyticrich infiltrate, hemorrhage, and ectopic endometrial glands consistent with cutaneous endometriosis (CE)(Figure). Cutaneous endometriosis is a rare condition that typically affects females of reproductive potential and is characterized by endometrial glands and stroma within the dermis and hypodermis. Cutaneous endometriosis is classified as primary or secondary. There is no surgical history of the abdomen or pelvis in primary CE. In contrast, a history of abdominopelvic surgery is the defining characteristic of secondary CE, which is more common than primary CE and typically manifests as painful red, brown, or purple papules along preexisting surgical scars of the umbilicus, lower abdomen, or pelvic region.1 Our patient may have developed secondary CE related to the laparoscopic cholecystectomy performed 10 years prior. Surgical excision is considered the definitive treatment for CE, and hormonal therapy with danazol or leuprolide may help ameliorate symptoms.1 Our patient deferred any hormonal or surgical interventions to undergo fertility treatments for pregnancy.
Cyclical bleeding and pain that coincides with menstruation is consistent with CE; however, cyclical symptoms are not always present, which can lead to delayed or incorrect diagnosis. Biopsy and histopathologic analysis are required for definitive diagnosis and are critical for distinguishing CE from other conditions. The differential diagnosis in our patient included pyogenic granuloma, dermatofibrosarcoma protuberans, keloid, and cutaneous metastasis of a primary malignancy. Vascular lesions such as pyogenic granuloma can manifest with bleeding but have a characteristic histopathologic lobular capillary arrangement that was not present in our patient.
Dermatofibrosarcoma protuberans is a rare, slow-growing, malignant soft-tissue sarcoma that most commonly manifests on the trunk, arms, and legs.2 It is characterized by a slow-growing, indurated plaque that often is present for years and may suddenly progress into a smooth, red-brown, multinodular mass. Histopathology typically shows spindle cells infiltrating the dermis and subcutaneous tissue in storiform or whorled pattern with variations based on the tumor stage, as well as diffuse CD34 immunoreactivity.2
Keloids are dense, raised, hyperpigmented, fibrous nodules—sometimes with accompanying telangiectasias—that typically grow secondary to trauma and project past the boundaries of the initial trauma site.1 Keloids are more commonly seen in individuals with darker skin types and tend to grow larger in this population. Histopathology reveals thickened hyalinized collagen bundles, which were not seen in our patient.1
Metastatic skin lesions of the umbilicus are rare but can arise from internal malignancies including cancers of the lung, colon, and breast.3 We considered Sister Mary Joseph nodule, which is caused most commonly by metastasis of a primary gastrointestinal cancer and signifies poor prognosis. The histopathology of metastatic lesions would reveal the presence of atypical cells with cancer-specific markers. Histopathology along with the patient’s personal and family history, a comprehensive review of symptoms, and cancer screening may help with reaching the correct diagnosis.
The average duration between abdominopelvic surgery and onset of secondary CE symptoms is 3.7 to 5.3 years.4 Our patient presented 10 years post surgery and after cessation of oral contraception, which may suggest a potential role of hormonal contraception in delayed CE onset. Diagnosis of CE can be challenging due to atypical signs or symptoms, delayed onset, and lack of awareness among health care professionals. Patients with delayed diagnosis may endure multiple procedures, prolonged physical pain, and emotional distress. Furthermore, 30% to 50% of females with endometriosis experience infertility. Delayed diagnosis of CE compounded with associated age-related increase in oocyte atresia could potentially worsen fecundity as patients age.5 It is important to consider CE in the differential diagnosis of females of reproductive age who present with cyclical bleeding and abdominal or umbilical nodules.
- James WD, Elston D, Treat JR, et al. Andrews Diseases of the Skin: Clinical Dermatology. 13th ed. Elsevier; 2019. Accessed March 19, 2024. https://search.worldcat.org/title/1084979207
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Komurcugil I, Arslan Z, Bal ZI, et al. Cutaneous metastases different clinical presentations: case series and review of the literature. Dermatol Reports. 2022;15:9553.
- Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Published online September 16, 2019. Eur J Obstet Gynecol Reprod Biol X.
- Missmer SA, Hankinson SE, Spiegelman D, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784-796.
- James WD, Elston D, Treat JR, et al. Andrews Diseases of the Skin: Clinical Dermatology. 13th ed. Elsevier; 2019. Accessed March 19, 2024. https://search.worldcat.org/title/1084979207
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Komurcugil I, Arslan Z, Bal ZI, et al. Cutaneous metastases different clinical presentations: case series and review of the literature. Dermatol Reports. 2022;15:9553.
- Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Published online September 16, 2019. Eur J Obstet Gynecol Reprod Biol X.
- Missmer SA, Hankinson SE, Spiegelman D, et al. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784-796.
Cyclically Bleeding Umbilical Papules
Cyclically Bleeding Umbilical Papules
A 38-year-old nulligravid female with menorrhagia and dysmenorrhea presented with cyclical umbilical bleeding of 1 year’s duration. Shortly before the onset of symptoms, the patient had discontinued oral contraceptive therapy with the intent to become pregnant. She had an uncomplicated laparoscopic cholecystectomy 10 years prior, but her medical history was otherwise unremarkable. At the current presentation, physical examination revealed multilobular brown papules with serosanguineous crusting in the umbilicus.
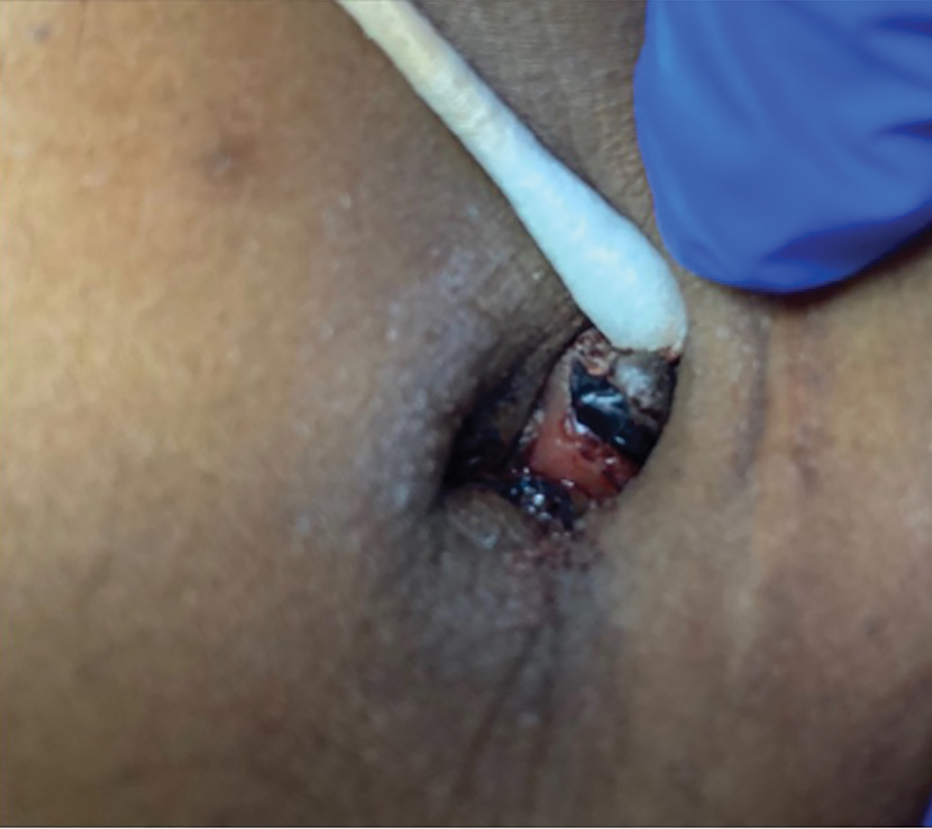

Dupilumab in the Treatment of Pemphigoid Gestationis
Dupilumab in the Treatment of Pemphigoid Gestationis
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
Pemphigoid gestationis (PG), which manifests in the second or third trimester of pregnancy, is thought to result from an excessive type 2 inflammatory response that leads to the formation of antibodies primarily targeting BP180 antigens with resultant damage to the skin basement membrane.1 Maternal antibodies can be transferred to the fetus, resulting in neonatal pemphigoid with the development of widespread vesicles and bullae.2 Maternal morbidity from placental insufficiency, intrauterine growth restriction, and premature labor are common comorbidities of PG, underscoring the critical need for safe and effective treatments for this condition.3
Systemic corticosteroids currently are the first-line treatment for moderate to severe PG but carry considerable risks to both the mother and fetus, including preterm labor and intrauterine growth restriction.4,5 Dupilumab is approved by the US Food and Drug Administration for moderate to severe atopic dermatitis in children aged 6 months and older. Dupilumab inhibits downstream signaling of IL-4Rα, reducing IL-4 and IL-13. Use of dupilumab to target the type 2 inflammatory response has shown significant promise in the treatment of BP, where it met primary and secondary endpoints in adults with moderate to severe disease, but studies in PG are limited.6-8 There are multiple reports in the literature demonstrating the safety of dupilumab in pregnancy and postpartum,9-27 including a pharmacovigilance report that found no adverse drug reactions from dupilumab reported during pregnancy.9 There also are 4 reports of pregnant patients who were diagnosed with PG and treated with dupilumab, all of whom were initially started on prednisone prior to treatment initiation.9-12 In this article, we report 2 additional cases of dupilumab treatment in patients with PG.
Case Reports
Patient 1—A 39-year-old G5P1 woman presented to the dermatology department at 27.5 weeks’ gestation with a widespread eruption of erythematous, annular, urticarial, edematous papules and plaques on the abdomen of 4 weeks’ duration (Figure 1A). Direct immunofluorescence was positive, indirect immunofluorescence confirmed an IgG-positive epidermal pattern, and serum BP180 levels were elevated, supporting a diagnosis of PG. The patient was prescribed prednisone (60 mg/d) but developed type 1 diabetes mellitus after 1 week of treatment. Following insurance approval, dupilumab therapy was initiated 3 weeks later at a dose of 300 mg subcutaneously every 2 weeks. Rapid and complete resolution of papules and plaques as well as symptomatic relief from pruritus was noted within 2 weeks of treatment (Figure 1B). The prednisone dose was tapered to 2.5 mg every other day at 6 weeks prior to induction of labor; the diabetes resolved 7 weeks after initiation of dupilumab.
At the recommendation of the patient’s high-risk maternal-fetal medicine team, 100 mg of stress-dose hydrocortisone was administered intravenously just prior to delivery to prevent flaring of PG. She delivered a healthy infant at 37 weeks and 3 days’ gestation without bullous disease and was discharged from the hospital the day after delivery on a prednisone dose of 2.5 mg every other day.
The patient subsequently developed localized pruritic papules on the hands and feet at 2 weeks postpartum. Based on shared decision-making and the patient’s concern for the severity of the previous pruritic eruption, prednisone was increased to 10 mg daily for 5 days and then was tapered over 2 weeks without flaring. Dupilumab was continued until 12 weeks postpartum with complete resolution of PG and no further sequelae.
Patient 2—A 30-year-old G1P0 woman presented to the dermatology department at 25 weeks’ gestation with a widespread eruption of 1 week’s duration on the abdomen, hands, thighs, legs, buttocks, and feet that was clinically consistent with PG (Figure 2A). Direct immunofluorescence was positive, indirect immunofluorescence showed an IgG-positive epidermal pattern, and an enzyme-linked immunosorbent assay for BP180 was elevated, confirming a diagnosis of PG. The patient was started on 40 mg of prednisone and topical steroids daily, with improvement of the pruritus but persistence of the eruption after 3 to 4 days. Five days after the initial presentation following expedited insurance approval, dupilumab 300 mg was initiated subcutaneously every 2 weeks along with a slow taper of prednisone to 5 mg, with complete clearance of the eruption within 4 weeks (Figure 2B). She delivered a healthy infant at 38 weeks’ gestation without bullous disease.
In contrast to patient 1, this patient did not receive corticosteroids at the time of delivery and did not experience flaring of her disease. The patient remained on dupilumab 5 weeks postpartum without subsequent recurrence after treatment discontinuation.
Comment
Although a myriad of effective treatments exist for bullous pemphigoid, there are very few options for PG due to the need for treatment during pregnancy. Systemic corticosteroids—the treatment of choice in severe PG disease—are not without risk in pregnancy and complicate assessment of morbidity, as both PG and chronic steroid exposure are associated with preterm labor and intrauterine growth restriction.3
Dupilumab currently is undergoing phase III trials (Clinicaltrials.gov identifiers NCT02277743 and NCT02277769) for the treatment of bullous pemphigoid, with interim reports suggesting efficacy across all primary and key secondary endpoints in moderate to severe disease, including notable steroid-sparing effects.8 In our patients, treatment with dupilumab resulted in resolution of cutaneous disease and was well tolerated, facilitating the tapering of corticosteroids and resolution of type 1 diabetes in patient 1. Although the response to dupilumab in both cases may have been confounded by concomitant steroid administration, which was started due to the severity of symptoms and uncertainty regarding insurance approval, the dose was tapered in both patients after initiation of dupilumab. Patient 1 was given a stress dose of hydrocortisone during delivery and developed a mild flare following delivery, consistent with previous literature.28, 29 Because the flare was localized to the hands and feet, she might have responded to clobetasol in addition to dupilumab, but given the severity of disease at presentation and her concern that it might worsen, low-dose prednisone was added with resolution of the flare within 2 weeks.
Dupilumab dosing regimens have not been studied in a controlled prospective manner for PG. We acknowledge that dupilumab (at least using the conventional atopic dermatitis dosing regimen) may be insufficient as monotherapy to control PG, as both patients received steroids prior to initiation of dupilumab, in part due to concern that the insurance might delay or deny approval. Previous World Health Organization vigilance reporting has suggested that dupilumab appears safe during pregnancy although it lacks pregnancy categorization in the United States due to limited studies in this population.9-28 This observation supports the conclusion that, like bullous pemphigoid, PG also is driven by Th2–mediated inflammation. Treatment with dupilumab may be safe and effective in pregnancy, reducing maternal complications from long-term corticosteroids. Additional studies are needed to confirm these hypotheses.
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
- Vičić M, MarinoviĆ B. Autoimmune bullous diseases in pregnancy: an overview of pathogenesis, clinical presentations, diagnostics and available therapies. Ital J Dermatol Venerol. 2023;158:99-109. doi:10.23736/ S2784-8671.23.07553-9
- Aoyama Y, Asai K, Hioki K, et al. Herpes gestationis in a mother and newborn: immunoclinical perspectives based on a weekly follow-up of the enzyme-linked immunosorbent assay index of a bullous pemphigoid antigen noncollagenous domain. Arch Dermatol. 2007;143:1168- 1172. doi:10.1001/archderm.143.9.1168
- Patsatsi A, Marinovic B, Murrell D. Autoimmune bullous diseases during pregnancy: solving common and uncommon issues. Int J Womens Dermatol. 2019;5:166-170. doi:10.1016/j.ijwd.2019.01.003
- Genovese G, Derlino F, Cerri A, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med (Lausanne). 2020;7:604945. doi:10.3389/fmed.2020.604945
- Tavakolpour S, Mirsafaei HS, Delshad S. Management of pemphigus disease in pregnancy. Am J Reprod Immunol. 2017;77. doi:10.1111/aji.12601
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621. doi:10.3389/fimmu.2022.928621
- Zhang Y, Xu Q, Chen L, et al. Efficacy and safety of dupilumab in moderate- to-severe bullous pemphigoid. Front Immunol. 2021;12: 738907. doi:10.3389/fimmu.2021.738907
- Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study. News release. Sanofi. September 11, 2024. Accessed February 17, 2025. https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237
- Khamisy-Farah R, Damiani G, Kong JD, et al. Safety profile of dupilumab during pregnancy: a data mining and disproportionality analysis of over 37,000 reports from the WHO individual case safety reporting database (VigiBase™). Eur Rev Med Pharmacol Sci. 2021;25:5448-5451. doi:10.26355/eurrev_202109_26652
- Avallone G, Cavallo F, Tancredi A, et al. Association between maternal dupilumab exposure and pregnancy outcomes in patients with moderate-to-severe atopic dermatitis: a nationwide retrospective cohort study. J Eur Acad Dermatol Venereol. 2024;38:1799 -1808. doi:10.1111/jdv.19794
- Chen RE, Yokoyama CC, Anadkat MJ. Pemphigoid gestationis treated with dupilumab. JAAD Case Rep. 2023;41:10-12. doi:10.1016/ j.jdcr.2023.08.013
- Liu Y, Yuan J, Xia Y, et al. A case of pemphigoid gestationis successfully treated with dupilumab. J Eur Acad Dermatol Venereol. 2023;37:E1164-E1165. doi:10.1111/jdv.19171
- Alvarez Martinez D, Russo G, Fontao L, et al. Successful therapy of pemphigoid gestationis with dupilumab—a new case. J Eur Acad Dermatol Venereol. 2023;37:E752-E753. doi:10.1111/jdv.18911
- Riquelme-Mc Loughlin C, Mascaró JM Jr. Treatment of pemphigoid gestationis with dupilumab. Clin Exp Dermatol. 2021;46:1578-1579. doi:10.1111/ced.14765
- Adam DN, Gooderham MJ, Beecker JR, et al. Expert consensus on the systemic treatment of atopic dermatitis in special populations. J Eur Acad Dermatol Venereol. 2023;37:1135-1148. doi:10.1111/jdv.18922
- Akhtar NH, Khosravi-Hafshejani T, Akhtar D, et al. The use of dupilumab in severe atopic dermatitis during pregnancy: a case report. Allergy Asthma Clin Immunol. 2022;18:9. doi:10.1186 /s13223-022-00650-w
- Bosma AL, Gerbens LAA, Middelkamp-Hup MA, et al. Paternal and maternal use of dupilumab in patients with atopic dermatitis: a case series. Clin Exp Dermatol. 2021;46:1089-1092. doi:10.1111 /ced.14725
- Chan TC, Wu NL, Wong LS, et al. Taiwanese dermatological association consensus for the management of atopic dermatitis: a 2020 update. J Formos Med Assoc. 2021;120:429-442. doi:10.101 6/j.jfma.2020.06.008
- Costley M, Murphy B. Severe atopic dermatitis treated successfully with dupilumab throughout pregnancy. Clin Exp Dermatol. 2022;47:960-961. doi:10.1111/ced.15049
- Gracia-Darder I, Pons De Ves J, Reyero Cortina M, et al. Patient with atopic dermatitis, hyper IgE syndrome and ulcerative colitis, treated successfully with dupilumab during pregnancy. Dermatol Ther. 2022;35:E15237. doi:10.1111/dth.15237
- Heilskov S, Deleuran MS, Vestergaard C. Immunosuppressive and immunomodulating therapy for atopic dermatitis in pregnancy: an appraisal of the literature. Dermatol Ther (Heidelb). 2020;10:1215-1228. doi:10.1007/s13555-020-00457-w
- Kage P, Simon JC, Treudler R. A case of atopic eczema treated safely with dupilumab during pregnancy and lactation. J Eur Acad Dermatol Venereol. 2020;34:E256-E257. doi:10.1111/jdv.16235
- Kage P, Simon JC, Treudler R. Case of atopic eczema treated with dupilumab throughout conception, pregnancy, and lactation. J Dermatol. 2021;48:E484-E485. doi:10.1111/1346-8138.16033
- Lobo Y, Lee RC, Spelman L. Atopic dermatitis treated safely with dupilumab during pregnancy: a case report and review of the literature. Case Rep Dermatol. 2021;13:248-256. doi:10.1159/000515246
- Mian M, Dunlap R, Simpson E. Dupilumab for the treatment of severe atopic dermatitis in a pregnant patient: a case report. JAAD Case Rep. 2020;6:1051-1052. doi:10.1016/j.jdcr.2020.08.001
- Napolitano M, Ruggiero A, Fontanella G, et al. New emergent therapies for atopic dermatitis: a review of safety profile with respect to female fertility, pregnancy, and breastfeeding. Dermatol Ther. 2021;34:E14475. doi:10.1111/dth.14475
- Vestergaard C, Wollenberg A, Barbarot S, et al. European task force on atopic dermatitis position paper: treatment of parental atopic dermatitis during preconception, pregnancy and lactation period. J Eur Acad Dermatol Venereol. 2019;33:1644-1659. doi:10.1111/jdv.15709
- Minakawa S, Kaneko T, Rokunohe D, et al. Pemphigoid gestationis with prepartum flare. J Dermatol. 2014;41:850-851. doi:10.1111 /1346-8138.12576
- Baxi LV, Kovilam OP, Collins MH, et al. Recurrent herpes gestationis with postpartum flare: a case report. Am J Obstet Gynecol. 1991;164: 778-780. doi:10.1016/0002-9378(91)90514-r
Dupilumab in the Treatment of Pemphigoid Gestationis
Dupilumab in the Treatment of Pemphigoid Gestationis
PRACTICE POINTS
- Dupilumab inhibits the IL-4Rα subunit, which is bound by IL‐4 and IL‐13, thereby reducing type 2 inflammation associated with pemphigoid gestationis (PG).
- Dupilumab may reduce the dose and duration of systemic corticosteroid therapy for PG, and its use in the second and third trimesters of pregnancy has been supported by emerging safety data.

Managing Contact Dermatitis Related to Amputee Care
Managing Contact Dermatitis Related to Amputee Care
Amputees who use prosthetic devices are particularly susceptible to contact dermatitis due to moisture, irritation, and prolonged contact with components of the device. Contact dermatitis accounts for approximately one-third of the dermatoses encountered by amputees who wear a prosthesis.1 Diagnosing allergic contact dermatitis (ACD) and irritant contact dermatitis (ICD) is challenging due to errors of omission from the differential and the substantial clinical overlap with other eczematous dermatoses. Diagnosis relies on patient history, clinical examination, exposure assessment, diagnostic testing, and a high index of suspicion. Conventionally, ACD comprises approximately 20% of all contact dermatitis cases, whereas ICD accounts for 80%.2 Symptoms vary between the 2 conditions, with pruritus more common in ACD and burning and soreness more common in ICD.3 Onset of dermatitis relative to exposure is crucial, with ICD often manifesting more quickly and ACD requiring an initial sensitization phase.4 Additionally, the complexity of ICD as a condition with variable features adds to the diagnostic difficulty, especially when allergens also have irritant effects.
Understanding these 2 primary types of contact dermatitis is crucial for effective management and prevention strategies in amputees who use prosthetics. In this article, we describe common causes of ACD and ICD related to amputee prosthetics and propose a tailored patch testing panel in order to better diagnose ACD in this patient population.
Allergic Contact Dermatitis
Allergic contact dermatitis occurs when the skin comes into contact with a substance to which the individual is sensitized. In amputees who use prosthetics, the socket and sock liner materials are frequent culprits for triggering allergic reactions. Components such as rubber, metals (eg, nickel), adhesives, and various plastic monomers can induce ACD in susceptible individuals. Additionally, chronic friction and sweat augment hapten penetration, increasing the risk of developing ACD.5
Contact allergens (typically small molecules under 500 Da) penetrate the skin, engage dendritic cells, activate T lymphocytes, and trigger the immune response and memory.6 The skin contains a substantial population of memory T cells, with CD8+ T cells in the epidermis and CD4+ T cells in the dermis, expressing markers that facilitate skin reactivity. The balance between effector and regulatory T cells, which can produce suppressive cytokines such as IL-10, promotes clinical tolerance to allergens such as nickel.
Textile-driven ACD presents with a distinct clinical pattern, often manifesting as patchy generalized dermatitis that coincides with sites where garments fit most snugly. This presentation can mimic other forms of dermatitis, such as nummular or asteatotic dermatitis. The skin beneath undergarments such as underwear or prosthetic socks may be spared, as these act as shields from contact allergens. Notably, the face and hands typically are spared unless the patient has a cross-reaction to formaldehyde-based preservatives found in personal care products.4
Allergy to Components of the Prosthetic Socket and Sock Liner
A prosthesis consists of several key components, including a socket, sleeve, liner, and stump shrinker (eFigure 1). The prosthetic socket, custom-made to fit the residual limb, is the upper part of the prosthesis, while the lower part consists of prosthetic components such as joints and terminal devices ordered to meet individual needs. Prosthetic sleeves provide suspension by securely holding the prosthetic limb in place, while liners offer cushioning and protection to the residual limb, enhancing comfort and reducing friction. Stump shrinkers aid in reducing swelling and shaping the residual limb, facilitating a better fit for the prosthetic socket. Together, these components work in harmony to optimize stability, comfort, and functionality for the user, enabling them to navigate daily activities with greater ease and confidence. Common allergens found in components of the socket and sock liner include rubbers and other elastomers, metals, plastics, adhesives, and textiles.

Rubbers and Other Elastomers—Consumables, including liners, knee sleeves, and socks, are tailored to each client and utilize materials such as silicone and natural and synthetic rubbers for comfort and secure fit. Allergic reactions to natural rubber latex, more commonly used in earlier prosthetics, are associated with both type I and type IV hypersensitivity reactions.4 Proteins inherent to natural rubber are overwhelmingly associated with an immediate urticarial eruption, whereas chemical additives used to produce latex are mostly linked to delayed hypersensitivity reactions, manifesting as allergic reactions ranging from mild itching to severe skin blistering.4
Vulcanization is the process of using heat and other accelerators to manufacture rubber. Common rubber accelerators include thiurams (the most common allergen associated with rubbers and other elastomers), carbamates/carba mix, 1,3-diphenylguanidine, and mercaptobenzothiazole.4 Thiourea is an implicated cause of ACD to neoprene rubber.7 These sensitizing chemicals are all included in the North American 80 Comprehensive Series; only thiuram mix, carba mix, and mercaptobenzothiazole are available in the T.R.U.E. TEST (SmartPractice). Sensitization often occurs due to repeated exposure, particularly in individuals who have undergone multiple prosthetic fittings. Many modern prospective liners utilize a medical-grade silicone as an elastomer for its high flexibility; silicone is considered biologically nonreactive and generally is considered a rare cause of ACD.8
Metals—Nickel, a ubiquitous allergen found in metal alloys used in prosthetic hardware, can cause localized itching, redness, and even blistering upon contact with the skin. Other metals, such as cobalt and chromium, also may trigger allergic reactions in susceptible individuals. Though many elastic fitting prosthetic socks contain silver fibers to reduce odors and friction-causing blisters, pure silver used in clothing or jewelry rarely causes dermatitis.4
Plastics and Adhesives—Leg prosthesis sockets typically are finished with the application of varnish, plastics, and/or resins—all potential allergens—to improve the appearance of the device and protect it from external agents.9 Polyester plastics themselves can cause ICD, only rarely leading to ACD.4 Incomplete curing during their manufacture may result in inadvertent exposure to epoxy resins or other phenol- formaldehyde resins such as 4-tert-butylcatechol and 4-tert-butylphenol formaldehyde, demonstrated causes of ACD in amputees.10 Adhesives used in sock liners or tapes to secure prosthetic devices can contain ingredients such as acrylates (a well-known cause of nail allergens) and other formaldehyde resins.4 Additionally, benzophenone commonly is added to paints and rubbers as a UV light absorber, reducing UV degradation and enhancing the material’s durability under light exposure.11
Textiles—Cotton, a common component in prosthetic sock liners, is almost 100% cellulose and typically does not cause ACD; however, synthetic fibers such as polypropylene and elastane (spandex) can elicit allergic reactions.4 Allergy to textiles often is driven by the chemicals used in the manufacturing process, particularly textile finishes, dyes, and formaldehyde resins, which are commonly used as fabric treatments. Disperse dyes are another common cause of allergic reactions. Para-phenylenediamine, a dye found in permanent hair dye and other darkly colored fabrics, is a potent sensitizer that may cross-react with other compounds that also contain similar amine groups, such as ester anesthetics, sunscreens containing para-aminobenzoic acid, other para dyes, and sulfonamides.12 Sweat can exacerbate these reactions by causing allergens to leach out of textiles, increasing skin exposure. Additionally, prosthetics containing leather may trigger allergies to potassium dichromate and other chromium compounds used in the leather-tanning process.12
Allergy to Personal Care Products
Skin protectants and prosthetic cleansers are crucial in dermatologic care for amputees, working together to safeguard the skin and maintain prosthetic hygiene. Skin protectants form a barrier against irritation, friction, and moisture, protecting the residual limb from damage and enhancing comfort and mobility. Meanwhile, prosthetic cleansers remove sweat, oils, and bacteria from the prosthetic socket, reducing the risk of infections and odors and ensuring the longevity and optimal function of the prosthetic device. Together, they support skin health, comfort, and overall quality of life for amputees.
The socket should be cleaned with warm water prior to use, but more importantly, immediately after removing the prosthesis. If cleaning products are used at night, residual haptens may remain on the device, increasing the risk of sensitization. Common contact irritants found in personal care products utilized in amputee care include sulfates, surfactants, preservatives, and fragrances (eTable 1).4 Additionally, common household cleaners and disinfectants can damage the prosthesis, leading to breakdown and the release of the monomers, precipitating ACD.
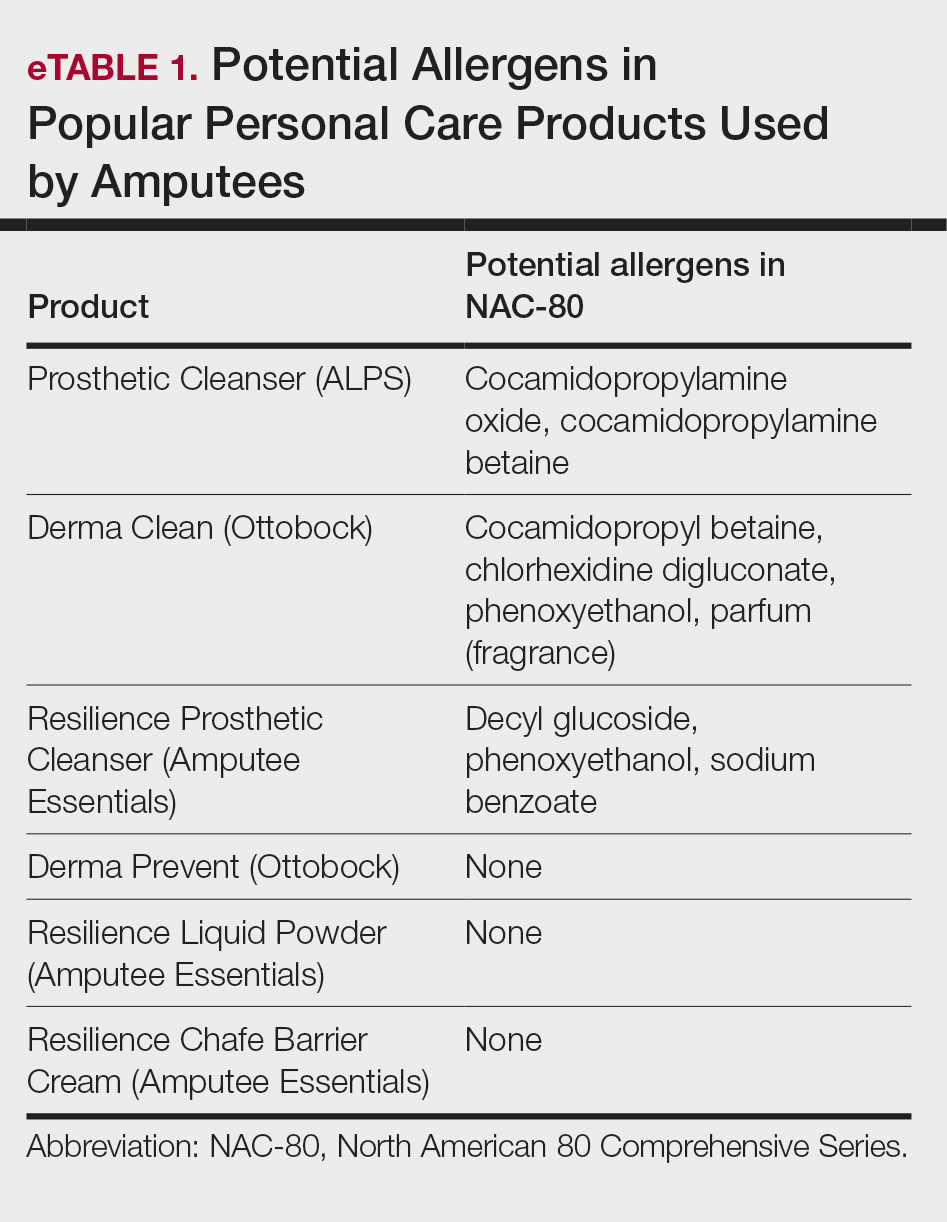
Patch Testing to Identify Causative Allergens
Patch testing is a valuable tool for identifying specific allergens responsible for ACD in amputees. This procedure involves applying small amounts of suspected allergens to the patient’s skin under occlusion and leaving the patches in place for 48 hours. After removal, the skin is assessed for reactions at 48 hours, with additional assessments conducted according to International Contact Dermatitis Research Group guidelines, typically at 72 and 96 hours, to identify delayed responses. This diagnostic approach helps pinpoint the substances to which the individual is allergic, enabling targeted avoidance strategies and treatment recommendations. Two widely used patch tests—the T.R.U.E. TEST, a preassembled patch test encompassing 35 allergens, and the North American 80 Comprehensive Series, which includes 80 allergens—demonstrate a sensitivity range between 70% and 80%.13,14 eTable 2 shows a recommended custom contact dermatitis panel to assess the most common causes of ACD related to amputee care.

Irritant Contact Dermatitis
Irritant contact dermatitis occurs when the skin’s protective barrier is damaged by repeated exposure to a particular irritant. In amputees, perspiration, friction, and pressure from prosthetic devices can exacerbate irritant reactions, leading to skin maceration, breakdown, and increased transepidermal penetration. Sweat accumulation within the prosthetic socket creates a moist environment conducive to ICD. The combination of sweat and friction can strip the skin of its natural oils, leading to dryness, chafing, and maceration. Continuous exposure to moisture also can exacerbate existing dermatitis and compromise skin integrity.4 Additionally, chronic irritation may increase transepidermal penetration of haptens, potentiating the development of ACD.15
Management of ICD in amputees involves a combination of treatments aimed at reducing friction, reducing sweating, and restoring barrier protection. Strategies to minimize mechanical trauma to the skin include ensuring proper socket fit, managing moisture, and protecting the skin. Using moisture-wicking sock liners and breathable prosthetic materials can help keep the skin dry. Topical antiperspirants containing aluminum chloride or similar compounds that help to block sweat glands often are the first line of treatment. Oral anticholinergics may be prescribed to reduce overall sweating, though they can have systemic side effects. Iontophoresis, a procedure where the affected area is exposed to a mild electrical current, can also be effective, especially for sweating of the hands and feet, though its application in amputees might be more limited.14
Recently, 2 treatments have emerged as options for managing excessive sweating (hyperhidrosis) in amputees: botulinum toxin injections and laser hair removal. By inhibiting the release of acetylcholine from sweat glands, botulinum toxin effectively reduces sweat production, thereby alleviating perspiration-induced skin irritation. Approximately 2 to 3 units of botulinum toxin at a dilution of 100 units in 1 mL of bacteriostatic saline 0.9% are injected transdermally at 1-cm intervals in a circumferential pattern on the skin covered by the prosthesis socket (typically a total of 300-500 units are utilized in the procedure)(eFigure 2).16 Laser hair removal can assist amputees with hyperhidrosis by reducing hair in the residual limb area, which decreases sweat retention and the potential for skin irritation due to friction.
Final Thoughts
In amputee dermatologic care, individuals with limb loss are particularly prone to contact dermatitis due to moisture, friction, and prolonged contact with prosthetic components. Diagnosing ACD and ICD is challenging due to overlapping symptoms and the potential for simultaneous occurrence. Distinguishing between these conditions is crucial for effective management. Understanding their causes, particularly in relation to prosthetic use, is essential for developing targeted prevention and treatment strategies, including the use of tailored patch testing panels to better diagnose ACD in amputees.
- Lyon CC, Kulkarni J, Zimersonc E, et al. Skin disorders in amputees. J Am Acad Dermatol. 2000;42:501-507.
- Bains SN, Nash P, Fonacier L. Irritant contact dermatitis. Clin Rev Allergy Immunol. 2018;56:99-109.
- Angelini G, Bonamonte D, Foti C, eds. Clinical Contact Dermatitis: A Practical Approach. Springer; 2021:57-92.
- Fisher AA, Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis. BC Decker Inc; 2008.
- Johansen JD, Frosch PJ, Lepoittevin JP. Contact Dermatitis. Springer; 2010:43-90.
- Eisen HN, Orris L, Belman S. Elicitation of delayed allergic skin reactions with haptens: the dependence of elicitation on hapten combination with protein. J Exp Med. 1952;95:473-487.
- Johnson R. Wrist dermatitis: contact allergy to neoprene in a keyboard wrist rest. Am J Contact Dermat. 1997;8:172-174.
- Adams RM. Occupational Skin Disease. WB Saunders; 1999:501-551.
- Requena L, Vázquez F, Requena C, et al. Epoxy dermatitis of an amputation stump. Contact Dermatitis. 1986;14:320.
- Freeman S. Contact dermatitis of a limb stump caused by p-tertiary butyl catechol in the artificial limb. Contact Dermatitis. 1986;14:68-69.
- Heurung AR, Raju SI, Warshaw EM. Benzophenones. Dermatitis. 2014;25:3-10.
- Manneschi V, Palmerio B, Pauluzzi P, et al. Contact dermatitis from myoelectric prostheses. Contact Dermatitis. 1989;21:116-117.
- Heinrich D, Altmeyer P, Brasch J. “New” techniques for more sensitive patch testing? J Dtsch Dermatol Ges. 2011;9:889-896.
- James WD. Contact dermatitis update. Presented at: Walter Reed National Military Medical Center; April 18, 2024.
- Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clin Exp Dermatol. 2002;27:138-146.
- Lannan FM, Powell J, Kim GM, et al. Hyperhidrosis of the residual limb: a narrative review of the measurement and treatment of excess perspiration affecting individuals with amputation. Prosthet Orthot Int. 2021;45:477-486.
Amputees who use prosthetic devices are particularly susceptible to contact dermatitis due to moisture, irritation, and prolonged contact with components of the device. Contact dermatitis accounts for approximately one-third of the dermatoses encountered by amputees who wear a prosthesis.1 Diagnosing allergic contact dermatitis (ACD) and irritant contact dermatitis (ICD) is challenging due to errors of omission from the differential and the substantial clinical overlap with other eczematous dermatoses. Diagnosis relies on patient history, clinical examination, exposure assessment, diagnostic testing, and a high index of suspicion. Conventionally, ACD comprises approximately 20% of all contact dermatitis cases, whereas ICD accounts for 80%.2 Symptoms vary between the 2 conditions, with pruritus more common in ACD and burning and soreness more common in ICD.3 Onset of dermatitis relative to exposure is crucial, with ICD often manifesting more quickly and ACD requiring an initial sensitization phase.4 Additionally, the complexity of ICD as a condition with variable features adds to the diagnostic difficulty, especially when allergens also have irritant effects.
Understanding these 2 primary types of contact dermatitis is crucial for effective management and prevention strategies in amputees who use prosthetics. In this article, we describe common causes of ACD and ICD related to amputee prosthetics and propose a tailored patch testing panel in order to better diagnose ACD in this patient population.
Allergic Contact Dermatitis
Allergic contact dermatitis occurs when the skin comes into contact with a substance to which the individual is sensitized. In amputees who use prosthetics, the socket and sock liner materials are frequent culprits for triggering allergic reactions. Components such as rubber, metals (eg, nickel), adhesives, and various plastic monomers can induce ACD in susceptible individuals. Additionally, chronic friction and sweat augment hapten penetration, increasing the risk of developing ACD.5
Contact allergens (typically small molecules under 500 Da) penetrate the skin, engage dendritic cells, activate T lymphocytes, and trigger the immune response and memory.6 The skin contains a substantial population of memory T cells, with CD8+ T cells in the epidermis and CD4+ T cells in the dermis, expressing markers that facilitate skin reactivity. The balance between effector and regulatory T cells, which can produce suppressive cytokines such as IL-10, promotes clinical tolerance to allergens such as nickel.
Textile-driven ACD presents with a distinct clinical pattern, often manifesting as patchy generalized dermatitis that coincides with sites where garments fit most snugly. This presentation can mimic other forms of dermatitis, such as nummular or asteatotic dermatitis. The skin beneath undergarments such as underwear or prosthetic socks may be spared, as these act as shields from contact allergens. Notably, the face and hands typically are spared unless the patient has a cross-reaction to formaldehyde-based preservatives found in personal care products.4
Allergy to Components of the Prosthetic Socket and Sock Liner
A prosthesis consists of several key components, including a socket, sleeve, liner, and stump shrinker (eFigure 1). The prosthetic socket, custom-made to fit the residual limb, is the upper part of the prosthesis, while the lower part consists of prosthetic components such as joints and terminal devices ordered to meet individual needs. Prosthetic sleeves provide suspension by securely holding the prosthetic limb in place, while liners offer cushioning and protection to the residual limb, enhancing comfort and reducing friction. Stump shrinkers aid in reducing swelling and shaping the residual limb, facilitating a better fit for the prosthetic socket. Together, these components work in harmony to optimize stability, comfort, and functionality for the user, enabling them to navigate daily activities with greater ease and confidence. Common allergens found in components of the socket and sock liner include rubbers and other elastomers, metals, plastics, adhesives, and textiles.
Rubbers and Other Elastomers—Consumables, including liners, knee sleeves, and socks, are tailored to each client and utilize materials such as silicone and natural and synthetic rubbers for comfort and secure fit. Allergic reactions to natural rubber latex, more commonly used in earlier prosthetics, are associated with both type I and type IV hypersensitivity reactions.4 Proteins inherent to natural rubber are overwhelmingly associated with an immediate urticarial eruption, whereas chemical additives used to produce latex are mostly linked to delayed hypersensitivity reactions, manifesting as allergic reactions ranging from mild itching to severe skin blistering.4
Vulcanization is the process of using heat and other accelerators to manufacture rubber. Common rubber accelerators include thiurams (the most common allergen associated with rubbers and other elastomers), carbamates/carba mix, 1,3-diphenylguanidine, and mercaptobenzothiazole.4 Thiourea is an implicated cause of ACD to neoprene rubber.7 These sensitizing chemicals are all included in the North American 80 Comprehensive Series; only thiuram mix, carba mix, and mercaptobenzothiazole are available in the T.R.U.E. TEST (SmartPractice). Sensitization often occurs due to repeated exposure, particularly in individuals who have undergone multiple prosthetic fittings. Many modern prospective liners utilize a medical-grade silicone as an elastomer for its high flexibility; silicone is considered biologically nonreactive and generally is considered a rare cause of ACD.8
Metals—Nickel, a ubiquitous allergen found in metal alloys used in prosthetic hardware, can cause localized itching, redness, and even blistering upon contact with the skin. Other metals, such as cobalt and chromium, also may trigger allergic reactions in susceptible individuals. Though many elastic fitting prosthetic socks contain silver fibers to reduce odors and friction-causing blisters, pure silver used in clothing or jewelry rarely causes dermatitis.4
Plastics and Adhesives—Leg prosthesis sockets typically are finished with the application of varnish, plastics, and/or resins—all potential allergens—to improve the appearance of the device and protect it from external agents.9 Polyester plastics themselves can cause ICD, only rarely leading to ACD.4 Incomplete curing during their manufacture may result in inadvertent exposure to epoxy resins or other phenol- formaldehyde resins such as 4-tert-butylcatechol and 4-tert-butylphenol formaldehyde, demonstrated causes of ACD in amputees.10 Adhesives used in sock liners or tapes to secure prosthetic devices can contain ingredients such as acrylates (a well-known cause of nail allergens) and other formaldehyde resins.4 Additionally, benzophenone commonly is added to paints and rubbers as a UV light absorber, reducing UV degradation and enhancing the material’s durability under light exposure.11
Textiles—Cotton, a common component in prosthetic sock liners, is almost 100% cellulose and typically does not cause ACD; however, synthetic fibers such as polypropylene and elastane (spandex) can elicit allergic reactions.4 Allergy to textiles often is driven by the chemicals used in the manufacturing process, particularly textile finishes, dyes, and formaldehyde resins, which are commonly used as fabric treatments. Disperse dyes are another common cause of allergic reactions. Para-phenylenediamine, a dye found in permanent hair dye and other darkly colored fabrics, is a potent sensitizer that may cross-react with other compounds that also contain similar amine groups, such as ester anesthetics, sunscreens containing para-aminobenzoic acid, other para dyes, and sulfonamides.12 Sweat can exacerbate these reactions by causing allergens to leach out of textiles, increasing skin exposure. Additionally, prosthetics containing leather may trigger allergies to potassium dichromate and other chromium compounds used in the leather-tanning process.12
Allergy to Personal Care Products
Skin protectants and prosthetic cleansers are crucial in dermatologic care for amputees, working together to safeguard the skin and maintain prosthetic hygiene. Skin protectants form a barrier against irritation, friction, and moisture, protecting the residual limb from damage and enhancing comfort and mobility. Meanwhile, prosthetic cleansers remove sweat, oils, and bacteria from the prosthetic socket, reducing the risk of infections and odors and ensuring the longevity and optimal function of the prosthetic device. Together, they support skin health, comfort, and overall quality of life for amputees.
The socket should be cleaned with warm water prior to use, but more importantly, immediately after removing the prosthesis. If cleaning products are used at night, residual haptens may remain on the device, increasing the risk of sensitization. Common contact irritants found in personal care products utilized in amputee care include sulfates, surfactants, preservatives, and fragrances (eTable 1).4 Additionally, common household cleaners and disinfectants can damage the prosthesis, leading to breakdown and the release of the monomers, precipitating ACD.
Patch Testing to Identify Causative Allergens
Patch testing is a valuable tool for identifying specific allergens responsible for ACD in amputees. This procedure involves applying small amounts of suspected allergens to the patient’s skin under occlusion and leaving the patches in place for 48 hours. After removal, the skin is assessed for reactions at 48 hours, with additional assessments conducted according to International Contact Dermatitis Research Group guidelines, typically at 72 and 96 hours, to identify delayed responses. This diagnostic approach helps pinpoint the substances to which the individual is allergic, enabling targeted avoidance strategies and treatment recommendations. Two widely used patch tests—the T.R.U.E. TEST, a preassembled patch test encompassing 35 allergens, and the North American 80 Comprehensive Series, which includes 80 allergens—demonstrate a sensitivity range between 70% and 80%.13,14 eTable 2 shows a recommended custom contact dermatitis panel to assess the most common causes of ACD related to amputee care.
Irritant Contact Dermatitis
Irritant contact dermatitis occurs when the skin’s protective barrier is damaged by repeated exposure to a particular irritant. In amputees, perspiration, friction, and pressure from prosthetic devices can exacerbate irritant reactions, leading to skin maceration, breakdown, and increased transepidermal penetration. Sweat accumulation within the prosthetic socket creates a moist environment conducive to ICD. The combination of sweat and friction can strip the skin of its natural oils, leading to dryness, chafing, and maceration. Continuous exposure to moisture also can exacerbate existing dermatitis and compromise skin integrity.4 Additionally, chronic irritation may increase transepidermal penetration of haptens, potentiating the development of ACD.15
Management of ICD in amputees involves a combination of treatments aimed at reducing friction, reducing sweating, and restoring barrier protection. Strategies to minimize mechanical trauma to the skin include ensuring proper socket fit, managing moisture, and protecting the skin. Using moisture-wicking sock liners and breathable prosthetic materials can help keep the skin dry. Topical antiperspirants containing aluminum chloride or similar compounds that help to block sweat glands often are the first line of treatment. Oral anticholinergics may be prescribed to reduce overall sweating, though they can have systemic side effects. Iontophoresis, a procedure where the affected area is exposed to a mild electrical current, can also be effective, especially for sweating of the hands and feet, though its application in amputees might be more limited.14
Recently, 2 treatments have emerged as options for managing excessive sweating (hyperhidrosis) in amputees: botulinum toxin injections and laser hair removal. By inhibiting the release of acetylcholine from sweat glands, botulinum toxin effectively reduces sweat production, thereby alleviating perspiration-induced skin irritation. Approximately 2 to 3 units of botulinum toxin at a dilution of 100 units in 1 mL of bacteriostatic saline 0.9% are injected transdermally at 1-cm intervals in a circumferential pattern on the skin covered by the prosthesis socket (typically a total of 300-500 units are utilized in the procedure)(eFigure 2).16 Laser hair removal can assist amputees with hyperhidrosis by reducing hair in the residual limb area, which decreases sweat retention and the potential for skin irritation due to friction.
Final Thoughts
In amputee dermatologic care, individuals with limb loss are particularly prone to contact dermatitis due to moisture, friction, and prolonged contact with prosthetic components. Diagnosing ACD and ICD is challenging due to overlapping symptoms and the potential for simultaneous occurrence. Distinguishing between these conditions is crucial for effective management. Understanding their causes, particularly in relation to prosthetic use, is essential for developing targeted prevention and treatment strategies, including the use of tailored patch testing panels to better diagnose ACD in amputees.
Amputees who use prosthetic devices are particularly susceptible to contact dermatitis due to moisture, irritation, and prolonged contact with components of the device. Contact dermatitis accounts for approximately one-third of the dermatoses encountered by amputees who wear a prosthesis.1 Diagnosing allergic contact dermatitis (ACD) and irritant contact dermatitis (ICD) is challenging due to errors of omission from the differential and the substantial clinical overlap with other eczematous dermatoses. Diagnosis relies on patient history, clinical examination, exposure assessment, diagnostic testing, and a high index of suspicion. Conventionally, ACD comprises approximately 20% of all contact dermatitis cases, whereas ICD accounts for 80%.2 Symptoms vary between the 2 conditions, with pruritus more common in ACD and burning and soreness more common in ICD.3 Onset of dermatitis relative to exposure is crucial, with ICD often manifesting more quickly and ACD requiring an initial sensitization phase.4 Additionally, the complexity of ICD as a condition with variable features adds to the diagnostic difficulty, especially when allergens also have irritant effects.
Understanding these 2 primary types of contact dermatitis is crucial for effective management and prevention strategies in amputees who use prosthetics. In this article, we describe common causes of ACD and ICD related to amputee prosthetics and propose a tailored patch testing panel in order to better diagnose ACD in this patient population.
Allergic Contact Dermatitis
Allergic contact dermatitis occurs when the skin comes into contact with a substance to which the individual is sensitized. In amputees who use prosthetics, the socket and sock liner materials are frequent culprits for triggering allergic reactions. Components such as rubber, metals (eg, nickel), adhesives, and various plastic monomers can induce ACD in susceptible individuals. Additionally, chronic friction and sweat augment hapten penetration, increasing the risk of developing ACD.5
Contact allergens (typically small molecules under 500 Da) penetrate the skin, engage dendritic cells, activate T lymphocytes, and trigger the immune response and memory.6 The skin contains a substantial population of memory T cells, with CD8+ T cells in the epidermis and CD4+ T cells in the dermis, expressing markers that facilitate skin reactivity. The balance between effector and regulatory T cells, which can produce suppressive cytokines such as IL-10, promotes clinical tolerance to allergens such as nickel.
Textile-driven ACD presents with a distinct clinical pattern, often manifesting as patchy generalized dermatitis that coincides with sites where garments fit most snugly. This presentation can mimic other forms of dermatitis, such as nummular or asteatotic dermatitis. The skin beneath undergarments such as underwear or prosthetic socks may be spared, as these act as shields from contact allergens. Notably, the face and hands typically are spared unless the patient has a cross-reaction to formaldehyde-based preservatives found in personal care products.4
Allergy to Components of the Prosthetic Socket and Sock Liner
A prosthesis consists of several key components, including a socket, sleeve, liner, and stump shrinker (eFigure 1). The prosthetic socket, custom-made to fit the residual limb, is the upper part of the prosthesis, while the lower part consists of prosthetic components such as joints and terminal devices ordered to meet individual needs. Prosthetic sleeves provide suspension by securely holding the prosthetic limb in place, while liners offer cushioning and protection to the residual limb, enhancing comfort and reducing friction. Stump shrinkers aid in reducing swelling and shaping the residual limb, facilitating a better fit for the prosthetic socket. Together, these components work in harmony to optimize stability, comfort, and functionality for the user, enabling them to navigate daily activities with greater ease and confidence. Common allergens found in components of the socket and sock liner include rubbers and other elastomers, metals, plastics, adhesives, and textiles.
Rubbers and Other Elastomers—Consumables, including liners, knee sleeves, and socks, are tailored to each client and utilize materials such as silicone and natural and synthetic rubbers for comfort and secure fit. Allergic reactions to natural rubber latex, more commonly used in earlier prosthetics, are associated with both type I and type IV hypersensitivity reactions.4 Proteins inherent to natural rubber are overwhelmingly associated with an immediate urticarial eruption, whereas chemical additives used to produce latex are mostly linked to delayed hypersensitivity reactions, manifesting as allergic reactions ranging from mild itching to severe skin blistering.4
Vulcanization is the process of using heat and other accelerators to manufacture rubber. Common rubber accelerators include thiurams (the most common allergen associated with rubbers and other elastomers), carbamates/carba mix, 1,3-diphenylguanidine, and mercaptobenzothiazole.4 Thiourea is an implicated cause of ACD to neoprene rubber.7 These sensitizing chemicals are all included in the North American 80 Comprehensive Series; only thiuram mix, carba mix, and mercaptobenzothiazole are available in the T.R.U.E. TEST (SmartPractice). Sensitization often occurs due to repeated exposure, particularly in individuals who have undergone multiple prosthetic fittings. Many modern prospective liners utilize a medical-grade silicone as an elastomer for its high flexibility; silicone is considered biologically nonreactive and generally is considered a rare cause of ACD.8
Metals—Nickel, a ubiquitous allergen found in metal alloys used in prosthetic hardware, can cause localized itching, redness, and even blistering upon contact with the skin. Other metals, such as cobalt and chromium, also may trigger allergic reactions in susceptible individuals. Though many elastic fitting prosthetic socks contain silver fibers to reduce odors and friction-causing blisters, pure silver used in clothing or jewelry rarely causes dermatitis.4
Plastics and Adhesives—Leg prosthesis sockets typically are finished with the application of varnish, plastics, and/or resins—all potential allergens—to improve the appearance of the device and protect it from external agents.9 Polyester plastics themselves can cause ICD, only rarely leading to ACD.4 Incomplete curing during their manufacture may result in inadvertent exposure to epoxy resins or other phenol- formaldehyde resins such as 4-tert-butylcatechol and 4-tert-butylphenol formaldehyde, demonstrated causes of ACD in amputees.10 Adhesives used in sock liners or tapes to secure prosthetic devices can contain ingredients such as acrylates (a well-known cause of nail allergens) and other formaldehyde resins.4 Additionally, benzophenone commonly is added to paints and rubbers as a UV light absorber, reducing UV degradation and enhancing the material’s durability under light exposure.11
Textiles—Cotton, a common component in prosthetic sock liners, is almost 100% cellulose and typically does not cause ACD; however, synthetic fibers such as polypropylene and elastane (spandex) can elicit allergic reactions.4 Allergy to textiles often is driven by the chemicals used in the manufacturing process, particularly textile finishes, dyes, and formaldehyde resins, which are commonly used as fabric treatments. Disperse dyes are another common cause of allergic reactions. Para-phenylenediamine, a dye found in permanent hair dye and other darkly colored fabrics, is a potent sensitizer that may cross-react with other compounds that also contain similar amine groups, such as ester anesthetics, sunscreens containing para-aminobenzoic acid, other para dyes, and sulfonamides.12 Sweat can exacerbate these reactions by causing allergens to leach out of textiles, increasing skin exposure. Additionally, prosthetics containing leather may trigger allergies to potassium dichromate and other chromium compounds used in the leather-tanning process.12
Allergy to Personal Care Products
Skin protectants and prosthetic cleansers are crucial in dermatologic care for amputees, working together to safeguard the skin and maintain prosthetic hygiene. Skin protectants form a barrier against irritation, friction, and moisture, protecting the residual limb from damage and enhancing comfort and mobility. Meanwhile, prosthetic cleansers remove sweat, oils, and bacteria from the prosthetic socket, reducing the risk of infections and odors and ensuring the longevity and optimal function of the prosthetic device. Together, they support skin health, comfort, and overall quality of life for amputees.
The socket should be cleaned with warm water prior to use, but more importantly, immediately after removing the prosthesis. If cleaning products are used at night, residual haptens may remain on the device, increasing the risk of sensitization. Common contact irritants found in personal care products utilized in amputee care include sulfates, surfactants, preservatives, and fragrances (eTable 1).4 Additionally, common household cleaners and disinfectants can damage the prosthesis, leading to breakdown and the release of the monomers, precipitating ACD.
Patch Testing to Identify Causative Allergens
Patch testing is a valuable tool for identifying specific allergens responsible for ACD in amputees. This procedure involves applying small amounts of suspected allergens to the patient’s skin under occlusion and leaving the patches in place for 48 hours. After removal, the skin is assessed for reactions at 48 hours, with additional assessments conducted according to International Contact Dermatitis Research Group guidelines, typically at 72 and 96 hours, to identify delayed responses. This diagnostic approach helps pinpoint the substances to which the individual is allergic, enabling targeted avoidance strategies and treatment recommendations. Two widely used patch tests—the T.R.U.E. TEST, a preassembled patch test encompassing 35 allergens, and the North American 80 Comprehensive Series, which includes 80 allergens—demonstrate a sensitivity range between 70% and 80%.13,14 eTable 2 shows a recommended custom contact dermatitis panel to assess the most common causes of ACD related to amputee care.
Irritant Contact Dermatitis
Irritant contact dermatitis occurs when the skin’s protective barrier is damaged by repeated exposure to a particular irritant. In amputees, perspiration, friction, and pressure from prosthetic devices can exacerbate irritant reactions, leading to skin maceration, breakdown, and increased transepidermal penetration. Sweat accumulation within the prosthetic socket creates a moist environment conducive to ICD. The combination of sweat and friction can strip the skin of its natural oils, leading to dryness, chafing, and maceration. Continuous exposure to moisture also can exacerbate existing dermatitis and compromise skin integrity.4 Additionally, chronic irritation may increase transepidermal penetration of haptens, potentiating the development of ACD.15
Management of ICD in amputees involves a combination of treatments aimed at reducing friction, reducing sweating, and restoring barrier protection. Strategies to minimize mechanical trauma to the skin include ensuring proper socket fit, managing moisture, and protecting the skin. Using moisture-wicking sock liners and breathable prosthetic materials can help keep the skin dry. Topical antiperspirants containing aluminum chloride or similar compounds that help to block sweat glands often are the first line of treatment. Oral anticholinergics may be prescribed to reduce overall sweating, though they can have systemic side effects. Iontophoresis, a procedure where the affected area is exposed to a mild electrical current, can also be effective, especially for sweating of the hands and feet, though its application in amputees might be more limited.14
Recently, 2 treatments have emerged as options for managing excessive sweating (hyperhidrosis) in amputees: botulinum toxin injections and laser hair removal. By inhibiting the release of acetylcholine from sweat glands, botulinum toxin effectively reduces sweat production, thereby alleviating perspiration-induced skin irritation. Approximately 2 to 3 units of botulinum toxin at a dilution of 100 units in 1 mL of bacteriostatic saline 0.9% are injected transdermally at 1-cm intervals in a circumferential pattern on the skin covered by the prosthesis socket (typically a total of 300-500 units are utilized in the procedure)(eFigure 2).16 Laser hair removal can assist amputees with hyperhidrosis by reducing hair in the residual limb area, which decreases sweat retention and the potential for skin irritation due to friction.
Final Thoughts
In amputee dermatologic care, individuals with limb loss are particularly prone to contact dermatitis due to moisture, friction, and prolonged contact with prosthetic components. Diagnosing ACD and ICD is challenging due to overlapping symptoms and the potential for simultaneous occurrence. Distinguishing between these conditions is crucial for effective management. Understanding their causes, particularly in relation to prosthetic use, is essential for developing targeted prevention and treatment strategies, including the use of tailored patch testing panels to better diagnose ACD in amputees.
- Lyon CC, Kulkarni J, Zimersonc E, et al. Skin disorders in amputees. J Am Acad Dermatol. 2000;42:501-507.
- Bains SN, Nash P, Fonacier L. Irritant contact dermatitis. Clin Rev Allergy Immunol. 2018;56:99-109.
- Angelini G, Bonamonte D, Foti C, eds. Clinical Contact Dermatitis: A Practical Approach. Springer; 2021:57-92.
- Fisher AA, Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis. BC Decker Inc; 2008.
- Johansen JD, Frosch PJ, Lepoittevin JP. Contact Dermatitis. Springer; 2010:43-90.
- Eisen HN, Orris L, Belman S. Elicitation of delayed allergic skin reactions with haptens: the dependence of elicitation on hapten combination with protein. J Exp Med. 1952;95:473-487.
- Johnson R. Wrist dermatitis: contact allergy to neoprene in a keyboard wrist rest. Am J Contact Dermat. 1997;8:172-174.
- Adams RM. Occupational Skin Disease. WB Saunders; 1999:501-551.
- Requena L, Vázquez F, Requena C, et al. Epoxy dermatitis of an amputation stump. Contact Dermatitis. 1986;14:320.
- Freeman S. Contact dermatitis of a limb stump caused by p-tertiary butyl catechol in the artificial limb. Contact Dermatitis. 1986;14:68-69.
- Heurung AR, Raju SI, Warshaw EM. Benzophenones. Dermatitis. 2014;25:3-10.
- Manneschi V, Palmerio B, Pauluzzi P, et al. Contact dermatitis from myoelectric prostheses. Contact Dermatitis. 1989;21:116-117.
- Heinrich D, Altmeyer P, Brasch J. “New” techniques for more sensitive patch testing? J Dtsch Dermatol Ges. 2011;9:889-896.
- James WD. Contact dermatitis update. Presented at: Walter Reed National Military Medical Center; April 18, 2024.
- Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clin Exp Dermatol. 2002;27:138-146.
- Lannan FM, Powell J, Kim GM, et al. Hyperhidrosis of the residual limb: a narrative review of the measurement and treatment of excess perspiration affecting individuals with amputation. Prosthet Orthot Int. 2021;45:477-486.
- Lyon CC, Kulkarni J, Zimersonc E, et al. Skin disorders in amputees. J Am Acad Dermatol. 2000;42:501-507.
- Bains SN, Nash P, Fonacier L. Irritant contact dermatitis. Clin Rev Allergy Immunol. 2018;56:99-109.
- Angelini G, Bonamonte D, Foti C, eds. Clinical Contact Dermatitis: A Practical Approach. Springer; 2021:57-92.
- Fisher AA, Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis. BC Decker Inc; 2008.
- Johansen JD, Frosch PJ, Lepoittevin JP. Contact Dermatitis. Springer; 2010:43-90.
- Eisen HN, Orris L, Belman S. Elicitation of delayed allergic skin reactions with haptens: the dependence of elicitation on hapten combination with protein. J Exp Med. 1952;95:473-487.
- Johnson R. Wrist dermatitis: contact allergy to neoprene in a keyboard wrist rest. Am J Contact Dermat. 1997;8:172-174.
- Adams RM. Occupational Skin Disease. WB Saunders; 1999:501-551.
- Requena L, Vázquez F, Requena C, et al. Epoxy dermatitis of an amputation stump. Contact Dermatitis. 1986;14:320.
- Freeman S. Contact dermatitis of a limb stump caused by p-tertiary butyl catechol in the artificial limb. Contact Dermatitis. 1986;14:68-69.
- Heurung AR, Raju SI, Warshaw EM. Benzophenones. Dermatitis. 2014;25:3-10.
- Manneschi V, Palmerio B, Pauluzzi P, et al. Contact dermatitis from myoelectric prostheses. Contact Dermatitis. 1989;21:116-117.
- Heinrich D, Altmeyer P, Brasch J. “New” techniques for more sensitive patch testing? J Dtsch Dermatol Ges. 2011;9:889-896.
- James WD. Contact dermatitis update. Presented at: Walter Reed National Military Medical Center; April 18, 2024.
- Smith HR, Basketter DA, McFadden JP. Irritant dermatitis, irritancy and its role in allergic contact dermatitis. Clin Exp Dermatol. 2002;27:138-146.
- Lannan FM, Powell J, Kim GM, et al. Hyperhidrosis of the residual limb: a narrative review of the measurement and treatment of excess perspiration affecting individuals with amputation. Prosthet Orthot Int. 2021;45:477-486.
Managing Contact Dermatitis Related to Amputee Care
Managing Contact Dermatitis Related to Amputee Care
PRACTICE POINTS
- Incorporating a tailored patch testing panel that includes common prosthetic-related allergens (eg, rubber, metals, adhesives) can greatly improve the diagnosis and treatment of allergic vs irritant contact dermatitis in amputees.
- Effective management of irritant contact dermatitis in amputees involves reducing moisture and friction in the prosthetic socket with moisture-wicking liners, ensuring proper fit, and utilizing treatments such as topical antiperspirants and botulinum toxin injections.
