User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Pigmented Pruritic Macules in the Genital Area
The Diagnosis: Pediculosis Pubis
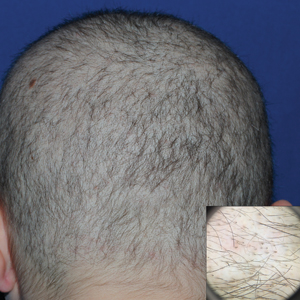
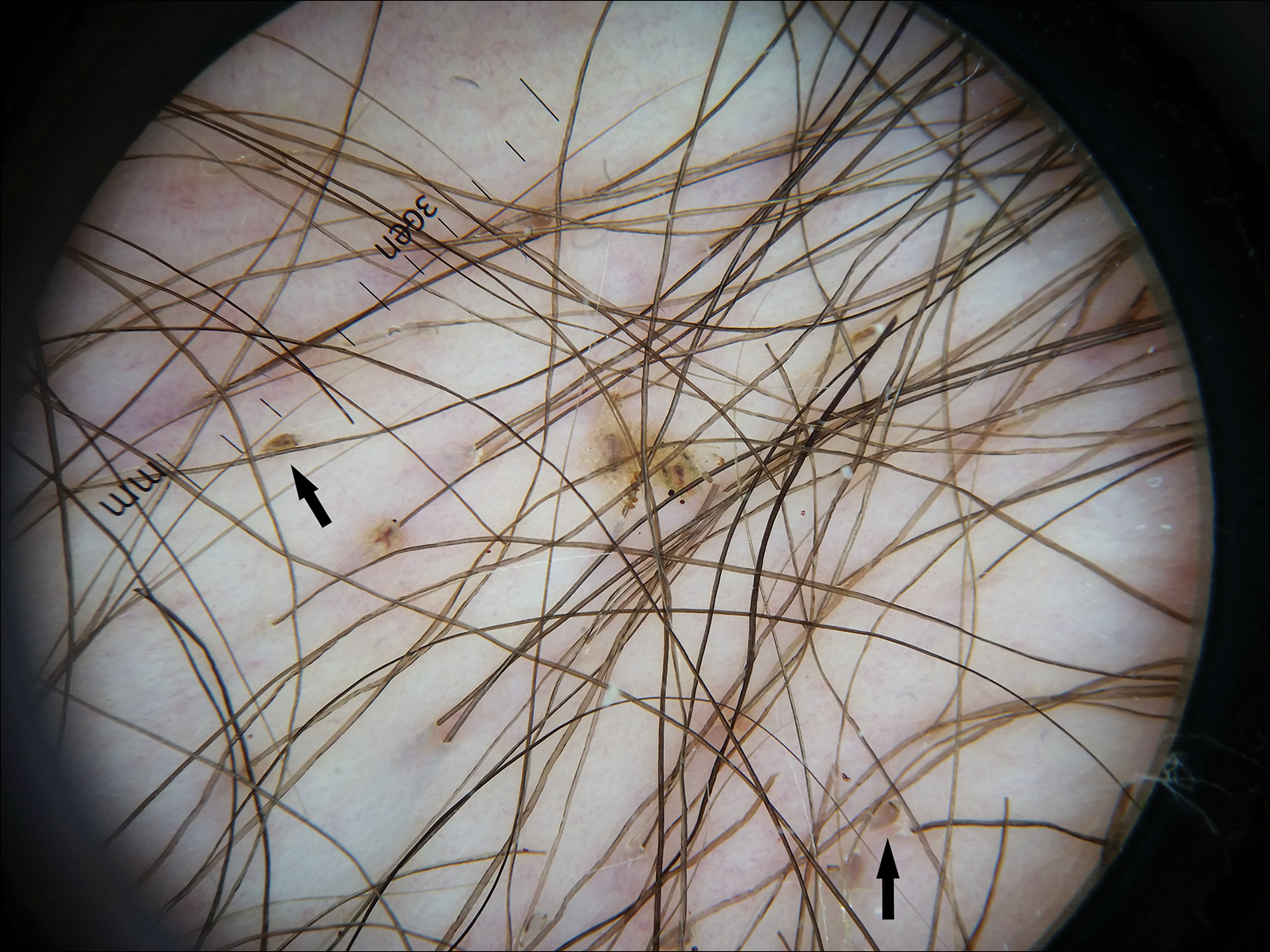
Dermoscopy of the pubic hair demonstrated a louse clutching multiple shafts of hairs (Figure) as well as scattered nits, confirming the presence of Phthirus pubis and diagnosis of pediculosis pubis. The key clinical diagnostic feature in this case was severe itching of the pubic area, with visible nits present on pubic hairs. Itching and infestation also can involve other hair-bearing areas such as the chest, legs, and axillae. The patient was treated with permethrin cream 5% applied to the pubic area and chest. Symptoms and infestation were resolved at 1-week follow-up.
Pediculosis pubis is an infestation of pubic hairs by the pubic (crab) louse P pubis, which feeds on host blood. Other body areas covered with dense hair also may be involved; 60% of patients are infested in at least 2 different sites.1 Pediculosis pubis is most commonly sexually transmitted through direct contact.2 Worldwide prevalence has been estimated at approximately 2% of the adult population, and a survey of 817 US college students in 2009 indicated a lifetime prevalence of 1.3%.3 The prevalence is slightly higher in men, highest in men who have sex with men, and rare in individuals with shaved pubic hair.1 The most common symptom is pruritus of the genital area. Infested patients also may develop asymptomatic bluish gray macules (maculae ceruleae) secondary to hemosiderin deposition from louse bites.4
The diagnosis of pediculosis pubis is made by identification of P pubis, either by examination with the naked eye or confirmation with dermoscopy or microscopy. Although the 0.8- to 1.2-mm lice are visible to the naked eye, they can be difficult to see if not filled with blood, and nits on the hairs can be mistaken for white piedra (fungal infection of the hair shafts) or trichomycosis pubis (bacterial infection of the hair shafts).4 Scabies and tinea cruris do not present with attachments to the hairs. Scabies may present with papules and burrows and tinea cruris with scaly erythematous plaques. Small numbers of lice and nits may be missed by the naked eye or a traditional magnifying glass.5 The use of dermoscopy allows for fast and accurate identification of the characteristic lice and nits, even in these more challenging cases.5 Accurate diagnosis is important, as approximately 31% of infested patients have other concurrent sexually transmitted infections that warrant screening.6
The Centers for Disease Control and Prevention recommends first-line treatment with permethrin cream (1% or 5%) or pyrethrin with piperonyl butoxide applied to all affected areas and washed off after 10 minutes.7 Patients should be reevaluated after 1 week if symptoms persist and re-treated if lice are found on examination.7,8 Malathion lotion 0.5% (applied and washed off after 8-12 hours) or oral ivermectin (250 µg/kg, repeated after 2 weeks) may be used for alternative therapies or cases of permethrin or pyrethrin resistance.7 Ivermectin also is effective for involvement of eyelashes where topical insecticides should not be used.9 Sexual partners should be treated to prevent repeat transmission.7,8 Bedding and clothing can be decontaminated by machine wash on hot cycle or isolating from body contact for 72 hours.7 Patients also should be screened for other sexually transmitted infections, including human immunodeficiency virus.6
- Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier/Saunders; 2012:1429-1430.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592-600.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12; quiz 13-14.
- Chuh A, Lee A, Wong W, et al. Diagnosis of pediculosis pubis: a novel application of digital epiluminescence dermatoscopy. J Eur Acad Dermatol Venereol. 2007;21:837-838.
- Chapel TA, Katta T, Kuszmar T, et al. Pediculosis pubis in a clinic for treatment of sexually transmitted diseases. Sex Transm Dis. 1979;6:257-260.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- Leone PA. Scabies and pediculosis pubis: an update of treatment regimens and general review. Clin Infect Dis. 2007;44(suppl 3):S153-S159.
- Burkhart CN, Burkhart CG. Oral ivermectin therapy for phthiriasis palpebrum. Arch Ophthalmol. 2000;118:134-135.
The Diagnosis: Pediculosis Pubis
Dermoscopy of the pubic hair demonstrated a louse clutching multiple shafts of hairs (Figure) as well as scattered nits, confirming the presence of Phthirus pubis and diagnosis of pediculosis pubis. The key clinical diagnostic feature in this case was severe itching of the pubic area, with visible nits present on pubic hairs. Itching and infestation also can involve other hair-bearing areas such as the chest, legs, and axillae. The patient was treated with permethrin cream 5% applied to the pubic area and chest. Symptoms and infestation were resolved at 1-week follow-up.
Pediculosis pubis is an infestation of pubic hairs by the pubic (crab) louse P pubis, which feeds on host blood. Other body areas covered with dense hair also may be involved; 60% of patients are infested in at least 2 different sites.1 Pediculosis pubis is most commonly sexually transmitted through direct contact.2 Worldwide prevalence has been estimated at approximately 2% of the adult population, and a survey of 817 US college students in 2009 indicated a lifetime prevalence of 1.3%.3 The prevalence is slightly higher in men, highest in men who have sex with men, and rare in individuals with shaved pubic hair.1 The most common symptom is pruritus of the genital area. Infested patients also may develop asymptomatic bluish gray macules (maculae ceruleae) secondary to hemosiderin deposition from louse bites.4
The diagnosis of pediculosis pubis is made by identification of P pubis, either by examination with the naked eye or confirmation with dermoscopy or microscopy. Although the 0.8- to 1.2-mm lice are visible to the naked eye, they can be difficult to see if not filled with blood, and nits on the hairs can be mistaken for white piedra (fungal infection of the hair shafts) or trichomycosis pubis (bacterial infection of the hair shafts).4 Scabies and tinea cruris do not present with attachments to the hairs. Scabies may present with papules and burrows and tinea cruris with scaly erythematous plaques. Small numbers of lice and nits may be missed by the naked eye or a traditional magnifying glass.5 The use of dermoscopy allows for fast and accurate identification of the characteristic lice and nits, even in these more challenging cases.5 Accurate diagnosis is important, as approximately 31% of infested patients have other concurrent sexually transmitted infections that warrant screening.6
The Centers for Disease Control and Prevention recommends first-line treatment with permethrin cream (1% or 5%) or pyrethrin with piperonyl butoxide applied to all affected areas and washed off after 10 minutes.7 Patients should be reevaluated after 1 week if symptoms persist and re-treated if lice are found on examination.7,8 Malathion lotion 0.5% (applied and washed off after 8-12 hours) or oral ivermectin (250 µg/kg, repeated after 2 weeks) may be used for alternative therapies or cases of permethrin or pyrethrin resistance.7 Ivermectin also is effective for involvement of eyelashes where topical insecticides should not be used.9 Sexual partners should be treated to prevent repeat transmission.7,8 Bedding and clothing can be decontaminated by machine wash on hot cycle or isolating from body contact for 72 hours.7 Patients also should be screened for other sexually transmitted infections, including human immunodeficiency virus.6
The Diagnosis: Pediculosis Pubis
Dermoscopy of the pubic hair demonstrated a louse clutching multiple shafts of hairs (Figure) as well as scattered nits, confirming the presence of Phthirus pubis and diagnosis of pediculosis pubis. The key clinical diagnostic feature in this case was severe itching of the pubic area, with visible nits present on pubic hairs. Itching and infestation also can involve other hair-bearing areas such as the chest, legs, and axillae. The patient was treated with permethrin cream 5% applied to the pubic area and chest. Symptoms and infestation were resolved at 1-week follow-up.
Pediculosis pubis is an infestation of pubic hairs by the pubic (crab) louse P pubis, which feeds on host blood. Other body areas covered with dense hair also may be involved; 60% of patients are infested in at least 2 different sites.1 Pediculosis pubis is most commonly sexually transmitted through direct contact.2 Worldwide prevalence has been estimated at approximately 2% of the adult population, and a survey of 817 US college students in 2009 indicated a lifetime prevalence of 1.3%.3 The prevalence is slightly higher in men, highest in men who have sex with men, and rare in individuals with shaved pubic hair.1 The most common symptom is pruritus of the genital area. Infested patients also may develop asymptomatic bluish gray macules (maculae ceruleae) secondary to hemosiderin deposition from louse bites.4
The diagnosis of pediculosis pubis is made by identification of P pubis, either by examination with the naked eye or confirmation with dermoscopy or microscopy. Although the 0.8- to 1.2-mm lice are visible to the naked eye, they can be difficult to see if not filled with blood, and nits on the hairs can be mistaken for white piedra (fungal infection of the hair shafts) or trichomycosis pubis (bacterial infection of the hair shafts).4 Scabies and tinea cruris do not present with attachments to the hairs. Scabies may present with papules and burrows and tinea cruris with scaly erythematous plaques. Small numbers of lice and nits may be missed by the naked eye or a traditional magnifying glass.5 The use of dermoscopy allows for fast and accurate identification of the characteristic lice and nits, even in these more challenging cases.5 Accurate diagnosis is important, as approximately 31% of infested patients have other concurrent sexually transmitted infections that warrant screening.6
The Centers for Disease Control and Prevention recommends first-line treatment with permethrin cream (1% or 5%) or pyrethrin with piperonyl butoxide applied to all affected areas and washed off after 10 minutes.7 Patients should be reevaluated after 1 week if symptoms persist and re-treated if lice are found on examination.7,8 Malathion lotion 0.5% (applied and washed off after 8-12 hours) or oral ivermectin (250 µg/kg, repeated after 2 weeks) may be used for alternative therapies or cases of permethrin or pyrethrin resistance.7 Ivermectin also is effective for involvement of eyelashes where topical insecticides should not be used.9 Sexual partners should be treated to prevent repeat transmission.7,8 Bedding and clothing can be decontaminated by machine wash on hot cycle or isolating from body contact for 72 hours.7 Patients also should be screened for other sexually transmitted infections, including human immunodeficiency virus.6
- Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier/Saunders; 2012:1429-1430.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592-600.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12; quiz 13-14.
- Chuh A, Lee A, Wong W, et al. Diagnosis of pediculosis pubis: a novel application of digital epiluminescence dermatoscopy. J Eur Acad Dermatol Venereol. 2007;21:837-838.
- Chapel TA, Katta T, Kuszmar T, et al. Pediculosis pubis in a clinic for treatment of sexually transmitted diseases. Sex Transm Dis. 1979;6:257-260.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- Leone PA. Scabies and pediculosis pubis: an update of treatment regimens and general review. Clin Infect Dis. 2007;44(suppl 3):S153-S159.
- Burkhart CN, Burkhart CG. Oral ivermectin therapy for phthiriasis palpebrum. Arch Ophthalmol. 2000;118:134-135.
- Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier/Saunders; 2012:1429-1430.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592-600.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12; quiz 13-14.
- Chuh A, Lee A, Wong W, et al. Diagnosis of pediculosis pubis: a novel application of digital epiluminescence dermatoscopy. J Eur Acad Dermatol Venereol. 2007;21:837-838.
- Chapel TA, Katta T, Kuszmar T, et al. Pediculosis pubis in a clinic for treatment of sexually transmitted diseases. Sex Transm Dis. 1979;6:257-260.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- Leone PA. Scabies and pediculosis pubis: an update of treatment regimens and general review. Clin Infect Dis. 2007;44(suppl 3):S153-S159.
- Burkhart CN, Burkhart CG. Oral ivermectin therapy for phthiriasis palpebrum. Arch Ophthalmol. 2000;118:134-135.
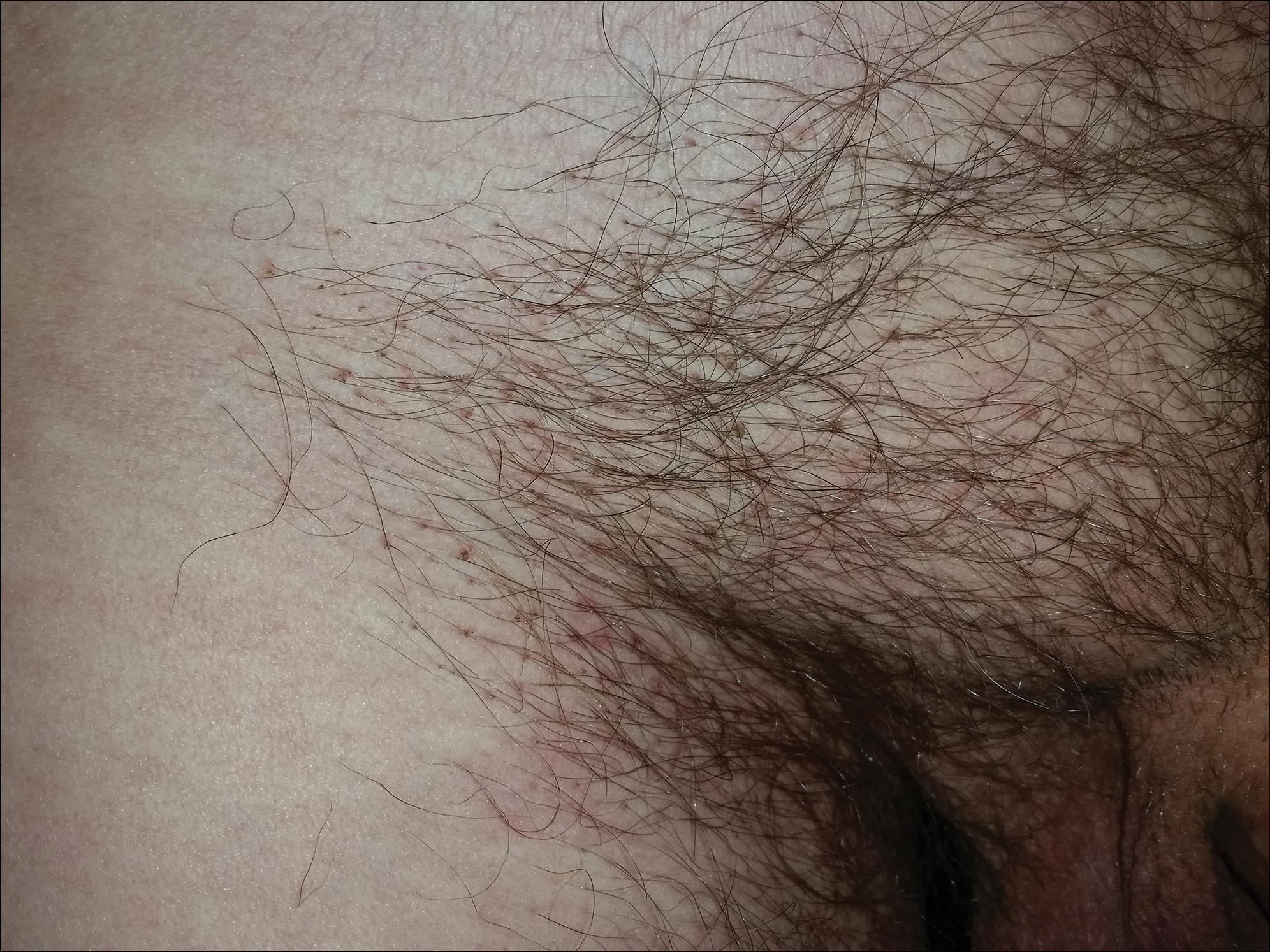
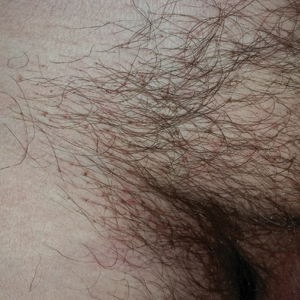
A 50-year-old man with a history of cerebrovascular accident presented with severe itching along the inguinal folds and over the chest of 2 months' duration. His last sexual encounter was 5 months prior. He had previously seen a primary care physician who told him he needed to clean the hair better. Examination of the genital area revealed pigmented macules and overlying particles among the pubic hair.
Innovations in Dermatology: Antibiotic Stewardship in Acne Therapy


Dermatology Residency Match: Data on Who Matched in 2018


Cross-contamination of Pathology Specimens: A Cautionary Tale
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
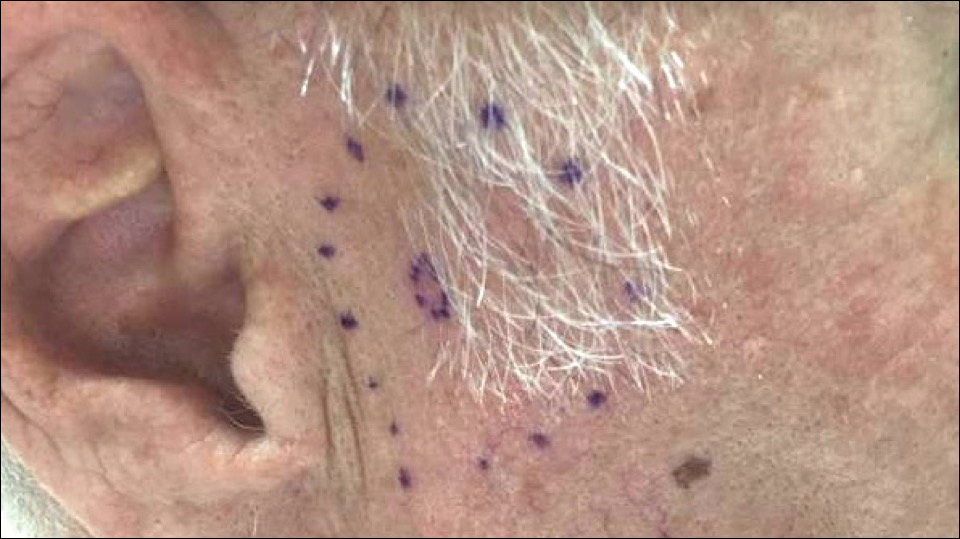
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
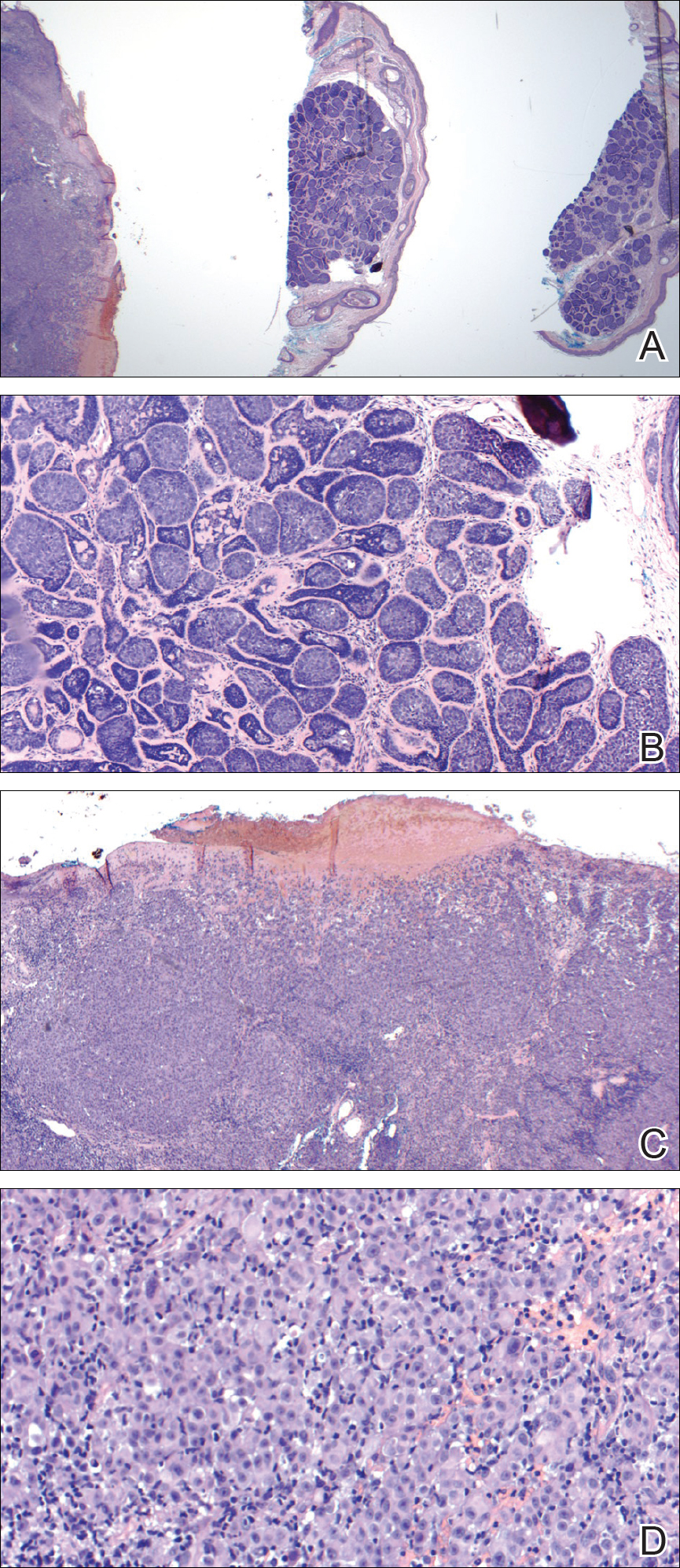
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
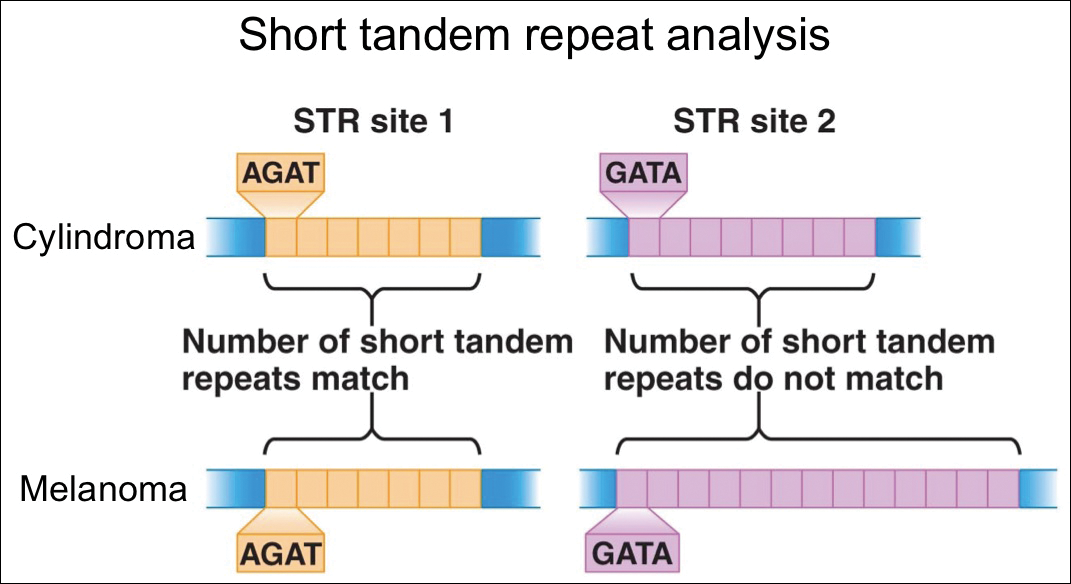
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
Cross-contamination of pathology specimens is a rare but nonnegligible source of potential morbidity in clinical practice. Contaminant tissue fragments, colloquially referred to as floaters, typically are readily identifiable based on obvious cytomorphologic differences, especially if the tissues arise from different organs; however, one cannot rely on such distinctions in a pathology laboratory dedicated to a single organ system (eg, dermatopathology). The inability to identify quickly and confidently the presence of a contaminant puts the patient at risk for misdiagnosis, which can lead to unnecessary morbidity or even mortality in the case of cancer misdiagnosis. Studies that have been conducted to estimate the incidence of this type of error have suggested an overall incidence rate between approximately 1% and 3%.1,2 Awareness of this phenomenon and careful scrutiny when the histopathologic evidence diverges considerably from the clinical impression is critical for minimizing the negative outcomes that could result from the presence of contaminant tissue. We present a case in which cross-contamination of a pathology specimen led to an initial erroneous diagnosis of an aggressive cutaneous melanoma in a patient with a benign adnexal neoplasm.
Case Report
A 72-year-old man was referred to the Pigmented Lesion and Melanoma Program at Stanford University Medical Center and Cancer Institute (Palo Alto, California) for evaluation and treatment of a presumed stage IIB melanoma on the right preauricular cheek based on a shave biopsy that had been performed (<1 month prior) by his local dermatology provider and subsequently read by an affiliated out-of-state dermatopathology laboratory. Per the clinical history that was gathered at the current presentation, neither the patient nor his wife had noticed the lesion prior to his dermatology provider pointing it out on the day of the biopsy. Additionally, he denied associated pain, bleeding, or ulceration. According to outside medical records, the referring dermatology provider described the lesion as a 4-mm pink pearly papule with telangiectasia favoring a diagnosis of basal cell carcinoma, and a diagnostic shave biopsy was performed. On presentation to our clinic, physical examination of the right preauricular cheek revealed a 4×3-mm depressed erythematous scar with no evidence of residual pigmentation or nodularity (Figure 1). There was no clinically appreciable regional lymphadenopathy.
The original dermatopathology report indicated an invasive melanoma with the following pathologic characteristics: superficial spreading type, Breslow depth of at least 2.16 mm, ulceration, and a mitotic index of 8 mitotic figures/mm2 with transection of the invasive component at the peripheral and deep margins. There was no evidence of regression, perineural invasion, lymphovascular invasion, or microsatellites. Interestingly, the report indicated that there also was a basaloid proliferation with features of cylindroma in the same pathology slide adjacent to the aggressive invasive melanoma that was described. Given the complexity of cases referred to our academic center, the standard of care includes internal dermatopathology review of all outside pathology specimens. This review proved critical to this patient’s care in light of the considerable divergence of the initial pathologic diagnosis and the reported clinical features of the lesion.
Internal review of the single pathology slide received from the referring provider showed a total of 4 sections, 3 of which are shown here (Figure 2A). Three sections, including the one not shown, were all consistent with a diagnosis of cylindroma and showed no evidence of a melanocytic proliferation (Figure 2B). However, the fourth section demonstrated marked morphologic dissimilarity compared to the other 3 sections. This outlier section showed a thick cutaneous melanoma with a Breslow depth of at least 2.1 mm, ulceration, a mitotic rate of 12 mitotic figures/mm2, and broad transection of the invasive component at the peripheral and deep margins (Figures 2C and 2D). Correlation with the gross description of tissue processing on the original pathology report indicating that the specimen had been trisected raised suspicion that the fourth and very dissimilar section could be a contaminant from another source that was incorporated into our patient’s histologic sections during processing. Taken together, these discrepancies made the diagnosis of cylindroma alone far more likely than cutaneous melanoma, but we needed conclusive evidence given the dramatic difference in prognosis and management between a cylindroma and an aggressive cutaneous melanoma.
For further diagnostic clarification, we performed polymorphic short tandem repeat (STR) analysis, a well-described forensic pathology technique, to determine if the melanoma and cylindroma specimens derived from different patients, as we hypothesized. This analysis revealed differences in all but one DNA locus tested between the cylindroma specimen and the melanoma specimen, confirming our hypothesis (Figure 3). Subsequent discussion of the case with staff from the dermatopathology laboratory that processed this specimen provided further support for our suspicion that the invasive melanoma specimen was part of a case processed prior to our patient’s benign lesion. Therefore, the wide local excision for treatment of the suspected melanoma fortunately was canceled, and the patient did not require further treatment of the benign cylindroma. The patient expressed relief and gratitude for this critical clarification and change in management.
Comment
Shah et al3 reported a similar case in which a benign granuloma of the lung masqueraded as a squamous cell carcinoma due to histopathologic contamination. Although few similar cases have been described in the literature, the risk posed by such contamination is remarkable, regardless of whether it occurs during specimen grossing, embedding, sectioning, or staining.1,4,5 This risk is amplified in facilities that process specimens originating predominantly from a single organ system or tissue type, as is often the case in dedicated dermatopathology laboratories. In this scenario, it is unlikely that one could use the presence of tissues from 2 different organ systems on a single slide as a way of easily recognizing the presence of a contaminant and rectifying the error. Additionally, the presence of malignant cells in the contaminant further complicates the problem and requires an investigation that can conclusively distinguish the contaminant from the patient’s actual tissue.
In our case, our dermatology and dermatopathology teams partnered with our molecular pathology team to find a solution. Polymorphic STR analysis via polymerase chain reaction amplification is a sensitive method employed commonly in forensic DNA laboratories for determining whether a sample submitted as evidence belongs to a given suspect.6 Although much more commonly used in forensics, STR analysis does have known roles in clinical medicine, such as chimerism testing after bone marrow or allogeneic stem cell transplantation.7 Given the relatively short period of time it takes along with the convenience of commercially available kits, a high discriminative ability, and well-validated interpretation procedures, STR analysis is an excellent method for determining if a given tissue sample came from a given patient, which is what was needed in our case.
The combined clinical, histopathologic, and molecular data in our case allowed for confident clarification of our patient’s diagnosis, sparing him the morbidity of wide local excision on the face, sentinel lymph node biopsy, and emotional distress associated with a diagnosis of aggressive cutaneous melanoma. Our case highlights the critical importance of internal review of pathology specimens in ensuring proper diagnosis and management and reminds us that, though rare, accidental contamination during processing of pathology specimens is a potential adverse event that must be considered, especially when a pathologic finding diverges considerably from what is anticipated based on the patient’s history and physical examination.
Acknowledgment
The authors express gratitude to the patient described herein who graciously provided permission for us to publish his case and clinical photography.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
- Gephardt GN, Zarbo RJ. Extraneous tissue in surgical pathology: a College of American Pathologists Q-Probes study of 275 laboratories. Arch Pathol Lab Med. 1996;120:1009-1014.
- Alam M, Shah AD, Ali S, et al. Floaters in Mohs micrographic surgery [published online June 27, 2013]. Dermatol Surg. 2013;39:1317-1322.
- Shah PA, Prat MP, Hostler DC. Benign granuloma masquerading as squamous cell carcinoma due to a “floater.” Hawaii J Med Public Health. 2017;76(11, suppl 2):19-21.
- Platt E, Sommer P, McDonald L, et al. Tissue floaters and contaminants in the histology laboratory. Arch Pathol Lab Med. 2009;133:973-978.
- Layfield LJ, Witt BL, Metzger KG, et al. Extraneous tissue: a potential source for diagnostic error in surgical pathology. Am J Clin Pathol. 2011;136:767-772.
- Butler JM. Forensic DNA testing. Cold Spring Harb Protoc. 2011;2011:1438-1450.
- Manasatienkij C, Ra-ngabpai C. Clinical application of forensic DNA analysis: a literature review. J Med Assoc Thai. 2012;95:1357-1363.
Resident Pearl
- Although cross-contamination of pathology specimens is rare, it does occur and can impact diagnosis and management if detected early.
Solitary Exophytic Plaque on the Left Groin
The Diagnosis: Pemphigus Vegetans
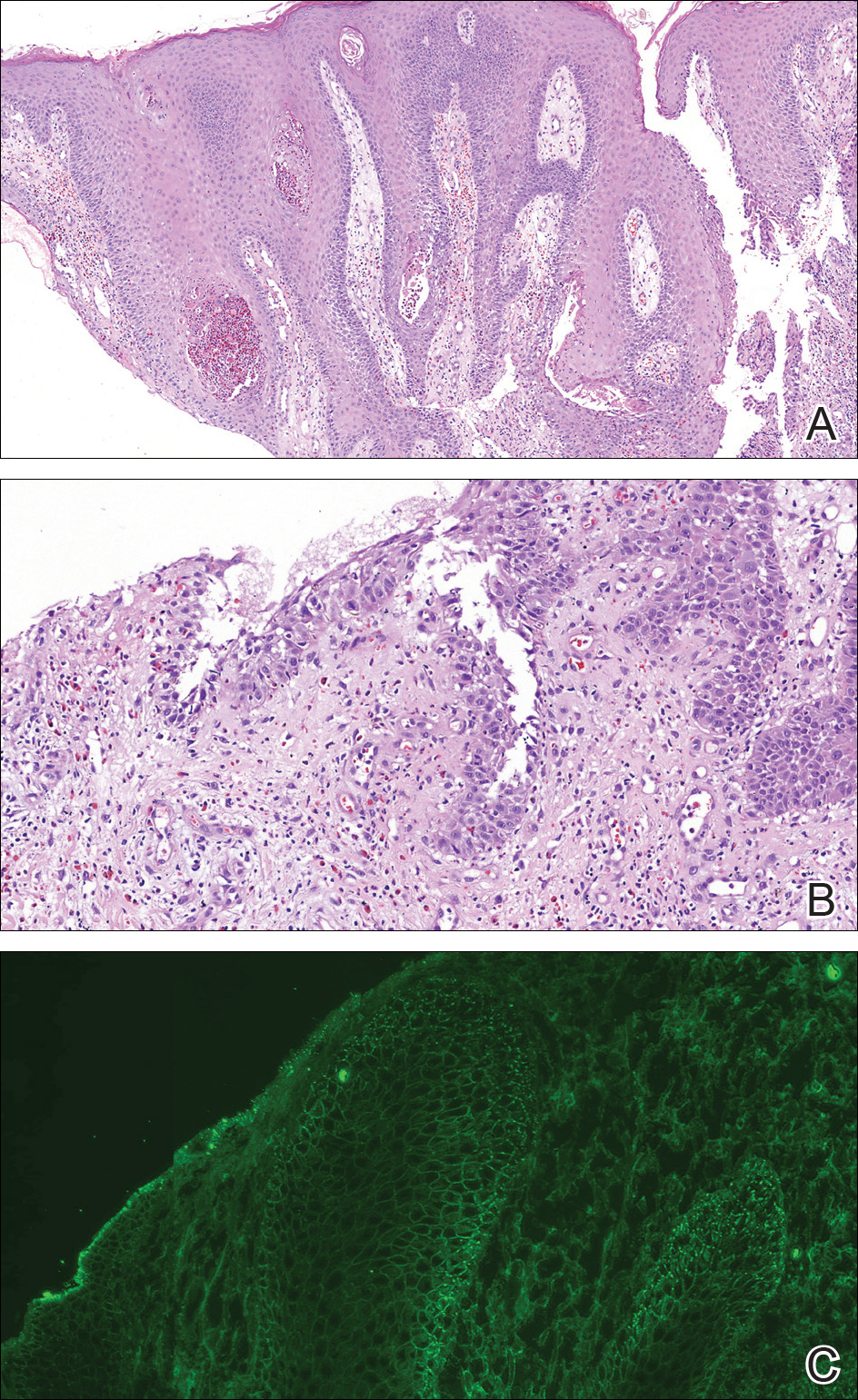
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
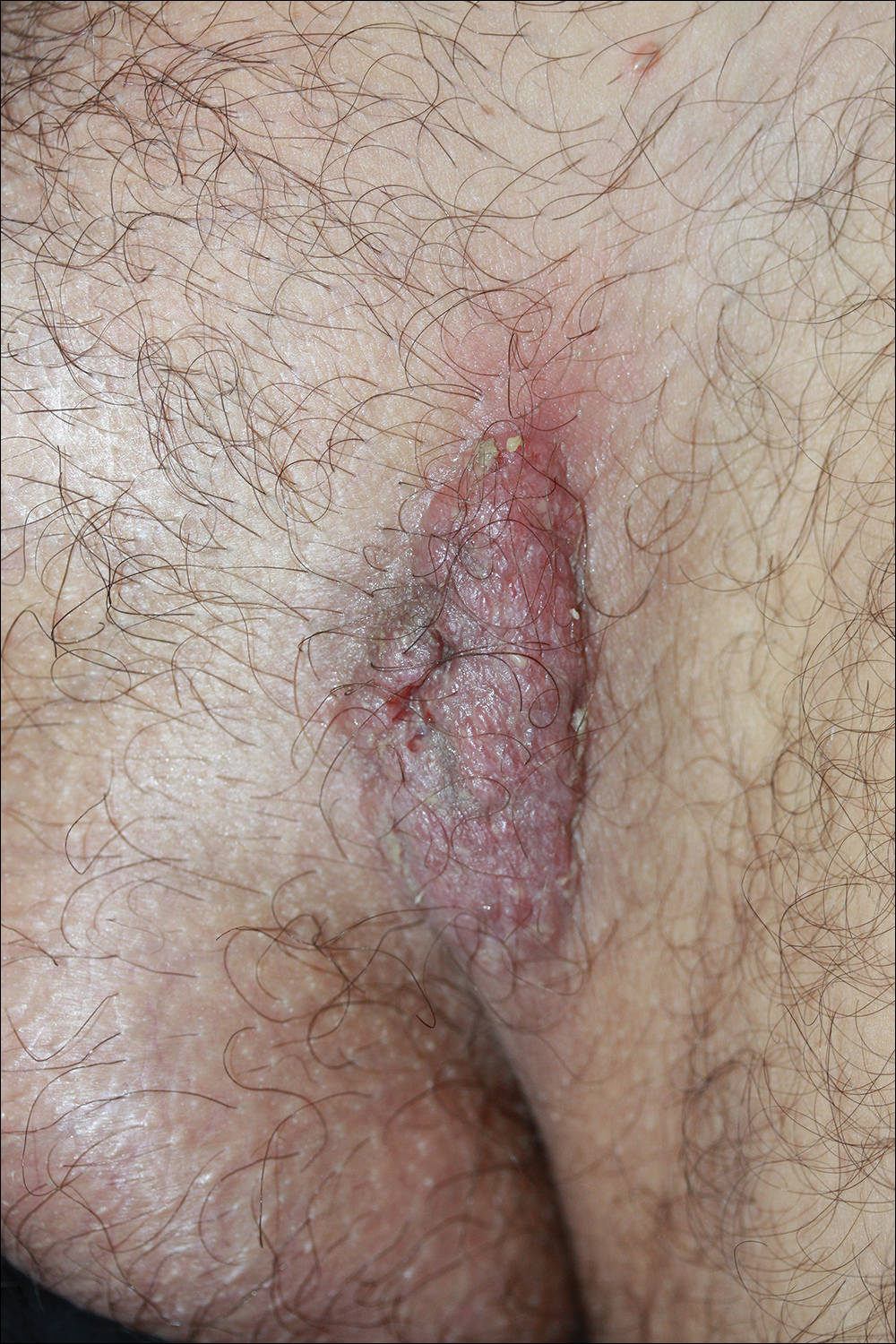
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
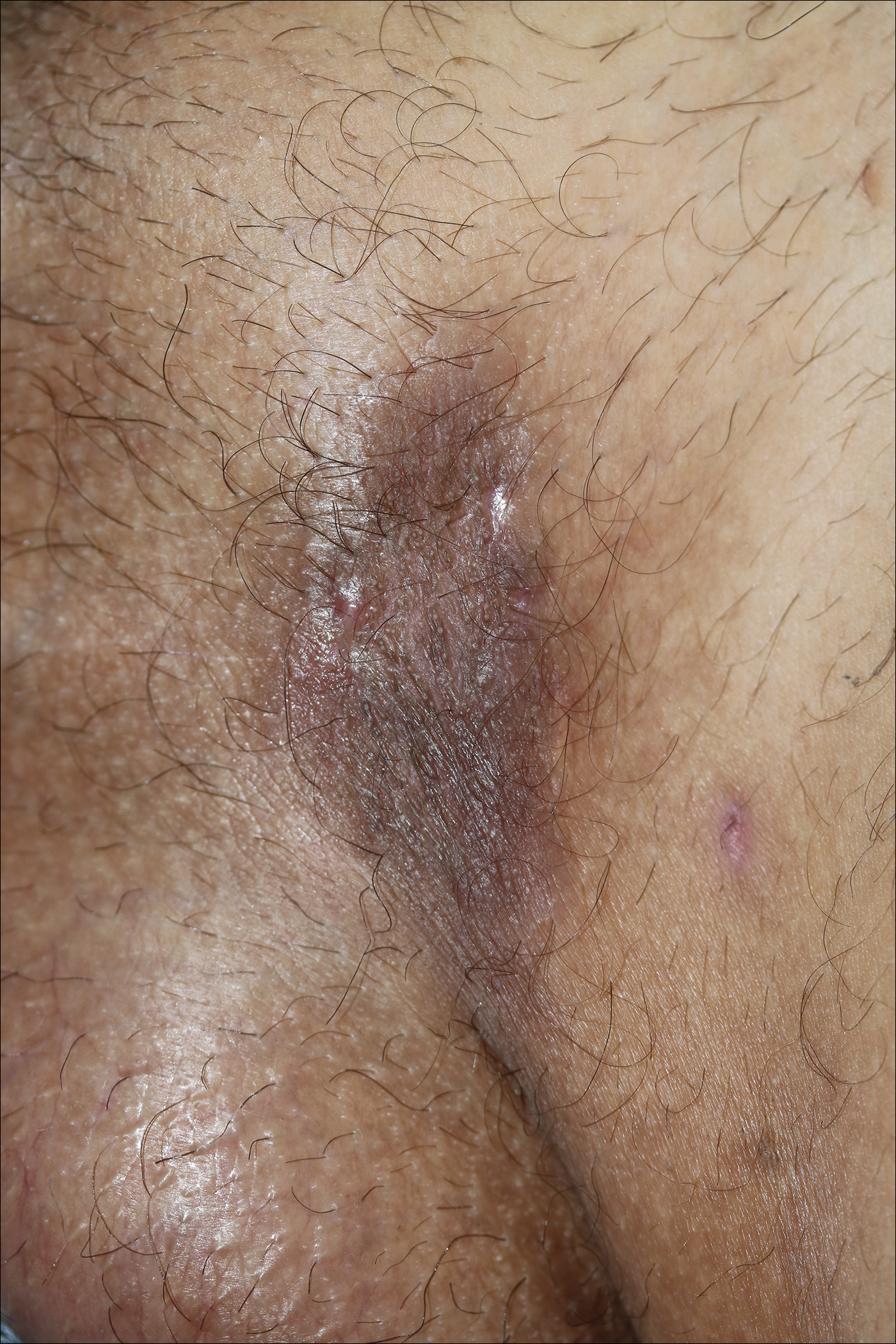
A 40-year-old otherwise healthy man presented with an exophytic plaque on the left groin of 1 month's duration. The lesion reportedly emerged as pustules that slowly expanded and coalesced. At an outside institution, cryotherapy was planned for a presumed diagnosis of condyloma acuminatum; however, the patient decided to get a second opinion. He denied recent intake of new drugs. Six months prior he had traveled to China and engaged in unprotected sexual intercourse. Physical examination revealed an approximately 4×2-cm exophytic plaque with a partially eroded and exudative surface on the left inguinal fold. Dermatologic examination, including the oral mucosa, was otherwise normal. Complete blood cell count and sexually transmitted disease panel were unremarkable.
Pediatric Skin Care: Survey of the Cutis Editorial Board
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on pediatric skin care. Here’s what we found.
Do you recommend sunscreen in babies younger than 6 months?
More than half (64%) of dermatologists we surveyed do not recommend using sunscreens in babies younger than 6 months; they should stay out of the sun. They recommended sun-protective clothing, hats, and sunglasses in this age group.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Babies younger than 6 months are still developing barrier functionality and have a higher surface area to body weight ratio compared to older children and adults. There is decreased UV light barrier protection and increased risk for systemic drug absorption. Therefore, it is best to avoid sunscreen in babies younger than 6 months. Instead, I recommend avoiding sunlight during peak hours, keeping babies in the shade, and dressing them in sun-protective clothing and hats.
Next page: Bathing and eczema
What advice do you give parents/guardians on bathing for babies with eczema?
Two-thirds of dermatologists (68%) indicated that moisturizers should be applied after bathing babies with eczema. More than half (64%) suggested using fragrance-free cleansers. Results varied on the frequency of bathing; 32% said bathe once daily for short periods with warm water, and 41% suggested to reduce bathing to a few nights a week.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Dry skin is a characteristic feature of atopic dermatitis. We now know that skin barrier dysfunction, such as filaggrin deficiency, contributes to the pathophysiology in some patients. In addition, a paucity of stratum corneum and intercellular lipids increases transepidermal water loss. Therefore, emollients containing humectants to augment stratum corneum hydration and occludents to reduce evaporation are extremely helpful in maintenance treatment of atopic dermatitis.
Next page: Nonsteroidal agents
Do you prescribe nonsteroidal agents for children younger than 2 years?
Almost two-thirds (64%) of dermatologists do prescribe nonsteroidal agents for children younger than 2 years.
If yes, which nonsteroidal agents do you prescribe for children?
Of those dermatologists who indicated that they do prescribe nonsteroidal agents for children younger than 2 years, almost half (45%) prescribe pimecrolimus, while 36% prescribe crisaborole or tacrolimus.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
We have limited options for US Food and Drug Administration (FDA)–approved therapies for atopic dermatitis in children younger than 2 years. The risks of adverse effects also are higher in younger children compared to older children and adults, particularly hypothalamic-pituitary-adrenal suppression with corticosteroids. Pimecrolimus was the most common nonsteroidal prescribed in this group. It should be noted that pimecrolimus is not FDA approved for use in children younger than 2 years; however, 2 phase 3 studies were conducted with 436 infants aged 3 months to 23 months and they demonstrated safety and efficacy.
Next page: Procedures in adolescents
Which procedures do you perform on adolescents?
Forty-one percent of dermatologists surveyed indicated that they do not perform procedures on adolescents. Of those that do, 32% use laser hair removal or chemical peels in adolescents, while only 23% each use light therapy for acne or onabotulinumtoxinA for hyperhidrosis. Pulsed dye laser use was reported in only 18% of dermatologists.

Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The results of this survey are a reflection of the relatively recent trends in dermatology training. Now that there is board certification in pediatric dermatology, many dermatologists now refer to pediatric dermatologists for children who need or want procedures.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Keep the regimen as simple as possible and make sure your skin care advice is culturally relevant.—Craig Burkhart, MD (Chapel Hill, North Carolina)
Please, avoid direct sun exposure as much as possible by wearing protective garments and finding shades in the outside.—Jisun Cha, MD (New Brunswick, New Jersey)
Good habits start early. Teach children how to care for their skin and they will carry that practice with them over the course of their lifetime, and hopefully pass it on.—James Q. Del Rosso, DO (Las Vegas, Nevada)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from September 13, 2018, to October 4, 2018. A total of 22 usable responses were received.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on pediatric skin care. Here’s what we found.
Do you recommend sunscreen in babies younger than 6 months?
More than half (64%) of dermatologists we surveyed do not recommend using sunscreens in babies younger than 6 months; they should stay out of the sun. They recommended sun-protective clothing, hats, and sunglasses in this age group.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Babies younger than 6 months are still developing barrier functionality and have a higher surface area to body weight ratio compared to older children and adults. There is decreased UV light barrier protection and increased risk for systemic drug absorption. Therefore, it is best to avoid sunscreen in babies younger than 6 months. Instead, I recommend avoiding sunlight during peak hours, keeping babies in the shade, and dressing them in sun-protective clothing and hats.
Next page: Bathing and eczema
What advice do you give parents/guardians on bathing for babies with eczema?
Two-thirds of dermatologists (68%) indicated that moisturizers should be applied after bathing babies with eczema. More than half (64%) suggested using fragrance-free cleansers. Results varied on the frequency of bathing; 32% said bathe once daily for short periods with warm water, and 41% suggested to reduce bathing to a few nights a week.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Dry skin is a characteristic feature of atopic dermatitis. We now know that skin barrier dysfunction, such as filaggrin deficiency, contributes to the pathophysiology in some patients. In addition, a paucity of stratum corneum and intercellular lipids increases transepidermal water loss. Therefore, emollients containing humectants to augment stratum corneum hydration and occludents to reduce evaporation are extremely helpful in maintenance treatment of atopic dermatitis.
Next page: Nonsteroidal agents
Do you prescribe nonsteroidal agents for children younger than 2 years?
Almost two-thirds (64%) of dermatologists do prescribe nonsteroidal agents for children younger than 2 years.
If yes, which nonsteroidal agents do you prescribe for children?
Of those dermatologists who indicated that they do prescribe nonsteroidal agents for children younger than 2 years, almost half (45%) prescribe pimecrolimus, while 36% prescribe crisaborole or tacrolimus.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
We have limited options for US Food and Drug Administration (FDA)–approved therapies for atopic dermatitis in children younger than 2 years. The risks of adverse effects also are higher in younger children compared to older children and adults, particularly hypothalamic-pituitary-adrenal suppression with corticosteroids. Pimecrolimus was the most common nonsteroidal prescribed in this group. It should be noted that pimecrolimus is not FDA approved for use in children younger than 2 years; however, 2 phase 3 studies were conducted with 436 infants aged 3 months to 23 months and they demonstrated safety and efficacy.
Next page: Procedures in adolescents
Which procedures do you perform on adolescents?
Forty-one percent of dermatologists surveyed indicated that they do not perform procedures on adolescents. Of those that do, 32% use laser hair removal or chemical peels in adolescents, while only 23% each use light therapy for acne or onabotulinumtoxinA for hyperhidrosis. Pulsed dye laser use was reported in only 18% of dermatologists.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The results of this survey are a reflection of the relatively recent trends in dermatology training. Now that there is board certification in pediatric dermatology, many dermatologists now refer to pediatric dermatologists for children who need or want procedures.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Keep the regimen as simple as possible and make sure your skin care advice is culturally relevant.—Craig Burkhart, MD (Chapel Hill, North Carolina)
Please, avoid direct sun exposure as much as possible by wearing protective garments and finding shades in the outside.—Jisun Cha, MD (New Brunswick, New Jersey)
Good habits start early. Teach children how to care for their skin and they will carry that practice with them over the course of their lifetime, and hopefully pass it on.—James Q. Del Rosso, DO (Las Vegas, Nevada)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from September 13, 2018, to October 4, 2018. A total of 22 usable responses were received.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on pediatric skin care. Here’s what we found.
Do you recommend sunscreen in babies younger than 6 months?
More than half (64%) of dermatologists we surveyed do not recommend using sunscreens in babies younger than 6 months; they should stay out of the sun. They recommended sun-protective clothing, hats, and sunglasses in this age group.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Babies younger than 6 months are still developing barrier functionality and have a higher surface area to body weight ratio compared to older children and adults. There is decreased UV light barrier protection and increased risk for systemic drug absorption. Therefore, it is best to avoid sunscreen in babies younger than 6 months. Instead, I recommend avoiding sunlight during peak hours, keeping babies in the shade, and dressing them in sun-protective clothing and hats.
Next page: Bathing and eczema
What advice do you give parents/guardians on bathing for babies with eczema?
Two-thirds of dermatologists (68%) indicated that moisturizers should be applied after bathing babies with eczema. More than half (64%) suggested using fragrance-free cleansers. Results varied on the frequency of bathing; 32% said bathe once daily for short periods with warm water, and 41% suggested to reduce bathing to a few nights a week.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Dry skin is a characteristic feature of atopic dermatitis. We now know that skin barrier dysfunction, such as filaggrin deficiency, contributes to the pathophysiology in some patients. In addition, a paucity of stratum corneum and intercellular lipids increases transepidermal water loss. Therefore, emollients containing humectants to augment stratum corneum hydration and occludents to reduce evaporation are extremely helpful in maintenance treatment of atopic dermatitis.
Next page: Nonsteroidal agents
Do you prescribe nonsteroidal agents for children younger than 2 years?
Almost two-thirds (64%) of dermatologists do prescribe nonsteroidal agents for children younger than 2 years.
If yes, which nonsteroidal agents do you prescribe for children?
Of those dermatologists who indicated that they do prescribe nonsteroidal agents for children younger than 2 years, almost half (45%) prescribe pimecrolimus, while 36% prescribe crisaborole or tacrolimus.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
We have limited options for US Food and Drug Administration (FDA)–approved therapies for atopic dermatitis in children younger than 2 years. The risks of adverse effects also are higher in younger children compared to older children and adults, particularly hypothalamic-pituitary-adrenal suppression with corticosteroids. Pimecrolimus was the most common nonsteroidal prescribed in this group. It should be noted that pimecrolimus is not FDA approved for use in children younger than 2 years; however, 2 phase 3 studies were conducted with 436 infants aged 3 months to 23 months and they demonstrated safety and efficacy.
Next page: Procedures in adolescents
Which procedures do you perform on adolescents?
Forty-one percent of dermatologists surveyed indicated that they do not perform procedures on adolescents. Of those that do, 32% use laser hair removal or chemical peels in adolescents, while only 23% each use light therapy for acne or onabotulinumtoxinA for hyperhidrosis. Pulsed dye laser use was reported in only 18% of dermatologists.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The results of this survey are a reflection of the relatively recent trends in dermatology training. Now that there is board certification in pediatric dermatology, many dermatologists now refer to pediatric dermatologists for children who need or want procedures.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Keep the regimen as simple as possible and make sure your skin care advice is culturally relevant.—Craig Burkhart, MD (Chapel Hill, North Carolina)
Please, avoid direct sun exposure as much as possible by wearing protective garments and finding shades in the outside.—Jisun Cha, MD (New Brunswick, New Jersey)
Good habits start early. Teach children how to care for their skin and they will carry that practice with them over the course of their lifetime, and hopefully pass it on.—James Q. Del Rosso, DO (Las Vegas, Nevada)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from September 13, 2018, to October 4, 2018. A total of 22 usable responses were received.
Cutaneous Angiosarcoma of the Lower Leg
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.

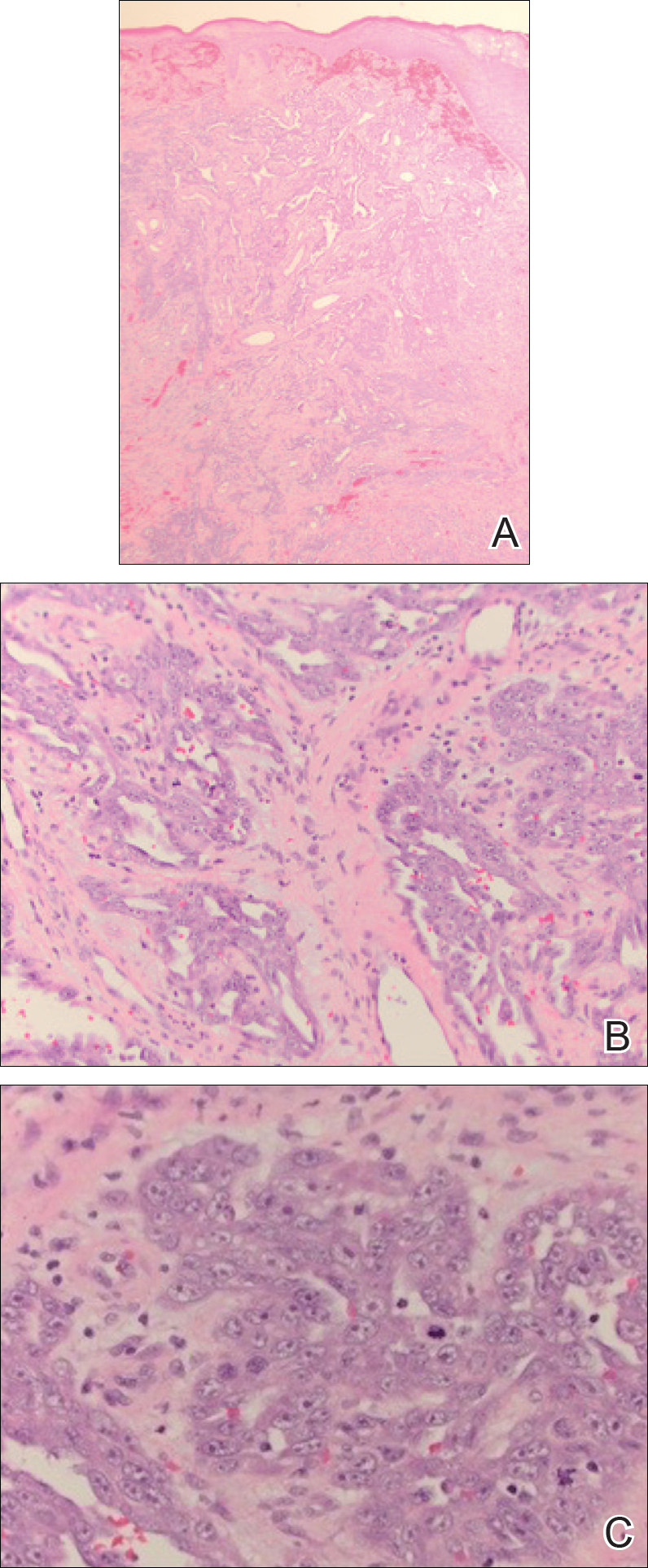
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.

Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.
Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.
Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
Practice Points
- Cutaneous angiosarcoma is a rare malignant vascular neoplasm typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically in the setting of chronic lymphedema following mastectomy (Stewart-Treves syndrome).
- There should be a low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy.
- Histologic analysis of angiosarcoma shows positive staining for CD34 and vimentin with poorly differentiated endothelial atypia with mitotic figures.
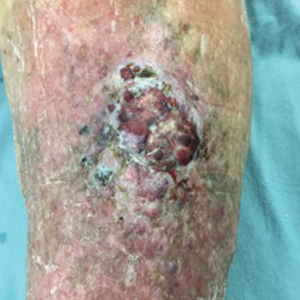
What Would I Tell My Intern-Year Self?
The training path to dermatology can seem interminable. From getting good grades in college to seeking out the “right” extracurricular activities and cramming for the MCAT, just getting into medical school was a huge challenge. In medical school, you may recognize the same chaos as you begin to prepare for US Medical Licensing Examination Step 1, try to volunteer, and publish original research. Dermatology is undeniably a competitive specialty. The 2018 data released by the National Resident Match Program (also called The Match) showed that only 83% of 412 US seniors who applied were matched to dermatology.1 The average Step 1 score for those who matched was 249 versus 241 for those who did not match. In addition, they had an average of 5.2 research experiences, 9.1 volunteer experiences, and 49.1 were members of Alpha Omega Alpha.1
After studying and working to meet these targets, it is not surprising that the transition to residency is a big change. As a dermatology preliminary intern, or“prelim,” our experience differs compared to other specialties, as other interns are jumping into their area of practice right away.
During my intern year, I had a tremendous amount of anxiety about 2 things: (1) being a subpar medical intern and (2) being unprepared for the beginning of my dermatology residency. This anxiety drove me to read a tremendous amount of medical and dermatological literature in an effort to do everything. Although hindsight is always 20/20, I will share some thoughts of my own as well as some from friends and colleagues.
First, enjoy intern year. I know that may sound ridiculous, but there were many aspects of intern year that I loved! When your pager beeps, it’s for YOU! You are no longer a subintern, running every decision past your intern or explaining your student status to the patients! Proudly introduce yourself as Dr. So-and-So. You earned it! I loved the camaraderie of working with my co-interns and senior residents. Going through the challenges of intern year together is a deep bonding experience, and I absolutely made lifelong friendships. It also does not hurt that I met my boyfriend (now husband), which has changed my life in a big way.
When it comes to learning internal medicine, pediatrics, or surgery (depending on your intern year), prepare for rounds, read about your patients, and pay attention in Grand Rounds. You can even consider taking the dermatologic cases that may be on your team, just for fun. I am always grateful for my internal medicine knowledge when managing complex medical dermatology patients and rounding on our consultation service on the wards. However, do not burden yourself with excessive studying. Enjoy your time off: spend it with family and friends or rediscover a hobby that has been neglected while you have been working toward your achievements.
When it comes to learning dermatology, do not rush it! You have 3 years and a ton of studying ahead of you! You will learn all of it. When July 1 of your first year of dermatology finally starts, immerse yourself in this new world:
- Attend conferences. Even if they are on topics you might not be interested in—from cosmetics to psoriasis—they provide a real-world perspective and often have great lecturers sharing their knowledge.
- Get involved. There are many dermatologic societies to take part in, and dues are waived or reduced when you sign up as a resident. Many of them provide great resources from study materials to journals, and they are always a great way to network when there are events.
- Volunteer. Many of the dermatologic societies sponsor volunteer events such as skin cancer screenings. It can be a fun way to network while also giving back to the community.
- Spend time figuring out what you really enjoy. This step may seem self-evident, but after many years of fulfilling the necessary criteria to get into medical school and residency, it can be habitual to start fulfilling the same criteria all over again. Explore all aspects of dermatology and see what truly interests you. Consider how you expect your life after residency to be and think what learning opportunities might be helpful down the road. Reach out to attendings you would like to work with, both in dermatology and in other specialties. I personally enjoyed working in wound and oncology clinics, learning how other specialties approach clinical dilemmas that we see in dermatology.
As I embark on my final year of dermatology residency, I am truly grateful for the wisdom that has been shared with me on this journey. Many people have provided key pieces of information that have helped shape my training and my plans for the future, and I hope that sharing it will help others!
- National Resident Matching Program, Charting Outcomes in the Match: U.S. Allopathic Seniors, 2018. Washington, DC: National Resident Matching Program; 2018. http://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf. Accessed September 20, 2018.
The training path to dermatology can seem interminable. From getting good grades in college to seeking out the “right” extracurricular activities and cramming for the MCAT, just getting into medical school was a huge challenge. In medical school, you may recognize the same chaos as you begin to prepare for US Medical Licensing Examination Step 1, try to volunteer, and publish original research. Dermatology is undeniably a competitive specialty. The 2018 data released by the National Resident Match Program (also called The Match) showed that only 83% of 412 US seniors who applied were matched to dermatology.1 The average Step 1 score for those who matched was 249 versus 241 for those who did not match. In addition, they had an average of 5.2 research experiences, 9.1 volunteer experiences, and 49.1 were members of Alpha Omega Alpha.1
After studying and working to meet these targets, it is not surprising that the transition to residency is a big change. As a dermatology preliminary intern, or“prelim,” our experience differs compared to other specialties, as other interns are jumping into their area of practice right away.
During my intern year, I had a tremendous amount of anxiety about 2 things: (1) being a subpar medical intern and (2) being unprepared for the beginning of my dermatology residency. This anxiety drove me to read a tremendous amount of medical and dermatological literature in an effort to do everything. Although hindsight is always 20/20, I will share some thoughts of my own as well as some from friends and colleagues.
First, enjoy intern year. I know that may sound ridiculous, but there were many aspects of intern year that I loved! When your pager beeps, it’s for YOU! You are no longer a subintern, running every decision past your intern or explaining your student status to the patients! Proudly introduce yourself as Dr. So-and-So. You earned it! I loved the camaraderie of working with my co-interns and senior residents. Going through the challenges of intern year together is a deep bonding experience, and I absolutely made lifelong friendships. It also does not hurt that I met my boyfriend (now husband), which has changed my life in a big way.
When it comes to learning internal medicine, pediatrics, or surgery (depending on your intern year), prepare for rounds, read about your patients, and pay attention in Grand Rounds. You can even consider taking the dermatologic cases that may be on your team, just for fun. I am always grateful for my internal medicine knowledge when managing complex medical dermatology patients and rounding on our consultation service on the wards. However, do not burden yourself with excessive studying. Enjoy your time off: spend it with family and friends or rediscover a hobby that has been neglected while you have been working toward your achievements.
When it comes to learning dermatology, do not rush it! You have 3 years and a ton of studying ahead of you! You will learn all of it. When July 1 of your first year of dermatology finally starts, immerse yourself in this new world:
- Attend conferences. Even if they are on topics you might not be interested in—from cosmetics to psoriasis—they provide a real-world perspective and often have great lecturers sharing their knowledge.
- Get involved. There are many dermatologic societies to take part in, and dues are waived or reduced when you sign up as a resident. Many of them provide great resources from study materials to journals, and they are always a great way to network when there are events.
- Volunteer. Many of the dermatologic societies sponsor volunteer events such as skin cancer screenings. It can be a fun way to network while also giving back to the community.
- Spend time figuring out what you really enjoy. This step may seem self-evident, but after many years of fulfilling the necessary criteria to get into medical school and residency, it can be habitual to start fulfilling the same criteria all over again. Explore all aspects of dermatology and see what truly interests you. Consider how you expect your life after residency to be and think what learning opportunities might be helpful down the road. Reach out to attendings you would like to work with, both in dermatology and in other specialties. I personally enjoyed working in wound and oncology clinics, learning how other specialties approach clinical dilemmas that we see in dermatology.
As I embark on my final year of dermatology residency, I am truly grateful for the wisdom that has been shared with me on this journey. Many people have provided key pieces of information that have helped shape my training and my plans for the future, and I hope that sharing it will help others!
The training path to dermatology can seem interminable. From getting good grades in college to seeking out the “right” extracurricular activities and cramming for the MCAT, just getting into medical school was a huge challenge. In medical school, you may recognize the same chaos as you begin to prepare for US Medical Licensing Examination Step 1, try to volunteer, and publish original research. Dermatology is undeniably a competitive specialty. The 2018 data released by the National Resident Match Program (also called The Match) showed that only 83% of 412 US seniors who applied were matched to dermatology.1 The average Step 1 score for those who matched was 249 versus 241 for those who did not match. In addition, they had an average of 5.2 research experiences, 9.1 volunteer experiences, and 49.1 were members of Alpha Omega Alpha.1
After studying and working to meet these targets, it is not surprising that the transition to residency is a big change. As a dermatology preliminary intern, or“prelim,” our experience differs compared to other specialties, as other interns are jumping into their area of practice right away.
During my intern year, I had a tremendous amount of anxiety about 2 things: (1) being a subpar medical intern and (2) being unprepared for the beginning of my dermatology residency. This anxiety drove me to read a tremendous amount of medical and dermatological literature in an effort to do everything. Although hindsight is always 20/20, I will share some thoughts of my own as well as some from friends and colleagues.
First, enjoy intern year. I know that may sound ridiculous, but there were many aspects of intern year that I loved! When your pager beeps, it’s for YOU! You are no longer a subintern, running every decision past your intern or explaining your student status to the patients! Proudly introduce yourself as Dr. So-and-So. You earned it! I loved the camaraderie of working with my co-interns and senior residents. Going through the challenges of intern year together is a deep bonding experience, and I absolutely made lifelong friendships. It also does not hurt that I met my boyfriend (now husband), which has changed my life in a big way.
When it comes to learning internal medicine, pediatrics, or surgery (depending on your intern year), prepare for rounds, read about your patients, and pay attention in Grand Rounds. You can even consider taking the dermatologic cases that may be on your team, just for fun. I am always grateful for my internal medicine knowledge when managing complex medical dermatology patients and rounding on our consultation service on the wards. However, do not burden yourself with excessive studying. Enjoy your time off: spend it with family and friends or rediscover a hobby that has been neglected while you have been working toward your achievements.
When it comes to learning dermatology, do not rush it! You have 3 years and a ton of studying ahead of you! You will learn all of it. When July 1 of your first year of dermatology finally starts, immerse yourself in this new world:
- Attend conferences. Even if they are on topics you might not be interested in—from cosmetics to psoriasis—they provide a real-world perspective and often have great lecturers sharing their knowledge.
- Get involved. There are many dermatologic societies to take part in, and dues are waived or reduced when you sign up as a resident. Many of them provide great resources from study materials to journals, and they are always a great way to network when there are events.
- Volunteer. Many of the dermatologic societies sponsor volunteer events such as skin cancer screenings. It can be a fun way to network while also giving back to the community.
- Spend time figuring out what you really enjoy. This step may seem self-evident, but after many years of fulfilling the necessary criteria to get into medical school and residency, it can be habitual to start fulfilling the same criteria all over again. Explore all aspects of dermatology and see what truly interests you. Consider how you expect your life after residency to be and think what learning opportunities might be helpful down the road. Reach out to attendings you would like to work with, both in dermatology and in other specialties. I personally enjoyed working in wound and oncology clinics, learning how other specialties approach clinical dilemmas that we see in dermatology.
As I embark on my final year of dermatology residency, I am truly grateful for the wisdom that has been shared with me on this journey. Many people have provided key pieces of information that have helped shape my training and my plans for the future, and I hope that sharing it will help others!
- National Resident Matching Program, Charting Outcomes in the Match: U.S. Allopathic Seniors, 2018. Washington, DC: National Resident Matching Program; 2018. http://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf. Accessed September 20, 2018.
- National Resident Matching Program, Charting Outcomes in the Match: U.S. Allopathic Seniors, 2018. Washington, DC: National Resident Matching Program; 2018. http://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf. Accessed September 20, 2018.

Diffuse Nonscarring Alopecia
The Diagnosis: Trichotillomania
A scalp punch biopsy revealed pigmented hair casts, an increase in catagen and telogen follicles, and a lack of perifollicular inflammation (Figure). Based on the clinical and histopathological findings, a diagnosis of trichotillomania (TTM) was established.
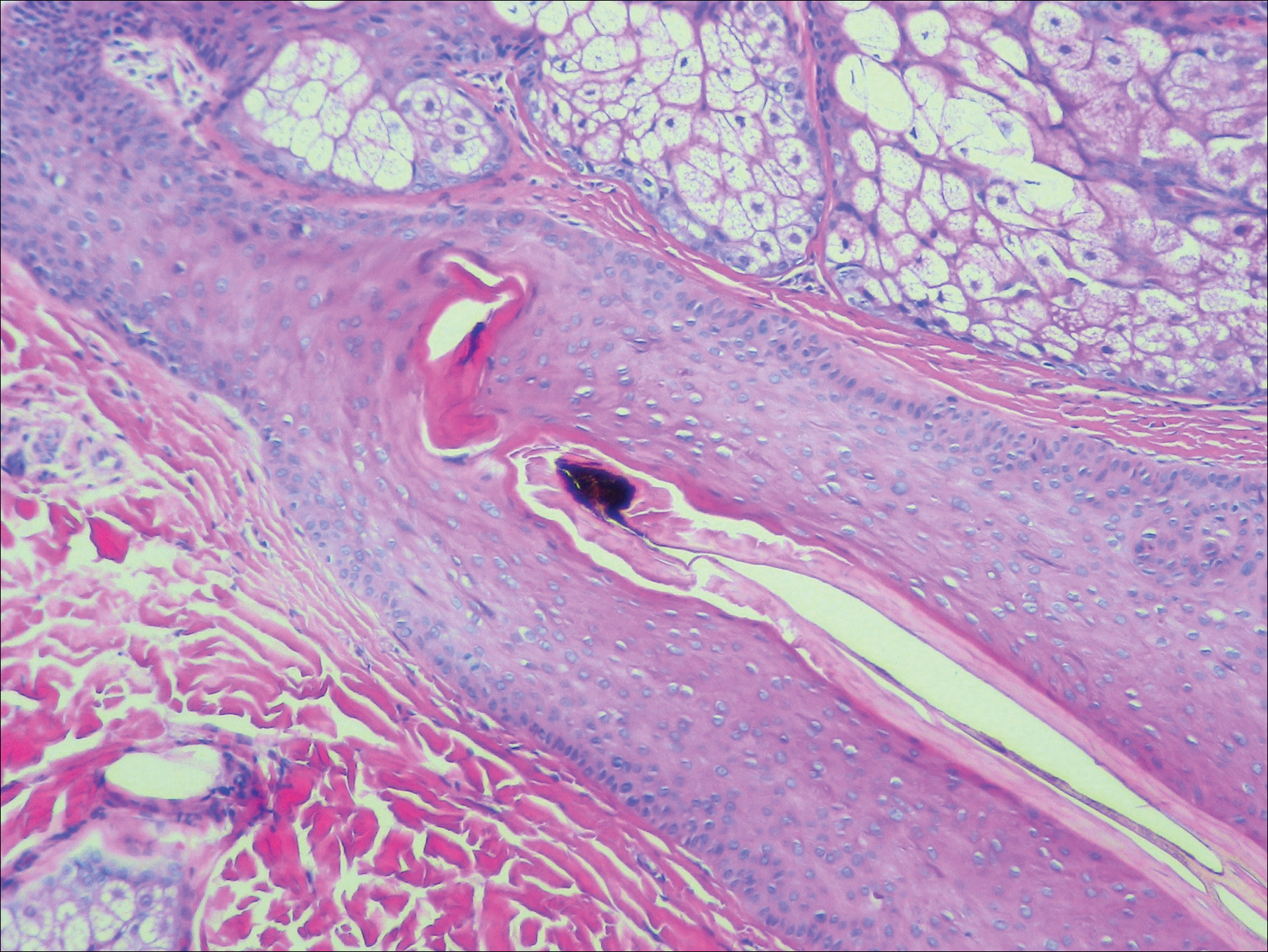
Trichotillomania is a hairpulling disorder with notable dermatologic and psychiatric overlap. Although previously considered an impulse control disorder, the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) reclassified it within obsessive-compulsive and related disorders, which also include body dysmorphic disorder and excoriation (skin-picking) disorder. Diagnostic criteria for TTM include the following: the patient must have recurrent pulling out of his/her hair resulting in hair loss despite repeated attempts to stop; underlying medical conditions and other psychiatric diagnoses must be excluded; and the patient must experience distress or impairment in social, occupational, or other areas of functioning from the hairpulling.1 Trichotillomania mainly occurs in children and young adults, with a lifetime prevalence of approximately 1% to 2%.2 The coexistence of a mood or anxiety disorder is common, as seen in our patient.
The diagnosis of TTM requires strong clinical suspicion because patients and their parents/guardians usually deny hairpulling. The main clinical differential diagnosis often is alopecia areata (AA) because both conditions can present as well-defined patches of nonscarring hair loss. Trichoscopy provides an invaluable noninvasive diagnostic tool that can be particularly useful in pediatric patients who may be reluctant to have a scalp biopsy. There are many overlapping trichoscopic findings of TTM and AA, including yellow dots, black dots, broken hairs, coiled hairs, and exclamation mark hairs.3 More specific trichoscopy findings for TTM include flame hairs (wavy proximal hair residue), V-sign (2 shafts within 1 follicle broken at the same length), and tulip hairs (dark, tulip-shaped ends of broken hairs).4 Hair breakage of varying lengths and trichoptilosis (split ends) can be better visualized using trichoscopy and support a diagnosis of TTM over AA.
Androgenetic alopecia (female pattern hair loss) presents with gradual thinning around the part line of the frontal and parietal scalp with trichoscopy showing miniaturization of hairs and decreased follicle density. The moth-eaten-like appearance of alopecia due to secondary syphilis may mimic alopecia areata clinically, but serologic testing can confirm the diagnosis of syphilis. Telogen effluvium does not have the trichoscopic features that are seen in TTM and is clinically distinguished by hair shedding and a positive hair pull test.
Biopsy can provide objective yet nonspecific support for the diagnosis, demonstrating trichomalacia, pigmented hair casts, empty follicles, and an increase in catagen hairs with a lack of inflammation. Normal and damaged hair follicles may be seen in close proximity, and hemorrhage may be seen secondary to trauma. Pigmented hair casts are not specific to TTM and are present in other traumatic hair disorders, such as traction alopecia; therefore, clinical correlation is essential for diagnosis.
Habit reversal training is the most effective treatment of TTM and involves 3 major components: awareness training with self-monitoring, stimulus control, and competing response procedures.5 Although numerous pharmacotherapies have been reported as effective treatments for TTM, a 2013 Cochrane review of 8 randomized controlled trials concluded that no medication has demonstrated reliable efficacy. Reported therapies included selective serotonin reuptake inhibitors, naltrexone, olanzapine, N-acetylcysteine, and clomipramine.6
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
- Schumer MC, Panza KE, Mulqueen JM, et al. Long-term outcome in pediatric trichotillomania. Depress Anxiety. 2015;32:737-743.
- Lencastre A, Tosti A. Role of trichoscopy on children's scalp and hair disorders. Pediatr Dermatol. 2013;30:674-682.
- Rakowska A, Slowinska M, Olszewska M, et al. New trichoscopy findings in trichotillomania: flame hairs, V-sign, hook hairs, hair powder, tulip hairs. Acta Derm Venereol. 2014;94:303-306.
- Morris S, Zickgraf H, Dingfelder H, et al. Habit reversal training in trichotillomania: guide for the clinician. Expert Rev Neurother. 2013;13:1069-1177.
- Rothbart R, Amos T, Siegfried N, et al. Pharmacotherapy for trichotillomania. Cochrane Database Syst Rev. 2013;11:CD007662.
The Diagnosis: Trichotillomania
A scalp punch biopsy revealed pigmented hair casts, an increase in catagen and telogen follicles, and a lack of perifollicular inflammation (Figure). Based on the clinical and histopathological findings, a diagnosis of trichotillomania (TTM) was established.
Trichotillomania is a hairpulling disorder with notable dermatologic and psychiatric overlap. Although previously considered an impulse control disorder, the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) reclassified it within obsessive-compulsive and related disorders, which also include body dysmorphic disorder and excoriation (skin-picking) disorder. Diagnostic criteria for TTM include the following: the patient must have recurrent pulling out of his/her hair resulting in hair loss despite repeated attempts to stop; underlying medical conditions and other psychiatric diagnoses must be excluded; and the patient must experience distress or impairment in social, occupational, or other areas of functioning from the hairpulling.1 Trichotillomania mainly occurs in children and young adults, with a lifetime prevalence of approximately 1% to 2%.2 The coexistence of a mood or anxiety disorder is common, as seen in our patient.
The diagnosis of TTM requires strong clinical suspicion because patients and their parents/guardians usually deny hairpulling. The main clinical differential diagnosis often is alopecia areata (AA) because both conditions can present as well-defined patches of nonscarring hair loss. Trichoscopy provides an invaluable noninvasive diagnostic tool that can be particularly useful in pediatric patients who may be reluctant to have a scalp biopsy. There are many overlapping trichoscopic findings of TTM and AA, including yellow dots, black dots, broken hairs, coiled hairs, and exclamation mark hairs.3 More specific trichoscopy findings for TTM include flame hairs (wavy proximal hair residue), V-sign (2 shafts within 1 follicle broken at the same length), and tulip hairs (dark, tulip-shaped ends of broken hairs).4 Hair breakage of varying lengths and trichoptilosis (split ends) can be better visualized using trichoscopy and support a diagnosis of TTM over AA.
Androgenetic alopecia (female pattern hair loss) presents with gradual thinning around the part line of the frontal and parietal scalp with trichoscopy showing miniaturization of hairs and decreased follicle density. The moth-eaten-like appearance of alopecia due to secondary syphilis may mimic alopecia areata clinically, but serologic testing can confirm the diagnosis of syphilis. Telogen effluvium does not have the trichoscopic features that are seen in TTM and is clinically distinguished by hair shedding and a positive hair pull test.
Biopsy can provide objective yet nonspecific support for the diagnosis, demonstrating trichomalacia, pigmented hair casts, empty follicles, and an increase in catagen hairs with a lack of inflammation. Normal and damaged hair follicles may be seen in close proximity, and hemorrhage may be seen secondary to trauma. Pigmented hair casts are not specific to TTM and are present in other traumatic hair disorders, such as traction alopecia; therefore, clinical correlation is essential for diagnosis.
Habit reversal training is the most effective treatment of TTM and involves 3 major components: awareness training with self-monitoring, stimulus control, and competing response procedures.5 Although numerous pharmacotherapies have been reported as effective treatments for TTM, a 2013 Cochrane review of 8 randomized controlled trials concluded that no medication has demonstrated reliable efficacy. Reported therapies included selective serotonin reuptake inhibitors, naltrexone, olanzapine, N-acetylcysteine, and clomipramine.6
The Diagnosis: Trichotillomania
A scalp punch biopsy revealed pigmented hair casts, an increase in catagen and telogen follicles, and a lack of perifollicular inflammation (Figure). Based on the clinical and histopathological findings, a diagnosis of trichotillomania (TTM) was established.
Trichotillomania is a hairpulling disorder with notable dermatologic and psychiatric overlap. Although previously considered an impulse control disorder, the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) reclassified it within obsessive-compulsive and related disorders, which also include body dysmorphic disorder and excoriation (skin-picking) disorder. Diagnostic criteria for TTM include the following: the patient must have recurrent pulling out of his/her hair resulting in hair loss despite repeated attempts to stop; underlying medical conditions and other psychiatric diagnoses must be excluded; and the patient must experience distress or impairment in social, occupational, or other areas of functioning from the hairpulling.1 Trichotillomania mainly occurs in children and young adults, with a lifetime prevalence of approximately 1% to 2%.2 The coexistence of a mood or anxiety disorder is common, as seen in our patient.
The diagnosis of TTM requires strong clinical suspicion because patients and their parents/guardians usually deny hairpulling. The main clinical differential diagnosis often is alopecia areata (AA) because both conditions can present as well-defined patches of nonscarring hair loss. Trichoscopy provides an invaluable noninvasive diagnostic tool that can be particularly useful in pediatric patients who may be reluctant to have a scalp biopsy. There are many overlapping trichoscopic findings of TTM and AA, including yellow dots, black dots, broken hairs, coiled hairs, and exclamation mark hairs.3 More specific trichoscopy findings for TTM include flame hairs (wavy proximal hair residue), V-sign (2 shafts within 1 follicle broken at the same length), and tulip hairs (dark, tulip-shaped ends of broken hairs).4 Hair breakage of varying lengths and trichoptilosis (split ends) can be better visualized using trichoscopy and support a diagnosis of TTM over AA.
Androgenetic alopecia (female pattern hair loss) presents with gradual thinning around the part line of the frontal and parietal scalp with trichoscopy showing miniaturization of hairs and decreased follicle density. The moth-eaten-like appearance of alopecia due to secondary syphilis may mimic alopecia areata clinically, but serologic testing can confirm the diagnosis of syphilis. Telogen effluvium does not have the trichoscopic features that are seen in TTM and is clinically distinguished by hair shedding and a positive hair pull test.
Biopsy can provide objective yet nonspecific support for the diagnosis, demonstrating trichomalacia, pigmented hair casts, empty follicles, and an increase in catagen hairs with a lack of inflammation. Normal and damaged hair follicles may be seen in close proximity, and hemorrhage may be seen secondary to trauma. Pigmented hair casts are not specific to TTM and are present in other traumatic hair disorders, such as traction alopecia; therefore, clinical correlation is essential for diagnosis.
Habit reversal training is the most effective treatment of TTM and involves 3 major components: awareness training with self-monitoring, stimulus control, and competing response procedures.5 Although numerous pharmacotherapies have been reported as effective treatments for TTM, a 2013 Cochrane review of 8 randomized controlled trials concluded that no medication has demonstrated reliable efficacy. Reported therapies included selective serotonin reuptake inhibitors, naltrexone, olanzapine, N-acetylcysteine, and clomipramine.6
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
- Schumer MC, Panza KE, Mulqueen JM, et al. Long-term outcome in pediatric trichotillomania. Depress Anxiety. 2015;32:737-743.
- Lencastre A, Tosti A. Role of trichoscopy on children's scalp and hair disorders. Pediatr Dermatol. 2013;30:674-682.
- Rakowska A, Slowinska M, Olszewska M, et al. New trichoscopy findings in trichotillomania: flame hairs, V-sign, hook hairs, hair powder, tulip hairs. Acta Derm Venereol. 2014;94:303-306.
- Morris S, Zickgraf H, Dingfelder H, et al. Habit reversal training in trichotillomania: guide for the clinician. Expert Rev Neurother. 2013;13:1069-1177.
- Rothbart R, Amos T, Siegfried N, et al. Pharmacotherapy for trichotillomania. Cochrane Database Syst Rev. 2013;11:CD007662.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
- Schumer MC, Panza KE, Mulqueen JM, et al. Long-term outcome in pediatric trichotillomania. Depress Anxiety. 2015;32:737-743.
- Lencastre A, Tosti A. Role of trichoscopy on children's scalp and hair disorders. Pediatr Dermatol. 2013;30:674-682.
- Rakowska A, Slowinska M, Olszewska M, et al. New trichoscopy findings in trichotillomania: flame hairs, V-sign, hook hairs, hair powder, tulip hairs. Acta Derm Venereol. 2014;94:303-306.
- Morris S, Zickgraf H, Dingfelder H, et al. Habit reversal training in trichotillomania: guide for the clinician. Expert Rev Neurother. 2013;13:1069-1177.
- Rothbart R, Amos T, Siegfried N, et al. Pharmacotherapy for trichotillomania. Cochrane Database Syst Rev. 2013;11:CD007662.
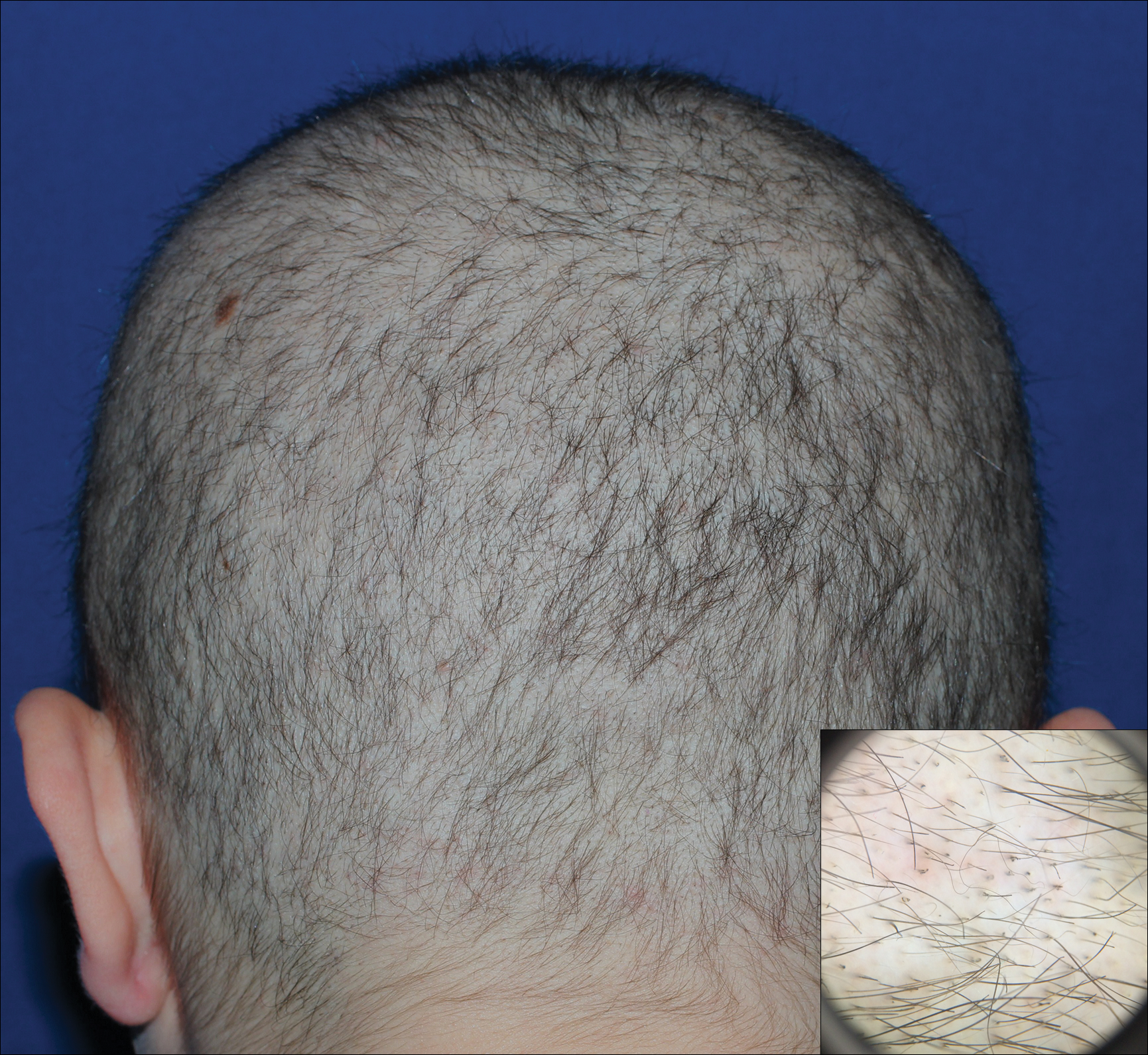
A 19-year-old woman with attention deficit hyperactivity disorder and an anxiety disorder presented with hair loss of 2 years' duration. She initially had small circular bald areas throughout the scalp that had progressed to diffuse hair loss of the entire scalp. She denied recent hairpulling but admitted to a remote prior history of eyelash and eyebrow pulling. She denied any voice changes, acne, or menstrual irregularities. Physical examination revealed short hairs of varying lengths throughout the scalp with no loss of follicles, erythema, scale, or exclamation point hairs. Eyebrows and eyelashes were normal. A hair-pull test was negative. Trichoscopy illuminated variation in hair shaft diameters, as well as short, irregularly broken hairs of different lengths (inset).