User login
Mycobacterium haemophilum: A Challenging Treatment Dilemma in an Immunocompromised Patient
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
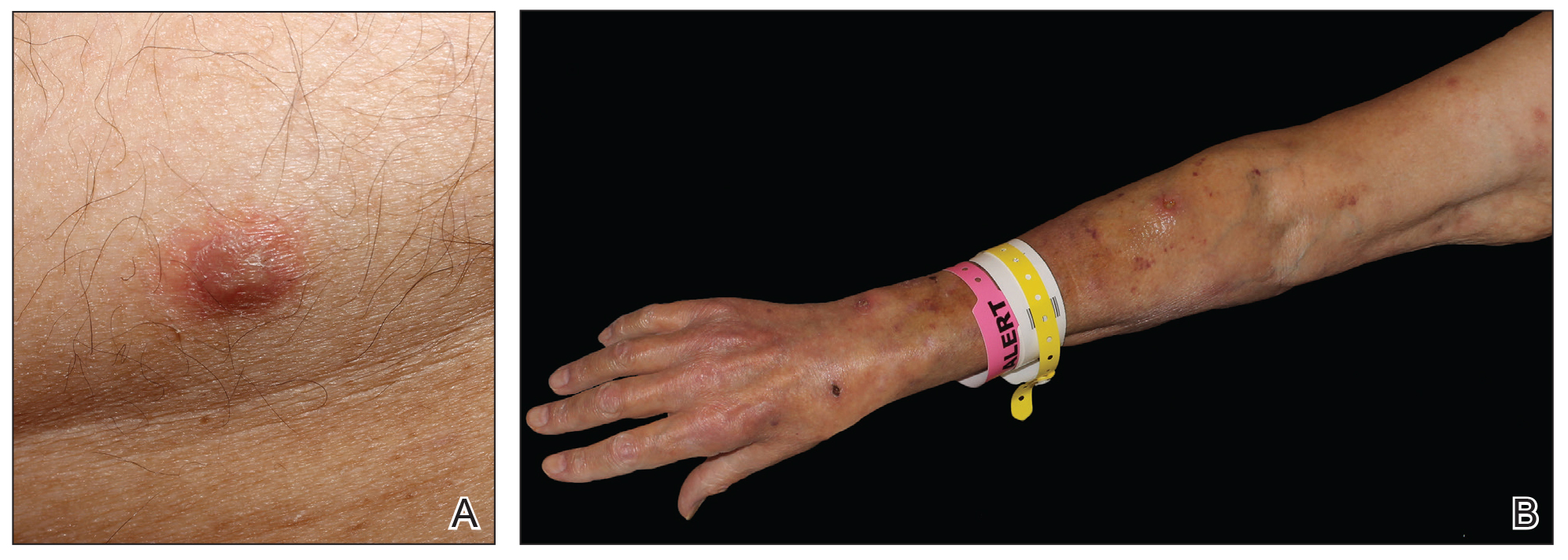
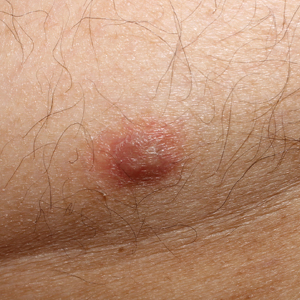
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.

The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
Practice Points
- Mycobacterium haemophilum is a slow-growing acid-fast bacillus that requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. Because these requirements for growth are not standard for acid-fast bacteria cultures, M haemophilum infection may be underrecognized and underreported.
- There are no species-specific treatment guidelines, but extended course of treatment with multiple active antibacterials typically is recommended.
Atrophodermalike Guttate Morphea
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
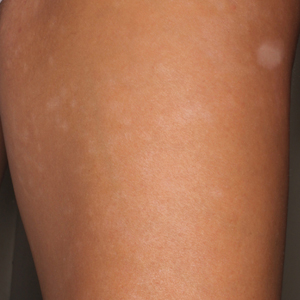
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
 as well as hypopigmented macules and a minimally depressed patch on the left thigh (B).")
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
(H&E, original magnification ×40)...")
(Verhoeff-van Gieson, original magnification ×40). Normal elastin network in the lower reticular dermis; note the normal size of elastin fibers (B)(Verhoeff-van Gieson, original magnification ×200).")
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
Practice Points
- Atrophodermalike guttate morphea is a potentially underreported or undescribed entity consisting of a combination of clinicopathologic features.
- Widespread hypopigmented macules on the trunk and extremities marked by thinned collagen, fibroplasia, and altered fragmented elastin in the papillary dermis and upper reticular dermis are the key features.
- Atrophoderma, morphea, and lichen sclerosus et atrophicus should be ruled out during clinical workup.