User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Low-Dose Oral Naltrexone for Darier Disease
To the Editor:
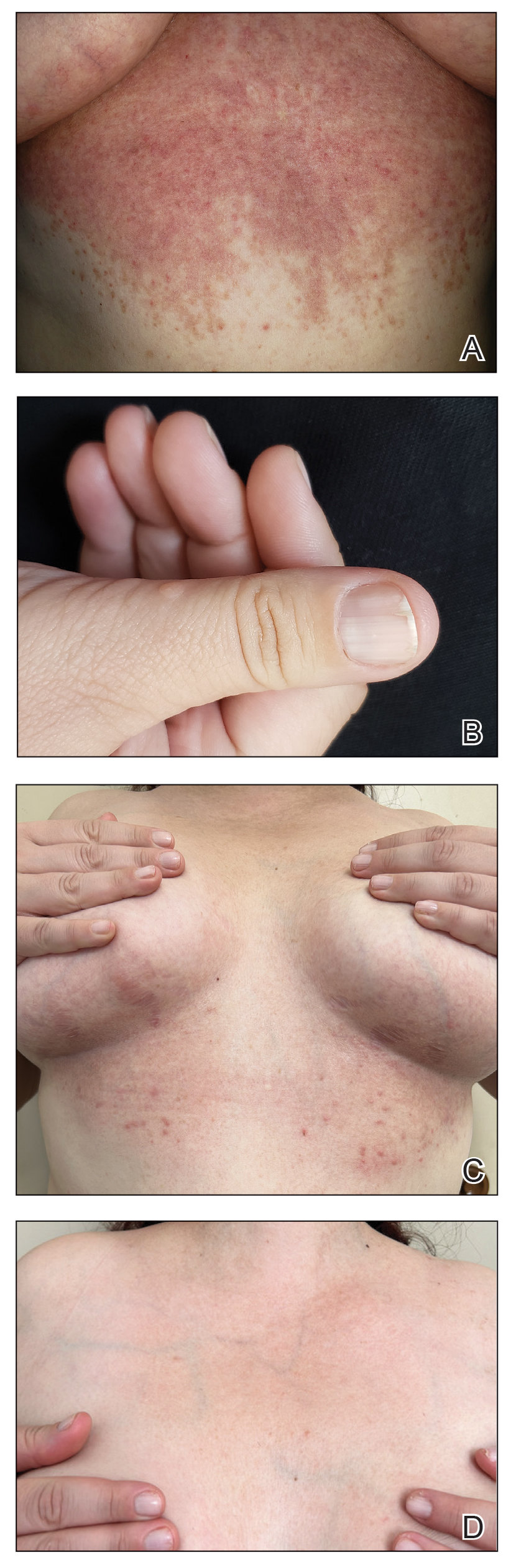
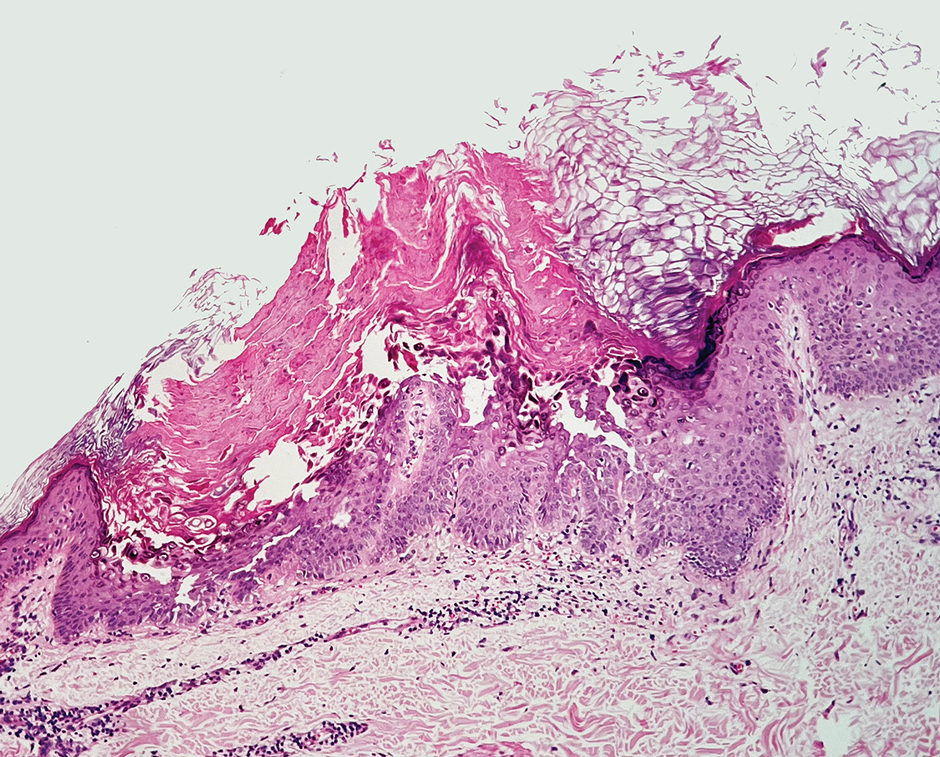
A 34-year-old Brazilian woman presented to the dermatology department with pruritic lesions on the neck and chest that had been present since adolescence. She reported a family history of Darier disease in her father. Physical examination revealed erythematous follicular papules on the neck, inframammary region, and abdomen (Figure 1A), as well as longitudinal bandlike leukonychia and distal nail splits on the fingernails (Figure 1B). Histopathology of a lesion on the back revealed compact hyperkeratosis and parakeratosis above an acantholytic cleft accompanied by dyskeratotic keratinocytes, including some corps ronds and grains, which supported the clinical impression of Darier disease (Figure 2). The typical clinical presentation along with the family history and histopathology confirmed the diagnosis. After therapeutic failure with topical corticosteroids and oral antibiotics for 3 months, low-dose oral naltrexone (4.5 mg/d) as monotherapy noticeably improved the lesions and pruritus within 2 months, with near-complete regression at 6 months, achieving disease stability (Figures 1C and 1D). The patient remained stable with no recurrence after 1 year of follow-up.
Darier disease is an autosomal-dominant genodermatosis caused by a mutation in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum calcium ATPase, leading to defective intracellular calcium signaling and alterations in epidermal adhesion and keratinization.1 Darier disease typically begins in adolescence and is aggravated by exposure to heat and friction. It is characterized by seborrheic distribution of painful and pruritic red-brown keratotic papules. Nail manifestations include longitudinal ridges—erythronychia and/or leukonychia—and grooves that end in a V-shaped notch. The differential diagnosis includes Hailey-Hailey disease, psoriasis, and pityriasis rubra pilaris.1,2 The diagnosis is clinical and is confirmed by histopathology, which reveals suprabasal cleavage, acantholytic dyskeratosis, corps ronds, and grains. Treatment options are limited and include corticosteroids, oral and/or topical antibiotics, and systemic retinoids.2
Oral naltrexone has been used in Darier disease based on its observed effectiveness in Hailey-Hailey disease, considering the histopathologic similarities and alterations in calcium homeostasis in both conditions. Low-dose oral naltrexone (1-5 mg/d) increases the expression of opioid receptors (δ, μ, κ), enhancing its immunomodulatory and antinociceptive effects. The δ opioid receptor regulates the expression of desmoglein, improving epidermal differentiation and wound healing.3 Activation of the δ and μ receptors increases intracellular calcium through the inositol phosphate pathway, which contributes to calcium homeostasis.4 Naltrexone blocks the nonopioid toll-like receptor 4 found in keratinocytes and macrophages, exerting an anti-inflammatory effect by reducing proinflammatory cytokines.3 Adverse events associated with low-dose naltrexone are minimal, mostly mild, and often related to sleep disorders3,5; however, patients should undergo screening for prior opioid dependence, recent opioid usage, and signs of opioid withdrawal before initiating naltrexone treatment.5
Boehmer et al6 used naltrexone (4.5 mg/d) and oral magnesium (200 mg/d) in 6 patients with inconsistent results, except for 1 case that concurrently used acitretin (25 mg/d) with satisfactory improvement. Pessoa et al7 added naltrexone (4.5 mg/d) to oral isotretinoin (0.5 mg/kg/d) in 1 patient, resulting in notable improvement of lesions within 3 months.
In our patient with Darier disease, low-dose naltrexone demonstrated a substantial response as monotherapy after 2 months of treatment and nearly complete regression of lesions within 6 months, with no reported side effects after 1 year of follow-up. The use of low-dose naltrexone could be a promising and safe treatment option as monotherapy or in combination with conventional therapy for Darier disease; however, further studies are needed.
Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271-277. doi:10.1038/6784
Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50. doi:10.1016/0190-9622(92)70154-8
Lee B, Elston DM. The uses of naltrexone in dermatologic conditions. Am Acad Dermatol. 2019;80:1746-1752. doi:10.1016/j.jaad.2018.12.031
Samways DSK, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151-161. doi:10.1016/j.cellsig.2005.08.005
Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236. doi:10.1001/jamadermatol.2018.4093
Boehmer D, Eyerich K, Darsow U, et al. Variable response to low‐dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol. 2019;33:950-953. doi:10.1111/jdv.15457
Pessoa T, Rebelo C, Gabriela Marques Pinto, et al. Combination of naltrexone and isotretinoin for the treatment of Darier disease. Cureus. 2023;15:E33321. doi:10.7759/cureus.33321
To the Editor:
A 34-year-old Brazilian woman presented to the dermatology department with pruritic lesions on the neck and chest that had been present since adolescence. She reported a family history of Darier disease in her father. Physical examination revealed erythematous follicular papules on the neck, inframammary region, and abdomen (Figure 1A), as well as longitudinal bandlike leukonychia and distal nail splits on the fingernails (Figure 1B). Histopathology of a lesion on the back revealed compact hyperkeratosis and parakeratosis above an acantholytic cleft accompanied by dyskeratotic keratinocytes, including some corps ronds and grains, which supported the clinical impression of Darier disease (Figure 2). The typical clinical presentation along with the family history and histopathology confirmed the diagnosis. After therapeutic failure with topical corticosteroids and oral antibiotics for 3 months, low-dose oral naltrexone (4.5 mg/d) as monotherapy noticeably improved the lesions and pruritus within 2 months, with near-complete regression at 6 months, achieving disease stability (Figures 1C and 1D). The patient remained stable with no recurrence after 1 year of follow-up.
Darier disease is an autosomal-dominant genodermatosis caused by a mutation in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum calcium ATPase, leading to defective intracellular calcium signaling and alterations in epidermal adhesion and keratinization.1 Darier disease typically begins in adolescence and is aggravated by exposure to heat and friction. It is characterized by seborrheic distribution of painful and pruritic red-brown keratotic papules. Nail manifestations include longitudinal ridges—erythronychia and/or leukonychia—and grooves that end in a V-shaped notch. The differential diagnosis includes Hailey-Hailey disease, psoriasis, and pityriasis rubra pilaris.1,2 The diagnosis is clinical and is confirmed by histopathology, which reveals suprabasal cleavage, acantholytic dyskeratosis, corps ronds, and grains. Treatment options are limited and include corticosteroids, oral and/or topical antibiotics, and systemic retinoids.2
Oral naltrexone has been used in Darier disease based on its observed effectiveness in Hailey-Hailey disease, considering the histopathologic similarities and alterations in calcium homeostasis in both conditions. Low-dose oral naltrexone (1-5 mg/d) increases the expression of opioid receptors (δ, μ, κ), enhancing its immunomodulatory and antinociceptive effects. The δ opioid receptor regulates the expression of desmoglein, improving epidermal differentiation and wound healing.3 Activation of the δ and μ receptors increases intracellular calcium through the inositol phosphate pathway, which contributes to calcium homeostasis.4 Naltrexone blocks the nonopioid toll-like receptor 4 found in keratinocytes and macrophages, exerting an anti-inflammatory effect by reducing proinflammatory cytokines.3 Adverse events associated with low-dose naltrexone are minimal, mostly mild, and often related to sleep disorders3,5; however, patients should undergo screening for prior opioid dependence, recent opioid usage, and signs of opioid withdrawal before initiating naltrexone treatment.5
Boehmer et al6 used naltrexone (4.5 mg/d) and oral magnesium (200 mg/d) in 6 patients with inconsistent results, except for 1 case that concurrently used acitretin (25 mg/d) with satisfactory improvement. Pessoa et al7 added naltrexone (4.5 mg/d) to oral isotretinoin (0.5 mg/kg/d) in 1 patient, resulting in notable improvement of lesions within 3 months.
In our patient with Darier disease, low-dose naltrexone demonstrated a substantial response as monotherapy after 2 months of treatment and nearly complete regression of lesions within 6 months, with no reported side effects after 1 year of follow-up. The use of low-dose naltrexone could be a promising and safe treatment option as monotherapy or in combination with conventional therapy for Darier disease; however, further studies are needed.
To the Editor:
A 34-year-old Brazilian woman presented to the dermatology department with pruritic lesions on the neck and chest that had been present since adolescence. She reported a family history of Darier disease in her father. Physical examination revealed erythematous follicular papules on the neck, inframammary region, and abdomen (Figure 1A), as well as longitudinal bandlike leukonychia and distal nail splits on the fingernails (Figure 1B). Histopathology of a lesion on the back revealed compact hyperkeratosis and parakeratosis above an acantholytic cleft accompanied by dyskeratotic keratinocytes, including some corps ronds and grains, which supported the clinical impression of Darier disease (Figure 2). The typical clinical presentation along with the family history and histopathology confirmed the diagnosis. After therapeutic failure with topical corticosteroids and oral antibiotics for 3 months, low-dose oral naltrexone (4.5 mg/d) as monotherapy noticeably improved the lesions and pruritus within 2 months, with near-complete regression at 6 months, achieving disease stability (Figures 1C and 1D). The patient remained stable with no recurrence after 1 year of follow-up.
Darier disease is an autosomal-dominant genodermatosis caused by a mutation in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum calcium ATPase, leading to defective intracellular calcium signaling and alterations in epidermal adhesion and keratinization.1 Darier disease typically begins in adolescence and is aggravated by exposure to heat and friction. It is characterized by seborrheic distribution of painful and pruritic red-brown keratotic papules. Nail manifestations include longitudinal ridges—erythronychia and/or leukonychia—and grooves that end in a V-shaped notch. The differential diagnosis includes Hailey-Hailey disease, psoriasis, and pityriasis rubra pilaris.1,2 The diagnosis is clinical and is confirmed by histopathology, which reveals suprabasal cleavage, acantholytic dyskeratosis, corps ronds, and grains. Treatment options are limited and include corticosteroids, oral and/or topical antibiotics, and systemic retinoids.2
Oral naltrexone has been used in Darier disease based on its observed effectiveness in Hailey-Hailey disease, considering the histopathologic similarities and alterations in calcium homeostasis in both conditions. Low-dose oral naltrexone (1-5 mg/d) increases the expression of opioid receptors (δ, μ, κ), enhancing its immunomodulatory and antinociceptive effects. The δ opioid receptor regulates the expression of desmoglein, improving epidermal differentiation and wound healing.3 Activation of the δ and μ receptors increases intracellular calcium through the inositol phosphate pathway, which contributes to calcium homeostasis.4 Naltrexone blocks the nonopioid toll-like receptor 4 found in keratinocytes and macrophages, exerting an anti-inflammatory effect by reducing proinflammatory cytokines.3 Adverse events associated with low-dose naltrexone are minimal, mostly mild, and often related to sleep disorders3,5; however, patients should undergo screening for prior opioid dependence, recent opioid usage, and signs of opioid withdrawal before initiating naltrexone treatment.5
Boehmer et al6 used naltrexone (4.5 mg/d) and oral magnesium (200 mg/d) in 6 patients with inconsistent results, except for 1 case that concurrently used acitretin (25 mg/d) with satisfactory improvement. Pessoa et al7 added naltrexone (4.5 mg/d) to oral isotretinoin (0.5 mg/kg/d) in 1 patient, resulting in notable improvement of lesions within 3 months.
In our patient with Darier disease, low-dose naltrexone demonstrated a substantial response as monotherapy after 2 months of treatment and nearly complete regression of lesions within 6 months, with no reported side effects after 1 year of follow-up. The use of low-dose naltrexone could be a promising and safe treatment option as monotherapy or in combination with conventional therapy for Darier disease; however, further studies are needed.
Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271-277. doi:10.1038/6784
Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50. doi:10.1016/0190-9622(92)70154-8
Lee B, Elston DM. The uses of naltrexone in dermatologic conditions. Am Acad Dermatol. 2019;80:1746-1752. doi:10.1016/j.jaad.2018.12.031
Samways DSK, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151-161. doi:10.1016/j.cellsig.2005.08.005
Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236. doi:10.1001/jamadermatol.2018.4093
Boehmer D, Eyerich K, Darsow U, et al. Variable response to low‐dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol. 2019;33:950-953. doi:10.1111/jdv.15457
Pessoa T, Rebelo C, Gabriela Marques Pinto, et al. Combination of naltrexone and isotretinoin for the treatment of Darier disease. Cureus. 2023;15:E33321. doi:10.7759/cureus.33321
Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999;21:271-277. doi:10.1038/6784
Burge SM, Wilkinson JD. Darier-White disease: a review of the clinical features in 163 patients. J Am Acad Dermatol. 1992;27:40-50. doi:10.1016/0190-9622(92)70154-8
Lee B, Elston DM. The uses of naltrexone in dermatologic conditions. Am Acad Dermatol. 2019;80:1746-1752. doi:10.1016/j.jaad.2018.12.031
Samways DSK, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151-161. doi:10.1016/j.cellsig.2005.08.005
Ekelem C, Juhasz M, Khera P, et al. Utility of naltrexone treatment for chronic inflammatory dermatologic conditions: a systematic review. JAMA Dermatol. 2019;155:229-236. doi:10.1001/jamadermatol.2018.4093
Boehmer D, Eyerich K, Darsow U, et al. Variable response to low‐dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol. 2019;33:950-953. doi:10.1111/jdv.15457
Pessoa T, Rebelo C, Gabriela Marques Pinto, et al. Combination of naltrexone and isotretinoin for the treatment of Darier disease. Cureus. 2023;15:E33321. doi:10.7759/cureus.33321
Practice Points
- Consider low-dose naltrexone as a potential treatment option for patients with Darier disease, as it regulates opioid receptors and has shown benefits in enhancing epidermal differentiation, wound healing, and anti-inflammatory effects.
- Further research is needed to validate the efficacy and safety of low-dose naltrexone in treating Darier disease considering its observed clinical improvement in this single patient case.
Evaluating Factors Impacting Hidradenitis Suppurativa Disease Severity in Patients With Darker Skin Types
Evaluating Factors Impacting Hidradenitis Suppurativa Disease Severity in Patients With Darker Skin Types
Hidradenitis suppurativa (HS) is a debilitating chronic skin disease that often affects apocrinebearing regions of the skin such as the axillae, perineum, and groin.1 Although current research on the etiology and pathogenesis of HS is limited, the disease is known to have a considerable psychosocial impact on patient quality of life.
Clinically, HS lesions manifest as tender subcutaneous nodules that rupture to form painful and deep dermal abscesses.2 These lesions typically develop due to hair follicle occlusion, followed by a cyclic process of inflammation, healing, re-inflammation, and scarring. Often, they are mistaken for cysts or a simple abscess in the early stages of the disease, leading to a delay in diagnosis.1 Disease severity is categorized based on Hurley staging: stage 1 involves abscess formation without scarring; stage 2 involves limited sinus tracts and recurrent abscesses with scarring and/or multiple separated lesions; and stage 3 is the most advanced stage, with diffuse involvement or multiple interconnected sinus tracts across an area with scarring. The condition primarily is medically managed with antibiotics and immunomodulators, but patients who have refractory disease can benefit from surgical excision.1,2
The prevalence of HS in the United States ranges from 0.77% to 1.19%, and individuals who self-identify as Black have 3-fold higher odds of having this condition compared with all other racial groups.3-5 Black patients also are thought to have a greater number and size of apocrine glands compared with patients who self-identify as White, suggesting an anatomic predisposition to developing HS and greater disease severity.6 However, despite HS disproportionately impacting individuals with skin of color (SOC), the majority of published HS research includes predominantly White patient cohorts.5 There is insufficient research assessing HS epidemiology, comorbidities, and treatment responses in patients with SOC.
A 2020 review reported the notable lack of clinical trials that sufficiently examine systemic medication treatment response in HS patients with SOC.7 Of the 15 HS treatment trials published from 2000 to 2019, only 16.4% (138/840) of the patient population were of African descent.7 Clinical trials investigating the efficacy of adalimumab in reducing HS burden also did not adequately evaluate clinical response in patients with SOC. One clinical trial did not include any Black patients as part of the cohort,8 and in 3 other studies, 80% to 85% of the study participants self-identified as White.9 The current literature does not reflect the patient populations most affected by HS, as several studies have reported that 65% of patients diagnosed with HS in the United States annually are Black.5,7 These results emphasize the underrepresentation of SOC populations in the current HS literature and the need for more research that investigates the disease processes, comorbidities, and treatment outcomes of the diverse patient population impacted by HS.
Methods
Study Population and Data Extraction—Following a protocol reviewed and approved by the MedStar Health/Georgetown University institutional review board (IRB #00006783), a retrospective chart review of 31 adult patients with HS who underwent surgery at a regional verified burn center from April 2014 to April 2023 was conducted. The following variables were collected from the electronic medical record (EMR): baseline demographics including age, sex, body mass index (BMI), obesity status, race, ethnicity, Fitzpatrick skin type, smoking status, substance use, employment status, and family history of HS; HS-specific details including Hurley staging, affected areas, and age at initial diagnosis; comorbidities such as dermatologic conditions, autoimmune disorders, infectious diseases, cardiovascular and associated diseases, ovarian disorders, gastrointestinal diseases, and othother common chronic comorbidities (psychiatric illness, kidney disease, type 2 diabetes [T2D], asthma, allergies, lymphedema, and inflammatory eye disease); and use of pharmacologics such as topical medications, oral antibiotics, immunomodulators, and steroids.
Study Definitions—Obesity was defined as both a continuous and categorical variable. Each patient’s BMI at the surgery date was recorded from the EMR as a continuous variable. Patients with obesity also had this condition listed under their complaints and problem list in the EMR, which was recorded as a categorical variable. Race and ethnicity were self-reported by patients. Comorbidity data, including T2D and hyperlipidemia, were defined by previously diagnosed diseases listed in the EMR. Pharmacologic medication data were included in the study if a patient was recommended/prescribed a medication and they had confirmed use of the medication in a subsequent office visit.
Statistical Analysis—Descriptive statistics were calculated for demographics, HS characteristics (eg, location, Hurley stage), and comorbidities. Continuous variables were presented as mean and standard deviation or median and interquartile range and were evaluated using a t test or Mann-Whitney U test when appropriate. Categorical variables were presented as frequencies and percentages and tested for associations using the X2 or Fisher exact test. Data analyses were performed using SAS software version 9.4 (SAS Institute Inc.).
Results
Thirty-one patients (17 females, 14 males; mean age, 40.9 years) were included in the study. Twenty-nine (93.5%) patients identified as Black. All study patients had at least 1 comorbidity. Obesity was diagnosed in 22 (71.0%) patients (mean BMI, 35.5 kg/m2). A total of 16 (51.6%) patients were current smokers, 3 (9.7%) were past smokers, 22 (71%) reported alcohol use, and 17 (54.8%) were active marijuana users. Only 3 (9.7%) patients had a family history of HS (Table 1).
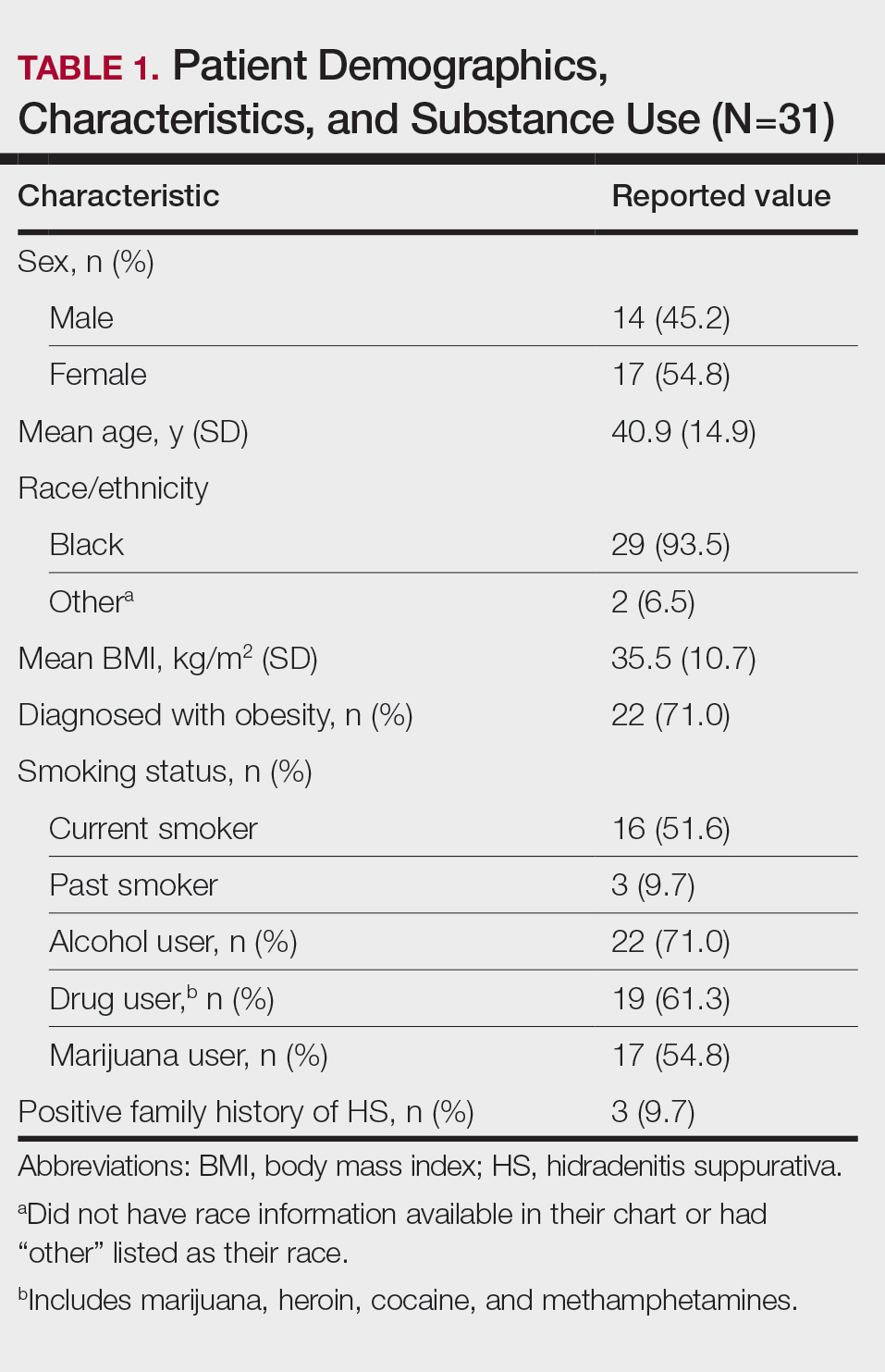
Other common comorbidities associated with HS were anemia (64.5% [20/31]), a non–inflammatory bowel disease gastrointestinal disease (61.3% [19/31]), allergies (54.8% [17/31]), hypertension (41.9% [13/31]), cardiovascular disease (41.9% [13/31]), T2D (32.3% [10/31]), asthma (32.3% [10/31]), kidney disease (29.0% [9/31]), and atopic dermatitis (25.8% [8/31]). More than half (54.8% [17/31]) of patients were diagnosed with psychiatric illnesses, including depression, anxiety, bipolar depression, psychosis, anorexia, impulsive anger, hallucinations, delusion, attention deficit-hyperactivity disorder, and panic disorder (Table 2). Depression was diagnosed in 38.7% (12/31) of patients, and 22.6% (7/31) were diagnosed with anxiety.
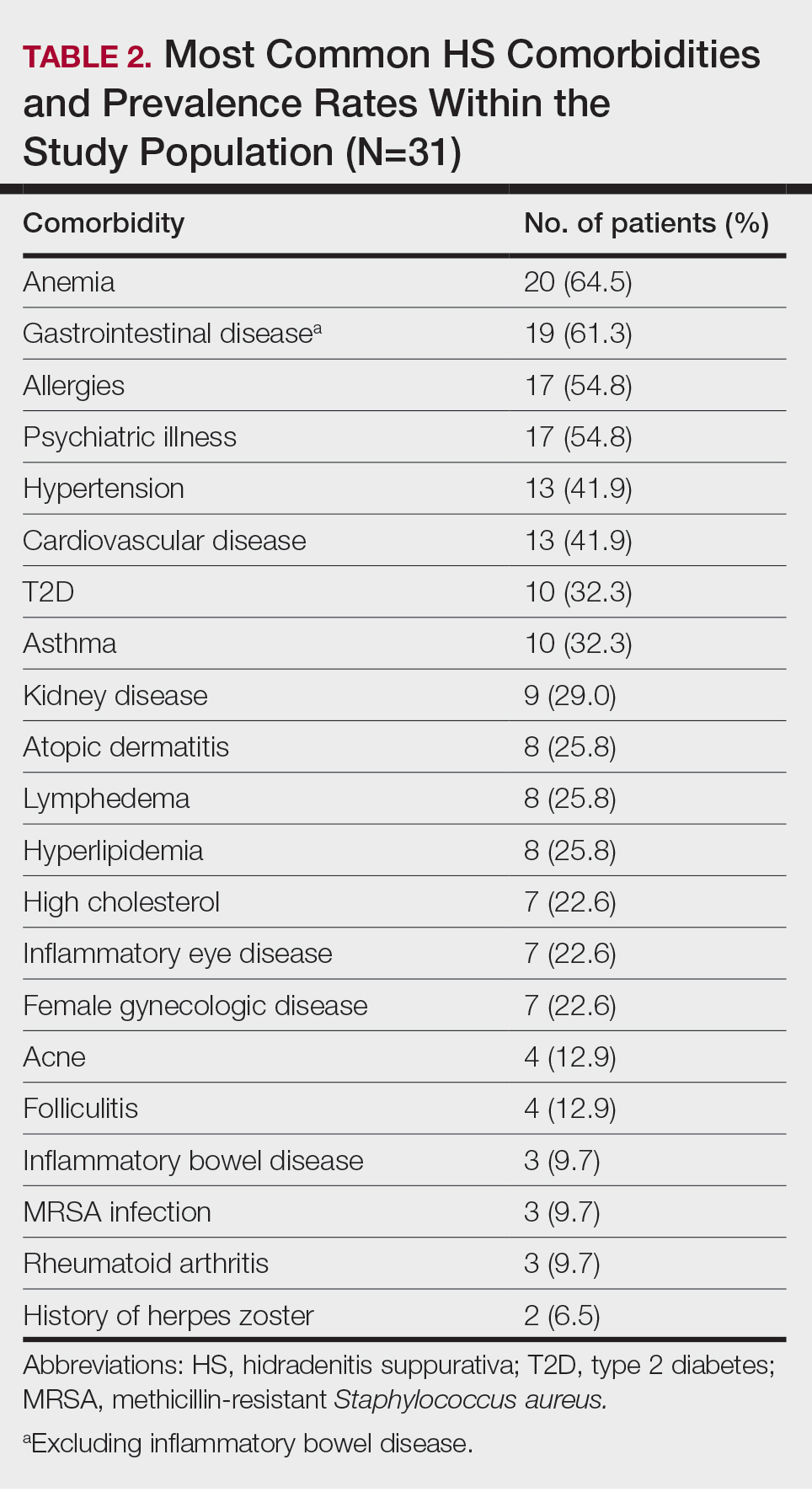
The most common anatomic locations for HS were the right axilla (74.2% [23/31]), left axilla (74.2% [23/31]), groin (71% [22/31]), perineum (61.3% [19/31]), buttocks (41.9% [13/31]), and thigh (41.9% [13/31]). Other locations included the breast, lower back, posterior neck, dorsal foot, and scalp (all 3.2% [1/31])(Table 3). Twenty (64.5%) patients had Hurley staging recorded in the EMR. Seventeen (54.8%) were categorized as Hurley stage 3, and 3 (9.7%) were categorized as Hurley stage 2.

Twenty-nine (93.5%) patients were prescribed an oral antibiotic regimen. The most common oral antibiotics were clindamycin (35.5% [11/31]), doxycycline (35.5% [11/31]), rifampin (29% [9/31]), trimethoprim/sulfamethoxazole (22.6% [7/31]), and cephalexin (22.6% [7/31]). Of the patients who were prescribed rifampin, 87.5% (8/9) also were prescribed an adjunct oral clindamycin regimen. Twenty-nine percent (9/31) of patients were prescribed a biologic regimen; 22.6% (7/31) were prescribed adalimumab, 3.2% (1/31) were prescribed secukinumab, and 3.2% (1/31) were prescribed ustekinumab (Table 4).
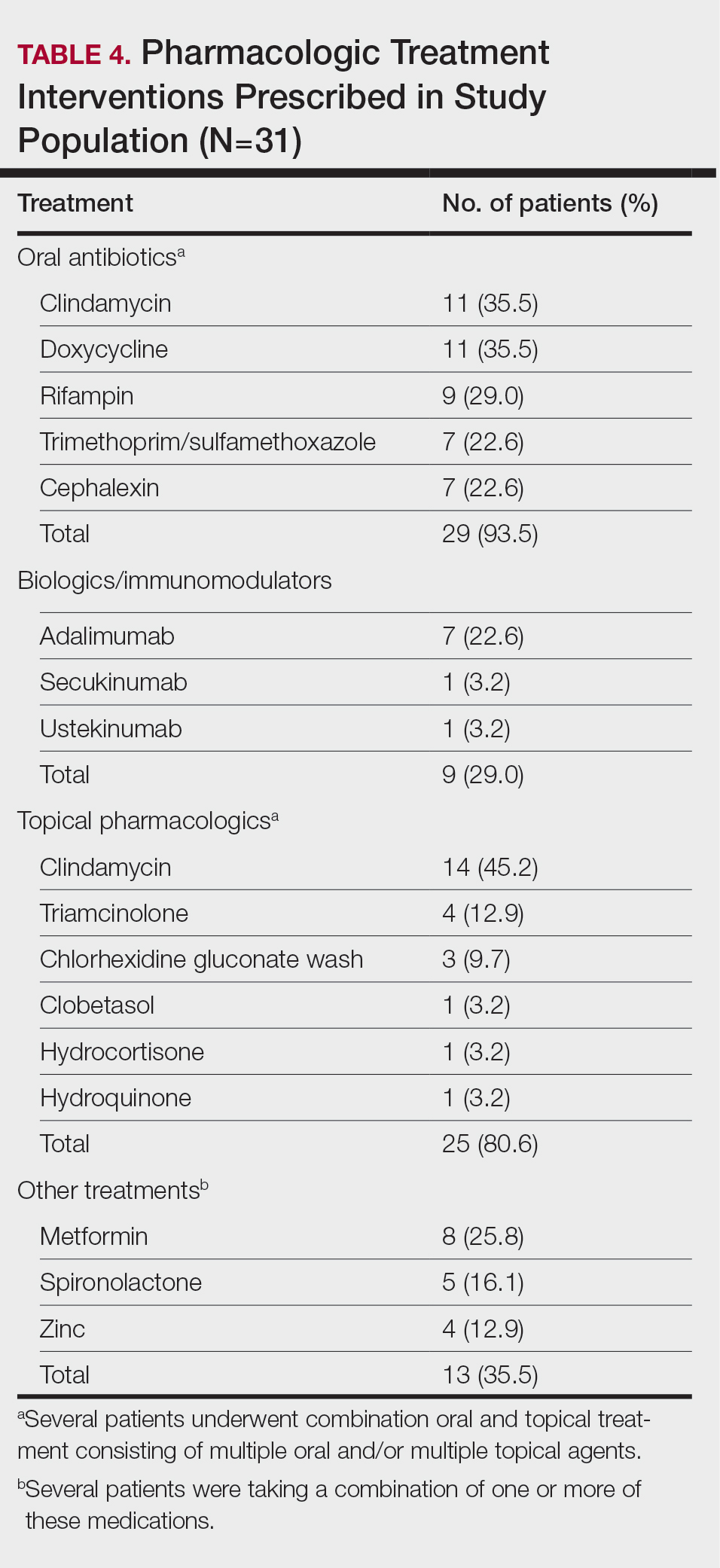
Twenty-five (80.6%) patients were prescribed a topical treatment regimen, the most common being topical clindamycin (45.2% [14/31]). Other topical medications included triamcinolone (12.9% [4/31]), chlorhexidine gluconate wash (9.7% [3/31]), clobetasol (3.2% [1/31]), hydrocortisone (3.2% [1/31]), and hydroquinone (3.2% [1/31])(Table 4).
Other medical treatments for HS included metformin (25.8% [8/31]), spironolactone (16.1% [5/31]), and zinc supplements (12.9% [4/31]). Four patients (12.9%) were prescribed clindamycin plus rifampin as well as a combination of metformin, spironolactone, and/or zinc (Table 4).
Twenty-two (71.0%) patients had a history of receiving incision and drainage procedures as treatment for HS. All 31 patients underwent excisional surgery followed by appropriate reconstruction. The total number of excisional surgeries a single patient underwent for HS treatment ranged from 1 to 9, with a mean of 2 excisional surgeries per patient.
Comment
Our regional verified burn center in Washington, DC, serves a large population of patients with SOC, making it a unique and important sample to study for HS. Our results suggest that Black patients with HS may be at a higher risk for depression and anxiety. Twelve (38.7%) of our patients were diagnosed with depression, which is substantially higher than the 17% to 21% depression prevalence rate among all HS patients reported in meta-analyses.10,11 Additionally, 22.6% (7/31) of our patients were diagnosed with anxiety, which is higher than the 5% to 12% prevalence rate of anxiety among HS patients reported in meta-analyses.10,11 The stress of chronic disease management, psychosocial impact of living with HS, social stigma, sexual dysfunction, pain, and financial concerns make mental illness a debilitating yet common comorbidity for patients with HS. The results of our study suggest that anxiety and depression are highly prevalent among Black patients with HS. It is important to identify if this finding is due to the interplay of health care disparities and social determinants of health; the cause likely is multifactorial, as race and ethnicity may be potential predictors for increased disease severity. Hidradenitis suppurativa is known to be a major economic burden on patients, and race-dependent structural and societal inequalities may be influencing the increased prevalence of anxiety and depression among Black patients with HS.12 Therefore, clinicians must be vigilant for the signs and symptoms of mental illnesses to refer patients for psychiatric treatment when appropriate. Implementing self-report Patient Health Questionnaire-9, General Anxiety Disorder-7 depression and anxiety screening tools, and Dermatology Life Quality Index questionnaires at primary care and dermatology office visits may be a beneficial step toward identifying patients who could benefit from additional mental health resources.13
The patients included in our study predominantly self-identified as Black, and the current smoker prevalence rate was 51.6% (16/31). This percentage is lower than the smoking rates of other published HS studies conducted in predominantly White patient populations, which report up to a 76.5% smoking prevalence rate.14-16 One review article published in 2022 reported that approximately 90% of HS patients are current or former smokers.17 Additionally, a retrospective cohort analysis identifying HS cases among 3,924,310 tobacco smokers in the United States reported that tobacco smokers diagnosed with HS most commonly racially self-identified as White (66.2%).18 Tobacco chemicals and smoke can increase inflammatory cytokine levels, and the activation of nicotinic acetylcholine receptors surrounding pilosebaceous-apocrine units can increase follicular occlusion.14 While several studies1-3,14,19,20 support the strong correlation between tobacco smoking and HS, there are very few that specifically investigate the association between smoking and HS disease in SOC populations. It is possible that smoking rates may be lower in Black patients with HS compared with White patients with HS, which would suggest a multifactorial nature of HS disease pathophysiology. Future large, multicenter studies are needed that investigate smoking rates and HS disease severity in patients across various racial groups.
Prior research has shown a strong correlation between cigarette smoking and HS, but there is minimal data on the role of use of marijuana and other illicit drugs in HS disease pathophysiology.21 A total of 54.8% of our patients were active marijuana users with daily or weekly usage. Further research is needed to investigate whether marijuana use is linked with HS disease pathophysiology and severity or if patients with HS may be using marijuana to relieve pain, anxiety, and depression. Additional studies that survey the method of marijuana use (eg, joint, vape devices, or edibles) would clarify the relationship between not only HS and marijuana but also a potential link between disease severity and the process of inhaling large amounts of smoke vs a link with the active ingredients in the marijuana plant itself.
Approximately 61% (19/31) of our patients were diagnosed with a gastrointestinal disease in addition to HS. Current research reports the link between HS and inflammatory bowel disease, but few studies have investigated if a relationship exists between the gut microbiome and HS, as well as the incidence of general gastrointestinal disease among Black patients with HS.14,22 Our patients were diagnosed with gastrointestinal conditions such as colonic polyps, gastroesophageal reflux disease, benign neoplasms of the cecum and sigmoid colons, small bowel obstruction and perforation, biliary tract diseases, ileus, abdominal hernia, peritonitis, and diverticulosis. Further research is warranted to identify if there is a true relationship between gastrointestinal disease, the gut microbiome, and skin conditions such as HS.22 Biochemical research on the common genetic and inflammatory cytokine pathways involved in HS and gastrointestinal manifestations could help predict disease severity and management in HS patients with SOC.
Several research studies have reported the association between obesity and HS, likely due to adipose cells producing increased estrogen and leading to an estrogen-dominant hormone profile and increased local androgen production in adipose tissue.14,23,24 Antiandrogenic drugs such as finasteride and spironolactone lead to positive results in HS treatment compared to oral antibiotics alone.24 While 71.9% (22/31) of our patients were diagnosed with obesity, only 16.1% (5/31) were prescribed antiandrogen therapy such as spironolactone. It is unclear if this result reflects a health disparity due to insufficient insurance coverage and low prescribing rates or if there is patient hesitancy to taking antiandrogen medications. Additional clinical trials are needed to investigate the efficacy of antiandrogen therapies for HS. If proven to be efficacious, providers should consider adding these medications to the pharmacologic regimen of HS patients with SOC prior to recommending wide-excision surgeries. Furthermore, in addition to antiandrogen medication, weight-management interventions may be helpful in reducing HS disease. The results of a survey conducted in 35 HS patients who underwent bariatric surgery reported 48.6% (17/35) experienced complete disease remission after more than a 15% weight reduction.25,26 Investigating the impact of weight-management practices on disease severity would be helpful in outlining nonpharmacologic treatments for patients with HS.
Limitations
Our study was limited by the constraints of a retrospective chart review and small sample size. Retrospective chart reviews are susceptible to recall bias, variability in providers’ charting practices, and human error from data collectors. We acknowledge that a control group of non-HS patients should be the next step in furthering our research on HS disease comorbidities. Also, since 35.5% (11/31) of our patients did not have Hurley staging recorded in the EMR, it would be beneficial to conduct a future study comprehensive of all 3 Hurley stages. Since 93.5% (29/31) of the patients in our study racially identified as Black, having a control group of racially diverse HS patients would help further our understanding of HS pathophysiology. Lastly, since the inclusion criteria required patients to have undergone excisional surgery for HS, future studies that consider comorbidities among both surgical and nonsurgical patients with HS will aid in our understanding of HS patients with SOC.
Conclusion
The results of our study demonstrate a descriptive analysis of the demographics, most common comorbidities, lesion sites, pharmacologic treatments, and surgical profiles in patients with SOC who underwent surgical treatment for HS. Our data show that HS patients with SOC may be more likely to experience anxiety, depression, and gastrointestinal disease than other HS patients. Additionally, our patients had a high prevalence of marijuana use but lower prevalence of current cigarette use compared to studies conducted in predominantly White HS patient populations, emphasizing the multifactorial nature of HS pathophysiology. Furthermore, despite published research on the efficacy of immunomodulator therapy for HS, most of our HS patients with SOC underwent surgical intervention without first attempting biologic treatment regimens, indicating possible gaps in health care access for minority patients that may be impacting disease severity and outcomes. Studies such as this one that investigate disease pathophysiology and risk factors in SOC patient populations with HS are imperative in minimizing the health care disparity gap, improving disease outcomes, and providing more equitable health care for all patients.
- Wieczorek M, Walecka I. Hidradenitis suppurativa—known and unknown disease. Reumatologia. 2018;56:337-339. doi:10.5114/reum.2018.80709
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-563. doi:10.1016/j. jaad.2008.11.911
- Garg A, Lavian J, Lin G, et al. Incidence of hidradenitis suppurativa in the United States: a sex- and age-adjusted population analysis. J Am Acad Dermatol. 2017;77:118-122. doi:10.1016/j.jaad.2017.02.005
- Ingram JR, Jenkins-Jones S, Knipe DW, et al. Population-based Clinical Practice Research Datalink study using algorithm modelling to identify the true burden of hidradenitis suppurativa. Br J Dermatol. 2018;178:917-924. doi:10.1111/bjd.16101
- Lee DE, Clark AK, Shi VY. Hidradenitis suppurativa: disease burden and etiology in skin of color. Dermatology. 2017;233:456-461. doi:10.1159/000486741
- Brown-Korsah JB, McKenzie S, Omar D, et al. Variations in genetics, biology, and phenotype of cutaneous disorders in skin of color—part I: genetic, biologic, and structural differences in skin of color. J Am Acad Dermatol. 2022;87:1239-1258. doi:10.1016/j.jaad.2022.06.1193
- Narla S, Lyons AB, Hamzavi IH. The most recent advances in understanding and managing hidradenitis suppurativa. F1000Res. 2020;9:F1000 Faculty Rev-1049. doi:10.12688/f1000research.26083.1
- Arenbergerova M, Gkalpakiotis S, Arenberger P. Effective long-term control of refractory hidradenitis suppurativa with adalimumab after failure of conventional therapy. Int J Dermatol. 2010;49:1445-1449. doi:10.1111/j.1365-4632.2010.04638.x
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434. doi:10.1056/NEJMoa1504370
- Jalenques I, Ciortianu L, Pereira B, et al. The prevalence and odds of anxiety and depression in children and adults with hidradenitis suppurativa: systematic review and meta-analysis. J Am Acad Dermatol. 2020;83:542-553. doi:10.1016/j.jaad.2020.03.041
- Machado MO, Stergiopoulos V, Maes M, et al. Depression and anxiety in adults with hidradenitis suppurativa: a systematic review and meta-analysis. JAMA Dermatol. 2019;155:939-945. doi:10.1001 /jamadermatol.2019.0759
- Kilgour JM, Li S, Sarin KY. Hidradenitis suppurativa in patients of color is associated with increased disease severity and healthcare utilization: a retrospective analysis of 2 U.S. cohorts. JAAD Int. 2021;3:42-52. doi:10.1016/j.jdin.2021.01.007
- Rymaszewska JE, Krajewski PK, Szcze² ch J, et al. Depression and anxiety in hidradenitis suppurativa patients: a cross-sectional study among Polish patients. Postep Dermatol Alergol. 2023;40:35-39. doi:10.5114ada.2022.119080
- Johnston LA, Alhusayen R, Bourcier M, et al. Practical guidelines for managing patients with hidradenitis suppurativa: an update. J Cutan Med Surg. 2022;26(2 suppl):2S-24S. doi:10.1177/12034754221116115
- Vazquez BG, Alikhan A, Weaver AL, et al. Incidence of hidradenitis suppurativa and associated factors: a population-based study of Olmsted County, Minnesota. J Invest Dermatol. 2013;133:97-103. doi:10.1038/jid.2012.255
- Seyed Jafari SM, Knüsel E, Cazzaniga S, et al. A retrospective cohort study on patients with hidradenitis suppurativa. Dermatology. 2018;234:71-78. doi:10.1159/000488344
- Lewandowski M, S´ wierczewska Z, Baran´ ska-Rybak W. Hidradenitis suppurativa: a review of current treatment options. Int J Dermatol. 2022;61:1152-1164. doi:10.1111/ijd.16115
- Garg A, Papagermanos V, Midura M, et al. Incidence of hidradenitis suppurativa among tobacco smokers: a population-based retrospective analysis in the U.S.A. Br J Dermatol. 2018;178:709-714. doi:10.1111/bjd.15939
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2022;86:1092-1101. doi:10.1016/j.jaad.2021.01.059
- Tzellos T, Zouboulis CC. Which hidradenitis suppurativa comorbidities should I take into account? Exp Dermatol. 2022;31(suppl 1):29-32. doi:10.1111/exd.14633
- Metko D, Mehta S, Piguet V. Cannabis usage among patients with hidradenitis suppurativa: a scoping review. J Cutan Med Surg. 2024;28:307-308. doi:10.1177/12034754241238719
- Mahmud MR, Akter S, Tamanna SK, et al. Impact of gut microbiome on skin health: gut-skin axis observed through the lenses of therapeutics and skin diseases. Gut Microbes. 2022;14:2096995. doi:10.1080/194 90976.2022.2096995
- Mair KM, Gaw R, MacLean MR. Obesity, estrogens and adipose tissue dysfunction—implications for pulmonary arterial hypertension. Pulm Circ. 2020;10:2045894020952019. doi:10.1177/2045894020952023
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016 /j.jaad.2019.02.067
- Choi ECE, Phan PHC, Oon HH. Hidradenitis suppurativa: racial and socioeconomic considerations in management. Int J Dermatol. 2022;61:1452-1457. doi:10.1111/ijd.16163
Hidradenitis suppurativa (HS) is a debilitating chronic skin disease that often affects apocrinebearing regions of the skin such as the axillae, perineum, and groin.1 Although current research on the etiology and pathogenesis of HS is limited, the disease is known to have a considerable psychosocial impact on patient quality of life.
Clinically, HS lesions manifest as tender subcutaneous nodules that rupture to form painful and deep dermal abscesses.2 These lesions typically develop due to hair follicle occlusion, followed by a cyclic process of inflammation, healing, re-inflammation, and scarring. Often, they are mistaken for cysts or a simple abscess in the early stages of the disease, leading to a delay in diagnosis.1 Disease severity is categorized based on Hurley staging: stage 1 involves abscess formation without scarring; stage 2 involves limited sinus tracts and recurrent abscesses with scarring and/or multiple separated lesions; and stage 3 is the most advanced stage, with diffuse involvement or multiple interconnected sinus tracts across an area with scarring. The condition primarily is medically managed with antibiotics and immunomodulators, but patients who have refractory disease can benefit from surgical excision.1,2
The prevalence of HS in the United States ranges from 0.77% to 1.19%, and individuals who self-identify as Black have 3-fold higher odds of having this condition compared with all other racial groups.3-5 Black patients also are thought to have a greater number and size of apocrine glands compared with patients who self-identify as White, suggesting an anatomic predisposition to developing HS and greater disease severity.6 However, despite HS disproportionately impacting individuals with skin of color (SOC), the majority of published HS research includes predominantly White patient cohorts.5 There is insufficient research assessing HS epidemiology, comorbidities, and treatment responses in patients with SOC.
A 2020 review reported the notable lack of clinical trials that sufficiently examine systemic medication treatment response in HS patients with SOC.7 Of the 15 HS treatment trials published from 2000 to 2019, only 16.4% (138/840) of the patient population were of African descent.7 Clinical trials investigating the efficacy of adalimumab in reducing HS burden also did not adequately evaluate clinical response in patients with SOC. One clinical trial did not include any Black patients as part of the cohort,8 and in 3 other studies, 80% to 85% of the study participants self-identified as White.9 The current literature does not reflect the patient populations most affected by HS, as several studies have reported that 65% of patients diagnosed with HS in the United States annually are Black.5,7 These results emphasize the underrepresentation of SOC populations in the current HS literature and the need for more research that investigates the disease processes, comorbidities, and treatment outcomes of the diverse patient population impacted by HS.
Methods
Study Population and Data Extraction—Following a protocol reviewed and approved by the MedStar Health/Georgetown University institutional review board (IRB #00006783), a retrospective chart review of 31 adult patients with HS who underwent surgery at a regional verified burn center from April 2014 to April 2023 was conducted. The following variables were collected from the electronic medical record (EMR): baseline demographics including age, sex, body mass index (BMI), obesity status, race, ethnicity, Fitzpatrick skin type, smoking status, substance use, employment status, and family history of HS; HS-specific details including Hurley staging, affected areas, and age at initial diagnosis; comorbidities such as dermatologic conditions, autoimmune disorders, infectious diseases, cardiovascular and associated diseases, ovarian disorders, gastrointestinal diseases, and othother common chronic comorbidities (psychiatric illness, kidney disease, type 2 diabetes [T2D], asthma, allergies, lymphedema, and inflammatory eye disease); and use of pharmacologics such as topical medications, oral antibiotics, immunomodulators, and steroids.
Study Definitions—Obesity was defined as both a continuous and categorical variable. Each patient’s BMI at the surgery date was recorded from the EMR as a continuous variable. Patients with obesity also had this condition listed under their complaints and problem list in the EMR, which was recorded as a categorical variable. Race and ethnicity were self-reported by patients. Comorbidity data, including T2D and hyperlipidemia, were defined by previously diagnosed diseases listed in the EMR. Pharmacologic medication data were included in the study if a patient was recommended/prescribed a medication and they had confirmed use of the medication in a subsequent office visit.
Statistical Analysis—Descriptive statistics were calculated for demographics, HS characteristics (eg, location, Hurley stage), and comorbidities. Continuous variables were presented as mean and standard deviation or median and interquartile range and were evaluated using a t test or Mann-Whitney U test when appropriate. Categorical variables were presented as frequencies and percentages and tested for associations using the X2 or Fisher exact test. Data analyses were performed using SAS software version 9.4 (SAS Institute Inc.).
Results
Thirty-one patients (17 females, 14 males; mean age, 40.9 years) were included in the study. Twenty-nine (93.5%) patients identified as Black. All study patients had at least 1 comorbidity. Obesity was diagnosed in 22 (71.0%) patients (mean BMI, 35.5 kg/m2). A total of 16 (51.6%) patients were current smokers, 3 (9.7%) were past smokers, 22 (71%) reported alcohol use, and 17 (54.8%) were active marijuana users. Only 3 (9.7%) patients had a family history of HS (Table 1).
Other common comorbidities associated with HS were anemia (64.5% [20/31]), a non–inflammatory bowel disease gastrointestinal disease (61.3% [19/31]), allergies (54.8% [17/31]), hypertension (41.9% [13/31]), cardiovascular disease (41.9% [13/31]), T2D (32.3% [10/31]), asthma (32.3% [10/31]), kidney disease (29.0% [9/31]), and atopic dermatitis (25.8% [8/31]). More than half (54.8% [17/31]) of patients were diagnosed with psychiatric illnesses, including depression, anxiety, bipolar depression, psychosis, anorexia, impulsive anger, hallucinations, delusion, attention deficit-hyperactivity disorder, and panic disorder (Table 2). Depression was diagnosed in 38.7% (12/31) of patients, and 22.6% (7/31) were diagnosed with anxiety.
The most common anatomic locations for HS were the right axilla (74.2% [23/31]), left axilla (74.2% [23/31]), groin (71% [22/31]), perineum (61.3% [19/31]), buttocks (41.9% [13/31]), and thigh (41.9% [13/31]). Other locations included the breast, lower back, posterior neck, dorsal foot, and scalp (all 3.2% [1/31])(Table 3). Twenty (64.5%) patients had Hurley staging recorded in the EMR. Seventeen (54.8%) were categorized as Hurley stage 3, and 3 (9.7%) were categorized as Hurley stage 2.
Twenty-nine (93.5%) patients were prescribed an oral antibiotic regimen. The most common oral antibiotics were clindamycin (35.5% [11/31]), doxycycline (35.5% [11/31]), rifampin (29% [9/31]), trimethoprim/sulfamethoxazole (22.6% [7/31]), and cephalexin (22.6% [7/31]). Of the patients who were prescribed rifampin, 87.5% (8/9) also were prescribed an adjunct oral clindamycin regimen. Twenty-nine percent (9/31) of patients were prescribed a biologic regimen; 22.6% (7/31) were prescribed adalimumab, 3.2% (1/31) were prescribed secukinumab, and 3.2% (1/31) were prescribed ustekinumab (Table 4).
Twenty-five (80.6%) patients were prescribed a topical treatment regimen, the most common being topical clindamycin (45.2% [14/31]). Other topical medications included triamcinolone (12.9% [4/31]), chlorhexidine gluconate wash (9.7% [3/31]), clobetasol (3.2% [1/31]), hydrocortisone (3.2% [1/31]), and hydroquinone (3.2% [1/31])(Table 4).
Other medical treatments for HS included metformin (25.8% [8/31]), spironolactone (16.1% [5/31]), and zinc supplements (12.9% [4/31]). Four patients (12.9%) were prescribed clindamycin plus rifampin as well as a combination of metformin, spironolactone, and/or zinc (Table 4).
Twenty-two (71.0%) patients had a history of receiving incision and drainage procedures as treatment for HS. All 31 patients underwent excisional surgery followed by appropriate reconstruction. The total number of excisional surgeries a single patient underwent for HS treatment ranged from 1 to 9, with a mean of 2 excisional surgeries per patient.
Comment
Our regional verified burn center in Washington, DC, serves a large population of patients with SOC, making it a unique and important sample to study for HS. Our results suggest that Black patients with HS may be at a higher risk for depression and anxiety. Twelve (38.7%) of our patients were diagnosed with depression, which is substantially higher than the 17% to 21% depression prevalence rate among all HS patients reported in meta-analyses.10,11 Additionally, 22.6% (7/31) of our patients were diagnosed with anxiety, which is higher than the 5% to 12% prevalence rate of anxiety among HS patients reported in meta-analyses.10,11 The stress of chronic disease management, psychosocial impact of living with HS, social stigma, sexual dysfunction, pain, and financial concerns make mental illness a debilitating yet common comorbidity for patients with HS. The results of our study suggest that anxiety and depression are highly prevalent among Black patients with HS. It is important to identify if this finding is due to the interplay of health care disparities and social determinants of health; the cause likely is multifactorial, as race and ethnicity may be potential predictors for increased disease severity. Hidradenitis suppurativa is known to be a major economic burden on patients, and race-dependent structural and societal inequalities may be influencing the increased prevalence of anxiety and depression among Black patients with HS.12 Therefore, clinicians must be vigilant for the signs and symptoms of mental illnesses to refer patients for psychiatric treatment when appropriate. Implementing self-report Patient Health Questionnaire-9, General Anxiety Disorder-7 depression and anxiety screening tools, and Dermatology Life Quality Index questionnaires at primary care and dermatology office visits may be a beneficial step toward identifying patients who could benefit from additional mental health resources.13
The patients included in our study predominantly self-identified as Black, and the current smoker prevalence rate was 51.6% (16/31). This percentage is lower than the smoking rates of other published HS studies conducted in predominantly White patient populations, which report up to a 76.5% smoking prevalence rate.14-16 One review article published in 2022 reported that approximately 90% of HS patients are current or former smokers.17 Additionally, a retrospective cohort analysis identifying HS cases among 3,924,310 tobacco smokers in the United States reported that tobacco smokers diagnosed with HS most commonly racially self-identified as White (66.2%).18 Tobacco chemicals and smoke can increase inflammatory cytokine levels, and the activation of nicotinic acetylcholine receptors surrounding pilosebaceous-apocrine units can increase follicular occlusion.14 While several studies1-3,14,19,20 support the strong correlation between tobacco smoking and HS, there are very few that specifically investigate the association between smoking and HS disease in SOC populations. It is possible that smoking rates may be lower in Black patients with HS compared with White patients with HS, which would suggest a multifactorial nature of HS disease pathophysiology. Future large, multicenter studies are needed that investigate smoking rates and HS disease severity in patients across various racial groups.
Prior research has shown a strong correlation between cigarette smoking and HS, but there is minimal data on the role of use of marijuana and other illicit drugs in HS disease pathophysiology.21 A total of 54.8% of our patients were active marijuana users with daily or weekly usage. Further research is needed to investigate whether marijuana use is linked with HS disease pathophysiology and severity or if patients with HS may be using marijuana to relieve pain, anxiety, and depression. Additional studies that survey the method of marijuana use (eg, joint, vape devices, or edibles) would clarify the relationship between not only HS and marijuana but also a potential link between disease severity and the process of inhaling large amounts of smoke vs a link with the active ingredients in the marijuana plant itself.
Approximately 61% (19/31) of our patients were diagnosed with a gastrointestinal disease in addition to HS. Current research reports the link between HS and inflammatory bowel disease, but few studies have investigated if a relationship exists between the gut microbiome and HS, as well as the incidence of general gastrointestinal disease among Black patients with HS.14,22 Our patients were diagnosed with gastrointestinal conditions such as colonic polyps, gastroesophageal reflux disease, benign neoplasms of the cecum and sigmoid colons, small bowel obstruction and perforation, biliary tract diseases, ileus, abdominal hernia, peritonitis, and diverticulosis. Further research is warranted to identify if there is a true relationship between gastrointestinal disease, the gut microbiome, and skin conditions such as HS.22 Biochemical research on the common genetic and inflammatory cytokine pathways involved in HS and gastrointestinal manifestations could help predict disease severity and management in HS patients with SOC.
Several research studies have reported the association between obesity and HS, likely due to adipose cells producing increased estrogen and leading to an estrogen-dominant hormone profile and increased local androgen production in adipose tissue.14,23,24 Antiandrogenic drugs such as finasteride and spironolactone lead to positive results in HS treatment compared to oral antibiotics alone.24 While 71.9% (22/31) of our patients were diagnosed with obesity, only 16.1% (5/31) were prescribed antiandrogen therapy such as spironolactone. It is unclear if this result reflects a health disparity due to insufficient insurance coverage and low prescribing rates or if there is patient hesitancy to taking antiandrogen medications. Additional clinical trials are needed to investigate the efficacy of antiandrogen therapies for HS. If proven to be efficacious, providers should consider adding these medications to the pharmacologic regimen of HS patients with SOC prior to recommending wide-excision surgeries. Furthermore, in addition to antiandrogen medication, weight-management interventions may be helpful in reducing HS disease. The results of a survey conducted in 35 HS patients who underwent bariatric surgery reported 48.6% (17/35) experienced complete disease remission after more than a 15% weight reduction.25,26 Investigating the impact of weight-management practices on disease severity would be helpful in outlining nonpharmacologic treatments for patients with HS.
Limitations
Our study was limited by the constraints of a retrospective chart review and small sample size. Retrospective chart reviews are susceptible to recall bias, variability in providers’ charting practices, and human error from data collectors. We acknowledge that a control group of non-HS patients should be the next step in furthering our research on HS disease comorbidities. Also, since 35.5% (11/31) of our patients did not have Hurley staging recorded in the EMR, it would be beneficial to conduct a future study comprehensive of all 3 Hurley stages. Since 93.5% (29/31) of the patients in our study racially identified as Black, having a control group of racially diverse HS patients would help further our understanding of HS pathophysiology. Lastly, since the inclusion criteria required patients to have undergone excisional surgery for HS, future studies that consider comorbidities among both surgical and nonsurgical patients with HS will aid in our understanding of HS patients with SOC.
Conclusion
The results of our study demonstrate a descriptive analysis of the demographics, most common comorbidities, lesion sites, pharmacologic treatments, and surgical profiles in patients with SOC who underwent surgical treatment for HS. Our data show that HS patients with SOC may be more likely to experience anxiety, depression, and gastrointestinal disease than other HS patients. Additionally, our patients had a high prevalence of marijuana use but lower prevalence of current cigarette use compared to studies conducted in predominantly White HS patient populations, emphasizing the multifactorial nature of HS pathophysiology. Furthermore, despite published research on the efficacy of immunomodulator therapy for HS, most of our HS patients with SOC underwent surgical intervention without first attempting biologic treatment regimens, indicating possible gaps in health care access for minority patients that may be impacting disease severity and outcomes. Studies such as this one that investigate disease pathophysiology and risk factors in SOC patient populations with HS are imperative in minimizing the health care disparity gap, improving disease outcomes, and providing more equitable health care for all patients.
Hidradenitis suppurativa (HS) is a debilitating chronic skin disease that often affects apocrinebearing regions of the skin such as the axillae, perineum, and groin.1 Although current research on the etiology and pathogenesis of HS is limited, the disease is known to have a considerable psychosocial impact on patient quality of life.
Clinically, HS lesions manifest as tender subcutaneous nodules that rupture to form painful and deep dermal abscesses.2 These lesions typically develop due to hair follicle occlusion, followed by a cyclic process of inflammation, healing, re-inflammation, and scarring. Often, they are mistaken for cysts or a simple abscess in the early stages of the disease, leading to a delay in diagnosis.1 Disease severity is categorized based on Hurley staging: stage 1 involves abscess formation without scarring; stage 2 involves limited sinus tracts and recurrent abscesses with scarring and/or multiple separated lesions; and stage 3 is the most advanced stage, with diffuse involvement or multiple interconnected sinus tracts across an area with scarring. The condition primarily is medically managed with antibiotics and immunomodulators, but patients who have refractory disease can benefit from surgical excision.1,2
The prevalence of HS in the United States ranges from 0.77% to 1.19%, and individuals who self-identify as Black have 3-fold higher odds of having this condition compared with all other racial groups.3-5 Black patients also are thought to have a greater number and size of apocrine glands compared with patients who self-identify as White, suggesting an anatomic predisposition to developing HS and greater disease severity.6 However, despite HS disproportionately impacting individuals with skin of color (SOC), the majority of published HS research includes predominantly White patient cohorts.5 There is insufficient research assessing HS epidemiology, comorbidities, and treatment responses in patients with SOC.
A 2020 review reported the notable lack of clinical trials that sufficiently examine systemic medication treatment response in HS patients with SOC.7 Of the 15 HS treatment trials published from 2000 to 2019, only 16.4% (138/840) of the patient population were of African descent.7 Clinical trials investigating the efficacy of adalimumab in reducing HS burden also did not adequately evaluate clinical response in patients with SOC. One clinical trial did not include any Black patients as part of the cohort,8 and in 3 other studies, 80% to 85% of the study participants self-identified as White.9 The current literature does not reflect the patient populations most affected by HS, as several studies have reported that 65% of patients diagnosed with HS in the United States annually are Black.5,7 These results emphasize the underrepresentation of SOC populations in the current HS literature and the need for more research that investigates the disease processes, comorbidities, and treatment outcomes of the diverse patient population impacted by HS.
Methods
Study Population and Data Extraction—Following a protocol reviewed and approved by the MedStar Health/Georgetown University institutional review board (IRB #00006783), a retrospective chart review of 31 adult patients with HS who underwent surgery at a regional verified burn center from April 2014 to April 2023 was conducted. The following variables were collected from the electronic medical record (EMR): baseline demographics including age, sex, body mass index (BMI), obesity status, race, ethnicity, Fitzpatrick skin type, smoking status, substance use, employment status, and family history of HS; HS-specific details including Hurley staging, affected areas, and age at initial diagnosis; comorbidities such as dermatologic conditions, autoimmune disorders, infectious diseases, cardiovascular and associated diseases, ovarian disorders, gastrointestinal diseases, and othother common chronic comorbidities (psychiatric illness, kidney disease, type 2 diabetes [T2D], asthma, allergies, lymphedema, and inflammatory eye disease); and use of pharmacologics such as topical medications, oral antibiotics, immunomodulators, and steroids.
Study Definitions—Obesity was defined as both a continuous and categorical variable. Each patient’s BMI at the surgery date was recorded from the EMR as a continuous variable. Patients with obesity also had this condition listed under their complaints and problem list in the EMR, which was recorded as a categorical variable. Race and ethnicity were self-reported by patients. Comorbidity data, including T2D and hyperlipidemia, were defined by previously diagnosed diseases listed in the EMR. Pharmacologic medication data were included in the study if a patient was recommended/prescribed a medication and they had confirmed use of the medication in a subsequent office visit.
Statistical Analysis—Descriptive statistics were calculated for demographics, HS characteristics (eg, location, Hurley stage), and comorbidities. Continuous variables were presented as mean and standard deviation or median and interquartile range and were evaluated using a t test or Mann-Whitney U test when appropriate. Categorical variables were presented as frequencies and percentages and tested for associations using the X2 or Fisher exact test. Data analyses were performed using SAS software version 9.4 (SAS Institute Inc.).
Results
Thirty-one patients (17 females, 14 males; mean age, 40.9 years) were included in the study. Twenty-nine (93.5%) patients identified as Black. All study patients had at least 1 comorbidity. Obesity was diagnosed in 22 (71.0%) patients (mean BMI, 35.5 kg/m2). A total of 16 (51.6%) patients were current smokers, 3 (9.7%) were past smokers, 22 (71%) reported alcohol use, and 17 (54.8%) were active marijuana users. Only 3 (9.7%) patients had a family history of HS (Table 1).
Other common comorbidities associated with HS were anemia (64.5% [20/31]), a non–inflammatory bowel disease gastrointestinal disease (61.3% [19/31]), allergies (54.8% [17/31]), hypertension (41.9% [13/31]), cardiovascular disease (41.9% [13/31]), T2D (32.3% [10/31]), asthma (32.3% [10/31]), kidney disease (29.0% [9/31]), and atopic dermatitis (25.8% [8/31]). More than half (54.8% [17/31]) of patients were diagnosed with psychiatric illnesses, including depression, anxiety, bipolar depression, psychosis, anorexia, impulsive anger, hallucinations, delusion, attention deficit-hyperactivity disorder, and panic disorder (Table 2). Depression was diagnosed in 38.7% (12/31) of patients, and 22.6% (7/31) were diagnosed with anxiety.
The most common anatomic locations for HS were the right axilla (74.2% [23/31]), left axilla (74.2% [23/31]), groin (71% [22/31]), perineum (61.3% [19/31]), buttocks (41.9% [13/31]), and thigh (41.9% [13/31]). Other locations included the breast, lower back, posterior neck, dorsal foot, and scalp (all 3.2% [1/31])(Table 3). Twenty (64.5%) patients had Hurley staging recorded in the EMR. Seventeen (54.8%) were categorized as Hurley stage 3, and 3 (9.7%) were categorized as Hurley stage 2.
Twenty-nine (93.5%) patients were prescribed an oral antibiotic regimen. The most common oral antibiotics were clindamycin (35.5% [11/31]), doxycycline (35.5% [11/31]), rifampin (29% [9/31]), trimethoprim/sulfamethoxazole (22.6% [7/31]), and cephalexin (22.6% [7/31]). Of the patients who were prescribed rifampin, 87.5% (8/9) also were prescribed an adjunct oral clindamycin regimen. Twenty-nine percent (9/31) of patients were prescribed a biologic regimen; 22.6% (7/31) were prescribed adalimumab, 3.2% (1/31) were prescribed secukinumab, and 3.2% (1/31) were prescribed ustekinumab (Table 4).
Twenty-five (80.6%) patients were prescribed a topical treatment regimen, the most common being topical clindamycin (45.2% [14/31]). Other topical medications included triamcinolone (12.9% [4/31]), chlorhexidine gluconate wash (9.7% [3/31]), clobetasol (3.2% [1/31]), hydrocortisone (3.2% [1/31]), and hydroquinone (3.2% [1/31])(Table 4).
Other medical treatments for HS included metformin (25.8% [8/31]), spironolactone (16.1% [5/31]), and zinc supplements (12.9% [4/31]). Four patients (12.9%) were prescribed clindamycin plus rifampin as well as a combination of metformin, spironolactone, and/or zinc (Table 4).
Twenty-two (71.0%) patients had a history of receiving incision and drainage procedures as treatment for HS. All 31 patients underwent excisional surgery followed by appropriate reconstruction. The total number of excisional surgeries a single patient underwent for HS treatment ranged from 1 to 9, with a mean of 2 excisional surgeries per patient.
Comment
Our regional verified burn center in Washington, DC, serves a large population of patients with SOC, making it a unique and important sample to study for HS. Our results suggest that Black patients with HS may be at a higher risk for depression and anxiety. Twelve (38.7%) of our patients were diagnosed with depression, which is substantially higher than the 17% to 21% depression prevalence rate among all HS patients reported in meta-analyses.10,11 Additionally, 22.6% (7/31) of our patients were diagnosed with anxiety, which is higher than the 5% to 12% prevalence rate of anxiety among HS patients reported in meta-analyses.10,11 The stress of chronic disease management, psychosocial impact of living with HS, social stigma, sexual dysfunction, pain, and financial concerns make mental illness a debilitating yet common comorbidity for patients with HS. The results of our study suggest that anxiety and depression are highly prevalent among Black patients with HS. It is important to identify if this finding is due to the interplay of health care disparities and social determinants of health; the cause likely is multifactorial, as race and ethnicity may be potential predictors for increased disease severity. Hidradenitis suppurativa is known to be a major economic burden on patients, and race-dependent structural and societal inequalities may be influencing the increased prevalence of anxiety and depression among Black patients with HS.12 Therefore, clinicians must be vigilant for the signs and symptoms of mental illnesses to refer patients for psychiatric treatment when appropriate. Implementing self-report Patient Health Questionnaire-9, General Anxiety Disorder-7 depression and anxiety screening tools, and Dermatology Life Quality Index questionnaires at primary care and dermatology office visits may be a beneficial step toward identifying patients who could benefit from additional mental health resources.13
The patients included in our study predominantly self-identified as Black, and the current smoker prevalence rate was 51.6% (16/31). This percentage is lower than the smoking rates of other published HS studies conducted in predominantly White patient populations, which report up to a 76.5% smoking prevalence rate.14-16 One review article published in 2022 reported that approximately 90% of HS patients are current or former smokers.17 Additionally, a retrospective cohort analysis identifying HS cases among 3,924,310 tobacco smokers in the United States reported that tobacco smokers diagnosed with HS most commonly racially self-identified as White (66.2%).18 Tobacco chemicals and smoke can increase inflammatory cytokine levels, and the activation of nicotinic acetylcholine receptors surrounding pilosebaceous-apocrine units can increase follicular occlusion.14 While several studies1-3,14,19,20 support the strong correlation between tobacco smoking and HS, there are very few that specifically investigate the association between smoking and HS disease in SOC populations. It is possible that smoking rates may be lower in Black patients with HS compared with White patients with HS, which would suggest a multifactorial nature of HS disease pathophysiology. Future large, multicenter studies are needed that investigate smoking rates and HS disease severity in patients across various racial groups.
Prior research has shown a strong correlation between cigarette smoking and HS, but there is minimal data on the role of use of marijuana and other illicit drugs in HS disease pathophysiology.21 A total of 54.8% of our patients were active marijuana users with daily or weekly usage. Further research is needed to investigate whether marijuana use is linked with HS disease pathophysiology and severity or if patients with HS may be using marijuana to relieve pain, anxiety, and depression. Additional studies that survey the method of marijuana use (eg, joint, vape devices, or edibles) would clarify the relationship between not only HS and marijuana but also a potential link between disease severity and the process of inhaling large amounts of smoke vs a link with the active ingredients in the marijuana plant itself.
Approximately 61% (19/31) of our patients were diagnosed with a gastrointestinal disease in addition to HS. Current research reports the link between HS and inflammatory bowel disease, but few studies have investigated if a relationship exists between the gut microbiome and HS, as well as the incidence of general gastrointestinal disease among Black patients with HS.14,22 Our patients were diagnosed with gastrointestinal conditions such as colonic polyps, gastroesophageal reflux disease, benign neoplasms of the cecum and sigmoid colons, small bowel obstruction and perforation, biliary tract diseases, ileus, abdominal hernia, peritonitis, and diverticulosis. Further research is warranted to identify if there is a true relationship between gastrointestinal disease, the gut microbiome, and skin conditions such as HS.22 Biochemical research on the common genetic and inflammatory cytokine pathways involved in HS and gastrointestinal manifestations could help predict disease severity and management in HS patients with SOC.
Several research studies have reported the association between obesity and HS, likely due to adipose cells producing increased estrogen and leading to an estrogen-dominant hormone profile and increased local androgen production in adipose tissue.14,23,24 Antiandrogenic drugs such as finasteride and spironolactone lead to positive results in HS treatment compared to oral antibiotics alone.24 While 71.9% (22/31) of our patients were diagnosed with obesity, only 16.1% (5/31) were prescribed antiandrogen therapy such as spironolactone. It is unclear if this result reflects a health disparity due to insufficient insurance coverage and low prescribing rates or if there is patient hesitancy to taking antiandrogen medications. Additional clinical trials are needed to investigate the efficacy of antiandrogen therapies for HS. If proven to be efficacious, providers should consider adding these medications to the pharmacologic regimen of HS patients with SOC prior to recommending wide-excision surgeries. Furthermore, in addition to antiandrogen medication, weight-management interventions may be helpful in reducing HS disease. The results of a survey conducted in 35 HS patients who underwent bariatric surgery reported 48.6% (17/35) experienced complete disease remission after more than a 15% weight reduction.25,26 Investigating the impact of weight-management practices on disease severity would be helpful in outlining nonpharmacologic treatments for patients with HS.
Limitations
Our study was limited by the constraints of a retrospective chart review and small sample size. Retrospective chart reviews are susceptible to recall bias, variability in providers’ charting practices, and human error from data collectors. We acknowledge that a control group of non-HS patients should be the next step in furthering our research on HS disease comorbidities. Also, since 35.5% (11/31) of our patients did not have Hurley staging recorded in the EMR, it would be beneficial to conduct a future study comprehensive of all 3 Hurley stages. Since 93.5% (29/31) of the patients in our study racially identified as Black, having a control group of racially diverse HS patients would help further our understanding of HS pathophysiology. Lastly, since the inclusion criteria required patients to have undergone excisional surgery for HS, future studies that consider comorbidities among both surgical and nonsurgical patients with HS will aid in our understanding of HS patients with SOC.
Conclusion
The results of our study demonstrate a descriptive analysis of the demographics, most common comorbidities, lesion sites, pharmacologic treatments, and surgical profiles in patients with SOC who underwent surgical treatment for HS. Our data show that HS patients with SOC may be more likely to experience anxiety, depression, and gastrointestinal disease than other HS patients. Additionally, our patients had a high prevalence of marijuana use but lower prevalence of current cigarette use compared to studies conducted in predominantly White HS patient populations, emphasizing the multifactorial nature of HS pathophysiology. Furthermore, despite published research on the efficacy of immunomodulator therapy for HS, most of our HS patients with SOC underwent surgical intervention without first attempting biologic treatment regimens, indicating possible gaps in health care access for minority patients that may be impacting disease severity and outcomes. Studies such as this one that investigate disease pathophysiology and risk factors in SOC patient populations with HS are imperative in minimizing the health care disparity gap, improving disease outcomes, and providing more equitable health care for all patients.
- Wieczorek M, Walecka I. Hidradenitis suppurativa—known and unknown disease. Reumatologia. 2018;56:337-339. doi:10.5114/reum.2018.80709
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-563. doi:10.1016/j. jaad.2008.11.911
- Garg A, Lavian J, Lin G, et al. Incidence of hidradenitis suppurativa in the United States: a sex- and age-adjusted population analysis. J Am Acad Dermatol. 2017;77:118-122. doi:10.1016/j.jaad.2017.02.005
- Ingram JR, Jenkins-Jones S, Knipe DW, et al. Population-based Clinical Practice Research Datalink study using algorithm modelling to identify the true burden of hidradenitis suppurativa. Br J Dermatol. 2018;178:917-924. doi:10.1111/bjd.16101
- Lee DE, Clark AK, Shi VY. Hidradenitis suppurativa: disease burden and etiology in skin of color. Dermatology. 2017;233:456-461. doi:10.1159/000486741
- Brown-Korsah JB, McKenzie S, Omar D, et al. Variations in genetics, biology, and phenotype of cutaneous disorders in skin of color—part I: genetic, biologic, and structural differences in skin of color. J Am Acad Dermatol. 2022;87:1239-1258. doi:10.1016/j.jaad.2022.06.1193
- Narla S, Lyons AB, Hamzavi IH. The most recent advances in understanding and managing hidradenitis suppurativa. F1000Res. 2020;9:F1000 Faculty Rev-1049. doi:10.12688/f1000research.26083.1
- Arenbergerova M, Gkalpakiotis S, Arenberger P. Effective long-term control of refractory hidradenitis suppurativa with adalimumab after failure of conventional therapy. Int J Dermatol. 2010;49:1445-1449. doi:10.1111/j.1365-4632.2010.04638.x
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434. doi:10.1056/NEJMoa1504370
- Jalenques I, Ciortianu L, Pereira B, et al. The prevalence and odds of anxiety and depression in children and adults with hidradenitis suppurativa: systematic review and meta-analysis. J Am Acad Dermatol. 2020;83:542-553. doi:10.1016/j.jaad.2020.03.041
- Machado MO, Stergiopoulos V, Maes M, et al. Depression and anxiety in adults with hidradenitis suppurativa: a systematic review and meta-analysis. JAMA Dermatol. 2019;155:939-945. doi:10.1001 /jamadermatol.2019.0759
- Kilgour JM, Li S, Sarin KY. Hidradenitis suppurativa in patients of color is associated with increased disease severity and healthcare utilization: a retrospective analysis of 2 U.S. cohorts. JAAD Int. 2021;3:42-52. doi:10.1016/j.jdin.2021.01.007
- Rymaszewska JE, Krajewski PK, Szcze² ch J, et al. Depression and anxiety in hidradenitis suppurativa patients: a cross-sectional study among Polish patients. Postep Dermatol Alergol. 2023;40:35-39. doi:10.5114ada.2022.119080
- Johnston LA, Alhusayen R, Bourcier M, et al. Practical guidelines for managing patients with hidradenitis suppurativa: an update. J Cutan Med Surg. 2022;26(2 suppl):2S-24S. doi:10.1177/12034754221116115
- Vazquez BG, Alikhan A, Weaver AL, et al. Incidence of hidradenitis suppurativa and associated factors: a population-based study of Olmsted County, Minnesota. J Invest Dermatol. 2013;133:97-103. doi:10.1038/jid.2012.255
- Seyed Jafari SM, Knüsel E, Cazzaniga S, et al. A retrospective cohort study on patients with hidradenitis suppurativa. Dermatology. 2018;234:71-78. doi:10.1159/000488344
- Lewandowski M, S´ wierczewska Z, Baran´ ska-Rybak W. Hidradenitis suppurativa: a review of current treatment options. Int J Dermatol. 2022;61:1152-1164. doi:10.1111/ijd.16115
- Garg A, Papagermanos V, Midura M, et al. Incidence of hidradenitis suppurativa among tobacco smokers: a population-based retrospective analysis in the U.S.A. Br J Dermatol. 2018;178:709-714. doi:10.1111/bjd.15939
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2022;86:1092-1101. doi:10.1016/j.jaad.2021.01.059
- Tzellos T, Zouboulis CC. Which hidradenitis suppurativa comorbidities should I take into account? Exp Dermatol. 2022;31(suppl 1):29-32. doi:10.1111/exd.14633
- Metko D, Mehta S, Piguet V. Cannabis usage among patients with hidradenitis suppurativa: a scoping review. J Cutan Med Surg. 2024;28:307-308. doi:10.1177/12034754241238719
- Mahmud MR, Akter S, Tamanna SK, et al. Impact of gut microbiome on skin health: gut-skin axis observed through the lenses of therapeutics and skin diseases. Gut Microbes. 2022;14:2096995. doi:10.1080/194 90976.2022.2096995
- Mair KM, Gaw R, MacLean MR. Obesity, estrogens and adipose tissue dysfunction—implications for pulmonary arterial hypertension. Pulm Circ. 2020;10:2045894020952019. doi:10.1177/2045894020952023
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016 /j.jaad.2019.02.067
- Choi ECE, Phan PHC, Oon HH. Hidradenitis suppurativa: racial and socioeconomic considerations in management. Int J Dermatol. 2022;61:1452-1457. doi:10.1111/ijd.16163
- Wieczorek M, Walecka I. Hidradenitis suppurativa—known and unknown disease. Reumatologia. 2018;56:337-339. doi:10.5114/reum.2018.80709
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-563. doi:10.1016/j. jaad.2008.11.911
- Garg A, Lavian J, Lin G, et al. Incidence of hidradenitis suppurativa in the United States: a sex- and age-adjusted population analysis. J Am Acad Dermatol. 2017;77:118-122. doi:10.1016/j.jaad.2017.02.005
- Ingram JR, Jenkins-Jones S, Knipe DW, et al. Population-based Clinical Practice Research Datalink study using algorithm modelling to identify the true burden of hidradenitis suppurativa. Br J Dermatol. 2018;178:917-924. doi:10.1111/bjd.16101
- Lee DE, Clark AK, Shi VY. Hidradenitis suppurativa: disease burden and etiology in skin of color. Dermatology. 2017;233:456-461. doi:10.1159/000486741
- Brown-Korsah JB, McKenzie S, Omar D, et al. Variations in genetics, biology, and phenotype of cutaneous disorders in skin of color—part I: genetic, biologic, and structural differences in skin of color. J Am Acad Dermatol. 2022;87:1239-1258. doi:10.1016/j.jaad.2022.06.1193
- Narla S, Lyons AB, Hamzavi IH. The most recent advances in understanding and managing hidradenitis suppurativa. F1000Res. 2020;9:F1000 Faculty Rev-1049. doi:10.12688/f1000research.26083.1
- Arenbergerova M, Gkalpakiotis S, Arenberger P. Effective long-term control of refractory hidradenitis suppurativa with adalimumab after failure of conventional therapy. Int J Dermatol. 2010;49:1445-1449. doi:10.1111/j.1365-4632.2010.04638.x
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434. doi:10.1056/NEJMoa1504370
- Jalenques I, Ciortianu L, Pereira B, et al. The prevalence and odds of anxiety and depression in children and adults with hidradenitis suppurativa: systematic review and meta-analysis. J Am Acad Dermatol. 2020;83:542-553. doi:10.1016/j.jaad.2020.03.041
- Machado MO, Stergiopoulos V, Maes M, et al. Depression and anxiety in adults with hidradenitis suppurativa: a systematic review and meta-analysis. JAMA Dermatol. 2019;155:939-945. doi:10.1001 /jamadermatol.2019.0759
- Kilgour JM, Li S, Sarin KY. Hidradenitis suppurativa in patients of color is associated with increased disease severity and healthcare utilization: a retrospective analysis of 2 U.S. cohorts. JAAD Int. 2021;3:42-52. doi:10.1016/j.jdin.2021.01.007
- Rymaszewska JE, Krajewski PK, Szcze² ch J, et al. Depression and anxiety in hidradenitis suppurativa patients: a cross-sectional study among Polish patients. Postep Dermatol Alergol. 2023;40:35-39. doi:10.5114ada.2022.119080
- Johnston LA, Alhusayen R, Bourcier M, et al. Practical guidelines for managing patients with hidradenitis suppurativa: an update. J Cutan Med Surg. 2022;26(2 suppl):2S-24S. doi:10.1177/12034754221116115
- Vazquez BG, Alikhan A, Weaver AL, et al. Incidence of hidradenitis suppurativa and associated factors: a population-based study of Olmsted County, Minnesota. J Invest Dermatol. 2013;133:97-103. doi:10.1038/jid.2012.255
- Seyed Jafari SM, Knüsel E, Cazzaniga S, et al. A retrospective cohort study on patients with hidradenitis suppurativa. Dermatology. 2018;234:71-78. doi:10.1159/000488344
- Lewandowski M, S´ wierczewska Z, Baran´ ska-Rybak W. Hidradenitis suppurativa: a review of current treatment options. Int J Dermatol. 2022;61:1152-1164. doi:10.1111/ijd.16115
- Garg A, Papagermanos V, Midura M, et al. Incidence of hidradenitis suppurativa among tobacco smokers: a population-based retrospective analysis in the U.S.A. Br J Dermatol. 2018;178:709-714. doi:10.1111/bjd.15939
- Garg A, Malviya N, Strunk A, et al. Comorbidity screening in hidradenitis suppurativa: evidence-based recommendations from the US and Canadian Hidradenitis Suppurativa Foundations. J Am Acad Dermatol. 2022;86:1092-1101. doi:10.1016/j.jaad.2021.01.059
- Tzellos T, Zouboulis CC. Which hidradenitis suppurativa comorbidities should I take into account? Exp Dermatol. 2022;31(suppl 1):29-32. doi:10.1111/exd.14633
- Metko D, Mehta S, Piguet V. Cannabis usage among patients with hidradenitis suppurativa: a scoping review. J Cutan Med Surg. 2024;28:307-308. doi:10.1177/12034754241238719
- Mahmud MR, Akter S, Tamanna SK, et al. Impact of gut microbiome on skin health: gut-skin axis observed through the lenses of therapeutics and skin diseases. Gut Microbes. 2022;14:2096995. doi:10.1080/194 90976.2022.2096995
- Mair KM, Gaw R, MacLean MR. Obesity, estrogens and adipose tissue dysfunction—implications for pulmonary arterial hypertension. Pulm Circ. 2020;10:2045894020952019. doi:10.1177/2045894020952023
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016 /j.jaad.2019.02.067
- Choi ECE, Phan PHC, Oon HH. Hidradenitis suppurativa: racial and socioeconomic considerations in management. Int J Dermatol. 2022;61:1452-1457. doi:10.1111/ijd.16163
Evaluating Factors Impacting Hidradenitis Suppurativa Disease Severity in Patients With Darker Skin Types
Evaluating Factors Impacting Hidradenitis Suppurativa Disease Severity in Patients With Darker Skin Types
PRACTICE POINTS
- Anxiety and depression are highly prevalent among Black patients with hidradenitis suppurativa (HS). Implementing self-report questionnaires at medical office visits are crucial to identifying patients who could benefit from additional psychiatric resources.
- Hidradenitis suppurativa patients with skin of color may have a higher incidence of comorbid gastrointestinal disease than other HS patients.
- Investigating the impact of weight-management practices on disease severity would be helpful in outlining nonpharmacologic treatments for patients with HS.
- The patient cohort described here had a high prevalence of marijuana use but lower prevalence of current cigarette use compared to studies conducted in predominantly White HS patient populations, emphasizing the multifactorial nature of HS pathophysiology.
Evaluating Access to Full-Body Skin Examinations in Los Angeles County, California
Evaluating Access to Full-Body Skin Examinations in Los Angeles County, California
To the Editor:
Early skin cancer detection improves patient outcomes1; however, socioeconomic and racial disparities may impact access to dermatologic care.2 Although non-Hispanic White individuals have a high incidence of skin cancer, they experience higher melanoma-specific survival rates than non-White patients, who often receive later-stage diagnoses and experience higher mortality.2 Furthermore, racial/ ethnic minorities often face longer surgery wait times after diagnosis and have lower socioeconomic status (SES) and less favorable health insurance coverage, contributing to poorer outcomes.2,3
To examine access to full-body skin examinations (FBSEs) by board-certified dermatologists in Los Angeles (LA) County, California, we analyzed the availability of FBSEs based on racial demographics, income, and insurance type (Medicaid [Medi-Cal] vs private [Blue Cross Blue Shield (BCBS)]). Demographic data by zip code were obtained from the US Census Bureau.4 This validated metric highlights socioeconomic disparities and minimizes data gaps5,6 and was used to assess health care access among different population subgroups. Dermatologists’ contact information was obtained from the Find a Dermatologist page on the American Academy of Dermatology website and the listed phone numbers of their practice were used to contact them. Practices with board-certified dermatologists accepting new patients were included in the study; practices were not included if they had exclusive insurance plans; were pediatric, cosmetic, or research only; or were nonresponsive to calls. From August 2022 to September 2022, each practice was called twice within a 36-hour period—once by a simulated patient with Medi-Cal and once by a simulated patient with BCBS—and were asked about availability for new patient FBSE appointments and accepted insurance types. Data were analyzed using SAS software (SAS Institute Inc.).
Los Angeles County comprises 269 zip codes, of which 82 (30.5%) have dermatology practices. Of 213 total dermatologists in LA County listed on the American Academy of Dermatology website, 193 (90.6%) met preliminary criteria, and 169 (79.3%) were successfully contacted. Almost all (94.6% [160/169]) accepted new patients for FBSEs; of those, 63.1% (101/160) accepted only private insurance, 16.9% (27/160) accepted both private insurance and Medi-Cal, and 16.2% (26/160) did not accept any insurance. Racial predominance for each dermatology practice was analyzed by zip code (Table). Dermatologists included in our study were significantly more concentrated in predominantly non- Hispanic White areas of LA County vs predominantly Hispanic areas (P<.0001). Notably, the average income in predominantly non-Hispanic White zip codes ($114,757.74) was significantly higher than in predominantly Hispanic areas ($58,278.54)(P=.001)(Table).4
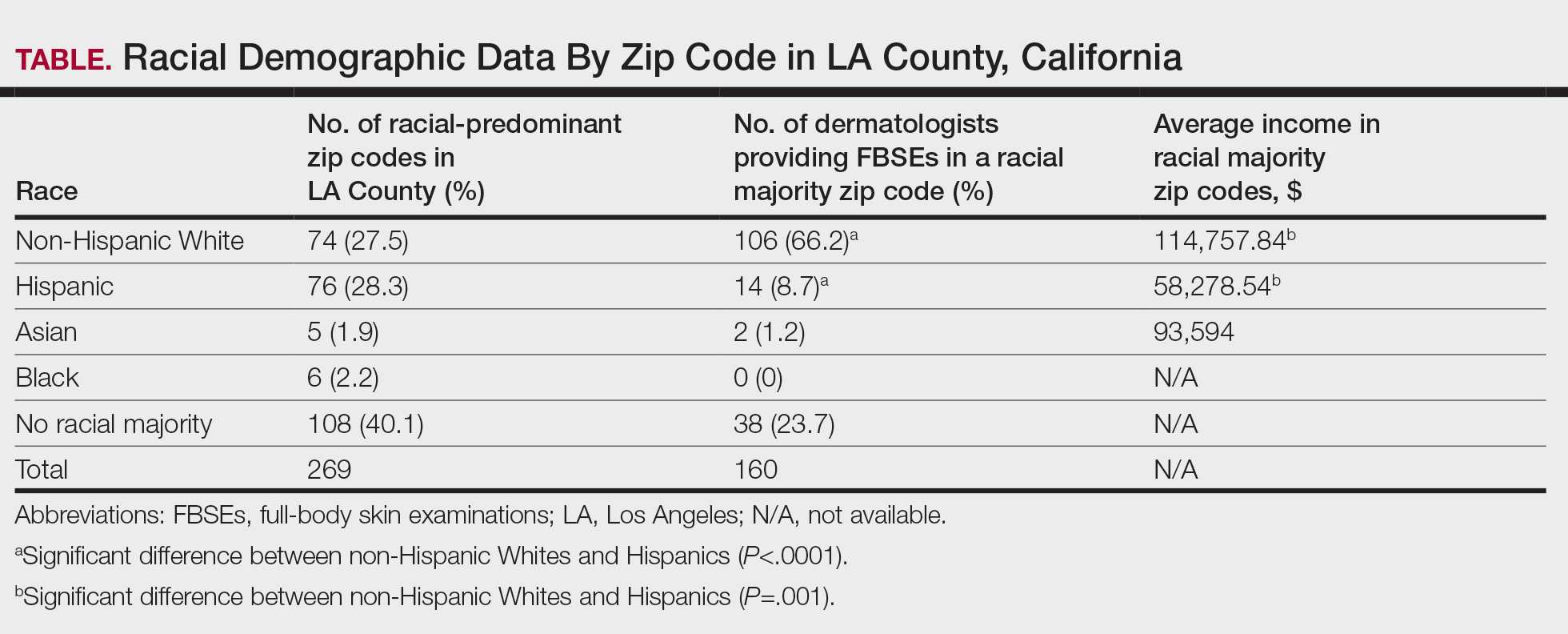
In LA County, 40.1% (108/269) of zip codes have no racial majority, 28.2% (76/269) are predominantly Hispanic, 27.5% (74/269) are predominantly non-Hispanic White, 2.2% (6/269) are predominantly Black, and 1.9% (5/269) are predominantly Asian.4 There are no dermatologists in predominantly Black zip codes, 2 in predominantly Asian zip codes, 14 in predominantly Hispanic zip codes, 38 in zip codes with no racial majority, and 106 in predominantly non-Hispanic White zip codes. There are significantly more dermatologists in predominantly non-Hispanic White zip codes compared to predominantly Hispanic zip codes (P<.0001). In LA County, the average income in predominantly Asian, non-Hispanic White, and Hispanic zip codes was $93,594, $114,757.84, and $58,278.54, respectively, in 2021.4 The average income in predominantly non-Hispanic White zip codes was significantly higher than in predominantly Hispanic zip codes (P=.001). There were no income data available for predominantly Black zip codes or zip codes with no racial majority.
The results from our study revealed potential barriers to FBSEs for racial and ethnic minorities in LA County, which supports previous research on the impact of SES, race, and insurance on access to dermatologic care.2,3 Predominantly Hispanic zip codes have significantly lower income (P<.0001) and fewer dermatologists (P=.001) compared to zip codes that are predominantly non-Hispanic White, reflecting how lower SES correlates with worse health outcomes and higher melanoma mortality. Conversely, predominantly non-Hispanic White areas with higher income have better access to dermatologists, which may contribute to the improved melanoma survival rates among White patients. Additionally, most dermatologists accept only private insurance, further highlighting the disparity in FBSE access for non-White patients across LA County. While our study focused on FBSE access, our findings may point to a wider barrier to dermatologic care, especially in zip codes with fewer dermatologists. Further studies are needed to determine whether these areas also face barriers to accessing primary care.
Our study was limited by the exclusion of nonphysician providers (eg, nurse practitioners, physician assistants), a small sample size, and lack of available economic data for predominantly Black zip codes.4 Additionally, the exclusion of practices with exclusive insurance plans (eg, Kaiser Permanente) limited the generalizability of our findings, as our results did not account for the populations served by these practices. Furthermore, our analysis did not account for variations in practice size or the proportion of care provided to patients with different insurance types, which could impact overall accessibility. Additional studies are needed to explore the impact of these factors on access to general dermatologic care and not just FBSEs.
Racial/ethnic minorities and lower SES populations face major barriers to FBSE access in LA County, such as difficulty finding a dermatologist in their area or one who accepts Medi-Cal. Addressing these disparities is crucial for improving skin cancer outcomes. Further research is needed to develop strategies to eliminate these barriers to dermatologic care, such as increasing access to teledermatology, offering mobile dermatology clinics, and improving insurance coverage.
- Chiaravalloti AJ, Laduca JR. Melanoma screening by means of complete skin exams for all patients in a dermatology practice reduces the thickness of primary melanomas at diagnosis. J Clin Aesthet Dermatol. 2014;7:18-22.
- Qian Y, Johannet P, Sawyers A, et al. The ongoing racial disparities in melanoma: an analysis of the Surveillance, Epidemiology, and End Results database (1975-2016). J Am Acad Dermatol. 2021;84:1585-1593.
- Baranowski MLH, Yeung H, Chen SC, et al. Factors associated with time to surgery in melanoma: an analysis of the National Cancer Database. J Am Acad Dermatol. 2019;81:908-916.
- United States Census Bureau. Explore census data. Accessed March 17, 2025. https://data.census.gov/all?q=los+angeles+county
- Berkowitz SA, Traore CY, Singer DE, et al. Evaluating area-based socioeconomic status indicators for monitoring disparities within health care systems: results from a primary care network. Health Serv Res. 2015;50:398-417.
- Jacobs B, Ir P, Bigdeli M, et al. Addressing access barriers to health services: an analytical framework for selecting appropriate interventions in lowincome Asian countries. Health Policy Plan. 2012;27:288-300.
To the Editor:
Early skin cancer detection improves patient outcomes1; however, socioeconomic and racial disparities may impact access to dermatologic care.2 Although non-Hispanic White individuals have a high incidence of skin cancer, they experience higher melanoma-specific survival rates than non-White patients, who often receive later-stage diagnoses and experience higher mortality.2 Furthermore, racial/ ethnic minorities often face longer surgery wait times after diagnosis and have lower socioeconomic status (SES) and less favorable health insurance coverage, contributing to poorer outcomes.2,3
To examine access to full-body skin examinations (FBSEs) by board-certified dermatologists in Los Angeles (LA) County, California, we analyzed the availability of FBSEs based on racial demographics, income, and insurance type (Medicaid [Medi-Cal] vs private [Blue Cross Blue Shield (BCBS)]). Demographic data by zip code were obtained from the US Census Bureau.4 This validated metric highlights socioeconomic disparities and minimizes data gaps5,6 and was used to assess health care access among different population subgroups. Dermatologists’ contact information was obtained from the Find a Dermatologist page on the American Academy of Dermatology website and the listed phone numbers of their practice were used to contact them. Practices with board-certified dermatologists accepting new patients were included in the study; practices were not included if they had exclusive insurance plans; were pediatric, cosmetic, or research only; or were nonresponsive to calls. From August 2022 to September 2022, each practice was called twice within a 36-hour period—once by a simulated patient with Medi-Cal and once by a simulated patient with BCBS—and were asked about availability for new patient FBSE appointments and accepted insurance types. Data were analyzed using SAS software (SAS Institute Inc.).
Los Angeles County comprises 269 zip codes, of which 82 (30.5%) have dermatology practices. Of 213 total dermatologists in LA County listed on the American Academy of Dermatology website, 193 (90.6%) met preliminary criteria, and 169 (79.3%) were successfully contacted. Almost all (94.6% [160/169]) accepted new patients for FBSEs; of those, 63.1% (101/160) accepted only private insurance, 16.9% (27/160) accepted both private insurance and Medi-Cal, and 16.2% (26/160) did not accept any insurance. Racial predominance for each dermatology practice was analyzed by zip code (Table). Dermatologists included in our study were significantly more concentrated in predominantly non- Hispanic White areas of LA County vs predominantly Hispanic areas (P<.0001). Notably, the average income in predominantly non-Hispanic White zip codes ($114,757.74) was significantly higher than in predominantly Hispanic areas ($58,278.54)(P=.001)(Table).4
In LA County, 40.1% (108/269) of zip codes have no racial majority, 28.2% (76/269) are predominantly Hispanic, 27.5% (74/269) are predominantly non-Hispanic White, 2.2% (6/269) are predominantly Black, and 1.9% (5/269) are predominantly Asian.4 There are no dermatologists in predominantly Black zip codes, 2 in predominantly Asian zip codes, 14 in predominantly Hispanic zip codes, 38 in zip codes with no racial majority, and 106 in predominantly non-Hispanic White zip codes. There are significantly more dermatologists in predominantly non-Hispanic White zip codes compared to predominantly Hispanic zip codes (P<.0001). In LA County, the average income in predominantly Asian, non-Hispanic White, and Hispanic zip codes was $93,594, $114,757.84, and $58,278.54, respectively, in 2021.4 The average income in predominantly non-Hispanic White zip codes was significantly higher than in predominantly Hispanic zip codes (P=.001). There were no income data available for predominantly Black zip codes or zip codes with no racial majority.
The results from our study revealed potential barriers to FBSEs for racial and ethnic minorities in LA County, which supports previous research on the impact of SES, race, and insurance on access to dermatologic care.2,3 Predominantly Hispanic zip codes have significantly lower income (P<.0001) and fewer dermatologists (P=.001) compared to zip codes that are predominantly non-Hispanic White, reflecting how lower SES correlates with worse health outcomes and higher melanoma mortality. Conversely, predominantly non-Hispanic White areas with higher income have better access to dermatologists, which may contribute to the improved melanoma survival rates among White patients. Additionally, most dermatologists accept only private insurance, further highlighting the disparity in FBSE access for non-White patients across LA County. While our study focused on FBSE access, our findings may point to a wider barrier to dermatologic care, especially in zip codes with fewer dermatologists. Further studies are needed to determine whether these areas also face barriers to accessing primary care.
Our study was limited by the exclusion of nonphysician providers (eg, nurse practitioners, physician assistants), a small sample size, and lack of available economic data for predominantly Black zip codes.4 Additionally, the exclusion of practices with exclusive insurance plans (eg, Kaiser Permanente) limited the generalizability of our findings, as our results did not account for the populations served by these practices. Furthermore, our analysis did not account for variations in practice size or the proportion of care provided to patients with different insurance types, which could impact overall accessibility. Additional studies are needed to explore the impact of these factors on access to general dermatologic care and not just FBSEs.
Racial/ethnic minorities and lower SES populations face major barriers to FBSE access in LA County, such as difficulty finding a dermatologist in their area or one who accepts Medi-Cal. Addressing these disparities is crucial for improving skin cancer outcomes. Further research is needed to develop strategies to eliminate these barriers to dermatologic care, such as increasing access to teledermatology, offering mobile dermatology clinics, and improving insurance coverage.
To the Editor:
Early skin cancer detection improves patient outcomes1; however, socioeconomic and racial disparities may impact access to dermatologic care.2 Although non-Hispanic White individuals have a high incidence of skin cancer, they experience higher melanoma-specific survival rates than non-White patients, who often receive later-stage diagnoses and experience higher mortality.2 Furthermore, racial/ ethnic minorities often face longer surgery wait times after diagnosis and have lower socioeconomic status (SES) and less favorable health insurance coverage, contributing to poorer outcomes.2,3
To examine access to full-body skin examinations (FBSEs) by board-certified dermatologists in Los Angeles (LA) County, California, we analyzed the availability of FBSEs based on racial demographics, income, and insurance type (Medicaid [Medi-Cal] vs private [Blue Cross Blue Shield (BCBS)]). Demographic data by zip code were obtained from the US Census Bureau.4 This validated metric highlights socioeconomic disparities and minimizes data gaps5,6 and was used to assess health care access among different population subgroups. Dermatologists’ contact information was obtained from the Find a Dermatologist page on the American Academy of Dermatology website and the listed phone numbers of their practice were used to contact them. Practices with board-certified dermatologists accepting new patients were included in the study; practices were not included if they had exclusive insurance plans; were pediatric, cosmetic, or research only; or were nonresponsive to calls. From August 2022 to September 2022, each practice was called twice within a 36-hour period—once by a simulated patient with Medi-Cal and once by a simulated patient with BCBS—and were asked about availability for new patient FBSE appointments and accepted insurance types. Data were analyzed using SAS software (SAS Institute Inc.).
Los Angeles County comprises 269 zip codes, of which 82 (30.5%) have dermatology practices. Of 213 total dermatologists in LA County listed on the American Academy of Dermatology website, 193 (90.6%) met preliminary criteria, and 169 (79.3%) were successfully contacted. Almost all (94.6% [160/169]) accepted new patients for FBSEs; of those, 63.1% (101/160) accepted only private insurance, 16.9% (27/160) accepted both private insurance and Medi-Cal, and 16.2% (26/160) did not accept any insurance. Racial predominance for each dermatology practice was analyzed by zip code (Table). Dermatologists included in our study were significantly more concentrated in predominantly non- Hispanic White areas of LA County vs predominantly Hispanic areas (P<.0001). Notably, the average income in predominantly non-Hispanic White zip codes ($114,757.74) was significantly higher than in predominantly Hispanic areas ($58,278.54)(P=.001)(Table).4
In LA County, 40.1% (108/269) of zip codes have no racial majority, 28.2% (76/269) are predominantly Hispanic, 27.5% (74/269) are predominantly non-Hispanic White, 2.2% (6/269) are predominantly Black, and 1.9% (5/269) are predominantly Asian.4 There are no dermatologists in predominantly Black zip codes, 2 in predominantly Asian zip codes, 14 in predominantly Hispanic zip codes, 38 in zip codes with no racial majority, and 106 in predominantly non-Hispanic White zip codes. There are significantly more dermatologists in predominantly non-Hispanic White zip codes compared to predominantly Hispanic zip codes (P<.0001). In LA County, the average income in predominantly Asian, non-Hispanic White, and Hispanic zip codes was $93,594, $114,757.84, and $58,278.54, respectively, in 2021.4 The average income in predominantly non-Hispanic White zip codes was significantly higher than in predominantly Hispanic zip codes (P=.001). There were no income data available for predominantly Black zip codes or zip codes with no racial majority.
The results from our study revealed potential barriers to FBSEs for racial and ethnic minorities in LA County, which supports previous research on the impact of SES, race, and insurance on access to dermatologic care.2,3 Predominantly Hispanic zip codes have significantly lower income (P<.0001) and fewer dermatologists (P=.001) compared to zip codes that are predominantly non-Hispanic White, reflecting how lower SES correlates with worse health outcomes and higher melanoma mortality. Conversely, predominantly non-Hispanic White areas with higher income have better access to dermatologists, which may contribute to the improved melanoma survival rates among White patients. Additionally, most dermatologists accept only private insurance, further highlighting the disparity in FBSE access for non-White patients across LA County. While our study focused on FBSE access, our findings may point to a wider barrier to dermatologic care, especially in zip codes with fewer dermatologists. Further studies are needed to determine whether these areas also face barriers to accessing primary care.
Our study was limited by the exclusion of nonphysician providers (eg, nurse practitioners, physician assistants), a small sample size, and lack of available economic data for predominantly Black zip codes.4 Additionally, the exclusion of practices with exclusive insurance plans (eg, Kaiser Permanente) limited the generalizability of our findings, as our results did not account for the populations served by these practices. Furthermore, our analysis did not account for variations in practice size or the proportion of care provided to patients with different insurance types, which could impact overall accessibility. Additional studies are needed to explore the impact of these factors on access to general dermatologic care and not just FBSEs.
Racial/ethnic minorities and lower SES populations face major barriers to FBSE access in LA County, such as difficulty finding a dermatologist in their area or one who accepts Medi-Cal. Addressing these disparities is crucial for improving skin cancer outcomes. Further research is needed to develop strategies to eliminate these barriers to dermatologic care, such as increasing access to teledermatology, offering mobile dermatology clinics, and improving insurance coverage.
- Chiaravalloti AJ, Laduca JR. Melanoma screening by means of complete skin exams for all patients in a dermatology practice reduces the thickness of primary melanomas at diagnosis. J Clin Aesthet Dermatol. 2014;7:18-22.
- Qian Y, Johannet P, Sawyers A, et al. The ongoing racial disparities in melanoma: an analysis of the Surveillance, Epidemiology, and End Results database (1975-2016). J Am Acad Dermatol. 2021;84:1585-1593.
- Baranowski MLH, Yeung H, Chen SC, et al. Factors associated with time to surgery in melanoma: an analysis of the National Cancer Database. J Am Acad Dermatol. 2019;81:908-916.
- United States Census Bureau. Explore census data. Accessed March 17, 2025. https://data.census.gov/all?q=los+angeles+county
- Berkowitz SA, Traore CY, Singer DE, et al. Evaluating area-based socioeconomic status indicators for monitoring disparities within health care systems: results from a primary care network. Health Serv Res. 2015;50:398-417.
- Jacobs B, Ir P, Bigdeli M, et al. Addressing access barriers to health services: an analytical framework for selecting appropriate interventions in lowincome Asian countries. Health Policy Plan. 2012;27:288-300.
- Chiaravalloti AJ, Laduca JR. Melanoma screening by means of complete skin exams for all patients in a dermatology practice reduces the thickness of primary melanomas at diagnosis. J Clin Aesthet Dermatol. 2014;7:18-22.
- Qian Y, Johannet P, Sawyers A, et al. The ongoing racial disparities in melanoma: an analysis of the Surveillance, Epidemiology, and End Results database (1975-2016). J Am Acad Dermatol. 2021;84:1585-1593.
- Baranowski MLH, Yeung H, Chen SC, et al. Factors associated with time to surgery in melanoma: an analysis of the National Cancer Database. J Am Acad Dermatol. 2019;81:908-916.
- United States Census Bureau. Explore census data. Accessed March 17, 2025. https://data.census.gov/all?q=los+angeles+county
- Berkowitz SA, Traore CY, Singer DE, et al. Evaluating area-based socioeconomic status indicators for monitoring disparities within health care systems: results from a primary care network. Health Serv Res. 2015;50:398-417.
- Jacobs B, Ir P, Bigdeli M, et al. Addressing access barriers to health services: an analytical framework for selecting appropriate interventions in lowincome Asian countries. Health Policy Plan. 2012;27:288-300.
Evaluating Access to Full-Body Skin Examinations in Los Angeles County, California
Evaluating Access to Full-Body Skin Examinations in Los Angeles County, California
PRACTICE POINTS
- Socioeconomic and racial disparities impact access to full-body skin examinations (FBSEs) in Los Angeles County.
- Most dermatologists included in this study were accepting new patients for a FBSE.
- There are significantly more dermatologists in predominantly non-Hispanic White zip codes than in predominantly Hispanic zip codes in Los Angeles County.
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
To the Editor:
Basal cell carcinoma (BCC), which is the most common type of skin cancer, typically arises on sun-damaged skin as a result of long-term exposure to UV radiation. Another known risk factor for BCC is exposure to ionizing radiation, though this is less commonly encountered.1 We present a unique case of a BCC arising at the site of an involuted infantile hemangioma that had been treated with implanted and retained gold radon seeds more than 7 decades prior. This case highlights the importance of obtaining a detailed history of radiation exposures to better counsel patients about skin cancer risk and manage disease in complex skin locations.
A 75-year-old woman presented to an outside dermatologist for evaluation of a pink papule on the right upper cutaneous lip that had enlarged over several months (Figure 1). The patient’s medical history was remarkable for an infantile hemangioma present since shortly after birth in the same location that had been treated with 10 implanted gold radon seeds when she was 6 years old. Over her lifetime, several seeds had self-extruded from the area, but some remained within the subcutaneous tissue as confirmed by dental radiographs. A shave biopsy of the papule demonstrated a superficial BCC, and the patient was referred to our institution for Mohs micrographic surgery.
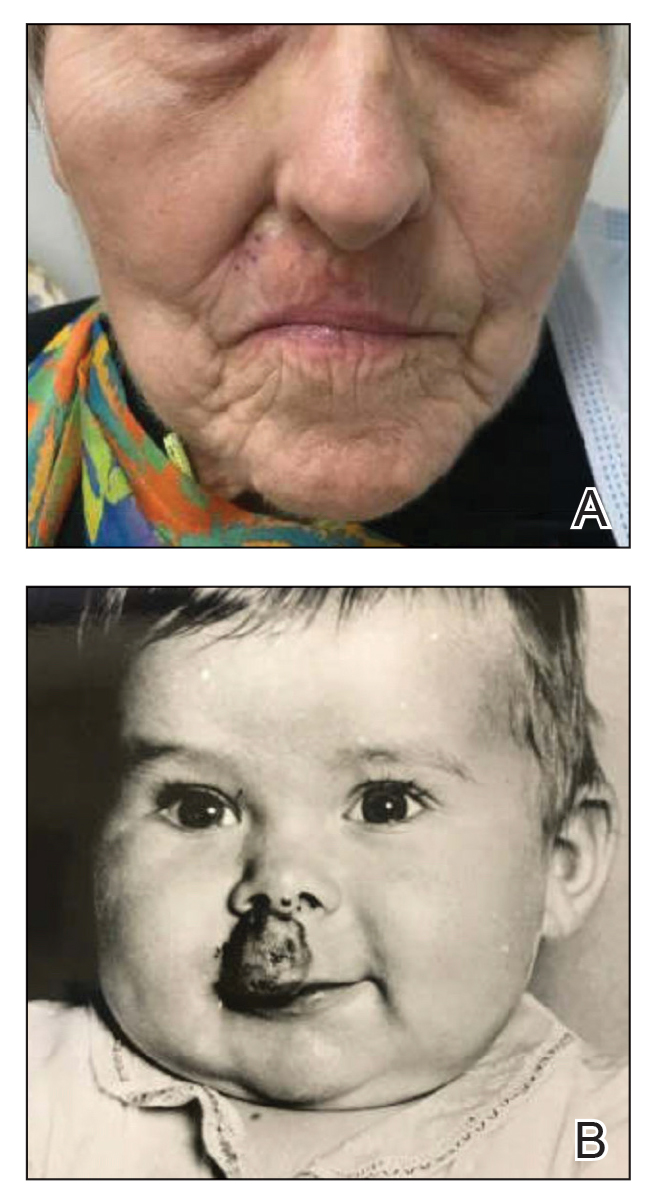
Intraoperative frozen sections revealed both superficial and nodular BCC, and the tumor was cleared in 3 stages. During surgery, a gold radon seed was visualized at the base of the excised BCC and was removed from the subcutaneous tissue (Figure 2). The primary defect on the upper lip was closed with a rotation flap. The patient returned for follow-up 2 months later and showed good healing and cosmetic outcome.
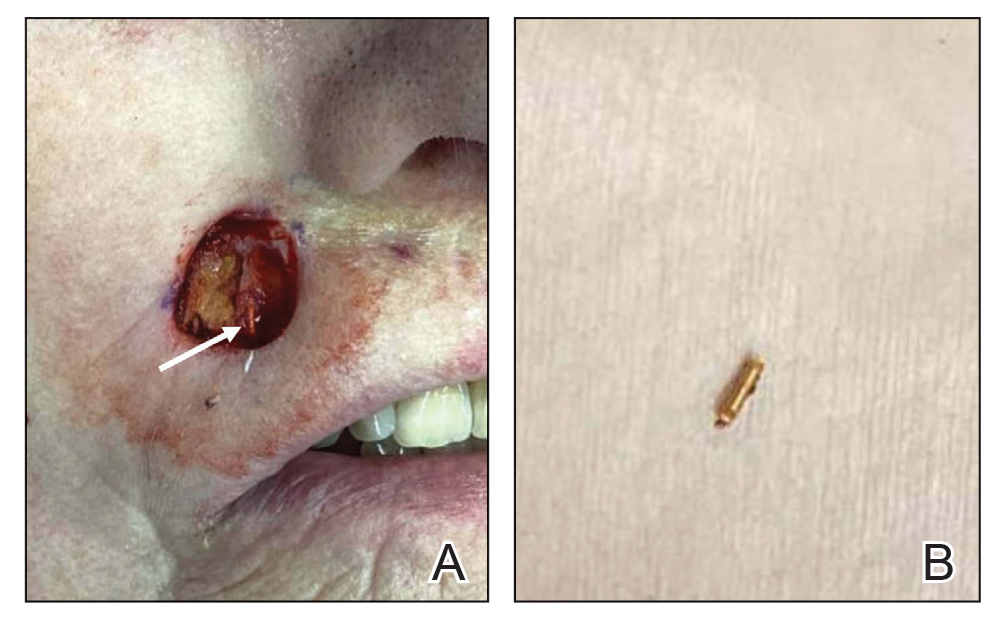
Although not commonly encountered, ionizing radiation is a known risk factor for BCC.1 Basal cell carcinoma arising from implanted gold radon seeds represents a minority of reported cases.2,3 Radium was first used to treat skin disease in the early 1900s.1 The radioactive decay of radium produced tissue destruction via alpha, beta, and gamma particles, which slowly released over weeks when radium was packaged into a capsule.4 Following implantation of the capsule, DNA damage occurred due to double-stranded breaks, chromosomal aberrations, and generation of reactive oxygen species. The downstream effect of these cellular insults resulted in cell-cycle shortening, apoptosis, and carcinogenesis.5
Gold radon seeds were used to treat infantile hemangiomas in the United States and Europe from the early 1940s to the 1960s; their use declined dramatically in the 1950s due to adverse effects and discovery of the potential for future malignancies as well as the development of safer and more effective treatments.1,3 Our patient received a substantial dose of ionizing radiation from the implantation of gold radon seeds at the site of the infantile hemangioma, which dramatically increased her risk for BCC in this location.
Infantile hemangiomas are the most common vascular tumors in children. Most infantile hemangiomas regress spontaneously and are stably involuted by about 5 or 6 years of age.6 Treatment is indicated for rapidly growing hemangiomas that are at risk for ulceration or are located by critical structures (eg, the eyes or airway). Hemangiomas located on or near the lips should be treated to avoid disfigurement and loss of function as a consequence of rapid growth and involution.7 The treatment of choice for large or high-risk infantile hemangiomas over the past 10 to 15 years has been beta blockers.6-8 Propranolol hydrochloride, a systemic beta blocker, was approved by the US Food and Drug Administration in 2014 for the treatment of infantile hemangiomas and has demonstrated safety and effectiveness in promoting involution in these lesions.8 Unlike radiation therapy from implanted gold radon seeds, propranolol does not increase the risk for BCC. Although other risk factors such as skin type and cumulative UV exposure contribute to the development of BCC, the exact location of the BCC overlying the residual gold radon seeds was highly suggestive of ionizing radiation playing a major role in the carcinogenesis of the tumor in our patient.
Our case highlights the importance of screening elderly patients for exposures that may increase the risk for skin carcinogenesis. Dermatologists are accustomed to asking about history of UV exposure, sunburns, and use of sun-protective measures; however, direct questioning about less common sources of radiation exposure also may help stratify a patient’s risk for developing BCC. Although the US Preventive Services Task Force 2023 guidelines determined there is insufficient evidence to recommend visual skin cancer screening examinations in asymptomatic adults,9 we advocate for verbal screening of radiation exposure in both primary care and dermatology office settings. At a time when access to care, particularly dermatology services, is challenging, determining the appropriate interval for follow-up based on the patient’s skin cancer risk is imperative.
- Fürst CJ, Lundell M, Holm LE. Radiation therapy of hemangiomas, 1909- 1959. a cohort based on 50 years of clinical practice at Radiumhemmet, Stockholm. Acta Oncol. 1987;26:33-36. doi:10.3109/02841868709092974
- Bräuner EV, Loft S, Sørensen M, et al. Residential radon exposure and skin cancer incidence in a prospective Danish cohort. PLoS ONE. 2015;10:E0135642. doi:10.1371/journal.pone.0135642
- Weiss E, Sukal SA, Zimbler MS, et al. Basal cell carcinoma arising 57 years after interstitial radiotherapy of a nasal hemangioma. Dermatol Surg. 2008;34:1137-1140. doi:10.1111/j.1524-4725.2008.34229.x
- Lavery MJ, Lorenzelli D, Crema J. A radon seed identified during skin surgery: an unusual finding. Clin Exp Dermatol. 2021;46:604-606. doi:10.1111/ced.14454
- Robertson A, Allen J, Laney R, et al. The cellular and molecular carcinogenic effects of radon exposure: a review. Int J Mol Sci. 2013;14:14024-14063. doi:10.3390/ijms140714024
- Rodríguez Bandera AI, Sebaratnam DF, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392. doi:10.1016 /j.jaad.2021.08.019
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475. doi:10.1542/peds.2018-3475
- Sebaratnam DF, Rodríguez Bandera AL, Wong LF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85: 1395-1404. doi:10.1016/j.jaad.2021.08.020
- US Preventive Services Task Force, Mangione CM, Barry MJ, Nicholson WK, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2023;329:1290-1295. doi:10.1001/jama.2023.4342
To the Editor:
Basal cell carcinoma (BCC), which is the most common type of skin cancer, typically arises on sun-damaged skin as a result of long-term exposure to UV radiation. Another known risk factor for BCC is exposure to ionizing radiation, though this is less commonly encountered.1 We present a unique case of a BCC arising at the site of an involuted infantile hemangioma that had been treated with implanted and retained gold radon seeds more than 7 decades prior. This case highlights the importance of obtaining a detailed history of radiation exposures to better counsel patients about skin cancer risk and manage disease in complex skin locations.
A 75-year-old woman presented to an outside dermatologist for evaluation of a pink papule on the right upper cutaneous lip that had enlarged over several months (Figure 1). The patient’s medical history was remarkable for an infantile hemangioma present since shortly after birth in the same location that had been treated with 10 implanted gold radon seeds when she was 6 years old. Over her lifetime, several seeds had self-extruded from the area, but some remained within the subcutaneous tissue as confirmed by dental radiographs. A shave biopsy of the papule demonstrated a superficial BCC, and the patient was referred to our institution for Mohs micrographic surgery.
Intraoperative frozen sections revealed both superficial and nodular BCC, and the tumor was cleared in 3 stages. During surgery, a gold radon seed was visualized at the base of the excised BCC and was removed from the subcutaneous tissue (Figure 2). The primary defect on the upper lip was closed with a rotation flap. The patient returned for follow-up 2 months later and showed good healing and cosmetic outcome.
Although not commonly encountered, ionizing radiation is a known risk factor for BCC.1 Basal cell carcinoma arising from implanted gold radon seeds represents a minority of reported cases.2,3 Radium was first used to treat skin disease in the early 1900s.1 The radioactive decay of radium produced tissue destruction via alpha, beta, and gamma particles, which slowly released over weeks when radium was packaged into a capsule.4 Following implantation of the capsule, DNA damage occurred due to double-stranded breaks, chromosomal aberrations, and generation of reactive oxygen species. The downstream effect of these cellular insults resulted in cell-cycle shortening, apoptosis, and carcinogenesis.5
Gold radon seeds were used to treat infantile hemangiomas in the United States and Europe from the early 1940s to the 1960s; their use declined dramatically in the 1950s due to adverse effects and discovery of the potential for future malignancies as well as the development of safer and more effective treatments.1,3 Our patient received a substantial dose of ionizing radiation from the implantation of gold radon seeds at the site of the infantile hemangioma, which dramatically increased her risk for BCC in this location.
Infantile hemangiomas are the most common vascular tumors in children. Most infantile hemangiomas regress spontaneously and are stably involuted by about 5 or 6 years of age.6 Treatment is indicated for rapidly growing hemangiomas that are at risk for ulceration or are located by critical structures (eg, the eyes or airway). Hemangiomas located on or near the lips should be treated to avoid disfigurement and loss of function as a consequence of rapid growth and involution.7 The treatment of choice for large or high-risk infantile hemangiomas over the past 10 to 15 years has been beta blockers.6-8 Propranolol hydrochloride, a systemic beta blocker, was approved by the US Food and Drug Administration in 2014 for the treatment of infantile hemangiomas and has demonstrated safety and effectiveness in promoting involution in these lesions.8 Unlike radiation therapy from implanted gold radon seeds, propranolol does not increase the risk for BCC. Although other risk factors such as skin type and cumulative UV exposure contribute to the development of BCC, the exact location of the BCC overlying the residual gold radon seeds was highly suggestive of ionizing radiation playing a major role in the carcinogenesis of the tumor in our patient.
Our case highlights the importance of screening elderly patients for exposures that may increase the risk for skin carcinogenesis. Dermatologists are accustomed to asking about history of UV exposure, sunburns, and use of sun-protective measures; however, direct questioning about less common sources of radiation exposure also may help stratify a patient’s risk for developing BCC. Although the US Preventive Services Task Force 2023 guidelines determined there is insufficient evidence to recommend visual skin cancer screening examinations in asymptomatic adults,9 we advocate for verbal screening of radiation exposure in both primary care and dermatology office settings. At a time when access to care, particularly dermatology services, is challenging, determining the appropriate interval for follow-up based on the patient’s skin cancer risk is imperative.
To the Editor:
Basal cell carcinoma (BCC), which is the most common type of skin cancer, typically arises on sun-damaged skin as a result of long-term exposure to UV radiation. Another known risk factor for BCC is exposure to ionizing radiation, though this is less commonly encountered.1 We present a unique case of a BCC arising at the site of an involuted infantile hemangioma that had been treated with implanted and retained gold radon seeds more than 7 decades prior. This case highlights the importance of obtaining a detailed history of radiation exposures to better counsel patients about skin cancer risk and manage disease in complex skin locations.
A 75-year-old woman presented to an outside dermatologist for evaluation of a pink papule on the right upper cutaneous lip that had enlarged over several months (Figure 1). The patient’s medical history was remarkable for an infantile hemangioma present since shortly after birth in the same location that had been treated with 10 implanted gold radon seeds when she was 6 years old. Over her lifetime, several seeds had self-extruded from the area, but some remained within the subcutaneous tissue as confirmed by dental radiographs. A shave biopsy of the papule demonstrated a superficial BCC, and the patient was referred to our institution for Mohs micrographic surgery.
Intraoperative frozen sections revealed both superficial and nodular BCC, and the tumor was cleared in 3 stages. During surgery, a gold radon seed was visualized at the base of the excised BCC and was removed from the subcutaneous tissue (Figure 2). The primary defect on the upper lip was closed with a rotation flap. The patient returned for follow-up 2 months later and showed good healing and cosmetic outcome.
Although not commonly encountered, ionizing radiation is a known risk factor for BCC.1 Basal cell carcinoma arising from implanted gold radon seeds represents a minority of reported cases.2,3 Radium was first used to treat skin disease in the early 1900s.1 The radioactive decay of radium produced tissue destruction via alpha, beta, and gamma particles, which slowly released over weeks when radium was packaged into a capsule.4 Following implantation of the capsule, DNA damage occurred due to double-stranded breaks, chromosomal aberrations, and generation of reactive oxygen species. The downstream effect of these cellular insults resulted in cell-cycle shortening, apoptosis, and carcinogenesis.5
Gold radon seeds were used to treat infantile hemangiomas in the United States and Europe from the early 1940s to the 1960s; their use declined dramatically in the 1950s due to adverse effects and discovery of the potential for future malignancies as well as the development of safer and more effective treatments.1,3 Our patient received a substantial dose of ionizing radiation from the implantation of gold radon seeds at the site of the infantile hemangioma, which dramatically increased her risk for BCC in this location.
Infantile hemangiomas are the most common vascular tumors in children. Most infantile hemangiomas regress spontaneously and are stably involuted by about 5 or 6 years of age.6 Treatment is indicated for rapidly growing hemangiomas that are at risk for ulceration or are located by critical structures (eg, the eyes or airway). Hemangiomas located on or near the lips should be treated to avoid disfigurement and loss of function as a consequence of rapid growth and involution.7 The treatment of choice for large or high-risk infantile hemangiomas over the past 10 to 15 years has been beta blockers.6-8 Propranolol hydrochloride, a systemic beta blocker, was approved by the US Food and Drug Administration in 2014 for the treatment of infantile hemangiomas and has demonstrated safety and effectiveness in promoting involution in these lesions.8 Unlike radiation therapy from implanted gold radon seeds, propranolol does not increase the risk for BCC. Although other risk factors such as skin type and cumulative UV exposure contribute to the development of BCC, the exact location of the BCC overlying the residual gold radon seeds was highly suggestive of ionizing radiation playing a major role in the carcinogenesis of the tumor in our patient.
Our case highlights the importance of screening elderly patients for exposures that may increase the risk for skin carcinogenesis. Dermatologists are accustomed to asking about history of UV exposure, sunburns, and use of sun-protective measures; however, direct questioning about less common sources of radiation exposure also may help stratify a patient’s risk for developing BCC. Although the US Preventive Services Task Force 2023 guidelines determined there is insufficient evidence to recommend visual skin cancer screening examinations in asymptomatic adults,9 we advocate for verbal screening of radiation exposure in both primary care and dermatology office settings. At a time when access to care, particularly dermatology services, is challenging, determining the appropriate interval for follow-up based on the patient’s skin cancer risk is imperative.
- Fürst CJ, Lundell M, Holm LE. Radiation therapy of hemangiomas, 1909- 1959. a cohort based on 50 years of clinical practice at Radiumhemmet, Stockholm. Acta Oncol. 1987;26:33-36. doi:10.3109/02841868709092974
- Bräuner EV, Loft S, Sørensen M, et al. Residential radon exposure and skin cancer incidence in a prospective Danish cohort. PLoS ONE. 2015;10:E0135642. doi:10.1371/journal.pone.0135642
- Weiss E, Sukal SA, Zimbler MS, et al. Basal cell carcinoma arising 57 years after interstitial radiotherapy of a nasal hemangioma. Dermatol Surg. 2008;34:1137-1140. doi:10.1111/j.1524-4725.2008.34229.x
- Lavery MJ, Lorenzelli D, Crema J. A radon seed identified during skin surgery: an unusual finding. Clin Exp Dermatol. 2021;46:604-606. doi:10.1111/ced.14454
- Robertson A, Allen J, Laney R, et al. The cellular and molecular carcinogenic effects of radon exposure: a review. Int J Mol Sci. 2013;14:14024-14063. doi:10.3390/ijms140714024
- Rodríguez Bandera AI, Sebaratnam DF, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392. doi:10.1016 /j.jaad.2021.08.019
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475. doi:10.1542/peds.2018-3475
- Sebaratnam DF, Rodríguez Bandera AL, Wong LF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85: 1395-1404. doi:10.1016/j.jaad.2021.08.020
- US Preventive Services Task Force, Mangione CM, Barry MJ, Nicholson WK, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2023;329:1290-1295. doi:10.1001/jama.2023.4342
- Fürst CJ, Lundell M, Holm LE. Radiation therapy of hemangiomas, 1909- 1959. a cohort based on 50 years of clinical practice at Radiumhemmet, Stockholm. Acta Oncol. 1987;26:33-36. doi:10.3109/02841868709092974
- Bräuner EV, Loft S, Sørensen M, et al. Residential radon exposure and skin cancer incidence in a prospective Danish cohort. PLoS ONE. 2015;10:E0135642. doi:10.1371/journal.pone.0135642
- Weiss E, Sukal SA, Zimbler MS, et al. Basal cell carcinoma arising 57 years after interstitial radiotherapy of a nasal hemangioma. Dermatol Surg. 2008;34:1137-1140. doi:10.1111/j.1524-4725.2008.34229.x
- Lavery MJ, Lorenzelli D, Crema J. A radon seed identified during skin surgery: an unusual finding. Clin Exp Dermatol. 2021;46:604-606. doi:10.1111/ced.14454
- Robertson A, Allen J, Laney R, et al. The cellular and molecular carcinogenic effects of radon exposure: a review. Int J Mol Sci. 2013;14:14024-14063. doi:10.3390/ijms140714024
- Rodríguez Bandera AI, Sebaratnam DF, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392. doi:10.1016 /j.jaad.2021.08.019
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475. doi:10.1542/peds.2018-3475
- Sebaratnam DF, Rodríguez Bandera AL, Wong LF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85: 1395-1404. doi:10.1016/j.jaad.2021.08.020
- US Preventive Services Task Force, Mangione CM, Barry MJ, Nicholson WK, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2023;329:1290-1295. doi:10.1001/jama.2023.4342
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
Basal Cell Carcinoma Arising From an Infantile Hemangioma Treated With Gold Radon Seeds
PRACTICE POINTS
- Historical use of ionizing radiation to treat skin disease is a risk factor for basal cell carcinoma (BCC).
- Mohs micrographic surgery is the treatment of choice for BCC in high-risk areas such as the nose, eyelids, and lips, where tissue conservation and complete margin control are essential.
- Elderly patients should be screened for less common sources of radiation exposure for better risk stratification and to determine appropriate intervals for follow-up with a dermatologist.

Multiple Firm Papules on the Wrists and Forearms
Multiple Firm Papules on the Wrists and Forearms
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
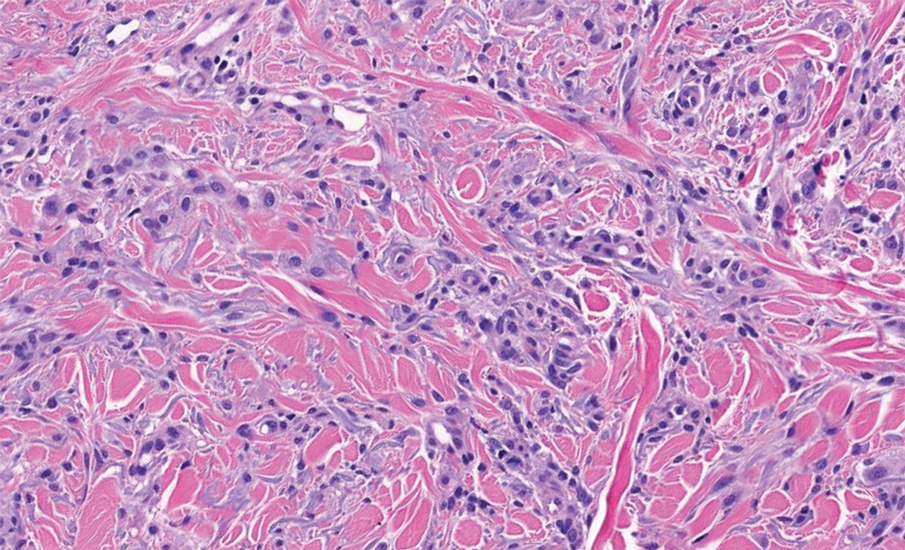
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
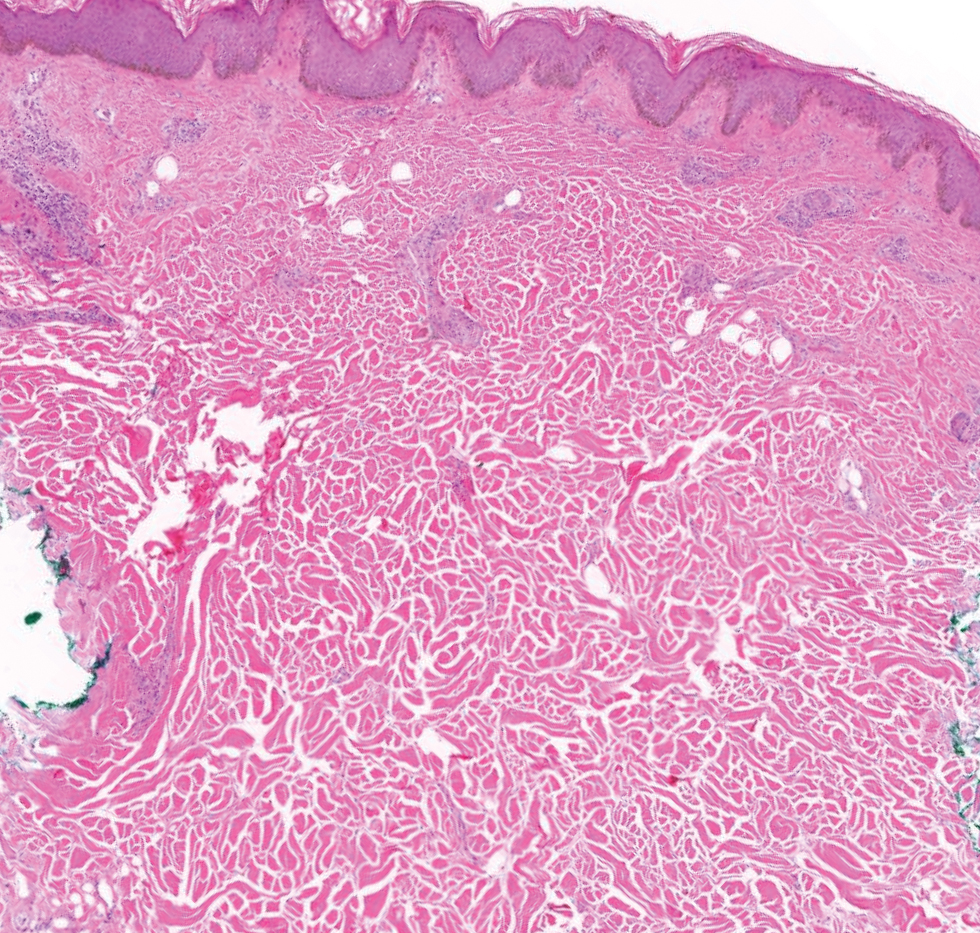
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
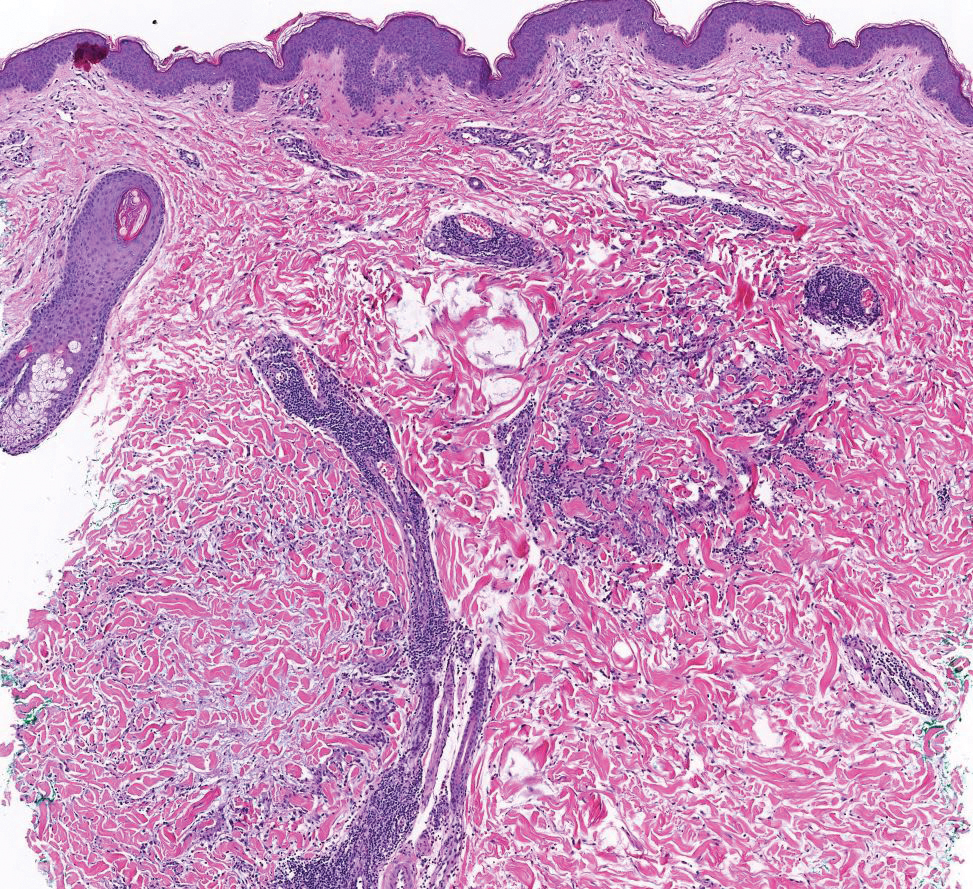
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
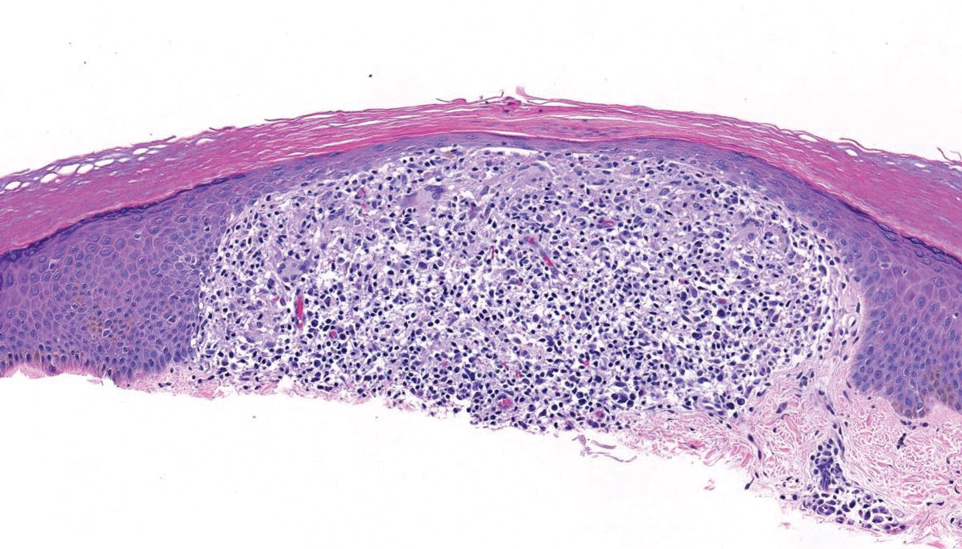
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
THE DIAGNOSIS: Acral Persistent Papular Mucinosis
Histopathologic analysis revealed conspicuous interstitial mucin deposition throughout the upper to mid reticular dermis in the absence of a cellular infiltrate or fibroplasia. Colloidal iron staining confirmed the presence of mucin. In correlation with the clinical presentation, a diagnosis of acral persistent papular mucinosis (APPM) was made. The patient was counseled on the benign disease course and lack of associated comorbidities, and additional treatment was not pursued.
Acral persistent papular mucinosis is a rare distinct subtype of cutaneous mucinosis that initially was described by Rongioletti et al1 in 1986. As a localized form of lichen myxedematosus, APPM is characterized by mucin deposition in the dermis with no systemic involvement. The precise pathogenesis remains unclear, although some investigators have suggested that cytokine-mediated stimulation of glycosaminoglycan production may contribute to increased mucin accumulation in the dermis.2 Acral persistent papular mucinosis predominantly affects middle-aged women with a 5:1 female-to-male predominance.3 Clinically, patients present with discrete, nonfollicular, waxy papules that typically measure 2 to 5 mm and are distributed symmetrically on the extensor surfaces of the wrists and forearms. While the lesions generally are asymptomatic, some patients may report mild pruritus. The condition is chronic, with lesions seldom resolving and often increasing in number over time.3
Histologically, APPM is characterized by focal deposits of mucin in the upper reticular dermis with no evidence of increased fibroblast proliferation or fibrosis.4 This feature is pivotal in differentiating APPM from other subtypes of localized lichen myxedematosus and similar dermatoses. Diagnosis of APPM requires exclusion of systemic involvement, including thyroid abnormalities and monoclonal gammopathy, aligning with its classification as a purely cutaneous condition.5 Management of APPM is unclear due to its rarity. Reassurance for patients of its benign nature as well as clinical observation are recommended, though some reports cite benefits of treatment with topical corticosteroids or calcineurin inhibitors.6,7 The long-term prognosis for patients with APPM is favorable, although the persistence of and potential increase in lesions over time can be a cosmetic concern.
The differential diagnoses for APPM include scleromyxedema, scleredema, and other cutaneous eruptions that manifest as smooth flesh-colored papules, such as granuloma annulare and lichen nitidus.3 Scleromyxedema is a systemic cutaneous mucinosis that is part of the same disease spectrum as lichen myxedematosus. The papular eruption of scleromyxedema is much more widespread, and coalescing of the lesions may lead to characteristic skin thickening, creating leonine facies and deep furrowing over the trunk.8 Extracutaneous manifestations are frequent in scleromyxedema, and up to 90% of patients exhibit evidence of an underlying plasma cell dyscrasia.2 Histopathologically, scleromyxedema shows extensive fibroblast proliferation and fibrosis, in contrast to the findings of APPM (Figure 1).
The histopathology of APPM is most similar to scleredema, a rare fibromucinous disorder of the skin associated with diabetes, infection (especially poststreptococcal), or monoclonal gammopathy.9 Biopsy evaluation of scleredema reveals a normal epidermis with mucin deposition between collagen bundles predominantly in the deep reticular dermis as well as absent fibroblast proliferation (Figure 2). Unlike APPM, scleredema manifests with diffuse woody induration with erythema and hyperpigmentation on the posterior neck and upper back.9 On physical examination, the distinct clinical features of scleredema distinguish this condition from APPM and scleromyxedema.
Papular granuloma annulare also was considered in our patient due to the presence of small flesh-colored papules. Histologically, granuloma annulare is characterized by palisading granulomas and mucin deposition in the dermis.10 However, the pattern of mucin deposition differs from that seen in APPM. In granuloma annulare, mucin is observed around foci of degenerated collagen (Figure 3), which was not observed in our patient.10 Additionally, the absence of an inflammatory infiltrate in our patient further ruled out this diagnosis.
Lichen nitidus also could be considered in the differential diagnosis for ACCM. It typically manifests with minute, clustered, monomorphous papules with a predilection for the chest, abdomen, flexural forearms, and genitalia. The histology of lichen nitidus is distinct, showing a well-circumscribed lymphohistiocytic infiltrate in the papillary dermis bordered by epidermal ridges, resembling a ball and clutch appearance (Figure 4).11
Although the clinical differential diagnosis in our patient was broad, histopathologic evaluation played a crucial role in confirming the diagnosis of APPM. This benign condition could be overlooked by patients and physicians; thorough clinical evaluation is necessary to rule out systemic mucinoses, which are associated with higher risks of morbidity and mortality.
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
- Rongioletti F, Rebora A. Acral persistent papular mucinosis: a new entity. Arch Dermatol. 1986;122:1237-1239. doi:10.1001 /archderm.1986.01660230027002
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosus (LM)(discrete papular type). Dermatol Online J. 2017;23:13030/qt3xp109qd.
- Rongioletti F, Ferreli C, Atzori L. Acral persistent papular mucinosis. Clin Dermatol. 2021;39:211-214. doi:10.1016/j.clindermatol.2020.10.001
- Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-267. doi:10.1097/00000372- 200106000-00022
- Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-104. doi:10.1016/j.sder.2006.04.001
- Jun JY, Oh SH, Shim JH, et al. Acral persistent papular mucinosis with partial response to tacrolimus ointment. Ann Dermatol. 2016;28:517-519. doi:10.5021/ad.2016.28.4.517
- Rongioletti F, Zaccaria E, Cozzani E, et al. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-532. doi:10.1016/j.jaad.2006.10.021
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72. doi:10.1016 /j.jaad.2013.01.007
- Rongioletti F, Kaiser F, Cinotti E, et al. Scleredema. a multicentre study of characteristics, comorbidities, course and therapy in 44 patients. J Eur Acad Dermatol Venereol. 2015;29:2399-2404. doi:10.1111/jdv.13272
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465. doi:10.1016/j.jaad.2015.03.054
- Al-Mutairi N, Hassanein A, Nour-Eldin O, et al. Generalized lichen nitidus. Pediatr Dermatol. 2005;22:158-160. doi:10.1111 /j.1525-1470.2005.22215.x
Multiple Firm Papules on the Wrists and Forearms
Multiple Firm Papules on the Wrists and Forearms
A 69-year-old woman presented to the dermatology department with persistent asymptomatic skin lesions on the wrists and forearms of several months’ duration. The lesions had slowly grown in number over the past few months with no identifiable triggers. The patient reported no known history of injury or trauma to the affected sites and was not taking any prescription medications other than daily vitamins. She denied any family history of similar lesions and was otherwise healthy. Physical examination revealed multiple waxy, firm, hypopigmented, 3- to 5-mm papules located exclusively on the dorsal wrists and forearms. No extracutaneous involvement was observed. A 4-mm punch biopsy from the forearm was obtained.
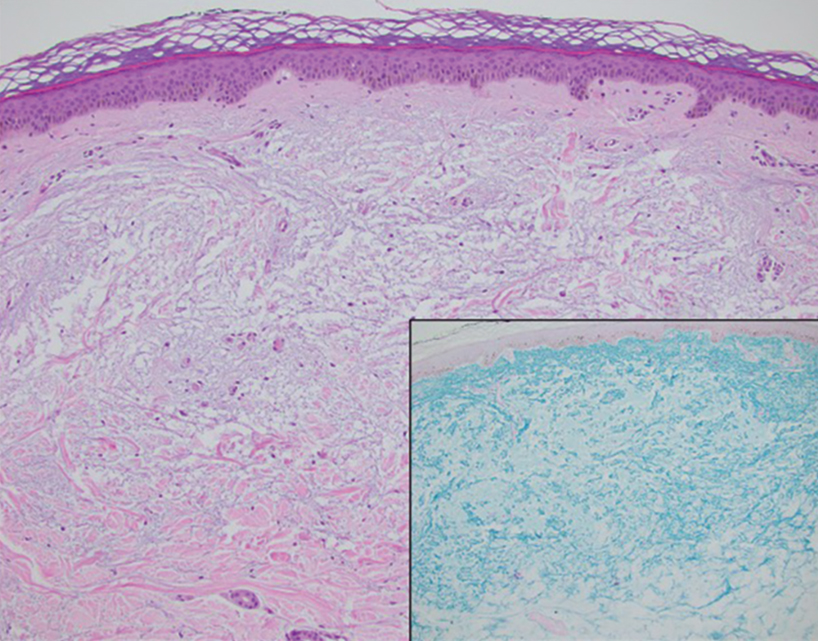
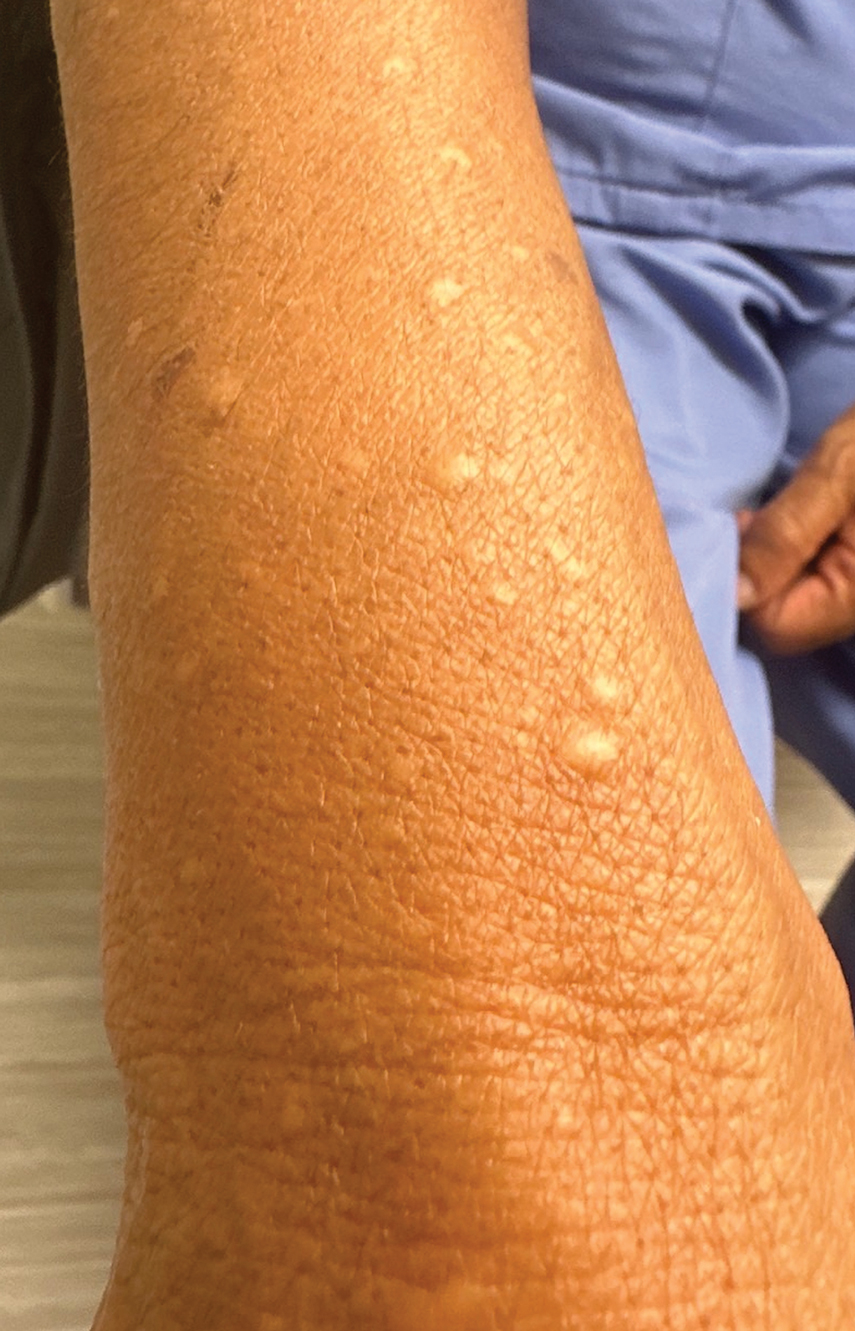

Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
Skin cancer is a major health concern for military service members, who experience notably higher incidence rates than the general population.1 Active-duty military personnel are particularly vulnerable to prolonged sun exposure due to deployments, specialized training, and everyday outdoor duties.1 Despite skin cancer being the most commonly diagnosed malignancy in active-duty service members,2 tracking and documenting the quantity and diversity of these risk factors remain limited. This knowledge gap comes at high cost, simultaneously impairing military medicine preventive measures while burdening the military health care system with substantial expenditures.3 These findings underscore the critical need for targeted surveillance, early-detection programs, and policy-driven interventions to mitigate these medical and economic concerns.
Skin cancer has been recognized as a major health risk to the military population for decades, yet incidence and prevalence remain high. This phenomenon is closely linked to the inherent responsibilities and expectations of active-duty military members, including outdoor physical training, field exercises, standing in formation, and outdoor working environments—all of which can occur during peak sunlight hours. These risks are further elevated at duty stations in geographic regions with high levels of UV exposure, such as those in tropical and arid regions of the world. Certain military occupational specialties and missions may further introduce unique risk factors; for instance, pilots with frequent high-altitude missions experience heightened UV exposure and melanoma risk.4 Secondary to compounding determinants, the aviation, diving, and nuclear subgroups of the military community are particularly vulnerable to skin cancer.5
Despite well-documented risks, considerable gaps remain in quantifying and analyzing variations in UV exposure across military occupations, duty locations, and operational roles. Factors such as the existence of over 150 distinct military occupational specialties, frequent geographic relocations, and routine work in austere environments contribute to a wide range of UV exposure profiles that remain insufficiently characterized. This lack of comprehensive exposure data hinders the development of large-scale, targeted skin cancer prevention strategies. Initial approaches to addressing these challenges include enhanced surveillance, education, and policy initiatives. The Table presents practical recommendations for military leadership to consider in implementing preventive measures for skin cancer. Herein, we outline broader systemic strategies to bridge knowledge gaps and address underrecognized occupational risk factors for skin cancer in military service members; these elements include proposed modifications to the electronic Periodic Health Assessment (ePHA) and the development of standardized, military-specific screening and prevention guidelines to support early detection and resource optimization.
Skin Cancer Education for Service Members
Sunscreen and Signage—Diligent primary prevention offers a promising avenue for mitigating skin cancer incidence in military service members. Basic education and precautionary messaging on photoprotection can be widely implemented to simultaneously educate service members on the dangers of sun exposure while reinforcing healthy behaviors in real time. Simple low-cost initiatives such as strategically placed visual signage reminding service members to apply sunscreen in high UV environments can support consistent sun-safe practices. Educational efforts also should emphasize proper sunscreen use, including application on high-risk anatomic sites (eg, the face, neck, scalp, dorsal hands, and ears) and the essentiality of using sufficient quantities of broad-spectrum sunscreen for effective protection. Incorporating this guidance into training materials, briefings, and visual reminders allow seamless integration of photoprotection into service members’ daily routines without compromising operational efficiency.6 Younger service members, who may be less likely to prioritize preventive behaviors, may be particularly responsive to sun safety reminders in training areas, bases, and deployment zones.7 Health fairs and orientation briefs in high-UV regions also offer potential opportunities for targeted education.
Resources for Sun Protection in the Military
Sunscreen—Although sunscreen is critical in minimizing the risk for UV-induced skin cancer, its widespread use in the military is hindered by practical challenges related to accessibility and the need for consistent reapplication; for instance, providing free sunscreen dispensers at institutions for staff working under intense or prolonged UV exposure may improve sunscreen accessibility and use.8 Including sunscreen in standard-issue gear offers another logical way to embed its use into operational readiness as part of the routine protective measures.
Uniform Modifications—Adapting military uniforms and practices to improve sun protection plays a critical role in reducing skin cancer risk. Targeted protective gear for commonly sun-exposed areas can help mitigate UV exposure. One practical option is the use of wide-brimmed headgear (eg, boonie hats), which provide more face and neck coverage than standard-issue military caps, or covers. The wide-brimmed headgear currently is only selectively authorized during specific scenarios, such as field operations and training exercises, or at the discretion of unit-level leadership. Wide-brimmed headgear, already used by many service members, has been associated with up to a 17% reduction in UV exposure to inadequately protected areas, potentially lowering skin cancer risk.9,10 Similarly, a “sleeves-down” policy—requiring sleeves to remain unrolled and covering the forearms during outdoor activities—offers a simple way to minimize sun exposure without necessitating additional gear. Other specialized clothing items, including UV-blocking neck gaiters, photoprotective clothing, and lightweight gloves, also may be appropriate for high-risk groups and can be implemented in a relatively straightforward manner.
Shade Structures and UV Index Monitoring—Aside from uniform adaptation, physical barrier intervention can further complement skin cancer prevention efforts in the military. Shade structures offer a straightforward way to reduce UV exposure during prolonged outdoor activities. Incorporating daily UV index monitoring into operational guidance can help inform adjustments to training schedules and guide the implementation of additional sun protection measures, such as mandatory sunscreen application, use of wide-brimmed hats, or increased access to shaded rest areas during heavy sunlight hours. Currently, outdoor physical training is restricted during periods of high heat index, measured via Wet Bulb Globe Temperature, to reduce heat-related injuries. We argue that avoidance of nonoperational outdoor activity during peak UV index hours also should be incorporated into standardized policies. This intervention is of particular benefit to service members stationed in regions with a high UV index year-round, such as those stationed in the Middle East, Guam, Okinawa, and southern coastal United States bases.
Policy Changes to Support Photoprotective Measures
Annual Risk Factor Screening‐Screening—Effective secondary prevention efforts by military dermatologists remain an important measure in reducing the burden of skin cancer among military personnel; however, these efforts have become increasingly challenging due to 2 main factors—the diversity of military occupational specialties and their associated unique occupational risks as well as the limited availability of military dermatologists across all branches (approximately 100 active-duty dermatologists for nearly 3 million service members).11 Therefore, targeted interventions that enhance risk assessment, refined screening protocols, and leveraging of existing military health networks can improve early skin cancer detection while optimizing resource allocation.
The ePHA is an online screening tool used annually by all service members to evaluate their overall health. Presently, the ePHA lacks specific questions to assess sun exposure and skin cancer risks. Integrating annual skin cancer risk factor assessments into the ePHA would offer a practical and straightforward approach to identifying at-risk individuals, as suggested by Newnam et al12 in 2022. Skin cancer risk factor assessments allow for targeted data collection related to sun exposure history, family history, and personal risk factors, which can be used to determine individualized risk stratification to assess the need for early secondary prevention measures and specialist referral. These ePHA data can also support population-based analyses to inform preventive strategies and address knowledge gaps related to high-risk exposures, such as extended field exercises or assignments in high-UV regions, that may impede effective skin cancer prevention.
Development of Military-Specific Screening Guidelines—Given the limited number of military dermatologists, a standardized risk-assessment tool could enhance early detection of skin cancer and streamline the referral process. We propose a military-specific skin cancer screening algorithm or risk nomogram that could help to consolidate risk factors into a clear and actionable framework for more efficient triage and appropriate allocation of dermatologic resources and manpower. This nomogram could be developed by military dermatologists and then implemented on a command level, affording primary care providers a useful tool to expedite evaluation of individuals at higher risk for skin cancer while simultaneously promoting judicious use of limited dermatology resources.
Although the United States Preventive Services Task Force does not universally recommend routine skin cancer screenings for asymptomatic adults, military service members are exposed to higher occupational risks than the general population, as previously mentioned. Currently, there is no standardized screening guideline across all military services due to the unique nature and exposure risks for each branch of service and their varied occupations; however, we propose the development of basic standardized screening guidelines by adapting the framework of the United States Preventive Services Task Force and adjusting for military-specific UV exposure and occupational risks to improve early detection of skin cancer. These guidelines could be updated and tailored appropriately when additional population-based data are collected and analyzed through ePHA.
Critiques and Limitations of Implementation
Several challenges and limitations must be considered when attempting to integrate large-scale preventive measures for skin cancer within the US military. A primary concern is the extent to which military resources should be allocated to prevention when off-duty sun exposure remains largely beyond institutional control. Although military health initiatives can address workplace risk through education and policy, individual decisions during both work and leisure time remain a major variable that cannot be feasibly controlled. Cultural and operational barriers also pose challenges; for instance, the US Marine Corps maintains a strong cultural identity tied to uniform appearance, making it difficult to implement widespread changes to clothing-based sun-protection measures. Institutional changes, particularly those involving uniforms, likely will face substantial administrative resistance and potential operational limitations. When broad uniform modifications are unattainable, a more feasible approach may be to encourage unit-level leadership to authorize and promote the frequent use of nonuniform protective measures.
Furthermore, integrating additional skin cancer risk questions into the already extensive ePHA means extra time required to complete the assessment; this adds to service members’ administrative burden, potentially leading to reduced timely compliance, rushed responses, and survey fatigue, which threaten data quality. If new items are to be included, they should be carefully selected for efficiency and clinical relevance. Existing validated questionnaires such as those from the study by Lyford et al7 published in 2021 can serve as a foundation.
Another critical limitation is access to dermatologic care for active-duty service members. Raising awareness of skin cancer risk without ensuring adequate resources may create ethical concerns, particularly in high-risk environments such as the Middle East and Indo-Pacific. Additionally, because skin cancer often develops years or decades after exposure, securing early buy-in from service members and their leaders can be challenging. These concerns make it clear that, while skin cancer prevention is important, implementing widespread measures is not straightforward and requires a practical and balanced approach.
Final Thoughts
Implementing prevention strategies for skin cancer in the military requires balancing evidence-based recommendations with the practical realities of military culture, resource limitations, and operational demands. Challenges remain for dermatologists in providing targeted recommendations due to the multifaceted nature of military roles, including over 150 Navy Military Occupational Specialties, limited familiarity with the unique UV exposure risks associated with each occupation, and variability in local and regional policies on uniform wear, physical training requirements, and other operational practices. Although targeted prevention measures are difficult to establish in the setting of these knowledge gaps, leveraging unit-level leadership to align with existing screening guidelines and optimizing primary prevention measures can be meaningful steps toward reducing skin cancer risk for military service members while maintaining mission readiness.
- Riemenschneider K, Liu J, Powers JG. Skin cancer in the military: a systematic review of melanoma and nonmelanoma skin cancerincidence, prevention, and screening among active duty and veteran personnel. J Am Acad Dermatol. 2018;78:1185-1192. doi:10.1016/j.jaad.2017.11.062
- Lee T, Taubman SB, Williams VF. Incident diagnoses of non-melanoma skin cancer, active component, U.S. Armed Forces, 2005-2014. MSMR. 2016;23:2-6.
- Krivda KR, Watson NL, Lyford WH, et al. The burden of skin cancer in the military health system, 2017-2022. Cutis. 2024;113:200-215. doi:10.12788/cutis.1015
- Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2015;151:51-58. doi:10.1001/jamadermatol.2014.1077
- Brundage JF, Williams VF, Stahlman S, et al. Incidence rates of malignant melanoma in relation to years of military service, overall and in selected military occupational groups, active component, U.S. Armed Forces, 2001-2015. MMSR. 2017;24:8-14.
- Subramaniam P, Olsen CM, Thompson BS, et al, for the QSkin Sun and Health Study Investigators. Anatomical distributions of basal cell carcinoma and squamous cell carcinoma in a population-based study in Queensland, Australia. JAMA Dermatol. 2017;153:175-182. doi:10.1001/jamadermatol.2016.4070
- Lyford WH, Crotty A, Logemann NF. Sun exposure prevention practices within U.S. naval aviation. Mil Med. 2021;186:1169-1175. doi:10.1093/milmed/usab099
- Wood M, Raisanen T, Polcari I. Observational study of free public sunscreen dispenser use at a major US outdoor event. J Am Acad Dermatol. 2017;77:164-166.
- Schissel D. Operation shadow warrior: a quantitative analysis of the ultraviolet radiation protection demonstrated by various headgear. Mil Med. 2001;166:783-785.
- Milch JM, Logemann NF. Photoprotection prevents skin cancer: let’s make it fashionable to wear sun-protective clothing. Cutis. 2017;99:89-92.
- Association of Military Dermatologists. (n.d.). Military dermatology. https://militaryderm.org/military-dermatology/
- Newnam R, Le-Jenkins U, Rutledge C, et al. The association of skin cancer prevention knowledge, sun-protective attitudes, and sunprotective behaviors in a Navy population. Mil Med. 2024;189:1-7. doi:10.1093/milmed/usac285
Skin cancer is a major health concern for military service members, who experience notably higher incidence rates than the general population.1 Active-duty military personnel are particularly vulnerable to prolonged sun exposure due to deployments, specialized training, and everyday outdoor duties.1 Despite skin cancer being the most commonly diagnosed malignancy in active-duty service members,2 tracking and documenting the quantity and diversity of these risk factors remain limited. This knowledge gap comes at high cost, simultaneously impairing military medicine preventive measures while burdening the military health care system with substantial expenditures.3 These findings underscore the critical need for targeted surveillance, early-detection programs, and policy-driven interventions to mitigate these medical and economic concerns.
Skin cancer has been recognized as a major health risk to the military population for decades, yet incidence and prevalence remain high. This phenomenon is closely linked to the inherent responsibilities and expectations of active-duty military members, including outdoor physical training, field exercises, standing in formation, and outdoor working environments—all of which can occur during peak sunlight hours. These risks are further elevated at duty stations in geographic regions with high levels of UV exposure, such as those in tropical and arid regions of the world. Certain military occupational specialties and missions may further introduce unique risk factors; for instance, pilots with frequent high-altitude missions experience heightened UV exposure and melanoma risk.4 Secondary to compounding determinants, the aviation, diving, and nuclear subgroups of the military community are particularly vulnerable to skin cancer.5
Despite well-documented risks, considerable gaps remain in quantifying and analyzing variations in UV exposure across military occupations, duty locations, and operational roles. Factors such as the existence of over 150 distinct military occupational specialties, frequent geographic relocations, and routine work in austere environments contribute to a wide range of UV exposure profiles that remain insufficiently characterized. This lack of comprehensive exposure data hinders the development of large-scale, targeted skin cancer prevention strategies. Initial approaches to addressing these challenges include enhanced surveillance, education, and policy initiatives. The Table presents practical recommendations for military leadership to consider in implementing preventive measures for skin cancer. Herein, we outline broader systemic strategies to bridge knowledge gaps and address underrecognized occupational risk factors for skin cancer in military service members; these elements include proposed modifications to the electronic Periodic Health Assessment (ePHA) and the development of standardized, military-specific screening and prevention guidelines to support early detection and resource optimization.
Skin Cancer Education for Service Members
Sunscreen and Signage—Diligent primary prevention offers a promising avenue for mitigating skin cancer incidence in military service members. Basic education and precautionary messaging on photoprotection can be widely implemented to simultaneously educate service members on the dangers of sun exposure while reinforcing healthy behaviors in real time. Simple low-cost initiatives such as strategically placed visual signage reminding service members to apply sunscreen in high UV environments can support consistent sun-safe practices. Educational efforts also should emphasize proper sunscreen use, including application on high-risk anatomic sites (eg, the face, neck, scalp, dorsal hands, and ears) and the essentiality of using sufficient quantities of broad-spectrum sunscreen for effective protection. Incorporating this guidance into training materials, briefings, and visual reminders allow seamless integration of photoprotection into service members’ daily routines without compromising operational efficiency.6 Younger service members, who may be less likely to prioritize preventive behaviors, may be particularly responsive to sun safety reminders in training areas, bases, and deployment zones.7 Health fairs and orientation briefs in high-UV regions also offer potential opportunities for targeted education.
Resources for Sun Protection in the Military
Sunscreen—Although sunscreen is critical in minimizing the risk for UV-induced skin cancer, its widespread use in the military is hindered by practical challenges related to accessibility and the need for consistent reapplication; for instance, providing free sunscreen dispensers at institutions for staff working under intense or prolonged UV exposure may improve sunscreen accessibility and use.8 Including sunscreen in standard-issue gear offers another logical way to embed its use into operational readiness as part of the routine protective measures.
Uniform Modifications—Adapting military uniforms and practices to improve sun protection plays a critical role in reducing skin cancer risk. Targeted protective gear for commonly sun-exposed areas can help mitigate UV exposure. One practical option is the use of wide-brimmed headgear (eg, boonie hats), which provide more face and neck coverage than standard-issue military caps, or covers. The wide-brimmed headgear currently is only selectively authorized during specific scenarios, such as field operations and training exercises, or at the discretion of unit-level leadership. Wide-brimmed headgear, already used by many service members, has been associated with up to a 17% reduction in UV exposure to inadequately protected areas, potentially lowering skin cancer risk.9,10 Similarly, a “sleeves-down” policy—requiring sleeves to remain unrolled and covering the forearms during outdoor activities—offers a simple way to minimize sun exposure without necessitating additional gear. Other specialized clothing items, including UV-blocking neck gaiters, photoprotective clothing, and lightweight gloves, also may be appropriate for high-risk groups and can be implemented in a relatively straightforward manner.
Shade Structures and UV Index Monitoring—Aside from uniform adaptation, physical barrier intervention can further complement skin cancer prevention efforts in the military. Shade structures offer a straightforward way to reduce UV exposure during prolonged outdoor activities. Incorporating daily UV index monitoring into operational guidance can help inform adjustments to training schedules and guide the implementation of additional sun protection measures, such as mandatory sunscreen application, use of wide-brimmed hats, or increased access to shaded rest areas during heavy sunlight hours. Currently, outdoor physical training is restricted during periods of high heat index, measured via Wet Bulb Globe Temperature, to reduce heat-related injuries. We argue that avoidance of nonoperational outdoor activity during peak UV index hours also should be incorporated into standardized policies. This intervention is of particular benefit to service members stationed in regions with a high UV index year-round, such as those stationed in the Middle East, Guam, Okinawa, and southern coastal United States bases.
Policy Changes to Support Photoprotective Measures
Annual Risk Factor Screening‐Screening—Effective secondary prevention efforts by military dermatologists remain an important measure in reducing the burden of skin cancer among military personnel; however, these efforts have become increasingly challenging due to 2 main factors—the diversity of military occupational specialties and their associated unique occupational risks as well as the limited availability of military dermatologists across all branches (approximately 100 active-duty dermatologists for nearly 3 million service members).11 Therefore, targeted interventions that enhance risk assessment, refined screening protocols, and leveraging of existing military health networks can improve early skin cancer detection while optimizing resource allocation.
The ePHA is an online screening tool used annually by all service members to evaluate their overall health. Presently, the ePHA lacks specific questions to assess sun exposure and skin cancer risks. Integrating annual skin cancer risk factor assessments into the ePHA would offer a practical and straightforward approach to identifying at-risk individuals, as suggested by Newnam et al12 in 2022. Skin cancer risk factor assessments allow for targeted data collection related to sun exposure history, family history, and personal risk factors, which can be used to determine individualized risk stratification to assess the need for early secondary prevention measures and specialist referral. These ePHA data can also support population-based analyses to inform preventive strategies and address knowledge gaps related to high-risk exposures, such as extended field exercises or assignments in high-UV regions, that may impede effective skin cancer prevention.
Development of Military-Specific Screening Guidelines—Given the limited number of military dermatologists, a standardized risk-assessment tool could enhance early detection of skin cancer and streamline the referral process. We propose a military-specific skin cancer screening algorithm or risk nomogram that could help to consolidate risk factors into a clear and actionable framework for more efficient triage and appropriate allocation of dermatologic resources and manpower. This nomogram could be developed by military dermatologists and then implemented on a command level, affording primary care providers a useful tool to expedite evaluation of individuals at higher risk for skin cancer while simultaneously promoting judicious use of limited dermatology resources.
Although the United States Preventive Services Task Force does not universally recommend routine skin cancer screenings for asymptomatic adults, military service members are exposed to higher occupational risks than the general population, as previously mentioned. Currently, there is no standardized screening guideline across all military services due to the unique nature and exposure risks for each branch of service and their varied occupations; however, we propose the development of basic standardized screening guidelines by adapting the framework of the United States Preventive Services Task Force and adjusting for military-specific UV exposure and occupational risks to improve early detection of skin cancer. These guidelines could be updated and tailored appropriately when additional population-based data are collected and analyzed through ePHA.
Critiques and Limitations of Implementation
Several challenges and limitations must be considered when attempting to integrate large-scale preventive measures for skin cancer within the US military. A primary concern is the extent to which military resources should be allocated to prevention when off-duty sun exposure remains largely beyond institutional control. Although military health initiatives can address workplace risk through education and policy, individual decisions during both work and leisure time remain a major variable that cannot be feasibly controlled. Cultural and operational barriers also pose challenges; for instance, the US Marine Corps maintains a strong cultural identity tied to uniform appearance, making it difficult to implement widespread changes to clothing-based sun-protection measures. Institutional changes, particularly those involving uniforms, likely will face substantial administrative resistance and potential operational limitations. When broad uniform modifications are unattainable, a more feasible approach may be to encourage unit-level leadership to authorize and promote the frequent use of nonuniform protective measures.
Furthermore, integrating additional skin cancer risk questions into the already extensive ePHA means extra time required to complete the assessment; this adds to service members’ administrative burden, potentially leading to reduced timely compliance, rushed responses, and survey fatigue, which threaten data quality. If new items are to be included, they should be carefully selected for efficiency and clinical relevance. Existing validated questionnaires such as those from the study by Lyford et al7 published in 2021 can serve as a foundation.
Another critical limitation is access to dermatologic care for active-duty service members. Raising awareness of skin cancer risk without ensuring adequate resources may create ethical concerns, particularly in high-risk environments such as the Middle East and Indo-Pacific. Additionally, because skin cancer often develops years or decades after exposure, securing early buy-in from service members and their leaders can be challenging. These concerns make it clear that, while skin cancer prevention is important, implementing widespread measures is not straightforward and requires a practical and balanced approach.
Final Thoughts
Implementing prevention strategies for skin cancer in the military requires balancing evidence-based recommendations with the practical realities of military culture, resource limitations, and operational demands. Challenges remain for dermatologists in providing targeted recommendations due to the multifaceted nature of military roles, including over 150 Navy Military Occupational Specialties, limited familiarity with the unique UV exposure risks associated with each occupation, and variability in local and regional policies on uniform wear, physical training requirements, and other operational practices. Although targeted prevention measures are difficult to establish in the setting of these knowledge gaps, leveraging unit-level leadership to align with existing screening guidelines and optimizing primary prevention measures can be meaningful steps toward reducing skin cancer risk for military service members while maintaining mission readiness.
Skin cancer is a major health concern for military service members, who experience notably higher incidence rates than the general population.1 Active-duty military personnel are particularly vulnerable to prolonged sun exposure due to deployments, specialized training, and everyday outdoor duties.1 Despite skin cancer being the most commonly diagnosed malignancy in active-duty service members,2 tracking and documenting the quantity and diversity of these risk factors remain limited. This knowledge gap comes at high cost, simultaneously impairing military medicine preventive measures while burdening the military health care system with substantial expenditures.3 These findings underscore the critical need for targeted surveillance, early-detection programs, and policy-driven interventions to mitigate these medical and economic concerns.
Skin cancer has been recognized as a major health risk to the military population for decades, yet incidence and prevalence remain high. This phenomenon is closely linked to the inherent responsibilities and expectations of active-duty military members, including outdoor physical training, field exercises, standing in formation, and outdoor working environments—all of which can occur during peak sunlight hours. These risks are further elevated at duty stations in geographic regions with high levels of UV exposure, such as those in tropical and arid regions of the world. Certain military occupational specialties and missions may further introduce unique risk factors; for instance, pilots with frequent high-altitude missions experience heightened UV exposure and melanoma risk.4 Secondary to compounding determinants, the aviation, diving, and nuclear subgroups of the military community are particularly vulnerable to skin cancer.5
Despite well-documented risks, considerable gaps remain in quantifying and analyzing variations in UV exposure across military occupations, duty locations, and operational roles. Factors such as the existence of over 150 distinct military occupational specialties, frequent geographic relocations, and routine work in austere environments contribute to a wide range of UV exposure profiles that remain insufficiently characterized. This lack of comprehensive exposure data hinders the development of large-scale, targeted skin cancer prevention strategies. Initial approaches to addressing these challenges include enhanced surveillance, education, and policy initiatives. The Table presents practical recommendations for military leadership to consider in implementing preventive measures for skin cancer. Herein, we outline broader systemic strategies to bridge knowledge gaps and address underrecognized occupational risk factors for skin cancer in military service members; these elements include proposed modifications to the electronic Periodic Health Assessment (ePHA) and the development of standardized, military-specific screening and prevention guidelines to support early detection and resource optimization.
Skin Cancer Education for Service Members
Sunscreen and Signage—Diligent primary prevention offers a promising avenue for mitigating skin cancer incidence in military service members. Basic education and precautionary messaging on photoprotection can be widely implemented to simultaneously educate service members on the dangers of sun exposure while reinforcing healthy behaviors in real time. Simple low-cost initiatives such as strategically placed visual signage reminding service members to apply sunscreen in high UV environments can support consistent sun-safe practices. Educational efforts also should emphasize proper sunscreen use, including application on high-risk anatomic sites (eg, the face, neck, scalp, dorsal hands, and ears) and the essentiality of using sufficient quantities of broad-spectrum sunscreen for effective protection. Incorporating this guidance into training materials, briefings, and visual reminders allow seamless integration of photoprotection into service members’ daily routines without compromising operational efficiency.6 Younger service members, who may be less likely to prioritize preventive behaviors, may be particularly responsive to sun safety reminders in training areas, bases, and deployment zones.7 Health fairs and orientation briefs in high-UV regions also offer potential opportunities for targeted education.
Resources for Sun Protection in the Military
Sunscreen—Although sunscreen is critical in minimizing the risk for UV-induced skin cancer, its widespread use in the military is hindered by practical challenges related to accessibility and the need for consistent reapplication; for instance, providing free sunscreen dispensers at institutions for staff working under intense or prolonged UV exposure may improve sunscreen accessibility and use.8 Including sunscreen in standard-issue gear offers another logical way to embed its use into operational readiness as part of the routine protective measures.
Uniform Modifications—Adapting military uniforms and practices to improve sun protection plays a critical role in reducing skin cancer risk. Targeted protective gear for commonly sun-exposed areas can help mitigate UV exposure. One practical option is the use of wide-brimmed headgear (eg, boonie hats), which provide more face and neck coverage than standard-issue military caps, or covers. The wide-brimmed headgear currently is only selectively authorized during specific scenarios, such as field operations and training exercises, or at the discretion of unit-level leadership. Wide-brimmed headgear, already used by many service members, has been associated with up to a 17% reduction in UV exposure to inadequately protected areas, potentially lowering skin cancer risk.9,10 Similarly, a “sleeves-down” policy—requiring sleeves to remain unrolled and covering the forearms during outdoor activities—offers a simple way to minimize sun exposure without necessitating additional gear. Other specialized clothing items, including UV-blocking neck gaiters, photoprotective clothing, and lightweight gloves, also may be appropriate for high-risk groups and can be implemented in a relatively straightforward manner.
Shade Structures and UV Index Monitoring—Aside from uniform adaptation, physical barrier intervention can further complement skin cancer prevention efforts in the military. Shade structures offer a straightforward way to reduce UV exposure during prolonged outdoor activities. Incorporating daily UV index monitoring into operational guidance can help inform adjustments to training schedules and guide the implementation of additional sun protection measures, such as mandatory sunscreen application, use of wide-brimmed hats, or increased access to shaded rest areas during heavy sunlight hours. Currently, outdoor physical training is restricted during periods of high heat index, measured via Wet Bulb Globe Temperature, to reduce heat-related injuries. We argue that avoidance of nonoperational outdoor activity during peak UV index hours also should be incorporated into standardized policies. This intervention is of particular benefit to service members stationed in regions with a high UV index year-round, such as those stationed in the Middle East, Guam, Okinawa, and southern coastal United States bases.
Policy Changes to Support Photoprotective Measures
Annual Risk Factor Screening‐Screening—Effective secondary prevention efforts by military dermatologists remain an important measure in reducing the burden of skin cancer among military personnel; however, these efforts have become increasingly challenging due to 2 main factors—the diversity of military occupational specialties and their associated unique occupational risks as well as the limited availability of military dermatologists across all branches (approximately 100 active-duty dermatologists for nearly 3 million service members).11 Therefore, targeted interventions that enhance risk assessment, refined screening protocols, and leveraging of existing military health networks can improve early skin cancer detection while optimizing resource allocation.
The ePHA is an online screening tool used annually by all service members to evaluate their overall health. Presently, the ePHA lacks specific questions to assess sun exposure and skin cancer risks. Integrating annual skin cancer risk factor assessments into the ePHA would offer a practical and straightforward approach to identifying at-risk individuals, as suggested by Newnam et al12 in 2022. Skin cancer risk factor assessments allow for targeted data collection related to sun exposure history, family history, and personal risk factors, which can be used to determine individualized risk stratification to assess the need for early secondary prevention measures and specialist referral. These ePHA data can also support population-based analyses to inform preventive strategies and address knowledge gaps related to high-risk exposures, such as extended field exercises or assignments in high-UV regions, that may impede effective skin cancer prevention.
Development of Military-Specific Screening Guidelines—Given the limited number of military dermatologists, a standardized risk-assessment tool could enhance early detection of skin cancer and streamline the referral process. We propose a military-specific skin cancer screening algorithm or risk nomogram that could help to consolidate risk factors into a clear and actionable framework for more efficient triage and appropriate allocation of dermatologic resources and manpower. This nomogram could be developed by military dermatologists and then implemented on a command level, affording primary care providers a useful tool to expedite evaluation of individuals at higher risk for skin cancer while simultaneously promoting judicious use of limited dermatology resources.
Although the United States Preventive Services Task Force does not universally recommend routine skin cancer screenings for asymptomatic adults, military service members are exposed to higher occupational risks than the general population, as previously mentioned. Currently, there is no standardized screening guideline across all military services due to the unique nature and exposure risks for each branch of service and their varied occupations; however, we propose the development of basic standardized screening guidelines by adapting the framework of the United States Preventive Services Task Force and adjusting for military-specific UV exposure and occupational risks to improve early detection of skin cancer. These guidelines could be updated and tailored appropriately when additional population-based data are collected and analyzed through ePHA.
Critiques and Limitations of Implementation
Several challenges and limitations must be considered when attempting to integrate large-scale preventive measures for skin cancer within the US military. A primary concern is the extent to which military resources should be allocated to prevention when off-duty sun exposure remains largely beyond institutional control. Although military health initiatives can address workplace risk through education and policy, individual decisions during both work and leisure time remain a major variable that cannot be feasibly controlled. Cultural and operational barriers also pose challenges; for instance, the US Marine Corps maintains a strong cultural identity tied to uniform appearance, making it difficult to implement widespread changes to clothing-based sun-protection measures. Institutional changes, particularly those involving uniforms, likely will face substantial administrative resistance and potential operational limitations. When broad uniform modifications are unattainable, a more feasible approach may be to encourage unit-level leadership to authorize and promote the frequent use of nonuniform protective measures.
Furthermore, integrating additional skin cancer risk questions into the already extensive ePHA means extra time required to complete the assessment; this adds to service members’ administrative burden, potentially leading to reduced timely compliance, rushed responses, and survey fatigue, which threaten data quality. If new items are to be included, they should be carefully selected for efficiency and clinical relevance. Existing validated questionnaires such as those from the study by Lyford et al7 published in 2021 can serve as a foundation.
Another critical limitation is access to dermatologic care for active-duty service members. Raising awareness of skin cancer risk without ensuring adequate resources may create ethical concerns, particularly in high-risk environments such as the Middle East and Indo-Pacific. Additionally, because skin cancer often develops years or decades after exposure, securing early buy-in from service members and their leaders can be challenging. These concerns make it clear that, while skin cancer prevention is important, implementing widespread measures is not straightforward and requires a practical and balanced approach.
Final Thoughts
Implementing prevention strategies for skin cancer in the military requires balancing evidence-based recommendations with the practical realities of military culture, resource limitations, and operational demands. Challenges remain for dermatologists in providing targeted recommendations due to the multifaceted nature of military roles, including over 150 Navy Military Occupational Specialties, limited familiarity with the unique UV exposure risks associated with each occupation, and variability in local and regional policies on uniform wear, physical training requirements, and other operational practices. Although targeted prevention measures are difficult to establish in the setting of these knowledge gaps, leveraging unit-level leadership to align with existing screening guidelines and optimizing primary prevention measures can be meaningful steps toward reducing skin cancer risk for military service members while maintaining mission readiness.
- Riemenschneider K, Liu J, Powers JG. Skin cancer in the military: a systematic review of melanoma and nonmelanoma skin cancerincidence, prevention, and screening among active duty and veteran personnel. J Am Acad Dermatol. 2018;78:1185-1192. doi:10.1016/j.jaad.2017.11.062
- Lee T, Taubman SB, Williams VF. Incident diagnoses of non-melanoma skin cancer, active component, U.S. Armed Forces, 2005-2014. MSMR. 2016;23:2-6.
- Krivda KR, Watson NL, Lyford WH, et al. The burden of skin cancer in the military health system, 2017-2022. Cutis. 2024;113:200-215. doi:10.12788/cutis.1015
- Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2015;151:51-58. doi:10.1001/jamadermatol.2014.1077
- Brundage JF, Williams VF, Stahlman S, et al. Incidence rates of malignant melanoma in relation to years of military service, overall and in selected military occupational groups, active component, U.S. Armed Forces, 2001-2015. MMSR. 2017;24:8-14.
- Subramaniam P, Olsen CM, Thompson BS, et al, for the QSkin Sun and Health Study Investigators. Anatomical distributions of basal cell carcinoma and squamous cell carcinoma in a population-based study in Queensland, Australia. JAMA Dermatol. 2017;153:175-182. doi:10.1001/jamadermatol.2016.4070
- Lyford WH, Crotty A, Logemann NF. Sun exposure prevention practices within U.S. naval aviation. Mil Med. 2021;186:1169-1175. doi:10.1093/milmed/usab099
- Wood M, Raisanen T, Polcari I. Observational study of free public sunscreen dispenser use at a major US outdoor event. J Am Acad Dermatol. 2017;77:164-166.
- Schissel D. Operation shadow warrior: a quantitative analysis of the ultraviolet radiation protection demonstrated by various headgear. Mil Med. 2001;166:783-785.
- Milch JM, Logemann NF. Photoprotection prevents skin cancer: let’s make it fashionable to wear sun-protective clothing. Cutis. 2017;99:89-92.
- Association of Military Dermatologists. (n.d.). Military dermatology. https://militaryderm.org/military-dermatology/
- Newnam R, Le-Jenkins U, Rutledge C, et al. The association of skin cancer prevention knowledge, sun-protective attitudes, and sunprotective behaviors in a Navy population. Mil Med. 2024;189:1-7. doi:10.1093/milmed/usac285
- Riemenschneider K, Liu J, Powers JG. Skin cancer in the military: a systematic review of melanoma and nonmelanoma skin cancerincidence, prevention, and screening among active duty and veteran personnel. J Am Acad Dermatol. 2018;78:1185-1192. doi:10.1016/j.jaad.2017.11.062
- Lee T, Taubman SB, Williams VF. Incident diagnoses of non-melanoma skin cancer, active component, U.S. Armed Forces, 2005-2014. MSMR. 2016;23:2-6.
- Krivda KR, Watson NL, Lyford WH, et al. The burden of skin cancer in the military health system, 2017-2022. Cutis. 2024;113:200-215. doi:10.12788/cutis.1015
- Sanlorenzo M, Wehner MR, Linos E, et al. The risk of melanoma in airline pilots and cabin crew: a meta-analysis. JAMA Dermatol. 2015;151:51-58. doi:10.1001/jamadermatol.2014.1077
- Brundage JF, Williams VF, Stahlman S, et al. Incidence rates of malignant melanoma in relation to years of military service, overall and in selected military occupational groups, active component, U.S. Armed Forces, 2001-2015. MMSR. 2017;24:8-14.
- Subramaniam P, Olsen CM, Thompson BS, et al, for the QSkin Sun and Health Study Investigators. Anatomical distributions of basal cell carcinoma and squamous cell carcinoma in a population-based study in Queensland, Australia. JAMA Dermatol. 2017;153:175-182. doi:10.1001/jamadermatol.2016.4070
- Lyford WH, Crotty A, Logemann NF. Sun exposure prevention practices within U.S. naval aviation. Mil Med. 2021;186:1169-1175. doi:10.1093/milmed/usab099
- Wood M, Raisanen T, Polcari I. Observational study of free public sunscreen dispenser use at a major US outdoor event. J Am Acad Dermatol. 2017;77:164-166.
- Schissel D. Operation shadow warrior: a quantitative analysis of the ultraviolet radiation protection demonstrated by various headgear. Mil Med. 2001;166:783-785.
- Milch JM, Logemann NF. Photoprotection prevents skin cancer: let’s make it fashionable to wear sun-protective clothing. Cutis. 2017;99:89-92.
- Association of Military Dermatologists. (n.d.). Military dermatology. https://militaryderm.org/military-dermatology/
- Newnam R, Le-Jenkins U, Rutledge C, et al. The association of skin cancer prevention knowledge, sun-protective attitudes, and sunprotective behaviors in a Navy population. Mil Med. 2024;189:1-7. doi:10.1093/milmed/usac285
Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
Bridging the Knowledge-Action Gap in Skin Cancer Prevention Among US Military Personnel
PRACTICE POINTS
- Military personnel face elevated skin cancer risks due to prolonged occupational UV exposure.
- Medical providers can partner with unit-level leadership to implement low-cost interventions such as shade structures and uniform modifications.
- Annual sun exposure risk assessments should be integrated into the military Electronic Periodic Health Assessment for targeted screening and early intervention of risk factors.
- Photoprotective gear and signage in high—UV index areas can improve service member awareness and adherence to preventive measures.
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
Worldwide, it is estimated that up to 1 in 5 individuals will experience a dermatophyte infection (commonly called ringworm or tinea infection) in their lifetime.1 Historically, dermatophyte infections have been considered relatively minor conditions usually treated with short courses of topical antifungals.2 Oral antifungals historically were needed only for patients with nail or hair shaft infections or extensive cutaneous fungal infections, which typically occurred in immunosuppressed patients.2 However, the landscape is changing rapidly due to the global emergence of severe dermatophyte infections that frequently are resistant to first-line antifungal medications.3-5 In this article, we aimed to review the epidemiology of emerging dermatophyte infections and provide dermatologists with information needed for effective diagnosis and management.
Emergence of Trichophyton indotineae
In recent decades, public health officials and dermatologists have noted with concern the spread of the recently emerged dermatophyte species Trichophyton indotineae in South Asia.3,6 This species (previously known as Trichophyton mentagrophytes genotype VIII) usually is transmitted from person to person, either through direct skin-to-skin contact or by fomites.4,6 Potential sexual transmission of T indotineae infections also has been reported,7 and it is possible that animals may serve as reservoirs for this pathogen, although there are no known reports of direct spread from animals to humans.8,9 Major outbreaks of T indotineae are ongoing in South Asia, and cases have been documented in 6 continents.10-12 In the United States, most but not all cases have occurred in immigrants from or recently returned travelers to South Asia.6,13 The emergence and spread of T indotineae is hypothesized to be promoted by the misuse and overuse of topical antifungal products, particularly those containing combinations of potent corticosteroids with other antimicrobial drugs.14,15
Cutaneous manifestations of T indotineae infections tend to cover large body surface areas, recur frequently, and pose substantial treatment challenges.6,13,16 Several clinical presentations have been documented, including erythematous, scaly concentric plaques; papulosquamous lesions; pustular forms; and corticosteroid-modified disease (Figure 1).6,16 Affected patients seldom are immunocompromised and often have a history of multiple failed courses of topical or oral antifungals, including oral terbinafine.13 Many also have been prescribed topical corticosteroids or have used over-the-counter topical corticosteroids, which worsen the rash.17
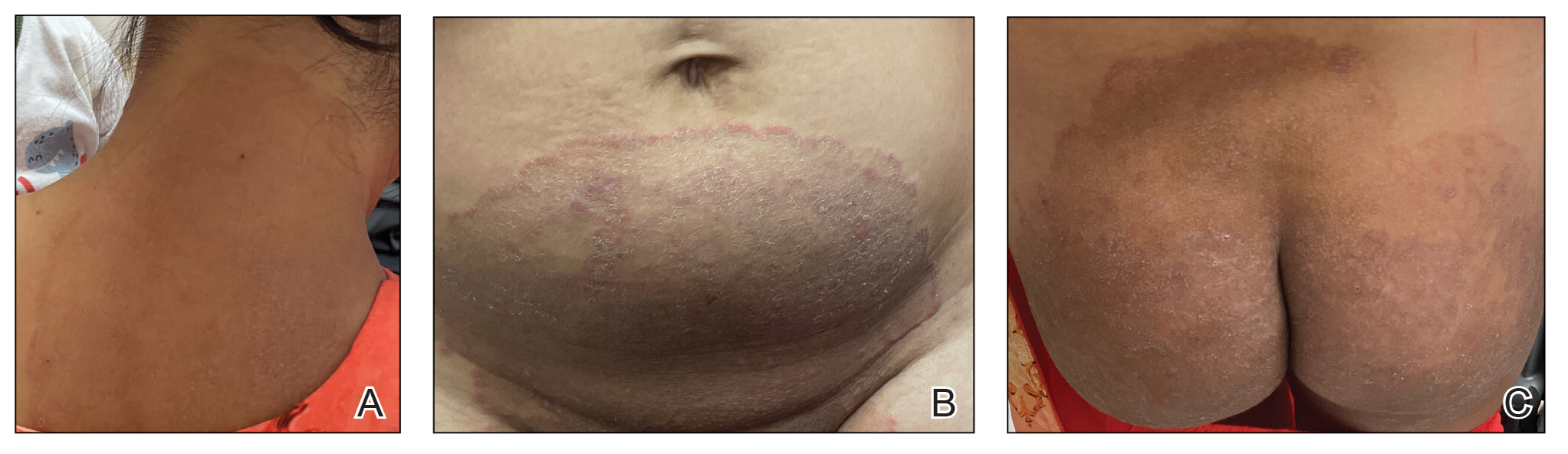
Direct microscopy with potassium hydroxide could be used to confirm the diagnosis of dermatophyte infection, but it does not distinguish T indotineae from other dermatophyte species.2,6 Importantly, culture-based testing usually will misidentify T indotineae as other Trichophyton species such as the more common T mentagrophytes or Trichophyton interdigitale. Definitive identification of T indotineae requires advanced molecular techniques that are available only at select laboratories.6 Unfortunately, availability of such testing is limited (Table), and results may take several weeks; therefore, it is suggested that dermatologists who suspect T indotineae infections based on the patient’s history and clinical presentation begin antifungal treatment after confirmation of dermatophyte infection but not wait for definitive confirmation of the causative organism.16
Itraconazole is considered the first-line therapy for T indotineae infection, as terbinafine usually is ineffective due to mutations in the squalene epoxidase gene.16 Dermatologists should be aware that itraconazole is available in different formulations that can affect absorption. The oral solution has greater bioavailability and should be taken on an empty stomach, whereas the capsules are required to be taken with food for effective absorption; the capsules also should be taken with an acidic beverage such as orange juice. Dermatologists should carefully assess for drug-drug interactions when prescribing itraconazole, given its extensive interaction profile with numerous other medications. Patients may require treatment with itraconazole (100 mg/d or 200 mg/d) for a minimum of 6 to 8 weeks until complete clearance has been achieved and ideally a negative potassium hydroxide preparation of skin scrapings has been obtained. A longer treatment period (eg, ≥3 months) frequently is needed, and relapses are common.6,16,18 Regular follow-up is needed to monitor for infection clearance and recurrences. It is important to note that cases of itraconazole resistance have been reported, although this currently appears to be uncommon.19,20
Other Emerging Dermatophytes to Watch
Trichophyton rubrum is the most common cause of dermatophyte infections among humans,21 and cases of terbinafine-resistant T rubrum infections have been reported increasingly in the United States and Canada.5,22-24 Onychomycosis caused by terbinafine-resistant T rubrum has been documented, and patients may have infections that do not respond to terbinafine given at the standard dose and duration.22,23 Case reports have indicated successful treatment using itraconazole 200 mg/d and posaconazole 300 mg/d.5,23
Trichophyton mentagrophytes genotype VII (TMVII) is an emerging dermatophyte that recently has been reported as a cause of sexually transmitted dermatophyte infections in Europe and the United States primarily affecting men who have sex with men.25-27 Patients may present with pruritic, annular, scaly patches and plaques involving the trunk, groin, genital region, or face (Figure 2). Although closely related to T indotineae, TMVII differs in that it more often affects the genital region, generally is susceptible to terbinafine, and in the United States and Europe usually is not related to travel or immigration involving South Asia.26 Although TMVII has not been associated with antifungal resistance, awareness among dermatologists is important because patients may experience inflamed, painful, and persistent rashes that can lead to secondary bacterial infection or scarring, and physicians might mistake it for mimics including eczema or psoriasis.25,26
Importance of Judicious Antifungal Use
Optimizing the use of antifungals is critical to improving patient outcomes and preserving available treatment options.28,29 A retrospective analysis of commercial health insurance data estimated that topical antifungal prescriptions were potentially unnecessary for more than half of the more than 560,000 patients who were prescribed these medications in 2023. In this study, it also was observed that only 16% of patients prescribed a topical antifungal had received diagnostic testing, with low rates across specialties.30 This is concerning because even among board-certified dermatologists, incorrect diagnosis of suspected fungal skin infections can occur; in one survey-based study of board-certified dermatologists who were presented with dermatomycosis images, respondents categorized cases with greater than 75% accuracy in only 31% (4/13) of instances.31 Clotrimazole-betamethasone is among the most commonly prescribed topical antifungals in the United States,14,32 and 2 recent retrospective analyses highlighted that the majority of patients prescribed this medication did not receive any fungal diagnostic testing.33,34
Final Thoughts
In an era of emerging antifungal-resistant dermatophyte infections, it is important for dermatologists to educate nondermatologists about the importance of using diagnostic testing for suspected dermatophyte infections.14,28 Dermatologists also can educate nondermatologist colleagues on the importance of avoiding the use of topical combination antifungal/corticosteroid medications and referring for dermatologic evaluation when diagnoses are uncertain.33,34 Strategies for education by dermatologists could include giving workshops, creating educational materials, and fostering open communication about optimal treatment practices and referral parameters for suspected dermatophyte infections.
- Noble SL, Forbes RC, Stamm PL. Diagnosis and management of common tinea infections. Am Fam Physician. 1998;58:163-174, 177-168.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Uhrlaß S, Verma SB, Gräser Y, et al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: I. epidemiology, risk factors and clinical features. Indian J Dermatol Venereol Leprol. 2021;87:154-175.
- Chen E, Ghannoum M, Elewski BE. Treatment]resistant tinea corporis, a potential public health issue. Br J Dermatol. 2021;184:164-165.
- Caplan AS. Notes from the field: first reported US cases of tinea caused by Trichophyton indotineae—New York City, December 2021–March 2023. MMWR Morbidity and Mortality Weekly Report. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Spivack S, Gold JA, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807.
- Jabet A, Brun S, Normand AC, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233.
- Thakur S, Spruijtenburg B, Abhishek, et al. Whole genome sequence analysis of terbinafine resistant and susceptible Trichophyton isolates from human and animal origin. Mycopathologia. 2025;190:13.
- Lockhart SR, Chowdhary A, Gold JA. The rapid emergence of antifungal-resistant human-pathogenic fungi. Nat Rev Microbiol. 2023;21:818-832.
- Mosam A, Shuping L, Naicker S, et al. A case of antifungal-resistant ringworm infection in KwaZulu-Natal Province, South Africa, caused by Trichophyton indotineae. Public Health Bulletin South Africa. Accessed April 4, 2025. https://www.phbsa.ac.za/wp-content/uploads/2023/12PHBSA-Ringworm-Article-2023.pdf
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:E0056223
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709. doi:10.1001/jamadermatol.2024.1126
- Benedict K. Topical antifungal prescribing for Medicare Part D beneficiaries—United States, 2021. MMWR Morb Mortal Wkly Rep. 2024;73:1-5.
- Verma SB. Emergence of recalcitrant dermatophytosis in India. Lancet Infect Dis. 2018;18:718-719.
- Khurana A, Sharath S, Sardana K, et al. Clinico-mycological and therapeutic updates on cutaneous dermatophytic infections in the era of Trichophyton indotineae. J Am Acad Dermatol. 2024;91:315-323. doi:10.1016/j.jaad.2024.03.024
- Verma S. Steroid modified tinea. BMJ. 2017;356:j973.
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278.
- Burmester A, Hipler UC, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180.
- Bhuiyan MSI, Verma SB, Illigner GM, et al. Trichophyton mentagrophytes ITS genotype VIII/Trichophyton indotineae infection and antifungal resistance in Bangladesh. J Fungi (Basel). 2024;10:768. doi:10.3390 /jof10110768
- Hay RJ. Chapter 82: superficial mycoses. In: Ryan ET, Hill DR, Solomon T, et al, eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 10th ed. Elsevier; 2020:648-652.
- Gupta AK, Cooper EA, Wang T, et al. Detection of squalene epoxidase mutations in United States patients with onychomycosis: implications for management. J Invest Dermatol. 2023;143:2476-2483.E2477.
- Hwang JK, Bakotic WL, Gold JA, et al. Isolation of terbinafine-resistant Trichophyton rubrum from onychomycosis patients who failed treatment at an academic center in New York, United States. J Fungi. 2023;9:710.
- Gu D, Hatch M, Ghannoum M, et al. Treatment-resistant dermatophytosis: a representative case highlighting an emerging public health threat. JAAD Case Rep. 2020;6:1153-1155.
- Jabet A, Dellière S, Seang S, et al. Sexually transmitted Trichophyton mentagrophytes genotype VII infection among men who have sex with men. Emerg Infect Dis. 2023;29:1411-1414.
- Zucker J, Caplan AS, Gunaratne SH, et al. Notes from the field: Trichophyton mentagrophytes genotype VII—New York City, April-July 2024. MMWR Morb Mortal Wkly Rep. 2024;73:985-988.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Caplan AS, Gold JA, Smith DJ, et al. Improving antifungal stewardship in dermatology in an era of emerging dermatophyte resistance. JAAD International. 2024;15:168-169.
- Elewski B. A call for antifungal stewardship. Br J Dermatol. 2020; 183:798-799.
- Gold JAW, Benedict K, Caplan AS, et al. High rates of potentially unnecessary topical antifungal prescribing in a large commercial health insurance claims database, United States. J Am Acad Dermatol. 2025:S0190-9622(25)00098-2. doi:10.1016/j.jaad.2025.01.022
- Yadgar RJ, Bhatia N, Friedman A. Cutaneous fungal infections are commonly misdiagnosed: a survey-based study. J Am Acad Dermatol. 2017;76:562-563.
- Flint ND, Rhoads JLW, Carlisle R, et al. The continued inappropriate use and overuse of combination topical clotrimazole-betamethasone. Dermatol Online J. 2021;27. doi:10.5070/D327854686
- Currie DW, Caplan AS, Benedict K, et al. Prescribing of clotrimazolebetamethasone dipropionate, a topical combination corticosteroidantifungal product, for Medicare part D beneficiaries, United States, 2016–2022. Antimicrob Steward Healthc Epidemiol. 2024;4:E174.
- Gold JA, Caplan AS, Benedict K, et al. Clotrimazole-betamethasone dipropionate prescribing for nonfungal skin conditions. JAMA Network Open. 2024;7:E2411721-E2411721.
Worldwide, it is estimated that up to 1 in 5 individuals will experience a dermatophyte infection (commonly called ringworm or tinea infection) in their lifetime.1 Historically, dermatophyte infections have been considered relatively minor conditions usually treated with short courses of topical antifungals.2 Oral antifungals historically were needed only for patients with nail or hair shaft infections or extensive cutaneous fungal infections, which typically occurred in immunosuppressed patients.2 However, the landscape is changing rapidly due to the global emergence of severe dermatophyte infections that frequently are resistant to first-line antifungal medications.3-5 In this article, we aimed to review the epidemiology of emerging dermatophyte infections and provide dermatologists with information needed for effective diagnosis and management.
Emergence of Trichophyton indotineae
In recent decades, public health officials and dermatologists have noted with concern the spread of the recently emerged dermatophyte species Trichophyton indotineae in South Asia.3,6 This species (previously known as Trichophyton mentagrophytes genotype VIII) usually is transmitted from person to person, either through direct skin-to-skin contact or by fomites.4,6 Potential sexual transmission of T indotineae infections also has been reported,7 and it is possible that animals may serve as reservoirs for this pathogen, although there are no known reports of direct spread from animals to humans.8,9 Major outbreaks of T indotineae are ongoing in South Asia, and cases have been documented in 6 continents.10-12 In the United States, most but not all cases have occurred in immigrants from or recently returned travelers to South Asia.6,13 The emergence and spread of T indotineae is hypothesized to be promoted by the misuse and overuse of topical antifungal products, particularly those containing combinations of potent corticosteroids with other antimicrobial drugs.14,15
Cutaneous manifestations of T indotineae infections tend to cover large body surface areas, recur frequently, and pose substantial treatment challenges.6,13,16 Several clinical presentations have been documented, including erythematous, scaly concentric plaques; papulosquamous lesions; pustular forms; and corticosteroid-modified disease (Figure 1).6,16 Affected patients seldom are immunocompromised and often have a history of multiple failed courses of topical or oral antifungals, including oral terbinafine.13 Many also have been prescribed topical corticosteroids or have used over-the-counter topical corticosteroids, which worsen the rash.17
Direct microscopy with potassium hydroxide could be used to confirm the diagnosis of dermatophyte infection, but it does not distinguish T indotineae from other dermatophyte species.2,6 Importantly, culture-based testing usually will misidentify T indotineae as other Trichophyton species such as the more common T mentagrophytes or Trichophyton interdigitale. Definitive identification of T indotineae requires advanced molecular techniques that are available only at select laboratories.6 Unfortunately, availability of such testing is limited (Table), and results may take several weeks; therefore, it is suggested that dermatologists who suspect T indotineae infections based on the patient’s history and clinical presentation begin antifungal treatment after confirmation of dermatophyte infection but not wait for definitive confirmation of the causative organism.16
Itraconazole is considered the first-line therapy for T indotineae infection, as terbinafine usually is ineffective due to mutations in the squalene epoxidase gene.16 Dermatologists should be aware that itraconazole is available in different formulations that can affect absorption. The oral solution has greater bioavailability and should be taken on an empty stomach, whereas the capsules are required to be taken with food for effective absorption; the capsules also should be taken with an acidic beverage such as orange juice. Dermatologists should carefully assess for drug-drug interactions when prescribing itraconazole, given its extensive interaction profile with numerous other medications. Patients may require treatment with itraconazole (100 mg/d or 200 mg/d) for a minimum of 6 to 8 weeks until complete clearance has been achieved and ideally a negative potassium hydroxide preparation of skin scrapings has been obtained. A longer treatment period (eg, ≥3 months) frequently is needed, and relapses are common.6,16,18 Regular follow-up is needed to monitor for infection clearance and recurrences. It is important to note that cases of itraconazole resistance have been reported, although this currently appears to be uncommon.19,20
Other Emerging Dermatophytes to Watch
Trichophyton rubrum is the most common cause of dermatophyte infections among humans,21 and cases of terbinafine-resistant T rubrum infections have been reported increasingly in the United States and Canada.5,22-24 Onychomycosis caused by terbinafine-resistant T rubrum has been documented, and patients may have infections that do not respond to terbinafine given at the standard dose and duration.22,23 Case reports have indicated successful treatment using itraconazole 200 mg/d and posaconazole 300 mg/d.5,23
Trichophyton mentagrophytes genotype VII (TMVII) is an emerging dermatophyte that recently has been reported as a cause of sexually transmitted dermatophyte infections in Europe and the United States primarily affecting men who have sex with men.25-27 Patients may present with pruritic, annular, scaly patches and plaques involving the trunk, groin, genital region, or face (Figure 2). Although closely related to T indotineae, TMVII differs in that it more often affects the genital region, generally is susceptible to terbinafine, and in the United States and Europe usually is not related to travel or immigration involving South Asia.26 Although TMVII has not been associated with antifungal resistance, awareness among dermatologists is important because patients may experience inflamed, painful, and persistent rashes that can lead to secondary bacterial infection or scarring, and physicians might mistake it for mimics including eczema or psoriasis.25,26
Importance of Judicious Antifungal Use
Optimizing the use of antifungals is critical to improving patient outcomes and preserving available treatment options.28,29 A retrospective analysis of commercial health insurance data estimated that topical antifungal prescriptions were potentially unnecessary for more than half of the more than 560,000 patients who were prescribed these medications in 2023. In this study, it also was observed that only 16% of patients prescribed a topical antifungal had received diagnostic testing, with low rates across specialties.30 This is concerning because even among board-certified dermatologists, incorrect diagnosis of suspected fungal skin infections can occur; in one survey-based study of board-certified dermatologists who were presented with dermatomycosis images, respondents categorized cases with greater than 75% accuracy in only 31% (4/13) of instances.31 Clotrimazole-betamethasone is among the most commonly prescribed topical antifungals in the United States,14,32 and 2 recent retrospective analyses highlighted that the majority of patients prescribed this medication did not receive any fungal diagnostic testing.33,34
Final Thoughts
In an era of emerging antifungal-resistant dermatophyte infections, it is important for dermatologists to educate nondermatologists about the importance of using diagnostic testing for suspected dermatophyte infections.14,28 Dermatologists also can educate nondermatologist colleagues on the importance of avoiding the use of topical combination antifungal/corticosteroid medications and referring for dermatologic evaluation when diagnoses are uncertain.33,34 Strategies for education by dermatologists could include giving workshops, creating educational materials, and fostering open communication about optimal treatment practices and referral parameters for suspected dermatophyte infections.
Worldwide, it is estimated that up to 1 in 5 individuals will experience a dermatophyte infection (commonly called ringworm or tinea infection) in their lifetime.1 Historically, dermatophyte infections have been considered relatively minor conditions usually treated with short courses of topical antifungals.2 Oral antifungals historically were needed only for patients with nail or hair shaft infections or extensive cutaneous fungal infections, which typically occurred in immunosuppressed patients.2 However, the landscape is changing rapidly due to the global emergence of severe dermatophyte infections that frequently are resistant to first-line antifungal medications.3-5 In this article, we aimed to review the epidemiology of emerging dermatophyte infections and provide dermatologists with information needed for effective diagnosis and management.
Emergence of Trichophyton indotineae
In recent decades, public health officials and dermatologists have noted with concern the spread of the recently emerged dermatophyte species Trichophyton indotineae in South Asia.3,6 This species (previously known as Trichophyton mentagrophytes genotype VIII) usually is transmitted from person to person, either through direct skin-to-skin contact or by fomites.4,6 Potential sexual transmission of T indotineae infections also has been reported,7 and it is possible that animals may serve as reservoirs for this pathogen, although there are no known reports of direct spread from animals to humans.8,9 Major outbreaks of T indotineae are ongoing in South Asia, and cases have been documented in 6 continents.10-12 In the United States, most but not all cases have occurred in immigrants from or recently returned travelers to South Asia.6,13 The emergence and spread of T indotineae is hypothesized to be promoted by the misuse and overuse of topical antifungal products, particularly those containing combinations of potent corticosteroids with other antimicrobial drugs.14,15
Cutaneous manifestations of T indotineae infections tend to cover large body surface areas, recur frequently, and pose substantial treatment challenges.6,13,16 Several clinical presentations have been documented, including erythematous, scaly concentric plaques; papulosquamous lesions; pustular forms; and corticosteroid-modified disease (Figure 1).6,16 Affected patients seldom are immunocompromised and often have a history of multiple failed courses of topical or oral antifungals, including oral terbinafine.13 Many also have been prescribed topical corticosteroids or have used over-the-counter topical corticosteroids, which worsen the rash.17
Direct microscopy with potassium hydroxide could be used to confirm the diagnosis of dermatophyte infection, but it does not distinguish T indotineae from other dermatophyte species.2,6 Importantly, culture-based testing usually will misidentify T indotineae as other Trichophyton species such as the more common T mentagrophytes or Trichophyton interdigitale. Definitive identification of T indotineae requires advanced molecular techniques that are available only at select laboratories.6 Unfortunately, availability of such testing is limited (Table), and results may take several weeks; therefore, it is suggested that dermatologists who suspect T indotineae infections based on the patient’s history and clinical presentation begin antifungal treatment after confirmation of dermatophyte infection but not wait for definitive confirmation of the causative organism.16
Itraconazole is considered the first-line therapy for T indotineae infection, as terbinafine usually is ineffective due to mutations in the squalene epoxidase gene.16 Dermatologists should be aware that itraconazole is available in different formulations that can affect absorption. The oral solution has greater bioavailability and should be taken on an empty stomach, whereas the capsules are required to be taken with food for effective absorption; the capsules also should be taken with an acidic beverage such as orange juice. Dermatologists should carefully assess for drug-drug interactions when prescribing itraconazole, given its extensive interaction profile with numerous other medications. Patients may require treatment with itraconazole (100 mg/d or 200 mg/d) for a minimum of 6 to 8 weeks until complete clearance has been achieved and ideally a negative potassium hydroxide preparation of skin scrapings has been obtained. A longer treatment period (eg, ≥3 months) frequently is needed, and relapses are common.6,16,18 Regular follow-up is needed to monitor for infection clearance and recurrences. It is important to note that cases of itraconazole resistance have been reported, although this currently appears to be uncommon.19,20
Other Emerging Dermatophytes to Watch
Trichophyton rubrum is the most common cause of dermatophyte infections among humans,21 and cases of terbinafine-resistant T rubrum infections have been reported increasingly in the United States and Canada.5,22-24 Onychomycosis caused by terbinafine-resistant T rubrum has been documented, and patients may have infections that do not respond to terbinafine given at the standard dose and duration.22,23 Case reports have indicated successful treatment using itraconazole 200 mg/d and posaconazole 300 mg/d.5,23
Trichophyton mentagrophytes genotype VII (TMVII) is an emerging dermatophyte that recently has been reported as a cause of sexually transmitted dermatophyte infections in Europe and the United States primarily affecting men who have sex with men.25-27 Patients may present with pruritic, annular, scaly patches and plaques involving the trunk, groin, genital region, or face (Figure 2). Although closely related to T indotineae, TMVII differs in that it more often affects the genital region, generally is susceptible to terbinafine, and in the United States and Europe usually is not related to travel or immigration involving South Asia.26 Although TMVII has not been associated with antifungal resistance, awareness among dermatologists is important because patients may experience inflamed, painful, and persistent rashes that can lead to secondary bacterial infection or scarring, and physicians might mistake it for mimics including eczema or psoriasis.25,26
Importance of Judicious Antifungal Use
Optimizing the use of antifungals is critical to improving patient outcomes and preserving available treatment options.28,29 A retrospective analysis of commercial health insurance data estimated that topical antifungal prescriptions were potentially unnecessary for more than half of the more than 560,000 patients who were prescribed these medications in 2023. In this study, it also was observed that only 16% of patients prescribed a topical antifungal had received diagnostic testing, with low rates across specialties.30 This is concerning because even among board-certified dermatologists, incorrect diagnosis of suspected fungal skin infections can occur; in one survey-based study of board-certified dermatologists who were presented with dermatomycosis images, respondents categorized cases with greater than 75% accuracy in only 31% (4/13) of instances.31 Clotrimazole-betamethasone is among the most commonly prescribed topical antifungals in the United States,14,32 and 2 recent retrospective analyses highlighted that the majority of patients prescribed this medication did not receive any fungal diagnostic testing.33,34
Final Thoughts
In an era of emerging antifungal-resistant dermatophyte infections, it is important for dermatologists to educate nondermatologists about the importance of using diagnostic testing for suspected dermatophyte infections.14,28 Dermatologists also can educate nondermatologist colleagues on the importance of avoiding the use of topical combination antifungal/corticosteroid medications and referring for dermatologic evaluation when diagnoses are uncertain.33,34 Strategies for education by dermatologists could include giving workshops, creating educational materials, and fostering open communication about optimal treatment practices and referral parameters for suspected dermatophyte infections.
- Noble SL, Forbes RC, Stamm PL. Diagnosis and management of common tinea infections. Am Fam Physician. 1998;58:163-174, 177-168.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Uhrlaß S, Verma SB, Gräser Y, et al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: I. epidemiology, risk factors and clinical features. Indian J Dermatol Venereol Leprol. 2021;87:154-175.
- Chen E, Ghannoum M, Elewski BE. Treatment]resistant tinea corporis, a potential public health issue. Br J Dermatol. 2021;184:164-165.
- Caplan AS. Notes from the field: first reported US cases of tinea caused by Trichophyton indotineae—New York City, December 2021–March 2023. MMWR Morbidity and Mortality Weekly Report. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Spivack S, Gold JA, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807.
- Jabet A, Brun S, Normand AC, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233.
- Thakur S, Spruijtenburg B, Abhishek, et al. Whole genome sequence analysis of terbinafine resistant and susceptible Trichophyton isolates from human and animal origin. Mycopathologia. 2025;190:13.
- Lockhart SR, Chowdhary A, Gold JA. The rapid emergence of antifungal-resistant human-pathogenic fungi. Nat Rev Microbiol. 2023;21:818-832.
- Mosam A, Shuping L, Naicker S, et al. A case of antifungal-resistant ringworm infection in KwaZulu-Natal Province, South Africa, caused by Trichophyton indotineae. Public Health Bulletin South Africa. Accessed April 4, 2025. https://www.phbsa.ac.za/wp-content/uploads/2023/12PHBSA-Ringworm-Article-2023.pdf
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:E0056223
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709. doi:10.1001/jamadermatol.2024.1126
- Benedict K. Topical antifungal prescribing for Medicare Part D beneficiaries—United States, 2021. MMWR Morb Mortal Wkly Rep. 2024;73:1-5.
- Verma SB. Emergence of recalcitrant dermatophytosis in India. Lancet Infect Dis. 2018;18:718-719.
- Khurana A, Sharath S, Sardana K, et al. Clinico-mycological and therapeutic updates on cutaneous dermatophytic infections in the era of Trichophyton indotineae. J Am Acad Dermatol. 2024;91:315-323. doi:10.1016/j.jaad.2024.03.024
- Verma S. Steroid modified tinea. BMJ. 2017;356:j973.
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278.
- Burmester A, Hipler UC, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180.
- Bhuiyan MSI, Verma SB, Illigner GM, et al. Trichophyton mentagrophytes ITS genotype VIII/Trichophyton indotineae infection and antifungal resistance in Bangladesh. J Fungi (Basel). 2024;10:768. doi:10.3390 /jof10110768
- Hay RJ. Chapter 82: superficial mycoses. In: Ryan ET, Hill DR, Solomon T, et al, eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 10th ed. Elsevier; 2020:648-652.
- Gupta AK, Cooper EA, Wang T, et al. Detection of squalene epoxidase mutations in United States patients with onychomycosis: implications for management. J Invest Dermatol. 2023;143:2476-2483.E2477.
- Hwang JK, Bakotic WL, Gold JA, et al. Isolation of terbinafine-resistant Trichophyton rubrum from onychomycosis patients who failed treatment at an academic center in New York, United States. J Fungi. 2023;9:710.
- Gu D, Hatch M, Ghannoum M, et al. Treatment-resistant dermatophytosis: a representative case highlighting an emerging public health threat. JAAD Case Rep. 2020;6:1153-1155.
- Jabet A, Dellière S, Seang S, et al. Sexually transmitted Trichophyton mentagrophytes genotype VII infection among men who have sex with men. Emerg Infect Dis. 2023;29:1411-1414.
- Zucker J, Caplan AS, Gunaratne SH, et al. Notes from the field: Trichophyton mentagrophytes genotype VII—New York City, April-July 2024. MMWR Morb Mortal Wkly Rep. 2024;73:985-988.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Caplan AS, Gold JA, Smith DJ, et al. Improving antifungal stewardship in dermatology in an era of emerging dermatophyte resistance. JAAD International. 2024;15:168-169.
- Elewski B. A call for antifungal stewardship. Br J Dermatol. 2020; 183:798-799.
- Gold JAW, Benedict K, Caplan AS, et al. High rates of potentially unnecessary topical antifungal prescribing in a large commercial health insurance claims database, United States. J Am Acad Dermatol. 2025:S0190-9622(25)00098-2. doi:10.1016/j.jaad.2025.01.022
- Yadgar RJ, Bhatia N, Friedman A. Cutaneous fungal infections are commonly misdiagnosed: a survey-based study. J Am Acad Dermatol. 2017;76:562-563.
- Flint ND, Rhoads JLW, Carlisle R, et al. The continued inappropriate use and overuse of combination topical clotrimazole-betamethasone. Dermatol Online J. 2021;27. doi:10.5070/D327854686
- Currie DW, Caplan AS, Benedict K, et al. Prescribing of clotrimazolebetamethasone dipropionate, a topical combination corticosteroidantifungal product, for Medicare part D beneficiaries, United States, 2016–2022. Antimicrob Steward Healthc Epidemiol. 2024;4:E174.
- Gold JA, Caplan AS, Benedict K, et al. Clotrimazole-betamethasone dipropionate prescribing for nonfungal skin conditions. JAMA Network Open. 2024;7:E2411721-E2411721.
- Noble SL, Forbes RC, Stamm PL. Diagnosis and management of common tinea infections. Am Fam Physician. 1998;58:163-174, 177-168.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Uhrlaß S, Verma SB, Gräser Y, et al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: I. epidemiology, risk factors and clinical features. Indian J Dermatol Venereol Leprol. 2021;87:154-175.
- Chen E, Ghannoum M, Elewski BE. Treatment]resistant tinea corporis, a potential public health issue. Br J Dermatol. 2021;184:164-165.
- Caplan AS. Notes from the field: first reported US cases of tinea caused by Trichophyton indotineae—New York City, December 2021–March 2023. MMWR Morbidity and Mortality Weekly Report. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Spivack S, Gold JA, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807.
- Jabet A, Brun S, Normand AC, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233.
- Thakur S, Spruijtenburg B, Abhishek, et al. Whole genome sequence analysis of terbinafine resistant and susceptible Trichophyton isolates from human and animal origin. Mycopathologia. 2025;190:13.
- Lockhart SR, Chowdhary A, Gold JA. The rapid emergence of antifungal-resistant human-pathogenic fungi. Nat Rev Microbiol. 2023;21:818-832.
- Mosam A, Shuping L, Naicker S, et al. A case of antifungal-resistant ringworm infection in KwaZulu-Natal Province, South Africa, caused by Trichophyton indotineae. Public Health Bulletin South Africa. Accessed April 4, 2025. https://www.phbsa.ac.za/wp-content/uploads/2023/12PHBSA-Ringworm-Article-2023.pdf
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:E0056223
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. 2024;160:701-709. doi:10.1001/jamadermatol.2024.1126
- Benedict K. Topical antifungal prescribing for Medicare Part D beneficiaries—United States, 2021. MMWR Morb Mortal Wkly Rep. 2024;73:1-5.
- Verma SB. Emergence of recalcitrant dermatophytosis in India. Lancet Infect Dis. 2018;18:718-719.
- Khurana A, Sharath S, Sardana K, et al. Clinico-mycological and therapeutic updates on cutaneous dermatophytic infections in the era of Trichophyton indotineae. J Am Acad Dermatol. 2024;91:315-323. doi:10.1016/j.jaad.2024.03.024
- Verma S. Steroid modified tinea. BMJ. 2017;356:j973.
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278.
- Burmester A, Hipler UC, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180.
- Bhuiyan MSI, Verma SB, Illigner GM, et al. Trichophyton mentagrophytes ITS genotype VIII/Trichophyton indotineae infection and antifungal resistance in Bangladesh. J Fungi (Basel). 2024;10:768. doi:10.3390 /jof10110768
- Hay RJ. Chapter 82: superficial mycoses. In: Ryan ET, Hill DR, Solomon T, et al, eds. Hunter’s Tropical Medicine and Emerging Infectious Diseases. 10th ed. Elsevier; 2020:648-652.
- Gupta AK, Cooper EA, Wang T, et al. Detection of squalene epoxidase mutations in United States patients with onychomycosis: implications for management. J Invest Dermatol. 2023;143:2476-2483.E2477.
- Hwang JK, Bakotic WL, Gold JA, et al. Isolation of terbinafine-resistant Trichophyton rubrum from onychomycosis patients who failed treatment at an academic center in New York, United States. J Fungi. 2023;9:710.
- Gu D, Hatch M, Ghannoum M, et al. Treatment-resistant dermatophytosis: a representative case highlighting an emerging public health threat. JAAD Case Rep. 2020;6:1153-1155.
- Jabet A, Dellière S, Seang S, et al. Sexually transmitted Trichophyton mentagrophytes genotype VII infection among men who have sex with men. Emerg Infect Dis. 2023;29:1411-1414.
- Zucker J, Caplan AS, Gunaratne SH, et al. Notes from the field: Trichophyton mentagrophytes genotype VII—New York City, April-July 2024. MMWR Morb Mortal Wkly Rep. 2024;73:985-988.
- Jabet A, Bérot V, Chiarabini T, et al. Trichophyton mentagrophytes ITS genotype VII infections among men who have sex with men in France: an ongoing phenomenon. J Eur Acad Dermatol Venereol. 2025;39:407-415.
- Caplan AS, Gold JA, Smith DJ, et al. Improving antifungal stewardship in dermatology in an era of emerging dermatophyte resistance. JAAD International. 2024;15:168-169.
- Elewski B. A call for antifungal stewardship. Br J Dermatol. 2020; 183:798-799.
- Gold JAW, Benedict K, Caplan AS, et al. High rates of potentially unnecessary topical antifungal prescribing in a large commercial health insurance claims database, United States. J Am Acad Dermatol. 2025:S0190-9622(25)00098-2. doi:10.1016/j.jaad.2025.01.022
- Yadgar RJ, Bhatia N, Friedman A. Cutaneous fungal infections are commonly misdiagnosed: a survey-based study. J Am Acad Dermatol. 2017;76:562-563.
- Flint ND, Rhoads JLW, Carlisle R, et al. The continued inappropriate use and overuse of combination topical clotrimazole-betamethasone. Dermatol Online J. 2021;27. doi:10.5070/D327854686
- Currie DW, Caplan AS, Benedict K, et al. Prescribing of clotrimazolebetamethasone dipropionate, a topical combination corticosteroidantifungal product, for Medicare part D beneficiaries, United States, 2016–2022. Antimicrob Steward Healthc Epidemiol. 2024;4:E174.
- Gold JA, Caplan AS, Benedict K, et al. Clotrimazole-betamethasone dipropionate prescribing for nonfungal skin conditions. JAMA Network Open. 2024;7:E2411721-E2411721.
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
The Rise of Antifungal-Resistant Dermatophyte Infections: What Dermatologists Need to Know
PRACTICE POINTS
- Recently emerged dermatophyte species pose a global public health concern because of infection severity, frequent resistance to terbinafine, and easy person-to-person transmission.
- Prolonged itraconazole therapy is considered the firstline treatment for infections caused by Trichophyton indotineae, a globally emerging and frequently terbinafine-resistant dermatophyte.
- Dermatologists can educate nondermatologists on the importance of mycologic confirmation and avoidance of the use of topical antifungal/ corticosteroid products, which are hypothesized to contribute to emergence and spread of resistance.

Training Lifeguards to Assist in Skin Cancer Prevention
Training Lifeguards to Assist in Skin Cancer Prevention
Lifeguards play a crucial role in ensuring water safety, but they also are uniquely positioned to promote skin cancer prevention and proper sunscreen use.1,2 There are several benefits and challenges to offering skin cancer prevention training for lifeguards.3 We examine the advantages of training, highlight the role lifeguards can play in larger public skin cancer prevention efforts, and address practical techniques for developing lifeguardfocused skin cancer education programs. By providing this knowledge to lifeguards, we can improve community health outcomes and encourage sun-safe behaviors in high-risk outdoor locations.
Benefits of Skin Cancer Prevention Training for Lifeguards
Research has shown that lifeguards are at an elevated risk for basal cell carcinoma, squamous cell carcinoma, and melanoma due to frequent prolonged occupational sun exposure.1,2,4-6 Therefore, comprehensive education on skin cancer prevention—including instruction on proper sunscreen application techniques and the importance of regular reapplication as well as how to recognize suspicious skin lesions—should be incorporated into lifeguard certification programs. One study evaluating the effectiveness of a skin cancer prevention program for lifeguards found that many of the participants lacked a thorough understanding of the different types of skin cancer.5 Another study found that lifeguards at pools in areas where societal norms supporting sun safety are stronger exhibited noticeably more sun protection practices, with regression estimates of 0.22 (95% CI, 0.17-0.26).7 Empowering lifeguards with valuable health knowledge during their regular training could potentially reduce their risk for skin cancer,4 as they may be more inclined to use sunscreen appropriately and reach out to a dermatologist for regular skin checks and evaluation of suspicious lesions.
Role of Lifeguards in Public Skin Cancer Prevention Efforts
Once trained on skin cancer prevention, lifeguards also can play a pivotal role in promoting sunscreen use among the public. Despite the widespread availability of high-quality sunscreens, many swimmers and beachgoers neglect to regularly apply or reapply sunscreen, especially on commonly exposed areas such as the back, shoulders, and face.8 Educating lifeguards on skin cancer prevention could enhance health outcomes by increasing early detection rates and promoting sun-safe behaviors among the general public.9 However, additional training requirements might increase the cost and time commitment for lifeguard certification, potentially leading to staffing shortages.3,7 There also is a risk of lifeguards overstepping their role and providing inaccurate medical advice, which could cause distress or even lead to liability issues.7 Balancing these factors will be crucial in developing effective and sustainable skin cancer prevention programs for lifeguards.
Implementing Lifeguard Skin Cancer Training
Implementing skin cancer prevention training programs for lifeguards requires strategic collaboration between dermatologists, and lifeguard training organizations to ensure that the participants receive consistent and comprehensive training.10 Additionally, public health campaigns can support these efforts by raising awareness about the importance of sun safety and regular skin checks.6 Tailored training modules/materials, ongoing technical assistance, and active, multicomponent approaches that account for both individual and environmental factors can increase program implementation in a variety of community settings.
Final Thoughts
Through effective education, lifeguards can potentially have a substantial impact on skin cancer prevention, both among lifeguards themselves and the general public. By promoting proper sunscreen use, lifeguards can help reduce the incidence and mortality associated with skin cancers. Future studies should focus on developing and implementing targeted education initiatives for lifeguards, fostering collaboration between relevant stakeholders, and raising public awareness about the importance of sun safety and early skin cancer detection. These efforts ultimately could lead to improved public health outcomes and reduced skin cancer rates, particularly in high-risk populations that frequently are exposed to UV radiation.
- Enos CW, Rey S, Slocum J, et al. Sun-protection behaviors among active members of the United States Lifesaving Association. J Clin Aesthet Dermatol. 2021;14:14-20.
- Verma K, Lewis DJ, Siddiqui FS, et al. Mohs micrographic surgery management of melanoma and melanoma in situ. StatPearls. Updated August 28, 2024. Accessed April 15, 2025. https://www.ncbi.nlm.nih.gov/books/NBK606123/
- Verma KK, Joshi TP, Lewis DJ, et al. Nail technicians as partners in early melanoma detection: bridging the knowledge gap. Arch Dermatol Res. 2024;316:586. doi:10.1007/s00403-024-03342-0
- Geller AC, Glanz K, Shigaki D, et al. Impact of skin cancer prevention on outdoor aquatics staff: the Pool Cool program in Hawaii and Massachusetts. Prev Med. 2001;33:155-161. doi:10.1006/pmed.2001.0870
- Hiemstra M, Glanz K, Nehl E. Changes in sunburn and tanning attitudes among lifeguards over a summer season. J Am Acad Dermatol. 2012;66:430-437. doi:10.1016/j.jaad.2010.11.050
- Verma KK, Ahmad N, Friedmann DP, et al. Melanoma in tattooed skin: diagnostic challenges and the potential for tattoo artists in early detection. Arch Dermatol Res. 2024;316:690. doi:10.1007/s00403-024-03415-0
- Hall DM, McCarty F, Elliott T, et al. Lifeguards’ sun protection habits and sunburns: association with sun-safe environments and skin cancer prevention program participation. Arch Dermatol. 2009;145:139-144. doi:10.1001/archdermatol.2008.553
- Emmons KM, Geller AC, Puleo E, et al. Skin cancer education and early detection at the beach: a randomized trial of dermatologist examination and biometric feedback. J Am Acad Dermatol. 2011;64:282-289. doi:10.1016/j.jaad.2010.01.040
- Rabin BA, Nehl E, Elliott T, et al. Individual and setting level predictors of the implementation of a skin cancer prevention program: a multilevel analysis. Implement Sci. 2010;5:40. doi:10.1186/1748-5908-5-40
- Walkosz BJ, Buller D, Buller M, et al. Sun safe workplaces: effect of an occupational skin cancer prevention program on employee sun safety practices. J Occup Environ Med. 2018;60:900-997. doi:10.1097 /JOM.0000000000001427
Lifeguards play a crucial role in ensuring water safety, but they also are uniquely positioned to promote skin cancer prevention and proper sunscreen use.1,2 There are several benefits and challenges to offering skin cancer prevention training for lifeguards.3 We examine the advantages of training, highlight the role lifeguards can play in larger public skin cancer prevention efforts, and address practical techniques for developing lifeguardfocused skin cancer education programs. By providing this knowledge to lifeguards, we can improve community health outcomes and encourage sun-safe behaviors in high-risk outdoor locations.
Benefits of Skin Cancer Prevention Training for Lifeguards
Research has shown that lifeguards are at an elevated risk for basal cell carcinoma, squamous cell carcinoma, and melanoma due to frequent prolonged occupational sun exposure.1,2,4-6 Therefore, comprehensive education on skin cancer prevention—including instruction on proper sunscreen application techniques and the importance of regular reapplication as well as how to recognize suspicious skin lesions—should be incorporated into lifeguard certification programs. One study evaluating the effectiveness of a skin cancer prevention program for lifeguards found that many of the participants lacked a thorough understanding of the different types of skin cancer.5 Another study found that lifeguards at pools in areas where societal norms supporting sun safety are stronger exhibited noticeably more sun protection practices, with regression estimates of 0.22 (95% CI, 0.17-0.26).7 Empowering lifeguards with valuable health knowledge during their regular training could potentially reduce their risk for skin cancer,4 as they may be more inclined to use sunscreen appropriately and reach out to a dermatologist for regular skin checks and evaluation of suspicious lesions.
Role of Lifeguards in Public Skin Cancer Prevention Efforts
Once trained on skin cancer prevention, lifeguards also can play a pivotal role in promoting sunscreen use among the public. Despite the widespread availability of high-quality sunscreens, many swimmers and beachgoers neglect to regularly apply or reapply sunscreen, especially on commonly exposed areas such as the back, shoulders, and face.8 Educating lifeguards on skin cancer prevention could enhance health outcomes by increasing early detection rates and promoting sun-safe behaviors among the general public.9 However, additional training requirements might increase the cost and time commitment for lifeguard certification, potentially leading to staffing shortages.3,7 There also is a risk of lifeguards overstepping their role and providing inaccurate medical advice, which could cause distress or even lead to liability issues.7 Balancing these factors will be crucial in developing effective and sustainable skin cancer prevention programs for lifeguards.
Implementing Lifeguard Skin Cancer Training
Implementing skin cancer prevention training programs for lifeguards requires strategic collaboration between dermatologists, and lifeguard training organizations to ensure that the participants receive consistent and comprehensive training.10 Additionally, public health campaigns can support these efforts by raising awareness about the importance of sun safety and regular skin checks.6 Tailored training modules/materials, ongoing technical assistance, and active, multicomponent approaches that account for both individual and environmental factors can increase program implementation in a variety of community settings.
Final Thoughts
Through effective education, lifeguards can potentially have a substantial impact on skin cancer prevention, both among lifeguards themselves and the general public. By promoting proper sunscreen use, lifeguards can help reduce the incidence and mortality associated with skin cancers. Future studies should focus on developing and implementing targeted education initiatives for lifeguards, fostering collaboration between relevant stakeholders, and raising public awareness about the importance of sun safety and early skin cancer detection. These efforts ultimately could lead to improved public health outcomes and reduced skin cancer rates, particularly in high-risk populations that frequently are exposed to UV radiation.
Lifeguards play a crucial role in ensuring water safety, but they also are uniquely positioned to promote skin cancer prevention and proper sunscreen use.1,2 There are several benefits and challenges to offering skin cancer prevention training for lifeguards.3 We examine the advantages of training, highlight the role lifeguards can play in larger public skin cancer prevention efforts, and address practical techniques for developing lifeguardfocused skin cancer education programs. By providing this knowledge to lifeguards, we can improve community health outcomes and encourage sun-safe behaviors in high-risk outdoor locations.
Benefits of Skin Cancer Prevention Training for Lifeguards
Research has shown that lifeguards are at an elevated risk for basal cell carcinoma, squamous cell carcinoma, and melanoma due to frequent prolonged occupational sun exposure.1,2,4-6 Therefore, comprehensive education on skin cancer prevention—including instruction on proper sunscreen application techniques and the importance of regular reapplication as well as how to recognize suspicious skin lesions—should be incorporated into lifeguard certification programs. One study evaluating the effectiveness of a skin cancer prevention program for lifeguards found that many of the participants lacked a thorough understanding of the different types of skin cancer.5 Another study found that lifeguards at pools in areas where societal norms supporting sun safety are stronger exhibited noticeably more sun protection practices, with regression estimates of 0.22 (95% CI, 0.17-0.26).7 Empowering lifeguards with valuable health knowledge during their regular training could potentially reduce their risk for skin cancer,4 as they may be more inclined to use sunscreen appropriately and reach out to a dermatologist for regular skin checks and evaluation of suspicious lesions.
Role of Lifeguards in Public Skin Cancer Prevention Efforts
Once trained on skin cancer prevention, lifeguards also can play a pivotal role in promoting sunscreen use among the public. Despite the widespread availability of high-quality sunscreens, many swimmers and beachgoers neglect to regularly apply or reapply sunscreen, especially on commonly exposed areas such as the back, shoulders, and face.8 Educating lifeguards on skin cancer prevention could enhance health outcomes by increasing early detection rates and promoting sun-safe behaviors among the general public.9 However, additional training requirements might increase the cost and time commitment for lifeguard certification, potentially leading to staffing shortages.3,7 There also is a risk of lifeguards overstepping their role and providing inaccurate medical advice, which could cause distress or even lead to liability issues.7 Balancing these factors will be crucial in developing effective and sustainable skin cancer prevention programs for lifeguards.
Implementing Lifeguard Skin Cancer Training
Implementing skin cancer prevention training programs for lifeguards requires strategic collaboration between dermatologists, and lifeguard training organizations to ensure that the participants receive consistent and comprehensive training.10 Additionally, public health campaigns can support these efforts by raising awareness about the importance of sun safety and regular skin checks.6 Tailored training modules/materials, ongoing technical assistance, and active, multicomponent approaches that account for both individual and environmental factors can increase program implementation in a variety of community settings.
Final Thoughts
Through effective education, lifeguards can potentially have a substantial impact on skin cancer prevention, both among lifeguards themselves and the general public. By promoting proper sunscreen use, lifeguards can help reduce the incidence and mortality associated with skin cancers. Future studies should focus on developing and implementing targeted education initiatives for lifeguards, fostering collaboration between relevant stakeholders, and raising public awareness about the importance of sun safety and early skin cancer detection. These efforts ultimately could lead to improved public health outcomes and reduced skin cancer rates, particularly in high-risk populations that frequently are exposed to UV radiation.
- Enos CW, Rey S, Slocum J, et al. Sun-protection behaviors among active members of the United States Lifesaving Association. J Clin Aesthet Dermatol. 2021;14:14-20.
- Verma K, Lewis DJ, Siddiqui FS, et al. Mohs micrographic surgery management of melanoma and melanoma in situ. StatPearls. Updated August 28, 2024. Accessed April 15, 2025. https://www.ncbi.nlm.nih.gov/books/NBK606123/
- Verma KK, Joshi TP, Lewis DJ, et al. Nail technicians as partners in early melanoma detection: bridging the knowledge gap. Arch Dermatol Res. 2024;316:586. doi:10.1007/s00403-024-03342-0
- Geller AC, Glanz K, Shigaki D, et al. Impact of skin cancer prevention on outdoor aquatics staff: the Pool Cool program in Hawaii and Massachusetts. Prev Med. 2001;33:155-161. doi:10.1006/pmed.2001.0870
- Hiemstra M, Glanz K, Nehl E. Changes in sunburn and tanning attitudes among lifeguards over a summer season. J Am Acad Dermatol. 2012;66:430-437. doi:10.1016/j.jaad.2010.11.050
- Verma KK, Ahmad N, Friedmann DP, et al. Melanoma in tattooed skin: diagnostic challenges and the potential for tattoo artists in early detection. Arch Dermatol Res. 2024;316:690. doi:10.1007/s00403-024-03415-0
- Hall DM, McCarty F, Elliott T, et al. Lifeguards’ sun protection habits and sunburns: association with sun-safe environments and skin cancer prevention program participation. Arch Dermatol. 2009;145:139-144. doi:10.1001/archdermatol.2008.553
- Emmons KM, Geller AC, Puleo E, et al. Skin cancer education and early detection at the beach: a randomized trial of dermatologist examination and biometric feedback. J Am Acad Dermatol. 2011;64:282-289. doi:10.1016/j.jaad.2010.01.040
- Rabin BA, Nehl E, Elliott T, et al. Individual and setting level predictors of the implementation of a skin cancer prevention program: a multilevel analysis. Implement Sci. 2010;5:40. doi:10.1186/1748-5908-5-40
- Walkosz BJ, Buller D, Buller M, et al. Sun safe workplaces: effect of an occupational skin cancer prevention program on employee sun safety practices. J Occup Environ Med. 2018;60:900-997. doi:10.1097 /JOM.0000000000001427
- Enos CW, Rey S, Slocum J, et al. Sun-protection behaviors among active members of the United States Lifesaving Association. J Clin Aesthet Dermatol. 2021;14:14-20.
- Verma K, Lewis DJ, Siddiqui FS, et al. Mohs micrographic surgery management of melanoma and melanoma in situ. StatPearls. Updated August 28, 2024. Accessed April 15, 2025. https://www.ncbi.nlm.nih.gov/books/NBK606123/
- Verma KK, Joshi TP, Lewis DJ, et al. Nail technicians as partners in early melanoma detection: bridging the knowledge gap. Arch Dermatol Res. 2024;316:586. doi:10.1007/s00403-024-03342-0
- Geller AC, Glanz K, Shigaki D, et al. Impact of skin cancer prevention on outdoor aquatics staff: the Pool Cool program in Hawaii and Massachusetts. Prev Med. 2001;33:155-161. doi:10.1006/pmed.2001.0870
- Hiemstra M, Glanz K, Nehl E. Changes in sunburn and tanning attitudes among lifeguards over a summer season. J Am Acad Dermatol. 2012;66:430-437. doi:10.1016/j.jaad.2010.11.050
- Verma KK, Ahmad N, Friedmann DP, et al. Melanoma in tattooed skin: diagnostic challenges and the potential for tattoo artists in early detection. Arch Dermatol Res. 2024;316:690. doi:10.1007/s00403-024-03415-0
- Hall DM, McCarty F, Elliott T, et al. Lifeguards’ sun protection habits and sunburns: association with sun-safe environments and skin cancer prevention program participation. Arch Dermatol. 2009;145:139-144. doi:10.1001/archdermatol.2008.553
- Emmons KM, Geller AC, Puleo E, et al. Skin cancer education and early detection at the beach: a randomized trial of dermatologist examination and biometric feedback. J Am Acad Dermatol. 2011;64:282-289. doi:10.1016/j.jaad.2010.01.040
- Rabin BA, Nehl E, Elliott T, et al. Individual and setting level predictors of the implementation of a skin cancer prevention program: a multilevel analysis. Implement Sci. 2010;5:40. doi:10.1186/1748-5908-5-40
- Walkosz BJ, Buller D, Buller M, et al. Sun safe workplaces: effect of an occupational skin cancer prevention program on employee sun safety practices. J Occup Environ Med. 2018;60:900-997. doi:10.1097 /JOM.0000000000001427
Training Lifeguards to Assist in Skin Cancer Prevention
Training Lifeguards to Assist in Skin Cancer Prevention

Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
To the Editor:
The incidence of nonmelanoma skin cancer (NMSC) is rapidly increasing worldwide. Due to its highly curable nature when treated early, accurate diagnosis is the cornerstone to good patient outcomes.1 Accurate diagnosis of skin cancer and subsequent treatment decisions rely heavily on the congruence between clinical observations and histopathologic assessments. Clinical misdiagnosis of a malignant lesion can lead to delayed and suboptimal treatment, which may contribute to serious complications such as metastasis or even mortality. In this study, data from clinically diagnosed basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) were compared to their identified histopathologic subtype classifications. The accuracy of the clinical diagnosis of these NMSCs was assessed by determining the rate of misdiagnosis and the respective positive predictive value (PPV).
A retrospective review of medical records from a private dermatology practice in Lubbock, Texas, was conducted to identify patients diagnosed with NMSC from January 1, 2017, through December 31, 2021. A total of 11,229 NMSCs were diagnosed and treated in 5877 patients. Of the NMSCs diagnosed, 11,145 were identified as keratinocyte carcinomas and were classified as BCCs or SCCs. The accuracy of the clinical diagnoses was determined by comparison to the histologic subtype identified via biopsy of the lesion. Although the use of a dermatoscope during the clinical encounter was not formally recorded, reports from the examining dermatologists indicated it was not used in the majority of cases.
If a lesion was clinically diagnosed as a BCC but was identified as a subtype of SCC on histology (or vice versa), the lesion was considered to be mismatched. The number of mismatched lesions and the mismatch rate for each lesion type/subtype is recorded in the Table. Of the total 11,145 keratinocyte carcinomas included in our study, there was an overall 10.63% mismatch rate, with 1185 of the malignancies having a differing clinical diagnosis (eg, BCC vs SCC) from the histologic findings. The clinical mismatch rate was notably higher for SCC compared to BCC (15.83% vs 7.03%, respectively).
The Table provides a breakdown of the BCC subtypes identified by histology with their computed mismatch rate and PPV. It is worth clarifying that lesions classified as more than one BCC subtype per the histologic findings were diagnosed as mixed BCC; these were further classified as mixed-aggressive BCC (if at least one aggressive BCC subtype was present) and mixed nonaggressive BCC (if no aggressive BCC subtype was present). Overall, BCCs were less likely to be misdiagnosed, with an average PPV of 92.97% compared to 84.17% for SCCs. Basosquamous BCC was the BCC subtype with the highest mismatch rate (25.48%), while sclerosing BCC has the lowest overall mismatch rate (1.33%). The most common malignancy was BCC, with nodular BCC being the most common subtype.
The Table also breaks down the SCC subtypes, reporting the most commonly misdiagnosed of any BCC or SCC subtype to be poorly differentiated SCC (mismatch rate, 38.46%). The lowest mismatch rate of the SCC subtypes was 5.97% for well-differentiated SCC.
There was an overall PPV of 89.37% in clinically evaluated malignancies and their respective histologic subtypes. Basal cell carcinoma had a lower overall mismatch rate of 7.03% compared to 15.83% in SCC. The most common misdiagnosis was attributed to poorly differentiated SCC (mismatch rate, 38.46%), while the least common misdiagnosed malignancy was sclerosing BCC (1.33%). The high mismatch rate of poorly differentiated SCC may be due to its diverging presentation from a typical SCC as a flat lesion with the absence of scaling, keratin, or bleeding, leading to the misdiagnosis of BCC.2
Accurate clinical diagnosis of NMSCs is the basis for further evaluation and treatment that should ensue in a timely manner; however, accurately identifying BCCs vs SCCs solely based on clinical examination can be challenging due to variable manifestations and overlapping features. Basal cell carcinoma commonly presents as a shiny pink/flesh-colored nodule, macule, or patch with surface telangiectasia, sometimes appearing with ulceration or crusting.3 Alternatively, SCC typically appears as a firm, sharply demarcated, red nodule with a thick overlying scale.4 Definitive diagnoses can be difficult upon clinical examination since these features can be shared between the 2 subtypes. To aid in these uncertainties, a growing number of clinicians are implementing the use of dermoscopy in their everyday practice.
Dermoscopy is an extremely useful tool in improving the diagnostic accuracy of skin cancers compared to examination with the naked eye, as it provides detailed visualization of specific structures and patterns in skin cancer lesions.5 The dermoscopic appearance of BCC is characterized by pearly blue-gray or translucent globules with arborizing vessels, spoke-wheel structures, and leaflike areas.5,6 Conversely, dermoscopic features of SCC may include a milky-red globule with a scaly, sharply demarcated, crusted lesion with polymorphous vasculature, sometimes resembling a persistent sore or nonhealing wound.4,5 Though the use of dermoscopy can aid in diagnosis upon initial examination, certain factors such as trauma, ulceration, and previous treatments that distorted the lesion’s architecture may lead to misdiagnosis. Furthermore, the distinct vascular patterns found in BCC and SCC may be mistaken for each other and therefore lead to misdiagnosis upon examination.7 Other variables that may complicate diagnosis include the location of the lesion, its size, and the presence of other skin conditions or nearby lesions.
The primary limitation of the current study was the limited scope of the data, as they were derived from patients seen at one private dermatology practice, preventing the generalizability of our findings. However, our results show trends similar to those observed in other studies analyzing the clinical accuracy of skin cancer diagnoses, with higher PPVs for BCC compared to SCC. A study by Ahnlide and Bjellerup8 was based in a hospital dermatology department and demonstrated a PPV of 85.5% for BCC compared to 92.97% in our study; for SCC, the PPV was 67.3% compared to 84.17% in our study. In another study by Heal et al,9 data were collected from an Australian registry that included records of all histologically confirmed skin cancers from December 1996 to October 1999 from 202 general practitioners and 42 specialists, including 1 dermatologist. The PPVs for BCC and SCC were 72.7% and 49.4%, respectively. Although our results indicated higher PPVs compared to these 2 studies, some of the discrepancies can be accounted for by the differences in clinical setting as well as the lack of expertise of nondermatologist physicians in identifying skin malignancies in the study by Heal et al.9
The current study was further limited by the lack of data quantifying the number of lesions clinically suspected to be malignant but found to be histologically benign. It is typical for clinicians to have a low threshold to biopsy a suspicious lesion with atypical features (eg, rapid evolution and growth, bleeding, crusting). Furthermore, the identification of risk factors in the patient’s medical and family history (eg, exposure to radiation, personal or family history of skin cancers) can heavily influence a clinician’s decision to biopsy a lesion with an atypical appearance.10 Many benign lesions are biopsied to avoid missing a diagnosis of malignancy. Consequently, our results suggest a high degree of clinical misdiagnosis of BCCs and SCCs. Obtaining data on the number of lesions suspected to be BCC or SCC that were found to be histologically benign would be a valuable addition to our study, as it would provide a measurable insight into the sensitivity of clinicians’ decision-making to identify a lesion as suspicious and warranting biopsy.
While clinical diagnosis plays a vital role in identifying suspected NMSCs such as BCC and SCC, its accuracy can be limited even with the use of dermoscopy. Overall, our data have shown a high rate of diagnostic accuracy upon suspicion of malignancy, but the different variables that affect clinical presentation promote histologic diagnosis to prevail as the gold standard.
- Seyed Ahadi M, Firooz A, Rahimi H, et al. Clinical diagnosis has a high negative predictive value in evaluation of malignant skin lesions. Dermatol Res Pract. 2021;2021:6618990. doi:10.1155/2021/6618990
- Lallas A, Pyne J, Kyrgidis A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015;172:1308- 1315. doi:10.1111/bjd.13510
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. September 19, 2022. In: StatPearls. StatPearls Publishing; 2023.
- Suárez AL, Louis P, Kitts J, et al. Clinical and dermoscopic features of combined cutaneous squamous cell carcinoma (SCC)/neuroendocrine [Merkel cell] carcinoma (MCC). J Am Acad Dermatol. 2015;73:968-975. doi:10.1016/j.jaad.2015.08.041
- Wolner ZJ, Yélamos O, Liopyris K, et al. Enhancing skin cancer diagnosis with dermoscopy. Dermatol Clin. 2017;35:417-437. doi:10.1016/j.det.2017.06.003
- Reiter O, Mimouni I, Dusza S, et al. Dermoscopic features of basal cell carcinoma and its subtypes: a systematic review. J Am Acad Dermatol. 2021;85:653-664. doi:10.1016/j.jaad.2019.11.008
- Pruneda C, Ramesh M, Hope L, et al. Nonmelanoma skin cancers: diagnostic accuracy of midlevel providers versus dermatologists. The Dermatologist. March 2023. Accessed March 18, 2025. https://www.hmpgloballearningnetwork.com/site/thederm/feature-story/nonmelanoma-skin-cancers-diagnostic-accuracy-midlevel-providers-vs
- Ahnlide I, Bjellerup M. Accuracy of clinical skin tumour diagnosis in a dermatological setting. Acta Derm Venereol. 2013;93:305-308. doi:10.2340/00015555-1560
- Heal CF, Raasch BA, Buettner PG, et al. Accuracy of clinical diagnosis of skin lesions. Br J Dermatol. 2008;159:661-668.
- Fu S, Kim S, Wasko C. Dermatological guide for primary care physicians: full body skin checks, skin cancer detection, and patient education on self-skin checks and sun protection. Proc (Bayl Univ Med Cent). 2024;37:647-654. doi:10.1080/08998280.2024.2351751
To the Editor:
The incidence of nonmelanoma skin cancer (NMSC) is rapidly increasing worldwide. Due to its highly curable nature when treated early, accurate diagnosis is the cornerstone to good patient outcomes.1 Accurate diagnosis of skin cancer and subsequent treatment decisions rely heavily on the congruence between clinical observations and histopathologic assessments. Clinical misdiagnosis of a malignant lesion can lead to delayed and suboptimal treatment, which may contribute to serious complications such as metastasis or even mortality. In this study, data from clinically diagnosed basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) were compared to their identified histopathologic subtype classifications. The accuracy of the clinical diagnosis of these NMSCs was assessed by determining the rate of misdiagnosis and the respective positive predictive value (PPV).
A retrospective review of medical records from a private dermatology practice in Lubbock, Texas, was conducted to identify patients diagnosed with NMSC from January 1, 2017, through December 31, 2021. A total of 11,229 NMSCs were diagnosed and treated in 5877 patients. Of the NMSCs diagnosed, 11,145 were identified as keratinocyte carcinomas and were classified as BCCs or SCCs. The accuracy of the clinical diagnoses was determined by comparison to the histologic subtype identified via biopsy of the lesion. Although the use of a dermatoscope during the clinical encounter was not formally recorded, reports from the examining dermatologists indicated it was not used in the majority of cases.
If a lesion was clinically diagnosed as a BCC but was identified as a subtype of SCC on histology (or vice versa), the lesion was considered to be mismatched. The number of mismatched lesions and the mismatch rate for each lesion type/subtype is recorded in the Table. Of the total 11,145 keratinocyte carcinomas included in our study, there was an overall 10.63% mismatch rate, with 1185 of the malignancies having a differing clinical diagnosis (eg, BCC vs SCC) from the histologic findings. The clinical mismatch rate was notably higher for SCC compared to BCC (15.83% vs 7.03%, respectively).
The Table provides a breakdown of the BCC subtypes identified by histology with their computed mismatch rate and PPV. It is worth clarifying that lesions classified as more than one BCC subtype per the histologic findings were diagnosed as mixed BCC; these were further classified as mixed-aggressive BCC (if at least one aggressive BCC subtype was present) and mixed nonaggressive BCC (if no aggressive BCC subtype was present). Overall, BCCs were less likely to be misdiagnosed, with an average PPV of 92.97% compared to 84.17% for SCCs. Basosquamous BCC was the BCC subtype with the highest mismatch rate (25.48%), while sclerosing BCC has the lowest overall mismatch rate (1.33%). The most common malignancy was BCC, with nodular BCC being the most common subtype.
The Table also breaks down the SCC subtypes, reporting the most commonly misdiagnosed of any BCC or SCC subtype to be poorly differentiated SCC (mismatch rate, 38.46%). The lowest mismatch rate of the SCC subtypes was 5.97% for well-differentiated SCC.
There was an overall PPV of 89.37% in clinically evaluated malignancies and their respective histologic subtypes. Basal cell carcinoma had a lower overall mismatch rate of 7.03% compared to 15.83% in SCC. The most common misdiagnosis was attributed to poorly differentiated SCC (mismatch rate, 38.46%), while the least common misdiagnosed malignancy was sclerosing BCC (1.33%). The high mismatch rate of poorly differentiated SCC may be due to its diverging presentation from a typical SCC as a flat lesion with the absence of scaling, keratin, or bleeding, leading to the misdiagnosis of BCC.2
Accurate clinical diagnosis of NMSCs is the basis for further evaluation and treatment that should ensue in a timely manner; however, accurately identifying BCCs vs SCCs solely based on clinical examination can be challenging due to variable manifestations and overlapping features. Basal cell carcinoma commonly presents as a shiny pink/flesh-colored nodule, macule, or patch with surface telangiectasia, sometimes appearing with ulceration or crusting.3 Alternatively, SCC typically appears as a firm, sharply demarcated, red nodule with a thick overlying scale.4 Definitive diagnoses can be difficult upon clinical examination since these features can be shared between the 2 subtypes. To aid in these uncertainties, a growing number of clinicians are implementing the use of dermoscopy in their everyday practice.
Dermoscopy is an extremely useful tool in improving the diagnostic accuracy of skin cancers compared to examination with the naked eye, as it provides detailed visualization of specific structures and patterns in skin cancer lesions.5 The dermoscopic appearance of BCC is characterized by pearly blue-gray or translucent globules with arborizing vessels, spoke-wheel structures, and leaflike areas.5,6 Conversely, dermoscopic features of SCC may include a milky-red globule with a scaly, sharply demarcated, crusted lesion with polymorphous vasculature, sometimes resembling a persistent sore or nonhealing wound.4,5 Though the use of dermoscopy can aid in diagnosis upon initial examination, certain factors such as trauma, ulceration, and previous treatments that distorted the lesion’s architecture may lead to misdiagnosis. Furthermore, the distinct vascular patterns found in BCC and SCC may be mistaken for each other and therefore lead to misdiagnosis upon examination.7 Other variables that may complicate diagnosis include the location of the lesion, its size, and the presence of other skin conditions or nearby lesions.
The primary limitation of the current study was the limited scope of the data, as they were derived from patients seen at one private dermatology practice, preventing the generalizability of our findings. However, our results show trends similar to those observed in other studies analyzing the clinical accuracy of skin cancer diagnoses, with higher PPVs for BCC compared to SCC. A study by Ahnlide and Bjellerup8 was based in a hospital dermatology department and demonstrated a PPV of 85.5% for BCC compared to 92.97% in our study; for SCC, the PPV was 67.3% compared to 84.17% in our study. In another study by Heal et al,9 data were collected from an Australian registry that included records of all histologically confirmed skin cancers from December 1996 to October 1999 from 202 general practitioners and 42 specialists, including 1 dermatologist. The PPVs for BCC and SCC were 72.7% and 49.4%, respectively. Although our results indicated higher PPVs compared to these 2 studies, some of the discrepancies can be accounted for by the differences in clinical setting as well as the lack of expertise of nondermatologist physicians in identifying skin malignancies in the study by Heal et al.9
The current study was further limited by the lack of data quantifying the number of lesions clinically suspected to be malignant but found to be histologically benign. It is typical for clinicians to have a low threshold to biopsy a suspicious lesion with atypical features (eg, rapid evolution and growth, bleeding, crusting). Furthermore, the identification of risk factors in the patient’s medical and family history (eg, exposure to radiation, personal or family history of skin cancers) can heavily influence a clinician’s decision to biopsy a lesion with an atypical appearance.10 Many benign lesions are biopsied to avoid missing a diagnosis of malignancy. Consequently, our results suggest a high degree of clinical misdiagnosis of BCCs and SCCs. Obtaining data on the number of lesions suspected to be BCC or SCC that were found to be histologically benign would be a valuable addition to our study, as it would provide a measurable insight into the sensitivity of clinicians’ decision-making to identify a lesion as suspicious and warranting biopsy.
While clinical diagnosis plays a vital role in identifying suspected NMSCs such as BCC and SCC, its accuracy can be limited even with the use of dermoscopy. Overall, our data have shown a high rate of diagnostic accuracy upon suspicion of malignancy, but the different variables that affect clinical presentation promote histologic diagnosis to prevail as the gold standard.
To the Editor:
The incidence of nonmelanoma skin cancer (NMSC) is rapidly increasing worldwide. Due to its highly curable nature when treated early, accurate diagnosis is the cornerstone to good patient outcomes.1 Accurate diagnosis of skin cancer and subsequent treatment decisions rely heavily on the congruence between clinical observations and histopathologic assessments. Clinical misdiagnosis of a malignant lesion can lead to delayed and suboptimal treatment, which may contribute to serious complications such as metastasis or even mortality. In this study, data from clinically diagnosed basal cell carcinomas (BCCs) and squamous cell carcinomas (SCCs) were compared to their identified histopathologic subtype classifications. The accuracy of the clinical diagnosis of these NMSCs was assessed by determining the rate of misdiagnosis and the respective positive predictive value (PPV).
A retrospective review of medical records from a private dermatology practice in Lubbock, Texas, was conducted to identify patients diagnosed with NMSC from January 1, 2017, through December 31, 2021. A total of 11,229 NMSCs were diagnosed and treated in 5877 patients. Of the NMSCs diagnosed, 11,145 were identified as keratinocyte carcinomas and were classified as BCCs or SCCs. The accuracy of the clinical diagnoses was determined by comparison to the histologic subtype identified via biopsy of the lesion. Although the use of a dermatoscope during the clinical encounter was not formally recorded, reports from the examining dermatologists indicated it was not used in the majority of cases.
If a lesion was clinically diagnosed as a BCC but was identified as a subtype of SCC on histology (or vice versa), the lesion was considered to be mismatched. The number of mismatched lesions and the mismatch rate for each lesion type/subtype is recorded in the Table. Of the total 11,145 keratinocyte carcinomas included in our study, there was an overall 10.63% mismatch rate, with 1185 of the malignancies having a differing clinical diagnosis (eg, BCC vs SCC) from the histologic findings. The clinical mismatch rate was notably higher for SCC compared to BCC (15.83% vs 7.03%, respectively).
The Table provides a breakdown of the BCC subtypes identified by histology with their computed mismatch rate and PPV. It is worth clarifying that lesions classified as more than one BCC subtype per the histologic findings were diagnosed as mixed BCC; these were further classified as mixed-aggressive BCC (if at least one aggressive BCC subtype was present) and mixed nonaggressive BCC (if no aggressive BCC subtype was present). Overall, BCCs were less likely to be misdiagnosed, with an average PPV of 92.97% compared to 84.17% for SCCs. Basosquamous BCC was the BCC subtype with the highest mismatch rate (25.48%), while sclerosing BCC has the lowest overall mismatch rate (1.33%). The most common malignancy was BCC, with nodular BCC being the most common subtype.
The Table also breaks down the SCC subtypes, reporting the most commonly misdiagnosed of any BCC or SCC subtype to be poorly differentiated SCC (mismatch rate, 38.46%). The lowest mismatch rate of the SCC subtypes was 5.97% for well-differentiated SCC.
There was an overall PPV of 89.37% in clinically evaluated malignancies and their respective histologic subtypes. Basal cell carcinoma had a lower overall mismatch rate of 7.03% compared to 15.83% in SCC. The most common misdiagnosis was attributed to poorly differentiated SCC (mismatch rate, 38.46%), while the least common misdiagnosed malignancy was sclerosing BCC (1.33%). The high mismatch rate of poorly differentiated SCC may be due to its diverging presentation from a typical SCC as a flat lesion with the absence of scaling, keratin, or bleeding, leading to the misdiagnosis of BCC.2
Accurate clinical diagnosis of NMSCs is the basis for further evaluation and treatment that should ensue in a timely manner; however, accurately identifying BCCs vs SCCs solely based on clinical examination can be challenging due to variable manifestations and overlapping features. Basal cell carcinoma commonly presents as a shiny pink/flesh-colored nodule, macule, or patch with surface telangiectasia, sometimes appearing with ulceration or crusting.3 Alternatively, SCC typically appears as a firm, sharply demarcated, red nodule with a thick overlying scale.4 Definitive diagnoses can be difficult upon clinical examination since these features can be shared between the 2 subtypes. To aid in these uncertainties, a growing number of clinicians are implementing the use of dermoscopy in their everyday practice.
Dermoscopy is an extremely useful tool in improving the diagnostic accuracy of skin cancers compared to examination with the naked eye, as it provides detailed visualization of specific structures and patterns in skin cancer lesions.5 The dermoscopic appearance of BCC is characterized by pearly blue-gray or translucent globules with arborizing vessels, spoke-wheel structures, and leaflike areas.5,6 Conversely, dermoscopic features of SCC may include a milky-red globule with a scaly, sharply demarcated, crusted lesion with polymorphous vasculature, sometimes resembling a persistent sore or nonhealing wound.4,5 Though the use of dermoscopy can aid in diagnosis upon initial examination, certain factors such as trauma, ulceration, and previous treatments that distorted the lesion’s architecture may lead to misdiagnosis. Furthermore, the distinct vascular patterns found in BCC and SCC may be mistaken for each other and therefore lead to misdiagnosis upon examination.7 Other variables that may complicate diagnosis include the location of the lesion, its size, and the presence of other skin conditions or nearby lesions.
The primary limitation of the current study was the limited scope of the data, as they were derived from patients seen at one private dermatology practice, preventing the generalizability of our findings. However, our results show trends similar to those observed in other studies analyzing the clinical accuracy of skin cancer diagnoses, with higher PPVs for BCC compared to SCC. A study by Ahnlide and Bjellerup8 was based in a hospital dermatology department and demonstrated a PPV of 85.5% for BCC compared to 92.97% in our study; for SCC, the PPV was 67.3% compared to 84.17% in our study. In another study by Heal et al,9 data were collected from an Australian registry that included records of all histologically confirmed skin cancers from December 1996 to October 1999 from 202 general practitioners and 42 specialists, including 1 dermatologist. The PPVs for BCC and SCC were 72.7% and 49.4%, respectively. Although our results indicated higher PPVs compared to these 2 studies, some of the discrepancies can be accounted for by the differences in clinical setting as well as the lack of expertise of nondermatologist physicians in identifying skin malignancies in the study by Heal et al.9
The current study was further limited by the lack of data quantifying the number of lesions clinically suspected to be malignant but found to be histologically benign. It is typical for clinicians to have a low threshold to biopsy a suspicious lesion with atypical features (eg, rapid evolution and growth, bleeding, crusting). Furthermore, the identification of risk factors in the patient’s medical and family history (eg, exposure to radiation, personal or family history of skin cancers) can heavily influence a clinician’s decision to biopsy a lesion with an atypical appearance.10 Many benign lesions are biopsied to avoid missing a diagnosis of malignancy. Consequently, our results suggest a high degree of clinical misdiagnosis of BCCs and SCCs. Obtaining data on the number of lesions suspected to be BCC or SCC that were found to be histologically benign would be a valuable addition to our study, as it would provide a measurable insight into the sensitivity of clinicians’ decision-making to identify a lesion as suspicious and warranting biopsy.
While clinical diagnosis plays a vital role in identifying suspected NMSCs such as BCC and SCC, its accuracy can be limited even with the use of dermoscopy. Overall, our data have shown a high rate of diagnostic accuracy upon suspicion of malignancy, but the different variables that affect clinical presentation promote histologic diagnosis to prevail as the gold standard.
- Seyed Ahadi M, Firooz A, Rahimi H, et al. Clinical diagnosis has a high negative predictive value in evaluation of malignant skin lesions. Dermatol Res Pract. 2021;2021:6618990. doi:10.1155/2021/6618990
- Lallas A, Pyne J, Kyrgidis A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015;172:1308- 1315. doi:10.1111/bjd.13510
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. September 19, 2022. In: StatPearls. StatPearls Publishing; 2023.
- Suárez AL, Louis P, Kitts J, et al. Clinical and dermoscopic features of combined cutaneous squamous cell carcinoma (SCC)/neuroendocrine [Merkel cell] carcinoma (MCC). J Am Acad Dermatol. 2015;73:968-975. doi:10.1016/j.jaad.2015.08.041
- Wolner ZJ, Yélamos O, Liopyris K, et al. Enhancing skin cancer diagnosis with dermoscopy. Dermatol Clin. 2017;35:417-437. doi:10.1016/j.det.2017.06.003
- Reiter O, Mimouni I, Dusza S, et al. Dermoscopic features of basal cell carcinoma and its subtypes: a systematic review. J Am Acad Dermatol. 2021;85:653-664. doi:10.1016/j.jaad.2019.11.008
- Pruneda C, Ramesh M, Hope L, et al. Nonmelanoma skin cancers: diagnostic accuracy of midlevel providers versus dermatologists. The Dermatologist. March 2023. Accessed March 18, 2025. https://www.hmpgloballearningnetwork.com/site/thederm/feature-story/nonmelanoma-skin-cancers-diagnostic-accuracy-midlevel-providers-vs
- Ahnlide I, Bjellerup M. Accuracy of clinical skin tumour diagnosis in a dermatological setting. Acta Derm Venereol. 2013;93:305-308. doi:10.2340/00015555-1560
- Heal CF, Raasch BA, Buettner PG, et al. Accuracy of clinical diagnosis of skin lesions. Br J Dermatol. 2008;159:661-668.
- Fu S, Kim S, Wasko C. Dermatological guide for primary care physicians: full body skin checks, skin cancer detection, and patient education on self-skin checks and sun protection. Proc (Bayl Univ Med Cent). 2024;37:647-654. doi:10.1080/08998280.2024.2351751
- Seyed Ahadi M, Firooz A, Rahimi H, et al. Clinical diagnosis has a high negative predictive value in evaluation of malignant skin lesions. Dermatol Res Pract. 2021;2021:6618990. doi:10.1155/2021/6618990
- Lallas A, Pyne J, Kyrgidis A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015;172:1308- 1315. doi:10.1111/bjd.13510
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. September 19, 2022. In: StatPearls. StatPearls Publishing; 2023.
- Suárez AL, Louis P, Kitts J, et al. Clinical and dermoscopic features of combined cutaneous squamous cell carcinoma (SCC)/neuroendocrine [Merkel cell] carcinoma (MCC). J Am Acad Dermatol. 2015;73:968-975. doi:10.1016/j.jaad.2015.08.041
- Wolner ZJ, Yélamos O, Liopyris K, et al. Enhancing skin cancer diagnosis with dermoscopy. Dermatol Clin. 2017;35:417-437. doi:10.1016/j.det.2017.06.003
- Reiter O, Mimouni I, Dusza S, et al. Dermoscopic features of basal cell carcinoma and its subtypes: a systematic review. J Am Acad Dermatol. 2021;85:653-664. doi:10.1016/j.jaad.2019.11.008
- Pruneda C, Ramesh M, Hope L, et al. Nonmelanoma skin cancers: diagnostic accuracy of midlevel providers versus dermatologists. The Dermatologist. March 2023. Accessed March 18, 2025. https://www.hmpgloballearningnetwork.com/site/thederm/feature-story/nonmelanoma-skin-cancers-diagnostic-accuracy-midlevel-providers-vs
- Ahnlide I, Bjellerup M. Accuracy of clinical skin tumour diagnosis in a dermatological setting. Acta Derm Venereol. 2013;93:305-308. doi:10.2340/00015555-1560
- Heal CF, Raasch BA, Buettner PG, et al. Accuracy of clinical diagnosis of skin lesions. Br J Dermatol. 2008;159:661-668.
- Fu S, Kim S, Wasko C. Dermatological guide for primary care physicians: full body skin checks, skin cancer detection, and patient education on self-skin checks and sun protection. Proc (Bayl Univ Med Cent). 2024;37:647-654. doi:10.1080/08998280.2024.2351751
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
Clinical Accuracy of Skin Cancer Diagnosis: Investigation of Keratinocyte Carcinoma Mismatch Rates
PRACTICE POINTS
- Malignant lesions may be misdiagnosed when assessments are guided by clinical features that align with typical presentations of other lesion types, potentially leading to diagnostic errors among experienced clinicians.
- Although dermoscopy is a beneficial tool in examining potential skin cancers, clinical observations should not bypass the gold standard of histopathologic examination.
Repair of a Large Full-Thickness Conchal Bowl Defect
Repair of a Large Full-Thickness Conchal Bowl Defect
Practice Gap
Large full-thickness conchal bowl defects often pose a reconstructive challenge. Maintaining the shape and structural integrity of the concha is fundamental for optimal cosmetic and functional outcomes. Prior reports have suggested wedge excisions, composite grafts, interpolation flaps with or without cartilage struts, and hinge flaps as possible options for reconstruction.1-3 However, patients with large defects who prefer single-stage reconstruction procedures present a unique challenge. Herein, we describe a single-stage full-thickness hinge flap technique for a large conchal bowl defect.
The Technique
A 77-year-old man was referred to our dermatology clinic by an outside dermatologist for Mohs micrographic surgery of a biopsy-proven cutaneous squamous cell carcinoma on the right conchal bowl measuring 1.1×2.1 cm and extending to the edge of the external auditory canal (EAC). The excision was performed that same day and was completed in 2 stages, achieving negative margins and resulting in a full-thickness defect measuring 2.0×3.6 cm that included the posterior auricular sulcus, cavum, antitragus, and proximal EAC (Figure 1). The patient requested a single-stage procedure but emphasized that his main priority was an optimal cosmetic outcome.
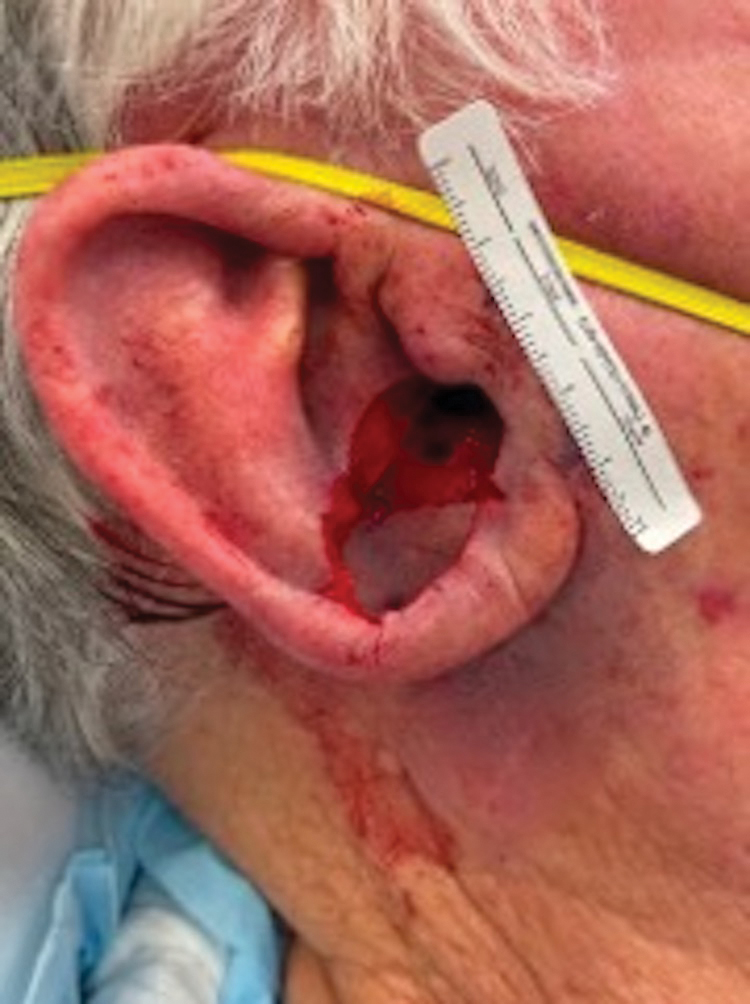
To repair this large defect, a full-thickness hinge flap with Burow graft was performed. The hinge-type flap was designed in a triangular fashion emanating at the posterior auricular sulcus adjacent to the posterior aspect of the defect and extending down the lateral neck (Figure 2). The flap was incised and the surrounding tissue was undermined, maintaining a robust pedicle in the center of its body on the superolateral neck. The flap was passed through the posterior aspect of the full-thickness defect and was secured in place with 4-0 polyglactin sutures in a buried interrupted fashion, thereby recreating the anterior portion of the defect. The superficial skin edges were reapproximated using 4-0 and 5-0 polypropylene sutures in a running interrupted fashion. The distal Burow triangle created from closure of the flap’s secondary defect was aggressively thinned and was utilized as a full-thickness graft for the residual postauricular groove defect (Figure 3). At 2 weeks’ follow-up, the patient was healing well with no postoperative issues and the sutures were removed (Figure 4).
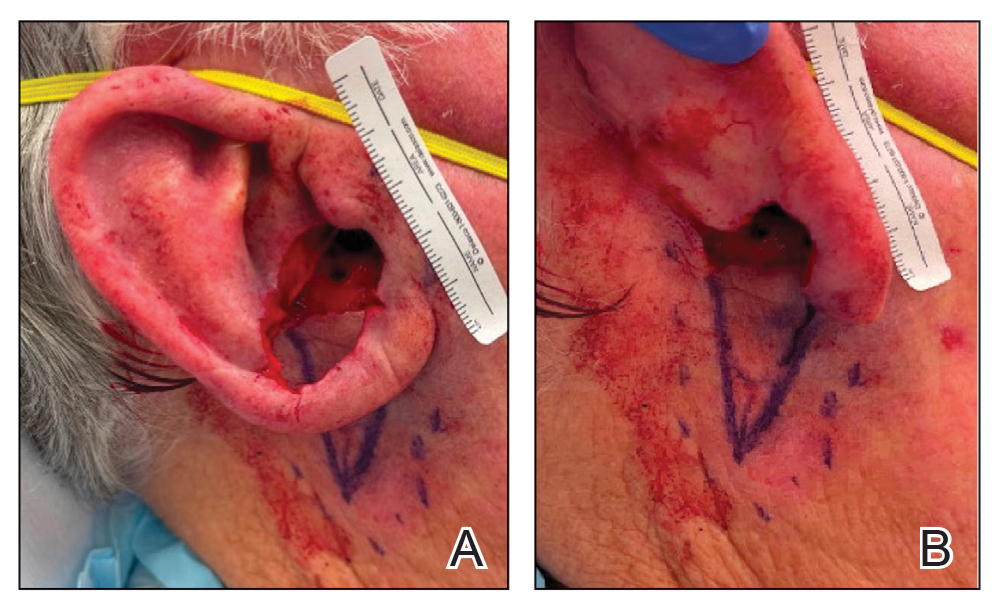
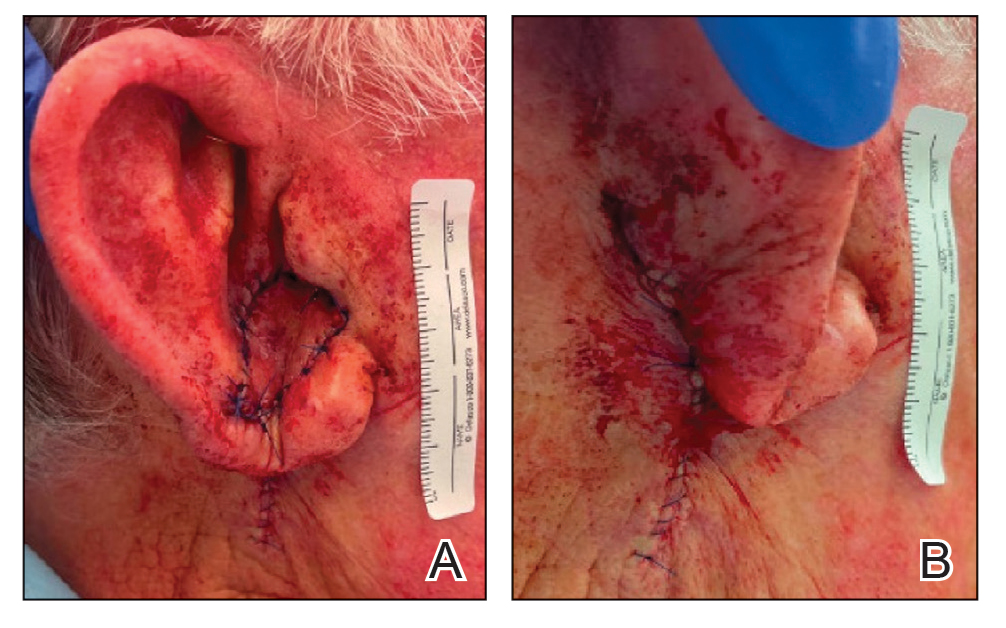
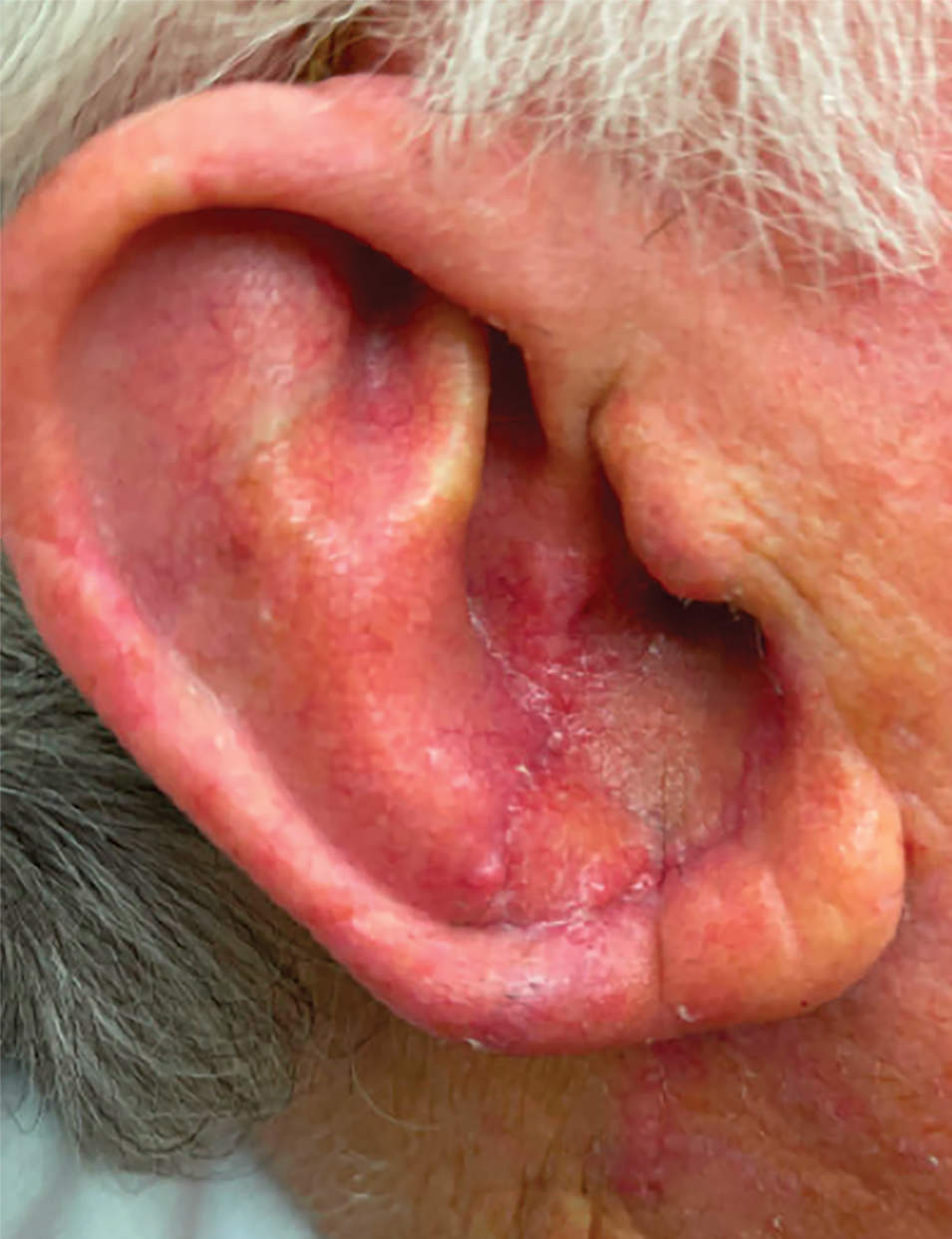
Practice Implications
There are many different reconstructive options for conchal bowl defects, including primary repair, wedge excision, composite graft and interpolation flaps with or without cartilage struts, and hinge flaps. Structural support, EAC patency, auricle symmetry, overall auricle size, and re-creation of natural contours were considered when designing the reconstruction of the defect in our patient; however, his main priority was achieving the greatest cosmetic outcome in a single-stage procedure, therefore limiting our reconstruction options.
Wedge excision, in which the residual lobule and inferior helical rim are removed, could have been considered in our patient but would have drastically altered the symmetry of the size of the ears. A folded postauricular flap, as described in the otolaryngology literature, is an interpolation flap based on the posterior auricular artery that was designed for full-thickness defects of the auricle to prevent any posterior pinning.1 This technique may have worked well in our case, but the patient preferred to avoid a multistage procedure. Additionally, the positional symmetry of the ears was maintained despite utilizing a hinge flap, which does not involve takedown of the pedicle. A composite graft from the contralateral ear could be considered for smaller conchal bowl defects but likely would have resulted in graft failure in our patient’s large defect due to its need for rich blood supply to heal and dependence on lateral wound edges. Cartilage struts in conjunction with a flap could have been considered in this scenario for greater structural support, but in our patient’s case, by maintaining the robust pedicle of our flap and having residual superior cartilage, further structural support was not necessary.
A prior case report described a partial and full-thickness defect in a similar location that was repaired with a retroauricular hinge flap, in which a portion of the flap was extensively de-epithelialized to address the varied thicknesses of the surgical defect.2 In our patient, the defect abutted the skin reservoir on the superolateral neck, and therefore no de-epithelialization was required as the entire epithelialized portion was utilized to recreate the anterior aspect of the defect. Postauricular hinge-type flaps are a reliable, single-stage surgical alternative to the 2-stage folded postauricular interpolation flap when reconstructing large conchal bowl defects. For small full-thickness defects of the ear, a composite graft may be considered; however, blood supply and other nutritional requirements limit this option for large full-thickness defects.
- Roche AM, Griffin M, Shelton R, et al. The folded postauricular flap: a novel approach to reconstruction of large full thickness defects of the conchal bowl. Am J Otolaryngol. 2017;38:706-709. doi:10.1016 /j.amjoto.2017.09.006
- Klein JC, Nijhawan RI. Retroauricular hinge flaps for full-thickness conchal bowl defects. J Am Acad Dermatol. 2024;90:E71-E72. doi:10.1016/j.jaad.2022.10.056
- Pickrell BB, Hughes CD, Maricevich RS. Partial ear defects. Semin Plast Surg. 2017 Aug;31:134-140. doi:10.1055/s-0037-1603968.
Practice Gap
Large full-thickness conchal bowl defects often pose a reconstructive challenge. Maintaining the shape and structural integrity of the concha is fundamental for optimal cosmetic and functional outcomes. Prior reports have suggested wedge excisions, composite grafts, interpolation flaps with or without cartilage struts, and hinge flaps as possible options for reconstruction.1-3 However, patients with large defects who prefer single-stage reconstruction procedures present a unique challenge. Herein, we describe a single-stage full-thickness hinge flap technique for a large conchal bowl defect.
The Technique
A 77-year-old man was referred to our dermatology clinic by an outside dermatologist for Mohs micrographic surgery of a biopsy-proven cutaneous squamous cell carcinoma on the right conchal bowl measuring 1.1×2.1 cm and extending to the edge of the external auditory canal (EAC). The excision was performed that same day and was completed in 2 stages, achieving negative margins and resulting in a full-thickness defect measuring 2.0×3.6 cm that included the posterior auricular sulcus, cavum, antitragus, and proximal EAC (Figure 1). The patient requested a single-stage procedure but emphasized that his main priority was an optimal cosmetic outcome.
To repair this large defect, a full-thickness hinge flap with Burow graft was performed. The hinge-type flap was designed in a triangular fashion emanating at the posterior auricular sulcus adjacent to the posterior aspect of the defect and extending down the lateral neck (Figure 2). The flap was incised and the surrounding tissue was undermined, maintaining a robust pedicle in the center of its body on the superolateral neck. The flap was passed through the posterior aspect of the full-thickness defect and was secured in place with 4-0 polyglactin sutures in a buried interrupted fashion, thereby recreating the anterior portion of the defect. The superficial skin edges were reapproximated using 4-0 and 5-0 polypropylene sutures in a running interrupted fashion. The distal Burow triangle created from closure of the flap’s secondary defect was aggressively thinned and was utilized as a full-thickness graft for the residual postauricular groove defect (Figure 3). At 2 weeks’ follow-up, the patient was healing well with no postoperative issues and the sutures were removed (Figure 4).
Practice Implications
There are many different reconstructive options for conchal bowl defects, including primary repair, wedge excision, composite graft and interpolation flaps with or without cartilage struts, and hinge flaps. Structural support, EAC patency, auricle symmetry, overall auricle size, and re-creation of natural contours were considered when designing the reconstruction of the defect in our patient; however, his main priority was achieving the greatest cosmetic outcome in a single-stage procedure, therefore limiting our reconstruction options.
Wedge excision, in which the residual lobule and inferior helical rim are removed, could have been considered in our patient but would have drastically altered the symmetry of the size of the ears. A folded postauricular flap, as described in the otolaryngology literature, is an interpolation flap based on the posterior auricular artery that was designed for full-thickness defects of the auricle to prevent any posterior pinning.1 This technique may have worked well in our case, but the patient preferred to avoid a multistage procedure. Additionally, the positional symmetry of the ears was maintained despite utilizing a hinge flap, which does not involve takedown of the pedicle. A composite graft from the contralateral ear could be considered for smaller conchal bowl defects but likely would have resulted in graft failure in our patient’s large defect due to its need for rich blood supply to heal and dependence on lateral wound edges. Cartilage struts in conjunction with a flap could have been considered in this scenario for greater structural support, but in our patient’s case, by maintaining the robust pedicle of our flap and having residual superior cartilage, further structural support was not necessary.
A prior case report described a partial and full-thickness defect in a similar location that was repaired with a retroauricular hinge flap, in which a portion of the flap was extensively de-epithelialized to address the varied thicknesses of the surgical defect.2 In our patient, the defect abutted the skin reservoir on the superolateral neck, and therefore no de-epithelialization was required as the entire epithelialized portion was utilized to recreate the anterior aspect of the defect. Postauricular hinge-type flaps are a reliable, single-stage surgical alternative to the 2-stage folded postauricular interpolation flap when reconstructing large conchal bowl defects. For small full-thickness defects of the ear, a composite graft may be considered; however, blood supply and other nutritional requirements limit this option for large full-thickness defects.
Practice Gap
Large full-thickness conchal bowl defects often pose a reconstructive challenge. Maintaining the shape and structural integrity of the concha is fundamental for optimal cosmetic and functional outcomes. Prior reports have suggested wedge excisions, composite grafts, interpolation flaps with or without cartilage struts, and hinge flaps as possible options for reconstruction.1-3 However, patients with large defects who prefer single-stage reconstruction procedures present a unique challenge. Herein, we describe a single-stage full-thickness hinge flap technique for a large conchal bowl defect.
The Technique
A 77-year-old man was referred to our dermatology clinic by an outside dermatologist for Mohs micrographic surgery of a biopsy-proven cutaneous squamous cell carcinoma on the right conchal bowl measuring 1.1×2.1 cm and extending to the edge of the external auditory canal (EAC). The excision was performed that same day and was completed in 2 stages, achieving negative margins and resulting in a full-thickness defect measuring 2.0×3.6 cm that included the posterior auricular sulcus, cavum, antitragus, and proximal EAC (Figure 1). The patient requested a single-stage procedure but emphasized that his main priority was an optimal cosmetic outcome.
To repair this large defect, a full-thickness hinge flap with Burow graft was performed. The hinge-type flap was designed in a triangular fashion emanating at the posterior auricular sulcus adjacent to the posterior aspect of the defect and extending down the lateral neck (Figure 2). The flap was incised and the surrounding tissue was undermined, maintaining a robust pedicle in the center of its body on the superolateral neck. The flap was passed through the posterior aspect of the full-thickness defect and was secured in place with 4-0 polyglactin sutures in a buried interrupted fashion, thereby recreating the anterior portion of the defect. The superficial skin edges were reapproximated using 4-0 and 5-0 polypropylene sutures in a running interrupted fashion. The distal Burow triangle created from closure of the flap’s secondary defect was aggressively thinned and was utilized as a full-thickness graft for the residual postauricular groove defect (Figure 3). At 2 weeks’ follow-up, the patient was healing well with no postoperative issues and the sutures were removed (Figure 4).
Practice Implications
There are many different reconstructive options for conchal bowl defects, including primary repair, wedge excision, composite graft and interpolation flaps with or without cartilage struts, and hinge flaps. Structural support, EAC patency, auricle symmetry, overall auricle size, and re-creation of natural contours were considered when designing the reconstruction of the defect in our patient; however, his main priority was achieving the greatest cosmetic outcome in a single-stage procedure, therefore limiting our reconstruction options.
Wedge excision, in which the residual lobule and inferior helical rim are removed, could have been considered in our patient but would have drastically altered the symmetry of the size of the ears. A folded postauricular flap, as described in the otolaryngology literature, is an interpolation flap based on the posterior auricular artery that was designed for full-thickness defects of the auricle to prevent any posterior pinning.1 This technique may have worked well in our case, but the patient preferred to avoid a multistage procedure. Additionally, the positional symmetry of the ears was maintained despite utilizing a hinge flap, which does not involve takedown of the pedicle. A composite graft from the contralateral ear could be considered for smaller conchal bowl defects but likely would have resulted in graft failure in our patient’s large defect due to its need for rich blood supply to heal and dependence on lateral wound edges. Cartilage struts in conjunction with a flap could have been considered in this scenario for greater structural support, but in our patient’s case, by maintaining the robust pedicle of our flap and having residual superior cartilage, further structural support was not necessary.
A prior case report described a partial and full-thickness defect in a similar location that was repaired with a retroauricular hinge flap, in which a portion of the flap was extensively de-epithelialized to address the varied thicknesses of the surgical defect.2 In our patient, the defect abutted the skin reservoir on the superolateral neck, and therefore no de-epithelialization was required as the entire epithelialized portion was utilized to recreate the anterior aspect of the defect. Postauricular hinge-type flaps are a reliable, single-stage surgical alternative to the 2-stage folded postauricular interpolation flap when reconstructing large conchal bowl defects. For small full-thickness defects of the ear, a composite graft may be considered; however, blood supply and other nutritional requirements limit this option for large full-thickness defects.
- Roche AM, Griffin M, Shelton R, et al. The folded postauricular flap: a novel approach to reconstruction of large full thickness defects of the conchal bowl. Am J Otolaryngol. 2017;38:706-709. doi:10.1016 /j.amjoto.2017.09.006
- Klein JC, Nijhawan RI. Retroauricular hinge flaps for full-thickness conchal bowl defects. J Am Acad Dermatol. 2024;90:E71-E72. doi:10.1016/j.jaad.2022.10.056
- Pickrell BB, Hughes CD, Maricevich RS. Partial ear defects. Semin Plast Surg. 2017 Aug;31:134-140. doi:10.1055/s-0037-1603968.
- Roche AM, Griffin M, Shelton R, et al. The folded postauricular flap: a novel approach to reconstruction of large full thickness defects of the conchal bowl. Am J Otolaryngol. 2017;38:706-709. doi:10.1016 /j.amjoto.2017.09.006
- Klein JC, Nijhawan RI. Retroauricular hinge flaps for full-thickness conchal bowl defects. J Am Acad Dermatol. 2024;90:E71-E72. doi:10.1016/j.jaad.2022.10.056
- Pickrell BB, Hughes CD, Maricevich RS. Partial ear defects. Semin Plast Surg. 2017 Aug;31:134-140. doi:10.1055/s-0037-1603968.
Repair of a Large Full-Thickness Conchal Bowl Defect
Repair of a Large Full-Thickness Conchal Bowl Defect
