User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Frontal Fibrosing Alopecia Demographics: A Survey of 29 Patients
Frontal fibrosing alopecia (FFA) is a form of lymphocytic cicatricial alopecia that presents as frontotemporal hairline recession, typically in postmenopausal women.1 The condition is considered to be a variant of lichen planopilaris (LPP) due to its similar histologic appearance.2 Loss of eyebrow1-11 and body5-11 hair also is commonly present in FFA, and histologic findings are identical to those for hair loss on the scalp,8,9 suggesting that FFA may be a form of generalized alopecia.
The pathogenesis of FFA is unknown, but several etiologies have been postulated. Some suggest that as a variant of LPP, FFA is a hair-specific autoimmune disorder characterized by a T cell–mediated immune reaction against epithelial hair follicle stem cells, leading to fibrosis and depletion of hair regeneration potential.12 In support of this theory, FFA has been associated with other autoimmune diseases including hypothyroidism,6,8,13-16 mucocutaneous lichen planus,8,15,17 vitiligo,15,18 Sjögren syndrome,19 and lichen sclerosus et atrophicus.15,20 Another hypothesis suggests that the proandrogenic state in postmenopausal women may be related to the disease process.1 This hypothesis is supported by the reported success of antiandrogen therapy with 5α-reductase inhibitors (5α-RIs) in stabilizing FFA.3-5,7 Finally, genetic16,21 and environmental factors related to smoking and socioeconomic status5 also have been postulated to be risk factors for FFA. A variety of treatments have shown varying success, including topical and intralesional corticosteroids, hydroxychloroquine, immunomodulators, antibiotics, and 5α-RIs.1,3-6,8,15,17,22 However, FFA is considered to be relatively difficult to treat and commonly progresses regardless of treatment before spontaneously stabilizing.2-4,6,8,10
Since its discovery in 1994,1 FFA has become increasingly prevalent, comprising 17% of new referrals for hair loss in one study (N=57).6 Although growing recognition of the condition likely plays a role in its increasing presentation, other unidentified factors may contribute to its expanding incidence. In this report, we describe the demographics, clinical features, and disease progression of 29 cases of FFA treated within our division using a series of surveys and chart reviews.
Methods
Upon receiving approval for the project from the institutional review board, we identified 29 patients who met the criteria for diagnosis of FFA through a chart review of all patients being treated for hair loss by clinics within the Washington University Division of Dermatology (St. Louis, Missouri). Diagnostic criteria for FFA included scarring alopecia in the frontotemporal distribution with associated perifollicular erythema or papules and, if performed, a scalp biopsy of the involved area of alopecia showing lymphocytic cicatricial alopecia, compatible with LPP. The diagnosis was confirmed by biopsy in 18 patients (62%), while the remainder of the diagnoses were made clinically. Most biopsy specimens were diagnosed by board-certified dermatopathologists at Washington University, with the remainder diagnosed by outside pathologists if the patient was initially diagnosed at another institution.
Patients meeting criteria for FFA were mailed a study consent form, as well as a 2-page survey to assess demographics, clinical features of hair loss, medical histories, social and family histories, and treatments utilized. After receiving consent from patients, survey results were collected and summarized. If there was any need for clarification of answers, follow-up questions were conducted via email prior to any data analysis that was performed.
For analysis of treatment response, patients were asked what treatments they had utilized and about the progression of their hair loss. Patients reporting stabilization of hair loss or hair regrowth were classified as treatment responsive. Patients who underwent multiple treatments were included in the analyses for each of those treatments. Physician records for treatment response were not correlated with patient responses due to inconsistent documentation, care received outside of our medical system, and prolonged or loss to follow-up. Physician-reported data were only used to identify qualifying patients and their biopsy results, as described above.
Results
Patient Demographic
Between October 2013 and May 2014, 29 patients with FFA were recruited into the study. Patients were diagnosed between January 2006 and December 2013. There were 28 female patients (97%) and 1 male patient (3%). The average age of disease onset was 55.4 years (range, 29–75 years). Twenty-five patients (86%) self-identified as non-Hispanic white, 3 patients (10%) as Asian, and 1 patient (3%) as black. Patients also appeared to be a more affluent group than the general St. Louis County population, with a median household income between $75,000 and $100,000. In comparison, the median household income reported in St. Louis County from 2008 to 2012 was $58,485.23 The patient population was primarily composed of nonsmokers, with 22 (76%) patients who had never smoked, 6 (21%) who were present smokers, and 1 (3%) smoked in the past. These results were comparable to the reported number of female smokers in Missouri.24
Clinicopathologic Features
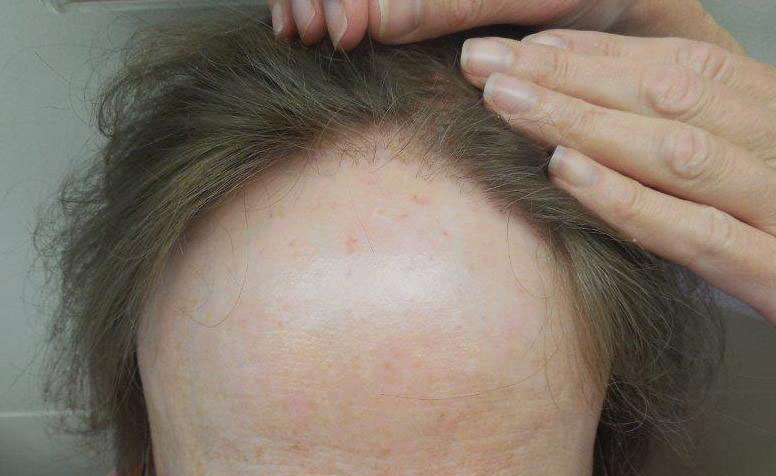
The clinical features of FFA are described in Table 1. All patients had frontotemporal recession of the hairline with some degree of scarring and perifollicular erythema (Figure 1). Most patients also reported hair loss at other sites, including 25 patients (86%) with eyebrow hair loss, 18 (62%) with limb hair loss, 11 (38%) with axillary hair loss, 11 (38%) with pubic hair loss, and 1 (3%) with eyelash hair loss. Patients also frequently reported inflammatory symptoms, including 19 patients (66%) with itching, 18 (62%) with redness, 3 (10%) with pain, 2 (7%) with papular lesions, and 1 (3%) with sores and erosions on the skin. Regarding progression of hair loss over time, 16 patients (55%) reported stabilization of hair loss, 11 (38%) reported progressive hair loss, and 2 (7%) reported some hair regrowth. Thirteen patients (45%) identified some inciting event that they believed to have triggered the disease. Ten patients (35%) identified stress as the inciting event, and 5 patients (17%) specifically referred to health-related stressors, including hip-replacement surgery, new diagnoses of systemic diseases, starting new medications, and stopping hormone replacement therapy. Furthermore, 2 (7%) patients reported exposure to chemicals and pesticides as suspected triggers.

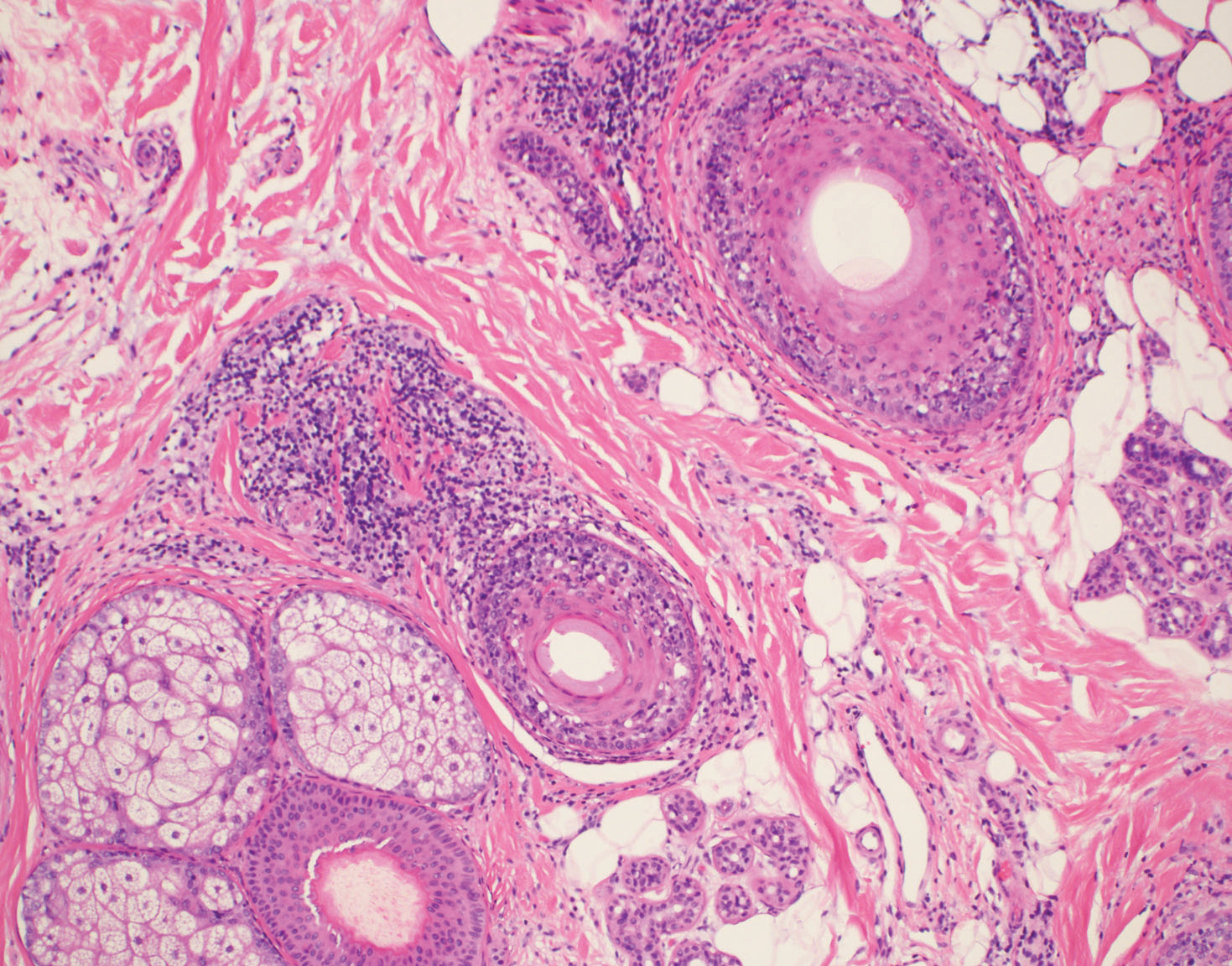
Typical biopsy results showed a perifollicular lymphocytic infiltrate and fibrosis surrounding the infundibulum and isthmus of hair follicles (Figure 2). There were associated vacuolar changes in the basal layer and scattered dyskeratosis throughout the follicular epithelium. As the disease progressed to end-stage scarring, there was marked reduction in the number of hair follicles, which were replaced by fibrous tracts, and a disappearance of the previous inflammatory infiltrate.
Medical History
Of the 26 female patients who provided data about menopause status at time of disease onset, 16 (62%) were postmenopausal, 5 (19%) were menopausal, and 5 (19%) were premenopausal. Of the 28 female patients in the study, 8 (29%) had a history of hysterectomy and 2 (7%) also had surgically induced menopause through bilateral surgical oophorectomy. Twenty-four patients (86%) had a childbearing history, with an average of 2.3 children. Twelve patients (43%) reported use of hormone replacement therapy after menopause. Twelve patients (43%) also reported a history of oral contraceptive use.
Table 2 describes the comorbidities of all 29 patients. A history of autoimmune disease was prominent, found in 16 patients (55%). Thirteen patients (45%) reported thyroid disease, including 10 patients (35%) with hypothyroidism. Additionally, 8 patients (28%) had a history of mucocutaneous lichen planus, 2 (7%) of psoriasis/psoriatic arthritis, 1 (3%) of vitiligo, 1 (3%) of systemic lupus erythematosus, 1 (3%) of iritis, 1 (3%) of Sjögren syndrome, and 1 (3%) of ulcerative colitis. Six patients (21%) also reported a history of breast cancer.

A dental history was obtained in 24 patients. All 24 patients reported having some dental implant or filling placed. Twenty-four patients (100%) had a history of metal amalgam implants, 8 (33%) had gold alloy implants, 4 (17%) had composite resin implants, and 3 (13%) had porcelain implants. Two patients had metal amalgam implants that had since been replaced by nonmetal implants. Both patients reported no change in their clinical conditions with removal of the metal implants. Six of 8 patients (75%) with mucocutaneous lichen planus reported having dental implants. Of them, all 6 patients (100%) reported having metal amalgam implants, and 3 patients (50%) additionally reported having gold alloy implants.
Treatments
On average, patients were treated with 3 different therapies for FFA (range, 0–14). The treatments utilized are listed in Table 3, and responses to treatments are summarized in Table 4. Topical steroids were the most popular treatment modality and were used by 21 patients (72%). Approximately half of those patients reported treatment response with stabilization of hair loss or regrowth (n=11; 52%). Hydroxychloroquine was the second most commonly used modality (16 patients [55%]), with 10 of those patients (63%) reporting treatment response. Intralesional steroids were used in 11 patients (38%), with a treatment response in 36% (4/11) of those patients. Topical pimecrolimus and tacrolimus were used by 6 patients (21%), with 5 of those patients (83%) reporting treatment response. UVB excimer laser therapy was used on 3 patients (10%) with 100% treatment response.
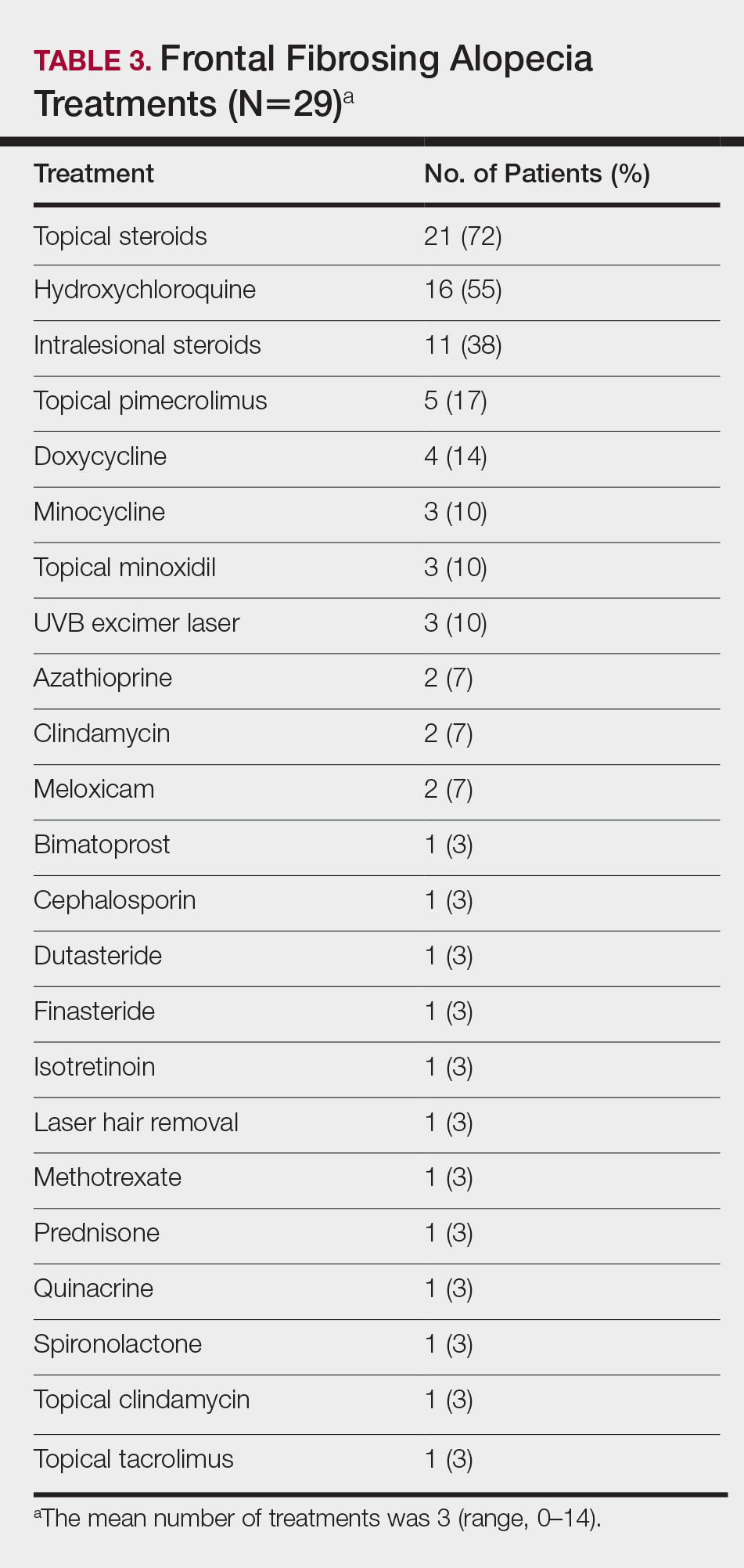
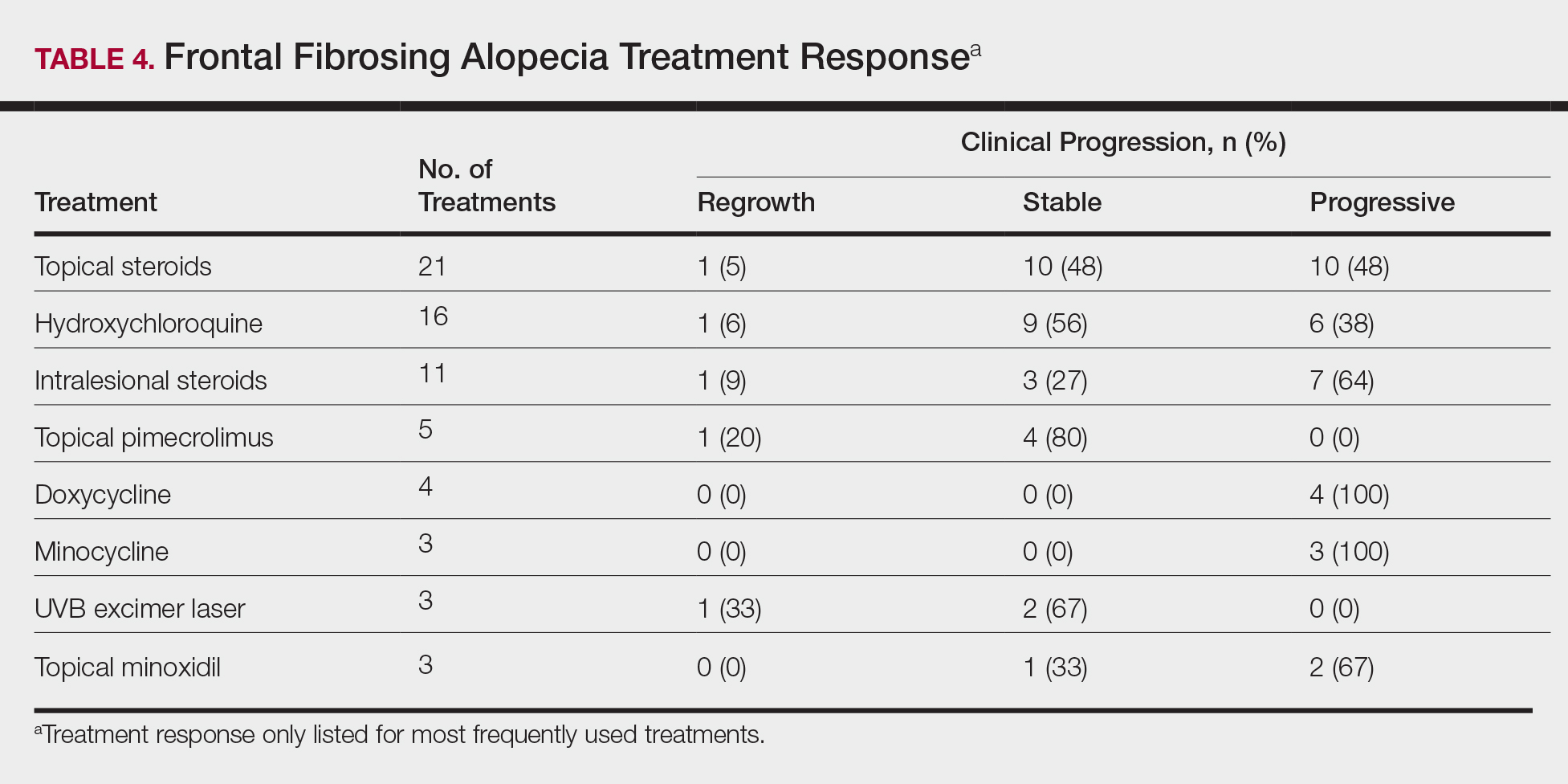
Treatments with little or no treatment response to hair loss include doxycycline, minocycline, and topical minoxidil. Seven patients (24%) were treated with doxycycline or minocycline, all of whom reported no clinical response. Topical minoxidil was used by 3 patients (10%), with only 1 patient (33%) reporting stabilization of hair loss but no regrowth of hair. 5α-reductase inhibitors such as finasteride and dutasteride were only used by 1 patient (3%), who reported no treatment response. Other treatments that were rarely used include meloxicam (n=2), azathioprine (n=2), oral clindamycin (n=2), bimatoprost (n=1), quinacrine (n=1), cephalosporin (n=1), prednisone (n=1), isotretinoin (n=1), methotrexate (n=1), spironolactone (n=1), topical clindamycin (n=1), and laser hair removal (n=1). Of these, only meloxicam and quinacrine were anecdotally associated with stabilization of hair loss, while the rest of the treatments were associated with progressive hair loss despite therapy.
Comment
Frontal fibrosing alopecia is a form of cicatricial alopecia considered to be a clinical subset of LPP. Although the pathogeneses of both diseases are poorly understood, LPP is the better-studied model and is generally considered to be an autoimmune disease specific to the hair follicle, involving a cell-mediated inflammatory response to epithelial hair follicle stem cells.12 In support of this hypothesis, FFA and LPP have been frequently associated with autoimmune diseases, particularly with hypothyroidism.6,13-15 We found that 55% of our patients had a history of autoimmune disease, including 35% with hypothyroidism, 28% with mucocutaneous lichen planus, 7% with psoriasis, 3% with vitiligo, 3% with systemic lupus erythematosus, 3% with iritis, 3% with Sjögren syndrome, and 3% with ulcerative colitis. The link between FFA and hypothyroidism has been the best studied, with a large study by Atanskova Mesinkovska et al14 finding that 34% of 166 patients with LPP and FFA have some kind of thyroid disease and 29% have hypothyroidism. Fron
Although FFA has been classically described to affect postmenopausal women, recent studies have consistently identified that premenopausal women4-6,8,16,17 and men14,16 also can be affected by the condition. In our patient cohort, there was 1 male patient (3%), and a substantial number of the female patients were premenopausal (19%) and menopausal (19%) at the time of disease onset. Most of the patients studied were white; Asian and black patients were a consistent minority across FFA studies,5,13-16,25 highlighting the importance of screening for FFA in all demographics.
In our study, FFA patients also appeared to be more affluent than the general population and were predominantly nonsmokers (76%). These statistics are consistent with the United Kingdom population studied by MacDonald et al,6 which demonstrated a higher socioeconomic status and higher incidence of nonsmoking in their cases of FFA. Another large retrospective study of FFA patients in Spain found that 87% of their FFA cases (N=355) were nonsmokers, though they did not note a difference from the general unaffected population.15 In our study, we replicated these trends, finding an above average affluence level and a high but not statistically significant incidence of nonsmokers. Although it is not clear how socioeconomic status or smoking factors into the pathology of FFA, these studies may show a general trend in the environmental demographics of the disease.
Clinically, patients with FFA typically present with hair loss of the scalp as well as other sites. The eyebrows are the most common site to be affected outside of the scalp, affecting 86% of our patients, whereas eyelashes are the least commonly affected, presenting in only 3% of our patients. Body hair loss also is common, with almost two-thirds of our cohort reporting hair loss on the limbs and more than one-third reporting loss of axillary and pubic hair. These findings are consistent with those of other studies.3-6,8,13,15 Eyelash loss, body hair loss, and facial papules have been found to be associated with more severe forms of FFA,15 though we did not investigate these forms in our study. Inflammatory symptoms are common, with pruritus affecting 66% of our patients and pain affecting 10% of patients, consistent with the published literature.3,13,15,17
Multiple studies have shown that female FFA patients have a higher incidence of hysterectomies in their medical history.5,8,15 This observation has been used to further support the hypothesis that a change in sex hormone balance may trigger the initial onset of disease.5,8,15 A considerable number of the female patients in our study had also undergone hysterectomies (29%). Only 2 patients (7%) underwent premature surgical menopause through bilateral removal of the ovaries, and neither of these patients had abnormally early onset of FFA (age at onset, 52 and 65 years). Many patients in our study also reported a history of pharmacologic manipulation of sex hormones with hormone replacement therapy (43%) and oral contraceptive use (43%). However, patients with FFA have not been identified to have abnormal hormone levels compared to unaffected postmenopausal women.1 Additionally, the disease does not exclusively affect androgen-dependent hair, as indicated by the high prevalence of eyebrow hair loss. We hypothesize that the link between increased prevalence of hysterectomy and FFA is not due to hormonal changes but rather from the stresses related to the hysterectomy or associated conditions that required the surgery. In our study, 35% of patients identified stress as the inciting event prior to their onset of hair loss, with 17% specifically referring to health-related stress such as surgery or new diagnoses as the cause. Although this pattern is purely observational, it is valuable to consider that stress could contribute to the initial onset of FFA as with alopecia areata.26
A dental history was obtained in 24 patients to explore the possibility of FFA as a manifestation of contact allergy secondary to exposure to metal dental implants. Contact allergies to metal amalgam and gold alloy dental implants/fillings frequently have been described as presenting as oral lichen planus in the literature.27-34 Given the histologic overlap between oral lichen planus and LPP/FFA, it is worth exploring the possibility that LPP and FFA are other manifestations of contact allergic response. In our study, 100% of the patients who provided a dental history had metal amalgam implants and 33% had gold alloy implants. It is an interesting observation, but it should be noted that none of the patients in our study had undergone patch testing for contact allergies to the metals in their dental implants, and further studies are required to explore this hypothesis.
Frontal fibrosing alopecia is a difficult condition to treat. In our study, patients tried an average of 3 different treatments, the most common being topical steroids (72%), hydroxychloroquine (55%), and intralesional steroids (38%).
A PubMed search of articles indexed for MEDLINE using the terms randomized control trial and frontal fibrosing alopecia yielded no randomized controlled trials that have been performed to demonstrate the most efficacious treatments of FFA. However, one systematic review of 114 patients found 5α-RIs, antimalarials, and intralesional corticosteroids to yield the best responses in treating FFA.22 Another large, multicenter, retrospective study of 355 patients also demonstrated that 5α-RIs and intralesional corticosteroids minimized hair loss most effectively across treatment modalities.15 One treatment that was not discussed in either study but was utilized in ours was the UVB excimer laser, which has been demonstrated to induce T-cell apoptosis and decrease inflammation in psoriasis but has been infrequently studied in the use of FFA or LPP. In one study of 13 patients with LPP, excimer laser treatment was successful in reducing inflammatory symptoms and improving hair loss.35 Our results reaffirm that laser therapy could be considered more frequently as a treatment of FFA.
This study is subject to several limitations. The study size was comprised of a relatively small number of patients with the condition. Additionally, only one-third of patients contacted agreed to participate in the study, and therefore the responses received may not be completely representative of all FFA patients. With a retrospective study, there is potential for recall bias in the data that are collected. Physician chart correlation to patient responses could not be reliably performed due to inconsistent documentation, care received outside our medical system, and prolonged or loss to follow-up. Another concern is that not all diagnoses of FFA in this study were biopsy confirmed. In one patient with systemic lupus erythematous who declined biopsy, it cannot be confirmed that her etiology of scarring alopecia was FFA rather than discoid lupus erythematous. Finally, because patients were treated with multiple medications, often concurrently, it was difficult to parse out which medications were efficacious and which were not. Despite these limitations, the findings in the study add to the growing literature about a rare but increasingly prevalent presentation.
Conclusion
Frontal fibrosing alopecia is a condition that predominantly affects white postmenopausal women but should not be overlooked in other demographics; higher socioeconomic status and nonsmoking are consistent with cases of FFA worldwide. Alopecia frequently involves other body hair, particularly the eyebrows, and is commonly associated with pruritus and pain. Many patients can identify an inciting event, usually stress, a health crisis, or new external exposures that they believe to have triggered the event. Consistent with data about LPP, FFA is frequently associated with autoimmune conditions, particularly hypothyroidism. A substantial portion of patients with FFA have had metal amalgam or gold alloy dental implants placed, though no patch testing was done to confirm that these patients have a contact allergy to these metals. Treatment for the condition is difficult, but topical and intralesional steroids, hydroxychloroquine, calcineurin inhibitors, and excimer laser therapy are efficacious in a large proportion of patients. Nevertheless, further research through prospective randomized trials is necessary to determine the best treatment modalities for FFA. Frontal fibrosing alopecia is a scarring form of hair loss that causes substantial emotional distress; therefore, it is critical to continue to investigate its etiology and treatments to improve patient care.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Tosti A, Piraccini BM, Iorizzo M, et al. Frontal fibrosing alopecia in postmenopausal women. J Am Acad Dermatol. 2005;52:55-60.
- Moreno-Ramírez D, Camacho Martínez F. Frontal fibrosing alopecia: a survey in 16 patients. J Eur Acad Dermatol Venereol. 2005;19:700-705.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Georgala S, Katoulis AC, Befon A, et al. Treatment of postmenopausal frontal fibrosing alopecia with oral dutasteride. J Am Acad Dermatol. 2009;61:157-158.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Chew AL, Bashir SJ, Wain EM, et al. Expanding the spectrum of frontal fibrosing alopecia: a unifying concept. J Am Acad Dermatol. 2010;63:653-660.
- Miteva M, Camacho I, Romanelli P, et al. Acute hair loss on the limbs in frontal fibrosing alopecia: a clinicopathological study of two cases. Br J Dermatol. 2010;163:426-428.
- Abbas O, Chedraoui A, Ghosn S. Frontal fibrosing alopecia presenting with components of Piccardi-Lassueur-Graham-Little syndrome. J Am Acad Dermatol. 2007;57(2 suppl):S15-S18.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle’s epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Dlova NC, Jordaan HF, Skenjane A, et al. Frontal fibrosing alopecia: a clinical review of 20 black patients from South Africa. Br J Dermatol. 2013;169:939-941.
- Atanaskova Mesinkovska N, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Dlova N, Goh CL, Tosti A. Familial frontal fibrosing alopecia. Br J Dermatol. 2013;168:220-222.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Miteva M, Aber C, Torres F, et al. Frontal fibrosing alopecia occurring on scalp vitiligo: report of four cases. Br J Dermatol. 2011;165:445-447.
- Sato M, Saga K, Takahashi H. Postmenopausal frontal fibrosing alopecia in a Japanese woman with Sjögren’s syndrome. J Dermatol. 2008;35:729-731.
- Feldmann R, Harms M, Saurat JH. Postmenopausal frontal fibrosing alopecia. Hautarzt. 1996;47:533-536.
- Junqueira Ribeiro Pereira AF, Vincenzi C, Tosti A. Frontal fibrosing alopecia in two sisters. Br J Dermatol. 2010;162:1154-1155.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
- QuickFacts: St. Louis County, Missouri. United States Census Bureau website. https://www.census.gov/quickfacts/fact/table/stlouiscountymissouri/PST045217. Accessed February 6, 2019.
- State tobacco activities tracking and evaluation (STATE) system. State highlights. Centers for Disease Control and Prevention website. https://nccd.cdc.gov/STATESystem/rdPage.aspx?rdReport=OSH_STATE.Highlights. Accessed February 6, 2019.
- Miteva M, Whiting D, Harries M, et al. Frontal fibrosing alopecia in black patients. Br J Dermatol. 2012;167:208-210.
- Willemsen R, Vanderlinden J, Roseeuw D, et al. Increased history of childhood and lifetime traumatic events among adults with alopecia areata. J Am Acad Dermatol. 2009;60:388-393.
- Segura-Egea JJ, Bullón-Fernández P. Lichenoid reaction associated to amalgam restoration. Med Oral Patol Oral Cir Bucal. 2004;9:421-424.
- Laeijendecker R, van Joost T. Oral manifestations of gold allergy. J Am Acad Dermatol. 1994;30:205-209.
- Marcusson JA. Contact allergies to nickel sulfate, gold sodium thiosulfate and palladium chloride in patients claiming side-effects from dental alloy components. Contact Dermatitis. 1996;34:320-323.
- Nordlind K, Lidén S. Patch test reactions to metal salts in patients with oral mucosal lesions associated with amalgam restorations. Contact Dermatitis. 1992;27:157-160.
- Koch P, Bahmer FA. Oral lichenoid lesions, mercury hypersensitivity and combined hypersensitivity to mercury and other metals: histologically-proven reproduction of the reaction by patch testing with metal salts. Contact Dermatitis. 1995;33:323-328.
- Laine J, Kalimo K, Happonen RP. Contact allergy to dental restorative materials in patients with oral lichenoid lesions. Contact Dermatitis. 1997;36:141-146.
- Yiannias JA, el-Azhary RA, Hand JH, et al. Relevant contact sensitivities in patients with the diagnosis of oral lichen planus. J Am Acad Dermatol. 2000;42:177-182.
- Scalf LA, Fowler JF Jr, Morgan KW, et al. Dental metal allergy in patients with oral, cutaneous, and genital lichenoid reactions. Am J Contact Dermat. 2001;12:146-150.
- Navarini AA, Kolios AG, Prinz-Vavricka BM, et al. Low-dose excimer 308-nm laser for treatment of lichen planopilaris. Arch Dermatol. 2011;147:1325-1326.
Frontal fibrosing alopecia (FFA) is a form of lymphocytic cicatricial alopecia that presents as frontotemporal hairline recession, typically in postmenopausal women.1 The condition is considered to be a variant of lichen planopilaris (LPP) due to its similar histologic appearance.2 Loss of eyebrow1-11 and body5-11 hair also is commonly present in FFA, and histologic findings are identical to those for hair loss on the scalp,8,9 suggesting that FFA may be a form of generalized alopecia.
The pathogenesis of FFA is unknown, but several etiologies have been postulated. Some suggest that as a variant of LPP, FFA is a hair-specific autoimmune disorder characterized by a T cell–mediated immune reaction against epithelial hair follicle stem cells, leading to fibrosis and depletion of hair regeneration potential.12 In support of this theory, FFA has been associated with other autoimmune diseases including hypothyroidism,6,8,13-16 mucocutaneous lichen planus,8,15,17 vitiligo,15,18 Sjögren syndrome,19 and lichen sclerosus et atrophicus.15,20 Another hypothesis suggests that the proandrogenic state in postmenopausal women may be related to the disease process.1 This hypothesis is supported by the reported success of antiandrogen therapy with 5α-reductase inhibitors (5α-RIs) in stabilizing FFA.3-5,7 Finally, genetic16,21 and environmental factors related to smoking and socioeconomic status5 also have been postulated to be risk factors for FFA. A variety of treatments have shown varying success, including topical and intralesional corticosteroids, hydroxychloroquine, immunomodulators, antibiotics, and 5α-RIs.1,3-6,8,15,17,22 However, FFA is considered to be relatively difficult to treat and commonly progresses regardless of treatment before spontaneously stabilizing.2-4,6,8,10
Since its discovery in 1994,1 FFA has become increasingly prevalent, comprising 17% of new referrals for hair loss in one study (N=57).6 Although growing recognition of the condition likely plays a role in its increasing presentation, other unidentified factors may contribute to its expanding incidence. In this report, we describe the demographics, clinical features, and disease progression of 29 cases of FFA treated within our division using a series of surveys and chart reviews.
Methods
Upon receiving approval for the project from the institutional review board, we identified 29 patients who met the criteria for diagnosis of FFA through a chart review of all patients being treated for hair loss by clinics within the Washington University Division of Dermatology (St. Louis, Missouri). Diagnostic criteria for FFA included scarring alopecia in the frontotemporal distribution with associated perifollicular erythema or papules and, if performed, a scalp biopsy of the involved area of alopecia showing lymphocytic cicatricial alopecia, compatible with LPP. The diagnosis was confirmed by biopsy in 18 patients (62%), while the remainder of the diagnoses were made clinically. Most biopsy specimens were diagnosed by board-certified dermatopathologists at Washington University, with the remainder diagnosed by outside pathologists if the patient was initially diagnosed at another institution.
Patients meeting criteria for FFA were mailed a study consent form, as well as a 2-page survey to assess demographics, clinical features of hair loss, medical histories, social and family histories, and treatments utilized. After receiving consent from patients, survey results were collected and summarized. If there was any need for clarification of answers, follow-up questions were conducted via email prior to any data analysis that was performed.
For analysis of treatment response, patients were asked what treatments they had utilized and about the progression of their hair loss. Patients reporting stabilization of hair loss or hair regrowth were classified as treatment responsive. Patients who underwent multiple treatments were included in the analyses for each of those treatments. Physician records for treatment response were not correlated with patient responses due to inconsistent documentation, care received outside of our medical system, and prolonged or loss to follow-up. Physician-reported data were only used to identify qualifying patients and their biopsy results, as described above.
Results
Patient Demographic
Between October 2013 and May 2014, 29 patients with FFA were recruited into the study. Patients were diagnosed between January 2006 and December 2013. There were 28 female patients (97%) and 1 male patient (3%). The average age of disease onset was 55.4 years (range, 29–75 years). Twenty-five patients (86%) self-identified as non-Hispanic white, 3 patients (10%) as Asian, and 1 patient (3%) as black. Patients also appeared to be a more affluent group than the general St. Louis County population, with a median household income between $75,000 and $100,000. In comparison, the median household income reported in St. Louis County from 2008 to 2012 was $58,485.23 The patient population was primarily composed of nonsmokers, with 22 (76%) patients who had never smoked, 6 (21%) who were present smokers, and 1 (3%) smoked in the past. These results were comparable to the reported number of female smokers in Missouri.24
Clinicopathologic Features
The clinical features of FFA are described in Table 1. All patients had frontotemporal recession of the hairline with some degree of scarring and perifollicular erythema (Figure 1). Most patients also reported hair loss at other sites, including 25 patients (86%) with eyebrow hair loss, 18 (62%) with limb hair loss, 11 (38%) with axillary hair loss, 11 (38%) with pubic hair loss, and 1 (3%) with eyelash hair loss. Patients also frequently reported inflammatory symptoms, including 19 patients (66%) with itching, 18 (62%) with redness, 3 (10%) with pain, 2 (7%) with papular lesions, and 1 (3%) with sores and erosions on the skin. Regarding progression of hair loss over time, 16 patients (55%) reported stabilization of hair loss, 11 (38%) reported progressive hair loss, and 2 (7%) reported some hair regrowth. Thirteen patients (45%) identified some inciting event that they believed to have triggered the disease. Ten patients (35%) identified stress as the inciting event, and 5 patients (17%) specifically referred to health-related stressors, including hip-replacement surgery, new diagnoses of systemic diseases, starting new medications, and stopping hormone replacement therapy. Furthermore, 2 (7%) patients reported exposure to chemicals and pesticides as suspected triggers.
Typical biopsy results showed a perifollicular lymphocytic infiltrate and fibrosis surrounding the infundibulum and isthmus of hair follicles (Figure 2). There were associated vacuolar changes in the basal layer and scattered dyskeratosis throughout the follicular epithelium. As the disease progressed to end-stage scarring, there was marked reduction in the number of hair follicles, which were replaced by fibrous tracts, and a disappearance of the previous inflammatory infiltrate.
Medical History
Of the 26 female patients who provided data about menopause status at time of disease onset, 16 (62%) were postmenopausal, 5 (19%) were menopausal, and 5 (19%) were premenopausal. Of the 28 female patients in the study, 8 (29%) had a history of hysterectomy and 2 (7%) also had surgically induced menopause through bilateral surgical oophorectomy. Twenty-four patients (86%) had a childbearing history, with an average of 2.3 children. Twelve patients (43%) reported use of hormone replacement therapy after menopause. Twelve patients (43%) also reported a history of oral contraceptive use.
Table 2 describes the comorbidities of all 29 patients. A history of autoimmune disease was prominent, found in 16 patients (55%). Thirteen patients (45%) reported thyroid disease, including 10 patients (35%) with hypothyroidism. Additionally, 8 patients (28%) had a history of mucocutaneous lichen planus, 2 (7%) of psoriasis/psoriatic arthritis, 1 (3%) of vitiligo, 1 (3%) of systemic lupus erythematosus, 1 (3%) of iritis, 1 (3%) of Sjögren syndrome, and 1 (3%) of ulcerative colitis. Six patients (21%) also reported a history of breast cancer.
A dental history was obtained in 24 patients. All 24 patients reported having some dental implant or filling placed. Twenty-four patients (100%) had a history of metal amalgam implants, 8 (33%) had gold alloy implants, 4 (17%) had composite resin implants, and 3 (13%) had porcelain implants. Two patients had metal amalgam implants that had since been replaced by nonmetal implants. Both patients reported no change in their clinical conditions with removal of the metal implants. Six of 8 patients (75%) with mucocutaneous lichen planus reported having dental implants. Of them, all 6 patients (100%) reported having metal amalgam implants, and 3 patients (50%) additionally reported having gold alloy implants.
Treatments
On average, patients were treated with 3 different therapies for FFA (range, 0–14). The treatments utilized are listed in Table 3, and responses to treatments are summarized in Table 4. Topical steroids were the most popular treatment modality and were used by 21 patients (72%). Approximately half of those patients reported treatment response with stabilization of hair loss or regrowth (n=11; 52%). Hydroxychloroquine was the second most commonly used modality (16 patients [55%]), with 10 of those patients (63%) reporting treatment response. Intralesional steroids were used in 11 patients (38%), with a treatment response in 36% (4/11) of those patients. Topical pimecrolimus and tacrolimus were used by 6 patients (21%), with 5 of those patients (83%) reporting treatment response. UVB excimer laser therapy was used on 3 patients (10%) with 100% treatment response.
Treatments with little or no treatment response to hair loss include doxycycline, minocycline, and topical minoxidil. Seven patients (24%) were treated with doxycycline or minocycline, all of whom reported no clinical response. Topical minoxidil was used by 3 patients (10%), with only 1 patient (33%) reporting stabilization of hair loss but no regrowth of hair. 5α-reductase inhibitors such as finasteride and dutasteride were only used by 1 patient (3%), who reported no treatment response. Other treatments that were rarely used include meloxicam (n=2), azathioprine (n=2), oral clindamycin (n=2), bimatoprost (n=1), quinacrine (n=1), cephalosporin (n=1), prednisone (n=1), isotretinoin (n=1), methotrexate (n=1), spironolactone (n=1), topical clindamycin (n=1), and laser hair removal (n=1). Of these, only meloxicam and quinacrine were anecdotally associated with stabilization of hair loss, while the rest of the treatments were associated with progressive hair loss despite therapy.
Comment
Frontal fibrosing alopecia is a form of cicatricial alopecia considered to be a clinical subset of LPP. Although the pathogeneses of both diseases are poorly understood, LPP is the better-studied model and is generally considered to be an autoimmune disease specific to the hair follicle, involving a cell-mediated inflammatory response to epithelial hair follicle stem cells.12 In support of this hypothesis, FFA and LPP have been frequently associated with autoimmune diseases, particularly with hypothyroidism.6,13-15 We found that 55% of our patients had a history of autoimmune disease, including 35% with hypothyroidism, 28% with mucocutaneous lichen planus, 7% with psoriasis, 3% with vitiligo, 3% with systemic lupus erythematosus, 3% with iritis, 3% with Sjögren syndrome, and 3% with ulcerative colitis. The link between FFA and hypothyroidism has been the best studied, with a large study by Atanskova Mesinkovska et al14 finding that 34% of 166 patients with LPP and FFA have some kind of thyroid disease and 29% have hypothyroidism. Fron
Although FFA has been classically described to affect postmenopausal women, recent studies have consistently identified that premenopausal women4-6,8,16,17 and men14,16 also can be affected by the condition. In our patient cohort, there was 1 male patient (3%), and a substantial number of the female patients were premenopausal (19%) and menopausal (19%) at the time of disease onset. Most of the patients studied were white; Asian and black patients were a consistent minority across FFA studies,5,13-16,25 highlighting the importance of screening for FFA in all demographics.
In our study, FFA patients also appeared to be more affluent than the general population and were predominantly nonsmokers (76%). These statistics are consistent with the United Kingdom population studied by MacDonald et al,6 which demonstrated a higher socioeconomic status and higher incidence of nonsmoking in their cases of FFA. Another large retrospective study of FFA patients in Spain found that 87% of their FFA cases (N=355) were nonsmokers, though they did not note a difference from the general unaffected population.15 In our study, we replicated these trends, finding an above average affluence level and a high but not statistically significant incidence of nonsmokers. Although it is not clear how socioeconomic status or smoking factors into the pathology of FFA, these studies may show a general trend in the environmental demographics of the disease.
Clinically, patients with FFA typically present with hair loss of the scalp as well as other sites. The eyebrows are the most common site to be affected outside of the scalp, affecting 86% of our patients, whereas eyelashes are the least commonly affected, presenting in only 3% of our patients. Body hair loss also is common, with almost two-thirds of our cohort reporting hair loss on the limbs and more than one-third reporting loss of axillary and pubic hair. These findings are consistent with those of other studies.3-6,8,13,15 Eyelash loss, body hair loss, and facial papules have been found to be associated with more severe forms of FFA,15 though we did not investigate these forms in our study. Inflammatory symptoms are common, with pruritus affecting 66% of our patients and pain affecting 10% of patients, consistent with the published literature.3,13,15,17
Multiple studies have shown that female FFA patients have a higher incidence of hysterectomies in their medical history.5,8,15 This observation has been used to further support the hypothesis that a change in sex hormone balance may trigger the initial onset of disease.5,8,15 A considerable number of the female patients in our study had also undergone hysterectomies (29%). Only 2 patients (7%) underwent premature surgical menopause through bilateral removal of the ovaries, and neither of these patients had abnormally early onset of FFA (age at onset, 52 and 65 years). Many patients in our study also reported a history of pharmacologic manipulation of sex hormones with hormone replacement therapy (43%) and oral contraceptive use (43%). However, patients with FFA have not been identified to have abnormal hormone levels compared to unaffected postmenopausal women.1 Additionally, the disease does not exclusively affect androgen-dependent hair, as indicated by the high prevalence of eyebrow hair loss. We hypothesize that the link between increased prevalence of hysterectomy and FFA is not due to hormonal changes but rather from the stresses related to the hysterectomy or associated conditions that required the surgery. In our study, 35% of patients identified stress as the inciting event prior to their onset of hair loss, with 17% specifically referring to health-related stress such as surgery or new diagnoses as the cause. Although this pattern is purely observational, it is valuable to consider that stress could contribute to the initial onset of FFA as with alopecia areata.26
A dental history was obtained in 24 patients to explore the possibility of FFA as a manifestation of contact allergy secondary to exposure to metal dental implants. Contact allergies to metal amalgam and gold alloy dental implants/fillings frequently have been described as presenting as oral lichen planus in the literature.27-34 Given the histologic overlap between oral lichen planus and LPP/FFA, it is worth exploring the possibility that LPP and FFA are other manifestations of contact allergic response. In our study, 100% of the patients who provided a dental history had metal amalgam implants and 33% had gold alloy implants. It is an interesting observation, but it should be noted that none of the patients in our study had undergone patch testing for contact allergies to the metals in their dental implants, and further studies are required to explore this hypothesis.
Frontal fibrosing alopecia is a difficult condition to treat. In our study, patients tried an average of 3 different treatments, the most common being topical steroids (72%), hydroxychloroquine (55%), and intralesional steroids (38%).
A PubMed search of articles indexed for MEDLINE using the terms randomized control trial and frontal fibrosing alopecia yielded no randomized controlled trials that have been performed to demonstrate the most efficacious treatments of FFA. However, one systematic review of 114 patients found 5α-RIs, antimalarials, and intralesional corticosteroids to yield the best responses in treating FFA.22 Another large, multicenter, retrospective study of 355 patients also demonstrated that 5α-RIs and intralesional corticosteroids minimized hair loss most effectively across treatment modalities.15 One treatment that was not discussed in either study but was utilized in ours was the UVB excimer laser, which has been demonstrated to induce T-cell apoptosis and decrease inflammation in psoriasis but has been infrequently studied in the use of FFA or LPP. In one study of 13 patients with LPP, excimer laser treatment was successful in reducing inflammatory symptoms and improving hair loss.35 Our results reaffirm that laser therapy could be considered more frequently as a treatment of FFA.
This study is subject to several limitations. The study size was comprised of a relatively small number of patients with the condition. Additionally, only one-third of patients contacted agreed to participate in the study, and therefore the responses received may not be completely representative of all FFA patients. With a retrospective study, there is potential for recall bias in the data that are collected. Physician chart correlation to patient responses could not be reliably performed due to inconsistent documentation, care received outside our medical system, and prolonged or loss to follow-up. Another concern is that not all diagnoses of FFA in this study were biopsy confirmed. In one patient with systemic lupus erythematous who declined biopsy, it cannot be confirmed that her etiology of scarring alopecia was FFA rather than discoid lupus erythematous. Finally, because patients were treated with multiple medications, often concurrently, it was difficult to parse out which medications were efficacious and which were not. Despite these limitations, the findings in the study add to the growing literature about a rare but increasingly prevalent presentation.
Conclusion
Frontal fibrosing alopecia is a condition that predominantly affects white postmenopausal women but should not be overlooked in other demographics; higher socioeconomic status and nonsmoking are consistent with cases of FFA worldwide. Alopecia frequently involves other body hair, particularly the eyebrows, and is commonly associated with pruritus and pain. Many patients can identify an inciting event, usually stress, a health crisis, or new external exposures that they believe to have triggered the event. Consistent with data about LPP, FFA is frequently associated with autoimmune conditions, particularly hypothyroidism. A substantial portion of patients with FFA have had metal amalgam or gold alloy dental implants placed, though no patch testing was done to confirm that these patients have a contact allergy to these metals. Treatment for the condition is difficult, but topical and intralesional steroids, hydroxychloroquine, calcineurin inhibitors, and excimer laser therapy are efficacious in a large proportion of patients. Nevertheless, further research through prospective randomized trials is necessary to determine the best treatment modalities for FFA. Frontal fibrosing alopecia is a scarring form of hair loss that causes substantial emotional distress; therefore, it is critical to continue to investigate its etiology and treatments to improve patient care.
Frontal fibrosing alopecia (FFA) is a form of lymphocytic cicatricial alopecia that presents as frontotemporal hairline recession, typically in postmenopausal women.1 The condition is considered to be a variant of lichen planopilaris (LPP) due to its similar histologic appearance.2 Loss of eyebrow1-11 and body5-11 hair also is commonly present in FFA, and histologic findings are identical to those for hair loss on the scalp,8,9 suggesting that FFA may be a form of generalized alopecia.
The pathogenesis of FFA is unknown, but several etiologies have been postulated. Some suggest that as a variant of LPP, FFA is a hair-specific autoimmune disorder characterized by a T cell–mediated immune reaction against epithelial hair follicle stem cells, leading to fibrosis and depletion of hair regeneration potential.12 In support of this theory, FFA has been associated with other autoimmune diseases including hypothyroidism,6,8,13-16 mucocutaneous lichen planus,8,15,17 vitiligo,15,18 Sjögren syndrome,19 and lichen sclerosus et atrophicus.15,20 Another hypothesis suggests that the proandrogenic state in postmenopausal women may be related to the disease process.1 This hypothesis is supported by the reported success of antiandrogen therapy with 5α-reductase inhibitors (5α-RIs) in stabilizing FFA.3-5,7 Finally, genetic16,21 and environmental factors related to smoking and socioeconomic status5 also have been postulated to be risk factors for FFA. A variety of treatments have shown varying success, including topical and intralesional corticosteroids, hydroxychloroquine, immunomodulators, antibiotics, and 5α-RIs.1,3-6,8,15,17,22 However, FFA is considered to be relatively difficult to treat and commonly progresses regardless of treatment before spontaneously stabilizing.2-4,6,8,10
Since its discovery in 1994,1 FFA has become increasingly prevalent, comprising 17% of new referrals for hair loss in one study (N=57).6 Although growing recognition of the condition likely plays a role in its increasing presentation, other unidentified factors may contribute to its expanding incidence. In this report, we describe the demographics, clinical features, and disease progression of 29 cases of FFA treated within our division using a series of surveys and chart reviews.
Methods
Upon receiving approval for the project from the institutional review board, we identified 29 patients who met the criteria for diagnosis of FFA through a chart review of all patients being treated for hair loss by clinics within the Washington University Division of Dermatology (St. Louis, Missouri). Diagnostic criteria for FFA included scarring alopecia in the frontotemporal distribution with associated perifollicular erythema or papules and, if performed, a scalp biopsy of the involved area of alopecia showing lymphocytic cicatricial alopecia, compatible with LPP. The diagnosis was confirmed by biopsy in 18 patients (62%), while the remainder of the diagnoses were made clinically. Most biopsy specimens were diagnosed by board-certified dermatopathologists at Washington University, with the remainder diagnosed by outside pathologists if the patient was initially diagnosed at another institution.
Patients meeting criteria for FFA were mailed a study consent form, as well as a 2-page survey to assess demographics, clinical features of hair loss, medical histories, social and family histories, and treatments utilized. After receiving consent from patients, survey results were collected and summarized. If there was any need for clarification of answers, follow-up questions were conducted via email prior to any data analysis that was performed.
For analysis of treatment response, patients were asked what treatments they had utilized and about the progression of their hair loss. Patients reporting stabilization of hair loss or hair regrowth were classified as treatment responsive. Patients who underwent multiple treatments were included in the analyses for each of those treatments. Physician records for treatment response were not correlated with patient responses due to inconsistent documentation, care received outside of our medical system, and prolonged or loss to follow-up. Physician-reported data were only used to identify qualifying patients and their biopsy results, as described above.
Results
Patient Demographic
Between October 2013 and May 2014, 29 patients with FFA were recruited into the study. Patients were diagnosed between January 2006 and December 2013. There were 28 female patients (97%) and 1 male patient (3%). The average age of disease onset was 55.4 years (range, 29–75 years). Twenty-five patients (86%) self-identified as non-Hispanic white, 3 patients (10%) as Asian, and 1 patient (3%) as black. Patients also appeared to be a more affluent group than the general St. Louis County population, with a median household income between $75,000 and $100,000. In comparison, the median household income reported in St. Louis County from 2008 to 2012 was $58,485.23 The patient population was primarily composed of nonsmokers, with 22 (76%) patients who had never smoked, 6 (21%) who were present smokers, and 1 (3%) smoked in the past. These results were comparable to the reported number of female smokers in Missouri.24
Clinicopathologic Features
The clinical features of FFA are described in Table 1. All patients had frontotemporal recession of the hairline with some degree of scarring and perifollicular erythema (Figure 1). Most patients also reported hair loss at other sites, including 25 patients (86%) with eyebrow hair loss, 18 (62%) with limb hair loss, 11 (38%) with axillary hair loss, 11 (38%) with pubic hair loss, and 1 (3%) with eyelash hair loss. Patients also frequently reported inflammatory symptoms, including 19 patients (66%) with itching, 18 (62%) with redness, 3 (10%) with pain, 2 (7%) with papular lesions, and 1 (3%) with sores and erosions on the skin. Regarding progression of hair loss over time, 16 patients (55%) reported stabilization of hair loss, 11 (38%) reported progressive hair loss, and 2 (7%) reported some hair regrowth. Thirteen patients (45%) identified some inciting event that they believed to have triggered the disease. Ten patients (35%) identified stress as the inciting event, and 5 patients (17%) specifically referred to health-related stressors, including hip-replacement surgery, new diagnoses of systemic diseases, starting new medications, and stopping hormone replacement therapy. Furthermore, 2 (7%) patients reported exposure to chemicals and pesticides as suspected triggers.
Typical biopsy results showed a perifollicular lymphocytic infiltrate and fibrosis surrounding the infundibulum and isthmus of hair follicles (Figure 2). There were associated vacuolar changes in the basal layer and scattered dyskeratosis throughout the follicular epithelium. As the disease progressed to end-stage scarring, there was marked reduction in the number of hair follicles, which were replaced by fibrous tracts, and a disappearance of the previous inflammatory infiltrate.
Medical History
Of the 26 female patients who provided data about menopause status at time of disease onset, 16 (62%) were postmenopausal, 5 (19%) were menopausal, and 5 (19%) were premenopausal. Of the 28 female patients in the study, 8 (29%) had a history of hysterectomy and 2 (7%) also had surgically induced menopause through bilateral surgical oophorectomy. Twenty-four patients (86%) had a childbearing history, with an average of 2.3 children. Twelve patients (43%) reported use of hormone replacement therapy after menopause. Twelve patients (43%) also reported a history of oral contraceptive use.
Table 2 describes the comorbidities of all 29 patients. A history of autoimmune disease was prominent, found in 16 patients (55%). Thirteen patients (45%) reported thyroid disease, including 10 patients (35%) with hypothyroidism. Additionally, 8 patients (28%) had a history of mucocutaneous lichen planus, 2 (7%) of psoriasis/psoriatic arthritis, 1 (3%) of vitiligo, 1 (3%) of systemic lupus erythematosus, 1 (3%) of iritis, 1 (3%) of Sjögren syndrome, and 1 (3%) of ulcerative colitis. Six patients (21%) also reported a history of breast cancer.
A dental history was obtained in 24 patients. All 24 patients reported having some dental implant or filling placed. Twenty-four patients (100%) had a history of metal amalgam implants, 8 (33%) had gold alloy implants, 4 (17%) had composite resin implants, and 3 (13%) had porcelain implants. Two patients had metal amalgam implants that had since been replaced by nonmetal implants. Both patients reported no change in their clinical conditions with removal of the metal implants. Six of 8 patients (75%) with mucocutaneous lichen planus reported having dental implants. Of them, all 6 patients (100%) reported having metal amalgam implants, and 3 patients (50%) additionally reported having gold alloy implants.
Treatments
On average, patients were treated with 3 different therapies for FFA (range, 0–14). The treatments utilized are listed in Table 3, and responses to treatments are summarized in Table 4. Topical steroids were the most popular treatment modality and were used by 21 patients (72%). Approximately half of those patients reported treatment response with stabilization of hair loss or regrowth (n=11; 52%). Hydroxychloroquine was the second most commonly used modality (16 patients [55%]), with 10 of those patients (63%) reporting treatment response. Intralesional steroids were used in 11 patients (38%), with a treatment response in 36% (4/11) of those patients. Topical pimecrolimus and tacrolimus were used by 6 patients (21%), with 5 of those patients (83%) reporting treatment response. UVB excimer laser therapy was used on 3 patients (10%) with 100% treatment response.
Treatments with little or no treatment response to hair loss include doxycycline, minocycline, and topical minoxidil. Seven patients (24%) were treated with doxycycline or minocycline, all of whom reported no clinical response. Topical minoxidil was used by 3 patients (10%), with only 1 patient (33%) reporting stabilization of hair loss but no regrowth of hair. 5α-reductase inhibitors such as finasteride and dutasteride were only used by 1 patient (3%), who reported no treatment response. Other treatments that were rarely used include meloxicam (n=2), azathioprine (n=2), oral clindamycin (n=2), bimatoprost (n=1), quinacrine (n=1), cephalosporin (n=1), prednisone (n=1), isotretinoin (n=1), methotrexate (n=1), spironolactone (n=1), topical clindamycin (n=1), and laser hair removal (n=1). Of these, only meloxicam and quinacrine were anecdotally associated with stabilization of hair loss, while the rest of the treatments were associated with progressive hair loss despite therapy.
Comment
Frontal fibrosing alopecia is a form of cicatricial alopecia considered to be a clinical subset of LPP. Although the pathogeneses of both diseases are poorly understood, LPP is the better-studied model and is generally considered to be an autoimmune disease specific to the hair follicle, involving a cell-mediated inflammatory response to epithelial hair follicle stem cells.12 In support of this hypothesis, FFA and LPP have been frequently associated with autoimmune diseases, particularly with hypothyroidism.6,13-15 We found that 55% of our patients had a history of autoimmune disease, including 35% with hypothyroidism, 28% with mucocutaneous lichen planus, 7% with psoriasis, 3% with vitiligo, 3% with systemic lupus erythematosus, 3% with iritis, 3% with Sjögren syndrome, and 3% with ulcerative colitis. The link between FFA and hypothyroidism has been the best studied, with a large study by Atanskova Mesinkovska et al14 finding that 34% of 166 patients with LPP and FFA have some kind of thyroid disease and 29% have hypothyroidism. Fron
Although FFA has been classically described to affect postmenopausal women, recent studies have consistently identified that premenopausal women4-6,8,16,17 and men14,16 also can be affected by the condition. In our patient cohort, there was 1 male patient (3%), and a substantial number of the female patients were premenopausal (19%) and menopausal (19%) at the time of disease onset. Most of the patients studied were white; Asian and black patients were a consistent minority across FFA studies,5,13-16,25 highlighting the importance of screening for FFA in all demographics.
In our study, FFA patients also appeared to be more affluent than the general population and were predominantly nonsmokers (76%). These statistics are consistent with the United Kingdom population studied by MacDonald et al,6 which demonstrated a higher socioeconomic status and higher incidence of nonsmoking in their cases of FFA. Another large retrospective study of FFA patients in Spain found that 87% of their FFA cases (N=355) were nonsmokers, though they did not note a difference from the general unaffected population.15 In our study, we replicated these trends, finding an above average affluence level and a high but not statistically significant incidence of nonsmokers. Although it is not clear how socioeconomic status or smoking factors into the pathology of FFA, these studies may show a general trend in the environmental demographics of the disease.
Clinically, patients with FFA typically present with hair loss of the scalp as well as other sites. The eyebrows are the most common site to be affected outside of the scalp, affecting 86% of our patients, whereas eyelashes are the least commonly affected, presenting in only 3% of our patients. Body hair loss also is common, with almost two-thirds of our cohort reporting hair loss on the limbs and more than one-third reporting loss of axillary and pubic hair. These findings are consistent with those of other studies.3-6,8,13,15 Eyelash loss, body hair loss, and facial papules have been found to be associated with more severe forms of FFA,15 though we did not investigate these forms in our study. Inflammatory symptoms are common, with pruritus affecting 66% of our patients and pain affecting 10% of patients, consistent with the published literature.3,13,15,17
Multiple studies have shown that female FFA patients have a higher incidence of hysterectomies in their medical history.5,8,15 This observation has been used to further support the hypothesis that a change in sex hormone balance may trigger the initial onset of disease.5,8,15 A considerable number of the female patients in our study had also undergone hysterectomies (29%). Only 2 patients (7%) underwent premature surgical menopause through bilateral removal of the ovaries, and neither of these patients had abnormally early onset of FFA (age at onset, 52 and 65 years). Many patients in our study also reported a history of pharmacologic manipulation of sex hormones with hormone replacement therapy (43%) and oral contraceptive use (43%). However, patients with FFA have not been identified to have abnormal hormone levels compared to unaffected postmenopausal women.1 Additionally, the disease does not exclusively affect androgen-dependent hair, as indicated by the high prevalence of eyebrow hair loss. We hypothesize that the link between increased prevalence of hysterectomy and FFA is not due to hormonal changes but rather from the stresses related to the hysterectomy or associated conditions that required the surgery. In our study, 35% of patients identified stress as the inciting event prior to their onset of hair loss, with 17% specifically referring to health-related stress such as surgery or new diagnoses as the cause. Although this pattern is purely observational, it is valuable to consider that stress could contribute to the initial onset of FFA as with alopecia areata.26
A dental history was obtained in 24 patients to explore the possibility of FFA as a manifestation of contact allergy secondary to exposure to metal dental implants. Contact allergies to metal amalgam and gold alloy dental implants/fillings frequently have been described as presenting as oral lichen planus in the literature.27-34 Given the histologic overlap between oral lichen planus and LPP/FFA, it is worth exploring the possibility that LPP and FFA are other manifestations of contact allergic response. In our study, 100% of the patients who provided a dental history had metal amalgam implants and 33% had gold alloy implants. It is an interesting observation, but it should be noted that none of the patients in our study had undergone patch testing for contact allergies to the metals in their dental implants, and further studies are required to explore this hypothesis.
Frontal fibrosing alopecia is a difficult condition to treat. In our study, patients tried an average of 3 different treatments, the most common being topical steroids (72%), hydroxychloroquine (55%), and intralesional steroids (38%).
A PubMed search of articles indexed for MEDLINE using the terms randomized control trial and frontal fibrosing alopecia yielded no randomized controlled trials that have been performed to demonstrate the most efficacious treatments of FFA. However, one systematic review of 114 patients found 5α-RIs, antimalarials, and intralesional corticosteroids to yield the best responses in treating FFA.22 Another large, multicenter, retrospective study of 355 patients also demonstrated that 5α-RIs and intralesional corticosteroids minimized hair loss most effectively across treatment modalities.15 One treatment that was not discussed in either study but was utilized in ours was the UVB excimer laser, which has been demonstrated to induce T-cell apoptosis and decrease inflammation in psoriasis but has been infrequently studied in the use of FFA or LPP. In one study of 13 patients with LPP, excimer laser treatment was successful in reducing inflammatory symptoms and improving hair loss.35 Our results reaffirm that laser therapy could be considered more frequently as a treatment of FFA.
This study is subject to several limitations. The study size was comprised of a relatively small number of patients with the condition. Additionally, only one-third of patients contacted agreed to participate in the study, and therefore the responses received may not be completely representative of all FFA patients. With a retrospective study, there is potential for recall bias in the data that are collected. Physician chart correlation to patient responses could not be reliably performed due to inconsistent documentation, care received outside our medical system, and prolonged or loss to follow-up. Another concern is that not all diagnoses of FFA in this study were biopsy confirmed. In one patient with systemic lupus erythematous who declined biopsy, it cannot be confirmed that her etiology of scarring alopecia was FFA rather than discoid lupus erythematous. Finally, because patients were treated with multiple medications, often concurrently, it was difficult to parse out which medications were efficacious and which were not. Despite these limitations, the findings in the study add to the growing literature about a rare but increasingly prevalent presentation.
Conclusion
Frontal fibrosing alopecia is a condition that predominantly affects white postmenopausal women but should not be overlooked in other demographics; higher socioeconomic status and nonsmoking are consistent with cases of FFA worldwide. Alopecia frequently involves other body hair, particularly the eyebrows, and is commonly associated with pruritus and pain. Many patients can identify an inciting event, usually stress, a health crisis, or new external exposures that they believe to have triggered the event. Consistent with data about LPP, FFA is frequently associated with autoimmune conditions, particularly hypothyroidism. A substantial portion of patients with FFA have had metal amalgam or gold alloy dental implants placed, though no patch testing was done to confirm that these patients have a contact allergy to these metals. Treatment for the condition is difficult, but topical and intralesional steroids, hydroxychloroquine, calcineurin inhibitors, and excimer laser therapy are efficacious in a large proportion of patients. Nevertheless, further research through prospective randomized trials is necessary to determine the best treatment modalities for FFA. Frontal fibrosing alopecia is a scarring form of hair loss that causes substantial emotional distress; therefore, it is critical to continue to investigate its etiology and treatments to improve patient care.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Tosti A, Piraccini BM, Iorizzo M, et al. Frontal fibrosing alopecia in postmenopausal women. J Am Acad Dermatol. 2005;52:55-60.
- Moreno-Ramírez D, Camacho Martínez F. Frontal fibrosing alopecia: a survey in 16 patients. J Eur Acad Dermatol Venereol. 2005;19:700-705.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Georgala S, Katoulis AC, Befon A, et al. Treatment of postmenopausal frontal fibrosing alopecia with oral dutasteride. J Am Acad Dermatol. 2009;61:157-158.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Chew AL, Bashir SJ, Wain EM, et al. Expanding the spectrum of frontal fibrosing alopecia: a unifying concept. J Am Acad Dermatol. 2010;63:653-660.
- Miteva M, Camacho I, Romanelli P, et al. Acute hair loss on the limbs in frontal fibrosing alopecia: a clinicopathological study of two cases. Br J Dermatol. 2010;163:426-428.
- Abbas O, Chedraoui A, Ghosn S. Frontal fibrosing alopecia presenting with components of Piccardi-Lassueur-Graham-Little syndrome. J Am Acad Dermatol. 2007;57(2 suppl):S15-S18.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle’s epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Dlova NC, Jordaan HF, Skenjane A, et al. Frontal fibrosing alopecia: a clinical review of 20 black patients from South Africa. Br J Dermatol. 2013;169:939-941.
- Atanaskova Mesinkovska N, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Dlova N, Goh CL, Tosti A. Familial frontal fibrosing alopecia. Br J Dermatol. 2013;168:220-222.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Miteva M, Aber C, Torres F, et al. Frontal fibrosing alopecia occurring on scalp vitiligo: report of four cases. Br J Dermatol. 2011;165:445-447.
- Sato M, Saga K, Takahashi H. Postmenopausal frontal fibrosing alopecia in a Japanese woman with Sjögren’s syndrome. J Dermatol. 2008;35:729-731.
- Feldmann R, Harms M, Saurat JH. Postmenopausal frontal fibrosing alopecia. Hautarzt. 1996;47:533-536.
- Junqueira Ribeiro Pereira AF, Vincenzi C, Tosti A. Frontal fibrosing alopecia in two sisters. Br J Dermatol. 2010;162:1154-1155.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
- QuickFacts: St. Louis County, Missouri. United States Census Bureau website. https://www.census.gov/quickfacts/fact/table/stlouiscountymissouri/PST045217. Accessed February 6, 2019.
- State tobacco activities tracking and evaluation (STATE) system. State highlights. Centers for Disease Control and Prevention website. https://nccd.cdc.gov/STATESystem/rdPage.aspx?rdReport=OSH_STATE.Highlights. Accessed February 6, 2019.
- Miteva M, Whiting D, Harries M, et al. Frontal fibrosing alopecia in black patients. Br J Dermatol. 2012;167:208-210.
- Willemsen R, Vanderlinden J, Roseeuw D, et al. Increased history of childhood and lifetime traumatic events among adults with alopecia areata. J Am Acad Dermatol. 2009;60:388-393.
- Segura-Egea JJ, Bullón-Fernández P. Lichenoid reaction associated to amalgam restoration. Med Oral Patol Oral Cir Bucal. 2004;9:421-424.
- Laeijendecker R, van Joost T. Oral manifestations of gold allergy. J Am Acad Dermatol. 1994;30:205-209.
- Marcusson JA. Contact allergies to nickel sulfate, gold sodium thiosulfate and palladium chloride in patients claiming side-effects from dental alloy components. Contact Dermatitis. 1996;34:320-323.
- Nordlind K, Lidén S. Patch test reactions to metal salts in patients with oral mucosal lesions associated with amalgam restorations. Contact Dermatitis. 1992;27:157-160.
- Koch P, Bahmer FA. Oral lichenoid lesions, mercury hypersensitivity and combined hypersensitivity to mercury and other metals: histologically-proven reproduction of the reaction by patch testing with metal salts. Contact Dermatitis. 1995;33:323-328.
- Laine J, Kalimo K, Happonen RP. Contact allergy to dental restorative materials in patients with oral lichenoid lesions. Contact Dermatitis. 1997;36:141-146.
- Yiannias JA, el-Azhary RA, Hand JH, et al. Relevant contact sensitivities in patients with the diagnosis of oral lichen planus. J Am Acad Dermatol. 2000;42:177-182.
- Scalf LA, Fowler JF Jr, Morgan KW, et al. Dental metal allergy in patients with oral, cutaneous, and genital lichenoid reactions. Am J Contact Dermat. 2001;12:146-150.
- Navarini AA, Kolios AG, Prinz-Vavricka BM, et al. Low-dose excimer 308-nm laser for treatment of lichen planopilaris. Arch Dermatol. 2011;147:1325-1326.
- Kossard S. Postmenopausal frontal fibrosing alopecia: scarring alopecia in a pattern distribution. Arch Dermatol. 1994;130:770-774.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Tosti A, Piraccini BM, Iorizzo M, et al. Frontal fibrosing alopecia in postmenopausal women. J Am Acad Dermatol. 2005;52:55-60.
- Moreno-Ramírez D, Camacho Martínez F. Frontal fibrosing alopecia: a survey in 16 patients. J Eur Acad Dermatol Venereol. 2005;19:700-705.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755.
- MacDonald A, Clark C, Holmes S. Frontal fibrosing alopecia: a review of 60 cases. J Am Acad Dermatol. 2012;67:955-961.
- Georgala S, Katoulis AC, Befon A, et al. Treatment of postmenopausal frontal fibrosing alopecia with oral dutasteride. J Am Acad Dermatol. 2009;61:157-158.
- Tan KT, Messenger AG. Frontal fibrosing alopecia: clinical presentations and prognosis. Br J Dermatol. 2009;160:75-79.
- Chew AL, Bashir SJ, Wain EM, et al. Expanding the spectrum of frontal fibrosing alopecia: a unifying concept. J Am Acad Dermatol. 2010;63:653-660.
- Miteva M, Camacho I, Romanelli P, et al. Acute hair loss on the limbs in frontal fibrosing alopecia: a clinicopathological study of two cases. Br J Dermatol. 2010;163:426-428.
- Abbas O, Chedraoui A, Ghosn S. Frontal fibrosing alopecia presenting with components of Piccardi-Lassueur-Graham-Little syndrome. J Am Acad Dermatol. 2007;57(2 suppl):S15-S18.
- Harries MJ, Meyer K, Chaudhry I, et al. Lichen planopilaris is characterized by immune privilege collapse of the hair follicle’s epithelial stem cell niche. J Pathol. 2013;231:236-247.
- Dlova NC, Jordaan HF, Skenjane A, et al. Frontal fibrosing alopecia: a clinical review of 20 black patients from South Africa. Br J Dermatol. 2013;169:939-941.
- Atanaskova Mesinkovska N, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Vañó-Galván S, Molina-Ruiz AM, Serrano-Falcón C, et al. Frontal fibrosing alopecia: a multicenter review of 355 patients. J Am Acad Dermatol. 2014;70:670-678.
- Dlova N, Goh CL, Tosti A. Familial frontal fibrosing alopecia. Br J Dermatol. 2013;168:220-222.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Miteva M, Aber C, Torres F, et al. Frontal fibrosing alopecia occurring on scalp vitiligo: report of four cases. Br J Dermatol. 2011;165:445-447.
- Sato M, Saga K, Takahashi H. Postmenopausal frontal fibrosing alopecia in a Japanese woman with Sjögren’s syndrome. J Dermatol. 2008;35:729-731.
- Feldmann R, Harms M, Saurat JH. Postmenopausal frontal fibrosing alopecia. Hautarzt. 1996;47:533-536.
- Junqueira Ribeiro Pereira AF, Vincenzi C, Tosti A. Frontal fibrosing alopecia in two sisters. Br J Dermatol. 2010;162:1154-1155.
- Rácz E, Gho C, Moorman PW, et al. Treatment of frontal fibrosing alopecia and lichen planopilaris: a systematic review. J Eur Acad Dermatol Venereol. 2013;27:1461-1470.
- QuickFacts: St. Louis County, Missouri. United States Census Bureau website. https://www.census.gov/quickfacts/fact/table/stlouiscountymissouri/PST045217. Accessed February 6, 2019.
- State tobacco activities tracking and evaluation (STATE) system. State highlights. Centers for Disease Control and Prevention website. https://nccd.cdc.gov/STATESystem/rdPage.aspx?rdReport=OSH_STATE.Highlights. Accessed February 6, 2019.
- Miteva M, Whiting D, Harries M, et al. Frontal fibrosing alopecia in black patients. Br J Dermatol. 2012;167:208-210.
- Willemsen R, Vanderlinden J, Roseeuw D, et al. Increased history of childhood and lifetime traumatic events among adults with alopecia areata. J Am Acad Dermatol. 2009;60:388-393.
- Segura-Egea JJ, Bullón-Fernández P. Lichenoid reaction associated to amalgam restoration. Med Oral Patol Oral Cir Bucal. 2004;9:421-424.
- Laeijendecker R, van Joost T. Oral manifestations of gold allergy. J Am Acad Dermatol. 1994;30:205-209.
- Marcusson JA. Contact allergies to nickel sulfate, gold sodium thiosulfate and palladium chloride in patients claiming side-effects from dental alloy components. Contact Dermatitis. 1996;34:320-323.
- Nordlind K, Lidén S. Patch test reactions to metal salts in patients with oral mucosal lesions associated with amalgam restorations. Contact Dermatitis. 1992;27:157-160.
- Koch P, Bahmer FA. Oral lichenoid lesions, mercury hypersensitivity and combined hypersensitivity to mercury and other metals: histologically-proven reproduction of the reaction by patch testing with metal salts. Contact Dermatitis. 1995;33:323-328.
- Laine J, Kalimo K, Happonen RP. Contact allergy to dental restorative materials in patients with oral lichenoid lesions. Contact Dermatitis. 1997;36:141-146.
- Yiannias JA, el-Azhary RA, Hand JH, et al. Relevant contact sensitivities in patients with the diagnosis of oral lichen planus. J Am Acad Dermatol. 2000;42:177-182.
- Scalf LA, Fowler JF Jr, Morgan KW, et al. Dental metal allergy in patients with oral, cutaneous, and genital lichenoid reactions. Am J Contact Dermat. 2001;12:146-150.
- Navarini AA, Kolios AG, Prinz-Vavricka BM, et al. Low-dose excimer 308-nm laser for treatment of lichen planopilaris. Arch Dermatol. 2011;147:1325-1326.
Practice Points
- Frontal fibrosing alopecia (FFA) may be associated with other autoimmune conditions, and patients should be screened accordingly.
- The most efficacious treatments for FFA include topical and intralesional steroids, hydroxychloroquine, calcineurin inhibitors, and excimer laser therapy.
- A stressful precipitating event or metal dental implants/fillings are 2 possible environmental triggers for this condition.
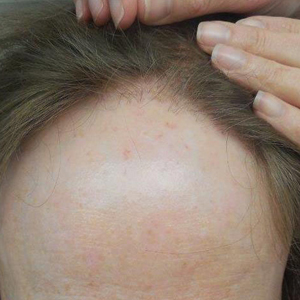
New Topical Treatments for Psoriasis

Debunking Acne Myths: Patients Need Photoprotection, Not a Tan
Myth: Getting a Tan Helps Improve Acne
Acne has a multifaceted impact on patients, and facial acne in particular can impair self-image, psychological well-being, and ability to develop relationships. Patients cope with the clinical presentation of the disease in various ways, such as wearing makeup to cover blemishes, changing their hair color or diet, or getting regular facials. A common misconception is that a tan will help resolve acne.
A 2014 study on the burden of adult female acne (N=208) found that 5.3% of patients go to tanning salons or lay out in the sun to cope with their acne and 17% use self-tanning products to make their acne less visible. Many patients (40%) also believed that sunscreen exacerbates acne. Furthermore, a study of adolescents in Stockholm reported that those with acne, eczema, or psoriasis used sunbeds more than others without skin diseases.
The risk of developing skin cancer from sun exposure or UV light has been well established, and there is no evidence that UV light helps improve acne. A 2015 review of the literature on tanning bed use and phototherapy associated with treatment of conditions such as acne reported that experimental trials have been conducted for various light source therapies (eg, blue light, red-blue light, photodynamic therapy); however, there is no direct evidence for UV light.
In fact, acne patients should be counseled on the importance of photoprotection. Many acne therapies leave patients predisposed to UV damage, and UV damage generates free radical formation, which has been implicated in acne flares.
Expert Commentary
Often I hear from patients they feel tanning helps improve their acne. I tell them there is no evidence this is at all true. Just like cream makeup can camouflage acne, so too can tanning. But, trying to hide your blemishes does not actually help nor treat them. And moreover, tanning is so dangerous for potential skin cancer later in life.
—Lawrence J. Green, MD (Washington, DC)
Boldeman C, Beitner H, Jansson B, et al. Sunbed use in relation to phenotype, erythema, sunscreen use and skin diseases. a questionnaire survey among Swedish adolescents. Br J Dermatol. 1996;135:712-726.
Bowe WP, Kircik LH. The importance of photoprotection and moisturization in treating acne vulgaris. J Drugs Dermatol. 2014;13:S89-S94.
Radack KP, Farhangian ME, Anderson KL, et al. A review of the use of tanning beds as a dermatological treatment. Dermatol Ther (Heidelb). 2015;5:37-51.
Tanghetti EA, Kawata AK, Daniels SR, et al. Understanding the burden of adult female acne. J Clin Aesthet Dermatol. 2014;7:22-30.
Myth: Getting a Tan Helps Improve Acne
Acne has a multifaceted impact on patients, and facial acne in particular can impair self-image, psychological well-being, and ability to develop relationships. Patients cope with the clinical presentation of the disease in various ways, such as wearing makeup to cover blemishes, changing their hair color or diet, or getting regular facials. A common misconception is that a tan will help resolve acne.
A 2014 study on the burden of adult female acne (N=208) found that 5.3% of patients go to tanning salons or lay out in the sun to cope with their acne and 17% use self-tanning products to make their acne less visible. Many patients (40%) also believed that sunscreen exacerbates acne. Furthermore, a study of adolescents in Stockholm reported that those with acne, eczema, or psoriasis used sunbeds more than others without skin diseases.
The risk of developing skin cancer from sun exposure or UV light has been well established, and there is no evidence that UV light helps improve acne. A 2015 review of the literature on tanning bed use and phototherapy associated with treatment of conditions such as acne reported that experimental trials have been conducted for various light source therapies (eg, blue light, red-blue light, photodynamic therapy); however, there is no direct evidence for UV light.
In fact, acne patients should be counseled on the importance of photoprotection. Many acne therapies leave patients predisposed to UV damage, and UV damage generates free radical formation, which has been implicated in acne flares.
Expert Commentary
Often I hear from patients they feel tanning helps improve their acne. I tell them there is no evidence this is at all true. Just like cream makeup can camouflage acne, so too can tanning. But, trying to hide your blemishes does not actually help nor treat them. And moreover, tanning is so dangerous for potential skin cancer later in life.
—Lawrence J. Green, MD (Washington, DC)
Myth: Getting a Tan Helps Improve Acne
Acne has a multifaceted impact on patients, and facial acne in particular can impair self-image, psychological well-being, and ability to develop relationships. Patients cope with the clinical presentation of the disease in various ways, such as wearing makeup to cover blemishes, changing their hair color or diet, or getting regular facials. A common misconception is that a tan will help resolve acne.
A 2014 study on the burden of adult female acne (N=208) found that 5.3% of patients go to tanning salons or lay out in the sun to cope with their acne and 17% use self-tanning products to make their acne less visible. Many patients (40%) also believed that sunscreen exacerbates acne. Furthermore, a study of adolescents in Stockholm reported that those with acne, eczema, or psoriasis used sunbeds more than others without skin diseases.
The risk of developing skin cancer from sun exposure or UV light has been well established, and there is no evidence that UV light helps improve acne. A 2015 review of the literature on tanning bed use and phototherapy associated with treatment of conditions such as acne reported that experimental trials have been conducted for various light source therapies (eg, blue light, red-blue light, photodynamic therapy); however, there is no direct evidence for UV light.
In fact, acne patients should be counseled on the importance of photoprotection. Many acne therapies leave patients predisposed to UV damage, and UV damage generates free radical formation, which has been implicated in acne flares.
Expert Commentary
Often I hear from patients they feel tanning helps improve their acne. I tell them there is no evidence this is at all true. Just like cream makeup can camouflage acne, so too can tanning. But, trying to hide your blemishes does not actually help nor treat them. And moreover, tanning is so dangerous for potential skin cancer later in life.
—Lawrence J. Green, MD (Washington, DC)
Boldeman C, Beitner H, Jansson B, et al. Sunbed use in relation to phenotype, erythema, sunscreen use and skin diseases. a questionnaire survey among Swedish adolescents. Br J Dermatol. 1996;135:712-726.
Bowe WP, Kircik LH. The importance of photoprotection and moisturization in treating acne vulgaris. J Drugs Dermatol. 2014;13:S89-S94.
Radack KP, Farhangian ME, Anderson KL, et al. A review of the use of tanning beds as a dermatological treatment. Dermatol Ther (Heidelb). 2015;5:37-51.
Tanghetti EA, Kawata AK, Daniels SR, et al. Understanding the burden of adult female acne. J Clin Aesthet Dermatol. 2014;7:22-30.
Boldeman C, Beitner H, Jansson B, et al. Sunbed use in relation to phenotype, erythema, sunscreen use and skin diseases. a questionnaire survey among Swedish adolescents. Br J Dermatol. 1996;135:712-726.
Bowe WP, Kircik LH. The importance of photoprotection and moisturization in treating acne vulgaris. J Drugs Dermatol. 2014;13:S89-S94.
Radack KP, Farhangian ME, Anderson KL, et al. A review of the use of tanning beds as a dermatological treatment. Dermatol Ther (Heidelb). 2015;5:37-51.
Tanghetti EA, Kawata AK, Daniels SR, et al. Understanding the burden of adult female acne. J Clin Aesthet Dermatol. 2014;7:22-30.

Growing Painful Nodule on the Lower Lip
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
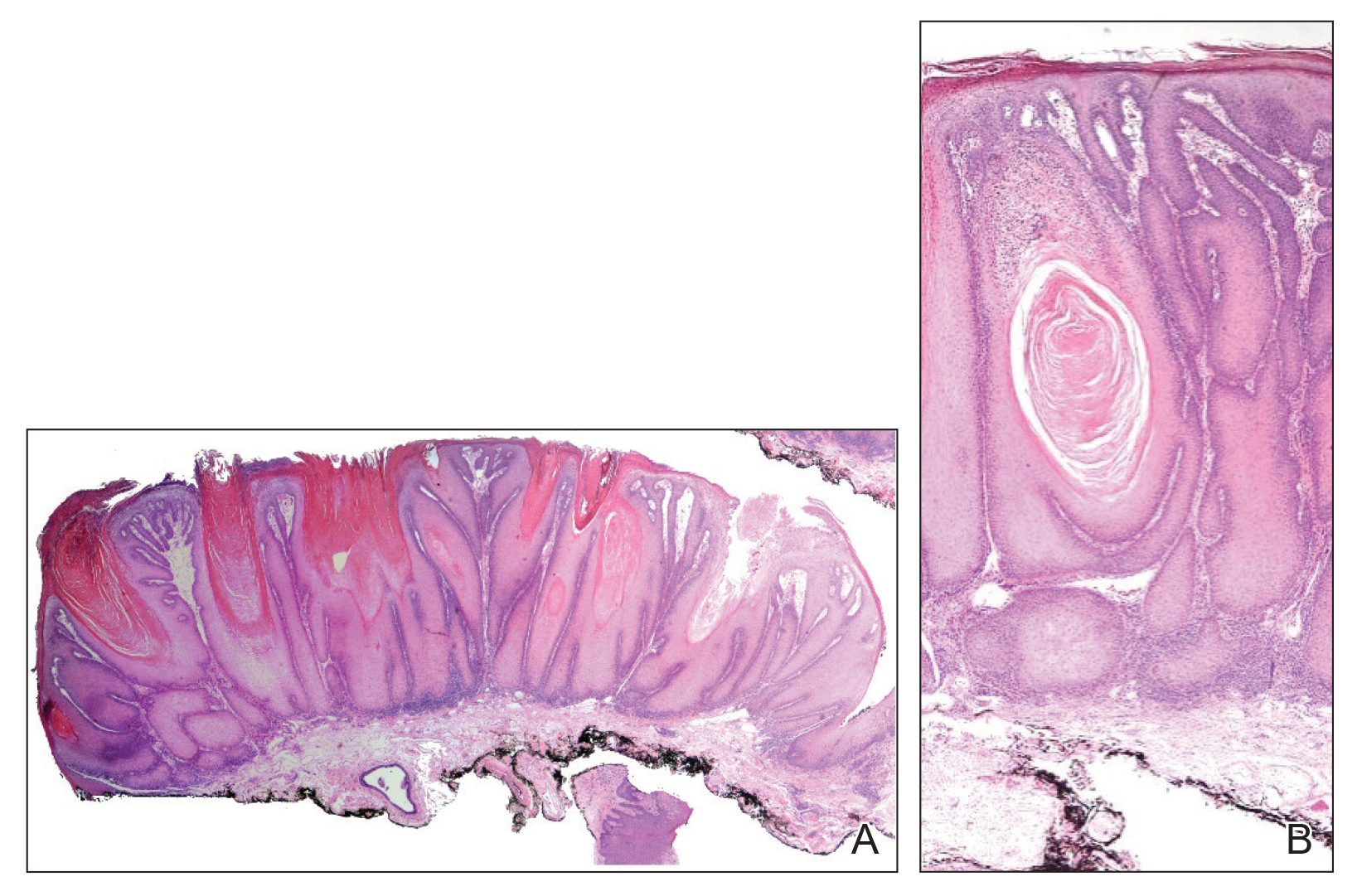
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
Laser Hair Removal: Survey of the Cutis Editorial Board
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on laser hair removal. Here’s what we found.
Do you perform laser hair removal in your practice?

More than half (58%) of dermatologists perform laser hair removal, while 12% have a PA/NP or aesthetician who performs this procedure on patients. Almost one-third (31%) of respondents do not perform laser hair removal.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Lasers are an important part of dermatology residency training and not a formal part of any other residency program. Therefore, dermatologists are best equipped to treat patients who are interested in removing unwanted hair safely and effectively. Dermatologists should advocate use of both a mask and a vacuum when performing these procedures to protect patients, themselves, residents, and staff from the resulting plume.
Next page: Incidence of treatment
Has the number of patients getting laser hair removal changed over the last 5 years?

Fewer patients are getting laser hair removal now vs 5 years ago, according to half of dermatologists; 42% reported that roughly the same number of patients are getting it done. Only 8% reported that more patients are getting laser hair removal.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Unfortunately, many patients often undergo hair laser treatments at spas by practitioners with limited laser training with sometimes adverse effects, including burns and scars. Therefore, we have a duty to educate our patients about laser safety and encourage them to seek treatment from a board-certified dermatologist.
Next page: Treatment areas
What area do you treat most often in women?
What area do you treat most often in men?
The majority of dermatologists (64%) treat the face most often in women, followed by the bikini area and legs (18% each). In men, half (52%) of dermatologists treat the back most often in men, followed by the neck (43%) and chest (5%).
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Before undergoing laser hair procedures, patients should be counseled that multiple treatments are often necessary, with the goal being reduction in hair density. Some hairs may still remain even after sufficient treatments. Some patients may be more comfortable with a topical numbing agent.
Next page: Lasers for darker skin types
What laser or device do you prefer to use for darker skin types?

Most dermatologists (79%) prefer to use the Nd:YAG 1064-nm laser for laser hair removal in darker skin types; 11% each prefer intense pulsed light or the alexandrite 755-nm laser.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The alexandrite 755-nm laser can be used safely in lighter skin types, while the Nd:YAG 1064-nm laser is preferred for darker skin types. It is also highly recommended to perform test spots in darker-skinned individuals.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Laser hair removal appears to be a safe and effective adjunctive therapy for adolescent hidradenitis patients. This has greatly increased the amount of laser hair removal treatments I perform as a pediatric dermatologist over the past 5 years.—Craig Burkhart, MD, MS, MPH (Chapel Hill, North Carolina)
Curbing unrealistic expectations is essential. It isn't magic. You won't have silky smooth, hairless skin after 1 treatment, or 2, or maybe ever. Discoloration, dyspigmentation, and scarring are possible. Making all of that clear in advance—in writing—will preempt 95% of postoperative complaints and angry phone calls.—Joseph Eastern, MD (Belleville, New Jersey)
In some states, laser hair removal is performed in medical spas without any dermatologist supervision. The lasers used in laser hair removal can be very harmful if used by nonphysicians who are not supervised.—Lawrence J. Green, MD (Washington, DC)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from January 7, 2019, to January 29, 2019. A total of 26 usable responses were received.
Georgesen C, Lipner SR. Surgical smoke: risk assessment and mitigation strategies. J Am Acad Dermatol. 2018;79:746-755.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on laser hair removal. Here’s what we found.
Do you perform laser hair removal in your practice?
More than half (58%) of dermatologists perform laser hair removal, while 12% have a PA/NP or aesthetician who performs this procedure on patients. Almost one-third (31%) of respondents do not perform laser hair removal.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Lasers are an important part of dermatology residency training and not a formal part of any other residency program. Therefore, dermatologists are best equipped to treat patients who are interested in removing unwanted hair safely and effectively. Dermatologists should advocate use of both a mask and a vacuum when performing these procedures to protect patients, themselves, residents, and staff from the resulting plume.
Next page: Incidence of treatment
Has the number of patients getting laser hair removal changed over the last 5 years?
Fewer patients are getting laser hair removal now vs 5 years ago, according to half of dermatologists; 42% reported that roughly the same number of patients are getting it done. Only 8% reported that more patients are getting laser hair removal.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Unfortunately, many patients often undergo hair laser treatments at spas by practitioners with limited laser training with sometimes adverse effects, including burns and scars. Therefore, we have a duty to educate our patients about laser safety and encourage them to seek treatment from a board-certified dermatologist.
Next page: Treatment areas
What area do you treat most often in women?
What area do you treat most often in men?
The majority of dermatologists (64%) treat the face most often in women, followed by the bikini area and legs (18% each). In men, half (52%) of dermatologists treat the back most often in men, followed by the neck (43%) and chest (5%).
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Before undergoing laser hair procedures, patients should be counseled that multiple treatments are often necessary, with the goal being reduction in hair density. Some hairs may still remain even after sufficient treatments. Some patients may be more comfortable with a topical numbing agent.
Next page: Lasers for darker skin types
What laser or device do you prefer to use for darker skin types?
Most dermatologists (79%) prefer to use the Nd:YAG 1064-nm laser for laser hair removal in darker skin types; 11% each prefer intense pulsed light or the alexandrite 755-nm laser.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The alexandrite 755-nm laser can be used safely in lighter skin types, while the Nd:YAG 1064-nm laser is preferred for darker skin types. It is also highly recommended to perform test spots in darker-skinned individuals.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Laser hair removal appears to be a safe and effective adjunctive therapy for adolescent hidradenitis patients. This has greatly increased the amount of laser hair removal treatments I perform as a pediatric dermatologist over the past 5 years.—Craig Burkhart, MD, MS, MPH (Chapel Hill, North Carolina)
Curbing unrealistic expectations is essential. It isn't magic. You won't have silky smooth, hairless skin after 1 treatment, or 2, or maybe ever. Discoloration, dyspigmentation, and scarring are possible. Making all of that clear in advance—in writing—will preempt 95% of postoperative complaints and angry phone calls.—Joseph Eastern, MD (Belleville, New Jersey)
In some states, laser hair removal is performed in medical spas without any dermatologist supervision. The lasers used in laser hair removal can be very harmful if used by nonphysicians who are not supervised.—Lawrence J. Green, MD (Washington, DC)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from January 7, 2019, to January 29, 2019. A total of 26 usable responses were received.
To improve patient care and outcomes, leading dermatologists from the Cutis Editorial Board answered 5 questions on laser hair removal. Here’s what we found.
Do you perform laser hair removal in your practice?
More than half (58%) of dermatologists perform laser hair removal, while 12% have a PA/NP or aesthetician who performs this procedure on patients. Almost one-third (31%) of respondents do not perform laser hair removal.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Lasers are an important part of dermatology residency training and not a formal part of any other residency program. Therefore, dermatologists are best equipped to treat patients who are interested in removing unwanted hair safely and effectively. Dermatologists should advocate use of both a mask and a vacuum when performing these procedures to protect patients, themselves, residents, and staff from the resulting plume.
Next page: Incidence of treatment
Has the number of patients getting laser hair removal changed over the last 5 years?
Fewer patients are getting laser hair removal now vs 5 years ago, according to half of dermatologists; 42% reported that roughly the same number of patients are getting it done. Only 8% reported that more patients are getting laser hair removal.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Unfortunately, many patients often undergo hair laser treatments at spas by practitioners with limited laser training with sometimes adverse effects, including burns and scars. Therefore, we have a duty to educate our patients about laser safety and encourage them to seek treatment from a board-certified dermatologist.
Next page: Treatment areas
What area do you treat most often in women?
What area do you treat most often in men?
The majority of dermatologists (64%) treat the face most often in women, followed by the bikini area and legs (18% each). In men, half (52%) of dermatologists treat the back most often in men, followed by the neck (43%) and chest (5%).
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
Before undergoing laser hair procedures, patients should be counseled that multiple treatments are often necessary, with the goal being reduction in hair density. Some hairs may still remain even after sufficient treatments. Some patients may be more comfortable with a topical numbing agent.
Next page: Lasers for darker skin types
What laser or device do you prefer to use for darker skin types?
Most dermatologists (79%) prefer to use the Nd:YAG 1064-nm laser for laser hair removal in darker skin types; 11% each prefer intense pulsed light or the alexandrite 755-nm laser.
Expert Commentary
Provided by Shari R. Lipner, MD, PhD (New York, New York)
The alexandrite 755-nm laser can be used safely in lighter skin types, while the Nd:YAG 1064-nm laser is preferred for darker skin types. It is also highly recommended to perform test spots in darker-skinned individuals.
Next page: More tips from derms
More Tips From Dermatologists
The dermatologists we polled had the following advice for their peers:
Laser hair removal appears to be a safe and effective adjunctive therapy for adolescent hidradenitis patients. This has greatly increased the amount of laser hair removal treatments I perform as a pediatric dermatologist over the past 5 years.—Craig Burkhart, MD, MS, MPH (Chapel Hill, North Carolina)
Curbing unrealistic expectations is essential. It isn't magic. You won't have silky smooth, hairless skin after 1 treatment, or 2, or maybe ever. Discoloration, dyspigmentation, and scarring are possible. Making all of that clear in advance—in writing—will preempt 95% of postoperative complaints and angry phone calls.—Joseph Eastern, MD (Belleville, New Jersey)
In some states, laser hair removal is performed in medical spas without any dermatologist supervision. The lasers used in laser hair removal can be very harmful if used by nonphysicians who are not supervised.—Lawrence J. Green, MD (Washington, DC)
About This Survey
The survey was fielded electronically to Cutis Editorial Board Members within the United States from January 7, 2019, to January 29, 2019. A total of 26 usable responses were received.
Georgesen C, Lipner SR. Surgical smoke: risk assessment and mitigation strategies. J Am Acad Dermatol. 2018;79:746-755.
Georgesen C, Lipner SR. Surgical smoke: risk assessment and mitigation strategies. J Am Acad Dermatol. 2018;79:746-755.
Chronic Lymphocytic Leukemia and Infiltrates Seen During Excision of Nonmelanoma Skin Cancer
To the Editor:
Specific characteristics of a lymphocytic infiltrate noted on frozen section histologic examination during Mohs micrographic surgery (MMS) tumor excision should raise suspicion of an underlying chronic lymphocytic leukemia (CLL). This infiltrate may be the presenting sign of the underlying leukemia and has variable presentation that may mimic aggressive features. The following 3 cases highlight this phenomenon.
A 74-year-old man (patient 1) with a medical history of multiple nonmelanoma skin cancers (NMSCs) presented for definitive treatment of a biopsy-proven infiltrative basal cell carcinoma involving the right infra-auricular region. Mohs section histologic evaluation revealed patches of lymphocytic infiltrates so dense they obscured the tumor margins. The lymphocytic infiltrates persisted even after 3 MMS stages, though they were moderately less dense compared to the initial MMS stage. Clinical interpretation determined no relationship between the lymphocytic infiltrates and residual tumor. Due to concerns that this lymphocytic infiltrate may indicate an underlying leukemic process, preoperative laboratory tests were ordered prior to closure of the surgical wound, which demonstrated an elevated white blood cell count of 65,000/µL (reference range, 4500–11,000/µL) with 93% lymphocytes. A follow-up complete blood cell count (CBC) and blood smear confirmed the diagnosis of CLL. The patient was referred to a hematologist/oncologist.
An 84-year old man (patient 2) with a medical history of numerous precancerous lesions and 1 squamous cell carcinoma (SCC) presented for a biopsy, which determined moderately differentiated SCC. Mohs micrographic surgery was performed. The initial stage of MMS histologic examination demonstrated basosquamous carcinoma in the specimen margins, including perineural growth, with an extensive lymphoid infiltrate surrounding the tumor (Figure 1). A second stage of MMS was performed, and although margins appeared to be clear of the basosquamous histology, complete assessment was difficult due to areas of dense inflammatory infiltrate (Figure 2), including perineural infiltration that remained and appeared to extend deeper into the tissues. Pathology was consulted and it was determined that the perineural infiltration was unlikely related to tumor spread but rather secondary to an unknown cause. Further investigation of the patient’s medical history revealed previously diagnosed CLL, which had been omitted by the patient, as he had forgotten this diagnosis and denied a history of cancer, lymphoma, and even leukemia. A query to the patient’s primary care physician found the most recent CBC demonstrated an elevated white blood cell count of 37,600/µL with 78% lymphocytes.
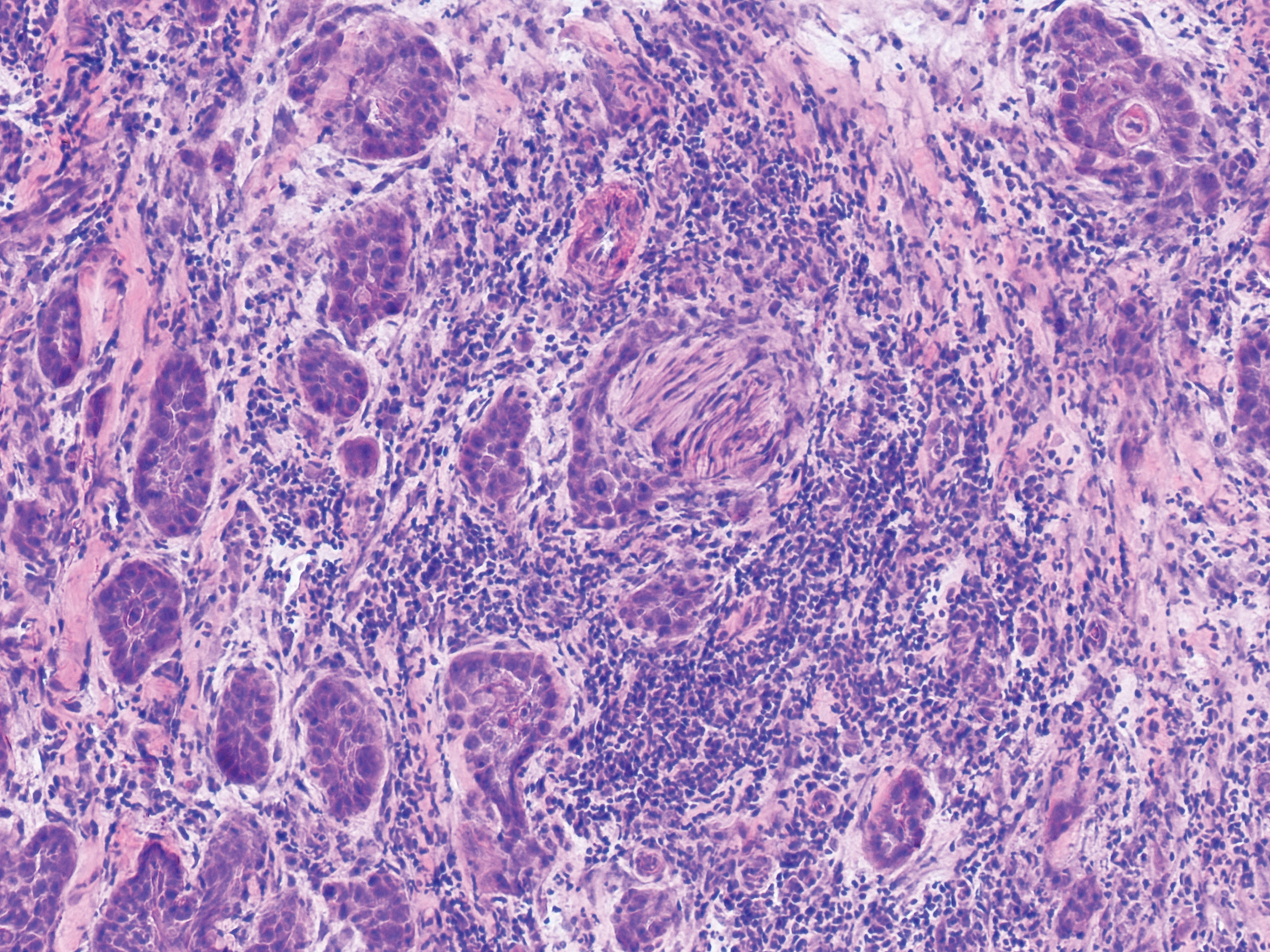
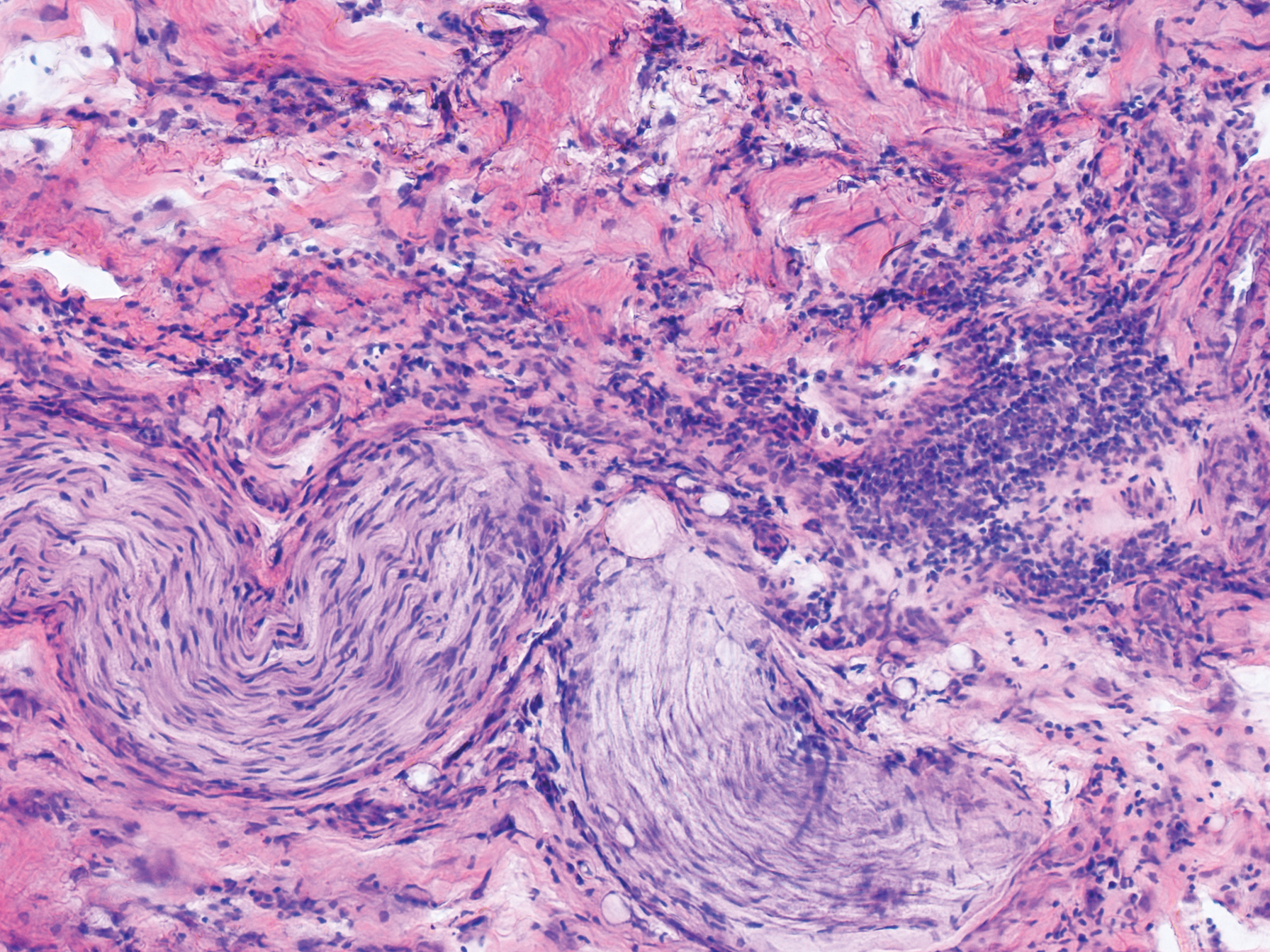
An 84-year-old man (patient 3) with a known history of CLL was referred for MMS excision of a 3.5×4.0-cm SCC on the right anterior temple extending onto the lateral upper and lower eyelids. Mohs frozen section histologic examination of excised tissue revealed patches of heavy lymphocytic infiltrates not found exclusively around the residual tumor but additionally around superficial and deep neurovascular bundles. The second stage of MMS appeared to be clear of tumor cells, but lymphocytic infiltrates remained. Because this patient had a clear history of CLL, the decision was made in conjunction with a dermatopathologist to conclude the surgery at this point. However, secondary to the aggressive, deeply invasive growth of this SCC, the patient was referred for adjunctive radiation therapy to the surgical site after wound reconstruction.
Chronic lymphocytic leukemia is the most common leukemia in the Western world1 and is estimated to account for 27% of all new cases of leukemia. An individual’s lifetime risk is 0.5%. Chronic lymphocytic leukemia is predominantly a disease of the elderly, with an average age at diagnosis of 71 years. It is more common among males, North American and European populations, and those with a positive family history. Although epidemiologic factors including farming, prolonged pesticide exposure, and contact with Agent Orange have tentative links to CLL, the relationships are poorly established.2
Symptoms associated with acute leukemia only rarely manifest in patients with CLL.3 If present, symptoms are vague and include weakness, fatigue, weight loss, fever, night sweats, and a feeling of abdominal fullness.2,3 On clinical examination, patients also may have lymphadenopathy, splenomegaly, or hepatomegaly. Increasing severity of symptoms at time of presentation directly correlates with the severity and staging at the time of diagnosis.4 Not only do patients with CLL have a greater incidence of NMSCs with more notable subclinical tumor extension than the average person, but these individuals also have a greatly increased risk for skin cancer recurrence posttreatment.5,6
Although tissue pathology is not routinely part of the diagnosis of patients with CLL, findings can add to clinical suspicion. Smudge cells, which are cell debris, are characteristic morphologic features found in CLL. Most CLL cells are characteristically small mature lymphocytes with a dense nucleus.3 The presence of aggregates of these cells may obscure tumor margins during resection of NMSCs.7 This infiltrate is present in more than one-third of patients with CLL, as described in one retrospective cohort. This study simultaneously demonstrated the relationship between CLL and a 2-fold increase in postoperative defect size, which was attributed to either subclinical tumor spread or extra tissue removal to ensure clearance due to the leukemic infiltrates themselves.8 The presence of perineural tumor growth, which can occur with aggressive SCC and basal cell carcinoma, may be mimicked by perineural involvement of CLL cells rather than the reactive inflammation associated with continued tumor margins.7
When evaluating a patient with suspected CLL, laboratory tests should include a CBC with differential and examination of the peripheral smear. If abnormal, immunophenotyping of lymphocytes by flow cytometry will rule out other lymphoproliferative diseases and verify CLL as the diagnosis.3 Diagnosis of CLL requires the presence of monoclonal B lymphocytes (≥5×109/L) in the peripheral blood as confirmed by flow cytometry.3 Clonality of circulating B lymphocytes must be confirmed, and immunophenotyping will establish a diagnosis with leukemic cells having positive expression of CD20 (Figure 3A) and CD23 (Figure 3B)(characteristic of B-cell lineage) with coexpression of CD43 and CD5 (Figure 3C)(characteristic of T-cell lineage).7,9 This pattern of immunohistochemical markers can be differentiated from the normal immune response to cutaneous malignancies, which have the pattern of being CD3+, CD5+, and CD43+ with absence of B-cell markers (ie, CD20, CD23)(Table).7

The pathogenesis of this peritumoral infiltrate is unknown, though multiple theories exist. One theory is that the neoplastic lymphocytes are responding as a dysfunctional arm of the immune system to tumor-specific antigens. In patients with CLL, leukemic lymphocytes comprise a large portion of the circulating leukocyte population and this peritumoral infiltrate may simply be a reflection of the circulating leukocytic population. Another theory contends that neoplastic lymphocytes are simply nonspecific aggregations secondary to tumor neovascularization and increased vascular permeability.10
This neoplastic infiltrate seen incidentally during MMS excision of NMSCs not only provides a unique opportunity to diagnose and intervene in those with unknown CLL but also to be aware of complicating features that can spare the patient from unnecessary tissue removal, thereby maximizing the benefit of MMS. This infiltrate can obscure tumor margins; is unusually dense and patchy, with or without infiltrating perineural or perivascular components; and persists beyond what would seem to be an adequate margin to clear a tumor. These cases show these findings, which exemplify the peritumoral infiltrate of CLL and should prompt further workup.
- Rozman C, Monserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052-1057.
- What are the risk factors for chronic lymphocytic leukemia? American Cancer Society website. https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/causes-risks-prevention/risk-factors.html. Revised May 10, 2018. Accessed February 11, 2019.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-5456.
- Rai KR, Wasil T, Iqbal U, et al. Clinical staging and prognostic markers in chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2004;18:795-805, vii.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol Surg. 2005;31:38-42.
- Brewer JD, Shanafelt TD, Khezri F, et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: a Rochester epidemiology project population-based study in Minnesota. J Am Acad Dermatol. 2015;72:302-309.
- Wilson ML, Elston DM, Tyler WB, et al. Dense lymphocytic infiltrates associated with non-melanoma skin cancer in patients with chronic lymphocytic leukemia. Dermatol Online J. 2010;16:4.
- Mehrany K, Byrd DR, Roenigk RK, et al. Lymphocytic infiltrates and subclinical epithelial tumor extension in patients with chronic leukemia and solid-organ transplantation. Dermatol Surg. 2003;29:129-134.
- Khandelwal A, Seilstad KH, Magro CM. Subclinical chronic lymphocytic leukaemia associated with a 13q deletion presenting initially in the skin: apropos of a case. J Cutan Pathol. 2006;33:256-259.
- Padgett JK, Parlette HL, English JC. A diagnosis of chronic lymphocytic leukemia prompted by cutaneous lymphocytic infiltrates present in mohs micrographic surgery frozen sections. Dermatol Surg. 2003;29:769-771.
To the Editor:
Specific characteristics of a lymphocytic infiltrate noted on frozen section histologic examination during Mohs micrographic surgery (MMS) tumor excision should raise suspicion of an underlying chronic lymphocytic leukemia (CLL). This infiltrate may be the presenting sign of the underlying leukemia and has variable presentation that may mimic aggressive features. The following 3 cases highlight this phenomenon.
A 74-year-old man (patient 1) with a medical history of multiple nonmelanoma skin cancers (NMSCs) presented for definitive treatment of a biopsy-proven infiltrative basal cell carcinoma involving the right infra-auricular region. Mohs section histologic evaluation revealed patches of lymphocytic infiltrates so dense they obscured the tumor margins. The lymphocytic infiltrates persisted even after 3 MMS stages, though they were moderately less dense compared to the initial MMS stage. Clinical interpretation determined no relationship between the lymphocytic infiltrates and residual tumor. Due to concerns that this lymphocytic infiltrate may indicate an underlying leukemic process, preoperative laboratory tests were ordered prior to closure of the surgical wound, which demonstrated an elevated white blood cell count of 65,000/µL (reference range, 4500–11,000/µL) with 93% lymphocytes. A follow-up complete blood cell count (CBC) and blood smear confirmed the diagnosis of CLL. The patient was referred to a hematologist/oncologist.
An 84-year old man (patient 2) with a medical history of numerous precancerous lesions and 1 squamous cell carcinoma (SCC) presented for a biopsy, which determined moderately differentiated SCC. Mohs micrographic surgery was performed. The initial stage of MMS histologic examination demonstrated basosquamous carcinoma in the specimen margins, including perineural growth, with an extensive lymphoid infiltrate surrounding the tumor (Figure 1). A second stage of MMS was performed, and although margins appeared to be clear of the basosquamous histology, complete assessment was difficult due to areas of dense inflammatory infiltrate (Figure 2), including perineural infiltration that remained and appeared to extend deeper into the tissues. Pathology was consulted and it was determined that the perineural infiltration was unlikely related to tumor spread but rather secondary to an unknown cause. Further investigation of the patient’s medical history revealed previously diagnosed CLL, which had been omitted by the patient, as he had forgotten this diagnosis and denied a history of cancer, lymphoma, and even leukemia. A query to the patient’s primary care physician found the most recent CBC demonstrated an elevated white blood cell count of 37,600/µL with 78% lymphocytes.
An 84-year-old man (patient 3) with a known history of CLL was referred for MMS excision of a 3.5×4.0-cm SCC on the right anterior temple extending onto the lateral upper and lower eyelids. Mohs frozen section histologic examination of excised tissue revealed patches of heavy lymphocytic infiltrates not found exclusively around the residual tumor but additionally around superficial and deep neurovascular bundles. The second stage of MMS appeared to be clear of tumor cells, but lymphocytic infiltrates remained. Because this patient had a clear history of CLL, the decision was made in conjunction with a dermatopathologist to conclude the surgery at this point. However, secondary to the aggressive, deeply invasive growth of this SCC, the patient was referred for adjunctive radiation therapy to the surgical site after wound reconstruction.
Chronic lymphocytic leukemia is the most common leukemia in the Western world1 and is estimated to account for 27% of all new cases of leukemia. An individual’s lifetime risk is 0.5%. Chronic lymphocytic leukemia is predominantly a disease of the elderly, with an average age at diagnosis of 71 years. It is more common among males, North American and European populations, and those with a positive family history. Although epidemiologic factors including farming, prolonged pesticide exposure, and contact with Agent Orange have tentative links to CLL, the relationships are poorly established.2
Symptoms associated with acute leukemia only rarely manifest in patients with CLL.3 If present, symptoms are vague and include weakness, fatigue, weight loss, fever, night sweats, and a feeling of abdominal fullness.2,3 On clinical examination, patients also may have lymphadenopathy, splenomegaly, or hepatomegaly. Increasing severity of symptoms at time of presentation directly correlates with the severity and staging at the time of diagnosis.4 Not only do patients with CLL have a greater incidence of NMSCs with more notable subclinical tumor extension than the average person, but these individuals also have a greatly increased risk for skin cancer recurrence posttreatment.5,6
Although tissue pathology is not routinely part of the diagnosis of patients with CLL, findings can add to clinical suspicion. Smudge cells, which are cell debris, are characteristic morphologic features found in CLL. Most CLL cells are characteristically small mature lymphocytes with a dense nucleus.3 The presence of aggregates of these cells may obscure tumor margins during resection of NMSCs.7 This infiltrate is present in more than one-third of patients with CLL, as described in one retrospective cohort. This study simultaneously demonstrated the relationship between CLL and a 2-fold increase in postoperative defect size, which was attributed to either subclinical tumor spread or extra tissue removal to ensure clearance due to the leukemic infiltrates themselves.8 The presence of perineural tumor growth, which can occur with aggressive SCC and basal cell carcinoma, may be mimicked by perineural involvement of CLL cells rather than the reactive inflammation associated with continued tumor margins.7
When evaluating a patient with suspected CLL, laboratory tests should include a CBC with differential and examination of the peripheral smear. If abnormal, immunophenotyping of lymphocytes by flow cytometry will rule out other lymphoproliferative diseases and verify CLL as the diagnosis.3 Diagnosis of CLL requires the presence of monoclonal B lymphocytes (≥5×109/L) in the peripheral blood as confirmed by flow cytometry.3 Clonality of circulating B lymphocytes must be confirmed, and immunophenotyping will establish a diagnosis with leukemic cells having positive expression of CD20 (Figure 3A) and CD23 (Figure 3B)(characteristic of B-cell lineage) with coexpression of CD43 and CD5 (Figure 3C)(characteristic of T-cell lineage).7,9 This pattern of immunohistochemical markers can be differentiated from the normal immune response to cutaneous malignancies, which have the pattern of being CD3+, CD5+, and CD43+ with absence of B-cell markers (ie, CD20, CD23)(Table).7
The pathogenesis of this peritumoral infiltrate is unknown, though multiple theories exist. One theory is that the neoplastic lymphocytes are responding as a dysfunctional arm of the immune system to tumor-specific antigens. In patients with CLL, leukemic lymphocytes comprise a large portion of the circulating leukocyte population and this peritumoral infiltrate may simply be a reflection of the circulating leukocytic population. Another theory contends that neoplastic lymphocytes are simply nonspecific aggregations secondary to tumor neovascularization and increased vascular permeability.10
This neoplastic infiltrate seen incidentally during MMS excision of NMSCs not only provides a unique opportunity to diagnose and intervene in those with unknown CLL but also to be aware of complicating features that can spare the patient from unnecessary tissue removal, thereby maximizing the benefit of MMS. This infiltrate can obscure tumor margins; is unusually dense and patchy, with or without infiltrating perineural or perivascular components; and persists beyond what would seem to be an adequate margin to clear a tumor. These cases show these findings, which exemplify the peritumoral infiltrate of CLL and should prompt further workup.
To the Editor:
Specific characteristics of a lymphocytic infiltrate noted on frozen section histologic examination during Mohs micrographic surgery (MMS) tumor excision should raise suspicion of an underlying chronic lymphocytic leukemia (CLL). This infiltrate may be the presenting sign of the underlying leukemia and has variable presentation that may mimic aggressive features. The following 3 cases highlight this phenomenon.
A 74-year-old man (patient 1) with a medical history of multiple nonmelanoma skin cancers (NMSCs) presented for definitive treatment of a biopsy-proven infiltrative basal cell carcinoma involving the right infra-auricular region. Mohs section histologic evaluation revealed patches of lymphocytic infiltrates so dense they obscured the tumor margins. The lymphocytic infiltrates persisted even after 3 MMS stages, though they were moderately less dense compared to the initial MMS stage. Clinical interpretation determined no relationship between the lymphocytic infiltrates and residual tumor. Due to concerns that this lymphocytic infiltrate may indicate an underlying leukemic process, preoperative laboratory tests were ordered prior to closure of the surgical wound, which demonstrated an elevated white blood cell count of 65,000/µL (reference range, 4500–11,000/µL) with 93% lymphocytes. A follow-up complete blood cell count (CBC) and blood smear confirmed the diagnosis of CLL. The patient was referred to a hematologist/oncologist.
An 84-year old man (patient 2) with a medical history of numerous precancerous lesions and 1 squamous cell carcinoma (SCC) presented for a biopsy, which determined moderately differentiated SCC. Mohs micrographic surgery was performed. The initial stage of MMS histologic examination demonstrated basosquamous carcinoma in the specimen margins, including perineural growth, with an extensive lymphoid infiltrate surrounding the tumor (Figure 1). A second stage of MMS was performed, and although margins appeared to be clear of the basosquamous histology, complete assessment was difficult due to areas of dense inflammatory infiltrate (Figure 2), including perineural infiltration that remained and appeared to extend deeper into the tissues. Pathology was consulted and it was determined that the perineural infiltration was unlikely related to tumor spread but rather secondary to an unknown cause. Further investigation of the patient’s medical history revealed previously diagnosed CLL, which had been omitted by the patient, as he had forgotten this diagnosis and denied a history of cancer, lymphoma, and even leukemia. A query to the patient’s primary care physician found the most recent CBC demonstrated an elevated white blood cell count of 37,600/µL with 78% lymphocytes.
An 84-year-old man (patient 3) with a known history of CLL was referred for MMS excision of a 3.5×4.0-cm SCC on the right anterior temple extending onto the lateral upper and lower eyelids. Mohs frozen section histologic examination of excised tissue revealed patches of heavy lymphocytic infiltrates not found exclusively around the residual tumor but additionally around superficial and deep neurovascular bundles. The second stage of MMS appeared to be clear of tumor cells, but lymphocytic infiltrates remained. Because this patient had a clear history of CLL, the decision was made in conjunction with a dermatopathologist to conclude the surgery at this point. However, secondary to the aggressive, deeply invasive growth of this SCC, the patient was referred for adjunctive radiation therapy to the surgical site after wound reconstruction.
Chronic lymphocytic leukemia is the most common leukemia in the Western world1 and is estimated to account for 27% of all new cases of leukemia. An individual’s lifetime risk is 0.5%. Chronic lymphocytic leukemia is predominantly a disease of the elderly, with an average age at diagnosis of 71 years. It is more common among males, North American and European populations, and those with a positive family history. Although epidemiologic factors including farming, prolonged pesticide exposure, and contact with Agent Orange have tentative links to CLL, the relationships are poorly established.2
Symptoms associated with acute leukemia only rarely manifest in patients with CLL.3 If present, symptoms are vague and include weakness, fatigue, weight loss, fever, night sweats, and a feeling of abdominal fullness.2,3 On clinical examination, patients also may have lymphadenopathy, splenomegaly, or hepatomegaly. Increasing severity of symptoms at time of presentation directly correlates with the severity and staging at the time of diagnosis.4 Not only do patients with CLL have a greater incidence of NMSCs with more notable subclinical tumor extension than the average person, but these individuals also have a greatly increased risk for skin cancer recurrence posttreatment.5,6
Although tissue pathology is not routinely part of the diagnosis of patients with CLL, findings can add to clinical suspicion. Smudge cells, which are cell debris, are characteristic morphologic features found in CLL. Most CLL cells are characteristically small mature lymphocytes with a dense nucleus.3 The presence of aggregates of these cells may obscure tumor margins during resection of NMSCs.7 This infiltrate is present in more than one-third of patients with CLL, as described in one retrospective cohort. This study simultaneously demonstrated the relationship between CLL and a 2-fold increase in postoperative defect size, which was attributed to either subclinical tumor spread or extra tissue removal to ensure clearance due to the leukemic infiltrates themselves.8 The presence of perineural tumor growth, which can occur with aggressive SCC and basal cell carcinoma, may be mimicked by perineural involvement of CLL cells rather than the reactive inflammation associated with continued tumor margins.7
When evaluating a patient with suspected CLL, laboratory tests should include a CBC with differential and examination of the peripheral smear. If abnormal, immunophenotyping of lymphocytes by flow cytometry will rule out other lymphoproliferative diseases and verify CLL as the diagnosis.3 Diagnosis of CLL requires the presence of monoclonal B lymphocytes (≥5×109/L) in the peripheral blood as confirmed by flow cytometry.3 Clonality of circulating B lymphocytes must be confirmed, and immunophenotyping will establish a diagnosis with leukemic cells having positive expression of CD20 (Figure 3A) and CD23 (Figure 3B)(characteristic of B-cell lineage) with coexpression of CD43 and CD5 (Figure 3C)(characteristic of T-cell lineage).7,9 This pattern of immunohistochemical markers can be differentiated from the normal immune response to cutaneous malignancies, which have the pattern of being CD3+, CD5+, and CD43+ with absence of B-cell markers (ie, CD20, CD23)(Table).7
The pathogenesis of this peritumoral infiltrate is unknown, though multiple theories exist. One theory is that the neoplastic lymphocytes are responding as a dysfunctional arm of the immune system to tumor-specific antigens. In patients with CLL, leukemic lymphocytes comprise a large portion of the circulating leukocyte population and this peritumoral infiltrate may simply be a reflection of the circulating leukocytic population. Another theory contends that neoplastic lymphocytes are simply nonspecific aggregations secondary to tumor neovascularization and increased vascular permeability.10
This neoplastic infiltrate seen incidentally during MMS excision of NMSCs not only provides a unique opportunity to diagnose and intervene in those with unknown CLL but also to be aware of complicating features that can spare the patient from unnecessary tissue removal, thereby maximizing the benefit of MMS. This infiltrate can obscure tumor margins; is unusually dense and patchy, with or without infiltrating perineural or perivascular components; and persists beyond what would seem to be an adequate margin to clear a tumor. These cases show these findings, which exemplify the peritumoral infiltrate of CLL and should prompt further workup.
- Rozman C, Monserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052-1057.
- What are the risk factors for chronic lymphocytic leukemia? American Cancer Society website. https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/causes-risks-prevention/risk-factors.html. Revised May 10, 2018. Accessed February 11, 2019.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-5456.
- Rai KR, Wasil T, Iqbal U, et al. Clinical staging and prognostic markers in chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2004;18:795-805, vii.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol Surg. 2005;31:38-42.
- Brewer JD, Shanafelt TD, Khezri F, et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: a Rochester epidemiology project population-based study in Minnesota. J Am Acad Dermatol. 2015;72:302-309.
- Wilson ML, Elston DM, Tyler WB, et al. Dense lymphocytic infiltrates associated with non-melanoma skin cancer in patients with chronic lymphocytic leukemia. Dermatol Online J. 2010;16:4.
- Mehrany K, Byrd DR, Roenigk RK, et al. Lymphocytic infiltrates and subclinical epithelial tumor extension in patients with chronic leukemia and solid-organ transplantation. Dermatol Surg. 2003;29:129-134.
- Khandelwal A, Seilstad KH, Magro CM. Subclinical chronic lymphocytic leukaemia associated with a 13q deletion presenting initially in the skin: apropos of a case. J Cutan Pathol. 2006;33:256-259.
- Padgett JK, Parlette HL, English JC. A diagnosis of chronic lymphocytic leukemia prompted by cutaneous lymphocytic infiltrates present in mohs micrographic surgery frozen sections. Dermatol Surg. 2003;29:769-771.
- Rozman C, Monserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333:1052-1057.
- What are the risk factors for chronic lymphocytic leukemia? American Cancer Society website. https://www.cancer.org/cancer/chronic-lymphocytic-leukemia/causes-risks-prevention/risk-factors.html. Revised May 10, 2018. Accessed February 11, 2019.
- Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-5456.
- Rai KR, Wasil T, Iqbal U, et al. Clinical staging and prognostic markers in chronic lymphocytic leukemia. Hematol Oncol Clin North Am. 2004;18:795-805, vii.
- Mehrany K, Weenig RH, Pittelkow MR, et al. High recurrence rates of squamous cell carcinoma after Mohs’ surgery in patients with chronic lymphocytic leukemia. Dermatol Surg. 2005;31:38-42.
- Brewer JD, Shanafelt TD, Khezri F, et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: a Rochester epidemiology project population-based study in Minnesota. J Am Acad Dermatol. 2015;72:302-309.
- Wilson ML, Elston DM, Tyler WB, et al. Dense lymphocytic infiltrates associated with non-melanoma skin cancer in patients with chronic lymphocytic leukemia. Dermatol Online J. 2010;16:4.
- Mehrany K, Byrd DR, Roenigk RK, et al. Lymphocytic infiltrates and subclinical epithelial tumor extension in patients with chronic leukemia and solid-organ transplantation. Dermatol Surg. 2003;29:129-134.
- Khandelwal A, Seilstad KH, Magro CM. Subclinical chronic lymphocytic leukaemia associated with a 13q deletion presenting initially in the skin: apropos of a case. J Cutan Pathol. 2006;33:256-259.
- Padgett JK, Parlette HL, English JC. A diagnosis of chronic lymphocytic leukemia prompted by cutaneous lymphocytic infiltrates present in mohs micrographic surgery frozen sections. Dermatol Surg. 2003;29:769-771.
Practice Points
- Chronic lymphocytic leukemia (CLL) may be seen during histologic examination of specimens during Mohs micrographic surgery as a monomorphic infiltrate of small mature lymphocytes with dense nuclei. Patients may be unaware of their diagnosis, which can be the presenting feature.
- An infiltrate of CLL may mimic aggressive behavior of nonmelanoma skin cancers including perineural invasion. A leukemic infiltrate may appear more dense and monomorphic. If needed, immunohistochemical staining of leukemic cells will show CD5 and CD23 positivity.
- Anecdotally, patients with CLL may not remember this pertinent medical history. Whether due to its asymptomatic nature or lack of treatment in early stages, direct questioning about CLL may be warranted if this characteristic infiltrate is encountered.
Irregularly Hyperpigmented Plaque on the Right Heel
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
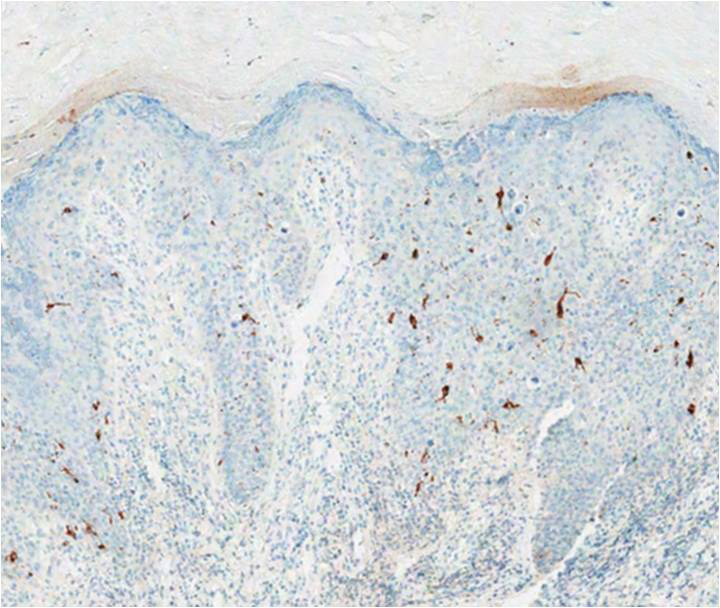
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
A 56-year-old woman presented with an asymptomatic plaque on the right heel that had grown
steadily over the last year. Pigmented lesions were not appreciated on other sites, and lymph nodes were not enlarged. Her medical history was otherwise normal, except for bilateral hearing loss due to encephalitis at the age of 5 years. None of her family members had similar symptoms. Physical examination revealed a well-defined, irregularly hyperpigmented plaque on the right heel.
Cutaneous Collagenous Vasculopathy
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
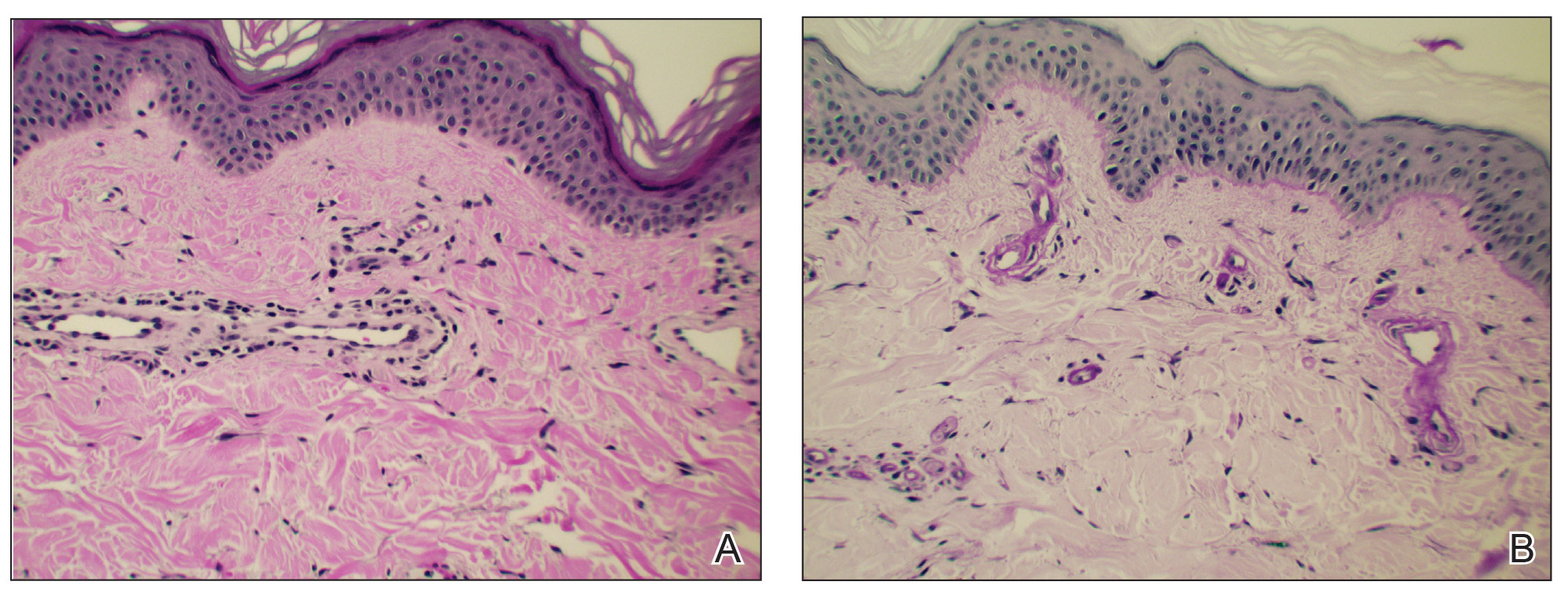
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
To the Editor:
Cutaneous microangiopathy describes pathology of the small blood vessels within the dermis.
We report a case of CCV in a 41-year-old woman who presented for evaluation of a rash on the bilateral lower extremities of 7 to 8 months’ duration. The eruption had started on the left ankle and spread over several weeks to the bilateral dorsal feet followed by the ankles and shins. The patient noted associated swelling and a pressure like dysesthesia of the lower legs. She was otherwise in good health, though she had started an oral contraceptive 1 year prior for heavy menstrual bleeding. A review of systems was negative for deep vein thrombosis, pulmonary embolus, and other thromboembolic phenomena, and the patient had no history of hepatic or renal dysfunction, cancer, or heart disease. Her family history was negative for clotting disorders or bleeding diatheses.
On physical examination, telangiectatic matting was present on the bilateral ankles and dorsal feet with an associated blanchable erythema (Figure 1). The matting extended into a fine, mottled, pretibial telangiectasia associated with Schamberg purpura. She had no pitting edema, and both dorsalis pedis and posterior popliteal pulses were intact and symmetric bilaterally. No popliteal lymphadenopathy or palpable cords were present.
Two punch biopsies taken from the erythematous telangiectatic area on the left foot and metatarsal region demonstrated an unremarkable epidermis without interface change, thickening of the epidermal basement membrane, or single-cell dyskeratosis. There was mild dilatation of blood vessels within the superficial dermis with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes. Leukocytoclastic debris, fibrinoid necrosis of vessel walls, and endothelial cell necrosis were not seen. As is classic in CCV, the vessel walls appeared thickened by eosinophilic hyaline material, which was periodic acid–Schiff positive and diastase resistant (Figure 2). Sclerotic thickening of collagen bundles or absence of periadnexal adipose tissue was not seen. CD34 immunohistochemical staining demonstrated normal retained CD34 interstitial dermal positivity, which excluded morphea. Additionally, direct immunofluorescence testing was negative for IgG, IgA, IgM, C3, fibrin, and C1q. Nodular reduplication of vessels or other changes of stasis were not seen. Fibrin thrombi or neoplastic cells were not identified. The clinical and histopathologic findings were suggestive of CCV.
Prior case reports of CCV have described a similar clinical manifestation with blanching macules that occur symmetrically on the lower extremities and spread cephalically.1-6 A distinction from hereditary hemorrhagic telangiectasia is the noninvolvement of mucous membranes and nails. The etiology of this rare microangiopathy has not been elucidated, though disease concurrence with local trauma, stressful events such as childbirth, and diabetes mellitus has been documented.6 As the body of literature continues to grow, more research regarding the etiology, mechanism, prognosis, and treatment options will enhance our understanding of CCV.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Lloyd BM, Pruden SJ, Lind AC, et al. Cutaneous collagenous vasculopathy: report of the first pediatric case. Pediatr Dermatol. 2011;28:598-599.
- Salama S, Chorneyko K, Belovic B. Cutaneous collagenous vasculopathy associated with intravascular occlusive fibrin thrombi. J Cutan Pathol. 2014;41:386-393.
- Perez A, Wain ME, Robson A, et al. Cutaneous collagenous vasculopathy with generalized telangiectasia in two female patients. J Am Acad Dermatol. 2010;63:882-885.
- Burdick LM, Losher S, Somach SC, et al. Cutaneous collagenous vasculopathy: a rare cutaneous microangiopathy. J Cutan Pathol. 2012;39:741-746.
Practice Points
- In cutaneous collagenous vasculopathy (CCV), skin biopsy may demonstrate eosinophilic hyaline thickening of superficial dermal blood vessels with mild perivascular lymphocytic inflammation and rare extravasated erythrocytes.
- Lack of mucous membrane and nail involvement differentiates CCV from hereditary hemorrhagic telangiectasia.
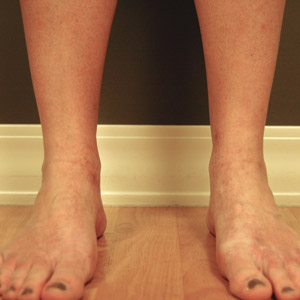
Elephantiasis Nostras Verrucosa Secondary to Scleroderma
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).

A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).
A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
To the Editor:
Elephantiasis nostras verrucosa (ENV) is a skin disorder caused by marked underlying lymphedema that leads to hyperkeratosis, papillomatosis, and verrucous growths on the epidermis.1 The pathophysiology of ENV relates to noninfectious lymphatic obstruction and lymphatic fibrosis secondary to venous stasis, malignancy, radiation therapy, or trauma.2 We present an unusual case of lymphedema and subsequent ENV limited to the arms and hands in a patient with scleroderma, an autoimmune fibrosing disorder.
A 54-year-old woman with a 5-year history of scleroderma presented to our dermatology clinic for treatment of progressive skin changes including pruritus, tightness, finger ulcerations, and pus exuding from papules on the dorsal arms and hands. She had been experiencing several systemic symptoms including dysphagia and lung involvement, necessitating oxygen therapy and a continuous positive airway pressure device for pulmonary arterial hypertension. A computed tomography scan of the lungs demonstrated an increase in ground-glass infiltrates in the right lower lobe and an air-fluid level in the esophagus. At the time of presentation, she was being treated with bosentan and sildenafil for pulmonary arterial hypertension, in addition to prednisone, venlafaxine, lansoprazole, metoclopramide, levothyroxine, temazepam, aspirin, and oxycodone. In the 2 years prior to presentation, she had been treated with intravenous cyclophosphamide once monthly for 6 months, adalimumab for 1 year, and 1 session of photodynamic therapy to the arms, all without benefit.
Physical examination showed cutaneous signs of scleroderma including marked sclerosis of the skin on the face, hands, V of the neck, proximal arms, and mid and proximal thighs. Excoriated papules with overlying crusting and pustulation were superimposed on the sclerotic skin of the arms (Figure 1).
A superinfection was diagnosed and treated with cephalexin 500 mg 4 times daily for 2 weeks; thereafter, mupirocin cream twice daily was used as needed. She was prescribed fexofenadine 180 mg twice daily and doxepin 20 mg at bedtime for pruritus.
At 3-week follow-up, a trial of narrowband UVB therapy was recommended for control of pruritus. Two weeks later, a modified wet-wrap regimen using clobetasol ointment 0.5% twice daily covered with wet gauze followed by a self-adherent dressing was initiated only on the right arm for comparison purposes. This treatment was not successful. A biopsy taken from the left arm showed lymphedema with perivascular fibroplasia and epidermal hyperplasia consistent with ENV (Figure 2).
Two months after her initial presentation, we instituted treatment with tazarotene gel 0.1% twice daily to the arms as well as a water-based topical emulsion to the finger ulcerations and a healing ointment to the hands. A month later, the patient reported no benefit with tazarotene. She desired more flexibility in her arms and hands; therefore, after a discussion with her rheumatologist, biweekly psoralen plus UVA (PUVA) therapy was initiated. Five months after presentation, methotrexate (MTX) 15 mg once weekly with folic acid 1 mg once daily was added. The PUVA therapy and MTX were stopped 3 months later due to lack of treatment benefit.
The patient was referred to vascular medicine for possible compression therapy. It was determined that her vasculature was intact, but compression therapy was contraindicated due to underlying systemic sclerosis. She was subsequently prescribed mycophenolate mofetil 1000 mg twice daily by her rheumatologist. The options of serial excisions or laser resurfacing were presented, but she declined.
Elephantiasis nostras verrucosa is differentiated from elephantiasis tropica, which is caused by a filarial infection of the lymphatic system. The chronic obstructive lymphedema characteristic of ENV can present as a result of various primary or secondary etiologies including trauma, malignancy, venous stasis, inflammation, or infection.3 In systemic sclerosis, extravascular fibrosis theoretically can lead to lymphatic obstruction and subsequent lymphatic stasis. In turn, the pathophysiology of dermal and subcutaneous fibrosis likely reflects autoantibodies (eg, anticardiolipin antibodies) that can damage lymphatic and nonlymphatic vessels.4,5
As the underlying mechanism of ENV, fibrosis of lymphatic vessels in systemic sclerosis is not well documented. Characteristic features of systemic sclerosis include extensive fibrosis, fibroproliferative vasculopathy, and inflammation, which are all possible mechanisms for the internal lymphatic obstruction resulting in the skin changes observed in ENV.6 It seemed unusual that the fibrotic changes of lymphatic vessels in our patient were extensive enough to cause ENV of the upper extremities; lower extremity involvement is the more common presentation because of the greater likelihood of lymphedema manifesting in the legs and feet. Lower extremity ENV has been reported in association with scleroderma.7,8
Regarding therapeutic options, Boyd et al9 reported a good response in a patient with ENV of the abdomen who was treated with topical tazarotene. Additionally, PUVA and MTX have been reported to be beneficial for the progressive skin changes of systemic sclerosis.10 Mycophenolate mofetil has been used in patients who fail MTX therapy because of its antifibrotic properties without the side-effect profiles of other immunosuppressives, such as imatinib.10,11 In our patient, skin lesions persisted following these varied approaches, and compression therapy was not advised due to the underlying sclerosis.
Because options for medical treatment of severe ENV are limited, surgical debridement of the affected limb often remains the only viable option in advanced cases.12 A PubMed search of articles indexed for MEDLINE using the terms elephantiasis (MeSH terms) or elephantiasis (all fields) and scleroderma, systemic (MeSH terms) or scleroderma (all fields) and systemic (all fields) or systemic scleroderma (all fields) or scleroderma (all fields) or scleroderma, localized (MeSH terms) or scleroderma (all fields) and localized (all fields) or localized scleroderma (all fields) yielded only 1 other case report of lower extremity ENV in a patient with systemic sclerosis who ultimately required bilateral leg amputation.8 When possible, avoiding lymphostasis through compression and control of any underlying infections is important in the treatment and prevention of ENV.3
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
- Sisto K, Khachemoune A. Elephantiasis nostras verrucosa: a review. Am J Clin Dermatol. 2008;9:141-146.
- Schissel DJ, Hivnor C, Elston DM. Elephantiasis nostras verrucosa. Cutis. 1998;62:77-80.
- Duckworth A, Husain J, DeHeer P. Elephantiasis nostras verrucosa or ‘mossy foot lesions’ in lymphedema praecox. J Am Podiatr Med Assoc. 2008;98:66-69.
- Assous N, Allanore Y, Batteaux F, et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin Exp Rheumatol. 2005;23:199-204.
- Derrett-Smith EC, Dooley A, Gilbane AJ, et al. Endothelial injury in a transforming growth-factor-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65:2928-2939.
- Pattanaik M, Brown M, Postlethwaite A. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105-125.
- Kerchner K, Fleischer A, Yosipovitch G. Lower extremity lymphedema update: pathophysiology, diagnosis and treatment guidelines. J Am Acad Dermatol. 2008;59:324-331.
- Chatterjee S, Karai L. Elephantiasis nostras verrucosa in a patient with systemic sclerosis. Clin Exp Dermatol. 2009;34:e696-e698.
- Boyd J, Sloan S, Meffert J. Elephantiasis nostrum verrucosa of the abdomen: clinical results with tazarotene. J Drugs Dermatol. 2004;3:446-448.
- Fett, N. Scleroderma: nomenclature, etiology, pathogenesis, prognosis, and treatments: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63:e102-e104.
- Iwao F, Sato-Matsumura KC, Sawamura D, et al. Elephantiasis nostras verrucosa successfully treated by surgical debridement. Dermatol Surg. 2004;30:939-941.
Practice Points
- Scleroderma rarely may lead to elephantiasis nostras verrucosa (ENV) of the upper extremities.
- Avoiding lymphostasis through compression and control of concomitant skin and soft tissue infections is important in the treatment and prevention of ENV.
Neurofibromatosis Type 1 in the Setting of Systemic Lupus Erythematosus
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
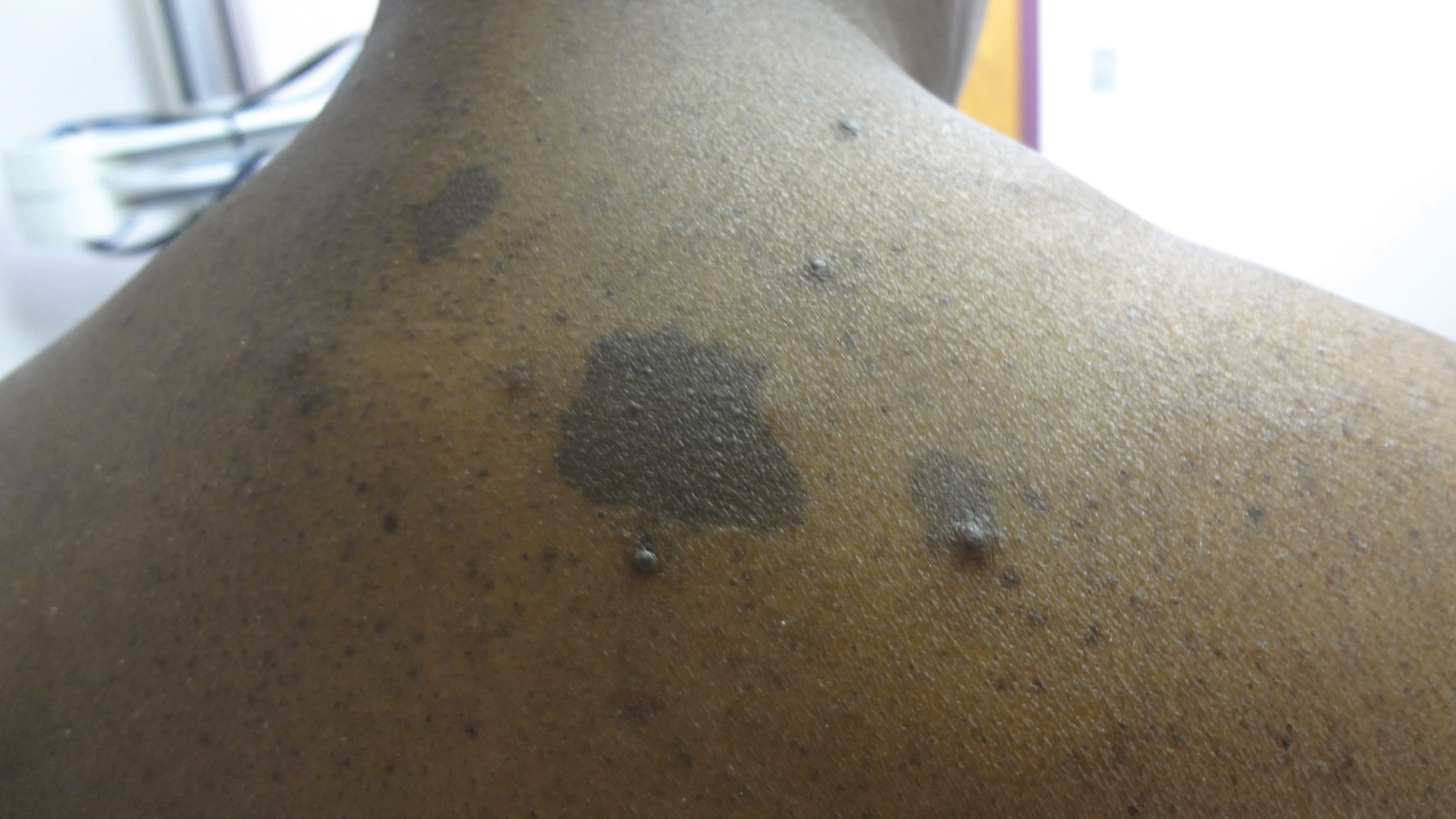
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.

We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
To the Editor:
Patients with concurrent neurofibromatosis type 1 (NF-1) and systemic lupus erythematosus (SLE) rarely have been reported in the literature. Neurofibromatosis type 1 is one of the most common genetic disorders, with a worldwide birth incidence of 1 in 2500 individuals and prevalence of 1 in 4000 individuals.1 The incidence and prevalence of SLE varies widely depending on race and geographic location. Estimated incidence rates for SLE range from 1 to 25 per 100,000 individuals annually in North America, South America, Europe, and Asia.2,3 The reported worldwide prevalence is 20 to 150 cases per 100,000 individuals annually.2,4,5
Given the high prevalence of both conditions, the association between SLE and NF-1 likely is underrecognized; therefore, identifying more patients with concurrent SLE and NF-1 and describing the interplay between the 2 conditions may have important therapeutic implications. We present the case of a middle-aged woman with a history of SLE who had cutaneous lesions characteristic of NF-1 to further the understanding of these concurrent conditions.
A middle-aged woman presented to our academic dermatology clinic for evaluation and removal of dark spots that had been present diffusely on the trunk and extremities since birth. She reported a history of SLE with lupus nephritis, hypertension, and a nodular goiter following a partial thyroidectomy. She noted that she did not seek treatment for the skin findings sooner because she was more concerned about her other medical conditions; however, because she felt these conditions were now stable, she decided to seek treatment for the “rash.” Physical examination revealed hundreds of café au lait macules and numerous neurofibromas diffusely distributed on the trunk and extremities (Figure 1) as well as bilateral axillary freckling. A clinical diagnosis of NF-1 was made.
When questioned, the patient reported that she may have been diagnosed with NF-1 in the past by another physician, but she did not recall it specifically. The patient was advised that there were no treatments for the café au lait macules. We notified her other physicians of the NF-1 diagnosis so she could be monitored for systemic conditions related to NF-1, including optic gliomas, pheochromocytoma, renal artery stenosis, and internal neurofibromas. We also referred the patient for genetic counseling; of note, the patient reported she had 4 children without any evidence of similar skin lesions or chronic health problems.
A PubMed search of articles indexed for MEDLINE using the terms systemic lupus and neurofibromatosis yielded 8 cases of patients having both SLE and NF-1 (including our case).6-11 Our patient reported having multiple lesions since birth, decades before the onset and diagnosis of SLE. In 3 other cases, patients were diagnosed with SLE and then presented with neurofibromas, leading to NF-1 diagnosis.In the discussion of those 3 cases, it was proposed that immune system alterations caused by SLE leading to viral illness may have predisposed the patients to the development of tumors and other collagen diseases, or it could be coincidental.6,7 In another case, a patient with NF-1 developed SLE, which was thought to be coincidental.8 Akyuz et al9 described the case of a pediatric patient with NF-1 who subsequently was diagnosed with SLE. The authors suggested that the lack of neurofibromin contributed to the development of SLE, an autoimmune condition. Under normal circumstances, neurofibromin acts as a guanosine triphosphatase–activating protein for RAS in T cells.10 CD8+ T-cell function also is impaired in patients with SLE.9 Additionally, it has been reported that anti–double-stranded DNA antibodies and immune complexes were present in NF-1 patients, even though there were low titers.12 Thus, the authors proposed that the lack of neurofibromin led to dysregulation of the RAS pathway and impairment of T cells, creating an immune milieu that predisposed the patient to development of SLE. Our case gives additional credence to this theory, as our patient had a similar clinical course: the café au lait macules were present since birth and the symptoms of SLE surfaced much later in her late 20s and 30s. Another case by Makino and Tampo10 described a patient with a history of SLE who was later diagnosed with NF-1 based on choroidal findings highly specific for NF-1 but did not have other classic findings of NF-1. The authors mentioned that there might be a potential relationship between these two disorders but did not speculate any theory in particular for their case.10
The interplay between an autoimmune condition such as SLE and NF-1, a condition traditionally thought to be due to a genetic mutation, may have greater clinical and therapeutic implications beyond just these two disorders. Although it is well established that RAS pathway disruption causes NF-1, it has been uncovered that dysfunction in the RAS pathway also can contribute to melanoma oncogenesis.13,14 These insights have led to the development of and approval of targeted drugs designed to inhibit the RAS pathway (eg, vemurafenib, dabrafenib, trametinib).14-17 Melanoma also is considered a “model” tumor for studying the relationship between the immune system and cancer.18
Our case also is instructive in another point: our patient had never sought treatment for her skin lesions, as she said she had other more serious health conditions. Closer evaluation of her skin condition may have led to earlier diagnosis of NF-1, which has important health implications. The average lifespan of patients with NF-1 is 10 to 15 years lower than the general population, with cancer being the leading cause of death.20 Malignant peripheral nerve sheath tumors are the most common malignant tumors observed in such patients.21-23 Other cancers that are associated with NF-1 include rhabdomyosarcomas, gastrointestinal stromal tumors, neuroectodermal tumors, pheochromocytomas, and breast carcinomas.23
To make a clinical diagnosis of NF-1, a patient must have 2 of 7 cardinal clinical features as defined by the National Institutes of Health (Table).24 In our patient with hundreds of café au lait macules and dozens of neurofibromas, the diagnosis was clear; however, in other patients, the skin findings of NF-1 may not be as prominent. A patient could meet criteria for NF-1 diagnosis with the inconspicuous presentation of 6 café au lait macules and either 1 plexiform neurofibroma or 2 neurofibromas (of any type) on the entire body.
We recommend that patients with SLE undergo skin examinations to look for more subtle presentations of NF-1. Earlier diagnosis will help to initiate close monitoring of the disorder’s associated systemic health risks. In addition, the identification of more patients with both NF-1 and SLE may help shed light on the etiology of both conditions.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
- Carey JC, Baty BJ, Johnson JP, et al. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45-56.
- Pons-Estel GJ, Alarcón GS, Scofield L, et al. Understanding the epidemiology and progression of systemic lupus erythematosus. Semin Arthritis Rheum. 2010;39:257-268.
- Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: a comparison of worldwide disease burden. Lupus. 2006;15:308-318.
- Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778-799.
- Chakravarty EF, Bush TM, Manzi S, et al. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092-2094.
- Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
- Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
- Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. a matter of coincidence? Clin Rhematol. 2003;22:496-497.
- Akyuz SG, Caltik A, Bulbul M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheumatol Int. 2012;32:2345-47.
- Makino S, Tampo H. Rare and unusual choroidal abnormalities in a patient with systemic lupus erythematosus. Case Rep Ophthalmol. 2013;4:81-86.
- Galvan JM, Hofkamp MP. Usefulness of intrapartum magnetic resonance imaging for a parturient with neurofibromatosis type I during induction of labor for preeclampsia. Proc (Bayl Univ Med Cent). 2018;31:92-93.
- Gerosa PL, Vai C, Bizzozer L, et al. Immunological and clinical surveillance in Recklinghausen’s neurofibromatosis (NF1). Panminerva Med. 1993;35:80-85.
- Busca R, Abbe P, Mantoux F, et al. RAS mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J. 2000;19:2900-2910.
- Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373-2379.
- Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;12:988-1004.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.
- Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23:(suppl 8):viii10-4.
- Zmajkovicova K, Jesenberger V, Catalanotti F, et al. MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol Cell. 2013;50:43-55.
- Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101-116.
- Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590-1605.
- Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33-40.
- Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247-253.
- Neurofibromatosis. conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575-78.
Practice Points
- Patients with neurofibromatosis type 1 (NF-1) benefit from early diagnosis and long-term follow-up.
- Patients with systemic lupus erythematosus (SLE) may develop different malignancies given the immune dysregulation. We recommend that patients with SLE undergo detailed skin examinations to check for subtle clues for NF-1.
- Similarly, patients with NF-1 can develop SLE later in life.