User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
nav[contains(@class, 'nav-ce-stack nav-ce-stack__large-screen')]
header[@id='header']
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
Turmeric may be as effective as omeprazole for dyspepsia
TOPLINE:
METHODOLOGY:
- The researchers randomly assigned 206 patients to receive curcumin – the active ingredient in turmeric – alone; omeprazole alone; or curcumin plus omeprazole for 28 days. A total of 151 patients completed the study.
- Doses were two 250-mg curcumin pills four times daily, plus one placebo pill; one 20-mg omeprazole pill daily, plus two placebo pills four times daily; or two 250-mg curcumin pills four times daily, plus one 20-mg omeprazole pill once daily.
- Symptoms of functional dyspepsia were assessed on days 28 and 56 using the Severity of Dyspepsia Assessment (SODA) score.
TAKEAWAY:
- In the combined group, the curcumin-alone group, and the omeprazole-alone group, SODA scores for pain severity declined significantly by day 28 (–4.83, –5.46, and –6.22, respectively), as did scores for severity of other symptoms (–2.22, –2.32, and –2.31, respectively).
- Symptom improvements were even stronger by day 56 for pain (–7.19, –8.07, –8.85) and other symptoms (–4.09, –4.12, –3.71) in the same groups.
- Curcumin was safe and well tolerated, but satisfaction scores did not change significantly over time among those taking it, suggesting the possible need for improvement in its taste or smell.
- There was no synergistic effect between omeprazole and curcumin.
IN PRACTICE:
“The new findings from our study may justify considering curcumin in clinical practice. This multicenter, randomized, controlled trial provides highly reliable evidence for the treatment of functional dyspepsia,” the authors wrote.
SOURCE:
Pradermchai Kongkam, MD, of Chulalongkorn University, Bangkok, and Wichittra Khongkha of Chao Phraya Abhaibhubejhr Hospital, Prachin Buri, Thailand, are joint first authors. The study was published online in BMJ Evidence-Based Medicine.
LIMITATIONS:
A small number of participants in each group were lost to follow-up, and the follow-up period was short (less than 2 months) for all.
DISCLOSURES:
The study was funded by the Thai Traditional and Alternative Medicine Fund. The authors have disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The researchers randomly assigned 206 patients to receive curcumin – the active ingredient in turmeric – alone; omeprazole alone; or curcumin plus omeprazole for 28 days. A total of 151 patients completed the study.
- Doses were two 250-mg curcumin pills four times daily, plus one placebo pill; one 20-mg omeprazole pill daily, plus two placebo pills four times daily; or two 250-mg curcumin pills four times daily, plus one 20-mg omeprazole pill once daily.
- Symptoms of functional dyspepsia were assessed on days 28 and 56 using the Severity of Dyspepsia Assessment (SODA) score.
TAKEAWAY:
- In the combined group, the curcumin-alone group, and the omeprazole-alone group, SODA scores for pain severity declined significantly by day 28 (–4.83, –5.46, and –6.22, respectively), as did scores for severity of other symptoms (–2.22, –2.32, and –2.31, respectively).
- Symptom improvements were even stronger by day 56 for pain (–7.19, –8.07, –8.85) and other symptoms (–4.09, –4.12, –3.71) in the same groups.
- Curcumin was safe and well tolerated, but satisfaction scores did not change significantly over time among those taking it, suggesting the possible need for improvement in its taste or smell.
- There was no synergistic effect between omeprazole and curcumin.
IN PRACTICE:
“The new findings from our study may justify considering curcumin in clinical practice. This multicenter, randomized, controlled trial provides highly reliable evidence for the treatment of functional dyspepsia,” the authors wrote.
SOURCE:
Pradermchai Kongkam, MD, of Chulalongkorn University, Bangkok, and Wichittra Khongkha of Chao Phraya Abhaibhubejhr Hospital, Prachin Buri, Thailand, are joint first authors. The study was published online in BMJ Evidence-Based Medicine.
LIMITATIONS:
A small number of participants in each group were lost to follow-up, and the follow-up period was short (less than 2 months) for all.
DISCLOSURES:
The study was funded by the Thai Traditional and Alternative Medicine Fund. The authors have disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The researchers randomly assigned 206 patients to receive curcumin – the active ingredient in turmeric – alone; omeprazole alone; or curcumin plus omeprazole for 28 days. A total of 151 patients completed the study.
- Doses were two 250-mg curcumin pills four times daily, plus one placebo pill; one 20-mg omeprazole pill daily, plus two placebo pills four times daily; or two 250-mg curcumin pills four times daily, plus one 20-mg omeprazole pill once daily.
- Symptoms of functional dyspepsia were assessed on days 28 and 56 using the Severity of Dyspepsia Assessment (SODA) score.
TAKEAWAY:
- In the combined group, the curcumin-alone group, and the omeprazole-alone group, SODA scores for pain severity declined significantly by day 28 (–4.83, –5.46, and –6.22, respectively), as did scores for severity of other symptoms (–2.22, –2.32, and –2.31, respectively).
- Symptom improvements were even stronger by day 56 for pain (–7.19, –8.07, –8.85) and other symptoms (–4.09, –4.12, –3.71) in the same groups.
- Curcumin was safe and well tolerated, but satisfaction scores did not change significantly over time among those taking it, suggesting the possible need for improvement in its taste or smell.
- There was no synergistic effect between omeprazole and curcumin.
IN PRACTICE:
“The new findings from our study may justify considering curcumin in clinical practice. This multicenter, randomized, controlled trial provides highly reliable evidence for the treatment of functional dyspepsia,” the authors wrote.
SOURCE:
Pradermchai Kongkam, MD, of Chulalongkorn University, Bangkok, and Wichittra Khongkha of Chao Phraya Abhaibhubejhr Hospital, Prachin Buri, Thailand, are joint first authors. The study was published online in BMJ Evidence-Based Medicine.
LIMITATIONS:
A small number of participants in each group were lost to follow-up, and the follow-up period was short (less than 2 months) for all.
DISCLOSURES:
The study was funded by the Thai Traditional and Alternative Medicine Fund. The authors have disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
FROM BMJ EVIDENCE-BASED MEDICINE
Company submits supplemental NDA for topical atopic dermatitis treatment
in adults and children aged 6 years and older.
Roflumilast cream 0.3% (Zoryve) is currently approved by the FDA for the topical treatment of plaque psoriasis, including intertriginous areas, in patients 12 years of age and older. Submission of the sNDA is based on positive results from the Interventional Trial Evaluating Roflumilast Cream for the Treatment of Atopic Dermatitis (INTEGUMENT-1 and INTEGUMENT-2) trials; two identical Phase 3, vehicle-controlled trials in which roflumilast cream 0.15% or vehicle was applied once daily for 4 weeks to individuals 6 years of age and older with mild to moderate AD involving at least 3% body surface area. Roflumilast is a phosphodiesterase-4 (PDE-4) inhibitor.
According to a press release from Arcutis, both studies met the primary endpoint of IGA Success, which was defined as a validated Investigator Global Assessment – Atopic Dermatitis (vIGA-AD) score of ‘clear’ or ‘almost clear’ plus a 2-grade improvement from baseline at week 4. In INTEGUMENT-1 this endpoint was achieved by 32.0% of subjects in the roflumilast cream group vs. 15.2% of those in the vehicle group (P < .0001). In INTEGUMENT-2, this endpoint was achieved by 28.9% of subjects in the roflumilast cream group vs. 12.0% of those in the vehicle group (P < .0001). The most common adverse reactions based on data from the combined trials were headache (2.9%), nausea (1.9%), application-site pain (1.5%), diarrhea (1.5%), and vomiting (1.5%).
in adults and children aged 6 years and older.
Roflumilast cream 0.3% (Zoryve) is currently approved by the FDA for the topical treatment of plaque psoriasis, including intertriginous areas, in patients 12 years of age and older. Submission of the sNDA is based on positive results from the Interventional Trial Evaluating Roflumilast Cream for the Treatment of Atopic Dermatitis (INTEGUMENT-1 and INTEGUMENT-2) trials; two identical Phase 3, vehicle-controlled trials in which roflumilast cream 0.15% or vehicle was applied once daily for 4 weeks to individuals 6 years of age and older with mild to moderate AD involving at least 3% body surface area. Roflumilast is a phosphodiesterase-4 (PDE-4) inhibitor.
According to a press release from Arcutis, both studies met the primary endpoint of IGA Success, which was defined as a validated Investigator Global Assessment – Atopic Dermatitis (vIGA-AD) score of ‘clear’ or ‘almost clear’ plus a 2-grade improvement from baseline at week 4. In INTEGUMENT-1 this endpoint was achieved by 32.0% of subjects in the roflumilast cream group vs. 15.2% of those in the vehicle group (P < .0001). In INTEGUMENT-2, this endpoint was achieved by 28.9% of subjects in the roflumilast cream group vs. 12.0% of those in the vehicle group (P < .0001). The most common adverse reactions based on data from the combined trials were headache (2.9%), nausea (1.9%), application-site pain (1.5%), diarrhea (1.5%), and vomiting (1.5%).
in adults and children aged 6 years and older.
Roflumilast cream 0.3% (Zoryve) is currently approved by the FDA for the topical treatment of plaque psoriasis, including intertriginous areas, in patients 12 years of age and older. Submission of the sNDA is based on positive results from the Interventional Trial Evaluating Roflumilast Cream for the Treatment of Atopic Dermatitis (INTEGUMENT-1 and INTEGUMENT-2) trials; two identical Phase 3, vehicle-controlled trials in which roflumilast cream 0.15% or vehicle was applied once daily for 4 weeks to individuals 6 years of age and older with mild to moderate AD involving at least 3% body surface area. Roflumilast is a phosphodiesterase-4 (PDE-4) inhibitor.
According to a press release from Arcutis, both studies met the primary endpoint of IGA Success, which was defined as a validated Investigator Global Assessment – Atopic Dermatitis (vIGA-AD) score of ‘clear’ or ‘almost clear’ plus a 2-grade improvement from baseline at week 4. In INTEGUMENT-1 this endpoint was achieved by 32.0% of subjects in the roflumilast cream group vs. 15.2% of those in the vehicle group (P < .0001). In INTEGUMENT-2, this endpoint was achieved by 28.9% of subjects in the roflumilast cream group vs. 12.0% of those in the vehicle group (P < .0001). The most common adverse reactions based on data from the combined trials were headache (2.9%), nausea (1.9%), application-site pain (1.5%), diarrhea (1.5%), and vomiting (1.5%).
Diabetes patients satisfied with continuous glucose monitors
TOPLINE:
However, significant proportions also reported concerns about accuracy under certain circumstances and about skin problems.
METHODOLOGY:
Researchers did an online survey of 504 people with type 1 diabetes from the T1D Exchange and 101 with type 2 diabetes from the Dynata database.
TAKEAWAY:
- The Dexcom G6 device was used by 60.7% of all current CGM users, including 69% of those with type 1 diabetes vs. 12% with type 2 diabetes.
- People with type 2 diabetes were more likely to use older Dexcom versions (G4/G5) (32%) or Abbott’s FreeStyle Libre systems (35%).
- Overall, 90% agreed that most sensors were accurate, but just 79% and 78%, respectively, were satisfied with sensor performance on the first and last day of wear.
- Moreover, 42% suspected variations in accuracy from sensor to sensor, and 32% continue to perform finger-stick monitoring more than six times a week.
- Individuals with type 2 diabetes were more likely than those with type 1 diabetes to be concerned about poor sensor performance affecting confidence in making diabetes management decisions (52% vs. 19%).
- Over half reported skin reactions and/or pain with the sensors (53.7% and 55.4%, respectively).
- Concerns about medications affecting sensor accuracy were more common among those with type 2 vs. type 1 diabetes (65% vs. 29%).
- Among overall concerns about substances or situations affecting sensor accuracy, the top choice (47%) was dehydration (despite a lack of supportive published literature), followed by pain medications (43%), cold/flu medications (32%), and coffee (24%).
- Inaccurate/false alarms negatively affected daily life for 36% of participants and diabetes management for 34%.
IN PRACTICE:
“CGM is a game-changing technology and has evolved in the past decade to overcome many technical and usability obstacles. Our survey suggests that there remain areas for further improvement ... Mistrust in CGM performance was more common than expected.”
SOURCE:
The study was done by Elizabeth Holt, of LifeScan, and colleagues. It was published in Clinical Diabetes.
LIMITATIONS:
- The databases used to recruit study participants may not be representative of the entire respective patient populations.
- Exercise wasn’t given as an option for affecting CGM accuracy, which might partly explain the dehydration finding.
DISCLOSURES:
Funding for this study and preparation of the manuscript were provided by LifeScan Inc. Two authors are LifeScan employees, and two others currently work for the T1D Exchange.
A version of this article first appeared on Medscape.com.
TOPLINE:
However, significant proportions also reported concerns about accuracy under certain circumstances and about skin problems.
METHODOLOGY:
Researchers did an online survey of 504 people with type 1 diabetes from the T1D Exchange and 101 with type 2 diabetes from the Dynata database.
TAKEAWAY:
- The Dexcom G6 device was used by 60.7% of all current CGM users, including 69% of those with type 1 diabetes vs. 12% with type 2 diabetes.
- People with type 2 diabetes were more likely to use older Dexcom versions (G4/G5) (32%) or Abbott’s FreeStyle Libre systems (35%).
- Overall, 90% agreed that most sensors were accurate, but just 79% and 78%, respectively, were satisfied with sensor performance on the first and last day of wear.
- Moreover, 42% suspected variations in accuracy from sensor to sensor, and 32% continue to perform finger-stick monitoring more than six times a week.
- Individuals with type 2 diabetes were more likely than those with type 1 diabetes to be concerned about poor sensor performance affecting confidence in making diabetes management decisions (52% vs. 19%).
- Over half reported skin reactions and/or pain with the sensors (53.7% and 55.4%, respectively).
- Concerns about medications affecting sensor accuracy were more common among those with type 2 vs. type 1 diabetes (65% vs. 29%).
- Among overall concerns about substances or situations affecting sensor accuracy, the top choice (47%) was dehydration (despite a lack of supportive published literature), followed by pain medications (43%), cold/flu medications (32%), and coffee (24%).
- Inaccurate/false alarms negatively affected daily life for 36% of participants and diabetes management for 34%.
IN PRACTICE:
“CGM is a game-changing technology and has evolved in the past decade to overcome many technical and usability obstacles. Our survey suggests that there remain areas for further improvement ... Mistrust in CGM performance was more common than expected.”
SOURCE:
The study was done by Elizabeth Holt, of LifeScan, and colleagues. It was published in Clinical Diabetes.
LIMITATIONS:
- The databases used to recruit study participants may not be representative of the entire respective patient populations.
- Exercise wasn’t given as an option for affecting CGM accuracy, which might partly explain the dehydration finding.
DISCLOSURES:
Funding for this study and preparation of the manuscript were provided by LifeScan Inc. Two authors are LifeScan employees, and two others currently work for the T1D Exchange.
A version of this article first appeared on Medscape.com.
TOPLINE:
However, significant proportions also reported concerns about accuracy under certain circumstances and about skin problems.
METHODOLOGY:
Researchers did an online survey of 504 people with type 1 diabetes from the T1D Exchange and 101 with type 2 diabetes from the Dynata database.
TAKEAWAY:
- The Dexcom G6 device was used by 60.7% of all current CGM users, including 69% of those with type 1 diabetes vs. 12% with type 2 diabetes.
- People with type 2 diabetes were more likely to use older Dexcom versions (G4/G5) (32%) or Abbott’s FreeStyle Libre systems (35%).
- Overall, 90% agreed that most sensors were accurate, but just 79% and 78%, respectively, were satisfied with sensor performance on the first and last day of wear.
- Moreover, 42% suspected variations in accuracy from sensor to sensor, and 32% continue to perform finger-stick monitoring more than six times a week.
- Individuals with type 2 diabetes were more likely than those with type 1 diabetes to be concerned about poor sensor performance affecting confidence in making diabetes management decisions (52% vs. 19%).
- Over half reported skin reactions and/or pain with the sensors (53.7% and 55.4%, respectively).
- Concerns about medications affecting sensor accuracy were more common among those with type 2 vs. type 1 diabetes (65% vs. 29%).
- Among overall concerns about substances or situations affecting sensor accuracy, the top choice (47%) was dehydration (despite a lack of supportive published literature), followed by pain medications (43%), cold/flu medications (32%), and coffee (24%).
- Inaccurate/false alarms negatively affected daily life for 36% of participants and diabetes management for 34%.
IN PRACTICE:
“CGM is a game-changing technology and has evolved in the past decade to overcome many technical and usability obstacles. Our survey suggests that there remain areas for further improvement ... Mistrust in CGM performance was more common than expected.”
SOURCE:
The study was done by Elizabeth Holt, of LifeScan, and colleagues. It was published in Clinical Diabetes.
LIMITATIONS:
- The databases used to recruit study participants may not be representative of the entire respective patient populations.
- Exercise wasn’t given as an option for affecting CGM accuracy, which might partly explain the dehydration finding.
DISCLOSURES:
Funding for this study and preparation of the manuscript were provided by LifeScan Inc. Two authors are LifeScan employees, and two others currently work for the T1D Exchange.
A version of this article first appeared on Medscape.com.
Steady VKA therapy beats switch to NOAC in frail AFib patients: FRAIL-AF
Switching frail patients with atrial fibrillation (AFib) from anticoagulation therapy with vitamin K antagonists (VKAs) to a novel oral anticoagulant (NOAC) resulted in more bleeding without any reduction in thromboembolic complications or all-cause mortality, randomized trial results show.
The study, FRAIL-AF, is the first randomized NOAC trial to exclusively include frail older patients, said lead author Linda P.T. Joosten, MD, Julius Center for Health Sciences and Primary Care in Utrecht, the Netherlands, and these unexpected findings provide evidence that goes beyond what is currently available.
“Data from the FRAIL-AF trial showed that switching from a VKA to a NOAC should not be considered without a clear indication in frail older patients with AF[ib], as switching to a NOAC leads to 69% more bleeding,” she concluded, without any benefit on secondary clinical endpoints, including thromboembolic events and all-cause mortality.
“The results turned out different than we expected,” Dr. Joosten said. “The hypothesis of this superiority trial was that switching from VKA therapy to a NOAC would result in less bleeding. However, we observed the opposite. After the interim analysis, the data and safety monitoring board advised to stop inclusion because switching from a VKA to a NOAC was clearly contraindicated with a hazard ratio of 1.69 and a highly significant P value of .001.”
Results of FRAIL-AF were presented at the annual congress of the European Society of Cardiology and published online in the journal Circulation.
Session moderator Renate B. Schnabel, MD, interventional cardiologist with University Heart & Vascular Center Hamburg (Germany), congratulated the researchers on these “astonishing” data.
“The thing I want to emphasize here is that, in the absence of randomized controlled trial data, we should be very cautious in extrapolating data from the landmark trials to populations not enrolled in those, and to rely on observational data only,” Dr. Schnabel told Dr. Joosten. “We need randomized controlled trials that sometimes give astonishing results.”
Frailty a clinical syndrome
Frailty is “a lot more than just aging, multiple comorbidities and polypharmacy,” Dr. Joosten explained. “It’s really a clinical syndrome, with people with a high biological vulnerability, dependency on significant others, and a reduced capacity to resist stressors, all leading to a reduced homeostatic reserve.”
Frailty is common in the community, with a prevalence of about 12%, she noted, “and even more important, AF[ib] in frail older people is very common, with a prevalence of 18%. And “without any doubt, we have to adequately anticoagulate frail AF[ib] patients, as they have a high stroke risk, with an incidence of 12.4% per year,” Dr. Joosten noted, compared with 3.9% per year among nonfrail AFib patients.
NOACs are preferred over VKAs in nonfrail AFib patients, after four major trials, RE-LY with dabigatran, ROCKET-AF with rivaroxaban, ARISTOTLE with apixaban, and ENGAGE-AF with edoxaban, showed that NOAC treatment resulted in less major bleeding while stroke risk was comparable with treatment with warfarin, she noted.
The 2023 European Heart Rhythm Association consensus document on management of arrhythmias in frailty syndrome concludes that the advantages of NOACs relative to VKAs are “likely consistent” in frail and nonfrail AFib patients, but the level of evidence is low.
So it’s unknown if NOACs are preferred over VKAs in frail AFib patients, “and it’s even more questionable whether patients on VKAs should switch to NOAC therapy,” Dr. Joosten said.
This new trial aimed to answer the question of whether switching frail AFib patients currently managed on a VKA to a NOAC would reduce bleeding. FRAIL-AF was a pragmatic, multicenter, open-label, randomized, controlled superiority trial.
Older AFib patients were deemed frail if they were aged 75 years or older and had a score of 3 or more on the validated Groningen Frailty Indicator (GFI). Patients with a glomerular filtration rate of less than 30 mL/min per 1.73 m2 or with valvular AFib were excluded.
Eligible patients were then assigned randomly to switch from their international normalized ratio (INR)–guided VKA treatment with either 1 mg acenocoumarol or 3 mg phenprocoumon, to a NOAC, or to continue VKA treatment. They were followed for 12 months for the primary outcome – major bleeding or clinically relevant nonmajor bleeding complication, whichever came first – accounting for death as a competing risk.
A total of 1,330 patients were randomly assigned between January 2018 and June 2022. Their mean age was 83 years, and they had a median GFI of 4. After randomization, 6 patients in the switch-to-NOAC arm, and 1 in the continue-VKA arm were found to have exclusion criteria, so in the end, 662 patients were switched from a VKA to NOAC, while 661 continued on VKA therapy. The choice of NOAC was made by the treating physician.
Major bleeding was defined as a fatal bleeding; bleeding in a critical area or organ; bleeding leading to transfusion; and/or bleeding leading to a fall in hemoglobin level of 2 g/dL (1.24 mmol/L) or more. Nonmajor bleeding was bleeding not considered major but requiring face-to-face consultation, hospitalization or increased level of care, or medical intervention.
After a prespecified futility analysis planned after 163 primary outcome events, the trial was halted when it was seen that there were 101 primary outcome events in the switch arm compared to 62 in the continue arm, Dr. Joosten said. The difference appeared to be driven by clinically relevant nonmajor bleeding.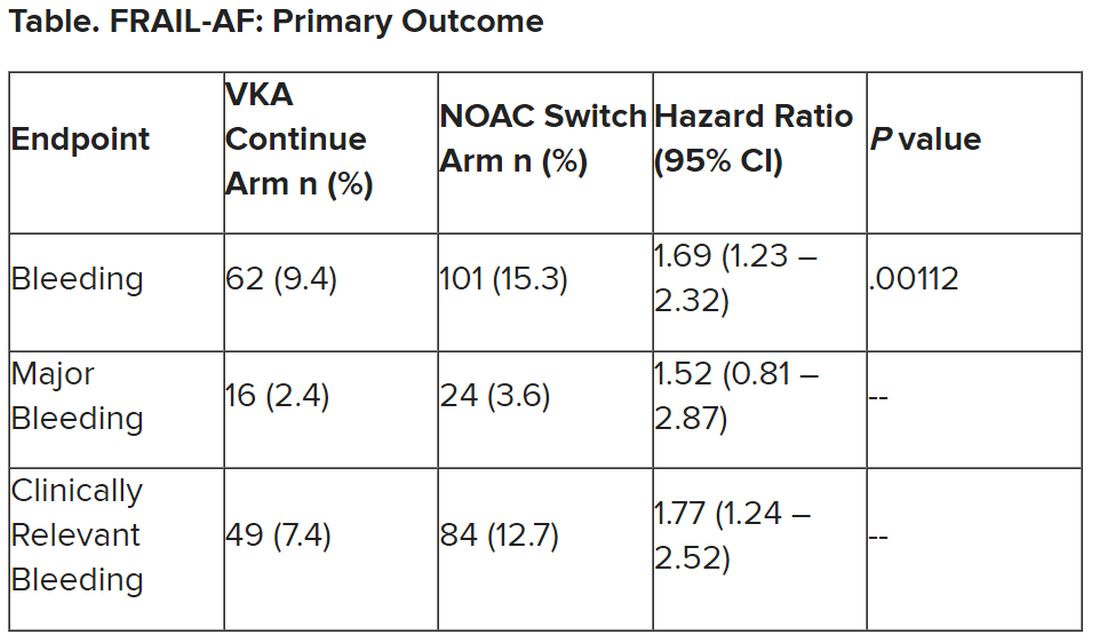
Secondary outcomes of thromboembolic events and all-cause mortality were similar between the groups.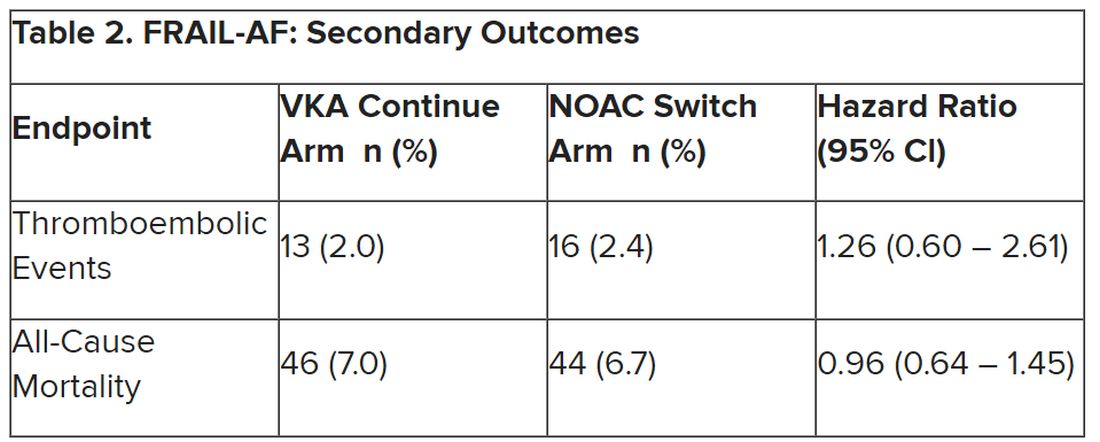
Completely different patients
Discussant at the meeting for the presentation was Isabelle C. Van Gelder, MD, University Medical Centre Groningen (the Netherlands). She said the results are important and relevant because it “provides data on an important gap of knowledge in our AF[ib] guidelines, and a note for all the cardiologists – this study was not done in the hospital. This trial was done in general practitioner practices, so that’s important to consider.”
Comparing FRAIL-AF patients with those of the four previous NOAC trials, “you see that enormous difference in age,” with an average age of 83 years versus 70-73 years in those trials. “These are completely different patients than have been included previously,” she said.
That GFI score of 4 or more includes patients on four or more different types of medication, as well as memory complaints, an inability to walk around the house, and problems with vision or hearing.
The finding of a 69% increase in bleeding with NOACs in FRAIL-AF was “completely unexpected, and I think that we as cardiologists and as NOAC believers did not expect it at all, but it is as clear as it is.” The curves don’t diverge immediately, but rather after 3 months or thereafter, “so it has nothing to do with the switching process. So why did it occur?”
The Netherlands has dedicated thrombosis services that might improve time in therapeutic range for VKA patients, but there is no real difference in TTRs in FRAIL-AF versus the other NOAC trials, Dr. Van Gelder noted.
The most likely suspect in her view is frailty itself, in particular the tendency for patients to be on a high number of medications. A previous study showed, for example, that polypharmacy could be used as a proxy for the effect of frailty on bleeding risk; patients on 10 or more medications had a higher risk for bleeding on treatment with rivaroxaban versus those on 4 or fewer medications.
“Therefore, in my view, why was there such a high risk of bleeding? It’s because these are other patients than we are normally used to treat, we as cardiologists,” although general practitioners see these patients all the time. “It’s all about frailty.”
NOACs are still relatively new drugs, with possible unknown interactions, she added. Because of their frailty and polypharmacy, these patients may benefit from INR control, Dr. Van Gelder speculated. “Therefore, I agree with them that we should be careful; if such old, frail patients survive on VKA, do not change medications and do not switch!”
The study was supported by the Dutch government with additional and unrestricted educational grants from Boehringer Ingelheim, BMS-Pfizer, Bayer, and Daiichi Sankyo. Dr. Joosten reported no relevant financial relationships. Dr. Van Gelder reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Switching frail patients with atrial fibrillation (AFib) from anticoagulation therapy with vitamin K antagonists (VKAs) to a novel oral anticoagulant (NOAC) resulted in more bleeding without any reduction in thromboembolic complications or all-cause mortality, randomized trial results show.
The study, FRAIL-AF, is the first randomized NOAC trial to exclusively include frail older patients, said lead author Linda P.T. Joosten, MD, Julius Center for Health Sciences and Primary Care in Utrecht, the Netherlands, and these unexpected findings provide evidence that goes beyond what is currently available.
“Data from the FRAIL-AF trial showed that switching from a VKA to a NOAC should not be considered without a clear indication in frail older patients with AF[ib], as switching to a NOAC leads to 69% more bleeding,” she concluded, without any benefit on secondary clinical endpoints, including thromboembolic events and all-cause mortality.
“The results turned out different than we expected,” Dr. Joosten said. “The hypothesis of this superiority trial was that switching from VKA therapy to a NOAC would result in less bleeding. However, we observed the opposite. After the interim analysis, the data and safety monitoring board advised to stop inclusion because switching from a VKA to a NOAC was clearly contraindicated with a hazard ratio of 1.69 and a highly significant P value of .001.”
Results of FRAIL-AF were presented at the annual congress of the European Society of Cardiology and published online in the journal Circulation.
Session moderator Renate B. Schnabel, MD, interventional cardiologist with University Heart & Vascular Center Hamburg (Germany), congratulated the researchers on these “astonishing” data.
“The thing I want to emphasize here is that, in the absence of randomized controlled trial data, we should be very cautious in extrapolating data from the landmark trials to populations not enrolled in those, and to rely on observational data only,” Dr. Schnabel told Dr. Joosten. “We need randomized controlled trials that sometimes give astonishing results.”
Frailty a clinical syndrome
Frailty is “a lot more than just aging, multiple comorbidities and polypharmacy,” Dr. Joosten explained. “It’s really a clinical syndrome, with people with a high biological vulnerability, dependency on significant others, and a reduced capacity to resist stressors, all leading to a reduced homeostatic reserve.”
Frailty is common in the community, with a prevalence of about 12%, she noted, “and even more important, AF[ib] in frail older people is very common, with a prevalence of 18%. And “without any doubt, we have to adequately anticoagulate frail AF[ib] patients, as they have a high stroke risk, with an incidence of 12.4% per year,” Dr. Joosten noted, compared with 3.9% per year among nonfrail AFib patients.
NOACs are preferred over VKAs in nonfrail AFib patients, after four major trials, RE-LY with dabigatran, ROCKET-AF with rivaroxaban, ARISTOTLE with apixaban, and ENGAGE-AF with edoxaban, showed that NOAC treatment resulted in less major bleeding while stroke risk was comparable with treatment with warfarin, she noted.
The 2023 European Heart Rhythm Association consensus document on management of arrhythmias in frailty syndrome concludes that the advantages of NOACs relative to VKAs are “likely consistent” in frail and nonfrail AFib patients, but the level of evidence is low.
So it’s unknown if NOACs are preferred over VKAs in frail AFib patients, “and it’s even more questionable whether patients on VKAs should switch to NOAC therapy,” Dr. Joosten said.
This new trial aimed to answer the question of whether switching frail AFib patients currently managed on a VKA to a NOAC would reduce bleeding. FRAIL-AF was a pragmatic, multicenter, open-label, randomized, controlled superiority trial.
Older AFib patients were deemed frail if they were aged 75 years or older and had a score of 3 or more on the validated Groningen Frailty Indicator (GFI). Patients with a glomerular filtration rate of less than 30 mL/min per 1.73 m2 or with valvular AFib were excluded.
Eligible patients were then assigned randomly to switch from their international normalized ratio (INR)–guided VKA treatment with either 1 mg acenocoumarol or 3 mg phenprocoumon, to a NOAC, or to continue VKA treatment. They were followed for 12 months for the primary outcome – major bleeding or clinically relevant nonmajor bleeding complication, whichever came first – accounting for death as a competing risk.
A total of 1,330 patients were randomly assigned between January 2018 and June 2022. Their mean age was 83 years, and they had a median GFI of 4. After randomization, 6 patients in the switch-to-NOAC arm, and 1 in the continue-VKA arm were found to have exclusion criteria, so in the end, 662 patients were switched from a VKA to NOAC, while 661 continued on VKA therapy. The choice of NOAC was made by the treating physician.
Major bleeding was defined as a fatal bleeding; bleeding in a critical area or organ; bleeding leading to transfusion; and/or bleeding leading to a fall in hemoglobin level of 2 g/dL (1.24 mmol/L) or more. Nonmajor bleeding was bleeding not considered major but requiring face-to-face consultation, hospitalization or increased level of care, or medical intervention.
After a prespecified futility analysis planned after 163 primary outcome events, the trial was halted when it was seen that there were 101 primary outcome events in the switch arm compared to 62 in the continue arm, Dr. Joosten said. The difference appeared to be driven by clinically relevant nonmajor bleeding.
Secondary outcomes of thromboembolic events and all-cause mortality were similar between the groups.
Completely different patients
Discussant at the meeting for the presentation was Isabelle C. Van Gelder, MD, University Medical Centre Groningen (the Netherlands). She said the results are important and relevant because it “provides data on an important gap of knowledge in our AF[ib] guidelines, and a note for all the cardiologists – this study was not done in the hospital. This trial was done in general practitioner practices, so that’s important to consider.”
Comparing FRAIL-AF patients with those of the four previous NOAC trials, “you see that enormous difference in age,” with an average age of 83 years versus 70-73 years in those trials. “These are completely different patients than have been included previously,” she said.
That GFI score of 4 or more includes patients on four or more different types of medication, as well as memory complaints, an inability to walk around the house, and problems with vision or hearing.
The finding of a 69% increase in bleeding with NOACs in FRAIL-AF was “completely unexpected, and I think that we as cardiologists and as NOAC believers did not expect it at all, but it is as clear as it is.” The curves don’t diverge immediately, but rather after 3 months or thereafter, “so it has nothing to do with the switching process. So why did it occur?”
The Netherlands has dedicated thrombosis services that might improve time in therapeutic range for VKA patients, but there is no real difference in TTRs in FRAIL-AF versus the other NOAC trials, Dr. Van Gelder noted.
The most likely suspect in her view is frailty itself, in particular the tendency for patients to be on a high number of medications. A previous study showed, for example, that polypharmacy could be used as a proxy for the effect of frailty on bleeding risk; patients on 10 or more medications had a higher risk for bleeding on treatment with rivaroxaban versus those on 4 or fewer medications.
“Therefore, in my view, why was there such a high risk of bleeding? It’s because these are other patients than we are normally used to treat, we as cardiologists,” although general practitioners see these patients all the time. “It’s all about frailty.”
NOACs are still relatively new drugs, with possible unknown interactions, she added. Because of their frailty and polypharmacy, these patients may benefit from INR control, Dr. Van Gelder speculated. “Therefore, I agree with them that we should be careful; if such old, frail patients survive on VKA, do not change medications and do not switch!”
The study was supported by the Dutch government with additional and unrestricted educational grants from Boehringer Ingelheim, BMS-Pfizer, Bayer, and Daiichi Sankyo. Dr. Joosten reported no relevant financial relationships. Dr. Van Gelder reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Switching frail patients with atrial fibrillation (AFib) from anticoagulation therapy with vitamin K antagonists (VKAs) to a novel oral anticoagulant (NOAC) resulted in more bleeding without any reduction in thromboembolic complications or all-cause mortality, randomized trial results show.
The study, FRAIL-AF, is the first randomized NOAC trial to exclusively include frail older patients, said lead author Linda P.T. Joosten, MD, Julius Center for Health Sciences and Primary Care in Utrecht, the Netherlands, and these unexpected findings provide evidence that goes beyond what is currently available.
“Data from the FRAIL-AF trial showed that switching from a VKA to a NOAC should not be considered without a clear indication in frail older patients with AF[ib], as switching to a NOAC leads to 69% more bleeding,” she concluded, without any benefit on secondary clinical endpoints, including thromboembolic events and all-cause mortality.
“The results turned out different than we expected,” Dr. Joosten said. “The hypothesis of this superiority trial was that switching from VKA therapy to a NOAC would result in less bleeding. However, we observed the opposite. After the interim analysis, the data and safety monitoring board advised to stop inclusion because switching from a VKA to a NOAC was clearly contraindicated with a hazard ratio of 1.69 and a highly significant P value of .001.”
Results of FRAIL-AF were presented at the annual congress of the European Society of Cardiology and published online in the journal Circulation.
Session moderator Renate B. Schnabel, MD, interventional cardiologist with University Heart & Vascular Center Hamburg (Germany), congratulated the researchers on these “astonishing” data.
“The thing I want to emphasize here is that, in the absence of randomized controlled trial data, we should be very cautious in extrapolating data from the landmark trials to populations not enrolled in those, and to rely on observational data only,” Dr. Schnabel told Dr. Joosten. “We need randomized controlled trials that sometimes give astonishing results.”
Frailty a clinical syndrome
Frailty is “a lot more than just aging, multiple comorbidities and polypharmacy,” Dr. Joosten explained. “It’s really a clinical syndrome, with people with a high biological vulnerability, dependency on significant others, and a reduced capacity to resist stressors, all leading to a reduced homeostatic reserve.”
Frailty is common in the community, with a prevalence of about 12%, she noted, “and even more important, AF[ib] in frail older people is very common, with a prevalence of 18%. And “without any doubt, we have to adequately anticoagulate frail AF[ib] patients, as they have a high stroke risk, with an incidence of 12.4% per year,” Dr. Joosten noted, compared with 3.9% per year among nonfrail AFib patients.
NOACs are preferred over VKAs in nonfrail AFib patients, after four major trials, RE-LY with dabigatran, ROCKET-AF with rivaroxaban, ARISTOTLE with apixaban, and ENGAGE-AF with edoxaban, showed that NOAC treatment resulted in less major bleeding while stroke risk was comparable with treatment with warfarin, she noted.
The 2023 European Heart Rhythm Association consensus document on management of arrhythmias in frailty syndrome concludes that the advantages of NOACs relative to VKAs are “likely consistent” in frail and nonfrail AFib patients, but the level of evidence is low.
So it’s unknown if NOACs are preferred over VKAs in frail AFib patients, “and it’s even more questionable whether patients on VKAs should switch to NOAC therapy,” Dr. Joosten said.
This new trial aimed to answer the question of whether switching frail AFib patients currently managed on a VKA to a NOAC would reduce bleeding. FRAIL-AF was a pragmatic, multicenter, open-label, randomized, controlled superiority trial.
Older AFib patients were deemed frail if they were aged 75 years or older and had a score of 3 or more on the validated Groningen Frailty Indicator (GFI). Patients with a glomerular filtration rate of less than 30 mL/min per 1.73 m2 or with valvular AFib were excluded.
Eligible patients were then assigned randomly to switch from their international normalized ratio (INR)–guided VKA treatment with either 1 mg acenocoumarol or 3 mg phenprocoumon, to a NOAC, or to continue VKA treatment. They were followed for 12 months for the primary outcome – major bleeding or clinically relevant nonmajor bleeding complication, whichever came first – accounting for death as a competing risk.
A total of 1,330 patients were randomly assigned between January 2018 and June 2022. Their mean age was 83 years, and they had a median GFI of 4. After randomization, 6 patients in the switch-to-NOAC arm, and 1 in the continue-VKA arm were found to have exclusion criteria, so in the end, 662 patients were switched from a VKA to NOAC, while 661 continued on VKA therapy. The choice of NOAC was made by the treating physician.
Major bleeding was defined as a fatal bleeding; bleeding in a critical area or organ; bleeding leading to transfusion; and/or bleeding leading to a fall in hemoglobin level of 2 g/dL (1.24 mmol/L) or more. Nonmajor bleeding was bleeding not considered major but requiring face-to-face consultation, hospitalization or increased level of care, or medical intervention.
After a prespecified futility analysis planned after 163 primary outcome events, the trial was halted when it was seen that there were 101 primary outcome events in the switch arm compared to 62 in the continue arm, Dr. Joosten said. The difference appeared to be driven by clinically relevant nonmajor bleeding.
Secondary outcomes of thromboembolic events and all-cause mortality were similar between the groups.
Completely different patients
Discussant at the meeting for the presentation was Isabelle C. Van Gelder, MD, University Medical Centre Groningen (the Netherlands). She said the results are important and relevant because it “provides data on an important gap of knowledge in our AF[ib] guidelines, and a note for all the cardiologists – this study was not done in the hospital. This trial was done in general practitioner practices, so that’s important to consider.”
Comparing FRAIL-AF patients with those of the four previous NOAC trials, “you see that enormous difference in age,” with an average age of 83 years versus 70-73 years in those trials. “These are completely different patients than have been included previously,” she said.
That GFI score of 4 or more includes patients on four or more different types of medication, as well as memory complaints, an inability to walk around the house, and problems with vision or hearing.
The finding of a 69% increase in bleeding with NOACs in FRAIL-AF was “completely unexpected, and I think that we as cardiologists and as NOAC believers did not expect it at all, but it is as clear as it is.” The curves don’t diverge immediately, but rather after 3 months or thereafter, “so it has nothing to do with the switching process. So why did it occur?”
The Netherlands has dedicated thrombosis services that might improve time in therapeutic range for VKA patients, but there is no real difference in TTRs in FRAIL-AF versus the other NOAC trials, Dr. Van Gelder noted.
The most likely suspect in her view is frailty itself, in particular the tendency for patients to be on a high number of medications. A previous study showed, for example, that polypharmacy could be used as a proxy for the effect of frailty on bleeding risk; patients on 10 or more medications had a higher risk for bleeding on treatment with rivaroxaban versus those on 4 or fewer medications.
“Therefore, in my view, why was there such a high risk of bleeding? It’s because these are other patients than we are normally used to treat, we as cardiologists,” although general practitioners see these patients all the time. “It’s all about frailty.”
NOACs are still relatively new drugs, with possible unknown interactions, she added. Because of their frailty and polypharmacy, these patients may benefit from INR control, Dr. Van Gelder speculated. “Therefore, I agree with them that we should be careful; if such old, frail patients survive on VKA, do not change medications and do not switch!”
The study was supported by the Dutch government with additional and unrestricted educational grants from Boehringer Ingelheim, BMS-Pfizer, Bayer, and Daiichi Sankyo. Dr. Joosten reported no relevant financial relationships. Dr. Van Gelder reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE ESC CONGRESS 2023
‘New dawn’ for aldosterone as drug target in hypertension?
Once-daily treatment with the selective aldosterone synthase inhibitor lorundrostat (Mineralys Therapeutics) safely and significantly reduced blood pressure in adults with uncontrolled hypertension in a phase 2, randomized, controlled trial.
Eight weeks after adding lorundrostat (50 mg or 100 mg once daily) or placebo to background therapy, the medication lowered seated automated office systolic BP significantly more than placebo (−9.6 mm Hg with 50 mg; −7.8 mm Hg with 100 mg), with the greatest effects seen in adults with obesity.
“We need new drugs for treatment-resistant hypertension,” study investigator Steven Nissen, MD, chief academic officer at the Heart Vascular & Thoracic Institute at the Cleveland Clinic, said in an interview. Lorundrostat represents a “new class” of antihypertensive that “looks to be safe and we’re seeing very large reductions in blood pressure.”
Results of the Target-HTN trial were published online in JAMA to coincide with presentation at the Hypertension Scientific Sessions, sponsored by the American Heart Association.
Aldosterone’s contribution ‘vastly underappreciated’
Excess aldosterone production contributes to uncontrolled BP in patients with obesity and other associated diseases, such as obstructive sleep apnea and metabolic syndrome.
“Aldosterone’s contribution to uncontrolled hypertension is vastly underappreciated,” first author and study presenter Luke Laffin, MD, also with the Cleveland Clinic, said in an interview.
Aldosterone synthase inhibitors are a novel class of BP-lowering medications that decrease aldosterone production. Lorundrostat is one of two such agents in advanced clinical development. The other is baxdrostat (CinCor Pharma/AstraZeneca).
The Target-HTN randomized, placebo-controlled, dose-ranging trial enrolled 200 adults (mean age, 66 years; 60% women) with uncontrolled hypertension while taking two or more antihypertensive medications; 42% of participants were taking three or more antihypertensive medications, 48% were obese and 40% had diabetes.
The study population was divided into two cohorts: an initial cohort of 163 adults with suppressed plasma renin activity at baseline (PRA ≤ 1.0 ng/mL per hour) and elevated plasma aldosterone (≥ 1.0 ng/dL) and a second cohort of 37 adults with PRA greater than 1.0 ng/mL per hour.
Participants were randomly assigned to placebo or one of five doses of lorundrostat in the initial cohort (12.5 mg, 50 mg, or 100 mg once daily or 12.5 mg or 25 mg twice daily).
In the second cohort, participants were randomly assigned (1:6) to placebo or lorundrostat 100 mg once daily. The primary endpoint was change in automated office systolic BP from baseline to week 8.
Among participants with suppressed PRA, following 8 weeks of treatment, changes in office systolic BP of −14.1, −13.2, and −6.9 mm Hg were observed with 100 mg, 50 mg, and 12.5 mg once-daily lorundrostat, respectively, compared with a change of −4.1 mm Hg with placebo.
Reductions in systolic BP in individuals receiving twice-daily doses of 25 mg and 12.5 mg of lorundrostat were −10.1 and −13.8 mm Hg, respectively.
Among participants without suppressed PRA, lorundrostat 100 mg once daily decreased systolic BP by 11.4 mm Hg, similar to BP reduction in those with suppressed PRA receiving the same dose.
A prespecified subgroup analysis showed that participants with obesity demonstrated greater BP lowering in response to lorundrostat.
No instances of cortisol insufficiency occurred. Six participants had increases in serum potassium above 6.0 mEq/L (6.0 mmol/L) that corrected with dose reduction or drug discontinuation.
The increase in serum potassium is “expected and manageable,” Dr. Laffin said in an interview. “Anytime you disrupt aldosterone production, you’re going to have to have an increase in serum potassium, but it’s very manageable and not something that is worrisome.”
A phase 2 trial in 300 adults with uncontrolled hypertension is currently underway. The trial will evaluate the BP-lowering effects of lorundrostat, administered on a background of a standardized antihypertensive medication regimen. A larger phase 3 study will start before the end of the year.
‘New dawn’ for therapies targeting aldosterone
The author of an editorial in JAMA noted that more 70 years after the first isolation of aldosterone, then called electrocortin, “there is a new dawn for therapies targeting aldosterone.”
“There is now real potential to provide better-targeted treatment for patients in whom aldosterone excess is known to contribute to their clinical condition and influence their clinical outcome, notably those with difficult-to-control hypertension, obesity, heart failure, chronic kidney disease, and the many with yet-to-be-diagnosed primary aldosteronism,” said Bryan Williams, MD, University College London.
The trial was funded by Mineralys Therapeutics, which is developing lorundrostat. Dr. Laffin reported that the Cleveland Clinic, his employer, was a study site for the Target-HTN trial and that C5Research, the academic research organization of the Cleveland Clinic, receives payment for services related to other Mineralys clinical trials. Dr. Laffin also reported receipt of personal fees from Medtronic, Lilly, and Crispr Therapeutics, grants from AstraZeneca, and stock options for LucidAct Health and Gordy Health. Dr. Nissen reported receipt of grants from Mineralys during the conduct of the study and grants from AbbVie, AstraZeneca, Amgen, Bristol-Myers Squibb, Lilly, Esperion Therapeutics, Medtronic, grants from MyoKardia, New Amsterdam Pharmaceuticals, Novartis, and Silence Therapeutics. Dr. Williams reported being the unremunerated chair of the steering committee designing a phase 3 trial of the aldosterone synthase inhibitor baxdrostat for AstraZeneca.
A version of this article first appeared on Medscape.com.
Once-daily treatment with the selective aldosterone synthase inhibitor lorundrostat (Mineralys Therapeutics) safely and significantly reduced blood pressure in adults with uncontrolled hypertension in a phase 2, randomized, controlled trial.
Eight weeks after adding lorundrostat (50 mg or 100 mg once daily) or placebo to background therapy, the medication lowered seated automated office systolic BP significantly more than placebo (−9.6 mm Hg with 50 mg; −7.8 mm Hg with 100 mg), with the greatest effects seen in adults with obesity.
“We need new drugs for treatment-resistant hypertension,” study investigator Steven Nissen, MD, chief academic officer at the Heart Vascular & Thoracic Institute at the Cleveland Clinic, said in an interview. Lorundrostat represents a “new class” of antihypertensive that “looks to be safe and we’re seeing very large reductions in blood pressure.”
Results of the Target-HTN trial were published online in JAMA to coincide with presentation at the Hypertension Scientific Sessions, sponsored by the American Heart Association.
Aldosterone’s contribution ‘vastly underappreciated’
Excess aldosterone production contributes to uncontrolled BP in patients with obesity and other associated diseases, such as obstructive sleep apnea and metabolic syndrome.
“Aldosterone’s contribution to uncontrolled hypertension is vastly underappreciated,” first author and study presenter Luke Laffin, MD, also with the Cleveland Clinic, said in an interview.
Aldosterone synthase inhibitors are a novel class of BP-lowering medications that decrease aldosterone production. Lorundrostat is one of two such agents in advanced clinical development. The other is baxdrostat (CinCor Pharma/AstraZeneca).
The Target-HTN randomized, placebo-controlled, dose-ranging trial enrolled 200 adults (mean age, 66 years; 60% women) with uncontrolled hypertension while taking two or more antihypertensive medications; 42% of participants were taking three or more antihypertensive medications, 48% were obese and 40% had diabetes.
The study population was divided into two cohorts: an initial cohort of 163 adults with suppressed plasma renin activity at baseline (PRA ≤ 1.0 ng/mL per hour) and elevated plasma aldosterone (≥ 1.0 ng/dL) and a second cohort of 37 adults with PRA greater than 1.0 ng/mL per hour.
Participants were randomly assigned to placebo or one of five doses of lorundrostat in the initial cohort (12.5 mg, 50 mg, or 100 mg once daily or 12.5 mg or 25 mg twice daily).
In the second cohort, participants were randomly assigned (1:6) to placebo or lorundrostat 100 mg once daily. The primary endpoint was change in automated office systolic BP from baseline to week 8.
Among participants with suppressed PRA, following 8 weeks of treatment, changes in office systolic BP of −14.1, −13.2, and −6.9 mm Hg were observed with 100 mg, 50 mg, and 12.5 mg once-daily lorundrostat, respectively, compared with a change of −4.1 mm Hg with placebo.
Reductions in systolic BP in individuals receiving twice-daily doses of 25 mg and 12.5 mg of lorundrostat were −10.1 and −13.8 mm Hg, respectively.
Among participants without suppressed PRA, lorundrostat 100 mg once daily decreased systolic BP by 11.4 mm Hg, similar to BP reduction in those with suppressed PRA receiving the same dose.
A prespecified subgroup analysis showed that participants with obesity demonstrated greater BP lowering in response to lorundrostat.
No instances of cortisol insufficiency occurred. Six participants had increases in serum potassium above 6.0 mEq/L (6.0 mmol/L) that corrected with dose reduction or drug discontinuation.
The increase in serum potassium is “expected and manageable,” Dr. Laffin said in an interview. “Anytime you disrupt aldosterone production, you’re going to have to have an increase in serum potassium, but it’s very manageable and not something that is worrisome.”
A phase 2 trial in 300 adults with uncontrolled hypertension is currently underway. The trial will evaluate the BP-lowering effects of lorundrostat, administered on a background of a standardized antihypertensive medication regimen. A larger phase 3 study will start before the end of the year.
‘New dawn’ for therapies targeting aldosterone
The author of an editorial in JAMA noted that more 70 years after the first isolation of aldosterone, then called electrocortin, “there is a new dawn for therapies targeting aldosterone.”
“There is now real potential to provide better-targeted treatment for patients in whom aldosterone excess is known to contribute to their clinical condition and influence their clinical outcome, notably those with difficult-to-control hypertension, obesity, heart failure, chronic kidney disease, and the many with yet-to-be-diagnosed primary aldosteronism,” said Bryan Williams, MD, University College London.
The trial was funded by Mineralys Therapeutics, which is developing lorundrostat. Dr. Laffin reported that the Cleveland Clinic, his employer, was a study site for the Target-HTN trial and that C5Research, the academic research organization of the Cleveland Clinic, receives payment for services related to other Mineralys clinical trials. Dr. Laffin also reported receipt of personal fees from Medtronic, Lilly, and Crispr Therapeutics, grants from AstraZeneca, and stock options for LucidAct Health and Gordy Health. Dr. Nissen reported receipt of grants from Mineralys during the conduct of the study and grants from AbbVie, AstraZeneca, Amgen, Bristol-Myers Squibb, Lilly, Esperion Therapeutics, Medtronic, grants from MyoKardia, New Amsterdam Pharmaceuticals, Novartis, and Silence Therapeutics. Dr. Williams reported being the unremunerated chair of the steering committee designing a phase 3 trial of the aldosterone synthase inhibitor baxdrostat for AstraZeneca.
A version of this article first appeared on Medscape.com.
Once-daily treatment with the selective aldosterone synthase inhibitor lorundrostat (Mineralys Therapeutics) safely and significantly reduced blood pressure in adults with uncontrolled hypertension in a phase 2, randomized, controlled trial.
Eight weeks after adding lorundrostat (50 mg or 100 mg once daily) or placebo to background therapy, the medication lowered seated automated office systolic BP significantly more than placebo (−9.6 mm Hg with 50 mg; −7.8 mm Hg with 100 mg), with the greatest effects seen in adults with obesity.
“We need new drugs for treatment-resistant hypertension,” study investigator Steven Nissen, MD, chief academic officer at the Heart Vascular & Thoracic Institute at the Cleveland Clinic, said in an interview. Lorundrostat represents a “new class” of antihypertensive that “looks to be safe and we’re seeing very large reductions in blood pressure.”
Results of the Target-HTN trial were published online in JAMA to coincide with presentation at the Hypertension Scientific Sessions, sponsored by the American Heart Association.
Aldosterone’s contribution ‘vastly underappreciated’
Excess aldosterone production contributes to uncontrolled BP in patients with obesity and other associated diseases, such as obstructive sleep apnea and metabolic syndrome.
“Aldosterone’s contribution to uncontrolled hypertension is vastly underappreciated,” first author and study presenter Luke Laffin, MD, also with the Cleveland Clinic, said in an interview.
Aldosterone synthase inhibitors are a novel class of BP-lowering medications that decrease aldosterone production. Lorundrostat is one of two such agents in advanced clinical development. The other is baxdrostat (CinCor Pharma/AstraZeneca).
The Target-HTN randomized, placebo-controlled, dose-ranging trial enrolled 200 adults (mean age, 66 years; 60% women) with uncontrolled hypertension while taking two or more antihypertensive medications; 42% of participants were taking three or more antihypertensive medications, 48% were obese and 40% had diabetes.
The study population was divided into two cohorts: an initial cohort of 163 adults with suppressed plasma renin activity at baseline (PRA ≤ 1.0 ng/mL per hour) and elevated plasma aldosterone (≥ 1.0 ng/dL) and a second cohort of 37 adults with PRA greater than 1.0 ng/mL per hour.
Participants were randomly assigned to placebo or one of five doses of lorundrostat in the initial cohort (12.5 mg, 50 mg, or 100 mg once daily or 12.5 mg or 25 mg twice daily).
In the second cohort, participants were randomly assigned (1:6) to placebo or lorundrostat 100 mg once daily. The primary endpoint was change in automated office systolic BP from baseline to week 8.
Among participants with suppressed PRA, following 8 weeks of treatment, changes in office systolic BP of −14.1, −13.2, and −6.9 mm Hg were observed with 100 mg, 50 mg, and 12.5 mg once-daily lorundrostat, respectively, compared with a change of −4.1 mm Hg with placebo.
Reductions in systolic BP in individuals receiving twice-daily doses of 25 mg and 12.5 mg of lorundrostat were −10.1 and −13.8 mm Hg, respectively.
Among participants without suppressed PRA, lorundrostat 100 mg once daily decreased systolic BP by 11.4 mm Hg, similar to BP reduction in those with suppressed PRA receiving the same dose.
A prespecified subgroup analysis showed that participants with obesity demonstrated greater BP lowering in response to lorundrostat.
No instances of cortisol insufficiency occurred. Six participants had increases in serum potassium above 6.0 mEq/L (6.0 mmol/L) that corrected with dose reduction or drug discontinuation.
The increase in serum potassium is “expected and manageable,” Dr. Laffin said in an interview. “Anytime you disrupt aldosterone production, you’re going to have to have an increase in serum potassium, but it’s very manageable and not something that is worrisome.”
A phase 2 trial in 300 adults with uncontrolled hypertension is currently underway. The trial will evaluate the BP-lowering effects of lorundrostat, administered on a background of a standardized antihypertensive medication regimen. A larger phase 3 study will start before the end of the year.
‘New dawn’ for therapies targeting aldosterone
The author of an editorial in JAMA noted that more 70 years after the first isolation of aldosterone, then called electrocortin, “there is a new dawn for therapies targeting aldosterone.”
“There is now real potential to provide better-targeted treatment for patients in whom aldosterone excess is known to contribute to their clinical condition and influence their clinical outcome, notably those with difficult-to-control hypertension, obesity, heart failure, chronic kidney disease, and the many with yet-to-be-diagnosed primary aldosteronism,” said Bryan Williams, MD, University College London.
The trial was funded by Mineralys Therapeutics, which is developing lorundrostat. Dr. Laffin reported that the Cleveland Clinic, his employer, was a study site for the Target-HTN trial and that C5Research, the academic research organization of the Cleveland Clinic, receives payment for services related to other Mineralys clinical trials. Dr. Laffin also reported receipt of personal fees from Medtronic, Lilly, and Crispr Therapeutics, grants from AstraZeneca, and stock options for LucidAct Health and Gordy Health. Dr. Nissen reported receipt of grants from Mineralys during the conduct of the study and grants from AbbVie, AstraZeneca, Amgen, Bristol-Myers Squibb, Lilly, Esperion Therapeutics, Medtronic, grants from MyoKardia, New Amsterdam Pharmaceuticals, Novartis, and Silence Therapeutics. Dr. Williams reported being the unremunerated chair of the steering committee designing a phase 3 trial of the aldosterone synthase inhibitor baxdrostat for AstraZeneca.
A version of this article first appeared on Medscape.com.
FROM HYPERTENSION 2023
New COVID vaccines force bivalents out
COVID vaccines will have a new formulation in 2023, according to a decision announced by the U.S. Food and Drug Administration, that will focus efforts on circulating variants. The move pushes last year’s bivalent vaccines out of circulation because they will no longer be authorized for use in the United States.
The updated mRNA vaccines for 2023-2024 are being revised to include a single component that corresponds to the Omicron variant XBB.1.5. Like the bivalents offered before, the new monovalents are being manufactured by Moderna and Pfizer.
The new vaccines are authorized for use in individuals age 6 months and older. And the new options are being developed using a similar process as previous formulations, according to the FDA.
Targeting circulating variants
In recent studies, regulators point out the extent of neutralization observed by the updated vaccines against currently circulating viral variants causing COVID-19, including EG.5, BA.2.86, appears to be of a similar magnitude to the extent of neutralization observed with previous versions of the vaccines against corresponding prior variants.
“This suggests that the vaccines are a good match for protecting against the currently circulating COVID-19 variants,” according to the report.
Hundreds of millions of people in the United States have already received previously approved mRNA COVID vaccines, according to regulators who say the benefit-to-risk profile is well understood as they move forward with new formulations.
“Vaccination remains critical to public health and continued protection against serious consequences of COVID-19, including hospitalization and death,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in a statement. “The public can be assured that these updated vaccines have met the agency’s rigorous scientific standards for safety, effectiveness, and manufacturing quality. We very much encourage those who are eligible to consider getting vaccinated.”
Timing the effort
On Sept. 12 the U.S. Centers for Disease Control and Prevention recommended that everyone 6 months and older get an updated COVID-19 vaccine. Updated vaccines from Pfizer-BioNTech and Moderna will be available later this week, according to the agency.
This article was updated 9/14/23.
A version of this article appeared on Medscape.com.
COVID vaccines will have a new formulation in 2023, according to a decision announced by the U.S. Food and Drug Administration, that will focus efforts on circulating variants. The move pushes last year’s bivalent vaccines out of circulation because they will no longer be authorized for use in the United States.
The updated mRNA vaccines for 2023-2024 are being revised to include a single component that corresponds to the Omicron variant XBB.1.5. Like the bivalents offered before, the new monovalents are being manufactured by Moderna and Pfizer.
The new vaccines are authorized for use in individuals age 6 months and older. And the new options are being developed using a similar process as previous formulations, according to the FDA.
Targeting circulating variants
In recent studies, regulators point out the extent of neutralization observed by the updated vaccines against currently circulating viral variants causing COVID-19, including EG.5, BA.2.86, appears to be of a similar magnitude to the extent of neutralization observed with previous versions of the vaccines against corresponding prior variants.
“This suggests that the vaccines are a good match for protecting against the currently circulating COVID-19 variants,” according to the report.
Hundreds of millions of people in the United States have already received previously approved mRNA COVID vaccines, according to regulators who say the benefit-to-risk profile is well understood as they move forward with new formulations.
“Vaccination remains critical to public health and continued protection against serious consequences of COVID-19, including hospitalization and death,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in a statement. “The public can be assured that these updated vaccines have met the agency’s rigorous scientific standards for safety, effectiveness, and manufacturing quality. We very much encourage those who are eligible to consider getting vaccinated.”
Timing the effort
On Sept. 12 the U.S. Centers for Disease Control and Prevention recommended that everyone 6 months and older get an updated COVID-19 vaccine. Updated vaccines from Pfizer-BioNTech and Moderna will be available later this week, according to the agency.
This article was updated 9/14/23.
A version of this article appeared on Medscape.com.
COVID vaccines will have a new formulation in 2023, according to a decision announced by the U.S. Food and Drug Administration, that will focus efforts on circulating variants. The move pushes last year’s bivalent vaccines out of circulation because they will no longer be authorized for use in the United States.
The updated mRNA vaccines for 2023-2024 are being revised to include a single component that corresponds to the Omicron variant XBB.1.5. Like the bivalents offered before, the new monovalents are being manufactured by Moderna and Pfizer.
The new vaccines are authorized for use in individuals age 6 months and older. And the new options are being developed using a similar process as previous formulations, according to the FDA.
Targeting circulating variants
In recent studies, regulators point out the extent of neutralization observed by the updated vaccines against currently circulating viral variants causing COVID-19, including EG.5, BA.2.86, appears to be of a similar magnitude to the extent of neutralization observed with previous versions of the vaccines against corresponding prior variants.
“This suggests that the vaccines are a good match for protecting against the currently circulating COVID-19 variants,” according to the report.
Hundreds of millions of people in the United States have already received previously approved mRNA COVID vaccines, according to regulators who say the benefit-to-risk profile is well understood as they move forward with new formulations.
“Vaccination remains critical to public health and continued protection against serious consequences of COVID-19, including hospitalization and death,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in a statement. “The public can be assured that these updated vaccines have met the agency’s rigorous scientific standards for safety, effectiveness, and manufacturing quality. We very much encourage those who are eligible to consider getting vaccinated.”
Timing the effort
On Sept. 12 the U.S. Centers for Disease Control and Prevention recommended that everyone 6 months and older get an updated COVID-19 vaccine. Updated vaccines from Pfizer-BioNTech and Moderna will be available later this week, according to the agency.
This article was updated 9/14/23.
A version of this article appeared on Medscape.com.
Night owls have higher risk of developing type 2 diabetes
, compared with their “early bird” counterparts, according to a new study, published in Annals of Internal Medicine.
The work focused on participants’ self-assessed chronotype – an individuals’ circadian preference, or natural preference to sleep and wake up earlier or later, commonly known as being an early bird or a night owl.
Analyzing the self-reported lifestyle behaviors and sleeping habits of more than 60,000 middle-aged female nurses, researchers from Brigham and Women’s Hospital and Harvard Medical School, both in Boston, found that those with a preference for waking up later had a 72% higher risk for diabetes and were 54% more likely to have unhealthy lifestyle behaviors, compared with participants who tended to wake up earlier.
After adjustment for six lifestyle factors – diet, alcohol use, body mass index (BMI), physical activity, smoking status, and sleep duration – the association between diabetes risk and evening chronotype weakened to a 19% higher risk of developing type 2 diabetes.
In a subgroup analysis, this association was stronger among women who either had had no night shifts over the previous 2 years or had worked night shifts for less than 10 years in their careers. For nurses who had worked night shifts recently, the study found no association between evening chronotype and diabetes risk.
The participants, drawn from the Nurses’ Health Study II, were between 45 and 62 years age, with no history of cancer, cardiovascular disease, or diabetes. Researchers followed the group from 2009 until 2017.
Is there a mismatch between natural circadian rhythm and work schedule?
The authors, led by Sina Kianersi, DVM, PhD, of Harvard Medical School, Boston, suggest that their results may be linked to a mismatch between a person’s circadian rhythm and their physical and social environment – for example, if someone lives on a schedule opposite to their circadian preference.
In one 2015 study, female nurses who had worked daytime shifts for more than 10 years but had an evening chronotype had the highest diabetes risk, compared with early chronotypes (51% more likely to develop type 2 diabetes).
In a 2022 study, an evening chronotype was associated with a 30% elevated risk for type 2 diabetes. The authors speculated that circadian misalignment could be to blame – for example, being a night owl but working early morning – which can disrupt glycemic and lipid metabolism.
Previous studies have found that shorter or irregular sleep habits are associated with a higher risk of type 2 diabetes. Other studies have also found that people with an evening chronotype are more likely than early birds to have unhealthy eating habits, have lower levels of physical activity, and smoke and drink.
This new study did not find that an evening chronotype was associated with unhealthy drinking, which the authors defined as having one or more drinks per day.
In an accompanying editorial, two physicians from the Harvard T.H. Chan School of Public Health in Boston caution that the statistical design of the study limits its ability to establish causation.
“Chronotype could change later, which might correlate with lifestyle changes,” write Kehuan Lin, MS, Mingyang Song, MBBS, and Edward Giovannucci, MD. “Experimental trials are required to determine whether chronotype is a marker of unhealthy lifestyle or an independent determinant.”
They also suggest that psychological factors and the type of work being performed by the participants could be potential confounders.
The authors of the study note that their findings might not be generalizable to groups other than middle-aged White female nurses. The study population also had a relatively high level of education and were socioeconomically advantaged.
Self-reporting chronotypes with a single question could also result in misclassification and measurement error, the authors acknowledge.
The findings underscore the value of assessing an individuals’ chronotype for scheduling shift work – for example, assigning night owls to night shifts may improve their metabolic health and sleeping habits, according to the authors of the study.
“Given the importance of lifestyle modification in diabetes prevention, future research is warranted to investigate whether improving lifestyle behaviors could effectively reduce diabetes risk in persons with an evening chronotype,” the authors conclude.
The study was supported by grants from the National Institutes of Health and the European Research Council.
A version of this article first appeared on Medscape.com.
, compared with their “early bird” counterparts, according to a new study, published in Annals of Internal Medicine.
The work focused on participants’ self-assessed chronotype – an individuals’ circadian preference, or natural preference to sleep and wake up earlier or later, commonly known as being an early bird or a night owl.
Analyzing the self-reported lifestyle behaviors and sleeping habits of more than 60,000 middle-aged female nurses, researchers from Brigham and Women’s Hospital and Harvard Medical School, both in Boston, found that those with a preference for waking up later had a 72% higher risk for diabetes and were 54% more likely to have unhealthy lifestyle behaviors, compared with participants who tended to wake up earlier.
After adjustment for six lifestyle factors – diet, alcohol use, body mass index (BMI), physical activity, smoking status, and sleep duration – the association between diabetes risk and evening chronotype weakened to a 19% higher risk of developing type 2 diabetes.
In a subgroup analysis, this association was stronger among women who either had had no night shifts over the previous 2 years or had worked night shifts for less than 10 years in their careers. For nurses who had worked night shifts recently, the study found no association between evening chronotype and diabetes risk.
The participants, drawn from the Nurses’ Health Study II, were between 45 and 62 years age, with no history of cancer, cardiovascular disease, or diabetes. Researchers followed the group from 2009 until 2017.
Is there a mismatch between natural circadian rhythm and work schedule?
The authors, led by Sina Kianersi, DVM, PhD, of Harvard Medical School, Boston, suggest that their results may be linked to a mismatch between a person’s circadian rhythm and their physical and social environment – for example, if someone lives on a schedule opposite to their circadian preference.
In one 2015 study, female nurses who had worked daytime shifts for more than 10 years but had an evening chronotype had the highest diabetes risk, compared with early chronotypes (51% more likely to develop type 2 diabetes).
In a 2022 study, an evening chronotype was associated with a 30% elevated risk for type 2 diabetes. The authors speculated that circadian misalignment could be to blame – for example, being a night owl but working early morning – which can disrupt glycemic and lipid metabolism.
Previous studies have found that shorter or irregular sleep habits are associated with a higher risk of type 2 diabetes. Other studies have also found that people with an evening chronotype are more likely than early birds to have unhealthy eating habits, have lower levels of physical activity, and smoke and drink.
This new study did not find that an evening chronotype was associated with unhealthy drinking, which the authors defined as having one or more drinks per day.
In an accompanying editorial, two physicians from the Harvard T.H. Chan School of Public Health in Boston caution that the statistical design of the study limits its ability to establish causation.
“Chronotype could change later, which might correlate with lifestyle changes,” write Kehuan Lin, MS, Mingyang Song, MBBS, and Edward Giovannucci, MD. “Experimental trials are required to determine whether chronotype is a marker of unhealthy lifestyle or an independent determinant.”
They also suggest that psychological factors and the type of work being performed by the participants could be potential confounders.
The authors of the study note that their findings might not be generalizable to groups other than middle-aged White female nurses. The study population also had a relatively high level of education and were socioeconomically advantaged.
Self-reporting chronotypes with a single question could also result in misclassification and measurement error, the authors acknowledge.
The findings underscore the value of assessing an individuals’ chronotype for scheduling shift work – for example, assigning night owls to night shifts may improve their metabolic health and sleeping habits, according to the authors of the study.
“Given the importance of lifestyle modification in diabetes prevention, future research is warranted to investigate whether improving lifestyle behaviors could effectively reduce diabetes risk in persons with an evening chronotype,” the authors conclude.
The study was supported by grants from the National Institutes of Health and the European Research Council.
A version of this article first appeared on Medscape.com.
, compared with their “early bird” counterparts, according to a new study, published in Annals of Internal Medicine.
The work focused on participants’ self-assessed chronotype – an individuals’ circadian preference, or natural preference to sleep and wake up earlier or later, commonly known as being an early bird or a night owl.
Analyzing the self-reported lifestyle behaviors and sleeping habits of more than 60,000 middle-aged female nurses, researchers from Brigham and Women’s Hospital and Harvard Medical School, both in Boston, found that those with a preference for waking up later had a 72% higher risk for diabetes and were 54% more likely to have unhealthy lifestyle behaviors, compared with participants who tended to wake up earlier.
After adjustment for six lifestyle factors – diet, alcohol use, body mass index (BMI), physical activity, smoking status, and sleep duration – the association between diabetes risk and evening chronotype weakened to a 19% higher risk of developing type 2 diabetes.
In a subgroup analysis, this association was stronger among women who either had had no night shifts over the previous 2 years or had worked night shifts for less than 10 years in their careers. For nurses who had worked night shifts recently, the study found no association between evening chronotype and diabetes risk.
The participants, drawn from the Nurses’ Health Study II, were between 45 and 62 years age, with no history of cancer, cardiovascular disease, or diabetes. Researchers followed the group from 2009 until 2017.
Is there a mismatch between natural circadian rhythm and work schedule?
The authors, led by Sina Kianersi, DVM, PhD, of Harvard Medical School, Boston, suggest that their results may be linked to a mismatch between a person’s circadian rhythm and their physical and social environment – for example, if someone lives on a schedule opposite to their circadian preference.
In one 2015 study, female nurses who had worked daytime shifts for more than 10 years but had an evening chronotype had the highest diabetes risk, compared with early chronotypes (51% more likely to develop type 2 diabetes).
In a 2022 study, an evening chronotype was associated with a 30% elevated risk for type 2 diabetes. The authors speculated that circadian misalignment could be to blame – for example, being a night owl but working early morning – which can disrupt glycemic and lipid metabolism.
Previous studies have found that shorter or irregular sleep habits are associated with a higher risk of type 2 diabetes. Other studies have also found that people with an evening chronotype are more likely than early birds to have unhealthy eating habits, have lower levels of physical activity, and smoke and drink.
This new study did not find that an evening chronotype was associated with unhealthy drinking, which the authors defined as having one or more drinks per day.
In an accompanying editorial, two physicians from the Harvard T.H. Chan School of Public Health in Boston caution that the statistical design of the study limits its ability to establish causation.
“Chronotype could change later, which might correlate with lifestyle changes,” write Kehuan Lin, MS, Mingyang Song, MBBS, and Edward Giovannucci, MD. “Experimental trials are required to determine whether chronotype is a marker of unhealthy lifestyle or an independent determinant.”
They also suggest that psychological factors and the type of work being performed by the participants could be potential confounders.
The authors of the study note that their findings might not be generalizable to groups other than middle-aged White female nurses. The study population also had a relatively high level of education and were socioeconomically advantaged.
Self-reporting chronotypes with a single question could also result in misclassification and measurement error, the authors acknowledge.
The findings underscore the value of assessing an individuals’ chronotype for scheduling shift work – for example, assigning night owls to night shifts may improve their metabolic health and sleeping habits, according to the authors of the study.
“Given the importance of lifestyle modification in diabetes prevention, future research is warranted to investigate whether improving lifestyle behaviors could effectively reduce diabetes risk in persons with an evening chronotype,” the authors conclude.
The study was supported by grants from the National Institutes of Health and the European Research Council.
A version of this article first appeared on Medscape.com.
FROM ANNALS OF INTERNAL MEDICINE
Colchicine still needed to prevent gout flares in allopurinol ‘start-low, go-slow’ strategy
The use of colchicine prophylaxis during the process of up-titrating allopurinol slowly to reach target serum urate levels for patients with gout proved necessary to reduce the risk of flares in a randomized, double-blind, placebo-controlled noninferiority trial conducted by researchers in New Zealand.
Use of colchicine led to fewer flares during the first 6 months after initiating treatment with allopurinol, but when colchicine and placebo were stopped at 6 months, the number of flares rose, and there was no overall difference in the average number of flares between the placebo and colchicine groups over 12 months.
“Gout flares are common when starting urate-lowering therapy, although may be less frequent using the newer allopurinol ‘start-low, go-slow’ dose escalation strategy,” Lisa Stamp, MBChB, PhD, of the University of Otago, Christchurch, New Zealand, and colleagues write in Annals of the Rheumatic Diseases. “This study was undertaken to determine whether colchicine prophylaxis is required with the more gradual dose escalation of allopurinol to achieve target serum urate.”
The study was published online in the BMJ.
The researchers randomly assigned 200 participants to receive colchicine 0.5 mg daily or placebo. At the same time, allopurinol 50 mg daily was initially given to those with an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2, and 100 mg daily was given to those with eGFR greater than or equal to 60 mL/min/1.73 m2; doses were increased monthly by 50 mg daily for those with eGFR less than 60 and by 100 mg daily for those with eGFR greater than or equal to 60 until the target urate level of less than 0.36 mmol/L (about < 6 mg/dL) was achieved. In total, 93% of patients were male, and 69% were White. Patients were enrolled from February 2019 through December 2021. The final study visit was in January 2023. Researchers tracked the mean number of patient-reported gout flares per month with a prespecified noninferiority margin of 0.12 gout flares/month. After the first 6 months, colchicine and placebo were stopped, and researchers continued to track outcomes up to month 12.
From baseline to 6 months, there were more gout flares per month in the placebo group (0.61) than in the colchicine group (0.35). From 6 to 12 months, there were more gout flares among patients who previously received colchicine, compared with those who received placebo. While 22.8% of patients in the colchicine group experienced flares at month 6, the following month – after stopping the drug – 41.2% experienced flares. For comparison, 30.8% of the placebo group experienced flares at month 6, and in month 7, 23.5% experienced flares. The spike in flares in the colchicine group began to decline after month 9.
Among the colchicine and placebo groups, reductions in serum urate at 6 and 12 months were similar, and mean serum urate levels declined below 0.36 mmol/L at 4 months and thereafter. Both groups reached a mean allopurinol dose of 280 mg/d at 6 months.
It is not clear what caused this rise in flares in the colchicine group, Stamp noted in an email to this news organization. Overall, there was no difference in the average number of gout flares between either group over the entire 12-month study period.
The study findings make a case for continuing colchicine prophylaxis considerably beyond the point at which the target urate serum level is reached, said Robert Terkeltaub, MD, professor emeritus of medicine in the division of rheumatology, allergy, and immunology at the University of California, San Diego. He was not involved with the research.
“The flare burden with this really conservative allopurinol dosing regimen is really stunning,” he said in an interview. Despite this “start low and go slow” strategy, flares remained a problem, he noted. “It argues that you need at least a year to get out of the woods and have a lower flare burden, even if colchicine prophylaxis is used for the first 6 months.”
The current standard of care in rheumatology for gout treatment includes flare prophylaxis when starting urate-lowering therapy. In daily practice, “this can (and often does) mean continuing flare prophylaxis for longer than the recommended minimum duration from [established] gout treatment guidelines for an individual patient,” explained Elizabeth R. Graef, DO, assistant professor of medicine at Boston University and a rheumatologist at Boston Medical Center.
There are many different factors to consider when determining a patient’s flare risk beyond serum urate levels and date of last flare, she noted. Flare severity, concurrent diuretic use, dose changes, tophus count, and additional health conditions such as diabetes can all contribute to individual patient outcomes.
“Gout is an erosive arthritis, and every time someone flares, there’s a risk of permanent erosive damage, so I tend to be more aggressive with prophylaxis,” she said.
The study was funded by the Health Research Council of New Zealand. Dr. Graef disclosed no relevant financial relationships. Dr. Terkeltaub consults for LG Chem, Fortress/Urica, Selecta Biosciences, Horizon Therapeutics, Atom Biosciences, Acquist Therapeutics, Generate Biomedicines, AstraZeneca, and Synlogic. Dr. Terkeltaub serves as the nonsalaried president of the Gout, Hyperuricemia, and Crystal-Associated Disease Network, which annually receives unrestricted arms-length grant support from pharma donors.
A version of this article first appeared on Medscape.com.
The use of colchicine prophylaxis during the process of up-titrating allopurinol slowly to reach target serum urate levels for patients with gout proved necessary to reduce the risk of flares in a randomized, double-blind, placebo-controlled noninferiority trial conducted by researchers in New Zealand.
Use of colchicine led to fewer flares during the first 6 months after initiating treatment with allopurinol, but when colchicine and placebo were stopped at 6 months, the number of flares rose, and there was no overall difference in the average number of flares between the placebo and colchicine groups over 12 months.
“Gout flares are common when starting urate-lowering therapy, although may be less frequent using the newer allopurinol ‘start-low, go-slow’ dose escalation strategy,” Lisa Stamp, MBChB, PhD, of the University of Otago, Christchurch, New Zealand, and colleagues write in Annals of the Rheumatic Diseases. “This study was undertaken to determine whether colchicine prophylaxis is required with the more gradual dose escalation of allopurinol to achieve target serum urate.”
The study was published online in the BMJ.
The researchers randomly assigned 200 participants to receive colchicine 0.5 mg daily or placebo. At the same time, allopurinol 50 mg daily was initially given to those with an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2, and 100 mg daily was given to those with eGFR greater than or equal to 60 mL/min/1.73 m2; doses were increased monthly by 50 mg daily for those with eGFR less than 60 and by 100 mg daily for those with eGFR greater than or equal to 60 until the target urate level of less than 0.36 mmol/L (about < 6 mg/dL) was achieved. In total, 93% of patients were male, and 69% were White. Patients were enrolled from February 2019 through December 2021. The final study visit was in January 2023. Researchers tracked the mean number of patient-reported gout flares per month with a prespecified noninferiority margin of 0.12 gout flares/month. After the first 6 months, colchicine and placebo were stopped, and researchers continued to track outcomes up to month 12.
From baseline to 6 months, there were more gout flares per month in the placebo group (0.61) than in the colchicine group (0.35). From 6 to 12 months, there were more gout flares among patients who previously received colchicine, compared with those who received placebo. While 22.8% of patients in the colchicine group experienced flares at month 6, the following month – after stopping the drug – 41.2% experienced flares. For comparison, 30.8% of the placebo group experienced flares at month 6, and in month 7, 23.5% experienced flares. The spike in flares in the colchicine group began to decline after month 9.
Among the colchicine and placebo groups, reductions in serum urate at 6 and 12 months were similar, and mean serum urate levels declined below 0.36 mmol/L at 4 months and thereafter. Both groups reached a mean allopurinol dose of 280 mg/d at 6 months.
It is not clear what caused this rise in flares in the colchicine group, Stamp noted in an email to this news organization. Overall, there was no difference in the average number of gout flares between either group over the entire 12-month study period.
The study findings make a case for continuing colchicine prophylaxis considerably beyond the point at which the target urate serum level is reached, said Robert Terkeltaub, MD, professor emeritus of medicine in the division of rheumatology, allergy, and immunology at the University of California, San Diego. He was not involved with the research.
“The flare burden with this really conservative allopurinol dosing regimen is really stunning,” he said in an interview. Despite this “start low and go slow” strategy, flares remained a problem, he noted. “It argues that you need at least a year to get out of the woods and have a lower flare burden, even if colchicine prophylaxis is used for the first 6 months.”
The current standard of care in rheumatology for gout treatment includes flare prophylaxis when starting urate-lowering therapy. In daily practice, “this can (and often does) mean continuing flare prophylaxis for longer than the recommended minimum duration from [established] gout treatment guidelines for an individual patient,” explained Elizabeth R. Graef, DO, assistant professor of medicine at Boston University and a rheumatologist at Boston Medical Center.
There are many different factors to consider when determining a patient’s flare risk beyond serum urate levels and date of last flare, she noted. Flare severity, concurrent diuretic use, dose changes, tophus count, and additional health conditions such as diabetes can all contribute to individual patient outcomes.
“Gout is an erosive arthritis, and every time someone flares, there’s a risk of permanent erosive damage, so I tend to be more aggressive with prophylaxis,” she said.
The study was funded by the Health Research Council of New Zealand. Dr. Graef disclosed no relevant financial relationships. Dr. Terkeltaub consults for LG Chem, Fortress/Urica, Selecta Biosciences, Horizon Therapeutics, Atom Biosciences, Acquist Therapeutics, Generate Biomedicines, AstraZeneca, and Synlogic. Dr. Terkeltaub serves as the nonsalaried president of the Gout, Hyperuricemia, and Crystal-Associated Disease Network, which annually receives unrestricted arms-length grant support from pharma donors.
A version of this article first appeared on Medscape.com.
The use of colchicine prophylaxis during the process of up-titrating allopurinol slowly to reach target serum urate levels for patients with gout proved necessary to reduce the risk of flares in a randomized, double-blind, placebo-controlled noninferiority trial conducted by researchers in New Zealand.
Use of colchicine led to fewer flares during the first 6 months after initiating treatment with allopurinol, but when colchicine and placebo were stopped at 6 months, the number of flares rose, and there was no overall difference in the average number of flares between the placebo and colchicine groups over 12 months.
“Gout flares are common when starting urate-lowering therapy, although may be less frequent using the newer allopurinol ‘start-low, go-slow’ dose escalation strategy,” Lisa Stamp, MBChB, PhD, of the University of Otago, Christchurch, New Zealand, and colleagues write in Annals of the Rheumatic Diseases. “This study was undertaken to determine whether colchicine prophylaxis is required with the more gradual dose escalation of allopurinol to achieve target serum urate.”
The study was published online in the BMJ.
The researchers randomly assigned 200 participants to receive colchicine 0.5 mg daily or placebo. At the same time, allopurinol 50 mg daily was initially given to those with an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2, and 100 mg daily was given to those with eGFR greater than or equal to 60 mL/min/1.73 m2; doses were increased monthly by 50 mg daily for those with eGFR less than 60 and by 100 mg daily for those with eGFR greater than or equal to 60 until the target urate level of less than 0.36 mmol/L (about < 6 mg/dL) was achieved. In total, 93% of patients were male, and 69% were White. Patients were enrolled from February 2019 through December 2021. The final study visit was in January 2023. Researchers tracked the mean number of patient-reported gout flares per month with a prespecified noninferiority margin of 0.12 gout flares/month. After the first 6 months, colchicine and placebo were stopped, and researchers continued to track outcomes up to month 12.
From baseline to 6 months, there were more gout flares per month in the placebo group (0.61) than in the colchicine group (0.35). From 6 to 12 months, there were more gout flares among patients who previously received colchicine, compared with those who received placebo. While 22.8% of patients in the colchicine group experienced flares at month 6, the following month – after stopping the drug – 41.2% experienced flares. For comparison, 30.8% of the placebo group experienced flares at month 6, and in month 7, 23.5% experienced flares. The spike in flares in the colchicine group began to decline after month 9.
Among the colchicine and placebo groups, reductions in serum urate at 6 and 12 months were similar, and mean serum urate levels declined below 0.36 mmol/L at 4 months and thereafter. Both groups reached a mean allopurinol dose of 280 mg/d at 6 months.
It is not clear what caused this rise in flares in the colchicine group, Stamp noted in an email to this news organization. Overall, there was no difference in the average number of gout flares between either group over the entire 12-month study period.
The study findings make a case for continuing colchicine prophylaxis considerably beyond the point at which the target urate serum level is reached, said Robert Terkeltaub, MD, professor emeritus of medicine in the division of rheumatology, allergy, and immunology at the University of California, San Diego. He was not involved with the research.
“The flare burden with this really conservative allopurinol dosing regimen is really stunning,” he said in an interview. Despite this “start low and go slow” strategy, flares remained a problem, he noted. “It argues that you need at least a year to get out of the woods and have a lower flare burden, even if colchicine prophylaxis is used for the first 6 months.”
The current standard of care in rheumatology for gout treatment includes flare prophylaxis when starting urate-lowering therapy. In daily practice, “this can (and often does) mean continuing flare prophylaxis for longer than the recommended minimum duration from [established] gout treatment guidelines for an individual patient,” explained Elizabeth R. Graef, DO, assistant professor of medicine at Boston University and a rheumatologist at Boston Medical Center.
There are many different factors to consider when determining a patient’s flare risk beyond serum urate levels and date of last flare, she noted. Flare severity, concurrent diuretic use, dose changes, tophus count, and additional health conditions such as diabetes can all contribute to individual patient outcomes.
“Gout is an erosive arthritis, and every time someone flares, there’s a risk of permanent erosive damage, so I tend to be more aggressive with prophylaxis,” she said.
The study was funded by the Health Research Council of New Zealand. Dr. Graef disclosed no relevant financial relationships. Dr. Terkeltaub consults for LG Chem, Fortress/Urica, Selecta Biosciences, Horizon Therapeutics, Atom Biosciences, Acquist Therapeutics, Generate Biomedicines, AstraZeneca, and Synlogic. Dr. Terkeltaub serves as the nonsalaried president of the Gout, Hyperuricemia, and Crystal-Associated Disease Network, which annually receives unrestricted arms-length grant support from pharma donors.
A version of this article first appeared on Medscape.com.
Navigating chronic cough in primary care
Chronic cough took center stage at the European Respiratory Society Congress session titled “Conditions We Are Just Dealing With the Tip of the Iceberg in Primary Care: Frequently Mismanaged Conditions in Primary Health Care.”
“When it comes to chronic cough, general practitioners often feel lost,” Miguel Román Rodríguez, family doctor and an associate professor of family medicine at the University of the Balearic Islands, Palma, Mallorca, Spain, and one of the chairs of the session, said to this news organization.
“GPs are central in diagnosing conditions like chronic cough. We bring something that the specialists don’t bring: the knowledge of the context, of the family, the longitudinal history,” echoed the second chair of the session, Hilary Pinnock, family physician and professor of primary care respiratory medicine at the University of Edinburgh.
Understanding the multifaceted nature of chronic cough
Imran Satia, an assistant professor at McMaster University, Hamilton, Ont., guided attendees at the Milan, Italy, meeting through a comprehensive exploration of chronic cough. The first issue he addressed was the definition of the condition, emphasizing that it is defined by its duration, with chronic cough typically lasting for more than 8 weeks. Prof. Satia pointed out common associations of chronic cough, including asthma, nasal disease, and reflux disease.
Delving into epidemiology, he cited a meta-analysis indicating a global prevalence of approximately 10% in the adult population, with significant regional variability: from 18.1% in Australia to 2.3% in Africa. Notably, the Canadian Longitudinal Study on Aging found an overall prevalence of 16% at baseline. “The most common risk factor was smoke, but even in nonsmokers the prevalence reached 10%,” Prof. Satia added, highlighting that it increased with age and changed depending on location. “The most common associated comorbidities were heart failure and hypertension, but also conditions related to chronic pain, mood, and anxiety,” he explained.
Mental health was identified as a crucial factor in chronic cough, with psychological distress and depressive symptoms emerging as risk factors for developing chronic cough over the next 3 years, contributing to a 20% increased risk.
Effective management strategies
Prof. Satia proposed the use of algorithms to aid in the management of patients with chronic cough in primary care. He introduced a Canadian algorithm that offers specific recommendations for both primary and secondary care.
The algorithm’s primary care assessment, step 1, includes a comprehensive evaluation of the cough history (duration, severity, triggers, nature, location); cardiorespiratory, gastrointestinal, and nasal symptoms; and use of angiotensin-converting enzyme inhibitors and smoking status. Essential diagnostic tests, such as chest radiography (to check for structural disease), complete blood cell count, and spirometry (with or without bronchodilator reversibility), were emphasized. Urgent referral criteria encompassed symptoms like hemoptysis, weight loss, fever, or abnormal chest radiography findings.
“When checking for cough history, GPs should always consider factors like the presence of dry or productive cough, mental health, presence of chronic pain, stroke, and swallowing,” said Prof. Satia, stressing the importance of documenting the impact of chronic cough on quality of life, work life, social life, and family life. “This is something that doctors sometimes do not ask about. They may think that these are not major problems, but acknowledging their importance can help the patient,” he added.
Step 2 of the algorithm focuses on treatment options tailored to specific diagnoses, such as asthma or chronic obstructive pulmonary disease. Prof. Satia urged caution, emphasizing that treatment should only be initiated when evidence of these conditions is present. Additionally, he encouraged early consideration of cough hypersensitivity syndrome when patients exhibit coughing in response to low levels of mechanical stimulation.
Current treatments and future prospects
Prof. Satia presented an overview of existing treatments for chronic cough, outlining their respective advantages and disadvantages. For instance, speech therapy is a patient-led approach with no side effects but entails challenges related to access, costs, and patient motivation. On the other hand, low-dose morphine offers rapid relief but is associated with issues like nausea, stigma, and constipation.
Looking ahead, Prof. Satia shared the results of COUGH-1 and COUGH-2, pivotal phase 3 trials evaluating the oral, peripherally acting P2X3-receptor antagonist gefapixant. This drug, currently approved in Switzerland and Japan, demonstrated a significant reduction in cough frequency, compared with placebo, with rapid and sustained effects. “The estimated relative reduction for 45 mg was 18.45% in COUGH-1 (12 weeks) and 14.64% in COUGH-2 (24 weeks). Of note, cough reduction is very quick and sustained with gefapixant, but a 40% reduction is observed in the placebo arm,” commented Prof. Satia.
Experts unanimously stressed the importance for specialists and GPs of effective communication in managing chronic cough, involving both patients and their families.
“As GPs, we are crucial to manage the common problems, but we are also crucial to spot the needle in the haystack: this is extremely difficult and challenging, and we need support from our colleagues,” Dr. Pinnock concluded.
Prof. Satia reported funding from Merck MSD, AstraZeneca, and GSK; consulting fees from Merck MSD, Genentech, and Respiplus; and speaker fees from AstraZeneca, GSK, and Merck MSD.
A version of this article first appeared on Medscape.com.
Chronic cough took center stage at the European Respiratory Society Congress session titled “Conditions We Are Just Dealing With the Tip of the Iceberg in Primary Care: Frequently Mismanaged Conditions in Primary Health Care.”
“When it comes to chronic cough, general practitioners often feel lost,” Miguel Román Rodríguez, family doctor and an associate professor of family medicine at the University of the Balearic Islands, Palma, Mallorca, Spain, and one of the chairs of the session, said to this news organization.
“GPs are central in diagnosing conditions like chronic cough. We bring something that the specialists don’t bring: the knowledge of the context, of the family, the longitudinal history,” echoed the second chair of the session, Hilary Pinnock, family physician and professor of primary care respiratory medicine at the University of Edinburgh.
Understanding the multifaceted nature of chronic cough
Imran Satia, an assistant professor at McMaster University, Hamilton, Ont., guided attendees at the Milan, Italy, meeting through a comprehensive exploration of chronic cough. The first issue he addressed was the definition of the condition, emphasizing that it is defined by its duration, with chronic cough typically lasting for more than 8 weeks. Prof. Satia pointed out common associations of chronic cough, including asthma, nasal disease, and reflux disease.
Delving into epidemiology, he cited a meta-analysis indicating a global prevalence of approximately 10% in the adult population, with significant regional variability: from 18.1% in Australia to 2.3% in Africa. Notably, the Canadian Longitudinal Study on Aging found an overall prevalence of 16% at baseline. “The most common risk factor was smoke, but even in nonsmokers the prevalence reached 10%,” Prof. Satia added, highlighting that it increased with age and changed depending on location. “The most common associated comorbidities were heart failure and hypertension, but also conditions related to chronic pain, mood, and anxiety,” he explained.
Mental health was identified as a crucial factor in chronic cough, with psychological distress and depressive symptoms emerging as risk factors for developing chronic cough over the next 3 years, contributing to a 20% increased risk.
Effective management strategies
Prof. Satia proposed the use of algorithms to aid in the management of patients with chronic cough in primary care. He introduced a Canadian algorithm that offers specific recommendations for both primary and secondary care.
The algorithm’s primary care assessment, step 1, includes a comprehensive evaluation of the cough history (duration, severity, triggers, nature, location); cardiorespiratory, gastrointestinal, and nasal symptoms; and use of angiotensin-converting enzyme inhibitors and smoking status. Essential diagnostic tests, such as chest radiography (to check for structural disease), complete blood cell count, and spirometry (with or without bronchodilator reversibility), were emphasized. Urgent referral criteria encompassed symptoms like hemoptysis, weight loss, fever, or abnormal chest radiography findings.
“When checking for cough history, GPs should always consider factors like the presence of dry or productive cough, mental health, presence of chronic pain, stroke, and swallowing,” said Prof. Satia, stressing the importance of documenting the impact of chronic cough on quality of life, work life, social life, and family life. “This is something that doctors sometimes do not ask about. They may think that these are not major problems, but acknowledging their importance can help the patient,” he added.
Step 2 of the algorithm focuses on treatment options tailored to specific diagnoses, such as asthma or chronic obstructive pulmonary disease. Prof. Satia urged caution, emphasizing that treatment should only be initiated when evidence of these conditions is present. Additionally, he encouraged early consideration of cough hypersensitivity syndrome when patients exhibit coughing in response to low levels of mechanical stimulation.
Current treatments and future prospects
Prof. Satia presented an overview of existing treatments for chronic cough, outlining their respective advantages and disadvantages. For instance, speech therapy is a patient-led approach with no side effects but entails challenges related to access, costs, and patient motivation. On the other hand, low-dose morphine offers rapid relief but is associated with issues like nausea, stigma, and constipation.
Looking ahead, Prof. Satia shared the results of COUGH-1 and COUGH-2, pivotal phase 3 trials evaluating the oral, peripherally acting P2X3-receptor antagonist gefapixant. This drug, currently approved in Switzerland and Japan, demonstrated a significant reduction in cough frequency, compared with placebo, with rapid and sustained effects. “The estimated relative reduction for 45 mg was 18.45% in COUGH-1 (12 weeks) and 14.64% in COUGH-2 (24 weeks). Of note, cough reduction is very quick and sustained with gefapixant, but a 40% reduction is observed in the placebo arm,” commented Prof. Satia.
Experts unanimously stressed the importance for specialists and GPs of effective communication in managing chronic cough, involving both patients and their families.
“As GPs, we are crucial to manage the common problems, but we are also crucial to spot the needle in the haystack: this is extremely difficult and challenging, and we need support from our colleagues,” Dr. Pinnock concluded.
Prof. Satia reported funding from Merck MSD, AstraZeneca, and GSK; consulting fees from Merck MSD, Genentech, and Respiplus; and speaker fees from AstraZeneca, GSK, and Merck MSD.
A version of this article first appeared on Medscape.com.
Chronic cough took center stage at the European Respiratory Society Congress session titled “Conditions We Are Just Dealing With the Tip of the Iceberg in Primary Care: Frequently Mismanaged Conditions in Primary Health Care.”
“When it comes to chronic cough, general practitioners often feel lost,” Miguel Román Rodríguez, family doctor and an associate professor of family medicine at the University of the Balearic Islands, Palma, Mallorca, Spain, and one of the chairs of the session, said to this news organization.
“GPs are central in diagnosing conditions like chronic cough. We bring something that the specialists don’t bring: the knowledge of the context, of the family, the longitudinal history,” echoed the second chair of the session, Hilary Pinnock, family physician and professor of primary care respiratory medicine at the University of Edinburgh.
Understanding the multifaceted nature of chronic cough
Imran Satia, an assistant professor at McMaster University, Hamilton, Ont., guided attendees at the Milan, Italy, meeting through a comprehensive exploration of chronic cough. The first issue he addressed was the definition of the condition, emphasizing that it is defined by its duration, with chronic cough typically lasting for more than 8 weeks. Prof. Satia pointed out common associations of chronic cough, including asthma, nasal disease, and reflux disease.
Delving into epidemiology, he cited a meta-analysis indicating a global prevalence of approximately 10% in the adult population, with significant regional variability: from 18.1% in Australia to 2.3% in Africa. Notably, the Canadian Longitudinal Study on Aging found an overall prevalence of 16% at baseline. “The most common risk factor was smoke, but even in nonsmokers the prevalence reached 10%,” Prof. Satia added, highlighting that it increased with age and changed depending on location. “The most common associated comorbidities were heart failure and hypertension, but also conditions related to chronic pain, mood, and anxiety,” he explained.
Mental health was identified as a crucial factor in chronic cough, with psychological distress and depressive symptoms emerging as risk factors for developing chronic cough over the next 3 years, contributing to a 20% increased risk.
Effective management strategies
Prof. Satia proposed the use of algorithms to aid in the management of patients with chronic cough in primary care. He introduced a Canadian algorithm that offers specific recommendations for both primary and secondary care.
The algorithm’s primary care assessment, step 1, includes a comprehensive evaluation of the cough history (duration, severity, triggers, nature, location); cardiorespiratory, gastrointestinal, and nasal symptoms; and use of angiotensin-converting enzyme inhibitors and smoking status. Essential diagnostic tests, such as chest radiography (to check for structural disease), complete blood cell count, and spirometry (with or without bronchodilator reversibility), were emphasized. Urgent referral criteria encompassed symptoms like hemoptysis, weight loss, fever, or abnormal chest radiography findings.
“When checking for cough history, GPs should always consider factors like the presence of dry or productive cough, mental health, presence of chronic pain, stroke, and swallowing,” said Prof. Satia, stressing the importance of documenting the impact of chronic cough on quality of life, work life, social life, and family life. “This is something that doctors sometimes do not ask about. They may think that these are not major problems, but acknowledging their importance can help the patient,” he added.
Step 2 of the algorithm focuses on treatment options tailored to specific diagnoses, such as asthma or chronic obstructive pulmonary disease. Prof. Satia urged caution, emphasizing that treatment should only be initiated when evidence of these conditions is present. Additionally, he encouraged early consideration of cough hypersensitivity syndrome when patients exhibit coughing in response to low levels of mechanical stimulation.
Current treatments and future prospects
Prof. Satia presented an overview of existing treatments for chronic cough, outlining their respective advantages and disadvantages. For instance, speech therapy is a patient-led approach with no side effects but entails challenges related to access, costs, and patient motivation. On the other hand, low-dose morphine offers rapid relief but is associated with issues like nausea, stigma, and constipation.
Looking ahead, Prof. Satia shared the results of COUGH-1 and COUGH-2, pivotal phase 3 trials evaluating the oral, peripherally acting P2X3-receptor antagonist gefapixant. This drug, currently approved in Switzerland and Japan, demonstrated a significant reduction in cough frequency, compared with placebo, with rapid and sustained effects. “The estimated relative reduction for 45 mg was 18.45% in COUGH-1 (12 weeks) and 14.64% in COUGH-2 (24 weeks). Of note, cough reduction is very quick and sustained with gefapixant, but a 40% reduction is observed in the placebo arm,” commented Prof. Satia.
Experts unanimously stressed the importance for specialists and GPs of effective communication in managing chronic cough, involving both patients and their families.
“As GPs, we are crucial to manage the common problems, but we are also crucial to spot the needle in the haystack: this is extremely difficult and challenging, and we need support from our colleagues,” Dr. Pinnock concluded.
Prof. Satia reported funding from Merck MSD, AstraZeneca, and GSK; consulting fees from Merck MSD, Genentech, and Respiplus; and speaker fees from AstraZeneca, GSK, and Merck MSD.
A version of this article first appeared on Medscape.com.
FROM ERS 2023
Positive topline results for antihypertensive zilebesiran
Zilebesiran (Alnylam Pharmaceuticals), an investigational, subcutaneously administered small-interfering RNA (siRNA) therapeutic in development for the treatment of hypertension, met the primary and secondary endpoints, with an “encouraging” safety profile in the phase 2 KARDIA-1 study, the company announced.
KARDIA-1 is a phase 2 randomized, double-blind, placebo-controlled, dose-ranging study evaluating the efficacy and safety of zilebesiran as monotherapy in 394 adults with mild to moderate untreated hypertension or on stable therapy with one or more antihypertensive drugs.
Patients were randomly assigned to one of five treatment arms during a 12-month double-blind period and double-blind extension period: 150 mg or 300 mg zilebesiran subcutaneously once every 6 months, 300 mg or 600 mg zilebesiran subcutaneously once every 3 months, or placebo. Patients taking placebo were randomly assigned to one of the four initial zilebesiran dose regimens beginning at month 6.
The primary endpoint was change from baseline in systolic blood pressure (SBP) at 3 months assessed by 24-hour ambulatory blood pressure monitoring.
Topline data show a dose-dependent, clinically significant reduction in 24-hour mean SBP, with a placebo-subtracted reduction greater than 15 mm Hg (P < .0001) with both the 300 mg and 600 mg doses.
The study also met key secondary endpoints, showing “consistent and sustained reductions” in SBP at 6 months, which supports quarterly or biannual dosing, the company said.
There was one death due to cardiopulmonary arrest in a zilebesiran-treated patient that was considered unrelated to the drug. Serious adverse events were reported in 3.6% of zilebesiran-treated patients and 6.7% of placebo-treated patients. None was considered related to the study drug.
Adverse events occurring in 5% or more of zilebesiran-treated patients in any dose arm included COVID-19, injection-site reaction, hyperkalemia, hypertension, upper respiratory tract infection, arthralgia, and headache.
“As a physician, I believe these KARDIA-1 results, which demonstrate clinically significant reductions in systolic blood pressure of greater than 15 mm Hg, along with the ability to achieve durable tonic blood pressure control, provide hope that we may one day have access to a novel therapy with the potential to address the significant unmet needs of patients with uncontrolled hypertension who are at high risk of future cardiovascular events,” study investigator George L. Bakris, MD, director, American Heart Association Comprehensive Hypertension Center, University of Chicago Medicine, said in a statement.
The phase 2 results “further validate” the phase 1 results, published in the New England Journal of Medicine, Simon Fox, PhD, vice president, zilebesiran program lead at Alnylam, said in the statement.
The full KARDIA-1 results will be reported at an upcoming scientific conference, the statement notes. Topline results from the KARDIA-2 phase 2 study of zilebesiran in combination with one of three standard classes of antihypertensive medications in patients with mild to moderate hypertension are expected in early 2024.
A version of this article first appeared on Medscape.com.
Zilebesiran (Alnylam Pharmaceuticals), an investigational, subcutaneously administered small-interfering RNA (siRNA) therapeutic in development for the treatment of hypertension, met the primary and secondary endpoints, with an “encouraging” safety profile in the phase 2 KARDIA-1 study, the company announced.
KARDIA-1 is a phase 2 randomized, double-blind, placebo-controlled, dose-ranging study evaluating the efficacy and safety of zilebesiran as monotherapy in 394 adults with mild to moderate untreated hypertension or on stable therapy with one or more antihypertensive drugs.
Patients were randomly assigned to one of five treatment arms during a 12-month double-blind period and double-blind extension period: 150 mg or 300 mg zilebesiran subcutaneously once every 6 months, 300 mg or 600 mg zilebesiran subcutaneously once every 3 months, or placebo. Patients taking placebo were randomly assigned to one of the four initial zilebesiran dose regimens beginning at month 6.
The primary endpoint was change from baseline in systolic blood pressure (SBP) at 3 months assessed by 24-hour ambulatory blood pressure monitoring.
Topline data show a dose-dependent, clinically significant reduction in 24-hour mean SBP, with a placebo-subtracted reduction greater than 15 mm Hg (P < .0001) with both the 300 mg and 600 mg doses.
The study also met key secondary endpoints, showing “consistent and sustained reductions” in SBP at 6 months, which supports quarterly or biannual dosing, the company said.
There was one death due to cardiopulmonary arrest in a zilebesiran-treated patient that was considered unrelated to the drug. Serious adverse events were reported in 3.6% of zilebesiran-treated patients and 6.7% of placebo-treated patients. None was considered related to the study drug.
Adverse events occurring in 5% or more of zilebesiran-treated patients in any dose arm included COVID-19, injection-site reaction, hyperkalemia, hypertension, upper respiratory tract infection, arthralgia, and headache.
“As a physician, I believe these KARDIA-1 results, which demonstrate clinically significant reductions in systolic blood pressure of greater than 15 mm Hg, along with the ability to achieve durable tonic blood pressure control, provide hope that we may one day have access to a novel therapy with the potential to address the significant unmet needs of patients with uncontrolled hypertension who are at high risk of future cardiovascular events,” study investigator George L. Bakris, MD, director, American Heart Association Comprehensive Hypertension Center, University of Chicago Medicine, said in a statement.
The phase 2 results “further validate” the phase 1 results, published in the New England Journal of Medicine, Simon Fox, PhD, vice president, zilebesiran program lead at Alnylam, said in the statement.
The full KARDIA-1 results will be reported at an upcoming scientific conference, the statement notes. Topline results from the KARDIA-2 phase 2 study of zilebesiran in combination with one of three standard classes of antihypertensive medications in patients with mild to moderate hypertension are expected in early 2024.
A version of this article first appeared on Medscape.com.
Zilebesiran (Alnylam Pharmaceuticals), an investigational, subcutaneously administered small-interfering RNA (siRNA) therapeutic in development for the treatment of hypertension, met the primary and secondary endpoints, with an “encouraging” safety profile in the phase 2 KARDIA-1 study, the company announced.
KARDIA-1 is a phase 2 randomized, double-blind, placebo-controlled, dose-ranging study evaluating the efficacy and safety of zilebesiran as monotherapy in 394 adults with mild to moderate untreated hypertension or on stable therapy with one or more antihypertensive drugs.
Patients were randomly assigned to one of five treatment arms during a 12-month double-blind period and double-blind extension period: 150 mg or 300 mg zilebesiran subcutaneously once every 6 months, 300 mg or 600 mg zilebesiran subcutaneously once every 3 months, or placebo. Patients taking placebo were randomly assigned to one of the four initial zilebesiran dose regimens beginning at month 6.
The primary endpoint was change from baseline in systolic blood pressure (SBP) at 3 months assessed by 24-hour ambulatory blood pressure monitoring.
Topline data show a dose-dependent, clinically significant reduction in 24-hour mean SBP, with a placebo-subtracted reduction greater than 15 mm Hg (P < .0001) with both the 300 mg and 600 mg doses.
The study also met key secondary endpoints, showing “consistent and sustained reductions” in SBP at 6 months, which supports quarterly or biannual dosing, the company said.
There was one death due to cardiopulmonary arrest in a zilebesiran-treated patient that was considered unrelated to the drug. Serious adverse events were reported in 3.6% of zilebesiran-treated patients and 6.7% of placebo-treated patients. None was considered related to the study drug.
Adverse events occurring in 5% or more of zilebesiran-treated patients in any dose arm included COVID-19, injection-site reaction, hyperkalemia, hypertension, upper respiratory tract infection, arthralgia, and headache.
“As a physician, I believe these KARDIA-1 results, which demonstrate clinically significant reductions in systolic blood pressure of greater than 15 mm Hg, along with the ability to achieve durable tonic blood pressure control, provide hope that we may one day have access to a novel therapy with the potential to address the significant unmet needs of patients with uncontrolled hypertension who are at high risk of future cardiovascular events,” study investigator George L. Bakris, MD, director, American Heart Association Comprehensive Hypertension Center, University of Chicago Medicine, said in a statement.
The phase 2 results “further validate” the phase 1 results, published in the New England Journal of Medicine, Simon Fox, PhD, vice president, zilebesiran program lead at Alnylam, said in the statement.
The full KARDIA-1 results will be reported at an upcoming scientific conference, the statement notes. Topline results from the KARDIA-2 phase 2 study of zilebesiran in combination with one of three standard classes of antihypertensive medications in patients with mild to moderate hypertension are expected in early 2024.
A version of this article first appeared on Medscape.com.