User login
Approach to Diagnosing and Managing Implantation Mycoses
Approach to Diagnosing and Managing Implantation Mycoses
Implantation mycoses such as chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma are a diverse group of fungal diseases that occur when a break in the skin allows the entry of the causative fungus. These diseases disproportionately affect individuals in low- and middle-income countries causing substantial disability, decreased quality of life, and severe social stigma.1-3 Timely diagnosis and appropriate treatment are critical.
Chromoblastomycosis and mycetoma are designated as neglected tropical diseases, but research to improve their management is sparse, even compared to other neglected tropical diseases.4,5 Since there are no global diagnostic and treatment guidelines to date, we outline steps to diagnose and manage chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma.
Chromoblastomycosis
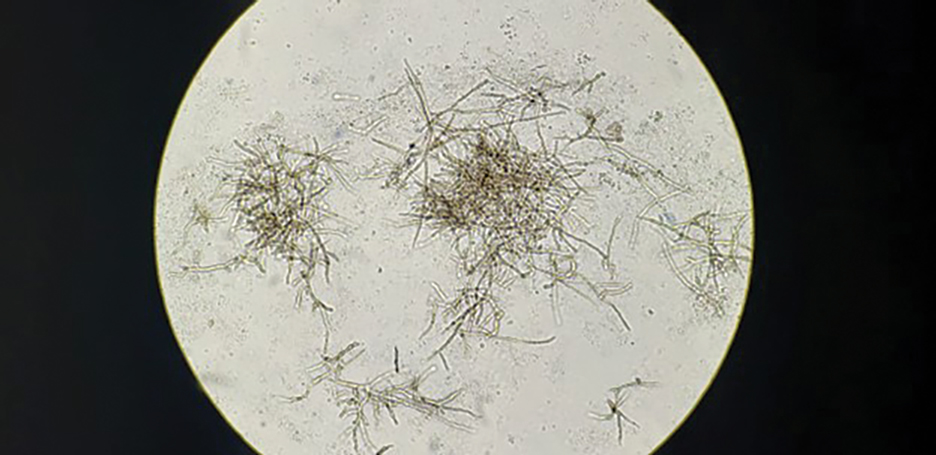
Chromoblastomycosis is caused by dematiaceous fungi that typically affect the skin and subcutaneous tissue. Chromoblastomycosis is distinguished from subcutaneous phaeohyphomycosis by microscopically visualizing the characteristic thick-walled, single, or multicellular clusters of pigmented fungal cells (also known as medlar bodies, muriform cells, or sclerotic bodies).6 In phaeohyphomycosis, short hyphae and pseudohyphae plus some single cells typically are seen.
Epidemiology—Globally, the distribution and burden of chromoblastomycosis are relatively unknown. Infections are more common in tropical and subtropical areas but can be acquired anywhere. A literature review conducted in 2021 identified 7740 cases of chromoblastomycosis, mostly reported in South America, Africa, Central America and Mexico, and Asia.7 Most of the patients were male, and the median age was 52 years. One study found an incidence of 14.7 per 1,000,000 patients in the United States for both chromoblastomycosis and phaeohyphomycotic abscesses (which included both skin and brain abscesses).8 Most patients were aged 65 years or older, with a higher incidence in males. Geographically, the incidence was highest in the Northeast followed by the South; patients in rural areas also had higher incidence of disease.8
Causative Organisms—Causative species cannot reliably distinguish between chromoblastomycosis and subcutaneous phaeohyphomycosis, as some species overlap. Cladophialophora carrionii, Fonsecaea species, Phialophora verrucosa species complex, and Rhinocladiella aquaspersa most commonly cause chromoblastomycosis.9,10
Clinical Manifestations—Chromoblastomycosis initially manifests as a solitary erythematous macule at a site of trauma (often not recalled by the patient) that can evolve to a smooth pink papule and may progress to 1 of 5 morphologies: nodular, verrucous, tumorous, cicatricial, or plaque.6 Patients may present with more than one morphology, particularly in long-standing or advanced disease. Lesions commonly manifest on the arms and legs in otherwise healthy individuals in environments (eg, rural, agricultural) that have more opportunities for injury and exposure to the causative fungi. Affected individuals often have small black specks on the lesion surface that are visible with the naked eye.6
Diagnosis—Common differential diagnoses include cutaneous blastomycosis, fixed sporotrichosis, warty tuberculosis nocardiosis, cutaneous leishmaniasis, human papillomavirus (HPV) infection, podoconiosis, lymphatic filariasis, cutaneous tuberculosis, and psoriasis.6 Squamous cell carcinoma is both a differential diagnosis as well as a potential complication of the disease.11
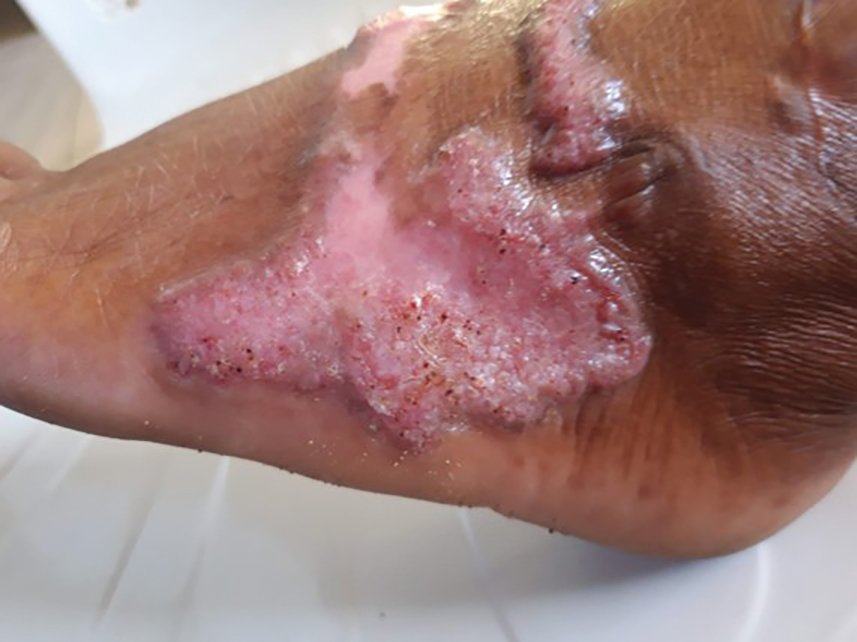
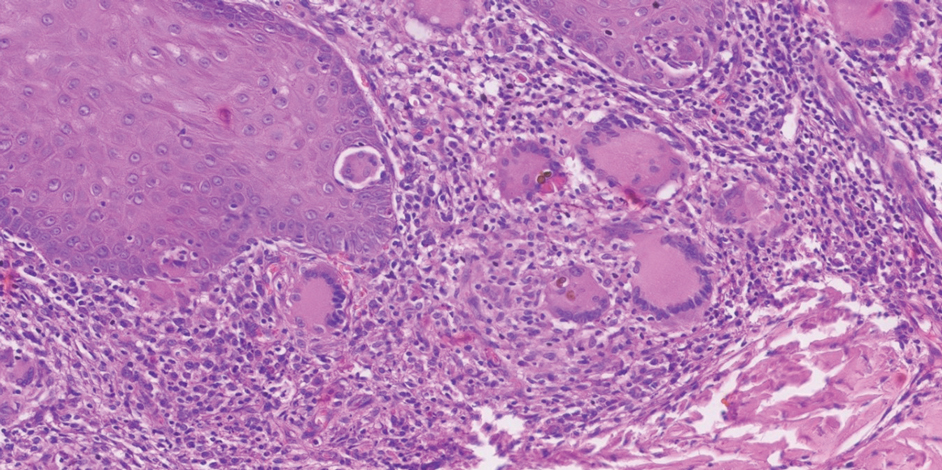
Potassium hydroxide preparation with skin scapings or a biopsy from the lesion has high sensitivity and quick turnaround times. There often is a background histopathologic reaction of pseudoepitheliomatous hyperplasia. Examining samples taken from areas with the visible small black dots on the skin surface can increase the likelihood of detecting fungal elements (Figure 1). Clinicians also may choose to obtain a 6- to 8-mm deep skin biopsy from the lesion and splice it in half, with one sample sent for histopathology and the other for culture (Figure 2). Skin scrapings can be sent for culture instead. In the case of verrucous lesions, biopsy is preferred if feasible.
Treatment should not be delayed while awaiting the culture results if infection is otherwise confirmed by direct microscopy or histopathology. The treatment approach remains similar regardless of the causative species. If the culture results are positive, the causative genus can be identified by the microscopic morphology; however, molecular diagnostic tools are needed for accurate species identification.12,13
Antifungal Susceptibility Testing—For most dematiaceous fungi, interpreting minimum inhibitory concentrations (MICs) is challenging due to a lack of data from multicenter studies. One report examined sequential isolates of Fonsecaea pedrosoi and demonstrated both high MIC values and clinical resistance to itraconazole in some cases, likely from treatment pressure.14 Clinical Laboratory Standards Institute–approved epidemiologic cutoff values (ECVs) are established for F pedrosoi for commonly used antifungals including itraconazole (0.5 µg/mL), terbinafine (0.25 µg/mL), and posaconazole (0.5 µg/mL).15 Clinicians may choose to obtain sequential isolates for any causative fungi in recalcitrant disease to monitor for increases in MIC.
Management—In early-stage disease, excision of the skin nodule may be curative, although concomitant treatment for several months with an antifungal is advised. If antifungals are needed, itraconazole is the most commonly prescribed agent, typically at a dose of 100 to 200 mg twice daily. Terbinafine also has been used first-line at a dose of 250 to 500 mg per day. Voriconazole and posaconazole also may be suitable options for first-line or for refractory disease treatment. Fluconazole does not have good activity against dematiaceous fungi and should be avoided.16 Topical antifungals will not reach the site of infection in adequate concentrations. Topical corticosteroids can make the disease worse and should be avoided. The duration of therapy usually is several months, but many patients require years of therapy until resolution of lesions.
Clinicians can consider combination therapy with an antifungal and a topical immunomodulator such as imiquimod (applied topically 3 times per week); this combination can be considered in refractory disease and even upon initial diagnosis, especially in severe disease.17,18 Nonpharmacologic interventions such as cryotherapy, heat, and light-based therapies have been used, but outcome data are scarce.19-23
Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis also is caused by dematiaceous fungi that typically affect the skin and subcutaneous tissue. Subcutaneous phaeohyphomycosis is distinguished from chromoblastomycosis by short hyphae and hyphal fragments usually seen microscopically instead of visualizing thick-walled, single, or multicellular clusters of pigmented fungal cells.6
Epidemiology—Globally, the burden and distribution of phaeohyphomycosis, including its cutaneous manifestations, are not well understood. Infections are more common in tropical and subtropical areas but can be acquired anywhere. Phaeohyphomycosis is a generic term used to describe infections caused by pigmented hyphal fungi that can manifest on the skin (subcutaneous phaeohyphomycosis) but also can affect deep structures including the brain (systemic phaeohyphomycosis).24
Causative Organisms—Alternaria, Bipolaris, Cladosporium, Curvularia, Exophiala, and Exserohilum species most commonly cause subcutaneous phaeohyphomycosis. Alternaria infections manifesting with skin lesions often are referred to as cutaneous alternariosis.25
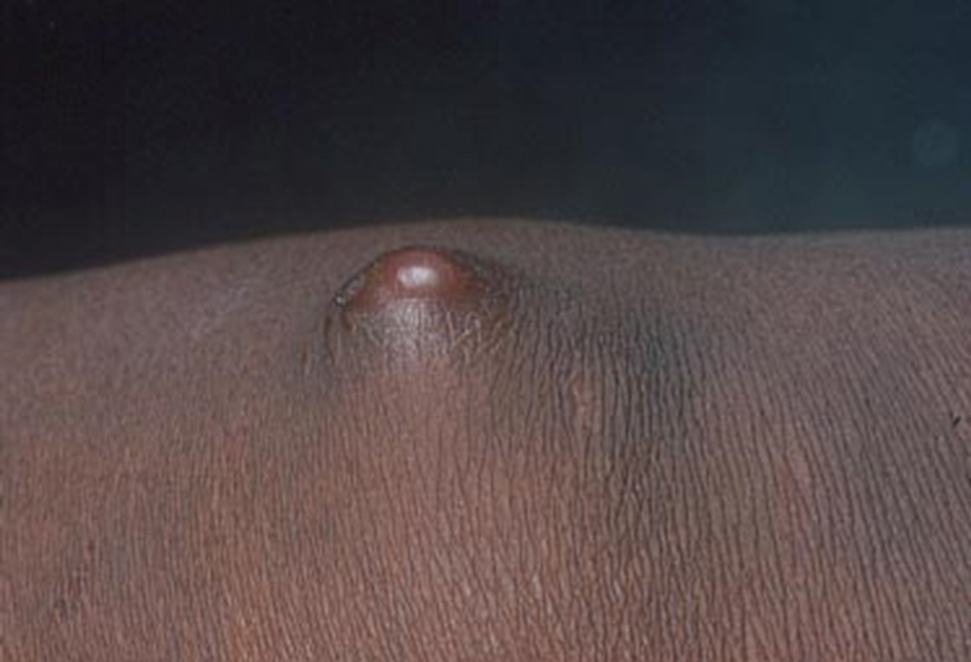
Clinical Manifestations—The most common skin manifestation of phaeohyphomycosis is a subcutaneous cyst (cystic phaeohyphomycosis)(Figure 2). Subcutaneous phaeohyphomycosis also may manifest with nodules or plaques (Figure 3). Phaeohyphomycosis appears to occur more commonly in individuals who are immunosuppressed, those in whom T-cell function is affected, in congenital immunodeficiency states (eg, individuals with CARD9 mutations).26
Diagnosis—Culture is the gold standard for confirming phaeohyphomycosis.27 For cystic phaeohyphomycosis, clinicians can consider aspiration of the cyst for direct microscopic examination and culture. Histopathology may be utilized but can have lower sensitivity in showing dematiaceous hyphae and granulomatous inflammation; using the Masson-Fontana stain for melanin can be helpful. Molecular diagnostic tools including metagenomics applied directly to the tissue may be useful but are likely to have lower sensitivity than culture and require specialist diagnostic facilities.
Management—The approaches to managing chromoblastomycosis and subcutaneous phaeohyphomycosis are similar, though the preferred agents often differ. In early-stage disease, excision of the skin nodule may be curative, although concomitant treatment for several months with an antifungal is advised. In localized forms, itraconazole usually is used, but in those cases associated with immunodeficiency states, voriconazole may be necessary. Fluconazole does not have good activity against dematiaceous fungi and should be avoided.16 Topical antifungals will not reach the site of infection in adequate concentrations. Topical corticosteroids can make the disease worse and should be avoided. The duration of therapy may be substantially longer for chromoblastomycosis (months to years) compared to subcutaneous phaeohyphomycosis (weeks to months), although in immunocompromised individuals treatment may be even more prolonged.
Mycetoma
Mycetoma is caused by one of several different types of fungi (eumycetoma) and bacteria (actinomycetoma) that lead to progressively debilitating yet painless subcutaneous tumorlike lesions. The lesions usually manifest on the arms and legs but can occur anywhere.
Epidemiology—Little is known about the true global burden of mycetoma, but it occurs more frequently in low-income communities in rural areas.28 A retrospective review identified 19,494 cases published from 1876 to 2019, with cases reported in 102 countries.29 The countries with the highest numbers of cases are Sudan and Mexico, where there is more information on the distribution of the disease. Cases often are reported in what is known as the mycetoma belt (between latitudes 15° south and 30° north) but are increasingly identified outside this region.28 Young men aged 20 to 40 years are most commonly affected.
In the United States, mycetoma is uncommon, but clinicians can encounter locally acquired and travel-associated cases; hence, taking a good travel history is essential. One study specifically evaluating eumycetoma found a prevalence of 5.2 per 1,000,000 patients.8 Women and those aged 65 years or older had a higher incidence. Incidence was similar across US regions, but a higher incidence was reported in nonrural areas.8
Causative Organisms—More than 60 different species of fungi can cause eumycetoma; most cases are caused by Madurella mycetomatis, Trematosphaeria grisea (formerly Madurella grisea); Pseudallescheria boydii species complex, and Falciformispora (formerly Leptosphaeria) senegalensis.30 Actinomycetoma commonly is caused by Nocardia species (Nocardia brasiliensis, Nocardia asteroides, Nocardia otitidiscaviarum, Nocardia transvalensis, Nocardia harenae, and Nocardia takedensis), Streptomyces somaliensis, and Actinomadura species (Actinomadura madurae, Actinomadura pelletieri).31
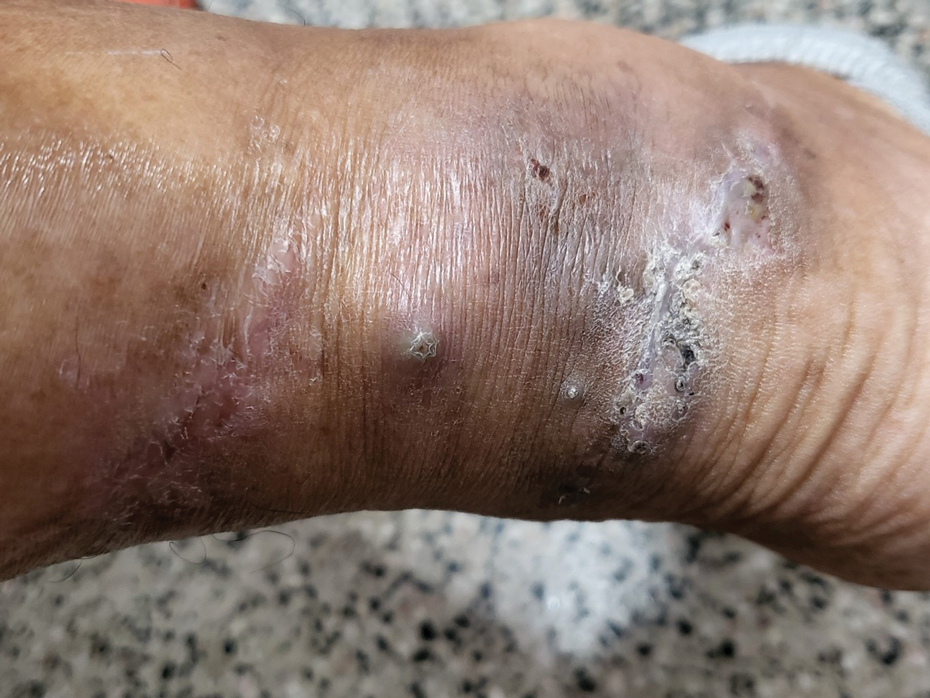
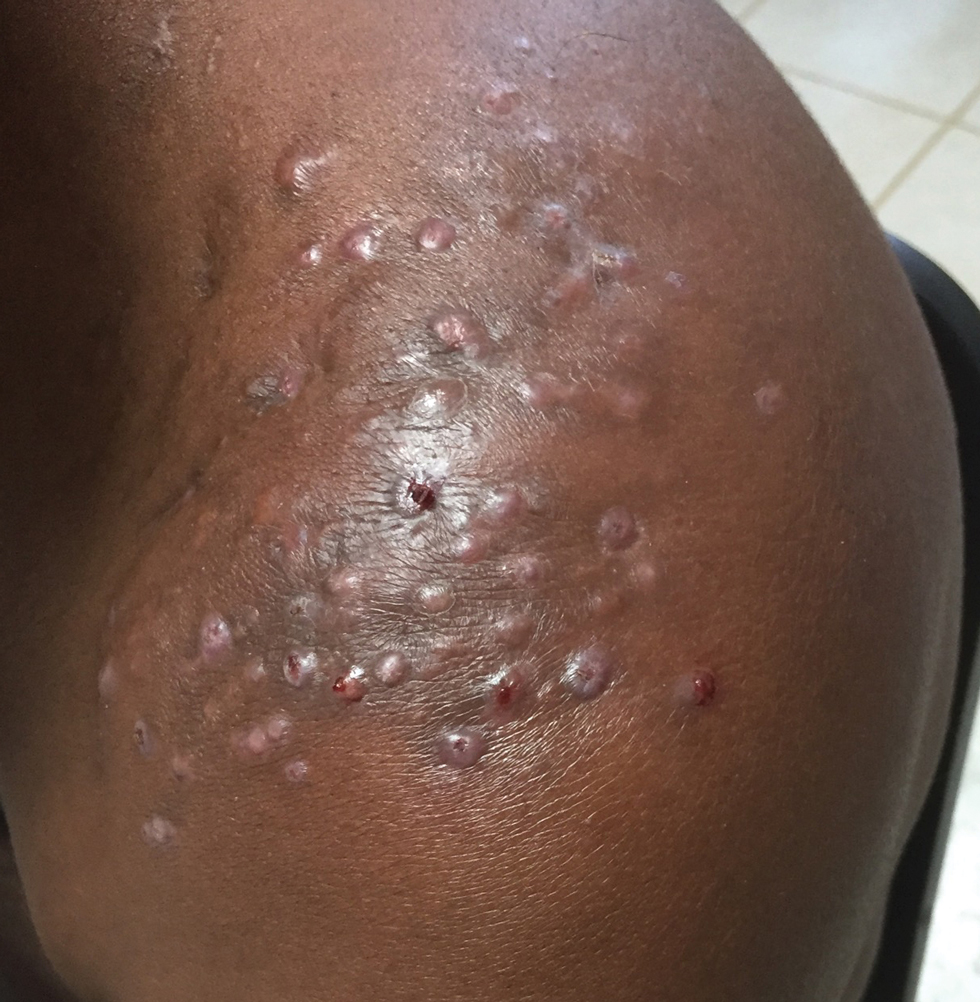
Clinical Manifestations—Mycetoma is a chronic granulomatous disease with a progressive inflammatory reaction (Figures 4 and 5). Over the course of years, mycetoma progresses from small nodules to large, bone-invasive, mutilating lesions. Mycetoma manifests as a triad of painless firm subcutaneous masses, formation of multiple sinuses within the masses, and a purulent or seropurulent discharge containing sandlike visible particles (grains) that can be white, yellow, red, or black.28 Lesions usually are painless in early disease and are slowly progressive. Large lesion size, bone destruction, secondary bacterial infections, and actinomycetoma may lead to higher likelihood of pain.32
Diagnosis—Other conditions that could manifest with the same triad seen in mycetoma such as botryomycosis should be included in the differential. Other differential diagnoses include foreign body granuloma, filariasis, mycobacterial infection, skeletal tuberculosis, and yaws.
Proper treatment requires an accurate diagnosis that distinguishes actinomycetoma from eumycetoma.33 Culturing of grains obtained from deep lesion aspirates enables identification of the causative organism (Figure 6). The color of the grains may provide clues to their etiology: black grains are caused by fungus, red grains by a bacterium (A pelletieri), and pale (yellow or white) grains can be caused by either one.31Nocardia mycetoma grains are very small and usually cannot be appreciated with the naked eye. Histopathology of deep biopsy specimens (biopsy needle or surgical biopsy) stained with hematoxylin and eosin can diagnose actinomycetoma and eumycetoma. Punch biopsies often are not helpful, as the inflammatory mass is too deeply located. Deep surgical biopsy is preferred; however, species identification cannot be made without culture. Molecular tests for certain causative organisms of mycetoma have been developed but are not readily available.34,35 Currently, no serologic tests can diagnose mycetoma reliably. Ultrasonography can be used to diagnose mycetoma and, with appropriate training, distinguish between actinomycetoma and eumycetoma; it also can be combined with needle aspiration for taking grain samples.36
Treatment—Treatment of mycetoma depends on identification of the causal etiology and requires long-term and expensive drug regimens. It is not possible to determine the causative organism clinically. Actinomycetoma generally responds to medical treatment, and surgery rarely is needed. The current first-line treatment is co-trimoxazole (trimethoprim/sulfamethoxazole) in combination with amoxicillin and clavulanate acid or co-trimoxazole and amikacin for refractory disease; linezolid also may be a promising option for refractory disease.37
Eumycetoma is less responsive to medical therapies, and recurrence is common. Current recommended therapy is itraconazole for 9 to 12 months; however, cure rates ranging from 26% to 75% in combination with surgery have been reported, and fungi often can still be cultured from lesions posttreatment.38,39 Surgical excision often is used following 6 months of treatment with itraconazole to obtain better outcomes. Amputation may be required if the combination of antifungals and surgical excision fails. Fosravuconazole has shown promise in one clinical trial, but it is not approved in most countries, including the United States.39
Final Thoughts
Chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma can cause devastating disease. Patients with these conditions often are unable to carry out daily activities and experience stigma and discrimination. Limited diagnostic and treatment options hamper the ability of clinicians to respond appropriately to suspect and confirmed disease. Effectively examining the skin is the starting point for diagnosing and managing these diseases and can help clinicians to care for patients and prevent severe disease.
- Smith DJ, Soebono H, Parajuli N, et al. South-east Asia regional neglected tropical disease framework: improving control of mycetoma, chromoblastomycosis, and sporotrichosis. Lancet Reg Health Southeast Asia. 2025;35:100561. doi:10.1016/j.lansea.2025.100561
- Abbas M, Scolding PS, Yosif AA, et al. The disabling consequences of mycetoma. PLoS Negl Trop Dis. 2018;12:E0007019. doi:10.1371/journal.pntd.0007019
- Siregar GO, Harianja M, Rinonce HT, et al. Chromoblastomycosis: a case series from Sumba, eastern Indonesia. Clin Exp Dermatol. Published online March 8, 2025. doi:10.1093/ced/llaf111
- World Health Organization. Ending the neglect to attain the Sustainable Development Goals: a road map for neglected tropical diseases 2021-2030. Published January 28, 2021. Accessed May 5, 2024. https://www.who.int/publications/i/item/9789240010352
- Impact Global Health. The G-FINDER 2024 neglected disease R&D report. Impact Global Health. Published January 30, 2025. Accessed January 12, 2025. https://cdn.impactglobalhealth.org/media/G-FINDER%202024_Full%20report-1.pdf
- Queiroz-Telles F, de Hoog S, Santos DWCL, et al. Chromoblastomycosis. Clin Microbiol Rev. 2017;30:233-276. doi:10.1128/CMR.00032-16
- Santos DWCL, de Azevedo CMPS, Vicente VA, et al. The global burden of chromoblastomycosis. PLoS Negl Trop Dis. 2021;15:E0009611. doi:10.1371/journal.pntd.0009611
- Gold JAW, Smith DJ, Benedict K, et al. Epidemiology of implantation mycoses in the United States: an analysis of commercial insurance claims data, 2017 to 2021. J Am Acad Dermatol. 2023;89:427-430. doi:10.1016/j.jaad.2023.04.048
- Smith DJ, Queiroz-Telles F, Rabenja FR, et al. A global chromoblastomycosis strategy and development of the global chromoblastomycosis working group. PLoS Negl Trop Dis. 2024;18:e0012562. doi:10.1371/journal.pntd.0012562
- Heath CP, Sharma PC, Sontakke S, et al. The brief case: hidden in plain sight—exophiala jeanselmei subcutaneous phaeohyphomycosis of hand masquerading as a hematoma. J Clin Microbiol. 2024;62:E01068-24. doi:10.1128/jcm.01068-24
- Azevedo CMPS, Marques SG, Santos DWCL, et al. Squamous cell carcinoma derived from chronic chromoblastomycosis in Brazil. Clin Infect Dis. 2015;60:1500-1504. doi:10.1093/cid/civ104
- Sun J, Najafzadeh MJ, Gerrits van den Ende AHG, et al. Molecular characterization of pathogenic members of the genus Fonsecaea using multilocus analysis. PloS One. 2012;7:E41512. doi:10.1371/journal.pone.0041512
- Najafzadeh MJ, Sun J, Vicente V, et al. Fonsecaea nubica sp. nov, a new agent of human chromoblastomycosis revealed using molecular data. Med Mycol. 2010;48:800-806. doi:10.3109/13693780903503081
- Andrade TS, Castro LGM, Nunes RS, et al. Susceptibility of sequential Fonsecaea pedrosoi isolates from chromoblastomycosis patients to antifungal agents. Mycoses. 2004;47:216-221. doi:10.1111/j.1439-0507.2004.00984.x
- Smith DJ, Melhem MSC, Dirven J, et al. Establishment of epidemiological cutoff values for Fonsecaea pedrosoi, the primary etiologic agent of chromoblastomycosis, and eight antifungal medications. J Clin Microbiol. Published online April 4, 2025. doi:10.1128/jcm.01903-24
- Revankar SG, Sutton DA. Melanized fungi in human disease. Clin Microbiol Rev. 2010;23:884-928. doi:10.1128/CMR.00019-10
- de Sousa M da GT, Belda W, Spina R, et al. Topical application of imiquimod as a treatment for chromoblastomycosis. Clin Infect Dis. 2014;58:1734-1737. doi:10.1093/cid/ciu168
- Logan C, Singh M, Fox N, et al. Chromoblastomycosis treated with posaconazole and adjunctive imiquimod: lending innate immunity a helping hand. Open Forum Infect Dis. Published online March 14, 2023. doi:10.1093/ofid/ofad124
- Castro LGM, Pimentel ERA, Lacaz CS. Treatment of chromomycosis by cryosurgery with liquid nitrogen: 15 years’ experience. Int J Dermatol. 2003;42:408-412. doi:10.1046/j.1365-4362.2003.01532.x
- Tagami H, Ohi M, Aoshima T, et al. Topical heat therapy for cutaneous chromomycosis. Arch Dermatol. 1979;115:740-741.
- Lyon JP, Pedroso e Silva Azevedo C de M, Moreira LM, et al. Photodynamic antifungal therapy against chromoblastomycosis. Mycopathologia. 2011;172:293-297. doi:10.1007/s11046-011-9434-6
- Kinbara T, Fukushiro R, Eryu Y. Chromomycosis—report of two cases successfully treated with local heat therapy. Mykosen. 1982;25:689-694. doi:10.1111/j.1439-0507.1982.tb01944.x
- Yang Y, Hu Y, Zhang J, et al. A refractory case of chromoblastomycosis due to Fonsecaea monophora with improvement by photodynamic therapy. Med Mycol. 2012;50:649-653. doi:10.3109/13693786.2012.655258
- Sánchez-Cárdenas CD, Isa-Pimentel M, Arenas R. Phaeohyphomycosis: a review. Microbiol Res. 2023;14:1751-1763. doi:10.3390/microbiolres14040120
- Guillet J, Berkaoui I, Gargala G, et al. Cutaneous alternariosis. Mycopathologia. 2024;189:81. doi:10.1007/s11046-024-00888-5
- Wang X, Wang W, Lin Z, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol. 2014;133:905-908. doi:10.1016/j.jaci.2013.09.033
- Revankar SG, Baddley JW, Chen SCA, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200. doi:10.1093/ofid/ofx200
- Zijlstra EE, van de Sande WWJ, Welsh O, et al. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016;16:100-112. doi:10.1016/S1473-3099(15)00359-X
- Emery D, Denning DW. The global distribution of actinomycetoma and eumycetoma. PLoS Negl Trop Dis. 2020;14:E0008397. doi:10.1371/journal.pntd.0008397
- van de Sande WWJ, Fahal AH. An updated list of eumycetoma causative agents and their differences in grain formation and treatment response. Clin Microbiol Rev. Published online May 2024. doi:10.1128/cmr.00034-23
- Nenoff P, van de Sande WWJ, Fahal AH, et al. Eumycetoma and actinomycetoma—an update on causative agents, epidemiology, pathogenesis, diagnostics and therapy. J Eur Acad Dermatol Venereol. 2015;29:1873-1883. doi:10.1111/jdv.13008
- El-Amin SO, El-Amin RO, El-Amin SM, et al. Painful mycetoma: a study to understand the risk factors in patients visiting the Mycetoma Research Centre (MRC) in Khartoum, Sudan. Trans R Soc Trop Med Hyg. 2025;119:145-151. doi:10.1093/trstmh/trae093
- Ahmed AA, van de Sande W, Fahal AH. Mycetoma laboratory diagnosis: review article. PLoS Negl Trop Dis. 2017;11:e0005638. doi:10.1371/journal.pntd.0005638
- Siddig EE, Ahmed A, Hassan OB, et al. Using a Madurella mycetomatis specific PCR on grains obtained via noninvasive fine needle aspirated material is more accurate than cytology. Mycoses. Published online February 5, 2023. doi:10.1111/myc.13572
- Konings M, Siddig E, Eadie K, et al. The development of a multiplex recombinase polymerase amplification reaction to detect the most common causative agents of eumycetoma. Eur J Clin Microbiol Infect Dis. Published online April 30, 2025. doi:10.1007/s10096-025-05134-4
- Siddig EE, El Had Bakhait O, El nour Hussein Bahar M, et al. Ultrasound-guided fine-needle aspiration cytology significantly improved mycetoma diagnosis. J Eur Acad Dermatol Venereol. 2022;36:1845-1850. doi:10.1111/jdv.18363
- Bonifaz A, García-Sotelo RS, Lumbán-Ramirez F, et al. Update on actinomycetoma treatment: linezolid in the treatment of actinomycetomas due to Nocardia spp and Actinomadura madurae resistant to conventional treatments. Expert Rev Anti Infect Ther. 2025;23:79-89. doi:10.1080/14787210.2024.2448723
- Chandler DJ, Bonifaz A, van de Sande WWJ. An update on the development of novel antifungal agents for eumycetoma. Front Pharmacol. 2023;14:1165273. doi:10.3389/fphar.2023.1165273
- Fahal AH, Siddig Ahmed E, Mubarak Bakhiet S, et al. Two dose levels of once-weekly fosravuconazole versus daily itraconazole, in combination with surgery, in patients with eumycetoma in Sudan: a randomised, double-blind, phase 2, proof-of-concept superiority trial. Lancet Infect Dis. 2024;24:1254-1265. doi:10.1016/S1473-3099(24)00404-3
Implantation mycoses such as chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma are a diverse group of fungal diseases that occur when a break in the skin allows the entry of the causative fungus. These diseases disproportionately affect individuals in low- and middle-income countries causing substantial disability, decreased quality of life, and severe social stigma.1-3 Timely diagnosis and appropriate treatment are critical.
Chromoblastomycosis and mycetoma are designated as neglected tropical diseases, but research to improve their management is sparse, even compared to other neglected tropical diseases.4,5 Since there are no global diagnostic and treatment guidelines to date, we outline steps to diagnose and manage chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma.
Chromoblastomycosis
Chromoblastomycosis is caused by dematiaceous fungi that typically affect the skin and subcutaneous tissue. Chromoblastomycosis is distinguished from subcutaneous phaeohyphomycosis by microscopically visualizing the characteristic thick-walled, single, or multicellular clusters of pigmented fungal cells (also known as medlar bodies, muriform cells, or sclerotic bodies).6 In phaeohyphomycosis, short hyphae and pseudohyphae plus some single cells typically are seen.
Epidemiology—Globally, the distribution and burden of chromoblastomycosis are relatively unknown. Infections are more common in tropical and subtropical areas but can be acquired anywhere. A literature review conducted in 2021 identified 7740 cases of chromoblastomycosis, mostly reported in South America, Africa, Central America and Mexico, and Asia.7 Most of the patients were male, and the median age was 52 years. One study found an incidence of 14.7 per 1,000,000 patients in the United States for both chromoblastomycosis and phaeohyphomycotic abscesses (which included both skin and brain abscesses).8 Most patients were aged 65 years or older, with a higher incidence in males. Geographically, the incidence was highest in the Northeast followed by the South; patients in rural areas also had higher incidence of disease.8
Causative Organisms—Causative species cannot reliably distinguish between chromoblastomycosis and subcutaneous phaeohyphomycosis, as some species overlap. Cladophialophora carrionii, Fonsecaea species, Phialophora verrucosa species complex, and Rhinocladiella aquaspersa most commonly cause chromoblastomycosis.9,10
Clinical Manifestations—Chromoblastomycosis initially manifests as a solitary erythematous macule at a site of trauma (often not recalled by the patient) that can evolve to a smooth pink papule and may progress to 1 of 5 morphologies: nodular, verrucous, tumorous, cicatricial, or plaque.6 Patients may present with more than one morphology, particularly in long-standing or advanced disease. Lesions commonly manifest on the arms and legs in otherwise healthy individuals in environments (eg, rural, agricultural) that have more opportunities for injury and exposure to the causative fungi. Affected individuals often have small black specks on the lesion surface that are visible with the naked eye.6
Diagnosis—Common differential diagnoses include cutaneous blastomycosis, fixed sporotrichosis, warty tuberculosis nocardiosis, cutaneous leishmaniasis, human papillomavirus (HPV) infection, podoconiosis, lymphatic filariasis, cutaneous tuberculosis, and psoriasis.6 Squamous cell carcinoma is both a differential diagnosis as well as a potential complication of the disease.11
Potassium hydroxide preparation with skin scapings or a biopsy from the lesion has high sensitivity and quick turnaround times. There often is a background histopathologic reaction of pseudoepitheliomatous hyperplasia. Examining samples taken from areas with the visible small black dots on the skin surface can increase the likelihood of detecting fungal elements (Figure 1). Clinicians also may choose to obtain a 6- to 8-mm deep skin biopsy from the lesion and splice it in half, with one sample sent for histopathology and the other for culture (Figure 2). Skin scrapings can be sent for culture instead. In the case of verrucous lesions, biopsy is preferred if feasible.
Treatment should not be delayed while awaiting the culture results if infection is otherwise confirmed by direct microscopy or histopathology. The treatment approach remains similar regardless of the causative species. If the culture results are positive, the causative genus can be identified by the microscopic morphology; however, molecular diagnostic tools are needed for accurate species identification.12,13
Antifungal Susceptibility Testing—For most dematiaceous fungi, interpreting minimum inhibitory concentrations (MICs) is challenging due to a lack of data from multicenter studies. One report examined sequential isolates of Fonsecaea pedrosoi and demonstrated both high MIC values and clinical resistance to itraconazole in some cases, likely from treatment pressure.14 Clinical Laboratory Standards Institute–approved epidemiologic cutoff values (ECVs) are established for F pedrosoi for commonly used antifungals including itraconazole (0.5 µg/mL), terbinafine (0.25 µg/mL), and posaconazole (0.5 µg/mL).15 Clinicians may choose to obtain sequential isolates for any causative fungi in recalcitrant disease to monitor for increases in MIC.
Management—In early-stage disease, excision of the skin nodule may be curative, although concomitant treatment for several months with an antifungal is advised. If antifungals are needed, itraconazole is the most commonly prescribed agent, typically at a dose of 100 to 200 mg twice daily. Terbinafine also has been used first-line at a dose of 250 to 500 mg per day. Voriconazole and posaconazole also may be suitable options for first-line or for refractory disease treatment. Fluconazole does not have good activity against dematiaceous fungi and should be avoided.16 Topical antifungals will not reach the site of infection in adequate concentrations. Topical corticosteroids can make the disease worse and should be avoided. The duration of therapy usually is several months, but many patients require years of therapy until resolution of lesions.
Clinicians can consider combination therapy with an antifungal and a topical immunomodulator such as imiquimod (applied topically 3 times per week); this combination can be considered in refractory disease and even upon initial diagnosis, especially in severe disease.17,18 Nonpharmacologic interventions such as cryotherapy, heat, and light-based therapies have been used, but outcome data are scarce.19-23
Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis also is caused by dematiaceous fungi that typically affect the skin and subcutaneous tissue. Subcutaneous phaeohyphomycosis is distinguished from chromoblastomycosis by short hyphae and hyphal fragments usually seen microscopically instead of visualizing thick-walled, single, or multicellular clusters of pigmented fungal cells.6
Epidemiology—Globally, the burden and distribution of phaeohyphomycosis, including its cutaneous manifestations, are not well understood. Infections are more common in tropical and subtropical areas but can be acquired anywhere. Phaeohyphomycosis is a generic term used to describe infections caused by pigmented hyphal fungi that can manifest on the skin (subcutaneous phaeohyphomycosis) but also can affect deep structures including the brain (systemic phaeohyphomycosis).24
Causative Organisms—Alternaria, Bipolaris, Cladosporium, Curvularia, Exophiala, and Exserohilum species most commonly cause subcutaneous phaeohyphomycosis. Alternaria infections manifesting with skin lesions often are referred to as cutaneous alternariosis.25
Clinical Manifestations—The most common skin manifestation of phaeohyphomycosis is a subcutaneous cyst (cystic phaeohyphomycosis)(Figure 2). Subcutaneous phaeohyphomycosis also may manifest with nodules or plaques (Figure 3). Phaeohyphomycosis appears to occur more commonly in individuals who are immunosuppressed, those in whom T-cell function is affected, in congenital immunodeficiency states (eg, individuals with CARD9 mutations).26
Diagnosis—Culture is the gold standard for confirming phaeohyphomycosis.27 For cystic phaeohyphomycosis, clinicians can consider aspiration of the cyst for direct microscopic examination and culture. Histopathology may be utilized but can have lower sensitivity in showing dematiaceous hyphae and granulomatous inflammation; using the Masson-Fontana stain for melanin can be helpful. Molecular diagnostic tools including metagenomics applied directly to the tissue may be useful but are likely to have lower sensitivity than culture and require specialist diagnostic facilities.
Management—The approaches to managing chromoblastomycosis and subcutaneous phaeohyphomycosis are similar, though the preferred agents often differ. In early-stage disease, excision of the skin nodule may be curative, although concomitant treatment for several months with an antifungal is advised. In localized forms, itraconazole usually is used, but in those cases associated with immunodeficiency states, voriconazole may be necessary. Fluconazole does not have good activity against dematiaceous fungi and should be avoided.16 Topical antifungals will not reach the site of infection in adequate concentrations. Topical corticosteroids can make the disease worse and should be avoided. The duration of therapy may be substantially longer for chromoblastomycosis (months to years) compared to subcutaneous phaeohyphomycosis (weeks to months), although in immunocompromised individuals treatment may be even more prolonged.
Mycetoma
Mycetoma is caused by one of several different types of fungi (eumycetoma) and bacteria (actinomycetoma) that lead to progressively debilitating yet painless subcutaneous tumorlike lesions. The lesions usually manifest on the arms and legs but can occur anywhere.
Epidemiology—Little is known about the true global burden of mycetoma, but it occurs more frequently in low-income communities in rural areas.28 A retrospective review identified 19,494 cases published from 1876 to 2019, with cases reported in 102 countries.29 The countries with the highest numbers of cases are Sudan and Mexico, where there is more information on the distribution of the disease. Cases often are reported in what is known as the mycetoma belt (between latitudes 15° south and 30° north) but are increasingly identified outside this region.28 Young men aged 20 to 40 years are most commonly affected.
In the United States, mycetoma is uncommon, but clinicians can encounter locally acquired and travel-associated cases; hence, taking a good travel history is essential. One study specifically evaluating eumycetoma found a prevalence of 5.2 per 1,000,000 patients.8 Women and those aged 65 years or older had a higher incidence. Incidence was similar across US regions, but a higher incidence was reported in nonrural areas.8
Causative Organisms—More than 60 different species of fungi can cause eumycetoma; most cases are caused by Madurella mycetomatis, Trematosphaeria grisea (formerly Madurella grisea); Pseudallescheria boydii species complex, and Falciformispora (formerly Leptosphaeria) senegalensis.30 Actinomycetoma commonly is caused by Nocardia species (Nocardia brasiliensis, Nocardia asteroides, Nocardia otitidiscaviarum, Nocardia transvalensis, Nocardia harenae, and Nocardia takedensis), Streptomyces somaliensis, and Actinomadura species (Actinomadura madurae, Actinomadura pelletieri).31
Clinical Manifestations—Mycetoma is a chronic granulomatous disease with a progressive inflammatory reaction (Figures 4 and 5). Over the course of years, mycetoma progresses from small nodules to large, bone-invasive, mutilating lesions. Mycetoma manifests as a triad of painless firm subcutaneous masses, formation of multiple sinuses within the masses, and a purulent or seropurulent discharge containing sandlike visible particles (grains) that can be white, yellow, red, or black.28 Lesions usually are painless in early disease and are slowly progressive. Large lesion size, bone destruction, secondary bacterial infections, and actinomycetoma may lead to higher likelihood of pain.32
Diagnosis—Other conditions that could manifest with the same triad seen in mycetoma such as botryomycosis should be included in the differential. Other differential diagnoses include foreign body granuloma, filariasis, mycobacterial infection, skeletal tuberculosis, and yaws.
Proper treatment requires an accurate diagnosis that distinguishes actinomycetoma from eumycetoma.33 Culturing of grains obtained from deep lesion aspirates enables identification of the causative organism (Figure 6). The color of the grains may provide clues to their etiology: black grains are caused by fungus, red grains by a bacterium (A pelletieri), and pale (yellow or white) grains can be caused by either one.31Nocardia mycetoma grains are very small and usually cannot be appreciated with the naked eye. Histopathology of deep biopsy specimens (biopsy needle or surgical biopsy) stained with hematoxylin and eosin can diagnose actinomycetoma and eumycetoma. Punch biopsies often are not helpful, as the inflammatory mass is too deeply located. Deep surgical biopsy is preferred; however, species identification cannot be made without culture. Molecular tests for certain causative organisms of mycetoma have been developed but are not readily available.34,35 Currently, no serologic tests can diagnose mycetoma reliably. Ultrasonography can be used to diagnose mycetoma and, with appropriate training, distinguish between actinomycetoma and eumycetoma; it also can be combined with needle aspiration for taking grain samples.36
Treatment—Treatment of mycetoma depends on identification of the causal etiology and requires long-term and expensive drug regimens. It is not possible to determine the causative organism clinically. Actinomycetoma generally responds to medical treatment, and surgery rarely is needed. The current first-line treatment is co-trimoxazole (trimethoprim/sulfamethoxazole) in combination with amoxicillin and clavulanate acid or co-trimoxazole and amikacin for refractory disease; linezolid also may be a promising option for refractory disease.37
Eumycetoma is less responsive to medical therapies, and recurrence is common. Current recommended therapy is itraconazole for 9 to 12 months; however, cure rates ranging from 26% to 75% in combination with surgery have been reported, and fungi often can still be cultured from lesions posttreatment.38,39 Surgical excision often is used following 6 months of treatment with itraconazole to obtain better outcomes. Amputation may be required if the combination of antifungals and surgical excision fails. Fosravuconazole has shown promise in one clinical trial, but it is not approved in most countries, including the United States.39
Final Thoughts
Chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma can cause devastating disease. Patients with these conditions often are unable to carry out daily activities and experience stigma and discrimination. Limited diagnostic and treatment options hamper the ability of clinicians to respond appropriately to suspect and confirmed disease. Effectively examining the skin is the starting point for diagnosing and managing these diseases and can help clinicians to care for patients and prevent severe disease.
Implantation mycoses such as chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma are a diverse group of fungal diseases that occur when a break in the skin allows the entry of the causative fungus. These diseases disproportionately affect individuals in low- and middle-income countries causing substantial disability, decreased quality of life, and severe social stigma.1-3 Timely diagnosis and appropriate treatment are critical.
Chromoblastomycosis and mycetoma are designated as neglected tropical diseases, but research to improve their management is sparse, even compared to other neglected tropical diseases.4,5 Since there are no global diagnostic and treatment guidelines to date, we outline steps to diagnose and manage chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma.
Chromoblastomycosis
Chromoblastomycosis is caused by dematiaceous fungi that typically affect the skin and subcutaneous tissue. Chromoblastomycosis is distinguished from subcutaneous phaeohyphomycosis by microscopically visualizing the characteristic thick-walled, single, or multicellular clusters of pigmented fungal cells (also known as medlar bodies, muriform cells, or sclerotic bodies).6 In phaeohyphomycosis, short hyphae and pseudohyphae plus some single cells typically are seen.
Epidemiology—Globally, the distribution and burden of chromoblastomycosis are relatively unknown. Infections are more common in tropical and subtropical areas but can be acquired anywhere. A literature review conducted in 2021 identified 7740 cases of chromoblastomycosis, mostly reported in South America, Africa, Central America and Mexico, and Asia.7 Most of the patients were male, and the median age was 52 years. One study found an incidence of 14.7 per 1,000,000 patients in the United States for both chromoblastomycosis and phaeohyphomycotic abscesses (which included both skin and brain abscesses).8 Most patients were aged 65 years or older, with a higher incidence in males. Geographically, the incidence was highest in the Northeast followed by the South; patients in rural areas also had higher incidence of disease.8
Causative Organisms—Causative species cannot reliably distinguish between chromoblastomycosis and subcutaneous phaeohyphomycosis, as some species overlap. Cladophialophora carrionii, Fonsecaea species, Phialophora verrucosa species complex, and Rhinocladiella aquaspersa most commonly cause chromoblastomycosis.9,10
Clinical Manifestations—Chromoblastomycosis initially manifests as a solitary erythematous macule at a site of trauma (often not recalled by the patient) that can evolve to a smooth pink papule and may progress to 1 of 5 morphologies: nodular, verrucous, tumorous, cicatricial, or plaque.6 Patients may present with more than one morphology, particularly in long-standing or advanced disease. Lesions commonly manifest on the arms and legs in otherwise healthy individuals in environments (eg, rural, agricultural) that have more opportunities for injury and exposure to the causative fungi. Affected individuals often have small black specks on the lesion surface that are visible with the naked eye.6
Diagnosis—Common differential diagnoses include cutaneous blastomycosis, fixed sporotrichosis, warty tuberculosis nocardiosis, cutaneous leishmaniasis, human papillomavirus (HPV) infection, podoconiosis, lymphatic filariasis, cutaneous tuberculosis, and psoriasis.6 Squamous cell carcinoma is both a differential diagnosis as well as a potential complication of the disease.11
Potassium hydroxide preparation with skin scapings or a biopsy from the lesion has high sensitivity and quick turnaround times. There often is a background histopathologic reaction of pseudoepitheliomatous hyperplasia. Examining samples taken from areas with the visible small black dots on the skin surface can increase the likelihood of detecting fungal elements (Figure 1). Clinicians also may choose to obtain a 6- to 8-mm deep skin biopsy from the lesion and splice it in half, with one sample sent for histopathology and the other for culture (Figure 2). Skin scrapings can be sent for culture instead. In the case of verrucous lesions, biopsy is preferred if feasible.
Treatment should not be delayed while awaiting the culture results if infection is otherwise confirmed by direct microscopy or histopathology. The treatment approach remains similar regardless of the causative species. If the culture results are positive, the causative genus can be identified by the microscopic morphology; however, molecular diagnostic tools are needed for accurate species identification.12,13
Antifungal Susceptibility Testing—For most dematiaceous fungi, interpreting minimum inhibitory concentrations (MICs) is challenging due to a lack of data from multicenter studies. One report examined sequential isolates of Fonsecaea pedrosoi and demonstrated both high MIC values and clinical resistance to itraconazole in some cases, likely from treatment pressure.14 Clinical Laboratory Standards Institute–approved epidemiologic cutoff values (ECVs) are established for F pedrosoi for commonly used antifungals including itraconazole (0.5 µg/mL), terbinafine (0.25 µg/mL), and posaconazole (0.5 µg/mL).15 Clinicians may choose to obtain sequential isolates for any causative fungi in recalcitrant disease to monitor for increases in MIC.
Management—In early-stage disease, excision of the skin nodule may be curative, although concomitant treatment for several months with an antifungal is advised. If antifungals are needed, itraconazole is the most commonly prescribed agent, typically at a dose of 100 to 200 mg twice daily. Terbinafine also has been used first-line at a dose of 250 to 500 mg per day. Voriconazole and posaconazole also may be suitable options for first-line or for refractory disease treatment. Fluconazole does not have good activity against dematiaceous fungi and should be avoided.16 Topical antifungals will not reach the site of infection in adequate concentrations. Topical corticosteroids can make the disease worse and should be avoided. The duration of therapy usually is several months, but many patients require years of therapy until resolution of lesions.
Clinicians can consider combination therapy with an antifungal and a topical immunomodulator such as imiquimod (applied topically 3 times per week); this combination can be considered in refractory disease and even upon initial diagnosis, especially in severe disease.17,18 Nonpharmacologic interventions such as cryotherapy, heat, and light-based therapies have been used, but outcome data are scarce.19-23
Subcutaneous Phaeohyphomycosis
Subcutaneous phaeohyphomycosis also is caused by dematiaceous fungi that typically affect the skin and subcutaneous tissue. Subcutaneous phaeohyphomycosis is distinguished from chromoblastomycosis by short hyphae and hyphal fragments usually seen microscopically instead of visualizing thick-walled, single, or multicellular clusters of pigmented fungal cells.6
Epidemiology—Globally, the burden and distribution of phaeohyphomycosis, including its cutaneous manifestations, are not well understood. Infections are more common in tropical and subtropical areas but can be acquired anywhere. Phaeohyphomycosis is a generic term used to describe infections caused by pigmented hyphal fungi that can manifest on the skin (subcutaneous phaeohyphomycosis) but also can affect deep structures including the brain (systemic phaeohyphomycosis).24
Causative Organisms—Alternaria, Bipolaris, Cladosporium, Curvularia, Exophiala, and Exserohilum species most commonly cause subcutaneous phaeohyphomycosis. Alternaria infections manifesting with skin lesions often are referred to as cutaneous alternariosis.25
Clinical Manifestations—The most common skin manifestation of phaeohyphomycosis is a subcutaneous cyst (cystic phaeohyphomycosis)(Figure 2). Subcutaneous phaeohyphomycosis also may manifest with nodules or plaques (Figure 3). Phaeohyphomycosis appears to occur more commonly in individuals who are immunosuppressed, those in whom T-cell function is affected, in congenital immunodeficiency states (eg, individuals with CARD9 mutations).26
Diagnosis—Culture is the gold standard for confirming phaeohyphomycosis.27 For cystic phaeohyphomycosis, clinicians can consider aspiration of the cyst for direct microscopic examination and culture. Histopathology may be utilized but can have lower sensitivity in showing dematiaceous hyphae and granulomatous inflammation; using the Masson-Fontana stain for melanin can be helpful. Molecular diagnostic tools including metagenomics applied directly to the tissue may be useful but are likely to have lower sensitivity than culture and require specialist diagnostic facilities.
Management—The approaches to managing chromoblastomycosis and subcutaneous phaeohyphomycosis are similar, though the preferred agents often differ. In early-stage disease, excision of the skin nodule may be curative, although concomitant treatment for several months with an antifungal is advised. In localized forms, itraconazole usually is used, but in those cases associated with immunodeficiency states, voriconazole may be necessary. Fluconazole does not have good activity against dematiaceous fungi and should be avoided.16 Topical antifungals will not reach the site of infection in adequate concentrations. Topical corticosteroids can make the disease worse and should be avoided. The duration of therapy may be substantially longer for chromoblastomycosis (months to years) compared to subcutaneous phaeohyphomycosis (weeks to months), although in immunocompromised individuals treatment may be even more prolonged.
Mycetoma
Mycetoma is caused by one of several different types of fungi (eumycetoma) and bacteria (actinomycetoma) that lead to progressively debilitating yet painless subcutaneous tumorlike lesions. The lesions usually manifest on the arms and legs but can occur anywhere.
Epidemiology—Little is known about the true global burden of mycetoma, but it occurs more frequently in low-income communities in rural areas.28 A retrospective review identified 19,494 cases published from 1876 to 2019, with cases reported in 102 countries.29 The countries with the highest numbers of cases are Sudan and Mexico, where there is more information on the distribution of the disease. Cases often are reported in what is known as the mycetoma belt (between latitudes 15° south and 30° north) but are increasingly identified outside this region.28 Young men aged 20 to 40 years are most commonly affected.
In the United States, mycetoma is uncommon, but clinicians can encounter locally acquired and travel-associated cases; hence, taking a good travel history is essential. One study specifically evaluating eumycetoma found a prevalence of 5.2 per 1,000,000 patients.8 Women and those aged 65 years or older had a higher incidence. Incidence was similar across US regions, but a higher incidence was reported in nonrural areas.8
Causative Organisms—More than 60 different species of fungi can cause eumycetoma; most cases are caused by Madurella mycetomatis, Trematosphaeria grisea (formerly Madurella grisea); Pseudallescheria boydii species complex, and Falciformispora (formerly Leptosphaeria) senegalensis.30 Actinomycetoma commonly is caused by Nocardia species (Nocardia brasiliensis, Nocardia asteroides, Nocardia otitidiscaviarum, Nocardia transvalensis, Nocardia harenae, and Nocardia takedensis), Streptomyces somaliensis, and Actinomadura species (Actinomadura madurae, Actinomadura pelletieri).31
Clinical Manifestations—Mycetoma is a chronic granulomatous disease with a progressive inflammatory reaction (Figures 4 and 5). Over the course of years, mycetoma progresses from small nodules to large, bone-invasive, mutilating lesions. Mycetoma manifests as a triad of painless firm subcutaneous masses, formation of multiple sinuses within the masses, and a purulent or seropurulent discharge containing sandlike visible particles (grains) that can be white, yellow, red, or black.28 Lesions usually are painless in early disease and are slowly progressive. Large lesion size, bone destruction, secondary bacterial infections, and actinomycetoma may lead to higher likelihood of pain.32
Diagnosis—Other conditions that could manifest with the same triad seen in mycetoma such as botryomycosis should be included in the differential. Other differential diagnoses include foreign body granuloma, filariasis, mycobacterial infection, skeletal tuberculosis, and yaws.
Proper treatment requires an accurate diagnosis that distinguishes actinomycetoma from eumycetoma.33 Culturing of grains obtained from deep lesion aspirates enables identification of the causative organism (Figure 6). The color of the grains may provide clues to their etiology: black grains are caused by fungus, red grains by a bacterium (A pelletieri), and pale (yellow or white) grains can be caused by either one.31Nocardia mycetoma grains are very small and usually cannot be appreciated with the naked eye. Histopathology of deep biopsy specimens (biopsy needle or surgical biopsy) stained with hematoxylin and eosin can diagnose actinomycetoma and eumycetoma. Punch biopsies often are not helpful, as the inflammatory mass is too deeply located. Deep surgical biopsy is preferred; however, species identification cannot be made without culture. Molecular tests for certain causative organisms of mycetoma have been developed but are not readily available.34,35 Currently, no serologic tests can diagnose mycetoma reliably. Ultrasonography can be used to diagnose mycetoma and, with appropriate training, distinguish between actinomycetoma and eumycetoma; it also can be combined with needle aspiration for taking grain samples.36
Treatment—Treatment of mycetoma depends on identification of the causal etiology and requires long-term and expensive drug regimens. It is not possible to determine the causative organism clinically. Actinomycetoma generally responds to medical treatment, and surgery rarely is needed. The current first-line treatment is co-trimoxazole (trimethoprim/sulfamethoxazole) in combination with amoxicillin and clavulanate acid or co-trimoxazole and amikacin for refractory disease; linezolid also may be a promising option for refractory disease.37
Eumycetoma is less responsive to medical therapies, and recurrence is common. Current recommended therapy is itraconazole for 9 to 12 months; however, cure rates ranging from 26% to 75% in combination with surgery have been reported, and fungi often can still be cultured from lesions posttreatment.38,39 Surgical excision often is used following 6 months of treatment with itraconazole to obtain better outcomes. Amputation may be required if the combination of antifungals and surgical excision fails. Fosravuconazole has shown promise in one clinical trial, but it is not approved in most countries, including the United States.39
Final Thoughts
Chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma can cause devastating disease. Patients with these conditions often are unable to carry out daily activities and experience stigma and discrimination. Limited diagnostic and treatment options hamper the ability of clinicians to respond appropriately to suspect and confirmed disease. Effectively examining the skin is the starting point for diagnosing and managing these diseases and can help clinicians to care for patients and prevent severe disease.
- Smith DJ, Soebono H, Parajuli N, et al. South-east Asia regional neglected tropical disease framework: improving control of mycetoma, chromoblastomycosis, and sporotrichosis. Lancet Reg Health Southeast Asia. 2025;35:100561. doi:10.1016/j.lansea.2025.100561
- Abbas M, Scolding PS, Yosif AA, et al. The disabling consequences of mycetoma. PLoS Negl Trop Dis. 2018;12:E0007019. doi:10.1371/journal.pntd.0007019
- Siregar GO, Harianja M, Rinonce HT, et al. Chromoblastomycosis: a case series from Sumba, eastern Indonesia. Clin Exp Dermatol. Published online March 8, 2025. doi:10.1093/ced/llaf111
- World Health Organization. Ending the neglect to attain the Sustainable Development Goals: a road map for neglected tropical diseases 2021-2030. Published January 28, 2021. Accessed May 5, 2024. https://www.who.int/publications/i/item/9789240010352
- Impact Global Health. The G-FINDER 2024 neglected disease R&D report. Impact Global Health. Published January 30, 2025. Accessed January 12, 2025. https://cdn.impactglobalhealth.org/media/G-FINDER%202024_Full%20report-1.pdf
- Queiroz-Telles F, de Hoog S, Santos DWCL, et al. Chromoblastomycosis. Clin Microbiol Rev. 2017;30:233-276. doi:10.1128/CMR.00032-16
- Santos DWCL, de Azevedo CMPS, Vicente VA, et al. The global burden of chromoblastomycosis. PLoS Negl Trop Dis. 2021;15:E0009611. doi:10.1371/journal.pntd.0009611
- Gold JAW, Smith DJ, Benedict K, et al. Epidemiology of implantation mycoses in the United States: an analysis of commercial insurance claims data, 2017 to 2021. J Am Acad Dermatol. 2023;89:427-430. doi:10.1016/j.jaad.2023.04.048
- Smith DJ, Queiroz-Telles F, Rabenja FR, et al. A global chromoblastomycosis strategy and development of the global chromoblastomycosis working group. PLoS Negl Trop Dis. 2024;18:e0012562. doi:10.1371/journal.pntd.0012562
- Heath CP, Sharma PC, Sontakke S, et al. The brief case: hidden in plain sight—exophiala jeanselmei subcutaneous phaeohyphomycosis of hand masquerading as a hematoma. J Clin Microbiol. 2024;62:E01068-24. doi:10.1128/jcm.01068-24
- Azevedo CMPS, Marques SG, Santos DWCL, et al. Squamous cell carcinoma derived from chronic chromoblastomycosis in Brazil. Clin Infect Dis. 2015;60:1500-1504. doi:10.1093/cid/civ104
- Sun J, Najafzadeh MJ, Gerrits van den Ende AHG, et al. Molecular characterization of pathogenic members of the genus Fonsecaea using multilocus analysis. PloS One. 2012;7:E41512. doi:10.1371/journal.pone.0041512
- Najafzadeh MJ, Sun J, Vicente V, et al. Fonsecaea nubica sp. nov, a new agent of human chromoblastomycosis revealed using molecular data. Med Mycol. 2010;48:800-806. doi:10.3109/13693780903503081
- Andrade TS, Castro LGM, Nunes RS, et al. Susceptibility of sequential Fonsecaea pedrosoi isolates from chromoblastomycosis patients to antifungal agents. Mycoses. 2004;47:216-221. doi:10.1111/j.1439-0507.2004.00984.x
- Smith DJ, Melhem MSC, Dirven J, et al. Establishment of epidemiological cutoff values for Fonsecaea pedrosoi, the primary etiologic agent of chromoblastomycosis, and eight antifungal medications. J Clin Microbiol. Published online April 4, 2025. doi:10.1128/jcm.01903-24
- Revankar SG, Sutton DA. Melanized fungi in human disease. Clin Microbiol Rev. 2010;23:884-928. doi:10.1128/CMR.00019-10
- de Sousa M da GT, Belda W, Spina R, et al. Topical application of imiquimod as a treatment for chromoblastomycosis. Clin Infect Dis. 2014;58:1734-1737. doi:10.1093/cid/ciu168
- Logan C, Singh M, Fox N, et al. Chromoblastomycosis treated with posaconazole and adjunctive imiquimod: lending innate immunity a helping hand. Open Forum Infect Dis. Published online March 14, 2023. doi:10.1093/ofid/ofad124
- Castro LGM, Pimentel ERA, Lacaz CS. Treatment of chromomycosis by cryosurgery with liquid nitrogen: 15 years’ experience. Int J Dermatol. 2003;42:408-412. doi:10.1046/j.1365-4362.2003.01532.x
- Tagami H, Ohi M, Aoshima T, et al. Topical heat therapy for cutaneous chromomycosis. Arch Dermatol. 1979;115:740-741.
- Lyon JP, Pedroso e Silva Azevedo C de M, Moreira LM, et al. Photodynamic antifungal therapy against chromoblastomycosis. Mycopathologia. 2011;172:293-297. doi:10.1007/s11046-011-9434-6
- Kinbara T, Fukushiro R, Eryu Y. Chromomycosis—report of two cases successfully treated with local heat therapy. Mykosen. 1982;25:689-694. doi:10.1111/j.1439-0507.1982.tb01944.x
- Yang Y, Hu Y, Zhang J, et al. A refractory case of chromoblastomycosis due to Fonsecaea monophora with improvement by photodynamic therapy. Med Mycol. 2012;50:649-653. doi:10.3109/13693786.2012.655258
- Sánchez-Cárdenas CD, Isa-Pimentel M, Arenas R. Phaeohyphomycosis: a review. Microbiol Res. 2023;14:1751-1763. doi:10.3390/microbiolres14040120
- Guillet J, Berkaoui I, Gargala G, et al. Cutaneous alternariosis. Mycopathologia. 2024;189:81. doi:10.1007/s11046-024-00888-5
- Wang X, Wang W, Lin Z, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol. 2014;133:905-908. doi:10.1016/j.jaci.2013.09.033
- Revankar SG, Baddley JW, Chen SCA, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200. doi:10.1093/ofid/ofx200
- Zijlstra EE, van de Sande WWJ, Welsh O, et al. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016;16:100-112. doi:10.1016/S1473-3099(15)00359-X
- Emery D, Denning DW. The global distribution of actinomycetoma and eumycetoma. PLoS Negl Trop Dis. 2020;14:E0008397. doi:10.1371/journal.pntd.0008397
- van de Sande WWJ, Fahal AH. An updated list of eumycetoma causative agents and their differences in grain formation and treatment response. Clin Microbiol Rev. Published online May 2024. doi:10.1128/cmr.00034-23
- Nenoff P, van de Sande WWJ, Fahal AH, et al. Eumycetoma and actinomycetoma—an update on causative agents, epidemiology, pathogenesis, diagnostics and therapy. J Eur Acad Dermatol Venereol. 2015;29:1873-1883. doi:10.1111/jdv.13008
- El-Amin SO, El-Amin RO, El-Amin SM, et al. Painful mycetoma: a study to understand the risk factors in patients visiting the Mycetoma Research Centre (MRC) in Khartoum, Sudan. Trans R Soc Trop Med Hyg. 2025;119:145-151. doi:10.1093/trstmh/trae093
- Ahmed AA, van de Sande W, Fahal AH. Mycetoma laboratory diagnosis: review article. PLoS Negl Trop Dis. 2017;11:e0005638. doi:10.1371/journal.pntd.0005638
- Siddig EE, Ahmed A, Hassan OB, et al. Using a Madurella mycetomatis specific PCR on grains obtained via noninvasive fine needle aspirated material is more accurate than cytology. Mycoses. Published online February 5, 2023. doi:10.1111/myc.13572
- Konings M, Siddig E, Eadie K, et al. The development of a multiplex recombinase polymerase amplification reaction to detect the most common causative agents of eumycetoma. Eur J Clin Microbiol Infect Dis. Published online April 30, 2025. doi:10.1007/s10096-025-05134-4
- Siddig EE, El Had Bakhait O, El nour Hussein Bahar M, et al. Ultrasound-guided fine-needle aspiration cytology significantly improved mycetoma diagnosis. J Eur Acad Dermatol Venereol. 2022;36:1845-1850. doi:10.1111/jdv.18363
- Bonifaz A, García-Sotelo RS, Lumbán-Ramirez F, et al. Update on actinomycetoma treatment: linezolid in the treatment of actinomycetomas due to Nocardia spp and Actinomadura madurae resistant to conventional treatments. Expert Rev Anti Infect Ther. 2025;23:79-89. doi:10.1080/14787210.2024.2448723
- Chandler DJ, Bonifaz A, van de Sande WWJ. An update on the development of novel antifungal agents for eumycetoma. Front Pharmacol. 2023;14:1165273. doi:10.3389/fphar.2023.1165273
- Fahal AH, Siddig Ahmed E, Mubarak Bakhiet S, et al. Two dose levels of once-weekly fosravuconazole versus daily itraconazole, in combination with surgery, in patients with eumycetoma in Sudan: a randomised, double-blind, phase 2, proof-of-concept superiority trial. Lancet Infect Dis. 2024;24:1254-1265. doi:10.1016/S1473-3099(24)00404-3
- Smith DJ, Soebono H, Parajuli N, et al. South-east Asia regional neglected tropical disease framework: improving control of mycetoma, chromoblastomycosis, and sporotrichosis. Lancet Reg Health Southeast Asia. 2025;35:100561. doi:10.1016/j.lansea.2025.100561
- Abbas M, Scolding PS, Yosif AA, et al. The disabling consequences of mycetoma. PLoS Negl Trop Dis. 2018;12:E0007019. doi:10.1371/journal.pntd.0007019
- Siregar GO, Harianja M, Rinonce HT, et al. Chromoblastomycosis: a case series from Sumba, eastern Indonesia. Clin Exp Dermatol. Published online March 8, 2025. doi:10.1093/ced/llaf111
- World Health Organization. Ending the neglect to attain the Sustainable Development Goals: a road map for neglected tropical diseases 2021-2030. Published January 28, 2021. Accessed May 5, 2024. https://www.who.int/publications/i/item/9789240010352
- Impact Global Health. The G-FINDER 2024 neglected disease R&D report. Impact Global Health. Published January 30, 2025. Accessed January 12, 2025. https://cdn.impactglobalhealth.org/media/G-FINDER%202024_Full%20report-1.pdf
- Queiroz-Telles F, de Hoog S, Santos DWCL, et al. Chromoblastomycosis. Clin Microbiol Rev. 2017;30:233-276. doi:10.1128/CMR.00032-16
- Santos DWCL, de Azevedo CMPS, Vicente VA, et al. The global burden of chromoblastomycosis. PLoS Negl Trop Dis. 2021;15:E0009611. doi:10.1371/journal.pntd.0009611
- Gold JAW, Smith DJ, Benedict K, et al. Epidemiology of implantation mycoses in the United States: an analysis of commercial insurance claims data, 2017 to 2021. J Am Acad Dermatol. 2023;89:427-430. doi:10.1016/j.jaad.2023.04.048
- Smith DJ, Queiroz-Telles F, Rabenja FR, et al. A global chromoblastomycosis strategy and development of the global chromoblastomycosis working group. PLoS Negl Trop Dis. 2024;18:e0012562. doi:10.1371/journal.pntd.0012562
- Heath CP, Sharma PC, Sontakke S, et al. The brief case: hidden in plain sight—exophiala jeanselmei subcutaneous phaeohyphomycosis of hand masquerading as a hematoma. J Clin Microbiol. 2024;62:E01068-24. doi:10.1128/jcm.01068-24
- Azevedo CMPS, Marques SG, Santos DWCL, et al. Squamous cell carcinoma derived from chronic chromoblastomycosis in Brazil. Clin Infect Dis. 2015;60:1500-1504. doi:10.1093/cid/civ104
- Sun J, Najafzadeh MJ, Gerrits van den Ende AHG, et al. Molecular characterization of pathogenic members of the genus Fonsecaea using multilocus analysis. PloS One. 2012;7:E41512. doi:10.1371/journal.pone.0041512
- Najafzadeh MJ, Sun J, Vicente V, et al. Fonsecaea nubica sp. nov, a new agent of human chromoblastomycosis revealed using molecular data. Med Mycol. 2010;48:800-806. doi:10.3109/13693780903503081
- Andrade TS, Castro LGM, Nunes RS, et al. Susceptibility of sequential Fonsecaea pedrosoi isolates from chromoblastomycosis patients to antifungal agents. Mycoses. 2004;47:216-221. doi:10.1111/j.1439-0507.2004.00984.x
- Smith DJ, Melhem MSC, Dirven J, et al. Establishment of epidemiological cutoff values for Fonsecaea pedrosoi, the primary etiologic agent of chromoblastomycosis, and eight antifungal medications. J Clin Microbiol. Published online April 4, 2025. doi:10.1128/jcm.01903-24
- Revankar SG, Sutton DA. Melanized fungi in human disease. Clin Microbiol Rev. 2010;23:884-928. doi:10.1128/CMR.00019-10
- de Sousa M da GT, Belda W, Spina R, et al. Topical application of imiquimod as a treatment for chromoblastomycosis. Clin Infect Dis. 2014;58:1734-1737. doi:10.1093/cid/ciu168
- Logan C, Singh M, Fox N, et al. Chromoblastomycosis treated with posaconazole and adjunctive imiquimod: lending innate immunity a helping hand. Open Forum Infect Dis. Published online March 14, 2023. doi:10.1093/ofid/ofad124
- Castro LGM, Pimentel ERA, Lacaz CS. Treatment of chromomycosis by cryosurgery with liquid nitrogen: 15 years’ experience. Int J Dermatol. 2003;42:408-412. doi:10.1046/j.1365-4362.2003.01532.x
- Tagami H, Ohi M, Aoshima T, et al. Topical heat therapy for cutaneous chromomycosis. Arch Dermatol. 1979;115:740-741.
- Lyon JP, Pedroso e Silva Azevedo C de M, Moreira LM, et al. Photodynamic antifungal therapy against chromoblastomycosis. Mycopathologia. 2011;172:293-297. doi:10.1007/s11046-011-9434-6
- Kinbara T, Fukushiro R, Eryu Y. Chromomycosis—report of two cases successfully treated with local heat therapy. Mykosen. 1982;25:689-694. doi:10.1111/j.1439-0507.1982.tb01944.x
- Yang Y, Hu Y, Zhang J, et al. A refractory case of chromoblastomycosis due to Fonsecaea monophora with improvement by photodynamic therapy. Med Mycol. 2012;50:649-653. doi:10.3109/13693786.2012.655258
- Sánchez-Cárdenas CD, Isa-Pimentel M, Arenas R. Phaeohyphomycosis: a review. Microbiol Res. 2023;14:1751-1763. doi:10.3390/microbiolres14040120
- Guillet J, Berkaoui I, Gargala G, et al. Cutaneous alternariosis. Mycopathologia. 2024;189:81. doi:10.1007/s11046-024-00888-5
- Wang X, Wang W, Lin Z, et al. CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J Allergy Clin Immunol. 2014;133:905-908. doi:10.1016/j.jaci.2013.09.033
- Revankar SG, Baddley JW, Chen SCA, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200. doi:10.1093/ofid/ofx200
- Zijlstra EE, van de Sande WWJ, Welsh O, et al. Mycetoma: a unique neglected tropical disease. Lancet Infect Dis. 2016;16:100-112. doi:10.1016/S1473-3099(15)00359-X
- Emery D, Denning DW. The global distribution of actinomycetoma and eumycetoma. PLoS Negl Trop Dis. 2020;14:E0008397. doi:10.1371/journal.pntd.0008397
- van de Sande WWJ, Fahal AH. An updated list of eumycetoma causative agents and their differences in grain formation and treatment response. Clin Microbiol Rev. Published online May 2024. doi:10.1128/cmr.00034-23
- Nenoff P, van de Sande WWJ, Fahal AH, et al. Eumycetoma and actinomycetoma—an update on causative agents, epidemiology, pathogenesis, diagnostics and therapy. J Eur Acad Dermatol Venereol. 2015;29:1873-1883. doi:10.1111/jdv.13008
- El-Amin SO, El-Amin RO, El-Amin SM, et al. Painful mycetoma: a study to understand the risk factors in patients visiting the Mycetoma Research Centre (MRC) in Khartoum, Sudan. Trans R Soc Trop Med Hyg. 2025;119:145-151. doi:10.1093/trstmh/trae093
- Ahmed AA, van de Sande W, Fahal AH. Mycetoma laboratory diagnosis: review article. PLoS Negl Trop Dis. 2017;11:e0005638. doi:10.1371/journal.pntd.0005638
- Siddig EE, Ahmed A, Hassan OB, et al. Using a Madurella mycetomatis specific PCR on grains obtained via noninvasive fine needle aspirated material is more accurate than cytology. Mycoses. Published online February 5, 2023. doi:10.1111/myc.13572
- Konings M, Siddig E, Eadie K, et al. The development of a multiplex recombinase polymerase amplification reaction to detect the most common causative agents of eumycetoma. Eur J Clin Microbiol Infect Dis. Published online April 30, 2025. doi:10.1007/s10096-025-05134-4
- Siddig EE, El Had Bakhait O, El nour Hussein Bahar M, et al. Ultrasound-guided fine-needle aspiration cytology significantly improved mycetoma diagnosis. J Eur Acad Dermatol Venereol. 2022;36:1845-1850. doi:10.1111/jdv.18363
- Bonifaz A, García-Sotelo RS, Lumbán-Ramirez F, et al. Update on actinomycetoma treatment: linezolid in the treatment of actinomycetomas due to Nocardia spp and Actinomadura madurae resistant to conventional treatments. Expert Rev Anti Infect Ther. 2025;23:79-89. doi:10.1080/14787210.2024.2448723
- Chandler DJ, Bonifaz A, van de Sande WWJ. An update on the development of novel antifungal agents for eumycetoma. Front Pharmacol. 2023;14:1165273. doi:10.3389/fphar.2023.1165273
- Fahal AH, Siddig Ahmed E, Mubarak Bakhiet S, et al. Two dose levels of once-weekly fosravuconazole versus daily itraconazole, in combination with surgery, in patients with eumycetoma in Sudan: a randomised, double-blind, phase 2, proof-of-concept superiority trial. Lancet Infect Dis. 2024;24:1254-1265. doi:10.1016/S1473-3099(24)00404-3
Approach to Diagnosing and Managing Implantation Mycoses
Approach to Diagnosing and Managing Implantation Mycoses
Practice Points
- Chromoblastomycosis, subcutaneous phaeohyphomycosis, and mycetoma are implantation mycoses that cause substantial morbidity, decreased quality of life, and social stigma.
- Consider obtaining a biopsy of suspected chromoblastomycosis and subcutaneous phaeohyphomycosis to confirm infection while sending half of the sample for culture for organism identification.
- Distinguishing between actinomycetoma (caused by filamentous bacteria) and eumycetoma (caused by fungi) is critical for appropriate mycetoma treatment.

From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
Prurigo nodularis (PN) is a chronic, severely pruritic neuroimmunologic skin disorder characterized by multiple firm hyperkeratotic nodules and intense pruritus, often leading to considerable impairment in quality of life and increased rates of depression and anxiety.1 It is considered difficult to treat due to its complex pathogenesis, the severity and chronicity of pruritus, and the limited efficacy of conventional therapies.2,3 The disease is driven by a self-perpetuating itch-scratch cycle, underpinned by dysregulation of both immune and neural pathways including type 2 (interleukin [IL] 4, IL-13, IL-31), Th17, and Th22 cytokines as well as neuropeptides and altered cutaneous nerve architecture.1,3 This results in persistent severe pruritus and nodular lesions that are highly refractory to standard treatments.1 Conventional therapies (eg, locally acting agents, phototherapy, and systemic immunomodulators and neuromodulators) have varied efficacy and notable adverse effect profiles.3 While the approval of targeted biologics has transformed the therapeutic landscape, several other treatment options also are being explored in clinical trials. Herein, we review all recently approved therapies as well as emerging treatments currently under investigation.
Dupilumab
Dupilumab, the first therapy for PN approved by the US Food and Drug Administration (FDA) in 2022—is a monoclonal antibody that inhibits signaling of IL-4 and IL-13, key drivers of type 2 inflammation implicated in PN pathogenesis.4,5 In 2 pivotal phase 3 randomized controlled trials (LIBERTY-PN PRIME and PRIME2),5 dupilumab demonstrated notable efficacy in adults with moderate to severe PN. A reduction of 4 points or more on the Worst Itch Numeric Rating Scale (WI-NRS) was achieved by 60.0% (45/75) of patients treated with dupilumab at week 24 compared with 18.4% (14/76) receiving placebo in the PRIME trial. In PRIME2, the same outcome was achieved by 37.2% (29/78) of patients receiving dupilumab at week 12 compared with 22.0% (18/82) of patients receiving placebo.5 Dupilumab also led to a greater proportion of patients achieving a substantial reduction in nodule count (≤5 nodules) and improved quality of life compared with placebo.5,6 The safety profile of dupilumab for treatment of PN was favorable and consistent with prior experience in atopic dermatitis; conjunctivitis was the most common adverse event.5,6
Nemolizumab
Nemolizumab, an IL-31 receptor A antagonist, is the most recent agent approved by the FDA for PN in 2024.7 In the OLYMPIA 1 and OLYMPIA 2 phase 3 trials,8 nemolizumab produced a clinically meaningful reduction in itch (defined as a ≥4-point improvement in the Peak Pruritus Numerical Rating Scale score) in 56.3% (103/183) of patients at week 16 compared with 20.9% (19/91) receiving placebo. Additionally, 37.7% (69/183) of patients receiving nemolizumab achieved clear or almost clear skin (Investigator’s Global Assessment score of 0 or 1 with a ≥2-point reduction) vs 11.0% with placebo (both P<.001). Benefits were observed as early as week 4, including rapid improvements in itch, sleep disturbance, and nodule count.8 Nemolizumab also improved quality of life and reduced symptoms of anxiety and depression. The safety profile was favorable, with headache and atopic dermatitis the most common adverse events; serious adverse events were infrequent and similar between groups.8
Abrocitinib
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is an investigational therapy for PN and currently has not been approved by the FDA for this indication. In a phase 2 open-label trial, abrocitinib 200 mg daily for 12 weeks led to a 78.3% reduction in weekly Peak Pruritus Numerical Rating Scale scores in PN, with 80.0% (8/10) of patients achieving a clinically meaningful improvement of 4 points or higher. Nodule counts and quality of life also improved, with an onset of itch relief as early as week 2. The safety profile was favorable, with acneform eruptions the most common adverse event and no serious adverse events reported9; however, these results are based on small, nonrandomized studies and require confirmation in larger randomized controlled trials before abrocitinib can be considered a standard therapy for PN.
Cryosim-1
Transient receptor potential melastatin 8 (TRPM8) is a cold-sensing ion channel found in unmyelinated sensory neurons within the dorsal root and trigeminal ganglia.10 It is activated by cool temperatures (15-28 °C) and compounds such as menthol, leading to calcium influx and a cooling sensation. In a randomized, double-blind, vehicle-controlled trial, researchers investigated the efficacy of cryosim-1 (a synthetic TRPM8 agonist) in treating PN.10 Thirty patients were enrolled, with 18 (60.0%) receiving cryosim-1 and 12 (40.0%) receiving placebo over 8 weeks. By week 8, cryosim-1 significantly reduced itch severity (mean numerical rating scale score postapplication, 2.8 vs 4.3; P=.031) and improved sleep disturbances (2.2 vs 4.2; P=.031) compared to placebo. Patients reported higher satisfaction with itch relief, and no adverse effects were observed. The study concluded that cryosim-1 is a safe, effective topical therapy for PN, likely working by interrupting the itch-scratch cycle and potentially modulating inflammatory pathways involved in chronic itch.10
Nalbuphine
Nalbuphine is a κ opioid receptor agonist and μ opioid receptor antagonist that has been investigated for the treatment of PN.11 In a phase 2 randomized controlled trial, oral nalbuphine extended release (NAL-ER) 162 mg twice daily provided measurable antipruritic efficacy, with 44.4% (8/18) of patients achieving at least a 30% reduction in 7-day WI-NRS at week 10 compared with 36.4% (8/22) in the placebo group. Among those who completed the study, 66.7% (8/12) of patients receiving NAL-ER 162 mg achieved significant itch reduction vs 40% (8/20) receiving placebo (P=.03). At least a 50% reduction in WI-NRS was achieved by 33.3% (6/18) of patients receiving NAL-ER 162 mg twice daily. Extended open-label treatment was associated with further improvements in itch and lesion activity. Adverse events were mostly mild to moderate (eg, nausea, dizziness, headache, and fatigue) and occurred during dose titration. Physiologic opioid withdrawal symptoms were limited and resolved within a few days of discontinuing the medication.11
Final Thoughts
In conclusion, PN remains one of the most challenging chronic dermatologic conditions to manage and is driven by a complex interplay of neuroimmune mechanisms and resistance to many conventional therapies. The approval of dupilumab and nemolizumab has marked a pivotal shift in the therapeutic landscape, offering hope to patients who previously had limited options5,8; however, the burden of PN remains substantial, and many patients continue to experience relentless itch, poor sleep, and reduced quality of life.1 Emerging therapies such as TRPM8 agonists, Janus kinase inhibitors, and opioid modulators represent promising additions to the treatment options, targeting novel pathways beyond traditional immunosuppression.9-11
- Williams KA, Huang AH, Belzberg M, et al. Prurigo nodularis: pathogenesis and management. J Am Acad Dermatol. 2020;83:1567-1575. doi:10.1016/j.jaad.2020.04.182
- Gründel S, Pereira MP, Storck M, et al. Analysis of 325 patients with chronic nodular prurigo: clinics, burden of disease and course of treatment. Acta Derm Venereol. 2020;100:adv00269. doi:10.2340/00015555-3571
- Liao V, Cornman HL, Ma E, et al. Prurigo nodularis: new insights into pathogenesis and novel therapeutics. Br J Dermatol. 2024;190:798-810. doi:10.1093/bjd/ljae052
- Elmariah SB, Tao L, Valdes-Rodriguez R, et al. Individual article: management of prurigo nodularis. J Drugs Dermatol. 2023;22:SF365502s15-SF365502s22. doi:10.36849/JDD.SF365502
- Yosipovitch G, Mollanazar N, Ständer S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29:1180-1190. doi:10.1038/s41591-023-02320-9
- Cao P, Xu W, Jiang S, et al. Dupilumab for the treatment of prurigo nodularis: a systematic review. Front Immunol. 2023;14:1092685. doi:10.3389/fimmu.2023.1092685
- Dagenet CB, Saadi C, Phillips MA, et al. Landscape of prurigo nodularis clinical trials. JAAD Rev. 2024;2:127-136. doi:10.1016/j.jdrv.2024.09.006
- Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 trial of nemolizumab in patients with prurigo nodularis. N Engl J Med. 2023;389:1579-1589. doi:10.1056/NEJMoa2301333
- Kwatra SG, Bordeaux ZA, Parthasarathy V, et al. Efficacy and safety of abrocitinib in prurigo nodularis and chronic pruritus of unknown origin: a nonrandomized controlled trial. JAMA Dermatol. 2024;160:717-724. doi:10.1001/jamadermatol.2024.1464
- Choi ME, Lee JH, Jung CJ, et al. A randomized, double-blinded, vehicle-controlled clinical trial of topical cryosim-1, a synthetic TRPM8 agonist, in prurigo nodularis. J Cosmet Dermatol. 2024;23:931-937. doi:10.1111/jocd.16079
- Weisshaar E, Szepietowski JC, Bernhard JD, et al. Efficacy and safety of oral nalbuphine extended release in prurigo nodularis: results of a phase 2 randomized controlled trial with an open-label extension phase. J Eur Acad Dermatol Venereol. 2022;36:453-461. doi:10.1111/jdv.17816
Prurigo nodularis (PN) is a chronic, severely pruritic neuroimmunologic skin disorder characterized by multiple firm hyperkeratotic nodules and intense pruritus, often leading to considerable impairment in quality of life and increased rates of depression and anxiety.1 It is considered difficult to treat due to its complex pathogenesis, the severity and chronicity of pruritus, and the limited efficacy of conventional therapies.2,3 The disease is driven by a self-perpetuating itch-scratch cycle, underpinned by dysregulation of both immune and neural pathways including type 2 (interleukin [IL] 4, IL-13, IL-31), Th17, and Th22 cytokines as well as neuropeptides and altered cutaneous nerve architecture.1,3 This results in persistent severe pruritus and nodular lesions that are highly refractory to standard treatments.1 Conventional therapies (eg, locally acting agents, phototherapy, and systemic immunomodulators and neuromodulators) have varied efficacy and notable adverse effect profiles.3 While the approval of targeted biologics has transformed the therapeutic landscape, several other treatment options also are being explored in clinical trials. Herein, we review all recently approved therapies as well as emerging treatments currently under investigation.
Dupilumab
Dupilumab, the first therapy for PN approved by the US Food and Drug Administration (FDA) in 2022—is a monoclonal antibody that inhibits signaling of IL-4 and IL-13, key drivers of type 2 inflammation implicated in PN pathogenesis.4,5 In 2 pivotal phase 3 randomized controlled trials (LIBERTY-PN PRIME and PRIME2),5 dupilumab demonstrated notable efficacy in adults with moderate to severe PN. A reduction of 4 points or more on the Worst Itch Numeric Rating Scale (WI-NRS) was achieved by 60.0% (45/75) of patients treated with dupilumab at week 24 compared with 18.4% (14/76) receiving placebo in the PRIME trial. In PRIME2, the same outcome was achieved by 37.2% (29/78) of patients receiving dupilumab at week 12 compared with 22.0% (18/82) of patients receiving placebo.5 Dupilumab also led to a greater proportion of patients achieving a substantial reduction in nodule count (≤5 nodules) and improved quality of life compared with placebo.5,6 The safety profile of dupilumab for treatment of PN was favorable and consistent with prior experience in atopic dermatitis; conjunctivitis was the most common adverse event.5,6
Nemolizumab
Nemolizumab, an IL-31 receptor A antagonist, is the most recent agent approved by the FDA for PN in 2024.7 In the OLYMPIA 1 and OLYMPIA 2 phase 3 trials,8 nemolizumab produced a clinically meaningful reduction in itch (defined as a ≥4-point improvement in the Peak Pruritus Numerical Rating Scale score) in 56.3% (103/183) of patients at week 16 compared with 20.9% (19/91) receiving placebo. Additionally, 37.7% (69/183) of patients receiving nemolizumab achieved clear or almost clear skin (Investigator’s Global Assessment score of 0 or 1 with a ≥2-point reduction) vs 11.0% with placebo (both P<.001). Benefits were observed as early as week 4, including rapid improvements in itch, sleep disturbance, and nodule count.8 Nemolizumab also improved quality of life and reduced symptoms of anxiety and depression. The safety profile was favorable, with headache and atopic dermatitis the most common adverse events; serious adverse events were infrequent and similar between groups.8
Abrocitinib
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is an investigational therapy for PN and currently has not been approved by the FDA for this indication. In a phase 2 open-label trial, abrocitinib 200 mg daily for 12 weeks led to a 78.3% reduction in weekly Peak Pruritus Numerical Rating Scale scores in PN, with 80.0% (8/10) of patients achieving a clinically meaningful improvement of 4 points or higher. Nodule counts and quality of life also improved, with an onset of itch relief as early as week 2. The safety profile was favorable, with acneform eruptions the most common adverse event and no serious adverse events reported9; however, these results are based on small, nonrandomized studies and require confirmation in larger randomized controlled trials before abrocitinib can be considered a standard therapy for PN.
Cryosim-1
Transient receptor potential melastatin 8 (TRPM8) is a cold-sensing ion channel found in unmyelinated sensory neurons within the dorsal root and trigeminal ganglia.10 It is activated by cool temperatures (15-28 °C) and compounds such as menthol, leading to calcium influx and a cooling sensation. In a randomized, double-blind, vehicle-controlled trial, researchers investigated the efficacy of cryosim-1 (a synthetic TRPM8 agonist) in treating PN.10 Thirty patients were enrolled, with 18 (60.0%) receiving cryosim-1 and 12 (40.0%) receiving placebo over 8 weeks. By week 8, cryosim-1 significantly reduced itch severity (mean numerical rating scale score postapplication, 2.8 vs 4.3; P=.031) and improved sleep disturbances (2.2 vs 4.2; P=.031) compared to placebo. Patients reported higher satisfaction with itch relief, and no adverse effects were observed. The study concluded that cryosim-1 is a safe, effective topical therapy for PN, likely working by interrupting the itch-scratch cycle and potentially modulating inflammatory pathways involved in chronic itch.10
Nalbuphine
Nalbuphine is a κ opioid receptor agonist and μ opioid receptor antagonist that has been investigated for the treatment of PN.11 In a phase 2 randomized controlled trial, oral nalbuphine extended release (NAL-ER) 162 mg twice daily provided measurable antipruritic efficacy, with 44.4% (8/18) of patients achieving at least a 30% reduction in 7-day WI-NRS at week 10 compared with 36.4% (8/22) in the placebo group. Among those who completed the study, 66.7% (8/12) of patients receiving NAL-ER 162 mg achieved significant itch reduction vs 40% (8/20) receiving placebo (P=.03). At least a 50% reduction in WI-NRS was achieved by 33.3% (6/18) of patients receiving NAL-ER 162 mg twice daily. Extended open-label treatment was associated with further improvements in itch and lesion activity. Adverse events were mostly mild to moderate (eg, nausea, dizziness, headache, and fatigue) and occurred during dose titration. Physiologic opioid withdrawal symptoms were limited and resolved within a few days of discontinuing the medication.11
Final Thoughts
In conclusion, PN remains one of the most challenging chronic dermatologic conditions to manage and is driven by a complex interplay of neuroimmune mechanisms and resistance to many conventional therapies. The approval of dupilumab and nemolizumab has marked a pivotal shift in the therapeutic landscape, offering hope to patients who previously had limited options5,8; however, the burden of PN remains substantial, and many patients continue to experience relentless itch, poor sleep, and reduced quality of life.1 Emerging therapies such as TRPM8 agonists, Janus kinase inhibitors, and opioid modulators represent promising additions to the treatment options, targeting novel pathways beyond traditional immunosuppression.9-11
Prurigo nodularis (PN) is a chronic, severely pruritic neuroimmunologic skin disorder characterized by multiple firm hyperkeratotic nodules and intense pruritus, often leading to considerable impairment in quality of life and increased rates of depression and anxiety.1 It is considered difficult to treat due to its complex pathogenesis, the severity and chronicity of pruritus, and the limited efficacy of conventional therapies.2,3 The disease is driven by a self-perpetuating itch-scratch cycle, underpinned by dysregulation of both immune and neural pathways including type 2 (interleukin [IL] 4, IL-13, IL-31), Th17, and Th22 cytokines as well as neuropeptides and altered cutaneous nerve architecture.1,3 This results in persistent severe pruritus and nodular lesions that are highly refractory to standard treatments.1 Conventional therapies (eg, locally acting agents, phototherapy, and systemic immunomodulators and neuromodulators) have varied efficacy and notable adverse effect profiles.3 While the approval of targeted biologics has transformed the therapeutic landscape, several other treatment options also are being explored in clinical trials. Herein, we review all recently approved therapies as well as emerging treatments currently under investigation.
Dupilumab
Dupilumab, the first therapy for PN approved by the US Food and Drug Administration (FDA) in 2022—is a monoclonal antibody that inhibits signaling of IL-4 and IL-13, key drivers of type 2 inflammation implicated in PN pathogenesis.4,5 In 2 pivotal phase 3 randomized controlled trials (LIBERTY-PN PRIME and PRIME2),5 dupilumab demonstrated notable efficacy in adults with moderate to severe PN. A reduction of 4 points or more on the Worst Itch Numeric Rating Scale (WI-NRS) was achieved by 60.0% (45/75) of patients treated with dupilumab at week 24 compared with 18.4% (14/76) receiving placebo in the PRIME trial. In PRIME2, the same outcome was achieved by 37.2% (29/78) of patients receiving dupilumab at week 12 compared with 22.0% (18/82) of patients receiving placebo.5 Dupilumab also led to a greater proportion of patients achieving a substantial reduction in nodule count (≤5 nodules) and improved quality of life compared with placebo.5,6 The safety profile of dupilumab for treatment of PN was favorable and consistent with prior experience in atopic dermatitis; conjunctivitis was the most common adverse event.5,6
Nemolizumab
Nemolizumab, an IL-31 receptor A antagonist, is the most recent agent approved by the FDA for PN in 2024.7 In the OLYMPIA 1 and OLYMPIA 2 phase 3 trials,8 nemolizumab produced a clinically meaningful reduction in itch (defined as a ≥4-point improvement in the Peak Pruritus Numerical Rating Scale score) in 56.3% (103/183) of patients at week 16 compared with 20.9% (19/91) receiving placebo. Additionally, 37.7% (69/183) of patients receiving nemolizumab achieved clear or almost clear skin (Investigator’s Global Assessment score of 0 or 1 with a ≥2-point reduction) vs 11.0% with placebo (both P<.001). Benefits were observed as early as week 4, including rapid improvements in itch, sleep disturbance, and nodule count.8 Nemolizumab also improved quality of life and reduced symptoms of anxiety and depression. The safety profile was favorable, with headache and atopic dermatitis the most common adverse events; serious adverse events were infrequent and similar between groups.8
Abrocitinib
Abrocitinib, an oral selective Janus kinase 1 inhibitor, is an investigational therapy for PN and currently has not been approved by the FDA for this indication. In a phase 2 open-label trial, abrocitinib 200 mg daily for 12 weeks led to a 78.3% reduction in weekly Peak Pruritus Numerical Rating Scale scores in PN, with 80.0% (8/10) of patients achieving a clinically meaningful improvement of 4 points or higher. Nodule counts and quality of life also improved, with an onset of itch relief as early as week 2. The safety profile was favorable, with acneform eruptions the most common adverse event and no serious adverse events reported9; however, these results are based on small, nonrandomized studies and require confirmation in larger randomized controlled trials before abrocitinib can be considered a standard therapy for PN.
Cryosim-1
Transient receptor potential melastatin 8 (TRPM8) is a cold-sensing ion channel found in unmyelinated sensory neurons within the dorsal root and trigeminal ganglia.10 It is activated by cool temperatures (15-28 °C) and compounds such as menthol, leading to calcium influx and a cooling sensation. In a randomized, double-blind, vehicle-controlled trial, researchers investigated the efficacy of cryosim-1 (a synthetic TRPM8 agonist) in treating PN.10 Thirty patients were enrolled, with 18 (60.0%) receiving cryosim-1 and 12 (40.0%) receiving placebo over 8 weeks. By week 8, cryosim-1 significantly reduced itch severity (mean numerical rating scale score postapplication, 2.8 vs 4.3; P=.031) and improved sleep disturbances (2.2 vs 4.2; P=.031) compared to placebo. Patients reported higher satisfaction with itch relief, and no adverse effects were observed. The study concluded that cryosim-1 is a safe, effective topical therapy for PN, likely working by interrupting the itch-scratch cycle and potentially modulating inflammatory pathways involved in chronic itch.10
Nalbuphine
Nalbuphine is a κ opioid receptor agonist and μ opioid receptor antagonist that has been investigated for the treatment of PN.11 In a phase 2 randomized controlled trial, oral nalbuphine extended release (NAL-ER) 162 mg twice daily provided measurable antipruritic efficacy, with 44.4% (8/18) of patients achieving at least a 30% reduction in 7-day WI-NRS at week 10 compared with 36.4% (8/22) in the placebo group. Among those who completed the study, 66.7% (8/12) of patients receiving NAL-ER 162 mg achieved significant itch reduction vs 40% (8/20) receiving placebo (P=.03). At least a 50% reduction in WI-NRS was achieved by 33.3% (6/18) of patients receiving NAL-ER 162 mg twice daily. Extended open-label treatment was associated with further improvements in itch and lesion activity. Adverse events were mostly mild to moderate (eg, nausea, dizziness, headache, and fatigue) and occurred during dose titration. Physiologic opioid withdrawal symptoms were limited and resolved within a few days of discontinuing the medication.11
Final Thoughts
In conclusion, PN remains one of the most challenging chronic dermatologic conditions to manage and is driven by a complex interplay of neuroimmune mechanisms and resistance to many conventional therapies. The approval of dupilumab and nemolizumab has marked a pivotal shift in the therapeutic landscape, offering hope to patients who previously had limited options5,8; however, the burden of PN remains substantial, and many patients continue to experience relentless itch, poor sleep, and reduced quality of life.1 Emerging therapies such as TRPM8 agonists, Janus kinase inhibitors, and opioid modulators represent promising additions to the treatment options, targeting novel pathways beyond traditional immunosuppression.9-11
- Williams KA, Huang AH, Belzberg M, et al. Prurigo nodularis: pathogenesis and management. J Am Acad Dermatol. 2020;83:1567-1575. doi:10.1016/j.jaad.2020.04.182
- Gründel S, Pereira MP, Storck M, et al. Analysis of 325 patients with chronic nodular prurigo: clinics, burden of disease and course of treatment. Acta Derm Venereol. 2020;100:adv00269. doi:10.2340/00015555-3571
- Liao V, Cornman HL, Ma E, et al. Prurigo nodularis: new insights into pathogenesis and novel therapeutics. Br J Dermatol. 2024;190:798-810. doi:10.1093/bjd/ljae052
- Elmariah SB, Tao L, Valdes-Rodriguez R, et al. Individual article: management of prurigo nodularis. J Drugs Dermatol. 2023;22:SF365502s15-SF365502s22. doi:10.36849/JDD.SF365502
- Yosipovitch G, Mollanazar N, Ständer S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29:1180-1190. doi:10.1038/s41591-023-02320-9
- Cao P, Xu W, Jiang S, et al. Dupilumab for the treatment of prurigo nodularis: a systematic review. Front Immunol. 2023;14:1092685. doi:10.3389/fimmu.2023.1092685
- Dagenet CB, Saadi C, Phillips MA, et al. Landscape of prurigo nodularis clinical trials. JAAD Rev. 2024;2:127-136. doi:10.1016/j.jdrv.2024.09.006
- Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 trial of nemolizumab in patients with prurigo nodularis. N Engl J Med. 2023;389:1579-1589. doi:10.1056/NEJMoa2301333
- Kwatra SG, Bordeaux ZA, Parthasarathy V, et al. Efficacy and safety of abrocitinib in prurigo nodularis and chronic pruritus of unknown origin: a nonrandomized controlled trial. JAMA Dermatol. 2024;160:717-724. doi:10.1001/jamadermatol.2024.1464
- Choi ME, Lee JH, Jung CJ, et al. A randomized, double-blinded, vehicle-controlled clinical trial of topical cryosim-1, a synthetic TRPM8 agonist, in prurigo nodularis. J Cosmet Dermatol. 2024;23:931-937. doi:10.1111/jocd.16079
- Weisshaar E, Szepietowski JC, Bernhard JD, et al. Efficacy and safety of oral nalbuphine extended release in prurigo nodularis: results of a phase 2 randomized controlled trial with an open-label extension phase. J Eur Acad Dermatol Venereol. 2022;36:453-461. doi:10.1111/jdv.17816
- Williams KA, Huang AH, Belzberg M, et al. Prurigo nodularis: pathogenesis and management. J Am Acad Dermatol. 2020;83:1567-1575. doi:10.1016/j.jaad.2020.04.182
- Gründel S, Pereira MP, Storck M, et al. Analysis of 325 patients with chronic nodular prurigo: clinics, burden of disease and course of treatment. Acta Derm Venereol. 2020;100:adv00269. doi:10.2340/00015555-3571
- Liao V, Cornman HL, Ma E, et al. Prurigo nodularis: new insights into pathogenesis and novel therapeutics. Br J Dermatol. 2024;190:798-810. doi:10.1093/bjd/ljae052
- Elmariah SB, Tao L, Valdes-Rodriguez R, et al. Individual article: management of prurigo nodularis. J Drugs Dermatol. 2023;22:SF365502s15-SF365502s22. doi:10.36849/JDD.SF365502
- Yosipovitch G, Mollanazar N, Ständer S, et al. Dupilumab in patients with prurigo nodularis: two randomized, double-blind, placebo-controlled phase 3 trials. Nat Med. 2023;29:1180-1190. doi:10.1038/s41591-023-02320-9
- Cao P, Xu W, Jiang S, et al. Dupilumab for the treatment of prurigo nodularis: a systematic review. Front Immunol. 2023;14:1092685. doi:10.3389/fimmu.2023.1092685
- Dagenet CB, Saadi C, Phillips MA, et al. Landscape of prurigo nodularis clinical trials. JAAD Rev. 2024;2:127-136. doi:10.1016/j.jdrv.2024.09.006
- Kwatra SG, Yosipovitch G, Legat FJ, et al. Phase 3 trial of nemolizumab in patients with prurigo nodularis. N Engl J Med. 2023;389:1579-1589. doi:10.1056/NEJMoa2301333
- Kwatra SG, Bordeaux ZA, Parthasarathy V, et al. Efficacy and safety of abrocitinib in prurigo nodularis and chronic pruritus of unknown origin: a nonrandomized controlled trial. JAMA Dermatol. 2024;160:717-724. doi:10.1001/jamadermatol.2024.1464
- Choi ME, Lee JH, Jung CJ, et al. A randomized, double-blinded, vehicle-controlled clinical trial of topical cryosim-1, a synthetic TRPM8 agonist, in prurigo nodularis. J Cosmet Dermatol. 2024;23:931-937. doi:10.1111/jocd.16079
- Weisshaar E, Szepietowski JC, Bernhard JD, et al. Efficacy and safety of oral nalbuphine extended release in prurigo nodularis: results of a phase 2 randomized controlled trial with an open-label extension phase. J Eur Acad Dermatol Venereol. 2022;36:453-461. doi:10.1111/jdv.17816
From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
From Refractory to Responsive: The Expanding Therapeutic Landscape of Prurigo Nodularis
Type VII Collagen Disorders Simplified
Type VII Collagen Disorders Simplified
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
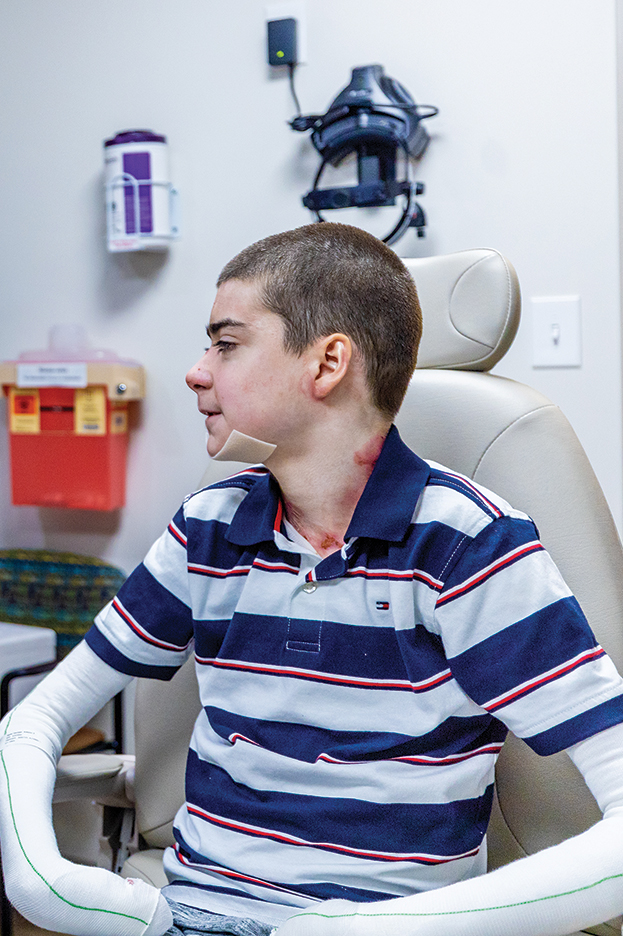
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
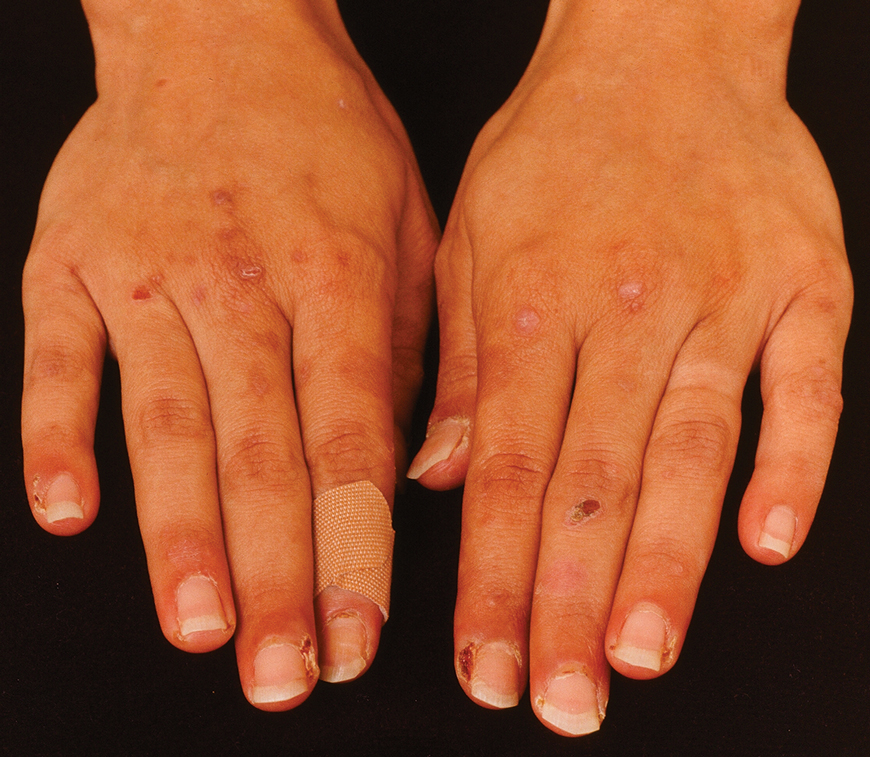
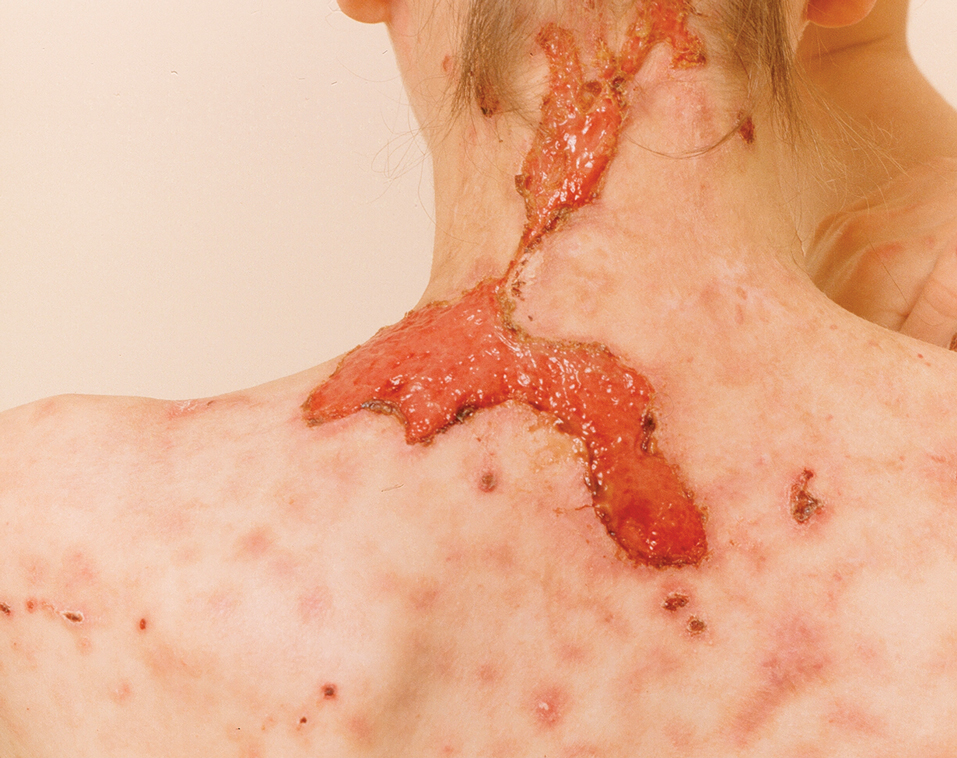
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
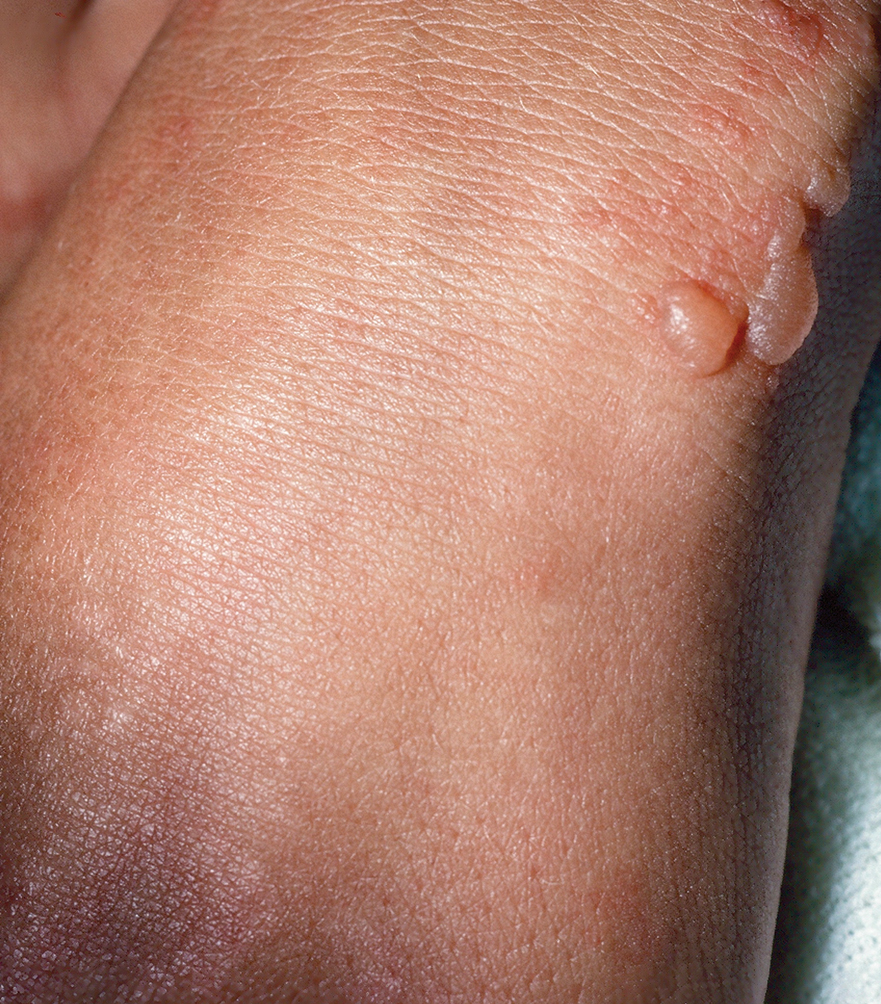
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
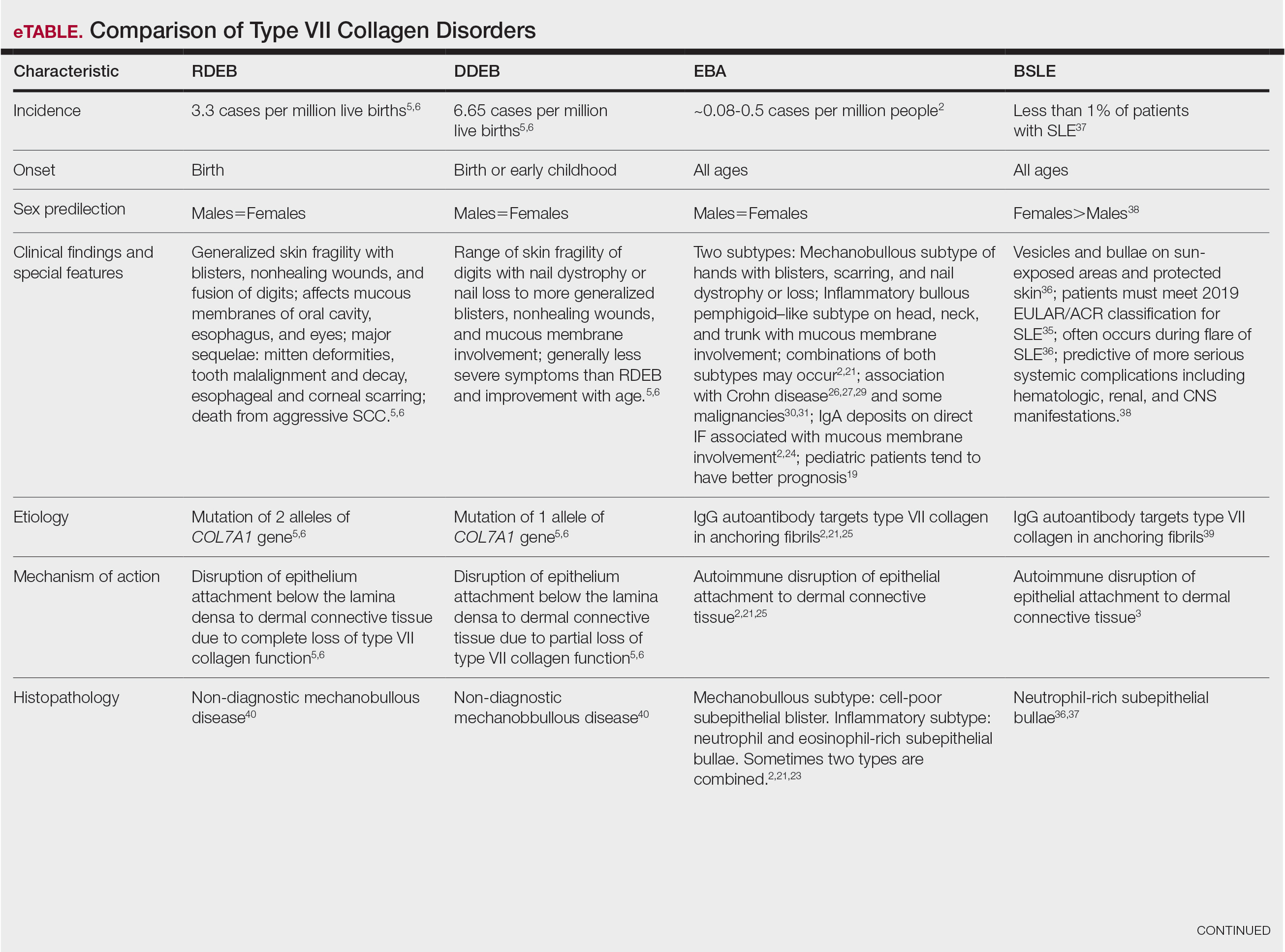
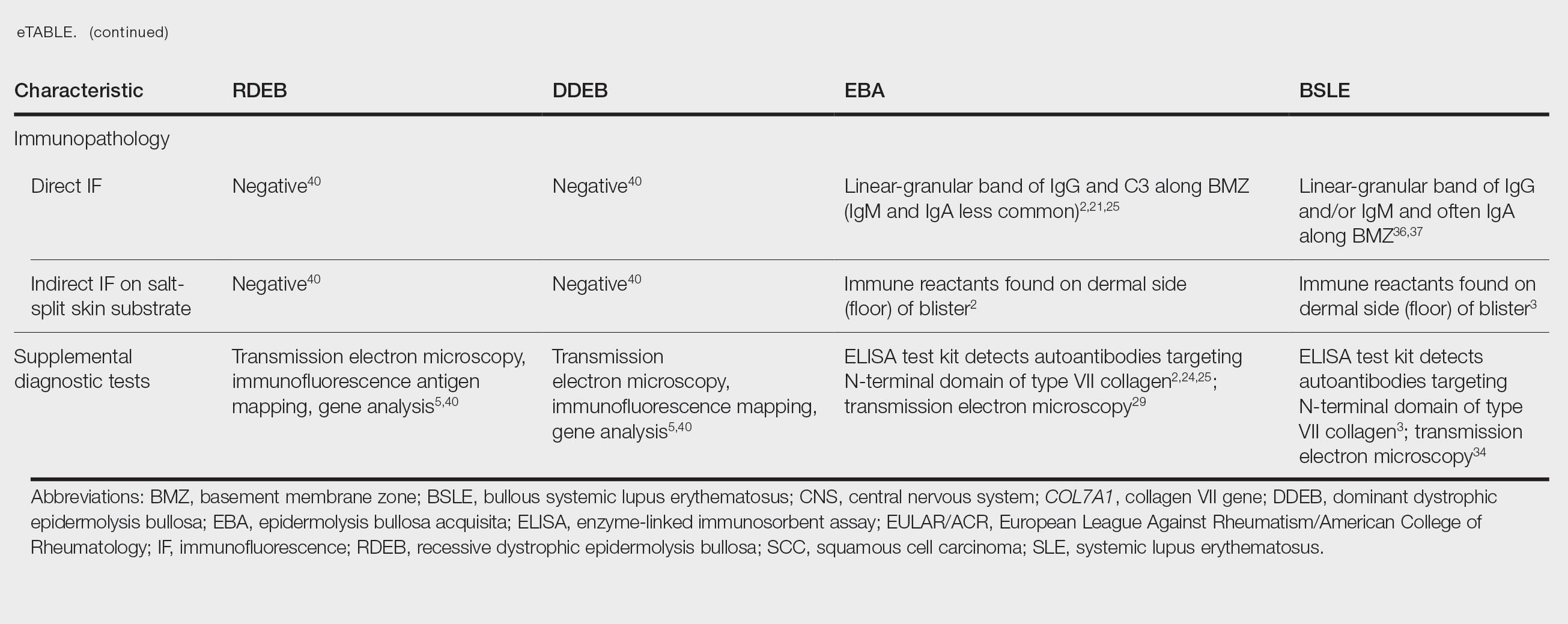
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
There are 3 uncommon types of mechanobullous skin diseases caused by relative reduction or complete loss of functional type VII collagen, which is the main component of anchoring fibrils in the lamina densa of the basement membrane zone (BMZ) of the skin and mucous membrane epithelium.1 The function of the anchoring fibrils is to maintain adherence of the basement membrane of the epithelium to the connective tissue of the papillary dermis and submucosa.1 The mechanism of action of the loss of type VII collagen function is via autoimmunity in epidermolysis bullosa acquisita (EBA)2 and
Epidermolysis Bullosa
Epidermolysis bullosa consists of a heterogeneous family of 4 major genetic mechanobullous diseases that affect the skin and mucous membranes with more than 30 subtypes.1 Dystrophic EB is caused by mutations in the COL7A1 gene, which encodes for the α-1 chain of collagen type VII. Classically, EB is divided into 4 main variants based on the location of the cleavage plane or split occurring in the epithelium, which in turn helps to predict the severity of the illness.
Epidermolysis bullosa may be inherited in an autosomal-dominant or autosomal-recessive fashion, or it may occur as a spontaneous mutation. All sexes and races are affected equally. Patients present at birth or in early childhood with fragile skin and mucous membranes that may develop blisters, erosions, and ulcerations after minor trauma.7 These lesions are marked by slow healing and scar formation and often are associated with itching and pain.
Dystrophic Epidermolysis Bullosa
Dystrophic EB accounts for approximately 25%6 of all EB cases in the United States and may be inherited as either a dominant or recessive trait. Hundreds of different pathogenic mutations have been discovered in the COL7A1 gene in the subtypes of DEB.4,8 Dominant DEB tends to cause milder disease because the patients retain one normal COL7A1 allele and produce some type VII collagen (Figure 1), whereas patients with recessive DEB lack type VII collagen completely.9 The cleavage plane is between the lamina densa and the superficial dermis or submucosa. Severity is variable and ranges from localization to the hands and feet to severe generalized blistering and painful ulcerations depending on which of the many possible gene mutations have been inherited. Sequelae include mitten deformities, malalignment and tooth decay, and the development of early aggressive squamous cell carcinomas, which may be fatal. The most severe cases of recessive DEB also may have internal organ involvement.
Epidermolysis Bullosa Simplex
Epidermolysis bullosa simplex is the most common variant, comprising approximately 70%of EB cases in the United States.6 Epidermolysis bullosa simplex usually is inherited as autosomal-dominant mutations in the keratin 5 or keratin 14 genes,10 not COL7A1. Skin blistering results from cleavage within the basal cell layer where the keratin genes are primarily expressed. Blisters tend to occur in acral areas such as hands and feet and may heal without scarring in the localized form of epidermolysis bullosa simplex (Figure 2).
Junctional Epidermolysis Bullosa and Kindler Syndrome
Junctional epidermolysis bullosa (JEB) and Kindler syndrome11 are the rarest of the autosomal-recessive EB variants.6 The plane of cleavage in JEB is through the lamina lucida of the BMZ. Junctional epidermolysis bullosa is caused by mutations of the genes that encode for the 3 chains of laminin 332 protein and type XVII collagen,5,12 not to be confused with type VII collagen. As with DEB, there is a wide range of severity in JEB, from localized effects on the eyes, oral cavity, and tooth enamel to widespread blistering and skin cancers. In JEB cases involving newborns, nonhealing wounds on the face, buttocks, fingers, and toes may be seen, with devastating complications in the oral cavity, esophagus, and larynx. Life expectancy is limited to 2 years or less.6 There have only been approximately 40013 cases of Kindler syndrome reported worldwide6 and there is clinical overlap with DEB. Patients also may demonstrate poikiloderma and photosensitivity. Kindler syndrome is caused by mutations in the FERMT1 gene which encodes for kindlin-1. This protein mediates anchorage between the actin cytoskeleton and the extracellular matrix.5,11 Loss of function produces variable cleavage planes around the dermoepidermal junction.
Clinical management of all EB variants, especially the severe recessive types, traditionally has been limited to the prevention of trauma to the skin and mucous membranes and supportive care, including dressing changes to erosions and ulcerations, antibiotic ointments as needed, and amelioration of pain and pruritus. Bone marrow and pluripotential stem cell transplants have been attempted.12 Complications of EB, such as deformities of the hands and feet caused by excessive scarring, esophageal strictures, poor dentition, and squamous cell carcinomas, must be addressed by a multidisciplinary team of specialists, including plastic surgery, gastroenterology, dentistry/oral surgery, ophthalmology, and dermatology/Mohs surgery.
Until recently, there were no medications approved by the US Food and Drug Administration (FDA) specifically indicated for EB. In 2023, topical gene therapy was approved by the FDA for both recessive and dominant forms of DEB. Normal COL7A1 sequences are delivered by an attenuated herpes simplex virus 1 vector (beremagene geperpavec) in a gel applied directly to the wounds of patients with DEB. In a clinical trial, matching wounds on 31 patients (62 wounds total) were treated with the active agent or placebo gel. After 6 months, complete wound closure was observed in 67% (21/31) of those treated with the active agent and 22% (7/31) of those treated with placebo (P=.002).14 In a single case report, a patient with recessive DEB and cicatrizing conjunctivitis (Figure 3) was given ophthalmic beremagene geperpavec after surgery and had improved visual acuity.15 A topical gel consisting of birch triterpenes to promote healing of partial-thickness wounds also was approved for patients with DEB and JEB by the FDA and the European Commission. In a study of 223 patients, 41% of those using active gel and 29% of those using placebo gel achieved the primary end point of percentage of target wounds that had first complete closure at 45 days.16
The most recent FDA approval for DEB involves transferring the functional COL7A1 gene to the patient’s skin cells, then expanding the gene-corrected cells into sheets of keratinocytes that can be surgically applied to the chronic wound sites. In a phase 3 trial of prademagene zamikeracel (pz-cel), 11 patients with 86 matched wounds were randomized to receive pz-cel (50%) or standard wound care (50%). After 24 weeks, 35 wounds treated with pz-cel were at least 50% healed compared to 7 control wounds.17 The results for healing and reduction of pain were statistically significant (P<.0001 and P<.0002, respectively).17 Recombinant collagen VII as replacement therapy also is under study to be given by intravenous infusion to increase tissue collagen VII where it is lacking. This treatment has shown early biologic and therapeutic effects.9,18 Larger long-term follow-up studies are necessary to confirm persistence of the gene-corrected skin cells, the functionality of the replacement collagen VII, and the potential risk for the development of autoantibodies to type VII collagen.
Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita is a rare autoimmune subepithelial bullous disease that primarily affects middle-aged adults but also has been reported in children.19 Epidermolysis bullosa acquisita is caused by circulating pathogenic IgG autoantibodies that target and bind to type VII collagen in the anchoring fibrils,20-22 thereby disrupting the attachment of the epithelium to its underlying connective tissue.
The 2 major clinical manifestations of EBA include a mechanobullous disease resembling inherited forms of DEB (Figure 4) and an inflammatory bullous pemphigoid (BP)–like disease,23 as well as a combination of both types of skin lesions (Figure 5). The skin and mucous membranes of the oral cavity, esophagus, eyes, and urogenital areas are affected in both types; scarring may cause functional disabilities. In the mechanobullous type of EBA, it is common for blisters and erosions to develop in trauma-prone areas such as the hands, feet, elbows, and knees. The blisters tend to heal with scarring and milia formation as might be seen in porphyria cutanea tarda or cicatricial pemphigoid, which are in the differential diagnosis. Dystrophy of the fingernails or complete nail loss may be observed, resembling DEB. In the BP-like presentation, tense blisters arise upon inflamed or urticarial skin and mucous membranes, which may then become generalized.
Histopathology in both forms of EBA demonstrates subepithelial separation as clefts or blisters. The mechanobullous type shows a sparse inflammatory infiltrate compared to large collections of neutrophils and eosinophils in the blister cavity and in the superficial dermis in the BP-like cases. The final diagnosis rests on the results of immunopathology testing.24 Direct immunofluorescence of perilesional skin and mucosa shows a linear-granular band of IgG and C3 and other conjugates along the BMZ. Deposits of IgA alone in EBA occur in only about 2.4% of cases and are observed more often when there is mucous membrane involvement.2 Indirect immunofluorescence of sera against salt-split skin substrates detects immunoreactants in the floor of the blister rather than in the roof, as would be seen in BP. Highly specific and sensitive enzyme-linked immunosorbent assay (ELISA) kits now are commercially available and can detect autoantibodies against the N-terminal domain of type VII collagen in more than 90% of cases of EBA.25
Inflammatory bowel disease (IBD), particularly Crohn disease (CD), precedes the onset of EBA in approximately 25% of cases.26,27 Ulcerative colitis is much less common. Type VII collagen is normally present in the basement membrane of intestinal epithelium. In a survey of patients with IBD, 68% of those with CD and 13% of those with ulcerative colitis had circulating anti–type VII collagen antibodies detected by ELISA without having symptoms of EBA.28 A case report of a patient with both well-proven EBA and CD highlighted the clinical difficulty of controlling EBA: treatment with prednisolone and sulfasalazine improved the CD but had little effect on the skin blisters.29 A variety of malignancies have been reported in association with EBA, including cancers of the uterine cervix,30 thyroid, and pancreas,31 lymphoma, and chronic lymphatic leukemia. Some of these cases have met the criteria for classification as paraneoplastic, whereas others may have been coincidental.
Treatment for chronic EBA generally has been limited.2,24 Putative antineutrophil drugs such as dapsone and colchicine combined with systemic corticosteroids may be useful in milder or juvenile cases, which tend to have a better prognosis than adult cases.19 In more severe EBA, systemic corticosteroids and/or immunosuppressive drugs such as azathioprine,23 cyclophosphamide,23 mycophenolate mofetil,31 methotrexate,23 cyclosporine,33 and infliximab23 have been used. More recently, rituximab infusion monotherapy33 and rituximab combined with intravenous immunoglobulin or
Bullous Systemic Lupus Erythematosus
Bullous systemic lupus erythematosus is a rare and specific autoimmune skin complication that mostly is seen in patients with an established diagnosis of systemic lupus erythematosus (SLE) who are experiencing a disease flare. Although more common in women, it has been reported in all sexes and races as well as in children. Vesicles and bullae may arise on sun-exposed (Figure 6) and sun-protected areas of skin.
Histopathology shows subepidermal separation with collections of neutrophils and nuclear fragments in the blister cavity. The differential diagnosis of BSLE includes EBA, BP, dermatitis herpetiformis, and linear IgA bullous dermatosis. Direct immunofluorescence testing shows linear-granular deposits of IgG and/or IgM and IgA along the BMZ.34 When utilizing the indirect immunofluorescence split-skin assay, the autoantibody to type VII collagen would be detected in the floor of the blister if the serum titer was sufficiently high.3 Proposed criteria for the diagnosis of BSLE have been published: 1) diagnosis of SLE now based on the 2019 European League Against Rheumatism/American College of Rheumatology classification35; 2) vesicles and bullae arising upon but not limited to sun-exposed skin; 3) histopathology featuring neutrophil-rich subepithelial bullae; 4) positive indirect immunofluorescence for circulating BMZ antibodies using separated human skin as substrate; 5) and direct immunofluorescence showing IgG and/or IgM and often IgA at the BMZ.36 Using ELISA to detect circulating antibodies against type VII collagen24 should now be added to the criteria. The new criteria for SLE34 do not include BSLE, perhaps because it occurs in less than 1% of patients with SLE.37
Further investigation by Gammon et al3 confirmed that the autoantibodies in BSLE are identical to those found in EBA (ie, directed against type VII collagen in the lamina densa). Bullous systemic lupus erythematosus is not considered to be the coexistence of EBA with SLE but rather a specific entity wherein type VII collagen autoantibodies are expressed in the autoimmune spectrum of SLE. It is especially important to make the diagnosis of BSLE because it is predictive of more serious systemic complications of SLE (eg, hematologic and renal disease is found in up to 90% of cases).38
The natural course of BSLE is variable. Treatments include systemic corticosteroids, dapsone, and immunosuppressive drugs such as azathioprine, methotrexate, mycophenolate mofetil, and cyclophosphamide, especially in cases with nephritis.37 There may be spontaneous resolution of the rash as the inflammatory activity of SLE subsides. Rituximab has been used effectively in several refractory cases of BSLE that failed to respond to all other conventional treatments.39
Conclusion
Anchoring fibrils are composed primarily of type VII collagen. Their role is to maintain the attachment of epithelium to the upper dermis and submucosa. The reduction or complete loss of type VII collagen caused by mutations of the COL7A1 gene results in dominant DEB or recessive DEB, respectively. Two distinct non-heritable immunobullous diseases, EBA and BSLE, are caused by autoantibodies that target type VII collagen. A comparison of the 4 type VII collagen disorders can be found in the eTable.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
- Bardhan A, Bruckner-Tuderman L, Chapple ILC, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6:78. doi:10.1038/s41572-020-0210-0
- Miyamoto D, Gordilho JO, Santi CG, et al. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409-423. doi:10.1016/j.abd.2021.09.010.
- Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. doi:10.1111/1523-1747.ep12272402.
- Yadav RS, Jaswal A, Shrestha S, et al. Dystrophic epidermolysis bullosa. J Nepal Med Assoc. 2018;56:879-882. doi:10.31729/jnma.3791
- Mariath LM, Santin JT, Schuler-Faccini L, et al. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551-569. doi:10.1016/j.abd.2020.05.001
- Understanding epidermolysis bullosa (EB). DEBRA website. Accessed August 17, 2025. https://www.debra.org/about-eb/understanding-epidermolysis-bullosa-eb
- Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: pediatric perspectives. Curr Pediatr Rev. 2022;18:182-190. doi:10.2174/1573396317666210525161252
- Dang N, Klingberg S, Marr P, et al. Review of collagen VII sequence variants found in Australasian patients with dystrophic epidermolysis bullosa reveals nine COL7A1 variants. J Dermatol Sci. 2007;46:169-178. doi:10.1016/j.jdermsci.2007.02.006
- Payne AS. Topical gene therapy for epidermolysis bullosa. N Engl J Med. 2022;387:2281-2284. doi:10.1056/NEJMe2213203
- Khani P, Ghazi F, Zekri A, et al. Keratins and epidermolysis bullosa simplex. J Cell Physiol. 2018;234:289-297. doi:10.1002/jcp.26898
- Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009;160:233-242. doi:10.1111/j.1365-2133.2008.08976.x
- Hou P-C, Wang H-T, Abhee S, et al. Investigational treatments for epidermolysis bullosa. Am J Clin Dermatol. 2021;22:801-817. doi:10.1007/s40257-021-00626-3
- Youseffian L, Vahidnezhad H, Uitto J. Kindler Syndrome. GeneReviews [Internet]. Updated January 6, 2022. Accessed August 21, 2025.
- Guide SV, Gonzalez ME, Bagci S, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387:2211-2219. doi:10.1056/NEJMoa2206663
- Vetencourt AT, Sayed-Ahmed I, Gomez J, et al. Ocular gene therapy in a patient with dystrophic epidermolysis bullosa. N Engl J Med. 2024;390:530-535. doi:10.1056/NEJMoa2301244
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188:12-21. doi:10.1093/bjd/ljac001
- Tang JY, Marinkovich MP, Wiss K, et al. Prademagene zamikeracel for recessive dystrophic epidermolysis bullosa wounds (VIITAL): a two-centre, randomized, open-label, intrapatient-controlled phase 3 trial. Lancet. 2025;406:163-173. doi:10.1016/S0140-6736(25)00778-0
- Gretzmeier C, Pin D, Kern JS, et al. Systemic collagen VII replacement therapy for advanced recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2022;142:1094-1102. doi:10.1016/j.jid.2021.09.008
- Hignett E, Sami N. Pediatric epidermolysis bullosa acquisita. A review. Pediatr Dermatol. 2021;38:1047-1050. doi:10.1111/pde.14722
- Chen M, Kim GH, Prakash L, et al. Autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91-101. doi:10.1007/s12016-007-0027-6.
- Kridin K, Kneiber D, Kowalski EH, et al. Epidermolysis bullosa acquisita: a comprehensive review. Autoimmun Rev. 2019;18:786-795. doi:10.1016/j.autrev.2019.06.007
- Hofmann SC, Weidinger A. Epidermolysis bullosa acquisita. Hautarzt. 2019;70:265-270. doi:10.1007/s00105-019-4387-7
- Ishi N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230. doi:10.1111/j.1346-8138.2009.00799.x
- Iwata H, Vorobyev A, Koga H, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Dis. 2018;13:153. doi:10.1186/s13023-018-0896-1
- Komorowski L, Muller R, Vorobyev A, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol. 2013;68:e89-95. doi:10.1016/j.jaad.2011.12.032
- Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10:364-390. doi:10.3390/jcm10020364
- Bezzio C, Della Corte C, Vernero M, et al. Inflammatory bowel disease and immune-mediated inflammatory diseases: looking at less frequent associations. Therap Adv Gastroenterol. 2022;15:17562848221115312. doi:10.1177/17562848221115312
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have antibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064. doi:10.1046/j.1523-1747.2002.01772.x
- Labeille B, Gineston JL, Denoeux JP, et al. Epidermolysis bullosa acquisita and Crohn’s disease. A case report with immunological and electron microscopic studies. Arch Intern Med. 1988;148:1457-1459.
- Etienne A, Ruffieux P, Didierjean L, et al. Epidermolysis bullosa acquisita and metastatic cancer of the uterine cervix. Ann Dermatol Venereol. 1998;125:321-323.
- Busch J-O, Sticherling M. Epidermolysis bullosa acquisita and neuroendocrine pancreatic cancer-Coincidence or patho-genetic relationship? J Dtsch Dermatol Ges. 2007;5:916-918. doi:10.111/j.1610-0387.2007.06338.x
- Bevans SL, Sami N. The use of rituximab in treatment of epidermolysis bullosa acquisita: three new cases and a review of the literature. Dermatol Ther. 2018;31:e12726. doi:10.1111/j.1610-0387.2007.06338.x
- Yang A, Kim M, Craig P, et al. A case report of the use of rituximab and the epidermolysis bullosa disease activity scoring index (EBDASI) in a patient with epidermolysis bullosa acquisita with extensive esophageal involvement. Arch Dermatovenerol Croat. 2018;26:325-328.
- Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. doi:10.1111/j.1365-2133.1993.tb00180.x
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheum Rep. 2020;22:18. doi:10.1007/s11926-020-00896-6
- Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. doi:10.1016/s0190-9622(88)70014-6
- Duan L, Chen L, Zhong S, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064. doi:10.1155/2015/167064
- Sprow G, Afarideh M, Dan J, et al. Bullous systemic lupus erythematosus in females. Int J Womens Dermatol. 2022;8:e034. doi:10.1097/JW9.0000000000000034
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524. doi:10.1007/s40257-014-0098-0
- Fine JD, Mellerio JE. Epidermolysis bullosa. In: Bolognia JL, Jorizzo JL, Schaffer JV (eds), Dermatology (ed 3), Elsevier Saunders; 2012: 501-513.
Type VII Collagen Disorders Simplified
Type VII Collagen Disorders Simplified
PRACTICE POINTS
- The full complement of type VII collagen is required for the normal assembly of anchoring fibrils, whose function is to adhere the basement membrane to the underlying connective tissue of skin and mucous membranes.
- In the heritable epidermolysis bullosa (EB) family of diseases, only dominant and recessive dystrophic epidermolysis bullosa are caused by partial or total loss of type VII collagen function.
- New treatments that have been approved for EB include topical gene therapy with COL7A1, topical birch triterpene gel, and skin cells from patients that are genetically corrected with a functional COL7A1 gene.
- Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus are rare distinct autoimmune subepithelial bullous diseases caused by IgG antibodies that target type VII collagen in the anchoring fibrils.

Sniffing Out Skin Disease: Odors in Dermatologic Conditions
Sniffing Out Skin Disease: Odors in Dermatologic Conditions
Humans possess the ability to recognize and distinguish a large range of odors that can be utilized in a wide range of applications. For example, sommeliers can classify more than 88 smells specific to the roughly 800 volatile organic compounds (VOCs) in wine. Thorough physical examination is essential in dermatology, and although sight and touch play the most important diagnostic roles, the sense of smell often is overlooked. Dermatologists are rigorously trained on the many visual aspects of skin disease and have a plethora of terms to describe these features while there is minimal characterization of odors. Research on odors and the role of olfaction in dermatologic practice is limited.1,2 We conducted a literature review of PubMed and Google Scholar for peer-reviewed articles discussing the role of odors in dermatologic diseases. Keywords included odor + dermatology, smell + dermatology, cutaneous odor, odor + diagnosis, and disease odor. Relevant studies were identified by screening their abstracts, followed by a full-text review. A total of 38 articles written in English that presented information on the odor associated with dermatologic diseases were included. Articles that were unrelated to the topic or written in a language other than English were excluded.
Common Skin Odors
The human body emits odorants—small VOCs—in various forms (skin/sweat, breath, urine, reproductive fluids). Human odor originates from the oxidation and bacterial metabolism of sweat and sebum on the skin.3 While many odors are physiologic and not cause for concern, others can signal underlying dermatologic pathologies.4 Odor-producing conditions can be categorized broadly into infectious diseases, disorders of keratinization and acantholysis, metabolic disorders, and organ dysfunction (Table). Infectious causes include bacterial infections and chronic wounds, which commonly emit characteristic offensive odors. For example, coryneform infections produce methanethiol, causing a cheesy odor of putrid fruit, and pseudomonal pyoderma infections emit a grape juice–like or mousy odor.
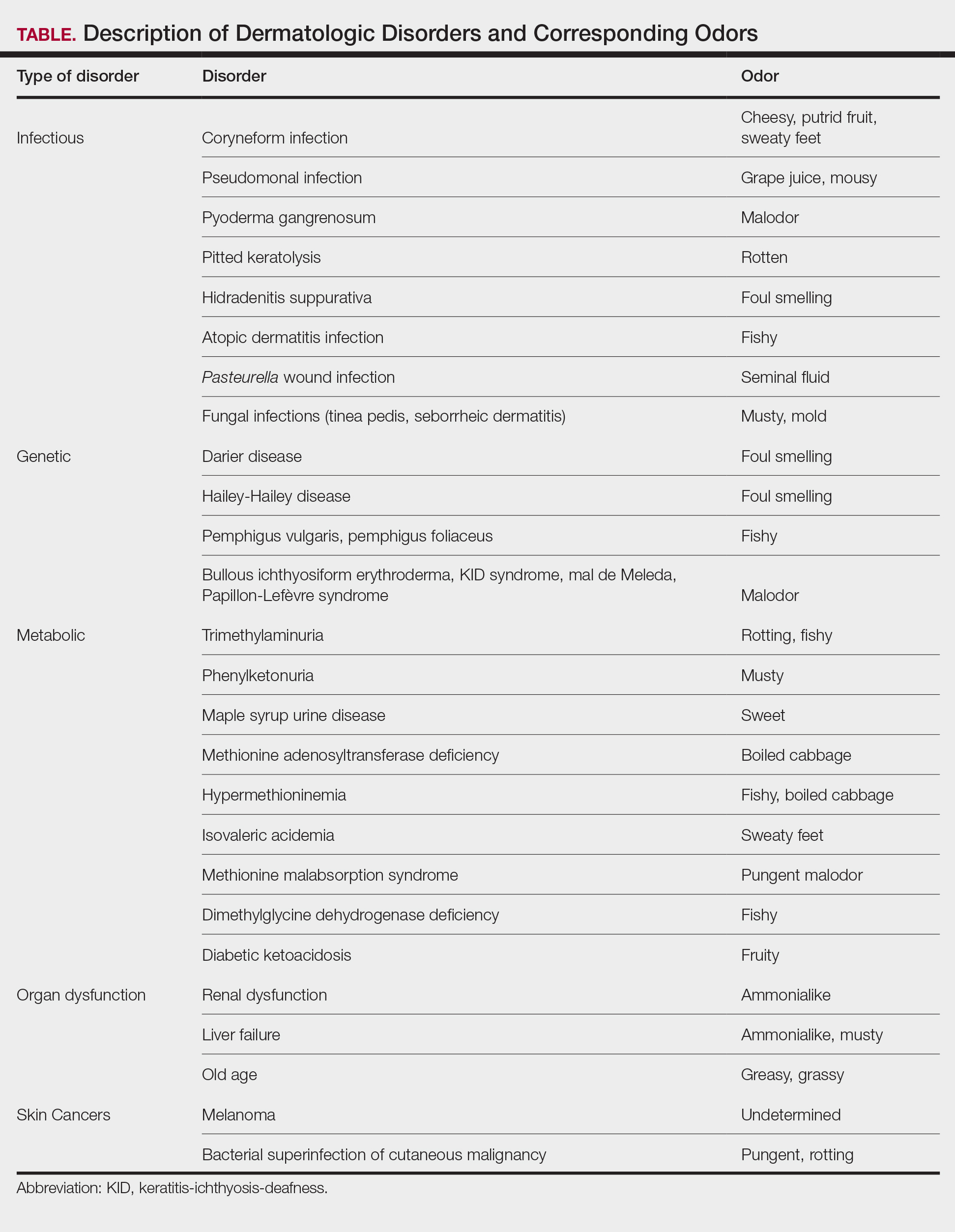
Bacterial and Fungal Infections
Bacterial and fungal infections often have distinct smells. Coryneform infections emit an odor of sweaty feet, pseudomonal infections emit a grape juice–like or mousy odor, and trichomycosis infections (caused by Corynebacterium tenuis) present with malodor.5 Pseudomonas can infect pyoderma gangrenosum lesions, producing a characteristic malodor.5 These smells can be clues for infectious etiology and guide further workup.
Pitted keratolysis, a malodorous pitted rash characterized by infection of the stratum corneum by Kytococcus sedentarius, Dermatophilus congolensis, or Corynebacterium species, is associated with a rotten smell. Its pungent odor, clinical location, and characteristic appearance often are enough to make a diagnosis. The amount of bacteria maintained in the stratum corneum is correlated with the extent of the lesion. Controlling excessive moisture in footwear, aluminum chloride, and topical microbial agents work together to eliminate the skin eruption.6
Hidradenitis suppurativa, a chronic inflammatory disease of apocrine gland–containing skin, can manifest with abscesses, draining sinuses, and nodules that produce a foul-smelling, purulent discharge. The disease can be debilitating, largely impacting patients’ quality of life, making early diagnosis and treatment critical.7,8 Therapy is dependent on disease severity and includes topical antibiotics, systemic therapies, and biologics.8
Patients with atopic dermatitis often experience bacterial superinfection with Staphylococcus aureus. A case report described a patient who developed a fishy odor in this setting that resolved with antibiotic treatment, implicating S aureus in the etiology of the smell.9
A seminal fluid odor has been reported in cases of Pasteurella wound infection. In such cases, Pasteurella multocida subspecies septica was identified in the wounds caused by a dog scratch and a cat bite. The seminal fluid–like odor was apparent hours after the inciting incident and resolved after treatment with antibiotics.10
Fungal infections frequently emit musty or moldy odors. Tinea pedis (athlete’s foot) is the most prevalent cutaneous fungal infection. The presence of tinea pedis is associated with an intense foul-smelling odor, itching, fissuring, scaling, or maceration of the interdigital regions. The rash and odor resolve with use of topical antifungal agents.11,12 Seborrheic dermatitis, a prevalent and chronic dermatosis, is characterized by yellow greasy scaling on an erythematous base. In severe cases, a greasy crust with an offensive odor can cover the entire scalp.13 The specific cause of this odor is unclear, but it is thought that sebum production and the immunological response to specific Malassezia yeast species may play a role.14
Genetic and Metabolic Disorders
An array of disorders of keratinization and acantholysis can manifest with distinctive smells that dermatologists frequently encounter. For example, Darier disease, characterized by keratotic papules progressing to crusted plaques, has a signature foul-smelling odor associated with cutaneous bacterial colonization.15 Similarly, Hailey-Hailey disease, an autosomal-dominant disorder with crusted erosions in skinfold areas, produces a distinct foul smell.16 Disorders such as pemphigus vulgaris and pemphigus foliaceus emit a peculiar fishy odor that can be helpful in making a diagnosis.17 Additionally, bullous ichthyosiform erythroderma, keratitis-ichthyosis-deafness syndrome, mal de Meleda, and Papillon-Lefèvre syndrome are all associated with malodor.5
Certain metabolic disorders can manifest and present initially with identifiable odors. Trimethylaminuria is a psychologically disabling disease known for its rotting fishy smell due to high amounts of trimethylamine appearing in affected individuals’ sweat, urine, and breath. Previously considered to be very rare, Messenger et al18 reported the disorder is likely underdiagnosed in those with idiopathic malodor production. Detection and treatment can greatly improve patient quality of life.
Phenylketonuria is an autosomal-recessive inborn error of phenylalanine metabolism that produces a musty body and urine odor as well as other neurologic and dermatologic symptoms.19,20 Patients can present with eczematous rashes, fair skin, and blue eyes. Phenylacetic acid produces the characteristic odor in the bodily fluids, and the disease is treated with a phenylalanine-free diet.21
Maple syrup urine disease is a disorder of the oxidative decarboxylation of valine, leucine, and isoleucine (branched-chain amino acids) characterized by urine that smells sweet, resembling maple syrup, in afflicted individuals. The odor also can be present in other bodily secretions, such as sweat. Patients present early in infancy with poor feeding and vomiting as well as neurologic symptoms, eventually leading to intellectual disability. These individuals must avoid the branched-chain amino acids in their diets.21
Other metabolic storage disorders linked with specific odors are methionine adenosyltransferase deficiency (boiled cabbage), hypermethioninemia (fishy, boiled cabbage), isovaleric acidemia (sweaty feet), methionine malabsorption syndrome (pungent malodor), and dimethylglycine dehydrogenase deficiency (fishy).5,21,22
In diabetic ketoacidosis, a life-threatening complication of diabetes, the excess of ketone bodies produced causes patients to have a distinct fruity breath and urine odor, as well as fatigue, polyuria, polydipsia, nausea, and vomiting.22 Although patients with type 1 diabetes typically comprise the cohort of patients presenting with diabetic ketoacidosis, patients with type 2 diabetes can exhibit cutaneous manifestations such as infection, xerosis, and inflammatory skin diseases.23,24
Organ Dysfunction
A peculiar body odor can be a sign of organ dysfunction. Renal dysfunction may present with both an odor and dermatologic manifestations. Patients with end-stage renal disease can have an ammonialike uremic breath odor as the result of excessive nitrogenous waste products and increased concentrations of urea in their saliva.4,22 These patients also can exhibit pruritus, xerosis, pigmentation changes, nail changes, other dermatoses, and rarely uremic frost with white urate crystals present on the skin.25,26
Liver failure has been associated with an ammonialike musty breath odor termed fetor hepaticus. Shimamoto et al27 reported notably higher levels of breath ammonia levels in patients with hepatic encephalopathy, indicating that excess ammonia is responsible for the odor. Fetor hepaticus has unique characteristics that can permit a diagnosis of liver disease, though it has been reported in cases in which a liver injury could not be identified.28
Aging patients typically have a distinctive smell. Haze et al29 analyzed the body odor of patients aged 26 to 75 years and discovered the compound 2-nonenal—an unsaturated aldehyde with a smell described as greasy and grassy—was found only in patients older than 40 years. The researchers’ analysis of skin-surface lipids also revealed that the presence of ω7 unsaturated fatty acids and lipid peroxides increased with age. They concluded that 2-nonenal is generated from the oxidative degradation of ω7 unsaturated fatty acids by lipid peroxides, suggesting that 2-nonenal may be a cause of the odor of old age.29
Cutaneous Malignancies
Research shows that the profiles of the body’s continuously released VOCs change in the presence of malignancy. Some studies suggest that melanoma may have a unique odor. Willis et al30 reported that after a 13-month training period, a dog was able to correctly identify melanoma and distinguish it from basal cell carcinoma, benign nevi, and healthy skin based on olfaction alone. Additional cases have been reported in which dogs have been able to identify melanoma based on smell, suggesting that canine olfactory detection of melanoma could possibly aid in the diagnosis of skin cancer, which warrants further investigation.31,32 There is limited evidence on the specific odors of other cutaneous malignancies, such as basal cell carcinoma and squamous cell carcinoma.
Bacterial superinfection of cutaneous malignancy can secrete pungent odors. An offensive rotting odor has been associated with necrotic malignant ulcers of the vagina. This malodor likely is a result of the formation of putrescine, cadaverine, short-chain fatty acids (isovaleric and butyric acids) and sulfur-containing compounds by bacteria.33 Recognition of similar smells may aid in management of these infections.
Diagnostic Techniques
Evaluating human skin odor is challenging, as the components of VOCs are complicated and typically found at trace levels. Studies indicate that gas chromatography–mass spectrometry is the most effective way to analyze human odor. This method separates, quantifies, and analyzes VOCs from samples containing odors.34 Gas chromatography–mass spectrometry, however, has limitations, as the time for analysis is lengthy, the equipment is large, and the process is expensive.3 Research supports the usefulness and validity of quantitative gas chromatography–olfactometry to detect odorants and evaluate odor activity of VOCs in various samples.35 With this technique, human assessors act in place of more conventional detectors, such as mass spectrometers. This method has been used to evaluate odorants in human urine with the goal of increasing understanding of metabolization and excretion processes.36 However, gas chromatography–olfactometry typically is used in the analysis of food and drink, and future research should be aimed at applying this method to medicine.
Zheng et al3 proposed a wearable electronic nose as a tool to identify human odor to emulate the odor recognition of a canine’s nose. They developed a sensor array based on the composites of carbon nanotubes and polymers able to examine and identify odors in the air. Study participants wore the electronic nose on the arm with the sensory array facing the armpits while they walked on a treadmill. Although many issues regarding odor measurement were not addressed in this study, the research suggests further studies are warranted to improve analysis of odor.3
Clinical Cases
Patient 1—Arseculeratne et al37 described a 41-year-old man who presented with a fishy odor that others had noticed since the age of 13 years but that the patient could not smell himself. Based on his presentation, he was worked up for trimethylaminuria and found to have elevated levels of urinary trimethylamine (TMA) with a raised TMA/TMA-oxidase ratio. These findings were consistent with a diagnosis of primary trimethylaminuria, and the patient was referred to a dietician for counseling on foods that contain low amounts of choline and lecithin. Initially his urinary TMA level fell but then rose again, indicating possible relaxation of his diet. He then took a 10-day course of metronidazole, which helped reduce some of the malodor. The authors reported that the most impactful therapy for the patient was being able to discuss the disorder with his friends and family members.37 This case highlighted the importance of confirming the diagnosis and early initiation of dietary and pharmacologic interventions in patients with trimethylaminuria. In patients reporting a persistent fishy body odor, trimethylaminuria should be on the differential.
Patient 2—In 1999, Schissel et al6 described a 20-year-old active-duty soldier who presented to the dermatology department with smelly trench foot and tinea pedis. The soldier reported having this malodorous pitted rash for more than 10 years. He also reported occasional interdigital burning and itching and noted no improvement despite using various topical antifungals. Physical examination revealed an “overpowering pungent odor” when the patient removed his shoes. He had many tender, white, and wet plaques with scalloped borders coalescing into shallow pits on the plantar surface of the feet and great toes. Potassium hydroxide preparation of the great toe plaques and interdigital web spaces were positive for fungal elements, and bacterial cultures isolated moderate coagulase-negative staphylococcal and Corynebacterium species. Additionally, fungal cultures identified Acremonium species. The patient was started on clotrimazole cream twice daily, clindamycin solution twice daily, and topical ammonium chloride nightly. Two weeks later, the patient reported resolution of symptoms, including the malodor.6 In pitted keratolysis, warm and wet environments within boots or shoes allow for the growth of bacteria and fungi. The extent of the lesions is related to the amount of bacteria within the stratum corneum. The diagnosis often is made based on odor, location, and appearance of the rash alone. The most common organisms implicated as causal agents in the condition are Kytococcus sedentarius, Dermatophilus congolensis, and species of Corynebacterium and Actinomyces. It is thought that these organisms release proteolytic enzymes that degrade the horny layer, releasing a mixture of thiols, thioesters, and sulfides, which cause the pungent odor. Familiarity with the characteristic odor aids in prompt diagnosis and treatment, which will ultimately heal the skin eruption.
Patient 3—Srivastava et al32 described a 43-year-old woman who presented with a nevus on the back since childhood. She noticed that it had changed and grown over the past few years and reported that her dog would often sniff the lesion and try to scratch and bite the lesion. This reaction from her dog led the patient to seek out evaluation from a dermatologist. The patient had no personal history of skin cancer, bad sunburns, tanning bed use, or use of immunosuppressants. She reported that her father had a history of basal cell carcinoma. Physical examination revealed a 1.2×1.5-cm brown patch with an ulcerated nodule located on the lower aspect of the lesion. The patient underwent a wide local excision and sentinel lymph node biopsy with pathology showing a 4-mm-thick melanoma with positive lymph nodes. She then underwent a right axillary lymphadenectomy and was diagnosed with stage IIIB malignant melanoma. Following the surgery, the patient’s dog would sniff the back and calmly rest his head in her lap. She has not had a recurrence and credits her dog for saving her life.32 Canine olfaction may play a role in detecting skin cancers, as evidenced by this case. Patients and dermatologists should pay attention to the behavior of dogs toward skin lesions. Harnessing this sense into a method to noninvasively screen for melanoma in humans should be further investigated.
Patient 4—Matthews et al38 described a 32-year-old woman who presented to an emergency eye clinic with a white “lump” on the left upper eyelid of 6 months’ duration. Physical examination revealed 3 nodular and cystic lesions oozing a thick yellow-white discharge. Cultures were taken, and the patient was started on chloramphenicol ointment once daily to the skin. At follow-up, the lesions had not changed, and the cultures were negative. The patient reported an intermittent malodorous discharge and noted multiple similar lesions on her body. Excisional biopsy demonstrated histologic findings including dyskeratosis, papillomatosis, and suprabasal acantholysis associated with focal underlying chronic inflammatory infiltrate. She was referred to a dermatologist and was diagnosed with Darier disease. She was started on clobetasone butyrate when necessary and adapalene nocte. Understanding the smell associated with Darier disease in conjunction with the cutaneous findings may aid in earlier diagnosis, improving outcomes for affected patients.38
Conclusion
The sense of smell may be an overlooked diagnostic tool that dermatologists innately possess. Odors detected when examining patients should be considered, as these odors may help guide a diagnosis. Early diagnosis and treatment are important in many dermatologic diseases, so it is imperative to consider all diagnostic clues. Although physician olfaction may aid in diagnosis, its utility remains challenging, as there is a lack of consensus and terminology regarding odor in disease. A limitation of training to identify disease-specific odors is the requirement of engaging in often unpleasant odors. Methods to objectively measure odor are expensive and still in the early stages of development. Further research and exploration of olfactory-based diagnostic techniques is warranted to potentially improve dermatologic diagnosis.
- Stitt WZ, Goldsmith A. Scratch and sniff: the dynamic duo. Arch Dermatol. 1995;131:997-999.
- Delahunty CM, Eyres G, Dufour JP. Gas chromatography-olfactometry. J Sep Sci. 2006;29:2107-2125.
- Zheng Y, Li H, Shen W, et al. Wearable electronic nose for human skin odor identification: a preliminary study. Sens Actuators A Phys. 2019;285:395-405.
- Mogilnicka I, Bogucki P, Ufnal M. Microbiota and malodor—etiology and management. Int J Mol Sci. 2020;21:2886. doi:10.3390/ijms21082886
- Ravindra K, Gandhi S, Sivuni A. Olfactory diagnosis in skin. Clin Derm Rev. 2018;2:38-40.
- Schissel DJ, Aydelotte J, Keller R. Road rash with a rotten odor. Mil Med. 1999;164:65-67.
- Buyukasik O, Osmanoglu CG, Polat Y, et al. A life-threatening multilocalized hidradenitis suppurativa case. MedGenMed. 2005;7:19.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Hon KLE, Leung AKC, Kong AYF, et al. Atopic dermatitis complicated by methicillin-resistant Staphylococcus aureus infection. J Natl Med Assoc. 2008;100:797-800.
- Arashima Y, Kumasaka K, Tutchiya T, et al. Two cases of pasteurellosis accompanied by exudate with semen-like odor from the wound. Article in Japanese. Kansenshogaku Zasshi. 1999;73:623-625.
- Goldstein AO, Smith KM, Ives TJ, et al. Mycotic infections. Effective management of conditions involving the skin, hair, and nails. Geriatrics. 2000;55:40-42, 45-47, 51-52.
- Kircik LH. Observational evaluation of sertaconazole nitrate cream 2% in the treatment of pruritus related to tinea pedis. Cutis. 2009;84:279-283.
- James WD, Elston DM, Treat JR, et al. Andrews’ Diseases of the Skin: Clinical Dermatology. Elsevier Health Sciences; 2019.
- Sameen K. A clinical study on the efficacy of homoeopathic medicines in the treatment of seborrhiec eczema. Int J Hom Sci. 2022;6:209-212.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
- Nanda KB, Saldanha CS, Jacintha M, et al. Hailey-Hailey disease responding to thalidomide. Indian J Dermatol. 2014;59:190-192.
- Kanwar AJ, Ghosh S, Dhar S, et al. Odor in pemphigus. Dermatology. 1992;185:215.
- Messenger J, Clark S, Massick S, et al. A review of trimethylaminuria: (fish odor syndrome). J Clin Aesthet Dermatol. 2013;6:45-48.
- Stone WL, Basit H, Los E. Phenylketonuria. StatPearls [Internet]. Updated August 8, 2023. Accessed August 12, 2025. https://www.ncbi.nlm.nih.gov/books/NBK535378/
- Williams RA, Mamotte CDS, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31-41.
- Cone TE Jr. Diagnosis and treatment: some diseases, syndromes, and conditions associated with an unusual odor. Pediatrics. 1968;41:993-995.
- Shirasu M, Touhara K. The scent of disease: volatile organic compounds of the human body related to disease and disorder. J Biochem. 2011;150:257-266.
- Ghimire P, Dhamoon AS. Ketoacidosis. StatPearls [Internet]. Updated August 8, 2023. Accessed August 12, 2025. https://www.ncbi.nlm.nih.gov/books/NBK534848/
- Duff M, Demidova O, Blackburn S, et al. Cutaneous manifestations of diabetes mellitus. Clin Diabetes. 2015;33:40-48.
- Raina S, Chauhan V, Sharma R, et al. Uremic frost. Indian Dermatol Online J. 2014;5(suppl 1):S58.
- Blaha T, Nigwekar S, Combs S, et al. Dermatologic manifestations in end stage renal disease. Hemodial Int. 2019;23:3-18.
- Shimamoto C, Hirata I, Katsu K. Breath and blood ammonia in liver cirrhosis. Hepatogastroenterology. 2000;47:443-445.
- Butt HR, Mason HL. Fetor hepaticus: its clinical significance and attempts at chemical isolation. Gastroenterology. 1954;26:829-845.
- Haze S, Gozu Y, Nakamura S, et al. 2-nonenal newly found in human body odor tends to increase with aging. J Invest Dermatol. 2001;116:520-524.
- Willis CM, Britton LE, Swindells MA, et al. Invasive melanoma in vivo can be distinguished from basal cell carcinoma, benign naevi and healthy skin by canine olfaction: a proof-of-principle study of differential volatile organic compound emission. Br J Dermatol. 2016;175:1020-1029.
- Campbell LF, Farmery L, George SMC, et al. Canine olfactory detection of malignant melanoma. BMJ Case Rep. 2013;2013:bcr2013008566. doi:10.1136/bcr-2013-008566
- Srivastava R, John JJ, Reilly C, et al. Sniffing out malignant melanoma: a case of canine olfactory detection. Cutis. 2019;104:E4-E6.
- Fleck CA. Fighting odor in wounds. Adv Skin Wound Care. 2006;19:242-244.
- Gallagher M, Wysocki CJ, Leyden JJ, et al. Analyses of volatile organic compounds from human skin. Br J Dermatol. 2008;159:780-791.
- Campo E, Ferreira V, Escudero A, et al. Quantitative gas chromatography–olfactometry and chemical quantitative study of the aroma of four Madeira wines. Anal Chim Acta. 2006;563:180-187.
- Wagenstaller M, Buettner A. Characterization of odorants in human urine using a combined chemo-analytical and human-sensory approach: a potential diagnostic strategy. Metabolomics. 2012;9:9-20.
- Arseculeratne G, Wong AKC, Goudie DR, et al. Trimethylaminuria (fish-odor syndrome): a case report. Arch Dermatol. 2007;143:81-84.
- Mathews D, Perera LP, Irion LD, et al. Darier disease: beware the cyst that smells. Ophthal Plast Reconstr Surg. 2010;26:206-207.
Humans possess the ability to recognize and distinguish a large range of odors that can be utilized in a wide range of applications. For example, sommeliers can classify more than 88 smells specific to the roughly 800 volatile organic compounds (VOCs) in wine. Thorough physical examination is essential in dermatology, and although sight and touch play the most important diagnostic roles, the sense of smell often is overlooked. Dermatologists are rigorously trained on the many visual aspects of skin disease and have a plethora of terms to describe these features while there is minimal characterization of odors. Research on odors and the role of olfaction in dermatologic practice is limited.1,2 We conducted a literature review of PubMed and Google Scholar for peer-reviewed articles discussing the role of odors in dermatologic diseases. Keywords included odor + dermatology, smell + dermatology, cutaneous odor, odor + diagnosis, and disease odor. Relevant studies were identified by screening their abstracts, followed by a full-text review. A total of 38 articles written in English that presented information on the odor associated with dermatologic diseases were included. Articles that were unrelated to the topic or written in a language other than English were excluded.
Common Skin Odors
The human body emits odorants—small VOCs—in various forms (skin/sweat, breath, urine, reproductive fluids). Human odor originates from the oxidation and bacterial metabolism of sweat and sebum on the skin.3 While many odors are physiologic and not cause for concern, others can signal underlying dermatologic pathologies.4 Odor-producing conditions can be categorized broadly into infectious diseases, disorders of keratinization and acantholysis, metabolic disorders, and organ dysfunction (Table). Infectious causes include bacterial infections and chronic wounds, which commonly emit characteristic offensive odors. For example, coryneform infections produce methanethiol, causing a cheesy odor of putrid fruit, and pseudomonal pyoderma infections emit a grape juice–like or mousy odor.
Bacterial and Fungal Infections
Bacterial and fungal infections often have distinct smells. Coryneform infections emit an odor of sweaty feet, pseudomonal infections emit a grape juice–like or mousy odor, and trichomycosis infections (caused by Corynebacterium tenuis) present with malodor.5 Pseudomonas can infect pyoderma gangrenosum lesions, producing a characteristic malodor.5 These smells can be clues for infectious etiology and guide further workup.
Pitted keratolysis, a malodorous pitted rash characterized by infection of the stratum corneum by Kytococcus sedentarius, Dermatophilus congolensis, or Corynebacterium species, is associated with a rotten smell. Its pungent odor, clinical location, and characteristic appearance often are enough to make a diagnosis. The amount of bacteria maintained in the stratum corneum is correlated with the extent of the lesion. Controlling excessive moisture in footwear, aluminum chloride, and topical microbial agents work together to eliminate the skin eruption.6
Hidradenitis suppurativa, a chronic inflammatory disease of apocrine gland–containing skin, can manifest with abscesses, draining sinuses, and nodules that produce a foul-smelling, purulent discharge. The disease can be debilitating, largely impacting patients’ quality of life, making early diagnosis and treatment critical.7,8 Therapy is dependent on disease severity and includes topical antibiotics, systemic therapies, and biologics.8
Patients with atopic dermatitis often experience bacterial superinfection with Staphylococcus aureus. A case report described a patient who developed a fishy odor in this setting that resolved with antibiotic treatment, implicating S aureus in the etiology of the smell.9
A seminal fluid odor has been reported in cases of Pasteurella wound infection. In such cases, Pasteurella multocida subspecies septica was identified in the wounds caused by a dog scratch and a cat bite. The seminal fluid–like odor was apparent hours after the inciting incident and resolved after treatment with antibiotics.10
Fungal infections frequently emit musty or moldy odors. Tinea pedis (athlete’s foot) is the most prevalent cutaneous fungal infection. The presence of tinea pedis is associated with an intense foul-smelling odor, itching, fissuring, scaling, or maceration of the interdigital regions. The rash and odor resolve with use of topical antifungal agents.11,12 Seborrheic dermatitis, a prevalent and chronic dermatosis, is characterized by yellow greasy scaling on an erythematous base. In severe cases, a greasy crust with an offensive odor can cover the entire scalp.13 The specific cause of this odor is unclear, but it is thought that sebum production and the immunological response to specific Malassezia yeast species may play a role.14
Genetic and Metabolic Disorders
An array of disorders of keratinization and acantholysis can manifest with distinctive smells that dermatologists frequently encounter. For example, Darier disease, characterized by keratotic papules progressing to crusted plaques, has a signature foul-smelling odor associated with cutaneous bacterial colonization.15 Similarly, Hailey-Hailey disease, an autosomal-dominant disorder with crusted erosions in skinfold areas, produces a distinct foul smell.16 Disorders such as pemphigus vulgaris and pemphigus foliaceus emit a peculiar fishy odor that can be helpful in making a diagnosis.17 Additionally, bullous ichthyosiform erythroderma, keratitis-ichthyosis-deafness syndrome, mal de Meleda, and Papillon-Lefèvre syndrome are all associated with malodor.5
Certain metabolic disorders can manifest and present initially with identifiable odors. Trimethylaminuria is a psychologically disabling disease known for its rotting fishy smell due to high amounts of trimethylamine appearing in affected individuals’ sweat, urine, and breath. Previously considered to be very rare, Messenger et al18 reported the disorder is likely underdiagnosed in those with idiopathic malodor production. Detection and treatment can greatly improve patient quality of life.
Phenylketonuria is an autosomal-recessive inborn error of phenylalanine metabolism that produces a musty body and urine odor as well as other neurologic and dermatologic symptoms.19,20 Patients can present with eczematous rashes, fair skin, and blue eyes. Phenylacetic acid produces the characteristic odor in the bodily fluids, and the disease is treated with a phenylalanine-free diet.21
Maple syrup urine disease is a disorder of the oxidative decarboxylation of valine, leucine, and isoleucine (branched-chain amino acids) characterized by urine that smells sweet, resembling maple syrup, in afflicted individuals. The odor also can be present in other bodily secretions, such as sweat. Patients present early in infancy with poor feeding and vomiting as well as neurologic symptoms, eventually leading to intellectual disability. These individuals must avoid the branched-chain amino acids in their diets.21
Other metabolic storage disorders linked with specific odors are methionine adenosyltransferase deficiency (boiled cabbage), hypermethioninemia (fishy, boiled cabbage), isovaleric acidemia (sweaty feet), methionine malabsorption syndrome (pungent malodor), and dimethylglycine dehydrogenase deficiency (fishy).5,21,22
In diabetic ketoacidosis, a life-threatening complication of diabetes, the excess of ketone bodies produced causes patients to have a distinct fruity breath and urine odor, as well as fatigue, polyuria, polydipsia, nausea, and vomiting.22 Although patients with type 1 diabetes typically comprise the cohort of patients presenting with diabetic ketoacidosis, patients with type 2 diabetes can exhibit cutaneous manifestations such as infection, xerosis, and inflammatory skin diseases.23,24
Organ Dysfunction
A peculiar body odor can be a sign of organ dysfunction. Renal dysfunction may present with both an odor and dermatologic manifestations. Patients with end-stage renal disease can have an ammonialike uremic breath odor as the result of excessive nitrogenous waste products and increased concentrations of urea in their saliva.4,22 These patients also can exhibit pruritus, xerosis, pigmentation changes, nail changes, other dermatoses, and rarely uremic frost with white urate crystals present on the skin.25,26
Liver failure has been associated with an ammonialike musty breath odor termed fetor hepaticus. Shimamoto et al27 reported notably higher levels of breath ammonia levels in patients with hepatic encephalopathy, indicating that excess ammonia is responsible for the odor. Fetor hepaticus has unique characteristics that can permit a diagnosis of liver disease, though it has been reported in cases in which a liver injury could not be identified.28
Aging patients typically have a distinctive smell. Haze et al29 analyzed the body odor of patients aged 26 to 75 years and discovered the compound 2-nonenal—an unsaturated aldehyde with a smell described as greasy and grassy—was found only in patients older than 40 years. The researchers’ analysis of skin-surface lipids also revealed that the presence of ω7 unsaturated fatty acids and lipid peroxides increased with age. They concluded that 2-nonenal is generated from the oxidative degradation of ω7 unsaturated fatty acids by lipid peroxides, suggesting that 2-nonenal may be a cause of the odor of old age.29
Cutaneous Malignancies
Research shows that the profiles of the body’s continuously released VOCs change in the presence of malignancy. Some studies suggest that melanoma may have a unique odor. Willis et al30 reported that after a 13-month training period, a dog was able to correctly identify melanoma and distinguish it from basal cell carcinoma, benign nevi, and healthy skin based on olfaction alone. Additional cases have been reported in which dogs have been able to identify melanoma based on smell, suggesting that canine olfactory detection of melanoma could possibly aid in the diagnosis of skin cancer, which warrants further investigation.31,32 There is limited evidence on the specific odors of other cutaneous malignancies, such as basal cell carcinoma and squamous cell carcinoma.
Bacterial superinfection of cutaneous malignancy can secrete pungent odors. An offensive rotting odor has been associated with necrotic malignant ulcers of the vagina. This malodor likely is a result of the formation of putrescine, cadaverine, short-chain fatty acids (isovaleric and butyric acids) and sulfur-containing compounds by bacteria.33 Recognition of similar smells may aid in management of these infections.
Diagnostic Techniques
Evaluating human skin odor is challenging, as the components of VOCs are complicated and typically found at trace levels. Studies indicate that gas chromatography–mass spectrometry is the most effective way to analyze human odor. This method separates, quantifies, and analyzes VOCs from samples containing odors.34 Gas chromatography–mass spectrometry, however, has limitations, as the time for analysis is lengthy, the equipment is large, and the process is expensive.3 Research supports the usefulness and validity of quantitative gas chromatography–olfactometry to detect odorants and evaluate odor activity of VOCs in various samples.35 With this technique, human assessors act in place of more conventional detectors, such as mass spectrometers. This method has been used to evaluate odorants in human urine with the goal of increasing understanding of metabolization and excretion processes.36 However, gas chromatography–olfactometry typically is used in the analysis of food and drink, and future research should be aimed at applying this method to medicine.
Zheng et al3 proposed a wearable electronic nose as a tool to identify human odor to emulate the odor recognition of a canine’s nose. They developed a sensor array based on the composites of carbon nanotubes and polymers able to examine and identify odors in the air. Study participants wore the electronic nose on the arm with the sensory array facing the armpits while they walked on a treadmill. Although many issues regarding odor measurement were not addressed in this study, the research suggests further studies are warranted to improve analysis of odor.3
Clinical Cases
Patient 1—Arseculeratne et al37 described a 41-year-old man who presented with a fishy odor that others had noticed since the age of 13 years but that the patient could not smell himself. Based on his presentation, he was worked up for trimethylaminuria and found to have elevated levels of urinary trimethylamine (TMA) with a raised TMA/TMA-oxidase ratio. These findings were consistent with a diagnosis of primary trimethylaminuria, and the patient was referred to a dietician for counseling on foods that contain low amounts of choline and lecithin. Initially his urinary TMA level fell but then rose again, indicating possible relaxation of his diet. He then took a 10-day course of metronidazole, which helped reduce some of the malodor. The authors reported that the most impactful therapy for the patient was being able to discuss the disorder with his friends and family members.37 This case highlighted the importance of confirming the diagnosis and early initiation of dietary and pharmacologic interventions in patients with trimethylaminuria. In patients reporting a persistent fishy body odor, trimethylaminuria should be on the differential.
Patient 2—In 1999, Schissel et al6 described a 20-year-old active-duty soldier who presented to the dermatology department with smelly trench foot and tinea pedis. The soldier reported having this malodorous pitted rash for more than 10 years. He also reported occasional interdigital burning and itching and noted no improvement despite using various topical antifungals. Physical examination revealed an “overpowering pungent odor” when the patient removed his shoes. He had many tender, white, and wet plaques with scalloped borders coalescing into shallow pits on the plantar surface of the feet and great toes. Potassium hydroxide preparation of the great toe plaques and interdigital web spaces were positive for fungal elements, and bacterial cultures isolated moderate coagulase-negative staphylococcal and Corynebacterium species. Additionally, fungal cultures identified Acremonium species. The patient was started on clotrimazole cream twice daily, clindamycin solution twice daily, and topical ammonium chloride nightly. Two weeks later, the patient reported resolution of symptoms, including the malodor.6 In pitted keratolysis, warm and wet environments within boots or shoes allow for the growth of bacteria and fungi. The extent of the lesions is related to the amount of bacteria within the stratum corneum. The diagnosis often is made based on odor, location, and appearance of the rash alone. The most common organisms implicated as causal agents in the condition are Kytococcus sedentarius, Dermatophilus congolensis, and species of Corynebacterium and Actinomyces. It is thought that these organisms release proteolytic enzymes that degrade the horny layer, releasing a mixture of thiols, thioesters, and sulfides, which cause the pungent odor. Familiarity with the characteristic odor aids in prompt diagnosis and treatment, which will ultimately heal the skin eruption.
Patient 3—Srivastava et al32 described a 43-year-old woman who presented with a nevus on the back since childhood. She noticed that it had changed and grown over the past few years and reported that her dog would often sniff the lesion and try to scratch and bite the lesion. This reaction from her dog led the patient to seek out evaluation from a dermatologist. The patient had no personal history of skin cancer, bad sunburns, tanning bed use, or use of immunosuppressants. She reported that her father had a history of basal cell carcinoma. Physical examination revealed a 1.2×1.5-cm brown patch with an ulcerated nodule located on the lower aspect of the lesion. The patient underwent a wide local excision and sentinel lymph node biopsy with pathology showing a 4-mm-thick melanoma with positive lymph nodes. She then underwent a right axillary lymphadenectomy and was diagnosed with stage IIIB malignant melanoma. Following the surgery, the patient’s dog would sniff the back and calmly rest his head in her lap. She has not had a recurrence and credits her dog for saving her life.32 Canine olfaction may play a role in detecting skin cancers, as evidenced by this case. Patients and dermatologists should pay attention to the behavior of dogs toward skin lesions. Harnessing this sense into a method to noninvasively screen for melanoma in humans should be further investigated.
Patient 4—Matthews et al38 described a 32-year-old woman who presented to an emergency eye clinic with a white “lump” on the left upper eyelid of 6 months’ duration. Physical examination revealed 3 nodular and cystic lesions oozing a thick yellow-white discharge. Cultures were taken, and the patient was started on chloramphenicol ointment once daily to the skin. At follow-up, the lesions had not changed, and the cultures were negative. The patient reported an intermittent malodorous discharge and noted multiple similar lesions on her body. Excisional biopsy demonstrated histologic findings including dyskeratosis, papillomatosis, and suprabasal acantholysis associated with focal underlying chronic inflammatory infiltrate. She was referred to a dermatologist and was diagnosed with Darier disease. She was started on clobetasone butyrate when necessary and adapalene nocte. Understanding the smell associated with Darier disease in conjunction with the cutaneous findings may aid in earlier diagnosis, improving outcomes for affected patients.38
Conclusion
The sense of smell may be an overlooked diagnostic tool that dermatologists innately possess. Odors detected when examining patients should be considered, as these odors may help guide a diagnosis. Early diagnosis and treatment are important in many dermatologic diseases, so it is imperative to consider all diagnostic clues. Although physician olfaction may aid in diagnosis, its utility remains challenging, as there is a lack of consensus and terminology regarding odor in disease. A limitation of training to identify disease-specific odors is the requirement of engaging in often unpleasant odors. Methods to objectively measure odor are expensive and still in the early stages of development. Further research and exploration of olfactory-based diagnostic techniques is warranted to potentially improve dermatologic diagnosis.
Humans possess the ability to recognize and distinguish a large range of odors that can be utilized in a wide range of applications. For example, sommeliers can classify more than 88 smells specific to the roughly 800 volatile organic compounds (VOCs) in wine. Thorough physical examination is essential in dermatology, and although sight and touch play the most important diagnostic roles, the sense of smell often is overlooked. Dermatologists are rigorously trained on the many visual aspects of skin disease and have a plethora of terms to describe these features while there is minimal characterization of odors. Research on odors and the role of olfaction in dermatologic practice is limited.1,2 We conducted a literature review of PubMed and Google Scholar for peer-reviewed articles discussing the role of odors in dermatologic diseases. Keywords included odor + dermatology, smell + dermatology, cutaneous odor, odor + diagnosis, and disease odor. Relevant studies were identified by screening their abstracts, followed by a full-text review. A total of 38 articles written in English that presented information on the odor associated with dermatologic diseases were included. Articles that were unrelated to the topic or written in a language other than English were excluded.
Common Skin Odors
The human body emits odorants—small VOCs—in various forms (skin/sweat, breath, urine, reproductive fluids). Human odor originates from the oxidation and bacterial metabolism of sweat and sebum on the skin.3 While many odors are physiologic and not cause for concern, others can signal underlying dermatologic pathologies.4 Odor-producing conditions can be categorized broadly into infectious diseases, disorders of keratinization and acantholysis, metabolic disorders, and organ dysfunction (Table). Infectious causes include bacterial infections and chronic wounds, which commonly emit characteristic offensive odors. For example, coryneform infections produce methanethiol, causing a cheesy odor of putrid fruit, and pseudomonal pyoderma infections emit a grape juice–like or mousy odor.
Bacterial and Fungal Infections
Bacterial and fungal infections often have distinct smells. Coryneform infections emit an odor of sweaty feet, pseudomonal infections emit a grape juice–like or mousy odor, and trichomycosis infections (caused by Corynebacterium tenuis) present with malodor.5 Pseudomonas can infect pyoderma gangrenosum lesions, producing a characteristic malodor.5 These smells can be clues for infectious etiology and guide further workup.
Pitted keratolysis, a malodorous pitted rash characterized by infection of the stratum corneum by Kytococcus sedentarius, Dermatophilus congolensis, or Corynebacterium species, is associated with a rotten smell. Its pungent odor, clinical location, and characteristic appearance often are enough to make a diagnosis. The amount of bacteria maintained in the stratum corneum is correlated with the extent of the lesion. Controlling excessive moisture in footwear, aluminum chloride, and topical microbial agents work together to eliminate the skin eruption.6
Hidradenitis suppurativa, a chronic inflammatory disease of apocrine gland–containing skin, can manifest with abscesses, draining sinuses, and nodules that produce a foul-smelling, purulent discharge. The disease can be debilitating, largely impacting patients’ quality of life, making early diagnosis and treatment critical.7,8 Therapy is dependent on disease severity and includes topical antibiotics, systemic therapies, and biologics.8
Patients with atopic dermatitis often experience bacterial superinfection with Staphylococcus aureus. A case report described a patient who developed a fishy odor in this setting that resolved with antibiotic treatment, implicating S aureus in the etiology of the smell.9
A seminal fluid odor has been reported in cases of Pasteurella wound infection. In such cases, Pasteurella multocida subspecies septica was identified in the wounds caused by a dog scratch and a cat bite. The seminal fluid–like odor was apparent hours after the inciting incident and resolved after treatment with antibiotics.10
Fungal infections frequently emit musty or moldy odors. Tinea pedis (athlete’s foot) is the most prevalent cutaneous fungal infection. The presence of tinea pedis is associated with an intense foul-smelling odor, itching, fissuring, scaling, or maceration of the interdigital regions. The rash and odor resolve with use of topical antifungal agents.11,12 Seborrheic dermatitis, a prevalent and chronic dermatosis, is characterized by yellow greasy scaling on an erythematous base. In severe cases, a greasy crust with an offensive odor can cover the entire scalp.13 The specific cause of this odor is unclear, but it is thought that sebum production and the immunological response to specific Malassezia yeast species may play a role.14
Genetic and Metabolic Disorders
An array of disorders of keratinization and acantholysis can manifest with distinctive smells that dermatologists frequently encounter. For example, Darier disease, characterized by keratotic papules progressing to crusted plaques, has a signature foul-smelling odor associated with cutaneous bacterial colonization.15 Similarly, Hailey-Hailey disease, an autosomal-dominant disorder with crusted erosions in skinfold areas, produces a distinct foul smell.16 Disorders such as pemphigus vulgaris and pemphigus foliaceus emit a peculiar fishy odor that can be helpful in making a diagnosis.17 Additionally, bullous ichthyosiform erythroderma, keratitis-ichthyosis-deafness syndrome, mal de Meleda, and Papillon-Lefèvre syndrome are all associated with malodor.5
Certain metabolic disorders can manifest and present initially with identifiable odors. Trimethylaminuria is a psychologically disabling disease known for its rotting fishy smell due to high amounts of trimethylamine appearing in affected individuals’ sweat, urine, and breath. Previously considered to be very rare, Messenger et al18 reported the disorder is likely underdiagnosed in those with idiopathic malodor production. Detection and treatment can greatly improve patient quality of life.
Phenylketonuria is an autosomal-recessive inborn error of phenylalanine metabolism that produces a musty body and urine odor as well as other neurologic and dermatologic symptoms.19,20 Patients can present with eczematous rashes, fair skin, and blue eyes. Phenylacetic acid produces the characteristic odor in the bodily fluids, and the disease is treated with a phenylalanine-free diet.21
Maple syrup urine disease is a disorder of the oxidative decarboxylation of valine, leucine, and isoleucine (branched-chain amino acids) characterized by urine that smells sweet, resembling maple syrup, in afflicted individuals. The odor also can be present in other bodily secretions, such as sweat. Patients present early in infancy with poor feeding and vomiting as well as neurologic symptoms, eventually leading to intellectual disability. These individuals must avoid the branched-chain amino acids in their diets.21
Other metabolic storage disorders linked with specific odors are methionine adenosyltransferase deficiency (boiled cabbage), hypermethioninemia (fishy, boiled cabbage), isovaleric acidemia (sweaty feet), methionine malabsorption syndrome (pungent malodor), and dimethylglycine dehydrogenase deficiency (fishy).5,21,22
In diabetic ketoacidosis, a life-threatening complication of diabetes, the excess of ketone bodies produced causes patients to have a distinct fruity breath and urine odor, as well as fatigue, polyuria, polydipsia, nausea, and vomiting.22 Although patients with type 1 diabetes typically comprise the cohort of patients presenting with diabetic ketoacidosis, patients with type 2 diabetes can exhibit cutaneous manifestations such as infection, xerosis, and inflammatory skin diseases.23,24
Organ Dysfunction
A peculiar body odor can be a sign of organ dysfunction. Renal dysfunction may present with both an odor and dermatologic manifestations. Patients with end-stage renal disease can have an ammonialike uremic breath odor as the result of excessive nitrogenous waste products and increased concentrations of urea in their saliva.4,22 These patients also can exhibit pruritus, xerosis, pigmentation changes, nail changes, other dermatoses, and rarely uremic frost with white urate crystals present on the skin.25,26
Liver failure has been associated with an ammonialike musty breath odor termed fetor hepaticus. Shimamoto et al27 reported notably higher levels of breath ammonia levels in patients with hepatic encephalopathy, indicating that excess ammonia is responsible for the odor. Fetor hepaticus has unique characteristics that can permit a diagnosis of liver disease, though it has been reported in cases in which a liver injury could not be identified.28
Aging patients typically have a distinctive smell. Haze et al29 analyzed the body odor of patients aged 26 to 75 years and discovered the compound 2-nonenal—an unsaturated aldehyde with a smell described as greasy and grassy—was found only in patients older than 40 years. The researchers’ analysis of skin-surface lipids also revealed that the presence of ω7 unsaturated fatty acids and lipid peroxides increased with age. They concluded that 2-nonenal is generated from the oxidative degradation of ω7 unsaturated fatty acids by lipid peroxides, suggesting that 2-nonenal may be a cause of the odor of old age.29
Cutaneous Malignancies
Research shows that the profiles of the body’s continuously released VOCs change in the presence of malignancy. Some studies suggest that melanoma may have a unique odor. Willis et al30 reported that after a 13-month training period, a dog was able to correctly identify melanoma and distinguish it from basal cell carcinoma, benign nevi, and healthy skin based on olfaction alone. Additional cases have been reported in which dogs have been able to identify melanoma based on smell, suggesting that canine olfactory detection of melanoma could possibly aid in the diagnosis of skin cancer, which warrants further investigation.31,32 There is limited evidence on the specific odors of other cutaneous malignancies, such as basal cell carcinoma and squamous cell carcinoma.
Bacterial superinfection of cutaneous malignancy can secrete pungent odors. An offensive rotting odor has been associated with necrotic malignant ulcers of the vagina. This malodor likely is a result of the formation of putrescine, cadaverine, short-chain fatty acids (isovaleric and butyric acids) and sulfur-containing compounds by bacteria.33 Recognition of similar smells may aid in management of these infections.
Diagnostic Techniques
Evaluating human skin odor is challenging, as the components of VOCs are complicated and typically found at trace levels. Studies indicate that gas chromatography–mass spectrometry is the most effective way to analyze human odor. This method separates, quantifies, and analyzes VOCs from samples containing odors.34 Gas chromatography–mass spectrometry, however, has limitations, as the time for analysis is lengthy, the equipment is large, and the process is expensive.3 Research supports the usefulness and validity of quantitative gas chromatography–olfactometry to detect odorants and evaluate odor activity of VOCs in various samples.35 With this technique, human assessors act in place of more conventional detectors, such as mass spectrometers. This method has been used to evaluate odorants in human urine with the goal of increasing understanding of metabolization and excretion processes.36 However, gas chromatography–olfactometry typically is used in the analysis of food and drink, and future research should be aimed at applying this method to medicine.
Zheng et al3 proposed a wearable electronic nose as a tool to identify human odor to emulate the odor recognition of a canine’s nose. They developed a sensor array based on the composites of carbon nanotubes and polymers able to examine and identify odors in the air. Study participants wore the electronic nose on the arm with the sensory array facing the armpits while they walked on a treadmill. Although many issues regarding odor measurement were not addressed in this study, the research suggests further studies are warranted to improve analysis of odor.3
Clinical Cases
Patient 1—Arseculeratne et al37 described a 41-year-old man who presented with a fishy odor that others had noticed since the age of 13 years but that the patient could not smell himself. Based on his presentation, he was worked up for trimethylaminuria and found to have elevated levels of urinary trimethylamine (TMA) with a raised TMA/TMA-oxidase ratio. These findings were consistent with a diagnosis of primary trimethylaminuria, and the patient was referred to a dietician for counseling on foods that contain low amounts of choline and lecithin. Initially his urinary TMA level fell but then rose again, indicating possible relaxation of his diet. He then took a 10-day course of metronidazole, which helped reduce some of the malodor. The authors reported that the most impactful therapy for the patient was being able to discuss the disorder with his friends and family members.37 This case highlighted the importance of confirming the diagnosis and early initiation of dietary and pharmacologic interventions in patients with trimethylaminuria. In patients reporting a persistent fishy body odor, trimethylaminuria should be on the differential.
Patient 2—In 1999, Schissel et al6 described a 20-year-old active-duty soldier who presented to the dermatology department with smelly trench foot and tinea pedis. The soldier reported having this malodorous pitted rash for more than 10 years. He also reported occasional interdigital burning and itching and noted no improvement despite using various topical antifungals. Physical examination revealed an “overpowering pungent odor” when the patient removed his shoes. He had many tender, white, and wet plaques with scalloped borders coalescing into shallow pits on the plantar surface of the feet and great toes. Potassium hydroxide preparation of the great toe plaques and interdigital web spaces were positive for fungal elements, and bacterial cultures isolated moderate coagulase-negative staphylococcal and Corynebacterium species. Additionally, fungal cultures identified Acremonium species. The patient was started on clotrimazole cream twice daily, clindamycin solution twice daily, and topical ammonium chloride nightly. Two weeks later, the patient reported resolution of symptoms, including the malodor.6 In pitted keratolysis, warm and wet environments within boots or shoes allow for the growth of bacteria and fungi. The extent of the lesions is related to the amount of bacteria within the stratum corneum. The diagnosis often is made based on odor, location, and appearance of the rash alone. The most common organisms implicated as causal agents in the condition are Kytococcus sedentarius, Dermatophilus congolensis, and species of Corynebacterium and Actinomyces. It is thought that these organisms release proteolytic enzymes that degrade the horny layer, releasing a mixture of thiols, thioesters, and sulfides, which cause the pungent odor. Familiarity with the characteristic odor aids in prompt diagnosis and treatment, which will ultimately heal the skin eruption.
Patient 3—Srivastava et al32 described a 43-year-old woman who presented with a nevus on the back since childhood. She noticed that it had changed and grown over the past few years and reported that her dog would often sniff the lesion and try to scratch and bite the lesion. This reaction from her dog led the patient to seek out evaluation from a dermatologist. The patient had no personal history of skin cancer, bad sunburns, tanning bed use, or use of immunosuppressants. She reported that her father had a history of basal cell carcinoma. Physical examination revealed a 1.2×1.5-cm brown patch with an ulcerated nodule located on the lower aspect of the lesion. The patient underwent a wide local excision and sentinel lymph node biopsy with pathology showing a 4-mm-thick melanoma with positive lymph nodes. She then underwent a right axillary lymphadenectomy and was diagnosed with stage IIIB malignant melanoma. Following the surgery, the patient’s dog would sniff the back and calmly rest his head in her lap. She has not had a recurrence and credits her dog for saving her life.32 Canine olfaction may play a role in detecting skin cancers, as evidenced by this case. Patients and dermatologists should pay attention to the behavior of dogs toward skin lesions. Harnessing this sense into a method to noninvasively screen for melanoma in humans should be further investigated.
Patient 4—Matthews et al38 described a 32-year-old woman who presented to an emergency eye clinic with a white “lump” on the left upper eyelid of 6 months’ duration. Physical examination revealed 3 nodular and cystic lesions oozing a thick yellow-white discharge. Cultures were taken, and the patient was started on chloramphenicol ointment once daily to the skin. At follow-up, the lesions had not changed, and the cultures were negative. The patient reported an intermittent malodorous discharge and noted multiple similar lesions on her body. Excisional biopsy demonstrated histologic findings including dyskeratosis, papillomatosis, and suprabasal acantholysis associated with focal underlying chronic inflammatory infiltrate. She was referred to a dermatologist and was diagnosed with Darier disease. She was started on clobetasone butyrate when necessary and adapalene nocte. Understanding the smell associated with Darier disease in conjunction with the cutaneous findings may aid in earlier diagnosis, improving outcomes for affected patients.38
Conclusion
The sense of smell may be an overlooked diagnostic tool that dermatologists innately possess. Odors detected when examining patients should be considered, as these odors may help guide a diagnosis. Early diagnosis and treatment are important in many dermatologic diseases, so it is imperative to consider all diagnostic clues. Although physician olfaction may aid in diagnosis, its utility remains challenging, as there is a lack of consensus and terminology regarding odor in disease. A limitation of training to identify disease-specific odors is the requirement of engaging in often unpleasant odors. Methods to objectively measure odor are expensive and still in the early stages of development. Further research and exploration of olfactory-based diagnostic techniques is warranted to potentially improve dermatologic diagnosis.
- Stitt WZ, Goldsmith A. Scratch and sniff: the dynamic duo. Arch Dermatol. 1995;131:997-999.
- Delahunty CM, Eyres G, Dufour JP. Gas chromatography-olfactometry. J Sep Sci. 2006;29:2107-2125.
- Zheng Y, Li H, Shen W, et al. Wearable electronic nose for human skin odor identification: a preliminary study. Sens Actuators A Phys. 2019;285:395-405.
- Mogilnicka I, Bogucki P, Ufnal M. Microbiota and malodor—etiology and management. Int J Mol Sci. 2020;21:2886. doi:10.3390/ijms21082886
- Ravindra K, Gandhi S, Sivuni A. Olfactory diagnosis in skin. Clin Derm Rev. 2018;2:38-40.
- Schissel DJ, Aydelotte J, Keller R. Road rash with a rotten odor. Mil Med. 1999;164:65-67.
- Buyukasik O, Osmanoglu CG, Polat Y, et al. A life-threatening multilocalized hidradenitis suppurativa case. MedGenMed. 2005;7:19.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Hon KLE, Leung AKC, Kong AYF, et al. Atopic dermatitis complicated by methicillin-resistant Staphylococcus aureus infection. J Natl Med Assoc. 2008;100:797-800.
- Arashima Y, Kumasaka K, Tutchiya T, et al. Two cases of pasteurellosis accompanied by exudate with semen-like odor from the wound. Article in Japanese. Kansenshogaku Zasshi. 1999;73:623-625.
- Goldstein AO, Smith KM, Ives TJ, et al. Mycotic infections. Effective management of conditions involving the skin, hair, and nails. Geriatrics. 2000;55:40-42, 45-47, 51-52.
- Kircik LH. Observational evaluation of sertaconazole nitrate cream 2% in the treatment of pruritus related to tinea pedis. Cutis. 2009;84:279-283.
- James WD, Elston DM, Treat JR, et al. Andrews’ Diseases of the Skin: Clinical Dermatology. Elsevier Health Sciences; 2019.
- Sameen K. A clinical study on the efficacy of homoeopathic medicines in the treatment of seborrhiec eczema. Int J Hom Sci. 2022;6:209-212.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
- Nanda KB, Saldanha CS, Jacintha M, et al. Hailey-Hailey disease responding to thalidomide. Indian J Dermatol. 2014;59:190-192.
- Kanwar AJ, Ghosh S, Dhar S, et al. Odor in pemphigus. Dermatology. 1992;185:215.
- Messenger J, Clark S, Massick S, et al. A review of trimethylaminuria: (fish odor syndrome). J Clin Aesthet Dermatol. 2013;6:45-48.
- Stone WL, Basit H, Los E. Phenylketonuria. StatPearls [Internet]. Updated August 8, 2023. Accessed August 12, 2025. https://www.ncbi.nlm.nih.gov/books/NBK535378/
- Williams RA, Mamotte CDS, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31-41.
- Cone TE Jr. Diagnosis and treatment: some diseases, syndromes, and conditions associated with an unusual odor. Pediatrics. 1968;41:993-995.
- Shirasu M, Touhara K. The scent of disease: volatile organic compounds of the human body related to disease and disorder. J Biochem. 2011;150:257-266.
- Ghimire P, Dhamoon AS. Ketoacidosis. StatPearls [Internet]. Updated August 8, 2023. Accessed August 12, 2025. https://www.ncbi.nlm.nih.gov/books/NBK534848/
- Duff M, Demidova O, Blackburn S, et al. Cutaneous manifestations of diabetes mellitus. Clin Diabetes. 2015;33:40-48.
- Raina S, Chauhan V, Sharma R, et al. Uremic frost. Indian Dermatol Online J. 2014;5(suppl 1):S58.
- Blaha T, Nigwekar S, Combs S, et al. Dermatologic manifestations in end stage renal disease. Hemodial Int. 2019;23:3-18.
- Shimamoto C, Hirata I, Katsu K. Breath and blood ammonia in liver cirrhosis. Hepatogastroenterology. 2000;47:443-445.
- Butt HR, Mason HL. Fetor hepaticus: its clinical significance and attempts at chemical isolation. Gastroenterology. 1954;26:829-845.
- Haze S, Gozu Y, Nakamura S, et al. 2-nonenal newly found in human body odor tends to increase with aging. J Invest Dermatol. 2001;116:520-524.
- Willis CM, Britton LE, Swindells MA, et al. Invasive melanoma in vivo can be distinguished from basal cell carcinoma, benign naevi and healthy skin by canine olfaction: a proof-of-principle study of differential volatile organic compound emission. Br J Dermatol. 2016;175:1020-1029.
- Campbell LF, Farmery L, George SMC, et al. Canine olfactory detection of malignant melanoma. BMJ Case Rep. 2013;2013:bcr2013008566. doi:10.1136/bcr-2013-008566
- Srivastava R, John JJ, Reilly C, et al. Sniffing out malignant melanoma: a case of canine olfactory detection. Cutis. 2019;104:E4-E6.
- Fleck CA. Fighting odor in wounds. Adv Skin Wound Care. 2006;19:242-244.
- Gallagher M, Wysocki CJ, Leyden JJ, et al. Analyses of volatile organic compounds from human skin. Br J Dermatol. 2008;159:780-791.
- Campo E, Ferreira V, Escudero A, et al. Quantitative gas chromatography–olfactometry and chemical quantitative study of the aroma of four Madeira wines. Anal Chim Acta. 2006;563:180-187.
- Wagenstaller M, Buettner A. Characterization of odorants in human urine using a combined chemo-analytical and human-sensory approach: a potential diagnostic strategy. Metabolomics. 2012;9:9-20.
- Arseculeratne G, Wong AKC, Goudie DR, et al. Trimethylaminuria (fish-odor syndrome): a case report. Arch Dermatol. 2007;143:81-84.
- Mathews D, Perera LP, Irion LD, et al. Darier disease: beware the cyst that smells. Ophthal Plast Reconstr Surg. 2010;26:206-207.
- Stitt WZ, Goldsmith A. Scratch and sniff: the dynamic duo. Arch Dermatol. 1995;131:997-999.
- Delahunty CM, Eyres G, Dufour JP. Gas chromatography-olfactometry. J Sep Sci. 2006;29:2107-2125.
- Zheng Y, Li H, Shen W, et al. Wearable electronic nose for human skin odor identification: a preliminary study. Sens Actuators A Phys. 2019;285:395-405.
- Mogilnicka I, Bogucki P, Ufnal M. Microbiota and malodor—etiology and management. Int J Mol Sci. 2020;21:2886. doi:10.3390/ijms21082886
- Ravindra K, Gandhi S, Sivuni A. Olfactory diagnosis in skin. Clin Derm Rev. 2018;2:38-40.
- Schissel DJ, Aydelotte J, Keller R. Road rash with a rotten odor. Mil Med. 1999;164:65-67.
- Buyukasik O, Osmanoglu CG, Polat Y, et al. A life-threatening multilocalized hidradenitis suppurativa case. MedGenMed. 2005;7:19.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Hon KLE, Leung AKC, Kong AYF, et al. Atopic dermatitis complicated by methicillin-resistant Staphylococcus aureus infection. J Natl Med Assoc. 2008;100:797-800.
- Arashima Y, Kumasaka K, Tutchiya T, et al. Two cases of pasteurellosis accompanied by exudate with semen-like odor from the wound. Article in Japanese. Kansenshogaku Zasshi. 1999;73:623-625.
- Goldstein AO, Smith KM, Ives TJ, et al. Mycotic infections. Effective management of conditions involving the skin, hair, and nails. Geriatrics. 2000;55:40-42, 45-47, 51-52.
- Kircik LH. Observational evaluation of sertaconazole nitrate cream 2% in the treatment of pruritus related to tinea pedis. Cutis. 2009;84:279-283.
- James WD, Elston DM, Treat JR, et al. Andrews’ Diseases of the Skin: Clinical Dermatology. Elsevier Health Sciences; 2019.
- Sameen K. A clinical study on the efficacy of homoeopathic medicines in the treatment of seborrhiec eczema. Int J Hom Sci. 2022;6:209-212.
- Burge S. Management of Darier’s disease. Clin Exp Dermatol. 1999;24:53-56.
- Nanda KB, Saldanha CS, Jacintha M, et al. Hailey-Hailey disease responding to thalidomide. Indian J Dermatol. 2014;59:190-192.
- Kanwar AJ, Ghosh S, Dhar S, et al. Odor in pemphigus. Dermatology. 1992;185:215.
- Messenger J, Clark S, Massick S, et al. A review of trimethylaminuria: (fish odor syndrome). J Clin Aesthet Dermatol. 2013;6:45-48.
- Stone WL, Basit H, Los E. Phenylketonuria. StatPearls [Internet]. Updated August 8, 2023. Accessed August 12, 2025. https://www.ncbi.nlm.nih.gov/books/NBK535378/
- Williams RA, Mamotte CDS, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29:31-41.
- Cone TE Jr. Diagnosis and treatment: some diseases, syndromes, and conditions associated with an unusual odor. Pediatrics. 1968;41:993-995.
- Shirasu M, Touhara K. The scent of disease: volatile organic compounds of the human body related to disease and disorder. J Biochem. 2011;150:257-266.
- Ghimire P, Dhamoon AS. Ketoacidosis. StatPearls [Internet]. Updated August 8, 2023. Accessed August 12, 2025. https://www.ncbi.nlm.nih.gov/books/NBK534848/
- Duff M, Demidova O, Blackburn S, et al. Cutaneous manifestations of diabetes mellitus. Clin Diabetes. 2015;33:40-48.
- Raina S, Chauhan V, Sharma R, et al. Uremic frost. Indian Dermatol Online J. 2014;5(suppl 1):S58.
- Blaha T, Nigwekar S, Combs S, et al. Dermatologic manifestations in end stage renal disease. Hemodial Int. 2019;23:3-18.
- Shimamoto C, Hirata I, Katsu K. Breath and blood ammonia in liver cirrhosis. Hepatogastroenterology. 2000;47:443-445.
- Butt HR, Mason HL. Fetor hepaticus: its clinical significance and attempts at chemical isolation. Gastroenterology. 1954;26:829-845.
- Haze S, Gozu Y, Nakamura S, et al. 2-nonenal newly found in human body odor tends to increase with aging. J Invest Dermatol. 2001;116:520-524.
- Willis CM, Britton LE, Swindells MA, et al. Invasive melanoma in vivo can be distinguished from basal cell carcinoma, benign naevi and healthy skin by canine olfaction: a proof-of-principle study of differential volatile organic compound emission. Br J Dermatol. 2016;175:1020-1029.
- Campbell LF, Farmery L, George SMC, et al. Canine olfactory detection of malignant melanoma. BMJ Case Rep. 2013;2013:bcr2013008566. doi:10.1136/bcr-2013-008566
- Srivastava R, John JJ, Reilly C, et al. Sniffing out malignant melanoma: a case of canine olfactory detection. Cutis. 2019;104:E4-E6.
- Fleck CA. Fighting odor in wounds. Adv Skin Wound Care. 2006;19:242-244.
- Gallagher M, Wysocki CJ, Leyden JJ, et al. Analyses of volatile organic compounds from human skin. Br J Dermatol. 2008;159:780-791.
- Campo E, Ferreira V, Escudero A, et al. Quantitative gas chromatography–olfactometry and chemical quantitative study of the aroma of four Madeira wines. Anal Chim Acta. 2006;563:180-187.
- Wagenstaller M, Buettner A. Characterization of odorants in human urine using a combined chemo-analytical and human-sensory approach: a potential diagnostic strategy. Metabolomics. 2012;9:9-20.
- Arseculeratne G, Wong AKC, Goudie DR, et al. Trimethylaminuria (fish-odor syndrome): a case report. Arch Dermatol. 2007;143:81-84.
- Mathews D, Perera LP, Irion LD, et al. Darier disease: beware the cyst that smells. Ophthal Plast Reconstr Surg. 2010;26:206-207.
Sniffing Out Skin Disease: Odors in Dermatologic Conditions
Sniffing Out Skin Disease: Odors in Dermatologic Conditions
PRACTICE POINTS
- Olfaction may be underutilized in making dermatologic diagnoses. Clinicians should include smell in their physical examination, as characteristic odors are associated with infectious disorders, disorders of keratinization and acantholysis, and metabolic disorders.
- Recognizing distinctive smells can help narrow the differential diagnosis and prompt targeted testing in dermatology.
- Canines and electronic noses have demonstrated the potential to detect certain malignancies, including melanoma, based on unique volatile organic compound profiles.
Demographic and Clinical Factors Associated With PD-L1 Testing of Veterans With Advanced Non-Small Cell Lung Cancer
Background
Programmed death-ligand 1 (PD-L1) checkpoint inhibitors revolutionized the treatment of advanced non-small cell lung cancer (aNSCLC) by improving overall survival compared to chemotherapy. PD-L1 biomarker testing is paramount for informing treatment decisions in aNSCLC. Real-world data describing patterns of PD-L1 testing within the Veteran Health Administration (VHA) are limited. This retrospective study seeks to evaluate demographic and clinical factors associated with PD-L1 testing in VHA.
Methods
Veterans diagnosed with aNSCLC from 2019-2022 were identified using VHA’s Corporate Data Warehouse. Wilcoxon Rank Sum and Chi- Square tests measured association between receipt of PD-L1 testing and patient demographic and clinical characteristics at aNSCLC diagnosis. Logistic regression assessed predictors of PD-L1 testing, and subgroup analyses were performed for significant interactions.
Results
Our study included 4575 patients with aNSCLC; 57.0% received PD-L1 testing. The likelihood of PD-L1 testing increased among patients diagnosed with aNSCLC after 2019 vs during 2019 (OR≥1.118, p≤0.035) and in Black vs White patients (OR=1.227, p=0.011). However, the following had decreased likelihood of PD-L1 testing: patients with stage IIIB vs IV cancer (OR=0.683, p=0.004); non vs current/former smokers (OR=0.733, p=0.039); squamous (OR=0.863, p=0.030) or NOS (OR=0.695,p=0.013) vs. adenocarcinoma histology. Interactions were observed between patient residential region and residential rurality (p=0.003), and region and receipt of oncology community care consults (OCCC) (p=0.030). Patients in rural Midwest (OR=0.445,p=0.004) and rural South (OR=0.566, p=0.032) were less likely to receive PD-L1 testing than Metropolitan patients. Across patients with OCCC, Western US patients were more likely to receive PD-L1 testing (OR=1.554, p=0.001) than patients in other regions. However, within Midwestern patients, those without a OCCC were more likely to receive PD-L1 testing (OR=1.724, p< 0.001) than those with a OCCC. High comorbidity index (CCI≥3) is associated with an increased likelihood of PD-L1 testing in a univariable model (OR=1.286 vs. CCI=0,p=0.009), but not in the multivariable model (p=0.278).
Conclusions
We identified demographic and clinical factors, including regional differences in rurality and OCCC patterns, associated with PD-L1 testing. These factors can focus ongoing efforts to improve PD-L1 testing and efforts to be more in line with recommended care.
Background
Programmed death-ligand 1 (PD-L1) checkpoint inhibitors revolutionized the treatment of advanced non-small cell lung cancer (aNSCLC) by improving overall survival compared to chemotherapy. PD-L1 biomarker testing is paramount for informing treatment decisions in aNSCLC. Real-world data describing patterns of PD-L1 testing within the Veteran Health Administration (VHA) are limited. This retrospective study seeks to evaluate demographic and clinical factors associated with PD-L1 testing in VHA.
Methods
Veterans diagnosed with aNSCLC from 2019-2022 were identified using VHA’s Corporate Data Warehouse. Wilcoxon Rank Sum and Chi- Square tests measured association between receipt of PD-L1 testing and patient demographic and clinical characteristics at aNSCLC diagnosis. Logistic regression assessed predictors of PD-L1 testing, and subgroup analyses were performed for significant interactions.
Results
Our study included 4575 patients with aNSCLC; 57.0% received PD-L1 testing. The likelihood of PD-L1 testing increased among patients diagnosed with aNSCLC after 2019 vs during 2019 (OR≥1.118, p≤0.035) and in Black vs White patients (OR=1.227, p=0.011). However, the following had decreased likelihood of PD-L1 testing: patients with stage IIIB vs IV cancer (OR=0.683, p=0.004); non vs current/former smokers (OR=0.733, p=0.039); squamous (OR=0.863, p=0.030) or NOS (OR=0.695,p=0.013) vs. adenocarcinoma histology. Interactions were observed between patient residential region and residential rurality (p=0.003), and region and receipt of oncology community care consults (OCCC) (p=0.030). Patients in rural Midwest (OR=0.445,p=0.004) and rural South (OR=0.566, p=0.032) were less likely to receive PD-L1 testing than Metropolitan patients. Across patients with OCCC, Western US patients were more likely to receive PD-L1 testing (OR=1.554, p=0.001) than patients in other regions. However, within Midwestern patients, those without a OCCC were more likely to receive PD-L1 testing (OR=1.724, p< 0.001) than those with a OCCC. High comorbidity index (CCI≥3) is associated with an increased likelihood of PD-L1 testing in a univariable model (OR=1.286 vs. CCI=0,p=0.009), but not in the multivariable model (p=0.278).
Conclusions
We identified demographic and clinical factors, including regional differences in rurality and OCCC patterns, associated with PD-L1 testing. These factors can focus ongoing efforts to improve PD-L1 testing and efforts to be more in line with recommended care.
Background
Programmed death-ligand 1 (PD-L1) checkpoint inhibitors revolutionized the treatment of advanced non-small cell lung cancer (aNSCLC) by improving overall survival compared to chemotherapy. PD-L1 biomarker testing is paramount for informing treatment decisions in aNSCLC. Real-world data describing patterns of PD-L1 testing within the Veteran Health Administration (VHA) are limited. This retrospective study seeks to evaluate demographic and clinical factors associated with PD-L1 testing in VHA.
Methods
Veterans diagnosed with aNSCLC from 2019-2022 were identified using VHA’s Corporate Data Warehouse. Wilcoxon Rank Sum and Chi- Square tests measured association between receipt of PD-L1 testing and patient demographic and clinical characteristics at aNSCLC diagnosis. Logistic regression assessed predictors of PD-L1 testing, and subgroup analyses were performed for significant interactions.
Results
Our study included 4575 patients with aNSCLC; 57.0% received PD-L1 testing. The likelihood of PD-L1 testing increased among patients diagnosed with aNSCLC after 2019 vs during 2019 (OR≥1.118, p≤0.035) and in Black vs White patients (OR=1.227, p=0.011). However, the following had decreased likelihood of PD-L1 testing: patients with stage IIIB vs IV cancer (OR=0.683, p=0.004); non vs current/former smokers (OR=0.733, p=0.039); squamous (OR=0.863, p=0.030) or NOS (OR=0.695,p=0.013) vs. adenocarcinoma histology. Interactions were observed between patient residential region and residential rurality (p=0.003), and region and receipt of oncology community care consults (OCCC) (p=0.030). Patients in rural Midwest (OR=0.445,p=0.004) and rural South (OR=0.566, p=0.032) were less likely to receive PD-L1 testing than Metropolitan patients. Across patients with OCCC, Western US patients were more likely to receive PD-L1 testing (OR=1.554, p=0.001) than patients in other regions. However, within Midwestern patients, those without a OCCC were more likely to receive PD-L1 testing (OR=1.724, p< 0.001) than those with a OCCC. High comorbidity index (CCI≥3) is associated with an increased likelihood of PD-L1 testing in a univariable model (OR=1.286 vs. CCI=0,p=0.009), but not in the multivariable model (p=0.278).
Conclusions
We identified demographic and clinical factors, including regional differences in rurality and OCCC patterns, associated with PD-L1 testing. These factors can focus ongoing efforts to improve PD-L1 testing and efforts to be more in line with recommended care.
Survival Outcomes of Skin Adnexal Tumors: A National Cancer Database Analysis
Purpose
Skin adnexal tumors (SAT) include a group of benign and malignant appendageal tumors that arise from hair follicles, sebaceous glands, or sweat glands. They typically appear as small, painless bumps or nodules on the skin, and are more common in men compared to women. The 5-year overall SAT survival rate ranges from 74-90%. To better understand the differences in survival outcomes based on subtypes of SAT, the National Cancer Database (NCDB) was analyzed.
Methods
A retrospective cohort study of 11,627 patients with histologically confirmed SAT between 2004 and 2021 was conducted across 1,500 Commission on Cancer facilities located in the US and Puerto Rico. Demographic factors such as sex, age, and race were analyzed using Pearson Chi-squared tests, and survival outcomes were analyzed by Kaplan- Meier survival analysis. P value < 0.05 was considered statistically significant.
Results
Most patients with SAT were male (57.3%). The average age at diagnosis was 65.9 (SD=14.4, range 0-90). Of the patient sample, 87.2% were White, 7.6% Black, 2.5% Asian, and 2.7% other. Several subtypes disproportionately affected Black individuals, including apocrine adenocarcinoma (15.7%) and hidradenocarcinoma (13.6%). The estimated 5-year survival of SAT was 74.9% with an overall survival of 135.8 months (SE=1.1). Sebaceous carcinoma (which accounts for 41.8% of all cases) had the lowest average survival time of 119.6 months (SE=1.8), while digital papillary adenocarcinoma had the highest survival at around 183.5 months (SE=4.6).
Conclusions
This study supports a higher frequency of SAT among men. While White patients were more likely to get SAT overall, including the most common sebaceous carcinoma, Black race were associated with higher frequency of rarer subtypes. The average age of diagnosis of SAT mimics other non-melanoma skin cancers, but has a lower overall survival rate. Future studies should consider other risk factors that may be impacting the differences in survival outcomes to guide treatment and address health disparities among the various subtypes.
Purpose
Skin adnexal tumors (SAT) include a group of benign and malignant appendageal tumors that arise from hair follicles, sebaceous glands, or sweat glands. They typically appear as small, painless bumps or nodules on the skin, and are more common in men compared to women. The 5-year overall SAT survival rate ranges from 74-90%. To better understand the differences in survival outcomes based on subtypes of SAT, the National Cancer Database (NCDB) was analyzed.
Methods
A retrospective cohort study of 11,627 patients with histologically confirmed SAT between 2004 and 2021 was conducted across 1,500 Commission on Cancer facilities located in the US and Puerto Rico. Demographic factors such as sex, age, and race were analyzed using Pearson Chi-squared tests, and survival outcomes were analyzed by Kaplan- Meier survival analysis. P value < 0.05 was considered statistically significant.
Results
Most patients with SAT were male (57.3%). The average age at diagnosis was 65.9 (SD=14.4, range 0-90). Of the patient sample, 87.2% were White, 7.6% Black, 2.5% Asian, and 2.7% other. Several subtypes disproportionately affected Black individuals, including apocrine adenocarcinoma (15.7%) and hidradenocarcinoma (13.6%). The estimated 5-year survival of SAT was 74.9% with an overall survival of 135.8 months (SE=1.1). Sebaceous carcinoma (which accounts for 41.8% of all cases) had the lowest average survival time of 119.6 months (SE=1.8), while digital papillary adenocarcinoma had the highest survival at around 183.5 months (SE=4.6).
Conclusions
This study supports a higher frequency of SAT among men. While White patients were more likely to get SAT overall, including the most common sebaceous carcinoma, Black race were associated with higher frequency of rarer subtypes. The average age of diagnosis of SAT mimics other non-melanoma skin cancers, but has a lower overall survival rate. Future studies should consider other risk factors that may be impacting the differences in survival outcomes to guide treatment and address health disparities among the various subtypes.
Purpose
Skin adnexal tumors (SAT) include a group of benign and malignant appendageal tumors that arise from hair follicles, sebaceous glands, or sweat glands. They typically appear as small, painless bumps or nodules on the skin, and are more common in men compared to women. The 5-year overall SAT survival rate ranges from 74-90%. To better understand the differences in survival outcomes based on subtypes of SAT, the National Cancer Database (NCDB) was analyzed.
Methods
A retrospective cohort study of 11,627 patients with histologically confirmed SAT between 2004 and 2021 was conducted across 1,500 Commission on Cancer facilities located in the US and Puerto Rico. Demographic factors such as sex, age, and race were analyzed using Pearson Chi-squared tests, and survival outcomes were analyzed by Kaplan- Meier survival analysis. P value < 0.05 was considered statistically significant.
Results
Most patients with SAT were male (57.3%). The average age at diagnosis was 65.9 (SD=14.4, range 0-90). Of the patient sample, 87.2% were White, 7.6% Black, 2.5% Asian, and 2.7% other. Several subtypes disproportionately affected Black individuals, including apocrine adenocarcinoma (15.7%) and hidradenocarcinoma (13.6%). The estimated 5-year survival of SAT was 74.9% with an overall survival of 135.8 months (SE=1.1). Sebaceous carcinoma (which accounts for 41.8% of all cases) had the lowest average survival time of 119.6 months (SE=1.8), while digital papillary adenocarcinoma had the highest survival at around 183.5 months (SE=4.6).
Conclusions
This study supports a higher frequency of SAT among men. While White patients were more likely to get SAT overall, including the most common sebaceous carcinoma, Black race were associated with higher frequency of rarer subtypes. The average age of diagnosis of SAT mimics other non-melanoma skin cancers, but has a lower overall survival rate. Future studies should consider other risk factors that may be impacting the differences in survival outcomes to guide treatment and address health disparities among the various subtypes.
Timeliness of Specialty Palliative Care for Veterans With Cancer: An Analysis of Administrative Data
Background
Studies show that early referral to Specialty Palliative Care (SPC) can improve patient- reported outcomes among Veterans with cancer; quality metrics include referral within 8 weeks of an advanced cancer diagnosis. In this study, we explored timeliness of specialty referrals and compared various factors.
Methods
We identified our cohort using Department of Veterans Affairs (VA) Corporate Data Warehouse (CDW). Eligibility criteria included active or history of cancer—using a peer-reviewed, in-house list of ICD-9 and ICD-10 codes—between 2013-2023. We stratified our cohort of Veterans using factors including cancer stage, rurality, and care assessment needs (CAN) scores. We performed survival analyses to look at time to SPC from initial diagnosis and peak CAN score. Predictors of utilization were evaluated using multinomial regression and Cox proportional hazards models through R.
Results
Using CDW’s oncology domain, we identified 475,775 Veterans. 28% received SPC. Most received it near the end of their life as evidenced by the mortality rates (79.5%) in the early period following SPC consultation. Median time to SPC was 515 days. There was a significant difference in utilization rates between urban and rural Veterans (Wilcoxon W-statistic = 2.31E+10, p < 0.001). Peak CAN scores ranged from 0 to 0.81, median peak of 0.057 and interquartile range of 0.1. Multinomial regression model indicated statistically significant associations of advanced cancer (Stages 3 and 4) with timing of SPC. Stage 4 cancer showed the strongest association with receipt of palliative care within 60 days of initial diagnosis (OR 4.8, 95% CI: 4.69-4.93, p < 0.001), suggesting higher stage disease increases the likelihood of palliative care referral and accelerates the timing of these referrals.
Conclusions
We found Veterans received SPC from a broad range of peak CAN scores (0 to 0.81), suggesting that absolute CAN scores may not be clinically actionable indicators but perhaps indicative of changes in condition warranting referral. Stage IV cancer at diagnosis was associated with early SPC. The significant differences in utilization rates between urban and rural patients highlight potential access barriers that should be addressed.
Background
Studies show that early referral to Specialty Palliative Care (SPC) can improve patient- reported outcomes among Veterans with cancer; quality metrics include referral within 8 weeks of an advanced cancer diagnosis. In this study, we explored timeliness of specialty referrals and compared various factors.
Methods
We identified our cohort using Department of Veterans Affairs (VA) Corporate Data Warehouse (CDW). Eligibility criteria included active or history of cancer—using a peer-reviewed, in-house list of ICD-9 and ICD-10 codes—between 2013-2023. We stratified our cohort of Veterans using factors including cancer stage, rurality, and care assessment needs (CAN) scores. We performed survival analyses to look at time to SPC from initial diagnosis and peak CAN score. Predictors of utilization were evaluated using multinomial regression and Cox proportional hazards models through R.
Results
Using CDW’s oncology domain, we identified 475,775 Veterans. 28% received SPC. Most received it near the end of their life as evidenced by the mortality rates (79.5%) in the early period following SPC consultation. Median time to SPC was 515 days. There was a significant difference in utilization rates between urban and rural Veterans (Wilcoxon W-statistic = 2.31E+10, p < 0.001). Peak CAN scores ranged from 0 to 0.81, median peak of 0.057 and interquartile range of 0.1. Multinomial regression model indicated statistically significant associations of advanced cancer (Stages 3 and 4) with timing of SPC. Stage 4 cancer showed the strongest association with receipt of palliative care within 60 days of initial diagnosis (OR 4.8, 95% CI: 4.69-4.93, p < 0.001), suggesting higher stage disease increases the likelihood of palliative care referral and accelerates the timing of these referrals.
Conclusions
We found Veterans received SPC from a broad range of peak CAN scores (0 to 0.81), suggesting that absolute CAN scores may not be clinically actionable indicators but perhaps indicative of changes in condition warranting referral. Stage IV cancer at diagnosis was associated with early SPC. The significant differences in utilization rates between urban and rural patients highlight potential access barriers that should be addressed.
Background
Studies show that early referral to Specialty Palliative Care (SPC) can improve patient- reported outcomes among Veterans with cancer; quality metrics include referral within 8 weeks of an advanced cancer diagnosis. In this study, we explored timeliness of specialty referrals and compared various factors.
Methods
We identified our cohort using Department of Veterans Affairs (VA) Corporate Data Warehouse (CDW). Eligibility criteria included active or history of cancer—using a peer-reviewed, in-house list of ICD-9 and ICD-10 codes—between 2013-2023. We stratified our cohort of Veterans using factors including cancer stage, rurality, and care assessment needs (CAN) scores. We performed survival analyses to look at time to SPC from initial diagnosis and peak CAN score. Predictors of utilization were evaluated using multinomial regression and Cox proportional hazards models through R.
Results
Using CDW’s oncology domain, we identified 475,775 Veterans. 28% received SPC. Most received it near the end of their life as evidenced by the mortality rates (79.5%) in the early period following SPC consultation. Median time to SPC was 515 days. There was a significant difference in utilization rates between urban and rural Veterans (Wilcoxon W-statistic = 2.31E+10, p < 0.001). Peak CAN scores ranged from 0 to 0.81, median peak of 0.057 and interquartile range of 0.1. Multinomial regression model indicated statistically significant associations of advanced cancer (Stages 3 and 4) with timing of SPC. Stage 4 cancer showed the strongest association with receipt of palliative care within 60 days of initial diagnosis (OR 4.8, 95% CI: 4.69-4.93, p < 0.001), suggesting higher stage disease increases the likelihood of palliative care referral and accelerates the timing of these referrals.
Conclusions
We found Veterans received SPC from a broad range of peak CAN scores (0 to 0.81), suggesting that absolute CAN scores may not be clinically actionable indicators but perhaps indicative of changes in condition warranting referral. Stage IV cancer at diagnosis was associated with early SPC. The significant differences in utilization rates between urban and rural patients highlight potential access barriers that should be addressed.
Uncovering Food Insecurity in Veterans with Cancer Distress
Background
To close the food insecurity gap by providing food assistance and increasing opportunities for screening in Veterans receiving cancer treatment at a VA outpatient cancer clinic. Food Insecurity is associated with chronic disease such as cancer given insufficient access to nutritious foods leading to nutritional deficiencies and worsening health outcomes. The rates of food insecurity among Veterans revealed 28% of female veterans and 16% overall in male Veterans were faced with limited or uncertain access to adequate food.
Methods
A pivotal distress screening occurs at time of education consult or cycle 1 day 1 of antineoplastic therapy. A positive screening for any practical concern generates a discussion about food insecurity. A positive distress screen triggers an oncology social work referral to complete a systematic screening assessing circumstances and offering resources for needs (ACORN).
Results
Root cause analysis uncovered 24% of Veterans with cancer screened positive for food insecurity in the 9E oncology outpatient clinic. Post-implementation of robust screenings and conversation initiatives identified 36 unique Veterans who received 251 meals from July to December 2024.
Sustainability/Scalability
Prospective screening of Veterans at the time of a cancer diagnosis and ongoing screening during cancer treatment is the first step toward uncovering food insecurity and addressing this social determinate of health. A standard operating procedure following VA guidance and distress management guidelines should be updated as required. Oversight of the cancer leadership team annually evaluates the distress process, and the findings are reported to the cancer committee.
Conclusions
Uncovering food insecurity in Veterans at time of diagnosis and during cancer treatment is critical to optimize treatment outcomes. A systematic and robust screening standard operating procedure is key to implement. Veterans are a unique population with a spectrum of socioeconomic needs. Case management conferences or weekly huddles to discuss the Veteran’s needs will ensure food insecurity is addressed. Collection and analysis of screening data will highlight a program’s food insecurity need and supports community partnerships to available food resources and the opportunity to create a cancer outpatient clinic food hub for Veterans receiving cancer treatment.
Background
To close the food insecurity gap by providing food assistance and increasing opportunities for screening in Veterans receiving cancer treatment at a VA outpatient cancer clinic. Food Insecurity is associated with chronic disease such as cancer given insufficient access to nutritious foods leading to nutritional deficiencies and worsening health outcomes. The rates of food insecurity among Veterans revealed 28% of female veterans and 16% overall in male Veterans were faced with limited or uncertain access to adequate food.
Methods
A pivotal distress screening occurs at time of education consult or cycle 1 day 1 of antineoplastic therapy. A positive screening for any practical concern generates a discussion about food insecurity. A positive distress screen triggers an oncology social work referral to complete a systematic screening assessing circumstances and offering resources for needs (ACORN).
Results
Root cause analysis uncovered 24% of Veterans with cancer screened positive for food insecurity in the 9E oncology outpatient clinic. Post-implementation of robust screenings and conversation initiatives identified 36 unique Veterans who received 251 meals from July to December 2024.
Sustainability/Scalability
Prospective screening of Veterans at the time of a cancer diagnosis and ongoing screening during cancer treatment is the first step toward uncovering food insecurity and addressing this social determinate of health. A standard operating procedure following VA guidance and distress management guidelines should be updated as required. Oversight of the cancer leadership team annually evaluates the distress process, and the findings are reported to the cancer committee.
Conclusions
Uncovering food insecurity in Veterans at time of diagnosis and during cancer treatment is critical to optimize treatment outcomes. A systematic and robust screening standard operating procedure is key to implement. Veterans are a unique population with a spectrum of socioeconomic needs. Case management conferences or weekly huddles to discuss the Veteran’s needs will ensure food insecurity is addressed. Collection and analysis of screening data will highlight a program’s food insecurity need and supports community partnerships to available food resources and the opportunity to create a cancer outpatient clinic food hub for Veterans receiving cancer treatment.
Background
To close the food insecurity gap by providing food assistance and increasing opportunities for screening in Veterans receiving cancer treatment at a VA outpatient cancer clinic. Food Insecurity is associated with chronic disease such as cancer given insufficient access to nutritious foods leading to nutritional deficiencies and worsening health outcomes. The rates of food insecurity among Veterans revealed 28% of female veterans and 16% overall in male Veterans were faced with limited or uncertain access to adequate food.
Methods
A pivotal distress screening occurs at time of education consult or cycle 1 day 1 of antineoplastic therapy. A positive screening for any practical concern generates a discussion about food insecurity. A positive distress screen triggers an oncology social work referral to complete a systematic screening assessing circumstances and offering resources for needs (ACORN).
Results
Root cause analysis uncovered 24% of Veterans with cancer screened positive for food insecurity in the 9E oncology outpatient clinic. Post-implementation of robust screenings and conversation initiatives identified 36 unique Veterans who received 251 meals from July to December 2024.
Sustainability/Scalability
Prospective screening of Veterans at the time of a cancer diagnosis and ongoing screening during cancer treatment is the first step toward uncovering food insecurity and addressing this social determinate of health. A standard operating procedure following VA guidance and distress management guidelines should be updated as required. Oversight of the cancer leadership team annually evaluates the distress process, and the findings are reported to the cancer committee.
Conclusions
Uncovering food insecurity in Veterans at time of diagnosis and during cancer treatment is critical to optimize treatment outcomes. A systematic and robust screening standard operating procedure is key to implement. Veterans are a unique population with a spectrum of socioeconomic needs. Case management conferences or weekly huddles to discuss the Veteran’s needs will ensure food insecurity is addressed. Collection and analysis of screening data will highlight a program’s food insecurity need and supports community partnerships to available food resources and the opportunity to create a cancer outpatient clinic food hub for Veterans receiving cancer treatment.
Enhancing Molecular Testing Documentation in Prostate Cancer
Background
Prostate cancer is the most common non-cutaneous malignancy at the Veterans Health Administration (VHA) and every year approximately 15,000 Veterans are diagnosed and treated. Many advanced prostate cancer cases harbor genetic mutations that significantly impact prognosis, treatment decisions, and familial screening. In February 2021, the Prostate Cancer Molecular Testing Pathway (PCMTP) flow map was developed to increase appropriate genetic testing.
Methods
VHA initiated the Oncology Clinical Pathways (OCP) program to standardize cancer care for Veterans. The PCMTP was developed by a multidisciplinary team that created interactive templates within the Computerized Patient Record System (CPRS), to facilitate identification of eligible Veterans for germline and comprehensive genomic profiling (CGP). Clinical decision-making for these tests is documented as Health Factors (HF), in CPRS, allowing for assessment of pathway adherence and overall uptake.
Results
The PCMTP has achieved success, as there is over 90% compliance to molecular testing among participating Veterans which exceeds the pathway benchmark of 80%. PCMTP has been utilized at 88 VA sites, by over 700 distinct VA providers, with over 7,000 Veterans participating. This implementation has yielded over 19,200 Health Factors within CPRS.
Conclusions
The PCMTP has markedly improved the documentation and application of germline and CGP testing among Veterans diagnosed with prostate cancer. By facilitating genomic testing in appropriate patients, the PCMTP aims to enhance patient outcomes and optimize the quality of care. Prior to PCMTP establishment, assessing the prevalence of germline and CGP testing in eligible Veterans posed significant challenges. Future work will concentrate on increasing PCMTP utilization, evaluating downstream outcomes from genomic testing, including the identification of pathogenic variants, utilization of genetic counseling services, referrals to clinical trials, and the genomic impact on treatment strategies.
Background
Prostate cancer is the most common non-cutaneous malignancy at the Veterans Health Administration (VHA) and every year approximately 15,000 Veterans are diagnosed and treated. Many advanced prostate cancer cases harbor genetic mutations that significantly impact prognosis, treatment decisions, and familial screening. In February 2021, the Prostate Cancer Molecular Testing Pathway (PCMTP) flow map was developed to increase appropriate genetic testing.
Methods
VHA initiated the Oncology Clinical Pathways (OCP) program to standardize cancer care for Veterans. The PCMTP was developed by a multidisciplinary team that created interactive templates within the Computerized Patient Record System (CPRS), to facilitate identification of eligible Veterans for germline and comprehensive genomic profiling (CGP). Clinical decision-making for these tests is documented as Health Factors (HF), in CPRS, allowing for assessment of pathway adherence and overall uptake.
Results
The PCMTP has achieved success, as there is over 90% compliance to molecular testing among participating Veterans which exceeds the pathway benchmark of 80%. PCMTP has been utilized at 88 VA sites, by over 700 distinct VA providers, with over 7,000 Veterans participating. This implementation has yielded over 19,200 Health Factors within CPRS.
Conclusions
The PCMTP has markedly improved the documentation and application of germline and CGP testing among Veterans diagnosed with prostate cancer. By facilitating genomic testing in appropriate patients, the PCMTP aims to enhance patient outcomes and optimize the quality of care. Prior to PCMTP establishment, assessing the prevalence of germline and CGP testing in eligible Veterans posed significant challenges. Future work will concentrate on increasing PCMTP utilization, evaluating downstream outcomes from genomic testing, including the identification of pathogenic variants, utilization of genetic counseling services, referrals to clinical trials, and the genomic impact on treatment strategies.
Background
Prostate cancer is the most common non-cutaneous malignancy at the Veterans Health Administration (VHA) and every year approximately 15,000 Veterans are diagnosed and treated. Many advanced prostate cancer cases harbor genetic mutations that significantly impact prognosis, treatment decisions, and familial screening. In February 2021, the Prostate Cancer Molecular Testing Pathway (PCMTP) flow map was developed to increase appropriate genetic testing.
Methods
VHA initiated the Oncology Clinical Pathways (OCP) program to standardize cancer care for Veterans. The PCMTP was developed by a multidisciplinary team that created interactive templates within the Computerized Patient Record System (CPRS), to facilitate identification of eligible Veterans for germline and comprehensive genomic profiling (CGP). Clinical decision-making for these tests is documented as Health Factors (HF), in CPRS, allowing for assessment of pathway adherence and overall uptake.
Results
The PCMTP has achieved success, as there is over 90% compliance to molecular testing among participating Veterans which exceeds the pathway benchmark of 80%. PCMTP has been utilized at 88 VA sites, by over 700 distinct VA providers, with over 7,000 Veterans participating. This implementation has yielded over 19,200 Health Factors within CPRS.
Conclusions
The PCMTP has markedly improved the documentation and application of germline and CGP testing among Veterans diagnosed with prostate cancer. By facilitating genomic testing in appropriate patients, the PCMTP aims to enhance patient outcomes and optimize the quality of care. Prior to PCMTP establishment, assessing the prevalence of germline and CGP testing in eligible Veterans posed significant challenges. Future work will concentrate on increasing PCMTP utilization, evaluating downstream outcomes from genomic testing, including the identification of pathogenic variants, utilization of genetic counseling services, referrals to clinical trials, and the genomic impact on treatment strategies.
Enhancing Veteran Health Research: A Quality Improvement Initiative to Optimize Biorepository Efficiency
Purpose
Biorepositories are critical to scientific research within the VA. They offer high-quality, well-characterized biospecimens linked to clinical, demographic, and molecular data. Biorepositories support studies on disease mechanisms, personalized therapies, and emerging infectious diseases by systematically collecting, processing, storing, and distributing biological materials, including tissue, blood, and DNA samples. Within the Department of Veterans Affairs (VA), biorepositories provide essential support to clinical and translational research on service- related conditions such as PTSD, traumatic brain injury, cancers, and toxic exposures. While the need for harmonized quality processes and resource allocation has long been acknowledged within the biorepository community (Siwek, 2015), each biorepository operates independently, limiting scalability and standardization. This quality improvement project describes a collaboration between two VA biorepository sites supporting a national genomic study investigating disease risk and treatment outcomes. The project aimed to expand capacity, improve processing times, and enhance quality control. Each site mirrors the other’s functions, including receiving, accessioning, processing, storing, and shipping biospecimens, and serves as a contingency site to strengthen operational resilience.
Methods
To address space limitations and improve processing efficiency, one site implemented a custom rack design, expanding storage capacity per freezer. Robotic workflows were optimized, reducing biospecimen processing time. An in-process quality control step was introduced to identify data discrepancies earlier in the workflow, reducing investigation time and supporting overall data integrity. Efficiency was measured by the increase in storage capacity and decreased processing time. Descriptive statistics were used to evaluate changes in performance. Metrics were monitored over twelve months and compared against baseline data.
Results
Following implementation, storage capacity per freezer increased by 20%, and specimen processing time decreased by 30%. The new quality control checkpoint reduced investigation times by 98%, resulting in a more streamlined workflow. These improvements enhanced coordination between sites and improved support for ongoing studies.
Conclusions
This effort demonstrates that collaboration between biorepositories can significantly enhance efficiency, reduce turnaround times, and support high-quality research. Strengthening infrastructure through joint initiatives enables more effective support of large-scale clinical studies and contributes to improved outcomes for Veterans. These findings may also inform process improvements at other VA research facilities.
Purpose
Biorepositories are critical to scientific research within the VA. They offer high-quality, well-characterized biospecimens linked to clinical, demographic, and molecular data. Biorepositories support studies on disease mechanisms, personalized therapies, and emerging infectious diseases by systematically collecting, processing, storing, and distributing biological materials, including tissue, blood, and DNA samples. Within the Department of Veterans Affairs (VA), biorepositories provide essential support to clinical and translational research on service- related conditions such as PTSD, traumatic brain injury, cancers, and toxic exposures. While the need for harmonized quality processes and resource allocation has long been acknowledged within the biorepository community (Siwek, 2015), each biorepository operates independently, limiting scalability and standardization. This quality improvement project describes a collaboration between two VA biorepository sites supporting a national genomic study investigating disease risk and treatment outcomes. The project aimed to expand capacity, improve processing times, and enhance quality control. Each site mirrors the other’s functions, including receiving, accessioning, processing, storing, and shipping biospecimens, and serves as a contingency site to strengthen operational resilience.
Methods
To address space limitations and improve processing efficiency, one site implemented a custom rack design, expanding storage capacity per freezer. Robotic workflows were optimized, reducing biospecimen processing time. An in-process quality control step was introduced to identify data discrepancies earlier in the workflow, reducing investigation time and supporting overall data integrity. Efficiency was measured by the increase in storage capacity and decreased processing time. Descriptive statistics were used to evaluate changes in performance. Metrics were monitored over twelve months and compared against baseline data.
Results
Following implementation, storage capacity per freezer increased by 20%, and specimen processing time decreased by 30%. The new quality control checkpoint reduced investigation times by 98%, resulting in a more streamlined workflow. These improvements enhanced coordination between sites and improved support for ongoing studies.
Conclusions
This effort demonstrates that collaboration between biorepositories can significantly enhance efficiency, reduce turnaround times, and support high-quality research. Strengthening infrastructure through joint initiatives enables more effective support of large-scale clinical studies and contributes to improved outcomes for Veterans. These findings may also inform process improvements at other VA research facilities.
Purpose
Biorepositories are critical to scientific research within the VA. They offer high-quality, well-characterized biospecimens linked to clinical, demographic, and molecular data. Biorepositories support studies on disease mechanisms, personalized therapies, and emerging infectious diseases by systematically collecting, processing, storing, and distributing biological materials, including tissue, blood, and DNA samples. Within the Department of Veterans Affairs (VA), biorepositories provide essential support to clinical and translational research on service- related conditions such as PTSD, traumatic brain injury, cancers, and toxic exposures. While the need for harmonized quality processes and resource allocation has long been acknowledged within the biorepository community (Siwek, 2015), each biorepository operates independently, limiting scalability and standardization. This quality improvement project describes a collaboration between two VA biorepository sites supporting a national genomic study investigating disease risk and treatment outcomes. The project aimed to expand capacity, improve processing times, and enhance quality control. Each site mirrors the other’s functions, including receiving, accessioning, processing, storing, and shipping biospecimens, and serves as a contingency site to strengthen operational resilience.
Methods
To address space limitations and improve processing efficiency, one site implemented a custom rack design, expanding storage capacity per freezer. Robotic workflows were optimized, reducing biospecimen processing time. An in-process quality control step was introduced to identify data discrepancies earlier in the workflow, reducing investigation time and supporting overall data integrity. Efficiency was measured by the increase in storage capacity and decreased processing time. Descriptive statistics were used to evaluate changes in performance. Metrics were monitored over twelve months and compared against baseline data.
Results
Following implementation, storage capacity per freezer increased by 20%, and specimen processing time decreased by 30%. The new quality control checkpoint reduced investigation times by 98%, resulting in a more streamlined workflow. These improvements enhanced coordination between sites and improved support for ongoing studies.
Conclusions
This effort demonstrates that collaboration between biorepositories can significantly enhance efficiency, reduce turnaround times, and support high-quality research. Strengthening infrastructure through joint initiatives enables more effective support of large-scale clinical studies and contributes to improved outcomes for Veterans. These findings may also inform process improvements at other VA research facilities.