User login
HIV: Just another chronic disease

In subsequent years we learned about HIV—the retrovirus, and the immune system that it cleverly and efficiently disables. For the most part, we matured professionally and moved past the social stigmas of the disease, although that was painful. We developed systems to keep acutely ill patients out of the hospital while providing them with “long-term” (weeks or months of) intravenous antibiotics and humane palliative care.
We learned about AZT and argued about when to use it. But mainly, we watched many, many young men (and some women) die in corner hospital rooms. For me, from the ′80s, there remain heartrending personal images, notes, and cassette tapes voicing thanks for my concern and time spent, but no notes of thanks like those I’ve received from my patients with chronic rheumatoid arthritis who, after years of care, are able to hold their nieces or grandchildren.
A few long-term survivors have raised the hope that immune systems could recover and exist in symbiosis with the virus, and that maybe a drug cocktail or vaccine could provide a cure or remission. Magic Johnson, known to be infected since at least 1991, is likely the most public example of a long-term survivor on highly active antiviral therapy—a hope in the flesh.
But did we ever expect a time when HIV would be viewed as a chronic disease, with patients warranting screening for coronary artery disease in order to decrease long-term coronary complications? Did we ever expect a time that we would be offering organ transplants to HIV-infected patients?
In this issue of the Journal, Drs. Malvestutto and Aberg discuss coronary issues that need to be recognized and managed in HIV-infected patients. This further complicates the management of these patients, and draws cardiologists and primary care providers back into management plans.
I can’t think of a management “complication” of a chronic illness that is more welcome—or more surprising.
In subsequent years we learned about HIV—the retrovirus, and the immune system that it cleverly and efficiently disables. For the most part, we matured professionally and moved past the social stigmas of the disease, although that was painful. We developed systems to keep acutely ill patients out of the hospital while providing them with “long-term” (weeks or months of) intravenous antibiotics and humane palliative care.
We learned about AZT and argued about when to use it. But mainly, we watched many, many young men (and some women) die in corner hospital rooms. For me, from the ′80s, there remain heartrending personal images, notes, and cassette tapes voicing thanks for my concern and time spent, but no notes of thanks like those I’ve received from my patients with chronic rheumatoid arthritis who, after years of care, are able to hold their nieces or grandchildren.
A few long-term survivors have raised the hope that immune systems could recover and exist in symbiosis with the virus, and that maybe a drug cocktail or vaccine could provide a cure or remission. Magic Johnson, known to be infected since at least 1991, is likely the most public example of a long-term survivor on highly active antiviral therapy—a hope in the flesh.
But did we ever expect a time when HIV would be viewed as a chronic disease, with patients warranting screening for coronary artery disease in order to decrease long-term coronary complications? Did we ever expect a time that we would be offering organ transplants to HIV-infected patients?
In this issue of the Journal, Drs. Malvestutto and Aberg discuss coronary issues that need to be recognized and managed in HIV-infected patients. This further complicates the management of these patients, and draws cardiologists and primary care providers back into management plans.
I can’t think of a management “complication” of a chronic illness that is more welcome—or more surprising.
In subsequent years we learned about HIV—the retrovirus, and the immune system that it cleverly and efficiently disables. For the most part, we matured professionally and moved past the social stigmas of the disease, although that was painful. We developed systems to keep acutely ill patients out of the hospital while providing them with “long-term” (weeks or months of) intravenous antibiotics and humane palliative care.
We learned about AZT and argued about when to use it. But mainly, we watched many, many young men (and some women) die in corner hospital rooms. For me, from the ′80s, there remain heartrending personal images, notes, and cassette tapes voicing thanks for my concern and time spent, but no notes of thanks like those I’ve received from my patients with chronic rheumatoid arthritis who, after years of care, are able to hold their nieces or grandchildren.
A few long-term survivors have raised the hope that immune systems could recover and exist in symbiosis with the virus, and that maybe a drug cocktail or vaccine could provide a cure or remission. Magic Johnson, known to be infected since at least 1991, is likely the most public example of a long-term survivor on highly active antiviral therapy—a hope in the flesh.
But did we ever expect a time when HIV would be viewed as a chronic disease, with patients warranting screening for coronary artery disease in order to decrease long-term coronary complications? Did we ever expect a time that we would be offering organ transplants to HIV-infected patients?
In this issue of the Journal, Drs. Malvestutto and Aberg discuss coronary issues that need to be recognized and managed in HIV-infected patients. This further complicates the management of these patients, and draws cardiologists and primary care providers back into management plans.
I can’t think of a management “complication” of a chronic illness that is more welcome—or more surprising.
Coronary heart disease in people infected with HIV
Widespread use of antiretroviral therapy has caused a remarkable decline in rates of morbidity and death related to acquired immunodeficiency syndrome (AIDS) and has effectively made human immunodeficiency virus (HIV) infection a manageable—although not yet curable— chronic condition. And as the HIV-infected population on antiretroviral therapy ages, the prevalence of chronic conditions (eg, cardiovascular disease, hepatic disease, pulmonary disease, non-AIDS cancers) and deaths attributable to these conditions have also increased.1
Many of the traditional risk factors for cardiovascular disease in the general population, including smoking, dyslipidemia, and diabetes, are common in HIV-infected patients, and HIV infection itself independently increases the risk of coronary heart disease. In addition, different antiretroviral combinations can contribute, in varying degrees, to changes in lipid levels and insulin resistance, further increasing coronary risk.
Ultimately, however, the immunologic benefits of antiretroviral therapy for individual patients far exceed the modest increase in cardiovascular risk associated with certain regimens. In most cases, careful selection of the initial antiretroviral regimen and the addition of lipid-lowering or glucose-controlling medications (with close attention to drug interactions) can effectively manage the metabolic changes associated with antiretroviral therapy and obviate any premature modification of virologically suppressive regimens.
TRADITIONAL CARDIAC RISK FACTORS IN HIV PATIENTS
The risk of coronary heart disease in HIV patients is influenced mostly by traditional factors such as age, smoking, diabetes, and dyslipidemia, including high levels of total cholesterol and low-density lipoprotein cholesterol (LDL-C) and low levels of high-density lipoprotein cholesterol (HDL-C).2
In various large cohorts, HIV-infected men had a higher prevalence of smoking,3 a lower mean HDL-C level,3,4 and a higher mean triglyceride level3,4 than men without HIV infection, placing them at greater risk of coronary heart disease. However, even after adjusting for traditional risk factors, rates of atherosclerosis are still higher in people who are infected with HIV than in those who are not.5
EFFECT OF HIV INFECTION ON CORONARY RISK
HIV infection has been shown to increase coronary risk.
In the Kaiser Permanente database,6 HIV-positive patients had a significantly higher rate of hospitalizations for coronary heart disease than did people who were not infected.
Similarly, in a cohort study of almost 4,000 HIV-infected patients and more than 1 million controls, the risk of acute myocardial infarction was 75% higher for HIV-positive patients than for HIV-negative patients, even after adjusting for sex, race, hypertension, diabetes, and dyslipidemia.5
The Fat Redistribution and Metabolism (FRAM) cross-sectional study7 showed that HIV infection was associated with greater carotid intima media thickness, an established marker of atherosclerosis, independently of traditional risk factors and to virtually the same degree as smoking and male sex.
Other studies of subclinical atherosclerosis in HIV patients have yielded disparate results, likely because of differences in study design, methods of measuring carotid thickness, and characteristics of the study populations (eg, prevalence of cardiovascular risk factors and stage of HIV disease). However, a meta-analysis of six prospective cohort studies, three case-control studies, and four cross-sectional studies confirmed that HIV patients had slightly but statistically significantly greater carotid intima media thickness than HIV-negative people.8
MECHANISMS BY WHICH HIV MAY PROMOTE CORONARY HEART DISEASE
The pathogenesis of coronary heart disease in HIV infection has not been fully elucidated, but the virus appears to contribute directly to the accelerated development of atherosclerosis. It may do so through direct effects on cholesterol processing and transport, attraction of monocytes to the intimal wall, and activation of monocytes to induce an inflammatory response and endothelial proliferation.
Effects on lipids
In early HIV infection, levels of total cholesterol and HDL-C are lower. In more advanced infection, lower CD4+ lymphocyte counts have been associated with lower levels of apolipoprotein B and with smaller LDL-C particles, suggesting that HIV affects lipid processing and delivery to vessel walls.9 HIV infection is also associated with reduced clearance of LDL-C.10 HIV appears to specifically inhibit the compensatory efflux of excess cholesterol from macrophages, thus promoting the formation of foam cells in atherosclerotic plaque.11
Attraction of monocytes to the vessel wall
In vitro studies also suggest that HIV enhances migration of monocytes into the vascular intima during atherosclerotic plaque development by promoting secretion of the chemokine monocyte chemoattractant protein 112 and the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.13
Inflammation
A recent study suggests that chronic inflammation may be a key contributor to the accelerated development of atherosclerosis in HIV patients. Hsue et al14 compared carotid intima media thickness and levels of C-reactive protein (a marker of systemic inflammation) in HIV-positive and HIV-negative patients. The carotid intima media thickness was greater in all groups of HIV patients, irrespective of level of viremia or exposure to antiretroviral therapy, than in healthy controls. In addition, C-reactive protein levels remained elevated in HIV-infected participants regardless of their level of viremia.
These findings suggest not only that HIV-associated atherosclerosis is determined by advanced immunodeficiency, high-level viremia, and exposure to antiretroviral drugs, but also that persistent inflammation due to HIV infection may play an important role in accelerated atherosclerosis.
EFFECT OF ANTIRETROVIRAL THERAPY ON CORONARY RISK
Antiretroviral therapy is associated with a small but significant increase in coronary risk.
Medi-Cal,15 a retrospective study of 28,513 patients, found antiretroviral therapy to be associated with coronary heart disease among patients 18 to 33 years of age (relative risk 2.06, P < .001).
The Data Collection on Adverse Events of Anti-HIV Drugs study16 prospectively followed 23,437 patients for 94,469 person-years. Adjusted for exposure to nonnucleoside reverse transcriptase inhibitors and for hypertension and diabetes, the relative risk of myocardial infarction per year of protease inhibitor exposure was 1.16 (95% confidence interval [CI] 1.10–1.23). The relative risk was lower after adjusting for serum lipid levels but remained significant at 1.10 (95% CI 1.04–1.18).
Reports have been mixed regarding a possible association between myocardial infarction and the nucleoside reverse transcriptase inhibitor abacavir (Ziagen): several studies found a statistically significant association,17–20 and others did not.21–23 Differences in study design (observational cohort studies vs prospective randomized clinical trials), populations studied (differing in age, cardiovascular risk factor prevalence, and whether the patients had already been exposed to treatment), and outcome definition probably contributed to the different conclusions.
On the other hand, several studies have shown that suppression of HIV with antiretroviral therapy actually improves some of the surrogate markers of cardiovascular disease. For example:
- Markers of endothelial function such as flow-mediated vasodilation improve significantly within 4 weeks of a patient’s starting antiretroviral therapy, regardless of the class of antiretroviral drug used.24
- After viral suppression is achieved, levels of the markers of endothelial activation VCAM-1 and P-selectin decline significantly, as do levels of the adipocyte activation marker leptin and the coagulation marker D-dimer.25,26
- Levels of the anti-inflammatory markers adiponectin and interleukin 10 increase. 25,26
Interrupting antiretroviral therapy may increase coronary risk
Not only is uncontrolled viral replication in untreated HIV infection associated with cardiovascular disease, but interrupting antiretroviral therapy may result in a supplementary increase in coronary risk.
In the 5,472-patient Strategies for Management of Antiretroviral Therapy (SMART) trial, the rate of cardiovascular disease events was higher if treatment was interrupted than with continuous treatment, with a hazard ratio of 1.57 (95% CI 1.0–2.46, P = .05).27
This association between treatment interruption and coronary events does not appear to be related to the level of viremia.28 Rather, development of cardiovascular disease in HIV-infected patients who interrupt antiretroviral therapy may be mediated, to a large extent, by chronic inflammation in the setting of viral replication. In the treatment-interruption group, levels of the inflammatory cytokine interleukin 6 (IL-6) and the coagulation marker D-dimer were significantly elevated 1 month after randomization, and these differences were strongly associated with death (odds ratio [OR] 12.6, P < .0001 for IL-6; OR 13.1, P < .0001 for D-dimer). Elevated IL-6 levels were also significantly associated with the development of cardiovascular disease (OR 2.8, P = .03).29
METABOLIC COMPLICATIONS OF ANTIRETROVIRAL THERAPY
Persons with HIV infection may experience metabolic complications that are due to HIV itself or to its treatment.
Cross-sectional studies that included HIV-negative patients as controls have demonstrated changes in lipid processing that are known to promote atherosclerosis. For example, persons with HIV infection have smaller LDL-C particles30 and higher levels of circulating oxidized LDL-C.31
In the Multicenter AIDS Cohort Study (MACS), after HIV seroconversion, nonfasting total cholesterol, LDL-C, and HDL-C levels declined, which is consistent with a chronic inflammatory state. After antiretroviral therapy was started, lipid levels returned to baseline levels or slightly higher except for HDL-C, which remained low.9 These changes may be due to a general “return to health,” or they may be direct medication effects.
Similar patterns were seen in the SMART study.28 Participants randomized to receive intermittent antiretroviral therapy had overall decreases in all lipid levels, with a marked reduction in HDL-C, while those randomized to receive continuous therapy had increased levels of all lipids, including HDL-C, at 12 months. Overall, the ratio of total cholesterol to HDL-C actually increased for participants on episodic therapy, while it decreased in the continuous-treatment group. Along with continued vascular inflammation, the low HDL-C may have contributed to the worse cardiovascular outcomes in patients who received intermittent antiretroviral therapy.
Some lipid changes associated with antiretroviral therapy may actually be beneficial. For example, nonnucleoside reverse transcriptase inhibitors may raise HDL-C levels. However, such increases alone do not necessarily offset the other lipid changes or translate to an observed improvement in coronary risk.32
The degree of dyslipidemia and specific lipid changes differ among the different classes of antiretroviral drugs and even among the individual drugs within each class. Furthermore, the magnitude of the observed lipid changes varies widely among patients on the same antiretroviral regimen, reflecting the likely important role of host genomics.
While the protease inhibitors and nonnucleoside reverse transcriptase inhibitors have well-described effects on lipids (described in greater detail in the following sections), there have been no reported significant changes in lipid profiles or cardiovascular risk associated with the newest classes, ie, fusion inhibitors such as enfuvirtide (Fuzeon), CC chemokine receptor type 5 (CCR5) receptor inhibitors such as maraviroc (Selzentry), or integrase inhibitors such as raltegravir (Isentress).
Impact of protease inhibitors on lipids
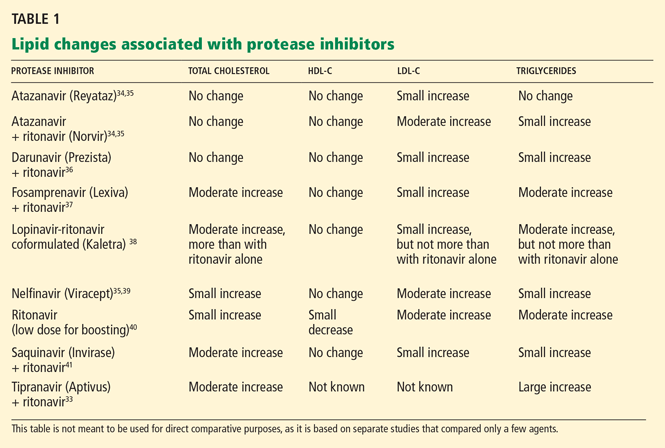
Ritonavir (Norvir) and ritonavir-boosted protease inhibitor combinations cause the most significant increases in lipids. Currently, ritonavir is used in low doses to boost the levels of most other protease inhibitors as the standard of care in protease inhibitor-based regimens. However, in most patients, giving ritonavir with protease inhibitors raises lipid levels, particularly triglycerides.
Most boosted protease inhibitor regimens have similar effects on lipid levels, with some exceptions.
Tipranavir (Aptivus) plus ritonavir, for example, markedly raises total cholesterol and triglyceride levels and would not be recommended for patients with dyslipidemia at baseline.33
Atazanavir (Reyataz)34,35 plus ritonavir and darunavir (Prezista)36 plus ritonavir cause more modest lipid changes. Unboosted atazanavir raises lipid levels only minimally, if at all,34,35 but it is no longer a preferred regimen according to US Department of Health and Human Services guidelines.42
Impact of nonnucleoside reverse transcriptase inhibitors on lipids

Efavirenz (Sustiva), a nonnucleoside reverse transcriptase inhibitor, when added to a regimen of two or three nucleoside reverse transcriptase inhibitors, resulted in modest increases in all lipids, including HDL-C (a potentially beneficial change) at 96 weeks compared with a regimen of three nucleoside reverse transcriptase inhibitors only.43
Nevirapine (Viramune), compared with efavirenz, results in a more favorable lipid profile in previously untreated patients, as shown by larger increases in HDL-C and smaller increases in triglycerides at 48 weeks.44
Etravirine (Intelence), the newest nonnucleoside reverse transcriptase inhibitor, does not appear to cause any further increase in lipids when added to a regimen containing darunavir-ritonavir and nucleoside agents.45
Impact of nucleoside reverse transcriptase inhibitors on lipids
As a class, nucleoside reverse transcriptase inhibitors have been associated with mitochondrial toxicity and insulin resistance,46 but the lipid changes associated with them are generally less significant than those caused by protease inhibitors or nonnucleoside reverse transcriptase inhibitors. Nevertheless, within the class, there is considerable variability in lipid changes associated with specific agents.
Stavudine (Zerit), for example, is associated with hypertriglyceridemia.
Tenofovir (Viread), for another example, in combination with emtricitabine (Emtriva) and the nonnucleoside reverse transcriptase inhibitor efavirenz (the three drugs are contained in a formulation called Atripla) was associated with a smaller increase in fasting total cholesterol than with zidovudine-lamivudine and efavirenz at 96 weeks.47
A recent placebo-controlled, crossover, pilot study of 17 HIV-infected patients suggested that tenofovir may actually have independent lipid-lowering properties.48
Abacavir, as discussed above, has been reported to be associated with a higher risk of myocardial infarction, but this is debatable.
MANAGING CORONARY RISK FACTORS IN HIV-INFECTED PATIENTS
Cardiovascular risk assessment
In HIV patients, cardiovascular risk can be assessed using models derived from large epidemiologic studies such as the Framingham Heart Study.49
Current guidelines from the Infectious Diseases Society of America and the AIDS Clinical Trials Group (ACTG) for evaluating and managing dyslipidemia in HIV-infected adults are based on the National Cholesterol Education Program Adult Treatment Panel III.50 They recommend obtaining a fasting lipid profile before starting antiretroviral therapy and within 3 to 6 months after starting a new regimen.
The guidelines also recommend stratifying risk by counting the number of cardiovascular risk factors, as is done for the general population. If the patient has more than two factors, the Framingham equation should be used to calculate the 10-year risk of myocardial infarction or cardiac death. Interventions should be offered for modifiable cardiovascular risk factors such as smoking, hypertension, physical inactivity, and diabetes mellitus. LDL-C goals should be determined, and lipid-lowering drugs should be initiated accordingly. If triglyceride levels are 200 to 500 mg/dL and levels of “non-HDL-C” (total cholesterol minus the HDL-C level) are high, a statin is recommended. If the triglyceride level is higher than 500 mg/dL, a fibrate should be started.51
Dyslipidemia management
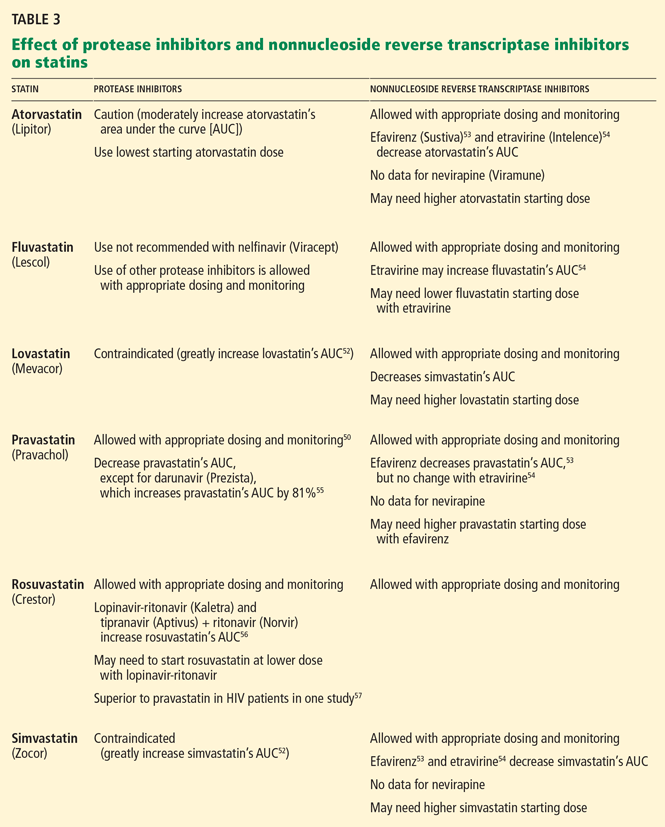
In HIV patients, statin and fibrate therapy must be considered cautiously, given the important drug interactions with protease inhibitors and especially ritonavir. Many statins are metabolized by cytochrome P3A4, which protease inhibitors inhibit.
Statins generally considered safe to use with most protease inhibitors:
- Pravastatin (Pravachol)
- Rosuvastatin (Crestor)
- Atorvastatin (Lipitor).
Exceptions and caveats:
- Pravastatin should not be prescribed with boosted darunavir.
- Data for fluvastatin (Lescol) in HIV-infected patients on antiretroviral therapy are limited.
- Lovastatin (Mevacor) and simvastatin (Zocor) are contraindicated with protease inhibitor therapy.52
- In contrast to the increase in statin levels seen with protease inhibitors, efavirenz lowers levels of simvastatin, pravastatin, and atorvastatin.53,54
Ezetimibe (Zetia), which is metabolized independently of the cytochrome P450 system, has been shown to be safe and effective when given to HIV-infected patients on antiretroviral therapy.58
Fenofibrate (Lofibra) is recommended by current guidelines for patients with elevated triglyceride levels (> 500 mg/dL).51 In the ACTG 5087 study, a combination of fenofibrate plus pravastatin was found to be safe and effective in improving lipid profiles.59
Long-acting niacin resulted in significant improvements in triglycerides, total cholesterol, HDL-C, and LDL-C after 48 weeks of use, although insulin sensitivity worsened.60
Fish oil has been shown to be an effective alternative to fibrates, or it can be used in combination with them.61
Switching antiretroviral agents vs adding lipid-lowering agents. In some patients with significant dyslipidemia, switching antiretro viral agents may lower lipid levels without compromising virologic control.62 However, due to the multifactorial nature of dyslipidemia in HIV patients on antiretroviral therapy, switching the HIV therapy alone may not result in sufficient improvement in the lipid profile45 and may be associated with virologic failure, particularly among patients who have underlying treatment-resistant HIV.63
In many cases, adding lipid-lowering agents may be more beneficial than switching the antiretroviral therapy. For example, a randomized trial in HIV-infected patients with hyperlipidemia found that adding a lipid-lowering agent such as pravastatin or bezafibrate to the unchanged antiretroviral regimen resulted in greater improvement in total cholesterol, LDL-C, and triglyceride levels than switching from a protease inhibitor to either nevirapine or efavirenz.64
Given the complexity of prescribing lipid-lowering therapies to patients on antiretroviral therapy, we recommend that providers check with a pharmacist or refer to package inserts and other medical literature if they are unfamiliar with these drug interactions and responses to lipid-lowering therapies.
Managing insulin resistance
Diabetes mellitus is a well-known risk factor for coronary heart disease. The Data Collection on Adverse Events of Anti-HIV Drugs study found a higher incidence of coronary heart disease in HIV-infected patients, with higher rates in those with longer duration of diabetes.65 The prevalence of diabetes in HIV-infected populations varies, depending on demographic characteristics,65,66 prevalence of coinfection with hepatitis C virus,66 and prevalence of exposure to antiretroviral drugs67 in the study population.
Drugs that lessen insulin resistance include the thiazolidinedione rosiglitazone (Avandia) and the biguanide metformin (Glucophage). In a randomized trial, both drugs, alone or in combination, improved insulin sensitivity in HIV-infected patients, but neither lessened the amount of visceral or subcutaneous fat.68
Smoking cessation
Smoking is another well-known modifiable risk factor for coronary heart disease.
The prevalence of smoking is usually higher in HIV patients than in HIV-negative people. For example, a French cohort study reported smoking prevalence rates of 56.6% in HIV-infected men vs 32.7% in HIV-negative men; in women, the rates were 58% vs 28.1%. The 5-year relative risk of coronary heart disease in HIV-infected vs HIV-negative persons was 1.20 for men and 1.59 for women. The estimated attributable risk due to smoking was 65% for men and 29% for women.3
Therefore, smoking cessation should be a top priority in managing cardiovascular risk in HIV-infected patients. In fact, control of modifiable risk factors through lifestyle changes such as smoking cessation, dietary changes, and exercise is likely to have a significant impact on cardiovascular risk in this population.
- Palella FJ, Baker RK, Moorman AC, et al; HIV Outpatient Study Investigators. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006; 43:27–34.
- Lichtenstein KA, Armon C, Buchacz K, Moorman AC, Wood KC, Brooks JT; HOPS investigators. Analysis of cardiovascular risk factors in the HIV Outpatient Study (HOPS) cohort. Presented at the 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO; 2006.
- Savès M, Chêne G, Ducimetière P, et al; French WHO MONICA Project and the APROCO (ANRS EP11) Study Group. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003; 37:292–298.
- Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007; 45:1074–1081.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Klein D, Hurley LB, Quesenberry CP, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr 2002; 30:471–477.
- Grunfeld C, Delaney JA, Wanke C, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS 2009; 23:1841–1849.
- Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009; 95:1826–1835.
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003; 289:2978–2982.
- Shahmanesh M, Das S, Stolinski M, et al. Antiretroviral treatment reduces very-low-density lipoprotein and intermediate-density lipoprotein apolipoprotein B fractional catabolic rate in human immunodeficiency virus-infected patients with mild dyslipidemia. J Clin Endocrinol Metab 2005; 90:755–760.
- Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365.
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 2001; 97:352–358.
- Fisher SD, Miller TL, Lipshultz SE. Impact of HIV and highly active antiretroviral therapy on leukocyte adhesion molecules, arterial inflammation, dyslipidemia, and atherosclerosis. Atherosclerosis 2006; 185:1–11.
- Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009; 23:1059–1067.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- DAD Study Group; Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007: 356:1723–1735.
- DAD Study Group; Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008; 371:1417–1426.
- Durand M, Sheehy O, Baril JG, Lelorier J, Tremblay C; GRUCHUM Research Center (Groupe de Recherche de l’UHRESS du Centre Hospitalier Universitaire de Montréal). Relation between use of nucleoside reverse transcriptase inhibitors (NRTI) and risk of myocardial infarction (MI): a nested case control study using Quebec’s public health insurance database (QPHID). Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 17–22, 2009.
- Lang S, Mary-Krause M, Cotte L, et al; the Clinical Epi Group of the French Hospital Database on HIV. Impact of specific NRTI and PI exposure on the risk of myocardial infarction: a case-control study nested within FHDH ANRS CO4. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Strategies for Management of Anti-Retroviral Therapy/INSIGHT. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS 2008; 22:F17–F24.
- Bedimo R, Westfall A, Drechsler H, Tebas P. Abacavir use and risk of acute myocardial infarction and cerebrovascular disease in the HAART era. Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 19–22, 2009.
- Brothers CH, Hernandez JE, Cutrell AG, et al. Risk of myocardial infarction and abacavir therapy: no increased risk across 52 GlaxoSmithKline-sponsored clinical trials in adult subjects. J Acquir Immune Defic Syndr 2009; 51:20–28.
- Benson C, Ribaudo H, Zheng E, et al; the ACTG A5001/ALLRT Protocol Team. No Association of Abacavir Use with Risk of Myocardial Infarction or Severe Cardiovascular Disease Events: Results from ACTG A5001. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Torriani FJ, Komarow L, Parker RA, et al; ACTG 5152s Study Team. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: The ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol 2008; 52:569–576.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- van Vonderen MG, Hassink EA, van Agtmael MA, et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis 2009; 199:1186–1194.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group; El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006; 355:2283–2296.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Kuller LH, Tracy R, Belloso WINSIGHT SMART Study Group. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203.
- Badiou S, De Boever CM, Dupuy AM, Baillat V, Cristol JP, Reynes J. Small dense LDL and atherogenic lipid profile in HIV-positive adults: influence of lopinavir/ritonavir-containing regimen. AIDS 2003; 17:772–774.
- Duong M, Petit JM, Martha B, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials 2006; 7:41–47.
- Fisac C, Fumero E, Crespo M, et al. Metabolic benefits 24 months after replacing a protease inhibitor with abacavir, efavirenz or nevirapine. AIDS 2005; 19:917–925.
- Hicks CB, Cahn P, Cooper DA, et al; RESIST investigator group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatmentexperienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 2006; 368:466–475.
- Malan DR, Krantz E, David N, Wirtz V, Hammond J, McGrath D; 089 Study Group. Efficacy and safety of atazanavir, with or without ritonavir, as part of once-daily highly active antiretroviral therapy regimens in antiretroviral-naive patients. J Acquir Immune Defic Syndr 2008; 47:161–167.
- Anastos K, Lu D, Shi Q, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr 2007; 45:34–42.
- Tomaka F, Lefebvre E, Sekar V, et al. Effects of ritonavir-boosted darunavir vs ritonavir-boosted atazanavir on lipid and glucose parameters in HIV-negative, healthy volunteers. HIV Med 2009; 10:318–327.
- Eron J, Yeni P, Gathe J, et al; KLEAN study team. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial. Lancet 2006; 368:476–482.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Kumar PN, Rodriguez-French A, Thompson MA, et al; ESS40002 Study Team. A prospective, 96-week study of the impact of trizivir, combivir/nelfinavir, and lamivudine/stavudine/nelfinavir on lipids, metabolic parameters and efficacy in antiretroviral-naive patients: effect of sex and ethnicity. HIV Med 2006; 7:85–98.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr 2009; 50:367–374.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents— A Working Group of the Office of AIDS Research Advisory Council (OARAC). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. December 1, 1009. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 29, 2010.
- Shikuma CM, Yang Y, Glesby MJ, et al. Metabolic effects of protease inhibitor-sparing antiretroviral regimens given as initial treatment of HIV-1 Infection (AIDS Clinical Trials Group Study A5095). J Acquir Immune Defic Syndr 2007; 44:540–550.
- van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapynaive patients infected with HIV-1. PLoS Med 2004; 1:e19.
- Katlama C, Haubrich R, Lalezari J, et al; DUET-1, DUET-2 study groups. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS 2009; 23:2289–2300.
- Hammond E, Nolan D, James I, Metcalf C, Mallal S. Reduction of mitochondrial DNA content and respiratory chain activity occurs in adipocytes within 6–12 months of commencing nucleoside reverse transcriptase inhibitor therapy. AIDS 2004; 18:815–817.
- Pozniak AL, Gallant JE, DeJesus E, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz versus fixed-dose zidovudine/lamivudine and efavirenz in antiretroviral-naive patients: virologic, immunologic, and morphologic changes—a 96-week analysis. J Acquir Immune Defic Syndr 2006; 43:535–540.
- Tungsiripat M, Kitch D, Glesby M, et al. A pilot study to determine the effect on dyslipidemia of the addition of tenofovir to stable background ART in HIV-infected subjects: results from the A5206 Study Team. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Law MG, Friis-Møller N, El-Sadr WM, et al; D:A:D Study Group. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med 2006; 7:218–230.
- Aberg JA. Cardiovascular complications in HIV management: past, present, and future. J Acquir Immune Defic Syndr 2009; 50:54–64.
- Dubé MP, Stein JH, Aberg JA, et al; Adult AIDS Clinical Trials Group Cardiovascular Subcommittee. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003; 37:613–627.
- Fichtenbaum CJ. Metabolic abnormalities associated with HIV infection and antiretroviral therapy. Curr Infect Dis Rep 2009; 11:84–92.
- Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al; AIDS Clinical Trials Group A5108 Team. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005; 39:307–312.
- Grennan T, Walmsley S. Etravirine for HIV-I: addressing the limitations of the nonnucleoside reverse transcriptase inhibitor class. J Int Assoc Physicians AIDS Care (Chic Ill) 2009; 8:354–363.
- Sekar V S-GS, Marien K. Pharmacokinetic drug-drug interaction between the new HIV protease inhibitor darunavir (TMC114) and the lipid-lowering agent pravastatin. Presented at the 8th International Workshop on Pharmacology of HIV Therapy; Budapest, Hungary, April 16–18, 2007.
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570–578.
- Aslangul E, Assoumou L, Bittar R, et al. Rosuvastatin versus pravastatin in dyslipidemic HIV-1-infected patients receiving protease inhibitors: a randomized trial. AIDS 2010; 24:77–83.
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009; 23:2133–2141.
- Aberg JA, Zackin RA, Brobst SW, et al; ACTG 5087 Study Team. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res Hum Retroviruses 2005; 21:757–767.
- Dubé MP, Wu JW, Aberg JA, et al; AIDS Clinical Trials Group A5148 Study Team. Safety and efficacy of extended-release niacin for the treatment of dyslipidaemia in patients with HIV infection: AIDS Clinical Trials Group Study A5148. Antivir Ther 2006; 11:1081–1089.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: results of ACTG A5186. J Acquir Immune Defic Syndr 2008; 47:459–466.
- Mallolas J, Podzamczer D, Milinkovic A, et al; ATAZIP Study Group. Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/rcontaining HAART: the ATAZIP study. J Acquir Immune Defic Syndr 2009; 51:29–36.
- Eron J, Andrade J, Zajdenverg R, et al. Switching from stable lopinavir/ritonavir-based to raltegravir-based combination ART resulted in a superior lipid profile at week 12 but did not demonstrate noninferior virologic efficacy at week 24. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005; 19:1051–1058.
- Worm SW, De Wit S, Weber R, et al. Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 2009; 119:805–811.
- Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005; 165:1179–1184.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, et al; Veterans Aging Cohort Study. HIV infection and the risk of diabetes mellitus. AIDS 2009; 23:1227–1234.
- Mulligan K, Yang Y, Wininger DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS 2007; 21:47–57.
Widespread use of antiretroviral therapy has caused a remarkable decline in rates of morbidity and death related to acquired immunodeficiency syndrome (AIDS) and has effectively made human immunodeficiency virus (HIV) infection a manageable—although not yet curable— chronic condition. And as the HIV-infected population on antiretroviral therapy ages, the prevalence of chronic conditions (eg, cardiovascular disease, hepatic disease, pulmonary disease, non-AIDS cancers) and deaths attributable to these conditions have also increased.1
Many of the traditional risk factors for cardiovascular disease in the general population, including smoking, dyslipidemia, and diabetes, are common in HIV-infected patients, and HIV infection itself independently increases the risk of coronary heart disease. In addition, different antiretroviral combinations can contribute, in varying degrees, to changes in lipid levels and insulin resistance, further increasing coronary risk.
Ultimately, however, the immunologic benefits of antiretroviral therapy for individual patients far exceed the modest increase in cardiovascular risk associated with certain regimens. In most cases, careful selection of the initial antiretroviral regimen and the addition of lipid-lowering or glucose-controlling medications (with close attention to drug interactions) can effectively manage the metabolic changes associated with antiretroviral therapy and obviate any premature modification of virologically suppressive regimens.
TRADITIONAL CARDIAC RISK FACTORS IN HIV PATIENTS
The risk of coronary heart disease in HIV patients is influenced mostly by traditional factors such as age, smoking, diabetes, and dyslipidemia, including high levels of total cholesterol and low-density lipoprotein cholesterol (LDL-C) and low levels of high-density lipoprotein cholesterol (HDL-C).2
In various large cohorts, HIV-infected men had a higher prevalence of smoking,3 a lower mean HDL-C level,3,4 and a higher mean triglyceride level3,4 than men without HIV infection, placing them at greater risk of coronary heart disease. However, even after adjusting for traditional risk factors, rates of atherosclerosis are still higher in people who are infected with HIV than in those who are not.5
EFFECT OF HIV INFECTION ON CORONARY RISK
HIV infection has been shown to increase coronary risk.
In the Kaiser Permanente database,6 HIV-positive patients had a significantly higher rate of hospitalizations for coronary heart disease than did people who were not infected.
Similarly, in a cohort study of almost 4,000 HIV-infected patients and more than 1 million controls, the risk of acute myocardial infarction was 75% higher for HIV-positive patients than for HIV-negative patients, even after adjusting for sex, race, hypertension, diabetes, and dyslipidemia.5
The Fat Redistribution and Metabolism (FRAM) cross-sectional study7 showed that HIV infection was associated with greater carotid intima media thickness, an established marker of atherosclerosis, independently of traditional risk factors and to virtually the same degree as smoking and male sex.
Other studies of subclinical atherosclerosis in HIV patients have yielded disparate results, likely because of differences in study design, methods of measuring carotid thickness, and characteristics of the study populations (eg, prevalence of cardiovascular risk factors and stage of HIV disease). However, a meta-analysis of six prospective cohort studies, three case-control studies, and four cross-sectional studies confirmed that HIV patients had slightly but statistically significantly greater carotid intima media thickness than HIV-negative people.8
MECHANISMS BY WHICH HIV MAY PROMOTE CORONARY HEART DISEASE
The pathogenesis of coronary heart disease in HIV infection has not been fully elucidated, but the virus appears to contribute directly to the accelerated development of atherosclerosis. It may do so through direct effects on cholesterol processing and transport, attraction of monocytes to the intimal wall, and activation of monocytes to induce an inflammatory response and endothelial proliferation.
Effects on lipids
In early HIV infection, levels of total cholesterol and HDL-C are lower. In more advanced infection, lower CD4+ lymphocyte counts have been associated with lower levels of apolipoprotein B and with smaller LDL-C particles, suggesting that HIV affects lipid processing and delivery to vessel walls.9 HIV infection is also associated with reduced clearance of LDL-C.10 HIV appears to specifically inhibit the compensatory efflux of excess cholesterol from macrophages, thus promoting the formation of foam cells in atherosclerotic plaque.11
Attraction of monocytes to the vessel wall
In vitro studies also suggest that HIV enhances migration of monocytes into the vascular intima during atherosclerotic plaque development by promoting secretion of the chemokine monocyte chemoattractant protein 112 and the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.13
Inflammation
A recent study suggests that chronic inflammation may be a key contributor to the accelerated development of atherosclerosis in HIV patients. Hsue et al14 compared carotid intima media thickness and levels of C-reactive protein (a marker of systemic inflammation) in HIV-positive and HIV-negative patients. The carotid intima media thickness was greater in all groups of HIV patients, irrespective of level of viremia or exposure to antiretroviral therapy, than in healthy controls. In addition, C-reactive protein levels remained elevated in HIV-infected participants regardless of their level of viremia.
These findings suggest not only that HIV-associated atherosclerosis is determined by advanced immunodeficiency, high-level viremia, and exposure to antiretroviral drugs, but also that persistent inflammation due to HIV infection may play an important role in accelerated atherosclerosis.
EFFECT OF ANTIRETROVIRAL THERAPY ON CORONARY RISK
Antiretroviral therapy is associated with a small but significant increase in coronary risk.
Medi-Cal,15 a retrospective study of 28,513 patients, found antiretroviral therapy to be associated with coronary heart disease among patients 18 to 33 years of age (relative risk 2.06, P < .001).
The Data Collection on Adverse Events of Anti-HIV Drugs study16 prospectively followed 23,437 patients for 94,469 person-years. Adjusted for exposure to nonnucleoside reverse transcriptase inhibitors and for hypertension and diabetes, the relative risk of myocardial infarction per year of protease inhibitor exposure was 1.16 (95% confidence interval [CI] 1.10–1.23). The relative risk was lower after adjusting for serum lipid levels but remained significant at 1.10 (95% CI 1.04–1.18).
Reports have been mixed regarding a possible association between myocardial infarction and the nucleoside reverse transcriptase inhibitor abacavir (Ziagen): several studies found a statistically significant association,17–20 and others did not.21–23 Differences in study design (observational cohort studies vs prospective randomized clinical trials), populations studied (differing in age, cardiovascular risk factor prevalence, and whether the patients had already been exposed to treatment), and outcome definition probably contributed to the different conclusions.
On the other hand, several studies have shown that suppression of HIV with antiretroviral therapy actually improves some of the surrogate markers of cardiovascular disease. For example:
- Markers of endothelial function such as flow-mediated vasodilation improve significantly within 4 weeks of a patient’s starting antiretroviral therapy, regardless of the class of antiretroviral drug used.24
- After viral suppression is achieved, levels of the markers of endothelial activation VCAM-1 and P-selectin decline significantly, as do levels of the adipocyte activation marker leptin and the coagulation marker D-dimer.25,26
- Levels of the anti-inflammatory markers adiponectin and interleukin 10 increase. 25,26
Interrupting antiretroviral therapy may increase coronary risk
Not only is uncontrolled viral replication in untreated HIV infection associated with cardiovascular disease, but interrupting antiretroviral therapy may result in a supplementary increase in coronary risk.
In the 5,472-patient Strategies for Management of Antiretroviral Therapy (SMART) trial, the rate of cardiovascular disease events was higher if treatment was interrupted than with continuous treatment, with a hazard ratio of 1.57 (95% CI 1.0–2.46, P = .05).27
This association between treatment interruption and coronary events does not appear to be related to the level of viremia.28 Rather, development of cardiovascular disease in HIV-infected patients who interrupt antiretroviral therapy may be mediated, to a large extent, by chronic inflammation in the setting of viral replication. In the treatment-interruption group, levels of the inflammatory cytokine interleukin 6 (IL-6) and the coagulation marker D-dimer were significantly elevated 1 month after randomization, and these differences were strongly associated with death (odds ratio [OR] 12.6, P < .0001 for IL-6; OR 13.1, P < .0001 for D-dimer). Elevated IL-6 levels were also significantly associated with the development of cardiovascular disease (OR 2.8, P = .03).29
METABOLIC COMPLICATIONS OF ANTIRETROVIRAL THERAPY
Persons with HIV infection may experience metabolic complications that are due to HIV itself or to its treatment.
Cross-sectional studies that included HIV-negative patients as controls have demonstrated changes in lipid processing that are known to promote atherosclerosis. For example, persons with HIV infection have smaller LDL-C particles30 and higher levels of circulating oxidized LDL-C.31
In the Multicenter AIDS Cohort Study (MACS), after HIV seroconversion, nonfasting total cholesterol, LDL-C, and HDL-C levels declined, which is consistent with a chronic inflammatory state. After antiretroviral therapy was started, lipid levels returned to baseline levels or slightly higher except for HDL-C, which remained low.9 These changes may be due to a general “return to health,” or they may be direct medication effects.
Similar patterns were seen in the SMART study.28 Participants randomized to receive intermittent antiretroviral therapy had overall decreases in all lipid levels, with a marked reduction in HDL-C, while those randomized to receive continuous therapy had increased levels of all lipids, including HDL-C, at 12 months. Overall, the ratio of total cholesterol to HDL-C actually increased for participants on episodic therapy, while it decreased in the continuous-treatment group. Along with continued vascular inflammation, the low HDL-C may have contributed to the worse cardiovascular outcomes in patients who received intermittent antiretroviral therapy.
Some lipid changes associated with antiretroviral therapy may actually be beneficial. For example, nonnucleoside reverse transcriptase inhibitors may raise HDL-C levels. However, such increases alone do not necessarily offset the other lipid changes or translate to an observed improvement in coronary risk.32
The degree of dyslipidemia and specific lipid changes differ among the different classes of antiretroviral drugs and even among the individual drugs within each class. Furthermore, the magnitude of the observed lipid changes varies widely among patients on the same antiretroviral regimen, reflecting the likely important role of host genomics.
While the protease inhibitors and nonnucleoside reverse transcriptase inhibitors have well-described effects on lipids (described in greater detail in the following sections), there have been no reported significant changes in lipid profiles or cardiovascular risk associated with the newest classes, ie, fusion inhibitors such as enfuvirtide (Fuzeon), CC chemokine receptor type 5 (CCR5) receptor inhibitors such as maraviroc (Selzentry), or integrase inhibitors such as raltegravir (Isentress).
Impact of protease inhibitors on lipids
Ritonavir (Norvir) and ritonavir-boosted protease inhibitor combinations cause the most significant increases in lipids. Currently, ritonavir is used in low doses to boost the levels of most other protease inhibitors as the standard of care in protease inhibitor-based regimens. However, in most patients, giving ritonavir with protease inhibitors raises lipid levels, particularly triglycerides.
Most boosted protease inhibitor regimens have similar effects on lipid levels, with some exceptions.
Tipranavir (Aptivus) plus ritonavir, for example, markedly raises total cholesterol and triglyceride levels and would not be recommended for patients with dyslipidemia at baseline.33
Atazanavir (Reyataz)34,35 plus ritonavir and darunavir (Prezista)36 plus ritonavir cause more modest lipid changes. Unboosted atazanavir raises lipid levels only minimally, if at all,34,35 but it is no longer a preferred regimen according to US Department of Health and Human Services guidelines.42
Impact of nonnucleoside reverse transcriptase inhibitors on lipids
Efavirenz (Sustiva), a nonnucleoside reverse transcriptase inhibitor, when added to a regimen of two or three nucleoside reverse transcriptase inhibitors, resulted in modest increases in all lipids, including HDL-C (a potentially beneficial change) at 96 weeks compared with a regimen of three nucleoside reverse transcriptase inhibitors only.43
Nevirapine (Viramune), compared with efavirenz, results in a more favorable lipid profile in previously untreated patients, as shown by larger increases in HDL-C and smaller increases in triglycerides at 48 weeks.44
Etravirine (Intelence), the newest nonnucleoside reverse transcriptase inhibitor, does not appear to cause any further increase in lipids when added to a regimen containing darunavir-ritonavir and nucleoside agents.45
Impact of nucleoside reverse transcriptase inhibitors on lipids
As a class, nucleoside reverse transcriptase inhibitors have been associated with mitochondrial toxicity and insulin resistance,46 but the lipid changes associated with them are generally less significant than those caused by protease inhibitors or nonnucleoside reverse transcriptase inhibitors. Nevertheless, within the class, there is considerable variability in lipid changes associated with specific agents.
Stavudine (Zerit), for example, is associated with hypertriglyceridemia.
Tenofovir (Viread), for another example, in combination with emtricitabine (Emtriva) and the nonnucleoside reverse transcriptase inhibitor efavirenz (the three drugs are contained in a formulation called Atripla) was associated with a smaller increase in fasting total cholesterol than with zidovudine-lamivudine and efavirenz at 96 weeks.47
A recent placebo-controlled, crossover, pilot study of 17 HIV-infected patients suggested that tenofovir may actually have independent lipid-lowering properties.48
Abacavir, as discussed above, has been reported to be associated with a higher risk of myocardial infarction, but this is debatable.
MANAGING CORONARY RISK FACTORS IN HIV-INFECTED PATIENTS
Cardiovascular risk assessment
In HIV patients, cardiovascular risk can be assessed using models derived from large epidemiologic studies such as the Framingham Heart Study.49
Current guidelines from the Infectious Diseases Society of America and the AIDS Clinical Trials Group (ACTG) for evaluating and managing dyslipidemia in HIV-infected adults are based on the National Cholesterol Education Program Adult Treatment Panel III.50 They recommend obtaining a fasting lipid profile before starting antiretroviral therapy and within 3 to 6 months after starting a new regimen.
The guidelines also recommend stratifying risk by counting the number of cardiovascular risk factors, as is done for the general population. If the patient has more than two factors, the Framingham equation should be used to calculate the 10-year risk of myocardial infarction or cardiac death. Interventions should be offered for modifiable cardiovascular risk factors such as smoking, hypertension, physical inactivity, and diabetes mellitus. LDL-C goals should be determined, and lipid-lowering drugs should be initiated accordingly. If triglyceride levels are 200 to 500 mg/dL and levels of “non-HDL-C” (total cholesterol minus the HDL-C level) are high, a statin is recommended. If the triglyceride level is higher than 500 mg/dL, a fibrate should be started.51
Dyslipidemia management
In HIV patients, statin and fibrate therapy must be considered cautiously, given the important drug interactions with protease inhibitors and especially ritonavir. Many statins are metabolized by cytochrome P3A4, which protease inhibitors inhibit.
Statins generally considered safe to use with most protease inhibitors:
- Pravastatin (Pravachol)
- Rosuvastatin (Crestor)
- Atorvastatin (Lipitor).
Exceptions and caveats:
- Pravastatin should not be prescribed with boosted darunavir.
- Data for fluvastatin (Lescol) in HIV-infected patients on antiretroviral therapy are limited.
- Lovastatin (Mevacor) and simvastatin (Zocor) are contraindicated with protease inhibitor therapy.52
- In contrast to the increase in statin levels seen with protease inhibitors, efavirenz lowers levels of simvastatin, pravastatin, and atorvastatin.53,54
Ezetimibe (Zetia), which is metabolized independently of the cytochrome P450 system, has been shown to be safe and effective when given to HIV-infected patients on antiretroviral therapy.58
Fenofibrate (Lofibra) is recommended by current guidelines for patients with elevated triglyceride levels (> 500 mg/dL).51 In the ACTG 5087 study, a combination of fenofibrate plus pravastatin was found to be safe and effective in improving lipid profiles.59
Long-acting niacin resulted in significant improvements in triglycerides, total cholesterol, HDL-C, and LDL-C after 48 weeks of use, although insulin sensitivity worsened.60
Fish oil has been shown to be an effective alternative to fibrates, or it can be used in combination with them.61
Switching antiretroviral agents vs adding lipid-lowering agents. In some patients with significant dyslipidemia, switching antiretro viral agents may lower lipid levels without compromising virologic control.62 However, due to the multifactorial nature of dyslipidemia in HIV patients on antiretroviral therapy, switching the HIV therapy alone may not result in sufficient improvement in the lipid profile45 and may be associated with virologic failure, particularly among patients who have underlying treatment-resistant HIV.63
In many cases, adding lipid-lowering agents may be more beneficial than switching the antiretroviral therapy. For example, a randomized trial in HIV-infected patients with hyperlipidemia found that adding a lipid-lowering agent such as pravastatin or bezafibrate to the unchanged antiretroviral regimen resulted in greater improvement in total cholesterol, LDL-C, and triglyceride levels than switching from a protease inhibitor to either nevirapine or efavirenz.64
Given the complexity of prescribing lipid-lowering therapies to patients on antiretroviral therapy, we recommend that providers check with a pharmacist or refer to package inserts and other medical literature if they are unfamiliar with these drug interactions and responses to lipid-lowering therapies.
Managing insulin resistance
Diabetes mellitus is a well-known risk factor for coronary heart disease. The Data Collection on Adverse Events of Anti-HIV Drugs study found a higher incidence of coronary heart disease in HIV-infected patients, with higher rates in those with longer duration of diabetes.65 The prevalence of diabetes in HIV-infected populations varies, depending on demographic characteristics,65,66 prevalence of coinfection with hepatitis C virus,66 and prevalence of exposure to antiretroviral drugs67 in the study population.
Drugs that lessen insulin resistance include the thiazolidinedione rosiglitazone (Avandia) and the biguanide metformin (Glucophage). In a randomized trial, both drugs, alone or in combination, improved insulin sensitivity in HIV-infected patients, but neither lessened the amount of visceral or subcutaneous fat.68
Smoking cessation
Smoking is another well-known modifiable risk factor for coronary heart disease.
The prevalence of smoking is usually higher in HIV patients than in HIV-negative people. For example, a French cohort study reported smoking prevalence rates of 56.6% in HIV-infected men vs 32.7% in HIV-negative men; in women, the rates were 58% vs 28.1%. The 5-year relative risk of coronary heart disease in HIV-infected vs HIV-negative persons was 1.20 for men and 1.59 for women. The estimated attributable risk due to smoking was 65% for men and 29% for women.3
Therefore, smoking cessation should be a top priority in managing cardiovascular risk in HIV-infected patients. In fact, control of modifiable risk factors through lifestyle changes such as smoking cessation, dietary changes, and exercise is likely to have a significant impact on cardiovascular risk in this population.
Widespread use of antiretroviral therapy has caused a remarkable decline in rates of morbidity and death related to acquired immunodeficiency syndrome (AIDS) and has effectively made human immunodeficiency virus (HIV) infection a manageable—although not yet curable— chronic condition. And as the HIV-infected population on antiretroviral therapy ages, the prevalence of chronic conditions (eg, cardiovascular disease, hepatic disease, pulmonary disease, non-AIDS cancers) and deaths attributable to these conditions have also increased.1
Many of the traditional risk factors for cardiovascular disease in the general population, including smoking, dyslipidemia, and diabetes, are common in HIV-infected patients, and HIV infection itself independently increases the risk of coronary heart disease. In addition, different antiretroviral combinations can contribute, in varying degrees, to changes in lipid levels and insulin resistance, further increasing coronary risk.
Ultimately, however, the immunologic benefits of antiretroviral therapy for individual patients far exceed the modest increase in cardiovascular risk associated with certain regimens. In most cases, careful selection of the initial antiretroviral regimen and the addition of lipid-lowering or glucose-controlling medications (with close attention to drug interactions) can effectively manage the metabolic changes associated with antiretroviral therapy and obviate any premature modification of virologically suppressive regimens.
TRADITIONAL CARDIAC RISK FACTORS IN HIV PATIENTS
The risk of coronary heart disease in HIV patients is influenced mostly by traditional factors such as age, smoking, diabetes, and dyslipidemia, including high levels of total cholesterol and low-density lipoprotein cholesterol (LDL-C) and low levels of high-density lipoprotein cholesterol (HDL-C).2
In various large cohorts, HIV-infected men had a higher prevalence of smoking,3 a lower mean HDL-C level,3,4 and a higher mean triglyceride level3,4 than men without HIV infection, placing them at greater risk of coronary heart disease. However, even after adjusting for traditional risk factors, rates of atherosclerosis are still higher in people who are infected with HIV than in those who are not.5
EFFECT OF HIV INFECTION ON CORONARY RISK
HIV infection has been shown to increase coronary risk.
In the Kaiser Permanente database,6 HIV-positive patients had a significantly higher rate of hospitalizations for coronary heart disease than did people who were not infected.
Similarly, in a cohort study of almost 4,000 HIV-infected patients and more than 1 million controls, the risk of acute myocardial infarction was 75% higher for HIV-positive patients than for HIV-negative patients, even after adjusting for sex, race, hypertension, diabetes, and dyslipidemia.5
The Fat Redistribution and Metabolism (FRAM) cross-sectional study7 showed that HIV infection was associated with greater carotid intima media thickness, an established marker of atherosclerosis, independently of traditional risk factors and to virtually the same degree as smoking and male sex.
Other studies of subclinical atherosclerosis in HIV patients have yielded disparate results, likely because of differences in study design, methods of measuring carotid thickness, and characteristics of the study populations (eg, prevalence of cardiovascular risk factors and stage of HIV disease). However, a meta-analysis of six prospective cohort studies, three case-control studies, and four cross-sectional studies confirmed that HIV patients had slightly but statistically significantly greater carotid intima media thickness than HIV-negative people.8
MECHANISMS BY WHICH HIV MAY PROMOTE CORONARY HEART DISEASE
The pathogenesis of coronary heart disease in HIV infection has not been fully elucidated, but the virus appears to contribute directly to the accelerated development of atherosclerosis. It may do so through direct effects on cholesterol processing and transport, attraction of monocytes to the intimal wall, and activation of monocytes to induce an inflammatory response and endothelial proliferation.
Effects on lipids
In early HIV infection, levels of total cholesterol and HDL-C are lower. In more advanced infection, lower CD4+ lymphocyte counts have been associated with lower levels of apolipoprotein B and with smaller LDL-C particles, suggesting that HIV affects lipid processing and delivery to vessel walls.9 HIV infection is also associated with reduced clearance of LDL-C.10 HIV appears to specifically inhibit the compensatory efflux of excess cholesterol from macrophages, thus promoting the formation of foam cells in atherosclerotic plaque.11
Attraction of monocytes to the vessel wall
In vitro studies also suggest that HIV enhances migration of monocytes into the vascular intima during atherosclerotic plaque development by promoting secretion of the chemokine monocyte chemoattractant protein 112 and the expression of endothelial cell adhesion molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1 (VCAM-1), and E-selectin.13
Inflammation
A recent study suggests that chronic inflammation may be a key contributor to the accelerated development of atherosclerosis in HIV patients. Hsue et al14 compared carotid intima media thickness and levels of C-reactive protein (a marker of systemic inflammation) in HIV-positive and HIV-negative patients. The carotid intima media thickness was greater in all groups of HIV patients, irrespective of level of viremia or exposure to antiretroviral therapy, than in healthy controls. In addition, C-reactive protein levels remained elevated in HIV-infected participants regardless of their level of viremia.
These findings suggest not only that HIV-associated atherosclerosis is determined by advanced immunodeficiency, high-level viremia, and exposure to antiretroviral drugs, but also that persistent inflammation due to HIV infection may play an important role in accelerated atherosclerosis.
EFFECT OF ANTIRETROVIRAL THERAPY ON CORONARY RISK
Antiretroviral therapy is associated with a small but significant increase in coronary risk.
Medi-Cal,15 a retrospective study of 28,513 patients, found antiretroviral therapy to be associated with coronary heart disease among patients 18 to 33 years of age (relative risk 2.06, P < .001).
The Data Collection on Adverse Events of Anti-HIV Drugs study16 prospectively followed 23,437 patients for 94,469 person-years. Adjusted for exposure to nonnucleoside reverse transcriptase inhibitors and for hypertension and diabetes, the relative risk of myocardial infarction per year of protease inhibitor exposure was 1.16 (95% confidence interval [CI] 1.10–1.23). The relative risk was lower after adjusting for serum lipid levels but remained significant at 1.10 (95% CI 1.04–1.18).
Reports have been mixed regarding a possible association between myocardial infarction and the nucleoside reverse transcriptase inhibitor abacavir (Ziagen): several studies found a statistically significant association,17–20 and others did not.21–23 Differences in study design (observational cohort studies vs prospective randomized clinical trials), populations studied (differing in age, cardiovascular risk factor prevalence, and whether the patients had already been exposed to treatment), and outcome definition probably contributed to the different conclusions.
On the other hand, several studies have shown that suppression of HIV with antiretroviral therapy actually improves some of the surrogate markers of cardiovascular disease. For example:
- Markers of endothelial function such as flow-mediated vasodilation improve significantly within 4 weeks of a patient’s starting antiretroviral therapy, regardless of the class of antiretroviral drug used.24
- After viral suppression is achieved, levels of the markers of endothelial activation VCAM-1 and P-selectin decline significantly, as do levels of the adipocyte activation marker leptin and the coagulation marker D-dimer.25,26
- Levels of the anti-inflammatory markers adiponectin and interleukin 10 increase. 25,26
Interrupting antiretroviral therapy may increase coronary risk
Not only is uncontrolled viral replication in untreated HIV infection associated with cardiovascular disease, but interrupting antiretroviral therapy may result in a supplementary increase in coronary risk.
In the 5,472-patient Strategies for Management of Antiretroviral Therapy (SMART) trial, the rate of cardiovascular disease events was higher if treatment was interrupted than with continuous treatment, with a hazard ratio of 1.57 (95% CI 1.0–2.46, P = .05).27
This association between treatment interruption and coronary events does not appear to be related to the level of viremia.28 Rather, development of cardiovascular disease in HIV-infected patients who interrupt antiretroviral therapy may be mediated, to a large extent, by chronic inflammation in the setting of viral replication. In the treatment-interruption group, levels of the inflammatory cytokine interleukin 6 (IL-6) and the coagulation marker D-dimer were significantly elevated 1 month after randomization, and these differences were strongly associated with death (odds ratio [OR] 12.6, P < .0001 for IL-6; OR 13.1, P < .0001 for D-dimer). Elevated IL-6 levels were also significantly associated with the development of cardiovascular disease (OR 2.8, P = .03).29
METABOLIC COMPLICATIONS OF ANTIRETROVIRAL THERAPY
Persons with HIV infection may experience metabolic complications that are due to HIV itself or to its treatment.
Cross-sectional studies that included HIV-negative patients as controls have demonstrated changes in lipid processing that are known to promote atherosclerosis. For example, persons with HIV infection have smaller LDL-C particles30 and higher levels of circulating oxidized LDL-C.31
In the Multicenter AIDS Cohort Study (MACS), after HIV seroconversion, nonfasting total cholesterol, LDL-C, and HDL-C levels declined, which is consistent with a chronic inflammatory state. After antiretroviral therapy was started, lipid levels returned to baseline levels or slightly higher except for HDL-C, which remained low.9 These changes may be due to a general “return to health,” or they may be direct medication effects.
Similar patterns were seen in the SMART study.28 Participants randomized to receive intermittent antiretroviral therapy had overall decreases in all lipid levels, with a marked reduction in HDL-C, while those randomized to receive continuous therapy had increased levels of all lipids, including HDL-C, at 12 months. Overall, the ratio of total cholesterol to HDL-C actually increased for participants on episodic therapy, while it decreased in the continuous-treatment group. Along with continued vascular inflammation, the low HDL-C may have contributed to the worse cardiovascular outcomes in patients who received intermittent antiretroviral therapy.
Some lipid changes associated with antiretroviral therapy may actually be beneficial. For example, nonnucleoside reverse transcriptase inhibitors may raise HDL-C levels. However, such increases alone do not necessarily offset the other lipid changes or translate to an observed improvement in coronary risk.32
The degree of dyslipidemia and specific lipid changes differ among the different classes of antiretroviral drugs and even among the individual drugs within each class. Furthermore, the magnitude of the observed lipid changes varies widely among patients on the same antiretroviral regimen, reflecting the likely important role of host genomics.
While the protease inhibitors and nonnucleoside reverse transcriptase inhibitors have well-described effects on lipids (described in greater detail in the following sections), there have been no reported significant changes in lipid profiles or cardiovascular risk associated with the newest classes, ie, fusion inhibitors such as enfuvirtide (Fuzeon), CC chemokine receptor type 5 (CCR5) receptor inhibitors such as maraviroc (Selzentry), or integrase inhibitors such as raltegravir (Isentress).
Impact of protease inhibitors on lipids
Ritonavir (Norvir) and ritonavir-boosted protease inhibitor combinations cause the most significant increases in lipids. Currently, ritonavir is used in low doses to boost the levels of most other protease inhibitors as the standard of care in protease inhibitor-based regimens. However, in most patients, giving ritonavir with protease inhibitors raises lipid levels, particularly triglycerides.
Most boosted protease inhibitor regimens have similar effects on lipid levels, with some exceptions.
Tipranavir (Aptivus) plus ritonavir, for example, markedly raises total cholesterol and triglyceride levels and would not be recommended for patients with dyslipidemia at baseline.33
Atazanavir (Reyataz)34,35 plus ritonavir and darunavir (Prezista)36 plus ritonavir cause more modest lipid changes. Unboosted atazanavir raises lipid levels only minimally, if at all,34,35 but it is no longer a preferred regimen according to US Department of Health and Human Services guidelines.42
Impact of nonnucleoside reverse transcriptase inhibitors on lipids
Efavirenz (Sustiva), a nonnucleoside reverse transcriptase inhibitor, when added to a regimen of two or three nucleoside reverse transcriptase inhibitors, resulted in modest increases in all lipids, including HDL-C (a potentially beneficial change) at 96 weeks compared with a regimen of three nucleoside reverse transcriptase inhibitors only.43
Nevirapine (Viramune), compared with efavirenz, results in a more favorable lipid profile in previously untreated patients, as shown by larger increases in HDL-C and smaller increases in triglycerides at 48 weeks.44
Etravirine (Intelence), the newest nonnucleoside reverse transcriptase inhibitor, does not appear to cause any further increase in lipids when added to a regimen containing darunavir-ritonavir and nucleoside agents.45
Impact of nucleoside reverse transcriptase inhibitors on lipids
As a class, nucleoside reverse transcriptase inhibitors have been associated with mitochondrial toxicity and insulin resistance,46 but the lipid changes associated with them are generally less significant than those caused by protease inhibitors or nonnucleoside reverse transcriptase inhibitors. Nevertheless, within the class, there is considerable variability in lipid changes associated with specific agents.
Stavudine (Zerit), for example, is associated with hypertriglyceridemia.
Tenofovir (Viread), for another example, in combination with emtricitabine (Emtriva) and the nonnucleoside reverse transcriptase inhibitor efavirenz (the three drugs are contained in a formulation called Atripla) was associated with a smaller increase in fasting total cholesterol than with zidovudine-lamivudine and efavirenz at 96 weeks.47
A recent placebo-controlled, crossover, pilot study of 17 HIV-infected patients suggested that tenofovir may actually have independent lipid-lowering properties.48
Abacavir, as discussed above, has been reported to be associated with a higher risk of myocardial infarction, but this is debatable.
MANAGING CORONARY RISK FACTORS IN HIV-INFECTED PATIENTS
Cardiovascular risk assessment
In HIV patients, cardiovascular risk can be assessed using models derived from large epidemiologic studies such as the Framingham Heart Study.49
Current guidelines from the Infectious Diseases Society of America and the AIDS Clinical Trials Group (ACTG) for evaluating and managing dyslipidemia in HIV-infected adults are based on the National Cholesterol Education Program Adult Treatment Panel III.50 They recommend obtaining a fasting lipid profile before starting antiretroviral therapy and within 3 to 6 months after starting a new regimen.
The guidelines also recommend stratifying risk by counting the number of cardiovascular risk factors, as is done for the general population. If the patient has more than two factors, the Framingham equation should be used to calculate the 10-year risk of myocardial infarction or cardiac death. Interventions should be offered for modifiable cardiovascular risk factors such as smoking, hypertension, physical inactivity, and diabetes mellitus. LDL-C goals should be determined, and lipid-lowering drugs should be initiated accordingly. If triglyceride levels are 200 to 500 mg/dL and levels of “non-HDL-C” (total cholesterol minus the HDL-C level) are high, a statin is recommended. If the triglyceride level is higher than 500 mg/dL, a fibrate should be started.51
Dyslipidemia management
In HIV patients, statin and fibrate therapy must be considered cautiously, given the important drug interactions with protease inhibitors and especially ritonavir. Many statins are metabolized by cytochrome P3A4, which protease inhibitors inhibit.
Statins generally considered safe to use with most protease inhibitors:
- Pravastatin (Pravachol)
- Rosuvastatin (Crestor)
- Atorvastatin (Lipitor).
Exceptions and caveats:
- Pravastatin should not be prescribed with boosted darunavir.
- Data for fluvastatin (Lescol) in HIV-infected patients on antiretroviral therapy are limited.
- Lovastatin (Mevacor) and simvastatin (Zocor) are contraindicated with protease inhibitor therapy.52
- In contrast to the increase in statin levels seen with protease inhibitors, efavirenz lowers levels of simvastatin, pravastatin, and atorvastatin.53,54
Ezetimibe (Zetia), which is metabolized independently of the cytochrome P450 system, has been shown to be safe and effective when given to HIV-infected patients on antiretroviral therapy.58
Fenofibrate (Lofibra) is recommended by current guidelines for patients with elevated triglyceride levels (> 500 mg/dL).51 In the ACTG 5087 study, a combination of fenofibrate plus pravastatin was found to be safe and effective in improving lipid profiles.59
Long-acting niacin resulted in significant improvements in triglycerides, total cholesterol, HDL-C, and LDL-C after 48 weeks of use, although insulin sensitivity worsened.60
Fish oil has been shown to be an effective alternative to fibrates, or it can be used in combination with them.61
Switching antiretroviral agents vs adding lipid-lowering agents. In some patients with significant dyslipidemia, switching antiretro viral agents may lower lipid levels without compromising virologic control.62 However, due to the multifactorial nature of dyslipidemia in HIV patients on antiretroviral therapy, switching the HIV therapy alone may not result in sufficient improvement in the lipid profile45 and may be associated with virologic failure, particularly among patients who have underlying treatment-resistant HIV.63
In many cases, adding lipid-lowering agents may be more beneficial than switching the antiretroviral therapy. For example, a randomized trial in HIV-infected patients with hyperlipidemia found that adding a lipid-lowering agent such as pravastatin or bezafibrate to the unchanged antiretroviral regimen resulted in greater improvement in total cholesterol, LDL-C, and triglyceride levels than switching from a protease inhibitor to either nevirapine or efavirenz.64
Given the complexity of prescribing lipid-lowering therapies to patients on antiretroviral therapy, we recommend that providers check with a pharmacist or refer to package inserts and other medical literature if they are unfamiliar with these drug interactions and responses to lipid-lowering therapies.
Managing insulin resistance
Diabetes mellitus is a well-known risk factor for coronary heart disease. The Data Collection on Adverse Events of Anti-HIV Drugs study found a higher incidence of coronary heart disease in HIV-infected patients, with higher rates in those with longer duration of diabetes.65 The prevalence of diabetes in HIV-infected populations varies, depending on demographic characteristics,65,66 prevalence of coinfection with hepatitis C virus,66 and prevalence of exposure to antiretroviral drugs67 in the study population.
Drugs that lessen insulin resistance include the thiazolidinedione rosiglitazone (Avandia) and the biguanide metformin (Glucophage). In a randomized trial, both drugs, alone or in combination, improved insulin sensitivity in HIV-infected patients, but neither lessened the amount of visceral or subcutaneous fat.68
Smoking cessation
Smoking is another well-known modifiable risk factor for coronary heart disease.
The prevalence of smoking is usually higher in HIV patients than in HIV-negative people. For example, a French cohort study reported smoking prevalence rates of 56.6% in HIV-infected men vs 32.7% in HIV-negative men; in women, the rates were 58% vs 28.1%. The 5-year relative risk of coronary heart disease in HIV-infected vs HIV-negative persons was 1.20 for men and 1.59 for women. The estimated attributable risk due to smoking was 65% for men and 29% for women.3
Therefore, smoking cessation should be a top priority in managing cardiovascular risk in HIV-infected patients. In fact, control of modifiable risk factors through lifestyle changes such as smoking cessation, dietary changes, and exercise is likely to have a significant impact on cardiovascular risk in this population.
- Palella FJ, Baker RK, Moorman AC, et al; HIV Outpatient Study Investigators. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006; 43:27–34.
- Lichtenstein KA, Armon C, Buchacz K, Moorman AC, Wood KC, Brooks JT; HOPS investigators. Analysis of cardiovascular risk factors in the HIV Outpatient Study (HOPS) cohort. Presented at the 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO; 2006.
- Savès M, Chêne G, Ducimetière P, et al; French WHO MONICA Project and the APROCO (ANRS EP11) Study Group. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003; 37:292–298.
- Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007; 45:1074–1081.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Klein D, Hurley LB, Quesenberry CP, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr 2002; 30:471–477.
- Grunfeld C, Delaney JA, Wanke C, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS 2009; 23:1841–1849.
- Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009; 95:1826–1835.
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003; 289:2978–2982.
- Shahmanesh M, Das S, Stolinski M, et al. Antiretroviral treatment reduces very-low-density lipoprotein and intermediate-density lipoprotein apolipoprotein B fractional catabolic rate in human immunodeficiency virus-infected patients with mild dyslipidemia. J Clin Endocrinol Metab 2005; 90:755–760.
- Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365.
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 2001; 97:352–358.
- Fisher SD, Miller TL, Lipshultz SE. Impact of HIV and highly active antiretroviral therapy on leukocyte adhesion molecules, arterial inflammation, dyslipidemia, and atherosclerosis. Atherosclerosis 2006; 185:1–11.
- Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009; 23:1059–1067.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- DAD Study Group; Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007: 356:1723–1735.
- DAD Study Group; Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008; 371:1417–1426.
- Durand M, Sheehy O, Baril JG, Lelorier J, Tremblay C; GRUCHUM Research Center (Groupe de Recherche de l’UHRESS du Centre Hospitalier Universitaire de Montréal). Relation between use of nucleoside reverse transcriptase inhibitors (NRTI) and risk of myocardial infarction (MI): a nested case control study using Quebec’s public health insurance database (QPHID). Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 17–22, 2009.
- Lang S, Mary-Krause M, Cotte L, et al; the Clinical Epi Group of the French Hospital Database on HIV. Impact of specific NRTI and PI exposure on the risk of myocardial infarction: a case-control study nested within FHDH ANRS CO4. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Strategies for Management of Anti-Retroviral Therapy/INSIGHT. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS 2008; 22:F17–F24.
- Bedimo R, Westfall A, Drechsler H, Tebas P. Abacavir use and risk of acute myocardial infarction and cerebrovascular disease in the HAART era. Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 19–22, 2009.
- Brothers CH, Hernandez JE, Cutrell AG, et al. Risk of myocardial infarction and abacavir therapy: no increased risk across 52 GlaxoSmithKline-sponsored clinical trials in adult subjects. J Acquir Immune Defic Syndr 2009; 51:20–28.
- Benson C, Ribaudo H, Zheng E, et al; the ACTG A5001/ALLRT Protocol Team. No Association of Abacavir Use with Risk of Myocardial Infarction or Severe Cardiovascular Disease Events: Results from ACTG A5001. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Torriani FJ, Komarow L, Parker RA, et al; ACTG 5152s Study Team. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: The ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol 2008; 52:569–576.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- van Vonderen MG, Hassink EA, van Agtmael MA, et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis 2009; 199:1186–1194.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group; El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006; 355:2283–2296.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Kuller LH, Tracy R, Belloso WINSIGHT SMART Study Group. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203.
- Badiou S, De Boever CM, Dupuy AM, Baillat V, Cristol JP, Reynes J. Small dense LDL and atherogenic lipid profile in HIV-positive adults: influence of lopinavir/ritonavir-containing regimen. AIDS 2003; 17:772–774.
- Duong M, Petit JM, Martha B, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials 2006; 7:41–47.
- Fisac C, Fumero E, Crespo M, et al. Metabolic benefits 24 months after replacing a protease inhibitor with abacavir, efavirenz or nevirapine. AIDS 2005; 19:917–925.
- Hicks CB, Cahn P, Cooper DA, et al; RESIST investigator group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatmentexperienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 2006; 368:466–475.
- Malan DR, Krantz E, David N, Wirtz V, Hammond J, McGrath D; 089 Study Group. Efficacy and safety of atazanavir, with or without ritonavir, as part of once-daily highly active antiretroviral therapy regimens in antiretroviral-naive patients. J Acquir Immune Defic Syndr 2008; 47:161–167.
- Anastos K, Lu D, Shi Q, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr 2007; 45:34–42.
- Tomaka F, Lefebvre E, Sekar V, et al. Effects of ritonavir-boosted darunavir vs ritonavir-boosted atazanavir on lipid and glucose parameters in HIV-negative, healthy volunteers. HIV Med 2009; 10:318–327.
- Eron J, Yeni P, Gathe J, et al; KLEAN study team. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial. Lancet 2006; 368:476–482.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Kumar PN, Rodriguez-French A, Thompson MA, et al; ESS40002 Study Team. A prospective, 96-week study of the impact of trizivir, combivir/nelfinavir, and lamivudine/stavudine/nelfinavir on lipids, metabolic parameters and efficacy in antiretroviral-naive patients: effect of sex and ethnicity. HIV Med 2006; 7:85–98.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr 2009; 50:367–374.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents— A Working Group of the Office of AIDS Research Advisory Council (OARAC). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. December 1, 1009. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 29, 2010.
- Shikuma CM, Yang Y, Glesby MJ, et al. Metabolic effects of protease inhibitor-sparing antiretroviral regimens given as initial treatment of HIV-1 Infection (AIDS Clinical Trials Group Study A5095). J Acquir Immune Defic Syndr 2007; 44:540–550.
- van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapynaive patients infected with HIV-1. PLoS Med 2004; 1:e19.
- Katlama C, Haubrich R, Lalezari J, et al; DUET-1, DUET-2 study groups. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS 2009; 23:2289–2300.
- Hammond E, Nolan D, James I, Metcalf C, Mallal S. Reduction of mitochondrial DNA content and respiratory chain activity occurs in adipocytes within 6–12 months of commencing nucleoside reverse transcriptase inhibitor therapy. AIDS 2004; 18:815–817.
- Pozniak AL, Gallant JE, DeJesus E, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz versus fixed-dose zidovudine/lamivudine and efavirenz in antiretroviral-naive patients: virologic, immunologic, and morphologic changes—a 96-week analysis. J Acquir Immune Defic Syndr 2006; 43:535–540.
- Tungsiripat M, Kitch D, Glesby M, et al. A pilot study to determine the effect on dyslipidemia of the addition of tenofovir to stable background ART in HIV-infected subjects: results from the A5206 Study Team. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Law MG, Friis-Møller N, El-Sadr WM, et al; D:A:D Study Group. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med 2006; 7:218–230.
- Aberg JA. Cardiovascular complications in HIV management: past, present, and future. J Acquir Immune Defic Syndr 2009; 50:54–64.
- Dubé MP, Stein JH, Aberg JA, et al; Adult AIDS Clinical Trials Group Cardiovascular Subcommittee. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003; 37:613–627.
- Fichtenbaum CJ. Metabolic abnormalities associated with HIV infection and antiretroviral therapy. Curr Infect Dis Rep 2009; 11:84–92.
- Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al; AIDS Clinical Trials Group A5108 Team. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005; 39:307–312.
- Grennan T, Walmsley S. Etravirine for HIV-I: addressing the limitations of the nonnucleoside reverse transcriptase inhibitor class. J Int Assoc Physicians AIDS Care (Chic Ill) 2009; 8:354–363.
- Sekar V S-GS, Marien K. Pharmacokinetic drug-drug interaction between the new HIV protease inhibitor darunavir (TMC114) and the lipid-lowering agent pravastatin. Presented at the 8th International Workshop on Pharmacology of HIV Therapy; Budapest, Hungary, April 16–18, 2007.
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570–578.
- Aslangul E, Assoumou L, Bittar R, et al. Rosuvastatin versus pravastatin in dyslipidemic HIV-1-infected patients receiving protease inhibitors: a randomized trial. AIDS 2010; 24:77–83.
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009; 23:2133–2141.
- Aberg JA, Zackin RA, Brobst SW, et al; ACTG 5087 Study Team. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res Hum Retroviruses 2005; 21:757–767.
- Dubé MP, Wu JW, Aberg JA, et al; AIDS Clinical Trials Group A5148 Study Team. Safety and efficacy of extended-release niacin for the treatment of dyslipidaemia in patients with HIV infection: AIDS Clinical Trials Group Study A5148. Antivir Ther 2006; 11:1081–1089.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: results of ACTG A5186. J Acquir Immune Defic Syndr 2008; 47:459–466.
- Mallolas J, Podzamczer D, Milinkovic A, et al; ATAZIP Study Group. Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/rcontaining HAART: the ATAZIP study. J Acquir Immune Defic Syndr 2009; 51:29–36.
- Eron J, Andrade J, Zajdenverg R, et al. Switching from stable lopinavir/ritonavir-based to raltegravir-based combination ART resulted in a superior lipid profile at week 12 but did not demonstrate noninferior virologic efficacy at week 24. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005; 19:1051–1058.
- Worm SW, De Wit S, Weber R, et al. Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 2009; 119:805–811.
- Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005; 165:1179–1184.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, et al; Veterans Aging Cohort Study. HIV infection and the risk of diabetes mellitus. AIDS 2009; 23:1227–1234.
- Mulligan K, Yang Y, Wininger DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS 2007; 21:47–57.
- Palella FJ, Baker RK, Moorman AC, et al; HIV Outpatient Study Investigators. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006; 43:27–34.
- Lichtenstein KA, Armon C, Buchacz K, Moorman AC, Wood KC, Brooks JT; HOPS investigators. Analysis of cardiovascular risk factors in the HIV Outpatient Study (HOPS) cohort. Presented at the 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO; 2006.
- Savès M, Chêne G, Ducimetière P, et al; French WHO MONICA Project and the APROCO (ANRS EP11) Study Group. Risk factors for coronary heart disease in patients treated for human immunodeficiency virus infection compared with the general population. Clin Infect Dis 2003; 37:292–298.
- Kaplan RC, Kingsley LA, Sharrett AR, et al. Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007; 45:1074–1081.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Klein D, Hurley LB, Quesenberry CP, Sidney S. Do protease inhibitors increase the risk for coronary heart disease in patients with HIV-1 infection? J Acquir Immune Defic Syndr 2002; 30:471–477.
- Grunfeld C, Delaney JA, Wanke C, et al. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. AIDS 2009; 23:1841–1849.
- Hulten E, Mitchell J, Scally J, Gibbs B, Villines TC. HIV positivity, protease inhibitor exposure and subclinical atherosclerosis: a systematic review and meta-analysis of observational studies. Heart 2009; 95:1826–1835.
- Riddler SA, Smit E, Cole SR, et al. Impact of HIV infection and HAART on serum lipids in men. JAMA 2003; 289:2978–2982.
- Shahmanesh M, Das S, Stolinski M, et al. Antiretroviral treatment reduces very-low-density lipoprotein and intermediate-density lipoprotein apolipoprotein B fractional catabolic rate in human immunodeficiency virus-infected patients with mild dyslipidemia. J Clin Endocrinol Metab 2005; 90:755–760.
- Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 2006; 4:e365.
- Park IW, Wang JF, Groopman JE. HIV-1 Tat promotes monocyte chemoattractant protein-1 secretion followed by transmigration of monocytes. Blood 2001; 97:352–358.
- Fisher SD, Miller TL, Lipshultz SE. Impact of HIV and highly active antiretroviral therapy on leukocyte adhesion molecules, arterial inflammation, dyslipidemia, and atherosclerosis. Atherosclerosis 2006; 185:1–11.
- Hsue PY, Hunt PW, Schnell A, et al. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009; 23:1059–1067.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- DAD Study Group; Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007: 356:1723–1735.
- DAD Study Group; Sabin CA, Worm SW, Weber R, et al. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients enrolled in the D:A:D study: a multi-cohort collaboration. Lancet 2008; 371:1417–1426.
- Durand M, Sheehy O, Baril JG, Lelorier J, Tremblay C; GRUCHUM Research Center (Groupe de Recherche de l’UHRESS du Centre Hospitalier Universitaire de Montréal). Relation between use of nucleoside reverse transcriptase inhibitors (NRTI) and risk of myocardial infarction (MI): a nested case control study using Quebec’s public health insurance database (QPHID). Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 17–22, 2009.
- Lang S, Mary-Krause M, Cotte L, et al; the Clinical Epi Group of the French Hospital Database on HIV. Impact of specific NRTI and PI exposure on the risk of myocardial infarction: a case-control study nested within FHDH ANRS CO4. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Strategies for Management of Anti-Retroviral Therapy/INSIGHT. Use of nucleoside reverse transcriptase inhibitors and risk of myocardial infarction in HIV-infected patients. AIDS 2008; 22:F17–F24.
- Bedimo R, Westfall A, Drechsler H, Tebas P. Abacavir use and risk of acute myocardial infarction and cerebrovascular disease in the HAART era. Presented at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention in Cape Town, South Africa, July 19–22, 2009.
- Brothers CH, Hernandez JE, Cutrell AG, et al. Risk of myocardial infarction and abacavir therapy: no increased risk across 52 GlaxoSmithKline-sponsored clinical trials in adult subjects. J Acquir Immune Defic Syndr 2009; 51:20–28.
- Benson C, Ribaudo H, Zheng E, et al; the ACTG A5001/ALLRT Protocol Team. No Association of Abacavir Use with Risk of Myocardial Infarction or Severe Cardiovascular Disease Events: Results from ACTG A5001. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Torriani FJ, Komarow L, Parker RA, et al; ACTG 5152s Study Team. Endothelial function in human immunodeficiency virus-infected antiretroviral-naive subjects before and after starting potent antiretroviral therapy: The ACTG (AIDS Clinical Trials Group) Study 5152s. J Am Coll Cardiol 2008; 52:569–576.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- van Vonderen MG, Hassink EA, van Agtmael MA, et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis 2009; 199:1186–1194.
- Strategies for Management of Antiretroviral Therapy (SMART) Study Group; El-Sadr WM, Lundgren JD, Neaton JD, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med 2006; 355:2283–2296.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Kuller LH, Tracy R, Belloso WINSIGHT SMART Study Group. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008; 5:e203.
- Badiou S, De Boever CM, Dupuy AM, Baillat V, Cristol JP, Reynes J. Small dense LDL and atherogenic lipid profile in HIV-positive adults: influence of lopinavir/ritonavir-containing regimen. AIDS 2003; 17:772–774.
- Duong M, Petit JM, Martha B, et al. Concentration of circulating oxidized LDL in HIV-infected patients treated with antiretroviral agents: relation to HIV-related lipodystrophy. HIV Clin Trials 2006; 7:41–47.
- Fisac C, Fumero E, Crespo M, et al. Metabolic benefits 24 months after replacing a protease inhibitor with abacavir, efavirenz or nevirapine. AIDS 2005; 19:917–925.
- Hicks CB, Cahn P, Cooper DA, et al; RESIST investigator group. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatmentexperienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 2006; 368:466–475.
- Malan DR, Krantz E, David N, Wirtz V, Hammond J, McGrath D; 089 Study Group. Efficacy and safety of atazanavir, with or without ritonavir, as part of once-daily highly active antiretroviral therapy regimens in antiretroviral-naive patients. J Acquir Immune Defic Syndr 2008; 47:161–167.
- Anastos K, Lu D, Shi Q, et al. Association of serum lipid levels with HIV serostatus, specific antiretroviral agents, and treatment regimens. J Acquir Immune Defic Syndr 2007; 45:34–42.
- Tomaka F, Lefebvre E, Sekar V, et al. Effects of ritonavir-boosted darunavir vs ritonavir-boosted atazanavir on lipid and glucose parameters in HIV-negative, healthy volunteers. HIV Med 2009; 10:318–327.
- Eron J, Yeni P, Gathe J, et al; KLEAN study team. The KLEAN study of fosamprenavir-ritonavir versus lopinavir-ritonavir, each in combination with abacavir-lamivudine, for initial treatment of HIV infection over 48 weeks: a randomised non-inferiority trial. Lancet 2006; 368:476–482.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Kumar PN, Rodriguez-French A, Thompson MA, et al; ESS40002 Study Team. A prospective, 96-week study of the impact of trizivir, combivir/nelfinavir, and lamivudine/stavudine/nelfinavir on lipids, metabolic parameters and efficacy in antiretroviral-naive patients: effect of sex and ethnicity. HIV Med 2006; 7:85–98.
- Shafran SD, Mashinter LD, Roberts SE. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med 2005; 6:421–425.
- Walmsley S, Avihingsanon A, Slim J, et al. Gemini: a noninferiority study of saquinavir/ritonavir versus lopinavir/ritonavir as initial HIV-1 therapy in adults. J Acquir Immune Defic Syndr 2009; 50:367–374.
- DHHS Panel on Antiretroviral Guidelines for Adults and Adolescents— A Working Group of the Office of AIDS Research Advisory Council (OARAC). Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. December 1, 1009. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 29, 2010.
- Shikuma CM, Yang Y, Glesby MJ, et al. Metabolic effects of protease inhibitor-sparing antiretroviral regimens given as initial treatment of HIV-1 Infection (AIDS Clinical Trials Group Study A5095). J Acquir Immune Defic Syndr 2007; 44:540–550.
- van Leth F, Phanuphak P, Stroes E, et al. Nevirapine and efavirenz elicit different changes in lipid profiles in antiretroviral-therapynaive patients infected with HIV-1. PLoS Med 2004; 1:e19.
- Katlama C, Haubrich R, Lalezari J, et al; DUET-1, DUET-2 study groups. Efficacy and safety of etravirine in treatment-experienced, HIV-1 patients: pooled 48 week analysis of two randomized, controlled trials. AIDS 2009; 23:2289–2300.
- Hammond E, Nolan D, James I, Metcalf C, Mallal S. Reduction of mitochondrial DNA content and respiratory chain activity occurs in adipocytes within 6–12 months of commencing nucleoside reverse transcriptase inhibitor therapy. AIDS 2004; 18:815–817.
- Pozniak AL, Gallant JE, DeJesus E, et al. Tenofovir disoproxil fumarate, emtricitabine, and efavirenz versus fixed-dose zidovudine/lamivudine and efavirenz in antiretroviral-naive patients: virologic, immunologic, and morphologic changes—a 96-week analysis. J Acquir Immune Defic Syndr 2006; 43:535–540.
- Tungsiripat M, Kitch D, Glesby M, et al. A pilot study to determine the effect on dyslipidemia of the addition of tenofovir to stable background ART in HIV-infected subjects: results from the A5206 Study Team. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Law MG, Friis-Møller N, El-Sadr WM, et al; D:A:D Study Group. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med 2006; 7:218–230.
- Aberg JA. Cardiovascular complications in HIV management: past, present, and future. J Acquir Immune Defic Syndr 2009; 50:54–64.
- Dubé MP, Stein JH, Aberg JA, et al; Adult AIDS Clinical Trials Group Cardiovascular Subcommittee. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clin Infect Dis 2003; 37:613–627.
- Fichtenbaum CJ. Metabolic abnormalities associated with HIV infection and antiretroviral therapy. Curr Infect Dis Rep 2009; 11:84–92.
- Gerber JG, Rosenkranz SL, Fichtenbaum CJ, et al; AIDS Clinical Trials Group A5108 Team. Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J Acquir Immune Defic Syndr 2005; 39:307–312.
- Grennan T, Walmsley S. Etravirine for HIV-I: addressing the limitations of the nonnucleoside reverse transcriptase inhibitor class. J Int Assoc Physicians AIDS Care (Chic Ill) 2009; 8:354–363.
- Sekar V S-GS, Marien K. Pharmacokinetic drug-drug interaction between the new HIV protease inhibitor darunavir (TMC114) and the lipid-lowering agent pravastatin. Presented at the 8th International Workshop on Pharmacology of HIV Therapy; Budapest, Hungary, April 16–18, 2007.
- Kiser JJ, Gerber JG, Predhomme JA, Wolfe P, Flynn DM, Hoody DW. Drug/drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 2008; 47:570–578.
- Aslangul E, Assoumou L, Bittar R, et al. Rosuvastatin versus pravastatin in dyslipidemic HIV-1-infected patients receiving protease inhibitors: a randomized trial. AIDS 2010; 24:77–83.
- Chow D, Chen H, Glesby MJ, et al. Short-term ezetimibe is well tolerated and effective in combination with statin therapy to treat elevated LDL cholesterol in HIV-infected patients. AIDS 2009; 23:2133–2141.
- Aberg JA, Zackin RA, Brobst SW, et al; ACTG 5087 Study Team. A randomized trial of the efficacy and safety of fenofibrate versus pravastatin in HIV-infected subjects with lipid abnormalities: AIDS Clinical Trials Group Study 5087. AIDS Res Hum Retroviruses 2005; 21:757–767.
- Dubé MP, Wu JW, Aberg JA, et al; AIDS Clinical Trials Group A5148 Study Team. Safety and efficacy of extended-release niacin for the treatment of dyslipidaemia in patients with HIV infection: AIDS Clinical Trials Group Study A5148. Antivir Ther 2006; 11:1081–1089.
- Gerber JG, Kitch DW, Fichtenbaum CJ, et al. Fish oil and fenofibrate for the treatment of hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: results of ACTG A5186. J Acquir Immune Defic Syndr 2008; 47:459–466.
- Mallolas J, Podzamczer D, Milinkovic A, et al; ATAZIP Study Group. Efficacy and safety of switching from boosted lopinavir to boosted atazanavir in patients with virological suppression receiving a LPV/rcontaining HAART: the ATAZIP study. J Acquir Immune Defic Syndr 2009; 51:29–36.
- Eron J, Andrade J, Zajdenverg R, et al. Switching from stable lopinavir/ritonavir-based to raltegravir-based combination ART resulted in a superior lipid profile at week 12 but did not demonstrate noninferior virologic efficacy at week 24. Presented at the 16th Conference on Retroviruses and Opportunistic Infections in Montreal, Canada, February 8–11, 2009.
- Calza L, Manfredi R, Colangeli V, et al. Substitution of nevirapine or efavirenz for protease inhibitor versus lipid-lowering therapy for the management of dyslipidaemia. AIDS 2005; 19:1051–1058.
- Worm SW, De Wit S, Weber R, et al. Diabetes mellitus, preexisting coronary heart disease, and the risk of subsequent coronary heart disease events in patients infected with human immunodeficiency virus: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D Study). Circulation 2009; 119:805–811.
- Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med 2005; 165:1179–1184.
- Butt AA, McGinnis K, Rodriguez-Barradas MC, et al; Veterans Aging Cohort Study. HIV infection and the risk of diabetes mellitus. AIDS 2009; 23:1227–1234.
- Mulligan K, Yang Y, Wininger DA, et al. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS 2007; 21:47–57.
KEY POINTS
- Traditional risk factors are the main contributors to cardiovascular disease in this population, although HIV infection is independently associated with increased cardiovascular risk.
- Antiretroviral therapy contributes modestly to the risk of coronary heart disease. Antiretroviral combinations that include protease inhibitors cause the most substantial deleterious changes in lipid levels.
- Most changes in lipids and insulin resistance can be managed by adding lipid-lowering and antiglycemic agents and may not require changes to the antiretroviral regimen.
- Close attention to drug interactions is important when selecting lipid-lowering medications for patients on antiretroviral therapy to avoid dangerous increases in the levels of certain statins.
- Addressing modifiable risk factors such as smoking, obesity, and sedentary lifestyle can have a far greater impact on cardiovascular risk than changes in antiretroviral therapy.
Coenzyme Q10: A therapy for hypertension and statin-induced myalgia?
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
KEY POINTS
- In some clinical trials, coenzyme Q10 supplements significantly lowered diastolic and systolic blood pressure.
- Statins may lower coenzyme Q10 serum levels, and some investigators have evaluated the relationship between coenzyme Q10 deficiency and statin-related myalgia, but more evidence is needed to support the use of coenzyme Q10 supplements to prevent or treat myalgia.
- Coenzyme Q10 supplementation appears to be relatively safe. Most clinical trials have not reported significant side effects that necessitated stopping therapy. Gastrointestinal effects include abdominal discomfort, nausea, vomiting, diarrhea, and anorexia. Allergic rash and headache have also been reported.
Incidence, outcomes, and management of bleeding in non-ST-elevation acute coronary syndromes
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?

Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13

BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
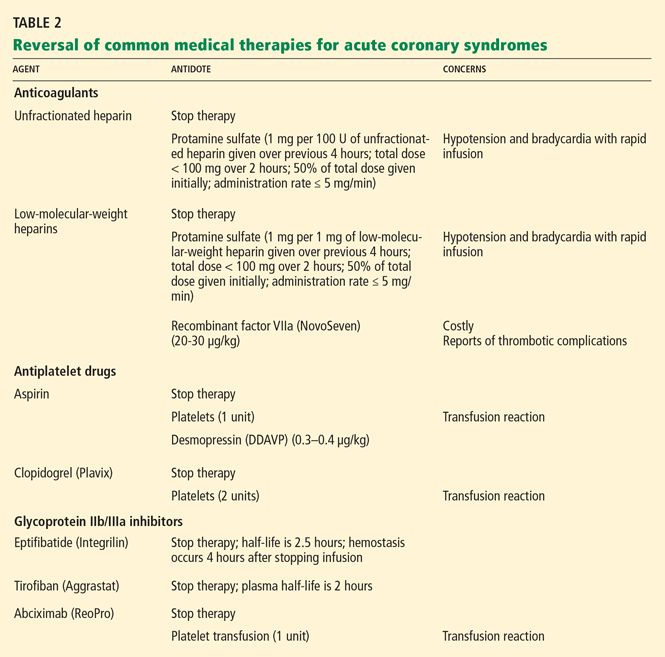
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?
Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13
BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?
Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13
BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
KEY POINTS
- The reported incidence of bleeding after treatment for non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%, depending on a number of factors.
- Bleeding is strongly associated with adverse outcomes, although a causal relationship has not been established.
- Patients should be assessed for risk of bleeding so that the antithrombotic and antiplatelet regimen can be adjusted, safer alternatives can be considered, and percutaneous interventions can be used less aggressively for those at high risk.
- If bleeding develops and the risk of continued bleeding outweighs the risk of recurrent ischemia, antithrombotic and antiplatelet drug therapy can be interrupted and other agents given to reverse the effects of these drugs.
LVH and hypertension: Is treating the pressure not enough?

In patients with “borderline” hypertension or those in whom the duration of blood pressure elevation is hard to ascertain, the finding of end-organ damage has traditionally been used as an argument to institute aggressive antihypertensive therapy. In this setting, retinal hypertensive disease, an S4 gallop, and left ventricular hypertrophy (LVH) are often specifically sought.
LVH and otherwise unexplained chronic kidney disease in patients with hypertension have generally been believed to be products of the elevated arterial pressure, and primary treatment has targeted pressure control. Bauml and Underwood, in this issue of the Journal, emphasize some published clinical trial data indicating that LVH may be an independent risk factor for poorer cardiovascular outcome. Even more provocative is the suggestion that LVH can be reversed, as can the associated increased risk of cardiovascular morbidity, independently of the hypertension.
Given our current understanding that LVH, under some conditions, can be induced by products of the renin-angiotensin system, this would suggest that pharmacologic blockade of this enzyme system should have extra benefit, above that seen from other antihypertensive agents. Conceivably, this may be true only in patients with LVH, and the time course of benefit may not directly parallel that seen with the control of hypertension. That theoretically may explain the lack of uniform advantage of angiotensin blockade over other effective antihypertensive approaches.
Since electrocardiography is a specific but not very sensitive test for LVH, the authors suggest that patients with hypertension be routinely screened for LVH using echocardiography. I am not sure the weight of the evidence supports this approach at present, particularly in the current frenzy of cost containment. Nonetheless, this concept warrants consideration, and at the least, large patient databases might be screened retrospectively to further validate or refute the concept that hypertension-associated LVH is an independent, reversible risk factor for cardiovascular morbidity.
In patients with “borderline” hypertension or those in whom the duration of blood pressure elevation is hard to ascertain, the finding of end-organ damage has traditionally been used as an argument to institute aggressive antihypertensive therapy. In this setting, retinal hypertensive disease, an S4 gallop, and left ventricular hypertrophy (LVH) are often specifically sought.
LVH and otherwise unexplained chronic kidney disease in patients with hypertension have generally been believed to be products of the elevated arterial pressure, and primary treatment has targeted pressure control. Bauml and Underwood, in this issue of the Journal, emphasize some published clinical trial data indicating that LVH may be an independent risk factor for poorer cardiovascular outcome. Even more provocative is the suggestion that LVH can be reversed, as can the associated increased risk of cardiovascular morbidity, independently of the hypertension.
Given our current understanding that LVH, under some conditions, can be induced by products of the renin-angiotensin system, this would suggest that pharmacologic blockade of this enzyme system should have extra benefit, above that seen from other antihypertensive agents. Conceivably, this may be true only in patients with LVH, and the time course of benefit may not directly parallel that seen with the control of hypertension. That theoretically may explain the lack of uniform advantage of angiotensin blockade over other effective antihypertensive approaches.
Since electrocardiography is a specific but not very sensitive test for LVH, the authors suggest that patients with hypertension be routinely screened for LVH using echocardiography. I am not sure the weight of the evidence supports this approach at present, particularly in the current frenzy of cost containment. Nonetheless, this concept warrants consideration, and at the least, large patient databases might be screened retrospectively to further validate or refute the concept that hypertension-associated LVH is an independent, reversible risk factor for cardiovascular morbidity.
In patients with “borderline” hypertension or those in whom the duration of blood pressure elevation is hard to ascertain, the finding of end-organ damage has traditionally been used as an argument to institute aggressive antihypertensive therapy. In this setting, retinal hypertensive disease, an S4 gallop, and left ventricular hypertrophy (LVH) are often specifically sought.
LVH and otherwise unexplained chronic kidney disease in patients with hypertension have generally been believed to be products of the elevated arterial pressure, and primary treatment has targeted pressure control. Bauml and Underwood, in this issue of the Journal, emphasize some published clinical trial data indicating that LVH may be an independent risk factor for poorer cardiovascular outcome. Even more provocative is the suggestion that LVH can be reversed, as can the associated increased risk of cardiovascular morbidity, independently of the hypertension.
Given our current understanding that LVH, under some conditions, can be induced by products of the renin-angiotensin system, this would suggest that pharmacologic blockade of this enzyme system should have extra benefit, above that seen from other antihypertensive agents. Conceivably, this may be true only in patients with LVH, and the time course of benefit may not directly parallel that seen with the control of hypertension. That theoretically may explain the lack of uniform advantage of angiotensin blockade over other effective antihypertensive approaches.
Since electrocardiography is a specific but not very sensitive test for LVH, the authors suggest that patients with hypertension be routinely screened for LVH using echocardiography. I am not sure the weight of the evidence supports this approach at present, particularly in the current frenzy of cost containment. Nonetheless, this concept warrants consideration, and at the least, large patient databases might be screened retrospectively to further validate or refute the concept that hypertension-associated LVH is an independent, reversible risk factor for cardiovascular morbidity.
Left ventricular hypertrophy: An overlooked cardiovascular risk factor
Left ventricular hypertrophy (LVH) strongly predicts cardiovascular morbidity and overall mortality in hypertensive patients. 1–7 Antihypertensive treatment that causes LVH to regress decreases the rates of adverse cardiovascular events and improves survival, independent of how much the blood pressure is lowered.8–11 It is clinically important to recognize that LVH is a modifiable risk factor and that management is more complex than just blood pressure control.
This paper reviews the definition of LVH, compares the diagnostic tests for it, and discusses the current evidence-based approach to managing this dangerous risk factor.
A CHRONICALLY ELEVATED CARDIAC WORKLOAD CAUSES LVH
LVH is an abnormal increase in the mass of the left ventricular myocardium caused by a chronically increased workload on the heart.12 This most commonly results from the heart pumping against an elevated afterload, as in hypertension and aortic stenosis. Another notable cause is increased filling of the left ventricle (ie, diastolic overload), which is the underlying mechanism for LVH in patients with aortic or mitral regurgitation and dilated cardiomyopathy. Coronary artery disease can also play a role in the pathogenesis of LVH, as the normal myocardium attempts to compensate for the ischemic or infarcted tissue.13
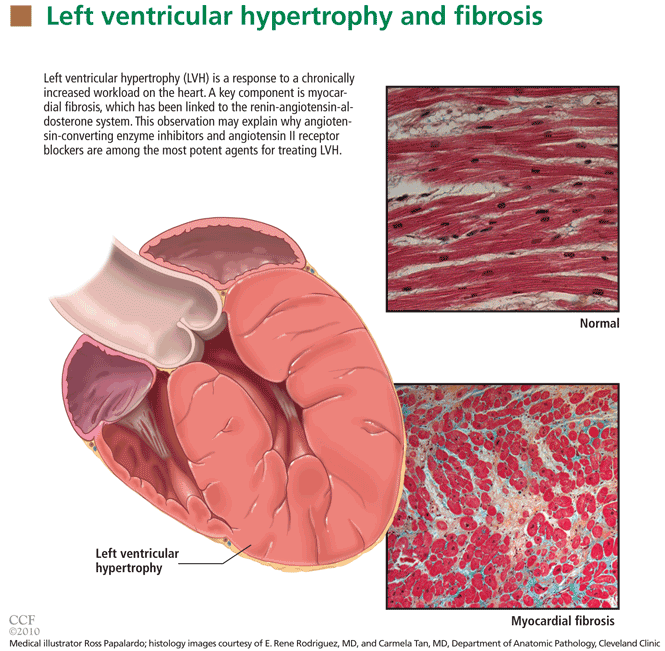
The development of myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system. Specifically, there is evidence that angiotensin II has a profibrotic effect on the myocardium of hypertensive patients.15 This may explain why angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are among the most potent agents for treating LVH, as we will discuss later in this review.
DIAGNOSIS BY ELECTROCARDIOGRAPHY, ECHOCARDIOGRAPHY, OR MRI
Many different criteria for electrocardiographic LVH have been proposed over the years. Most use the voltage in one or more leads, with or without additional factors such as QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. The most well known electrocardiographic criteria are the Cornell voltage,21 the Cornell product,22 the Sokolow-Lyon index,23 and the Romhilt-Estes point score system (Table 1).24

- Cornell voltage—median sensitivity 15%, median specificity 96%
- Cornell product—median sensitivity 19.5%, median specificity 91%
- Sokolow-Lyon voltage—median sensitivity 21%, median specificity 89%
- Romhilt-Estes point score—median sensitivity 17%, median specificity 95%.
Of note, the ranges of the published values were extremely broad. For example, the ranges in sensitivity were:
- Cornell voltage—2% to 41%
- Cornell product—8% to 32%
- Sokolow-Lyon voltage—4% to 51%
- Romhilt-Estes point score—0% to 41%.
While the studies with the extreme values may have had issues of small sample size or poor study quality, the wide range in values may primarily be the result of diverse study populations as well as different validation methods and cutoff values to define LVH. Regardless, the overall message of high specificity and low sensitivity is indisputable.
Electrocardiography is insensitive for diagnosing LVH because it relies on measuring the electrical activity of the heart by electrodes on the surface of the skin to predict the left ventricular mass. The intracardiac electrical activity is problematic to measure externally because the measurements are affected by everything between the myocardium and the electrodes, most notably fat, fluid, and air. Because of this effect, electrocardiography underdiagnoses LVH in patients with obesity, pleural effusions, pericardial effusions, anasarca, or chronic obstructive pulmonary disease. In addition, the diagnosis of LVH by electrocardiography is strongly influenced by age and ethnicity.25–26
While electrocardiography is not sensitive and cannot be used to rule out LVH, it still has a role in its diagnosis and management. In the landmark Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study, regression of LVH (diagnosed electrocardiographically by the Sokolow-Lyon index or the Cornell product criteria) in response to losartan (Cozaar) improved cardiovascular outcomes independent of blood pressure.10 Based on this, it is reasonable that all hypertensive patients and other patients at risk of LVH who undergo electrocardiography be screened with these two criteria.
Echocardiography is the test of choice
Echocardiography, if available, should be the test of choice to assess for LVH. It is much more sensitive than electrocardiography and can also detect other abnormalities such as left ventricular dysfunction and valvular disease.
This test uses transthoracic or transesophageal ultrasonography to measure the left ventricular end-diastolic diameter, posterior wall thickness, and interventricular septum thickness. From these measurements and the patient’s height and weight, the left ventricular mass index can be calculated.27
Several different cutoff values for the left ventricular mass index have been proposed; the LIFE study used values of > 104 g/m2 in women and > 116 g/m2 in men to define LVH.
When using echocardiography to assess for LVH, it is imperative that the left ventricular mass index be used and not just the left ventricular wall thickness, as often happens in clinical practice. This is necessary because diagnosis by wall thickness alone is not a good indicator of LVH, with a concordance between wall thickness and a left ventricular mass index of only 60%.28 In addition, wall thickness tends to underestimate LVH in women and overestimate it in men.
Is echocardiography cost-effective?
Despite its clear advantages, an important consideration about echocardiography as a screening test for all hypertensive patients is its cost.
A suggested way to reduce cost is to measure the left ventricular mass index only.29 A limited echocardiographic examination is much less expensive than a complete two-dimensional echocardiogram ($255 vs $431 per the 2009 Medicare Ambulatory Payment Classification30) and should be the examination performed if the patient has no other clinical indication for echocardiography.
Another way to control cost is to stratify patients by risk and to do echocardiography only in those who would benefit most from it. Based on the prevalence of LVH, one study concluded that echocardiography is most cost-effective in men 50 years or older.31
Further study is necessary to more precisely define the cost-effectiveness of echocardiographic screening for LVH in terms of potentially preventable cardiovascular morbidity and death.
Cardiac MRI: The costly gold standard
Cardiac MRI is the gold standard test for LVH, as it is even more accurate and reproducible than echocardiography.32 It can precisely estimate a patient's left ventricular mass and assess for other structural cardiac abnormalities.
MRI’s use, however, is severely restricted in clinical practice due to its high cost and limited availability. While it may never be used for general screening for LVH, it certainly has a role in clinical research and for assessing cardiac anatomy in special clinical situations.
TREATMENT SHOULD INCLUDE AN ACE INHIBITOR OR ARB
Once LVH has been diagnosed, the next step is to decide on an appropriate treatment plan.
While the choice of therapy will always depend on other comorbidities, a 2003 metaanalysis of antihypertensive medications in the treatment of LVH (controlling for the degree of blood pressure lowering) showed that ARBs were the most efficacious class of agents for reducing the left ventricular mass.33 Specifically, ARBs decreased the mass by 13%, followed by calcium-channel blockers at 11%, ACE inhibitors at 10%, diuretics at 8%, and beta-blockers at 6%. In pairwise comparison, ARBs, calcium-channel blockers, and ACE inhibitors were all significantly more effective in reducing the left ventricular mass than beta-blockers.
As previously discussed, LVH appears to be pathophysiologically linked to myocardial fibrosis and the renin-angiotensin-aldosterone system. For this reason and based on the data presented above regarding the degree of LVH regression, ACE inhibitors or ARBs should be used as the first-line agents for LVH unless they are contraindicated in the individual patient.
The LIFE study
The LIFE study offers the strongest evidence that treating LVH is beneficial. It showed that in hypertensive patients with electrocardiographic LVH by the Cornell product or Sokolow-Lyon criteria, treatment with antihypertensive drugs that resulted in less-severe LVH on electrocardiography was associated with lower rates of cardiovascular morbidity and death, independent of the blood pressure achieved or the drug used.10
The end point in this study was a composite of stroke, myocardial infarction, and cardiovascular death. Regression of electrocardiographic LVH in hypertensive patients has also been shown to decrease the incidence of diabetes mellitus,34 atrial fibrillation,35 and hospitalizations for heart failure.36
The LIFE study also examined the prognostic implications of treating LVH detected by echocardiography. In this prospective cohort substudy, patients who had a lower left ventricular mass index during treatment with antihypertensive drugs had lower rates of cardiovascular morbidity and all-cause mortality, independent of the effects of blood pressure and treatment used.11
These results suggest that there may be a role not only for treating LVH, but also for monitoring for a reduction in the left ventricular mass index as a goal of therapy (similar to the way hemoglobin A1c is used in diabetic patients). If the index is used in this way, one could potentially adjust the dose of current drugs, switch classes, or add an additional drug based on a persistently elevated left ventricular mass index in order to optimize the patient's overall cardiovascular risk. A randomized controlled trial of therapy directed by the mass index vs conventional therapy of LVH would be necessary to assess the clinical utility of this approach.
RECOMMENDATIONS
LVH is a common and potentially modifiable cardiovascular risk factor often overlooked in clinical practice. Ideally, all hypertensive patients should be screened with echocardiography to look for LVH, using the calculated left ventricular mass index rather than wall thickness alone to make the diagnosis. While electrocardiography is specific and also has prognostic implications, it is not sensitive enough to be used alone to screen for LVH.
Once the diagnosis of LVH is made, the initial therapy should be an ARB or an ACE inhibitor. Response to therapy can be assessed by monitoring for a reduction in left ventricular mass index or regression of electrocardiographic LVH.
Treatment-induced regression of LVH decreases adverse cardiovascular events and improves overall survival. When modifying medications in hypertensive patients, it is important to remember that the treatment of LVH is not synonymous with blood pressure control.
- Casale PN, Devereux RB, Milner M, et al. Value of echocardiographic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105:173–178.
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566.
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352.
- Verdecchia P, Carini G, Circo A, et al; MAVI (MAssa Ventricolare sinistra nell’Ipertensione) Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 2001; 38:1829–1835.
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998; 32:1454–1459.
- Verdecchia P, Porcellati C, Reboldi G, et al. Left ventricular hypertrophy as an independent predictor of acute cerebrovascular events in essential hypertension. Circulation 2001; 104:2039–2044.
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35:580–586.
- Verdecchia P, Schillaci G, Borgioni C, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation 1998; 97:48–54.
- Mathew J, Sleight P, Lonn E, et al; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621.
- Okin PM, Devereux RB, Jern S, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004; 292:2343–2349.
- Devereux RB, Wachtell K, Gerdts E, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292:2350–2356.
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470–479.
- Zabalgoitia M, Berning J, Koren MJ, et al; LIFE Study Investigators. Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 2001; 88:646–650.
- Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol 1990; 65:1G–7G.
- González A, López B, Querejeta R, Díez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 2002; 34:1585–1593.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320.
- Liebson PR, Grandits G, Prineas R, et al. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993; 87:476–486.
- Martinez MA, Sancho T, Armada E, et al; Vascular Risk Working Group Grupo Monitorizacíon Ambulatoria de la Presión Arterial (MAPA)-Madrid. Prevalence of left ventricular hypertrophy in patients with mild hypertension in primary care: impact of echocardiography on cardiovascular risk stratification. Am J Hypertens 2003; 16:556–563.
- Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ 2007; 335:711.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990; 81:1144–1146.
- Casale PN, Devereux RB, Kligfield P, et al. Electrocardiographic detection of left ventricular hypertrophy: development and prospective validation of improved criteria. J Am Coll Cardiol 1985; 6:572–580.
- Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol 1992; 20:1180–1186.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949; 37:161–186.
- Romhilt DW, Estes EH. A point-score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968; 75:752–758.
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation 1990; 81:815–820.
- Okin PM, Wright JT, Nieminen MS, et al. Ethnic differences in electrocardiographic criteria for left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint. Am J Hypertens 2002; 15:663–671.
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986; 57:450–458.
- Leibowitz D, Planer D, Ben-Ibgi F, Rott D, Weiss AT, Bursztyn M. Measurement of wall thickness alone does not accurately assess the presence of left ventricular hypertrophy. Clin Exp Hypertens 2007; 29:119–125.
- Black HR, Weltin G, Jaffe CC. The limited echocardiogram: a modification of standard echocardiography for use in the routine evaluation of patients with systemic hypertension. Am J Cardiol 1991; 67:1027–1030.
- American Society of Echocardiography Coding and Reimbursement Newsletter, January 2009. http://www.asecho.org/files/public/CodingnewsJan09.pdf. Accessed May 13, 2010.
- Cuspidi C, Meani S, Valerio C, Fusi V, Sala C, Zanchetti A. Left ventricular hypertrophy and cardiovascular risk stratification: impact and cost-effectiveness of echocardiography in recently diagnosed essential hypertensives. J Hypertens 2006; 24:1671–1677.
- Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, Gottdiener JS. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 1995; 8:221–228.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003; 115:41–46.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. In-treatment resolution or absence of electrocardiographic left ventricular hypertrophy is associated with decreased incidence of new-onset diabetes mellitus in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2007; 50:984–990.
- Okin PM, Wachtell K, Devereux RB, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006; 296:1242–1248.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Ann Intern Med 2007; 147:311–319.
Left ventricular hypertrophy (LVH) strongly predicts cardiovascular morbidity and overall mortality in hypertensive patients. 1–7 Antihypertensive treatment that causes LVH to regress decreases the rates of adverse cardiovascular events and improves survival, independent of how much the blood pressure is lowered.8–11 It is clinically important to recognize that LVH is a modifiable risk factor and that management is more complex than just blood pressure control.
This paper reviews the definition of LVH, compares the diagnostic tests for it, and discusses the current evidence-based approach to managing this dangerous risk factor.
A CHRONICALLY ELEVATED CARDIAC WORKLOAD CAUSES LVH
LVH is an abnormal increase in the mass of the left ventricular myocardium caused by a chronically increased workload on the heart.12 This most commonly results from the heart pumping against an elevated afterload, as in hypertension and aortic stenosis. Another notable cause is increased filling of the left ventricle (ie, diastolic overload), which is the underlying mechanism for LVH in patients with aortic or mitral regurgitation and dilated cardiomyopathy. Coronary artery disease can also play a role in the pathogenesis of LVH, as the normal myocardium attempts to compensate for the ischemic or infarcted tissue.13
The development of myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system. Specifically, there is evidence that angiotensin II has a profibrotic effect on the myocardium of hypertensive patients.15 This may explain why angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are among the most potent agents for treating LVH, as we will discuss later in this review.
DIAGNOSIS BY ELECTROCARDIOGRAPHY, ECHOCARDIOGRAPHY, OR MRI
Many different criteria for electrocardiographic LVH have been proposed over the years. Most use the voltage in one or more leads, with or without additional factors such as QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. The most well known electrocardiographic criteria are the Cornell voltage,21 the Cornell product,22 the Sokolow-Lyon index,23 and the Romhilt-Estes point score system (Table 1).24
- Cornell voltage—median sensitivity 15%, median specificity 96%
- Cornell product—median sensitivity 19.5%, median specificity 91%
- Sokolow-Lyon voltage—median sensitivity 21%, median specificity 89%
- Romhilt-Estes point score—median sensitivity 17%, median specificity 95%.
Of note, the ranges of the published values were extremely broad. For example, the ranges in sensitivity were:
- Cornell voltage—2% to 41%
- Cornell product—8% to 32%
- Sokolow-Lyon voltage—4% to 51%
- Romhilt-Estes point score—0% to 41%.
While the studies with the extreme values may have had issues of small sample size or poor study quality, the wide range in values may primarily be the result of diverse study populations as well as different validation methods and cutoff values to define LVH. Regardless, the overall message of high specificity and low sensitivity is indisputable.
Electrocardiography is insensitive for diagnosing LVH because it relies on measuring the electrical activity of the heart by electrodes on the surface of the skin to predict the left ventricular mass. The intracardiac electrical activity is problematic to measure externally because the measurements are affected by everything between the myocardium and the electrodes, most notably fat, fluid, and air. Because of this effect, electrocardiography underdiagnoses LVH in patients with obesity, pleural effusions, pericardial effusions, anasarca, or chronic obstructive pulmonary disease. In addition, the diagnosis of LVH by electrocardiography is strongly influenced by age and ethnicity.25–26
While electrocardiography is not sensitive and cannot be used to rule out LVH, it still has a role in its diagnosis and management. In the landmark Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study, regression of LVH (diagnosed electrocardiographically by the Sokolow-Lyon index or the Cornell product criteria) in response to losartan (Cozaar) improved cardiovascular outcomes independent of blood pressure.10 Based on this, it is reasonable that all hypertensive patients and other patients at risk of LVH who undergo electrocardiography be screened with these two criteria.
Echocardiography is the test of choice
Echocardiography, if available, should be the test of choice to assess for LVH. It is much more sensitive than electrocardiography and can also detect other abnormalities such as left ventricular dysfunction and valvular disease.
This test uses transthoracic or transesophageal ultrasonography to measure the left ventricular end-diastolic diameter, posterior wall thickness, and interventricular septum thickness. From these measurements and the patient’s height and weight, the left ventricular mass index can be calculated.27
Several different cutoff values for the left ventricular mass index have been proposed; the LIFE study used values of > 104 g/m2 in women and > 116 g/m2 in men to define LVH.
When using echocardiography to assess for LVH, it is imperative that the left ventricular mass index be used and not just the left ventricular wall thickness, as often happens in clinical practice. This is necessary because diagnosis by wall thickness alone is not a good indicator of LVH, with a concordance between wall thickness and a left ventricular mass index of only 60%.28 In addition, wall thickness tends to underestimate LVH in women and overestimate it in men.
Is echocardiography cost-effective?
Despite its clear advantages, an important consideration about echocardiography as a screening test for all hypertensive patients is its cost.
A suggested way to reduce cost is to measure the left ventricular mass index only.29 A limited echocardiographic examination is much less expensive than a complete two-dimensional echocardiogram ($255 vs $431 per the 2009 Medicare Ambulatory Payment Classification30) and should be the examination performed if the patient has no other clinical indication for echocardiography.
Another way to control cost is to stratify patients by risk and to do echocardiography only in those who would benefit most from it. Based on the prevalence of LVH, one study concluded that echocardiography is most cost-effective in men 50 years or older.31
Further study is necessary to more precisely define the cost-effectiveness of echocardiographic screening for LVH in terms of potentially preventable cardiovascular morbidity and death.
Cardiac MRI: The costly gold standard
Cardiac MRI is the gold standard test for LVH, as it is even more accurate and reproducible than echocardiography.32 It can precisely estimate a patient's left ventricular mass and assess for other structural cardiac abnormalities.
MRI’s use, however, is severely restricted in clinical practice due to its high cost and limited availability. While it may never be used for general screening for LVH, it certainly has a role in clinical research and for assessing cardiac anatomy in special clinical situations.
TREATMENT SHOULD INCLUDE AN ACE INHIBITOR OR ARB
Once LVH has been diagnosed, the next step is to decide on an appropriate treatment plan.
While the choice of therapy will always depend on other comorbidities, a 2003 metaanalysis of antihypertensive medications in the treatment of LVH (controlling for the degree of blood pressure lowering) showed that ARBs were the most efficacious class of agents for reducing the left ventricular mass.33 Specifically, ARBs decreased the mass by 13%, followed by calcium-channel blockers at 11%, ACE inhibitors at 10%, diuretics at 8%, and beta-blockers at 6%. In pairwise comparison, ARBs, calcium-channel blockers, and ACE inhibitors were all significantly more effective in reducing the left ventricular mass than beta-blockers.
As previously discussed, LVH appears to be pathophysiologically linked to myocardial fibrosis and the renin-angiotensin-aldosterone system. For this reason and based on the data presented above regarding the degree of LVH regression, ACE inhibitors or ARBs should be used as the first-line agents for LVH unless they are contraindicated in the individual patient.
The LIFE study
The LIFE study offers the strongest evidence that treating LVH is beneficial. It showed that in hypertensive patients with electrocardiographic LVH by the Cornell product or Sokolow-Lyon criteria, treatment with antihypertensive drugs that resulted in less-severe LVH on electrocardiography was associated with lower rates of cardiovascular morbidity and death, independent of the blood pressure achieved or the drug used.10
The end point in this study was a composite of stroke, myocardial infarction, and cardiovascular death. Regression of electrocardiographic LVH in hypertensive patients has also been shown to decrease the incidence of diabetes mellitus,34 atrial fibrillation,35 and hospitalizations for heart failure.36
The LIFE study also examined the prognostic implications of treating LVH detected by echocardiography. In this prospective cohort substudy, patients who had a lower left ventricular mass index during treatment with antihypertensive drugs had lower rates of cardiovascular morbidity and all-cause mortality, independent of the effects of blood pressure and treatment used.11
These results suggest that there may be a role not only for treating LVH, but also for monitoring for a reduction in the left ventricular mass index as a goal of therapy (similar to the way hemoglobin A1c is used in diabetic patients). If the index is used in this way, one could potentially adjust the dose of current drugs, switch classes, or add an additional drug based on a persistently elevated left ventricular mass index in order to optimize the patient's overall cardiovascular risk. A randomized controlled trial of therapy directed by the mass index vs conventional therapy of LVH would be necessary to assess the clinical utility of this approach.
RECOMMENDATIONS
LVH is a common and potentially modifiable cardiovascular risk factor often overlooked in clinical practice. Ideally, all hypertensive patients should be screened with echocardiography to look for LVH, using the calculated left ventricular mass index rather than wall thickness alone to make the diagnosis. While electrocardiography is specific and also has prognostic implications, it is not sensitive enough to be used alone to screen for LVH.
Once the diagnosis of LVH is made, the initial therapy should be an ARB or an ACE inhibitor. Response to therapy can be assessed by monitoring for a reduction in left ventricular mass index or regression of electrocardiographic LVH.
Treatment-induced regression of LVH decreases adverse cardiovascular events and improves overall survival. When modifying medications in hypertensive patients, it is important to remember that the treatment of LVH is not synonymous with blood pressure control.
Left ventricular hypertrophy (LVH) strongly predicts cardiovascular morbidity and overall mortality in hypertensive patients. 1–7 Antihypertensive treatment that causes LVH to regress decreases the rates of adverse cardiovascular events and improves survival, independent of how much the blood pressure is lowered.8–11 It is clinically important to recognize that LVH is a modifiable risk factor and that management is more complex than just blood pressure control.
This paper reviews the definition of LVH, compares the diagnostic tests for it, and discusses the current evidence-based approach to managing this dangerous risk factor.
A CHRONICALLY ELEVATED CARDIAC WORKLOAD CAUSES LVH
LVH is an abnormal increase in the mass of the left ventricular myocardium caused by a chronically increased workload on the heart.12 This most commonly results from the heart pumping against an elevated afterload, as in hypertension and aortic stenosis. Another notable cause is increased filling of the left ventricle (ie, diastolic overload), which is the underlying mechanism for LVH in patients with aortic or mitral regurgitation and dilated cardiomyopathy. Coronary artery disease can also play a role in the pathogenesis of LVH, as the normal myocardium attempts to compensate for the ischemic or infarcted tissue.13
The development of myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system. Specifically, there is evidence that angiotensin II has a profibrotic effect on the myocardium of hypertensive patients.15 This may explain why angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are among the most potent agents for treating LVH, as we will discuss later in this review.
DIAGNOSIS BY ELECTROCARDIOGRAPHY, ECHOCARDIOGRAPHY, OR MRI
Many different criteria for electrocardiographic LVH have been proposed over the years. Most use the voltage in one or more leads, with or without additional factors such as QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. The most well known electrocardiographic criteria are the Cornell voltage,21 the Cornell product,22 the Sokolow-Lyon index,23 and the Romhilt-Estes point score system (Table 1).24
- Cornell voltage—median sensitivity 15%, median specificity 96%
- Cornell product—median sensitivity 19.5%, median specificity 91%
- Sokolow-Lyon voltage—median sensitivity 21%, median specificity 89%
- Romhilt-Estes point score—median sensitivity 17%, median specificity 95%.
Of note, the ranges of the published values were extremely broad. For example, the ranges in sensitivity were:
- Cornell voltage—2% to 41%
- Cornell product—8% to 32%
- Sokolow-Lyon voltage—4% to 51%
- Romhilt-Estes point score—0% to 41%.
While the studies with the extreme values may have had issues of small sample size or poor study quality, the wide range in values may primarily be the result of diverse study populations as well as different validation methods and cutoff values to define LVH. Regardless, the overall message of high specificity and low sensitivity is indisputable.
Electrocardiography is insensitive for diagnosing LVH because it relies on measuring the electrical activity of the heart by electrodes on the surface of the skin to predict the left ventricular mass. The intracardiac electrical activity is problematic to measure externally because the measurements are affected by everything between the myocardium and the electrodes, most notably fat, fluid, and air. Because of this effect, electrocardiography underdiagnoses LVH in patients with obesity, pleural effusions, pericardial effusions, anasarca, or chronic obstructive pulmonary disease. In addition, the diagnosis of LVH by electrocardiography is strongly influenced by age and ethnicity.25–26
While electrocardiography is not sensitive and cannot be used to rule out LVH, it still has a role in its diagnosis and management. In the landmark Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study, regression of LVH (diagnosed electrocardiographically by the Sokolow-Lyon index or the Cornell product criteria) in response to losartan (Cozaar) improved cardiovascular outcomes independent of blood pressure.10 Based on this, it is reasonable that all hypertensive patients and other patients at risk of LVH who undergo electrocardiography be screened with these two criteria.
Echocardiography is the test of choice
Echocardiography, if available, should be the test of choice to assess for LVH. It is much more sensitive than electrocardiography and can also detect other abnormalities such as left ventricular dysfunction and valvular disease.
This test uses transthoracic or transesophageal ultrasonography to measure the left ventricular end-diastolic diameter, posterior wall thickness, and interventricular septum thickness. From these measurements and the patient’s height and weight, the left ventricular mass index can be calculated.27
Several different cutoff values for the left ventricular mass index have been proposed; the LIFE study used values of > 104 g/m2 in women and > 116 g/m2 in men to define LVH.
When using echocardiography to assess for LVH, it is imperative that the left ventricular mass index be used and not just the left ventricular wall thickness, as often happens in clinical practice. This is necessary because diagnosis by wall thickness alone is not a good indicator of LVH, with a concordance between wall thickness and a left ventricular mass index of only 60%.28 In addition, wall thickness tends to underestimate LVH in women and overestimate it in men.
Is echocardiography cost-effective?
Despite its clear advantages, an important consideration about echocardiography as a screening test for all hypertensive patients is its cost.
A suggested way to reduce cost is to measure the left ventricular mass index only.29 A limited echocardiographic examination is much less expensive than a complete two-dimensional echocardiogram ($255 vs $431 per the 2009 Medicare Ambulatory Payment Classification30) and should be the examination performed if the patient has no other clinical indication for echocardiography.
Another way to control cost is to stratify patients by risk and to do echocardiography only in those who would benefit most from it. Based on the prevalence of LVH, one study concluded that echocardiography is most cost-effective in men 50 years or older.31
Further study is necessary to more precisely define the cost-effectiveness of echocardiographic screening for LVH in terms of potentially preventable cardiovascular morbidity and death.
Cardiac MRI: The costly gold standard
Cardiac MRI is the gold standard test for LVH, as it is even more accurate and reproducible than echocardiography.32 It can precisely estimate a patient's left ventricular mass and assess for other structural cardiac abnormalities.
MRI’s use, however, is severely restricted in clinical practice due to its high cost and limited availability. While it may never be used for general screening for LVH, it certainly has a role in clinical research and for assessing cardiac anatomy in special clinical situations.
TREATMENT SHOULD INCLUDE AN ACE INHIBITOR OR ARB
Once LVH has been diagnosed, the next step is to decide on an appropriate treatment plan.
While the choice of therapy will always depend on other comorbidities, a 2003 metaanalysis of antihypertensive medications in the treatment of LVH (controlling for the degree of blood pressure lowering) showed that ARBs were the most efficacious class of agents for reducing the left ventricular mass.33 Specifically, ARBs decreased the mass by 13%, followed by calcium-channel blockers at 11%, ACE inhibitors at 10%, diuretics at 8%, and beta-blockers at 6%. In pairwise comparison, ARBs, calcium-channel blockers, and ACE inhibitors were all significantly more effective in reducing the left ventricular mass than beta-blockers.
As previously discussed, LVH appears to be pathophysiologically linked to myocardial fibrosis and the renin-angiotensin-aldosterone system. For this reason and based on the data presented above regarding the degree of LVH regression, ACE inhibitors or ARBs should be used as the first-line agents for LVH unless they are contraindicated in the individual patient.
The LIFE study
The LIFE study offers the strongest evidence that treating LVH is beneficial. It showed that in hypertensive patients with electrocardiographic LVH by the Cornell product or Sokolow-Lyon criteria, treatment with antihypertensive drugs that resulted in less-severe LVH on electrocardiography was associated with lower rates of cardiovascular morbidity and death, independent of the blood pressure achieved or the drug used.10
The end point in this study was a composite of stroke, myocardial infarction, and cardiovascular death. Regression of electrocardiographic LVH in hypertensive patients has also been shown to decrease the incidence of diabetes mellitus,34 atrial fibrillation,35 and hospitalizations for heart failure.36
The LIFE study also examined the prognostic implications of treating LVH detected by echocardiography. In this prospective cohort substudy, patients who had a lower left ventricular mass index during treatment with antihypertensive drugs had lower rates of cardiovascular morbidity and all-cause mortality, independent of the effects of blood pressure and treatment used.11
These results suggest that there may be a role not only for treating LVH, but also for monitoring for a reduction in the left ventricular mass index as a goal of therapy (similar to the way hemoglobin A1c is used in diabetic patients). If the index is used in this way, one could potentially adjust the dose of current drugs, switch classes, or add an additional drug based on a persistently elevated left ventricular mass index in order to optimize the patient's overall cardiovascular risk. A randomized controlled trial of therapy directed by the mass index vs conventional therapy of LVH would be necessary to assess the clinical utility of this approach.
RECOMMENDATIONS
LVH is a common and potentially modifiable cardiovascular risk factor often overlooked in clinical practice. Ideally, all hypertensive patients should be screened with echocardiography to look for LVH, using the calculated left ventricular mass index rather than wall thickness alone to make the diagnosis. While electrocardiography is specific and also has prognostic implications, it is not sensitive enough to be used alone to screen for LVH.
Once the diagnosis of LVH is made, the initial therapy should be an ARB or an ACE inhibitor. Response to therapy can be assessed by monitoring for a reduction in left ventricular mass index or regression of electrocardiographic LVH.
Treatment-induced regression of LVH decreases adverse cardiovascular events and improves overall survival. When modifying medications in hypertensive patients, it is important to remember that the treatment of LVH is not synonymous with blood pressure control.
- Casale PN, Devereux RB, Milner M, et al. Value of echocardiographic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105:173–178.
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566.
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352.
- Verdecchia P, Carini G, Circo A, et al; MAVI (MAssa Ventricolare sinistra nell’Ipertensione) Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 2001; 38:1829–1835.
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998; 32:1454–1459.
- Verdecchia P, Porcellati C, Reboldi G, et al. Left ventricular hypertrophy as an independent predictor of acute cerebrovascular events in essential hypertension. Circulation 2001; 104:2039–2044.
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35:580–586.
- Verdecchia P, Schillaci G, Borgioni C, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation 1998; 97:48–54.
- Mathew J, Sleight P, Lonn E, et al; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621.
- Okin PM, Devereux RB, Jern S, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004; 292:2343–2349.
- Devereux RB, Wachtell K, Gerdts E, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292:2350–2356.
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470–479.
- Zabalgoitia M, Berning J, Koren MJ, et al; LIFE Study Investigators. Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 2001; 88:646–650.
- Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol 1990; 65:1G–7G.
- González A, López B, Querejeta R, Díez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 2002; 34:1585–1593.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320.
- Liebson PR, Grandits G, Prineas R, et al. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993; 87:476–486.
- Martinez MA, Sancho T, Armada E, et al; Vascular Risk Working Group Grupo Monitorizacíon Ambulatoria de la Presión Arterial (MAPA)-Madrid. Prevalence of left ventricular hypertrophy in patients with mild hypertension in primary care: impact of echocardiography on cardiovascular risk stratification. Am J Hypertens 2003; 16:556–563.
- Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ 2007; 335:711.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990; 81:1144–1146.
- Casale PN, Devereux RB, Kligfield P, et al. Electrocardiographic detection of left ventricular hypertrophy: development and prospective validation of improved criteria. J Am Coll Cardiol 1985; 6:572–580.
- Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol 1992; 20:1180–1186.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949; 37:161–186.
- Romhilt DW, Estes EH. A point-score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968; 75:752–758.
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation 1990; 81:815–820.
- Okin PM, Wright JT, Nieminen MS, et al. Ethnic differences in electrocardiographic criteria for left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint. Am J Hypertens 2002; 15:663–671.
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986; 57:450–458.
- Leibowitz D, Planer D, Ben-Ibgi F, Rott D, Weiss AT, Bursztyn M. Measurement of wall thickness alone does not accurately assess the presence of left ventricular hypertrophy. Clin Exp Hypertens 2007; 29:119–125.
- Black HR, Weltin G, Jaffe CC. The limited echocardiogram: a modification of standard echocardiography for use in the routine evaluation of patients with systemic hypertension. Am J Cardiol 1991; 67:1027–1030.
- American Society of Echocardiography Coding and Reimbursement Newsletter, January 2009. http://www.asecho.org/files/public/CodingnewsJan09.pdf. Accessed May 13, 2010.
- Cuspidi C, Meani S, Valerio C, Fusi V, Sala C, Zanchetti A. Left ventricular hypertrophy and cardiovascular risk stratification: impact and cost-effectiveness of echocardiography in recently diagnosed essential hypertensives. J Hypertens 2006; 24:1671–1677.
- Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, Gottdiener JS. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 1995; 8:221–228.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003; 115:41–46.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. In-treatment resolution or absence of electrocardiographic left ventricular hypertrophy is associated with decreased incidence of new-onset diabetes mellitus in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2007; 50:984–990.
- Okin PM, Wachtell K, Devereux RB, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006; 296:1242–1248.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Ann Intern Med 2007; 147:311–319.
- Casale PN, Devereux RB, Milner M, et al. Value of echocardiographic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105:173–178.
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566.
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352.
- Verdecchia P, Carini G, Circo A, et al; MAVI (MAssa Ventricolare sinistra nell’Ipertensione) Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 2001; 38:1829–1835.
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998; 32:1454–1459.
- Verdecchia P, Porcellati C, Reboldi G, et al. Left ventricular hypertrophy as an independent predictor of acute cerebrovascular events in essential hypertension. Circulation 2001; 104:2039–2044.
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35:580–586.
- Verdecchia P, Schillaci G, Borgioni C, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation 1998; 97:48–54.
- Mathew J, Sleight P, Lonn E, et al; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621.
- Okin PM, Devereux RB, Jern S, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004; 292:2343–2349.
- Devereux RB, Wachtell K, Gerdts E, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292:2350–2356.
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470–479.
- Zabalgoitia M, Berning J, Koren MJ, et al; LIFE Study Investigators. Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 2001; 88:646–650.
- Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol 1990; 65:1G–7G.
- González A, López B, Querejeta R, Díez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 2002; 34:1585–1593.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320.
- Liebson PR, Grandits G, Prineas R, et al. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993; 87:476–486.
- Martinez MA, Sancho T, Armada E, et al; Vascular Risk Working Group Grupo Monitorizacíon Ambulatoria de la Presión Arterial (MAPA)-Madrid. Prevalence of left ventricular hypertrophy in patients with mild hypertension in primary care: impact of echocardiography on cardiovascular risk stratification. Am J Hypertens 2003; 16:556–563.
- Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ 2007; 335:711.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990; 81:1144–1146.
- Casale PN, Devereux RB, Kligfield P, et al. Electrocardiographic detection of left ventricular hypertrophy: development and prospective validation of improved criteria. J Am Coll Cardiol 1985; 6:572–580.
- Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol 1992; 20:1180–1186.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949; 37:161–186.
- Romhilt DW, Estes EH. A point-score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968; 75:752–758.
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation 1990; 81:815–820.
- Okin PM, Wright JT, Nieminen MS, et al. Ethnic differences in electrocardiographic criteria for left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint. Am J Hypertens 2002; 15:663–671.
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986; 57:450–458.
- Leibowitz D, Planer D, Ben-Ibgi F, Rott D, Weiss AT, Bursztyn M. Measurement of wall thickness alone does not accurately assess the presence of left ventricular hypertrophy. Clin Exp Hypertens 2007; 29:119–125.
- Black HR, Weltin G, Jaffe CC. The limited echocardiogram: a modification of standard echocardiography for use in the routine evaluation of patients with systemic hypertension. Am J Cardiol 1991; 67:1027–1030.
- American Society of Echocardiography Coding and Reimbursement Newsletter, January 2009. http://www.asecho.org/files/public/CodingnewsJan09.pdf. Accessed May 13, 2010.
- Cuspidi C, Meani S, Valerio C, Fusi V, Sala C, Zanchetti A. Left ventricular hypertrophy and cardiovascular risk stratification: impact and cost-effectiveness of echocardiography in recently diagnosed essential hypertensives. J Hypertens 2006; 24:1671–1677.
- Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, Gottdiener JS. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 1995; 8:221–228.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003; 115:41–46.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. In-treatment resolution or absence of electrocardiographic left ventricular hypertrophy is associated with decreased incidence of new-onset diabetes mellitus in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2007; 50:984–990.
- Okin PM, Wachtell K, Devereux RB, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006; 296:1242–1248.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Ann Intern Med 2007; 147:311–319.
KEY POINTS
- LVH is caused by a chronically increased cardiac workload, most commonly from hypertension.
- Ideally, all hypertensive patients should undergo echocardiography to screen for LVH, using the calculated left ventricular mass index.
- Electrocardiography is too insensitive to be used alone to screen for LVH.
- In hypertensive patients, initial therapy of LVH should consist of an angiotensin II receptor blocker or an angiotensin-converting enzyme inhibitor.
- Treatment-induced regression of LVH improves cardiovascular outcomes independent of blood pressure.
- Further study is necessary to examine the utility of following the left ventricular mass index as a treatment goal.
Preventing and treating orthostatic hypotension: As easy as A, B, C
Orthostatic hypotension is a chronic, debilitating illness associated with common neurologic conditions (eg, diabetic neuropathy, Parkinson disease). It is common in the elderly, especially in those who are institutionalized and are using multiple medications.
Treatment can be challenging, especially if the problem is neurogenic. This condition has no cure, symptoms vary in different circumstances, treatment is nonspecific, and aggressive treatment can lead to marked supine hypertension.
This review focuses on the prevention and treatment of neurogenic causes of orthostatic hypotension. We emphasize a simple but effective patient-oriented approach to management, using a combination of nonpharmacologic strategies and drugs clinically proven to be efficacious. The recommendations and their rationale are organized in a practical and easy-to-remember format for both physicians and patients.
WHAT HAPPENS WHEN WE STAND UP?
When we stand up, the blood goes down from the chest to the distensible venous capacitance system below the diaphragm. This fluid shift produces a decrease in venous return, ventricular filling, cardiac output, and blood pressure.1
This gravity-induced drop in blood pressure, detected by arterial baroreceptors in the aortic arch and carotid sinus, triggers a compensatory reflex tachycardia and vasoconstriction that restores normotension in the upright position. This compensatory mechanism is termed a baroreflex; it is mediated by afferent and efferent autonomic peripheral nerves and is integrated in autonomic centers in the brainstem.2
Orthostatic hypotension is the result of baroreflex failure (autonomic failure), end-organ dysfunction, or volume depletion. Injury to any limb of the baroreflex causes neurogenic orthostatic hypotension, although with afferent lesions alone, the hypotension tends to be modest and accompanied by wide fluctuations in blood pressure, including severe hypertension. Drugs can produce orthostatic hypotension by interfering with the autonomic pathways or their target end-organs or by affecting intravascular volume. Brain hypoperfusion, resulting from orthostatic hypotension from any cause, can lead to symptoms of orthostatic intolerance (eg, lightheadedness) and falls, and if the hypotension is severe, to syncope.
A DECREASE OF 20 MM HG SYSTOLIC OR 10 MM HG DIASTOLIC
The consensus definition of orthostatic hypotension is a reduction of systolic blood pressure of at least 20 mm Hg or a reduction of diastolic blood pressure of at least 10 mm Hg within 3 minutes of erect standing.3 A transient drop that occurs with abrupt standing and resolves rapidly suggests a benign condition, such as dehydration, rather than autonomic failure.
In the laboratory, patients are placed on a tilt table in the head-up position at an angle of at least 60 degrees to detect orthostatic changes in blood pressure. In the office, 1 minute of standing probably detects nearly all cases of orthostatic hypotension; however, standing beyond 2 minutes helps establish the severity (a further drop in blood pressure).4 Orthostatic hypotension developing after 3 minutes of standing is uncommon and may represent a reflex presyncope (eg, vasovagal) or a mild or early form of sympathetic adrenergic dysfunction.4, 5
NEUROGENIC AND NONNEUROGENIC CAUSES
Orthostatic hypotension may result from neurogenic and nonneurogenic causes.
Neurogenic orthostatic hypotension can be due to neuropathy (eg, diabetic or autoimmune neuropathies) or to central lesions (eg, Parkinson disease or multiple system atrophy). Its presence, severity, and temporal course can be important clues in diagnosing Parkinson disease and differentiating it from other parkinsonian syndromes with a more ominous prognosis, such as multiple system atrophy and Lewy body dementia.
Nonneurogenic causes include cardiac impairment (eg, from myocardial infarction or aortic stenosis), reduced intravascular volume (eg, from dehydration, adrenal insufficiency), and vasodilation (eg, from fever, systemic mastocytosis).
Common drugs that cause orthostatic hypotension are diuretics, alpha-adrenoceptor blockers for prostatic hypertrophy, antihypertensive drugs, and calcium channel blockers. Insulin, levodopa, and tricyclic antidepressants can also cause vasodilation and orthostatic hypotension in predisposed patients. Poon and Braun,6 in a retrospective study in elderly veterans, identified hydrochlorothiazide, lisinopril (Prinivil, Zestril), trazodone (Desyrel), furosemide (Lasix), and terazosin (Hytrin) as the most common culprits.
ORTHOSTATIC HYPOTENSION IS COMMON IN THE ELDERLY
The prevalence of orthostatic hypotension is high in the elderly and depends on the characteristics of the population studied, such as age, use of medications, and comorbidities known to be associated with this problem. Orthostatic hypotension is more common in institutionalized elderly people (up to 68%)7 than in those living in the community (6%).8 The high prevalence among institutionalized patients likely reflects multiple disease processes, including neurologic and cardiac conditions, as well as medications associated with orthostatic hypotension.
CLINICAL MANIFESTATIONS ARE DUE TO HYPOPERFUSION, OVERCOMPENSATION
Symptoms are related to cerebral hypoperfusion, with resulting lack of cerebral oxygenation (causing lightheadedness, dizziness, weakness, difficulty thinking, headache, syncope, or feeling faint) and a compensatory autonomic overreaction (causing palpitations, tremulousness, nausea, coldness of extremities, chest pain, and syncope).
Lightheadedness is a common symptom, but subtler issues such as difficulty thinking, weakness, and neck discomfort are also common in the elderly. Recurrent or unexplained falls in older adults may be a manifestation of syncope due to orthostatic hypotension.
PROGNOSIS DEPENDS ON CAUSE
Orthostatic hypotension is a syndrome, and its prognosis depends on its specific cause, its severity, and the distribution of its autonomic and nonautonomic involvement. In patients who have extrapyramidal and cerebellar disorders (eg, Parkinson disease, multiple system atrophy), the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis.9,10
In hypertensive patients with diabetes mellitus, the risk of death is higher if they have orthostatic hypotension.11 Diastolic orthostatic hypotension is associated with a higher risk of vascular death in older persons.12
MANAGEMENT: FROM A TO F
The goal of management of orthostatic hypotension is to raise the patient’s standing blood pressure without also raising his or her supine blood pressure, and specifically to reduce orthostatic symptoms, increase the time the patient can stand, and improve his or her ability to perform daily activities. No specific treatment is currently available that achieves all these goals, and drugs alone are never completely adequate.

Because the mainstays of treatment are volume expansion and vasoconstriction, it is difficult to improve the symptoms of orthostatic hypotension without inducing some degree of supine hypertension. Strategies to minimize nocturnal hypertension and to treat orthostatic hypotension in special circumstances are summarized in Table 1.


If hypovolemia is playing a major role, and the patient cannot ingest enough salt or plasma volume fails to increase despite salt supplementation, fludrocortisone (Florinef) should be considered. Untreated hypovolemia will decrease the efficacy of vasoconstrictor drugs.
Pyridostigmine (Mestinon) has a putative vasoconstrictor effect only during standing, but because its effect is modest it should be used in mild orthostatic hypotension that does not improve with nonpharmacologic measures and in moderate cases. Its effect can be enhanced with additional low doses of midodrine (ProAmatine). Midodrine with or without fludrocortisone should be used in severe orthostatic hypotension.

A: Abdominal compression
In conditions in which there is adrenergic denervation of vascular beds, there is an increase in vascular capacitance and peripheral venous pooling. Compression of capacitance beds (ie, the legs and abdomen) improves orthostatic symptoms.14 The improvement is due to a reduction of venous capacitance and an increase in total peripheral resistance.14
On standing, healthy adults experience an orthostatic shift of approximately 500 mL of blood to the lower extremities15 that, when added to an increased vascular capacitance in those with orthostatic hypotension, results in a relative state of hypovolemia.
Compression of the legs alone is not as beneficial as compression of the abdomen because the venous capacitance of the calves and thighs is relatively small compared with that of the splanchnic mesenteric bed, which accounts for 20% to 30% of total blood volume.16 Moreover, compression garments and stockings that are strong enough to produce a measurable effect on orthostatic hypotension are cumbersome to put on and uncomfortable to wear. Because some patients gain significant benefit from abdominal compression alone, this should be considered the first step in reducing venous capacitance.
In a laboratory experiment, Smit et al17 found that an elastic abdominal binder that exerted 15 to 20 mm Hg of pressure on the abdomen raised the standing blood pressure by about 11/6 mmHg, which was comparable to the effect of a gravity suit (such as those worn by fighter pilots to prevent syncope during violent aircraft maneuvers) inflated to 20 mm Hg—an increase of about 17/8 mm Hg. Higher gravity-suit pressures had a greater effect.
In practical terms, the binder should be tight enough to exert gentle pressure. It should be put on before rising from bed in the morning and taken off when lying supine, to avoid supine hypertension. Advantages are that a binder’s effects are immediate, its benefits can be easily assessed, and it can be used on an as-needed basis by patients who need it only during periods of prolonged orthostatic stress. Binders are also easy to fit and are available in most sporting good stores and on the Web (try searching for “abdominal binder”).
When abdominal compression alone is not enough, the addition of compression of the lower extremities can result in further benefits. This can be achieved by using compression garments that ideally extend to the waist or, at the least, to the proximal thigh.
B: Boluses of water
Rapidly drinking two 8-oz (500-mL) glasses of cold water helps expand plasma volume. It also, within a few minutes, elicits a significant pressor effect that is in part norepinephrine-mediated,18,19 increasing the standing systolic blood pressure by more than 20 mm Hg for about 2 hours and improving symptoms and orthostatic endurance.18,20 This easy technique can be used when prolonged standing is expected (eg, shopping).
B (continued): Bed up
The head of the bed of a patient with orthostatic hypotension should be elevated by 10 to 20 degrees or 4 inches (10 cm) to decrease nocturnal hypertension and nocturnal diuresis.21 During the day, adequate orthostatic stress, ie, upright activity, should be maintained. If patients are repeatedly tilted up, their orthostatic hypotension is gradually attenuated, presumably by increasing venomotor tone.22
C: Countermaneuvers
Physical countermaneuvers involve isometrically contracting the muscles below the waist for about 30 seconds at a time, which reduces venous capacitance, increases total peripheral resistance, and augments venous return to the heart.23,24 These countermeasures can help maintain blood pressure during daily activities and should be considered at the first symptoms of orthostatic intolerance and in situations of orthostatic stress (eg, standing for prolonged periods).
Specific techniques include23:
- Toe-raising
- Leg-crossing and contraction
- Thigh muscle co-contraction
- Bending at the waist
- Slow marching in place
- Leg elevation.
D: Drugs
Midodrine, a vasopressor, is effective and safe when used for treating neurogenic orthostatic hypotension.25 It has been shown to increase standing systolic blood pressure, reduce orthostatic lightheadedness, and increase standing and walking time.
A common starting dose is 5 mg three times a day; most patients respond best to 10 mg three times a day. As its duration of action is short (2 to 4 hours),25–27 it should be taken before arising in the morning, before lunch, and in the midafternoon. To avoid nocturnal supine hypertension, doses should not be taken after the midafternoon, and a dose should be omitted if the supine or sitting blood pressure is greater than 180/100 mm Hg.
Midodrine’s main side effects are supine hypertension, scalp paresthesias, and pilomotor reactions (goosebumps). Vasoconstrictors such as midodrine are ineffective when plasma volume is reduced.
Fludrocortisone is a synthetic mineralocorticoid that has a pressor effect as a result of its ability to expand plasma volume and increase vascular alpha-adrenoceptor sensitivity.28–30 This medication is helpful when plasma volume fails to adequately increase with salt supplementation31 and for patients who cannot ingest enough salt or do not respond adequately to midodrine.
The usual dose is 0.1 to 0.2 mg/day, but it may be increased to 0.4 to 0.6 mg/day in patients with refractory orthostatic hypotension.
If the patient gains 3 to 5 pounds (1.2–2.3 kg) and develops mild dependent edema, you can infer that the plasma volume has expanded adequately. However, in view of these effects, fludrocortisone is contraindicated in congestive heart failure and chronic renal failure. The potential risks are severe hypokalemia and excessive supine hypertension. Frequent monitoring of serum potassium, a diet high in potassium, and regular checks of supine blood pressure are advised, especially at higher doses, when added to midodrine, or in elderly patients who tend to poorly tolerate the medication.28,29,32
Pyridostigmine is a cholinesterase inhibitor that improves ganglionic neurotransmission in the sympathetic baroreflex pathway. Because this pathway is activated primarily during standing, this drug improves orthostatic hypotension and total peripheral resistance without aggravating supine hypertension. Because the pressor effect is modest, it is most adequate for patients with mild to moderate orthostatic hypotension.33,34
Dosing is started at 30 mg two to three times a day and is gradually increased to 60 mg three times a day. The drug’s effectiveness can be enhanced by combining each dose of pyridostigmine with 5 mg of midodrine without occurrence of supine hypertension.34 Mestinon Timespan, a 180-mg slow-release pyridostigmine tablet, can be taken once a day and may be a convenient alternative.
The main side effects are cholinergic (abdominal colic, diarrhea).
Review the patient’s medications. If he or she is taking any drug that may cause orthostatic hypotension, consider discontinuing it, substituting another drug, or changing the dosage (Table 2). In the elderly, antiparkinsonian, nitrate, antidepressant, diuretic, prostate, and antihypertensive medications35 may be particularly suspect.
E: Education
Education is probably the single most important factor in the proper control of orthostatic hypotension. A number of issues should be considered.
- Patients should be taught, in simple terms, the mechanisms that maintain postural normotension and how to recognize the onset of orthostatic symptoms.
- They must realize that there is no specific treatment of the underlying cause and that drug treatment alone is not adequate.
- They should be taught nonpharmacologic approaches and be aware that other drugs they start may worsen symptoms.
It is also important that the patient learn the conditions (and their mechanisms) that can lower blood pressure (Table 3). Such conditions include prolonged or motionless standing, alcohol ingestion (causing vasodilation), carbohydrate-heavy meals (causing postprandial orthostatic hypotension related to an increase in the splanchnic-mesenteric venous capacitance), early morning orthostatic hypotension related to nocturnal diuresis and arising from bed, physical activity sufficient to cause muscle vasodilation, heat exposure (eg, hot weather or a hot bath or shower) producing skin vessel vasodilation, sudden postural changes, and prolonged recumbency. Once these stressors are explained, patients have no difficulty recognizing them.
The patient should also be instructed in how to manage situations of increased orthostatic stress and periods of orthostatic decompensation, to minimize nocturnal hypertension, and to modify their activities of daily living. Keeping a log of supine and upright blood pressures (taken with an automated sphygmomanometer) during situations of orthostatic stress can help establish whether worsening symptoms are related to orthostatic hypotension or to another mechanism. Once patients discover that they can actively deal with these situations, they develop a great sense of empowerment.
E (continued): Exercise
Mild physical exercise improves orthostatic tolerance by reducing venous pooling and increasing plasma volume.36 Deconditioning from lack of exercise exacerbates orthostatic hypotension.37 Because upright exercise may increase the orthostatic drop in blood pressure, training in a supine or sitting position (eg, swimming, recumbent biking) is advisable. Isotonic exercise (eg, light weight-lifting) is recommended because the incorrect straining and breath-holding during isometric exercise (eg, holding weights in the same position) may decrease venous return.
F: Fluid and salt (volume expansion)
Maintaining an adequate plasma volume is crucial. Patients should drink five to eight 8-ounce glasses (1.25 to 2.5 L) of water or other fluid per day. Many elderly people do not take in this much. The patient should have at least 1 glass or cup of fluid with meals and at least twice at other times of each day to obtain 1 L/day.
Salt intake should be between 150 and 250 mmol of sodium (10 to 20 g of salt) per day. Sodium helps with retention of ingested fluids and should be maximized if tolerated. However, caution should be exercised in patients who have severe refractory supine hypertension, uncontrolled hypertension, or comorbidities characterized by insterstitial edema (eg, heart failure, liver failure). Some patients are very sensitive to sodium supplementation and can fine-tune their orthostatic control with salt alone. If salting food is not desired, prepared soups, pretzels, potato chips, and 0.5- or 1.0-g salt tablets can be an option.
Patients need to maintain a high-potassium diet, as the high sodium intake combined with fludrocortisone promotes potassium loss. Fruits (especially bananas) and vegetables have high potassium content.
The combination of fludrocortisone and a high-salt diet can also cause sustained supine hypertension, which can be minimized by the interventions noted in Table 2.
Appropriate salt supplementation and fluid intake leading to an adequate volume expansion can be verified by checking the 24-hour urinary sodium content: patients who excrete less than 170 mmol can be treated with 1 to 2 g of supplemental sodium three times a day.38
- Sjostrand T. The regulation of the blood distribution in man. Acta Physiol Scand 1952; 26:312–327.
- Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977; 296:293–297.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996; 46:1470.
- Gehrking JA, Hines SM, Benrud-Larson LM, Opher-Gehrking TL, Low PA. What is the minimum duration of head-up tilt necessary to detect orthostatic hypotension? Clin Auton Res 2005; 15:71–75.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005; 30:173–178.
- Weiss A, Grossman E, Beloosesky Y, Grinblat J. Orthostatic hypotension in acute geriatric ward: is it a consistent finding? Arch Intern Med 2002; 162:2369–2374.
- Mader SL, Josephson KR, Rubenstein LZ. Low prevalence of postural hypotension among community-dwelling elderly. JAMA 1987; 258:1511–1514.
- Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991; 1:147–155.
- Saito Y, Matsuoka Y, Takahashi A, Ohno Y. Survival of patients with multiple system atrophy. Intern Med 1994; 33:321–325.
- Davis BR, Langford HG, Blaufox MD, Curb JD, Polk BF, Shulman NB. The association of postural changes in systolic blood pressure and mortality in persons with hypertension: the Hypertension Detection and Follow-up Program experience. Circulation 1987; 75:340–346.
- Luukinen H, Koski K, Laippala P, Kivelä SL. Prognosis of diastolic and systolic orthostatic hypotension in older persons. Arch Intern Med 1999; 159:273–280.
- Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. N Engl J Med 1993; 329:611–615.
- Denq JC, Opfer-Gehrking TL, Giuliani M, Felten J, Convertino VA, Low PA. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clin Auton Res 1997; 7:321–326.
- Sjostrand T. Volume and distribution of blood and their significance in regulating the circulation. Physiol Rev 1953; 33:202–228.
- Rowell LB, Detry JM, Blackmon JR, Wyss C. Importance of the splanchnic vascular bed in human blood pressure regulation. J Appl Physiol 1972; 32:213–220.
- Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res 2004; 14:167–175.
- Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000; 101:504–509.
- Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002; 112:355–360.
- Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water [letter]. Lancet 1999; 353:723.
- MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia: treatment with the “head-up” bed. JAMA 1940; 115:2162–2167.
- Ector H, Reybrouck T, Heidbüchel H, Gewillig M, Van de Werf F. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol 1998; 21:193–196.
- Bouvette CM, McPhee BR, Opfer-Gehrking TL, Low PA. Role of physical countermaneuvers in the management of orthostatic hypotension: efficacy and biofeedback augmentation. Mayo Clin Proc 1996; 71:847–853.
- Ten Harkel AD, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci (Lond) 1994; 87:553–558.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993; 95:38–48.
- Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med 1995; 99:604–610.
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007; 7:63–70.
- Axelrod FB, Goldberg JD, Rolnitzky L, et al. Fludrocortisone in patients with familial dysautonomia—assessing effect on clinical parameters and gene expression. Clin Auton Res 2005; 15:284–291.
- Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301:68–73.
- van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. Clin Auton Res 2000; 10:35–42.
- Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart 1996; 76:507–509.
- Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry 2003; 74:1294–1298.
- Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63:513–518.
- Lipsitz LA, Pluchino FC, Wei JY, Rowe JW. Syncope in institutionalized elderly: the impact of multiple pathological conditions and situational stress. J Chronic Dis 1986; 39:619–630.
- Mtinangi BL, Hainsworth R. Effects of moderate exercise training on plasma volume, baroreceptor sensitivity and orthostatic tolerance in healthy subjects. Exp Physiol 1999; 84:121–130.
- Bonnin P, Ben Driss A, Benessiano J, Maillet A, Pavy le Traon A, Levy BI. Enhanced flow-dependent vasodilatation after bed rest, a possible mechanism for orthostatic intolerance in humans. Eur J Appl Physiol 2001; 85:420–426.
- El-Sayed H, Hainsworth R. Salt supplementation increases plasma volume and orthostatic tolerance in patients with unexplained syncope. Heart 1996; 75:134–140.
Orthostatic hypotension is a chronic, debilitating illness associated with common neurologic conditions (eg, diabetic neuropathy, Parkinson disease). It is common in the elderly, especially in those who are institutionalized and are using multiple medications.
Treatment can be challenging, especially if the problem is neurogenic. This condition has no cure, symptoms vary in different circumstances, treatment is nonspecific, and aggressive treatment can lead to marked supine hypertension.
This review focuses on the prevention and treatment of neurogenic causes of orthostatic hypotension. We emphasize a simple but effective patient-oriented approach to management, using a combination of nonpharmacologic strategies and drugs clinically proven to be efficacious. The recommendations and their rationale are organized in a practical and easy-to-remember format for both physicians and patients.
WHAT HAPPENS WHEN WE STAND UP?
When we stand up, the blood goes down from the chest to the distensible venous capacitance system below the diaphragm. This fluid shift produces a decrease in venous return, ventricular filling, cardiac output, and blood pressure.1
This gravity-induced drop in blood pressure, detected by arterial baroreceptors in the aortic arch and carotid sinus, triggers a compensatory reflex tachycardia and vasoconstriction that restores normotension in the upright position. This compensatory mechanism is termed a baroreflex; it is mediated by afferent and efferent autonomic peripheral nerves and is integrated in autonomic centers in the brainstem.2
Orthostatic hypotension is the result of baroreflex failure (autonomic failure), end-organ dysfunction, or volume depletion. Injury to any limb of the baroreflex causes neurogenic orthostatic hypotension, although with afferent lesions alone, the hypotension tends to be modest and accompanied by wide fluctuations in blood pressure, including severe hypertension. Drugs can produce orthostatic hypotension by interfering with the autonomic pathways or their target end-organs or by affecting intravascular volume. Brain hypoperfusion, resulting from orthostatic hypotension from any cause, can lead to symptoms of orthostatic intolerance (eg, lightheadedness) and falls, and if the hypotension is severe, to syncope.
A DECREASE OF 20 MM HG SYSTOLIC OR 10 MM HG DIASTOLIC
The consensus definition of orthostatic hypotension is a reduction of systolic blood pressure of at least 20 mm Hg or a reduction of diastolic blood pressure of at least 10 mm Hg within 3 minutes of erect standing.3 A transient drop that occurs with abrupt standing and resolves rapidly suggests a benign condition, such as dehydration, rather than autonomic failure.
In the laboratory, patients are placed on a tilt table in the head-up position at an angle of at least 60 degrees to detect orthostatic changes in blood pressure. In the office, 1 minute of standing probably detects nearly all cases of orthostatic hypotension; however, standing beyond 2 minutes helps establish the severity (a further drop in blood pressure).4 Orthostatic hypotension developing after 3 minutes of standing is uncommon and may represent a reflex presyncope (eg, vasovagal) or a mild or early form of sympathetic adrenergic dysfunction.4, 5
NEUROGENIC AND NONNEUROGENIC CAUSES
Orthostatic hypotension may result from neurogenic and nonneurogenic causes.
Neurogenic orthostatic hypotension can be due to neuropathy (eg, diabetic or autoimmune neuropathies) or to central lesions (eg, Parkinson disease or multiple system atrophy). Its presence, severity, and temporal course can be important clues in diagnosing Parkinson disease and differentiating it from other parkinsonian syndromes with a more ominous prognosis, such as multiple system atrophy and Lewy body dementia.
Nonneurogenic causes include cardiac impairment (eg, from myocardial infarction or aortic stenosis), reduced intravascular volume (eg, from dehydration, adrenal insufficiency), and vasodilation (eg, from fever, systemic mastocytosis).
Common drugs that cause orthostatic hypotension are diuretics, alpha-adrenoceptor blockers for prostatic hypertrophy, antihypertensive drugs, and calcium channel blockers. Insulin, levodopa, and tricyclic antidepressants can also cause vasodilation and orthostatic hypotension in predisposed patients. Poon and Braun,6 in a retrospective study in elderly veterans, identified hydrochlorothiazide, lisinopril (Prinivil, Zestril), trazodone (Desyrel), furosemide (Lasix), and terazosin (Hytrin) as the most common culprits.
ORTHOSTATIC HYPOTENSION IS COMMON IN THE ELDERLY
The prevalence of orthostatic hypotension is high in the elderly and depends on the characteristics of the population studied, such as age, use of medications, and comorbidities known to be associated with this problem. Orthostatic hypotension is more common in institutionalized elderly people (up to 68%)7 than in those living in the community (6%).8 The high prevalence among institutionalized patients likely reflects multiple disease processes, including neurologic and cardiac conditions, as well as medications associated with orthostatic hypotension.
CLINICAL MANIFESTATIONS ARE DUE TO HYPOPERFUSION, OVERCOMPENSATION
Symptoms are related to cerebral hypoperfusion, with resulting lack of cerebral oxygenation (causing lightheadedness, dizziness, weakness, difficulty thinking, headache, syncope, or feeling faint) and a compensatory autonomic overreaction (causing palpitations, tremulousness, nausea, coldness of extremities, chest pain, and syncope).
Lightheadedness is a common symptom, but subtler issues such as difficulty thinking, weakness, and neck discomfort are also common in the elderly. Recurrent or unexplained falls in older adults may be a manifestation of syncope due to orthostatic hypotension.
PROGNOSIS DEPENDS ON CAUSE
Orthostatic hypotension is a syndrome, and its prognosis depends on its specific cause, its severity, and the distribution of its autonomic and nonautonomic involvement. In patients who have extrapyramidal and cerebellar disorders (eg, Parkinson disease, multiple system atrophy), the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis.9,10
In hypertensive patients with diabetes mellitus, the risk of death is higher if they have orthostatic hypotension.11 Diastolic orthostatic hypotension is associated with a higher risk of vascular death in older persons.12
MANAGEMENT: FROM A TO F
The goal of management of orthostatic hypotension is to raise the patient’s standing blood pressure without also raising his or her supine blood pressure, and specifically to reduce orthostatic symptoms, increase the time the patient can stand, and improve his or her ability to perform daily activities. No specific treatment is currently available that achieves all these goals, and drugs alone are never completely adequate.
Because the mainstays of treatment are volume expansion and vasoconstriction, it is difficult to improve the symptoms of orthostatic hypotension without inducing some degree of supine hypertension. Strategies to minimize nocturnal hypertension and to treat orthostatic hypotension in special circumstances are summarized in Table 1.
If hypovolemia is playing a major role, and the patient cannot ingest enough salt or plasma volume fails to increase despite salt supplementation, fludrocortisone (Florinef) should be considered. Untreated hypovolemia will decrease the efficacy of vasoconstrictor drugs.
Pyridostigmine (Mestinon) has a putative vasoconstrictor effect only during standing, but because its effect is modest it should be used in mild orthostatic hypotension that does not improve with nonpharmacologic measures and in moderate cases. Its effect can be enhanced with additional low doses of midodrine (ProAmatine). Midodrine with or without fludrocortisone should be used in severe orthostatic hypotension.
A: Abdominal compression
In conditions in which there is adrenergic denervation of vascular beds, there is an increase in vascular capacitance and peripheral venous pooling. Compression of capacitance beds (ie, the legs and abdomen) improves orthostatic symptoms.14 The improvement is due to a reduction of venous capacitance and an increase in total peripheral resistance.14
On standing, healthy adults experience an orthostatic shift of approximately 500 mL of blood to the lower extremities15 that, when added to an increased vascular capacitance in those with orthostatic hypotension, results in a relative state of hypovolemia.
Compression of the legs alone is not as beneficial as compression of the abdomen because the venous capacitance of the calves and thighs is relatively small compared with that of the splanchnic mesenteric bed, which accounts for 20% to 30% of total blood volume.16 Moreover, compression garments and stockings that are strong enough to produce a measurable effect on orthostatic hypotension are cumbersome to put on and uncomfortable to wear. Because some patients gain significant benefit from abdominal compression alone, this should be considered the first step in reducing venous capacitance.
In a laboratory experiment, Smit et al17 found that an elastic abdominal binder that exerted 15 to 20 mm Hg of pressure on the abdomen raised the standing blood pressure by about 11/6 mmHg, which was comparable to the effect of a gravity suit (such as those worn by fighter pilots to prevent syncope during violent aircraft maneuvers) inflated to 20 mm Hg—an increase of about 17/8 mm Hg. Higher gravity-suit pressures had a greater effect.
In practical terms, the binder should be tight enough to exert gentle pressure. It should be put on before rising from bed in the morning and taken off when lying supine, to avoid supine hypertension. Advantages are that a binder’s effects are immediate, its benefits can be easily assessed, and it can be used on an as-needed basis by patients who need it only during periods of prolonged orthostatic stress. Binders are also easy to fit and are available in most sporting good stores and on the Web (try searching for “abdominal binder”).
When abdominal compression alone is not enough, the addition of compression of the lower extremities can result in further benefits. This can be achieved by using compression garments that ideally extend to the waist or, at the least, to the proximal thigh.
B: Boluses of water
Rapidly drinking two 8-oz (500-mL) glasses of cold water helps expand plasma volume. It also, within a few minutes, elicits a significant pressor effect that is in part norepinephrine-mediated,18,19 increasing the standing systolic blood pressure by more than 20 mm Hg for about 2 hours and improving symptoms and orthostatic endurance.18,20 This easy technique can be used when prolonged standing is expected (eg, shopping).
B (continued): Bed up
The head of the bed of a patient with orthostatic hypotension should be elevated by 10 to 20 degrees or 4 inches (10 cm) to decrease nocturnal hypertension and nocturnal diuresis.21 During the day, adequate orthostatic stress, ie, upright activity, should be maintained. If patients are repeatedly tilted up, their orthostatic hypotension is gradually attenuated, presumably by increasing venomotor tone.22
C: Countermaneuvers
Physical countermaneuvers involve isometrically contracting the muscles below the waist for about 30 seconds at a time, which reduces venous capacitance, increases total peripheral resistance, and augments venous return to the heart.23,24 These countermeasures can help maintain blood pressure during daily activities and should be considered at the first symptoms of orthostatic intolerance and in situations of orthostatic stress (eg, standing for prolonged periods).
Specific techniques include23:
- Toe-raising
- Leg-crossing and contraction
- Thigh muscle co-contraction
- Bending at the waist
- Slow marching in place
- Leg elevation.
D: Drugs
Midodrine, a vasopressor, is effective and safe when used for treating neurogenic orthostatic hypotension.25 It has been shown to increase standing systolic blood pressure, reduce orthostatic lightheadedness, and increase standing and walking time.
A common starting dose is 5 mg three times a day; most patients respond best to 10 mg three times a day. As its duration of action is short (2 to 4 hours),25–27 it should be taken before arising in the morning, before lunch, and in the midafternoon. To avoid nocturnal supine hypertension, doses should not be taken after the midafternoon, and a dose should be omitted if the supine or sitting blood pressure is greater than 180/100 mm Hg.
Midodrine’s main side effects are supine hypertension, scalp paresthesias, and pilomotor reactions (goosebumps). Vasoconstrictors such as midodrine are ineffective when plasma volume is reduced.
Fludrocortisone is a synthetic mineralocorticoid that has a pressor effect as a result of its ability to expand plasma volume and increase vascular alpha-adrenoceptor sensitivity.28–30 This medication is helpful when plasma volume fails to adequately increase with salt supplementation31 and for patients who cannot ingest enough salt or do not respond adequately to midodrine.
The usual dose is 0.1 to 0.2 mg/day, but it may be increased to 0.4 to 0.6 mg/day in patients with refractory orthostatic hypotension.
If the patient gains 3 to 5 pounds (1.2–2.3 kg) and develops mild dependent edema, you can infer that the plasma volume has expanded adequately. However, in view of these effects, fludrocortisone is contraindicated in congestive heart failure and chronic renal failure. The potential risks are severe hypokalemia and excessive supine hypertension. Frequent monitoring of serum potassium, a diet high in potassium, and regular checks of supine blood pressure are advised, especially at higher doses, when added to midodrine, or in elderly patients who tend to poorly tolerate the medication.28,29,32
Pyridostigmine is a cholinesterase inhibitor that improves ganglionic neurotransmission in the sympathetic baroreflex pathway. Because this pathway is activated primarily during standing, this drug improves orthostatic hypotension and total peripheral resistance without aggravating supine hypertension. Because the pressor effect is modest, it is most adequate for patients with mild to moderate orthostatic hypotension.33,34
Dosing is started at 30 mg two to three times a day and is gradually increased to 60 mg three times a day. The drug’s effectiveness can be enhanced by combining each dose of pyridostigmine with 5 mg of midodrine without occurrence of supine hypertension.34 Mestinon Timespan, a 180-mg slow-release pyridostigmine tablet, can be taken once a day and may be a convenient alternative.
The main side effects are cholinergic (abdominal colic, diarrhea).
Review the patient’s medications. If he or she is taking any drug that may cause orthostatic hypotension, consider discontinuing it, substituting another drug, or changing the dosage (Table 2). In the elderly, antiparkinsonian, nitrate, antidepressant, diuretic, prostate, and antihypertensive medications35 may be particularly suspect.
E: Education
Education is probably the single most important factor in the proper control of orthostatic hypotension. A number of issues should be considered.
- Patients should be taught, in simple terms, the mechanisms that maintain postural normotension and how to recognize the onset of orthostatic symptoms.
- They must realize that there is no specific treatment of the underlying cause and that drug treatment alone is not adequate.
- They should be taught nonpharmacologic approaches and be aware that other drugs they start may worsen symptoms.
It is also important that the patient learn the conditions (and their mechanisms) that can lower blood pressure (Table 3). Such conditions include prolonged or motionless standing, alcohol ingestion (causing vasodilation), carbohydrate-heavy meals (causing postprandial orthostatic hypotension related to an increase in the splanchnic-mesenteric venous capacitance), early morning orthostatic hypotension related to nocturnal diuresis and arising from bed, physical activity sufficient to cause muscle vasodilation, heat exposure (eg, hot weather or a hot bath or shower) producing skin vessel vasodilation, sudden postural changes, and prolonged recumbency. Once these stressors are explained, patients have no difficulty recognizing them.
The patient should also be instructed in how to manage situations of increased orthostatic stress and periods of orthostatic decompensation, to minimize nocturnal hypertension, and to modify their activities of daily living. Keeping a log of supine and upright blood pressures (taken with an automated sphygmomanometer) during situations of orthostatic stress can help establish whether worsening symptoms are related to orthostatic hypotension or to another mechanism. Once patients discover that they can actively deal with these situations, they develop a great sense of empowerment.
E (continued): Exercise
Mild physical exercise improves orthostatic tolerance by reducing venous pooling and increasing plasma volume.36 Deconditioning from lack of exercise exacerbates orthostatic hypotension.37 Because upright exercise may increase the orthostatic drop in blood pressure, training in a supine or sitting position (eg, swimming, recumbent biking) is advisable. Isotonic exercise (eg, light weight-lifting) is recommended because the incorrect straining and breath-holding during isometric exercise (eg, holding weights in the same position) may decrease venous return.
F: Fluid and salt (volume expansion)
Maintaining an adequate plasma volume is crucial. Patients should drink five to eight 8-ounce glasses (1.25 to 2.5 L) of water or other fluid per day. Many elderly people do not take in this much. The patient should have at least 1 glass or cup of fluid with meals and at least twice at other times of each day to obtain 1 L/day.
Salt intake should be between 150 and 250 mmol of sodium (10 to 20 g of salt) per day. Sodium helps with retention of ingested fluids and should be maximized if tolerated. However, caution should be exercised in patients who have severe refractory supine hypertension, uncontrolled hypertension, or comorbidities characterized by insterstitial edema (eg, heart failure, liver failure). Some patients are very sensitive to sodium supplementation and can fine-tune their orthostatic control with salt alone. If salting food is not desired, prepared soups, pretzels, potato chips, and 0.5- or 1.0-g salt tablets can be an option.
Patients need to maintain a high-potassium diet, as the high sodium intake combined with fludrocortisone promotes potassium loss. Fruits (especially bananas) and vegetables have high potassium content.
The combination of fludrocortisone and a high-salt diet can also cause sustained supine hypertension, which can be minimized by the interventions noted in Table 2.
Appropriate salt supplementation and fluid intake leading to an adequate volume expansion can be verified by checking the 24-hour urinary sodium content: patients who excrete less than 170 mmol can be treated with 1 to 2 g of supplemental sodium three times a day.38
Orthostatic hypotension is a chronic, debilitating illness associated with common neurologic conditions (eg, diabetic neuropathy, Parkinson disease). It is common in the elderly, especially in those who are institutionalized and are using multiple medications.
Treatment can be challenging, especially if the problem is neurogenic. This condition has no cure, symptoms vary in different circumstances, treatment is nonspecific, and aggressive treatment can lead to marked supine hypertension.
This review focuses on the prevention and treatment of neurogenic causes of orthostatic hypotension. We emphasize a simple but effective patient-oriented approach to management, using a combination of nonpharmacologic strategies and drugs clinically proven to be efficacious. The recommendations and their rationale are organized in a practical and easy-to-remember format for both physicians and patients.
WHAT HAPPENS WHEN WE STAND UP?
When we stand up, the blood goes down from the chest to the distensible venous capacitance system below the diaphragm. This fluid shift produces a decrease in venous return, ventricular filling, cardiac output, and blood pressure.1
This gravity-induced drop in blood pressure, detected by arterial baroreceptors in the aortic arch and carotid sinus, triggers a compensatory reflex tachycardia and vasoconstriction that restores normotension in the upright position. This compensatory mechanism is termed a baroreflex; it is mediated by afferent and efferent autonomic peripheral nerves and is integrated in autonomic centers in the brainstem.2
Orthostatic hypotension is the result of baroreflex failure (autonomic failure), end-organ dysfunction, or volume depletion. Injury to any limb of the baroreflex causes neurogenic orthostatic hypotension, although with afferent lesions alone, the hypotension tends to be modest and accompanied by wide fluctuations in blood pressure, including severe hypertension. Drugs can produce orthostatic hypotension by interfering with the autonomic pathways or their target end-organs or by affecting intravascular volume. Brain hypoperfusion, resulting from orthostatic hypotension from any cause, can lead to symptoms of orthostatic intolerance (eg, lightheadedness) and falls, and if the hypotension is severe, to syncope.
A DECREASE OF 20 MM HG SYSTOLIC OR 10 MM HG DIASTOLIC
The consensus definition of orthostatic hypotension is a reduction of systolic blood pressure of at least 20 mm Hg or a reduction of diastolic blood pressure of at least 10 mm Hg within 3 minutes of erect standing.3 A transient drop that occurs with abrupt standing and resolves rapidly suggests a benign condition, such as dehydration, rather than autonomic failure.
In the laboratory, patients are placed on a tilt table in the head-up position at an angle of at least 60 degrees to detect orthostatic changes in blood pressure. In the office, 1 minute of standing probably detects nearly all cases of orthostatic hypotension; however, standing beyond 2 minutes helps establish the severity (a further drop in blood pressure).4 Orthostatic hypotension developing after 3 minutes of standing is uncommon and may represent a reflex presyncope (eg, vasovagal) or a mild or early form of sympathetic adrenergic dysfunction.4, 5
NEUROGENIC AND NONNEUROGENIC CAUSES
Orthostatic hypotension may result from neurogenic and nonneurogenic causes.
Neurogenic orthostatic hypotension can be due to neuropathy (eg, diabetic or autoimmune neuropathies) or to central lesions (eg, Parkinson disease or multiple system atrophy). Its presence, severity, and temporal course can be important clues in diagnosing Parkinson disease and differentiating it from other parkinsonian syndromes with a more ominous prognosis, such as multiple system atrophy and Lewy body dementia.
Nonneurogenic causes include cardiac impairment (eg, from myocardial infarction or aortic stenosis), reduced intravascular volume (eg, from dehydration, adrenal insufficiency), and vasodilation (eg, from fever, systemic mastocytosis).
Common drugs that cause orthostatic hypotension are diuretics, alpha-adrenoceptor blockers for prostatic hypertrophy, antihypertensive drugs, and calcium channel blockers. Insulin, levodopa, and tricyclic antidepressants can also cause vasodilation and orthostatic hypotension in predisposed patients. Poon and Braun,6 in a retrospective study in elderly veterans, identified hydrochlorothiazide, lisinopril (Prinivil, Zestril), trazodone (Desyrel), furosemide (Lasix), and terazosin (Hytrin) as the most common culprits.
ORTHOSTATIC HYPOTENSION IS COMMON IN THE ELDERLY
The prevalence of orthostatic hypotension is high in the elderly and depends on the characteristics of the population studied, such as age, use of medications, and comorbidities known to be associated with this problem. Orthostatic hypotension is more common in institutionalized elderly people (up to 68%)7 than in those living in the community (6%).8 The high prevalence among institutionalized patients likely reflects multiple disease processes, including neurologic and cardiac conditions, as well as medications associated with orthostatic hypotension.
CLINICAL MANIFESTATIONS ARE DUE TO HYPOPERFUSION, OVERCOMPENSATION
Symptoms are related to cerebral hypoperfusion, with resulting lack of cerebral oxygenation (causing lightheadedness, dizziness, weakness, difficulty thinking, headache, syncope, or feeling faint) and a compensatory autonomic overreaction (causing palpitations, tremulousness, nausea, coldness of extremities, chest pain, and syncope).
Lightheadedness is a common symptom, but subtler issues such as difficulty thinking, weakness, and neck discomfort are also common in the elderly. Recurrent or unexplained falls in older adults may be a manifestation of syncope due to orthostatic hypotension.
PROGNOSIS DEPENDS ON CAUSE
Orthostatic hypotension is a syndrome, and its prognosis depends on its specific cause, its severity, and the distribution of its autonomic and nonautonomic involvement. In patients who have extrapyramidal and cerebellar disorders (eg, Parkinson disease, multiple system atrophy), the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis.9,10
In hypertensive patients with diabetes mellitus, the risk of death is higher if they have orthostatic hypotension.11 Diastolic orthostatic hypotension is associated with a higher risk of vascular death in older persons.12
MANAGEMENT: FROM A TO F
The goal of management of orthostatic hypotension is to raise the patient’s standing blood pressure without also raising his or her supine blood pressure, and specifically to reduce orthostatic symptoms, increase the time the patient can stand, and improve his or her ability to perform daily activities. No specific treatment is currently available that achieves all these goals, and drugs alone are never completely adequate.
Because the mainstays of treatment are volume expansion and vasoconstriction, it is difficult to improve the symptoms of orthostatic hypotension without inducing some degree of supine hypertension. Strategies to minimize nocturnal hypertension and to treat orthostatic hypotension in special circumstances are summarized in Table 1.
If hypovolemia is playing a major role, and the patient cannot ingest enough salt or plasma volume fails to increase despite salt supplementation, fludrocortisone (Florinef) should be considered. Untreated hypovolemia will decrease the efficacy of vasoconstrictor drugs.
Pyridostigmine (Mestinon) has a putative vasoconstrictor effect only during standing, but because its effect is modest it should be used in mild orthostatic hypotension that does not improve with nonpharmacologic measures and in moderate cases. Its effect can be enhanced with additional low doses of midodrine (ProAmatine). Midodrine with or without fludrocortisone should be used in severe orthostatic hypotension.
A: Abdominal compression
In conditions in which there is adrenergic denervation of vascular beds, there is an increase in vascular capacitance and peripheral venous pooling. Compression of capacitance beds (ie, the legs and abdomen) improves orthostatic symptoms.14 The improvement is due to a reduction of venous capacitance and an increase in total peripheral resistance.14
On standing, healthy adults experience an orthostatic shift of approximately 500 mL of blood to the lower extremities15 that, when added to an increased vascular capacitance in those with orthostatic hypotension, results in a relative state of hypovolemia.
Compression of the legs alone is not as beneficial as compression of the abdomen because the venous capacitance of the calves and thighs is relatively small compared with that of the splanchnic mesenteric bed, which accounts for 20% to 30% of total blood volume.16 Moreover, compression garments and stockings that are strong enough to produce a measurable effect on orthostatic hypotension are cumbersome to put on and uncomfortable to wear. Because some patients gain significant benefit from abdominal compression alone, this should be considered the first step in reducing venous capacitance.
In a laboratory experiment, Smit et al17 found that an elastic abdominal binder that exerted 15 to 20 mm Hg of pressure on the abdomen raised the standing blood pressure by about 11/6 mmHg, which was comparable to the effect of a gravity suit (such as those worn by fighter pilots to prevent syncope during violent aircraft maneuvers) inflated to 20 mm Hg—an increase of about 17/8 mm Hg. Higher gravity-suit pressures had a greater effect.
In practical terms, the binder should be tight enough to exert gentle pressure. It should be put on before rising from bed in the morning and taken off when lying supine, to avoid supine hypertension. Advantages are that a binder’s effects are immediate, its benefits can be easily assessed, and it can be used on an as-needed basis by patients who need it only during periods of prolonged orthostatic stress. Binders are also easy to fit and are available in most sporting good stores and on the Web (try searching for “abdominal binder”).
When abdominal compression alone is not enough, the addition of compression of the lower extremities can result in further benefits. This can be achieved by using compression garments that ideally extend to the waist or, at the least, to the proximal thigh.
B: Boluses of water
Rapidly drinking two 8-oz (500-mL) glasses of cold water helps expand plasma volume. It also, within a few minutes, elicits a significant pressor effect that is in part norepinephrine-mediated,18,19 increasing the standing systolic blood pressure by more than 20 mm Hg for about 2 hours and improving symptoms and orthostatic endurance.18,20 This easy technique can be used when prolonged standing is expected (eg, shopping).
B (continued): Bed up
The head of the bed of a patient with orthostatic hypotension should be elevated by 10 to 20 degrees or 4 inches (10 cm) to decrease nocturnal hypertension and nocturnal diuresis.21 During the day, adequate orthostatic stress, ie, upright activity, should be maintained. If patients are repeatedly tilted up, their orthostatic hypotension is gradually attenuated, presumably by increasing venomotor tone.22
C: Countermaneuvers
Physical countermaneuvers involve isometrically contracting the muscles below the waist for about 30 seconds at a time, which reduces venous capacitance, increases total peripheral resistance, and augments venous return to the heart.23,24 These countermeasures can help maintain blood pressure during daily activities and should be considered at the first symptoms of orthostatic intolerance and in situations of orthostatic stress (eg, standing for prolonged periods).
Specific techniques include23:
- Toe-raising
- Leg-crossing and contraction
- Thigh muscle co-contraction
- Bending at the waist
- Slow marching in place
- Leg elevation.
D: Drugs
Midodrine, a vasopressor, is effective and safe when used for treating neurogenic orthostatic hypotension.25 It has been shown to increase standing systolic blood pressure, reduce orthostatic lightheadedness, and increase standing and walking time.
A common starting dose is 5 mg three times a day; most patients respond best to 10 mg three times a day. As its duration of action is short (2 to 4 hours),25–27 it should be taken before arising in the morning, before lunch, and in the midafternoon. To avoid nocturnal supine hypertension, doses should not be taken after the midafternoon, and a dose should be omitted if the supine or sitting blood pressure is greater than 180/100 mm Hg.
Midodrine’s main side effects are supine hypertension, scalp paresthesias, and pilomotor reactions (goosebumps). Vasoconstrictors such as midodrine are ineffective when plasma volume is reduced.
Fludrocortisone is a synthetic mineralocorticoid that has a pressor effect as a result of its ability to expand plasma volume and increase vascular alpha-adrenoceptor sensitivity.28–30 This medication is helpful when plasma volume fails to adequately increase with salt supplementation31 and for patients who cannot ingest enough salt or do not respond adequately to midodrine.
The usual dose is 0.1 to 0.2 mg/day, but it may be increased to 0.4 to 0.6 mg/day in patients with refractory orthostatic hypotension.
If the patient gains 3 to 5 pounds (1.2–2.3 kg) and develops mild dependent edema, you can infer that the plasma volume has expanded adequately. However, in view of these effects, fludrocortisone is contraindicated in congestive heart failure and chronic renal failure. The potential risks are severe hypokalemia and excessive supine hypertension. Frequent monitoring of serum potassium, a diet high in potassium, and regular checks of supine blood pressure are advised, especially at higher doses, when added to midodrine, or in elderly patients who tend to poorly tolerate the medication.28,29,32
Pyridostigmine is a cholinesterase inhibitor that improves ganglionic neurotransmission in the sympathetic baroreflex pathway. Because this pathway is activated primarily during standing, this drug improves orthostatic hypotension and total peripheral resistance without aggravating supine hypertension. Because the pressor effect is modest, it is most adequate for patients with mild to moderate orthostatic hypotension.33,34
Dosing is started at 30 mg two to three times a day and is gradually increased to 60 mg three times a day. The drug’s effectiveness can be enhanced by combining each dose of pyridostigmine with 5 mg of midodrine without occurrence of supine hypertension.34 Mestinon Timespan, a 180-mg slow-release pyridostigmine tablet, can be taken once a day and may be a convenient alternative.
The main side effects are cholinergic (abdominal colic, diarrhea).
Review the patient’s medications. If he or she is taking any drug that may cause orthostatic hypotension, consider discontinuing it, substituting another drug, or changing the dosage (Table 2). In the elderly, antiparkinsonian, nitrate, antidepressant, diuretic, prostate, and antihypertensive medications35 may be particularly suspect.
E: Education
Education is probably the single most important factor in the proper control of orthostatic hypotension. A number of issues should be considered.
- Patients should be taught, in simple terms, the mechanisms that maintain postural normotension and how to recognize the onset of orthostatic symptoms.
- They must realize that there is no specific treatment of the underlying cause and that drug treatment alone is not adequate.
- They should be taught nonpharmacologic approaches and be aware that other drugs they start may worsen symptoms.
It is also important that the patient learn the conditions (and their mechanisms) that can lower blood pressure (Table 3). Such conditions include prolonged or motionless standing, alcohol ingestion (causing vasodilation), carbohydrate-heavy meals (causing postprandial orthostatic hypotension related to an increase in the splanchnic-mesenteric venous capacitance), early morning orthostatic hypotension related to nocturnal diuresis and arising from bed, physical activity sufficient to cause muscle vasodilation, heat exposure (eg, hot weather or a hot bath or shower) producing skin vessel vasodilation, sudden postural changes, and prolonged recumbency. Once these stressors are explained, patients have no difficulty recognizing them.
The patient should also be instructed in how to manage situations of increased orthostatic stress and periods of orthostatic decompensation, to minimize nocturnal hypertension, and to modify their activities of daily living. Keeping a log of supine and upright blood pressures (taken with an automated sphygmomanometer) during situations of orthostatic stress can help establish whether worsening symptoms are related to orthostatic hypotension or to another mechanism. Once patients discover that they can actively deal with these situations, they develop a great sense of empowerment.
E (continued): Exercise
Mild physical exercise improves orthostatic tolerance by reducing venous pooling and increasing plasma volume.36 Deconditioning from lack of exercise exacerbates orthostatic hypotension.37 Because upright exercise may increase the orthostatic drop in blood pressure, training in a supine or sitting position (eg, swimming, recumbent biking) is advisable. Isotonic exercise (eg, light weight-lifting) is recommended because the incorrect straining and breath-holding during isometric exercise (eg, holding weights in the same position) may decrease venous return.
F: Fluid and salt (volume expansion)
Maintaining an adequate plasma volume is crucial. Patients should drink five to eight 8-ounce glasses (1.25 to 2.5 L) of water or other fluid per day. Many elderly people do not take in this much. The patient should have at least 1 glass or cup of fluid with meals and at least twice at other times of each day to obtain 1 L/day.
Salt intake should be between 150 and 250 mmol of sodium (10 to 20 g of salt) per day. Sodium helps with retention of ingested fluids and should be maximized if tolerated. However, caution should be exercised in patients who have severe refractory supine hypertension, uncontrolled hypertension, or comorbidities characterized by insterstitial edema (eg, heart failure, liver failure). Some patients are very sensitive to sodium supplementation and can fine-tune their orthostatic control with salt alone. If salting food is not desired, prepared soups, pretzels, potato chips, and 0.5- or 1.0-g salt tablets can be an option.
Patients need to maintain a high-potassium diet, as the high sodium intake combined with fludrocortisone promotes potassium loss. Fruits (especially bananas) and vegetables have high potassium content.
The combination of fludrocortisone and a high-salt diet can also cause sustained supine hypertension, which can be minimized by the interventions noted in Table 2.
Appropriate salt supplementation and fluid intake leading to an adequate volume expansion can be verified by checking the 24-hour urinary sodium content: patients who excrete less than 170 mmol can be treated with 1 to 2 g of supplemental sodium three times a day.38
- Sjostrand T. The regulation of the blood distribution in man. Acta Physiol Scand 1952; 26:312–327.
- Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977; 296:293–297.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996; 46:1470.
- Gehrking JA, Hines SM, Benrud-Larson LM, Opher-Gehrking TL, Low PA. What is the minimum duration of head-up tilt necessary to detect orthostatic hypotension? Clin Auton Res 2005; 15:71–75.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005; 30:173–178.
- Weiss A, Grossman E, Beloosesky Y, Grinblat J. Orthostatic hypotension in acute geriatric ward: is it a consistent finding? Arch Intern Med 2002; 162:2369–2374.
- Mader SL, Josephson KR, Rubenstein LZ. Low prevalence of postural hypotension among community-dwelling elderly. JAMA 1987; 258:1511–1514.
- Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991; 1:147–155.
- Saito Y, Matsuoka Y, Takahashi A, Ohno Y. Survival of patients with multiple system atrophy. Intern Med 1994; 33:321–325.
- Davis BR, Langford HG, Blaufox MD, Curb JD, Polk BF, Shulman NB. The association of postural changes in systolic blood pressure and mortality in persons with hypertension: the Hypertension Detection and Follow-up Program experience. Circulation 1987; 75:340–346.
- Luukinen H, Koski K, Laippala P, Kivelä SL. Prognosis of diastolic and systolic orthostatic hypotension in older persons. Arch Intern Med 1999; 159:273–280.
- Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. N Engl J Med 1993; 329:611–615.
- Denq JC, Opfer-Gehrking TL, Giuliani M, Felten J, Convertino VA, Low PA. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clin Auton Res 1997; 7:321–326.
- Sjostrand T. Volume and distribution of blood and their significance in regulating the circulation. Physiol Rev 1953; 33:202–228.
- Rowell LB, Detry JM, Blackmon JR, Wyss C. Importance of the splanchnic vascular bed in human blood pressure regulation. J Appl Physiol 1972; 32:213–220.
- Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res 2004; 14:167–175.
- Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000; 101:504–509.
- Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002; 112:355–360.
- Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water [letter]. Lancet 1999; 353:723.
- MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia: treatment with the “head-up” bed. JAMA 1940; 115:2162–2167.
- Ector H, Reybrouck T, Heidbüchel H, Gewillig M, Van de Werf F. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol 1998; 21:193–196.
- Bouvette CM, McPhee BR, Opfer-Gehrking TL, Low PA. Role of physical countermaneuvers in the management of orthostatic hypotension: efficacy and biofeedback augmentation. Mayo Clin Proc 1996; 71:847–853.
- Ten Harkel AD, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci (Lond) 1994; 87:553–558.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993; 95:38–48.
- Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med 1995; 99:604–610.
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007; 7:63–70.
- Axelrod FB, Goldberg JD, Rolnitzky L, et al. Fludrocortisone in patients with familial dysautonomia—assessing effect on clinical parameters and gene expression. Clin Auton Res 2005; 15:284–291.
- Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301:68–73.
- van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. Clin Auton Res 2000; 10:35–42.
- Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart 1996; 76:507–509.
- Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry 2003; 74:1294–1298.
- Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63:513–518.
- Lipsitz LA, Pluchino FC, Wei JY, Rowe JW. Syncope in institutionalized elderly: the impact of multiple pathological conditions and situational stress. J Chronic Dis 1986; 39:619–630.
- Mtinangi BL, Hainsworth R. Effects of moderate exercise training on plasma volume, baroreceptor sensitivity and orthostatic tolerance in healthy subjects. Exp Physiol 1999; 84:121–130.
- Bonnin P, Ben Driss A, Benessiano J, Maillet A, Pavy le Traon A, Levy BI. Enhanced flow-dependent vasodilatation after bed rest, a possible mechanism for orthostatic intolerance in humans. Eur J Appl Physiol 2001; 85:420–426.
- El-Sayed H, Hainsworth R. Salt supplementation increases plasma volume and orthostatic tolerance in patients with unexplained syncope. Heart 1996; 75:134–140.
- Sjostrand T. The regulation of the blood distribution in man. Acta Physiol Scand 1952; 26:312–327.
- Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977; 296:293–297.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996; 46:1470.
- Gehrking JA, Hines SM, Benrud-Larson LM, Opher-Gehrking TL, Low PA. What is the minimum duration of head-up tilt necessary to detect orthostatic hypotension? Clin Auton Res 2005; 15:71–75.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005; 30:173–178.
- Weiss A, Grossman E, Beloosesky Y, Grinblat J. Orthostatic hypotension in acute geriatric ward: is it a consistent finding? Arch Intern Med 2002; 162:2369–2374.
- Mader SL, Josephson KR, Rubenstein LZ. Low prevalence of postural hypotension among community-dwelling elderly. JAMA 1987; 258:1511–1514.
- Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991; 1:147–155.
- Saito Y, Matsuoka Y, Takahashi A, Ohno Y. Survival of patients with multiple system atrophy. Intern Med 1994; 33:321–325.
- Davis BR, Langford HG, Blaufox MD, Curb JD, Polk BF, Shulman NB. The association of postural changes in systolic blood pressure and mortality in persons with hypertension: the Hypertension Detection and Follow-up Program experience. Circulation 1987; 75:340–346.
- Luukinen H, Koski K, Laippala P, Kivelä SL. Prognosis of diastolic and systolic orthostatic hypotension in older persons. Arch Intern Med 1999; 159:273–280.
- Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. N Engl J Med 1993; 329:611–615.
- Denq JC, Opfer-Gehrking TL, Giuliani M, Felten J, Convertino VA, Low PA. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clin Auton Res 1997; 7:321–326.
- Sjostrand T. Volume and distribution of blood and their significance in regulating the circulation. Physiol Rev 1953; 33:202–228.
- Rowell LB, Detry JM, Blackmon JR, Wyss C. Importance of the splanchnic vascular bed in human blood pressure regulation. J Appl Physiol 1972; 32:213–220.
- Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res 2004; 14:167–175.
- Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000; 101:504–509.
- Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002; 112:355–360.
- Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water [letter]. Lancet 1999; 353:723.
- MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia: treatment with the “head-up” bed. JAMA 1940; 115:2162–2167.
- Ector H, Reybrouck T, Heidbüchel H, Gewillig M, Van de Werf F. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol 1998; 21:193–196.
- Bouvette CM, McPhee BR, Opfer-Gehrking TL, Low PA. Role of physical countermaneuvers in the management of orthostatic hypotension: efficacy and biofeedback augmentation. Mayo Clin Proc 1996; 71:847–853.
- Ten Harkel AD, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci (Lond) 1994; 87:553–558.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993; 95:38–48.
- Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med 1995; 99:604–610.
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007; 7:63–70.
- Axelrod FB, Goldberg JD, Rolnitzky L, et al. Fludrocortisone in patients with familial dysautonomia—assessing effect on clinical parameters and gene expression. Clin Auton Res 2005; 15:284–291.
- Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301:68–73.
- van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. Clin Auton Res 2000; 10:35–42.
- Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart 1996; 76:507–509.
- Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry 2003; 74:1294–1298.
- Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63:513–518.
- Lipsitz LA, Pluchino FC, Wei JY, Rowe JW. Syncope in institutionalized elderly: the impact of multiple pathological conditions and situational stress. J Chronic Dis 1986; 39:619–630.
- Mtinangi BL, Hainsworth R. Effects of moderate exercise training on plasma volume, baroreceptor sensitivity and orthostatic tolerance in healthy subjects. Exp Physiol 1999; 84:121–130.
- Bonnin P, Ben Driss A, Benessiano J, Maillet A, Pavy le Traon A, Levy BI. Enhanced flow-dependent vasodilatation after bed rest, a possible mechanism for orthostatic intolerance in humans. Eur J Appl Physiol 2001; 85:420–426.
- El-Sayed H, Hainsworth R. Salt supplementation increases plasma volume and orthostatic tolerance in patients with unexplained syncope. Heart 1996; 75:134–140.
KEY POINTS
- Treatment is directed at increasing blood volume, decreasing venous pooling, and increasing vasoconstriction while minimizing supine hypertension.
- Patient education and nondrug strategies alone can be effective in mild cases. Examples: consuming extra fluids and salt, wearing an abdominal binder, drinking boluses of water, raising the head of the bed, and performing countermaneuvers and physical activity.
- Moderate and severe cases require additional drug treatment. Pyridostigmine (Mestinon) is helpful in moderate cases. Fludrocortisone (Florinef) and midodrine (ProAmatine) are indicated in more severe cases.
A rare complication of infective endocarditis
An 85-year-old woman presented to the emergency department with a 2-hour history of dyspnea, dizziness, generalized weakness, nausea, and diaphoresis. Her medical history included hypertension, end-stage renal disease with hemodialysis, and atrial fibrillation.
She had an arteriovenous fistula for dialysis access in her right upper arm, with erythema around the site.
Her creatine kinase level was 1,434 U/L (normal range 30–220), creatine kinase MB 143.4 ng/mL (0.0–8.8 ng/mL), and troponin T 0.1 ng/mL (0.0–0.1 ng/mL). She had ST elevation in leads I and aVL. She was taken for emergency cardiac catheterization.
Three blood cultures were drawn on the day of cardiac catheterization. Two grew gram-positive organisms: one grew coagulase-negative Staphylococcus, and the other grew gram-positive bacilli (anaerobic, non-sporeforming). On the basis of these findings, intravenous vancomycin (Vancocin) was started. Seventy-two hours later, one of two blood cultures again grew coagulase-negative Staphylococcus. Five days after the start of antibiotic treatment, blood cultures were negative, and the patient received intravenous vancomycin for 4 weeks (from the time the blood cultures became negative) for native mitral valve endocarditis.
EMBOLISM AND ENDOCARDITIS: KEY FEATURES
An embolic event occurs in 22% to 50% of cases of infective endocarditis and can involve the lungs, bowel, other organs, or extremities.1 The incidence of embolization of the coronary arteries in patients with infective endocarditis is unknown, but in one case series2 it occurred in 8 (7.5%) of 107 cases. The most common site of coronary embolism is the LAD.3
Myocardial infarction is a rare complication of coronary artery embolization.2 It was reported in 17 (2.9%) of 586 consecutive patients with infective endocarditis.4 In patients with infectious endocarditis complicated by myocardial infarction, the death rate was nearly double that seen in patients with infective endocarditis without myocardial infarction (64% vs 33%).4
TREATMENT
The best treatment for this complication of infective endocarditis is not known, as it has not been well studied. The high death rate in these patients makes restoration of coronary perfusion essential.
Thrombolytics are usually avoided in patients with septic embolization because of concerns about concurrent intracerebral mycotic aneurysms and the risk of hemorrhage.
Percutaneous transluminal angioplasty carries a risk of distal mobilization of emboli, development of mycotic aneurysm at the balloon dilation site, or reocclusion due to a mobile embolus.5 Stent placement may improve vessel patency but carries a theoretic risk of infection in bacteremic patients. Percutaneous embolectomy has also been used either prior to or instead of stent placement.6 Surgical options include embolectomy in patients who may require surgery, and coronary artery bypass grafting for patients with chronic embolization.7
- Baddour LM, Wilson WR, Bayer AS, et al; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease; Council on Cardiovascular Disease in the Young; Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia; American Heart Association; Infectious Diseases Society of America. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434.
- Garvey GJ, Neu HC. Infective endocarditis—an evolving disease. A review of endocarditis at Columbia-Presbyterian Medical Center, 1968–1973. Medicine (Baltimore) 1978; 57:105–127.
- Glazier JJ. Interventional treatment of septic coronary embolism: sailing into uncharted and dangerous waters. J Interv Cardiol 2002; 15:305–307.
- Manzano MC, Vilacosta I, San Roman JA, et al. Acute cornary syndrome in infective endocarditis. Rev Esp Cardiol 2007; 60:24–31.
- Khan F, Khakoo R, Failinger C. Managing embolic myocardial infarction in infective endocarditis: current options. J Infect 2005; 51:e101–105.
- Glazier JJ, McGinnity JG, Spears JR. Coronary embolism complicating aortic valve endocarditis: treatment with placement of an intracoronary stent. Clin Cardiol 1997; 20:885–888.
- Baek MJ, Kim HK, Yu CW, Na CY. Surgery with surgical embolectomy for mitral valve endocarditis complicated by septic coronary embolism. Eur J Cardiothorac Surg 2008; 33:116–118.
An 85-year-old woman presented to the emergency department with a 2-hour history of dyspnea, dizziness, generalized weakness, nausea, and diaphoresis. Her medical history included hypertension, end-stage renal disease with hemodialysis, and atrial fibrillation.
She had an arteriovenous fistula for dialysis access in her right upper arm, with erythema around the site.
Her creatine kinase level was 1,434 U/L (normal range 30–220), creatine kinase MB 143.4 ng/mL (0.0–8.8 ng/mL), and troponin T 0.1 ng/mL (0.0–0.1 ng/mL). She had ST elevation in leads I and aVL. She was taken for emergency cardiac catheterization.
Three blood cultures were drawn on the day of cardiac catheterization. Two grew gram-positive organisms: one grew coagulase-negative Staphylococcus, and the other grew gram-positive bacilli (anaerobic, non-sporeforming). On the basis of these findings, intravenous vancomycin (Vancocin) was started. Seventy-two hours later, one of two blood cultures again grew coagulase-negative Staphylococcus. Five days after the start of antibiotic treatment, blood cultures were negative, and the patient received intravenous vancomycin for 4 weeks (from the time the blood cultures became negative) for native mitral valve endocarditis.
EMBOLISM AND ENDOCARDITIS: KEY FEATURES
An embolic event occurs in 22% to 50% of cases of infective endocarditis and can involve the lungs, bowel, other organs, or extremities.1 The incidence of embolization of the coronary arteries in patients with infective endocarditis is unknown, but in one case series2 it occurred in 8 (7.5%) of 107 cases. The most common site of coronary embolism is the LAD.3
Myocardial infarction is a rare complication of coronary artery embolization.2 It was reported in 17 (2.9%) of 586 consecutive patients with infective endocarditis.4 In patients with infectious endocarditis complicated by myocardial infarction, the death rate was nearly double that seen in patients with infective endocarditis without myocardial infarction (64% vs 33%).4
TREATMENT
The best treatment for this complication of infective endocarditis is not known, as it has not been well studied. The high death rate in these patients makes restoration of coronary perfusion essential.
Thrombolytics are usually avoided in patients with septic embolization because of concerns about concurrent intracerebral mycotic aneurysms and the risk of hemorrhage.
Percutaneous transluminal angioplasty carries a risk of distal mobilization of emboli, development of mycotic aneurysm at the balloon dilation site, or reocclusion due to a mobile embolus.5 Stent placement may improve vessel patency but carries a theoretic risk of infection in bacteremic patients. Percutaneous embolectomy has also been used either prior to or instead of stent placement.6 Surgical options include embolectomy in patients who may require surgery, and coronary artery bypass grafting for patients with chronic embolization.7
An 85-year-old woman presented to the emergency department with a 2-hour history of dyspnea, dizziness, generalized weakness, nausea, and diaphoresis. Her medical history included hypertension, end-stage renal disease with hemodialysis, and atrial fibrillation.
She had an arteriovenous fistula for dialysis access in her right upper arm, with erythema around the site.
Her creatine kinase level was 1,434 U/L (normal range 30–220), creatine kinase MB 143.4 ng/mL (0.0–8.8 ng/mL), and troponin T 0.1 ng/mL (0.0–0.1 ng/mL). She had ST elevation in leads I and aVL. She was taken for emergency cardiac catheterization.
Three blood cultures were drawn on the day of cardiac catheterization. Two grew gram-positive organisms: one grew coagulase-negative Staphylococcus, and the other grew gram-positive bacilli (anaerobic, non-sporeforming). On the basis of these findings, intravenous vancomycin (Vancocin) was started. Seventy-two hours later, one of two blood cultures again grew coagulase-negative Staphylococcus. Five days after the start of antibiotic treatment, blood cultures were negative, and the patient received intravenous vancomycin for 4 weeks (from the time the blood cultures became negative) for native mitral valve endocarditis.
EMBOLISM AND ENDOCARDITIS: KEY FEATURES
An embolic event occurs in 22% to 50% of cases of infective endocarditis and can involve the lungs, bowel, other organs, or extremities.1 The incidence of embolization of the coronary arteries in patients with infective endocarditis is unknown, but in one case series2 it occurred in 8 (7.5%) of 107 cases. The most common site of coronary embolism is the LAD.3
Myocardial infarction is a rare complication of coronary artery embolization.2 It was reported in 17 (2.9%) of 586 consecutive patients with infective endocarditis.4 In patients with infectious endocarditis complicated by myocardial infarction, the death rate was nearly double that seen in patients with infective endocarditis without myocardial infarction (64% vs 33%).4
TREATMENT
The best treatment for this complication of infective endocarditis is not known, as it has not been well studied. The high death rate in these patients makes restoration of coronary perfusion essential.
Thrombolytics are usually avoided in patients with septic embolization because of concerns about concurrent intracerebral mycotic aneurysms and the risk of hemorrhage.
Percutaneous transluminal angioplasty carries a risk of distal mobilization of emboli, development of mycotic aneurysm at the balloon dilation site, or reocclusion due to a mobile embolus.5 Stent placement may improve vessel patency but carries a theoretic risk of infection in bacteremic patients. Percutaneous embolectomy has also been used either prior to or instead of stent placement.6 Surgical options include embolectomy in patients who may require surgery, and coronary artery bypass grafting for patients with chronic embolization.7
- Baddour LM, Wilson WR, Bayer AS, et al; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease; Council on Cardiovascular Disease in the Young; Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia; American Heart Association; Infectious Diseases Society of America. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434.
- Garvey GJ, Neu HC. Infective endocarditis—an evolving disease. A review of endocarditis at Columbia-Presbyterian Medical Center, 1968–1973. Medicine (Baltimore) 1978; 57:105–127.
- Glazier JJ. Interventional treatment of septic coronary embolism: sailing into uncharted and dangerous waters. J Interv Cardiol 2002; 15:305–307.
- Manzano MC, Vilacosta I, San Roman JA, et al. Acute cornary syndrome in infective endocarditis. Rev Esp Cardiol 2007; 60:24–31.
- Khan F, Khakoo R, Failinger C. Managing embolic myocardial infarction in infective endocarditis: current options. J Infect 2005; 51:e101–105.
- Glazier JJ, McGinnity JG, Spears JR. Coronary embolism complicating aortic valve endocarditis: treatment with placement of an intracoronary stent. Clin Cardiol 1997; 20:885–888.
- Baek MJ, Kim HK, Yu CW, Na CY. Surgery with surgical embolectomy for mitral valve endocarditis complicated by septic coronary embolism. Eur J Cardiothorac Surg 2008; 33:116–118.
- Baddour LM, Wilson WR, Bayer AS, et al; Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease; Council on Cardiovascular Disease in the Young; Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia; American Heart Association; Infectious Diseases Society of America. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434.
- Garvey GJ, Neu HC. Infective endocarditis—an evolving disease. A review of endocarditis at Columbia-Presbyterian Medical Center, 1968–1973. Medicine (Baltimore) 1978; 57:105–127.
- Glazier JJ. Interventional treatment of septic coronary embolism: sailing into uncharted and dangerous waters. J Interv Cardiol 2002; 15:305–307.
- Manzano MC, Vilacosta I, San Roman JA, et al. Acute cornary syndrome in infective endocarditis. Rev Esp Cardiol 2007; 60:24–31.
- Khan F, Khakoo R, Failinger C. Managing embolic myocardial infarction in infective endocarditis: current options. J Infect 2005; 51:e101–105.
- Glazier JJ, McGinnity JG, Spears JR. Coronary embolism complicating aortic valve endocarditis: treatment with placement of an intracoronary stent. Clin Cardiol 1997; 20:885–888.
- Baek MJ, Kim HK, Yu CW, Na CY. Surgery with surgical embolectomy for mitral valve endocarditis complicated by septic coronary embolism. Eur J Cardiothorac Surg 2008; 33:116–118.
Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival?
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE

The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
The pretravel visit: A ‘teaching moment’

Closer to home, general internists and other primary care providers can use the awareness of global health concerns to the health advantage of our patients, including the young and healthy, who generally eschew preventive health visits to their physicians, and busy traveling executives, who only see a doctor for (hopefully) quick resolution to intermittent problems.
In this issue of the Journal, Powell and Ford offer a general primer on travel medicine, highlighting specific concerns that should be addressed to facilitate our patients’ safe and uninterrupted travels. But often, a pretravel visit is also a good time to introduce concepts of preventive health to patients who might not otherwise be accessible or amenable.
Just as the preoperative medical consultation can provide a “teaching moment” to address smoking cessation or reversible cardiac risks to a captive audience, the visit regarding “What shots do I need to go to Thailand?” can open the door for talk about general vaccinations (see the article by Campos-Outcalt of this issue), venereal disease, air-travel-associated thrombosis, excessive alcohol use, and perhaps other wellness issues. Creating a travel advisory service within most practices will not supplant the benefits of having travelers review the CDC travel Web site or the need to refer some patients to travel medicine experts regarding specific diseases and vaccinations. But it may create the opportunity for interaction, dialogue, and even a blood pressure check with patients who might not otherwise have the time or see the need to schedule a visit with a physician in the absence of an acute medical concern.
Closer to home, general internists and other primary care providers can use the awareness of global health concerns to the health advantage of our patients, including the young and healthy, who generally eschew preventive health visits to their physicians, and busy traveling executives, who only see a doctor for (hopefully) quick resolution to intermittent problems.
In this issue of the Journal, Powell and Ford offer a general primer on travel medicine, highlighting specific concerns that should be addressed to facilitate our patients’ safe and uninterrupted travels. But often, a pretravel visit is also a good time to introduce concepts of preventive health to patients who might not otherwise be accessible or amenable.
Just as the preoperative medical consultation can provide a “teaching moment” to address smoking cessation or reversible cardiac risks to a captive audience, the visit regarding “What shots do I need to go to Thailand?” can open the door for talk about general vaccinations (see the article by Campos-Outcalt of this issue), venereal disease, air-travel-associated thrombosis, excessive alcohol use, and perhaps other wellness issues. Creating a travel advisory service within most practices will not supplant the benefits of having travelers review the CDC travel Web site or the need to refer some patients to travel medicine experts regarding specific diseases and vaccinations. But it may create the opportunity for interaction, dialogue, and even a blood pressure check with patients who might not otherwise have the time or see the need to schedule a visit with a physician in the absence of an acute medical concern.
Closer to home, general internists and other primary care providers can use the awareness of global health concerns to the health advantage of our patients, including the young and healthy, who generally eschew preventive health visits to their physicians, and busy traveling executives, who only see a doctor for (hopefully) quick resolution to intermittent problems.
In this issue of the Journal, Powell and Ford offer a general primer on travel medicine, highlighting specific concerns that should be addressed to facilitate our patients’ safe and uninterrupted travels. But often, a pretravel visit is also a good time to introduce concepts of preventive health to patients who might not otherwise be accessible or amenable.
Just as the preoperative medical consultation can provide a “teaching moment” to address smoking cessation or reversible cardiac risks to a captive audience, the visit regarding “What shots do I need to go to Thailand?” can open the door for talk about general vaccinations (see the article by Campos-Outcalt of this issue), venereal disease, air-travel-associated thrombosis, excessive alcohol use, and perhaps other wellness issues. Creating a travel advisory service within most practices will not supplant the benefits of having travelers review the CDC travel Web site or the need to refer some patients to travel medicine experts regarding specific diseases and vaccinations. But it may create the opportunity for interaction, dialogue, and even a blood pressure check with patients who might not otherwise have the time or see the need to schedule a visit with a physician in the absence of an acute medical concern.