User login
Risks of travel, benefits of a specialist consult
Before going abroad to areas that might pose a risk to their health, most people ought to visit their primary care physicians and many should be referred to a specialist in travel medicine.
In this article, we review the key elements of the pretravel consult as it relates to the prevention and self-treatment of the most common diseases that pose health risks for travelers. We also give guidelines for when to refer patients to a specialist.
WHY PRIMARY CARE PHYSICIANS NEED TO KNOW TRAVEL MEDICINE
International travel to exotic locations is becoming more popular. In 2008, one out of five Americans traveled abroad, and 38 million visits were to developing countries where there are significant health risks for travelers.1
One third to one half of travelers to developing countries experience some kind of illness while abroad, most commonly diarrhea or upper respiratory infections, which typically lead to 3 lost days during a 2-week trip.
These illnesses are often preventable and self-treatable.2 Unfortunately, studies suggest that most travelers do not seek adequate medical advice, and that when they do they often fail to complete courses of medication.3,4
All these factors point to the need for primary care providers to become proficient in the pretravel consult and, if necessary, to refer patients to travel specialists and clinics.
WHY REFER TO A TRAVEL CLINIC?
In one study of travelers to areas of high risk for malaria or hepatitis A, 42% of those who consulted only their family doctor became ill, in contrast to 22% of those who attended a travel medicine clinic.4
As a rule of thumb, anyone traveling to an area where malaria is endemic should be referred to a specialist, as should anyone at risk of yellow fever or typhoid fever. As many as 8 per 1,000 travelers may return from areas of risk infected with malaria.5
Long-term travelers and people who will spend time in urban slums or rural or remote regions have an even greater need for referral to a travel clinic, as they are at higher risk of exposure to Japanese encephalitis, cholera, epidemic meningitis, dengue fever, and rabies.6
THE PRETRAVEL CONSULT: ESSENTIALS
A pretravel consult ought to be scheduled 4 to 6 weeks in advance of the trip, since many vaccines require that much time to induce immunity, and some require a series of shots.
Unfortunately, many patients who think of arranging a travel consult make the appointment at the last minute, and some come with an incomplete knowledge of their travel plans. However, even without enough advance notice, a consult can be beneficial.
Travelers sometimes change their itineraries in-country or engage in unanticipated risky behaviors. A good travel medicine physician tries to anticipate even these unplanned risks and changes in itinerary.
Where is the traveler going? When? For how long?
The pretravel consult starts with a detailed discussion of the patient’s itinerary. It needs to include length, dates, and location of travel, as well as anticipated activities and accommodations.
A remarkable number of travelers come to consults not knowing the names of specific countries they will visit, perhaps saying only that they are going to Africa or South America. An accurate itinerary is indispensible, as appropriate medical advice is highly specific to country and region. The incidence and geographic distribution of many travelers’ diseases change over time, and this requires physicians to consult the most current information available.
Tropical countries, in general, are risky, but each pathogen has a unique distribution that may vary between urban and rural areas or by season. Detailed, up-to-date information is available from the US Centers for Disease Control and Prevention (CDC) (www.cdc.gov) for individual countries and for specific provinces and locations within those countries. Physicians should consult the CDC whenever advising a patient preparing to travel. 7
How is the traveler’s current health?

Several immunizations cannot be given to the very young, the elderly, or those who are immunocompromised.
The greatest risk of death to travelers is not from tropical diseases but from cardiovascular disease, which according to one study is responsible for half of deaths abroad.8 Patients with heart disease or other known health concerns need to be counseled to avoid activities that will put them at further risk. The advice applies especially in situations such as remote travel or even cruises, where prompt emergency medical care may be difficult or impossible to obtain.
People infected with human immunodeficiency virus (HIV) face discriminatory travel prohibitions in 74 countries.9
Foreign-born travelers who are visiting family and friends in developing countries may have lost their immunity to local pathogens and thus can be more at risk because they are not prepared to take necessary health precautions.
Also, a significant number of travelers become infected but develop illnesses only after they return, so a posttravel visit may be necessary.6
Prescription and even over-the-the-counter drugs may be difficult or impossible to obtain in foreign countries, and ample supplies should be brought along.
Is the traveler up to date on routine immunizations?
A number of infectious diseases that have been controlled or eradicated in North America through regular childhood immunizations are still endemic in many remote areas and developing countries. All travelers should be up to date on routine immunizations, including those for measles-mumps-rubella, tetanus, polio, meningitis, and hepatitis A and B.
Polio. A one-time polio booster is recommended for adults traveling to certain countries or areas of the world.
Meningitis vaccine is now routinely given to young people, but adult patients may need it before they travel.
Hepatitis A is contracted through fecal contamination of food and water. Common sources are foods prepared in an unhygienic manner, raw fruits and vegetables, shellfish, and contaminated water.

Hepatitis B vaccine is also now routinely given to young people, but it should be offered to travelers planning to stay more than 1 month and to long-term expatriates. This vaccine is also recommended for travelers who may be exposed to blood or body fluids, who are contemplating sexual activity or tattooing in the host country, or who may require medical or dental care while traveling, as well as for adventure travelers or travelers to remote regions.
The vaccination is given in a three-dose schedule at 0, 1, and 6 months. For protection against both hepatitis A and B, the vaccine Twinrix can be used on the same schedule as for hepatitis B. An accelerated schedule of 0, 7, and 21 days with a booster at 12 months allows completion of the entire series in 4 weeks, thus putting completion of vaccination before travel in the same time frame as other vaccines in a series, such as those for rabies and Japanese encephalitis.
PREVENTIVE COUNSELING
In addition, travelers going abroad should be advised on measures to avoid diarrhea, insectvector diseases, accidents, excessive exposure to the sun, altitude sickness, and other risks their itineraries may expose them to.
Avoiding traveler’s diarrhea
Traveler’s diarrhea is by far the most common health problem experienced abroad. It is prevalent in Mexico, where 20 million visits by Americans occur each year. A quarter to half of visitors to developing countries contract traveler’s diarrhea and, on average, lose 2 to 3 days of their business trip or vacation.3,10 The disease therefore imposes not only discomfort but also financial losses on travelers, especially business travelers.
Though many pathogens may be responsible, the most common one is Escherichia coli, usually transmitted by human fecal contamination of food or drink. Preventive measures against E coli are the same as for other foodborne and waterborne infections, such as hepatitis A, cholera, and typhoid fever.
The rule for avoiding traveler’s diarrhea may be summarized by the CDC-coined phrase, “boil it, cook it, peel it, or forget it.” Simple, written advice is most likely to be followed. 6 Thorough boiling or cooking kills bacteria in contaminated food, and food should be served steaming hot. Travelers should only eat foods they know have been well cooked, declining cold dishes like salsa or casseroles. They should avoid tap water for brushing teeth or in the form of ice cubes and should stick to drinking bottled beverages, preferably carbonated ones. No matter how appetizing a salad looks, travelers should avoid eating fresh fruits and vegetables unless they are sure that they were peeled under sanitary conditions. Simply eating at a high-priced restaurant is not a guarantee of uncontaminated food. Before meals or any hand-to-mouth contact, hands should be washed in soap and water or with sanitizers.
Travelers to remote areas may wish to acquire filtering devices, chlorine, or iodine for treating water. A combination of filtering and iodine treatment is most effective.
While this advice is undoubtedly wise, the evidence shows that, in practice, most travelers fail to take all precautions, and the benefits of this counseling have been difficult to demonstrate.11 Therefore, physicians should prescribe drugs for prophylaxis and self-treatment of traveler’s diarrhea during travel.
Bismuth subsalicylate (Pepto-Bismol) taken as two tablets or 2 oz of liquid 4 times a day while traveling may reduce the risk of diarrhea by one half, though it should be avoided by patients with contraindications to aspirin.3,6
Self-treating traveler’s diarrhea
Proper hydration is crucial, since dehydration can worsen and prolong symptoms.
Ciprofloxacin (Cipro) 500 mg orally two times daily for 3 to 5 days is effective.
Azithromycin (Zithromax) 500 mg daily for 3 to 5 days may be better in some areas of Southeast Asia, where fluoroquinolone-resistant bacteria are prevalent.
Rifaximin (Xifaxan), a nonsystemic antibiotic, is another option. The dosage is 200 mg three times a day for 3 days.
Avoiding insect bites
Malaria, yellow fever, tickborne encephalitis, and dengue fever are all transmitted by insect bites. Often the best protection is to avoid being bitten.
Bites can be avoided by using insect repellants containing diethyltoluamide (DEET) or picardin. If the traveler is going to be out in the sun, he or she should apply sunscreen first, then DEET on top of that. Anopheles, which transmits malaria, is a night-biting mosquito and may be avoided by staying in screened areas at dusk and dawn and by using bed netting. Permethrin, an insecticide, can be applied to clothing and mosquito netting.
Other things to avoid
Accidents are the second most common cause of death in travelers (after cardiovascular disease), accounting for as many as one-third of deaths.9 Several studies indicate road accidents are the major cause of accidental death, but also significant are drowning and air crashes. Travelers should be advised that transportation in developing countries is often more dangerous than at home. Seaside vacationers should be aware of the dangers of riptides and other threats to swimmers and should obey warnings posted at beaches.
Sexually transmitted diseases. When appropriate, physicians should warn travelers about the dangers of contracting HIV and other sexually transmitted diseases, especially in sub-Saharan Africa.
Sunburn, dehydration. Travelers should regularly use sunscreen and should remain hydrated.
Crimes against and involving tourists are a serious threat in many places, including some popular destinations. All of the 100,000 young people traveling to Mexico each year for spring break should read the US Department of State warnings against crime and possible arrest in that country.12 Travelers who are victims of crimes in foreign countries should contact their national consulate as soon as possible. The US Department of State issues advisories on countries where there is danger to travelers because of political turmoil, crime, or other causes.13
Motion sickness and jet lag can be ameliorated by proper hydration, avoiding caffeine, and using a scopolamine patch or dimenhydrinate (Dramamine).1
When traveling to wilderness areas
Wilderness and expedition medicine is a complex subset of travel medicine.14 All travelers need to understand the risks of whatever activities they undertake.
Mountain climbers and skiers have to contend with altitude sickness and frostbite. Scuba divers have the risks of decompression sickness, barotrauma, and hazardous marine life. Travelers on expeditions may have to deal with predatory animals, exotic parasites, and ethnic or political violence. People who participate in these activities should do so only when they are properly certified and educated in the associated health risks.
Ordinary tourists should enjoy safe adventures with well-established tour agencies and venues and should be cautioned against activities that expose them to dangers they may not be prepared to confront.
Insurance, evacuation, and emergency care
Health insurance often does not pay for preventive travel medicine. Unfortunately, cost can be a factor in immunizations and other health care. The cost of most travelers’ medications and vaccinations is generally comparable to that of other immunizations. The exceptions are two specialized vaccines—ie, for Japanese encephalitis ($1,000 or more for a full course) and for rabies, which can cost considerably more. Pricing by different providers can vary widely.
Travelers, especially those who are pregnant, elderly, disabled, or immunocompromised or who have preexisting diseases, need to review their insurance policies to make certain that care in foreign countries is covered. If not, evacuation insurance can be purchased at a relatively modest cost.

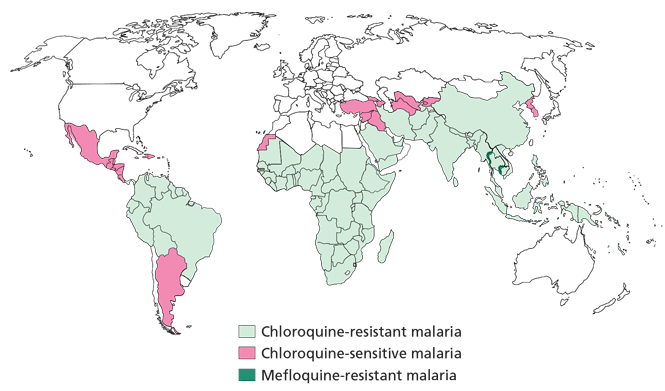
TRAVEL TO AREAS OF MALARIA
While many travelers can confidently consult their primary care provider, those traveling to places where malaria is prevalent should be referred to a physician with a thorough and current knowledge of the incidence of drugresistant strains of the disease and other complex issues in travel medicine. Short-term and long-term travelers are often approached differently, but a travel medicine consult should be obtained for any patient traveling to a region with malaria risk.
Malaria kills up to 3 million each year
Malaria, caused by the Plasmodium parasite, transmitted by the night-biting Anopheles mosquito, is responsible worldwide for between 1 and 3 million deaths annually, mostly of children in sub-Saharan Africa.15 Every year about 1,500 Americans are diagnosed with malaria and, on average, 10 die.6
Nearly all cases of malaria and deaths from it are preventable. Prophylaxis is imperative for travelers to affected areas, as is preventive counseling. Based on the patient’s itinerary, the physician needs to thoroughly research potential exposure to drug-resistant strains before choosing which antimalarial regimen to prescribe.
Malaria causes symptoms of anemia, fever, or nausea and, without treatment, can lead to coma and death. Because two of the five strains, P vivax and P ovale, can remain dormant in an infected person’s liver for up to 1 year and, in rare cases, up to 4 years after travel, it is imperative that a returned traveler who experiences flu-like symptoms seek medical attention and inform the treating physician of the need to screen for malarial infection. The primary means of diagnosis is through microscopic examination of the blood.
No malaria vaccine, but prophylactic drugs are available
Unlike many of the illnesses discussed below for which vaccines are available, malaria prophylaxis requires the active participation of the patient in completing a course of medication, so noncompliance becomes a risk.
A number of prophylactic drugs are available. The choice depends on the locally resistant strains.16
Chloroquine (Aralen), the traditional malarial prophylactic drug, is still effective against many strains, primarily in Central America and some areas of the Middle East. The dosage is 500 mg once a week, started 1 week before travel and continued for 4 weeks after return to the United States.
Mefloquine (Lariam) is dosed at 250 mg weekly. The patient should be carefully screened for depression, anxiety, and other mood disorders. Even the report of bad dreams or nightmares should make a patient be considered a poor candidate for this medication. The patient should start taking this drug 3 weeks before travel to provide time to assess for adverse effects and, if necessary, to change the antimalarial regimen. Mefloquine is taken weekly while traveling and is continued for 4 weeks after return.
Doxycycline (Vibramycin) is an antibiotic. As an antimalarial prophylactic, it is taken as 100 mg daily beginning 2 days prior to travel and continuing while travelling and for 4 weeks after return.
Atovaquone-proguanil (Malarone) prevents infection at the blood stage and in the liver. It is well tolerated and is begun 2 days before travel. It is taken daily while traveling and daily for 1 week after return.
Yellow fever
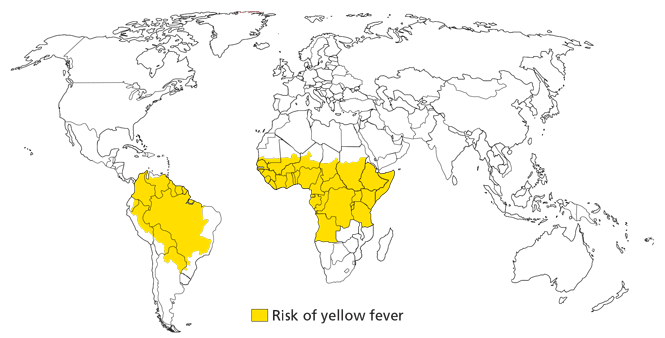
Immunization is required for entry to more than 20 African nations and is recommended for those traveling to most of South America. The only physicians who can give this vaccine are those who have approval from their state health department and have been issued an official stamp, used on the World Health Organization (WHO) yellow fever vaccination card. Several countries require the card for entry from places where yellow fever is present. For any multicountry travel involving at least one area where yellow fever is endemic, the entire itinerary needs to be reviewed to make sure all legal entry requirements are met. The WHO maintains a current list of these requirements.17 If there is any doubt, it is generally best to refer and certify the traveler.
Referral should be timely. The vaccine must be given 10 days prior to entry into a country where yellow fever is endemic; it is valid for 10 years.
The yellow fever vaccine is a live-attenuated vaccine and should not be given to infants younger than 9 months old, adults over age 60 who are not properly screened and informed, or pregnant women. Immunocompromised patients are excluded from receiving this vaccine, as are patients taking immunosuppressant drugs and patients with thymus disorders such as myasthenia gravis. Patients who have had chemotherapy must wait 3 months before being vaccinated. Those on steroids (eg, prednisone 20 mg or more daily) must wait until 2 weeks after cessation of steroids to receive this vaccine. Patients who cannot be vaccinated should be advised not to travel to areas with a high risk of yellow fever.
Women contemplating pregnancy should use contraception for 28 days after yellow fever vaccination. Children younger than 9 months and the elderly are at higher risk of adverse reactions from the vaccine, either neurotropic or viscerotropic disease that mimics yellow fever infection. It is possible for physicians to write a medical waiver of contraindication to vaccination for patients who should not be immunized.
Typhoid fever
Typhoid fever can occur anywhere in the world, but it is endemic in the tropics. Worldwide, an estimated 200,000 deaths occur each year from typhoid fever, and 400 cases are reported annually in the United States, most commonly acquired by travelers to the Indian subcontinent.18 One study indicates that 95% of infected travelers had not been vaccinated, and a significant number returned with drugresistant strains.19
Typhoid fever is caused by ingestion of Salmonella typhi bacteria. It causes a febrile illness with infection of the digestive tract and reticuloendothelial system.
Prevention is the same as for traveler’s diarrhea: drink no local water and eat nothing raw. Vaccination can be provided in an intramuscular shot or a series of oral capsules. The shot is well tolerated and is valid for 3 years. The capsule provides 5 years of immunity. Vaccination is recommended for people going to areas with a high prevalence of typhoid fever, such as India, and for people planning to spend more than 2 weeks in an area where typhoid is endemic, as well as for adventurous eaters.
SOME TRAVELERS NEED MORE PROTECTION
Some travelers need more preventive measures than typical tourists or other short-term visitors. Long-term visitors or travelers to remote or other high-risk areas (eg, adventure travelers, relief workers, mission workers) may need, in addition to the measures described above, measures against Japanese encephalitis, rabies, cholera, epidemic meningitis, and dengue fever.
Japanese encephalitis
Japanese encephalitis virus is transmitted by mosquito bite. The major regions where it is endemic are rural India and Southeast Asia, most typically in areas with rice paddies and pig farms. Travelers at risk are expatriates to these areas, those planning a long stay, and remote-adventure travelers.
The vaccine JE-VAX is given as a series of three shots, on days 0, 7, and 28. Another vaccine, Ixiaro, is given in a series of two shots, on days 0 and 28.
Patients who are allergic to bee or wasp stings should not be vaccinated. The patient should remain in the office for 30 minutes after each dose to permit observation for mild anaphylactic reactions such as angioedema and urticaria, and should complete the series 10 days before travel to allow for observation for delayed reactions. Patients must weigh the risk of contracting the disease against the high cost of the vaccine.
Rabies
Rabies is a potential risk anywhere in the world except in Western Europe and Australia. Because the vaccine is costly, it is generally not given for prophylaxis except for travelers certain to have contact with animals, especially the major vectors, ie, dogs, cats, bats, and monkeys.20 Counseling about vigilance in avoiding animal contact and not promoting interaction through feeding wild animals should be part of any pretravel consult. Rabies, once acquired, is fatal.
The patient should be instructed on proper care of a bite from a potential rabies source and told to halt travel and seek medical attention. The wound should be cleaned with soap and water for 15 minutes to remove any saliva and virus from the soft tissue; this has proven to be effective in animal experiments. A virucidal such as benzalkonium chloride (Zephiran) or aqueous iodine should then be put in the wound.
Preexposure vaccination is done in a three-dose series (given on days 0, 7, and 21– 28). The patient should complete the series and adhere to the dosing schedule as closely as possible. It may be necessary to find a source of vaccine for the patient once he or she has arrived in the destination country.
If bitten, travelers without preexposure vaccination must find a source of vaccine and human rabies immune globulin (HRIG) before continuing on their trip. Postexposure treatment is 20 IU/kg of HRIG infiltrated around the wound to wall off the virus inoculation site. If the wound is in a digit or small area and not all of the HRIG can be given, then the remaining HRIG is given intramuscularly at a site distant from the vaccine site. If the patient has multiple bites, the HRIG should be diluted so it can be infiltrated around all wounds. The HRIG should be given immediately or within 7 days of beginning the vaccine series once a source is located. Later treatment than this can interfere with the patient’s ability to mount an immune reaction.
Rabies vaccine is initiated at the same time as HRIG and is given on days 0, 3, 7, 14, and 28. The CDC may soon change the schedule to allow for only four postexposure shots, but this has not yet been done as of this writing.
The patient vaccinated before exposure requires only booster doses of rabies vaccine at days 0 and 3.
Cholera
Cholera is an epidemic gastrointestinal disease historically responsible for millions of deaths. It is endemic in most tropical countries, especially in Africa and southern and southeastern Asia.21
High-risk patients, most often those working with refugees and disaster victims in endemic areas, should receive the traveler’s diarrhea and cholera vaccine Dukoral, which immunizes against Vibrio cholera and enterotoxogenic E coli. The vaccine, which is not available in the United States but is available abroad, is given as two oral doses 1 week apart for adults and three oral doses for children ages 2 to 6, and the second dose must be given 7 days before travel; this provides protection for 6 months.
At various times, the above vaccines have been in short supply. Travel medicine consults should be obtained for proper identification of the at-risk traveler for efficient use of any possibly limited vaccine.
Epidemic meningitis
The vaccine against epidemic meningitis is now routinely given in the United States to adolescents at the age of 12 or upon entry to college or the military. The fatality rate from the disease is 10%.
Meningococcal disease transmission peaks in the sub-Saharan “meningitis belt” in the dry season of December through June. Travelers to these areas at these times should be immunized. Travelers planning close contact with the local population (eg, health care workers) should be immunized. Patients traveling to Saudi Arabia for Hajj in Mecca must be immunized for meningitis for entry to the country during this time. The vaccine must be given within 3 years of entering the country and not less than 10 days before.
Dengue fever
Dengue fever is a flavivirus transmitted through the Aedes aegypti mosquito. No vaccine is available for dengue fever, so for now the only advice is to avoid insect vectors.
There are four closely related but serologically different dengue viruses that provide only weak cross-protection. In fact, previous infection with one serotype in a traveler then infected with another poses a risk of dengue hemorrhagic fever.
Because of inattention to public sanitation, this virus and its mosquito vector have reemerged in areas where they were once eliminated. The viral infection is a risk for the traveler to both urban and rural areas in the Americas, Southeast Asia, and Africa. The Pan American Health Organization has seen the number of reported dengue cases increase from 66,000 in 1980 to 700,000 in 2003.22
- US Department of Commerce International Trade Administration. Outbound overview 2008. www.tinet.ita.doc.gov/outreachpages. Accessed October 27, 2009.
- Dick L. Travel medicine: helping patients prepare for trips abroad. Am Fam Physician 1998; 58:383–398, 401–402.
- Reed JM, McIntosh IB, Powers K. Travel illness and the family practitioner: a retrospective assessment of travel-induced illness in general practice and the effect of a travel illness clinic. J Travel Med 1994 1:192–198.
- Hamer DH, Connor BA. Travel health knowledge, attitudes, and practices among United States travelers. J Travel Med 2004; 11:23–26.
- Hill DR. Health problems in a large cohort of Americans traveling to developing countries. J Travel Med 2000; 7:259–266.
- Hill DR, Ericsson CD, et al., Infectious Diseases Society of America. The practice of travel medicine: guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2006; 43:1499–1539.
- US Centers for Disease Control and Prevention. Destinations. wwwn.cdc.gov/travel/destinations/list.aspx. Accessed February 11, 2010.
- Steffen R. Epidemiology: Morbidity and mortality in travelers. In:Keystone J, ed. Travel Medicine. Mosby: New York, 2004:5–12.
- Joint United Nations Programme on HIV/AIDS. HIV-related travel restrictions. www.unaids.org/en/KnowledgeCentre/Resources/FeatureStories/archive/2008/20080304_HIVrelated_travel_restrictions.asp. Accessed February 11, 2010.
- Brewster SJ, Taylor DN. Epidemiology of travelers’ diarrhea. In:Keystone J, ed. Travel Medicine. New York: Mosby, 2004:175–184.
- Ostrosky-Zeichner L, Ericsson CD. Prevention of travelers’ diarrhea. In:Keystone J, ed. Travel Medicine. New York: Mosby, 2004:185–189.
- US Department of State. Spring break in Mexico—“Know Before You Go!” http://travel.state.gov/travel/cis_pa_tw/spring_break_mexico/spring_break_mexico_2812.html. Accessed February 11, 2010.
- US Department of State. Current travel warnings. http://travel.state.gov/travel/cis_pa_tw/tw/tw_1764.html. Accessed February 11, 2010.
- Bledsoe GH, Manyak MJ, Townes DA, eds. Expedition and Wilderness Medicine. Cambridge University Press: New York, 2008.
- Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 2001; 64( suppl 1–2):97–106.
- US Centers for Disease Control and Prevention. The Pre-Travel Consultation: Malaria. wwwnc.cdc.gov/travel/yellowbook/2010/chapter-2/malaria.aspx. Accessed February 11, 2010.
- World Health Organization. Country list: yellow fever vaccination requirements and recommendations; and malaria situation. http://www.who.int/ith/ITH2009Countrylist.pdf. Accessed February 11, 2010.
- US Centers for Disease Control and Prevention. Typhoid Fever. www.cdc.gov/ncidod/dbmd/diseaseinfo/typhoidfever_t.htm. Accessed February 11, 2010.
- Lynch MF, Blanton EM, Bulens S, et al Typhoid fever in the United States, 1999–2006. JAMA 2009; 302:859–865.
- Plotkin SA. Rabies. Clin Infect Dis 2000; 30:4–12.
- Topps M. Oral cholera vaccine—for whom, when, and why? Travel Med Infect Dis 2006; 4:38–42.
- Petersen LR, Marfin AA. Shifting epidemiology of Flaviviridae. J Travel Med 2005; 12(suppl 1):S3–S11.
Before going abroad to areas that might pose a risk to their health, most people ought to visit their primary care physicians and many should be referred to a specialist in travel medicine.
In this article, we review the key elements of the pretravel consult as it relates to the prevention and self-treatment of the most common diseases that pose health risks for travelers. We also give guidelines for when to refer patients to a specialist.
WHY PRIMARY CARE PHYSICIANS NEED TO KNOW TRAVEL MEDICINE
International travel to exotic locations is becoming more popular. In 2008, one out of five Americans traveled abroad, and 38 million visits were to developing countries where there are significant health risks for travelers.1
One third to one half of travelers to developing countries experience some kind of illness while abroad, most commonly diarrhea or upper respiratory infections, which typically lead to 3 lost days during a 2-week trip.
These illnesses are often preventable and self-treatable.2 Unfortunately, studies suggest that most travelers do not seek adequate medical advice, and that when they do they often fail to complete courses of medication.3,4
All these factors point to the need for primary care providers to become proficient in the pretravel consult and, if necessary, to refer patients to travel specialists and clinics.
WHY REFER TO A TRAVEL CLINIC?
In one study of travelers to areas of high risk for malaria or hepatitis A, 42% of those who consulted only their family doctor became ill, in contrast to 22% of those who attended a travel medicine clinic.4
As a rule of thumb, anyone traveling to an area where malaria is endemic should be referred to a specialist, as should anyone at risk of yellow fever or typhoid fever. As many as 8 per 1,000 travelers may return from areas of risk infected with malaria.5
Long-term travelers and people who will spend time in urban slums or rural or remote regions have an even greater need for referral to a travel clinic, as they are at higher risk of exposure to Japanese encephalitis, cholera, epidemic meningitis, dengue fever, and rabies.6
THE PRETRAVEL CONSULT: ESSENTIALS
A pretravel consult ought to be scheduled 4 to 6 weeks in advance of the trip, since many vaccines require that much time to induce immunity, and some require a series of shots.
Unfortunately, many patients who think of arranging a travel consult make the appointment at the last minute, and some come with an incomplete knowledge of their travel plans. However, even without enough advance notice, a consult can be beneficial.
Travelers sometimes change their itineraries in-country or engage in unanticipated risky behaviors. A good travel medicine physician tries to anticipate even these unplanned risks and changes in itinerary.
Where is the traveler going? When? For how long?
The pretravel consult starts with a detailed discussion of the patient’s itinerary. It needs to include length, dates, and location of travel, as well as anticipated activities and accommodations.
A remarkable number of travelers come to consults not knowing the names of specific countries they will visit, perhaps saying only that they are going to Africa or South America. An accurate itinerary is indispensible, as appropriate medical advice is highly specific to country and region. The incidence and geographic distribution of many travelers’ diseases change over time, and this requires physicians to consult the most current information available.
Tropical countries, in general, are risky, but each pathogen has a unique distribution that may vary between urban and rural areas or by season. Detailed, up-to-date information is available from the US Centers for Disease Control and Prevention (CDC) (www.cdc.gov) for individual countries and for specific provinces and locations within those countries. Physicians should consult the CDC whenever advising a patient preparing to travel. 7
How is the traveler’s current health?
Several immunizations cannot be given to the very young, the elderly, or those who are immunocompromised.
The greatest risk of death to travelers is not from tropical diseases but from cardiovascular disease, which according to one study is responsible for half of deaths abroad.8 Patients with heart disease or other known health concerns need to be counseled to avoid activities that will put them at further risk. The advice applies especially in situations such as remote travel or even cruises, where prompt emergency medical care may be difficult or impossible to obtain.
People infected with human immunodeficiency virus (HIV) face discriminatory travel prohibitions in 74 countries.9
Foreign-born travelers who are visiting family and friends in developing countries may have lost their immunity to local pathogens and thus can be more at risk because they are not prepared to take necessary health precautions.
Also, a significant number of travelers become infected but develop illnesses only after they return, so a posttravel visit may be necessary.6
Prescription and even over-the-the-counter drugs may be difficult or impossible to obtain in foreign countries, and ample supplies should be brought along.
Is the traveler up to date on routine immunizations?
A number of infectious diseases that have been controlled or eradicated in North America through regular childhood immunizations are still endemic in many remote areas and developing countries. All travelers should be up to date on routine immunizations, including those for measles-mumps-rubella, tetanus, polio, meningitis, and hepatitis A and B.
Polio. A one-time polio booster is recommended for adults traveling to certain countries or areas of the world.
Meningitis vaccine is now routinely given to young people, but adult patients may need it before they travel.
Hepatitis A is contracted through fecal contamination of food and water. Common sources are foods prepared in an unhygienic manner, raw fruits and vegetables, shellfish, and contaminated water.
Hepatitis B vaccine is also now routinely given to young people, but it should be offered to travelers planning to stay more than 1 month and to long-term expatriates. This vaccine is also recommended for travelers who may be exposed to blood or body fluids, who are contemplating sexual activity or tattooing in the host country, or who may require medical or dental care while traveling, as well as for adventure travelers or travelers to remote regions.
The vaccination is given in a three-dose schedule at 0, 1, and 6 months. For protection against both hepatitis A and B, the vaccine Twinrix can be used on the same schedule as for hepatitis B. An accelerated schedule of 0, 7, and 21 days with a booster at 12 months allows completion of the entire series in 4 weeks, thus putting completion of vaccination before travel in the same time frame as other vaccines in a series, such as those for rabies and Japanese encephalitis.
PREVENTIVE COUNSELING
In addition, travelers going abroad should be advised on measures to avoid diarrhea, insectvector diseases, accidents, excessive exposure to the sun, altitude sickness, and other risks their itineraries may expose them to.
Avoiding traveler’s diarrhea
Traveler’s diarrhea is by far the most common health problem experienced abroad. It is prevalent in Mexico, where 20 million visits by Americans occur each year. A quarter to half of visitors to developing countries contract traveler’s diarrhea and, on average, lose 2 to 3 days of their business trip or vacation.3,10 The disease therefore imposes not only discomfort but also financial losses on travelers, especially business travelers.
Though many pathogens may be responsible, the most common one is Escherichia coli, usually transmitted by human fecal contamination of food or drink. Preventive measures against E coli are the same as for other foodborne and waterborne infections, such as hepatitis A, cholera, and typhoid fever.
The rule for avoiding traveler’s diarrhea may be summarized by the CDC-coined phrase, “boil it, cook it, peel it, or forget it.” Simple, written advice is most likely to be followed. 6 Thorough boiling or cooking kills bacteria in contaminated food, and food should be served steaming hot. Travelers should only eat foods they know have been well cooked, declining cold dishes like salsa or casseroles. They should avoid tap water for brushing teeth or in the form of ice cubes and should stick to drinking bottled beverages, preferably carbonated ones. No matter how appetizing a salad looks, travelers should avoid eating fresh fruits and vegetables unless they are sure that they were peeled under sanitary conditions. Simply eating at a high-priced restaurant is not a guarantee of uncontaminated food. Before meals or any hand-to-mouth contact, hands should be washed in soap and water or with sanitizers.
Travelers to remote areas may wish to acquire filtering devices, chlorine, or iodine for treating water. A combination of filtering and iodine treatment is most effective.
While this advice is undoubtedly wise, the evidence shows that, in practice, most travelers fail to take all precautions, and the benefits of this counseling have been difficult to demonstrate.11 Therefore, physicians should prescribe drugs for prophylaxis and self-treatment of traveler’s diarrhea during travel.
Bismuth subsalicylate (Pepto-Bismol) taken as two tablets or 2 oz of liquid 4 times a day while traveling may reduce the risk of diarrhea by one half, though it should be avoided by patients with contraindications to aspirin.3,6
Self-treating traveler’s diarrhea
Proper hydration is crucial, since dehydration can worsen and prolong symptoms.
Ciprofloxacin (Cipro) 500 mg orally two times daily for 3 to 5 days is effective.
Azithromycin (Zithromax) 500 mg daily for 3 to 5 days may be better in some areas of Southeast Asia, where fluoroquinolone-resistant bacteria are prevalent.
Rifaximin (Xifaxan), a nonsystemic antibiotic, is another option. The dosage is 200 mg three times a day for 3 days.
Avoiding insect bites
Malaria, yellow fever, tickborne encephalitis, and dengue fever are all transmitted by insect bites. Often the best protection is to avoid being bitten.
Bites can be avoided by using insect repellants containing diethyltoluamide (DEET) or picardin. If the traveler is going to be out in the sun, he or she should apply sunscreen first, then DEET on top of that. Anopheles, which transmits malaria, is a night-biting mosquito and may be avoided by staying in screened areas at dusk and dawn and by using bed netting. Permethrin, an insecticide, can be applied to clothing and mosquito netting.
Other things to avoid
Accidents are the second most common cause of death in travelers (after cardiovascular disease), accounting for as many as one-third of deaths.9 Several studies indicate road accidents are the major cause of accidental death, but also significant are drowning and air crashes. Travelers should be advised that transportation in developing countries is often more dangerous than at home. Seaside vacationers should be aware of the dangers of riptides and other threats to swimmers and should obey warnings posted at beaches.
Sexually transmitted diseases. When appropriate, physicians should warn travelers about the dangers of contracting HIV and other sexually transmitted diseases, especially in sub-Saharan Africa.
Sunburn, dehydration. Travelers should regularly use sunscreen and should remain hydrated.
Crimes against and involving tourists are a serious threat in many places, including some popular destinations. All of the 100,000 young people traveling to Mexico each year for spring break should read the US Department of State warnings against crime and possible arrest in that country.12 Travelers who are victims of crimes in foreign countries should contact their national consulate as soon as possible. The US Department of State issues advisories on countries where there is danger to travelers because of political turmoil, crime, or other causes.13
Motion sickness and jet lag can be ameliorated by proper hydration, avoiding caffeine, and using a scopolamine patch or dimenhydrinate (Dramamine).1
When traveling to wilderness areas
Wilderness and expedition medicine is a complex subset of travel medicine.14 All travelers need to understand the risks of whatever activities they undertake.
Mountain climbers and skiers have to contend with altitude sickness and frostbite. Scuba divers have the risks of decompression sickness, barotrauma, and hazardous marine life. Travelers on expeditions may have to deal with predatory animals, exotic parasites, and ethnic or political violence. People who participate in these activities should do so only when they are properly certified and educated in the associated health risks.
Ordinary tourists should enjoy safe adventures with well-established tour agencies and venues and should be cautioned against activities that expose them to dangers they may not be prepared to confront.
Insurance, evacuation, and emergency care
Health insurance often does not pay for preventive travel medicine. Unfortunately, cost can be a factor in immunizations and other health care. The cost of most travelers’ medications and vaccinations is generally comparable to that of other immunizations. The exceptions are two specialized vaccines—ie, for Japanese encephalitis ($1,000 or more for a full course) and for rabies, which can cost considerably more. Pricing by different providers can vary widely.
Travelers, especially those who are pregnant, elderly, disabled, or immunocompromised or who have preexisting diseases, need to review their insurance policies to make certain that care in foreign countries is covered. If not, evacuation insurance can be purchased at a relatively modest cost.
TRAVEL TO AREAS OF MALARIA
While many travelers can confidently consult their primary care provider, those traveling to places where malaria is prevalent should be referred to a physician with a thorough and current knowledge of the incidence of drugresistant strains of the disease and other complex issues in travel medicine. Short-term and long-term travelers are often approached differently, but a travel medicine consult should be obtained for any patient traveling to a region with malaria risk.
Malaria kills up to 3 million each year
Malaria, caused by the Plasmodium parasite, transmitted by the night-biting Anopheles mosquito, is responsible worldwide for between 1 and 3 million deaths annually, mostly of children in sub-Saharan Africa.15 Every year about 1,500 Americans are diagnosed with malaria and, on average, 10 die.6
Nearly all cases of malaria and deaths from it are preventable. Prophylaxis is imperative for travelers to affected areas, as is preventive counseling. Based on the patient’s itinerary, the physician needs to thoroughly research potential exposure to drug-resistant strains before choosing which antimalarial regimen to prescribe.
Malaria causes symptoms of anemia, fever, or nausea and, without treatment, can lead to coma and death. Because two of the five strains, P vivax and P ovale, can remain dormant in an infected person’s liver for up to 1 year and, in rare cases, up to 4 years after travel, it is imperative that a returned traveler who experiences flu-like symptoms seek medical attention and inform the treating physician of the need to screen for malarial infection. The primary means of diagnosis is through microscopic examination of the blood.
No malaria vaccine, but prophylactic drugs are available
Unlike many of the illnesses discussed below for which vaccines are available, malaria prophylaxis requires the active participation of the patient in completing a course of medication, so noncompliance becomes a risk.
A number of prophylactic drugs are available. The choice depends on the locally resistant strains.16
Chloroquine (Aralen), the traditional malarial prophylactic drug, is still effective against many strains, primarily in Central America and some areas of the Middle East. The dosage is 500 mg once a week, started 1 week before travel and continued for 4 weeks after return to the United States.
Mefloquine (Lariam) is dosed at 250 mg weekly. The patient should be carefully screened for depression, anxiety, and other mood disorders. Even the report of bad dreams or nightmares should make a patient be considered a poor candidate for this medication. The patient should start taking this drug 3 weeks before travel to provide time to assess for adverse effects and, if necessary, to change the antimalarial regimen. Mefloquine is taken weekly while traveling and is continued for 4 weeks after return.
Doxycycline (Vibramycin) is an antibiotic. As an antimalarial prophylactic, it is taken as 100 mg daily beginning 2 days prior to travel and continuing while travelling and for 4 weeks after return.
Atovaquone-proguanil (Malarone) prevents infection at the blood stage and in the liver. It is well tolerated and is begun 2 days before travel. It is taken daily while traveling and daily for 1 week after return.
Yellow fever
Immunization is required for entry to more than 20 African nations and is recommended for those traveling to most of South America. The only physicians who can give this vaccine are those who have approval from their state health department and have been issued an official stamp, used on the World Health Organization (WHO) yellow fever vaccination card. Several countries require the card for entry from places where yellow fever is present. For any multicountry travel involving at least one area where yellow fever is endemic, the entire itinerary needs to be reviewed to make sure all legal entry requirements are met. The WHO maintains a current list of these requirements.17 If there is any doubt, it is generally best to refer and certify the traveler.
Referral should be timely. The vaccine must be given 10 days prior to entry into a country where yellow fever is endemic; it is valid for 10 years.
The yellow fever vaccine is a live-attenuated vaccine and should not be given to infants younger than 9 months old, adults over age 60 who are not properly screened and informed, or pregnant women. Immunocompromised patients are excluded from receiving this vaccine, as are patients taking immunosuppressant drugs and patients with thymus disorders such as myasthenia gravis. Patients who have had chemotherapy must wait 3 months before being vaccinated. Those on steroids (eg, prednisone 20 mg or more daily) must wait until 2 weeks after cessation of steroids to receive this vaccine. Patients who cannot be vaccinated should be advised not to travel to areas with a high risk of yellow fever.
Women contemplating pregnancy should use contraception for 28 days after yellow fever vaccination. Children younger than 9 months and the elderly are at higher risk of adverse reactions from the vaccine, either neurotropic or viscerotropic disease that mimics yellow fever infection. It is possible for physicians to write a medical waiver of contraindication to vaccination for patients who should not be immunized.
Typhoid fever
Typhoid fever can occur anywhere in the world, but it is endemic in the tropics. Worldwide, an estimated 200,000 deaths occur each year from typhoid fever, and 400 cases are reported annually in the United States, most commonly acquired by travelers to the Indian subcontinent.18 One study indicates that 95% of infected travelers had not been vaccinated, and a significant number returned with drugresistant strains.19
Typhoid fever is caused by ingestion of Salmonella typhi bacteria. It causes a febrile illness with infection of the digestive tract and reticuloendothelial system.
Prevention is the same as for traveler’s diarrhea: drink no local water and eat nothing raw. Vaccination can be provided in an intramuscular shot or a series of oral capsules. The shot is well tolerated and is valid for 3 years. The capsule provides 5 years of immunity. Vaccination is recommended for people going to areas with a high prevalence of typhoid fever, such as India, and for people planning to spend more than 2 weeks in an area where typhoid is endemic, as well as for adventurous eaters.
SOME TRAVELERS NEED MORE PROTECTION
Some travelers need more preventive measures than typical tourists or other short-term visitors. Long-term visitors or travelers to remote or other high-risk areas (eg, adventure travelers, relief workers, mission workers) may need, in addition to the measures described above, measures against Japanese encephalitis, rabies, cholera, epidemic meningitis, and dengue fever.
Japanese encephalitis
Japanese encephalitis virus is transmitted by mosquito bite. The major regions where it is endemic are rural India and Southeast Asia, most typically in areas with rice paddies and pig farms. Travelers at risk are expatriates to these areas, those planning a long stay, and remote-adventure travelers.
The vaccine JE-VAX is given as a series of three shots, on days 0, 7, and 28. Another vaccine, Ixiaro, is given in a series of two shots, on days 0 and 28.
Patients who are allergic to bee or wasp stings should not be vaccinated. The patient should remain in the office for 30 minutes after each dose to permit observation for mild anaphylactic reactions such as angioedema and urticaria, and should complete the series 10 days before travel to allow for observation for delayed reactions. Patients must weigh the risk of contracting the disease against the high cost of the vaccine.
Rabies
Rabies is a potential risk anywhere in the world except in Western Europe and Australia. Because the vaccine is costly, it is generally not given for prophylaxis except for travelers certain to have contact with animals, especially the major vectors, ie, dogs, cats, bats, and monkeys.20 Counseling about vigilance in avoiding animal contact and not promoting interaction through feeding wild animals should be part of any pretravel consult. Rabies, once acquired, is fatal.
The patient should be instructed on proper care of a bite from a potential rabies source and told to halt travel and seek medical attention. The wound should be cleaned with soap and water for 15 minutes to remove any saliva and virus from the soft tissue; this has proven to be effective in animal experiments. A virucidal such as benzalkonium chloride (Zephiran) or aqueous iodine should then be put in the wound.
Preexposure vaccination is done in a three-dose series (given on days 0, 7, and 21– 28). The patient should complete the series and adhere to the dosing schedule as closely as possible. It may be necessary to find a source of vaccine for the patient once he or she has arrived in the destination country.
If bitten, travelers without preexposure vaccination must find a source of vaccine and human rabies immune globulin (HRIG) before continuing on their trip. Postexposure treatment is 20 IU/kg of HRIG infiltrated around the wound to wall off the virus inoculation site. If the wound is in a digit or small area and not all of the HRIG can be given, then the remaining HRIG is given intramuscularly at a site distant from the vaccine site. If the patient has multiple bites, the HRIG should be diluted so it can be infiltrated around all wounds. The HRIG should be given immediately or within 7 days of beginning the vaccine series once a source is located. Later treatment than this can interfere with the patient’s ability to mount an immune reaction.
Rabies vaccine is initiated at the same time as HRIG and is given on days 0, 3, 7, 14, and 28. The CDC may soon change the schedule to allow for only four postexposure shots, but this has not yet been done as of this writing.
The patient vaccinated before exposure requires only booster doses of rabies vaccine at days 0 and 3.
Cholera
Cholera is an epidemic gastrointestinal disease historically responsible for millions of deaths. It is endemic in most tropical countries, especially in Africa and southern and southeastern Asia.21
High-risk patients, most often those working with refugees and disaster victims in endemic areas, should receive the traveler’s diarrhea and cholera vaccine Dukoral, which immunizes against Vibrio cholera and enterotoxogenic E coli. The vaccine, which is not available in the United States but is available abroad, is given as two oral doses 1 week apart for adults and three oral doses for children ages 2 to 6, and the second dose must be given 7 days before travel; this provides protection for 6 months.
At various times, the above vaccines have been in short supply. Travel medicine consults should be obtained for proper identification of the at-risk traveler for efficient use of any possibly limited vaccine.
Epidemic meningitis
The vaccine against epidemic meningitis is now routinely given in the United States to adolescents at the age of 12 or upon entry to college or the military. The fatality rate from the disease is 10%.
Meningococcal disease transmission peaks in the sub-Saharan “meningitis belt” in the dry season of December through June. Travelers to these areas at these times should be immunized. Travelers planning close contact with the local population (eg, health care workers) should be immunized. Patients traveling to Saudi Arabia for Hajj in Mecca must be immunized for meningitis for entry to the country during this time. The vaccine must be given within 3 years of entering the country and not less than 10 days before.
Dengue fever
Dengue fever is a flavivirus transmitted through the Aedes aegypti mosquito. No vaccine is available for dengue fever, so for now the only advice is to avoid insect vectors.
There are four closely related but serologically different dengue viruses that provide only weak cross-protection. In fact, previous infection with one serotype in a traveler then infected with another poses a risk of dengue hemorrhagic fever.
Because of inattention to public sanitation, this virus and its mosquito vector have reemerged in areas where they were once eliminated. The viral infection is a risk for the traveler to both urban and rural areas in the Americas, Southeast Asia, and Africa. The Pan American Health Organization has seen the number of reported dengue cases increase from 66,000 in 1980 to 700,000 in 2003.22
Before going abroad to areas that might pose a risk to their health, most people ought to visit their primary care physicians and many should be referred to a specialist in travel medicine.
In this article, we review the key elements of the pretravel consult as it relates to the prevention and self-treatment of the most common diseases that pose health risks for travelers. We also give guidelines for when to refer patients to a specialist.
WHY PRIMARY CARE PHYSICIANS NEED TO KNOW TRAVEL MEDICINE
International travel to exotic locations is becoming more popular. In 2008, one out of five Americans traveled abroad, and 38 million visits were to developing countries where there are significant health risks for travelers.1
One third to one half of travelers to developing countries experience some kind of illness while abroad, most commonly diarrhea or upper respiratory infections, which typically lead to 3 lost days during a 2-week trip.
These illnesses are often preventable and self-treatable.2 Unfortunately, studies suggest that most travelers do not seek adequate medical advice, and that when they do they often fail to complete courses of medication.3,4
All these factors point to the need for primary care providers to become proficient in the pretravel consult and, if necessary, to refer patients to travel specialists and clinics.
WHY REFER TO A TRAVEL CLINIC?
In one study of travelers to areas of high risk for malaria or hepatitis A, 42% of those who consulted only their family doctor became ill, in contrast to 22% of those who attended a travel medicine clinic.4
As a rule of thumb, anyone traveling to an area where malaria is endemic should be referred to a specialist, as should anyone at risk of yellow fever or typhoid fever. As many as 8 per 1,000 travelers may return from areas of risk infected with malaria.5
Long-term travelers and people who will spend time in urban slums or rural or remote regions have an even greater need for referral to a travel clinic, as they are at higher risk of exposure to Japanese encephalitis, cholera, epidemic meningitis, dengue fever, and rabies.6
THE PRETRAVEL CONSULT: ESSENTIALS
A pretravel consult ought to be scheduled 4 to 6 weeks in advance of the trip, since many vaccines require that much time to induce immunity, and some require a series of shots.
Unfortunately, many patients who think of arranging a travel consult make the appointment at the last minute, and some come with an incomplete knowledge of their travel plans. However, even without enough advance notice, a consult can be beneficial.
Travelers sometimes change their itineraries in-country or engage in unanticipated risky behaviors. A good travel medicine physician tries to anticipate even these unplanned risks and changes in itinerary.
Where is the traveler going? When? For how long?
The pretravel consult starts with a detailed discussion of the patient’s itinerary. It needs to include length, dates, and location of travel, as well as anticipated activities and accommodations.
A remarkable number of travelers come to consults not knowing the names of specific countries they will visit, perhaps saying only that they are going to Africa or South America. An accurate itinerary is indispensible, as appropriate medical advice is highly specific to country and region. The incidence and geographic distribution of many travelers’ diseases change over time, and this requires physicians to consult the most current information available.
Tropical countries, in general, are risky, but each pathogen has a unique distribution that may vary between urban and rural areas or by season. Detailed, up-to-date information is available from the US Centers for Disease Control and Prevention (CDC) (www.cdc.gov) for individual countries and for specific provinces and locations within those countries. Physicians should consult the CDC whenever advising a patient preparing to travel. 7
How is the traveler’s current health?
Several immunizations cannot be given to the very young, the elderly, or those who are immunocompromised.
The greatest risk of death to travelers is not from tropical diseases but from cardiovascular disease, which according to one study is responsible for half of deaths abroad.8 Patients with heart disease or other known health concerns need to be counseled to avoid activities that will put them at further risk. The advice applies especially in situations such as remote travel or even cruises, where prompt emergency medical care may be difficult or impossible to obtain.
People infected with human immunodeficiency virus (HIV) face discriminatory travel prohibitions in 74 countries.9
Foreign-born travelers who are visiting family and friends in developing countries may have lost their immunity to local pathogens and thus can be more at risk because they are not prepared to take necessary health precautions.
Also, a significant number of travelers become infected but develop illnesses only after they return, so a posttravel visit may be necessary.6
Prescription and even over-the-the-counter drugs may be difficult or impossible to obtain in foreign countries, and ample supplies should be brought along.
Is the traveler up to date on routine immunizations?
A number of infectious diseases that have been controlled or eradicated in North America through regular childhood immunizations are still endemic in many remote areas and developing countries. All travelers should be up to date on routine immunizations, including those for measles-mumps-rubella, tetanus, polio, meningitis, and hepatitis A and B.
Polio. A one-time polio booster is recommended for adults traveling to certain countries or areas of the world.
Meningitis vaccine is now routinely given to young people, but adult patients may need it before they travel.
Hepatitis A is contracted through fecal contamination of food and water. Common sources are foods prepared in an unhygienic manner, raw fruits and vegetables, shellfish, and contaminated water.
Hepatitis B vaccine is also now routinely given to young people, but it should be offered to travelers planning to stay more than 1 month and to long-term expatriates. This vaccine is also recommended for travelers who may be exposed to blood or body fluids, who are contemplating sexual activity or tattooing in the host country, or who may require medical or dental care while traveling, as well as for adventure travelers or travelers to remote regions.
The vaccination is given in a three-dose schedule at 0, 1, and 6 months. For protection against both hepatitis A and B, the vaccine Twinrix can be used on the same schedule as for hepatitis B. An accelerated schedule of 0, 7, and 21 days with a booster at 12 months allows completion of the entire series in 4 weeks, thus putting completion of vaccination before travel in the same time frame as other vaccines in a series, such as those for rabies and Japanese encephalitis.
PREVENTIVE COUNSELING
In addition, travelers going abroad should be advised on measures to avoid diarrhea, insectvector diseases, accidents, excessive exposure to the sun, altitude sickness, and other risks their itineraries may expose them to.
Avoiding traveler’s diarrhea
Traveler’s diarrhea is by far the most common health problem experienced abroad. It is prevalent in Mexico, where 20 million visits by Americans occur each year. A quarter to half of visitors to developing countries contract traveler’s diarrhea and, on average, lose 2 to 3 days of their business trip or vacation.3,10 The disease therefore imposes not only discomfort but also financial losses on travelers, especially business travelers.
Though many pathogens may be responsible, the most common one is Escherichia coli, usually transmitted by human fecal contamination of food or drink. Preventive measures against E coli are the same as for other foodborne and waterborne infections, such as hepatitis A, cholera, and typhoid fever.
The rule for avoiding traveler’s diarrhea may be summarized by the CDC-coined phrase, “boil it, cook it, peel it, or forget it.” Simple, written advice is most likely to be followed. 6 Thorough boiling or cooking kills bacteria in contaminated food, and food should be served steaming hot. Travelers should only eat foods they know have been well cooked, declining cold dishes like salsa or casseroles. They should avoid tap water for brushing teeth or in the form of ice cubes and should stick to drinking bottled beverages, preferably carbonated ones. No matter how appetizing a salad looks, travelers should avoid eating fresh fruits and vegetables unless they are sure that they were peeled under sanitary conditions. Simply eating at a high-priced restaurant is not a guarantee of uncontaminated food. Before meals or any hand-to-mouth contact, hands should be washed in soap and water or with sanitizers.
Travelers to remote areas may wish to acquire filtering devices, chlorine, or iodine for treating water. A combination of filtering and iodine treatment is most effective.
While this advice is undoubtedly wise, the evidence shows that, in practice, most travelers fail to take all precautions, and the benefits of this counseling have been difficult to demonstrate.11 Therefore, physicians should prescribe drugs for prophylaxis and self-treatment of traveler’s diarrhea during travel.
Bismuth subsalicylate (Pepto-Bismol) taken as two tablets or 2 oz of liquid 4 times a day while traveling may reduce the risk of diarrhea by one half, though it should be avoided by patients with contraindications to aspirin.3,6
Self-treating traveler’s diarrhea
Proper hydration is crucial, since dehydration can worsen and prolong symptoms.
Ciprofloxacin (Cipro) 500 mg orally two times daily for 3 to 5 days is effective.
Azithromycin (Zithromax) 500 mg daily for 3 to 5 days may be better in some areas of Southeast Asia, where fluoroquinolone-resistant bacteria are prevalent.
Rifaximin (Xifaxan), a nonsystemic antibiotic, is another option. The dosage is 200 mg three times a day for 3 days.
Avoiding insect bites
Malaria, yellow fever, tickborne encephalitis, and dengue fever are all transmitted by insect bites. Often the best protection is to avoid being bitten.
Bites can be avoided by using insect repellants containing diethyltoluamide (DEET) or picardin. If the traveler is going to be out in the sun, he or she should apply sunscreen first, then DEET on top of that. Anopheles, which transmits malaria, is a night-biting mosquito and may be avoided by staying in screened areas at dusk and dawn and by using bed netting. Permethrin, an insecticide, can be applied to clothing and mosquito netting.
Other things to avoid
Accidents are the second most common cause of death in travelers (after cardiovascular disease), accounting for as many as one-third of deaths.9 Several studies indicate road accidents are the major cause of accidental death, but also significant are drowning and air crashes. Travelers should be advised that transportation in developing countries is often more dangerous than at home. Seaside vacationers should be aware of the dangers of riptides and other threats to swimmers and should obey warnings posted at beaches.
Sexually transmitted diseases. When appropriate, physicians should warn travelers about the dangers of contracting HIV and other sexually transmitted diseases, especially in sub-Saharan Africa.
Sunburn, dehydration. Travelers should regularly use sunscreen and should remain hydrated.
Crimes against and involving tourists are a serious threat in many places, including some popular destinations. All of the 100,000 young people traveling to Mexico each year for spring break should read the US Department of State warnings against crime and possible arrest in that country.12 Travelers who are victims of crimes in foreign countries should contact their national consulate as soon as possible. The US Department of State issues advisories on countries where there is danger to travelers because of political turmoil, crime, or other causes.13
Motion sickness and jet lag can be ameliorated by proper hydration, avoiding caffeine, and using a scopolamine patch or dimenhydrinate (Dramamine).1
When traveling to wilderness areas
Wilderness and expedition medicine is a complex subset of travel medicine.14 All travelers need to understand the risks of whatever activities they undertake.
Mountain climbers and skiers have to contend with altitude sickness and frostbite. Scuba divers have the risks of decompression sickness, barotrauma, and hazardous marine life. Travelers on expeditions may have to deal with predatory animals, exotic parasites, and ethnic or political violence. People who participate in these activities should do so only when they are properly certified and educated in the associated health risks.
Ordinary tourists should enjoy safe adventures with well-established tour agencies and venues and should be cautioned against activities that expose them to dangers they may not be prepared to confront.
Insurance, evacuation, and emergency care
Health insurance often does not pay for preventive travel medicine. Unfortunately, cost can be a factor in immunizations and other health care. The cost of most travelers’ medications and vaccinations is generally comparable to that of other immunizations. The exceptions are two specialized vaccines—ie, for Japanese encephalitis ($1,000 or more for a full course) and for rabies, which can cost considerably more. Pricing by different providers can vary widely.
Travelers, especially those who are pregnant, elderly, disabled, or immunocompromised or who have preexisting diseases, need to review their insurance policies to make certain that care in foreign countries is covered. If not, evacuation insurance can be purchased at a relatively modest cost.
TRAVEL TO AREAS OF MALARIA
While many travelers can confidently consult their primary care provider, those traveling to places where malaria is prevalent should be referred to a physician with a thorough and current knowledge of the incidence of drugresistant strains of the disease and other complex issues in travel medicine. Short-term and long-term travelers are often approached differently, but a travel medicine consult should be obtained for any patient traveling to a region with malaria risk.
Malaria kills up to 3 million each year
Malaria, caused by the Plasmodium parasite, transmitted by the night-biting Anopheles mosquito, is responsible worldwide for between 1 and 3 million deaths annually, mostly of children in sub-Saharan Africa.15 Every year about 1,500 Americans are diagnosed with malaria and, on average, 10 die.6
Nearly all cases of malaria and deaths from it are preventable. Prophylaxis is imperative for travelers to affected areas, as is preventive counseling. Based on the patient’s itinerary, the physician needs to thoroughly research potential exposure to drug-resistant strains before choosing which antimalarial regimen to prescribe.
Malaria causes symptoms of anemia, fever, or nausea and, without treatment, can lead to coma and death. Because two of the five strains, P vivax and P ovale, can remain dormant in an infected person’s liver for up to 1 year and, in rare cases, up to 4 years after travel, it is imperative that a returned traveler who experiences flu-like symptoms seek medical attention and inform the treating physician of the need to screen for malarial infection. The primary means of diagnosis is through microscopic examination of the blood.
No malaria vaccine, but prophylactic drugs are available
Unlike many of the illnesses discussed below for which vaccines are available, malaria prophylaxis requires the active participation of the patient in completing a course of medication, so noncompliance becomes a risk.
A number of prophylactic drugs are available. The choice depends on the locally resistant strains.16
Chloroquine (Aralen), the traditional malarial prophylactic drug, is still effective against many strains, primarily in Central America and some areas of the Middle East. The dosage is 500 mg once a week, started 1 week before travel and continued for 4 weeks after return to the United States.
Mefloquine (Lariam) is dosed at 250 mg weekly. The patient should be carefully screened for depression, anxiety, and other mood disorders. Even the report of bad dreams or nightmares should make a patient be considered a poor candidate for this medication. The patient should start taking this drug 3 weeks before travel to provide time to assess for adverse effects and, if necessary, to change the antimalarial regimen. Mefloquine is taken weekly while traveling and is continued for 4 weeks after return.
Doxycycline (Vibramycin) is an antibiotic. As an antimalarial prophylactic, it is taken as 100 mg daily beginning 2 days prior to travel and continuing while travelling and for 4 weeks after return.
Atovaquone-proguanil (Malarone) prevents infection at the blood stage and in the liver. It is well tolerated and is begun 2 days before travel. It is taken daily while traveling and daily for 1 week after return.
Yellow fever
Immunization is required for entry to more than 20 African nations and is recommended for those traveling to most of South America. The only physicians who can give this vaccine are those who have approval from their state health department and have been issued an official stamp, used on the World Health Organization (WHO) yellow fever vaccination card. Several countries require the card for entry from places where yellow fever is present. For any multicountry travel involving at least one area where yellow fever is endemic, the entire itinerary needs to be reviewed to make sure all legal entry requirements are met. The WHO maintains a current list of these requirements.17 If there is any doubt, it is generally best to refer and certify the traveler.
Referral should be timely. The vaccine must be given 10 days prior to entry into a country where yellow fever is endemic; it is valid for 10 years.
The yellow fever vaccine is a live-attenuated vaccine and should not be given to infants younger than 9 months old, adults over age 60 who are not properly screened and informed, or pregnant women. Immunocompromised patients are excluded from receiving this vaccine, as are patients taking immunosuppressant drugs and patients with thymus disorders such as myasthenia gravis. Patients who have had chemotherapy must wait 3 months before being vaccinated. Those on steroids (eg, prednisone 20 mg or more daily) must wait until 2 weeks after cessation of steroids to receive this vaccine. Patients who cannot be vaccinated should be advised not to travel to areas with a high risk of yellow fever.
Women contemplating pregnancy should use contraception for 28 days after yellow fever vaccination. Children younger than 9 months and the elderly are at higher risk of adverse reactions from the vaccine, either neurotropic or viscerotropic disease that mimics yellow fever infection. It is possible for physicians to write a medical waiver of contraindication to vaccination for patients who should not be immunized.
Typhoid fever
Typhoid fever can occur anywhere in the world, but it is endemic in the tropics. Worldwide, an estimated 200,000 deaths occur each year from typhoid fever, and 400 cases are reported annually in the United States, most commonly acquired by travelers to the Indian subcontinent.18 One study indicates that 95% of infected travelers had not been vaccinated, and a significant number returned with drugresistant strains.19
Typhoid fever is caused by ingestion of Salmonella typhi bacteria. It causes a febrile illness with infection of the digestive tract and reticuloendothelial system.
Prevention is the same as for traveler’s diarrhea: drink no local water and eat nothing raw. Vaccination can be provided in an intramuscular shot or a series of oral capsules. The shot is well tolerated and is valid for 3 years. The capsule provides 5 years of immunity. Vaccination is recommended for people going to areas with a high prevalence of typhoid fever, such as India, and for people planning to spend more than 2 weeks in an area where typhoid is endemic, as well as for adventurous eaters.
SOME TRAVELERS NEED MORE PROTECTION
Some travelers need more preventive measures than typical tourists or other short-term visitors. Long-term visitors or travelers to remote or other high-risk areas (eg, adventure travelers, relief workers, mission workers) may need, in addition to the measures described above, measures against Japanese encephalitis, rabies, cholera, epidemic meningitis, and dengue fever.
Japanese encephalitis
Japanese encephalitis virus is transmitted by mosquito bite. The major regions where it is endemic are rural India and Southeast Asia, most typically in areas with rice paddies and pig farms. Travelers at risk are expatriates to these areas, those planning a long stay, and remote-adventure travelers.
The vaccine JE-VAX is given as a series of three shots, on days 0, 7, and 28. Another vaccine, Ixiaro, is given in a series of two shots, on days 0 and 28.
Patients who are allergic to bee or wasp stings should not be vaccinated. The patient should remain in the office for 30 minutes after each dose to permit observation for mild anaphylactic reactions such as angioedema and urticaria, and should complete the series 10 days before travel to allow for observation for delayed reactions. Patients must weigh the risk of contracting the disease against the high cost of the vaccine.
Rabies
Rabies is a potential risk anywhere in the world except in Western Europe and Australia. Because the vaccine is costly, it is generally not given for prophylaxis except for travelers certain to have contact with animals, especially the major vectors, ie, dogs, cats, bats, and monkeys.20 Counseling about vigilance in avoiding animal contact and not promoting interaction through feeding wild animals should be part of any pretravel consult. Rabies, once acquired, is fatal.
The patient should be instructed on proper care of a bite from a potential rabies source and told to halt travel and seek medical attention. The wound should be cleaned with soap and water for 15 minutes to remove any saliva and virus from the soft tissue; this has proven to be effective in animal experiments. A virucidal such as benzalkonium chloride (Zephiran) or aqueous iodine should then be put in the wound.
Preexposure vaccination is done in a three-dose series (given on days 0, 7, and 21– 28). The patient should complete the series and adhere to the dosing schedule as closely as possible. It may be necessary to find a source of vaccine for the patient once he or she has arrived in the destination country.
If bitten, travelers without preexposure vaccination must find a source of vaccine and human rabies immune globulin (HRIG) before continuing on their trip. Postexposure treatment is 20 IU/kg of HRIG infiltrated around the wound to wall off the virus inoculation site. If the wound is in a digit or small area and not all of the HRIG can be given, then the remaining HRIG is given intramuscularly at a site distant from the vaccine site. If the patient has multiple bites, the HRIG should be diluted so it can be infiltrated around all wounds. The HRIG should be given immediately or within 7 days of beginning the vaccine series once a source is located. Later treatment than this can interfere with the patient’s ability to mount an immune reaction.
Rabies vaccine is initiated at the same time as HRIG and is given on days 0, 3, 7, 14, and 28. The CDC may soon change the schedule to allow for only four postexposure shots, but this has not yet been done as of this writing.
The patient vaccinated before exposure requires only booster doses of rabies vaccine at days 0 and 3.
Cholera
Cholera is an epidemic gastrointestinal disease historically responsible for millions of deaths. It is endemic in most tropical countries, especially in Africa and southern and southeastern Asia.21
High-risk patients, most often those working with refugees and disaster victims in endemic areas, should receive the traveler’s diarrhea and cholera vaccine Dukoral, which immunizes against Vibrio cholera and enterotoxogenic E coli. The vaccine, which is not available in the United States but is available abroad, is given as two oral doses 1 week apart for adults and three oral doses for children ages 2 to 6, and the second dose must be given 7 days before travel; this provides protection for 6 months.
At various times, the above vaccines have been in short supply. Travel medicine consults should be obtained for proper identification of the at-risk traveler for efficient use of any possibly limited vaccine.
Epidemic meningitis
The vaccine against epidemic meningitis is now routinely given in the United States to adolescents at the age of 12 or upon entry to college or the military. The fatality rate from the disease is 10%.
Meningococcal disease transmission peaks in the sub-Saharan “meningitis belt” in the dry season of December through June. Travelers to these areas at these times should be immunized. Travelers planning close contact with the local population (eg, health care workers) should be immunized. Patients traveling to Saudi Arabia for Hajj in Mecca must be immunized for meningitis for entry to the country during this time. The vaccine must be given within 3 years of entering the country and not less than 10 days before.
Dengue fever
Dengue fever is a flavivirus transmitted through the Aedes aegypti mosquito. No vaccine is available for dengue fever, so for now the only advice is to avoid insect vectors.
There are four closely related but serologically different dengue viruses that provide only weak cross-protection. In fact, previous infection with one serotype in a traveler then infected with another poses a risk of dengue hemorrhagic fever.
Because of inattention to public sanitation, this virus and its mosquito vector have reemerged in areas where they were once eliminated. The viral infection is a risk for the traveler to both urban and rural areas in the Americas, Southeast Asia, and Africa. The Pan American Health Organization has seen the number of reported dengue cases increase from 66,000 in 1980 to 700,000 in 2003.22
- US Department of Commerce International Trade Administration. Outbound overview 2008. www.tinet.ita.doc.gov/outreachpages. Accessed October 27, 2009.
- Dick L. Travel medicine: helping patients prepare for trips abroad. Am Fam Physician 1998; 58:383–398, 401–402.
- Reed JM, McIntosh IB, Powers K. Travel illness and the family practitioner: a retrospective assessment of travel-induced illness in general practice and the effect of a travel illness clinic. J Travel Med 1994 1:192–198.
- Hamer DH, Connor BA. Travel health knowledge, attitudes, and practices among United States travelers. J Travel Med 2004; 11:23–26.
- Hill DR. Health problems in a large cohort of Americans traveling to developing countries. J Travel Med 2000; 7:259–266.
- Hill DR, Ericsson CD, et al., Infectious Diseases Society of America. The practice of travel medicine: guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2006; 43:1499–1539.
- US Centers for Disease Control and Prevention. Destinations. wwwn.cdc.gov/travel/destinations/list.aspx. Accessed February 11, 2010.
- Steffen R. Epidemiology: Morbidity and mortality in travelers. In:Keystone J, ed. Travel Medicine. Mosby: New York, 2004:5–12.
- Joint United Nations Programme on HIV/AIDS. HIV-related travel restrictions. www.unaids.org/en/KnowledgeCentre/Resources/FeatureStories/archive/2008/20080304_HIVrelated_travel_restrictions.asp. Accessed February 11, 2010.
- Brewster SJ, Taylor DN. Epidemiology of travelers’ diarrhea. In:Keystone J, ed. Travel Medicine. New York: Mosby, 2004:175–184.
- Ostrosky-Zeichner L, Ericsson CD. Prevention of travelers’ diarrhea. In:Keystone J, ed. Travel Medicine. New York: Mosby, 2004:185–189.
- US Department of State. Spring break in Mexico—“Know Before You Go!” http://travel.state.gov/travel/cis_pa_tw/spring_break_mexico/spring_break_mexico_2812.html. Accessed February 11, 2010.
- US Department of State. Current travel warnings. http://travel.state.gov/travel/cis_pa_tw/tw/tw_1764.html. Accessed February 11, 2010.
- Bledsoe GH, Manyak MJ, Townes DA, eds. Expedition and Wilderness Medicine. Cambridge University Press: New York, 2008.
- Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 2001; 64( suppl 1–2):97–106.
- US Centers for Disease Control and Prevention. The Pre-Travel Consultation: Malaria. wwwnc.cdc.gov/travel/yellowbook/2010/chapter-2/malaria.aspx. Accessed February 11, 2010.
- World Health Organization. Country list: yellow fever vaccination requirements and recommendations; and malaria situation. http://www.who.int/ith/ITH2009Countrylist.pdf. Accessed February 11, 2010.
- US Centers for Disease Control and Prevention. Typhoid Fever. www.cdc.gov/ncidod/dbmd/diseaseinfo/typhoidfever_t.htm. Accessed February 11, 2010.
- Lynch MF, Blanton EM, Bulens S, et al Typhoid fever in the United States, 1999–2006. JAMA 2009; 302:859–865.
- Plotkin SA. Rabies. Clin Infect Dis 2000; 30:4–12.
- Topps M. Oral cholera vaccine—for whom, when, and why? Travel Med Infect Dis 2006; 4:38–42.
- Petersen LR, Marfin AA. Shifting epidemiology of Flaviviridae. J Travel Med 2005; 12(suppl 1):S3–S11.
- US Department of Commerce International Trade Administration. Outbound overview 2008. www.tinet.ita.doc.gov/outreachpages. Accessed October 27, 2009.
- Dick L. Travel medicine: helping patients prepare for trips abroad. Am Fam Physician 1998; 58:383–398, 401–402.
- Reed JM, McIntosh IB, Powers K. Travel illness and the family practitioner: a retrospective assessment of travel-induced illness in general practice and the effect of a travel illness clinic. J Travel Med 1994 1:192–198.
- Hamer DH, Connor BA. Travel health knowledge, attitudes, and practices among United States travelers. J Travel Med 2004; 11:23–26.
- Hill DR. Health problems in a large cohort of Americans traveling to developing countries. J Travel Med 2000; 7:259–266.
- Hill DR, Ericsson CD, et al., Infectious Diseases Society of America. The practice of travel medicine: guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2006; 43:1499–1539.
- US Centers for Disease Control and Prevention. Destinations. wwwn.cdc.gov/travel/destinations/list.aspx. Accessed February 11, 2010.
- Steffen R. Epidemiology: Morbidity and mortality in travelers. In:Keystone J, ed. Travel Medicine. Mosby: New York, 2004:5–12.
- Joint United Nations Programme on HIV/AIDS. HIV-related travel restrictions. www.unaids.org/en/KnowledgeCentre/Resources/FeatureStories/archive/2008/20080304_HIVrelated_travel_restrictions.asp. Accessed February 11, 2010.
- Brewster SJ, Taylor DN. Epidemiology of travelers’ diarrhea. In:Keystone J, ed. Travel Medicine. New York: Mosby, 2004:175–184.
- Ostrosky-Zeichner L, Ericsson CD. Prevention of travelers’ diarrhea. In:Keystone J, ed. Travel Medicine. New York: Mosby, 2004:185–189.
- US Department of State. Spring break in Mexico—“Know Before You Go!” http://travel.state.gov/travel/cis_pa_tw/spring_break_mexico/spring_break_mexico_2812.html. Accessed February 11, 2010.
- US Department of State. Current travel warnings. http://travel.state.gov/travel/cis_pa_tw/tw/tw_1764.html. Accessed February 11, 2010.
- Bledsoe GH, Manyak MJ, Townes DA, eds. Expedition and Wilderness Medicine. Cambridge University Press: New York, 2008.
- Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 2001; 64( suppl 1–2):97–106.
- US Centers for Disease Control and Prevention. The Pre-Travel Consultation: Malaria. wwwnc.cdc.gov/travel/yellowbook/2010/chapter-2/malaria.aspx. Accessed February 11, 2010.
- World Health Organization. Country list: yellow fever vaccination requirements and recommendations; and malaria situation. http://www.who.int/ith/ITH2009Countrylist.pdf. Accessed February 11, 2010.
- US Centers for Disease Control and Prevention. Typhoid Fever. www.cdc.gov/ncidod/dbmd/diseaseinfo/typhoidfever_t.htm. Accessed February 11, 2010.
- Lynch MF, Blanton EM, Bulens S, et al Typhoid fever in the United States, 1999–2006. JAMA 2009; 302:859–865.
- Plotkin SA. Rabies. Clin Infect Dis 2000; 30:4–12.
- Topps M. Oral cholera vaccine—for whom, when, and why? Travel Med Infect Dis 2006; 4:38–42.
- Petersen LR, Marfin AA. Shifting epidemiology of Flaviviridae. J Travel Med 2005; 12(suppl 1):S3–S11.
KEY POINTS
- Primary care physicians should be proficient in the basic pretravel consult, including advice on immunizations and travel-related health problems.
- Based on the traveler’s itinerary, the physician should consult current government recommendations for pretravel preparation at www.cdc.gov/travel/destinations/list.aspx.
- Drug-resistant malarial strains are on the rise in many areas of the world.
- To enter many African and South American countries, travelers need official certification by a specialist that they have been vaccinated against yellow fever.
Palpable purpura
Q: Which is the most likely diagnosis?
- Idiopathic thrombocytopenic purpura
- Vitamin C deficiency (scurvy)
- Kaposi sarcoma not related to human immunodeficiency virus (HIV) infection
- Henoch-Schönlein purpura
- Polyarteritis nodosa
A: The correct answer is Henoch-Schönlein purpura.
Idiopathic thrombocytopenic purpura is an autoimmune disease caused by specific antibodies against platelet-membrane glycoproteins. It is characterized by thrombocytopenia not explainable by contact with toxic substances or by other causes. Along with nonpalpable purpura, other common signs are epistaxis, gingival bleeding, menorrhagia, and retinal hemorrhage.
Scurvy is an uncommon deficiency of ascorbic acid (vitamin C). The elderly and alcoholics are at higher risk, as they do not take in enough vitamin C in the diet. Patients usually show perifollicular hemorrhages of the skin and mucous membranes, typically petechial hemorrhage or ecchymosis of the gums around the upper incisors. Other cutaneous signs are follicular hyperkeratosis on the forearms, small corkscrew hairs, and sicca syndrome, which is more common in adults.
Non-HIV Kaposi sarcoma usually affects elderly patients, with pink, red, or brown papules or nodules on the legs and, less commonly, on the head and neck. Histopathologic examination shows newly formed irregular blood vessels with an inflammatory infiltrate of plasma cells and lymphocytes; immunohistochemical human herpes virus staining is usually positive.
Polyarteritis nodosa is a systemic vasculitis that affects medium or small arteries with necrotizing inflammation; renal glomeruli and arterioles, capillaries, and venules are unaffected. Skin manifestations include palpable purpura, livedo reticularis, ulcers, and distal gangrene. The condition also usually affects the kidneys, the heart, and the musculoskeletal and nervous systems.
A SYSTEMIC VASCULITIS
Henoch-Schönlein purpura is a systemic vasculitis affecting the skin, gastrointestinal tract, kidneys, and joints. Palpable purpura and joint pain are the most common and consistent presenting symptoms. The kidneys are affected in about one-third of children and in 60% of adults, and this is the major factor determining the long-term outcome.1
In our patient, laboratory testing that included a complete blood cell count, biochemical testing (including IgA levels), urinary sediment, and coagulation studies showed no abnormalities except elevations of the erythrocyte sedimentation rate and the concentration of C-reactive protein (an acute-phase reactant). These can be normal in some patients. Renal involvement was also not present.
DIAGNOSIS
The diagnosis relies on clinical manifestations. Because Henoch-Schönlein purpura is less common in adults, biopsy plays a more important role in establishing the diagnosis in this age group, and it does this by demonstrating leukocytoclastic vasculitis with a predominance of IgA deposition under immunofluorescence. Recent studies in children showed that an elevated IgA concentration along with reduced IgM levels was associated with a higher rate of severe complications.2 However, depending on the age of the biopsied lesion, IgA may not be detected.
TREATMENT DIRECTED AT SYMPTOMS
Our patient received oral corticosteroids 0.5 mg/kg per day for 20 days, and the lesions resolved by 4 weeks.
Management of Henoch-Schönlein purpura is mainly directed at the symptoms, with oral hydration and nonsteroidal anti-inflammatory drugs. For severe cases, a short course of corticosteroids (0.5–1 mg/kg) may be used.
Although no controlled clinical trial has proven that Henoch-Schönlein purpura responds to corticosteroids, colchicine, or other drugs, corticosteroids are used most often, especially in patients with renal disease. Patients with severe renal insufficiency, abdominal pain, joint involvement, or bleeding should be hospitalized. Plasmapheresis3 has been used in severe cases.
HENOCH-SCHÖNLEIN PURPURA AND MALIGNANCY
During a follow-up evaluation 1 month later, our patient was diagnosed with adenocarcinoma of the breast. This highlights the value of a workup for cancer in adults with cutaneous vasculitis.
Cutaneous vasculitis can represent a paraneoplastic syndrome associated with a malignant tumor. The pathophysiology of this association is unclear, but one proposed mechanism is the exaggerated production of antibodies that react against tumor neoantigens, leading to the formation of immune complexes, or that occasionally recognize endothelial cells because of similarities with tumor antigens. Another theory is that abnormally high levels of inflammatory cytokines are produced by neoplastic cells or in response to decreased immune complex clearance.
Yet another theory is that hyperviscosity of the blood, seen in some cancers, increases the contact time for deposition of immune complexes and causes endothelial damage. Drugs used to treat cancer have also been reported to produce Henoch-Schönlein purpura.4
Although hematologic malignancy is three to five times more common than solid tumors in patients with small-vessel vasculitis, the disease has been associated with solid tumors of the liver, skin, colon, and breast in adults over age 40.5 An evaluation for neoplasm is therefore reasonable in adults with Henoch-Schönlein purpura, as is an evaluation for tumor recurrence or metastasis if the patient has been previously treated for a malignant tumor.
- Rieu P, Noël LH. Henoch-Schönlein nephritis in children and adults. Morphological features and clinicopathological correlations. Ann Med Interne (Paris) 1999; 150:151–159.
- Fretzayas A, Sionti I, Moustaki M, Nicolaidou P. Clinical impact of altered immunoglobulin levels in Henoch-Schönlein purpura. Pediatr Int 2009; 51:381–384.
- Donghi D, Schanz U, Sahrbacher U, et al. Life-threatening or organimpairing Henoch-Schönlein purpura: plasmapheresis may save lives and limit organ damage. Dermatology 2009; 219:167–170.
- Mitsui H, Shibagaki N, Kawamura T, Matsue H, Shimada S. A clinical study of Henoch-Schönlein purpura associated with malignancy. J Eur Acad Dermatol Venereol 2009; 23:394–401.
- Maestri A, Malacarne P, Santini A. Henoch-Schönlein syndrome associated with breast cancer. A case report. Angiology 1995; 46:625–627.
Q: Which is the most likely diagnosis?
- Idiopathic thrombocytopenic purpura
- Vitamin C deficiency (scurvy)
- Kaposi sarcoma not related to human immunodeficiency virus (HIV) infection
- Henoch-Schönlein purpura
- Polyarteritis nodosa
A: The correct answer is Henoch-Schönlein purpura.
Idiopathic thrombocytopenic purpura is an autoimmune disease caused by specific antibodies against platelet-membrane glycoproteins. It is characterized by thrombocytopenia not explainable by contact with toxic substances or by other causes. Along with nonpalpable purpura, other common signs are epistaxis, gingival bleeding, menorrhagia, and retinal hemorrhage.
Scurvy is an uncommon deficiency of ascorbic acid (vitamin C). The elderly and alcoholics are at higher risk, as they do not take in enough vitamin C in the diet. Patients usually show perifollicular hemorrhages of the skin and mucous membranes, typically petechial hemorrhage or ecchymosis of the gums around the upper incisors. Other cutaneous signs are follicular hyperkeratosis on the forearms, small corkscrew hairs, and sicca syndrome, which is more common in adults.
Non-HIV Kaposi sarcoma usually affects elderly patients, with pink, red, or brown papules or nodules on the legs and, less commonly, on the head and neck. Histopathologic examination shows newly formed irregular blood vessels with an inflammatory infiltrate of plasma cells and lymphocytes; immunohistochemical human herpes virus staining is usually positive.
Polyarteritis nodosa is a systemic vasculitis that affects medium or small arteries with necrotizing inflammation; renal glomeruli and arterioles, capillaries, and venules are unaffected. Skin manifestations include palpable purpura, livedo reticularis, ulcers, and distal gangrene. The condition also usually affects the kidneys, the heart, and the musculoskeletal and nervous systems.
A SYSTEMIC VASCULITIS
Henoch-Schönlein purpura is a systemic vasculitis affecting the skin, gastrointestinal tract, kidneys, and joints. Palpable purpura and joint pain are the most common and consistent presenting symptoms. The kidneys are affected in about one-third of children and in 60% of adults, and this is the major factor determining the long-term outcome.1
In our patient, laboratory testing that included a complete blood cell count, biochemical testing (including IgA levels), urinary sediment, and coagulation studies showed no abnormalities except elevations of the erythrocyte sedimentation rate and the concentration of C-reactive protein (an acute-phase reactant). These can be normal in some patients. Renal involvement was also not present.
DIAGNOSIS
The diagnosis relies on clinical manifestations. Because Henoch-Schönlein purpura is less common in adults, biopsy plays a more important role in establishing the diagnosis in this age group, and it does this by demonstrating leukocytoclastic vasculitis with a predominance of IgA deposition under immunofluorescence. Recent studies in children showed that an elevated IgA concentration along with reduced IgM levels was associated with a higher rate of severe complications.2 However, depending on the age of the biopsied lesion, IgA may not be detected.
TREATMENT DIRECTED AT SYMPTOMS
Our patient received oral corticosteroids 0.5 mg/kg per day for 20 days, and the lesions resolved by 4 weeks.
Management of Henoch-Schönlein purpura is mainly directed at the symptoms, with oral hydration and nonsteroidal anti-inflammatory drugs. For severe cases, a short course of corticosteroids (0.5–1 mg/kg) may be used.
Although no controlled clinical trial has proven that Henoch-Schönlein purpura responds to corticosteroids, colchicine, or other drugs, corticosteroids are used most often, especially in patients with renal disease. Patients with severe renal insufficiency, abdominal pain, joint involvement, or bleeding should be hospitalized. Plasmapheresis3 has been used in severe cases.
HENOCH-SCHÖNLEIN PURPURA AND MALIGNANCY
During a follow-up evaluation 1 month later, our patient was diagnosed with adenocarcinoma of the breast. This highlights the value of a workup for cancer in adults with cutaneous vasculitis.
Cutaneous vasculitis can represent a paraneoplastic syndrome associated with a malignant tumor. The pathophysiology of this association is unclear, but one proposed mechanism is the exaggerated production of antibodies that react against tumor neoantigens, leading to the formation of immune complexes, or that occasionally recognize endothelial cells because of similarities with tumor antigens. Another theory is that abnormally high levels of inflammatory cytokines are produced by neoplastic cells or in response to decreased immune complex clearance.
Yet another theory is that hyperviscosity of the blood, seen in some cancers, increases the contact time for deposition of immune complexes and causes endothelial damage. Drugs used to treat cancer have also been reported to produce Henoch-Schönlein purpura.4
Although hematologic malignancy is three to five times more common than solid tumors in patients with small-vessel vasculitis, the disease has been associated with solid tumors of the liver, skin, colon, and breast in adults over age 40.5 An evaluation for neoplasm is therefore reasonable in adults with Henoch-Schönlein purpura, as is an evaluation for tumor recurrence or metastasis if the patient has been previously treated for a malignant tumor.
Q: Which is the most likely diagnosis?
- Idiopathic thrombocytopenic purpura
- Vitamin C deficiency (scurvy)
- Kaposi sarcoma not related to human immunodeficiency virus (HIV) infection
- Henoch-Schönlein purpura
- Polyarteritis nodosa
A: The correct answer is Henoch-Schönlein purpura.
Idiopathic thrombocytopenic purpura is an autoimmune disease caused by specific antibodies against platelet-membrane glycoproteins. It is characterized by thrombocytopenia not explainable by contact with toxic substances or by other causes. Along with nonpalpable purpura, other common signs are epistaxis, gingival bleeding, menorrhagia, and retinal hemorrhage.
Scurvy is an uncommon deficiency of ascorbic acid (vitamin C). The elderly and alcoholics are at higher risk, as they do not take in enough vitamin C in the diet. Patients usually show perifollicular hemorrhages of the skin and mucous membranes, typically petechial hemorrhage or ecchymosis of the gums around the upper incisors. Other cutaneous signs are follicular hyperkeratosis on the forearms, small corkscrew hairs, and sicca syndrome, which is more common in adults.
Non-HIV Kaposi sarcoma usually affects elderly patients, with pink, red, or brown papules or nodules on the legs and, less commonly, on the head and neck. Histopathologic examination shows newly formed irregular blood vessels with an inflammatory infiltrate of plasma cells and lymphocytes; immunohistochemical human herpes virus staining is usually positive.
Polyarteritis nodosa is a systemic vasculitis that affects medium or small arteries with necrotizing inflammation; renal glomeruli and arterioles, capillaries, and venules are unaffected. Skin manifestations include palpable purpura, livedo reticularis, ulcers, and distal gangrene. The condition also usually affects the kidneys, the heart, and the musculoskeletal and nervous systems.
A SYSTEMIC VASCULITIS
Henoch-Schönlein purpura is a systemic vasculitis affecting the skin, gastrointestinal tract, kidneys, and joints. Palpable purpura and joint pain are the most common and consistent presenting symptoms. The kidneys are affected in about one-third of children and in 60% of adults, and this is the major factor determining the long-term outcome.1
In our patient, laboratory testing that included a complete blood cell count, biochemical testing (including IgA levels), urinary sediment, and coagulation studies showed no abnormalities except elevations of the erythrocyte sedimentation rate and the concentration of C-reactive protein (an acute-phase reactant). These can be normal in some patients. Renal involvement was also not present.
DIAGNOSIS
The diagnosis relies on clinical manifestations. Because Henoch-Schönlein purpura is less common in adults, biopsy plays a more important role in establishing the diagnosis in this age group, and it does this by demonstrating leukocytoclastic vasculitis with a predominance of IgA deposition under immunofluorescence. Recent studies in children showed that an elevated IgA concentration along with reduced IgM levels was associated with a higher rate of severe complications.2 However, depending on the age of the biopsied lesion, IgA may not be detected.
TREATMENT DIRECTED AT SYMPTOMS
Our patient received oral corticosteroids 0.5 mg/kg per day for 20 days, and the lesions resolved by 4 weeks.
Management of Henoch-Schönlein purpura is mainly directed at the symptoms, with oral hydration and nonsteroidal anti-inflammatory drugs. For severe cases, a short course of corticosteroids (0.5–1 mg/kg) may be used.
Although no controlled clinical trial has proven that Henoch-Schönlein purpura responds to corticosteroids, colchicine, or other drugs, corticosteroids are used most often, especially in patients with renal disease. Patients with severe renal insufficiency, abdominal pain, joint involvement, or bleeding should be hospitalized. Plasmapheresis3 has been used in severe cases.
HENOCH-SCHÖNLEIN PURPURA AND MALIGNANCY
During a follow-up evaluation 1 month later, our patient was diagnosed with adenocarcinoma of the breast. This highlights the value of a workup for cancer in adults with cutaneous vasculitis.
Cutaneous vasculitis can represent a paraneoplastic syndrome associated with a malignant tumor. The pathophysiology of this association is unclear, but one proposed mechanism is the exaggerated production of antibodies that react against tumor neoantigens, leading to the formation of immune complexes, or that occasionally recognize endothelial cells because of similarities with tumor antigens. Another theory is that abnormally high levels of inflammatory cytokines are produced by neoplastic cells or in response to decreased immune complex clearance.
Yet another theory is that hyperviscosity of the blood, seen in some cancers, increases the contact time for deposition of immune complexes and causes endothelial damage. Drugs used to treat cancer have also been reported to produce Henoch-Schönlein purpura.4
Although hematologic malignancy is three to five times more common than solid tumors in patients with small-vessel vasculitis, the disease has been associated with solid tumors of the liver, skin, colon, and breast in adults over age 40.5 An evaluation for neoplasm is therefore reasonable in adults with Henoch-Schönlein purpura, as is an evaluation for tumor recurrence or metastasis if the patient has been previously treated for a malignant tumor.
- Rieu P, Noël LH. Henoch-Schönlein nephritis in children and adults. Morphological features and clinicopathological correlations. Ann Med Interne (Paris) 1999; 150:151–159.
- Fretzayas A, Sionti I, Moustaki M, Nicolaidou P. Clinical impact of altered immunoglobulin levels in Henoch-Schönlein purpura. Pediatr Int 2009; 51:381–384.
- Donghi D, Schanz U, Sahrbacher U, et al. Life-threatening or organimpairing Henoch-Schönlein purpura: plasmapheresis may save lives and limit organ damage. Dermatology 2009; 219:167–170.
- Mitsui H, Shibagaki N, Kawamura T, Matsue H, Shimada S. A clinical study of Henoch-Schönlein purpura associated with malignancy. J Eur Acad Dermatol Venereol 2009; 23:394–401.
- Maestri A, Malacarne P, Santini A. Henoch-Schönlein syndrome associated with breast cancer. A case report. Angiology 1995; 46:625–627.
- Rieu P, Noël LH. Henoch-Schönlein nephritis in children and adults. Morphological features and clinicopathological correlations. Ann Med Interne (Paris) 1999; 150:151–159.
- Fretzayas A, Sionti I, Moustaki M, Nicolaidou P. Clinical impact of altered immunoglobulin levels in Henoch-Schönlein purpura. Pediatr Int 2009; 51:381–384.
- Donghi D, Schanz U, Sahrbacher U, et al. Life-threatening or organimpairing Henoch-Schönlein purpura: plasmapheresis may save lives and limit organ damage. Dermatology 2009; 219:167–170.
- Mitsui H, Shibagaki N, Kawamura T, Matsue H, Shimada S. A clinical study of Henoch-Schönlein purpura associated with malignancy. J Eur Acad Dermatol Venereol 2009; 23:394–401.
- Maestri A, Malacarne P, Santini A. Henoch-Schönlein syndrome associated with breast cancer. A case report. Angiology 1995; 46:625–627.
Interpreting The JUPITER Trial: Statins can prevent VTE, but more study is needed
A major placebo-controlled trial has found that a statin can reduce the risk of venous thromboembolism (VTE).1
We do not recommend prescribing this class of drugs for this purpose until much more research has been done, and we certainly do not recommend substituting a statin for anticoagulant therapy in a patient at risk of VTE.
Nevertheless, we are excited by the latest findings, and we find comfort in knowing that if a patient is taking a statin for an approved indication, ie, reducing the risk of cardiovascular disease in a patient with hyperlipidemia or a previous cardiovascular event, the drug will also reduce the risk of VTE.
In the pages that follow, we describe and comment on what is known about the effect of statins on the risk of VTE.
ARTERIAL AND VENOUS THROMBOSIS: HOW ARE THEY LINKED?
The causes of arterial thrombosis may not be entirely distinct from those of deep vein thrombosis and pulmonary embolism, collectively referred to as VTE. Some studies have found that risk factors for arterial thrombosis overlap with those for VTE.2–4 However, other studies have shown no association between venous and arterial events.5–10
Hyperlipidemia, in particular, has been evaluated to see if it is a risk factor for VTE. As with other risk factors for arterial thrombosis, the data have been mixed, with some reports favoring an association with VTE and others not.4,5,11 Even so, preventive strategies targeting arterial risk factors have shown promise in reducing VTE events.12
Although commonly used to treat hyperlipidemia, statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are believed to reduce the incidence of thrombosis by a number of mechanisms13:
- Decreasing platelet aggregation
- Inhibiting expression of tissue factor and plasminogen activator inhibitor 1
- Increasing expression of tissue plasminogen activator
- Increasing expression of thrombomodulin, which can activate protein C and prevent thrombin-induced platelet and factor V activation and fibrinogen clotting.
STATINS AND VTE IN OBSERVATIONAL AND CASE-CONTROL STUDIES
In view of the multiple effects of statins, several studies have looked at whether these drugs reduce the occurrence of both arterial thrombosis and VTE.14–19
Two prospective observational studies and four case-control studies found that statins reduced the risk of VTE by 20% to 60%.14–19 Interestingly, two of the case-control studies found that antiplatelet therapy did not reduce the risk of VTE.18,19
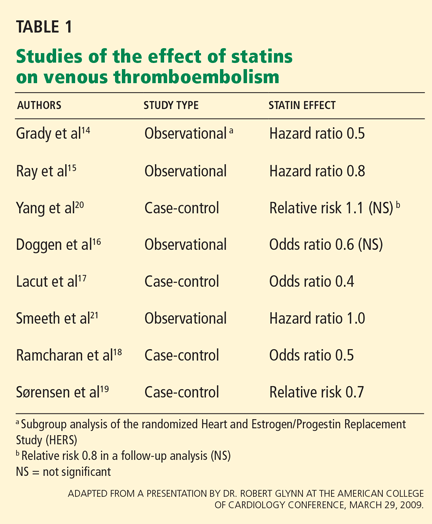
THE JUPITER STUDY
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study primarily sought to determine if rosuvastatin (Crestor) 20 mg/day, compared with placebo, would reduce the rate of first major cardiovascular events.22 A prespecified secondary end point of the trial was VTE, making JUPITER the first randomized, placebo-controlled trial to specifically test whether statins prevent VTE.1
Inclusion criteria: Normal LDL, high CRP
The study included men age 50 and older and women age 60 and older with no history of cardiovascular disease. In addition, their lowdensity lipoprotein (LDL) cholesterol levels had to be lower than 130 mg/dL (3.4 mmol/L), their triglyceride levels had to be lower than 500 mg/dL (5.6 mmol/L), and their highsensitivity C-reactive protein (hs-CRP) levels had to be 2.0 mg/L or higher.
Since high levels of hs-CRP, a marker of inflammation, predict cardiovascular events and since statins lower hs-CRP levels, the investigators hypothesized that people with elevated hs-CRP but without hyperlipidemia might benefit from statin treatment.21
Patients were excluded if they had received lipid-lowering therapy within 6 weeks of the trial screening, had diabetes mellitus or uncontrolled hypertension, were currently using postmenopausal hormone-replacement therapy, or had had cancer within the previous 5 years, except for certain skin cancers.
Candidates who complied well during a 4-week placebo run-in phase were randomly assigned to receive either rosuvastatin 20 mg daily (an intermediate dose) or a matching placebo. In all, 17,802 people were randomized. The two assigned groups appeared to be well matched.
Patients were to come in for visits twice a year for 60 months after randomization to be assessed for symptomatic deep venous thrombosis and pulmonary embolism. New cases of VTE were confirmed by imaging studies, by the initiation of anticoagulation therapy, or by death ascribed to pulmonary embolism.
Idiopathic VTE was classified as unprovoked if it occurred in the absence of trauma, hospitalization, or surgery within 3 months before the event, and in the absence of any diagnosed cancer within 3 months before and after the event. Provoked VTE events were those that occurred in a participant with cancer or when a precipitating event was associated with trauma, hospitalization, or surgery.
Rosuvastatin prevents heart attack, stroke
On the recommendation of the trial’s independent data and safety monitoring board, JUPITER was stopped early because the trial drug showed evidence of efficacy in preventing the combined primary end point of a first major cardiovascular event—ie, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from a cardiovascular cause.22 (The cardiovascular outcomes of the JUPITER study were reviewed by Shishehbor and Hazen23 in the January 2009 issue of the Cleveland Clinic Journal of Medicine; see doi:10.3949/ccjm.75a.08105).
Formal follow-up for the trial's primary and secondary efficacy end points ended then, but data on VTE continued to be collected until each patient’s closeout visit as part of a safety monitoring protocol. The last closeout visit occurred on August 20, 2008. The primary analysis focused on events occurring up to March 30, 2008, the date the study was stopped.
Secondary end point results: Rosuvastatin prevents VTE
At a median follow-up of 1.9 years, an episode of VTE had occurred in 94 (0.53%) of the 17,802 patients—34 in the rosuvastatin group and 60 in the placebo group.1 This translates to 0.18 and 0.32 events per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio for the rosuvastatin group 0.57, 95% confidence interval [CI] 0.37–0.86, P = .007).
Forty-four cases of VTE were classified as provoked and 50 cases were categorized as unprovoked. The risk reduction was statistically significant for provoked cases (hazard ratio 0.52, 95% CI 0.28–0.96, P = .03), but not for unprovoked events (hazard ratio 0.61, 95% CI 0.35–1.09, P = .09).
Subgroup analysis revealed no significant association between patient characteristics and the impact of rosuvastatin on the risk of a VTE event, but, as expected, more benefit was associated with higher baseline lipid levels.
STILL TOO SOON TO ADVISE ROUTINE STATIN USE TO PREVENT VTE
While the JUPITER trial data show an apparent benefit of statin use on the rate of VTE events, advising routine use of statins to prevent VTE is premature, for three main reasons.
Many must be treated to prevent one case of VTE. The number needed to treat (NNT) with rosuvastatin for 5 years to prevent either a case of VTE or a cardiovascular event was 21, and the NNT to prevent one cardiovascular event was 25. In a review of the two most recent case-control studies investigating the effects of statins on VTE,18,19 Cushman24 calculated that the NNT to prevent one VTE event each year was 333 for those age 75 and older. Though the Jupiter data did not provide the specific incidence of VTE at 1 year, except graphically, we can estimate that the NNT to prevent one VTE event at 1 year in the study is also very high.
Practically speaking, the perceived benefits of VTE prevention require large numbers to be treated, and the net clinical gain is still largely in preventing arterial events such as heart attack and stroke rather than VTE.
Statins, though safe, can still have adverse effects. The JUPITER study found a trend (albeit nonsignificant) toward more muscle complaints and elevations on liver function testing in apparently healthy persons taking a statin.22 Although severe complications of statin therapy such as rhabdomyolysis and elevations of creatine phosphokinase are rare, patients taking a statin have a 39% higher risk of an adverse event, most commonly myalgias or abnormalities on liver function testing.25 Were statins to be given routinely to even more people than they are now, more adverse outcomes would be likely.
More study is needed. The JUPITER study did not address a high risk of VTE. In fact, the investigators provided no information as to the VTE history of those enrolled.
Clearly, statins should not be substituted for proven prophylaxis and anticoagulation without further investigation, especially for patients with recurrent deep venous thrombosis, hospitalized patients, postoperative patients, and other patients prone to VTE.
OUR VIEW
The JUPITER study is an important leap forward in adding to our knowledge of how to prevent VTE. For people with another indication for taking a statin (eg, a previous cardiovascular event, hyperlipidemia), it is helpful to know that their risk of VTE may be reduced without exposure to the risks of other kinds of conventional thromboprophylaxis.
We look forward to additional studies to elaborate on the benefits of statins in both the prevention and treatment of VTE for averagerisk and VTE-prone populations.
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med 2009; 360:1851–1861.
- Prandoni P, Bilora F, Marchiori A, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med 2003; 348:1435–1441.
- Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost 2006; 4:1891–1896.
- Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Stormer J, Hansen JB. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost 2008; 6:1851–1857.
- Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med 2002; 162:1182–1189.
- van der Hagen PB, Folsom AR, Jenny NS, et al. Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost 2006; 4:1903–1908.
- Reich LM, Folsom AR, Key NS, et al. Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost 2006; 4:1909–1913.
- Huerta C, Johansson S, Wallander MA, Rodriguez LA. Risk of myocardial infarction and overall mortality in survivors of venous thromboembolism. Thromb J 2008; 6:10.
- Linnemann B, Schindewolf M, Zgouras D, Erbe M, Jarosch-Preusche M, Lindhoff-Last E. Are patients with thrombophilia and previous venous thromboembolism at higher risk to arterial thrombosis? Thromb Res 2008; 121:743–750.
- Schwaiger J, Kiechl S, Stockner H, et al. Burden of atherosclerosis and risk of venous thromboembolism in patients with migraine. Neurology 2008; 71:937–943.
- Linnemann B, Zgouras D, Schindewolf M, Schwonberg J, Jarosch-Preusche M, Lindhoff-Last E. Impact of sex and traditional cardiovascular risk factors on the risk of recurrent venous thromboembolism: results from the German MAISTHRO Registry. Blood Coagul Fibrinolysis 2008; 19:159–165.
- Steffen LM, Folsom AR, Cushman M, Jacobs DR, Rosamond WD. Greater fish, fruit, and vegetable intakes are related to lower incidence of venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology. Circulation 2007; 115:188–195.
- Arslan F, Pasterkamp G, de Kleijn DP. Unraveling pleiotropic effects of statins: bit by bit, a slow case with perspective. Circ Res 2008; 103:334–336.
- Grady D, Wenger NK, Herrington D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease. The Heart and Estrogen/progestin Replacement Study. Ann Intern Med 2000; 132:689–696.
- Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL, Laupacis A. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med 2001; 161:1405–1410.
- Doggen CJ, Lemaitre RN, Smith NL, Heckbert SR, Psaty BM. HMG CoA reductase inhibitors and the risk of venous thrombosis among postmenopausal women. J Thromb Haemost 2004; 2:700–701.
- Lacut K, Oger E, Le Gal G, et al. Statins but not fibrates are associated with a reduced risk of venous thromboembolism: a hospitalbased case-control study. Fundam Clin Pharmacol 2004; 18:477–482.
- Ramcharan AS, Van Stralen KJ, Snoep JD, Mantel-Teeuwisse AK, Rosendaal FR, Doggen CJ. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J Thromb Haemost 2009; 7:514–520.
- Sørensen HT, Horvath-Puho E, Sogaard KK, et al. Arterial cardiovascular events, statins, low-dose aspirin and subsequent risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost 2009; 7:521–528.
- Yang CC, Jick SS, Jick H. Statins and the risk of idiopathic venous thromboembolism. Br J Clin Pharmacol 2002; 53:101–105.
- Smeeth L, Douglas I, Hall AJ, Hubbard R, Evans S. Effect of statins on a wide range of health outcomes: a cohort study validated by comparison with randomized trials. Br J Clin Pharmacol 2009; 67:99–109.
- Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shishehbor MH, Hazen SL. JUPITER to Earth: A statin helps peole with normal LDL-C and high hs-CRP, but what does it mean? Cleve Clin J Med 2009; 76:37–44.
- Cushman M. A new indication for statins to prevent venous thromboembolism? Not yet. J Thromb Haemost 2009; 7:511–513.
- Silva MA, Swanson AC, Gandhi PJ, Tataronis GR. Statin-related adverse events: a meta-analysis. Clin Ther 2006; 28:26–35.
A major placebo-controlled trial has found that a statin can reduce the risk of venous thromboembolism (VTE).1
We do not recommend prescribing this class of drugs for this purpose until much more research has been done, and we certainly do not recommend substituting a statin for anticoagulant therapy in a patient at risk of VTE.
Nevertheless, we are excited by the latest findings, and we find comfort in knowing that if a patient is taking a statin for an approved indication, ie, reducing the risk of cardiovascular disease in a patient with hyperlipidemia or a previous cardiovascular event, the drug will also reduce the risk of VTE.
In the pages that follow, we describe and comment on what is known about the effect of statins on the risk of VTE.
ARTERIAL AND VENOUS THROMBOSIS: HOW ARE THEY LINKED?
The causes of arterial thrombosis may not be entirely distinct from those of deep vein thrombosis and pulmonary embolism, collectively referred to as VTE. Some studies have found that risk factors for arterial thrombosis overlap with those for VTE.2–4 However, other studies have shown no association between venous and arterial events.5–10
Hyperlipidemia, in particular, has been evaluated to see if it is a risk factor for VTE. As with other risk factors for arterial thrombosis, the data have been mixed, with some reports favoring an association with VTE and others not.4,5,11 Even so, preventive strategies targeting arterial risk factors have shown promise in reducing VTE events.12
Although commonly used to treat hyperlipidemia, statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are believed to reduce the incidence of thrombosis by a number of mechanisms13:
- Decreasing platelet aggregation
- Inhibiting expression of tissue factor and plasminogen activator inhibitor 1
- Increasing expression of tissue plasminogen activator
- Increasing expression of thrombomodulin, which can activate protein C and prevent thrombin-induced platelet and factor V activation and fibrinogen clotting.
STATINS AND VTE IN OBSERVATIONAL AND CASE-CONTROL STUDIES
In view of the multiple effects of statins, several studies have looked at whether these drugs reduce the occurrence of both arterial thrombosis and VTE.14–19
Two prospective observational studies and four case-control studies found that statins reduced the risk of VTE by 20% to 60%.14–19 Interestingly, two of the case-control studies found that antiplatelet therapy did not reduce the risk of VTE.18,19
THE JUPITER STUDY
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study primarily sought to determine if rosuvastatin (Crestor) 20 mg/day, compared with placebo, would reduce the rate of first major cardiovascular events.22 A prespecified secondary end point of the trial was VTE, making JUPITER the first randomized, placebo-controlled trial to specifically test whether statins prevent VTE.1
Inclusion criteria: Normal LDL, high CRP
The study included men age 50 and older and women age 60 and older with no history of cardiovascular disease. In addition, their lowdensity lipoprotein (LDL) cholesterol levels had to be lower than 130 mg/dL (3.4 mmol/L), their triglyceride levels had to be lower than 500 mg/dL (5.6 mmol/L), and their highsensitivity C-reactive protein (hs-CRP) levels had to be 2.0 mg/L or higher.
Since high levels of hs-CRP, a marker of inflammation, predict cardiovascular events and since statins lower hs-CRP levels, the investigators hypothesized that people with elevated hs-CRP but without hyperlipidemia might benefit from statin treatment.21
Patients were excluded if they had received lipid-lowering therapy within 6 weeks of the trial screening, had diabetes mellitus or uncontrolled hypertension, were currently using postmenopausal hormone-replacement therapy, or had had cancer within the previous 5 years, except for certain skin cancers.
Candidates who complied well during a 4-week placebo run-in phase were randomly assigned to receive either rosuvastatin 20 mg daily (an intermediate dose) or a matching placebo. In all, 17,802 people were randomized. The two assigned groups appeared to be well matched.
Patients were to come in for visits twice a year for 60 months after randomization to be assessed for symptomatic deep venous thrombosis and pulmonary embolism. New cases of VTE were confirmed by imaging studies, by the initiation of anticoagulation therapy, or by death ascribed to pulmonary embolism.
Idiopathic VTE was classified as unprovoked if it occurred in the absence of trauma, hospitalization, or surgery within 3 months before the event, and in the absence of any diagnosed cancer within 3 months before and after the event. Provoked VTE events were those that occurred in a participant with cancer or when a precipitating event was associated with trauma, hospitalization, or surgery.
Rosuvastatin prevents heart attack, stroke
On the recommendation of the trial’s independent data and safety monitoring board, JUPITER was stopped early because the trial drug showed evidence of efficacy in preventing the combined primary end point of a first major cardiovascular event—ie, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from a cardiovascular cause.22 (The cardiovascular outcomes of the JUPITER study were reviewed by Shishehbor and Hazen23 in the January 2009 issue of the Cleveland Clinic Journal of Medicine; see doi:10.3949/ccjm.75a.08105).
Formal follow-up for the trial's primary and secondary efficacy end points ended then, but data on VTE continued to be collected until each patient’s closeout visit as part of a safety monitoring protocol. The last closeout visit occurred on August 20, 2008. The primary analysis focused on events occurring up to March 30, 2008, the date the study was stopped.
Secondary end point results: Rosuvastatin prevents VTE
At a median follow-up of 1.9 years, an episode of VTE had occurred in 94 (0.53%) of the 17,802 patients—34 in the rosuvastatin group and 60 in the placebo group.1 This translates to 0.18 and 0.32 events per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio for the rosuvastatin group 0.57, 95% confidence interval [CI] 0.37–0.86, P = .007).
Forty-four cases of VTE were classified as provoked and 50 cases were categorized as unprovoked. The risk reduction was statistically significant for provoked cases (hazard ratio 0.52, 95% CI 0.28–0.96, P = .03), but not for unprovoked events (hazard ratio 0.61, 95% CI 0.35–1.09, P = .09).
Subgroup analysis revealed no significant association between patient characteristics and the impact of rosuvastatin on the risk of a VTE event, but, as expected, more benefit was associated with higher baseline lipid levels.
STILL TOO SOON TO ADVISE ROUTINE STATIN USE TO PREVENT VTE
While the JUPITER trial data show an apparent benefit of statin use on the rate of VTE events, advising routine use of statins to prevent VTE is premature, for three main reasons.
Many must be treated to prevent one case of VTE. The number needed to treat (NNT) with rosuvastatin for 5 years to prevent either a case of VTE or a cardiovascular event was 21, and the NNT to prevent one cardiovascular event was 25. In a review of the two most recent case-control studies investigating the effects of statins on VTE,18,19 Cushman24 calculated that the NNT to prevent one VTE event each year was 333 for those age 75 and older. Though the Jupiter data did not provide the specific incidence of VTE at 1 year, except graphically, we can estimate that the NNT to prevent one VTE event at 1 year in the study is also very high.
Practically speaking, the perceived benefits of VTE prevention require large numbers to be treated, and the net clinical gain is still largely in preventing arterial events such as heart attack and stroke rather than VTE.
Statins, though safe, can still have adverse effects. The JUPITER study found a trend (albeit nonsignificant) toward more muscle complaints and elevations on liver function testing in apparently healthy persons taking a statin.22 Although severe complications of statin therapy such as rhabdomyolysis and elevations of creatine phosphokinase are rare, patients taking a statin have a 39% higher risk of an adverse event, most commonly myalgias or abnormalities on liver function testing.25 Were statins to be given routinely to even more people than they are now, more adverse outcomes would be likely.
More study is needed. The JUPITER study did not address a high risk of VTE. In fact, the investigators provided no information as to the VTE history of those enrolled.
Clearly, statins should not be substituted for proven prophylaxis and anticoagulation without further investigation, especially for patients with recurrent deep venous thrombosis, hospitalized patients, postoperative patients, and other patients prone to VTE.
OUR VIEW
The JUPITER study is an important leap forward in adding to our knowledge of how to prevent VTE. For people with another indication for taking a statin (eg, a previous cardiovascular event, hyperlipidemia), it is helpful to know that their risk of VTE may be reduced without exposure to the risks of other kinds of conventional thromboprophylaxis.
We look forward to additional studies to elaborate on the benefits of statins in both the prevention and treatment of VTE for averagerisk and VTE-prone populations.
A major placebo-controlled trial has found that a statin can reduce the risk of venous thromboembolism (VTE).1
We do not recommend prescribing this class of drugs for this purpose until much more research has been done, and we certainly do not recommend substituting a statin for anticoagulant therapy in a patient at risk of VTE.
Nevertheless, we are excited by the latest findings, and we find comfort in knowing that if a patient is taking a statin for an approved indication, ie, reducing the risk of cardiovascular disease in a patient with hyperlipidemia or a previous cardiovascular event, the drug will also reduce the risk of VTE.
In the pages that follow, we describe and comment on what is known about the effect of statins on the risk of VTE.
ARTERIAL AND VENOUS THROMBOSIS: HOW ARE THEY LINKED?
The causes of arterial thrombosis may not be entirely distinct from those of deep vein thrombosis and pulmonary embolism, collectively referred to as VTE. Some studies have found that risk factors for arterial thrombosis overlap with those for VTE.2–4 However, other studies have shown no association between venous and arterial events.5–10
Hyperlipidemia, in particular, has been evaluated to see if it is a risk factor for VTE. As with other risk factors for arterial thrombosis, the data have been mixed, with some reports favoring an association with VTE and others not.4,5,11 Even so, preventive strategies targeting arterial risk factors have shown promise in reducing VTE events.12
Although commonly used to treat hyperlipidemia, statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) are believed to reduce the incidence of thrombosis by a number of mechanisms13:
- Decreasing platelet aggregation
- Inhibiting expression of tissue factor and plasminogen activator inhibitor 1
- Increasing expression of tissue plasminogen activator
- Increasing expression of thrombomodulin, which can activate protein C and prevent thrombin-induced platelet and factor V activation and fibrinogen clotting.
STATINS AND VTE IN OBSERVATIONAL AND CASE-CONTROL STUDIES
In view of the multiple effects of statins, several studies have looked at whether these drugs reduce the occurrence of both arterial thrombosis and VTE.14–19
Two prospective observational studies and four case-control studies found that statins reduced the risk of VTE by 20% to 60%.14–19 Interestingly, two of the case-control studies found that antiplatelet therapy did not reduce the risk of VTE.18,19
THE JUPITER STUDY
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study primarily sought to determine if rosuvastatin (Crestor) 20 mg/day, compared with placebo, would reduce the rate of first major cardiovascular events.22 A prespecified secondary end point of the trial was VTE, making JUPITER the first randomized, placebo-controlled trial to specifically test whether statins prevent VTE.1
Inclusion criteria: Normal LDL, high CRP
The study included men age 50 and older and women age 60 and older with no history of cardiovascular disease. In addition, their lowdensity lipoprotein (LDL) cholesterol levels had to be lower than 130 mg/dL (3.4 mmol/L), their triglyceride levels had to be lower than 500 mg/dL (5.6 mmol/L), and their highsensitivity C-reactive protein (hs-CRP) levels had to be 2.0 mg/L or higher.
Since high levels of hs-CRP, a marker of inflammation, predict cardiovascular events and since statins lower hs-CRP levels, the investigators hypothesized that people with elevated hs-CRP but without hyperlipidemia might benefit from statin treatment.21
Patients were excluded if they had received lipid-lowering therapy within 6 weeks of the trial screening, had diabetes mellitus or uncontrolled hypertension, were currently using postmenopausal hormone-replacement therapy, or had had cancer within the previous 5 years, except for certain skin cancers.
Candidates who complied well during a 4-week placebo run-in phase were randomly assigned to receive either rosuvastatin 20 mg daily (an intermediate dose) or a matching placebo. In all, 17,802 people were randomized. The two assigned groups appeared to be well matched.
Patients were to come in for visits twice a year for 60 months after randomization to be assessed for symptomatic deep venous thrombosis and pulmonary embolism. New cases of VTE were confirmed by imaging studies, by the initiation of anticoagulation therapy, or by death ascribed to pulmonary embolism.
Idiopathic VTE was classified as unprovoked if it occurred in the absence of trauma, hospitalization, or surgery within 3 months before the event, and in the absence of any diagnosed cancer within 3 months before and after the event. Provoked VTE events were those that occurred in a participant with cancer or when a precipitating event was associated with trauma, hospitalization, or surgery.
Rosuvastatin prevents heart attack, stroke
On the recommendation of the trial’s independent data and safety monitoring board, JUPITER was stopped early because the trial drug showed evidence of efficacy in preventing the combined primary end point of a first major cardiovascular event—ie, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from a cardiovascular cause.22 (The cardiovascular outcomes of the JUPITER study were reviewed by Shishehbor and Hazen23 in the January 2009 issue of the Cleveland Clinic Journal of Medicine; see doi:10.3949/ccjm.75a.08105).
Formal follow-up for the trial's primary and secondary efficacy end points ended then, but data on VTE continued to be collected until each patient’s closeout visit as part of a safety monitoring protocol. The last closeout visit occurred on August 20, 2008. The primary analysis focused on events occurring up to March 30, 2008, the date the study was stopped.
Secondary end point results: Rosuvastatin prevents VTE
At a median follow-up of 1.9 years, an episode of VTE had occurred in 94 (0.53%) of the 17,802 patients—34 in the rosuvastatin group and 60 in the placebo group.1 This translates to 0.18 and 0.32 events per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively (hazard ratio for the rosuvastatin group 0.57, 95% confidence interval [CI] 0.37–0.86, P = .007).
Forty-four cases of VTE were classified as provoked and 50 cases were categorized as unprovoked. The risk reduction was statistically significant for provoked cases (hazard ratio 0.52, 95% CI 0.28–0.96, P = .03), but not for unprovoked events (hazard ratio 0.61, 95% CI 0.35–1.09, P = .09).
Subgroup analysis revealed no significant association between patient characteristics and the impact of rosuvastatin on the risk of a VTE event, but, as expected, more benefit was associated with higher baseline lipid levels.
STILL TOO SOON TO ADVISE ROUTINE STATIN USE TO PREVENT VTE
While the JUPITER trial data show an apparent benefit of statin use on the rate of VTE events, advising routine use of statins to prevent VTE is premature, for three main reasons.
Many must be treated to prevent one case of VTE. The number needed to treat (NNT) with rosuvastatin for 5 years to prevent either a case of VTE or a cardiovascular event was 21, and the NNT to prevent one cardiovascular event was 25. In a review of the two most recent case-control studies investigating the effects of statins on VTE,18,19 Cushman24 calculated that the NNT to prevent one VTE event each year was 333 for those age 75 and older. Though the Jupiter data did not provide the specific incidence of VTE at 1 year, except graphically, we can estimate that the NNT to prevent one VTE event at 1 year in the study is also very high.
Practically speaking, the perceived benefits of VTE prevention require large numbers to be treated, and the net clinical gain is still largely in preventing arterial events such as heart attack and stroke rather than VTE.
Statins, though safe, can still have adverse effects. The JUPITER study found a trend (albeit nonsignificant) toward more muscle complaints and elevations on liver function testing in apparently healthy persons taking a statin.22 Although severe complications of statin therapy such as rhabdomyolysis and elevations of creatine phosphokinase are rare, patients taking a statin have a 39% higher risk of an adverse event, most commonly myalgias or abnormalities on liver function testing.25 Were statins to be given routinely to even more people than they are now, more adverse outcomes would be likely.
More study is needed. The JUPITER study did not address a high risk of VTE. In fact, the investigators provided no information as to the VTE history of those enrolled.
Clearly, statins should not be substituted for proven prophylaxis and anticoagulation without further investigation, especially for patients with recurrent deep venous thrombosis, hospitalized patients, postoperative patients, and other patients prone to VTE.
OUR VIEW
The JUPITER study is an important leap forward in adding to our knowledge of how to prevent VTE. For people with another indication for taking a statin (eg, a previous cardiovascular event, hyperlipidemia), it is helpful to know that their risk of VTE may be reduced without exposure to the risks of other kinds of conventional thromboprophylaxis.
We look forward to additional studies to elaborate on the benefits of statins in both the prevention and treatment of VTE for averagerisk and VTE-prone populations.
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med 2009; 360:1851–1861.
- Prandoni P, Bilora F, Marchiori A, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med 2003; 348:1435–1441.
- Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost 2006; 4:1891–1896.
- Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Stormer J, Hansen JB. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost 2008; 6:1851–1857.
- Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med 2002; 162:1182–1189.
- van der Hagen PB, Folsom AR, Jenny NS, et al. Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost 2006; 4:1903–1908.
- Reich LM, Folsom AR, Key NS, et al. Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost 2006; 4:1909–1913.
- Huerta C, Johansson S, Wallander MA, Rodriguez LA. Risk of myocardial infarction and overall mortality in survivors of venous thromboembolism. Thromb J 2008; 6:10.
- Linnemann B, Schindewolf M, Zgouras D, Erbe M, Jarosch-Preusche M, Lindhoff-Last E. Are patients with thrombophilia and previous venous thromboembolism at higher risk to arterial thrombosis? Thromb Res 2008; 121:743–750.
- Schwaiger J, Kiechl S, Stockner H, et al. Burden of atherosclerosis and risk of venous thromboembolism in patients with migraine. Neurology 2008; 71:937–943.
- Linnemann B, Zgouras D, Schindewolf M, Schwonberg J, Jarosch-Preusche M, Lindhoff-Last E. Impact of sex and traditional cardiovascular risk factors on the risk of recurrent venous thromboembolism: results from the German MAISTHRO Registry. Blood Coagul Fibrinolysis 2008; 19:159–165.
- Steffen LM, Folsom AR, Cushman M, Jacobs DR, Rosamond WD. Greater fish, fruit, and vegetable intakes are related to lower incidence of venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology. Circulation 2007; 115:188–195.
- Arslan F, Pasterkamp G, de Kleijn DP. Unraveling pleiotropic effects of statins: bit by bit, a slow case with perspective. Circ Res 2008; 103:334–336.
- Grady D, Wenger NK, Herrington D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease. The Heart and Estrogen/progestin Replacement Study. Ann Intern Med 2000; 132:689–696.
- Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL, Laupacis A. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med 2001; 161:1405–1410.
- Doggen CJ, Lemaitre RN, Smith NL, Heckbert SR, Psaty BM. HMG CoA reductase inhibitors and the risk of venous thrombosis among postmenopausal women. J Thromb Haemost 2004; 2:700–701.
- Lacut K, Oger E, Le Gal G, et al. Statins but not fibrates are associated with a reduced risk of venous thromboembolism: a hospitalbased case-control study. Fundam Clin Pharmacol 2004; 18:477–482.
- Ramcharan AS, Van Stralen KJ, Snoep JD, Mantel-Teeuwisse AK, Rosendaal FR, Doggen CJ. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J Thromb Haemost 2009; 7:514–520.
- Sørensen HT, Horvath-Puho E, Sogaard KK, et al. Arterial cardiovascular events, statins, low-dose aspirin and subsequent risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost 2009; 7:521–528.
- Yang CC, Jick SS, Jick H. Statins and the risk of idiopathic venous thromboembolism. Br J Clin Pharmacol 2002; 53:101–105.
- Smeeth L, Douglas I, Hall AJ, Hubbard R, Evans S. Effect of statins on a wide range of health outcomes: a cohort study validated by comparison with randomized trials. Br J Clin Pharmacol 2009; 67:99–109.
- Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shishehbor MH, Hazen SL. JUPITER to Earth: A statin helps peole with normal LDL-C and high hs-CRP, but what does it mean? Cleve Clin J Med 2009; 76:37–44.
- Cushman M. A new indication for statins to prevent venous thromboembolism? Not yet. J Thromb Haemost 2009; 7:511–513.
- Silva MA, Swanson AC, Gandhi PJ, Tataronis GR. Statin-related adverse events: a meta-analysis. Clin Ther 2006; 28:26–35.
- Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med 2009; 360:1851–1861.
- Prandoni P, Bilora F, Marchiori A, et al. An association between atherosclerosis and venous thrombosis. N Engl J Med 2003; 348:1435–1441.
- Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost 2006; 4:1891–1896.
- Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Stormer J, Hansen JB. Family history of myocardial infarction is an independent risk factor for venous thromboembolism: the Tromso study. J Thromb Haemost 2008; 6:1851–1857.
- Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: the longitudinal investigation of thromboembolism etiology. Arch Intern Med 2002; 162:1182–1189.
- van der Hagen PB, Folsom AR, Jenny NS, et al. Subclinical atherosclerosis and the risk of future venous thrombosis in the Cardiovascular Health Study. J Thromb Haemost 2006; 4:1903–1908.
- Reich LM, Folsom AR, Key NS, et al. Prospective study of subclinical atherosclerosis as a risk factor for venous thromboembolism. J Thromb Haemost 2006; 4:1909–1913.
- Huerta C, Johansson S, Wallander MA, Rodriguez LA. Risk of myocardial infarction and overall mortality in survivors of venous thromboembolism. Thromb J 2008; 6:10.
- Linnemann B, Schindewolf M, Zgouras D, Erbe M, Jarosch-Preusche M, Lindhoff-Last E. Are patients with thrombophilia and previous venous thromboembolism at higher risk to arterial thrombosis? Thromb Res 2008; 121:743–750.
- Schwaiger J, Kiechl S, Stockner H, et al. Burden of atherosclerosis and risk of venous thromboembolism in patients with migraine. Neurology 2008; 71:937–943.
- Linnemann B, Zgouras D, Schindewolf M, Schwonberg J, Jarosch-Preusche M, Lindhoff-Last E. Impact of sex and traditional cardiovascular risk factors on the risk of recurrent venous thromboembolism: results from the German MAISTHRO Registry. Blood Coagul Fibrinolysis 2008; 19:159–165.
- Steffen LM, Folsom AR, Cushman M, Jacobs DR, Rosamond WD. Greater fish, fruit, and vegetable intakes are related to lower incidence of venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology. Circulation 2007; 115:188–195.
- Arslan F, Pasterkamp G, de Kleijn DP. Unraveling pleiotropic effects of statins: bit by bit, a slow case with perspective. Circ Res 2008; 103:334–336.
- Grady D, Wenger NK, Herrington D, et al. Postmenopausal hormone therapy increases risk for venous thromboembolic disease. The Heart and Estrogen/progestin Replacement Study. Ann Intern Med 2000; 132:689–696.
- Ray JG, Mamdani M, Tsuyuki RT, Anderson DR, Yeo EL, Laupacis A. Use of statins and the subsequent development of deep vein thrombosis. Arch Intern Med 2001; 161:1405–1410.
- Doggen CJ, Lemaitre RN, Smith NL, Heckbert SR, Psaty BM. HMG CoA reductase inhibitors and the risk of venous thrombosis among postmenopausal women. J Thromb Haemost 2004; 2:700–701.
- Lacut K, Oger E, Le Gal G, et al. Statins but not fibrates are associated with a reduced risk of venous thromboembolism: a hospitalbased case-control study. Fundam Clin Pharmacol 2004; 18:477–482.
- Ramcharan AS, Van Stralen KJ, Snoep JD, Mantel-Teeuwisse AK, Rosendaal FR, Doggen CJ. HMG-CoA reductase inhibitors, other lipid-lowering medication, antiplatelet therapy, and the risk of venous thrombosis. J Thromb Haemost 2009; 7:514–520.
- Sørensen HT, Horvath-Puho E, Sogaard KK, et al. Arterial cardiovascular events, statins, low-dose aspirin and subsequent risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost 2009; 7:521–528.
- Yang CC, Jick SS, Jick H. Statins and the risk of idiopathic venous thromboembolism. Br J Clin Pharmacol 2002; 53:101–105.
- Smeeth L, Douglas I, Hall AJ, Hubbard R, Evans S. Effect of statins on a wide range of health outcomes: a cohort study validated by comparison with randomized trials. Br J Clin Pharmacol 2009; 67:99–109.
- Ridker PM, Danielson E, Fonseca FA, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shishehbor MH, Hazen SL. JUPITER to Earth: A statin helps peole with normal LDL-C and high hs-CRP, but what does it mean? Cleve Clin J Med 2009; 76:37–44.
- Cushman M. A new indication for statins to prevent venous thromboembolism? Not yet. J Thromb Haemost 2009; 7:511–513.
- Silva MA, Swanson AC, Gandhi PJ, Tataronis GR. Statin-related adverse events: a meta-analysis. Clin Ther 2006; 28:26–35.
KEY POINTS
- Risk factors for VTE overlap with those for arterial thrombosis, although the data are mixed.
- The statin drugs have a number of effects on factors other than lipid levels, notably on markers of inflammation and on clotting factors.
- In the JUPITER trial, the incidence of VTE in people taking rosuvastatin (Crestor) 20 mg/day was about half that in people taking placebo. This was a relatively healthy population, and the incidence in both groups was low.
- Further study is needed in patients at risk of VTE.
Stenting for atherosclerotic renal artery stenosis: One poorly designed trial after another
The role of stenting for atherosclerotic renal artery stenosis is hotly debated among different specialties.1,2 If we may generalize a bit, interventionalists (cardiologists, interventional radiologists, vascular surgeons, and vascular medicine specialists) have been in favor of liberal use of stenting, and nephrologists often favor medical therapy alone. And as with all controversial issues, each group feels rather strongly about its position.
Because few prospective randomized trials have been completed, the management of atherosclerotic renal artery stenosis has been guided by retrospective studies and case series. 3
In this issue of the Cleveland Clinic Journal of Medicine, Dr. James Simon4 provides an excellent overview of the prevalence, natural history, and clinical presentation of atherosclerotic renal artery stenosis. In addition, he does an admirable job of reviewing the available prospective randomized trials and providing editorial commentary about the role of the various specialists in the management of renal artery disease. And while the title of his paper says that it is “time to be less aggressive,” Dr. Simon ultimately comes to the same conclusions that we do5 on the indications for renal artery stenting (see Table 3 of Dr. Simon’s article), which are those of the multidisciplinary 2006 American College of Cardiology/American Heart Association guidelines on the management of peripheral artery disease.3
So what then is all the controversy about? We all agree that prospective randomized trials that provide class I, level A evidence impart the only unbiased scientific information on the best treatment strategy for patients with renal artery disease. The basic controversial issue is the interpretation of these trials. We contend that the three randomized trials of stenting vs medical therapy published so far6–8 (see below) are so seriously flawed that it is impossible to make treatment decisions based on their results.
Since these trials were published in wellrespected journals, their results are often taken as gospel. However, careful review of each of these will reveal the flaws in study design and implementation.
THE DRASTIC TRIAL
In the Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) trial,6 106 patients with renal artery stenosis and hypertension (diastolic blood pressure > 95 mm Hg) despite treatment with two antihypertensive medications were randomly assigned to either renal angioplasty (n = 56) or drug therapy (n = 50).
Authors’ conclusions
“In the treatment of patients with hypertension and renal-artery stenosis, angioplasty has little advantage over antihypertensive-drug therapy.”6
Four serious problems
As we discussed in a letter to the editor of the New England Journal of Medicine on August 10, 2000, this study had four serious problems that invalidate its authors’ conclusions.9
The sample size was insufficient to detect a significant difference between treatment groups. In other words, the chance of a type 2 statistical error is high.
Balloon angioplasty without stenting was used as the method of revascularization. Experts now recognize that stenting is required for renal artery intervention to have a durable result.3,5
Renal artery stenosis was defined as greater than 50% stenosis. This allowed a large number of patients to enter the trial who had hemodynamically and clinically insignificant lesions. Most clinicians believe that stenosis of less than 70% is not hemodynamically important.5,10,11
Twenty-two of the 50 patients randomized to medical therapy crossed over to the angioplasty group because their blood pressure became difficult to control. In other words, 44% of the patients in the medical group underwent angioplasty, an astounding percentage in an intention-to-treat analysis comparing one therapy with another.
Despite these serious flaws, the results of DRASTIC influenced therapy for years after its publication.
THE STAR TRIAL
In the Stent Placement in Patients With Atherosclerotic Renal Artery Stenosis and Impaired Renal Function (STAR) trial,7 140 patients with a creatinine clearance of less than 80 mL/min/1.73m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy (n = 64) or medical therapy alone (n = 76). The primary end point was a 20% or greater decrease in creatinine clearance. Secondary end points included measures of safety and cardiovascular morbidity and mortality.
Authors’ conclusions
“Stent placement with medical treatment had no clear effect on progression of impaired renal function but led to a small number of significant procedure-related complications. The study findings favor a conservative approach to patients with [atherosclerotic renal artery stenosis], focused on cardiovascular risk factor management and avoiding stenting.”7
Serious flaws
A number of serious flaws render this study uninterpretable.
Mild renal artery stenosis. At least 33% of the patients in the study had mild renal artery stenosis (50%–70%), and 12 (19%) of the 64 patients in the group randomized to stenting actually had stenosis of less than 50%. How can one expect there to be a benefit to stenting in patients with mild (and hemodynamically insignificant) renal artery stenosis? This is especially true when the primary end point is a change in renal function.
More than half of the patients had unilateral disease. It seems intuitive that if one were to plan a trial with a primary end point of a change in renal function, only patients with bilateral renal artery stenosis of greater than 70% or with stenosis of greater than 70% to a solitary functioning kidney would be included. One would not expect that patients with unilateral disease and a stenosis of less than 70% would derive any benefit from revascularization.
Not all “stent” patients received stents. All of the patients in the medical group received medication and there were no crossovers. However, only 46 (72%) of the 64 patients randomized to stenting actually received a stent, while 18 (28%) did not. There were two technical failures, and 12 patients should not have been randomized because they had less than 50% stenosis on angiography and thus were not stented. Yet all 64 patients were analyzed (by intention to treat) in the stent group. With these numbers, one could predict that the results would be negative.
Like DRASTIC, this trial was underpowered, meaning that the chance of a type 2 statistical error is high. In fact, the editors of the Annals of Internal Medicine, in a note accompanying the article, cautioned that the study “was underpowered to provide a definitive estimate of efficacy.”7 If the study was underpowered to answer the question at hand, why was it deemed worthy of publication?
High complication rates. The periprocedural complication and death rates were much higher than in many other reports on renal artery stenting (see details below).5
THE ASTRAL TRIAL
In the Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) trial,8 the primary outcome measure was the change in renal function over time as assessed by the mean slope of the reciprocal of the serum creatinine. In this trial, 806 patients with atherosclerotic renal artery stenosis were randomized to either stent-based revascularization combined with medical therapy or medical therapy alone.
Authors’ conclusions
“We found substantial risks but no evidence of a worthwhile clinical benefit from revascularization in patients with atherosclerotic renovascular disease.”8
Despite size, flaws remain
Unlike the other trials, ASTRAL had a sample size large enough to provide an answer. However, numerous flaws in study design and implementation invalidate its results for the overall population of patients with renal artery stenosis. The major flaws in ASTRAL were:
Selection bias. For a patient to be enrolled, the treating physician had to be undecided on whether the patient should undergo revascularization or medical management alone. However, the treatment of atherosclerotic renal artery stenosis is so controversial that physicians of different specialties cannot agree on the most effective treatment strategy for most patients.1,2 Therefore, to exclude patients when their physicians were sure they needed or did not need renal artery revascularization is incomprehensible and introduces considerable selection bias into the trial design.
Normal renal function at baseline. The primary outcome was a change in renal function over time. Yet 25% of patients had normal renal function at the outset of the trial. In addition, a significant number had unilateral disease, and 41% had a stenosis less than 70%. What made the investigators think that stent implantation could possibly be shown to be beneficial if they entered patients into a renal function study who had near-normal renal function, unilateral disease, and mild renal artery stenosis? These are patients whose condition would not be expected to worsen with medical therapy nor to improve with stenting. Most clinicians would not consider stenting a patient to preserve renal function if the patient has unilateral mild renal artery stenosis.
There was no core laboratory to adjudicate the interpretation of the imaging studies. To determine the degree of stenosis of an artery in an accurate and unbiased fashion, a core laboratory must be used.
The reason this is so important is that visual assessment of the degree of stenosis on angiography is not accurate and almost always overestimates the degree of stenosis.12,13 In a study assessing the physiologic importance of renal artery lesions, the lesion severity by visual estimation was 74.9% ± 11.5% (range 50%–90%), which exceeded the quantitative vascular angiographic lesion severity of 56.6% ± 10.8% (range 45%–76%).13
Therefore, in ASTRAL, some patients in the 50%–70% stenosis group (about 40% of patients entered) actually had a stenosis of less than 50%. And some patients in the group with stenosis greater than 70% had stenosis of less than 70%. This further illustrates that, for the most part, the patients in ASTRAL had mild to moderate renal artery stenosis.
A high adverse event rate. The major adverse event rate in the first 24 hours was 9%, whereas the usual rate is 2% or less.14–18 Of the 280 patients in the revascularization group for whom data on adverse events were available at 1 month, 55 (20%) suffered a serious adverse event (including two patients who died) between 24 hours and 1 month after the procedure. This is in contrast to a major complication rate of 1.99% in five reports involving 727 patients.5
The trial centers were not high-volume centers. During the 7 years of recruitment, 24 centers (42% of all participating centers) randomized between one and five patients, and 32 centers (61% of all participating centers) randomized nine patients or fewer. This means that many participating centers randomized, on average, less than one patient per year! This was not a group of high-volume operators.
WILL CORAL GIVE US THE ANSWER?
The CORAL (Cardiovascular Outcomes in Renal Atherosclerotic Lesions) trial is under way.19 Enrollment was to have ended on January 31, 2010, and it will be several years before the data are available for analysis.
CORAL, a multicenter study funded in 2004 by the National Institutes of Health, will have randomized more than 900 patients with greater than 60% stenosis to optimal medical therapy alone or optimal medical therapy plus renal artery stenting. Inclusion criteria are a documented history of hypertension on two or more antihypertensive drugs or renal dysfunction, defined as stage 3 or greater chronic kidney disease based on the National Kidney Foundation classification (estimated glomerular filtration rate < 60 mL/min/1.73 m2 calculated by the modified Modification of Diet in Renal Disease [MDRD] formula) and stenosis of 60% or greater but less than 100%, as assessed by a core laboratory. The primary end point is survival free of cardiovascular and renal adverse events, defined as a composite of cardiovascular or renal death, stroke, myocardial infarction, hospitalization for congestive heart failure, progressive renal insufficiency, or need for permanent renal replacement therapy.
We hope this trial will give us a clear answer to the question of whether renal artery stenting is beneficial in the patient population studied. One note of caution: recruitment for this trial was difficult and slow. Thus, there were a number of protocol amendments throughout the trial in order to make recruitment easier. Hopefully, this will not be a problem when analyzing the results.
WE ALL AGREE ON THE INDICATIONS FOR STENTING
So, are we really so far apart in our thinking? And is it really “time to be less aggressive” if we follow the precepts below?
All renal arteries with stenosis do not need to be (and should not be) stented.
There must be a good clinical indicationandhemodynamically significant stenosis. This means the degree of stenosis should be more than 70% on angiography or intravascular ultrasonography.
Indications for stenting. Until more data from compelling randomized trials become available, adherence to the American College of Cardiology/American Heart Association guidelines on indications for renal artery stenting is advised3:
- Hypertension: class IIa, level of evidence B. Percutaneous revascularization is reasonable for patients with hemodynamically significant renal artery stenosis and accelerated hypertension, resistant hypertension, and malignant hypertension.
- Preservation of renal function: class IIa, level of evidence B. Percutaneous revascularization is reasonable for patients with renal artery stenosis and progressive chronic kidney disease with bilateral renal artery stenosis or a stenosis to a solitary functioning kidney.
- Congestive heart failure: class I, level of evidence B. Percutaneous revascularization is indicated for patients with hemodynamically significant renal artery stenosis (ie, > 70% stenosis on angiography or intravascular ultrasonography) and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema.
- Cooper CJ, Murphy TP. Is renal artery stenting the correct treatment of renal artery stenosis? The case for renal artery stenting for treatment of renal artery stenosis. Circulation 2007; 115:263–269.
- Dworkin LD, Jamerson KA. Is renal artery stenting the correct treatment of renal artery stenosis? Case against angioplasty and stenting of atherosclerotic renal artery stenosis. Circulation 2007; 115:271–276.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic): A Collaborative Report from the American Association of Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Interventional Radiology, Society for Vascular Medicine and Biology and the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2006; 113:e463–e654.
- Simon JF. Stenting atherosclerotic renal arteries: time to be less aggressive. Cleve Clin J Med 2010; 77:178–189.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–841.
- Wheatley K, Ives N, Gray R, et al Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Tan WA, Wholey MH, Olin JW. The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis [letter]. N Engl J Med 2000; 343:438.
- Rocha-Singh KJ, Eisenhauer AC, Textor SC, et al Atherosclerotic Peripheral Vascular Disease Symposium II: intervention for renal artery disease. Circulation 2008; 118:2873–2878.
- Textor SC, Lerman L, McKusick M. The uncertain value of renal artery interventions: where are we now? JACC Cardiovasc Intervent 2009; 2:175–182.
- Topol EJ, Nissen SE. Our preoccupation with coronary luminology. The dissociation between clinical and angiographic findings in ischemic heart disease. Circulation 1995; 92:2333–2342.
- Subramanian R, White CJ, Rosenfield K, et al Renal fractional flow reserve: a hemodynamic evaluation of moderate renal artery stenoses. Catheter Cardiovasc Interv 2005; 64:480–486.
- Burket MW, Cooper CJ, Kennedy DJ, et al Renal artery angioplasty and stent placement: predictors of a favorable outcome. Am Heart J 2000; 139:64–71.
- Dorros G, Jaff M, Mathiak L, et al Four-year follow-up of Palmaz-Schatz stent revascularization as treatment for atherosclerotic renal artery stenosis. Circulation 1998; 98:642–647.
- Rocha-Singh K, Jaff MR, Rosenfield K. Evaluation of the safety and effectiveness of renal artery stenting after unsuccessful balloon angioplasty: the ASPIRE-2 study. J Am Coll Cardiol 2005; 46:776–783.
- Tuttle KR, Chouinard RF, Webber JT, et al Treatment of atherosclerotic ostial renal artery stenosis with the intravascular stent. Am J Kidney Dis 1998; 32:611–622.
- White CJ, Ramee SR, Collins TJ, Jenkins JS, Escobar A, Shaw D. Renal artery stent placement: utility in lesions difficult to treat with balloon angioplasty. J Am Coll Cardiol 1997; 30:1445–1450.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
The role of stenting for atherosclerotic renal artery stenosis is hotly debated among different specialties.1,2 If we may generalize a bit, interventionalists (cardiologists, interventional radiologists, vascular surgeons, and vascular medicine specialists) have been in favor of liberal use of stenting, and nephrologists often favor medical therapy alone. And as with all controversial issues, each group feels rather strongly about its position.
Because few prospective randomized trials have been completed, the management of atherosclerotic renal artery stenosis has been guided by retrospective studies and case series. 3
In this issue of the Cleveland Clinic Journal of Medicine, Dr. James Simon4 provides an excellent overview of the prevalence, natural history, and clinical presentation of atherosclerotic renal artery stenosis. In addition, he does an admirable job of reviewing the available prospective randomized trials and providing editorial commentary about the role of the various specialists in the management of renal artery disease. And while the title of his paper says that it is “time to be less aggressive,” Dr. Simon ultimately comes to the same conclusions that we do5 on the indications for renal artery stenting (see Table 3 of Dr. Simon’s article), which are those of the multidisciplinary 2006 American College of Cardiology/American Heart Association guidelines on the management of peripheral artery disease.3
So what then is all the controversy about? We all agree that prospective randomized trials that provide class I, level A evidence impart the only unbiased scientific information on the best treatment strategy for patients with renal artery disease. The basic controversial issue is the interpretation of these trials. We contend that the three randomized trials of stenting vs medical therapy published so far6–8 (see below) are so seriously flawed that it is impossible to make treatment decisions based on their results.
Since these trials were published in wellrespected journals, their results are often taken as gospel. However, careful review of each of these will reveal the flaws in study design and implementation.
THE DRASTIC TRIAL
In the Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) trial,6 106 patients with renal artery stenosis and hypertension (diastolic blood pressure > 95 mm Hg) despite treatment with two antihypertensive medications were randomly assigned to either renal angioplasty (n = 56) or drug therapy (n = 50).
Authors’ conclusions
“In the treatment of patients with hypertension and renal-artery stenosis, angioplasty has little advantage over antihypertensive-drug therapy.”6
Four serious problems
As we discussed in a letter to the editor of the New England Journal of Medicine on August 10, 2000, this study had four serious problems that invalidate its authors’ conclusions.9
The sample size was insufficient to detect a significant difference between treatment groups. In other words, the chance of a type 2 statistical error is high.
Balloon angioplasty without stenting was used as the method of revascularization. Experts now recognize that stenting is required for renal artery intervention to have a durable result.3,5
Renal artery stenosis was defined as greater than 50% stenosis. This allowed a large number of patients to enter the trial who had hemodynamically and clinically insignificant lesions. Most clinicians believe that stenosis of less than 70% is not hemodynamically important.5,10,11
Twenty-two of the 50 patients randomized to medical therapy crossed over to the angioplasty group because their blood pressure became difficult to control. In other words, 44% of the patients in the medical group underwent angioplasty, an astounding percentage in an intention-to-treat analysis comparing one therapy with another.
Despite these serious flaws, the results of DRASTIC influenced therapy for years after its publication.
THE STAR TRIAL
In the Stent Placement in Patients With Atherosclerotic Renal Artery Stenosis and Impaired Renal Function (STAR) trial,7 140 patients with a creatinine clearance of less than 80 mL/min/1.73m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy (n = 64) or medical therapy alone (n = 76). The primary end point was a 20% or greater decrease in creatinine clearance. Secondary end points included measures of safety and cardiovascular morbidity and mortality.
Authors’ conclusions
“Stent placement with medical treatment had no clear effect on progression of impaired renal function but led to a small number of significant procedure-related complications. The study findings favor a conservative approach to patients with [atherosclerotic renal artery stenosis], focused on cardiovascular risk factor management and avoiding stenting.”7
Serious flaws
A number of serious flaws render this study uninterpretable.
Mild renal artery stenosis. At least 33% of the patients in the study had mild renal artery stenosis (50%–70%), and 12 (19%) of the 64 patients in the group randomized to stenting actually had stenosis of less than 50%. How can one expect there to be a benefit to stenting in patients with mild (and hemodynamically insignificant) renal artery stenosis? This is especially true when the primary end point is a change in renal function.
More than half of the patients had unilateral disease. It seems intuitive that if one were to plan a trial with a primary end point of a change in renal function, only patients with bilateral renal artery stenosis of greater than 70% or with stenosis of greater than 70% to a solitary functioning kidney would be included. One would not expect that patients with unilateral disease and a stenosis of less than 70% would derive any benefit from revascularization.
Not all “stent” patients received stents. All of the patients in the medical group received medication and there were no crossovers. However, only 46 (72%) of the 64 patients randomized to stenting actually received a stent, while 18 (28%) did not. There were two technical failures, and 12 patients should not have been randomized because they had less than 50% stenosis on angiography and thus were not stented. Yet all 64 patients were analyzed (by intention to treat) in the stent group. With these numbers, one could predict that the results would be negative.
Like DRASTIC, this trial was underpowered, meaning that the chance of a type 2 statistical error is high. In fact, the editors of the Annals of Internal Medicine, in a note accompanying the article, cautioned that the study “was underpowered to provide a definitive estimate of efficacy.”7 If the study was underpowered to answer the question at hand, why was it deemed worthy of publication?
High complication rates. The periprocedural complication and death rates were much higher than in many other reports on renal artery stenting (see details below).5
THE ASTRAL TRIAL
In the Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) trial,8 the primary outcome measure was the change in renal function over time as assessed by the mean slope of the reciprocal of the serum creatinine. In this trial, 806 patients with atherosclerotic renal artery stenosis were randomized to either stent-based revascularization combined with medical therapy or medical therapy alone.
Authors’ conclusions
“We found substantial risks but no evidence of a worthwhile clinical benefit from revascularization in patients with atherosclerotic renovascular disease.”8
Despite size, flaws remain
Unlike the other trials, ASTRAL had a sample size large enough to provide an answer. However, numerous flaws in study design and implementation invalidate its results for the overall population of patients with renal artery stenosis. The major flaws in ASTRAL were:
Selection bias. For a patient to be enrolled, the treating physician had to be undecided on whether the patient should undergo revascularization or medical management alone. However, the treatment of atherosclerotic renal artery stenosis is so controversial that physicians of different specialties cannot agree on the most effective treatment strategy for most patients.1,2 Therefore, to exclude patients when their physicians were sure they needed or did not need renal artery revascularization is incomprehensible and introduces considerable selection bias into the trial design.
Normal renal function at baseline. The primary outcome was a change in renal function over time. Yet 25% of patients had normal renal function at the outset of the trial. In addition, a significant number had unilateral disease, and 41% had a stenosis less than 70%. What made the investigators think that stent implantation could possibly be shown to be beneficial if they entered patients into a renal function study who had near-normal renal function, unilateral disease, and mild renal artery stenosis? These are patients whose condition would not be expected to worsen with medical therapy nor to improve with stenting. Most clinicians would not consider stenting a patient to preserve renal function if the patient has unilateral mild renal artery stenosis.
There was no core laboratory to adjudicate the interpretation of the imaging studies. To determine the degree of stenosis of an artery in an accurate and unbiased fashion, a core laboratory must be used.
The reason this is so important is that visual assessment of the degree of stenosis on angiography is not accurate and almost always overestimates the degree of stenosis.12,13 In a study assessing the physiologic importance of renal artery lesions, the lesion severity by visual estimation was 74.9% ± 11.5% (range 50%–90%), which exceeded the quantitative vascular angiographic lesion severity of 56.6% ± 10.8% (range 45%–76%).13
Therefore, in ASTRAL, some patients in the 50%–70% stenosis group (about 40% of patients entered) actually had a stenosis of less than 50%. And some patients in the group with stenosis greater than 70% had stenosis of less than 70%. This further illustrates that, for the most part, the patients in ASTRAL had mild to moderate renal artery stenosis.
A high adverse event rate. The major adverse event rate in the first 24 hours was 9%, whereas the usual rate is 2% or less.14–18 Of the 280 patients in the revascularization group for whom data on adverse events were available at 1 month, 55 (20%) suffered a serious adverse event (including two patients who died) between 24 hours and 1 month after the procedure. This is in contrast to a major complication rate of 1.99% in five reports involving 727 patients.5
The trial centers were not high-volume centers. During the 7 years of recruitment, 24 centers (42% of all participating centers) randomized between one and five patients, and 32 centers (61% of all participating centers) randomized nine patients or fewer. This means that many participating centers randomized, on average, less than one patient per year! This was not a group of high-volume operators.
WILL CORAL GIVE US THE ANSWER?
The CORAL (Cardiovascular Outcomes in Renal Atherosclerotic Lesions) trial is under way.19 Enrollment was to have ended on January 31, 2010, and it will be several years before the data are available for analysis.
CORAL, a multicenter study funded in 2004 by the National Institutes of Health, will have randomized more than 900 patients with greater than 60% stenosis to optimal medical therapy alone or optimal medical therapy plus renal artery stenting. Inclusion criteria are a documented history of hypertension on two or more antihypertensive drugs or renal dysfunction, defined as stage 3 or greater chronic kidney disease based on the National Kidney Foundation classification (estimated glomerular filtration rate < 60 mL/min/1.73 m2 calculated by the modified Modification of Diet in Renal Disease [MDRD] formula) and stenosis of 60% or greater but less than 100%, as assessed by a core laboratory. The primary end point is survival free of cardiovascular and renal adverse events, defined as a composite of cardiovascular or renal death, stroke, myocardial infarction, hospitalization for congestive heart failure, progressive renal insufficiency, or need for permanent renal replacement therapy.
We hope this trial will give us a clear answer to the question of whether renal artery stenting is beneficial in the patient population studied. One note of caution: recruitment for this trial was difficult and slow. Thus, there were a number of protocol amendments throughout the trial in order to make recruitment easier. Hopefully, this will not be a problem when analyzing the results.
WE ALL AGREE ON THE INDICATIONS FOR STENTING
So, are we really so far apart in our thinking? And is it really “time to be less aggressive” if we follow the precepts below?
All renal arteries with stenosis do not need to be (and should not be) stented.
There must be a good clinical indicationandhemodynamically significant stenosis. This means the degree of stenosis should be more than 70% on angiography or intravascular ultrasonography.
Indications for stenting. Until more data from compelling randomized trials become available, adherence to the American College of Cardiology/American Heart Association guidelines on indications for renal artery stenting is advised3:
- Hypertension: class IIa, level of evidence B. Percutaneous revascularization is reasonable for patients with hemodynamically significant renal artery stenosis and accelerated hypertension, resistant hypertension, and malignant hypertension.
- Preservation of renal function: class IIa, level of evidence B. Percutaneous revascularization is reasonable for patients with renal artery stenosis and progressive chronic kidney disease with bilateral renal artery stenosis or a stenosis to a solitary functioning kidney.
- Congestive heart failure: class I, level of evidence B. Percutaneous revascularization is indicated for patients with hemodynamically significant renal artery stenosis (ie, > 70% stenosis on angiography or intravascular ultrasonography) and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema.
The role of stenting for atherosclerotic renal artery stenosis is hotly debated among different specialties.1,2 If we may generalize a bit, interventionalists (cardiologists, interventional radiologists, vascular surgeons, and vascular medicine specialists) have been in favor of liberal use of stenting, and nephrologists often favor medical therapy alone. And as with all controversial issues, each group feels rather strongly about its position.
Because few prospective randomized trials have been completed, the management of atherosclerotic renal artery stenosis has been guided by retrospective studies and case series. 3
In this issue of the Cleveland Clinic Journal of Medicine, Dr. James Simon4 provides an excellent overview of the prevalence, natural history, and clinical presentation of atherosclerotic renal artery stenosis. In addition, he does an admirable job of reviewing the available prospective randomized trials and providing editorial commentary about the role of the various specialists in the management of renal artery disease. And while the title of his paper says that it is “time to be less aggressive,” Dr. Simon ultimately comes to the same conclusions that we do5 on the indications for renal artery stenting (see Table 3 of Dr. Simon’s article), which are those of the multidisciplinary 2006 American College of Cardiology/American Heart Association guidelines on the management of peripheral artery disease.3
So what then is all the controversy about? We all agree that prospective randomized trials that provide class I, level A evidence impart the only unbiased scientific information on the best treatment strategy for patients with renal artery disease. The basic controversial issue is the interpretation of these trials. We contend that the three randomized trials of stenting vs medical therapy published so far6–8 (see below) are so seriously flawed that it is impossible to make treatment decisions based on their results.
Since these trials were published in wellrespected journals, their results are often taken as gospel. However, careful review of each of these will reveal the flaws in study design and implementation.
THE DRASTIC TRIAL
In the Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) trial,6 106 patients with renal artery stenosis and hypertension (diastolic blood pressure > 95 mm Hg) despite treatment with two antihypertensive medications were randomly assigned to either renal angioplasty (n = 56) or drug therapy (n = 50).
Authors’ conclusions
“In the treatment of patients with hypertension and renal-artery stenosis, angioplasty has little advantage over antihypertensive-drug therapy.”6
Four serious problems
As we discussed in a letter to the editor of the New England Journal of Medicine on August 10, 2000, this study had four serious problems that invalidate its authors’ conclusions.9
The sample size was insufficient to detect a significant difference between treatment groups. In other words, the chance of a type 2 statistical error is high.
Balloon angioplasty without stenting was used as the method of revascularization. Experts now recognize that stenting is required for renal artery intervention to have a durable result.3,5
Renal artery stenosis was defined as greater than 50% stenosis. This allowed a large number of patients to enter the trial who had hemodynamically and clinically insignificant lesions. Most clinicians believe that stenosis of less than 70% is not hemodynamically important.5,10,11
Twenty-two of the 50 patients randomized to medical therapy crossed over to the angioplasty group because their blood pressure became difficult to control. In other words, 44% of the patients in the medical group underwent angioplasty, an astounding percentage in an intention-to-treat analysis comparing one therapy with another.
Despite these serious flaws, the results of DRASTIC influenced therapy for years after its publication.
THE STAR TRIAL
In the Stent Placement in Patients With Atherosclerotic Renal Artery Stenosis and Impaired Renal Function (STAR) trial,7 140 patients with a creatinine clearance of less than 80 mL/min/1.73m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy (n = 64) or medical therapy alone (n = 76). The primary end point was a 20% or greater decrease in creatinine clearance. Secondary end points included measures of safety and cardiovascular morbidity and mortality.
Authors’ conclusions
“Stent placement with medical treatment had no clear effect on progression of impaired renal function but led to a small number of significant procedure-related complications. The study findings favor a conservative approach to patients with [atherosclerotic renal artery stenosis], focused on cardiovascular risk factor management and avoiding stenting.”7
Serious flaws
A number of serious flaws render this study uninterpretable.
Mild renal artery stenosis. At least 33% of the patients in the study had mild renal artery stenosis (50%–70%), and 12 (19%) of the 64 patients in the group randomized to stenting actually had stenosis of less than 50%. How can one expect there to be a benefit to stenting in patients with mild (and hemodynamically insignificant) renal artery stenosis? This is especially true when the primary end point is a change in renal function.
More than half of the patients had unilateral disease. It seems intuitive that if one were to plan a trial with a primary end point of a change in renal function, only patients with bilateral renal artery stenosis of greater than 70% or with stenosis of greater than 70% to a solitary functioning kidney would be included. One would not expect that patients with unilateral disease and a stenosis of less than 70% would derive any benefit from revascularization.
Not all “stent” patients received stents. All of the patients in the medical group received medication and there were no crossovers. However, only 46 (72%) of the 64 patients randomized to stenting actually received a stent, while 18 (28%) did not. There were two technical failures, and 12 patients should not have been randomized because they had less than 50% stenosis on angiography and thus were not stented. Yet all 64 patients were analyzed (by intention to treat) in the stent group. With these numbers, one could predict that the results would be negative.
Like DRASTIC, this trial was underpowered, meaning that the chance of a type 2 statistical error is high. In fact, the editors of the Annals of Internal Medicine, in a note accompanying the article, cautioned that the study “was underpowered to provide a definitive estimate of efficacy.”7 If the study was underpowered to answer the question at hand, why was it deemed worthy of publication?
High complication rates. The periprocedural complication and death rates were much higher than in many other reports on renal artery stenting (see details below).5
THE ASTRAL TRIAL
In the Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) trial,8 the primary outcome measure was the change in renal function over time as assessed by the mean slope of the reciprocal of the serum creatinine. In this trial, 806 patients with atherosclerotic renal artery stenosis were randomized to either stent-based revascularization combined with medical therapy or medical therapy alone.
Authors’ conclusions
“We found substantial risks but no evidence of a worthwhile clinical benefit from revascularization in patients with atherosclerotic renovascular disease.”8
Despite size, flaws remain
Unlike the other trials, ASTRAL had a sample size large enough to provide an answer. However, numerous flaws in study design and implementation invalidate its results for the overall population of patients with renal artery stenosis. The major flaws in ASTRAL were:
Selection bias. For a patient to be enrolled, the treating physician had to be undecided on whether the patient should undergo revascularization or medical management alone. However, the treatment of atherosclerotic renal artery stenosis is so controversial that physicians of different specialties cannot agree on the most effective treatment strategy for most patients.1,2 Therefore, to exclude patients when their physicians were sure they needed or did not need renal artery revascularization is incomprehensible and introduces considerable selection bias into the trial design.
Normal renal function at baseline. The primary outcome was a change in renal function over time. Yet 25% of patients had normal renal function at the outset of the trial. In addition, a significant number had unilateral disease, and 41% had a stenosis less than 70%. What made the investigators think that stent implantation could possibly be shown to be beneficial if they entered patients into a renal function study who had near-normal renal function, unilateral disease, and mild renal artery stenosis? These are patients whose condition would not be expected to worsen with medical therapy nor to improve with stenting. Most clinicians would not consider stenting a patient to preserve renal function if the patient has unilateral mild renal artery stenosis.
There was no core laboratory to adjudicate the interpretation of the imaging studies. To determine the degree of stenosis of an artery in an accurate and unbiased fashion, a core laboratory must be used.
The reason this is so important is that visual assessment of the degree of stenosis on angiography is not accurate and almost always overestimates the degree of stenosis.12,13 In a study assessing the physiologic importance of renal artery lesions, the lesion severity by visual estimation was 74.9% ± 11.5% (range 50%–90%), which exceeded the quantitative vascular angiographic lesion severity of 56.6% ± 10.8% (range 45%–76%).13
Therefore, in ASTRAL, some patients in the 50%–70% stenosis group (about 40% of patients entered) actually had a stenosis of less than 50%. And some patients in the group with stenosis greater than 70% had stenosis of less than 70%. This further illustrates that, for the most part, the patients in ASTRAL had mild to moderate renal artery stenosis.
A high adverse event rate. The major adverse event rate in the first 24 hours was 9%, whereas the usual rate is 2% or less.14–18 Of the 280 patients in the revascularization group for whom data on adverse events were available at 1 month, 55 (20%) suffered a serious adverse event (including two patients who died) between 24 hours and 1 month after the procedure. This is in contrast to a major complication rate of 1.99% in five reports involving 727 patients.5
The trial centers were not high-volume centers. During the 7 years of recruitment, 24 centers (42% of all participating centers) randomized between one and five patients, and 32 centers (61% of all participating centers) randomized nine patients or fewer. This means that many participating centers randomized, on average, less than one patient per year! This was not a group of high-volume operators.
WILL CORAL GIVE US THE ANSWER?
The CORAL (Cardiovascular Outcomes in Renal Atherosclerotic Lesions) trial is under way.19 Enrollment was to have ended on January 31, 2010, and it will be several years before the data are available for analysis.
CORAL, a multicenter study funded in 2004 by the National Institutes of Health, will have randomized more than 900 patients with greater than 60% stenosis to optimal medical therapy alone or optimal medical therapy plus renal artery stenting. Inclusion criteria are a documented history of hypertension on two or more antihypertensive drugs or renal dysfunction, defined as stage 3 or greater chronic kidney disease based on the National Kidney Foundation classification (estimated glomerular filtration rate < 60 mL/min/1.73 m2 calculated by the modified Modification of Diet in Renal Disease [MDRD] formula) and stenosis of 60% or greater but less than 100%, as assessed by a core laboratory. The primary end point is survival free of cardiovascular and renal adverse events, defined as a composite of cardiovascular or renal death, stroke, myocardial infarction, hospitalization for congestive heart failure, progressive renal insufficiency, or need for permanent renal replacement therapy.
We hope this trial will give us a clear answer to the question of whether renal artery stenting is beneficial in the patient population studied. One note of caution: recruitment for this trial was difficult and slow. Thus, there were a number of protocol amendments throughout the trial in order to make recruitment easier. Hopefully, this will not be a problem when analyzing the results.
WE ALL AGREE ON THE INDICATIONS FOR STENTING
So, are we really so far apart in our thinking? And is it really “time to be less aggressive” if we follow the precepts below?
All renal arteries with stenosis do not need to be (and should not be) stented.
There must be a good clinical indicationandhemodynamically significant stenosis. This means the degree of stenosis should be more than 70% on angiography or intravascular ultrasonography.
Indications for stenting. Until more data from compelling randomized trials become available, adherence to the American College of Cardiology/American Heart Association guidelines on indications for renal artery stenting is advised3:
- Hypertension: class IIa, level of evidence B. Percutaneous revascularization is reasonable for patients with hemodynamically significant renal artery stenosis and accelerated hypertension, resistant hypertension, and malignant hypertension.
- Preservation of renal function: class IIa, level of evidence B. Percutaneous revascularization is reasonable for patients with renal artery stenosis and progressive chronic kidney disease with bilateral renal artery stenosis or a stenosis to a solitary functioning kidney.
- Congestive heart failure: class I, level of evidence B. Percutaneous revascularization is indicated for patients with hemodynamically significant renal artery stenosis (ie, > 70% stenosis on angiography or intravascular ultrasonography) and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema.
- Cooper CJ, Murphy TP. Is renal artery stenting the correct treatment of renal artery stenosis? The case for renal artery stenting for treatment of renal artery stenosis. Circulation 2007; 115:263–269.
- Dworkin LD, Jamerson KA. Is renal artery stenting the correct treatment of renal artery stenosis? Case against angioplasty and stenting of atherosclerotic renal artery stenosis. Circulation 2007; 115:271–276.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic): A Collaborative Report from the American Association of Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Interventional Radiology, Society for Vascular Medicine and Biology and the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2006; 113:e463–e654.
- Simon JF. Stenting atherosclerotic renal arteries: time to be less aggressive. Cleve Clin J Med 2010; 77:178–189.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–841.
- Wheatley K, Ives N, Gray R, et al Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Tan WA, Wholey MH, Olin JW. The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis [letter]. N Engl J Med 2000; 343:438.
- Rocha-Singh KJ, Eisenhauer AC, Textor SC, et al Atherosclerotic Peripheral Vascular Disease Symposium II: intervention for renal artery disease. Circulation 2008; 118:2873–2878.
- Textor SC, Lerman L, McKusick M. The uncertain value of renal artery interventions: where are we now? JACC Cardiovasc Intervent 2009; 2:175–182.
- Topol EJ, Nissen SE. Our preoccupation with coronary luminology. The dissociation between clinical and angiographic findings in ischemic heart disease. Circulation 1995; 92:2333–2342.
- Subramanian R, White CJ, Rosenfield K, et al Renal fractional flow reserve: a hemodynamic evaluation of moderate renal artery stenoses. Catheter Cardiovasc Interv 2005; 64:480–486.
- Burket MW, Cooper CJ, Kennedy DJ, et al Renal artery angioplasty and stent placement: predictors of a favorable outcome. Am Heart J 2000; 139:64–71.
- Dorros G, Jaff M, Mathiak L, et al Four-year follow-up of Palmaz-Schatz stent revascularization as treatment for atherosclerotic renal artery stenosis. Circulation 1998; 98:642–647.
- Rocha-Singh K, Jaff MR, Rosenfield K. Evaluation of the safety and effectiveness of renal artery stenting after unsuccessful balloon angioplasty: the ASPIRE-2 study. J Am Coll Cardiol 2005; 46:776–783.
- Tuttle KR, Chouinard RF, Webber JT, et al Treatment of atherosclerotic ostial renal artery stenosis with the intravascular stent. Am J Kidney Dis 1998; 32:611–622.
- White CJ, Ramee SR, Collins TJ, Jenkins JS, Escobar A, Shaw D. Renal artery stent placement: utility in lesions difficult to treat with balloon angioplasty. J Am Coll Cardiol 1997; 30:1445–1450.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Cooper CJ, Murphy TP. Is renal artery stenting the correct treatment of renal artery stenosis? The case for renal artery stenting for treatment of renal artery stenosis. Circulation 2007; 115:263–269.
- Dworkin LD, Jamerson KA. Is renal artery stenting the correct treatment of renal artery stenosis? Case against angioplasty and stenting of atherosclerotic renal artery stenosis. Circulation 2007; 115:271–276.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA Guidelines for the Management of Patients with Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic): A Collaborative Report from the American Association of Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Interventional Radiology, Society for Vascular Medicine and Biology and the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2006; 113:e463–e654.
- Simon JF. Stenting atherosclerotic renal arteries: time to be less aggressive. Cleve Clin J Med 2010; 77:178–189.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–841.
- Wheatley K, Ives N, Gray R, et al Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Tan WA, Wholey MH, Olin JW. The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis [letter]. N Engl J Med 2000; 343:438.
- Rocha-Singh KJ, Eisenhauer AC, Textor SC, et al Atherosclerotic Peripheral Vascular Disease Symposium II: intervention for renal artery disease. Circulation 2008; 118:2873–2878.
- Textor SC, Lerman L, McKusick M. The uncertain value of renal artery interventions: where are we now? JACC Cardiovasc Intervent 2009; 2:175–182.
- Topol EJ, Nissen SE. Our preoccupation with coronary luminology. The dissociation between clinical and angiographic findings in ischemic heart disease. Circulation 1995; 92:2333–2342.
- Subramanian R, White CJ, Rosenfield K, et al Renal fractional flow reserve: a hemodynamic evaluation of moderate renal artery stenoses. Catheter Cardiovasc Interv 2005; 64:480–486.
- Burket MW, Cooper CJ, Kennedy DJ, et al Renal artery angioplasty and stent placement: predictors of a favorable outcome. Am Heart J 2000; 139:64–71.
- Dorros G, Jaff M, Mathiak L, et al Four-year follow-up of Palmaz-Schatz stent revascularization as treatment for atherosclerotic renal artery stenosis. Circulation 1998; 98:642–647.
- Rocha-Singh K, Jaff MR, Rosenfield K. Evaluation of the safety and effectiveness of renal artery stenting after unsuccessful balloon angioplasty: the ASPIRE-2 study. J Am Coll Cardiol 2005; 46:776–783.
- Tuttle KR, Chouinard RF, Webber JT, et al Treatment of atherosclerotic ostial renal artery stenosis with the intravascular stent. Am J Kidney Dis 1998; 32:611–622.
- White CJ, Ramee SR, Collins TJ, Jenkins JS, Escobar A, Shaw D. Renal artery stent placement: utility in lesions difficult to treat with balloon angioplasty. J Am Coll Cardiol 1997; 30:1445–1450.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
Stenting atherosclerotic renal arteries: Time to be less aggressive
Author’s note: Atherosclerosis accounts for about 90% of cases of renal artery stenosis in people over age 40.1 Fibromuscular dysplasia, the other major cause, is a separate topic; in this paper “renal artery stenosis” refers to atherosclerotic disease only.
Renal artery stenosis is very common, and the number of angioplasty-stenting procedures performed every year is on the rise. Yet there is no overwhelming evidence that intervention yields clinical benefits—ie, better blood pressure control or renal function— than does medical therapy.
Earlier randomized controlled trials comparing angioplasty without stents and medical management showed no impressive difference in blood pressure.2,3 The data on renal function were even more questionable, with some studies suggesting that, with stenting, the chance of worsening renal function is equal to that of improvement.4
Two large randomized trials comparing renal intervention with medical therapy failed to show any benefit of intervention.5–7 A third study is under way.8
It is time to strongly reconsider the current aggressive approach to revascularization of stenotic renal arteries and take a more coordinated, critical approach.
RENAL INTERVENTIONS ON THE RISE
Renal angioplasty began replacing surgical revascularization in the 1990s, as this less-invasive procedure became more readily available and was shown to have similar clinical outcomes.9 In the last decade, stent placement during angioplasty has become standard, improving the rates of technical success.
As these procedures have become easier to perform and their radiographic outcomes have become more consistent, interventionalists have become more likely, if they see stenosis in a renal artery, to intervene and insert a stent, regardless of proven benefit. In addition, interventionalists from at least three different specialties now compete for these procedures, often by looking at the renal arteries during angiography of other vascular beds (the “drive-by”).
As a result, the number of renal interventions has been rising. Medicare received 21,660 claims for renal artery interventions (surgery or angioplasty) in 2000, compared with 13,380 in 1996—an increase of 62%. However, the number of surgeries actually decreased by 45% during this time, while the number of percutaneous procedures increased by 240%. The number of endovascular claims for renal artery stenosis by cardiologists alone rose 390%.10 Since then, the reports on intervention have been mixed, with one report citing a continued increase in 2005 to 35,000 claims,11 and another suggesting a decrease back to 1997 levels.12
HOW COMMON IS RENAL ARTERY STENOSIS?
The prevalence of renal artery stenosis depends on the definition used and the population screened. It is more common in older patients who have risk factors for other vascular diseases than in the general population.
Renal Doppler ultrasonography can detect stenosis only if the artery is narrowed by more than 60%. Hansen et al13 used ultrasonography to screen 870 people over age 65 and found a lesion (a narrowing of more than 60%) in 6.8%.
Angiography (direct, computed tomographic, or magnetic resonance) can detect less-severe stenosis. Thus, most angiographic studies define renal artery stenosis as a narrowing of more than 50%, and severe disease as a narrowing of more than 70%. Many experts believe that unilateral stenosis needs to be more than 70% to pose a risk to the kidney.14,15
Angiographic prevalence studies have been performed only in patients who were undergoing angiography for another reason such as coronary or peripheral arterial disease that inherently places them at higher risk of renal artery stenosis. For instance, renal artery stenosis is found in 11% to 28% of patients undergoing diagnostic cardiac catheterization. 16
No studies of the prevalence of renal artery stenosis have been performed in the general population. Medicare data indicate that from 1999 to 2001 the incidence of diagnosed renal artery stenosis was 3.7 per 1,000 patientyears. 17 Holley et al,18 in an autopsy series, found renal artery stenosis of greater than 50% in 27% of patients over age 50 and in 56.4% of hypertensive patients. The prevalence was 10% in normotensive patients.
WHO IS AT RISK?
Factors associated with a higher risk of finding renal artery stenosis on a radiographic study include14:
- Older age
- Female sex
- Hypertension
- Three-vessel coronary artery disease
- Peripheral artery disease
- Chronic kidney disease
- Diabetes

- A low level of high-density lipoprotein cholesterol
- The use of at least two cardiovascular drugs.
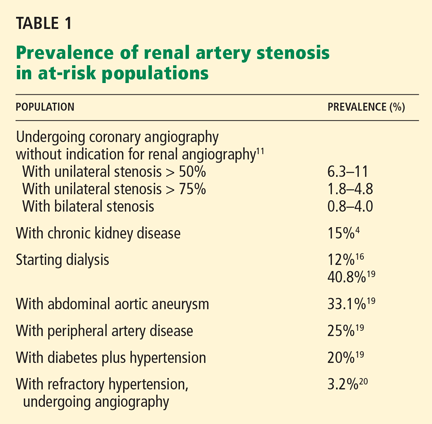
The prevalence of renal artery stenosis in at-risk populations ranges from 3% to 75% (Table 1).2,4,6,19,20
HOW OFTEN DOES STENOSIS PROGRESS?
The reported rates of progression of atherosclerotic renal artery lesions vary depending on the type of imaging test used and the reason for doing it.
In studies that used duplex ultrasonography, roughly half of lesions smaller than 60% grew to greater than 60% over 3 years.21,22 The risk of total occlusion of an artery was relatively low and depended on the severity of stenosis: 0.7% if the baseline stenosis was less than 60% and 2.3% to 7% if it was greater.21,22
In a seminal study in 1984, Schreiber and colleagues23 compared serial angiograms obtained a mean of 52 months apart in 85 patients who did not undergo intervention. Stenosis had progressed in 37 (44%), and to the point of total occlusion in 14 (16%). In contrast, a 1998 study found progression in 11.1% over 2.6 years, with older patients, women, and those with baseline coronary artery disease at higher risk.24
The the rates of progression differed in these two studies probably because the indications for screening were different (clinical suspicion23 vs routine screening during coronary angiography24), as was the severity of stenosis at the time of diagnosis. Also, when these studies were done, fewer people were taking statins. Thus, similar studies, if repeated, might show even lower rates of progression.
Finally, progression of renal artery stenosis has not been correlated with worsening renal function.
FOUR CLINICAL PRESENTATIONS OF RENAL ARTERY STENOSIS
Renal artery stenosis can present in one of four ways:
Clinically silent stenosis. Because renal artery stenosis is most often found in older patients, who are more likely to have essential hypertension and chronic kidney disease due to other causes, it can be an incidental finding that is completely clinically silent.16,25
Renovascular hypertension is defined as high blood pressure due to up-regulation of neurohormonal activity in response to decreased perfusion from renal artery stenosis. Renal artery stenosis is estimated to be the cause of hypertension in only 0.5% to 4.0% of hypertensive patients, or in 26% of patients with secondary hypertension.3
Ischemic nephropathy is more difficult to define because ischemia alone rarely explains the damage done to the kidneys. Activation of neurohormonal pathways and microvascular injury are thought to contribute to oxidative stress and fibrosis.26 These phenomena may explain why similar degrees of stenosis lead to varying degrees of kidney damage in different patients and why the severity of stenosis does not correlate with the degree of renal dysfunction.27
Furthermore, stenosis may lead to irreversible but stable kidney damage. It is therefore not surprising that, in studies in unselected populations (ie, studies that included patients with all presentations of renal artery stenosis, not just those more likely to benefit), up to two-thirds of renal interventions yielded no clinical benefit.25
As a result, if we define ischemic nephropathy as renal artery stenosis with renal dysfunction not attributable to another cause, we probably will overestimate the prevalence of ischemic nephropathy, leading to overly optimistic expectations about the response to revascularization.
Recurrent “flash” pulmonary edema is a less common presentation, usually occurring in patients with critical bilateral renal artery stenosis or unilateral stenosis in an artery supplying a solitary functioning kidney. Most have severe hypertension (average systolic blood pressure 174–207 mm Hg) and poor renal function.28–30
The association between pulmonary edema and bilateral renal artery stenosis was first noted in 1998 by Pickering et al,31 who in several case series showed that 82% to 92% of patients with recurrent pulmonary edema and renal artery stenosis had bilateral stenosis, compared with 20% to 65% of those with other presentations. Later case series corroborated this finding: 85% to 100% of patients with renal artery stenosis and pulmonary edema had bilateral stenosis.28–30
STENTING IS NOW STANDARD
Stenting has become standard in the endovascular treatment of renal artery stenosis.
Most atherosclerotic renal artery lesions are located in the ostium (ie, where the artery branches off from the aorta), and many are extensions of calcified aortic plaque.26,32 These hard lesions tend to rebound to their original shape more often with balloon angioplasty alone. Stenting provides the additional force needed to permanently disrupt the lesion, leading to a longer-lasting result.
Rates of technical success (dilating the artery during the intervention) are higher with stents than without them (98% vs 46%– 77%).33,34 If the lesion is ostial, this difference is even more impressive (75% vs 29%). In addition, restenosis rates at 6 months are lower with stents (14% vs 26%–48%).34
GOALS: LOWER THE BLOOD PRESSURE, SAVE THE KIDNEY
Because endovascular procedures pose some risk to the patient, it is critical to intervene only in patients most likely to respond clinically. The decision to intervene depends largely on the clinical goal, which should depend on the clinical presentation.

However, if renal artery stenosis is clinically silent, most of the evidence suggests that intervention has no benefit. Furthermore, although retrospective studies have indicated that intervention may improve survival rates,35,36 prospective studies have not. Similarly, studies have not shown that intervention generally improves cardiovascular outcomes, even though renal artery stenosis is associated with cardiovascular risk.
Hypertension plus stenosis is not necessarily renovascular hypertension
Essential hypertension and clinically silent renal artery stenosis often coexist, which is why blood pressure control often does not improve after stenting. Also, essential hypertension often coexists with renovascular hypertension.37 In this situation, stenting may not eliminate the need for antihypertensive drugs, although it may improve blood pressure control and decrease the drug burden.
Before stents came into use, several randomized controlled trials found that blood pressure was no better controlled after angioplasty, 2,3,38 except in cases of bilateral stenosis.2 This may be because stenosis tended to recur after angioplasty without stents.
The 2000 Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) study was the first randomized controlled trial to examine the effect of angioplasty on blood pressure control in renal artery stenosis.38 It had significant design flaws. For example, many patients crossed over from the medical management group to the intervention group because their hypertension was resistant to medical therapy. Overall, intervention (balloon angioplasty without stents in 54 of 56 patients, with stents in the other 2) carried no benefit. However, in subgroup analysis, the patients who crossed over because of resistant hypertension (failure of a three-drug regimen) were more likely to benefit from angioplasty. This suggested that risk stratification should take place early on, before proceeding with revascularization.
With stents, Zeller,39 in a prospective nonrandomized study, found that the mean arterial pressure decreased by 10 mm Hg. Randomized trials (see below) have failed to demonstrate such a benefit.
Stenting may not improve renal function
Coincidental renal artery stenosis in a patient with unrelated chronic kidney disease is very hard to differentiate from true ischemic nephropathy. Furthermore, most patients with ischemic nephropathy do not benefit from revascularization, making it challenging to identify those few whose renal function may respond.
Given that patients with chronic kidney disease tend to have a higher risk of cardiovascular disease, it is not surprising that 15% of them may have renal artery stenosis,4 most often incidental.
Chábová40 examined the outcomes of 68 patients who had chronic kidney disease and a renal artery lesion larger than 70% who did not undergo angioplasty. In only 10 (15%) of the patients did the glomerular filtration rate (GFR) decline by more than 50% of its baseline value during the study period of 3 years. Given the retrospective nature of the study, it cannot be determined (and is rather unlikely) that ischemic nephropathy was the cause of the decline in kidney function in all 10 patients.
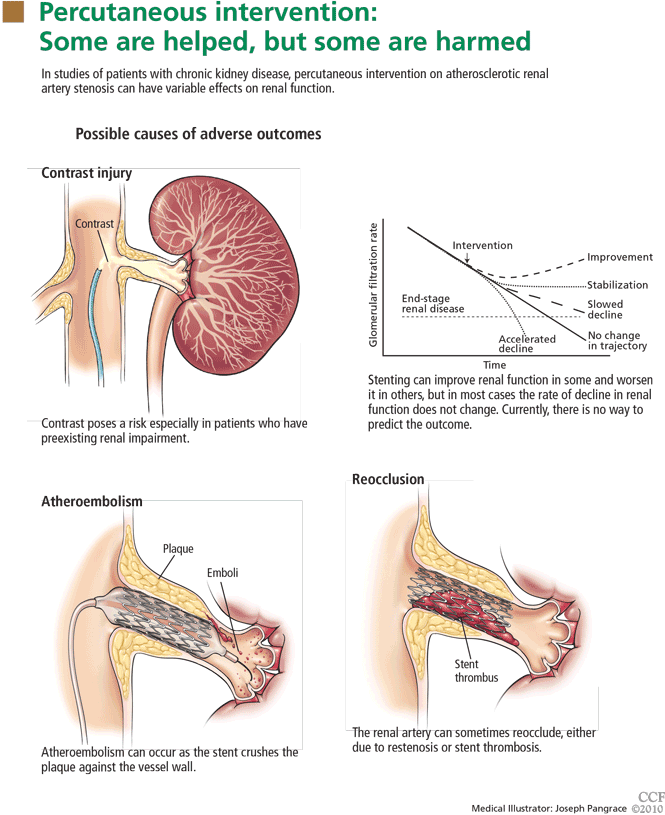
In a prospective cohort study in 304 patients with chronic kidney disease and renal artery stenosis who underwent surgical revascularization, Textor4 reported that the serum creatinine level showed a meaningful improvement afterward in 28%, worsened in 19.7%, and remained unchanged in 160 52.6%. (A “meaningful” change was defined as > 1.0 mg/dL.) Findings were similar in a cohort that underwent stenting.33
Davies et al41 found that 20% of patients who underwent renal stenting had a persistent increase in serum creatinine of 0.5 mg/dL or more. These patients were nearly three times more likely (19% vs 7%) to eventually require dialysis, and they had a lower 5-year survival rate (41% vs 71%).
Zeller et al39 found that renal function improved slightly in 52% of patients who received stents. The mean decrease in serum creatinine in this group was 0.22 mg/dL. However, the other 48% had a mean increase in serum creatinine of 1.1 mg/dL.
From these data we can conclude that, in an unselected population with renal artery stenosis, stenting provides no benefit to renal function, and that the risk of a worsening of renal function after intervention is roughly equal to the likelihood of achieving any benefit.
Other predictors of improvement in renal function have been proposed, but the evidence supporting them has not been consistent. For example, although Radermacher et al42 reported that a renal resistive index (which reflects arterial stiffness downstream of the stenosis) lower than 0.8 predicted a response in renal function, this finding has not been reliably reproduced.43,44 Similarly, while several studies suggest that patients with milder renal dysfunction have a higher likelihood of a renal response,45,46 other studies suggest either that the opposite is true39 or that baseline renal function alone has no impact on outcome.47
In addition, once significant renal atrophy occurs, revascularization may not help much, since irreversible sclerosis has developed. Thus, the goal is to identify kidneys being harmed by renal artery stenosis during the ischemic phase, when the tissue is still viable.
Unfortunately, we still lack a good renal stress test—eg, analogous to the cardiac stress test—to diagnose reversible ischemia in the kidney. The captopril renal scan has that capability but is not accurate in patients with bilateral stenosis or a GFR less than 50 mL/min, severely limiting its applicability.26 Newer technologies such as blood-oxygen-level-dependent (BOLD) magnetic resonance imaging are being investigated for such a role.48
Cohort studies in patients with declining renal function
In several case series, patients whose renal function had been declining before intervention had impressive rates of better renal function afterward.33,39,47,49–54 In a prospective cohort study by Muray et al,47 a rise in serum creatinine of more than 0.1 mg/mL/month before intervention seemed to predict an improvement in renal function afterward.
One would expect that, for renal function to respond to intervention, severe bilateral stenosis or unilateral stenosis to a solitary functioning kidney would need to be present. However, this was an inconsistent finding in these case series.33,39,47,52,53 The Angioplasty and Stent for Renal Artery Lesions (ASTRAL) trial,6,7 discussed later, sheds a bit more light on this.
Stenting usually improves flash pulmonary edema
Acute pulmonary edema in the setting of bilateral renal artery stenosis seems to be a unique case in which improvement in clinical status can be expected in most patients after intervention. Blood pressure improves in 94% to 100% of patients,28,31 renal function either improves or stabilizes in 77% to 91%,28–31 and pulmonary edema resolves without recurrence in 77% to 100%.28–30
NEW RANDOMIZED TRIALS: STAR, ASTRAL, AND CORAL
Despite the lack of evidence supporting revascularization of renal artery stenosis, many interventionalists practice under the assumption that the radiographic finding of renal artery stenosis alone is an indication for renal revascularization. Only three randomized controlled trials in the modern era attempt to examine this hypothesis: STAR, ASTRAL, and CORAL.
STAR: No clear benefit
The Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial5 was a European multicenter trial that enrolled 140 patients with ostial renal artery stenosis greater than 50%, blood pressure controlled to less than 140/90 mm Hg, and creatinine clearance 15 to 80 mL/min.
Patients were randomized to undergo stenting or medical therapy alone. High blood pressure was treated according to a protocol in which angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers were relegated to second-line use. All patients received a statin, regardless of lipid levels.
At 2 years, the primary end point (a decline in creatinine clearance of 20% or greater) had been reached in 10 (16%) of the 64 patients in the stent group and 16 (22%) of the 76 patients in the medication group; the difference was not statistically significant (hazard ratio 0.73, 95% confidence interval 0.33–1.61). No difference was seen in the secondary end points of the degree of blood pressure control or the rates of cardiovascular morbidity and death.5
ASTRAL: Also no clear benefit
In the international, multicenter ASTRAL trial,6,7 806 patients with at least one stenotic renal artery considered suitable for balloon angioplasty, stenting, or both7 were randomized to undergo intervention or medical management. Hypertension treatment was not specified by a protocol. The mean estimated GFR was 40 mL/min. Most patients (95%–96%) were on statin therapy. The primary outcome was the rate of decline of renal function over time. Secondary outcomes included blood pressure control, renal events, cardiovascular events, and death.
Results. At a mean follow-up of 33.6 months (range 1–4 years), no difference was noted between treatment groups in decline in renal function or blood pressure control at 1 year. Renal function worsened slightly in both groups.
The decline in renal function over time, calculated as the mean slope of the reciprocal of the serum creatinine level over time, was slightly slower in the revascularization group, but the difference was not statistically significant (−0.07 × 10−3 vs −0.13 × 10−3 L/μmol/year, P = .06). This difference did not appear until the last year of the study. There was no difference in the number of patients whose renal function improved or declined during the study.
There was no difference in the rate of any secondary outcome. The medical management group required a slightly higher number of antihypertensive drugs, reaching statistical but not clinical significance (2.97 vs 2.77 drugs, P = .03). More people in the revascularization group were taking ACE inhibitors or angiotensin receptor blockers. There was no difference in the number of patients on any antihypertensive therapy (97% vs 99%). Interestingly, amputations were more common in the revascularization group, occurring in 42 (12%) of the 386 patients in the revascularization group vs 29 (7%) of the 395 patients in the medical group (P = .04).
Seventeen percent of patients randomized to intervention did not have the procedure done. An as-treated analysis of the 317 (83%) patients randomized to revascularization who did receive it showed no differences in outcomes.
There were no differences in outcomes among specific, predefined subgroups based on severity of stenosis at baseline, renal length, baseline estimated GFR, baseline serum creatinine, and rate of progression of renal dysfunction before randomization.7
Comments. ASTRAL contradicts previous nonrandomized studies that suggested that rapidly declining renal function (loss of 20% in 1 year) predicts response to intervention. Considering the large number of patients with unilateral disease in the study, it would be interesting to see what effect stenting had on patients with both severe disease and declining renal function.
ASTRAL has been criticized because it lacked a central laboratory to interpret the severity of stenosis, it did not use a standardized intervention technique (5% of patients underwent angioplasty without stents, although this did not affect outcomes7), and patients were enrolled only if the clinician involved in the case was uncertain of the appropriate management.
This last issue raises the concern for selection bias toward inclusion of more difficult cases that may not respond to intervention. But these shortcomings are not serious enough to negate the fact that preliminary results from the largest randomized controlled trial to date confirm conclusions of other randomized trials, ie, that intervention in renal artery stenosis yields no benefits over medical management in most patients.
Based on the results of STAR and ASTRAL, the practice of indiscriminately revascularizing stenosed renal arteries without strong evidence that the procedure will provide a clinical benefit is no longer tenable. The challenge is to identify those few patients who will respond, and to intervene only on them. Unfortunately, none of the subgroups from ASTRAL helped characterize this population.
CORAL: A large trial is ongoing
The Cardiovascular Outcomes in Renal Artherosclerotic Lesions (CORAL) trial,8 an ongoing multicenter randomized controlled trial in the United States, may be of additional help.
Unlike ASTRAL, CORAL is studying patients who have difficult-to-control hypertension (systolic blood pressure ≥ 155 mm Hg on two or more drugs).8 Chronic kidney disease is not an exclusion criterion unless the serum creatinine concentration is greater than 3.0 mg/dL.
CORAL is using a standardized medical protocol to control blood pressure. In addition, use of embolic protection devices during stenting is encouraged. Hopefully, the large size (a goal of 1,080 patients) and the inclusion of patients with more marked hypertension will address the utility of intervention in higher-risk populations with renal artery stenosis.
RECOMMENDED APPROACH TO INTERVENTION IN RENAL ARTERY STENOSIS
As we wait for CORAL to be completed, we have two modern-era randomized controlled trials that leave us with fewer indications for renal intervention. Table 2 lists commonly cited indications for intervention in renal artery stenosis and the evidence to support them. As most of these are based on retrospective data or have conflicting support in the literature, their utility remains in question. At this point we can safely recommend:
- Patients with preserved or even decreased but stable renal function will not likely have a benefit in renal function after intervention.
- Patients with resistant hypertension may benefit.
- The best evidence supporting intervention is for bilateral stenosis with flash pulmonary edema, but the evidence is from retrospective studies.
- Stenting in bilateral disease without another indication has no apparent benefit.
- Declining renal function is not a guarantee of success.
- It is unclear if patients with severe bilateral stenosis or severe stenosis to a solitary functioning kidney with declining renal function will benefit. Anecdotally, they do respond more often, but as with many other indications for intervention that have gone by the wayside, this may not bear out when studied properly.

As the utility of intervention narrows, the scope of practice for such interventions should narrow accordingly. Attention should now be focusing on clinical, rather than radiographic, indications for intervening on renal artery stenosis.
Therefore, the decision to intervene must not be made solely by the interventionalist. A multidisciplinary approach should be adopted that at the very least includes the input of a nephrologist well versed in renal artery stenosis. In this way, the clinical risks and benefits of renal intervention can be discussed with the patient by providers who are likely to be involved in their care should renal function or hypertension fail to improve afterward.
RISK OF ATHEROEMBOLISM
While renal stenting yields improved technical success in the treatment of renal artery stenosis, it carries with it an increasingly common risk to kidney function: atheroembolism as the stent crushes the plaque against the vessel wall. This may lead to obstruction of the renal microvasculature, increasing the risk of irreversible damage to renal function.
Atheroembolic kidney disease can manifest as progressive renal failure occurring over weeks to months, commonly misdiagnosed as permanent damage from contrast nephropathy.55
Embolic protection devices, inserted downstream of the lesion before stenting to catch any debris that may break loose, have been developed to help address this problem.
Holden et al 57 prospectively studied 63 patients with renal artery stenosis and deteriorating renal function (undefined) who underwent stenting with an embolic protection device. At 6 months after the intervention, renal function had either improved or stabilized in 97% of patients, suggesting that many of the deleterious effects of stenting on renal function are related to atheroembolism.
The Prospective Randomized Study Comparing Renal Artery Stenting With or Without Distal Protection (RESIST) trial, in which renal dysfunction was mild and the GFR was not declining (average estimated GFR 59.3 mL/min), found contrary results.57 In a two-by-two factorial study, patients were randomized to undergo stenting alone, stenting with the antiplatelet agent abciximab (ReoPro), stenting with an embolic protection device, or stenting with both abciximab and an embolic protection device. Interestingly, renal function declined in the first three groups, but remained stable in the group that received both abciximab and an embolic protection device.
ANTIPLATELET THERAPY AFTER RENAL STENTING: HOW LONG?
We have no data on the optimal duration of antiplatelet therapy after renal stenting, and guidelines from professional societies do not comment on it.58 As a result, practice patterns vary significantly among practitioners.
While in-stent restenosis rates are acceptably low after renal stenting, the risks and side effects of antiplatelet therapy often lead to arbitrary withdrawal of these drugs. The effect on stent patency is yet to be determined.
FUTURE DEVELOPMENTS
Results of STAR and ASTRAL confirm the growing suspicion that the surge seen in the last decade in renal artery stenting should be coming to an end. We await results either from CORAL or possibly a post hoc analysis of ASTRAL that might identify potential high-risk groups that will benefit from renal intervention. And as embolic protection devices become more agile and suitable to different renal lesions, there remains the possibility that, due to lower rates of unidentified atheroembolic kidney disease, CORAL may demonstrate improved renal outcomes after stenting. If not, the search for the best means to predict who should have renal intervention will continue.
We know through experience that stenting does provide great benefits for some patients with renal artery stenosis. Furthermore, the clinical problem is too intriguing, and too profitable, to die altogether.
- Choncol M, Linas S. Diagnosis and management of ischemic nephropathy. Clin J Am Soc Nephrol 2006; 1:172–181.
- Webster J, Marshall F, Abdalla M, et al Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 1998; 12:329–335.
- Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 1998; 31:823–829.
- Textor SC. Revascularization in atherosclerotic renal artery disease. Kidney Int 1998; 53:799–811.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–848.
- Mistry S, Ives N, Harding J, et al Angioplasty and STent for Renal Artery Lesions (ASTRAL trial): rationale, methods and results so far. J Hum Hypertens 2007; 21:511–515.
- Wheatley K, Ives N, Kalra P, Moss J. Revascularization versus medical therapy for renal-artery stenosis (ASTRAL). N Engl J Med 2009; 361:1953–1962.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Galaria II, Surowiec SM, Rhodes JM, et al Percutaneous and open renal revascularizations have equivalent long-term functional outcomes. Ann Vasc Surg 2005; 19:218–228.
- Murphy TP, Soares G, Kim M. Increase in utilization of percutaneous renal artery interventions by Medicare beneficiaries 1996–2000. AJR Am J Roentgenol 2004; 183:561–568.
- Textor SC. Atherosclerotic renal artery stenosis: overtreated but underrated? J Am Soc Nephrol 2008; 19:656–659.
- Kalra PA, Guo H, Gilbertson DT, et al Atherosclerotic renovascular disease in the United States. Kidney Int 2010; 77:37–43.
- Hansen KJ, Edwards MS, Craven TE, et al Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Cohen MG, Pascua JA, Garcia-Ben M, et al A simple prediction rule for significant renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 2005; 150:1204–1211.
- Buller CE, Nogareda JG, Ramanathan K, et al The profile of cardiac patients with renal artery stenosis. J Am Coll Cardiol 2004; 43:1606–1613.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- Kalra PA, Guo H, Kausz AT, et al Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 69:293–301.
- Holley KE, Hunt JC, Brown AL, Kincaid OW, Sheps SG. Renal artery stenosis. A clinical-pathologic study in normotensive and hypertensive patients. Am J Med 1964; 37:14–22.
- de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systemic literature review. J Hypertens 2009; 27:1333–1340.
- Kuczera P, Włoszczynska E, Adamczak M, Pencak P, Chudek J, Wiecek A. Frequency of renal artery stenosis and variants of renal vascularization in hypertensive patients: analysis of 1550 angiographies in one centre. J Hum Hypertens 2009; 23:396–401.
- Caps MT, Perissinotto C, Zierler RE, et al Prospective study of atherosclerotic disease progression in the renal artery. Circulation 1998; 98:2866–2872.
- Zierler RE, Bergelin RO, Davidson RC, Cantwell-Gab K, Polissar NL, Strandness DE. A prospective study of disease progression in patients with atherosclerotic renal artery stenosis. Am J Hypertens 1996; 9:1055–1061.
- Schreiber MJ, Pohl MA, Novick AC. The natural history of atherosclerotic and fibrous renal artery disease. Urol Clin North Am 1984; 11:383–392.
- Crowley JJ, Santos RM, Peter RH, et al Progression of renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 1998; 136:913–918.
- Textor SC. Renovascular hypertension update. Curr Hypertens Rep 2006; 8:521–527.
- Textor SC. Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004; 15:1974–1982.
- Wright JR, Shurrab AE, Cheung C, et al A prospective study of the determinants of renal functional outcome and mortality in atherosclerotic renovascular disease. Am J Kidney Dis 2002; 39:1153–1161.
- Messina LM, Zelenock GB, Yao KA, Stanley JC. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg 1992; 15:73–80.
- Bloch MJ, Trost DW, Pickering TG, Sos TA, August P. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. Am J Hypertens 1999; 12:1–7.
- Gray BH, Olin JW, Childs MB, Sullivan TM, Bacharach JM. Clinical benefit of renal artery angioplasty with stenting for the control of recurrent and refractory congestive heart failure. Vasc Med 2002; 7:275–279.
- Pickering TG, Herman L, Devereux RB, et al Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet 1988; 2:551–552.
- Kennedy DJ, Colyer WR, Brewster PS, et al Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis 2003; 14:926–935.
- Beutler JJ, Van Ampting JM, Van De Ven PJ, et al Long-term effects of arterial stenting on kidney function for patients with ostial atherosclerotic renal artery stenosis and renal insufficiency. J Am Soc Nephrol 2001; 12:1475–1481.
- Van de Ven PJ, Kaatee R, Beutler JJ, et al Arterial stenting and balloon angioplasty in ostial atherosclerotic renovascular disease: a randomized trial. Lancet 1999; 353:282–286.
- Isles C, Main J, O’Connell J, et al Survival associated with renovascular disease in Glasgow and Newcastle: a collaborative study. Scott Med J 1990; 35:70–73.
- Hunt JC, Sheps SG, Harrison EG, Strong CG, Bernatz PE. Renal and renovascular hypertension. A reasoned approach to diagnosis and management. Arch Intern Med 1974; 133:988–999.
- Textor SC. Atherosclerotic renal artery stenosis: how big is the problem, and what happens if nothing is done? J Hypertens Suppl 2005; 23:S5–S13.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Zeller T, Frank U, Müller C, et al Predictors of improved renal function after percutaneous stent-supported angioplasty of severe atherosclerotic ostial renal artery stenosis. Circulation 2003; 108;2244–2249.
- Chábová V, Schirger A, Stanson AW, McKusick MA, Textor SC. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc 2000; 75:437–444.
- Davies MG, Saad WE, Peden EK, Mohiuddin IT, Naoum JJ, Lumsden AB. Implications of acute functional injury following percutaneous renal artery intervention. Ann Vasc Surg 2008; 22:783–789.
- Radermacher J, Chavin A, Bleck J, et al Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Eng J Med 2001; 344:410–417.
- García-Criado A, Gilabert R, Nicolau C, et al Value of Doppler sonography for predicting clinical outcome after renal artery revascularization in atherosclerotic renal artery stenosis. J Ultrasound Med 2005; 24:1641–1647.
- Zeller T, Müller C, Frank U, et al Stent angioplasty of severe atherosclerotic ostial renal artery stenosis in patients with diabetes mellitus and nephrosclerosis. Catheter Cardiovasc Interv 2003; 58:510–515.
- Harden PN, MacLeod MJ, Rodger RSC, et al Effect of renal-artery stenting on progression of renovascular renal failure. Lancet 1997; 349:1133–1136.
- Isles CG, Robertson S, Hill D. Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 1999; 92:159–167.
- Muray S, Martın M, Amoedo ML, et al Rapid decline in renal function reflects reversibility and predicts the outcome after angioplasty in renal artery stenosis. Am J Kidney Dis 2002; 39:60–66.
- Textor SC, Glockner JF, Lerman LO, et al The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 2008; 19:780–788.
- Paraskevas KI, Perrea D, Briana DD, Liapis CD. Management of atherosclerotic renovascular disease: the effect of renal artery stenting on renal function and blood pressure. Int Urol Nephrol 2006; 38:683–691.
- Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000; 102:1671–1677.
- Dean RH, Kieffer RW, Smith BM, et al Renovascular hypertension: anatomic and renal function changes during drug therapy. Arch Surg 1981; 116:1408–1415.
- Zhang Q, Shen W, Zhang R, Zhang J, Hu J, Zhang X. Effects of renal artery stenting on renal function and blood pressure in patients with atherosclerotic renovascular disease. Chin Med J (Engl) 2003; 116:1451–1454.
- Ramos F, Kotliar C, Alvarez D, et al Renal function and outcome of PTRA and stenting for atherosclerotic renal artery stenosis. Kidney Int 2003; 63:276–282.
- Rocha-Singh KJ, Ahuja RK, Sung CH, Rutherford J. Long-term renal function preservation after renal artery stenting in patients with progressive ischemic nephropathy. Catheter Cardiovasc Interv 2002; 57:135–141.
- Thadhani RI, Camargo CA, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore) 1995; 74:350–358.
- Holden A, Hill A, Jaff MR, Pilmore H. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int 2006; 70:948–955.
- Cooper CJ, Haller ST, Colyer W, et al Embolic protection and platelet inhibition during renal artery stenting. Circulation 2008; 117:2752–2760.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
Author’s note: Atherosclerosis accounts for about 90% of cases of renal artery stenosis in people over age 40.1 Fibromuscular dysplasia, the other major cause, is a separate topic; in this paper “renal artery stenosis” refers to atherosclerotic disease only.
Renal artery stenosis is very common, and the number of angioplasty-stenting procedures performed every year is on the rise. Yet there is no overwhelming evidence that intervention yields clinical benefits—ie, better blood pressure control or renal function— than does medical therapy.
Earlier randomized controlled trials comparing angioplasty without stents and medical management showed no impressive difference in blood pressure.2,3 The data on renal function were even more questionable, with some studies suggesting that, with stenting, the chance of worsening renal function is equal to that of improvement.4
Two large randomized trials comparing renal intervention with medical therapy failed to show any benefit of intervention.5–7 A third study is under way.8
It is time to strongly reconsider the current aggressive approach to revascularization of stenotic renal arteries and take a more coordinated, critical approach.
RENAL INTERVENTIONS ON THE RISE
Renal angioplasty began replacing surgical revascularization in the 1990s, as this less-invasive procedure became more readily available and was shown to have similar clinical outcomes.9 In the last decade, stent placement during angioplasty has become standard, improving the rates of technical success.
As these procedures have become easier to perform and their radiographic outcomes have become more consistent, interventionalists have become more likely, if they see stenosis in a renal artery, to intervene and insert a stent, regardless of proven benefit. In addition, interventionalists from at least three different specialties now compete for these procedures, often by looking at the renal arteries during angiography of other vascular beds (the “drive-by”).
As a result, the number of renal interventions has been rising. Medicare received 21,660 claims for renal artery interventions (surgery or angioplasty) in 2000, compared with 13,380 in 1996—an increase of 62%. However, the number of surgeries actually decreased by 45% during this time, while the number of percutaneous procedures increased by 240%. The number of endovascular claims for renal artery stenosis by cardiologists alone rose 390%.10 Since then, the reports on intervention have been mixed, with one report citing a continued increase in 2005 to 35,000 claims,11 and another suggesting a decrease back to 1997 levels.12
HOW COMMON IS RENAL ARTERY STENOSIS?
The prevalence of renal artery stenosis depends on the definition used and the population screened. It is more common in older patients who have risk factors for other vascular diseases than in the general population.
Renal Doppler ultrasonography can detect stenosis only if the artery is narrowed by more than 60%. Hansen et al13 used ultrasonography to screen 870 people over age 65 and found a lesion (a narrowing of more than 60%) in 6.8%.
Angiography (direct, computed tomographic, or magnetic resonance) can detect less-severe stenosis. Thus, most angiographic studies define renal artery stenosis as a narrowing of more than 50%, and severe disease as a narrowing of more than 70%. Many experts believe that unilateral stenosis needs to be more than 70% to pose a risk to the kidney.14,15
Angiographic prevalence studies have been performed only in patients who were undergoing angiography for another reason such as coronary or peripheral arterial disease that inherently places them at higher risk of renal artery stenosis. For instance, renal artery stenosis is found in 11% to 28% of patients undergoing diagnostic cardiac catheterization. 16
No studies of the prevalence of renal artery stenosis have been performed in the general population. Medicare data indicate that from 1999 to 2001 the incidence of diagnosed renal artery stenosis was 3.7 per 1,000 patientyears. 17 Holley et al,18 in an autopsy series, found renal artery stenosis of greater than 50% in 27% of patients over age 50 and in 56.4% of hypertensive patients. The prevalence was 10% in normotensive patients.
WHO IS AT RISK?
Factors associated with a higher risk of finding renal artery stenosis on a radiographic study include14:
- Older age
- Female sex
- Hypertension
- Three-vessel coronary artery disease
- Peripheral artery disease
- Chronic kidney disease
- Diabetes
- A low level of high-density lipoprotein cholesterol
- The use of at least two cardiovascular drugs.
The prevalence of renal artery stenosis in at-risk populations ranges from 3% to 75% (Table 1).2,4,6,19,20
HOW OFTEN DOES STENOSIS PROGRESS?
The reported rates of progression of atherosclerotic renal artery lesions vary depending on the type of imaging test used and the reason for doing it.
In studies that used duplex ultrasonography, roughly half of lesions smaller than 60% grew to greater than 60% over 3 years.21,22 The risk of total occlusion of an artery was relatively low and depended on the severity of stenosis: 0.7% if the baseline stenosis was less than 60% and 2.3% to 7% if it was greater.21,22
In a seminal study in 1984, Schreiber and colleagues23 compared serial angiograms obtained a mean of 52 months apart in 85 patients who did not undergo intervention. Stenosis had progressed in 37 (44%), and to the point of total occlusion in 14 (16%). In contrast, a 1998 study found progression in 11.1% over 2.6 years, with older patients, women, and those with baseline coronary artery disease at higher risk.24
The the rates of progression differed in these two studies probably because the indications for screening were different (clinical suspicion23 vs routine screening during coronary angiography24), as was the severity of stenosis at the time of diagnosis. Also, when these studies were done, fewer people were taking statins. Thus, similar studies, if repeated, might show even lower rates of progression.
Finally, progression of renal artery stenosis has not been correlated with worsening renal function.
FOUR CLINICAL PRESENTATIONS OF RENAL ARTERY STENOSIS
Renal artery stenosis can present in one of four ways:
Clinically silent stenosis. Because renal artery stenosis is most often found in older patients, who are more likely to have essential hypertension and chronic kidney disease due to other causes, it can be an incidental finding that is completely clinically silent.16,25
Renovascular hypertension is defined as high blood pressure due to up-regulation of neurohormonal activity in response to decreased perfusion from renal artery stenosis. Renal artery stenosis is estimated to be the cause of hypertension in only 0.5% to 4.0% of hypertensive patients, or in 26% of patients with secondary hypertension.3
Ischemic nephropathy is more difficult to define because ischemia alone rarely explains the damage done to the kidneys. Activation of neurohormonal pathways and microvascular injury are thought to contribute to oxidative stress and fibrosis.26 These phenomena may explain why similar degrees of stenosis lead to varying degrees of kidney damage in different patients and why the severity of stenosis does not correlate with the degree of renal dysfunction.27
Furthermore, stenosis may lead to irreversible but stable kidney damage. It is therefore not surprising that, in studies in unselected populations (ie, studies that included patients with all presentations of renal artery stenosis, not just those more likely to benefit), up to two-thirds of renal interventions yielded no clinical benefit.25
As a result, if we define ischemic nephropathy as renal artery stenosis with renal dysfunction not attributable to another cause, we probably will overestimate the prevalence of ischemic nephropathy, leading to overly optimistic expectations about the response to revascularization.
Recurrent “flash” pulmonary edema is a less common presentation, usually occurring in patients with critical bilateral renal artery stenosis or unilateral stenosis in an artery supplying a solitary functioning kidney. Most have severe hypertension (average systolic blood pressure 174–207 mm Hg) and poor renal function.28–30
The association between pulmonary edema and bilateral renal artery stenosis was first noted in 1998 by Pickering et al,31 who in several case series showed that 82% to 92% of patients with recurrent pulmonary edema and renal artery stenosis had bilateral stenosis, compared with 20% to 65% of those with other presentations. Later case series corroborated this finding: 85% to 100% of patients with renal artery stenosis and pulmonary edema had bilateral stenosis.28–30
STENTING IS NOW STANDARD
Stenting has become standard in the endovascular treatment of renal artery stenosis.
Most atherosclerotic renal artery lesions are located in the ostium (ie, where the artery branches off from the aorta), and many are extensions of calcified aortic plaque.26,32 These hard lesions tend to rebound to their original shape more often with balloon angioplasty alone. Stenting provides the additional force needed to permanently disrupt the lesion, leading to a longer-lasting result.
Rates of technical success (dilating the artery during the intervention) are higher with stents than without them (98% vs 46%– 77%).33,34 If the lesion is ostial, this difference is even more impressive (75% vs 29%). In addition, restenosis rates at 6 months are lower with stents (14% vs 26%–48%).34
GOALS: LOWER THE BLOOD PRESSURE, SAVE THE KIDNEY
Because endovascular procedures pose some risk to the patient, it is critical to intervene only in patients most likely to respond clinically. The decision to intervene depends largely on the clinical goal, which should depend on the clinical presentation.
However, if renal artery stenosis is clinically silent, most of the evidence suggests that intervention has no benefit. Furthermore, although retrospective studies have indicated that intervention may improve survival rates,35,36 prospective studies have not. Similarly, studies have not shown that intervention generally improves cardiovascular outcomes, even though renal artery stenosis is associated with cardiovascular risk.
Hypertension plus stenosis is not necessarily renovascular hypertension
Essential hypertension and clinically silent renal artery stenosis often coexist, which is why blood pressure control often does not improve after stenting. Also, essential hypertension often coexists with renovascular hypertension.37 In this situation, stenting may not eliminate the need for antihypertensive drugs, although it may improve blood pressure control and decrease the drug burden.
Before stents came into use, several randomized controlled trials found that blood pressure was no better controlled after angioplasty, 2,3,38 except in cases of bilateral stenosis.2 This may be because stenosis tended to recur after angioplasty without stents.
The 2000 Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) study was the first randomized controlled trial to examine the effect of angioplasty on blood pressure control in renal artery stenosis.38 It had significant design flaws. For example, many patients crossed over from the medical management group to the intervention group because their hypertension was resistant to medical therapy. Overall, intervention (balloon angioplasty without stents in 54 of 56 patients, with stents in the other 2) carried no benefit. However, in subgroup analysis, the patients who crossed over because of resistant hypertension (failure of a three-drug regimen) were more likely to benefit from angioplasty. This suggested that risk stratification should take place early on, before proceeding with revascularization.
With stents, Zeller,39 in a prospective nonrandomized study, found that the mean arterial pressure decreased by 10 mm Hg. Randomized trials (see below) have failed to demonstrate such a benefit.
Stenting may not improve renal function
Coincidental renal artery stenosis in a patient with unrelated chronic kidney disease is very hard to differentiate from true ischemic nephropathy. Furthermore, most patients with ischemic nephropathy do not benefit from revascularization, making it challenging to identify those few whose renal function may respond.
Given that patients with chronic kidney disease tend to have a higher risk of cardiovascular disease, it is not surprising that 15% of them may have renal artery stenosis,4 most often incidental.
Chábová40 examined the outcomes of 68 patients who had chronic kidney disease and a renal artery lesion larger than 70% who did not undergo angioplasty. In only 10 (15%) of the patients did the glomerular filtration rate (GFR) decline by more than 50% of its baseline value during the study period of 3 years. Given the retrospective nature of the study, it cannot be determined (and is rather unlikely) that ischemic nephropathy was the cause of the decline in kidney function in all 10 patients.
In a prospective cohort study in 304 patients with chronic kidney disease and renal artery stenosis who underwent surgical revascularization, Textor4 reported that the serum creatinine level showed a meaningful improvement afterward in 28%, worsened in 19.7%, and remained unchanged in 160 52.6%. (A “meaningful” change was defined as > 1.0 mg/dL.) Findings were similar in a cohort that underwent stenting.33
Davies et al41 found that 20% of patients who underwent renal stenting had a persistent increase in serum creatinine of 0.5 mg/dL or more. These patients were nearly three times more likely (19% vs 7%) to eventually require dialysis, and they had a lower 5-year survival rate (41% vs 71%).
Zeller et al39 found that renal function improved slightly in 52% of patients who received stents. The mean decrease in serum creatinine in this group was 0.22 mg/dL. However, the other 48% had a mean increase in serum creatinine of 1.1 mg/dL.
From these data we can conclude that, in an unselected population with renal artery stenosis, stenting provides no benefit to renal function, and that the risk of a worsening of renal function after intervention is roughly equal to the likelihood of achieving any benefit.
Other predictors of improvement in renal function have been proposed, but the evidence supporting them has not been consistent. For example, although Radermacher et al42 reported that a renal resistive index (which reflects arterial stiffness downstream of the stenosis) lower than 0.8 predicted a response in renal function, this finding has not been reliably reproduced.43,44 Similarly, while several studies suggest that patients with milder renal dysfunction have a higher likelihood of a renal response,45,46 other studies suggest either that the opposite is true39 or that baseline renal function alone has no impact on outcome.47
In addition, once significant renal atrophy occurs, revascularization may not help much, since irreversible sclerosis has developed. Thus, the goal is to identify kidneys being harmed by renal artery stenosis during the ischemic phase, when the tissue is still viable.
Unfortunately, we still lack a good renal stress test—eg, analogous to the cardiac stress test—to diagnose reversible ischemia in the kidney. The captopril renal scan has that capability but is not accurate in patients with bilateral stenosis or a GFR less than 50 mL/min, severely limiting its applicability.26 Newer technologies such as blood-oxygen-level-dependent (BOLD) magnetic resonance imaging are being investigated for such a role.48
Cohort studies in patients with declining renal function
In several case series, patients whose renal function had been declining before intervention had impressive rates of better renal function afterward.33,39,47,49–54 In a prospective cohort study by Muray et al,47 a rise in serum creatinine of more than 0.1 mg/mL/month before intervention seemed to predict an improvement in renal function afterward.
One would expect that, for renal function to respond to intervention, severe bilateral stenosis or unilateral stenosis to a solitary functioning kidney would need to be present. However, this was an inconsistent finding in these case series.33,39,47,52,53 The Angioplasty and Stent for Renal Artery Lesions (ASTRAL) trial,6,7 discussed later, sheds a bit more light on this.
Stenting usually improves flash pulmonary edema
Acute pulmonary edema in the setting of bilateral renal artery stenosis seems to be a unique case in which improvement in clinical status can be expected in most patients after intervention. Blood pressure improves in 94% to 100% of patients,28,31 renal function either improves or stabilizes in 77% to 91%,28–31 and pulmonary edema resolves without recurrence in 77% to 100%.28–30
NEW RANDOMIZED TRIALS: STAR, ASTRAL, AND CORAL
Despite the lack of evidence supporting revascularization of renal artery stenosis, many interventionalists practice under the assumption that the radiographic finding of renal artery stenosis alone is an indication for renal revascularization. Only three randomized controlled trials in the modern era attempt to examine this hypothesis: STAR, ASTRAL, and CORAL.
STAR: No clear benefit
The Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial5 was a European multicenter trial that enrolled 140 patients with ostial renal artery stenosis greater than 50%, blood pressure controlled to less than 140/90 mm Hg, and creatinine clearance 15 to 80 mL/min.
Patients were randomized to undergo stenting or medical therapy alone. High blood pressure was treated according to a protocol in which angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers were relegated to second-line use. All patients received a statin, regardless of lipid levels.
At 2 years, the primary end point (a decline in creatinine clearance of 20% or greater) had been reached in 10 (16%) of the 64 patients in the stent group and 16 (22%) of the 76 patients in the medication group; the difference was not statistically significant (hazard ratio 0.73, 95% confidence interval 0.33–1.61). No difference was seen in the secondary end points of the degree of blood pressure control or the rates of cardiovascular morbidity and death.5
ASTRAL: Also no clear benefit
In the international, multicenter ASTRAL trial,6,7 806 patients with at least one stenotic renal artery considered suitable for balloon angioplasty, stenting, or both7 were randomized to undergo intervention or medical management. Hypertension treatment was not specified by a protocol. The mean estimated GFR was 40 mL/min. Most patients (95%–96%) were on statin therapy. The primary outcome was the rate of decline of renal function over time. Secondary outcomes included blood pressure control, renal events, cardiovascular events, and death.
Results. At a mean follow-up of 33.6 months (range 1–4 years), no difference was noted between treatment groups in decline in renal function or blood pressure control at 1 year. Renal function worsened slightly in both groups.
The decline in renal function over time, calculated as the mean slope of the reciprocal of the serum creatinine level over time, was slightly slower in the revascularization group, but the difference was not statistically significant (−0.07 × 10−3 vs −0.13 × 10−3 L/μmol/year, P = .06). This difference did not appear until the last year of the study. There was no difference in the number of patients whose renal function improved or declined during the study.
There was no difference in the rate of any secondary outcome. The medical management group required a slightly higher number of antihypertensive drugs, reaching statistical but not clinical significance (2.97 vs 2.77 drugs, P = .03). More people in the revascularization group were taking ACE inhibitors or angiotensin receptor blockers. There was no difference in the number of patients on any antihypertensive therapy (97% vs 99%). Interestingly, amputations were more common in the revascularization group, occurring in 42 (12%) of the 386 patients in the revascularization group vs 29 (7%) of the 395 patients in the medical group (P = .04).
Seventeen percent of patients randomized to intervention did not have the procedure done. An as-treated analysis of the 317 (83%) patients randomized to revascularization who did receive it showed no differences in outcomes.
There were no differences in outcomes among specific, predefined subgroups based on severity of stenosis at baseline, renal length, baseline estimated GFR, baseline serum creatinine, and rate of progression of renal dysfunction before randomization.7
Comments. ASTRAL contradicts previous nonrandomized studies that suggested that rapidly declining renal function (loss of 20% in 1 year) predicts response to intervention. Considering the large number of patients with unilateral disease in the study, it would be interesting to see what effect stenting had on patients with both severe disease and declining renal function.
ASTRAL has been criticized because it lacked a central laboratory to interpret the severity of stenosis, it did not use a standardized intervention technique (5% of patients underwent angioplasty without stents, although this did not affect outcomes7), and patients were enrolled only if the clinician involved in the case was uncertain of the appropriate management.
This last issue raises the concern for selection bias toward inclusion of more difficult cases that may not respond to intervention. But these shortcomings are not serious enough to negate the fact that preliminary results from the largest randomized controlled trial to date confirm conclusions of other randomized trials, ie, that intervention in renal artery stenosis yields no benefits over medical management in most patients.
Based on the results of STAR and ASTRAL, the practice of indiscriminately revascularizing stenosed renal arteries without strong evidence that the procedure will provide a clinical benefit is no longer tenable. The challenge is to identify those few patients who will respond, and to intervene only on them. Unfortunately, none of the subgroups from ASTRAL helped characterize this population.
CORAL: A large trial is ongoing
The Cardiovascular Outcomes in Renal Artherosclerotic Lesions (CORAL) trial,8 an ongoing multicenter randomized controlled trial in the United States, may be of additional help.
Unlike ASTRAL, CORAL is studying patients who have difficult-to-control hypertension (systolic blood pressure ≥ 155 mm Hg on two or more drugs).8 Chronic kidney disease is not an exclusion criterion unless the serum creatinine concentration is greater than 3.0 mg/dL.
CORAL is using a standardized medical protocol to control blood pressure. In addition, use of embolic protection devices during stenting is encouraged. Hopefully, the large size (a goal of 1,080 patients) and the inclusion of patients with more marked hypertension will address the utility of intervention in higher-risk populations with renal artery stenosis.
RECOMMENDED APPROACH TO INTERVENTION IN RENAL ARTERY STENOSIS
As we wait for CORAL to be completed, we have two modern-era randomized controlled trials that leave us with fewer indications for renal intervention. Table 2 lists commonly cited indications for intervention in renal artery stenosis and the evidence to support them. As most of these are based on retrospective data or have conflicting support in the literature, their utility remains in question. At this point we can safely recommend:
- Patients with preserved or even decreased but stable renal function will not likely have a benefit in renal function after intervention.
- Patients with resistant hypertension may benefit.
- The best evidence supporting intervention is for bilateral stenosis with flash pulmonary edema, but the evidence is from retrospective studies.
- Stenting in bilateral disease without another indication has no apparent benefit.
- Declining renal function is not a guarantee of success.
- It is unclear if patients with severe bilateral stenosis or severe stenosis to a solitary functioning kidney with declining renal function will benefit. Anecdotally, they do respond more often, but as with many other indications for intervention that have gone by the wayside, this may not bear out when studied properly.
As the utility of intervention narrows, the scope of practice for such interventions should narrow accordingly. Attention should now be focusing on clinical, rather than radiographic, indications for intervening on renal artery stenosis.
Therefore, the decision to intervene must not be made solely by the interventionalist. A multidisciplinary approach should be adopted that at the very least includes the input of a nephrologist well versed in renal artery stenosis. In this way, the clinical risks and benefits of renal intervention can be discussed with the patient by providers who are likely to be involved in their care should renal function or hypertension fail to improve afterward.
RISK OF ATHEROEMBOLISM
While renal stenting yields improved technical success in the treatment of renal artery stenosis, it carries with it an increasingly common risk to kidney function: atheroembolism as the stent crushes the plaque against the vessel wall. This may lead to obstruction of the renal microvasculature, increasing the risk of irreversible damage to renal function.
Atheroembolic kidney disease can manifest as progressive renal failure occurring over weeks to months, commonly misdiagnosed as permanent damage from contrast nephropathy.55
Embolic protection devices, inserted downstream of the lesion before stenting to catch any debris that may break loose, have been developed to help address this problem.
Holden et al 57 prospectively studied 63 patients with renal artery stenosis and deteriorating renal function (undefined) who underwent stenting with an embolic protection device. At 6 months after the intervention, renal function had either improved or stabilized in 97% of patients, suggesting that many of the deleterious effects of stenting on renal function are related to atheroembolism.
The Prospective Randomized Study Comparing Renal Artery Stenting With or Without Distal Protection (RESIST) trial, in which renal dysfunction was mild and the GFR was not declining (average estimated GFR 59.3 mL/min), found contrary results.57 In a two-by-two factorial study, patients were randomized to undergo stenting alone, stenting with the antiplatelet agent abciximab (ReoPro), stenting with an embolic protection device, or stenting with both abciximab and an embolic protection device. Interestingly, renal function declined in the first three groups, but remained stable in the group that received both abciximab and an embolic protection device.
ANTIPLATELET THERAPY AFTER RENAL STENTING: HOW LONG?
We have no data on the optimal duration of antiplatelet therapy after renal stenting, and guidelines from professional societies do not comment on it.58 As a result, practice patterns vary significantly among practitioners.
While in-stent restenosis rates are acceptably low after renal stenting, the risks and side effects of antiplatelet therapy often lead to arbitrary withdrawal of these drugs. The effect on stent patency is yet to be determined.
FUTURE DEVELOPMENTS
Results of STAR and ASTRAL confirm the growing suspicion that the surge seen in the last decade in renal artery stenting should be coming to an end. We await results either from CORAL or possibly a post hoc analysis of ASTRAL that might identify potential high-risk groups that will benefit from renal intervention. And as embolic protection devices become more agile and suitable to different renal lesions, there remains the possibility that, due to lower rates of unidentified atheroembolic kidney disease, CORAL may demonstrate improved renal outcomes after stenting. If not, the search for the best means to predict who should have renal intervention will continue.
We know through experience that stenting does provide great benefits for some patients with renal artery stenosis. Furthermore, the clinical problem is too intriguing, and too profitable, to die altogether.
Author’s note: Atherosclerosis accounts for about 90% of cases of renal artery stenosis in people over age 40.1 Fibromuscular dysplasia, the other major cause, is a separate topic; in this paper “renal artery stenosis” refers to atherosclerotic disease only.
Renal artery stenosis is very common, and the number of angioplasty-stenting procedures performed every year is on the rise. Yet there is no overwhelming evidence that intervention yields clinical benefits—ie, better blood pressure control or renal function— than does medical therapy.
Earlier randomized controlled trials comparing angioplasty without stents and medical management showed no impressive difference in blood pressure.2,3 The data on renal function were even more questionable, with some studies suggesting that, with stenting, the chance of worsening renal function is equal to that of improvement.4
Two large randomized trials comparing renal intervention with medical therapy failed to show any benefit of intervention.5–7 A third study is under way.8
It is time to strongly reconsider the current aggressive approach to revascularization of stenotic renal arteries and take a more coordinated, critical approach.
RENAL INTERVENTIONS ON THE RISE
Renal angioplasty began replacing surgical revascularization in the 1990s, as this less-invasive procedure became more readily available and was shown to have similar clinical outcomes.9 In the last decade, stent placement during angioplasty has become standard, improving the rates of technical success.
As these procedures have become easier to perform and their radiographic outcomes have become more consistent, interventionalists have become more likely, if they see stenosis in a renal artery, to intervene and insert a stent, regardless of proven benefit. In addition, interventionalists from at least three different specialties now compete for these procedures, often by looking at the renal arteries during angiography of other vascular beds (the “drive-by”).
As a result, the number of renal interventions has been rising. Medicare received 21,660 claims for renal artery interventions (surgery or angioplasty) in 2000, compared with 13,380 in 1996—an increase of 62%. However, the number of surgeries actually decreased by 45% during this time, while the number of percutaneous procedures increased by 240%. The number of endovascular claims for renal artery stenosis by cardiologists alone rose 390%.10 Since then, the reports on intervention have been mixed, with one report citing a continued increase in 2005 to 35,000 claims,11 and another suggesting a decrease back to 1997 levels.12
HOW COMMON IS RENAL ARTERY STENOSIS?
The prevalence of renal artery stenosis depends on the definition used and the population screened. It is more common in older patients who have risk factors for other vascular diseases than in the general population.
Renal Doppler ultrasonography can detect stenosis only if the artery is narrowed by more than 60%. Hansen et al13 used ultrasonography to screen 870 people over age 65 and found a lesion (a narrowing of more than 60%) in 6.8%.
Angiography (direct, computed tomographic, or magnetic resonance) can detect less-severe stenosis. Thus, most angiographic studies define renal artery stenosis as a narrowing of more than 50%, and severe disease as a narrowing of more than 70%. Many experts believe that unilateral stenosis needs to be more than 70% to pose a risk to the kidney.14,15
Angiographic prevalence studies have been performed only in patients who were undergoing angiography for another reason such as coronary or peripheral arterial disease that inherently places them at higher risk of renal artery stenosis. For instance, renal artery stenosis is found in 11% to 28% of patients undergoing diagnostic cardiac catheterization. 16
No studies of the prevalence of renal artery stenosis have been performed in the general population. Medicare data indicate that from 1999 to 2001 the incidence of diagnosed renal artery stenosis was 3.7 per 1,000 patientyears. 17 Holley et al,18 in an autopsy series, found renal artery stenosis of greater than 50% in 27% of patients over age 50 and in 56.4% of hypertensive patients. The prevalence was 10% in normotensive patients.
WHO IS AT RISK?
Factors associated with a higher risk of finding renal artery stenosis on a radiographic study include14:
- Older age
- Female sex
- Hypertension
- Three-vessel coronary artery disease
- Peripheral artery disease
- Chronic kidney disease
- Diabetes
- A low level of high-density lipoprotein cholesterol
- The use of at least two cardiovascular drugs.
The prevalence of renal artery stenosis in at-risk populations ranges from 3% to 75% (Table 1).2,4,6,19,20
HOW OFTEN DOES STENOSIS PROGRESS?
The reported rates of progression of atherosclerotic renal artery lesions vary depending on the type of imaging test used and the reason for doing it.
In studies that used duplex ultrasonography, roughly half of lesions smaller than 60% grew to greater than 60% over 3 years.21,22 The risk of total occlusion of an artery was relatively low and depended on the severity of stenosis: 0.7% if the baseline stenosis was less than 60% and 2.3% to 7% if it was greater.21,22
In a seminal study in 1984, Schreiber and colleagues23 compared serial angiograms obtained a mean of 52 months apart in 85 patients who did not undergo intervention. Stenosis had progressed in 37 (44%), and to the point of total occlusion in 14 (16%). In contrast, a 1998 study found progression in 11.1% over 2.6 years, with older patients, women, and those with baseline coronary artery disease at higher risk.24
The the rates of progression differed in these two studies probably because the indications for screening were different (clinical suspicion23 vs routine screening during coronary angiography24), as was the severity of stenosis at the time of diagnosis. Also, when these studies were done, fewer people were taking statins. Thus, similar studies, if repeated, might show even lower rates of progression.
Finally, progression of renal artery stenosis has not been correlated with worsening renal function.
FOUR CLINICAL PRESENTATIONS OF RENAL ARTERY STENOSIS
Renal artery stenosis can present in one of four ways:
Clinically silent stenosis. Because renal artery stenosis is most often found in older patients, who are more likely to have essential hypertension and chronic kidney disease due to other causes, it can be an incidental finding that is completely clinically silent.16,25
Renovascular hypertension is defined as high blood pressure due to up-regulation of neurohormonal activity in response to decreased perfusion from renal artery stenosis. Renal artery stenosis is estimated to be the cause of hypertension in only 0.5% to 4.0% of hypertensive patients, or in 26% of patients with secondary hypertension.3
Ischemic nephropathy is more difficult to define because ischemia alone rarely explains the damage done to the kidneys. Activation of neurohormonal pathways and microvascular injury are thought to contribute to oxidative stress and fibrosis.26 These phenomena may explain why similar degrees of stenosis lead to varying degrees of kidney damage in different patients and why the severity of stenosis does not correlate with the degree of renal dysfunction.27
Furthermore, stenosis may lead to irreversible but stable kidney damage. It is therefore not surprising that, in studies in unselected populations (ie, studies that included patients with all presentations of renal artery stenosis, not just those more likely to benefit), up to two-thirds of renal interventions yielded no clinical benefit.25
As a result, if we define ischemic nephropathy as renal artery stenosis with renal dysfunction not attributable to another cause, we probably will overestimate the prevalence of ischemic nephropathy, leading to overly optimistic expectations about the response to revascularization.
Recurrent “flash” pulmonary edema is a less common presentation, usually occurring in patients with critical bilateral renal artery stenosis or unilateral stenosis in an artery supplying a solitary functioning kidney. Most have severe hypertension (average systolic blood pressure 174–207 mm Hg) and poor renal function.28–30
The association between pulmonary edema and bilateral renal artery stenosis was first noted in 1998 by Pickering et al,31 who in several case series showed that 82% to 92% of patients with recurrent pulmonary edema and renal artery stenosis had bilateral stenosis, compared with 20% to 65% of those with other presentations. Later case series corroborated this finding: 85% to 100% of patients with renal artery stenosis and pulmonary edema had bilateral stenosis.28–30
STENTING IS NOW STANDARD
Stenting has become standard in the endovascular treatment of renal artery stenosis.
Most atherosclerotic renal artery lesions are located in the ostium (ie, where the artery branches off from the aorta), and many are extensions of calcified aortic plaque.26,32 These hard lesions tend to rebound to their original shape more often with balloon angioplasty alone. Stenting provides the additional force needed to permanently disrupt the lesion, leading to a longer-lasting result.
Rates of technical success (dilating the artery during the intervention) are higher with stents than without them (98% vs 46%– 77%).33,34 If the lesion is ostial, this difference is even more impressive (75% vs 29%). In addition, restenosis rates at 6 months are lower with stents (14% vs 26%–48%).34
GOALS: LOWER THE BLOOD PRESSURE, SAVE THE KIDNEY
Because endovascular procedures pose some risk to the patient, it is critical to intervene only in patients most likely to respond clinically. The decision to intervene depends largely on the clinical goal, which should depend on the clinical presentation.
However, if renal artery stenosis is clinically silent, most of the evidence suggests that intervention has no benefit. Furthermore, although retrospective studies have indicated that intervention may improve survival rates,35,36 prospective studies have not. Similarly, studies have not shown that intervention generally improves cardiovascular outcomes, even though renal artery stenosis is associated with cardiovascular risk.
Hypertension plus stenosis is not necessarily renovascular hypertension
Essential hypertension and clinically silent renal artery stenosis often coexist, which is why blood pressure control often does not improve after stenting. Also, essential hypertension often coexists with renovascular hypertension.37 In this situation, stenting may not eliminate the need for antihypertensive drugs, although it may improve blood pressure control and decrease the drug burden.
Before stents came into use, several randomized controlled trials found that blood pressure was no better controlled after angioplasty, 2,3,38 except in cases of bilateral stenosis.2 This may be because stenosis tended to recur after angioplasty without stents.
The 2000 Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) study was the first randomized controlled trial to examine the effect of angioplasty on blood pressure control in renal artery stenosis.38 It had significant design flaws. For example, many patients crossed over from the medical management group to the intervention group because their hypertension was resistant to medical therapy. Overall, intervention (balloon angioplasty without stents in 54 of 56 patients, with stents in the other 2) carried no benefit. However, in subgroup analysis, the patients who crossed over because of resistant hypertension (failure of a three-drug regimen) were more likely to benefit from angioplasty. This suggested that risk stratification should take place early on, before proceeding with revascularization.
With stents, Zeller,39 in a prospective nonrandomized study, found that the mean arterial pressure decreased by 10 mm Hg. Randomized trials (see below) have failed to demonstrate such a benefit.
Stenting may not improve renal function
Coincidental renal artery stenosis in a patient with unrelated chronic kidney disease is very hard to differentiate from true ischemic nephropathy. Furthermore, most patients with ischemic nephropathy do not benefit from revascularization, making it challenging to identify those few whose renal function may respond.
Given that patients with chronic kidney disease tend to have a higher risk of cardiovascular disease, it is not surprising that 15% of them may have renal artery stenosis,4 most often incidental.
Chábová40 examined the outcomes of 68 patients who had chronic kidney disease and a renal artery lesion larger than 70% who did not undergo angioplasty. In only 10 (15%) of the patients did the glomerular filtration rate (GFR) decline by more than 50% of its baseline value during the study period of 3 years. Given the retrospective nature of the study, it cannot be determined (and is rather unlikely) that ischemic nephropathy was the cause of the decline in kidney function in all 10 patients.
In a prospective cohort study in 304 patients with chronic kidney disease and renal artery stenosis who underwent surgical revascularization, Textor4 reported that the serum creatinine level showed a meaningful improvement afterward in 28%, worsened in 19.7%, and remained unchanged in 160 52.6%. (A “meaningful” change was defined as > 1.0 mg/dL.) Findings were similar in a cohort that underwent stenting.33
Davies et al41 found that 20% of patients who underwent renal stenting had a persistent increase in serum creatinine of 0.5 mg/dL or more. These patients were nearly three times more likely (19% vs 7%) to eventually require dialysis, and they had a lower 5-year survival rate (41% vs 71%).
Zeller et al39 found that renal function improved slightly in 52% of patients who received stents. The mean decrease in serum creatinine in this group was 0.22 mg/dL. However, the other 48% had a mean increase in serum creatinine of 1.1 mg/dL.
From these data we can conclude that, in an unselected population with renal artery stenosis, stenting provides no benefit to renal function, and that the risk of a worsening of renal function after intervention is roughly equal to the likelihood of achieving any benefit.
Other predictors of improvement in renal function have been proposed, but the evidence supporting them has not been consistent. For example, although Radermacher et al42 reported that a renal resistive index (which reflects arterial stiffness downstream of the stenosis) lower than 0.8 predicted a response in renal function, this finding has not been reliably reproduced.43,44 Similarly, while several studies suggest that patients with milder renal dysfunction have a higher likelihood of a renal response,45,46 other studies suggest either that the opposite is true39 or that baseline renal function alone has no impact on outcome.47
In addition, once significant renal atrophy occurs, revascularization may not help much, since irreversible sclerosis has developed. Thus, the goal is to identify kidneys being harmed by renal artery stenosis during the ischemic phase, when the tissue is still viable.
Unfortunately, we still lack a good renal stress test—eg, analogous to the cardiac stress test—to diagnose reversible ischemia in the kidney. The captopril renal scan has that capability but is not accurate in patients with bilateral stenosis or a GFR less than 50 mL/min, severely limiting its applicability.26 Newer technologies such as blood-oxygen-level-dependent (BOLD) magnetic resonance imaging are being investigated for such a role.48
Cohort studies in patients with declining renal function
In several case series, patients whose renal function had been declining before intervention had impressive rates of better renal function afterward.33,39,47,49–54 In a prospective cohort study by Muray et al,47 a rise in serum creatinine of more than 0.1 mg/mL/month before intervention seemed to predict an improvement in renal function afterward.
One would expect that, for renal function to respond to intervention, severe bilateral stenosis or unilateral stenosis to a solitary functioning kidney would need to be present. However, this was an inconsistent finding in these case series.33,39,47,52,53 The Angioplasty and Stent for Renal Artery Lesions (ASTRAL) trial,6,7 discussed later, sheds a bit more light on this.
Stenting usually improves flash pulmonary edema
Acute pulmonary edema in the setting of bilateral renal artery stenosis seems to be a unique case in which improvement in clinical status can be expected in most patients after intervention. Blood pressure improves in 94% to 100% of patients,28,31 renal function either improves or stabilizes in 77% to 91%,28–31 and pulmonary edema resolves without recurrence in 77% to 100%.28–30
NEW RANDOMIZED TRIALS: STAR, ASTRAL, AND CORAL
Despite the lack of evidence supporting revascularization of renal artery stenosis, many interventionalists practice under the assumption that the radiographic finding of renal artery stenosis alone is an indication for renal revascularization. Only three randomized controlled trials in the modern era attempt to examine this hypothesis: STAR, ASTRAL, and CORAL.
STAR: No clear benefit
The Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial5 was a European multicenter trial that enrolled 140 patients with ostial renal artery stenosis greater than 50%, blood pressure controlled to less than 140/90 mm Hg, and creatinine clearance 15 to 80 mL/min.
Patients were randomized to undergo stenting or medical therapy alone. High blood pressure was treated according to a protocol in which angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers were relegated to second-line use. All patients received a statin, regardless of lipid levels.
At 2 years, the primary end point (a decline in creatinine clearance of 20% or greater) had been reached in 10 (16%) of the 64 patients in the stent group and 16 (22%) of the 76 patients in the medication group; the difference was not statistically significant (hazard ratio 0.73, 95% confidence interval 0.33–1.61). No difference was seen in the secondary end points of the degree of blood pressure control or the rates of cardiovascular morbidity and death.5
ASTRAL: Also no clear benefit
In the international, multicenter ASTRAL trial,6,7 806 patients with at least one stenotic renal artery considered suitable for balloon angioplasty, stenting, or both7 were randomized to undergo intervention or medical management. Hypertension treatment was not specified by a protocol. The mean estimated GFR was 40 mL/min. Most patients (95%–96%) were on statin therapy. The primary outcome was the rate of decline of renal function over time. Secondary outcomes included blood pressure control, renal events, cardiovascular events, and death.
Results. At a mean follow-up of 33.6 months (range 1–4 years), no difference was noted between treatment groups in decline in renal function or blood pressure control at 1 year. Renal function worsened slightly in both groups.
The decline in renal function over time, calculated as the mean slope of the reciprocal of the serum creatinine level over time, was slightly slower in the revascularization group, but the difference was not statistically significant (−0.07 × 10−3 vs −0.13 × 10−3 L/μmol/year, P = .06). This difference did not appear until the last year of the study. There was no difference in the number of patients whose renal function improved or declined during the study.
There was no difference in the rate of any secondary outcome. The medical management group required a slightly higher number of antihypertensive drugs, reaching statistical but not clinical significance (2.97 vs 2.77 drugs, P = .03). More people in the revascularization group were taking ACE inhibitors or angiotensin receptor blockers. There was no difference in the number of patients on any antihypertensive therapy (97% vs 99%). Interestingly, amputations were more common in the revascularization group, occurring in 42 (12%) of the 386 patients in the revascularization group vs 29 (7%) of the 395 patients in the medical group (P = .04).
Seventeen percent of patients randomized to intervention did not have the procedure done. An as-treated analysis of the 317 (83%) patients randomized to revascularization who did receive it showed no differences in outcomes.
There were no differences in outcomes among specific, predefined subgroups based on severity of stenosis at baseline, renal length, baseline estimated GFR, baseline serum creatinine, and rate of progression of renal dysfunction before randomization.7
Comments. ASTRAL contradicts previous nonrandomized studies that suggested that rapidly declining renal function (loss of 20% in 1 year) predicts response to intervention. Considering the large number of patients with unilateral disease in the study, it would be interesting to see what effect stenting had on patients with both severe disease and declining renal function.
ASTRAL has been criticized because it lacked a central laboratory to interpret the severity of stenosis, it did not use a standardized intervention technique (5% of patients underwent angioplasty without stents, although this did not affect outcomes7), and patients were enrolled only if the clinician involved in the case was uncertain of the appropriate management.
This last issue raises the concern for selection bias toward inclusion of more difficult cases that may not respond to intervention. But these shortcomings are not serious enough to negate the fact that preliminary results from the largest randomized controlled trial to date confirm conclusions of other randomized trials, ie, that intervention in renal artery stenosis yields no benefits over medical management in most patients.
Based on the results of STAR and ASTRAL, the practice of indiscriminately revascularizing stenosed renal arteries without strong evidence that the procedure will provide a clinical benefit is no longer tenable. The challenge is to identify those few patients who will respond, and to intervene only on them. Unfortunately, none of the subgroups from ASTRAL helped characterize this population.
CORAL: A large trial is ongoing
The Cardiovascular Outcomes in Renal Artherosclerotic Lesions (CORAL) trial,8 an ongoing multicenter randomized controlled trial in the United States, may be of additional help.
Unlike ASTRAL, CORAL is studying patients who have difficult-to-control hypertension (systolic blood pressure ≥ 155 mm Hg on two or more drugs).8 Chronic kidney disease is not an exclusion criterion unless the serum creatinine concentration is greater than 3.0 mg/dL.
CORAL is using a standardized medical protocol to control blood pressure. In addition, use of embolic protection devices during stenting is encouraged. Hopefully, the large size (a goal of 1,080 patients) and the inclusion of patients with more marked hypertension will address the utility of intervention in higher-risk populations with renal artery stenosis.
RECOMMENDED APPROACH TO INTERVENTION IN RENAL ARTERY STENOSIS
As we wait for CORAL to be completed, we have two modern-era randomized controlled trials that leave us with fewer indications for renal intervention. Table 2 lists commonly cited indications for intervention in renal artery stenosis and the evidence to support them. As most of these are based on retrospective data or have conflicting support in the literature, their utility remains in question. At this point we can safely recommend:
- Patients with preserved or even decreased but stable renal function will not likely have a benefit in renal function after intervention.
- Patients with resistant hypertension may benefit.
- The best evidence supporting intervention is for bilateral stenosis with flash pulmonary edema, but the evidence is from retrospective studies.
- Stenting in bilateral disease without another indication has no apparent benefit.
- Declining renal function is not a guarantee of success.
- It is unclear if patients with severe bilateral stenosis or severe stenosis to a solitary functioning kidney with declining renal function will benefit. Anecdotally, they do respond more often, but as with many other indications for intervention that have gone by the wayside, this may not bear out when studied properly.
As the utility of intervention narrows, the scope of practice for such interventions should narrow accordingly. Attention should now be focusing on clinical, rather than radiographic, indications for intervening on renal artery stenosis.
Therefore, the decision to intervene must not be made solely by the interventionalist. A multidisciplinary approach should be adopted that at the very least includes the input of a nephrologist well versed in renal artery stenosis. In this way, the clinical risks and benefits of renal intervention can be discussed with the patient by providers who are likely to be involved in their care should renal function or hypertension fail to improve afterward.
RISK OF ATHEROEMBOLISM
While renal stenting yields improved technical success in the treatment of renal artery stenosis, it carries with it an increasingly common risk to kidney function: atheroembolism as the stent crushes the plaque against the vessel wall. This may lead to obstruction of the renal microvasculature, increasing the risk of irreversible damage to renal function.
Atheroembolic kidney disease can manifest as progressive renal failure occurring over weeks to months, commonly misdiagnosed as permanent damage from contrast nephropathy.55
Embolic protection devices, inserted downstream of the lesion before stenting to catch any debris that may break loose, have been developed to help address this problem.
Holden et al 57 prospectively studied 63 patients with renal artery stenosis and deteriorating renal function (undefined) who underwent stenting with an embolic protection device. At 6 months after the intervention, renal function had either improved or stabilized in 97% of patients, suggesting that many of the deleterious effects of stenting on renal function are related to atheroembolism.
The Prospective Randomized Study Comparing Renal Artery Stenting With or Without Distal Protection (RESIST) trial, in which renal dysfunction was mild and the GFR was not declining (average estimated GFR 59.3 mL/min), found contrary results.57 In a two-by-two factorial study, patients were randomized to undergo stenting alone, stenting with the antiplatelet agent abciximab (ReoPro), stenting with an embolic protection device, or stenting with both abciximab and an embolic protection device. Interestingly, renal function declined in the first three groups, but remained stable in the group that received both abciximab and an embolic protection device.
ANTIPLATELET THERAPY AFTER RENAL STENTING: HOW LONG?
We have no data on the optimal duration of antiplatelet therapy after renal stenting, and guidelines from professional societies do not comment on it.58 As a result, practice patterns vary significantly among practitioners.
While in-stent restenosis rates are acceptably low after renal stenting, the risks and side effects of antiplatelet therapy often lead to arbitrary withdrawal of these drugs. The effect on stent patency is yet to be determined.
FUTURE DEVELOPMENTS
Results of STAR and ASTRAL confirm the growing suspicion that the surge seen in the last decade in renal artery stenting should be coming to an end. We await results either from CORAL or possibly a post hoc analysis of ASTRAL that might identify potential high-risk groups that will benefit from renal intervention. And as embolic protection devices become more agile and suitable to different renal lesions, there remains the possibility that, due to lower rates of unidentified atheroembolic kidney disease, CORAL may demonstrate improved renal outcomes after stenting. If not, the search for the best means to predict who should have renal intervention will continue.
We know through experience that stenting does provide great benefits for some patients with renal artery stenosis. Furthermore, the clinical problem is too intriguing, and too profitable, to die altogether.
- Choncol M, Linas S. Diagnosis and management of ischemic nephropathy. Clin J Am Soc Nephrol 2006; 1:172–181.
- Webster J, Marshall F, Abdalla M, et al Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 1998; 12:329–335.
- Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 1998; 31:823–829.
- Textor SC. Revascularization in atherosclerotic renal artery disease. Kidney Int 1998; 53:799–811.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–848.
- Mistry S, Ives N, Harding J, et al Angioplasty and STent for Renal Artery Lesions (ASTRAL trial): rationale, methods and results so far. J Hum Hypertens 2007; 21:511–515.
- Wheatley K, Ives N, Kalra P, Moss J. Revascularization versus medical therapy for renal-artery stenosis (ASTRAL). N Engl J Med 2009; 361:1953–1962.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Galaria II, Surowiec SM, Rhodes JM, et al Percutaneous and open renal revascularizations have equivalent long-term functional outcomes. Ann Vasc Surg 2005; 19:218–228.
- Murphy TP, Soares G, Kim M. Increase in utilization of percutaneous renal artery interventions by Medicare beneficiaries 1996–2000. AJR Am J Roentgenol 2004; 183:561–568.
- Textor SC. Atherosclerotic renal artery stenosis: overtreated but underrated? J Am Soc Nephrol 2008; 19:656–659.
- Kalra PA, Guo H, Gilbertson DT, et al Atherosclerotic renovascular disease in the United States. Kidney Int 2010; 77:37–43.
- Hansen KJ, Edwards MS, Craven TE, et al Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Cohen MG, Pascua JA, Garcia-Ben M, et al A simple prediction rule for significant renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 2005; 150:1204–1211.
- Buller CE, Nogareda JG, Ramanathan K, et al The profile of cardiac patients with renal artery stenosis. J Am Coll Cardiol 2004; 43:1606–1613.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- Kalra PA, Guo H, Kausz AT, et al Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 69:293–301.
- Holley KE, Hunt JC, Brown AL, Kincaid OW, Sheps SG. Renal artery stenosis. A clinical-pathologic study in normotensive and hypertensive patients. Am J Med 1964; 37:14–22.
- de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systemic literature review. J Hypertens 2009; 27:1333–1340.
- Kuczera P, Włoszczynska E, Adamczak M, Pencak P, Chudek J, Wiecek A. Frequency of renal artery stenosis and variants of renal vascularization in hypertensive patients: analysis of 1550 angiographies in one centre. J Hum Hypertens 2009; 23:396–401.
- Caps MT, Perissinotto C, Zierler RE, et al Prospective study of atherosclerotic disease progression in the renal artery. Circulation 1998; 98:2866–2872.
- Zierler RE, Bergelin RO, Davidson RC, Cantwell-Gab K, Polissar NL, Strandness DE. A prospective study of disease progression in patients with atherosclerotic renal artery stenosis. Am J Hypertens 1996; 9:1055–1061.
- Schreiber MJ, Pohl MA, Novick AC. The natural history of atherosclerotic and fibrous renal artery disease. Urol Clin North Am 1984; 11:383–392.
- Crowley JJ, Santos RM, Peter RH, et al Progression of renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 1998; 136:913–918.
- Textor SC. Renovascular hypertension update. Curr Hypertens Rep 2006; 8:521–527.
- Textor SC. Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004; 15:1974–1982.
- Wright JR, Shurrab AE, Cheung C, et al A prospective study of the determinants of renal functional outcome and mortality in atherosclerotic renovascular disease. Am J Kidney Dis 2002; 39:1153–1161.
- Messina LM, Zelenock GB, Yao KA, Stanley JC. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg 1992; 15:73–80.
- Bloch MJ, Trost DW, Pickering TG, Sos TA, August P. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. Am J Hypertens 1999; 12:1–7.
- Gray BH, Olin JW, Childs MB, Sullivan TM, Bacharach JM. Clinical benefit of renal artery angioplasty with stenting for the control of recurrent and refractory congestive heart failure. Vasc Med 2002; 7:275–279.
- Pickering TG, Herman L, Devereux RB, et al Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet 1988; 2:551–552.
- Kennedy DJ, Colyer WR, Brewster PS, et al Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis 2003; 14:926–935.
- Beutler JJ, Van Ampting JM, Van De Ven PJ, et al Long-term effects of arterial stenting on kidney function for patients with ostial atherosclerotic renal artery stenosis and renal insufficiency. J Am Soc Nephrol 2001; 12:1475–1481.
- Van de Ven PJ, Kaatee R, Beutler JJ, et al Arterial stenting and balloon angioplasty in ostial atherosclerotic renovascular disease: a randomized trial. Lancet 1999; 353:282–286.
- Isles C, Main J, O’Connell J, et al Survival associated with renovascular disease in Glasgow and Newcastle: a collaborative study. Scott Med J 1990; 35:70–73.
- Hunt JC, Sheps SG, Harrison EG, Strong CG, Bernatz PE. Renal and renovascular hypertension. A reasoned approach to diagnosis and management. Arch Intern Med 1974; 133:988–999.
- Textor SC. Atherosclerotic renal artery stenosis: how big is the problem, and what happens if nothing is done? J Hypertens Suppl 2005; 23:S5–S13.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Zeller T, Frank U, Müller C, et al Predictors of improved renal function after percutaneous stent-supported angioplasty of severe atherosclerotic ostial renal artery stenosis. Circulation 2003; 108;2244–2249.
- Chábová V, Schirger A, Stanson AW, McKusick MA, Textor SC. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc 2000; 75:437–444.
- Davies MG, Saad WE, Peden EK, Mohiuddin IT, Naoum JJ, Lumsden AB. Implications of acute functional injury following percutaneous renal artery intervention. Ann Vasc Surg 2008; 22:783–789.
- Radermacher J, Chavin A, Bleck J, et al Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Eng J Med 2001; 344:410–417.
- García-Criado A, Gilabert R, Nicolau C, et al Value of Doppler sonography for predicting clinical outcome after renal artery revascularization in atherosclerotic renal artery stenosis. J Ultrasound Med 2005; 24:1641–1647.
- Zeller T, Müller C, Frank U, et al Stent angioplasty of severe atherosclerotic ostial renal artery stenosis in patients with diabetes mellitus and nephrosclerosis. Catheter Cardiovasc Interv 2003; 58:510–515.
- Harden PN, MacLeod MJ, Rodger RSC, et al Effect of renal-artery stenting on progression of renovascular renal failure. Lancet 1997; 349:1133–1136.
- Isles CG, Robertson S, Hill D. Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 1999; 92:159–167.
- Muray S, Martın M, Amoedo ML, et al Rapid decline in renal function reflects reversibility and predicts the outcome after angioplasty in renal artery stenosis. Am J Kidney Dis 2002; 39:60–66.
- Textor SC, Glockner JF, Lerman LO, et al The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 2008; 19:780–788.
- Paraskevas KI, Perrea D, Briana DD, Liapis CD. Management of atherosclerotic renovascular disease: the effect of renal artery stenting on renal function and blood pressure. Int Urol Nephrol 2006; 38:683–691.
- Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000; 102:1671–1677.
- Dean RH, Kieffer RW, Smith BM, et al Renovascular hypertension: anatomic and renal function changes during drug therapy. Arch Surg 1981; 116:1408–1415.
- Zhang Q, Shen W, Zhang R, Zhang J, Hu J, Zhang X. Effects of renal artery stenting on renal function and blood pressure in patients with atherosclerotic renovascular disease. Chin Med J (Engl) 2003; 116:1451–1454.
- Ramos F, Kotliar C, Alvarez D, et al Renal function and outcome of PTRA and stenting for atherosclerotic renal artery stenosis. Kidney Int 2003; 63:276–282.
- Rocha-Singh KJ, Ahuja RK, Sung CH, Rutherford J. Long-term renal function preservation after renal artery stenting in patients with progressive ischemic nephropathy. Catheter Cardiovasc Interv 2002; 57:135–141.
- Thadhani RI, Camargo CA, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore) 1995; 74:350–358.
- Holden A, Hill A, Jaff MR, Pilmore H. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int 2006; 70:948–955.
- Cooper CJ, Haller ST, Colyer W, et al Embolic protection and platelet inhibition during renal artery stenting. Circulation 2008; 117:2752–2760.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Choncol M, Linas S. Diagnosis and management of ischemic nephropathy. Clin J Am Soc Nephrol 2006; 1:172–181.
- Webster J, Marshall F, Abdalla M, et al Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 1998; 12:329–335.
- Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 1998; 31:823–829.
- Textor SC. Revascularization in atherosclerotic renal artery disease. Kidney Int 1998; 53:799–811.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–848.
- Mistry S, Ives N, Harding J, et al Angioplasty and STent for Renal Artery Lesions (ASTRAL trial): rationale, methods and results so far. J Hum Hypertens 2007; 21:511–515.
- Wheatley K, Ives N, Kalra P, Moss J. Revascularization versus medical therapy for renal-artery stenosis (ASTRAL). N Engl J Med 2009; 361:1953–1962.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Galaria II, Surowiec SM, Rhodes JM, et al Percutaneous and open renal revascularizations have equivalent long-term functional outcomes. Ann Vasc Surg 2005; 19:218–228.
- Murphy TP, Soares G, Kim M. Increase in utilization of percutaneous renal artery interventions by Medicare beneficiaries 1996–2000. AJR Am J Roentgenol 2004; 183:561–568.
- Textor SC. Atherosclerotic renal artery stenosis: overtreated but underrated? J Am Soc Nephrol 2008; 19:656–659.
- Kalra PA, Guo H, Gilbertson DT, et al Atherosclerotic renovascular disease in the United States. Kidney Int 2010; 77:37–43.
- Hansen KJ, Edwards MS, Craven TE, et al Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Cohen MG, Pascua JA, Garcia-Ben M, et al A simple prediction rule for significant renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 2005; 150:1204–1211.
- Buller CE, Nogareda JG, Ramanathan K, et al The profile of cardiac patients with renal artery stenosis. J Am Coll Cardiol 2004; 43:1606–1613.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- Kalra PA, Guo H, Kausz AT, et al Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 69:293–301.
- Holley KE, Hunt JC, Brown AL, Kincaid OW, Sheps SG. Renal artery stenosis. A clinical-pathologic study in normotensive and hypertensive patients. Am J Med 1964; 37:14–22.
- de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systemic literature review. J Hypertens 2009; 27:1333–1340.
- Kuczera P, Włoszczynska E, Adamczak M, Pencak P, Chudek J, Wiecek A. Frequency of renal artery stenosis and variants of renal vascularization in hypertensive patients: analysis of 1550 angiographies in one centre. J Hum Hypertens 2009; 23:396–401.
- Caps MT, Perissinotto C, Zierler RE, et al Prospective study of atherosclerotic disease progression in the renal artery. Circulation 1998; 98:2866–2872.
- Zierler RE, Bergelin RO, Davidson RC, Cantwell-Gab K, Polissar NL, Strandness DE. A prospective study of disease progression in patients with atherosclerotic renal artery stenosis. Am J Hypertens 1996; 9:1055–1061.
- Schreiber MJ, Pohl MA, Novick AC. The natural history of atherosclerotic and fibrous renal artery disease. Urol Clin North Am 1984; 11:383–392.
- Crowley JJ, Santos RM, Peter RH, et al Progression of renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 1998; 136:913–918.
- Textor SC. Renovascular hypertension update. Curr Hypertens Rep 2006; 8:521–527.
- Textor SC. Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004; 15:1974–1982.
- Wright JR, Shurrab AE, Cheung C, et al A prospective study of the determinants of renal functional outcome and mortality in atherosclerotic renovascular disease. Am J Kidney Dis 2002; 39:1153–1161.
- Messina LM, Zelenock GB, Yao KA, Stanley JC. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg 1992; 15:73–80.
- Bloch MJ, Trost DW, Pickering TG, Sos TA, August P. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. Am J Hypertens 1999; 12:1–7.
- Gray BH, Olin JW, Childs MB, Sullivan TM, Bacharach JM. Clinical benefit of renal artery angioplasty with stenting for the control of recurrent and refractory congestive heart failure. Vasc Med 2002; 7:275–279.
- Pickering TG, Herman L, Devereux RB, et al Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet 1988; 2:551–552.
- Kennedy DJ, Colyer WR, Brewster PS, et al Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis 2003; 14:926–935.
- Beutler JJ, Van Ampting JM, Van De Ven PJ, et al Long-term effects of arterial stenting on kidney function for patients with ostial atherosclerotic renal artery stenosis and renal insufficiency. J Am Soc Nephrol 2001; 12:1475–1481.
- Van de Ven PJ, Kaatee R, Beutler JJ, et al Arterial stenting and balloon angioplasty in ostial atherosclerotic renovascular disease: a randomized trial. Lancet 1999; 353:282–286.
- Isles C, Main J, O’Connell J, et al Survival associated with renovascular disease in Glasgow and Newcastle: a collaborative study. Scott Med J 1990; 35:70–73.
- Hunt JC, Sheps SG, Harrison EG, Strong CG, Bernatz PE. Renal and renovascular hypertension. A reasoned approach to diagnosis and management. Arch Intern Med 1974; 133:988–999.
- Textor SC. Atherosclerotic renal artery stenosis: how big is the problem, and what happens if nothing is done? J Hypertens Suppl 2005; 23:S5–S13.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Zeller T, Frank U, Müller C, et al Predictors of improved renal function after percutaneous stent-supported angioplasty of severe atherosclerotic ostial renal artery stenosis. Circulation 2003; 108;2244–2249.
- Chábová V, Schirger A, Stanson AW, McKusick MA, Textor SC. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc 2000; 75:437–444.
- Davies MG, Saad WE, Peden EK, Mohiuddin IT, Naoum JJ, Lumsden AB. Implications of acute functional injury following percutaneous renal artery intervention. Ann Vasc Surg 2008; 22:783–789.
- Radermacher J, Chavin A, Bleck J, et al Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Eng J Med 2001; 344:410–417.
- García-Criado A, Gilabert R, Nicolau C, et al Value of Doppler sonography for predicting clinical outcome after renal artery revascularization in atherosclerotic renal artery stenosis. J Ultrasound Med 2005; 24:1641–1647.
- Zeller T, Müller C, Frank U, et al Stent angioplasty of severe atherosclerotic ostial renal artery stenosis in patients with diabetes mellitus and nephrosclerosis. Catheter Cardiovasc Interv 2003; 58:510–515.
- Harden PN, MacLeod MJ, Rodger RSC, et al Effect of renal-artery stenting on progression of renovascular renal failure. Lancet 1997; 349:1133–1136.
- Isles CG, Robertson S, Hill D. Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 1999; 92:159–167.
- Muray S, Martın M, Amoedo ML, et al Rapid decline in renal function reflects reversibility and predicts the outcome after angioplasty in renal artery stenosis. Am J Kidney Dis 2002; 39:60–66.
- Textor SC, Glockner JF, Lerman LO, et al The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 2008; 19:780–788.
- Paraskevas KI, Perrea D, Briana DD, Liapis CD. Management of atherosclerotic renovascular disease: the effect of renal artery stenting on renal function and blood pressure. Int Urol Nephrol 2006; 38:683–691.
- Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000; 102:1671–1677.
- Dean RH, Kieffer RW, Smith BM, et al Renovascular hypertension: anatomic and renal function changes during drug therapy. Arch Surg 1981; 116:1408–1415.
- Zhang Q, Shen W, Zhang R, Zhang J, Hu J, Zhang X. Effects of renal artery stenting on renal function and blood pressure in patients with atherosclerotic renovascular disease. Chin Med J (Engl) 2003; 116:1451–1454.
- Ramos F, Kotliar C, Alvarez D, et al Renal function and outcome of PTRA and stenting for atherosclerotic renal artery stenosis. Kidney Int 2003; 63:276–282.
- Rocha-Singh KJ, Ahuja RK, Sung CH, Rutherford J. Long-term renal function preservation after renal artery stenting in patients with progressive ischemic nephropathy. Catheter Cardiovasc Interv 2002; 57:135–141.
- Thadhani RI, Camargo CA, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore) 1995; 74:350–358.
- Holden A, Hill A, Jaff MR, Pilmore H. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int 2006; 70:948–955.
- Cooper CJ, Haller ST, Colyer W, et al Embolic protection and platelet inhibition during renal artery stenting. Circulation 2008; 117:2752–2760.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
KEY POINTS
- Two large randomized trials of intervention vs medical therapy showed negative results for intervention. A third trial is under way.
- Intervention is not recommended if renal function has remained stable over the past 6 to 12 months and if hypertension can be controlled medically.
- The best evidence supporting intervention is for bilateral stenosis with “flash” pulmonary edema, but the evidence is from retrospective studies.
- Stenosis by itself, even if bilateral, is not an indication for renal artery stenting.
Preventing venous thromboembolism in long-term care residents: Cautious advice based on limited data
Randomized trials that included more than 20,000 medical patients have shown that anticoagulant therapy is safe and effective in preventing venous thromboembolism (VTE), ie, deep vein thrombosis and pulmonary embolism.
However, these trials were done in hospitalized patients, who typically had an acute medical illness and who, if eligible, received a short (7- to 10-day) course of anticoagulant prophylaxis.
Little attention has been given to VTE prophylaxis in residents of long-term care facilities. These patients have risk profiles similar to those of hospitalized medical patients. Some of them may have been transferred from an acute care hospital. In addition, most are elderly, and many have reduced mobility and are at risk for illnesses such as stroke and cardiorespiratory insufficiency, which increase the risk of VTE.
VTE in residents of long-term care facilities is a growing concern. By some estimates, by the year 2030 more than 20% of the US population (70.2 million people) will be over 65 years of age.1 Of those who reached age 65 in 1990, an estimated 43% will enter a nursing home at least once before they die—32% for 3 months, 24% for at least a year, and 9% for at least 5 years.2
Against this background, the objectives of this review are to consider:
- The scope of the problem of VTE in long-term care residents
- Why VTE prophylaxis is often overlooked in medical patients
- Evidence—or lack of evidence—for the safety and efficacy of VTE prophylaxis in long-term care residents and other medical patients
- Available options for VTE prophylaxis
- Which long-term care residents should or should not be considered for prophylaxis.
THE TRUE SCOPE OF THE PROBLEM IS UNKNOWN
The incidence of acute VTE among nursing home residents is reported to be 1.3 events per 100 person-years.3 About 8% of cases of pulmonary embolism and 10% of cases of deep venous thrombosis in the elderly are in nursing home residents.4
However, only 20% of patients with VTE have typical symptoms such as leg pain and swelling or acute dyspnea and chest pain, while 80% have no symptoms.5
Furthermore, deep venous thrombosis is more likely to be clinically silent in patients whose mobility is impaired, such as nursing home residents, as the symptoms arising from obstruction of venous flow are more pronounced with walking.
Pulmonary embolism is also underdiagnosed in this group. An autopsy study of 234 nursing home residents found undiagnosed pulmonary embolism to be the cause of death in 8%, and 40% of cases of pulmonary embolism were not suspected before the patient died.6 Yet pulmonary embolism has a higher case-fatality rate in the elderly than in younger patients, particularly when elderly patients have comorbidities.7
A reason why the diagnosis is so often missed is that pulmonary embolism can present atypically in the elderly, with syncope being more common and tachycardia being less common than in younger patients.8
Since so many cases of VTE are clinically silent and most long-term care residents who die do not undergo autopsy, the true scope of VTE as a clinical problem in these patients is unknown. Consequently, the best way to diagnose, prevent, and treat VTE is also unclear.
WHY IS VTE PREVENTION SO OFTEN OVERLOOKED IN MEDICAL PATIENTS?
In general, nonsurgical patients receive suboptimal thromboprophylaxis. National and international chart audits and cross-sectional studies show that only 16% to 33% of hospitalized medical patients at risk for VTE receive appropriate anticoagulant prophylaxis.9 Though no audits in long-term care facilities have been published, the rate of appropriate prophylaxis is likely comparable to or possibly less than that in medical patients in the hospital. In contrast, in surgical patients the rate is much higher—up to 90%.10,11
Why is VTE prophylaxis so underused in medical patients?
One reason is that we do not really know the baseline risk of VTE in medical patients, particularly in those with chronic illness who require long-term care.12 This is relevant because, in the absence of data about patients’ baseline risk, anticoagulant prophylaxis should be ordered selectively, as it poses known risks of bleeding. The risk is greater in elderly people with comorbidities, as are the associated costs.
In addition, relatively few studies have assessed thromboprophylaxis in medical patients, especially in residents of long-term care facilities.
Another reason is that we lack practice guidelines for patients who need long-term care. The well-accepted guidelines from the American College of Chest Physicians (ACCP) cite advanced age and immobility as risk factors for VTE and strongly recommend prophylaxis in acutely ill medical patients who have limited mobility and an additional risk factor such as infection or cancer.13 Though elderly residents of long-term care facilities may share some of these risk factors, the ACCP guidelines make no specific recommendations for this group.
The attitudes of health care professionals may also pose a barrier. Lloyd et al (unpublished data, 2009) surveyed 1,601 health care professionals in Ontario, Canada, in 2007, to assess potential barriers to anticoagulant prophylaxis in hospitalized medical patients. Respondents cited concerns about the risk of bleeding from anticoagulants, lack of clear indications and contraindications for anticoagulant prophylaxis, and lack of time to consider VTE prophylaxis in every patient. (They did not, however, cite disagreement with guidelines or patient discomfort from subcutaneous anticoagulant injections as barriers.) It is reasonable to assume that these attitudes may also pose a problem in long-term care residents.
Finally, no randomized trials have evaluated the efficacy and safety of anticoagulant drugs or mechanical methods of prophylaxis in long-term care residents. Studies have shown that a short course (7–10 days) of an anticoagulant drug effectively prevents VTE in acutely ill patients, but the efficacy of an extended course in patients with chronic illness who require long-term care is not clear. Therefore, recommendations about thromboprophylaxis in long-term care residents should be made with the caveat that they are based on indirect evidence from other patient groups. This is a considerable limitation.
OPTIONS FOR THROMBOPROPHYLAXIS IN LONG-TERM CARE RESIDENTS
Options for thromboprophylaxis fall into two broad categories: anticoagulant drugs and mechanical devices.
Anticoagulant prophylactic drugs

These agents have been assessed in randomized trials in surgical or acutely ill medical patients, although fondaparinux and tinzaparin are not approved for use in medical patients. Furthermore, none of them has been evaluated in residents of long-term care facilities.
The choice of anticoagulant for prophylaxis is determined largely by clinical factors.
Low-molecular-weight heparins are popular both in and out of the hospital because they have predictable pharmacokinetic properties, they come in convenient prefilled syringes, and they can be given once daily. However, some of them may bioaccumulate in patients with impaired renal function, as they are cleared primarily by the kidney.
Unfractionated heparin is likely to be safer in patients with severe renal insufficiency (creatinine clearance < 30 mL/min), as it is cleared via nonrenal mechanisms.
However, a recent single-arm trial of dalteparin 5,000 IU once daily in critically ill patients with severe renal insufficiency found no evidence of an excessive anticoagulant effect or of drug bioaccumulation.15 Dalteparin may thus be an alternative to unfractionated heparin in medical patients with impaired renal function.
Fondaparinux, a newer anticoagulant, is also given once daily. It is the anticoagulant of choice in patients who have had heparin-induced thrombocytopenia because it is not derived from heparin and likely does not cross-react with heparin-induced thrombocytopenia antibodies.16,17
Limited data on benefit of prophylactic anticoagulant drugs
As mentioned, the trials that confirmed the efficacy and safety of anticoagulant prophylaxis were in surgical patients and hospitalized medical patients, not elderly long-term care residents. The poor evidence for anticoagulant prophylaxis in these patients may be strengthened if extended-duration, out-of-hospital prophylaxis were shown to be effective in medical patients. Long-term care residents could more reasonably be compared with medical patients discharged home with a chronic or resolving illness than with those who are hospitalized.
There is some evidence, although with caveats, that extended anticoagulant prophylaxis, started after an acute illness has resolved, confers a benefit. A recent randomized trial compared extended-duration and short-duration prophylaxis (5 weeks vs 10 days) with enoxaparin 40 mg once daily in 4,726 medical patients with impaired mobility.18 The risk of any VTE event was 44% lower with extended-duration prophylaxis (2.8% vs 4.9%; P = .001) and the risk of symptomatic VTE was 73% lower (0.3% vs 1.1%; P = .004), and this benefit persisted 2 months after treatment was stopped (3.0% vs 5.2%; P = .0015). However, extended treatment conferred a fourfold higher risk of major bleeding (0.6% vs 0.15%; P = .019).
These findings should also be considered in terms of absolute benefit and harm. Treating 1,000 patients for 5 weeks instead of 10 days would prevent eight episodes of symptomatic VTE (absolute risk reduction = 0.8%, number needed to treat = 125) at the cost of four to five episodes of major bleeding (absolute risk increase = 0.45%, number needed to harm = 222). This is a modest net therapeutic benefit.
The therapeutic benefit would be greater if we consider all episodes of VTE, both symptomatic and asymptomatic. Treating 1,000 patients for 5 weeks would prevent 20 episodes of symptomatic or asymptomatic VTE (absolute risk reduction = 2.1%, number needed to treat = 48). However, the clinical importance of asymptomatic VTE is questionable.
Given these considerations, if extended-duration anticoagulant prophylaxis is considered, it should be for patients at highest risk to optimize both its net therapeutic benefits and its cost-effectiveness.
Mechanical prophylaxis
Mechanical thromboprophylactic devices—graduated or elastic compression stockings and intermittent pneumatic compression devices—are effective when used by themselves in surgical patients.13 However, in a randomized controlled trial in patients with ischemic stroke, the rate of VTE was 10.0% with graduated compression stockings in addition to “usual care VTE prophylaxis” vs 10.5% with usual care alone, and patients in the stocking group had a fourfold higher risk of developing skin breaks, ulcers, blisters, or necrosis (5% vs 1%; odds ratio 4.18; 95% CI 2.4–7.3).19 Furthermore, improperly fitted stockings, especially those that are thigh-length, can be uncomfortable to wear and difficult to apply.
Overall, the role of mechanical thromboprophylaxis in long-term care facilities is not clear. If it is considered, there should be a compelling reason to use it—for example, for patients at high risk in whom anticoagulants are contraindicated because of ongoing bleeding or a higher risk of bleeding (eg, recent gastrointestinal bleeding, hemorrhagic stroke, coagulopathy, or thrombocytopenia). Furthermore, if stockings are used, they should be properly fitted and routinely monitored for adverse effects, since elderly patients are likely to be most susceptible to skin breakdown.
WHICH LONG-TERM CARE RESIDENTS SHOULD RECEIVE VTE PROPHYLAXIS?

Old age and immobility are not the only risk factors
The current ACCP guidelines suggest considering thromboprophylaxis for hospitalized medical patients over age 75 who cannot walk without assistance.13 However, we lack evidence to suggest a similar strategy in long-term care residents.
The ACCP guidelines are based on data on risk. Nearly 25% of elderly patients with confirmed pulmonary embolism had been immobile prior to their diagnosis.8 In addition, prolonged bed rest (> 14 days) has been reported to be the strongest independent risk factor for symptomatic deep venous thrombosis, increasing the risk more than fivefold.20 Advanced age is also considered a risk factor for VTE, as risk starts to increase at age 40 and doubles each decade of life thereafter.18
No study has assessed the impact of these factors on the risk of VTE in long-term care residents. Since most of such patients are elderly and have impaired mobility, we believe a more selective approach should be used in assigning VTE risk status, one that does not use advanced age and immobility as the only criteria for starting thromboprophylaxis.
Residents of long-term care facilities may be immobile because of underlying illness or disability, such as cognitive impairment, sensory impairment (eg, poor access to corrective lenses and hearing aids), or poor access to assist devices (eg, walkers, canes). In addition, iatrogenic factors that decrease mobility such as indwelling bladder catheters and physical restraints are also common in such patients.
Efforts to improve mobility should be encouraged. However, we recommend that thromboprophylaxis be considered only in patients who have both impaired mobility and an intercurrent acute medical illness such as an acute infection or acute inflammatory disease.13
A related issue is the difference between long-term care residents with a chronic but stable disease and those with acute disease. Patients with acute exacerbations of congestive heart failure or chronic obstructive lung disease may be considered for thromboprophylaxis, as they become more comparable to acutely ill medical patients in whom clinical trials have shown the effectiveness of anticoagulant prophylaxis. On the other hand, patients with these diseases who remain stable may not need prophylaxis.
This approach avoids giving long-term anticoagulant prophylaxis to patients who have irreversible diseases and limits the use of these drugs and devices to higher-risk periods.
Consider thromboprophylaxis if…

- An acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease
- Acute infection (eg, urosepsis, pneumonia, cellulitis, infectious diarrhea)
- An acute exacerbation of an inflammatory disease (eg, rheumatoid arthritis)
- Active cancer (eg, patient receiving radiation therapy or chemotherapy)
- Immobility and prior VTE.
Do not routinely consider prophylaxis if…
We also believe patients should not be routinely considered for anticoagulant VTE prophylaxis if they have:
- Chronic but stable cardiorespiratory disease
- Chronic but stable infectious or inflammatory disease
- Terminal cancer with very limited life expectancy
- Any contraindication to anticoagulants (eg, active bleeding, recent bleeding, coagulopathy, thrombocytopenia).
ANTICOAGULANT PROPHYLAXIS POSES RISKS IN LONG-TERM CARE RESIDENTS
Bleeding is the principal risk

The incidence of clinically important bleeding associated with anticoagulant prophylaxis is 0.2% to 5.6%, and the risk of fatal bleeding is 0.02% to 0.5%.21–24
As no randomized trial has examined anticoagulant prophylaxis in elderly long-term care residents, their bleeding risk with this therapy is unclear. However, older patients are likely to be at higher risk than younger patients because they have more comorbidities, take more drugs that could interact with heparin and potentiate bleeding, and have fragile skin, predisposing to injury from subcutaneous injections.
Also, renal function tends to decline with age. In a retrospective study of 854 outpatients over age 65, 29% had moderate renal insufficiency (creatinine clearance 30–50 mL/min), and 6% had severe renal insufficiency (creatinine clearance < 30 mL/min).25 Recent evidence suggests that some low-molecular-weight heparins (dalteparin and tinzaparin) do not bioaccumulate in patients with impaired renal function. However, enoxaparin and fondaparinux should be used with caution in patients with moderate to severe renal impairment.
Though much attention has recently been paid to increasing anticoagulant doses if the patient is obese, residents of long-term care facilities are more likely to be underweight. Dose adjustment should be considered when a low-molecular-weight heparin or fondaparinux is given to patients weighing less than 50 kg.
Heparin-induced thrombocytopenia
The other major risk of anticoagulant prophylaxis is heparin-induced thrombocytopenia, an infrequent but life-threatening complication caused by the formation of antibodies to the heparin-derived anticoagulant and a platelet surface antigen. It is associated with moderate thrombocytopenia and an incidence of venous or arterial thrombosis that is over 50%.26
No study has assessed the incidence of heparin-induced thrombocytopenia in long-term care residents. A meta-analysis reported that the risk with anticoagulant prophylaxis was 1.6% with unfractionated heparin (95% confidence interval [CI] 1.2%–2.1%) and 0.6% with low-molecular-weight heparin (95% CI 0.4%–0.9%), and that this risk increased with the duration of prophylaxis.27 If anticoagulant prophylaxis were given to all long-term care residents for extended durations (eg, for the duration of reduced mobility), the incidence and prevalence of heparin-induced thrombocytopenia would likely become a major concern.
Whenever anticoagulant prophylaxis is considered, the risks of both thrombosis and bleeding should be considered. Patients who are receiving anticoagulant prophylaxis should also be monitored for bleeding and heparin-induced thrombocytopenia. This is particularly true in long-term care residents, in whom the risks and benefits of anticoagulant prophylaxis are extrapolated from data from other populations.
MORE RESEARCH IS NEEDED
To date, we lack audits of thromboprophylaxis, clinical practice guidelines, and clear indications and contraindications for anticoagulant prophylaxis in long-term care residents. In the absence of such data, extrapolating the efficacy and safety of thromboprophylaxis from hospitalized patients to long-term care residents is difficult.
Clearly, additional research is needed to identify which long-term care residents would benefit most from thromboprophylaxis. In the meantime, a selective approach to identifying patients who should be considered for thromboprophylaxis should be adopted.
- Cornman JM. Questions for societies with “third age” populations. The Extension-of-Life Working Group, The Gerontological Society of America. Acad Med 1997; 72:856–862.
- Kemper P, Murtaugh CM. Lifetime use of nursing home care. N Engl J Med 1991; 324:595–600.
- Gomes JP, Shaheen WH, Truong SV, Brown EF, Beasley BW, Gajewski BJ. Incidence of venous thromboembolic events among nursing home residents. J Gen Intern Med 2003; 18:934–936.
- Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med 1994; 154:861–866.
- Bounameaux H. Integrating pharmacologic and mechanical prophylaxis of venous thromboembolism. Thromb Haemost 1999; 82:931–937.
- Gross JS, Neufeld RR, Libow LS, Gerber I, Rodstein M. Autopsy study of the elderly institutionalized patient. Review of 234 autopsies. Arch Intern Med 1988; 148:173–176.
- Spyropoulos AC, Merli G. Management of venous thromboembolism in the elderly. Drugs Aging 2006; 23:651–671.
- Punukollu H, Khan IA, Punukollu G, Gowda RM, Mendoza C, Sacchi TJ. Acute pulmonary embolism in elderly: clinical characteristics and outcome. Int J Cardiol 2005; 99:213–216.
- Douketis JD. Prevention of venous thromboembolism in hospitalized medical patients: addressing some practical questions. Curr Opin Pulm Med 2008; 14:381–388.
- Cohen AT, Tapson VF, Bergmann JF, et al; ENDORSE Investigators. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross-sectional study. Lancet 2008; 371:387–394.
- Kahn SR, Panju A, Geerts W, et al; CURVE study investigators. Multicenter evaluation of the use of venous thromboembolism prophylaxis in acutely ill medical patients in Canada. Thromb Res 2007; 119:145–155.
- Haas S, Spyropoulos AC. Primary prevention of venous thromboembolism in long-term care: identifying and managing the risk. Clin Appl Thromb Hemost 2008; 14:149–158.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133( suppl 6):381S–453S.
- Francis CW. Clinical practice. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med 2007; 356:1438–1444.
- Douketis J, Cook D, Meade M, et al; Canadian Critical Care Trials Group. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics: the DIRECT study. Arch Intern Med 2008; 168:1805–1812.
- Lobo B, Finch C, Howard A, Minhas S. Fondaparinux for the treatment of patients with acute heparin-induced thrombocytopenia. Thromb Haemost 2008; 99:208–214.
- Spinler SA. New concepts in heparin-induced thrombocytopenia: diagnosis and management. J Thromb Thrombolysis 2006; 21:17–21.
- Hull RD, Schellong SM, Tapson VF, et al. Extended-duration thromboprophylaxis in acutely ill medical patients with recent reduced mobility: methodology for the EXCLAIM study. J Thromb Thrombolysis 2006; 22:31–38.
- Dennis M, Sandercock PA, Reid J, et al; CLOTS Trials Collaboration Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009; 373:1958–1965.
- Weill-Engerer S, Meaume S, Lahlou A, et al. Risk factors for deep vein thrombosis in inpatients aged 65 and older: a case-control multicenter study. J Am Geriatr Soc 2004; 52:1299–1304.
- Dentali F, Douketis JD, Gianni M, Lim W, Crowther MA. Meta-analysis: anticoagulant prophylaxis to prevent symptomatic venous thromboembolism in hospitalized medical patients. Ann Intern Med 2007; 146:278–288.
- Douketis JD, Arneklev K, Goldhaber SZ, Spandorfer J, Halperin F, Horrow J. Comparison of bleeding in patients with nonvalvular atrial fibrillation treated with ximelagatran or warfarin: assessment of incidence, case-fatality rate, time course and sites of bleeding, and risk factors for bleeding. Arch Intern Med 2006; 166:853–859.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- Lloyd NS, Douketis JD, Moinuddin I, Lim W, Crowther MA. Anticoagulant prophylaxis to prevent asymptomatic deep vein thrombosis in hospitalized medical patients: a systematic review and meta-analysis. J Thromb Haemost 2008; 6:405–414.
- Swedko PJ, Clark HD, Paramsothy K, Akbari A. Serum creatinine is an inadequate screening test for renal failure in elderly patients. Arch Intern Med 2003; 163:356–360.
- Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005; 106:2710–2715.
- Stein PD, Hull RD, Matta F, Yaekoub AY, Liang J. Incidence of thrombocytopenia in hospitalized patients with venous thromboembolism. Am J Med 2009; 122:919–930.
Randomized trials that included more than 20,000 medical patients have shown that anticoagulant therapy is safe and effective in preventing venous thromboembolism (VTE), ie, deep vein thrombosis and pulmonary embolism.
However, these trials were done in hospitalized patients, who typically had an acute medical illness and who, if eligible, received a short (7- to 10-day) course of anticoagulant prophylaxis.
Little attention has been given to VTE prophylaxis in residents of long-term care facilities. These patients have risk profiles similar to those of hospitalized medical patients. Some of them may have been transferred from an acute care hospital. In addition, most are elderly, and many have reduced mobility and are at risk for illnesses such as stroke and cardiorespiratory insufficiency, which increase the risk of VTE.
VTE in residents of long-term care facilities is a growing concern. By some estimates, by the year 2030 more than 20% of the US population (70.2 million people) will be over 65 years of age.1 Of those who reached age 65 in 1990, an estimated 43% will enter a nursing home at least once before they die—32% for 3 months, 24% for at least a year, and 9% for at least 5 years.2
Against this background, the objectives of this review are to consider:
- The scope of the problem of VTE in long-term care residents
- Why VTE prophylaxis is often overlooked in medical patients
- Evidence—or lack of evidence—for the safety and efficacy of VTE prophylaxis in long-term care residents and other medical patients
- Available options for VTE prophylaxis
- Which long-term care residents should or should not be considered for prophylaxis.
THE TRUE SCOPE OF THE PROBLEM IS UNKNOWN
The incidence of acute VTE among nursing home residents is reported to be 1.3 events per 100 person-years.3 About 8% of cases of pulmonary embolism and 10% of cases of deep venous thrombosis in the elderly are in nursing home residents.4
However, only 20% of patients with VTE have typical symptoms such as leg pain and swelling or acute dyspnea and chest pain, while 80% have no symptoms.5
Furthermore, deep venous thrombosis is more likely to be clinically silent in patients whose mobility is impaired, such as nursing home residents, as the symptoms arising from obstruction of venous flow are more pronounced with walking.
Pulmonary embolism is also underdiagnosed in this group. An autopsy study of 234 nursing home residents found undiagnosed pulmonary embolism to be the cause of death in 8%, and 40% of cases of pulmonary embolism were not suspected before the patient died.6 Yet pulmonary embolism has a higher case-fatality rate in the elderly than in younger patients, particularly when elderly patients have comorbidities.7
A reason why the diagnosis is so often missed is that pulmonary embolism can present atypically in the elderly, with syncope being more common and tachycardia being less common than in younger patients.8
Since so many cases of VTE are clinically silent and most long-term care residents who die do not undergo autopsy, the true scope of VTE as a clinical problem in these patients is unknown. Consequently, the best way to diagnose, prevent, and treat VTE is also unclear.
WHY IS VTE PREVENTION SO OFTEN OVERLOOKED IN MEDICAL PATIENTS?
In general, nonsurgical patients receive suboptimal thromboprophylaxis. National and international chart audits and cross-sectional studies show that only 16% to 33% of hospitalized medical patients at risk for VTE receive appropriate anticoagulant prophylaxis.9 Though no audits in long-term care facilities have been published, the rate of appropriate prophylaxis is likely comparable to or possibly less than that in medical patients in the hospital. In contrast, in surgical patients the rate is much higher—up to 90%.10,11
Why is VTE prophylaxis so underused in medical patients?
One reason is that we do not really know the baseline risk of VTE in medical patients, particularly in those with chronic illness who require long-term care.12 This is relevant because, in the absence of data about patients’ baseline risk, anticoagulant prophylaxis should be ordered selectively, as it poses known risks of bleeding. The risk is greater in elderly people with comorbidities, as are the associated costs.
In addition, relatively few studies have assessed thromboprophylaxis in medical patients, especially in residents of long-term care facilities.
Another reason is that we lack practice guidelines for patients who need long-term care. The well-accepted guidelines from the American College of Chest Physicians (ACCP) cite advanced age and immobility as risk factors for VTE and strongly recommend prophylaxis in acutely ill medical patients who have limited mobility and an additional risk factor such as infection or cancer.13 Though elderly residents of long-term care facilities may share some of these risk factors, the ACCP guidelines make no specific recommendations for this group.
The attitudes of health care professionals may also pose a barrier. Lloyd et al (unpublished data, 2009) surveyed 1,601 health care professionals in Ontario, Canada, in 2007, to assess potential barriers to anticoagulant prophylaxis in hospitalized medical patients. Respondents cited concerns about the risk of bleeding from anticoagulants, lack of clear indications and contraindications for anticoagulant prophylaxis, and lack of time to consider VTE prophylaxis in every patient. (They did not, however, cite disagreement with guidelines or patient discomfort from subcutaneous anticoagulant injections as barriers.) It is reasonable to assume that these attitudes may also pose a problem in long-term care residents.
Finally, no randomized trials have evaluated the efficacy and safety of anticoagulant drugs or mechanical methods of prophylaxis in long-term care residents. Studies have shown that a short course (7–10 days) of an anticoagulant drug effectively prevents VTE in acutely ill patients, but the efficacy of an extended course in patients with chronic illness who require long-term care is not clear. Therefore, recommendations about thromboprophylaxis in long-term care residents should be made with the caveat that they are based on indirect evidence from other patient groups. This is a considerable limitation.
OPTIONS FOR THROMBOPROPHYLAXIS IN LONG-TERM CARE RESIDENTS
Options for thromboprophylaxis fall into two broad categories: anticoagulant drugs and mechanical devices.
Anticoagulant prophylactic drugs
These agents have been assessed in randomized trials in surgical or acutely ill medical patients, although fondaparinux and tinzaparin are not approved for use in medical patients. Furthermore, none of them has been evaluated in residents of long-term care facilities.
The choice of anticoagulant for prophylaxis is determined largely by clinical factors.
Low-molecular-weight heparins are popular both in and out of the hospital because they have predictable pharmacokinetic properties, they come in convenient prefilled syringes, and they can be given once daily. However, some of them may bioaccumulate in patients with impaired renal function, as they are cleared primarily by the kidney.
Unfractionated heparin is likely to be safer in patients with severe renal insufficiency (creatinine clearance < 30 mL/min), as it is cleared via nonrenal mechanisms.
However, a recent single-arm trial of dalteparin 5,000 IU once daily in critically ill patients with severe renal insufficiency found no evidence of an excessive anticoagulant effect or of drug bioaccumulation.15 Dalteparin may thus be an alternative to unfractionated heparin in medical patients with impaired renal function.
Fondaparinux, a newer anticoagulant, is also given once daily. It is the anticoagulant of choice in patients who have had heparin-induced thrombocytopenia because it is not derived from heparin and likely does not cross-react with heparin-induced thrombocytopenia antibodies.16,17
Limited data on benefit of prophylactic anticoagulant drugs
As mentioned, the trials that confirmed the efficacy and safety of anticoagulant prophylaxis were in surgical patients and hospitalized medical patients, not elderly long-term care residents. The poor evidence for anticoagulant prophylaxis in these patients may be strengthened if extended-duration, out-of-hospital prophylaxis were shown to be effective in medical patients. Long-term care residents could more reasonably be compared with medical patients discharged home with a chronic or resolving illness than with those who are hospitalized.
There is some evidence, although with caveats, that extended anticoagulant prophylaxis, started after an acute illness has resolved, confers a benefit. A recent randomized trial compared extended-duration and short-duration prophylaxis (5 weeks vs 10 days) with enoxaparin 40 mg once daily in 4,726 medical patients with impaired mobility.18 The risk of any VTE event was 44% lower with extended-duration prophylaxis (2.8% vs 4.9%; P = .001) and the risk of symptomatic VTE was 73% lower (0.3% vs 1.1%; P = .004), and this benefit persisted 2 months after treatment was stopped (3.0% vs 5.2%; P = .0015). However, extended treatment conferred a fourfold higher risk of major bleeding (0.6% vs 0.15%; P = .019).
These findings should also be considered in terms of absolute benefit and harm. Treating 1,000 patients for 5 weeks instead of 10 days would prevent eight episodes of symptomatic VTE (absolute risk reduction = 0.8%, number needed to treat = 125) at the cost of four to five episodes of major bleeding (absolute risk increase = 0.45%, number needed to harm = 222). This is a modest net therapeutic benefit.
The therapeutic benefit would be greater if we consider all episodes of VTE, both symptomatic and asymptomatic. Treating 1,000 patients for 5 weeks would prevent 20 episodes of symptomatic or asymptomatic VTE (absolute risk reduction = 2.1%, number needed to treat = 48). However, the clinical importance of asymptomatic VTE is questionable.
Given these considerations, if extended-duration anticoagulant prophylaxis is considered, it should be for patients at highest risk to optimize both its net therapeutic benefits and its cost-effectiveness.
Mechanical prophylaxis
Mechanical thromboprophylactic devices—graduated or elastic compression stockings and intermittent pneumatic compression devices—are effective when used by themselves in surgical patients.13 However, in a randomized controlled trial in patients with ischemic stroke, the rate of VTE was 10.0% with graduated compression stockings in addition to “usual care VTE prophylaxis” vs 10.5% with usual care alone, and patients in the stocking group had a fourfold higher risk of developing skin breaks, ulcers, blisters, or necrosis (5% vs 1%; odds ratio 4.18; 95% CI 2.4–7.3).19 Furthermore, improperly fitted stockings, especially those that are thigh-length, can be uncomfortable to wear and difficult to apply.
Overall, the role of mechanical thromboprophylaxis in long-term care facilities is not clear. If it is considered, there should be a compelling reason to use it—for example, for patients at high risk in whom anticoagulants are contraindicated because of ongoing bleeding or a higher risk of bleeding (eg, recent gastrointestinal bleeding, hemorrhagic stroke, coagulopathy, or thrombocytopenia). Furthermore, if stockings are used, they should be properly fitted and routinely monitored for adverse effects, since elderly patients are likely to be most susceptible to skin breakdown.
WHICH LONG-TERM CARE RESIDENTS SHOULD RECEIVE VTE PROPHYLAXIS?
Old age and immobility are not the only risk factors
The current ACCP guidelines suggest considering thromboprophylaxis for hospitalized medical patients over age 75 who cannot walk without assistance.13 However, we lack evidence to suggest a similar strategy in long-term care residents.
The ACCP guidelines are based on data on risk. Nearly 25% of elderly patients with confirmed pulmonary embolism had been immobile prior to their diagnosis.8 In addition, prolonged bed rest (> 14 days) has been reported to be the strongest independent risk factor for symptomatic deep venous thrombosis, increasing the risk more than fivefold.20 Advanced age is also considered a risk factor for VTE, as risk starts to increase at age 40 and doubles each decade of life thereafter.18
No study has assessed the impact of these factors on the risk of VTE in long-term care residents. Since most of such patients are elderly and have impaired mobility, we believe a more selective approach should be used in assigning VTE risk status, one that does not use advanced age and immobility as the only criteria for starting thromboprophylaxis.
Residents of long-term care facilities may be immobile because of underlying illness or disability, such as cognitive impairment, sensory impairment (eg, poor access to corrective lenses and hearing aids), or poor access to assist devices (eg, walkers, canes). In addition, iatrogenic factors that decrease mobility such as indwelling bladder catheters and physical restraints are also common in such patients.
Efforts to improve mobility should be encouraged. However, we recommend that thromboprophylaxis be considered only in patients who have both impaired mobility and an intercurrent acute medical illness such as an acute infection or acute inflammatory disease.13
A related issue is the difference between long-term care residents with a chronic but stable disease and those with acute disease. Patients with acute exacerbations of congestive heart failure or chronic obstructive lung disease may be considered for thromboprophylaxis, as they become more comparable to acutely ill medical patients in whom clinical trials have shown the effectiveness of anticoagulant prophylaxis. On the other hand, patients with these diseases who remain stable may not need prophylaxis.
This approach avoids giving long-term anticoagulant prophylaxis to patients who have irreversible diseases and limits the use of these drugs and devices to higher-risk periods.
Consider thromboprophylaxis if…
- An acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease
- Acute infection (eg, urosepsis, pneumonia, cellulitis, infectious diarrhea)
- An acute exacerbation of an inflammatory disease (eg, rheumatoid arthritis)
- Active cancer (eg, patient receiving radiation therapy or chemotherapy)
- Immobility and prior VTE.
Do not routinely consider prophylaxis if…
We also believe patients should not be routinely considered for anticoagulant VTE prophylaxis if they have:
- Chronic but stable cardiorespiratory disease
- Chronic but stable infectious or inflammatory disease
- Terminal cancer with very limited life expectancy
- Any contraindication to anticoagulants (eg, active bleeding, recent bleeding, coagulopathy, thrombocytopenia).
ANTICOAGULANT PROPHYLAXIS POSES RISKS IN LONG-TERM CARE RESIDENTS
Bleeding is the principal risk
The incidence of clinically important bleeding associated with anticoagulant prophylaxis is 0.2% to 5.6%, and the risk of fatal bleeding is 0.02% to 0.5%.21–24
As no randomized trial has examined anticoagulant prophylaxis in elderly long-term care residents, their bleeding risk with this therapy is unclear. However, older patients are likely to be at higher risk than younger patients because they have more comorbidities, take more drugs that could interact with heparin and potentiate bleeding, and have fragile skin, predisposing to injury from subcutaneous injections.
Also, renal function tends to decline with age. In a retrospective study of 854 outpatients over age 65, 29% had moderate renal insufficiency (creatinine clearance 30–50 mL/min), and 6% had severe renal insufficiency (creatinine clearance < 30 mL/min).25 Recent evidence suggests that some low-molecular-weight heparins (dalteparin and tinzaparin) do not bioaccumulate in patients with impaired renal function. However, enoxaparin and fondaparinux should be used with caution in patients with moderate to severe renal impairment.
Though much attention has recently been paid to increasing anticoagulant doses if the patient is obese, residents of long-term care facilities are more likely to be underweight. Dose adjustment should be considered when a low-molecular-weight heparin or fondaparinux is given to patients weighing less than 50 kg.
Heparin-induced thrombocytopenia
The other major risk of anticoagulant prophylaxis is heparin-induced thrombocytopenia, an infrequent but life-threatening complication caused by the formation of antibodies to the heparin-derived anticoagulant and a platelet surface antigen. It is associated with moderate thrombocytopenia and an incidence of venous or arterial thrombosis that is over 50%.26
No study has assessed the incidence of heparin-induced thrombocytopenia in long-term care residents. A meta-analysis reported that the risk with anticoagulant prophylaxis was 1.6% with unfractionated heparin (95% confidence interval [CI] 1.2%–2.1%) and 0.6% with low-molecular-weight heparin (95% CI 0.4%–0.9%), and that this risk increased with the duration of prophylaxis.27 If anticoagulant prophylaxis were given to all long-term care residents for extended durations (eg, for the duration of reduced mobility), the incidence and prevalence of heparin-induced thrombocytopenia would likely become a major concern.
Whenever anticoagulant prophylaxis is considered, the risks of both thrombosis and bleeding should be considered. Patients who are receiving anticoagulant prophylaxis should also be monitored for bleeding and heparin-induced thrombocytopenia. This is particularly true in long-term care residents, in whom the risks and benefits of anticoagulant prophylaxis are extrapolated from data from other populations.
MORE RESEARCH IS NEEDED
To date, we lack audits of thromboprophylaxis, clinical practice guidelines, and clear indications and contraindications for anticoagulant prophylaxis in long-term care residents. In the absence of such data, extrapolating the efficacy and safety of thromboprophylaxis from hospitalized patients to long-term care residents is difficult.
Clearly, additional research is needed to identify which long-term care residents would benefit most from thromboprophylaxis. In the meantime, a selective approach to identifying patients who should be considered for thromboprophylaxis should be adopted.
Randomized trials that included more than 20,000 medical patients have shown that anticoagulant therapy is safe and effective in preventing venous thromboembolism (VTE), ie, deep vein thrombosis and pulmonary embolism.
However, these trials were done in hospitalized patients, who typically had an acute medical illness and who, if eligible, received a short (7- to 10-day) course of anticoagulant prophylaxis.
Little attention has been given to VTE prophylaxis in residents of long-term care facilities. These patients have risk profiles similar to those of hospitalized medical patients. Some of them may have been transferred from an acute care hospital. In addition, most are elderly, and many have reduced mobility and are at risk for illnesses such as stroke and cardiorespiratory insufficiency, which increase the risk of VTE.
VTE in residents of long-term care facilities is a growing concern. By some estimates, by the year 2030 more than 20% of the US population (70.2 million people) will be over 65 years of age.1 Of those who reached age 65 in 1990, an estimated 43% will enter a nursing home at least once before they die—32% for 3 months, 24% for at least a year, and 9% for at least 5 years.2
Against this background, the objectives of this review are to consider:
- The scope of the problem of VTE in long-term care residents
- Why VTE prophylaxis is often overlooked in medical patients
- Evidence—or lack of evidence—for the safety and efficacy of VTE prophylaxis in long-term care residents and other medical patients
- Available options for VTE prophylaxis
- Which long-term care residents should or should not be considered for prophylaxis.
THE TRUE SCOPE OF THE PROBLEM IS UNKNOWN
The incidence of acute VTE among nursing home residents is reported to be 1.3 events per 100 person-years.3 About 8% of cases of pulmonary embolism and 10% of cases of deep venous thrombosis in the elderly are in nursing home residents.4
However, only 20% of patients with VTE have typical symptoms such as leg pain and swelling or acute dyspnea and chest pain, while 80% have no symptoms.5
Furthermore, deep venous thrombosis is more likely to be clinically silent in patients whose mobility is impaired, such as nursing home residents, as the symptoms arising from obstruction of venous flow are more pronounced with walking.
Pulmonary embolism is also underdiagnosed in this group. An autopsy study of 234 nursing home residents found undiagnosed pulmonary embolism to be the cause of death in 8%, and 40% of cases of pulmonary embolism were not suspected before the patient died.6 Yet pulmonary embolism has a higher case-fatality rate in the elderly than in younger patients, particularly when elderly patients have comorbidities.7
A reason why the diagnosis is so often missed is that pulmonary embolism can present atypically in the elderly, with syncope being more common and tachycardia being less common than in younger patients.8
Since so many cases of VTE are clinically silent and most long-term care residents who die do not undergo autopsy, the true scope of VTE as a clinical problem in these patients is unknown. Consequently, the best way to diagnose, prevent, and treat VTE is also unclear.
WHY IS VTE PREVENTION SO OFTEN OVERLOOKED IN MEDICAL PATIENTS?
In general, nonsurgical patients receive suboptimal thromboprophylaxis. National and international chart audits and cross-sectional studies show that only 16% to 33% of hospitalized medical patients at risk for VTE receive appropriate anticoagulant prophylaxis.9 Though no audits in long-term care facilities have been published, the rate of appropriate prophylaxis is likely comparable to or possibly less than that in medical patients in the hospital. In contrast, in surgical patients the rate is much higher—up to 90%.10,11
Why is VTE prophylaxis so underused in medical patients?
One reason is that we do not really know the baseline risk of VTE in medical patients, particularly in those with chronic illness who require long-term care.12 This is relevant because, in the absence of data about patients’ baseline risk, anticoagulant prophylaxis should be ordered selectively, as it poses known risks of bleeding. The risk is greater in elderly people with comorbidities, as are the associated costs.
In addition, relatively few studies have assessed thromboprophylaxis in medical patients, especially in residents of long-term care facilities.
Another reason is that we lack practice guidelines for patients who need long-term care. The well-accepted guidelines from the American College of Chest Physicians (ACCP) cite advanced age and immobility as risk factors for VTE and strongly recommend prophylaxis in acutely ill medical patients who have limited mobility and an additional risk factor such as infection or cancer.13 Though elderly residents of long-term care facilities may share some of these risk factors, the ACCP guidelines make no specific recommendations for this group.
The attitudes of health care professionals may also pose a barrier. Lloyd et al (unpublished data, 2009) surveyed 1,601 health care professionals in Ontario, Canada, in 2007, to assess potential barriers to anticoagulant prophylaxis in hospitalized medical patients. Respondents cited concerns about the risk of bleeding from anticoagulants, lack of clear indications and contraindications for anticoagulant prophylaxis, and lack of time to consider VTE prophylaxis in every patient. (They did not, however, cite disagreement with guidelines or patient discomfort from subcutaneous anticoagulant injections as barriers.) It is reasonable to assume that these attitudes may also pose a problem in long-term care residents.
Finally, no randomized trials have evaluated the efficacy and safety of anticoagulant drugs or mechanical methods of prophylaxis in long-term care residents. Studies have shown that a short course (7–10 days) of an anticoagulant drug effectively prevents VTE in acutely ill patients, but the efficacy of an extended course in patients with chronic illness who require long-term care is not clear. Therefore, recommendations about thromboprophylaxis in long-term care residents should be made with the caveat that they are based on indirect evidence from other patient groups. This is a considerable limitation.
OPTIONS FOR THROMBOPROPHYLAXIS IN LONG-TERM CARE RESIDENTS
Options for thromboprophylaxis fall into two broad categories: anticoagulant drugs and mechanical devices.
Anticoagulant prophylactic drugs
These agents have been assessed in randomized trials in surgical or acutely ill medical patients, although fondaparinux and tinzaparin are not approved for use in medical patients. Furthermore, none of them has been evaluated in residents of long-term care facilities.
The choice of anticoagulant for prophylaxis is determined largely by clinical factors.
Low-molecular-weight heparins are popular both in and out of the hospital because they have predictable pharmacokinetic properties, they come in convenient prefilled syringes, and they can be given once daily. However, some of them may bioaccumulate in patients with impaired renal function, as they are cleared primarily by the kidney.
Unfractionated heparin is likely to be safer in patients with severe renal insufficiency (creatinine clearance < 30 mL/min), as it is cleared via nonrenal mechanisms.
However, a recent single-arm trial of dalteparin 5,000 IU once daily in critically ill patients with severe renal insufficiency found no evidence of an excessive anticoagulant effect or of drug bioaccumulation.15 Dalteparin may thus be an alternative to unfractionated heparin in medical patients with impaired renal function.
Fondaparinux, a newer anticoagulant, is also given once daily. It is the anticoagulant of choice in patients who have had heparin-induced thrombocytopenia because it is not derived from heparin and likely does not cross-react with heparin-induced thrombocytopenia antibodies.16,17
Limited data on benefit of prophylactic anticoagulant drugs
As mentioned, the trials that confirmed the efficacy and safety of anticoagulant prophylaxis were in surgical patients and hospitalized medical patients, not elderly long-term care residents. The poor evidence for anticoagulant prophylaxis in these patients may be strengthened if extended-duration, out-of-hospital prophylaxis were shown to be effective in medical patients. Long-term care residents could more reasonably be compared with medical patients discharged home with a chronic or resolving illness than with those who are hospitalized.
There is some evidence, although with caveats, that extended anticoagulant prophylaxis, started after an acute illness has resolved, confers a benefit. A recent randomized trial compared extended-duration and short-duration prophylaxis (5 weeks vs 10 days) with enoxaparin 40 mg once daily in 4,726 medical patients with impaired mobility.18 The risk of any VTE event was 44% lower with extended-duration prophylaxis (2.8% vs 4.9%; P = .001) and the risk of symptomatic VTE was 73% lower (0.3% vs 1.1%; P = .004), and this benefit persisted 2 months after treatment was stopped (3.0% vs 5.2%; P = .0015). However, extended treatment conferred a fourfold higher risk of major bleeding (0.6% vs 0.15%; P = .019).
These findings should also be considered in terms of absolute benefit and harm. Treating 1,000 patients for 5 weeks instead of 10 days would prevent eight episodes of symptomatic VTE (absolute risk reduction = 0.8%, number needed to treat = 125) at the cost of four to five episodes of major bleeding (absolute risk increase = 0.45%, number needed to harm = 222). This is a modest net therapeutic benefit.
The therapeutic benefit would be greater if we consider all episodes of VTE, both symptomatic and asymptomatic. Treating 1,000 patients for 5 weeks would prevent 20 episodes of symptomatic or asymptomatic VTE (absolute risk reduction = 2.1%, number needed to treat = 48). However, the clinical importance of asymptomatic VTE is questionable.
Given these considerations, if extended-duration anticoagulant prophylaxis is considered, it should be for patients at highest risk to optimize both its net therapeutic benefits and its cost-effectiveness.
Mechanical prophylaxis
Mechanical thromboprophylactic devices—graduated or elastic compression stockings and intermittent pneumatic compression devices—are effective when used by themselves in surgical patients.13 However, in a randomized controlled trial in patients with ischemic stroke, the rate of VTE was 10.0% with graduated compression stockings in addition to “usual care VTE prophylaxis” vs 10.5% with usual care alone, and patients in the stocking group had a fourfold higher risk of developing skin breaks, ulcers, blisters, or necrosis (5% vs 1%; odds ratio 4.18; 95% CI 2.4–7.3).19 Furthermore, improperly fitted stockings, especially those that are thigh-length, can be uncomfortable to wear and difficult to apply.
Overall, the role of mechanical thromboprophylaxis in long-term care facilities is not clear. If it is considered, there should be a compelling reason to use it—for example, for patients at high risk in whom anticoagulants are contraindicated because of ongoing bleeding or a higher risk of bleeding (eg, recent gastrointestinal bleeding, hemorrhagic stroke, coagulopathy, or thrombocytopenia). Furthermore, if stockings are used, they should be properly fitted and routinely monitored for adverse effects, since elderly patients are likely to be most susceptible to skin breakdown.
WHICH LONG-TERM CARE RESIDENTS SHOULD RECEIVE VTE PROPHYLAXIS?
Old age and immobility are not the only risk factors
The current ACCP guidelines suggest considering thromboprophylaxis for hospitalized medical patients over age 75 who cannot walk without assistance.13 However, we lack evidence to suggest a similar strategy in long-term care residents.
The ACCP guidelines are based on data on risk. Nearly 25% of elderly patients with confirmed pulmonary embolism had been immobile prior to their diagnosis.8 In addition, prolonged bed rest (> 14 days) has been reported to be the strongest independent risk factor for symptomatic deep venous thrombosis, increasing the risk more than fivefold.20 Advanced age is also considered a risk factor for VTE, as risk starts to increase at age 40 and doubles each decade of life thereafter.18
No study has assessed the impact of these factors on the risk of VTE in long-term care residents. Since most of such patients are elderly and have impaired mobility, we believe a more selective approach should be used in assigning VTE risk status, one that does not use advanced age and immobility as the only criteria for starting thromboprophylaxis.
Residents of long-term care facilities may be immobile because of underlying illness or disability, such as cognitive impairment, sensory impairment (eg, poor access to corrective lenses and hearing aids), or poor access to assist devices (eg, walkers, canes). In addition, iatrogenic factors that decrease mobility such as indwelling bladder catheters and physical restraints are also common in such patients.
Efforts to improve mobility should be encouraged. However, we recommend that thromboprophylaxis be considered only in patients who have both impaired mobility and an intercurrent acute medical illness such as an acute infection or acute inflammatory disease.13
A related issue is the difference between long-term care residents with a chronic but stable disease and those with acute disease. Patients with acute exacerbations of congestive heart failure or chronic obstructive lung disease may be considered for thromboprophylaxis, as they become more comparable to acutely ill medical patients in whom clinical trials have shown the effectiveness of anticoagulant prophylaxis. On the other hand, patients with these diseases who remain stable may not need prophylaxis.
This approach avoids giving long-term anticoagulant prophylaxis to patients who have irreversible diseases and limits the use of these drugs and devices to higher-risk periods.
Consider thromboprophylaxis if…
- An acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease
- Acute infection (eg, urosepsis, pneumonia, cellulitis, infectious diarrhea)
- An acute exacerbation of an inflammatory disease (eg, rheumatoid arthritis)
- Active cancer (eg, patient receiving radiation therapy or chemotherapy)
- Immobility and prior VTE.
Do not routinely consider prophylaxis if…
We also believe patients should not be routinely considered for anticoagulant VTE prophylaxis if they have:
- Chronic but stable cardiorespiratory disease
- Chronic but stable infectious or inflammatory disease
- Terminal cancer with very limited life expectancy
- Any contraindication to anticoagulants (eg, active bleeding, recent bleeding, coagulopathy, thrombocytopenia).
ANTICOAGULANT PROPHYLAXIS POSES RISKS IN LONG-TERM CARE RESIDENTS
Bleeding is the principal risk
The incidence of clinically important bleeding associated with anticoagulant prophylaxis is 0.2% to 5.6%, and the risk of fatal bleeding is 0.02% to 0.5%.21–24
As no randomized trial has examined anticoagulant prophylaxis in elderly long-term care residents, their bleeding risk with this therapy is unclear. However, older patients are likely to be at higher risk than younger patients because they have more comorbidities, take more drugs that could interact with heparin and potentiate bleeding, and have fragile skin, predisposing to injury from subcutaneous injections.
Also, renal function tends to decline with age. In a retrospective study of 854 outpatients over age 65, 29% had moderate renal insufficiency (creatinine clearance 30–50 mL/min), and 6% had severe renal insufficiency (creatinine clearance < 30 mL/min).25 Recent evidence suggests that some low-molecular-weight heparins (dalteparin and tinzaparin) do not bioaccumulate in patients with impaired renal function. However, enoxaparin and fondaparinux should be used with caution in patients with moderate to severe renal impairment.
Though much attention has recently been paid to increasing anticoagulant doses if the patient is obese, residents of long-term care facilities are more likely to be underweight. Dose adjustment should be considered when a low-molecular-weight heparin or fondaparinux is given to patients weighing less than 50 kg.
Heparin-induced thrombocytopenia
The other major risk of anticoagulant prophylaxis is heparin-induced thrombocytopenia, an infrequent but life-threatening complication caused by the formation of antibodies to the heparin-derived anticoagulant and a platelet surface antigen. It is associated with moderate thrombocytopenia and an incidence of venous or arterial thrombosis that is over 50%.26
No study has assessed the incidence of heparin-induced thrombocytopenia in long-term care residents. A meta-analysis reported that the risk with anticoagulant prophylaxis was 1.6% with unfractionated heparin (95% confidence interval [CI] 1.2%–2.1%) and 0.6% with low-molecular-weight heparin (95% CI 0.4%–0.9%), and that this risk increased with the duration of prophylaxis.27 If anticoagulant prophylaxis were given to all long-term care residents for extended durations (eg, for the duration of reduced mobility), the incidence and prevalence of heparin-induced thrombocytopenia would likely become a major concern.
Whenever anticoagulant prophylaxis is considered, the risks of both thrombosis and bleeding should be considered. Patients who are receiving anticoagulant prophylaxis should also be monitored for bleeding and heparin-induced thrombocytopenia. This is particularly true in long-term care residents, in whom the risks and benefits of anticoagulant prophylaxis are extrapolated from data from other populations.
MORE RESEARCH IS NEEDED
To date, we lack audits of thromboprophylaxis, clinical practice guidelines, and clear indications and contraindications for anticoagulant prophylaxis in long-term care residents. In the absence of such data, extrapolating the efficacy and safety of thromboprophylaxis from hospitalized patients to long-term care residents is difficult.
Clearly, additional research is needed to identify which long-term care residents would benefit most from thromboprophylaxis. In the meantime, a selective approach to identifying patients who should be considered for thromboprophylaxis should be adopted.
- Cornman JM. Questions for societies with “third age” populations. The Extension-of-Life Working Group, The Gerontological Society of America. Acad Med 1997; 72:856–862.
- Kemper P, Murtaugh CM. Lifetime use of nursing home care. N Engl J Med 1991; 324:595–600.
- Gomes JP, Shaheen WH, Truong SV, Brown EF, Beasley BW, Gajewski BJ. Incidence of venous thromboembolic events among nursing home residents. J Gen Intern Med 2003; 18:934–936.
- Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med 1994; 154:861–866.
- Bounameaux H. Integrating pharmacologic and mechanical prophylaxis of venous thromboembolism. Thromb Haemost 1999; 82:931–937.
- Gross JS, Neufeld RR, Libow LS, Gerber I, Rodstein M. Autopsy study of the elderly institutionalized patient. Review of 234 autopsies. Arch Intern Med 1988; 148:173–176.
- Spyropoulos AC, Merli G. Management of venous thromboembolism in the elderly. Drugs Aging 2006; 23:651–671.
- Punukollu H, Khan IA, Punukollu G, Gowda RM, Mendoza C, Sacchi TJ. Acute pulmonary embolism in elderly: clinical characteristics and outcome. Int J Cardiol 2005; 99:213–216.
- Douketis JD. Prevention of venous thromboembolism in hospitalized medical patients: addressing some practical questions. Curr Opin Pulm Med 2008; 14:381–388.
- Cohen AT, Tapson VF, Bergmann JF, et al; ENDORSE Investigators. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross-sectional study. Lancet 2008; 371:387–394.
- Kahn SR, Panju A, Geerts W, et al; CURVE study investigators. Multicenter evaluation of the use of venous thromboembolism prophylaxis in acutely ill medical patients in Canada. Thromb Res 2007; 119:145–155.
- Haas S, Spyropoulos AC. Primary prevention of venous thromboembolism in long-term care: identifying and managing the risk. Clin Appl Thromb Hemost 2008; 14:149–158.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133( suppl 6):381S–453S.
- Francis CW. Clinical practice. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med 2007; 356:1438–1444.
- Douketis J, Cook D, Meade M, et al; Canadian Critical Care Trials Group. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics: the DIRECT study. Arch Intern Med 2008; 168:1805–1812.
- Lobo B, Finch C, Howard A, Minhas S. Fondaparinux for the treatment of patients with acute heparin-induced thrombocytopenia. Thromb Haemost 2008; 99:208–214.
- Spinler SA. New concepts in heparin-induced thrombocytopenia: diagnosis and management. J Thromb Thrombolysis 2006; 21:17–21.
- Hull RD, Schellong SM, Tapson VF, et al. Extended-duration thromboprophylaxis in acutely ill medical patients with recent reduced mobility: methodology for the EXCLAIM study. J Thromb Thrombolysis 2006; 22:31–38.
- Dennis M, Sandercock PA, Reid J, et al; CLOTS Trials Collaboration Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009; 373:1958–1965.
- Weill-Engerer S, Meaume S, Lahlou A, et al. Risk factors for deep vein thrombosis in inpatients aged 65 and older: a case-control multicenter study. J Am Geriatr Soc 2004; 52:1299–1304.
- Dentali F, Douketis JD, Gianni M, Lim W, Crowther MA. Meta-analysis: anticoagulant prophylaxis to prevent symptomatic venous thromboembolism in hospitalized medical patients. Ann Intern Med 2007; 146:278–288.
- Douketis JD, Arneklev K, Goldhaber SZ, Spandorfer J, Halperin F, Horrow J. Comparison of bleeding in patients with nonvalvular atrial fibrillation treated with ximelagatran or warfarin: assessment of incidence, case-fatality rate, time course and sites of bleeding, and risk factors for bleeding. Arch Intern Med 2006; 166:853–859.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- Lloyd NS, Douketis JD, Moinuddin I, Lim W, Crowther MA. Anticoagulant prophylaxis to prevent asymptomatic deep vein thrombosis in hospitalized medical patients: a systematic review and meta-analysis. J Thromb Haemost 2008; 6:405–414.
- Swedko PJ, Clark HD, Paramsothy K, Akbari A. Serum creatinine is an inadequate screening test for renal failure in elderly patients. Arch Intern Med 2003; 163:356–360.
- Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005; 106:2710–2715.
- Stein PD, Hull RD, Matta F, Yaekoub AY, Liang J. Incidence of thrombocytopenia in hospitalized patients with venous thromboembolism. Am J Med 2009; 122:919–930.
- Cornman JM. Questions for societies with “third age” populations. The Extension-of-Life Working Group, The Gerontological Society of America. Acad Med 1997; 72:856–862.
- Kemper P, Murtaugh CM. Lifetime use of nursing home care. N Engl J Med 1991; 324:595–600.
- Gomes JP, Shaheen WH, Truong SV, Brown EF, Beasley BW, Gajewski BJ. Incidence of venous thromboembolic events among nursing home residents. J Gen Intern Med 2003; 18:934–936.
- Kniffin WD, Baron JA, Barrett J, Birkmeyer JD, Anderson FA. The epidemiology of diagnosed pulmonary embolism and deep venous thrombosis in the elderly. Arch Intern Med 1994; 154:861–866.
- Bounameaux H. Integrating pharmacologic and mechanical prophylaxis of venous thromboembolism. Thromb Haemost 1999; 82:931–937.
- Gross JS, Neufeld RR, Libow LS, Gerber I, Rodstein M. Autopsy study of the elderly institutionalized patient. Review of 234 autopsies. Arch Intern Med 1988; 148:173–176.
- Spyropoulos AC, Merli G. Management of venous thromboembolism in the elderly. Drugs Aging 2006; 23:651–671.
- Punukollu H, Khan IA, Punukollu G, Gowda RM, Mendoza C, Sacchi TJ. Acute pulmonary embolism in elderly: clinical characteristics and outcome. Int J Cardiol 2005; 99:213–216.
- Douketis JD. Prevention of venous thromboembolism in hospitalized medical patients: addressing some practical questions. Curr Opin Pulm Med 2008; 14:381–388.
- Cohen AT, Tapson VF, Bergmann JF, et al; ENDORSE Investigators. Venous thromboembolism risk and prophylaxis in the acute hospital care setting (ENDORSE study): a multinational cross-sectional study. Lancet 2008; 371:387–394.
- Kahn SR, Panju A, Geerts W, et al; CURVE study investigators. Multicenter evaluation of the use of venous thromboembolism prophylaxis in acutely ill medical patients in Canada. Thromb Res 2007; 119:145–155.
- Haas S, Spyropoulos AC. Primary prevention of venous thromboembolism in long-term care: identifying and managing the risk. Clin Appl Thromb Hemost 2008; 14:149–158.
- Geerts WH, Bergqvist D, Pineo GF, et al; American College of Chest Physicians. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133( suppl 6):381S–453S.
- Francis CW. Clinical practice. Prophylaxis for thromboembolism in hospitalized medical patients. N Engl J Med 2007; 356:1438–1444.
- Douketis J, Cook D, Meade M, et al; Canadian Critical Care Trials Group. Prophylaxis against deep vein thrombosis in critically ill patients with severe renal insufficiency with the low-molecular-weight heparin dalteparin: an assessment of safety and pharmacodynamics: the DIRECT study. Arch Intern Med 2008; 168:1805–1812.
- Lobo B, Finch C, Howard A, Minhas S. Fondaparinux for the treatment of patients with acute heparin-induced thrombocytopenia. Thromb Haemost 2008; 99:208–214.
- Spinler SA. New concepts in heparin-induced thrombocytopenia: diagnosis and management. J Thromb Thrombolysis 2006; 21:17–21.
- Hull RD, Schellong SM, Tapson VF, et al. Extended-duration thromboprophylaxis in acutely ill medical patients with recent reduced mobility: methodology for the EXCLAIM study. J Thromb Thrombolysis 2006; 22:31–38.
- Dennis M, Sandercock PA, Reid J, et al; CLOTS Trials Collaboration Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009; 373:1958–1965.
- Weill-Engerer S, Meaume S, Lahlou A, et al. Risk factors for deep vein thrombosis in inpatients aged 65 and older: a case-control multicenter study. J Am Geriatr Soc 2004; 52:1299–1304.
- Dentali F, Douketis JD, Gianni M, Lim W, Crowther MA. Meta-analysis: anticoagulant prophylaxis to prevent symptomatic venous thromboembolism in hospitalized medical patients. Ann Intern Med 2007; 146:278–288.
- Douketis JD, Arneklev K, Goldhaber SZ, Spandorfer J, Halperin F, Horrow J. Comparison of bleeding in patients with nonvalvular atrial fibrillation treated with ximelagatran or warfarin: assessment of incidence, case-fatality rate, time course and sites of bleeding, and risk factors for bleeding. Arch Intern Med 2006; 166:853–859.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- Lloyd NS, Douketis JD, Moinuddin I, Lim W, Crowther MA. Anticoagulant prophylaxis to prevent asymptomatic deep vein thrombosis in hospitalized medical patients: a systematic review and meta-analysis. J Thromb Haemost 2008; 6:405–414.
- Swedko PJ, Clark HD, Paramsothy K, Akbari A. Serum creatinine is an inadequate screening test for renal failure in elderly patients. Arch Intern Med 2003; 163:356–360.
- Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood 2005; 106:2710–2715.
- Stein PD, Hull RD, Matta F, Yaekoub AY, Liang J. Incidence of thrombocytopenia in hospitalized patients with venous thromboembolism. Am J Med 2009; 122:919–930.
KEY POINTS
- Assessment of VTE risk and consideration of need for anticoagulant prophylaxis in long-term care residents are based on indirect data, derived primarily from studies of acutely ill hospitalized medical patients.
- Drugs and devices for thromboprophylaxis have been studied in medical and surgical populations, but issues of efficacy and safety are likely to also pertain to long-term care residents.
- Thromboprophylaxis should be considered for long-term care residents if they are definitely at increased risk of VTE—ie, if they have an acute exacerbation of congestive heart failure or chronic obstructive pulmonary disease; acute inflammatory disease; acute infection; active cancer; or immobility and prior VTE.
A 40-year-old man with spells of generalized weakness and paresthesias
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
Controversies in non-ST-elevation acute coronary syndromes and percutaneous coronary interventions
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
Despite all the attention paid to ST-segment-elevation myocardial infarction (MI), in terms of sheer numbers, non-ST-elevation MI and unstable angina are where the action is. Acute coronary syndromes account for 2.43 million hospital discharges per year. Of these, 0.46 million are for ST-elevation MI and 1.97 million are for non-ST-elevation MI and unstable angina.1,2
A number of recent studies have begun to answer some of the pressing questions about treating these types of acute coronary syndromes. In this article, I update the reader on these studies, along with recent findings regarding stenting and antiplatelet agents. As you will see, they are all interconnected.
TO CATHETERIZE IS BETTER THAN NOT TO CATHETERIZE
In the 1990s, a topic of debate was whether patients presenting with unstable angina or non-ST-elevation MI should routinely undergo catheterization or whether they would do just as well with a conservative approach, ie, undergoing catheterization only if they developed recurrent, spontaneous, or stress-induced ischemia. Now, the data are reasonably clear and favor an aggressive strategy.3
Mehta et al4 performed a meta-analysis of seven randomized controlled trials (N = 9,212 patients) of aggressive vs conservative angiography and revascularization for non-ST-elevation MI or unstable angina. The results favored the aggressive strategy. At 17 months of follow-up, death or MI had occurred in 7.4% of patients who received the aggressive therapy compared with 11.0% of those who received the conservative therapy, for an odds ratio of 0.82 (P = .001).
The CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes With Early Implemention of the ACC/AHA Guidelines?) Quality Improvement Initiative5 analyzed data from a registry of 17,926 patients with non-ST-elevation acute coronary syndrome who were at high risk because of positive cardiac markers or ischemic electrocardiographic changes. Overall, 2.0% of patients who received early invasive care (catheterization within the first 48 hours) died in the hospital compared with 6.2% of those who got no early invasive care, for an adjusted odds ratio of 0.63 (95% confidence interval [CI] 0.52–0.77).
The investigators also stratified the patients into those at low, medium, and high risk, using the criteria of the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) risk score. There were fewer deaths with early invasive therapy in each risk group, and the risk reduction was greatest in the high-risk group.5
Bavry et al6 performed an updated meta-analysis of randomized trials. At a mean follow-up of 24 months, the relative risk of death from any cause was 0.75 in patients who received early invasive therapy.
In another meta-analysis, O’Donoghue et al7 found that the odds ratio of death, MI, or rehospitalization with acute coronary syndromes was 0.73 (95% CI 0.55–0.98) in men who received invasive vs conservative therapy; in women it was 0.81 (95% CI 0.65–1.01). In women, the benefit was statistically significant in those who had elevations of creatine kinase MB or troponin but not in those who did not, though the benefit in men appeared to be less dependent on the presence of biomarker abnormalities.
MUST ANGIOGRAPHY BE DONE IN THE FIRST 24 HOURS?
Although a number of trials showed that a routine invasive strategy leads to better outcomes than a conservative strategy, until recently we had no information as to whether the catheterization needed to be done early (eg, within the first 24 hours) or if it could be delayed a day or two while the patient received medical therapy.
Mehta et al8 conducted a trial to find out: the Timing of Intervention in Acute Coronary Syndrome (TIMACS) trial. Patients were included if they had unstable angina or non-ST-elevation MI, presented to a hospital within 24 hours of the onset of symptoms, and had two of three high-risk features: age 60 years or older, elevated cardiac biomarkers, or electrocardiographic findings compatible with ischemia. All received standard medical therapy, and 3,031 were randomly assigned to undergo angiography either within 24 hours after randomization or 36 or more hours after randomization.
At 6 months, the primary outcome of death, new MI, or stroke had occurred in 9.6% of the patients in the early-intervention group and in 11.3% of those in the delayed-intervention group, but the difference was not statistically significant. However, the difference in the rate of a secondary end point, death, MI, or refractory ischemia, was statistically significant: 9.5% vs 12.9%, P = .003, owing mainly to less refractory ischemia with early intervention.
The patients were also stratified into two groups by baseline risk. The rate of the primary outcome was significantly lower with early intervention in high-risk patients, but not in those at intermediate or low risk. Thus, early intervention may be beneficial in patients at high risk, such as those with ongoing chest pain, but not necessarily in those at low risk.
LEAVE NO LESION BEHIND?
Coronary artery disease often affects more than one segment. Until recently, it was not known whether we should stent all stenotic segments in patients presenting with non-ST-elevation MI or unstable angina, or only the “culprit lesion.”
Shishehbor et al9 examined data from a Cleveland Clinic registry of 1,240 patients with acute coronary syndrome and multivessel coronary artery disease who underwent bare-metal stenting. The median follow-up was 2.3 years. Using a propensity model to match patients in the two groups with similar baseline characteristics, they found that the rate of repeat revascularization was less with multivessel intervention than with culprit-only stenting, as was the rate of the combined end point of death, MI, or revascularization, but not that of all-cause mortality or the composite of death or MI.
BARE-METAL VS DRUG-ELUTING STENTS: BALANCING THE RISKS AND BENEFITS
After a patient receives a stent, two bad things can happen: the artery can close up again either gradually, in a process called restenosis, or suddenly, via thrombosis.
Drug-eluting stents were invented to solve the problem of restenosis, and they work very well. Stone et al10 pooled the data from four double-blind trials of sirolimus (Rapamune) stents and five double-blind trials of paclitaxel (Taxol) stents and found that, at 4 years, the rates of target-lesion revascularization (for restenosis) were 7.8% with sirolimus stents vs 23.6% with bare-metal stents (P < .001), and 10.1% with paclitaxel stents vs 20.0% with bare-metal stents (P < .001).
Thrombosis was much less common in these studies, occurring in 1.2% of the sirolimus stent groups vs 0.6% of the bare-metal stent groups (P = .20), and in 1.3% of the paclitaxel stent groups vs 0.9% of the bare-metal stent groups (P = .30).10
However, drug-eluting stents appear to increase the risk of thrombosis later on, ie, after 1 year. Bavry et al,11 in a meta-analysis, calculated that when stent thrombosis occurred, the median time after implantation was 15.5 months with sirolimus stents vs 4 months with bare-metal stents (P = .0052), and 18 months with paclitaxel stents vs 3.5 months with bare-metal stents (P = .04). The absolute risk of very late stent thrombosis after 1 year was very low, with five events per 1,000 patients with drug-eluting stents vs no events with bare-metal stents (P = .02). Nevertheless, this finding has practical implications. How long must patients continue dual antiplatelet therapy? And what if a patient needs surgery a year later?
Restenosis is not always so gradual
Although stent thrombosis is serious and often fatal, bare-metal stent restenosis is not always benign either, despite the classic view that stent restenosis is a gradual process that results in exertional angina. Reviewing 1,186 cases of bare-metal stent restenosis in 984 patients at Cleveland Clinic, Chen et al12 reported that 9.5% of cases presented as acute MI (2.2% as ST-elevation MI and 7.3% as non-ST-elevation MI), and 26.4% as unstable angina requiring hospitalization.
A Mayo Clinic study13 corroborated these findings. The 10-year incidence of clinical bare-metal stent restenosis was 18.1%, and the incidence of MI was 2.1%. The 10-year rate of bare-metal stent thrombosis was 2%. Off-label use, primarily in saphenous vein grafts, increased the incidence; other correlates were prior MI, peripheral arterial disease, and ulcerated lesions.
Furthermore, bare-metal stent thrombosis can also occur later. We saw a case that occurred 13 years after the procedure, 3 days after the patient stopped taking aspirin because he was experiencing flu-like symptoms, ran out of aspirin, and felt too sick to go out and buy more. The presentation was with ST-elevation MI. The patient recovered after treatment with intracoronary abciximab (ReoPro), percutaneous thrombectomy, balloon angioplasty, and, eventually, bypass surgery.14
No difference in risk of death with drug-eluting vs bare-metal stents
Even though drug-eluting stents pose a slightly higher risk of thrombosis than bare-metal stents, the risk of death is no higher.15
I believe the reason is that there are competing risks, and that the higher risk of thrombosis with first-generation drug-eluting stents and the higher risk of restenosis with bare-metal stents essentially cancel each other out. For most patients, there is an absolute benefit with drug-eluting stents, which reduce the need for revascularization with no effect in terms of either increasing or decreasing the risk of MI or death. Second-generation drug-eluting stents may have advantages in reducing rates of death or MI compared with first-generation drug-eluting stents, though this remains to be proven conclusively.
The right revascularization for the right patient
Bavry and I16 developed an algorithm for deciding on revascularization, posing a series of questions:
- Does the patient need any form of revascularization?
- Is he or she at higher risk of both stent thrombosis and restenosis, as in patients with diabetes, diffuse multivessel disease with bifurcation lesions, or chronic total occlusions? If so, coronary artery bypass grafting remains an excellent option.
- Does he or she have a low risk of restenosis, as in patients without diabetes with focal lesions in large vessels? If so, one could consider a bare-metal stent, which would probably be more cost-effective than a drug-eluting stent in this situation.
- Does the patient have relative contraindications to drug-eluting stents? Examples are a history of noncompliance with medical therapy, financial issues such as lack of insurance that would make buying clopidogrel (Plavix) a problem, long-term anticoagulation, or anticipated need for surgery in the next few years.
If a drug-eluting stent is used, certain measures can help ensure that it is used optimally. It should often be placed under high pressure with a noncompliant balloon so that it achieves contact with the artery wall all around. One should consider intravascular ultrasonographic guidance to make sure the stent is well opposed if it is in a very calcified lesion. Dual antiplatelet therapy with clopidogrel and aspirin should be given for at least 1 year, and if there is no bleeding, perhaps longer, pending further data.16
LEAVE NO PLATELET ACTIVATED?
Platelets have several types of receptors that, when bound by their respective ligands, lead to platelet activation and aggregation and, ultimately, thrombus formation. Antagonists to some of these receptors are available or are being developed.17
For long-term therapy, blocking the process “upstream,” ie, preventing platelet activation, is better than blocking it “downstream,” ie, preventing aggregation. For example, clopidogrel, ticlopipine (Ticlid), and prasugrel (Effient) have active metabolites that bind to a subtype of the adenosine diphosphate receptor and prevent platelet activation, whereas the glycoprotein IIb/IIIa inhibitors such as abciximab work downstream, binding to a different receptor and preventing aggregation.18
Dual therapy for 1 year is the standard of care after acute coronary syndromes
The evidence for using dual antiplatelet therapy (ie, aspirin plus clopidogrel) in patients with acute coronary syndromes without ST-elevation is very well established.
The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial,19 published in 2001, found a 20% relative risk reduction and a 2% absolute risk reduction in the incidence of MI, stroke, or cardiovascular death in patients randomly assigned to receive clopidogrel plus aspirin for 1 year vs aspirin alone for 1 year (P < .001). In the subgroup of patients who underwent percutaneous coronary intervention, the relative risk reduction in the incidence of MI or cardiovascular death at 1 year of follow-up was 31% (P = .002).20
As a result of these findings, the cardiology society guidelines21 recommend a year of dual antiplatelet therapy after acute coronary syndromes, regardless of whether the patient is treated medically, percutaneously, or surgically.
But what happens after clopidogrel is withdrawn? Ho et al22 retrospectively analyzed data from Veterans Affairs hospitals and found a spike in the incidence of death or MI in the first 90 days after stopping clopidogrel treatment. This was true in medically treated patients as well as in those treated with percutaneous coronary interventions, in those with or without diabetes mellitus, in those who received a drug-eluting stent or a bare-metal stent, and in those treated longer than 9 months.
The investigators concluded that there might be a “clopidogrel rebound effect.” However, I believe that a true rebound effect, such as after withdrawal of heparin or warfarin, is biologically unlikely with clopidogrel, since clopidogrel irreversibly binds to its receptor for the 7- to 10-day life span of the platelet. Rather, I believe the phenomenon must be due to withdrawal of protection in patients at risk.
In stable patients, dual therapy is not as beneficial
Would dual antiplatelet therapy with clopidogrel and aspirin also benefit patients at risk of atherothrombotic events but without acute coronary syndromes?
The Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial23 included 15,603 patients with either clinically evident but stable cardiovascular disease or multiple risk factors for athero-thrombosis. They were randomly assigned to receive either clopidogrel 75 mg/day plus aspirin 75 to 162 mg/day or placebo plus aspirin. At a median of 28 months, the groups did not differ significantly in the rate of MI, stroke, or death from cardiovascular causes.
However, the subgroup of patients who had documented prior MI, ischemic stroke, or symptomatic peripheral arterial disease did appear to derive significant benefit from dual therapy.24 In this subgroup, the rate of MI, stroke, or cardiovascular death at a median follow-up of 27.6 months was 8.8% with placebo plus aspirin compared with 7.3% with clopidogrel plus aspirin, for a hazard ratio of 0.83 (95% CI 0.72–0.96, P = .01). Unstented patients with stable coronary artery disease but without prior MI derived no benefit.
Bleeding and thrombosis: The Scylla and Charybdis of antiplatelet therapy
However, with dual antiplatelet therapy, we steer between the Scylla of bleeding and the Charybdis of thrombosis.25
In the CHARISMA subgroup who had prior MI, ischemic stroke, or symptomatic peripheral arterial disease, the incidence of moderate or severe bleeding was higher with dual therapy than with aspirin alone, but the rates converged after about 1 year of treatment.24 Further, there was no difference in fatal bleeding or intracranial bleeding, although the rate of moderate bleeding (defined as the need for transfusion) was higher with dual therapy (2.0% vs 1.3%, P = .004).
I believe the data indicate that if a patient can tolerate dual antiplatelet therapy for 9 to 12 months without any bleeding issues, he or she is unlikely to have a major bleeding episode if dual therapy is continued beyond this time.
About half of bleeding events in patients on chronic antiplatelet therapy are gastrointestinal. To address this risk, in 2008 an expert committee from the American College of Cardiology, American College of Gastroenterology, and American Heart Association issued a consensus document26 in which they recommended assessing gastrointestinal risk factors in patients on antiplatelet therapy, such as history of ulcers (and testing for and treating Helicobacter pylori infection if present), history of gastrointestinal bleeding, concomitant anticoagulant therapy, and dual antiplatelet therapy. If any of these were present, the committee recommended considering a proton pump inhibitor. The committee also recommended a proton pump inhibitor for patients on antiplatelet therapy who have more than one of the following: age 60 years or more, corticosteroid use, or dyspepsia or gastroesophageal reflux symptoms.
Some ex vivo platelet studies and observational analyses have suggested that there might be an adverse interaction between clopidogrel and proton pump inhibitors due to a blunting of clopidogrel’s antiplatelet effect. A large randomized clinical trial was designed and launched to determine if a single-pill combination of the proton pump inhibitor omeprazole (Prilosec) and clopidogrel would be safer than clopidogrel alone when added to aspirin. Called COGENT-1 (Clopidogrel and the Optimization of GI Events Trial), it was halted early in 2009 when it lost its funding. However, preliminary data did not show an adverse interaction between clopidogrel and omeprazole.
What is the right dose of aspirin?
Steinhubl et al27 performed a post hoc observational analysis of data from the CHARISMA trial. Their findings suggested that higher doses of aspirin are not more effective than lower doses for chronic therapy. Furthermore, in the group receiving clopidogrel plus aspirin, the incidence of severe or life-threatening bleeding was significantly greater with aspirin doses higher than 100 mg than with doses lower than 100 mg, 2.6% vs 1.7%, P = .040.
A randomized, controlled trial called Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Optimal Antiplatelet Strategy for Interventions (CURRENT/OASIS 7)28 recently reported that higher-dose aspirin (ie, 325 mg) may be better than lower dose aspirin (ie, 81 mg) in patients with acute coronary syndromes undergoing percutaneous coronary intervention and receiving clopidogrel. During this 30-day study, there was no increase in overall bleeding with the higher dose of aspirin, though gastrointestinal bleeding was slightly increased.29 In a factorial design, the second part of this trial found that a higher-dose clopidogrel regimen reduced stent thrombosis.29
Should nonresponders get higher doses of clopidogrel?
In vitro, response to clopidogrel shows a normal bell-shaped distribution.30 In theory, therefore, patients who are hyperresponders may be at higher risk of bleeding, and those who are hyporesponders may be at risk of ischemic events.
A clinical trial is under way to examine whether hyporesponders should get higher doses. Called GRAVITAS (Gauging Responsiveness With a VerifyNow Assay Impact on Thrombosis and Safety), it will use a point-of-care platelet assay and then allocate patients to receive either standard therapy or double the dose of clopidogrel. The primary end point will be the rate of cardiovascular death, nonfatal MI, or stent thrombosis at 6 months.
Is prasugrel better than clopidogrel?
Prasugrel (Effient) is a new drug of the same class as clopidogrel, ie, a thienopyridine, with its active metabolite binding to the same platelet receptor as clopidogrel and inhibiting platelet aggregation more rapidly, more consistently, and to a greater extent than clopidogrel. Prasugrel was recently approved by the Food and Drug Administration. But is it better?31
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel–Thrombolysis in Myocardial Infarction (TRITON-TIMI 38) compared prasugrel and clopidogrel in 13,608 patients with moderate- to high-risk acute coronary syndromes who were scheduled to undergo percutaneous coronary intervention.32
Overall, prasugrel was better. At 15 months, the incidence of the primary end point (death from cardiovascular causes, nonfatal MI, or nonfatal stroke) was significantly lower with prasugrel therapy than with clopidogrel in the entire cohort (9.9% vs 12.1%, hazard ratio 0.81, 95% CI 0.73–0.90, P < .001), in the subgroup with ST-segment elevation MI, and in the subgroup with unstable angina or non-ST-elevation MI.
However, there was a price to pay. The rate of major bleeding was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% CI 1.03–1.68, P = .03). Assessing the balance between the risk and the benefit, the investigators identified three subgroups who did not derive a net clinical benefit from prasugrel: patients who had had a previous stroke or transient ischemic attack (this group actually had a net harm from prasugrel), patients 75 years of age or older, and patients weighing less than 60 kg (132 pounds).
More work is needed to determine which patients are best served by standard-dose clopidogrel, higher doses of clopidogrel, platelet-assay-guided dosing of clopidogrel, or prasugrel.24
Short-acting, potent intravenous platelet blockade with an agent such as cangrelor is theoretically appealing, but further research is necessary.33,34 Ticagrelor, a reversible adenosine diphosphate receptor antagonist, provides yet another potential option in antiplatelet therapy for acute coronary syndromes. In the recent PLATO trial (Study of Platelet Inhibition and Patient Outcomes), compared with clopidogrel, ticagrelor reduced the risk of ischemic events, including death.35,36 Here, too, there was more major bleeding (unrelated to coronary artery bypass grafting) with ticagrelor.
Thus, clinical assessment of an individual patient’s ischemic and bleeding risks will continue to be critical as therapeutic strategies evolve.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
- Wiviott SD, Morrow DA, Giugliano RP, et al. Performance of the Thrombolysis In Myocardial Infarction risk index for early acute coronary syndrome in the National Registry of Myocardial Infarction: a simple risk index predicts mortality in both ST and non-ST elevation myocardial infarction [abstract]. J Am Coll Cardiol 2003; 43( suppl 2):365A–366A.
- Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006; 113:e85–e151. Errata in Circulation 2006; 113:e696 and Circulation 2006 114:e630.
- Bhatt DL. To cath or not to cath. That is no longer the question. JAMA 2005; 293:2935–2937.
- Mehta SR, Cannon CP, Fox KA, et al. Routine vs selective invasive strategies in patients with acute coronary syndromes: a collaborative meta-analysis of randomized trials. JAMA 2005; 293:2908–2917.
- Bhatt DL, Roe MT, Peterson ED, et al; for the CRUSADE Investigators. Utilization of early invasive management strategies for high-risk patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE Quality Improvement Initiative. JAMA 2004; 292:2096–2104.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- O’Donoghue MO, Boden WE, Braunwald E, et al. Early invasive vs conservative treatment strategies in women and men with unstable angina and non-ST segment elevation myocardial infarction: a meta-analysis. JAMA 2008; 300:71–80.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Shishehbor MH, Lauer MS, Singh IM, et al. In unstable angina or non-ST-segment acute coronary syndrome, should patients with multivessel coronary artery disease undergo multivessel or culpritonly stenting? J Am Coll Cardiol 2007; 49:849–854.
- Stone GW, Moses JW, Ellis SG, et al. Safety and efficacy of sirolimus- and paclitaxel-eluting coronary stents. N Engl J Med 2007; 356:998–1008.
- Bavry AA, Kumbhani DJ, Helton TJ, Borek PP, Mood GR, Bhatt DL. Late thrombosis of drug-eluting stents: a meta-analysis of randomized clinical trials. Am J Med 2006; 119:1056–1061.
- Chen MS, John JM, Chew DP, Lee DS, Ellis SG, Bhatt DL. Bare metal stent restenosis is not a benign clinical entity. Am Heart J 2006; 151:1260–1264.
- Doyle B, Rihal CS, O’Sullivan CJ, et al. Outcomes of stent thrombosis and restenosis during extended follow-up of patients treated with bare-metal coronary stents. Circulation 2007; 116:2391–2398.
- Sarkees ML, Bavry AA, Galla JM, Bhatt DL. Bare metal stent thrombosis 13 years after implantation. Cardiovasc Revasc Med 2009; 10:58–91.
- Bavry AA, Bhatt DL. Appropriate use of drug-eluting stents: balancing the reduction in restenosis with the concern of late thrombosis. Lancet 2008; 371:2134–2143.
- Bavry AA, Bhatt DL. Drug-eluting stents: dual antiplatelet therapy for every survivor? Circulation 2007; 116:696–699.
- Meadows TA, Bhatt DL. Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 2007; 100:1261–1275.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov 2003; 2:15–28.
- Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK; Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial Investigators. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med 2001; 345:494–502. Errata in N Engl J Med 2001; 345:1506 and N Engl J Med 2001; 345:1716.
- Mehta SR, Yusuf S, Peters RJ, et al; Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet 2001; 358:527–533.
- Anderson JL, Adams CD, Antman EM, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction); american College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons; American Association of Cardiovascular and Pulmonary Rehabilitation; Society for Academic Emergency Medicine. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol 2007; 50:e1–e157.
- Ho PM, Peterson ED, Wang L, et al. Incidence of death and acute myocardial infarction associated with stopping clopidogrel after acute coronary syndrome. JAMA 2008; 299:532–539. Erratum in JAMA 2008; 299:2390.
- Bhatt DL, Fox KA, Hacke W, et al; CHARISMA Investigators. Clopidogrel and aspirin versus aspirin alone for the prevention of atherothrombotic events. N Engl J Med 2006; 354:1706–1717.
- Bhatt DL, Flather MD, Hacke W, et al; CHARISMA Investigators. Patients with prior myocardial infarction, stroke, or symptomatic peripheral arterial disease in the CHARISMA trial. J Am Coll Cardiol 2007; 49:1982–1988.
- Bhatt DL. Intensifying platelet inhibition—navigating between Scylla and Charybdis. N Engl J Med 2007; 357:2078–2081.
- Bhatt DL, Scheiman J, Abraham NS, et al; American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008; 118:1894–1909.
- Steinhubl SR, Bhatt DL, Brennan DM, et al; CHARISMA Investigators. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Ann Intern Med 2009; 150:379–386.
- Mehta SR, Bassand JP, Chrolavicius S, et al; CURRENT-OASIS 7 Steering Committee. Design and rationale of CURRENT-OASIS 7: a randomized, 2 x 2 factorial trial evaluating optimal dosing strategies for clopidogrel and aspirin in patients with ST and non-ST-elevation acute coronary syndromes managed with an early invasive strategy. Am Heart J 2008; 156:1080–1088.
- Mehta SR, Van de Werf F. A randomized comparison of a clopidogrel high loading and maintenance dose regimen versus standard dose and high versus low dose aspirin in 25,000 patients with acute coronary syndromes: results of the CURRENT OASIS 7 trial. Paper presented at the European Society of Cardiology Congress; August 30, 2009; Barcelona, Spain. Also available online at www.Escardio.org/congresses/esc-2009/congress-reports. Accessed December 12, 2009.
- Serebruany VL, Steinhubl SR, Berger PB, Malinin AT, Bhatt DL, Topol EJ. Variability in platelet responsiveness to clopidogrel among 544 individuals. J Am Coll Cardiol 2005; 45:246–251.
- Bhatt DL. Prasugrel in clinical practice [perspective]. N Engl J Med 2009; 361:940–942.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Bhatt DL, Lincoff AM, Gibson CM, et al; for the CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009 Nov 15(epub ahead of print).
- Harrington RA, Stone GW, McNulty S, et al. Platelet inhibition with cangrelor in patient sundergoing PCI. N Engl J Med 2009 Nov 17(epub ahead of print).
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- Bhatt DL. Ticagrelor in ACS—what does PLATO teach us? Nat Rev Cardiol 2009; 6:737–738.
KEY POINTS
- The data favor an aggressive strategy of routine catheterization, rather than a conservative strategy of catheterization only if a patient develops recurrent, spontaneous, or stress-induced ischemia.
- Early percutaneous intervention (within 24 hours) may be beneficial in patients at higher risk, but not necessarily in those at lower risk.
- Drug-eluting stents appear safe, assuming dual antiplatelet therapy is used. It is unclear how long this therapy needs to be continued.
- The choice of revascularization strategy—bypass surgery, bare-metal stent, or drug-eluting stent—should be individualized based on the risk of restenosis, thrombosis, and other factors.
Abdominal pain in a 20-year-old woman
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
An algorithm for managing warfarin resistance
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3

WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K

- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13

Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
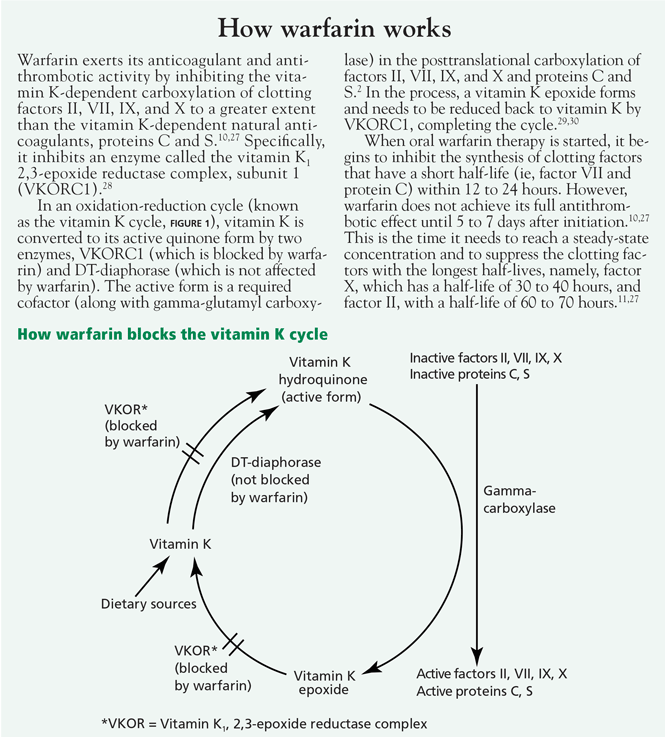
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.

TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3
WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K
- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13
Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.
TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3
WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K
- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13
Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.
TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
KEY POINTS
- The most common cause of warfarin resistance is noncompliance. Others include poor absorption, high vitamin K intake, hypersensitivity to vitamin K, and rapid drug deactivation.
- Patient education is necessary to improve compliance and to mitigate adverse effects of warfarin therapy, regardless of the dose.
- In time, it may be possible to individualize anticoagulant dosing on the basis of genetic testing for patients with warfarin resistance, although currently such tests are not routinely advocated and are usually done only in specialized laboratories.
- In true hereditary warfarin resistance, there are two approaches to treatment: increase the warfarin dosage (perhaps to as high as 100 mg/day or more), or switch to another anticoagulant.