User login
The blade, the flea, and the colon

As Elder et al point out in this issue of the Journal, the management of ischemic colitis presents an interesting clinical paradox: the internist makes the diagnosis of potentially life-threatening impending tissue necrosis, while the surgeon, consulted to act, tends to be a cheerleader for temperate observation.
Ischemic colitis may account for 1 in 1,000 hospitalizations. Many patients present with a combination of focal lower abdominal pain and some bloody diarrhea. The examiner often localizes the tender colon either by anterior palpation or by rectal examination, unlike the scenario of life-threatening small bowel ischemia, in which severe pain may be accompanied by a fairly “benign” examination.
Some cases of ischemic colitis require resection of a gangrenous colon or become chronic and lead to the development of a stricture. But far more often the ischemic episode resolves after several days of watchful waiting. The typical but not specific endoscopic findings and the thumb-printing and thickening seen on radiographic imaging resolve.
Whatever the assumed cause (a specific one is often not found), ischemic colitis gives the internist and the surgeon a chance to commiserate on the power of informed watchful waiting.
As Elder et al point out in this issue of the Journal, the management of ischemic colitis presents an interesting clinical paradox: the internist makes the diagnosis of potentially life-threatening impending tissue necrosis, while the surgeon, consulted to act, tends to be a cheerleader for temperate observation.
Ischemic colitis may account for 1 in 1,000 hospitalizations. Many patients present with a combination of focal lower abdominal pain and some bloody diarrhea. The examiner often localizes the tender colon either by anterior palpation or by rectal examination, unlike the scenario of life-threatening small bowel ischemia, in which severe pain may be accompanied by a fairly “benign” examination.
Some cases of ischemic colitis require resection of a gangrenous colon or become chronic and lead to the development of a stricture. But far more often the ischemic episode resolves after several days of watchful waiting. The typical but not specific endoscopic findings and the thumb-printing and thickening seen on radiographic imaging resolve.
Whatever the assumed cause (a specific one is often not found), ischemic colitis gives the internist and the surgeon a chance to commiserate on the power of informed watchful waiting.
As Elder et al point out in this issue of the Journal, the management of ischemic colitis presents an interesting clinical paradox: the internist makes the diagnosis of potentially life-threatening impending tissue necrosis, while the surgeon, consulted to act, tends to be a cheerleader for temperate observation.
Ischemic colitis may account for 1 in 1,000 hospitalizations. Many patients present with a combination of focal lower abdominal pain and some bloody diarrhea. The examiner often localizes the tender colon either by anterior palpation or by rectal examination, unlike the scenario of life-threatening small bowel ischemia, in which severe pain may be accompanied by a fairly “benign” examination.
Some cases of ischemic colitis require resection of a gangrenous colon or become chronic and lead to the development of a stricture. But far more often the ischemic episode resolves after several days of watchful waiting. The typical but not specific endoscopic findings and the thumb-printing and thickening seen on radiographic imaging resolve.
Whatever the assumed cause (a specific one is often not found), ischemic colitis gives the internist and the surgeon a chance to commiserate on the power of informed watchful waiting.
Clinical approach to colonic ischemia
Ischemic colitis is one of the diagnoses that should be considered when patients present with abdominal pain, diarrhea, and intestinal bleeding (others are infectious colitis, inflammatory bowel disease, diverticulitis, and colon cancer). Its incidence is difficult to determine, as many mild cases are transient and are either not reported or misdiagnosed. However, it is the most common type of intestinal ischemia1 and is responsible for an estimated 1 in 2,000 hospital admissions.2
In this review, we review the main causes of and risk factors for colonic ischemia and discuss how to diagnose and treat it.
BLOOD SUPPLY IS TENUOUS IN ‘WATERSHED’ AREAS
The superior and inferior mesenteric arteries have an extensive network of collateral blood vessels at both the base and border of the mesentery, called the arch of Riolan and the marginal artery of Drummond, respectively.
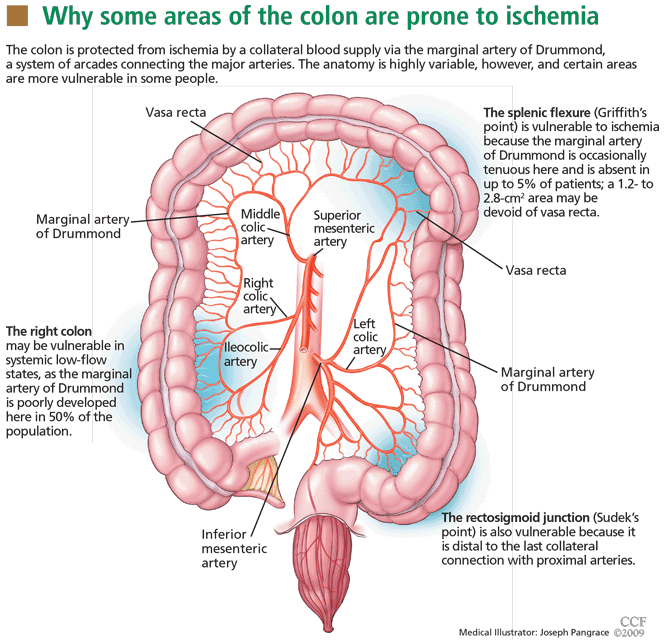
MANY POSSIBLE CAUSES AND FACTORS

Age. Ischemic colitis most often occurs in elderly people; the average age is 70 years.6 Binns and Isaacson7 suggest that age-related tortuosity of the colonic arteries increases vascular resistance and contributes to colonic ischemia in elderly patients.
Hypotension and hypovolemia are the most common mechanisms of colonic ischemia. Hypotension can be due to sepsis or impaired left ventricular function, and hypovolemia can be caused by dehydration or bleeding. These result in systemic hypoperfusion, triggering a mesenteric vasoconstrictive reflex. Once the hypoperfusion resolves and blood flow to the ulcerated portions resumes, bleeding develops from reperfusion injury.8
Cardiac thromboembolism can also contribute to colonic ischemia. Hourmand-Ollivier et al9 found a cardiac source of embolism in almost one-third of patients who had ischemic colitis, suggesting the need for routine screening with electrocardiography, Holter monitoring, and transthoracic echocardiography.
Myocardial infarction. Cappell10 found, upon colonoscopic examination, that about 14% of patients who developed hematochezia after a myocardial infarction had ischemic colitis. These patients had more complications and a worse in-hospital prognosis than did patients who had ischemic colitis due to other causes.11
Major vascular surgical procedures can disrupt the colonic blood supply, and in two case series,12,13 up to 7% of patients who underwent endoscopy after open aortoiliac reconstructive surgery had evidence of ischemic colitis. In other series,14,15 the segment most often affected was the distal left colon, and the cause was iatrogenic ligation of the inferior mesenteric artery or intraoperative hypoperfusion in patients with chronic occlusion of this artery. Endovascular repair of aortoiliac aneurysm also carries a risk of ischemic colitis, though this risk is smaller (< 2%).16
Hypercoagulable states. The role of acquired or hereditary hypercoagulable states in colonic ischemia has not been extensively investigated and remains poorly understood.
Conditions that increase clotting can cause thrombotic occlusion of small vessels that supply the colon, leading to ischemia. In small retrospective studies and case reports,17–26 28% to 74% of patients who had ischemic colitis had abnormal results on tests for protein C deficiency, protein S deficiency, antithrombin III deficiency, antiphospholipid antibodies, the factor V Leiden mutation, and the prothrombin G20210A mutation. However, in what percentage of cases the abnormality actually caused the ischemic colitis remains unknown.
Arnott et al27 reported that 9 of 24 patients with ischemic colitis had abnormal results on testing for hypercoagulable conditions. Three patients had mildly persistent elevation in anticardiolipin antibodies, but none had the factor V Leiden mutation or a deficiency of protein C, protein S, or antithrombin.
Koutroubakis et al20 reported significantly higher prevalences of antiphospholipid antibodies and heterogeneity for the factor V Leiden mutation in 35 patients with a history of ischemic colitis than in 18 patients with diverticulitis and 52 healthy controls (19.4% vs 0 and 1.9%, 22.2% vs 0 and 3.8%, respectively). Overall, 26 (72%) of 36 patients had at least one abnormal hypercoagulable test result.
Most patients with ischemic colitis are relatively old (over 60 years), and many have multiple concomitant vascular risk factors, suggesting that many factors contribute to ischemic colitis and that thrombophilia is not necessarily the direct cause. Hypercoagulable states may play a more important role in young, healthy patients who present with chronic or recurrent colonic ischemia.
Because no large clinical trials have been done and data are scarce and limited to case reports and small retrospective studies, a hypercoagulable evaluation is reserved for younger patients and those with recurrent, unexplained ischemic colitis.
Even if we detect thrombophilia, nobody yet knows what the appropriate medical treatment should be. Although some cases of ischemic colitis with associated thrombophilia have been successfully treated with anticoagulants,28,29 the benefit of diagnosing and treating a hypercoagulable state in ischemic colitis is still unproven. Therefore, oral anticoagulation should be used only in those in whom a hypercoagulable state is the most likely cause of severe or recurrent colonic ischemia.
There are no official guidelines on the duration of anticoagulation in such patients, but we treat for 6 months and consider indefinite treatment if the ischemic colitis recurs.
Medications that should always be considered as possible culprits include:
- Alosetron (Lotronex), which was temporarily withdrawn by the US Food and Drug Administration because of its association with ischemic colitis when prescribed to treat diarrhea-predominant irritable bowel syndrome.30 It was later reinstated, with some restrictions.
- Digitalis
- Diuretics
- Estrogens
- Danazol (Danocrine)
- Nonsteroidal anti-inflammatory drugs
- Tegaserod (Zelnorm)
- Paclitaxel (Abraxane)
- Carboplatin (Paraplatin)
- Sumatriptan (Imitrex)
- Simvastatin (Zocor)
- Interferon-ribavirin31
- Pseudoephedrine (eg, Sudafed).32
Endoscopic retrograde cholangiopancreatography can cause ischemic colitis if the rare life-threatening complication of mesenteric hematoma occurs.33
Chronic constipation can lead to colonic ischemia by increasing intraluminal pressure, which hinders blood flow and reduces the arteriovenous oxygen gradient in the colonic wall.34,35
Long-distance running can cause sustained bouts of ischemia, likely due to shunting of blood away from the splanchnic circulation, along with dehydration and electrolyte abnormalities. Affected runners present with lower abdominal pain and hematochezia. The colitis usually resolves without sequelae with rehydration and electrolyte correction.36
Vasospasm has been described as a cause of ischemia. During systemic hypoperfusion, vasoactive substances shunt blood from the gut to the brain through mesenteric vasoconstriction.37 This phenomenon can occur in dehydration-induced hypotension, heart failure, septic shock, or exposure to drugs such as antihypertensive medications, digoxin, or cocaine. Necrosis of the villous layer and transmural infarctions can occur with uninterrupted ischemia lasting more than 8 hours.38
Snake venom. The bite of Agkistrodon blomhoffii brevicaudus, a pit viper found in China and Korea, was recently reported to cause transient ischemic colitis due to disseminated intravascular coagulation. The condition resolved in about 10 days after treatment with polyvalent antivenom solution, transfusion of platelets and fresh frozen plasma, and empirically chosen antibiotics, ie, ampicillin-sulbactam (Unasyn) and metronidazole (Flagyl).39
CLINICAL MANIFESTATIONS
As stated above, ischemic colitis should be included in the differential diagnosis when assessing patients with abdominal pain, diarrhea, or bloody stools.
Typical presentation
The typical presentation of acute colonic ischemia includes:
- Rapid onset of mild abdominal pain
- Tenderness over the affected bowel area, usually on the left side near the splenic flexure or the rectosigmoid junction
- Mild to moderate hematochezia beginning within 1 day of the onset of abdominal pain. The bleeding is often not profuse and does not cause hemodynamic instability or require transfusion.40
Differentiate from mesenteric ischemia
It is important to differentiate between ischemic colitis and mesenteric ischemia, which is more serious and affects the small bowel.
Most patients with acute mesenteric ischemia complain of sudden onset of severe abdominal pain out of proportion to the tenderness on physical examination, they appear profoundly ill, and they do not have bloody stools until the late stages. They need urgent mesenteric angiography or another fast imaging test.4
In contrast, many patients with chronic mesenteric ischemia (or “abdominal angina”) report recurrent severe postprandial abdominal pain, leading to fear of food and weight loss.
Varies in severity
The severity of ischemic colitis varies widely, with hypoperfusion affecting as little as a single segment or as much as the entire colon. Over three-fourths of cases are the milder, nongangrenous form, which is temporary and rarely causes long-term complications such as persistent segmental colitis or strictures.41 In contrast, gangrenous colonic ischemia, which accounts for about 15% of cases, can be life-threatening.
Colonic ischemia can be categorized according to its severity and clinical presentation42:
- Reversible colonopathy (submucosal or intramural hemorrhage)
- Transient colitis (45% of cases were reversible or transient in a 1978 report by Boley et al43)
- Chronic colitis (19% of cases)
- Stricture (13%)
- Gangrene (19%)
- Fulminant universal colitis.
The resulting ischemic injury can range from superficial mucosal damage to mural or even full-thickness transmural infarction.44
Although most cases involve the left colon, about one-fourth involve the right. Right-sided colonic ischemia tends to be more severe: about 60% of patients require surgery (five times more than with colitis of other regions), and the death rate is twice as high (close to 23%).45
DIAGNOSIS DEPENDS ON SUSPICION
The diagnosis of colonic ischemia largely depends on clinical suspicion, especially since many other conditions (eg, infectious colitis, inflammatory bowel disease, diverticulitis, colon cancer) present with abdominal pain, diarrhea, and hematochezia. One study showed that a clinical presentation of lower abdominal pain or bleeding, or both, was 100% predictive of ischemic colitis when accompanied by four or more of the following risk factors: age over 60, hemodialysis, hypertension, hypoalbuminemia, diabetes mellitus, or drug-induced constipation. 46
Stool studies can identify organisms
Invasive infections with Salmonella, Shigella, and Campylobacter species and Eschericia coli O157:H7 should be identified early with stool studies if the patient presents as an outpatient or has been hospitalized less than 72 hours. Parasites such as Entamoeba histolytica and Angiostrongylus costaricensis and viruses such as cytomegalovirus should be considered in the differential diagnosis, as they can cause ischemic colitis.41,47Clostridium difficile should be excluded in those exposed to antibiotics.
Blood tests may indicate tissue injury
Although no laboratory marker is specific for ischemic colitis, elevated serum levels of lactate, lactate dehydrogenase, creatine kinase, or amylase may indicate tissue injury. The combination of abdominal pain, a white blood cell count greater than 20 × 109/L, and metabolic acidosis suggests intestinal ischemia and infarction.
Endoscopy is the test of choice
Endoscopy has become the diagnostic test of choice in establishing the diagnosis of ischemic colitis, although computed tomography (CT) can provide suggestive findings and exclude other illnesses. Colonoscopy has almost completely replaced radiography with bariumenema contrast as a diagnostic tool because it is more sensitive for detecting mucosal changes, it directly visualizes the mucosa, and it can be used to obtain biopsy specimens.48
Colonoscopy is performed without bowel preparation to prevent hypoperfusion caused by dehydrating cathartics. In addition, the scope should not be advanced beyond the affected area, and minimal air insufflation should be used to prevent perforation.
Endoscopic findings can help differentiate ischemic colitis from other, clinically similar diseases. For instance, unlike irritable bowel disease, ischemic colitis tends to affect a discrete segment with a clear delineation between affected and normal mucosa, it spares the rectum, the mucosa heals rapidly as seen on serial colonoscopic examinations, and a single linear ulcer, termed the “single-stripe” sign, runs along the longitudinal axis of the colon.49,50
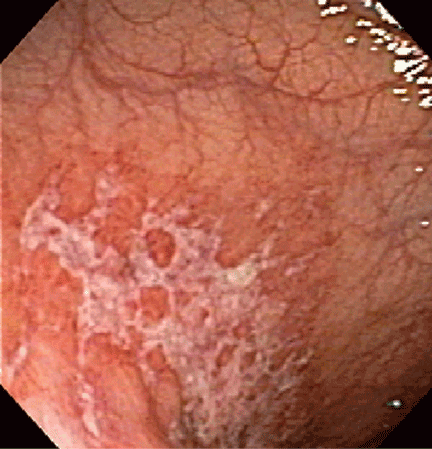
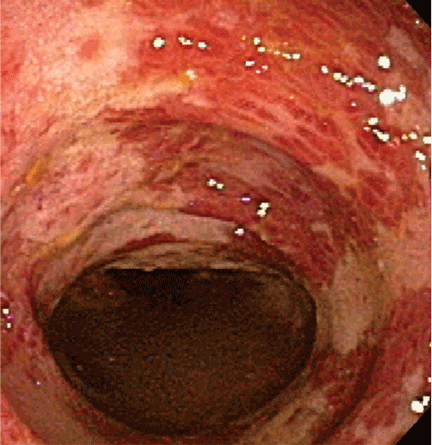
Biopsy features are not specific, as findings of hemorrhage, capillary thrombosis, granulation tissue with crypt abscesses, and pseudopolyps can also be seen in other conditions, such as Crohn disease.54,55
Imaging studies are not specific
Imaging studies are often used, but the findings lack specificity.
Plain abdominal radiography can help only in advanced ischemia, in which distention or pneumatosis can be seen.
CT with contrast can reveal thickening of the colon wall in a segmental pattern in ischemic colitis, but this finding also can be present in infectious and Crohn colitis. CT findings of colonic ischemia also include pericolic streakiness and free fluid. Pneumatosis coli often signifies infarcted bowel.56 However, CT findings can be completely normal in mild cases or if done early in the course.
Angiography in severe cases
Since colonic ischemia is most often transient, mesenteric angiography is not indicated in mild cases. Angiography is only considered in more severe cases, especially when only the right colon is involved, the diagnosis of colonic ischemia has not yet been determined, and acute mesenteric ischemia needs to be excluded. A focal lesion is often seen in mesenteric ischemia, but not often in colonic ischemia.
Looking for the underlying cause
Once the diagnosis of ischemic colitis is made, an effort should be made to identify the cause (Table 1). The initial step can be to remove or treat reversible causes such as medications or infections. As mentioned earlier, electrocardiography, Holter monitoring, and transthoracic echocardiography should be considered in patients with ischemic colitis to rule out cardiac embolic sources.9 A hypercoagulable workup can be done, but only in young patients without other clear causes or patients with recurrent events.
CONSERVATIVE TREATMENT IS ENOUGH FOR MOST
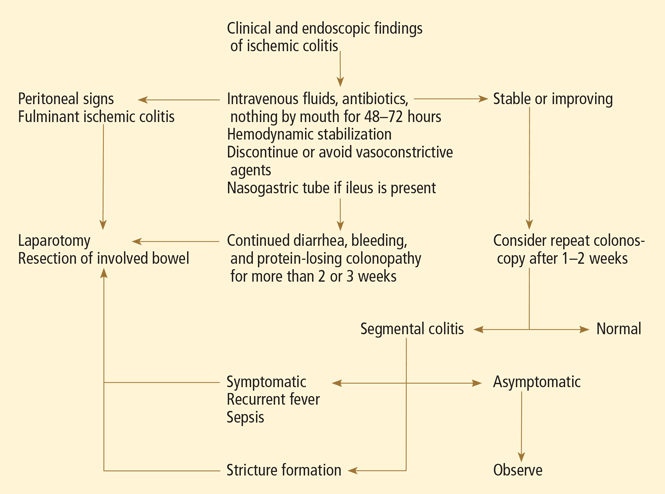
Empirically chosen broad-spectrum antibiotics that cover both aerobic and anaerobic coliform bacteria are reserved for patients with moderate to severe colitis to minimize bacterial translocation and sepsis.
Whenever symptomatic ileus is present, a nasogastric tube should be placed to alleviate vomiting and abdominal discomfort.
Antiplatelet agents have not been evaluated in treating ischemic colitis and are generally not used. As mentioned earlier, anticoagulation has been used in patients who have been proven to have hypercoagulable conditions,28,29 but its benefit is not yet proven. Currently, if the coagulation profile is abnormal, anticoagulation should be used only in cases of recurrent colonic ischemia or in young patients with severe cases in the absence of a clear cause. Anticoagulation should also be used in confirmed cases of cardiac embolization.
Surgery for some
Exploratory laparotomy with possible subtotal or segmental colectomy may be needed in acute, subacute, or chronic settings.42 Acute indications include peritoneal signs, massive bleeding, and fulminant ischemic colitis. Subacute indications are lack of resolution, with symptoms that persist for more than 2 or 3 weeks, or malnutrition or hypoalbuminemia due to protein-losing colonopathy. Colon stricture can be chronic and becomes an indication for surgery only when symptomatic, as some strictures resolve with time (months to years).
Right hemicolectomy and primary anastomosis of viable remaining colon is performed for right-sided colonic ischemia and necrosis, while left-sided colonic ischemia is managed with a proximal stoma and distal mucous fistula, or Hartmann procedure. Re-anastomosis and ostomy closure are usually done after 4 to 6 months.57 However, resection and primary anastomosis can also be an option for patients with isolated ischemia of the sigmoid colon.58 Transendoscopic dilation or stenting of short strictures can be an alternative to surgery, although experience with this is limited.59,60
THE PROGNOSIS IS USUALLY GOOD
The prognosis depends on the extent of injury and comorbidities. Transient, self-limited ischemia involving the mucosa and submucosa has a good prognosis, while fulminant ischemia with transmural infarction carries a poor one, as it can progress to necrosis and death.
Although up to 85% of cases of ischemic colitis managed conservatively improve within 1 or 2 days and resolve completely within 1 or 2 weeks, close to one-fifth of patients develop peritonitis or deteriorate clinically and require surgery.61,62 Surgical resection is required when irreversible ischemic injury and chronic colitis develop, as both can lead to bacteremia and sepsis, colonic stricture, persistent abdominal pain and bloody diarrhea, and protein-losing enteropathy.40
- Higgins PD, Davis KJ, Laine L. Systematic review: the epidemiology of ischaemic colitis. Aliment Pharmacol Ther 2004; 19:729–738.
- Feldman M, Friedman LS, Sleisenger MH, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 7th ed. Philadelphia, PA: Saunders; 2002.
- Gandhi SK, Hanson MM, Vernava AM, Kaminski DL, Longo WE. Ischemic colitis. Dis Colon Rectum 1996; 39:88–100.
- Greenwald DA, Brandt LJ, Reinus JF. Ischemic bowel disease in the elderly. Gastroenterol Clin North Am 2001; 30:445–473.
- Reeders JW, Tytgat GN, Rosenbusch G, et al. Ischaemic colitis. The Hague: Martinus Nijhoff, 1984;17.
- Brandt L, Boley S, Goldberg L, Mitsudo S, Berman A. Colitis in the elderly. A reappraisal. Am J Gastroenterol 1981; 76:239–245.
- Binns JC, Isaacson P. Age-related changes in the colonic blood supply: their relevance to ischaemic colitis. Gut 1978; 19:384–390.
- Zimmerman BJ, Granger DN. Reperfusion injury. Surg Clin North Am 1992; 72:65–83.
- Hourmand-Ollivier I, Bouin M, Saloux E, et al. Cardiac sources of embolism should be routinely screened in ischemic colitis. Am J Gastroenterol 2003; 98:1573–1577.
- Cappell MS. Safety and efficacy of colonoscopy after myocardial infarction: an analysis of 100 study patients and 100 control patients at two tertiary cardiac referral hospitals. Gastrointest Endosc 2004; 60:901–909.
- Cappell MS, Mahajan D, Kurupath V. Characterization of ischemic colitis associated with myocardial infarction: an analysis of 23 patients. Am J Med 2006; 119:527.e1–e9.
- Hagihara PF, Ernst CB, Griffen WO. Incidence of ischemic colitis following abdominal aortic reconstruction. Surg Gynecol Obstet 1979; 149:571–573.
- Brewster DC, Franklin DP, Cambria RP, et al. Intestinal ischemia complicating abdominal aortic surgery. Surgery 1991; 109:447–454.
- Piotrowski JJ, Ripepi AJ, Yuhas JP, Alexander JJ, Brandt CP. Colonic ischemia: the Achilles heel of ruptured aortic aneurysm repair. Am Surg 1996; 62:557–560.
- Ernst CB. Colonic ischemia following aortic reconstruction. In: Rutherford RB, editor. Vascular Surgery. 4th ed. Philadelphia, PA: WB Saunders; 1995:1312–1320.
- Geroghty PS, Sanchez LA, Rubin BG, et al. Overt ischemic colitis after endovascular repair of aortoiliac aneurysm. J Vasc Surg 2004; 40:413–418.
- Klestzick HN, McPhedran P, Cipolla D, Berry WA, DiCorato M, Denowitz J. The antiphospholipid syndrome and ischemic colitis. Gastroenterologist 1995; 3:249–256.
- Knot EA, ten Cate JW, Bruin T, Iburg AH, Tytgat GN. Antithrombin III metabolism in two colitis patients with acquired antithrombin III deficiency. Gastroenterology 1985; 89:421–425.
- Maloisel F. Role of coagulation disorders in mesenteric ischemia. J Chir (Paris) 1996; 133:442–447.
- Koutroubakis IE, Sfiridaki A, Theodoropoulou A, Kouroumalis EA. Role of acquired and hereditary thrombotic risk factors in colon ischemia of ambulatory patients. Gastroenterology 2001; 121:561–565.
- Midian-Singh R, Polen A, Durishin C, Crock RD, Whittier FC, Fahmy N. Ischemic colitis revisited: a prospective study identifying hypercoagulability as a risk factor. South Med J 2004; 97:120–123.
- Blanc P, Bories P, Donadio D, et al. Ischemic colitis and recurrent venous thrombosis caused by familial protein S deficiency. Gastroenterol Clin Biol 1989; 13:945.
- Verger P, Blanc C, Feydy P, Boey S. Ischemic colitis caused by protein S deficiency. Presse Med 1996; 25:1350.
- Ludwig D, Stahl M, David-Walek T, et al. Ischemic colitis, pulmonary embolism, and atrial thrombosis in a patient with inherited resistance to activated protein C. Dig Dis Sci 1998; 43:1362–1367.
- Yee NS, Guerry D, Lichtenstein GR. Ischemic colitis associated with factor V Leiden mutation. Ann Intern Med 2000; 132:595–596.
- Balian A, Veyradier A, Naveau S, et al. Prothrombin 20210G/A mutation in two patients with mesenteric ischemia. Dig Dis Sci 1999; 44:1910–1913.
- Arnott ID, Ghosh S, Ferguson A. The spectrum of ischaemic colitis. Eur J Gastroenterol Hepatol 1999; 11:295–303.
- Chin BW, Greenberg D, Wilson RB, Meredith CG. A case of ischemic colitis associated with factor V Leiden mutation: successful treatment with anticoagulation. Gastrointest Endosc 2007; 66:416–418.
- Heyn J, Buhmann S, Ladurner R, et al. Recurrent ischemic colitis in a patient with Leiden factor V mutation and systemic lupus erythematosus with antiphospholipid syndrome. Eur J Med Res 2008; 13:182–184.
- Chang L, Chey WD, Harris L, Olden K, Surawicz C, Schoenfeld P. Incidence of ischemic colitis and serious complications of constipation among patients using alosetron: systematic review of clinical trials and post-marketing surveillance data. Am J Gastroenterol 2006; 101:1069–1079.
- Punnam SR, Pothula VR, Gourineni N, Punnam A, Ranganathan V. Interferon-ribavirin-associated ischemic colitis. J Clin Gastroenterol 2008; 42:323–325.
- Dowd J, Bailey D, Moussa K, Nair S, Doyle R, Culpepper-Morgan JA. Ischemic colitis associated with pseudoephedrine: four cases. Am J Gastroenterol 1999; 94:2430–2434.
- Kingsley DD, Schermer CR, Jamal MM. Rare complications of endoscopic retrograde cholangiopancreatography: two case reports. JSLS 2001; 5:171–173.
- Boley SJ, Agrawal GP, Warren AR, et al. Pathophysiologic effects of bowel distension on intestinal blood flow. Am J Surg 1969; 117:228–234.
- Reinus JF, Brandt LJ, Boley SJ. Ischemic diseases of the bowel. Gastroenterol Clin North Am 1990; 19:319–343.
- Moses FM. Exercise-associated intestinal ischemia. Curr Sports Med Rep 2005; 4:91–95.
- Rosenblum JD, Boyle CM, Schwartz LB. The mesenteric circulation. Anatomy and physiology. Surg Clin North Am 1997; 77:289–306.
- Haglund U, Bulkley GB, Granger DN. On the pathophysiology of intestinal ischemic injury. Clinical review. Acta Chir Scand 1987; 153:321–324.
- Kim MK, Cho YS, Kim HK, Kim JS, Kim SS, Chae HS. Transient ischemic colitis after a pit viper bite (Agkistrodon blomhoffii brevicaudus). J Clin Gastroenterol 2008; 42:111–112.
- Cappell MS. Intestinal (mesenteric) vasculopathy. II. Ischemic colitis and chronic mesenteric ischemia. Gastroenterol Clin North Am 1998; 27:827–860.
- Greenwald DA, Brandt LJ. Colonic ischemia. J Clin Gastroenterol 1998; 27:122–128.
- Brandt LJ, Boley SJ. AGA technical review on intestinal ischemia. American Gastrointestinal Association. Gastroenterology 2000; 118:954–968.
- Boley SJ, Brandt LJ, Veith FJ. Ischemic disorders of the intestines. Curr Probl Surg 1978; 15:1–85.
- Schuler JG, Hudlin MM. Cecal necrosis: infrequent variant of ischemic colitis. Report of five cases. Dis Colon Rectum 2000; 43:708–712.
- Sotiriadis J, Brandt LJ, Behin DS, Southern WN. Ischemic colitis has a worse prognosis when isolated to the right side of the colon. Am J Gastroenterol 2007; 102:2247–2252.
- Park CJ, Jang MK, Shin WG, et al. Can we predict the development of ischemic colitis among patients with lower abdominal pain? Dis Colon Rectum 2007; 50:232–238.
- Su C, Brandt LJ, Sigal SH, et al. The immunohistological diagnosis of E. coli 0157:H7 colitis: possible association with colonic ischemia. Am J Gastroenterol 1998; 93:1055–1059.
- Scowcroft CW, Sanowski RA, Kozarek RA. Colonoscopy in ischemic colitis. Gastrointest Endosc 1981; 27:156–161.
- Rogers AI, David S. Intestinal blood flow and diseases of vascular impairment. In: Haubrich WS, Schaffner F, Berk JE, editors. Gastroenterology. 5th ed. Philadelphia: WB Saunders; 1995:1212–1234.
- Zuckerman GR, Prakash C, Merriman RB, Sawhney MS, DeSchryver-Kecskemeti K, Clouse RE. The colon single-stripe sign and its relationship to ischemic colitis. Am J Gastroenterol 2003; 98:2018–2022.
- Green BT, Tendler DA. Ischemic colitis: a clinical review. South Med J 2005; 98:217–222.
- Baixauli J, Kiran RP, Delaney CP. Investigation and management of ischemic colitis. Cleve Clin J Med 2003; 70:920–930.
- Habu Y, Tahashi Y, Kiyota K, et al. Reevaluation of clinical features of ischemic colitis: analysis of 68 consecutive cases diagnosed by early colonoscopy. Scand J Gastroenterol 1996; 31:881–886.
- Mitsudo S, Brandt LJ. Pathology of intestinal ischemia. Surg Clin North Am 1992; 72:43–63.
- Price AB. Ischaemic colitis. Curr Top Pathol 1990; 81:229–246.
- Balthazar EJ, Yen BC, Gordon RB. Ischemic colitis: CT evaluation of 54 cases. Radiology 1999; 211:381–388.
- Mosdell DM, Doberneck RC. Morbidity and mortality of ostomy closure. Am J Surg 1991; 162:633–636.
- Iqbal T, Zarin M, Iqbal A, et al. Results of primary closure in the management of gangrenous and viable sigmoid volvulus. Pak J Surg 2007; 23:118–121.
- Oz MC, Forde KA. Endoscopic alternatives in the management of colonic strictures. Surgery 1990; 108:513–519.
- Profili S, Bifulco V, Meloni GB, Demelas L, Niolu P, Manzoni MA. A case of ischemic stenosis of the colon-sigmoid treated with self-expandable uncoated metallic prosthesis. Radiol Med 1996; 91:665–667.
- Brandt LJ, Boley SJ. Colonic ischemia. Surg Clin North Am 1992; 72:203–229.
- Boley SJ. 1989 David H. Sun lecture. Colonic ischemia—25 years later. Am J Gastroenterol 1990; 85:931–934.
Ischemic colitis is one of the diagnoses that should be considered when patients present with abdominal pain, diarrhea, and intestinal bleeding (others are infectious colitis, inflammatory bowel disease, diverticulitis, and colon cancer). Its incidence is difficult to determine, as many mild cases are transient and are either not reported or misdiagnosed. However, it is the most common type of intestinal ischemia1 and is responsible for an estimated 1 in 2,000 hospital admissions.2
In this review, we review the main causes of and risk factors for colonic ischemia and discuss how to diagnose and treat it.
BLOOD SUPPLY IS TENUOUS IN ‘WATERSHED’ AREAS
The superior and inferior mesenteric arteries have an extensive network of collateral blood vessels at both the base and border of the mesentery, called the arch of Riolan and the marginal artery of Drummond, respectively.
MANY POSSIBLE CAUSES AND FACTORS
Age. Ischemic colitis most often occurs in elderly people; the average age is 70 years.6 Binns and Isaacson7 suggest that age-related tortuosity of the colonic arteries increases vascular resistance and contributes to colonic ischemia in elderly patients.
Hypotension and hypovolemia are the most common mechanisms of colonic ischemia. Hypotension can be due to sepsis or impaired left ventricular function, and hypovolemia can be caused by dehydration or bleeding. These result in systemic hypoperfusion, triggering a mesenteric vasoconstrictive reflex. Once the hypoperfusion resolves and blood flow to the ulcerated portions resumes, bleeding develops from reperfusion injury.8
Cardiac thromboembolism can also contribute to colonic ischemia. Hourmand-Ollivier et al9 found a cardiac source of embolism in almost one-third of patients who had ischemic colitis, suggesting the need for routine screening with electrocardiography, Holter monitoring, and transthoracic echocardiography.
Myocardial infarction. Cappell10 found, upon colonoscopic examination, that about 14% of patients who developed hematochezia after a myocardial infarction had ischemic colitis. These patients had more complications and a worse in-hospital prognosis than did patients who had ischemic colitis due to other causes.11
Major vascular surgical procedures can disrupt the colonic blood supply, and in two case series,12,13 up to 7% of patients who underwent endoscopy after open aortoiliac reconstructive surgery had evidence of ischemic colitis. In other series,14,15 the segment most often affected was the distal left colon, and the cause was iatrogenic ligation of the inferior mesenteric artery or intraoperative hypoperfusion in patients with chronic occlusion of this artery. Endovascular repair of aortoiliac aneurysm also carries a risk of ischemic colitis, though this risk is smaller (< 2%).16
Hypercoagulable states. The role of acquired or hereditary hypercoagulable states in colonic ischemia has not been extensively investigated and remains poorly understood.
Conditions that increase clotting can cause thrombotic occlusion of small vessels that supply the colon, leading to ischemia. In small retrospective studies and case reports,17–26 28% to 74% of patients who had ischemic colitis had abnormal results on tests for protein C deficiency, protein S deficiency, antithrombin III deficiency, antiphospholipid antibodies, the factor V Leiden mutation, and the prothrombin G20210A mutation. However, in what percentage of cases the abnormality actually caused the ischemic colitis remains unknown.
Arnott et al27 reported that 9 of 24 patients with ischemic colitis had abnormal results on testing for hypercoagulable conditions. Three patients had mildly persistent elevation in anticardiolipin antibodies, but none had the factor V Leiden mutation or a deficiency of protein C, protein S, or antithrombin.
Koutroubakis et al20 reported significantly higher prevalences of antiphospholipid antibodies and heterogeneity for the factor V Leiden mutation in 35 patients with a history of ischemic colitis than in 18 patients with diverticulitis and 52 healthy controls (19.4% vs 0 and 1.9%, 22.2% vs 0 and 3.8%, respectively). Overall, 26 (72%) of 36 patients had at least one abnormal hypercoagulable test result.
Most patients with ischemic colitis are relatively old (over 60 years), and many have multiple concomitant vascular risk factors, suggesting that many factors contribute to ischemic colitis and that thrombophilia is not necessarily the direct cause. Hypercoagulable states may play a more important role in young, healthy patients who present with chronic or recurrent colonic ischemia.
Because no large clinical trials have been done and data are scarce and limited to case reports and small retrospective studies, a hypercoagulable evaluation is reserved for younger patients and those with recurrent, unexplained ischemic colitis.
Even if we detect thrombophilia, nobody yet knows what the appropriate medical treatment should be. Although some cases of ischemic colitis with associated thrombophilia have been successfully treated with anticoagulants,28,29 the benefit of diagnosing and treating a hypercoagulable state in ischemic colitis is still unproven. Therefore, oral anticoagulation should be used only in those in whom a hypercoagulable state is the most likely cause of severe or recurrent colonic ischemia.
There are no official guidelines on the duration of anticoagulation in such patients, but we treat for 6 months and consider indefinite treatment if the ischemic colitis recurs.
Medications that should always be considered as possible culprits include:
- Alosetron (Lotronex), which was temporarily withdrawn by the US Food and Drug Administration because of its association with ischemic colitis when prescribed to treat diarrhea-predominant irritable bowel syndrome.30 It was later reinstated, with some restrictions.
- Digitalis
- Diuretics
- Estrogens
- Danazol (Danocrine)
- Nonsteroidal anti-inflammatory drugs
- Tegaserod (Zelnorm)
- Paclitaxel (Abraxane)
- Carboplatin (Paraplatin)
- Sumatriptan (Imitrex)
- Simvastatin (Zocor)
- Interferon-ribavirin31
- Pseudoephedrine (eg, Sudafed).32
Endoscopic retrograde cholangiopancreatography can cause ischemic colitis if the rare life-threatening complication of mesenteric hematoma occurs.33
Chronic constipation can lead to colonic ischemia by increasing intraluminal pressure, which hinders blood flow and reduces the arteriovenous oxygen gradient in the colonic wall.34,35
Long-distance running can cause sustained bouts of ischemia, likely due to shunting of blood away from the splanchnic circulation, along with dehydration and electrolyte abnormalities. Affected runners present with lower abdominal pain and hematochezia. The colitis usually resolves without sequelae with rehydration and electrolyte correction.36
Vasospasm has been described as a cause of ischemia. During systemic hypoperfusion, vasoactive substances shunt blood from the gut to the brain through mesenteric vasoconstriction.37 This phenomenon can occur in dehydration-induced hypotension, heart failure, septic shock, or exposure to drugs such as antihypertensive medications, digoxin, or cocaine. Necrosis of the villous layer and transmural infarctions can occur with uninterrupted ischemia lasting more than 8 hours.38
Snake venom. The bite of Agkistrodon blomhoffii brevicaudus, a pit viper found in China and Korea, was recently reported to cause transient ischemic colitis due to disseminated intravascular coagulation. The condition resolved in about 10 days after treatment with polyvalent antivenom solution, transfusion of platelets and fresh frozen plasma, and empirically chosen antibiotics, ie, ampicillin-sulbactam (Unasyn) and metronidazole (Flagyl).39
CLINICAL MANIFESTATIONS
As stated above, ischemic colitis should be included in the differential diagnosis when assessing patients with abdominal pain, diarrhea, or bloody stools.
Typical presentation
The typical presentation of acute colonic ischemia includes:
- Rapid onset of mild abdominal pain
- Tenderness over the affected bowel area, usually on the left side near the splenic flexure or the rectosigmoid junction
- Mild to moderate hematochezia beginning within 1 day of the onset of abdominal pain. The bleeding is often not profuse and does not cause hemodynamic instability or require transfusion.40
Differentiate from mesenteric ischemia
It is important to differentiate between ischemic colitis and mesenteric ischemia, which is more serious and affects the small bowel.
Most patients with acute mesenteric ischemia complain of sudden onset of severe abdominal pain out of proportion to the tenderness on physical examination, they appear profoundly ill, and they do not have bloody stools until the late stages. They need urgent mesenteric angiography or another fast imaging test.4
In contrast, many patients with chronic mesenteric ischemia (or “abdominal angina”) report recurrent severe postprandial abdominal pain, leading to fear of food and weight loss.
Varies in severity
The severity of ischemic colitis varies widely, with hypoperfusion affecting as little as a single segment or as much as the entire colon. Over three-fourths of cases are the milder, nongangrenous form, which is temporary and rarely causes long-term complications such as persistent segmental colitis or strictures.41 In contrast, gangrenous colonic ischemia, which accounts for about 15% of cases, can be life-threatening.
Colonic ischemia can be categorized according to its severity and clinical presentation42:
- Reversible colonopathy (submucosal or intramural hemorrhage)
- Transient colitis (45% of cases were reversible or transient in a 1978 report by Boley et al43)
- Chronic colitis (19% of cases)
- Stricture (13%)
- Gangrene (19%)
- Fulminant universal colitis.
The resulting ischemic injury can range from superficial mucosal damage to mural or even full-thickness transmural infarction.44
Although most cases involve the left colon, about one-fourth involve the right. Right-sided colonic ischemia tends to be more severe: about 60% of patients require surgery (five times more than with colitis of other regions), and the death rate is twice as high (close to 23%).45
DIAGNOSIS DEPENDS ON SUSPICION
The diagnosis of colonic ischemia largely depends on clinical suspicion, especially since many other conditions (eg, infectious colitis, inflammatory bowel disease, diverticulitis, colon cancer) present with abdominal pain, diarrhea, and hematochezia. One study showed that a clinical presentation of lower abdominal pain or bleeding, or both, was 100% predictive of ischemic colitis when accompanied by four or more of the following risk factors: age over 60, hemodialysis, hypertension, hypoalbuminemia, diabetes mellitus, or drug-induced constipation. 46
Stool studies can identify organisms
Invasive infections with Salmonella, Shigella, and Campylobacter species and Eschericia coli O157:H7 should be identified early with stool studies if the patient presents as an outpatient or has been hospitalized less than 72 hours. Parasites such as Entamoeba histolytica and Angiostrongylus costaricensis and viruses such as cytomegalovirus should be considered in the differential diagnosis, as they can cause ischemic colitis.41,47Clostridium difficile should be excluded in those exposed to antibiotics.
Blood tests may indicate tissue injury
Although no laboratory marker is specific for ischemic colitis, elevated serum levels of lactate, lactate dehydrogenase, creatine kinase, or amylase may indicate tissue injury. The combination of abdominal pain, a white blood cell count greater than 20 × 109/L, and metabolic acidosis suggests intestinal ischemia and infarction.
Endoscopy is the test of choice
Endoscopy has become the diagnostic test of choice in establishing the diagnosis of ischemic colitis, although computed tomography (CT) can provide suggestive findings and exclude other illnesses. Colonoscopy has almost completely replaced radiography with bariumenema contrast as a diagnostic tool because it is more sensitive for detecting mucosal changes, it directly visualizes the mucosa, and it can be used to obtain biopsy specimens.48
Colonoscopy is performed without bowel preparation to prevent hypoperfusion caused by dehydrating cathartics. In addition, the scope should not be advanced beyond the affected area, and minimal air insufflation should be used to prevent perforation.
Endoscopic findings can help differentiate ischemic colitis from other, clinically similar diseases. For instance, unlike irritable bowel disease, ischemic colitis tends to affect a discrete segment with a clear delineation between affected and normal mucosa, it spares the rectum, the mucosa heals rapidly as seen on serial colonoscopic examinations, and a single linear ulcer, termed the “single-stripe” sign, runs along the longitudinal axis of the colon.49,50
Biopsy features are not specific, as findings of hemorrhage, capillary thrombosis, granulation tissue with crypt abscesses, and pseudopolyps can also be seen in other conditions, such as Crohn disease.54,55
Imaging studies are not specific
Imaging studies are often used, but the findings lack specificity.
Plain abdominal radiography can help only in advanced ischemia, in which distention or pneumatosis can be seen.
CT with contrast can reveal thickening of the colon wall in a segmental pattern in ischemic colitis, but this finding also can be present in infectious and Crohn colitis. CT findings of colonic ischemia also include pericolic streakiness and free fluid. Pneumatosis coli often signifies infarcted bowel.56 However, CT findings can be completely normal in mild cases or if done early in the course.
Angiography in severe cases
Since colonic ischemia is most often transient, mesenteric angiography is not indicated in mild cases. Angiography is only considered in more severe cases, especially when only the right colon is involved, the diagnosis of colonic ischemia has not yet been determined, and acute mesenteric ischemia needs to be excluded. A focal lesion is often seen in mesenteric ischemia, but not often in colonic ischemia.
Looking for the underlying cause
Once the diagnosis of ischemic colitis is made, an effort should be made to identify the cause (Table 1). The initial step can be to remove or treat reversible causes such as medications or infections. As mentioned earlier, electrocardiography, Holter monitoring, and transthoracic echocardiography should be considered in patients with ischemic colitis to rule out cardiac embolic sources.9 A hypercoagulable workup can be done, but only in young patients without other clear causes or patients with recurrent events.
CONSERVATIVE TREATMENT IS ENOUGH FOR MOST
Empirically chosen broad-spectrum antibiotics that cover both aerobic and anaerobic coliform bacteria are reserved for patients with moderate to severe colitis to minimize bacterial translocation and sepsis.
Whenever symptomatic ileus is present, a nasogastric tube should be placed to alleviate vomiting and abdominal discomfort.
Antiplatelet agents have not been evaluated in treating ischemic colitis and are generally not used. As mentioned earlier, anticoagulation has been used in patients who have been proven to have hypercoagulable conditions,28,29 but its benefit is not yet proven. Currently, if the coagulation profile is abnormal, anticoagulation should be used only in cases of recurrent colonic ischemia or in young patients with severe cases in the absence of a clear cause. Anticoagulation should also be used in confirmed cases of cardiac embolization.
Surgery for some
Exploratory laparotomy with possible subtotal or segmental colectomy may be needed in acute, subacute, or chronic settings.42 Acute indications include peritoneal signs, massive bleeding, and fulminant ischemic colitis. Subacute indications are lack of resolution, with symptoms that persist for more than 2 or 3 weeks, or malnutrition or hypoalbuminemia due to protein-losing colonopathy. Colon stricture can be chronic and becomes an indication for surgery only when symptomatic, as some strictures resolve with time (months to years).
Right hemicolectomy and primary anastomosis of viable remaining colon is performed for right-sided colonic ischemia and necrosis, while left-sided colonic ischemia is managed with a proximal stoma and distal mucous fistula, or Hartmann procedure. Re-anastomosis and ostomy closure are usually done after 4 to 6 months.57 However, resection and primary anastomosis can also be an option for patients with isolated ischemia of the sigmoid colon.58 Transendoscopic dilation or stenting of short strictures can be an alternative to surgery, although experience with this is limited.59,60
THE PROGNOSIS IS USUALLY GOOD
The prognosis depends on the extent of injury and comorbidities. Transient, self-limited ischemia involving the mucosa and submucosa has a good prognosis, while fulminant ischemia with transmural infarction carries a poor one, as it can progress to necrosis and death.
Although up to 85% of cases of ischemic colitis managed conservatively improve within 1 or 2 days and resolve completely within 1 or 2 weeks, close to one-fifth of patients develop peritonitis or deteriorate clinically and require surgery.61,62 Surgical resection is required when irreversible ischemic injury and chronic colitis develop, as both can lead to bacteremia and sepsis, colonic stricture, persistent abdominal pain and bloody diarrhea, and protein-losing enteropathy.40
Ischemic colitis is one of the diagnoses that should be considered when patients present with abdominal pain, diarrhea, and intestinal bleeding (others are infectious colitis, inflammatory bowel disease, diverticulitis, and colon cancer). Its incidence is difficult to determine, as many mild cases are transient and are either not reported or misdiagnosed. However, it is the most common type of intestinal ischemia1 and is responsible for an estimated 1 in 2,000 hospital admissions.2
In this review, we review the main causes of and risk factors for colonic ischemia and discuss how to diagnose and treat it.
BLOOD SUPPLY IS TENUOUS IN ‘WATERSHED’ AREAS
The superior and inferior mesenteric arteries have an extensive network of collateral blood vessels at both the base and border of the mesentery, called the arch of Riolan and the marginal artery of Drummond, respectively.
MANY POSSIBLE CAUSES AND FACTORS
Age. Ischemic colitis most often occurs in elderly people; the average age is 70 years.6 Binns and Isaacson7 suggest that age-related tortuosity of the colonic arteries increases vascular resistance and contributes to colonic ischemia in elderly patients.
Hypotension and hypovolemia are the most common mechanisms of colonic ischemia. Hypotension can be due to sepsis or impaired left ventricular function, and hypovolemia can be caused by dehydration or bleeding. These result in systemic hypoperfusion, triggering a mesenteric vasoconstrictive reflex. Once the hypoperfusion resolves and blood flow to the ulcerated portions resumes, bleeding develops from reperfusion injury.8
Cardiac thromboembolism can also contribute to colonic ischemia. Hourmand-Ollivier et al9 found a cardiac source of embolism in almost one-third of patients who had ischemic colitis, suggesting the need for routine screening with electrocardiography, Holter monitoring, and transthoracic echocardiography.
Myocardial infarction. Cappell10 found, upon colonoscopic examination, that about 14% of patients who developed hematochezia after a myocardial infarction had ischemic colitis. These patients had more complications and a worse in-hospital prognosis than did patients who had ischemic colitis due to other causes.11
Major vascular surgical procedures can disrupt the colonic blood supply, and in two case series,12,13 up to 7% of patients who underwent endoscopy after open aortoiliac reconstructive surgery had evidence of ischemic colitis. In other series,14,15 the segment most often affected was the distal left colon, and the cause was iatrogenic ligation of the inferior mesenteric artery or intraoperative hypoperfusion in patients with chronic occlusion of this artery. Endovascular repair of aortoiliac aneurysm also carries a risk of ischemic colitis, though this risk is smaller (< 2%).16
Hypercoagulable states. The role of acquired or hereditary hypercoagulable states in colonic ischemia has not been extensively investigated and remains poorly understood.
Conditions that increase clotting can cause thrombotic occlusion of small vessels that supply the colon, leading to ischemia. In small retrospective studies and case reports,17–26 28% to 74% of patients who had ischemic colitis had abnormal results on tests for protein C deficiency, protein S deficiency, antithrombin III deficiency, antiphospholipid antibodies, the factor V Leiden mutation, and the prothrombin G20210A mutation. However, in what percentage of cases the abnormality actually caused the ischemic colitis remains unknown.
Arnott et al27 reported that 9 of 24 patients with ischemic colitis had abnormal results on testing for hypercoagulable conditions. Three patients had mildly persistent elevation in anticardiolipin antibodies, but none had the factor V Leiden mutation or a deficiency of protein C, protein S, or antithrombin.
Koutroubakis et al20 reported significantly higher prevalences of antiphospholipid antibodies and heterogeneity for the factor V Leiden mutation in 35 patients with a history of ischemic colitis than in 18 patients with diverticulitis and 52 healthy controls (19.4% vs 0 and 1.9%, 22.2% vs 0 and 3.8%, respectively). Overall, 26 (72%) of 36 patients had at least one abnormal hypercoagulable test result.
Most patients with ischemic colitis are relatively old (over 60 years), and many have multiple concomitant vascular risk factors, suggesting that many factors contribute to ischemic colitis and that thrombophilia is not necessarily the direct cause. Hypercoagulable states may play a more important role in young, healthy patients who present with chronic or recurrent colonic ischemia.
Because no large clinical trials have been done and data are scarce and limited to case reports and small retrospective studies, a hypercoagulable evaluation is reserved for younger patients and those with recurrent, unexplained ischemic colitis.
Even if we detect thrombophilia, nobody yet knows what the appropriate medical treatment should be. Although some cases of ischemic colitis with associated thrombophilia have been successfully treated with anticoagulants,28,29 the benefit of diagnosing and treating a hypercoagulable state in ischemic colitis is still unproven. Therefore, oral anticoagulation should be used only in those in whom a hypercoagulable state is the most likely cause of severe or recurrent colonic ischemia.
There are no official guidelines on the duration of anticoagulation in such patients, but we treat for 6 months and consider indefinite treatment if the ischemic colitis recurs.
Medications that should always be considered as possible culprits include:
- Alosetron (Lotronex), which was temporarily withdrawn by the US Food and Drug Administration because of its association with ischemic colitis when prescribed to treat diarrhea-predominant irritable bowel syndrome.30 It was later reinstated, with some restrictions.
- Digitalis
- Diuretics
- Estrogens
- Danazol (Danocrine)
- Nonsteroidal anti-inflammatory drugs
- Tegaserod (Zelnorm)
- Paclitaxel (Abraxane)
- Carboplatin (Paraplatin)
- Sumatriptan (Imitrex)
- Simvastatin (Zocor)
- Interferon-ribavirin31
- Pseudoephedrine (eg, Sudafed).32
Endoscopic retrograde cholangiopancreatography can cause ischemic colitis if the rare life-threatening complication of mesenteric hematoma occurs.33
Chronic constipation can lead to colonic ischemia by increasing intraluminal pressure, which hinders blood flow and reduces the arteriovenous oxygen gradient in the colonic wall.34,35
Long-distance running can cause sustained bouts of ischemia, likely due to shunting of blood away from the splanchnic circulation, along with dehydration and electrolyte abnormalities. Affected runners present with lower abdominal pain and hematochezia. The colitis usually resolves without sequelae with rehydration and electrolyte correction.36
Vasospasm has been described as a cause of ischemia. During systemic hypoperfusion, vasoactive substances shunt blood from the gut to the brain through mesenteric vasoconstriction.37 This phenomenon can occur in dehydration-induced hypotension, heart failure, septic shock, or exposure to drugs such as antihypertensive medications, digoxin, or cocaine. Necrosis of the villous layer and transmural infarctions can occur with uninterrupted ischemia lasting more than 8 hours.38
Snake venom. The bite of Agkistrodon blomhoffii brevicaudus, a pit viper found in China and Korea, was recently reported to cause transient ischemic colitis due to disseminated intravascular coagulation. The condition resolved in about 10 days after treatment with polyvalent antivenom solution, transfusion of platelets and fresh frozen plasma, and empirically chosen antibiotics, ie, ampicillin-sulbactam (Unasyn) and metronidazole (Flagyl).39
CLINICAL MANIFESTATIONS
As stated above, ischemic colitis should be included in the differential diagnosis when assessing patients with abdominal pain, diarrhea, or bloody stools.
Typical presentation
The typical presentation of acute colonic ischemia includes:
- Rapid onset of mild abdominal pain
- Tenderness over the affected bowel area, usually on the left side near the splenic flexure or the rectosigmoid junction
- Mild to moderate hematochezia beginning within 1 day of the onset of abdominal pain. The bleeding is often not profuse and does not cause hemodynamic instability or require transfusion.40
Differentiate from mesenteric ischemia
It is important to differentiate between ischemic colitis and mesenteric ischemia, which is more serious and affects the small bowel.
Most patients with acute mesenteric ischemia complain of sudden onset of severe abdominal pain out of proportion to the tenderness on physical examination, they appear profoundly ill, and they do not have bloody stools until the late stages. They need urgent mesenteric angiography or another fast imaging test.4
In contrast, many patients with chronic mesenteric ischemia (or “abdominal angina”) report recurrent severe postprandial abdominal pain, leading to fear of food and weight loss.
Varies in severity
The severity of ischemic colitis varies widely, with hypoperfusion affecting as little as a single segment or as much as the entire colon. Over three-fourths of cases are the milder, nongangrenous form, which is temporary and rarely causes long-term complications such as persistent segmental colitis or strictures.41 In contrast, gangrenous colonic ischemia, which accounts for about 15% of cases, can be life-threatening.
Colonic ischemia can be categorized according to its severity and clinical presentation42:
- Reversible colonopathy (submucosal or intramural hemorrhage)
- Transient colitis (45% of cases were reversible or transient in a 1978 report by Boley et al43)
- Chronic colitis (19% of cases)
- Stricture (13%)
- Gangrene (19%)
- Fulminant universal colitis.
The resulting ischemic injury can range from superficial mucosal damage to mural or even full-thickness transmural infarction.44
Although most cases involve the left colon, about one-fourth involve the right. Right-sided colonic ischemia tends to be more severe: about 60% of patients require surgery (five times more than with colitis of other regions), and the death rate is twice as high (close to 23%).45
DIAGNOSIS DEPENDS ON SUSPICION
The diagnosis of colonic ischemia largely depends on clinical suspicion, especially since many other conditions (eg, infectious colitis, inflammatory bowel disease, diverticulitis, colon cancer) present with abdominal pain, diarrhea, and hematochezia. One study showed that a clinical presentation of lower abdominal pain or bleeding, or both, was 100% predictive of ischemic colitis when accompanied by four or more of the following risk factors: age over 60, hemodialysis, hypertension, hypoalbuminemia, diabetes mellitus, or drug-induced constipation. 46
Stool studies can identify organisms
Invasive infections with Salmonella, Shigella, and Campylobacter species and Eschericia coli O157:H7 should be identified early with stool studies if the patient presents as an outpatient or has been hospitalized less than 72 hours. Parasites such as Entamoeba histolytica and Angiostrongylus costaricensis and viruses such as cytomegalovirus should be considered in the differential diagnosis, as they can cause ischemic colitis.41,47Clostridium difficile should be excluded in those exposed to antibiotics.
Blood tests may indicate tissue injury
Although no laboratory marker is specific for ischemic colitis, elevated serum levels of lactate, lactate dehydrogenase, creatine kinase, or amylase may indicate tissue injury. The combination of abdominal pain, a white blood cell count greater than 20 × 109/L, and metabolic acidosis suggests intestinal ischemia and infarction.
Endoscopy is the test of choice
Endoscopy has become the diagnostic test of choice in establishing the diagnosis of ischemic colitis, although computed tomography (CT) can provide suggestive findings and exclude other illnesses. Colonoscopy has almost completely replaced radiography with bariumenema contrast as a diagnostic tool because it is more sensitive for detecting mucosal changes, it directly visualizes the mucosa, and it can be used to obtain biopsy specimens.48
Colonoscopy is performed without bowel preparation to prevent hypoperfusion caused by dehydrating cathartics. In addition, the scope should not be advanced beyond the affected area, and minimal air insufflation should be used to prevent perforation.
Endoscopic findings can help differentiate ischemic colitis from other, clinically similar diseases. For instance, unlike irritable bowel disease, ischemic colitis tends to affect a discrete segment with a clear delineation between affected and normal mucosa, it spares the rectum, the mucosa heals rapidly as seen on serial colonoscopic examinations, and a single linear ulcer, termed the “single-stripe” sign, runs along the longitudinal axis of the colon.49,50
Biopsy features are not specific, as findings of hemorrhage, capillary thrombosis, granulation tissue with crypt abscesses, and pseudopolyps can also be seen in other conditions, such as Crohn disease.54,55
Imaging studies are not specific
Imaging studies are often used, but the findings lack specificity.
Plain abdominal radiography can help only in advanced ischemia, in which distention or pneumatosis can be seen.
CT with contrast can reveal thickening of the colon wall in a segmental pattern in ischemic colitis, but this finding also can be present in infectious and Crohn colitis. CT findings of colonic ischemia also include pericolic streakiness and free fluid. Pneumatosis coli often signifies infarcted bowel.56 However, CT findings can be completely normal in mild cases or if done early in the course.
Angiography in severe cases
Since colonic ischemia is most often transient, mesenteric angiography is not indicated in mild cases. Angiography is only considered in more severe cases, especially when only the right colon is involved, the diagnosis of colonic ischemia has not yet been determined, and acute mesenteric ischemia needs to be excluded. A focal lesion is often seen in mesenteric ischemia, but not often in colonic ischemia.
Looking for the underlying cause
Once the diagnosis of ischemic colitis is made, an effort should be made to identify the cause (Table 1). The initial step can be to remove or treat reversible causes such as medications or infections. As mentioned earlier, electrocardiography, Holter monitoring, and transthoracic echocardiography should be considered in patients with ischemic colitis to rule out cardiac embolic sources.9 A hypercoagulable workup can be done, but only in young patients without other clear causes or patients with recurrent events.
CONSERVATIVE TREATMENT IS ENOUGH FOR MOST
Empirically chosen broad-spectrum antibiotics that cover both aerobic and anaerobic coliform bacteria are reserved for patients with moderate to severe colitis to minimize bacterial translocation and sepsis.
Whenever symptomatic ileus is present, a nasogastric tube should be placed to alleviate vomiting and abdominal discomfort.
Antiplatelet agents have not been evaluated in treating ischemic colitis and are generally not used. As mentioned earlier, anticoagulation has been used in patients who have been proven to have hypercoagulable conditions,28,29 but its benefit is not yet proven. Currently, if the coagulation profile is abnormal, anticoagulation should be used only in cases of recurrent colonic ischemia or in young patients with severe cases in the absence of a clear cause. Anticoagulation should also be used in confirmed cases of cardiac embolization.
Surgery for some
Exploratory laparotomy with possible subtotal or segmental colectomy may be needed in acute, subacute, or chronic settings.42 Acute indications include peritoneal signs, massive bleeding, and fulminant ischemic colitis. Subacute indications are lack of resolution, with symptoms that persist for more than 2 or 3 weeks, or malnutrition or hypoalbuminemia due to protein-losing colonopathy. Colon stricture can be chronic and becomes an indication for surgery only when symptomatic, as some strictures resolve with time (months to years).
Right hemicolectomy and primary anastomosis of viable remaining colon is performed for right-sided colonic ischemia and necrosis, while left-sided colonic ischemia is managed with a proximal stoma and distal mucous fistula, or Hartmann procedure. Re-anastomosis and ostomy closure are usually done after 4 to 6 months.57 However, resection and primary anastomosis can also be an option for patients with isolated ischemia of the sigmoid colon.58 Transendoscopic dilation or stenting of short strictures can be an alternative to surgery, although experience with this is limited.59,60
THE PROGNOSIS IS USUALLY GOOD
The prognosis depends on the extent of injury and comorbidities. Transient, self-limited ischemia involving the mucosa and submucosa has a good prognosis, while fulminant ischemia with transmural infarction carries a poor one, as it can progress to necrosis and death.
Although up to 85% of cases of ischemic colitis managed conservatively improve within 1 or 2 days and resolve completely within 1 or 2 weeks, close to one-fifth of patients develop peritonitis or deteriorate clinically and require surgery.61,62 Surgical resection is required when irreversible ischemic injury and chronic colitis develop, as both can lead to bacteremia and sepsis, colonic stricture, persistent abdominal pain and bloody diarrhea, and protein-losing enteropathy.40
- Higgins PD, Davis KJ, Laine L. Systematic review: the epidemiology of ischaemic colitis. Aliment Pharmacol Ther 2004; 19:729–738.
- Feldman M, Friedman LS, Sleisenger MH, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 7th ed. Philadelphia, PA: Saunders; 2002.
- Gandhi SK, Hanson MM, Vernava AM, Kaminski DL, Longo WE. Ischemic colitis. Dis Colon Rectum 1996; 39:88–100.
- Greenwald DA, Brandt LJ, Reinus JF. Ischemic bowel disease in the elderly. Gastroenterol Clin North Am 2001; 30:445–473.
- Reeders JW, Tytgat GN, Rosenbusch G, et al. Ischaemic colitis. The Hague: Martinus Nijhoff, 1984;17.
- Brandt L, Boley S, Goldberg L, Mitsudo S, Berman A. Colitis in the elderly. A reappraisal. Am J Gastroenterol 1981; 76:239–245.
- Binns JC, Isaacson P. Age-related changes in the colonic blood supply: their relevance to ischaemic colitis. Gut 1978; 19:384–390.
- Zimmerman BJ, Granger DN. Reperfusion injury. Surg Clin North Am 1992; 72:65–83.
- Hourmand-Ollivier I, Bouin M, Saloux E, et al. Cardiac sources of embolism should be routinely screened in ischemic colitis. Am J Gastroenterol 2003; 98:1573–1577.
- Cappell MS. Safety and efficacy of colonoscopy after myocardial infarction: an analysis of 100 study patients and 100 control patients at two tertiary cardiac referral hospitals. Gastrointest Endosc 2004; 60:901–909.
- Cappell MS, Mahajan D, Kurupath V. Characterization of ischemic colitis associated with myocardial infarction: an analysis of 23 patients. Am J Med 2006; 119:527.e1–e9.
- Hagihara PF, Ernst CB, Griffen WO. Incidence of ischemic colitis following abdominal aortic reconstruction. Surg Gynecol Obstet 1979; 149:571–573.
- Brewster DC, Franklin DP, Cambria RP, et al. Intestinal ischemia complicating abdominal aortic surgery. Surgery 1991; 109:447–454.
- Piotrowski JJ, Ripepi AJ, Yuhas JP, Alexander JJ, Brandt CP. Colonic ischemia: the Achilles heel of ruptured aortic aneurysm repair. Am Surg 1996; 62:557–560.
- Ernst CB. Colonic ischemia following aortic reconstruction. In: Rutherford RB, editor. Vascular Surgery. 4th ed. Philadelphia, PA: WB Saunders; 1995:1312–1320.
- Geroghty PS, Sanchez LA, Rubin BG, et al. Overt ischemic colitis after endovascular repair of aortoiliac aneurysm. J Vasc Surg 2004; 40:413–418.
- Klestzick HN, McPhedran P, Cipolla D, Berry WA, DiCorato M, Denowitz J. The antiphospholipid syndrome and ischemic colitis. Gastroenterologist 1995; 3:249–256.
- Knot EA, ten Cate JW, Bruin T, Iburg AH, Tytgat GN. Antithrombin III metabolism in two colitis patients with acquired antithrombin III deficiency. Gastroenterology 1985; 89:421–425.
- Maloisel F. Role of coagulation disorders in mesenteric ischemia. J Chir (Paris) 1996; 133:442–447.
- Koutroubakis IE, Sfiridaki A, Theodoropoulou A, Kouroumalis EA. Role of acquired and hereditary thrombotic risk factors in colon ischemia of ambulatory patients. Gastroenterology 2001; 121:561–565.
- Midian-Singh R, Polen A, Durishin C, Crock RD, Whittier FC, Fahmy N. Ischemic colitis revisited: a prospective study identifying hypercoagulability as a risk factor. South Med J 2004; 97:120–123.
- Blanc P, Bories P, Donadio D, et al. Ischemic colitis and recurrent venous thrombosis caused by familial protein S deficiency. Gastroenterol Clin Biol 1989; 13:945.
- Verger P, Blanc C, Feydy P, Boey S. Ischemic colitis caused by protein S deficiency. Presse Med 1996; 25:1350.
- Ludwig D, Stahl M, David-Walek T, et al. Ischemic colitis, pulmonary embolism, and atrial thrombosis in a patient with inherited resistance to activated protein C. Dig Dis Sci 1998; 43:1362–1367.
- Yee NS, Guerry D, Lichtenstein GR. Ischemic colitis associated with factor V Leiden mutation. Ann Intern Med 2000; 132:595–596.
- Balian A, Veyradier A, Naveau S, et al. Prothrombin 20210G/A mutation in two patients with mesenteric ischemia. Dig Dis Sci 1999; 44:1910–1913.
- Arnott ID, Ghosh S, Ferguson A. The spectrum of ischaemic colitis. Eur J Gastroenterol Hepatol 1999; 11:295–303.
- Chin BW, Greenberg D, Wilson RB, Meredith CG. A case of ischemic colitis associated with factor V Leiden mutation: successful treatment with anticoagulation. Gastrointest Endosc 2007; 66:416–418.
- Heyn J, Buhmann S, Ladurner R, et al. Recurrent ischemic colitis in a patient with Leiden factor V mutation and systemic lupus erythematosus with antiphospholipid syndrome. Eur J Med Res 2008; 13:182–184.
- Chang L, Chey WD, Harris L, Olden K, Surawicz C, Schoenfeld P. Incidence of ischemic colitis and serious complications of constipation among patients using alosetron: systematic review of clinical trials and post-marketing surveillance data. Am J Gastroenterol 2006; 101:1069–1079.
- Punnam SR, Pothula VR, Gourineni N, Punnam A, Ranganathan V. Interferon-ribavirin-associated ischemic colitis. J Clin Gastroenterol 2008; 42:323–325.
- Dowd J, Bailey D, Moussa K, Nair S, Doyle R, Culpepper-Morgan JA. Ischemic colitis associated with pseudoephedrine: four cases. Am J Gastroenterol 1999; 94:2430–2434.
- Kingsley DD, Schermer CR, Jamal MM. Rare complications of endoscopic retrograde cholangiopancreatography: two case reports. JSLS 2001; 5:171–173.
- Boley SJ, Agrawal GP, Warren AR, et al. Pathophysiologic effects of bowel distension on intestinal blood flow. Am J Surg 1969; 117:228–234.
- Reinus JF, Brandt LJ, Boley SJ. Ischemic diseases of the bowel. Gastroenterol Clin North Am 1990; 19:319–343.
- Moses FM. Exercise-associated intestinal ischemia. Curr Sports Med Rep 2005; 4:91–95.
- Rosenblum JD, Boyle CM, Schwartz LB. The mesenteric circulation. Anatomy and physiology. Surg Clin North Am 1997; 77:289–306.
- Haglund U, Bulkley GB, Granger DN. On the pathophysiology of intestinal ischemic injury. Clinical review. Acta Chir Scand 1987; 153:321–324.
- Kim MK, Cho YS, Kim HK, Kim JS, Kim SS, Chae HS. Transient ischemic colitis after a pit viper bite (Agkistrodon blomhoffii brevicaudus). J Clin Gastroenterol 2008; 42:111–112.
- Cappell MS. Intestinal (mesenteric) vasculopathy. II. Ischemic colitis and chronic mesenteric ischemia. Gastroenterol Clin North Am 1998; 27:827–860.
- Greenwald DA, Brandt LJ. Colonic ischemia. J Clin Gastroenterol 1998; 27:122–128.
- Brandt LJ, Boley SJ. AGA technical review on intestinal ischemia. American Gastrointestinal Association. Gastroenterology 2000; 118:954–968.
- Boley SJ, Brandt LJ, Veith FJ. Ischemic disorders of the intestines. Curr Probl Surg 1978; 15:1–85.
- Schuler JG, Hudlin MM. Cecal necrosis: infrequent variant of ischemic colitis. Report of five cases. Dis Colon Rectum 2000; 43:708–712.
- Sotiriadis J, Brandt LJ, Behin DS, Southern WN. Ischemic colitis has a worse prognosis when isolated to the right side of the colon. Am J Gastroenterol 2007; 102:2247–2252.
- Park CJ, Jang MK, Shin WG, et al. Can we predict the development of ischemic colitis among patients with lower abdominal pain? Dis Colon Rectum 2007; 50:232–238.
- Su C, Brandt LJ, Sigal SH, et al. The immunohistological diagnosis of E. coli 0157:H7 colitis: possible association with colonic ischemia. Am J Gastroenterol 1998; 93:1055–1059.
- Scowcroft CW, Sanowski RA, Kozarek RA. Colonoscopy in ischemic colitis. Gastrointest Endosc 1981; 27:156–161.
- Rogers AI, David S. Intestinal blood flow and diseases of vascular impairment. In: Haubrich WS, Schaffner F, Berk JE, editors. Gastroenterology. 5th ed. Philadelphia: WB Saunders; 1995:1212–1234.
- Zuckerman GR, Prakash C, Merriman RB, Sawhney MS, DeSchryver-Kecskemeti K, Clouse RE. The colon single-stripe sign and its relationship to ischemic colitis. Am J Gastroenterol 2003; 98:2018–2022.
- Green BT, Tendler DA. Ischemic colitis: a clinical review. South Med J 2005; 98:217–222.
- Baixauli J, Kiran RP, Delaney CP. Investigation and management of ischemic colitis. Cleve Clin J Med 2003; 70:920–930.
- Habu Y, Tahashi Y, Kiyota K, et al. Reevaluation of clinical features of ischemic colitis: analysis of 68 consecutive cases diagnosed by early colonoscopy. Scand J Gastroenterol 1996; 31:881–886.
- Mitsudo S, Brandt LJ. Pathology of intestinal ischemia. Surg Clin North Am 1992; 72:43–63.
- Price AB. Ischaemic colitis. Curr Top Pathol 1990; 81:229–246.
- Balthazar EJ, Yen BC, Gordon RB. Ischemic colitis: CT evaluation of 54 cases. Radiology 1999; 211:381–388.
- Mosdell DM, Doberneck RC. Morbidity and mortality of ostomy closure. Am J Surg 1991; 162:633–636.
- Iqbal T, Zarin M, Iqbal A, et al. Results of primary closure in the management of gangrenous and viable sigmoid volvulus. Pak J Surg 2007; 23:118–121.
- Oz MC, Forde KA. Endoscopic alternatives in the management of colonic strictures. Surgery 1990; 108:513–519.
- Profili S, Bifulco V, Meloni GB, Demelas L, Niolu P, Manzoni MA. A case of ischemic stenosis of the colon-sigmoid treated with self-expandable uncoated metallic prosthesis. Radiol Med 1996; 91:665–667.
- Brandt LJ, Boley SJ. Colonic ischemia. Surg Clin North Am 1992; 72:203–229.
- Boley SJ. 1989 David H. Sun lecture. Colonic ischemia—25 years later. Am J Gastroenterol 1990; 85:931–934.
- Higgins PD, Davis KJ, Laine L. Systematic review: the epidemiology of ischaemic colitis. Aliment Pharmacol Ther 2004; 19:729–738.
- Feldman M, Friedman LS, Sleisenger MH, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 7th ed. Philadelphia, PA: Saunders; 2002.
- Gandhi SK, Hanson MM, Vernava AM, Kaminski DL, Longo WE. Ischemic colitis. Dis Colon Rectum 1996; 39:88–100.
- Greenwald DA, Brandt LJ, Reinus JF. Ischemic bowel disease in the elderly. Gastroenterol Clin North Am 2001; 30:445–473.
- Reeders JW, Tytgat GN, Rosenbusch G, et al. Ischaemic colitis. The Hague: Martinus Nijhoff, 1984;17.
- Brandt L, Boley S, Goldberg L, Mitsudo S, Berman A. Colitis in the elderly. A reappraisal. Am J Gastroenterol 1981; 76:239–245.
- Binns JC, Isaacson P. Age-related changes in the colonic blood supply: their relevance to ischaemic colitis. Gut 1978; 19:384–390.
- Zimmerman BJ, Granger DN. Reperfusion injury. Surg Clin North Am 1992; 72:65–83.
- Hourmand-Ollivier I, Bouin M, Saloux E, et al. Cardiac sources of embolism should be routinely screened in ischemic colitis. Am J Gastroenterol 2003; 98:1573–1577.
- Cappell MS. Safety and efficacy of colonoscopy after myocardial infarction: an analysis of 100 study patients and 100 control patients at two tertiary cardiac referral hospitals. Gastrointest Endosc 2004; 60:901–909.
- Cappell MS, Mahajan D, Kurupath V. Characterization of ischemic colitis associated with myocardial infarction: an analysis of 23 patients. Am J Med 2006; 119:527.e1–e9.
- Hagihara PF, Ernst CB, Griffen WO. Incidence of ischemic colitis following abdominal aortic reconstruction. Surg Gynecol Obstet 1979; 149:571–573.
- Brewster DC, Franklin DP, Cambria RP, et al. Intestinal ischemia complicating abdominal aortic surgery. Surgery 1991; 109:447–454.
- Piotrowski JJ, Ripepi AJ, Yuhas JP, Alexander JJ, Brandt CP. Colonic ischemia: the Achilles heel of ruptured aortic aneurysm repair. Am Surg 1996; 62:557–560.
- Ernst CB. Colonic ischemia following aortic reconstruction. In: Rutherford RB, editor. Vascular Surgery. 4th ed. Philadelphia, PA: WB Saunders; 1995:1312–1320.
- Geroghty PS, Sanchez LA, Rubin BG, et al. Overt ischemic colitis after endovascular repair of aortoiliac aneurysm. J Vasc Surg 2004; 40:413–418.
- Klestzick HN, McPhedran P, Cipolla D, Berry WA, DiCorato M, Denowitz J. The antiphospholipid syndrome and ischemic colitis. Gastroenterologist 1995; 3:249–256.
- Knot EA, ten Cate JW, Bruin T, Iburg AH, Tytgat GN. Antithrombin III metabolism in two colitis patients with acquired antithrombin III deficiency. Gastroenterology 1985; 89:421–425.
- Maloisel F. Role of coagulation disorders in mesenteric ischemia. J Chir (Paris) 1996; 133:442–447.
- Koutroubakis IE, Sfiridaki A, Theodoropoulou A, Kouroumalis EA. Role of acquired and hereditary thrombotic risk factors in colon ischemia of ambulatory patients. Gastroenterology 2001; 121:561–565.
- Midian-Singh R, Polen A, Durishin C, Crock RD, Whittier FC, Fahmy N. Ischemic colitis revisited: a prospective study identifying hypercoagulability as a risk factor. South Med J 2004; 97:120–123.
- Blanc P, Bories P, Donadio D, et al. Ischemic colitis and recurrent venous thrombosis caused by familial protein S deficiency. Gastroenterol Clin Biol 1989; 13:945.
- Verger P, Blanc C, Feydy P, Boey S. Ischemic colitis caused by protein S deficiency. Presse Med 1996; 25:1350.
- Ludwig D, Stahl M, David-Walek T, et al. Ischemic colitis, pulmonary embolism, and atrial thrombosis in a patient with inherited resistance to activated protein C. Dig Dis Sci 1998; 43:1362–1367.
- Yee NS, Guerry D, Lichtenstein GR. Ischemic colitis associated with factor V Leiden mutation. Ann Intern Med 2000; 132:595–596.
- Balian A, Veyradier A, Naveau S, et al. Prothrombin 20210G/A mutation in two patients with mesenteric ischemia. Dig Dis Sci 1999; 44:1910–1913.
- Arnott ID, Ghosh S, Ferguson A. The spectrum of ischaemic colitis. Eur J Gastroenterol Hepatol 1999; 11:295–303.
- Chin BW, Greenberg D, Wilson RB, Meredith CG. A case of ischemic colitis associated with factor V Leiden mutation: successful treatment with anticoagulation. Gastrointest Endosc 2007; 66:416–418.
- Heyn J, Buhmann S, Ladurner R, et al. Recurrent ischemic colitis in a patient with Leiden factor V mutation and systemic lupus erythematosus with antiphospholipid syndrome. Eur J Med Res 2008; 13:182–184.
- Chang L, Chey WD, Harris L, Olden K, Surawicz C, Schoenfeld P. Incidence of ischemic colitis and serious complications of constipation among patients using alosetron: systematic review of clinical trials and post-marketing surveillance data. Am J Gastroenterol 2006; 101:1069–1079.
- Punnam SR, Pothula VR, Gourineni N, Punnam A, Ranganathan V. Interferon-ribavirin-associated ischemic colitis. J Clin Gastroenterol 2008; 42:323–325.
- Dowd J, Bailey D, Moussa K, Nair S, Doyle R, Culpepper-Morgan JA. Ischemic colitis associated with pseudoephedrine: four cases. Am J Gastroenterol 1999; 94:2430–2434.
- Kingsley DD, Schermer CR, Jamal MM. Rare complications of endoscopic retrograde cholangiopancreatography: two case reports. JSLS 2001; 5:171–173.
- Boley SJ, Agrawal GP, Warren AR, et al. Pathophysiologic effects of bowel distension on intestinal blood flow. Am J Surg 1969; 117:228–234.
- Reinus JF, Brandt LJ, Boley SJ. Ischemic diseases of the bowel. Gastroenterol Clin North Am 1990; 19:319–343.
- Moses FM. Exercise-associated intestinal ischemia. Curr Sports Med Rep 2005; 4:91–95.
- Rosenblum JD, Boyle CM, Schwartz LB. The mesenteric circulation. Anatomy and physiology. Surg Clin North Am 1997; 77:289–306.
- Haglund U, Bulkley GB, Granger DN. On the pathophysiology of intestinal ischemic injury. Clinical review. Acta Chir Scand 1987; 153:321–324.
- Kim MK, Cho YS, Kim HK, Kim JS, Kim SS, Chae HS. Transient ischemic colitis after a pit viper bite (Agkistrodon blomhoffii brevicaudus). J Clin Gastroenterol 2008; 42:111–112.
- Cappell MS. Intestinal (mesenteric) vasculopathy. II. Ischemic colitis and chronic mesenteric ischemia. Gastroenterol Clin North Am 1998; 27:827–860.
- Greenwald DA, Brandt LJ. Colonic ischemia. J Clin Gastroenterol 1998; 27:122–128.
- Brandt LJ, Boley SJ. AGA technical review on intestinal ischemia. American Gastrointestinal Association. Gastroenterology 2000; 118:954–968.
- Boley SJ, Brandt LJ, Veith FJ. Ischemic disorders of the intestines. Curr Probl Surg 1978; 15:1–85.
- Schuler JG, Hudlin MM. Cecal necrosis: infrequent variant of ischemic colitis. Report of five cases. Dis Colon Rectum 2000; 43:708–712.
- Sotiriadis J, Brandt LJ, Behin DS, Southern WN. Ischemic colitis has a worse prognosis when isolated to the right side of the colon. Am J Gastroenterol 2007; 102:2247–2252.
- Park CJ, Jang MK, Shin WG, et al. Can we predict the development of ischemic colitis among patients with lower abdominal pain? Dis Colon Rectum 2007; 50:232–238.
- Su C, Brandt LJ, Sigal SH, et al. The immunohistological diagnosis of E. coli 0157:H7 colitis: possible association with colonic ischemia. Am J Gastroenterol 1998; 93:1055–1059.
- Scowcroft CW, Sanowski RA, Kozarek RA. Colonoscopy in ischemic colitis. Gastrointest Endosc 1981; 27:156–161.
- Rogers AI, David S. Intestinal blood flow and diseases of vascular impairment. In: Haubrich WS, Schaffner F, Berk JE, editors. Gastroenterology. 5th ed. Philadelphia: WB Saunders; 1995:1212–1234.
- Zuckerman GR, Prakash C, Merriman RB, Sawhney MS, DeSchryver-Kecskemeti K, Clouse RE. The colon single-stripe sign and its relationship to ischemic colitis. Am J Gastroenterol 2003; 98:2018–2022.
- Green BT, Tendler DA. Ischemic colitis: a clinical review. South Med J 2005; 98:217–222.
- Baixauli J, Kiran RP, Delaney CP. Investigation and management of ischemic colitis. Cleve Clin J Med 2003; 70:920–930.
- Habu Y, Tahashi Y, Kiyota K, et al. Reevaluation of clinical features of ischemic colitis: analysis of 68 consecutive cases diagnosed by early colonoscopy. Scand J Gastroenterol 1996; 31:881–886.
- Mitsudo S, Brandt LJ. Pathology of intestinal ischemia. Surg Clin North Am 1992; 72:43–63.
- Price AB. Ischaemic colitis. Curr Top Pathol 1990; 81:229–246.
- Balthazar EJ, Yen BC, Gordon RB. Ischemic colitis: CT evaluation of 54 cases. Radiology 1999; 211:381–388.
- Mosdell DM, Doberneck RC. Morbidity and mortality of ostomy closure. Am J Surg 1991; 162:633–636.
- Iqbal T, Zarin M, Iqbal A, et al. Results of primary closure in the management of gangrenous and viable sigmoid volvulus. Pak J Surg 2007; 23:118–121.
- Oz MC, Forde KA. Endoscopic alternatives in the management of colonic strictures. Surgery 1990; 108:513–519.
- Profili S, Bifulco V, Meloni GB, Demelas L, Niolu P, Manzoni MA. A case of ischemic stenosis of the colon-sigmoid treated with self-expandable uncoated metallic prosthesis. Radiol Med 1996; 91:665–667.
- Brandt LJ, Boley SJ. Colonic ischemia. Surg Clin North Am 1992; 72:203–229.
- Boley SJ. 1989 David H. Sun lecture. Colonic ischemia—25 years later. Am J Gastroenterol 1990; 85:931–934.
KEY POINTS
- The incidence of colonic ischemia is difficult to ascertain, as most cases are transient and either not reported or misdiagnosed.
- Most cases are in the elderly.
- The clinical presentation is not specific, as other conditions also present with abdominal pain and hematochezia.
- The most common mechanisms are hypotension and hypovolemia caused by dehydration or bleeding that results in systemic hypoperfusion.
- Endoscopy has become the diagnostic procedure of choice.
- Although most patients can be treated conservatively with intravenous fluids, bowel rest, and antibiotics, some develop peritonitis or clinically deteriorate and require surgery.
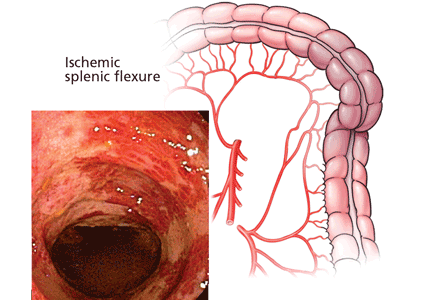
A 72-year-old man with a purpuric rash
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
A 72-year-old man whose medical history includes diabetes mellitus, hypertension, coronary artery disease, aortic valve replacement, atrial fibrillation, and chronic obstructive pulmonary disease was in his usual state of health until 2 weeks ago, when he developed a purpuric rash on his legs. His physician started him on prednisone 40 mg daily for the rash; however, 1 week later he presented to a hospital emergency room when his family found him confused and diaphoretic.
In the emergency room, he was found to be hypoglycemic, with a serum glucose level of 40 mg/dL, which was promptly treated. His mental status improved partially. In the hospital, the rash worsened and progressed upwards to his trunk and upper extremities. He was transferred to our institution for further workup and management.
A review of systems reveals occasional epistaxis in the summer, recent fatigue, cough, and shortness of breath on exertion. His medications at the time of transfer include warfarin (Coumadin), amlodipine (Norvasc), insulin, ipratropium and albuterol (Combivent) inhalers, and prednisone 40 mg daily. He has not undergone surgery recently.
PHYSICAL EXAMINATION
He is alert and oriented to person but not to time and place.
Vital signs. Oral temperature 101.1°F (38.4°C), pulse rate 108, blood pressure 108/79 mm/Hg, respiratory rate 22, oxygen saturation 93% by pulse oximetry on room air, weight 94 kg (207 lb).
Head, eyes, ears, nose, and throat. No pallor or icterus, pupils are equally reactive, nasal mucosa not inflamed or ulcerated, mucous membranes moist, no sinus tenderness.
Neck. No jugular venous distention and no cervical lymphadenopathy.
Cardiovascular. Tachycardia, irregularly irregular rhythm, prosthetic valve sounds, no murmurs, rubs, or gallops.
Respiratory. Bibasal crackles (right side more than the left). No wheezing.
Abdomen. Soft, nontender, nondistended, no palpable organomegaly, bowel sounds normal.
Extremities. No edema, good peripheral pulses.
Neurologic. No focal deficits noted.
Lymphatic. No enlarged lymph nodes.
Musculoskeletal. Traumatic right second distal interphalangeal amputation. Otherwise, no joint abnormality or restriction of movement.
Initial laboratory values:
- White blood cell count 15.78 × 109/L (normal 4.5–11.0)
- Absolute neutrophil count 13.3 × 109/L (4.0–11.0)
- Hemoglobin 13.3 g/dL (13.5–17.5)
- Platelet count 133 × 109/L (150–400)
- International normalized ratio (INR) 1.8
- Sodium 136 mmol/L (135–146)
- Potassium 4.6 mmol/L (3.5–5.0)
- Blood urea nitrogen 31 mg/dL (10–25)
- Creatinine 1.6 mg/dL (0.70–1.40)
- Glucose 62 mg/dL (65–100)
- Bicarbonate 23 mmol/L (23–32)
- Albumin 2.5 g/dL (3.5–5.0)
- Total protein 4.6 g/dL (6.0–8.4)
- Bilirubin 1.2 mg/dL (0.0–1.5)
- Aspartate aminotransferase 41 U/L (7–40)
- Alanine aminotransferase 74 U/L (5–50)
- Alkaline phosphatase 55 U/L (40–150)
- C-reactive protein 9.9 mg/dL (0.0–1.0).
Other studies
Electrocardiography shows atrial fibrillation and left ventricular hypertrophy, but no acute changes.
Computed tomography (CT) of the head shows no evidence of hemorrhage or infarction.
Blood cultures are sent at the time of hospital admission.
WHICH TEST IS NEXT?
1. Which is the most appropriate next step for this patient?
- Urinalysis
- CT of the chest
- Echocardiography
- Skin biopsy
The rash in Figure 1 is palpable purpura, which strongly suggests small-vessel cutaneous vasculitis, a condition that can occur in a broad range of settings. An underlying cause is identified in over 70% of cases. Cutaneous vasculitis may herald a primary small-vessel systemic vasculitis such as Wegener granulomatosis, microscopic polyangiitis, or Henoch-Schönlein purpura. It can also be secondary to a spectrum of underlying triggers or diseases that include medications, infections, malignancies such as lymphoproliferative disorders, cryoglobulinemia secondary to hepatitis C viral infection, and connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.
Infective endocarditis is associated with a secondary form of vasculitis and is a strong possibility in this patient, who has a prosthetic aortic valve, fever, and a high white blood cell count.
Thrombocytopenia should also prompt an assessment for any drugs the patient is taking that affect platelet function. However, thrombocytopenia typically results in nonpalpable purpura.
Idiopathic isolated cutaneous vasculitis, in which no underlying cause for the cutaneous vasculitis can be identified, is the diagnosis in less than 30% of cases.
A vasculitic disease process can involve multiple sites, which may be asymptomatic on presentation. Identifying these sites is important, not only to establish the diagnosis, but also to detect potentially life-threatening complications early.
Thus, in this patient, urinalysis should be done promptly to check for active sediment consisting of red cell casts, which would suggest renal involvement (glomerulonephritis). Also, a rising blood pressure and creatinine would point to renal involvement and warrant more aggressive initial therapy.
Chest radiography should be done to rule out pulmonary infiltrates, septic emboli, nodules, or cavities that could represent vasculitic or infectious involvement of the lungs. CT of the chest may be needed to further characterize abnormalities on chest radiography.
Echocardiography should certainly be pursued as part of the workup for endocarditis, but urinalysis is of the utmost importance in this patient at this point.
More diagnostic information is needed before considering skin biopsy.
Clues from the urinalysis and chest radiography
Our patient’s sedimentation rate is 24 mm/hour. The urinalysis is strongly positive for blood and a moderate amount of protein but negative for leukocyte esterase and nitrite. The urine sediment contains numerous red blood cell casts and 6 to 10 white blood cells per high-power field.
Pending return of blood cultures, ceftriaxone (Rocephin) and azithromycin (Zithromax) are started to treat possible community-acquired pneumonia. Vancomycin (Vancocin) is empirically added, given the possibility of prosthetic valve endocarditis. Gram stain on blood cultures shows gram-positive cocci.
Ground-glass attenuation implies focal or diffuse opacification of the lung that does not obscure the vascular structures, is not associated with air bronchograms, and appears as a “veil” across the lung parenchyma.1 It can be seen in acute diseases such as infection (including pneumonia from atypical bacteria, viruses, acid-fast bacilli, and Pneumocystis jiroveci), pulmonary hemorrhage of any cause, acute viral, eosinophilic, and interstitial pneumonias, and hypersensitivity pneumonitis. It can also be seen in chronic disease states such as interstitial lung disease, bronchoalveolar carcinoma, alveolar proteinosis, and sarcoidosis.
WHICH TEST WOULD NOT HELP?
2. Which of the following tests is least likely to help in the diagnostic evaluation at this point?
- Transthoracic echocardiography
- Transesophageal echocardiography
- Bronchoscopy
- Cystoscopy
In this case, the least likely to help is cystoscopy.
This patient’s vasculitic-appearing rash, ground-glass pulmonary infiltrates, and impaired renal function with red cell casts suggest a pulmonary-renal syndrome, and with this constellation of features, a systemic vasculitis is very likely. Therefore, the focus of the evaluation should be on any evidence to support a diagnosis of vasculitis, as well as other possible causes.
In a patient with diabetes, an artificial heart valve, and fever, the possibility of infection, especially endocarditis, remains high. Transthoracic echocardiography is warranted, and if it is negative for vegetations, transesophageal echocardiography would be a reasonable next option.
Bronchoscopy is warranted to determine if the infiltrates represent pulmonary hemorrhage, which can be seen in certain types of vasculitic and systemic disorders.
The finding of red cell casts in the urine indicates glomerulonephritis, and therefore the kidneys are the likely source of the urinary red blood cells, making cystoscopy of no utility in this current, acute setting.
Case continued: His condition worsens
Transthoracic echocardiography reveals a well-seated mechanical prosthetic aortic valve, trivial aortic regurgitation, a peak gradient of 23 mm Hg and a mean gradient of 12 mm Hg (normal values for his prosthetic valve), and no valvular vegetations. Transesophageal echocardiography confirms the absence of vegetations.
His oxygen requirement increases, and analysis of arterial blood gases reveals a pH of 7.37, Pco2 49 mm Hg (normal range 35–45), Po2 102 mm Hg (normal 80–100), and bicarbonate 28 mmol/L while breathing 100% supplemental oxygen by a nonrebreather face mask. He is taken to the medical intensive care unit for intubation and mechanical ventilation. Bronchoscopy performed while he is intubated confirms diffuse alveolar hemorrhage. Pulse intravenous methylprednisolone (Solu-Medrol) therapy is started.
DIFFUSE ALVEOLAR HEMORRHAGE
3. Which of the following is not true about diffuse alveolar hemorrhage?
- Its onset is usually abrupt or of short duration
- It is always associated with hemoptysis
- Radiography most commonly shows new patchy or diffuse alveolar opacities
- Pulmonary function testing shows increased diffusing capacity of the lung for carbon monoxide (Dlco)
Hemoptysis is absent at presentation in as many as 33% of patients.
The onset of diffuse alveolar hemorrhage is usually abrupt or of short duration, with initial symptoms of cough, fever, and dyspnea. Some patients, such as ours, can present with severe acute respiratory distress syndrome requiring mechanical ventilation. Radiography most often shows new patchy alveolar opacities, and CT may reveal a ground-glass appearance. On pulmonary function testing, the Dlco is high, owing to the hemoglobin within the alveoli.
ACUTE GLOMERULONEPHRITIS PLUS PULMONARY HEMORRHAGE EQUALS…?
4. Which disease could have manifestations consistent with acute glomerulonephritis and pulmonary hemorrhage?
- Antiglomerular basement membrane disease
- Wegener granulomatosis
- Microscopic polyangiitis
- Systemic lupus erythematosus
All of these are possible.
The combined presentation of acute glomerulonephritis and pulmonary hemorrhage (also called pulmonary-renal syndrome) is usually seen in antiglomerular basement membrane disease (Goodpasture syndrome) and small-vessel systemic vasculitides such as Wegener granulomatosis and microscopic polyangiitis.2,3 It can also be seen in patients with systemic lupus erythematosus.
Antiglomerular basement membrane disease
In antiglomerular basement membrane disease, circulating antibodies are directed towards an antigen intrinsic to the glomerular basement membrane, typically leading to acute glomerulonephritis associated with crescent formation. It may present as acute renal failure in which urinalysis shows proteinuria with sediment characterized by red cell casts. Pulmonary involvement, usually alveolar hemorrhage, is present in approximately 60% to 70% of cases.
The diagnosis requires demonstration of antiglomerular basement membrane antibodies in either the serum or the kidney. Renal biopsy is usually recommended because the accuracy of serum assays is variable.
A key histologic feature of the renal lesion in antiglomerular basement membrane disease is crescentic glomerulonephritis in which immunofluorescence microscopy demonstrates the virtually pathognomonic finding of linear deposition of immunoglobulin G along the glomerular capillaries.
The treatment of choice for antiglomerular basement membrane disease is plasmapheresis and immunosuppression with a combination of glucocorticoids and cyclophosphamide (Cytoxan). If the disease is high on the differential diagnosis, empiric plasmapheresis should be started while waiting for diagnostic studies, because the prognosis of untreated glomerulonephritis is poor.
Wegener granulomatosis
Wegener granulomatosis is a systemic vasculitis of the medium and small arteries, arterioles, and venules that classically involves the upper and lower respiratory tracts and the kidneys. Patients may present with persistent rhinorrhea and epistaxis, cough with chest radiographs showing nodules, fixed infiltrates, or cavities, and abnormal urinary sediment with microscopic hematuria with or without red cell casts.
From 75% to 90% of patients with active Wegener granulomatosis are positive for antineutrophil cytoplasmic antibody (ANCA). In 60% to 80% of cases, ANCA is directed against proteinase 3 (PR3), which produces a cytoplasmic standing pattern by immunofluorescence (cANCA), while 5% to 20% have ANCA directed against myeloper-oxidase, which produces a perinuclear staining pattern (pANCA). A small number of patients with Wegener granulomatosis are ANCA-negative.
The diagnosis is usually confirmed by tissue biopsy at the site of active disease, which shows necrotizing vasculitis with granulomatous inflammation. The renal lesion is typically that of a focal, segmental, necrotizing glomerulonephritis that has few to no immune complexes (pauci-immune glomerulonephritis).
The treatment of severe disease involves a combination of cyclophosphamide and glucocorticoids initially to achieve remission followed by maintenance therapy with methotrexate or azathioprine (Imuran).
Microscopic polyangiitis
Microscopic polyangiitis is a systemic vasculitis of the capillaries, venules, and arterioles, with little or no immune complex deposition. Nearly all patients have renal involvement, and 10% to 30% have lung involvement. In those with lung involvement, diffuse alveolar hemorrhage is the most common manifestation.
On histopathologic study, microscopic polyangiitis differs from Wegener granulomatosis in that it does not have granuloma formation. However, the renal lesion is that of a pauci-immune glomerulonephritis and is identical to that seen in Wegener granulomatosis. From 70% to 85% of patients with microscopic polyangiitis are ANCA-positive, and most of these have pANCA.
The management of active severe microscopic polyangiitis is identical to that of Wegener granulomatosis.
Systemic lupus erythematosus
Systemic lupus erythematosus is an autoimmune disease characterized by tissue-binding autoantibody and immune-complex-mediated organ damage. It can involve multiple organ systems, and the diagnosis is based on characteristic clinical features and autoantibodies. The sensitivity of antinuclear antibody for lupus is close to 100%, which makes it a good screening tool. Antibodies to dsDNA and Smith antigen have high specificity for lupus.
About 75% of patients have renal involvement at some point in their disease course. The different types of renal disease in systemic lupus are usually differentiated with a renal biopsy, with immune-complex-mediated glomerular diseases being the most common.
The most common pulmonary manifestation is pleuritis with or without pleural effusion. Life-threatening pulmonary manifestations include pulmonary hemorrhage and interstitial inflammation leading to fibrosis.
Lupus has great clinical variability and the treatment approach is based on the organ manifestations, disease activity, and severity.
CASE CONTINUED: ARRIVING AT THE DIAGNOSIS
We start our patient on cyclophosphamide 175 mg daily in view of possible Wegener granulomatosis.
Even though purpura is extremely rare in primary antiglomerular basement membrane disease, this patient has life-threatening pulmonary hemorrhage, a complication seen in over 50% of these patients. Therefore, plasmapheresis is started empirically.
On the second day of cyclophosphamide treatment, tests for ANCA, glomerular basement membrane antibody, and antinuclear antibody are reported as negative, and complement levels are normal. Bronchoalveolar lavage shows no infection. Follow-up blood cultures are negative.
To summarize the findings so far, this patient has a purpuric skin rash, active urine sediment with red cell casts indicating glomerulonephritis, acute renal failure, and severe pulmonary hemorrhage requiring mechanical ventilation. Although one set of blood cultures showed gram-positive cocci, no source of infection, particularly endocarditis, could be identified.
Antiglomerular basement membrane disease would still be high on the list of suspected diagnoses, given his diffuse alveolar hemorrhage. As mentioned earlier, renal biopsy is imperative to making a diagnosis, because serologic tests have variable accuracy. And making the correct diagnosis has therapeutic implications.
Renal biopsy is performed and shows immune-complex mesangiopathic glomerulonephritis with positive immunofluorescent staining in the mesangium for IgA. Only one glomerulus shows fibrinoid necrosis.
Skin biopsy results obtained earlier showed positive direct immunofluorescence for IgA. Both renal and skin biopsies suggested Henoch-Schönlein purpura.
IgA deposition in the kidney and skin has been associated with liver cirrhosis, celiac disease, and infections with agents such as human immunodeficiency virus, cytomegalovirus, Haemophilus parainfluenzae, and Staphylococcus aureus. In a Japanese study,4 renal biopsy specimens from 116 patients with IgA nephropathy and from 122 patients with other types of kidney disease were examined for the presence of S aureus antigen in the glomeruli. Although antigen was not detected in non-IgA disease, 68% of specimens from patients with IgA nephropathy had S aureus cell envelope antigen together with IgA antibody in the glomeruli. However, no single antigen has been consistently identified, so it seems more probable that the development of IgA deposition in kidneys is a consequence of aberrant IgA immune response rather than the antigen itself.
HENOCH-SCHÖNLEIN PURPURA
Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is characterized by the tissue deposition of IgA-containing immune complexes. It is predominantly a disease of children but it can be seen in adults. A UK study found the prevalence to be 20 per 100,000 children, with the highest prevalence between ages 4 and 7 (70 per 100,000).5
The four cardinal clinical features of Henoch-Schönlein purpura are purpuric rash, abdominal pain, arthralgia, and renal involvement. Almost all patients have purpuric rash at some point in their disease course. Arthralgia with or without arthritis is typically migratory, oligoarticular, and nondeforming, usually affecting the large joints of the lower extremities; involvement of the upper extremities is less common.
Skin biopsy typically shows leukocytoclastic vasculitis in postcapillary venules with IgA deposition, and these findings are pathognomonic of Henoch-Schönlein purpura.
Gastrointestinal involvement can range from mild symptoms such as nausea, vomiting, abdominal pain, and paralytic ileus to severe disease such as gastrointestinal hemorrhage, bowel infarction, bowel perforation, and intussusception.
Renal involvement is common and is important, as it can in rare cases progress to end-stage renal disease. The urinalysis usually shows mild proteinuria with active sediment with red cell casts. Most patients have relatively mild disease, characterized by asymptomatic hematuria with a normal or slightly elevated creatinine. However, severe involvement may occur, with nephrotic syndrome, hypertension, and acute renal failure.
Different presentation in adults vs children
Adults with Henoch-Schönlein purpura only rarely present with bowel intussusception, whereas some studies have found that adults are more likely than children to develop significant renal involvement, including end-stage renal disease.6,7
There is a general but not absolute correlation between the severity of clinical manifestations and the findings on renal biopsy. A poor prognosis (significant proteinuria, hypertension, renal insufficiency, or end-stage renal disease) is associated with crescent formation involving more than 50% of the glomeruli.8
Our current understanding of the longterm outcome of the renal disease in Henoch-Schönlein purpura is primarily derived from studies in children. In one study, complete recovery occurred in 94% of children and 89% of adults.7 A long-term study of 250 adults with Henoch-Schönlein purpura and renal involvement of sufficient severity to require biopsy reported that, at a median follow up of 15 years, 11% had become dialysis-dependent and 13% had severe renal failure (creatinine clearance < 30 mL/min).6 Recurrence is common, occurring in approximately one-third of patients, more likely in those with nephritis.8
The diagnosis of Henoch-Schönlein purpura is typically made on the basis of key clinical features. In patients such as ours who have an atypical presentation, biopsy of affected skin and renal biopsy can be essential in the diagnosis. Diffuse alveolar hemorrhage is exceedingly rare in Henoch-Schönlein purpura but can be seen, as in our patient.9,10 In this setting, the findings of IgA deposits in skin and renal biopsy specimens, together with the absence of other clinical, serologic, or histologic features of other more-common potential causes, secured the diagnosis in this patient.
Henoch-Schönlein purpura is usually self-limited and requires no specific therapy. Evidence suggests that glucocorticoids enhance the rate of resolution of the arthritis and abdominal pain but do not appear to prevent recurrent disease or lessen the likelihood of progression of renal disease.8 Patients with severe renal involvement with renal function impairment may benefit from pulse intravenous corticosteroid therapy (methylprednisolone 250–1,000 mg per day for 3 days), followed by oral steroids for 3 months.11
In anecdotal reports, renal function improved in 19 of 21 children with Henoch-Schönlein purpura and severe crescentic nephritis.12 Studies have evaluated cyclophosphamide13 and plasmapheresis,14 but their role remains uncertain. Renal transplantation is an option in patients who progress to end-stage renal disease.
OUR CASE CONTINUED
In our patient, plasmapheresis was discontinued. As the pulmonary hemorrhage had developed during treatment with prednisone, we decided to continue cyclophosphamide, given the life-threatening nature of his disease. His pulmonary status improved and he was extubated.
During his initial hospital stay, he was taking heparin for anticoagulation therapy. However, given the life-threatening diffuse alveolar hemorrhage, heparin was stopped during the course of his stay in the intensive care unit. Once he was stable and was transferred out of the intensive care unit, heparin was resumed, and his anticoagulation therapy was bridged to warfarin just before discharge. He was eventually discharged on a tapering dose of oral prednisone and cyclophosphamide for 3 months, after which he was switched to azathioprine for maintenance therapy. He was doing well 6 months later, with a serum creatinine level of 1.6 mg/dL, no red cell casts in the urine, and no rash.
TAKE-HOME POINT
In any case of suspected vasculitis that presents with skin disease, it is essential to look for other sites with potentially life-threatening involvement. Henoch-Schönlein purpura is a systemic vasculitis with a prominent cutaneous component. It is not always benign and can be associated with serious complications such as renal failure, gastrointestinal events, and, very rarely, diffuse alveolar hemorrhage.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
- Collins J, Stern EJ. Ground-glass opacity at CT: the ABCs. AJR Am J Roentgenol 1997; 169:355–367.
- Boyce NW, Holdsworth SR. Pulmonary manifestations of the clinical syndrome of acute glomerulonephritis and lung hemorrhage. Am J Kidney Dis 1986; 8:31–36.
- Gallagher H, Kwan JT, Jayne DR. Pulmonary renal syndrome: a 4-year, single-center experience. Am J Kidney Dis 2002; 39:42–47.
- Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int 2004; 66:121–132.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360:1197–1202.
- Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271–1278.
- Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome. Arthritis Rheum 1997; 40:859–864.
- Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78:395–409.
- Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc 2004; 79:1151–1157.
- Vats KR, Vats A, Kim Y, Dassenko D, Sinaiko AR. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol 1999; 13:530–534.
- Niaudet P, Habib R. Methylprednisolone pulse therapy in the treatment of severe forms of Schönlein-Henoch purpura nephritis. Pediatr Nephrol 1998; 12:238–243.
- Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schöenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49:9–14.
- Tarshish P, Bernstein J, Edelmann CM. Henoch-Schönlein purpura nephritis: course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19:51–56.
- Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33:427–433.
Salmonella-related mycotic pseudoaneurysm
A 74-year-old man is admitted to the hospital with a 7-day history of fever, rigors, chest pain, and general weakness. He underwent coronary artery bypass surgery 10 years ago.
After 2 weeks of intravenous ceftriaxone 2 g/day, the patient undergoes excision of the mycotic pseudoaneurysm of the descending aorta, with placement of an aortic homograft. Biopsy of the excised aortic segment shows calcified fibroatheromatous plaques with no evidence of cystic medial degeneration or granulomas.
DISCUSSION
Mycotic aneurysm is a localized and irreversible dilatation of an artery due to destruction of the vessel wall by an infection. The dilatation is at least one and one-half times the normal diameter of the affected artery. It may be a true aneurysm or a pseudoaneurysm, involving all or some layers of the arterial wall. It is a rare but life-threatening condition.
A mycotic aneurysm can develop from septic embolization to the vasa vasorum, hematogenous seeding of an existing aneurysm, or extension from a contiguous site of infection.1,2 Mycotic infections of the aorta show a preference for male patients already infected with S enteritidis or S typhimurium. 3 Predisposing factors include rheumatic deformity of the valves, a bicuspid valve, impaired immunity,4 self-induced or iatrogenic arterial trauma,5,6 atherosclerotic deposits and calcification of the endovascular structure,7 and, in elderly patients, Salmonella septicemia.8,9 Computed tomography is the most useful imaging modality.10,11 Surgical interventions, in addition to parenteral antibiotic therapy for at least 6 weeks,11,12 are required to:
- Confirm the diagnosis
- Reconstruct the arterial vasculature
- Manage the complications of sepsis
- Start preventive measures (ie, cholecystectomy).2
- Carreras M, Larena JA, Tabernero G, Langara E, Pena JM. Evolution of salmonella aortitis towards the formation of abdominal aneurysm. Eur Radiol 1997; 7:54–56.
- Cicconi V, Mannino S, Caminiti G, et al. Salmonella aortic aneurysm: suggestions for diagnosis and therapy based on personal experience. Angiology 2004; 55:701–705.
- Schneider S, Krulls-Munch J, Knorig J. A mycotic aneurysm of the ascending aorta and aortic arch induced by Salmonella enteritidis. Z Kardiol 2004; 93:964–967.
- Johnson JR, Ledgerwood AM, Lucas CE. Mycotic aneurysm. New concepts in therapy. Arch Surg 1983; 118:577–582.
- Qureshi T, Hawrych AB, Hopkins NF. Mycotic aneurysm after percutaneous transluminal femoral artery angioplasty. J R Soc Med 1999; 92:255–256.
- Samore MH, Wessolossky MA, Lewis SM, Shubrooks SJ, Karchmer AW. Frequency, risk factors, and outcome for bacteremia after percutaneous transluminal coronary angioplasty. Am J Cardiol 1997; 79:873–977.
- Carnevalini M, Faccenna F, Gabrielli R, et al. Abdominal aortic mycotic aneurysm, psoas abscess, and aorto-bisiliac graft infection due to Salmonella typhimurium. J Infect Chemother 2005; 11:297–299.
- Soravia-Dunand VA, Loo VG, Salit IE. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin Infect Dis 1999; 29:862–868.
- Malouf JF, Chandrasekaran K, Orszulak TA. Mycotic aneurysms of the thoracic aorta: a diagnostic challenge. Am J Med 2003; 115:489–496.
- G ufler H, Buitrago-Tellez CH, Nesbitt E, Hauenstein KH. Mycotic aneurysm rupture of the descending aorta. Eur Radiol 1998; 8:295–297.
- Lin CY, Hong GJ, Lee KC, Tsai CS. Successful treatment of Salmonella mycotic aneurysm of the descending thoracic aorta. Eur J Cardiothorac Surg 2003; 24:320–322.
- Schoevaerdts D, Hanon F, Vanpee D, et al. Prolonged survival of an elderly woman with Salmonella dublin aortitis and conservative treatment. J Am Geriatr Soc 2003; 51:1326–1328.
A 74-year-old man is admitted to the hospital with a 7-day history of fever, rigors, chest pain, and general weakness. He underwent coronary artery bypass surgery 10 years ago.
After 2 weeks of intravenous ceftriaxone 2 g/day, the patient undergoes excision of the mycotic pseudoaneurysm of the descending aorta, with placement of an aortic homograft. Biopsy of the excised aortic segment shows calcified fibroatheromatous plaques with no evidence of cystic medial degeneration or granulomas.
DISCUSSION
Mycotic aneurysm is a localized and irreversible dilatation of an artery due to destruction of the vessel wall by an infection. The dilatation is at least one and one-half times the normal diameter of the affected artery. It may be a true aneurysm or a pseudoaneurysm, involving all or some layers of the arterial wall. It is a rare but life-threatening condition.
A mycotic aneurysm can develop from septic embolization to the vasa vasorum, hematogenous seeding of an existing aneurysm, or extension from a contiguous site of infection.1,2 Mycotic infections of the aorta show a preference for male patients already infected with S enteritidis or S typhimurium. 3 Predisposing factors include rheumatic deformity of the valves, a bicuspid valve, impaired immunity,4 self-induced or iatrogenic arterial trauma,5,6 atherosclerotic deposits and calcification of the endovascular structure,7 and, in elderly patients, Salmonella septicemia.8,9 Computed tomography is the most useful imaging modality.10,11 Surgical interventions, in addition to parenteral antibiotic therapy for at least 6 weeks,11,12 are required to:
- Confirm the diagnosis
- Reconstruct the arterial vasculature
- Manage the complications of sepsis
- Start preventive measures (ie, cholecystectomy).2
A 74-year-old man is admitted to the hospital with a 7-day history of fever, rigors, chest pain, and general weakness. He underwent coronary artery bypass surgery 10 years ago.
After 2 weeks of intravenous ceftriaxone 2 g/day, the patient undergoes excision of the mycotic pseudoaneurysm of the descending aorta, with placement of an aortic homograft. Biopsy of the excised aortic segment shows calcified fibroatheromatous plaques with no evidence of cystic medial degeneration or granulomas.
DISCUSSION
Mycotic aneurysm is a localized and irreversible dilatation of an artery due to destruction of the vessel wall by an infection. The dilatation is at least one and one-half times the normal diameter of the affected artery. It may be a true aneurysm or a pseudoaneurysm, involving all or some layers of the arterial wall. It is a rare but life-threatening condition.
A mycotic aneurysm can develop from septic embolization to the vasa vasorum, hematogenous seeding of an existing aneurysm, or extension from a contiguous site of infection.1,2 Mycotic infections of the aorta show a preference for male patients already infected with S enteritidis or S typhimurium. 3 Predisposing factors include rheumatic deformity of the valves, a bicuspid valve, impaired immunity,4 self-induced or iatrogenic arterial trauma,5,6 atherosclerotic deposits and calcification of the endovascular structure,7 and, in elderly patients, Salmonella septicemia.8,9 Computed tomography is the most useful imaging modality.10,11 Surgical interventions, in addition to parenteral antibiotic therapy for at least 6 weeks,11,12 are required to:
- Confirm the diagnosis
- Reconstruct the arterial vasculature
- Manage the complications of sepsis
- Start preventive measures (ie, cholecystectomy).2
- Carreras M, Larena JA, Tabernero G, Langara E, Pena JM. Evolution of salmonella aortitis towards the formation of abdominal aneurysm. Eur Radiol 1997; 7:54–56.
- Cicconi V, Mannino S, Caminiti G, et al. Salmonella aortic aneurysm: suggestions for diagnosis and therapy based on personal experience. Angiology 2004; 55:701–705.
- Schneider S, Krulls-Munch J, Knorig J. A mycotic aneurysm of the ascending aorta and aortic arch induced by Salmonella enteritidis. Z Kardiol 2004; 93:964–967.
- Johnson JR, Ledgerwood AM, Lucas CE. Mycotic aneurysm. New concepts in therapy. Arch Surg 1983; 118:577–582.
- Qureshi T, Hawrych AB, Hopkins NF. Mycotic aneurysm after percutaneous transluminal femoral artery angioplasty. J R Soc Med 1999; 92:255–256.
- Samore MH, Wessolossky MA, Lewis SM, Shubrooks SJ, Karchmer AW. Frequency, risk factors, and outcome for bacteremia after percutaneous transluminal coronary angioplasty. Am J Cardiol 1997; 79:873–977.
- Carnevalini M, Faccenna F, Gabrielli R, et al. Abdominal aortic mycotic aneurysm, psoas abscess, and aorto-bisiliac graft infection due to Salmonella typhimurium. J Infect Chemother 2005; 11:297–299.
- Soravia-Dunand VA, Loo VG, Salit IE. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin Infect Dis 1999; 29:862–868.
- Malouf JF, Chandrasekaran K, Orszulak TA. Mycotic aneurysms of the thoracic aorta: a diagnostic challenge. Am J Med 2003; 115:489–496.
- G ufler H, Buitrago-Tellez CH, Nesbitt E, Hauenstein KH. Mycotic aneurysm rupture of the descending aorta. Eur Radiol 1998; 8:295–297.
- Lin CY, Hong GJ, Lee KC, Tsai CS. Successful treatment of Salmonella mycotic aneurysm of the descending thoracic aorta. Eur J Cardiothorac Surg 2003; 24:320–322.
- Schoevaerdts D, Hanon F, Vanpee D, et al. Prolonged survival of an elderly woman with Salmonella dublin aortitis and conservative treatment. J Am Geriatr Soc 2003; 51:1326–1328.
- Carreras M, Larena JA, Tabernero G, Langara E, Pena JM. Evolution of salmonella aortitis towards the formation of abdominal aneurysm. Eur Radiol 1997; 7:54–56.
- Cicconi V, Mannino S, Caminiti G, et al. Salmonella aortic aneurysm: suggestions for diagnosis and therapy based on personal experience. Angiology 2004; 55:701–705.
- Schneider S, Krulls-Munch J, Knorig J. A mycotic aneurysm of the ascending aorta and aortic arch induced by Salmonella enteritidis. Z Kardiol 2004; 93:964–967.
- Johnson JR, Ledgerwood AM, Lucas CE. Mycotic aneurysm. New concepts in therapy. Arch Surg 1983; 118:577–582.
- Qureshi T, Hawrych AB, Hopkins NF. Mycotic aneurysm after percutaneous transluminal femoral artery angioplasty. J R Soc Med 1999; 92:255–256.
- Samore MH, Wessolossky MA, Lewis SM, Shubrooks SJ, Karchmer AW. Frequency, risk factors, and outcome for bacteremia after percutaneous transluminal coronary angioplasty. Am J Cardiol 1997; 79:873–977.
- Carnevalini M, Faccenna F, Gabrielli R, et al. Abdominal aortic mycotic aneurysm, psoas abscess, and aorto-bisiliac graft infection due to Salmonella typhimurium. J Infect Chemother 2005; 11:297–299.
- Soravia-Dunand VA, Loo VG, Salit IE. Aortitis due to Salmonella: report of 10 cases and comprehensive review of the literature. Clin Infect Dis 1999; 29:862–868.
- Malouf JF, Chandrasekaran K, Orszulak TA. Mycotic aneurysms of the thoracic aorta: a diagnostic challenge. Am J Med 2003; 115:489–496.
- G ufler H, Buitrago-Tellez CH, Nesbitt E, Hauenstein KH. Mycotic aneurysm rupture of the descending aorta. Eur Radiol 1998; 8:295–297.
- Lin CY, Hong GJ, Lee KC, Tsai CS. Successful treatment of Salmonella mycotic aneurysm of the descending thoracic aorta. Eur J Cardiothorac Surg 2003; 24:320–322.
- Schoevaerdts D, Hanon F, Vanpee D, et al. Prolonged survival of an elderly woman with Salmonella dublin aortitis and conservative treatment. J Am Geriatr Soc 2003; 51:1326–1328.
A 43-year-old woman with chest pressure
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma

The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4

At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.

DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
Unilateral cotton wool spots: An important clue
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
- Hypertensive retinopathy
- Diabetic retinopathy
- Human immunodeficiency virus (HIV) retinopathy
- Retinal involvement of systemic autoimmune disease
- Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
- Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update 2007; 8:17–28.
- Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol 2001; 49:5–14.
- Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol 1988; 11:239–251.
- Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol 1989; 13:187–191.
- Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 1998; 339:1415–1425.
- Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated? J Neuroophthalmol 2005; 25:299–302.
- Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology 2003; 110:1683–1689.
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
- Hypertensive retinopathy
- Diabetic retinopathy
- Human immunodeficiency virus (HIV) retinopathy
- Retinal involvement of systemic autoimmune disease
- Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
A 54-year-old man presents with sudden visual loss in the left eye. The left eye and left periorbital area have been painful for the past 5 days.
Duplex ultrasonography of the carotid arteries shows total occlusion of the left internal carotid artery. Fluorescein angiography of the fundus reveals focal hyperfluorescence with delayed arteriovenous transit time in the left eye.
Q: Which of the following diagnoses is the most likely at this point in the evaluation?
- Hypertensive retinopathy
- Diabetic retinopathy
- Human immunodeficiency virus (HIV) retinopathy
- Retinal involvement of systemic autoimmune disease
- Ocular ischemic syndrome
A: The ocular symptoms of hypertension, diabetes mellitus, HIV infection, and other autoimmune diseases usually present bilaterally, and funduscopic examination often reveals other signs such as vessel tortuosity, venous dilation, microaneurysms, retinal hemorrhages, hard exudates, and new vessel formation, in addition to cotton wool spots. In this patient, the lack of these signs and the unilateral cotton wool spots combined with the delay in arteriovenous transit time on fluorescein angiography point to ocular ischemic syndrome.
Ocular ischemic syndrome is the result of hypoperfusion of the globe caused by obstruction of the carotid or the ophthalmic artery,1 most commonly from atherosclerosis. Retinal hypoperfusion is also caused by arteritis, external compression, dissection of the artery,2 and, rarely, cardiac failure.
USUAL SIGNS AND SYMPTOMS
Usually, the patient presents with visual loss that has progressed gradually over a period of weeks or months and is associated with dull aching in the eye or orbit (“ocular angina”).3 Cotton wool spots on funduscopic examination represent retinal nerve fiber layer infarcts, a sign of retinal hypoperfusion. Delays in the choroidal filling time and the arteriole-to-venule transit time on fluorescein angiography confirm the diagnosis.
Strong clue to underlying disease
Ocular ischemic syndrome is an important clue to underlying macrovascular atherosclerotic disease: 50% of patients with ocular ischemic syndrome have ischemic heart disease, 25% have a history of stroke, and 20% have severe peripheral vascular disease. Ocular complications of the syndrome are rubeosis iridis, neovascular glaucoma, and neovascularization of the optic disc and retina. Prompt diagnosis is very important because the death rate at 5 years is 40%.4
Recommended workup
The recommended workup is a thorough history and physical examination to identify underlying systemic disease such as diabetes, hypertension, or collagen vascular disease. When carotid artery disease is suspected, a noninvasive vascular workup with carotid duplex ultrasonography is mandatory to confirm carotid arterial disease, to establish its cause, and to assess the severity of the lesion.
CURRENT TREATMENT OPTIONS
Treatment focuses on the control of systemic risk factors and follow-up to monitor for systemic and ocular complications. The combination of aspirin and extended-release dipyridamole (Aggrenox) is currently considered the most effective antiplatelet strategy, as it reduces the risk of stroke by 37% compared with 25% with aspirin alone.5
Carotid endarterectomy has been shown to benefit symptomatic patients with nondisabling stroke, amaurosis fugax, and a hemispheric transient ischemic attack and who have carotid stenosis of 70% to 99%. The North American Symptomatic Carotid Endarterectomy Trial found a 2-year stroke rate of 9% in such patients who underwent endarterectomy vs 26% in those treated with antiplatelet therapy alone.6,7 Some improvement in visual outcomes was also noted, but the data so far are not conclusive.6
Bypass procedures such as anastomosis of the superficial temporal artery to the middle cerebral artery have been tried in patients with 100% obstruction of the carotid artery in whom a thrombus has propagated distally, thus precluding endarterectomy.
We continue to monitor our patient for the development of ocular complications. The development of retinal neovascularization may warrant panretinal photocoagulation with or without anterior retinal cryoablation. Panretinal photocoagulation decreases the retinal demand for oxygen and decreases the release of angiogenic factors, thereby arresting the growth of neovascularization and preventing complications such as vitreous hemorrhage and tractional retinal detachment. Although no studies have analyzed the benefit of panretinal photocoagulation in patients with ocular ischemia, its long-term benefit has been well documented in diabetic patients.8
- Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update 2007; 8:17–28.
- Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol 2001; 49:5–14.
- Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol 1988; 11:239–251.
- Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol 1989; 13:187–191.
- Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 1998; 339:1415–1425.
- Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated? J Neuroophthalmol 2005; 25:299–302.
- Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology 2003; 110:1683–1689.
- Chen CS, Miller NR. Ocular ischemic syndrome: review of clinical presentations, etiology, investigation, and management. Compr Ophthalmol Update 2007; 8:17–28.
- Hussain N, Falali S, Kaul S. Carotid artery disease and ocular vascular disorders. Indian J Ophthalmol 2001; 49:5–14.
- Brown GC, Magargal LE. The ocular ischemic syndrome. Clinical, fluorescein angiographic and carotid angiographic features. Int Ophthalmol 1988; 11:239–251.
- Sivalingham A, Brown GC, Magaragal LE, Menduke H. The ocular ischemic syndrome, II; mortality and systemic morbidity. Int Ophthalmol 1989; 13:187–191.
- Diener HC, Cundha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2: dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 1996; 143:1–13.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 1998; 339:1415–1425.
- Wolintz RJ. Carotid endarterectomy for ophthalmic manifestations: is it ever indicated? J Neuroophthalmol 2005; 25:299–302.
- Chew EY, Ferris FL, Csaky KG, et al. The long-term effects of laser photocoagulation treatment in patients with diabetic retinopathy: the Early Treatment Diabetic Retinopathy Follow-up Study. Ophthalmology 2003; 110:1683–1689.
The battle of the clot

In this issue of the Journal we review two special situations in which low-molecular-weight heparins have special advantages. Babu and Carman discuss patients with cancer and thromboembolic disease. These patients are particularly difficult to manage since they tend to have recurrent thrombosis, sometimes even while on anticoagulant therapy, and they tend to have more bleeding complications from warfarin therapy. Inanition, drug interactions, and organ dysfunction make warfarin titration problematic, and the possibility of vascular metastases is always a concern. Low-molecular-weight heparins —which, unlike warfarin, work primarily by antagonizing factor Xa activity—have proven to be as effective as warfarin in reversing the many hypercoagulable effects of malignancy, although it wasn’t obvious at first that they would be.
Gibson and Powrie review the issues we face when pregnant patients need anticoagulation. While drug interactions and organ dysfunction are rarely problems in this setting, warfarin is teratogenic and is therefore strongly contraindicated early in pregnancy, and its peripartum use has been associated with bleeding complications. Furthermore, unfractionated heparin is associated with the development of osteoporosis, and it requires frequent injections. The low-molecular-weight heparins thus have a definite niche in the management of pregnant women, but with a caveat: dosing of these agents by weight alone in this setting is fraught with the potential for underdosing. Catastrophic outcomes have been reported in pregnant patients with older mechanical cardiac valves who were switched from warfarin to low-molecular-weight heparin therapy. Plus, if the patient is to receive neuraxial regional anesthesia, low-molecular-weight heparins should be discontinued at least 12 hours before catheter placement if prophylactic doses have been given, or 24 hours if therapeutic doses have been given.
Low-molecular-weight heparins have greatly enhanced our ability to treat thromboembolic disease. But, as the authors of these two papers discuss, many management nuances still must be noted.
In this issue of the Journal we review two special situations in which low-molecular-weight heparins have special advantages. Babu and Carman discuss patients with cancer and thromboembolic disease. These patients are particularly difficult to manage since they tend to have recurrent thrombosis, sometimes even while on anticoagulant therapy, and they tend to have more bleeding complications from warfarin therapy. Inanition, drug interactions, and organ dysfunction make warfarin titration problematic, and the possibility of vascular metastases is always a concern. Low-molecular-weight heparins —which, unlike warfarin, work primarily by antagonizing factor Xa activity—have proven to be as effective as warfarin in reversing the many hypercoagulable effects of malignancy, although it wasn’t obvious at first that they would be.
Gibson and Powrie review the issues we face when pregnant patients need anticoagulation. While drug interactions and organ dysfunction are rarely problems in this setting, warfarin is teratogenic and is therefore strongly contraindicated early in pregnancy, and its peripartum use has been associated with bleeding complications. Furthermore, unfractionated heparin is associated with the development of osteoporosis, and it requires frequent injections. The low-molecular-weight heparins thus have a definite niche in the management of pregnant women, but with a caveat: dosing of these agents by weight alone in this setting is fraught with the potential for underdosing. Catastrophic outcomes have been reported in pregnant patients with older mechanical cardiac valves who were switched from warfarin to low-molecular-weight heparin therapy. Plus, if the patient is to receive neuraxial regional anesthesia, low-molecular-weight heparins should be discontinued at least 12 hours before catheter placement if prophylactic doses have been given, or 24 hours if therapeutic doses have been given.
Low-molecular-weight heparins have greatly enhanced our ability to treat thromboembolic disease. But, as the authors of these two papers discuss, many management nuances still must be noted.
In this issue of the Journal we review two special situations in which low-molecular-weight heparins have special advantages. Babu and Carman discuss patients with cancer and thromboembolic disease. These patients are particularly difficult to manage since they tend to have recurrent thrombosis, sometimes even while on anticoagulant therapy, and they tend to have more bleeding complications from warfarin therapy. Inanition, drug interactions, and organ dysfunction make warfarin titration problematic, and the possibility of vascular metastases is always a concern. Low-molecular-weight heparins —which, unlike warfarin, work primarily by antagonizing factor Xa activity—have proven to be as effective as warfarin in reversing the many hypercoagulable effects of malignancy, although it wasn’t obvious at first that they would be.
Gibson and Powrie review the issues we face when pregnant patients need anticoagulation. While drug interactions and organ dysfunction are rarely problems in this setting, warfarin is teratogenic and is therefore strongly contraindicated early in pregnancy, and its peripartum use has been associated with bleeding complications. Furthermore, unfractionated heparin is associated with the development of osteoporosis, and it requires frequent injections. The low-molecular-weight heparins thus have a definite niche in the management of pregnant women, but with a caveat: dosing of these agents by weight alone in this setting is fraught with the potential for underdosing. Catastrophic outcomes have been reported in pregnant patients with older mechanical cardiac valves who were switched from warfarin to low-molecular-weight heparin therapy. Plus, if the patient is to receive neuraxial regional anesthesia, low-molecular-weight heparins should be discontinued at least 12 hours before catheter placement if prophylactic doses have been given, or 24 hours if therapeutic doses have been given.
Low-molecular-weight heparins have greatly enhanced our ability to treat thromboembolic disease. But, as the authors of these two papers discuss, many management nuances still must be noted.
Cancer and clots: All cases of venous thromboembolism are not treated the same
Venous thromboembolism (VTE) has various differing causes, so its treatment is not necessarily the same in all cases. Most cases of VTE are related to an easily identified risk factor. In patients with an apparently idiopathic event, identifying an underlying cause may alter therapy. In particular, identification of a malignancy may affect the choice of therapy and the duration of treatment.
In this review, we explore the role of cancer screening in patients with idiopathic VTE, then highlight the treatment for VTE in patients with cancer.
‘IDIOPATHIC’ VTE CAN BE DUE TO CANCER
Most patients with venous thrombosis have one of the components of Virchow’s triad: a hypercoagulable state, venous injury, or venous stasis. Those without identifiable risk factors for VTE are considered to have idiopathic VTE. In these patients, a search for a contributing factor may be indicated.
In 1861, the astute clinician Dr. Armand Trousseau noted a link between deep venous thrombosis and pancreatic cancer, stating that if cancer of an internal organ is suspected but the diagnosis cannot be verified, the diagnosis may be confirmed by the sudden, spontaneous appearance of thrombophlebitis in a large vein.1
Today, from 2% to 25% of patients with idiopathic VTE are found to have cancer within 24 months of the diagnosis of VTE.2–11 The goals of cancer screening in idiopathic VTE are to detect cancer at an early, treatable stage and to optimize the VTE therapy to decrease the risks of recurrence and anticoagulation-associated complications in patients who are found to have cancer. However, several questions must be considered first:
- What are the risks and costs of the screening?
- Will discovering the cancer sooner benefit the patient in terms of survival?
- If cancer is found, what are the possible complications or risks of the additional procedures, interventions, or treatments required?
- What is the psychological impact of the screening?
EVIDENCE SUPPORTING CANCER SCREENING AFTER IDIOPATHIC VTE
Piccioli et al12 recently performed a randomized, controlled trial comparing cancer-related death rates in 99 patients with idiopathic VTE screened for malignancy vs 102 patients with idiopathic VTE who were not screened.
The screened group underwent:
- Abdominal and pelvic ultrasonography and computed tomography (CT)
- Gastroscopy or double-contrast barium-swallow evaluation
- Colonoscopy or sigmoidoscopy followed by barium enema
- Testing for fecal occult blood
- Sputum cytology
- Measurement of carcinoembryonic antigen, alpha-fetoprotein, and cancer antigen 125.
- Mammography and Papanicolaou smears (women)
- Ultrasonography of the prostate and prostate-specific antigen testing (men).
Patients were followed for 2 years. The screening uncovered cancer in 13 patients. Cancer developed in one other patient in the screening group during follow-up; in the control group, 10 patients developed symptomatic cancer during follow-up. Overall, the time to cancer diagnosis was 11.6 months in the unscreened group vs 1 month in the screened group (P < .001). Nine of the 14 patients with cancer in the screened group had T1 or T2 disease without local or distant metastasis vs 2 of the 10 control patients with cancer (P = .047). Unfortunately, this study did not have adequate power to detect the effect of screening on survival.
Di Nisio et al13 used data from this trial to perform a decision analysis for cancer screening. They calculated that abdominal and pelvic CT, with or without mammography and with or without sputum cytologic testing, would cost the least per life-year gained and would harm the fewest number of patients. They also suggested that substituting CT of the chest for sputum cytology may provide additional diagnostic benefit.
However, this strategy has not been clinically tested. Given the limited number of patients and the short follow-up in this initial trial, larger trials are needed to look at the cost-effectiveness of this screening model and whether it increases survival.
Our recommendations
Because the data are limited, our approach to looking for an early, treatable malignancy in patients with idiopathic VTE follows the current consensus:
- A thorough history and physical, including an extensive review of systems
- Basic laboratory testing with a complete blood cell count, comprehensive metabolic profile, and urinalysis
- Chest radiography
- Other age- and sex-specific cancer screening tests.
Adding CT of the abdomen, pelvis, or chest to this evaluation may be considered. However, tumor marker testing, which typically has high false-positive rates, is not routinely warranted.13 Additional investigation should be considered if abnormalities are detected during the initial evaluation or in patients with recurrent VTE during therapy.
While this strategy may be most cost-effective, Monreal et al14 suggest that it may miss up to half of cancers ultimately discovered.
MANAGING VTE IN PATIENTS WITH KNOWN CANCER
Managing VTE is far more complex in cancer patients than in patients without cancer. Also, cancer patients with VTE have lower rates of survival than cancer patients without VTE and are at greater risk of adverse outcomes such as anticoagulant-associated bleeding and recurrent venous thrombotic events.15–17
Up to 21.5% of patients with VTE have another event within 5 years,18 but the risk is two to three times higher if they also have cancer.16,18 The risk of recurrence may be linked to the location of the thrombus and to the extent of the malignancy.
In one study, the 3-month rate of recurrence was up to 5.1% if the clot was in the popliteal vein, 5.3% if in the femoral vein, and 11.8% if in the iliac vein.19
Prandoni et al16 found that the risks of VTE recurrence and bleeding were higher in patients with extensive cancer than in those with less-extensive cancer. In this study, major bleeding was documented in 12.4% of patients with cancer vs 4.9% of patients without cancer. Compared with patients without cancer, the hazard ratio for a major bleeding event was 4.8 in patients with extensive cancer and 0.5 in patients with less-extensive cancer.
In addition, not all patients with bleeding had excessive levels of anticoagulation, and not all patients with recurrent events had subtherapeutic levels.16,17 Therefore, treatment of venous thrombosis in cancer patients requires a careful, individualized risk-to-benefit decision analysis.
ACUTE THERAPY FOR VTE: PARENTERAL AGENTS
Treatment in the first several hours or days after a thromboembolic event is with short-acting parenteral agents: unfractionated heparin; one of the low-molecular-weight heparins (LMWHs), ie, dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep); or fondaparinux (Arixtra).
Before starting anticoagulation, consider:
- Does the patient have severe chronic kidney disease (ie, a creatinine clearance < 30 mL/min)? If so, unfractionated heparin may be better than an LMWH or fondaparinux, which are cleared by the kidney.
- Does he or she need inpatient care? If not, LMWH therapy at home may be appropriate.
- Are there concerns about the ease of anticoagulation administration (ie, whether the patient can give the injections or have a family member do it), the cost of the drugs, or the ability to reverse the anticoagulant effect, if necessary? If so, unfractionated heparin may be more appropriate.
For acute treatment, the 2008 guidelines of the American College of Chest Physicians20 (ACCP) recommend using an LMWH in a weight-based dose; unfractionated heparin given intravenously; unfractionated heparin given subcutaneously with monitoring and dosing adjustments; unfractionated heparin given subcutaneously at a fixed dose; or fondaparinux (grade 1A recommendation). The 2007 National Comprehensive Cancer Network (NCCN) guidelines21 recommend an LMWH, fondaparinux, or unfractionated heparin. Treatment should start promptly after the diagnosis of VTE is confirmed. However, if VTE is strongly suspected and a delay in diagnostic testing is anticipated, therapy should be started while awaiting the test results.
LONG-TERM THERAPY: LMWH OR WARFARIN
The ACCP and the NCCN guidelines recommend LMWH monotherapy for extended treatment of VTE in patients with active malignancy, when appropriate.20,21 However, if long-term LMWH is not appropriate, then oral anticoagulation with a vitamin K antagonist, such as the coumarin derivative warfarin (Coumadin), is an alternative and should be started on the same day as the heparin. The heparin and the warfarin therapy must overlap for a minimum of 4 or 5 days and until a stable, therapeutic level of anticoagulation is achieved, ie, an international normalized ratio (INR) of 2 to 3 for 2 consecutive days.20
The duration of anticoagulant therapy depends on comorbidities and the patient’s underlying predisposition for VTE. In patients with limited disease, the guidelines recommend continuing anticoagulation for a minimum of 3 to 6 months for deep venous thrombosis and pulmonary embolism.20–21 Patients with active malignancy, ongoing treatment for the cancer, or continued risk factors may need indefinite treatment. In some circumstances, such as catheter-associated deep venous thrombosis, anticoagulation should continue for as long as the catheter is in place and for 1 to 3 months after its removal.21
WARFARIN CAN BE DIFFICULT TO USE
In 1954, the US Food and Drug Administration (FDA) approved the vitamin K antagonist warfarin for medical use in humans. Experience has shown it to be effective in preventing and treating VTE. However, it can be somewhat difficult to use, for several reasons:
- A narrow therapeutic window
- Genetic polymorphisms and variability in dose response
- Drug interactions and dietary considerations
- The need for laboratory monitoring and dose adjustment
- Patient noncompliance or miscommunication between the patient and physician.22
In cancer patients, the response to warfarin may be unpredictable because of poor nutrition, interactions with chemotherapy and antibiotics, and comorbid conditions.22 Furthermore, its onset of action can be delayed and its clearance may be prolonged, further increasing the risk of complications, especially in patients prone to developing chemotherapy-related anemia or thrombocytopenia.22 Bleeding risk is the highest in the first 3 months of therapy. In addition, the risk of bleeding is higher in older patients, women, and patients with a history of gastrointestinal bleeding, stroke, recent myocardial infarction, diabetes, renal insufficiency, malignancy, or anemia.23,24
ADVANTAGES AND DISADVANTAGES OF LMWH
The advantages of the LMWHs over unfractionated heparin include a lower risk of heparin-induced thrombocytopenia, greater bioavailability when given subcutaneously (which also permits once-daily or twice-daily dosing), and no need for laboratory monitoring in most patients. LMWHs have a short half-life, so omitting one or two doses will adequately interrupt therapy. Also, LMWHs have been shown to be as safe and effective as unfractionated heparin in treating VTE. They can be given safely at home, thus enhancing quality of life.25–31
On the other hand, these drugs cost more than unfractionated heparin or warfarin, their dosage must be adjusted in patients with renal insufficiency, their anticoagulant effect can be reversed only to a limited extent, and their dose must be adjusted according to weight in morbidly obese or in very thin patients.32,33
LMWHs are expensive, but may be worth it
As initial therapy, the LMWHs are cost-effective compared with unfractionated heparin in patients with VTE.34,35 However, they cost more with extended use. A cost-effectiveness analysis comparing 6 months of LMWH therapy to standard warfarin concluded that LMWH therapy was more costly.35 However, the impact of fewer hospitalizations, probably fewer bleeding complications, and better quality of life are difficult to analyze in this decision model and should also be considered when deciding about therapy for an individual patient.35
LMWHs are cleared by the kidney
All LMWHs are renally cleared, so patients with significant renal insufficiency (creatinine clearance < 30 mL/min) are at greater risk of bleeding complications. The rate below which clearance is impaired varies among the different LMWHs. Only enoxaparin has approved dosing regimens for use in patients with renal impairment.
If the patient has renal insufficiency, the ACCP guidelines suggest using unfractionated heparin, or if using LMWH, monitoring anti-factor Xa levels to avoid drug accumulation and increased bleeding risk.25 If bleeding occurs, LMWHs have limited reversibility with protamine sulfate, which is estimated to neutralize about 60% of the anti-factor Xa activity of LMWHs.25
Adjusting LMWHs for body weight
In the Registro Informatizado de la Enfermedad Tromboembólica (RIETE),33 patients weighing less than 50 kg had a higher risk of bleeding than patients weighing 50 to 100 kg, so in thinner patients the risk of bleeding from LMWH vs oral anticoagulation must be considered carefully and monitored prudently.
Although there is little evidence to suggest a higher bleeding risk in morbidly obese patients (> 150 kg), they may be at risk of subtherapeutic treatment, and monitoring with anti-factor Xa assays is recommended.25,32,33
LMWH VS WARFARIN FOR VTE IN CANCER PATIENTS
LMWHs are the first-line treatment for VTE in cancer patients.20,21 Several randomized controlled trials compared the efficacy of LMWH vs warfarin in patients with cancer.
Meyer et al36 randomized patients to receive either warfarin for 3 months at an INR between 2 and 3, or enoxaparin 1.5 mg/kg subcutaneously daily. Seventy-one patients received warfarin and 67 received enoxaparin. Fifteen (21%, 95% confidence interval [CI] 12%–32%) of the 71 patients assigned to warfarin experienced one major outcome event, defined as major bleeding or recurrent VTE, compared with 7 (10.5%) of the 67 patients assigned to receive enoxaparin (95% CI 4%–20%, P = .09). Six patients in the warfarin group died of bleeding vs none of the patients in the enoxaparin group. Overall, the warfarin group had a higher rate of bleeding, although this did not reach statistical significance. Despite weekly INR measurements, only 41% of the measured values were within the therapeutic range during the 3 months of treatment.36
Lee et al37 randomized cancer patients with deep venous thrombosis, pulmonary embolism, or both to receive 6 months of dalteparin alone, dosed at 200 IU/kg daily for 1 month, then decreased to 75% to 80% of the original dose (150 IU/kg) daily for the duration of therapy, or dalteparin followed by warfarin. During the 6-month follow-up, 17.4% of patients in the warfarin group had a recurrent thromboembolic event vs 8.8% in the dalteparin group (P = .0017). No statistically significant difference was noted in rates of major bleeding, minor bleeding, or death.37
Hull et al38 reported statistically significantly fewer episodes of recurrent VTE at 12 months in cancer patients treated with once-daily tinzaparin vs warfarin. In the tinzaparin group the recurrence rate was 7%, vs 16% in the warfarin group (P = .044). No difference in rates of bleeding or death were found.
Deitcher et al39 compared enoxaparin with long-term warfarin in 102 patients. While this trial did not have the power to detect clinical differences in recurrent thromboembolic events or bleeding complications, at 180 days they noted 97% compliance with once-daily or twice-daily enoxaparin therapy.
Noble and Finlay,40 in another small study, found LMWH therapy to be qualitatively more acceptable for palliative-care cancer patients than oral therapy.
In general, long-term therapy with once-daily or twice-daily LMWH is well tolerated. Currently, dalteparin is the only LMWH approved by the FDA for extended monotherapy in cancer-related VTE.
DO LMWHS AFFECT CANCER?
In vitro and animal studies indicate that LMWH may have antimetastatic and antiangiogenic properties.41–44
Altinbas et al45 reported significantly better chemotherapy-induced tumor response rates and survival rates in patients with small cell lung cancer randomized to receive combination chemotherapy plus prophylactic dalteparin 5,000 IU daily compared with combination chemotherapy alone. However, as provocative as these results may be, we need to test the effects of LWMHs on different cancer types in a prospective clinical trial. For now, this area remains controversial.
It has been suggested that anticoagulants may improve survival in patients with nonmetastatic cancer. Supporting this observation, a post hoc analysis of the trial by Lee et al37 found a statistically significantly lower cancer-specific mortality rate in nonmetastatic cancer patients treated with dalteparin vs oral therapy with a coumarin derivative. In patients without metastatic disease, the death rate at 12 months was 36% in patients treated with oral therapy vs 20% in patients treated with dalteparin (P = .03).46
These findings are consistent with those of the Fragmin Advanced Malignancy Outcome Study (FAMOUS),47 the first randomized, placebo-controlled trial of dalteparin 5,000 IU daily in patients with advanced solid tumors and without evidence of underlying thrombosis. Overall, dalteparin prophylaxis did not increase survival. However, in a subgroup of patients with a better prognosis and who were alive 17 months after diagnosis, survival was statistically significantly longer in patients treated with dalteparin.
Another small trial showed similar survival benefits in cancer patients without VTE.48 The results may suggest a long-term favorable effect of LMWH on tumor cell biology, which could translate into a favorable outcome in some patients. It is important to note, however, that not all trials have shown this same clinical benefit.49
In general, the growing body of laboratory and clinical data indicates that LMWHs may suppress tumor growth and metastasis. However, definitive conclusions about these effects are not yet possible because of variations in study design, tumor type, and patient populations. Further investigations into the role of LMWHs in the treatment of VTE and in cancer progression are ongoing.
THE EVIDENCE IN PERSPECTIVE
Illness and the recurrence of VTE in patients with cancer depend on the location and extent of the underlying cancer. Rates of death are higher in VTE patients with cancer than in VTE patients without cancer. Patients with limited or localized disease may not die of the cancer itself but of complications of acute pulmonary embolism. Therefore, it is important to recognize the different options for and the potential side effects of treating VTE.
If patients are hospitalized for an acute thromboembolic event and unfractionated heparin is chosen as the initial anticoagulant, using a weight-based nomogram has been shown to achieve therapeutic levels within 24 hours and reduce the rates of recurrence of thromboembolic events.50
Warfarin treatment may pose a particular challenge for both cancer patients and physicians, since multiple drug interactions, anorexia, and comorbid conditions contribute to an unpredictable response.
The risk of bleeding is higher in cancer patients than in the general population, and the decision to start anticoagulants should be based on an individualized risk-benefit profile. Several trials have shown LMWH to be more effective and safer than warfarin in cancer patients.
These considerations, along with the other advantages of LMWHs (ease of use, less need for laboratory monitoring, and better patient tolerance), make LMWHs a good choice for initial therapy. Extended LMWH therapy is currently favored for initial management in patients with cancer. Trials are under way to further assess the antitumor properties and potential survival benefit in patients with selected solid tumors.
- Aron E. The 100th anniversary of the death of A. Trousseau. Presse Med 1967; 75:1429–1430.
- Hettiarachchi RJ, Lok J, Prins MH, Büller HR, Prandoni P. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998; 83:180–185.
- Baron JA, Gridley G, Weiderpass E, Nyrén O, Linet M. Venous thromboembolism and cancer. Lancet 1998; 351:1077–1080.
- Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 2000; 342:1953–1958.
- Sørensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998; 338:1169–1173.
- Monreal M, Lafoz E, Casals A, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer 1991; 67:541–545.
- Nordström M, Lindblad B, Anderson H, Bergqvist D, Kjellström T. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ 1994; 308:891–894.
- Prandoni P, Lensing AW, Büller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327:1128–1133.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Fennerty T. Screening for cancer in venous thromboembolic disease. BMJ 2001; 323:704–705.
- Bastounis EA, Karayiannakis AJ, Makri GG, Alexiou D, Papalambros EL. The incidence of occult cancer in patients with deep venous thrombosis: a prospective study. J Intern Med 1996; 239:153–156.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Di Nisio M, Otten HM, Piccioli A, et al. Decision analysis for cancer screening in idiopathic venous thromboembolism. J Thromb Haemost 2005; 3:2391–2396.
- Monreal M, Lensing AW, Prins MH, et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost 2004; 2:876–881.
- Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343:1846–1850.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hutten BA, Prins MH, Gent M, Ginsberg J, Tijssen JG, Büller HR. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078–3083.
- Hansson PO, Sörbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000; 160:769–774.
- Douketis JD, Crowther MA, Foster GA, Ginsberg JS. Does the location of thrombosis determine the risk of disease recurrence in patients with proximal deep vein thrombosis? Am J Med 2001; 110:515–519.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-based Clinical Practice Guidelines, 8th Edition. Chest 2008; 133 suppl 6:454S–545S.
- National Comprehensive Cancer Network. Venous Thromboembolic Disease Clinical Practice Guidelines in Oncology (V.1.2007). Available at www.nccn.org/professionals/physician_gls/PDF/vte.pdf. Accessed 01/02/2008.
- Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:204S–233S.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med 1999; 159:457–460.
- Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest 2004; 126 suppl 3:188S–203S.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Hettiarachchi RJ, Prins MH, Lensing AW, Büller HR. Low molecular weight heparin versus unfractionated heparin in the initial treatment of venous thromboembolism. Curr Opin Pulm Med 1998; 4:220–225.
- Gould MK, Dembitzer AD, Doyle RL, Hastie TJ, Garber AM. Low-molecular-weight heparins compared with unfractionated heparin for treatment of acute deep venous thrombosis. A meta-analysis of randomized, controlled trials. Ann Intern Med 1999; 130:800–809.
- Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with un-fractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med 2000; 160:181–188.
- van Dongen CJ, van den Belt AG, Prins MH, Lensing AW. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2004; 4:CD001100.
- Cook LM, Kahn SR, Goodwin J, Kovacs MJ. Frequency of renal impairment, advanced age, obesity, and cancer in venous thromboembolism patients in clinical practice. J Thromb Haemost 2007; 5:937–941.
- Barba R, Marco J, Martin-Alvarez H, et al. The influence of extreme body weight on clinical outcome of patients with venous thromboembolism: findings from a prospective registry (RIETE). J Thromb Haemost 2005; 3:856–862.
- Segal JB, Strieff MB, Hofmann LV, Thornton K, Bass EB. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007; 146:211–222.
- Aujesky D, Smith KJ, Cornuz J, Roberts MS. Cost-effectiveness of low-molecular-weight heparin for secondary prophylaxis of cancer-related venous thromboembolism. Thromb Haemost 2005; 93:592–599.
- Meyer G, Marjanovic Z, Valcke J, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162:1729–1735.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J ON-CENOX investigators. Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180-day period. Clin Appl Thromb Hemost 2006; 12:389–396.
- Noble SI, Finlay IG. Is long-term low-molecular-weight heparin acceptable to palliative care patients in the treatment of cancer related venous thromboembolism? A qualitative study. Palliat Med 2005; 19:197–201.
- Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastatic effect of tinzaparin, a low-molecular-weight heparin. J Thromb Haemost 2003; 1:1972–1976.
- Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep 2005; 14:99–104.
- Mousa SA, Mohamed S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: anti-cancer efficacy. Oncol Rep 2004; 12:683–688.
- Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004; 58:213–219.
- Altinbas M, Coskun HS, Er O, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost 2004; 2:1266–1271.
- Lee AY, Rickles FR, Julian JA, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23:2123–2129.
- Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the Fragmin Advanced Malignancy Outcome Study (FAMOUS). J Clin Oncol 2004; 22:1944–1948.
- Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23:2130–2135.
- Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a phase 3 clinical trial. Mayo Clin Proc 2006; 81:758–767.
- Bernardi E, Piccioli A, Oliboni G, Zuin R, Girolami A, Prandoni P. Nomograms for the administration of unfractionated heparin in the initial treatment of acute thromboembolism—an overview. Thromb Haemost 2000; 84:22–26.
Venous thromboembolism (VTE) has various differing causes, so its treatment is not necessarily the same in all cases. Most cases of VTE are related to an easily identified risk factor. In patients with an apparently idiopathic event, identifying an underlying cause may alter therapy. In particular, identification of a malignancy may affect the choice of therapy and the duration of treatment.
In this review, we explore the role of cancer screening in patients with idiopathic VTE, then highlight the treatment for VTE in patients with cancer.
‘IDIOPATHIC’ VTE CAN BE DUE TO CANCER
Most patients with venous thrombosis have one of the components of Virchow’s triad: a hypercoagulable state, venous injury, or venous stasis. Those without identifiable risk factors for VTE are considered to have idiopathic VTE. In these patients, a search for a contributing factor may be indicated.
In 1861, the astute clinician Dr. Armand Trousseau noted a link between deep venous thrombosis and pancreatic cancer, stating that if cancer of an internal organ is suspected but the diagnosis cannot be verified, the diagnosis may be confirmed by the sudden, spontaneous appearance of thrombophlebitis in a large vein.1
Today, from 2% to 25% of patients with idiopathic VTE are found to have cancer within 24 months of the diagnosis of VTE.2–11 The goals of cancer screening in idiopathic VTE are to detect cancer at an early, treatable stage and to optimize the VTE therapy to decrease the risks of recurrence and anticoagulation-associated complications in patients who are found to have cancer. However, several questions must be considered first:
- What are the risks and costs of the screening?
- Will discovering the cancer sooner benefit the patient in terms of survival?
- If cancer is found, what are the possible complications or risks of the additional procedures, interventions, or treatments required?
- What is the psychological impact of the screening?
EVIDENCE SUPPORTING CANCER SCREENING AFTER IDIOPATHIC VTE
Piccioli et al12 recently performed a randomized, controlled trial comparing cancer-related death rates in 99 patients with idiopathic VTE screened for malignancy vs 102 patients with idiopathic VTE who were not screened.
The screened group underwent:
- Abdominal and pelvic ultrasonography and computed tomography (CT)
- Gastroscopy or double-contrast barium-swallow evaluation
- Colonoscopy or sigmoidoscopy followed by barium enema
- Testing for fecal occult blood
- Sputum cytology
- Measurement of carcinoembryonic antigen, alpha-fetoprotein, and cancer antigen 125.
- Mammography and Papanicolaou smears (women)
- Ultrasonography of the prostate and prostate-specific antigen testing (men).
Patients were followed for 2 years. The screening uncovered cancer in 13 patients. Cancer developed in one other patient in the screening group during follow-up; in the control group, 10 patients developed symptomatic cancer during follow-up. Overall, the time to cancer diagnosis was 11.6 months in the unscreened group vs 1 month in the screened group (P < .001). Nine of the 14 patients with cancer in the screened group had T1 or T2 disease without local or distant metastasis vs 2 of the 10 control patients with cancer (P = .047). Unfortunately, this study did not have adequate power to detect the effect of screening on survival.
Di Nisio et al13 used data from this trial to perform a decision analysis for cancer screening. They calculated that abdominal and pelvic CT, with or without mammography and with or without sputum cytologic testing, would cost the least per life-year gained and would harm the fewest number of patients. They also suggested that substituting CT of the chest for sputum cytology may provide additional diagnostic benefit.
However, this strategy has not been clinically tested. Given the limited number of patients and the short follow-up in this initial trial, larger trials are needed to look at the cost-effectiveness of this screening model and whether it increases survival.
Our recommendations
Because the data are limited, our approach to looking for an early, treatable malignancy in patients with idiopathic VTE follows the current consensus:
- A thorough history and physical, including an extensive review of systems
- Basic laboratory testing with a complete blood cell count, comprehensive metabolic profile, and urinalysis
- Chest radiography
- Other age- and sex-specific cancer screening tests.
Adding CT of the abdomen, pelvis, or chest to this evaluation may be considered. However, tumor marker testing, which typically has high false-positive rates, is not routinely warranted.13 Additional investigation should be considered if abnormalities are detected during the initial evaluation or in patients with recurrent VTE during therapy.
While this strategy may be most cost-effective, Monreal et al14 suggest that it may miss up to half of cancers ultimately discovered.
MANAGING VTE IN PATIENTS WITH KNOWN CANCER
Managing VTE is far more complex in cancer patients than in patients without cancer. Also, cancer patients with VTE have lower rates of survival than cancer patients without VTE and are at greater risk of adverse outcomes such as anticoagulant-associated bleeding and recurrent venous thrombotic events.15–17
Up to 21.5% of patients with VTE have another event within 5 years,18 but the risk is two to three times higher if they also have cancer.16,18 The risk of recurrence may be linked to the location of the thrombus and to the extent of the malignancy.
In one study, the 3-month rate of recurrence was up to 5.1% if the clot was in the popliteal vein, 5.3% if in the femoral vein, and 11.8% if in the iliac vein.19
Prandoni et al16 found that the risks of VTE recurrence and bleeding were higher in patients with extensive cancer than in those with less-extensive cancer. In this study, major bleeding was documented in 12.4% of patients with cancer vs 4.9% of patients without cancer. Compared with patients without cancer, the hazard ratio for a major bleeding event was 4.8 in patients with extensive cancer and 0.5 in patients with less-extensive cancer.
In addition, not all patients with bleeding had excessive levels of anticoagulation, and not all patients with recurrent events had subtherapeutic levels.16,17 Therefore, treatment of venous thrombosis in cancer patients requires a careful, individualized risk-to-benefit decision analysis.
ACUTE THERAPY FOR VTE: PARENTERAL AGENTS
Treatment in the first several hours or days after a thromboembolic event is with short-acting parenteral agents: unfractionated heparin; one of the low-molecular-weight heparins (LMWHs), ie, dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep); or fondaparinux (Arixtra).
Before starting anticoagulation, consider:
- Does the patient have severe chronic kidney disease (ie, a creatinine clearance < 30 mL/min)? If so, unfractionated heparin may be better than an LMWH or fondaparinux, which are cleared by the kidney.
- Does he or she need inpatient care? If not, LMWH therapy at home may be appropriate.
- Are there concerns about the ease of anticoagulation administration (ie, whether the patient can give the injections or have a family member do it), the cost of the drugs, or the ability to reverse the anticoagulant effect, if necessary? If so, unfractionated heparin may be more appropriate.
For acute treatment, the 2008 guidelines of the American College of Chest Physicians20 (ACCP) recommend using an LMWH in a weight-based dose; unfractionated heparin given intravenously; unfractionated heparin given subcutaneously with monitoring and dosing adjustments; unfractionated heparin given subcutaneously at a fixed dose; or fondaparinux (grade 1A recommendation). The 2007 National Comprehensive Cancer Network (NCCN) guidelines21 recommend an LMWH, fondaparinux, or unfractionated heparin. Treatment should start promptly after the diagnosis of VTE is confirmed. However, if VTE is strongly suspected and a delay in diagnostic testing is anticipated, therapy should be started while awaiting the test results.
LONG-TERM THERAPY: LMWH OR WARFARIN
The ACCP and the NCCN guidelines recommend LMWH monotherapy for extended treatment of VTE in patients with active malignancy, when appropriate.20,21 However, if long-term LMWH is not appropriate, then oral anticoagulation with a vitamin K antagonist, such as the coumarin derivative warfarin (Coumadin), is an alternative and should be started on the same day as the heparin. The heparin and the warfarin therapy must overlap for a minimum of 4 or 5 days and until a stable, therapeutic level of anticoagulation is achieved, ie, an international normalized ratio (INR) of 2 to 3 for 2 consecutive days.20
The duration of anticoagulant therapy depends on comorbidities and the patient’s underlying predisposition for VTE. In patients with limited disease, the guidelines recommend continuing anticoagulation for a minimum of 3 to 6 months for deep venous thrombosis and pulmonary embolism.20–21 Patients with active malignancy, ongoing treatment for the cancer, or continued risk factors may need indefinite treatment. In some circumstances, such as catheter-associated deep venous thrombosis, anticoagulation should continue for as long as the catheter is in place and for 1 to 3 months after its removal.21
WARFARIN CAN BE DIFFICULT TO USE
In 1954, the US Food and Drug Administration (FDA) approved the vitamin K antagonist warfarin for medical use in humans. Experience has shown it to be effective in preventing and treating VTE. However, it can be somewhat difficult to use, for several reasons:
- A narrow therapeutic window
- Genetic polymorphisms and variability in dose response
- Drug interactions and dietary considerations
- The need for laboratory monitoring and dose adjustment
- Patient noncompliance or miscommunication between the patient and physician.22
In cancer patients, the response to warfarin may be unpredictable because of poor nutrition, interactions with chemotherapy and antibiotics, and comorbid conditions.22 Furthermore, its onset of action can be delayed and its clearance may be prolonged, further increasing the risk of complications, especially in patients prone to developing chemotherapy-related anemia or thrombocytopenia.22 Bleeding risk is the highest in the first 3 months of therapy. In addition, the risk of bleeding is higher in older patients, women, and patients with a history of gastrointestinal bleeding, stroke, recent myocardial infarction, diabetes, renal insufficiency, malignancy, or anemia.23,24
ADVANTAGES AND DISADVANTAGES OF LMWH
The advantages of the LMWHs over unfractionated heparin include a lower risk of heparin-induced thrombocytopenia, greater bioavailability when given subcutaneously (which also permits once-daily or twice-daily dosing), and no need for laboratory monitoring in most patients. LMWHs have a short half-life, so omitting one or two doses will adequately interrupt therapy. Also, LMWHs have been shown to be as safe and effective as unfractionated heparin in treating VTE. They can be given safely at home, thus enhancing quality of life.25–31
On the other hand, these drugs cost more than unfractionated heparin or warfarin, their dosage must be adjusted in patients with renal insufficiency, their anticoagulant effect can be reversed only to a limited extent, and their dose must be adjusted according to weight in morbidly obese or in very thin patients.32,33
LMWHs are expensive, but may be worth it
As initial therapy, the LMWHs are cost-effective compared with unfractionated heparin in patients with VTE.34,35 However, they cost more with extended use. A cost-effectiveness analysis comparing 6 months of LMWH therapy to standard warfarin concluded that LMWH therapy was more costly.35 However, the impact of fewer hospitalizations, probably fewer bleeding complications, and better quality of life are difficult to analyze in this decision model and should also be considered when deciding about therapy for an individual patient.35
LMWHs are cleared by the kidney
All LMWHs are renally cleared, so patients with significant renal insufficiency (creatinine clearance < 30 mL/min) are at greater risk of bleeding complications. The rate below which clearance is impaired varies among the different LMWHs. Only enoxaparin has approved dosing regimens for use in patients with renal impairment.
If the patient has renal insufficiency, the ACCP guidelines suggest using unfractionated heparin, or if using LMWH, monitoring anti-factor Xa levels to avoid drug accumulation and increased bleeding risk.25 If bleeding occurs, LMWHs have limited reversibility with protamine sulfate, which is estimated to neutralize about 60% of the anti-factor Xa activity of LMWHs.25
Adjusting LMWHs for body weight
In the Registro Informatizado de la Enfermedad Tromboembólica (RIETE),33 patients weighing less than 50 kg had a higher risk of bleeding than patients weighing 50 to 100 kg, so in thinner patients the risk of bleeding from LMWH vs oral anticoagulation must be considered carefully and monitored prudently.
Although there is little evidence to suggest a higher bleeding risk in morbidly obese patients (> 150 kg), they may be at risk of subtherapeutic treatment, and monitoring with anti-factor Xa assays is recommended.25,32,33
LMWH VS WARFARIN FOR VTE IN CANCER PATIENTS
LMWHs are the first-line treatment for VTE in cancer patients.20,21 Several randomized controlled trials compared the efficacy of LMWH vs warfarin in patients with cancer.
Meyer et al36 randomized patients to receive either warfarin for 3 months at an INR between 2 and 3, or enoxaparin 1.5 mg/kg subcutaneously daily. Seventy-one patients received warfarin and 67 received enoxaparin. Fifteen (21%, 95% confidence interval [CI] 12%–32%) of the 71 patients assigned to warfarin experienced one major outcome event, defined as major bleeding or recurrent VTE, compared with 7 (10.5%) of the 67 patients assigned to receive enoxaparin (95% CI 4%–20%, P = .09). Six patients in the warfarin group died of bleeding vs none of the patients in the enoxaparin group. Overall, the warfarin group had a higher rate of bleeding, although this did not reach statistical significance. Despite weekly INR measurements, only 41% of the measured values were within the therapeutic range during the 3 months of treatment.36
Lee et al37 randomized cancer patients with deep venous thrombosis, pulmonary embolism, or both to receive 6 months of dalteparin alone, dosed at 200 IU/kg daily for 1 month, then decreased to 75% to 80% of the original dose (150 IU/kg) daily for the duration of therapy, or dalteparin followed by warfarin. During the 6-month follow-up, 17.4% of patients in the warfarin group had a recurrent thromboembolic event vs 8.8% in the dalteparin group (P = .0017). No statistically significant difference was noted in rates of major bleeding, minor bleeding, or death.37
Hull et al38 reported statistically significantly fewer episodes of recurrent VTE at 12 months in cancer patients treated with once-daily tinzaparin vs warfarin. In the tinzaparin group the recurrence rate was 7%, vs 16% in the warfarin group (P = .044). No difference in rates of bleeding or death were found.
Deitcher et al39 compared enoxaparin with long-term warfarin in 102 patients. While this trial did not have the power to detect clinical differences in recurrent thromboembolic events or bleeding complications, at 180 days they noted 97% compliance with once-daily or twice-daily enoxaparin therapy.
Noble and Finlay,40 in another small study, found LMWH therapy to be qualitatively more acceptable for palliative-care cancer patients than oral therapy.
In general, long-term therapy with once-daily or twice-daily LMWH is well tolerated. Currently, dalteparin is the only LMWH approved by the FDA for extended monotherapy in cancer-related VTE.
DO LMWHS AFFECT CANCER?
In vitro and animal studies indicate that LMWH may have antimetastatic and antiangiogenic properties.41–44
Altinbas et al45 reported significantly better chemotherapy-induced tumor response rates and survival rates in patients with small cell lung cancer randomized to receive combination chemotherapy plus prophylactic dalteparin 5,000 IU daily compared with combination chemotherapy alone. However, as provocative as these results may be, we need to test the effects of LWMHs on different cancer types in a prospective clinical trial. For now, this area remains controversial.
It has been suggested that anticoagulants may improve survival in patients with nonmetastatic cancer. Supporting this observation, a post hoc analysis of the trial by Lee et al37 found a statistically significantly lower cancer-specific mortality rate in nonmetastatic cancer patients treated with dalteparin vs oral therapy with a coumarin derivative. In patients without metastatic disease, the death rate at 12 months was 36% in patients treated with oral therapy vs 20% in patients treated with dalteparin (P = .03).46
These findings are consistent with those of the Fragmin Advanced Malignancy Outcome Study (FAMOUS),47 the first randomized, placebo-controlled trial of dalteparin 5,000 IU daily in patients with advanced solid tumors and without evidence of underlying thrombosis. Overall, dalteparin prophylaxis did not increase survival. However, in a subgroup of patients with a better prognosis and who were alive 17 months after diagnosis, survival was statistically significantly longer in patients treated with dalteparin.
Another small trial showed similar survival benefits in cancer patients without VTE.48 The results may suggest a long-term favorable effect of LMWH on tumor cell biology, which could translate into a favorable outcome in some patients. It is important to note, however, that not all trials have shown this same clinical benefit.49
In general, the growing body of laboratory and clinical data indicates that LMWHs may suppress tumor growth and metastasis. However, definitive conclusions about these effects are not yet possible because of variations in study design, tumor type, and patient populations. Further investigations into the role of LMWHs in the treatment of VTE and in cancer progression are ongoing.
THE EVIDENCE IN PERSPECTIVE
Illness and the recurrence of VTE in patients with cancer depend on the location and extent of the underlying cancer. Rates of death are higher in VTE patients with cancer than in VTE patients without cancer. Patients with limited or localized disease may not die of the cancer itself but of complications of acute pulmonary embolism. Therefore, it is important to recognize the different options for and the potential side effects of treating VTE.
If patients are hospitalized for an acute thromboembolic event and unfractionated heparin is chosen as the initial anticoagulant, using a weight-based nomogram has been shown to achieve therapeutic levels within 24 hours and reduce the rates of recurrence of thromboembolic events.50
Warfarin treatment may pose a particular challenge for both cancer patients and physicians, since multiple drug interactions, anorexia, and comorbid conditions contribute to an unpredictable response.
The risk of bleeding is higher in cancer patients than in the general population, and the decision to start anticoagulants should be based on an individualized risk-benefit profile. Several trials have shown LMWH to be more effective and safer than warfarin in cancer patients.
These considerations, along with the other advantages of LMWHs (ease of use, less need for laboratory monitoring, and better patient tolerance), make LMWHs a good choice for initial therapy. Extended LMWH therapy is currently favored for initial management in patients with cancer. Trials are under way to further assess the antitumor properties and potential survival benefit in patients with selected solid tumors.
Venous thromboembolism (VTE) has various differing causes, so its treatment is not necessarily the same in all cases. Most cases of VTE are related to an easily identified risk factor. In patients with an apparently idiopathic event, identifying an underlying cause may alter therapy. In particular, identification of a malignancy may affect the choice of therapy and the duration of treatment.
In this review, we explore the role of cancer screening in patients with idiopathic VTE, then highlight the treatment for VTE in patients with cancer.
‘IDIOPATHIC’ VTE CAN BE DUE TO CANCER
Most patients with venous thrombosis have one of the components of Virchow’s triad: a hypercoagulable state, venous injury, or venous stasis. Those without identifiable risk factors for VTE are considered to have idiopathic VTE. In these patients, a search for a contributing factor may be indicated.
In 1861, the astute clinician Dr. Armand Trousseau noted a link between deep venous thrombosis and pancreatic cancer, stating that if cancer of an internal organ is suspected but the diagnosis cannot be verified, the diagnosis may be confirmed by the sudden, spontaneous appearance of thrombophlebitis in a large vein.1
Today, from 2% to 25% of patients with idiopathic VTE are found to have cancer within 24 months of the diagnosis of VTE.2–11 The goals of cancer screening in idiopathic VTE are to detect cancer at an early, treatable stage and to optimize the VTE therapy to decrease the risks of recurrence and anticoagulation-associated complications in patients who are found to have cancer. However, several questions must be considered first:
- What are the risks and costs of the screening?
- Will discovering the cancer sooner benefit the patient in terms of survival?
- If cancer is found, what are the possible complications or risks of the additional procedures, interventions, or treatments required?
- What is the psychological impact of the screening?
EVIDENCE SUPPORTING CANCER SCREENING AFTER IDIOPATHIC VTE
Piccioli et al12 recently performed a randomized, controlled trial comparing cancer-related death rates in 99 patients with idiopathic VTE screened for malignancy vs 102 patients with idiopathic VTE who were not screened.
The screened group underwent:
- Abdominal and pelvic ultrasonography and computed tomography (CT)
- Gastroscopy or double-contrast barium-swallow evaluation
- Colonoscopy or sigmoidoscopy followed by barium enema
- Testing for fecal occult blood
- Sputum cytology
- Measurement of carcinoembryonic antigen, alpha-fetoprotein, and cancer antigen 125.
- Mammography and Papanicolaou smears (women)
- Ultrasonography of the prostate and prostate-specific antigen testing (men).
Patients were followed for 2 years. The screening uncovered cancer in 13 patients. Cancer developed in one other patient in the screening group during follow-up; in the control group, 10 patients developed symptomatic cancer during follow-up. Overall, the time to cancer diagnosis was 11.6 months in the unscreened group vs 1 month in the screened group (P < .001). Nine of the 14 patients with cancer in the screened group had T1 or T2 disease without local or distant metastasis vs 2 of the 10 control patients with cancer (P = .047). Unfortunately, this study did not have adequate power to detect the effect of screening on survival.
Di Nisio et al13 used data from this trial to perform a decision analysis for cancer screening. They calculated that abdominal and pelvic CT, with or without mammography and with or without sputum cytologic testing, would cost the least per life-year gained and would harm the fewest number of patients. They also suggested that substituting CT of the chest for sputum cytology may provide additional diagnostic benefit.
However, this strategy has not been clinically tested. Given the limited number of patients and the short follow-up in this initial trial, larger trials are needed to look at the cost-effectiveness of this screening model and whether it increases survival.
Our recommendations
Because the data are limited, our approach to looking for an early, treatable malignancy in patients with idiopathic VTE follows the current consensus:
- A thorough history and physical, including an extensive review of systems
- Basic laboratory testing with a complete blood cell count, comprehensive metabolic profile, and urinalysis
- Chest radiography
- Other age- and sex-specific cancer screening tests.
Adding CT of the abdomen, pelvis, or chest to this evaluation may be considered. However, tumor marker testing, which typically has high false-positive rates, is not routinely warranted.13 Additional investigation should be considered if abnormalities are detected during the initial evaluation or in patients with recurrent VTE during therapy.
While this strategy may be most cost-effective, Monreal et al14 suggest that it may miss up to half of cancers ultimately discovered.
MANAGING VTE IN PATIENTS WITH KNOWN CANCER
Managing VTE is far more complex in cancer patients than in patients without cancer. Also, cancer patients with VTE have lower rates of survival than cancer patients without VTE and are at greater risk of adverse outcomes such as anticoagulant-associated bleeding and recurrent venous thrombotic events.15–17
Up to 21.5% of patients with VTE have another event within 5 years,18 but the risk is two to three times higher if they also have cancer.16,18 The risk of recurrence may be linked to the location of the thrombus and to the extent of the malignancy.
In one study, the 3-month rate of recurrence was up to 5.1% if the clot was in the popliteal vein, 5.3% if in the femoral vein, and 11.8% if in the iliac vein.19
Prandoni et al16 found that the risks of VTE recurrence and bleeding were higher in patients with extensive cancer than in those with less-extensive cancer. In this study, major bleeding was documented in 12.4% of patients with cancer vs 4.9% of patients without cancer. Compared with patients without cancer, the hazard ratio for a major bleeding event was 4.8 in patients with extensive cancer and 0.5 in patients with less-extensive cancer.
In addition, not all patients with bleeding had excessive levels of anticoagulation, and not all patients with recurrent events had subtherapeutic levels.16,17 Therefore, treatment of venous thrombosis in cancer patients requires a careful, individualized risk-to-benefit decision analysis.
ACUTE THERAPY FOR VTE: PARENTERAL AGENTS
Treatment in the first several hours or days after a thromboembolic event is with short-acting parenteral agents: unfractionated heparin; one of the low-molecular-weight heparins (LMWHs), ie, dalteparin (Fragmin), enoxaparin (Lovenox), or tinzaparin (Innohep); or fondaparinux (Arixtra).
Before starting anticoagulation, consider:
- Does the patient have severe chronic kidney disease (ie, a creatinine clearance < 30 mL/min)? If so, unfractionated heparin may be better than an LMWH or fondaparinux, which are cleared by the kidney.
- Does he or she need inpatient care? If not, LMWH therapy at home may be appropriate.
- Are there concerns about the ease of anticoagulation administration (ie, whether the patient can give the injections or have a family member do it), the cost of the drugs, or the ability to reverse the anticoagulant effect, if necessary? If so, unfractionated heparin may be more appropriate.
For acute treatment, the 2008 guidelines of the American College of Chest Physicians20 (ACCP) recommend using an LMWH in a weight-based dose; unfractionated heparin given intravenously; unfractionated heparin given subcutaneously with monitoring and dosing adjustments; unfractionated heparin given subcutaneously at a fixed dose; or fondaparinux (grade 1A recommendation). The 2007 National Comprehensive Cancer Network (NCCN) guidelines21 recommend an LMWH, fondaparinux, or unfractionated heparin. Treatment should start promptly after the diagnosis of VTE is confirmed. However, if VTE is strongly suspected and a delay in diagnostic testing is anticipated, therapy should be started while awaiting the test results.
LONG-TERM THERAPY: LMWH OR WARFARIN
The ACCP and the NCCN guidelines recommend LMWH monotherapy for extended treatment of VTE in patients with active malignancy, when appropriate.20,21 However, if long-term LMWH is not appropriate, then oral anticoagulation with a vitamin K antagonist, such as the coumarin derivative warfarin (Coumadin), is an alternative and should be started on the same day as the heparin. The heparin and the warfarin therapy must overlap for a minimum of 4 or 5 days and until a stable, therapeutic level of anticoagulation is achieved, ie, an international normalized ratio (INR) of 2 to 3 for 2 consecutive days.20
The duration of anticoagulant therapy depends on comorbidities and the patient’s underlying predisposition for VTE. In patients with limited disease, the guidelines recommend continuing anticoagulation for a minimum of 3 to 6 months for deep venous thrombosis and pulmonary embolism.20–21 Patients with active malignancy, ongoing treatment for the cancer, or continued risk factors may need indefinite treatment. In some circumstances, such as catheter-associated deep venous thrombosis, anticoagulation should continue for as long as the catheter is in place and for 1 to 3 months after its removal.21
WARFARIN CAN BE DIFFICULT TO USE
In 1954, the US Food and Drug Administration (FDA) approved the vitamin K antagonist warfarin for medical use in humans. Experience has shown it to be effective in preventing and treating VTE. However, it can be somewhat difficult to use, for several reasons:
- A narrow therapeutic window
- Genetic polymorphisms and variability in dose response
- Drug interactions and dietary considerations
- The need for laboratory monitoring and dose adjustment
- Patient noncompliance or miscommunication between the patient and physician.22
In cancer patients, the response to warfarin may be unpredictable because of poor nutrition, interactions with chemotherapy and antibiotics, and comorbid conditions.22 Furthermore, its onset of action can be delayed and its clearance may be prolonged, further increasing the risk of complications, especially in patients prone to developing chemotherapy-related anemia or thrombocytopenia.22 Bleeding risk is the highest in the first 3 months of therapy. In addition, the risk of bleeding is higher in older patients, women, and patients with a history of gastrointestinal bleeding, stroke, recent myocardial infarction, diabetes, renal insufficiency, malignancy, or anemia.23,24
ADVANTAGES AND DISADVANTAGES OF LMWH
The advantages of the LMWHs over unfractionated heparin include a lower risk of heparin-induced thrombocytopenia, greater bioavailability when given subcutaneously (which also permits once-daily or twice-daily dosing), and no need for laboratory monitoring in most patients. LMWHs have a short half-life, so omitting one or two doses will adequately interrupt therapy. Also, LMWHs have been shown to be as safe and effective as unfractionated heparin in treating VTE. They can be given safely at home, thus enhancing quality of life.25–31
On the other hand, these drugs cost more than unfractionated heparin or warfarin, their dosage must be adjusted in patients with renal insufficiency, their anticoagulant effect can be reversed only to a limited extent, and their dose must be adjusted according to weight in morbidly obese or in very thin patients.32,33
LMWHs are expensive, but may be worth it
As initial therapy, the LMWHs are cost-effective compared with unfractionated heparin in patients with VTE.34,35 However, they cost more with extended use. A cost-effectiveness analysis comparing 6 months of LMWH therapy to standard warfarin concluded that LMWH therapy was more costly.35 However, the impact of fewer hospitalizations, probably fewer bleeding complications, and better quality of life are difficult to analyze in this decision model and should also be considered when deciding about therapy for an individual patient.35
LMWHs are cleared by the kidney
All LMWHs are renally cleared, so patients with significant renal insufficiency (creatinine clearance < 30 mL/min) are at greater risk of bleeding complications. The rate below which clearance is impaired varies among the different LMWHs. Only enoxaparin has approved dosing regimens for use in patients with renal impairment.
If the patient has renal insufficiency, the ACCP guidelines suggest using unfractionated heparin, or if using LMWH, monitoring anti-factor Xa levels to avoid drug accumulation and increased bleeding risk.25 If bleeding occurs, LMWHs have limited reversibility with protamine sulfate, which is estimated to neutralize about 60% of the anti-factor Xa activity of LMWHs.25
Adjusting LMWHs for body weight
In the Registro Informatizado de la Enfermedad Tromboembólica (RIETE),33 patients weighing less than 50 kg had a higher risk of bleeding than patients weighing 50 to 100 kg, so in thinner patients the risk of bleeding from LMWH vs oral anticoagulation must be considered carefully and monitored prudently.
Although there is little evidence to suggest a higher bleeding risk in morbidly obese patients (> 150 kg), they may be at risk of subtherapeutic treatment, and monitoring with anti-factor Xa assays is recommended.25,32,33
LMWH VS WARFARIN FOR VTE IN CANCER PATIENTS
LMWHs are the first-line treatment for VTE in cancer patients.20,21 Several randomized controlled trials compared the efficacy of LMWH vs warfarin in patients with cancer.
Meyer et al36 randomized patients to receive either warfarin for 3 months at an INR between 2 and 3, or enoxaparin 1.5 mg/kg subcutaneously daily. Seventy-one patients received warfarin and 67 received enoxaparin. Fifteen (21%, 95% confidence interval [CI] 12%–32%) of the 71 patients assigned to warfarin experienced one major outcome event, defined as major bleeding or recurrent VTE, compared with 7 (10.5%) of the 67 patients assigned to receive enoxaparin (95% CI 4%–20%, P = .09). Six patients in the warfarin group died of bleeding vs none of the patients in the enoxaparin group. Overall, the warfarin group had a higher rate of bleeding, although this did not reach statistical significance. Despite weekly INR measurements, only 41% of the measured values were within the therapeutic range during the 3 months of treatment.36
Lee et al37 randomized cancer patients with deep venous thrombosis, pulmonary embolism, or both to receive 6 months of dalteparin alone, dosed at 200 IU/kg daily for 1 month, then decreased to 75% to 80% of the original dose (150 IU/kg) daily for the duration of therapy, or dalteparin followed by warfarin. During the 6-month follow-up, 17.4% of patients in the warfarin group had a recurrent thromboembolic event vs 8.8% in the dalteparin group (P = .0017). No statistically significant difference was noted in rates of major bleeding, minor bleeding, or death.37
Hull et al38 reported statistically significantly fewer episodes of recurrent VTE at 12 months in cancer patients treated with once-daily tinzaparin vs warfarin. In the tinzaparin group the recurrence rate was 7%, vs 16% in the warfarin group (P = .044). No difference in rates of bleeding or death were found.
Deitcher et al39 compared enoxaparin with long-term warfarin in 102 patients. While this trial did not have the power to detect clinical differences in recurrent thromboembolic events or bleeding complications, at 180 days they noted 97% compliance with once-daily or twice-daily enoxaparin therapy.
Noble and Finlay,40 in another small study, found LMWH therapy to be qualitatively more acceptable for palliative-care cancer patients than oral therapy.
In general, long-term therapy with once-daily or twice-daily LMWH is well tolerated. Currently, dalteparin is the only LMWH approved by the FDA for extended monotherapy in cancer-related VTE.
DO LMWHS AFFECT CANCER?
In vitro and animal studies indicate that LMWH may have antimetastatic and antiangiogenic properties.41–44
Altinbas et al45 reported significantly better chemotherapy-induced tumor response rates and survival rates in patients with small cell lung cancer randomized to receive combination chemotherapy plus prophylactic dalteparin 5,000 IU daily compared with combination chemotherapy alone. However, as provocative as these results may be, we need to test the effects of LWMHs on different cancer types in a prospective clinical trial. For now, this area remains controversial.
It has been suggested that anticoagulants may improve survival in patients with nonmetastatic cancer. Supporting this observation, a post hoc analysis of the trial by Lee et al37 found a statistically significantly lower cancer-specific mortality rate in nonmetastatic cancer patients treated with dalteparin vs oral therapy with a coumarin derivative. In patients without metastatic disease, the death rate at 12 months was 36% in patients treated with oral therapy vs 20% in patients treated with dalteparin (P = .03).46
These findings are consistent with those of the Fragmin Advanced Malignancy Outcome Study (FAMOUS),47 the first randomized, placebo-controlled trial of dalteparin 5,000 IU daily in patients with advanced solid tumors and without evidence of underlying thrombosis. Overall, dalteparin prophylaxis did not increase survival. However, in a subgroup of patients with a better prognosis and who were alive 17 months after diagnosis, survival was statistically significantly longer in patients treated with dalteparin.
Another small trial showed similar survival benefits in cancer patients without VTE.48 The results may suggest a long-term favorable effect of LMWH on tumor cell biology, which could translate into a favorable outcome in some patients. It is important to note, however, that not all trials have shown this same clinical benefit.49
In general, the growing body of laboratory and clinical data indicates that LMWHs may suppress tumor growth and metastasis. However, definitive conclusions about these effects are not yet possible because of variations in study design, tumor type, and patient populations. Further investigations into the role of LMWHs in the treatment of VTE and in cancer progression are ongoing.
THE EVIDENCE IN PERSPECTIVE
Illness and the recurrence of VTE in patients with cancer depend on the location and extent of the underlying cancer. Rates of death are higher in VTE patients with cancer than in VTE patients without cancer. Patients with limited or localized disease may not die of the cancer itself but of complications of acute pulmonary embolism. Therefore, it is important to recognize the different options for and the potential side effects of treating VTE.
If patients are hospitalized for an acute thromboembolic event and unfractionated heparin is chosen as the initial anticoagulant, using a weight-based nomogram has been shown to achieve therapeutic levels within 24 hours and reduce the rates of recurrence of thromboembolic events.50
Warfarin treatment may pose a particular challenge for both cancer patients and physicians, since multiple drug interactions, anorexia, and comorbid conditions contribute to an unpredictable response.
The risk of bleeding is higher in cancer patients than in the general population, and the decision to start anticoagulants should be based on an individualized risk-benefit profile. Several trials have shown LMWH to be more effective and safer than warfarin in cancer patients.
These considerations, along with the other advantages of LMWHs (ease of use, less need for laboratory monitoring, and better patient tolerance), make LMWHs a good choice for initial therapy. Extended LMWH therapy is currently favored for initial management in patients with cancer. Trials are under way to further assess the antitumor properties and potential survival benefit in patients with selected solid tumors.
- Aron E. The 100th anniversary of the death of A. Trousseau. Presse Med 1967; 75:1429–1430.
- Hettiarachchi RJ, Lok J, Prins MH, Büller HR, Prandoni P. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998; 83:180–185.
- Baron JA, Gridley G, Weiderpass E, Nyrén O, Linet M. Venous thromboembolism and cancer. Lancet 1998; 351:1077–1080.
- Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 2000; 342:1953–1958.
- Sørensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998; 338:1169–1173.
- Monreal M, Lafoz E, Casals A, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer 1991; 67:541–545.
- Nordström M, Lindblad B, Anderson H, Bergqvist D, Kjellström T. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ 1994; 308:891–894.
- Prandoni P, Lensing AW, Büller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327:1128–1133.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Fennerty T. Screening for cancer in venous thromboembolic disease. BMJ 2001; 323:704–705.
- Bastounis EA, Karayiannakis AJ, Makri GG, Alexiou D, Papalambros EL. The incidence of occult cancer in patients with deep venous thrombosis: a prospective study. J Intern Med 1996; 239:153–156.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Di Nisio M, Otten HM, Piccioli A, et al. Decision analysis for cancer screening in idiopathic venous thromboembolism. J Thromb Haemost 2005; 3:2391–2396.
- Monreal M, Lensing AW, Prins MH, et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost 2004; 2:876–881.
- Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343:1846–1850.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hutten BA, Prins MH, Gent M, Ginsberg J, Tijssen JG, Büller HR. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078–3083.
- Hansson PO, Sörbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000; 160:769–774.
- Douketis JD, Crowther MA, Foster GA, Ginsberg JS. Does the location of thrombosis determine the risk of disease recurrence in patients with proximal deep vein thrombosis? Am J Med 2001; 110:515–519.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-based Clinical Practice Guidelines, 8th Edition. Chest 2008; 133 suppl 6:454S–545S.
- National Comprehensive Cancer Network. Venous Thromboembolic Disease Clinical Practice Guidelines in Oncology (V.1.2007). Available at www.nccn.org/professionals/physician_gls/PDF/vte.pdf. Accessed 01/02/2008.
- Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:204S–233S.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med 1999; 159:457–460.
- Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest 2004; 126 suppl 3:188S–203S.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Hettiarachchi RJ, Prins MH, Lensing AW, Büller HR. Low molecular weight heparin versus unfractionated heparin in the initial treatment of venous thromboembolism. Curr Opin Pulm Med 1998; 4:220–225.
- Gould MK, Dembitzer AD, Doyle RL, Hastie TJ, Garber AM. Low-molecular-weight heparins compared with unfractionated heparin for treatment of acute deep venous thrombosis. A meta-analysis of randomized, controlled trials. Ann Intern Med 1999; 130:800–809.
- Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with un-fractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med 2000; 160:181–188.
- van Dongen CJ, van den Belt AG, Prins MH, Lensing AW. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2004; 4:CD001100.
- Cook LM, Kahn SR, Goodwin J, Kovacs MJ. Frequency of renal impairment, advanced age, obesity, and cancer in venous thromboembolism patients in clinical practice. J Thromb Haemost 2007; 5:937–941.
- Barba R, Marco J, Martin-Alvarez H, et al. The influence of extreme body weight on clinical outcome of patients with venous thromboembolism: findings from a prospective registry (RIETE). J Thromb Haemost 2005; 3:856–862.
- Segal JB, Strieff MB, Hofmann LV, Thornton K, Bass EB. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007; 146:211–222.
- Aujesky D, Smith KJ, Cornuz J, Roberts MS. Cost-effectiveness of low-molecular-weight heparin for secondary prophylaxis of cancer-related venous thromboembolism. Thromb Haemost 2005; 93:592–599.
- Meyer G, Marjanovic Z, Valcke J, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162:1729–1735.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J ON-CENOX investigators. Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180-day period. Clin Appl Thromb Hemost 2006; 12:389–396.
- Noble SI, Finlay IG. Is long-term low-molecular-weight heparin acceptable to palliative care patients in the treatment of cancer related venous thromboembolism? A qualitative study. Palliat Med 2005; 19:197–201.
- Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastatic effect of tinzaparin, a low-molecular-weight heparin. J Thromb Haemost 2003; 1:1972–1976.
- Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep 2005; 14:99–104.
- Mousa SA, Mohamed S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: anti-cancer efficacy. Oncol Rep 2004; 12:683–688.
- Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004; 58:213–219.
- Altinbas M, Coskun HS, Er O, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost 2004; 2:1266–1271.
- Lee AY, Rickles FR, Julian JA, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23:2123–2129.
- Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the Fragmin Advanced Malignancy Outcome Study (FAMOUS). J Clin Oncol 2004; 22:1944–1948.
- Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23:2130–2135.
- Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a phase 3 clinical trial. Mayo Clin Proc 2006; 81:758–767.
- Bernardi E, Piccioli A, Oliboni G, Zuin R, Girolami A, Prandoni P. Nomograms for the administration of unfractionated heparin in the initial treatment of acute thromboembolism—an overview. Thromb Haemost 2000; 84:22–26.
- Aron E. The 100th anniversary of the death of A. Trousseau. Presse Med 1967; 75:1429–1430.
- Hettiarachchi RJ, Lok J, Prins MH, Büller HR, Prandoni P. Undiagnosed malignancy in patients with deep vein thrombosis: incidence, risk indicators, and diagnosis. Cancer 1998; 83:180–185.
- Baron JA, Gridley G, Weiderpass E, Nyrén O, Linet M. Venous thromboembolism and cancer. Lancet 1998; 351:1077–1080.
- Schulman S, Lindmarker P. Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 2000; 342:1953–1958.
- Sørensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL. The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 1998; 338:1169–1173.
- Monreal M, Lafoz E, Casals A, et al. Occult cancer in patients with deep venous thrombosis. A systematic approach. Cancer 1991; 67:541–545.
- Nordström M, Lindblad B, Anderson H, Bergqvist D, Kjellström T. Deep venous thrombosis and occult malignancy: an epidemiological study. BMJ 1994; 308:891–894.
- Prandoni P, Lensing AW, Büller HR, et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327:1128–1133.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Fennerty T. Screening for cancer in venous thromboembolic disease. BMJ 2001; 323:704–705.
- Bastounis EA, Karayiannakis AJ, Makri GG, Alexiou D, Papalambros EL. The incidence of occult cancer in patients with deep venous thrombosis: a prospective study. J Intern Med 1996; 239:153–156.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Di Nisio M, Otten HM, Piccioli A, et al. Decision analysis for cancer screening in idiopathic venous thromboembolism. J Thromb Haemost 2005; 3:2391–2396.
- Monreal M, Lensing AW, Prins MH, et al. Screening for occult cancer in patients with acute deep vein thrombosis or pulmonary embolism. J Thromb Haemost 2004; 2:876–881.
- Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343:1846–1850.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hutten BA, Prins MH, Gent M, Ginsberg J, Tijssen JG, Büller HR. Incidence of recurrent thromboembolic and bleeding complications among patients with venous thromboembolism in relation to both malignancy and achieved international normalized ratio: a retrospective analysis. J Clin Oncol 2000; 18:3078–3083.
- Hansson PO, Sörbo J, Eriksson H. Recurrent venous thromboembolism after deep vein thrombosis: incidence and risk factors. Arch Intern Med 2000; 160:769–774.
- Douketis JD, Crowther MA, Foster GA, Ginsberg JS. Does the location of thrombosis determine the risk of disease recurrence in patients with proximal deep vein thrombosis? Am J Med 2001; 110:515–519.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-based Clinical Practice Guidelines, 8th Edition. Chest 2008; 133 suppl 6:454S–545S.
- National Comprehensive Cancer Network. Venous Thromboembolic Disease Clinical Practice Guidelines in Oncology (V.1.2007). Available at www.nccn.org/professionals/physician_gls/PDF/vte.pdf. Accessed 01/02/2008.
- Ansell J, Hirsh J, Poller L, Bussey H, Jacobson A, Hylek E. The pharmacology and management of the vitamin K antagonists: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:204S–233S.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med 1999; 159:457–460.
- Hirsh J, Raschke R. Heparin and low-molecular-weight heparin: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest 2004; 126 suppl 3:188S–203S.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Hettiarachchi RJ, Prins MH, Lensing AW, Büller HR. Low molecular weight heparin versus unfractionated heparin in the initial treatment of venous thromboembolism. Curr Opin Pulm Med 1998; 4:220–225.
- Gould MK, Dembitzer AD, Doyle RL, Hastie TJ, Garber AM. Low-molecular-weight heparins compared with unfractionated heparin for treatment of acute deep venous thrombosis. A meta-analysis of randomized, controlled trials. Ann Intern Med 1999; 130:800–809.
- Dolovich LR, Ginsberg JS, Douketis JD, Holbrook AM, Cheah G. A meta-analysis comparing low-molecular-weight heparins with un-fractionated heparin in the treatment of venous thromboembolism: examining some unanswered questions regarding location of treatment, product type, and dosing frequency. Arch Intern Med 2000; 160:181–188.
- van Dongen CJ, van den Belt AG, Prins MH, Lensing AW. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev 2004; 4:CD001100.
- Cook LM, Kahn SR, Goodwin J, Kovacs MJ. Frequency of renal impairment, advanced age, obesity, and cancer in venous thromboembolism patients in clinical practice. J Thromb Haemost 2007; 5:937–941.
- Barba R, Marco J, Martin-Alvarez H, et al. The influence of extreme body weight on clinical outcome of patients with venous thromboembolism: findings from a prospective registry (RIETE). J Thromb Haemost 2005; 3:856–862.
- Segal JB, Strieff MB, Hofmann LV, Thornton K, Bass EB. Management of venous thromboembolism: a systematic review for a practice guideline. Ann Intern Med 2007; 146:211–222.
- Aujesky D, Smith KJ, Cornuz J, Roberts MS. Cost-effectiveness of low-molecular-weight heparin for secondary prophylaxis of cancer-related venous thromboembolism. Thromb Haemost 2005; 93:592–599.
- Meyer G, Marjanovic Z, Valcke J, et al. Comparison of low-molecular-weight heparin and warfarin for the secondary prevention of venous thromboembolism in patients with cancer: a randomized controlled study. Arch Intern Med 2002; 162:1729–1735.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Deitcher SR, Kessler CM, Merli G, Rigas JR, Lyons RM, Fareed J ON-CENOX investigators. Secondary prevention of venous thromboembolic events in patients with active cancer: enoxaparin alone versus initial enoxaparin followed by warfarin for a 180-day period. Clin Appl Thromb Hemost 2006; 12:389–396.
- Noble SI, Finlay IG. Is long-term low-molecular-weight heparin acceptable to palliative care patients in the treatment of cancer related venous thromboembolism? A qualitative study. Palliat Med 2005; 19:197–201.
- Amirkhosravi A, Mousa SA, Amaya M, Francis JL. Antimetastatic effect of tinzaparin, a low-molecular-weight heparin. J Thromb Haemost 2003; 1:1972–1976.
- Kragh M, Binderup L, Vig Hjarnaa PJ, Bramm E, Johansen KB, Frimundt Petersen C. Non-anti-coagulant heparin inhibits metastasis but not primary tumor growth. Oncol Rep 2005; 14:99–104.
- Mousa SA, Mohamed S. Anti-angiogenic mechanisms and efficacy of the low molecular weight heparin, tinzaparin: anti-cancer efficacy. Oncol Rep 2004; 12:683–688.
- Bobek V, Kovarik J. Antitumor and antimetastatic effect of warfarin and heparins. Biomed Pharmacother 2004; 58:213–219.
- Altinbas M, Coskun HS, Er O, et al. A randomized clinical trial of combination chemotherapy with and without low-molecular-weight heparin in small cell lung cancer. J Thromb Haemost 2004; 2:1266–1271.
- Lee AY, Rickles FR, Julian JA, et al. Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol 2005; 23:2123–2129.
- Kakkar AK, Levine MN, Kadziola Z, et al. Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the Fragmin Advanced Malignancy Outcome Study (FAMOUS). J Clin Oncol 2004; 22:1944–1948.
- Klerk CP, Smorenburg SM, Otten HM, et al. The effect of low molecular weight heparin on survival in patients with advanced malignancy. J Clin Oncol 2005; 23:2130–2135.
- Sideras K, Schaefer PL, Okuno SH, et al. Low-molecular-weight heparin in patients with advanced cancer: a phase 3 clinical trial. Mayo Clin Proc 2006; 81:758–767.
- Bernardi E, Piccioli A, Oliboni G, Zuin R, Girolami A, Prandoni P. Nomograms for the administration of unfractionated heparin in the initial treatment of acute thromboembolism—an overview. Thromb Haemost 2000; 84:22–26.
KEY POINTS
- We recommend judiciously screening for cancer with age- and sex-specific tests in patients with idiopathic VTE.
- Patients with VTE and cancer have a higher risk of both VTE recurrence and bleeding complications of anticoagulant therapy than do VTE patients without cancer.
- Either unfractionated heparin or a low-molecular-weight heparin (LMWH) should be started as soon as VTE is confirmed or even strongly suspected, while still awaiting confirmation.
- The current (grade 1A) recommendations for treating VTE in cancer patients are to use LMWH monotherapy for at least 3 to 6 months. Anticoagulation is necessary indefinitely when there is ongoing cancer treatment or persistent risk of VTE.
Anticoagulants and pregnancy: When are they safe?
Anticoagulation is essential in a wide variety of conditions in women of child-bearing age. Some, such as venous thromboembolism, occur more often during pregnancy. Others, such as recurrent fetal loss in the setting of antiphospholipid antibodies, are specific to pregnancy.
While anticoagulants are useful in many circumstances, their use during pregnancy increases the risk of hemorrhage and other adverse effects on the mother and the fetus. Treatment with anticoagulants during pregnancy must therefore be carefully considered, with judicious selection of the agent, and with reflection on the physiologic changes of pregnancy to ensure appropriate dosing. In this article, we review these issues.
WHY IS THROMBOTIC RISK HIGHER DURING PREGNANCY?
Venous thromboembolism is among the leading causes of maternal death in developed countries.1–3 Modern care has dramatically reduced the risk of maternal death from hemorrhage, infection, and hypertension, but rates of morbidity and death from thrombosis have remained stable or increased in recent years.4

- Much higher levels of fibrinogen and factors VII, VIII, IX, and X
- Lower levels of protein S and increased resistance to activated protein C
- Impaired fibrinolysis, due to inhibitors derived from the placenta.
Acquired antithrombin deficiency may also occur in high-proteinuric states such as nephrotic syndrome or preeclampsia, further increasing thrombotic risk. Pooling of venous blood, caused by progesterone-mediated venous dilation and compounded by compression of the inferior vena cava by the uterus in later pregnancy, also increases thrombotic risk. And endothelial disruption of the pelvic vessels may occur during delivery, particularly during cesarean section.
Additional factors that increase thrombotic risk include immobilization, such as bed rest for pregnancy complications; surgery, including cesarean section; ovarian hyperstimulation during gonadotropin use for in vitro fertilization; trauma; malignancy; and hereditary or acquired hypercoagulable states.6 These hypercoagulable states include deficiencies of antithrombin or the intrinsic anticoagulant proteins C or S; resistance to activated protein C, usually due to the factor V Leiden mutation; the PT20210A mutation of the prothrombin gene; hyperhomocystinemia due to mutation of the methyltetrahydrofolate reductase (MTHFR) gene; and the sustained presence of antiphospholipid antibodies, including lupus anticoagulant antibodies, sometimes also with moderately high titers of anticardiolipin or beta-2-glycoprotein I antibodies.
Other conditions that increase thrombotic risk include hyperemesis gravidarum, obesity, inflammatory bowel disease, infection, smoking, and indwelling intravenous catheters.6 Given the multitude of risk factors, pregnant women have a risk of thrombotic complications three to five times higher than nonpregnant women.7
HEPARIN USE DURING PREGNANCY
Low-molecular-weight heparins (LMWHs)8 and unfractionated heparin bind to anti-thrombin and thus change the shape of the antithrombin molecule, dramatically increasing its interaction with the clotting factors Xa and prothrombin (factor II). The enhanced clearance of these procoagulant proteins leads to the anticoagulant effect. Unfractionated heparin has roughly equivalent interaction with factors Xa and II and prolongs the activated partial thromboplastin time (aPTT), which is therefore used to monitor the intensity of anticoagulation.
LMWHs, on the other hand, interact relatively little with factor II and do not predictably prolong the aPTT. Monitoring their effect is therefore more difficult and requires direct measurement of anti-factor-Xa activity. This test is widely available, but it is time-consuming (it takes several hours and results may not be available within 24 hours if the test is requested “after hours”), and therefore it is of limited use in the acute clinical setting. While weight-based dosing of LMWHs is reliable and safe in nonpregnant patients, it has not yet been validated for pregnant women.
Unfractionated heparin has been used for decades for many indications during pregnancy. It is a large molecule, so it does not cross the placenta and thus, in contrast to the coumarin derivatives, does not cause teratogenesis or toxic fetal effects. Its main limitations in pregnancy are its inconvenient dosing (at least twice daily when given subcutaneously) and its potential maternal adverse effects (mainly osteoporosis and heparin-induced thrombocytopenia).
Over the last 10 years LMWHs have become the preferred anticoagulants for treating and preventing thromboembolism in all patients. They are equivalent or superior to unfractionated heparin in efficacy and safety in the initial treatment of acute deep venous thrombosis9,10 and pulmonary embolism11,12 outside of pregnancy. While comparative data are much less robust in pregnant patients, several series have confirmed the safety and efficacy of LMWHs in pregnancy.13–15 LMWHs do not cross the placenta15–17 and thus have a fetal safety profile equivalent to that of unfractionated heparin.
Pregnancy alters metabolism of LMWHs
The physiologic changes of pregnancy alter the metabolism of LMWH, resulting in lower peak levels and a higher rate of clearance,18,19 and so a pregnant woman may need higher doses or more frequent dosing.
Recent evidence suggests that thromboprophylaxis can be done with lower, fixed, once-daily doses of LMWH throughout pregnancy,20 although some clinicians still prefer twice-daily dosing (particularly during the latter half of pregnancy).
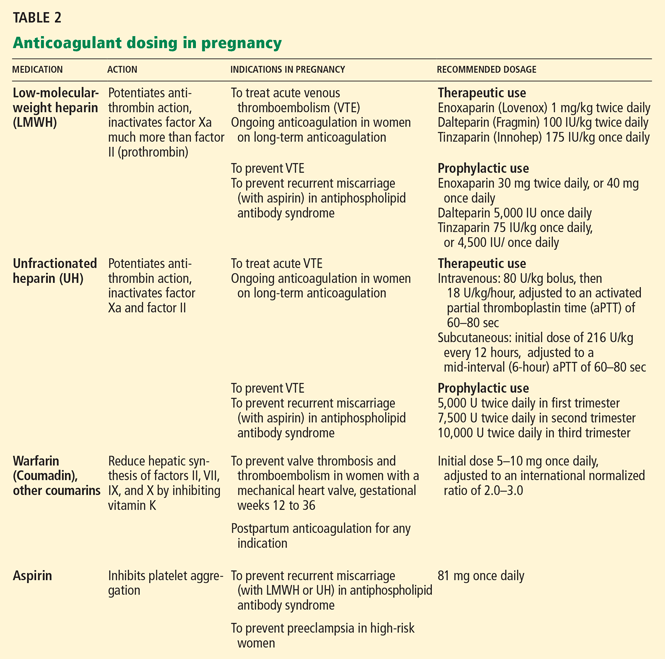
Pending more research on weight-based dosing of LMWH in pregnancy, anti-factor- Xa activity levels should be measured after treatment is started and every 1 to 3 months thereafter during pregnancy.21 Doses should be adjusted to keep the peak anti-Xa level (ie, 4 hours after the dose) at 0.5 to 1.2 U/mL.22
Heparin-induced thrombocytopenia
Type-2 heparin-induced thrombocytopenia is an uncommon but serious adverse effect of unfractionated heparin therapy (and, less commonly of LMWH), caused by heparin-dependent immunoglobulin G (IgG) antibodies that activate platelets via their Fc receptors, potentially precipitating life-threatening arterial or venous thrombosis.
In a trial in nonpregnant orthopedic patients,23 clinical heparin-induced thrombocytopenia occurred in 2.7% of patients receiving unfractionated heparin vs 0% of those receiving LMWH; heparin-dependent IgG was present in 7.8% vs 2.2%, respectively.
Fortunately, heparin-induced thrombocytopenia seems to be very rare in pregnancy: two recent prospective series evaluating prolonged LMWH use in pregnancy13,15 revealed no episodes of this disease. Nonetheless, it is reasonable to measure the platelet count once or twice weekly during the first few weeks of LMWH use and less often thereafter, unless symptoms of heparin-induced thrombocytopenia develop. In pregnant women with heparin-induced thrombocytopenia or heparin-related skin reactions, other anticoagulants must be considered24 (see discussion later).
Heparin-induced osteoporosis
Heparin-induced osteoporosis, a potential effect of prolonged heparin therapy, is of concern, given the prolonged duration and high doses of unfractionated heparin often needed to treat venous thromboembolism during pregnancy. Several studies found significant loss of bone mineral density in the proximal femur25 and lumbar spine26 during extended use of unfractionated heparin in pregnancy.
Fortunately, LMWH appears to be much safer with respect to bone loss. Three recent studies27–30 evaluated the use of LMWH for extended periods during pregnancy, and none found any greater loss of bone mineral density than that seen in normal pregnant controls. Giving supplemental calcium (1,000–1,500 mg/day) and vitamin D (400–1,000 IU/day) concomitantly with unfractionated heparin or LMWH in pregnancy is advisable to further reduce the risk.
Interrupt heparin to permit regional anesthesia
Heparin therapy should be temporarily stopped during the immediate peripartum interval to minimize the risk of hemorrhage and to permit regional anesthesia. Because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, many anesthetists will not perform neuraxial regional anesthesia in women who have recently received heparin.
Since unfractionated heparin has a relatively short duration of action, the American Society of Regional Anesthesia states that subcutaneous unfractionated heparin prophylaxis is not a contraindication to neuraxial regional anesthesia.31 However, LMWHs should be stopped for at least 12 to 24 hours before regional anesthesia can be considered safe. This issue is discussed in more detail in the section on peripartum and postpartum management of anticoagulation, below.
In summary, LMWH during pregnancy offers a number of advantages over unfractionated heparin: equivalent efficacy, once- or twice-daily dosing, lower risk of heparin-induced thrombocytopenia and osteoporosis, and less-intensive monitoring. Unfractionated heparin can be offered to women who cannot afford LMWH (which costs four to five times more), and it may be used peripartum to reduce hemorrhagic risk and to permit regional anesthesia.
COUMARINS
Coumarins are the mainstay of anticoagulant therapy in most nonpregnant women beyond the immediate thrombotic period.
Warfarin (Coumadin) is the most widely used coumarin because it has a predictable onset and duration of action and excellent bioavailability.32 Others, such as acenocoumarol (Sintrom) and phenprocoumon (Marcoumar), are used more outside the United States but can be ordered or brought into the United States.
Coumarins interfere with vitamin K metabolism, inhibiting the generation of vitamin-K-dependent procoagulant proteins (factors II, VII, IX, and X) and thereby preventing clotting. They also inhibit the formation of the vitamin-K-dependent intrinsic anticoagulant proteins C and S.
Major bleeding is the most significant side effect of coumarin therapy, occurring at a rate of 4% to 6% over 3 months when the prothrombin time is maintained at an international normalized ratio (INR) of 2 to 3,33 and more often if the INR is higher.
Other issues with warfarin are the effect of variations in dietary vitamin K intake on anticoagulation and potential drug interactions that may alter the anticoagulant effect. Thus, the INR needs to be monitored closely.
Risks to the fetus and the mother
Unlike the heparins, coumarins freely cross the placenta and thus pose a risk of teratogenicity. A cluster of fetal malformations including “warfarin embryopathy” (nasal bone hypoplasia and chondrodysplasia punctata) can occur when the drug is used between 6 and 12 weeks of gestation. Warfarin embryopathy may be avoided by stopping warfarin prior to 6 weeks from the onset of the last menstrual period (ie, 6-week “menstrual age” or 4-week gestational age34).
Later in pregnancy, warfarin is associated with potential fetal bleeding complications leading to central nervous system abnormalities, increased rates of intrauterine fetal death, and pregnancy loss. In pregnant women with mechanical cardiac valve prostheses who received oral anticoagulants throughout pregnancy, the incidence of congenital anomalies was 6.4% to 10.2%.35 Fetal demise (spontaneous abortion, stillbirth, neonatal death) was also very common (29.7% to 33.6% of pregnancies) in coumarin-treated women.
Severe maternal hemorrhage may also occur in pregnant women on oral anticoagulants, particularly those who remain fully anticoagulated around the time of labor and delivery.
General caveats to warfarin in pregnancy
Because of the many maternal and fetal concerns, oral anticoagulant use in pregnancy is largely restricted to women with older-generation prosthetic heart valves in whom the very high maternal thrombotic risk may outweigh the risk of maternal and fetal side effects.
While there are limited data on warfarin use in pregnant women with antiphospholipid syndrome,36 warfarin use in such patients should be considered only for those at highest risk and with careful informed consent. These issues are discussed further below in the section on mechanical heart valve prostheses.
ANTIPLATELET DRUGS
Aspirin is an antiplatelet agent rather than an anticoagulant. Although considered inadequate for preventing venous thrombosis in high-risk groups when used alone, aspirin can moderately reduce the risk of deep venous thrombosis and pulmonary embolism in nonpregnant patients.37 It also has a well-accepted role in preventing arterial thrombotic events, ie, coronary artery disease and stroke.38
Low-dose aspirin (≤ 100 mg/day) has been extensively evaluated during pregnancy39–41 and has been shown to be safe and effective in reducing the risk of preeclampsia in high-risk women39 and in treating women with antiphospholipid antibodies and recurrent pregnancy loss42 (in conjunction with prophylactic doses of heparin). Although higher doses of aspirin and other nonsteroidal anti-inflammatory drugs can be toxic to the fetus, low doses have been shown to be safe throughout pregnancy.43
Dipyridamole (Persantine) has been studied extensively in pregnancy, and while it appears to be safe, it has not found a well-defined therapeutic role.
Other antiplatelet drugs have been only rarely used, and data on their safety and efficacy during pregnancy are limited to case reports, for example, on ticlopidine44 (Ticlid) and clopidogrel45,46 (Plavix) given during pregnancy in women with cardiac disease. These drugs do not appear to be major teratogens or to cause specific fetal harm. Their use may be reasonable in some high-risk situations, such as recurrent thrombotic stroke despite aspirin therapy. They may be used alone or with other anticoagulants in women with a coronary or other vascular stent if fetal safety is uncertain or if there is an increased risk of maternal bleeding.
NEWER ANTICOAGULANTS
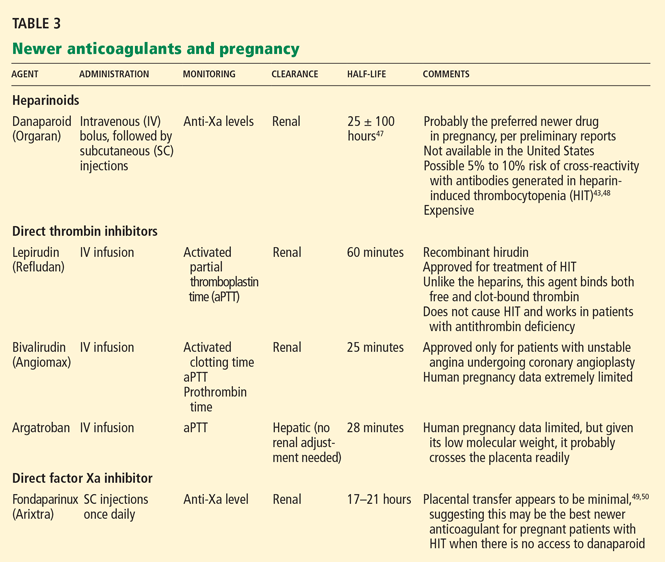
Danaparoid
The heparinoid danaparoid (Orgaran) is an LMWH, a combination of heparan, dermatan, and chondroitin sulfate. Since it is derived from heparin, in theory it can cross-react with antiheparin antibodies, but this is generally not a problem. Danaparoid inhibits factor Xa, and monitoring is via measurement of anti-factor-Xa activity levels. It has been shown to be safe and effective in nonpregnant patients with heparin-induced thrombocytopenia.51
Although no controlled study has been published on danaparoid in pregnancy, at least 51 pregnancies in 49 patients treated with danaparoid have been reported.52 Thirty-two of the patients received danaparoid because of heparin-induced thrombocytopenia and 19 because of heparin-induced skin intolerance. These reports suggest that danaparoid does not cross the placenta53 and that it may be effective and safe during pregnancy.54 For this reason, it is probably the preferred anticoagulant in pregnant patients with heparin-induced thrombocytopenia or other serious reactions to heparin.
Unfortunately, danaparoid has two major disadvantages. First, it has a prolonged half-life and no effective reversing agent, which makes its use problematic close to the time of delivery. Second, and perhaps more relevant to this discussion, it is not readily available in the United States; it was removed from the market by its manufacturer in April 2002 for business reasons rather than because of concerns over toxicity. It is still available in Canada and Europe, and it can be obtained in special circumstances in the United States via the US Food and Drug Administration (FDA); this may be worthwhile in pregnant patients who require a nonurgent alternative to heparin.
Direct thrombin inhibitors
Lepirudin (Refludan), bivalirudin (Angiomax), and argatroban are direct thrombin inhibitors and exert their anticoagulant effect independently of antithrombin. They are given by continuous intravenous infusion, and they have a very short half-life.
Lepirudin and argatroban are typically monitored via the aPTT. Bivalirudin can be monitored with the activated clotting time, partial thromboplastin time, or INR, depending on the circumstances. None of these agents generates or cross-reacts with antibodies generated in heparin-induced thrombocytopenia. None has an antidote, but the short half-life usually obviates the need for one.
Unfortunately, pregnancy data are very sparse for all three of these new agents. Argatroban has a low molecular weight and likely crosses the placenta. Also, because these agents are given intravenously, they are not practical for long-term use in pregnancy.
Fondaparinux
Fondaparinux (Arixtra), a direct factor Xa inhibitor, binds to antithrombin, causing an irreversible conformational change that increases antithrombin’s ability to inactivate factor Xa (as do the heparins). It has no effect on factor IIa (thrombin) and does not predictably affect the aPTT. Its half-life is 17 hours, and no agent is known to reverse its anticoagulant effect, although some experts would recommend a trial of high-dose recombinant factor VIIa (Novo-Seven) in uncontrolled hemorrhage.
While not FDA-approved for treating heparin-induced thrombocytopenia, it has been used for this in some patients.55–58 Animal studies and in vitro human placental perfusion studies suggest that fondaparinux does not cross the placenta in significant amounts.49 Since danaparoid is not available in the United States, fondaparinux would likely be the first choice among the newer anticoagulants when treating heparin-induced thrombocytopenia in pregnancy.
INDICATIONS FOR ANTICOAGULANTS DURING PREGNANCY
Acute deep venous thrombosis and pulmonary embolism
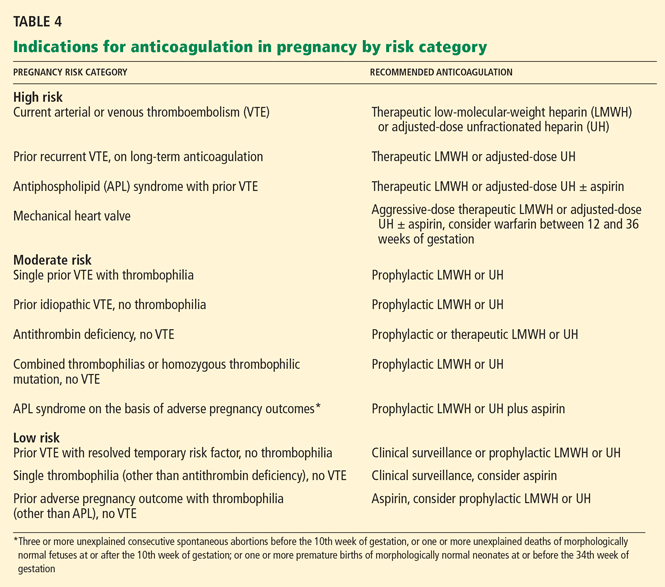
Anticoagulant therapy should begin as full doses of either LMWH or intravenous unfractionated heparin. We prefer starting with LMWH, as it can be started rapidly with less need for nursing care (eg, no need to start and maintain an intravenous line and monitor the aPTT) and has excellent safety. If LMWH is selected, initial dosing should be based on the current weight (Table 2). Subsequent monitoring of the peak anti-factor-Xa activity levels (ie, 4 hours after the dose) is recommended, with the first level drawn in the first few days of treatment, and repeat levels every 1 to 3 months for the rest of treatment. As mentioned earlier, weight-based dosing has not been systematically evaluated in pregnancy.
If unfractionated heparin is the initial agent, it should be given as a bolus followed by a continuous infusion, ideally utilizing a weight-based nomogram to estimate required doses, with adjustment of the infusion rate to maintain the aPTT at 1.5 to 2.5 times the baseline value (obtained during pregnancy). After several days, the heparin may be switched to LMWH in therapeutic doses (Table 2).
Alternatively, in women approaching term or who cannot afford LMWH, anticoagulation may be continued as adjusted-dose subcutaneous unfractionated heparin, ie, two or three large daily doses of subcutaneous heparin to provide therapeutic levels of anticoagulation. The starting dose can be calculated as the total units of heparin required to maintain full anticoagulation intravenously over 24 hours, given as two or three divided doses (Table 2). The aPTT at the mid-dosing interval (eg, 6 hours after the subcutaneous dose during every-12-hour dosing) should be monitored and the dose adjusted to maintain the aPTT at 1.5 to 2.5 times the baseline value.
A therapeutic level of anticoagulation should be maintained for at least 3 months after an acute thrombotic event during pregnancy, though many physicians prefer to continue full anticoagulation for a total of 6 months. Beyond this interval, if the woman is still pregnant, the anticoagulation may be reduced in intensity, perhaps even to a prophylactic level for the duration of the pregnancy (see discussion below on prior venous thromboembolic events) (Table 2). Peripartum and postpartum anticoagulation are discussed further below.
PRIOR VENOUS THROMBOEMBOLIC EVENT
While all pregnant women are at higher risk of venous thrombosis, the overall incidence of thromboembolism is only about one event per 1,000 pregnancies. Routine thromboprophylaxis in all pregnant women is therefore not justified. However, women who have previously had a venous thromboembolic event are at a substantially higher risk of recurrent thrombosis and should be considered for thromboprophylaxis in all subsequent high-risk situations, including pregnancy.
For women on indefinite therapeutic anticoagulation (ie, because of recurrent thrombosis), full therapeutic anticoagulation with LMWH or adjusted-dose unfractionated heparin should be maintained throughout pregnancy, as described above.
Which other women should receive prophylactic anticoagulation is a topic of ongoing debate and controversy.
How great is the risk of recurrent thromboembolism?
A small observational study59 examined the risk of recurrent venous thromboembolism during subsequent pregnancies in women with a prior thrombotic event. Anticoagulation was withheld during the antepartum period and restarted briefly after delivery. Among the 125 women enrolled, recurrent venous thromboembolism occurred in 4.8%, with half of the events occurring during the antepartum period. Among those with underlying thrombophilia, the rate of recurrent venous thromboembolism was 13% (95% confidence interval [CI] 1.7%–40.5%) to 20% (95% CI 2.5%–56.5%), and those with a prior idiopathic clot without thrombophilia had an event rate of 7.7% (95% CI 0.01%–25.1%). The subgroup with a prior reversible risk factor (at the time of their initial venous thromboembolic event) and without detectable thrombophilia had no recurrent events.
This study suggests that women with prior venous thromboembolism and thrombophilia or a prior idiopathic thrombotic event are at a substantial risk of recurrent thrombotic events during pregnancy. And other data confirm the high risk of recurrent venous thromboembolism in thrombophilic pregnant women.60 These women should all be offered active antepartum and postpartum thromboprophylaxis with LMWH or unfractionated heparin (Tables 2 and 4). Women without thrombophilia but with a history of venous thromboembolism related to pregnancy or oral contraceptive use also have a substantial risk of recurrent venous thrombosis and should be offered antepartum and postpartum thromboprophylaxis.61 In contrast, women with a prior “secondary” clot, no thrombophilia, and no additional current risk factors (Table 1) appear to be at low risk of recurrent venous thromboembolism.
The risks should be discussed with these women, with an option for close clinical surveillance during pregnancy (Table 4), but with a low threshold to investigate any worrisome symptoms. Such women may also elect to take LMWH or unfractionated heparin during pregnancy.
Which heparin to use?
Prophylactic anticoagulation during pregnancy can be with either LMWH or unfractionated heparin. For most women this involves “prophylactic” dosing with the goal of maintaining a mid-interval anti-factor-Xa activity level of approximately 0.05 to 0.2 U/mL. Thromboprophylaxis with LMWH can be with lower, fixed, once-daily doses throughout pregnancy20 (Table 2), although some clinicians still prefer twice-daily dosing. The heparin should be started as soon as pregnancy is confirmed, as the pregnancy-associated increase in thrombotic risk begins by the middle of the first trimester.
To maintain effective prophylactic levels, the dose of unfractionated heparin should be increased sequentially over the trimesters62,63: approximately 5,000 units subcutaneously twice daily in the first trimester, then 7,500 units twice daily in the second trimester, and 10,000 units twice daily in the third trimester for a woman of average size.
When to add low-dose aspirin
Women with antiphospholipid antibodies, particularly those with prior recurrent pregnancy loss or fetal demise, should receive aspirin 81 mg/day in addition to heparin.39 The aspirin may be started prior to conception or when pregnancy is confirmed.
Other measures
Women on anticoagulant therapy who are at risk of recurrent venous thromboembolism should be encouraged to wear elastic compression stockings. Intermittent pneumatic compression of the legs via automated devices may be considered for women hospitalized for any reason or on bedrest.
Whichever measures are used, a high index of suspicion and a low threshold for investigating for recurrent thrombosis should be maintained throughout pregnancy and the puerperium.
PERIPARTUM AND POSTPARTUM MANAGEMENT OF ANTICOAGULATION
Heparin therapy must be interrupted temporarily during the immediate peripartum interval to minimize the risk of hemorrhage and to allow for the option of regional anesthesia. As mentioned earlier, because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, the American Society of Regional Anesthesia guidelines advise waiting to insert the needle at least 10 to 12 hours after the last prophylactic dose of LMWH, and at least 24 hours after the last therapeutic dose.31
The guidelines state that neuraxial anesthesia is not contraindicated in patients on prophylactic unfractionated heparin.31
To facilitate use of regional anesthesia in these women, therefore, options include:
- Electively stopping LMWH 24 hours before planned induction of labor
- Electively stopping prophylactic-dose LMWH or unfractionated heparin at about 38 weeks of gestation, to await spontaneous labor, or
- Switching therapeutic or prophylactic LMWH to unfractionated heparin at about 36 weeks of gestation, with instructions to discontinue the injections in the earliest stages of spontaneous labor. This aims to shorten the heparin-free period required before neuraxial anesthesia while minimizing maternal thrombotic risk.
Additional advantages to using unfractionated heparin peripartum include the option of obtaining a rapid aPTT measurement to confirm the absence of a significant ongoing heparin effect prior to regional anesthesia or delivery, and the ability to completely reverse the heparin effect with protamine sulfate if major bleeding occurs. LMWHs are only partially reversible.64
Interrupting anticoagulation after an initial thrombotic event
If therapeutic anticoagulation must be interrupted for labor within 1 month of the initial thrombotic event, the risk of recurrent thrombotic complications is high65; these women must be observed very carefully and may benefit from intravenous heparin before and after delivery. They may even merit placement of a temporary vena cava filter (particularly if less than 2 weeks have elapsed since the venous thromboembolic event and in women with a large deep venous clot burden), a procedure that has been used safely but little studied in pregnant women.66
Fluoroscopic guidance may be needed for filter placement. This exposes the fetus to radiation, but the low-level exposure at this late gestational age is unlikely to pose a significant risk. The filter may be removed within 1 to 2 weeks postpartum, assuming there are no ongoing contraindications to anticoagulation.
In the rare woman with antithrombin deficiency and a recent or prior thrombotic event, giving antithrombin concentrate during the peripartum (heparin-free) interval has been described and may be considered under the guidance of a hematologist.67
Ongoing anticoagulation is essential postpartum, as the puerperium is the period of highest day-to-day risk of thromboembolic events: about one-third of pregnancy-associated events occur during these 6 to 12 weeks.2 Heparin should be resumed 6 to 12 hours after delivery, once hemostasis is confirmed.
Options for women requiring ongoing therapeutic anticoagulation include intravenous heparin started without a bolus, to minimize bleeding risk, with aPTT measured 12 hours later, or an initial prophylactic dose of LMWH 6 to 12 hours postpartum, with therapeutic dosing resumed on postpartum day 1. If prophylactic dosing is desired, unfractionated heparin or LMWH may be given subcutaneously starting at about 6 hours postpartum.
Warfarin in the puerperium
Women may subsequently be maintained on either LMWH or unfractionated heparin, or switched to an oral anticoagulant such as warfarin. Although warfarin may appear in minute amounts in breast milk, it has not been associated with adverse events in newborns and is considered compatible with breastfeeding.68 Heparin should be continued during the initial days of warfarin therapy, until the INR is at a therapeutic level for 24 hours. Some physicians prefer to delay warfarin for several days, giving LMWH alone in the immediate postpartum period, to allow wound-healing and to reduce bleeding risk.
Postpartum, anticoagulation should be continued for at least 6 to 12 weeks, at which point the physiologic changes in the coagulation system related to pregnancy will have returned to normal.
THROMBOPHILIA WITHOUT A PREVIOUS THROMBOEMBOLIC EVENT
Over the last 5 to 10 years, practitioners have been seeing many more young women with genetic or acquired thrombophilias who have never had a venous thromboembolic event. Physicians must advise these women about their risk of thromboembolic events during pregnancy and about the appropriateness of anticoagulant use.
Thrombophilias are often detected in women who develop venous thrombosis during pregnancy,69–71 but they are also very common in the general population (around 15%). While women with thrombophilia are at above-average risk of venous thromboembolism during pregnancy, the magnitude of risk in an individual patient is often difficult to estimate.
Data suggest that some types of thrombophilia confer greater thrombotic risk than others. McColl et al72 derived risk estimates for a primary event in women with several of the disorders: 0.23% in women heterozygous for the factor V Leiden mutation, 0.88% in women with protein C deficiency, and 2.4% to 35.7% in women with antithrombin deficiency. A case-control study70 found that all thrombophilic states were more common in women with pregnancy-associated venous thromboembolism than in healthy pregnant controls, except those with the MTHFR mutation and protein S deficiency. The estimated risk during pregnancy was 0.03% in women with no defect, 0.1% in women with protein C deficiency, 0.25% in women with the factor V Leiden mutation, 0.4% in those with antithrombin deficiency, 0.5% in those with the prothrombin gene mutation, and 4.6% in those with both factor V Leiden and prothrombin gene mutations.
Routine anticoagulation not advised in pregnant thrombophilic women
Because the risk of a primary venous thromboembolic event is less than 1% for most thrombophilic women, routine anticoagulant therapy does not seem prudent for this indication. Given the low absolute risk of venous thromboembolism, the cost and potential side effects of anticoagulant use are difficult to justify.
The women who seem at higher risk and in whom anticoagulation should be considered include those with antithrombin deficiency; those with high-titer anticardiolipin antibodies or a lupus anticoagulant antibody (treat with heparin and low-dose aspirin); those with combined thrombophilic defects or who are homozygotes for the factor V Leiden or prothrombin gene mutations; and those with multiple other current risk factors for venous thromboembolism (Table 1).
Since anticoagulants for primary prevention of adverse pregnancy outcomes in thrombophilic women have not yet been shown to have a definitive benefit, they are not recommended for this purpose.
ADVERSE PREGNANCY OUTCOMES IN WOMEN WITH THROMBOPHILIAS
Women with antiphospholipid antibodies and a previous poor obstetric outcome are clearly at increased risk of recurrent adverse pregnancy outcomes such as recurrent spontaneous abortion, unexplained fetal death, placental insufficiency, and early or severe preeclampsia. In such women who have both antiphospholipid antibodies and a history of venous thromboembolism or adverse pregnancy outcome, treatment during subsequent pregnancy with low-dose aspirin and prophylactic-dose LMWH or unfractionated heparin improves pregnancy outcomes.36–42 Women with antiphospholipid antibodies without previous thrombosis or pregnancy complications may also be at increased risk, but it is unclear whether thromboprophylaxis improves their outcomes.
Recent epidemiologic data reveal that women with other thrombophilic conditions also are at increased risk of early, severe preeclampsia73 as well as other pregnancy complications, including recurrent pregnancy loss, placental abruption, fetal growth restriction, and stillbirth.74 A recent meta-analysis75 looked at individual thrombophilias and found that factor V Leiden and prothrombin gene mutations were associated with recurrent fetal loss, stillbirth, and preeclampsia; that protein S deficiency was associated with recurrent fetal loss and stillbirth; that antiphospholipid antibodies were associated with recurrent pregnancy loss, preeclampsia, and intrauterine growth restriction; that the MTHFR mutation (homozygous) was associated with preeclampsia; and that protein C and antithrombin deficiencies were not significantly associated with adverse pregnancy outcomes. Data were scant for some of the rarer thrombophilias.75
Several recent small studies76–78 suggest that anticoagulants may improve pregnancy outcomes in women with genetic thrombophilias and recurrent pregnancy loss. These findings have not yet been confirmed in high-quality clinical trials, but such trials are under way. It is still unclear whether anticoagulants also reduce the risk of other adverse pregnancy outcomes associated with thrombophilias.
The current American College of Chest Physicians guidelines recommend testing of women with adverse pregnancy outcomes (recurrent pregnancy loss, prior severe or recurrent preeclampsia, abruptions, or otherwise unexplained intrauterine death) for congenital thrombophilias and antiphospholipid antibodies, and offering treatment to such women, if thrombophilic, with low-dose aspirin plus prophylactic heparin (unfractionated or LMWH).22 The authors of the guidelines admit that the evidence for this recommendation is weak, but they argue that the heparin will also serve as thromboprophylaxis in this high-risk group. Hopefully, the randomized clinical trials currently under way will provide clearer guidance regarding the most appropriate therapy in this difficult clinical situation.
MECHANICAL HEART VALVES
Internists may occasionally encounter a woman with a mechanical heart valve prosthesis who is either pregnant or is planning a pregnancy and therefore needs advice regarding optimal anticoagulant management. This should generally be undertaken in a multi-disciplinary fashion, with input from cardiology, hematology, and maternal-fetal medicine. The substantial maternal and fetal risks and the lack of definitive data on which to base treatment decisions make it a treacherous and stressful undertaking. Nonetheless, all internists should have a basic understanding of the complex issues regarding this management.
Outside of pregnancy, oral anticoagulants are the mainstay of therapy for patients with mechanical heart valves. Unfortunately, as discussed above, the use of these agents during pregnancy carries a risk of teratogenicity and toxic fetal effects and increases the risk of pregnancy loss and maternal hemorrhage. Heparins have been used in this setting for many years, but data on their efficacy and safety are very limited, and there are numerous reports of catastrophic maternal thrombotic complications.79,80
A systematic review of anticoagulation in pregnant women with prosthetic heart valves34 found very limited data on heparin use throughout pregnancy. Women maintained on warfarin vs heparin between pregnancy weeks 6 and 12 had higher rates of congenital anomalies (6.4% with warfarin vs 3.4% with heparin) and total fetal wastage (33.6% vs 26.5%). The warfarin group had fewer maternal thromboembolic complications (3.9% vs 9.2%), however, and a slightly lower rate of maternal death (1.8% vs 4.2%). Most of the women had higher-risk older-generation valves in the mitral position.
Recent data on LMWH consist mainly of case reports and case series,81 with a likely bias to publication of worse outcomes. Controlled trials in this area will be difficult to conduct. Still, aggressive anticoagulation with LMWH or unfractionated heparin, with close monitoring of the intensity of anticoagulation, may be safe and effective for pregnant women with newer-generation mechanical heart valves.82 A recent consensus statement22 suggested several regimens for pregnant women with mechanical heart valves:
- Twice-daily LMWH throughout pregnancy, with the dose adjusted either by weight, or to keep the 4-hour postinjection anti-factor-Xa activity level around 1.0 to 1.2 U/mL
- Aggressive adjusted-dose unfractionated heparin throughout pregnancy, given subcutaneously every 12 hours and adjusted to keep the mid-interval aPTT at least twice the control value or to attain a mid-interval anti-factor-Xa activity level of 0.35 to 0.70 U/mL
- Unfractionated heparin or LMWH (as above) until gestation week 13, then warfarin until the middle of the third trimester, and then heparin again.22
The authors also recommended adding low-dose aspirin (75–162 mg/day) in high-risk women.22
These options all seem reasonable, given our current knowledge, though warfarin use during pregnancy should be restricted to very-high-risk situations, such as women with older-generation mitral prostheses. LM-WHs may become the preferred therapy for this indication once further controlled data regarding their efficacy and safety become available.
- Chang J, Elam-Evans LD, Berg CJ, et al. Pregnancy-related mortality surveillance-United States, 1991–1999. MMWR Surveill Summ 2003; 52:1–8.
- Lewis G, Drife JO, Clutton-Brock T, et al. Why Mothers Die, 2000–2002. The Sixth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. London: RCOG Press, 2004.
- Health Canada. Special Report on Maternal Mortality and Severe Morbidity in Canada—Enhanced Surveillance: The Path to Prevention. Ottawa: Minister of Public Works and Government Services Canada, 2004. www.phac-aspc.gc.ca/rhs-ssg/srmm-rsmm/page1-eng.php. Accessed 11/26/2008.
- Stein PD, Hull RD, Kayali F, et al. Venous thromboembolism in pregnancy: 21-year trends. Am J Med 2004; 117:121–125.
- Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet 1999; 353:1258–1265.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353:1167–1173.
- Gherman RB, Goodwin TM, Leung B, et al. Incidence, clinical characteristics, and timing of objectively diagnosed venous thromboembolism during pregnancy. Obstet Gynecol 1999; 94:730–734.
- Weitz JI. Low-molecular-weight heparins. N Engl J Med 1997; 337:688–698.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Simonneau G, Sors H, Charbonnier B, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N Engl J Med 1997; 337:663–669.
- Hull RD, Raskob GE, Brant RF, et al. Low-molecular-weight heparin vs heparin in the treatment of patients with pulmonary embolism. American-Canadian Thrombosis Study Group. Arch Intern Med 2000; 160:229–236.
- Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost 1999; 81:668–672.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005; 106:401–407.
- Melissari E, Parker CJ, Wilson NV, et al. Use of low molecular weight heparin in pregnancy. Thromb Haemost 1992; 68:652–656.
- Forestier F, Daffos F, Capella-Pavlovsky M. Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 1984; 34:557–560.
- Forestier F, Daffos F, Rainaut M, Toulemonde F. Low molecular weight heparin (CY 216) does not cross the placenta during the third trimester of pregnancy. Thromb Haemost 1987; 57:234.
- Barbour LA, Oja JL, Schultz LK. A prospective trial that demonstrates that dalteparin requirements increase in pregnancy to maintain therapeutic levels of anticoagulation. Am J Obstet Gynecol 2004; 191:1024–1029.
- Smith MP, Norris LA, Steer PJ, Savidge GF, Bonnar J. Tinzaparin sodium for thrombosis treatment and prevention during pregnancy. Am J Obstet Gynecol 2004; 190:495–501.
- Ellison J, Walker ID, Greer IA. Antenatal use of enoxaparin for prevention and treatment of thromboembolism in pregnancy. BJOG 2000; 107:1116–1121.
- Sarig G, Brenner B. Monitoring of low molecular weight heparin (LMWH) in pregnancy. Thromb Res 2005; 115 suppl 1:84–86.
- Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:627S–644S.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995; 332:1330–1335.
- Hassell K. The management of patients with heparin-induced thrombocytopenia who require anticoagulant therapy. Chest 2005; 127 suppl 2:1S–8S.
- Barbour LA, Kick SD, Steiner JF, et al. A prospective study of heparin-induced osteoporosis in pregnancy using bone densitometry. Am J Obst Gynecol 1994; 170:862–869.
- Douketis JD, Ginsberg JS, Burrows RF, Duku EK, Webber CE, Brill-Edwards P. The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 1996; 75:254–257.
- Pettila V, Leinonen P, Markkola A, Hiilesmaa V, Kaaja R. Postpartum bone mineral density in women treated for thromboprophylaxis with unfractionated heparin or LMW heparin. Thromb Haemost 2002; 87:182–186.
- Carlin AJ, Farquharson RG, Quenby SM, Topping J, Fraser WD. Prospective observational study of bone mineral density during pregnancy: low molecular weight heparin versus control. Hum Reprod 2004; 19:1211–1214.
- Casele HL, Laifer SA. Prospective evaluation of bone density in pregnant women receiving the low molecular weight heparin enoxaparin sodium. J Matern Fetal Med 2000; 9:122–125.
- Casele H, Haney EI, James A, Rosene-Montella K, Carson M. Bone density changes in women who receive thromboprophylaxis in pregnancy. Am J Obstet Gynecol 2006; 195:1109–1113.
- Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the risks (the second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med 2003; 28:172–197.
- Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 suppl 1:8S–21S.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:287S–310S.
- Holmes LB. Teratogen-induced limb defects. Am J Med Genet 2002; 112:297–303.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Pauzner R, Dulitzki M, Langevitz P, Livneh A, Kenett R, Many A. Low molecular weight heparin and warfarin in the treatment of patients with antiphospholipid syndrome during pregnancy. Thromb Haemost 2001; 86:1379–1384.
- Pulmonary Embolism Prevention (PEP) Trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000; 355:1295–1302.
- Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:234S–264S.
- Duley L, Henderson-Smart DJ, Knight M, King JF. Antiplatelet agents for preventing preeclampsia and its complications. Cochrane Database Syst Rev. 2004; ( 1):CD004659.
- Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol 2003; 101:1319–1332.
- Caritis SN, Sibai BM, Hauth J, et al, and the National Institute of Child Health and Human Development Network of Maternal Fetal Medicine Units. Low-dose aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338:701–705.
- Rai R, Cohen H, Dave M, Regan L. Randomised controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage associated with phospholipid antibodies (or antiphospholipid antibodies). BMJ 1997; 314:253–257.
- Kozer E, Nikfar S, Costei A, Boskovic R, Nulman I, Koren G. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol 2002; 187:1623–1630.
- Sebastian C, Scherlag M, Kugelmass A, Schechter E. Primary stent implantation for acute myocardial infarction during pregnancy: use of abciximab, ticlopidine, and aspirin. Cathet Cardiovasc Diagn 1998; 45:275–249.
- Wilson AM, Boyle AJ, Fox P. Management of ischaemic heart disease in women of child-bearing age. Intern Med J 2004; 34:694–697.
- Klinzing P, Markert UR, Liesaus K, Peiker G. Case report: successful pregnancy and delivery after myocardial infarction and essential thrombocythemia treated with clopidogrel. Clin Exp Obstet Gynecol 2001; 28:215–216.
- Danhof M, de Boer A, Magnani HN, Stiekema JC. Pharmacokinetic considerations on Orgaran (Org 10172) therapy. Haemostasis 1992; 22:73–84.
- Tardy-Poncet B, Tardy B, Reynaud J, et al. Efficacy and safety of danaparoid sodium (ORG 10172) in critically ill patients with heparin-associated thrombocytopenia. Chest 1999; 115:1616–1620.
- Lagrange F, Vergnes C, Brun JL, et al. Absence of placental transfer of pentasaccharide (fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 2002; 87:831–835.
- Dempfle CE. Minor transplacental passge of fondapinux in vivo. N Engl J Med 2004; 350:1914.
- Magnani HN. Heparin-induced thrombocytopenia (HIT): an overview of 230 patients treated with orgaran (Org 10172). Thromb Haemost 1993; 70:554–561.
- Lindhoff-Last E, Kreutzenbeck HJ, Magnani HN. Treatment of 51 pregnancies with danaparoid because of heparin intolerance. Thromb Haemost 2005; 93:63–69.
- Greinacher A, Eckhardt T, Mussmann J, Mueller-Eckhardt C. Pregnancy complicated by heparin associated thrombocytopenia: management by a prospectively in vitro selected heparinoid (Org 10172). Thromb Res 1993; 71:123–126.
- Schindewolf M, Mosch G, Bauersachs RM, Lindhoff-Last E. Safe anticoagulation with danaparoid in pregnancy and lactation. Thromb Haemost 2004; 92:211.
- Harenberg J. Treatment of a woman with lupus and thromboembolism and cutaneous intolerance to heparins using fondaparinux during pregnancy. Thromb Res 2007; 119:385–388.
- Wijesiriwardana A, Lees DA, Lush C. Fondaparinux as anticoagulant in a pregnant woman with heparin allergy. Blood Coagul Fibrinolysis 2006; 17:147–149.
- Mazzolai L, Hohlfeld P, Spertini F, Hayoz D, Schapira M, Duchosal MA. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood 2006; 108:1569–1570.
- Hawkins D, Evans J. Minimizing the risk of heparin-induced osteoporosis during pregnancy. Expert Opin Drug Saf 2005; 4:583–590.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of clot in this pregnancy study group. N Engl J Med 2000; 343:1439–1444.
- Martinelli I, Legnani C, Bucciarelli P, Grandone E, De Stefano V, Mannucci PM. Risk of pregnancy-related venous thrombosis in carriers of severe inherited thrombophilia. Thromb Haemost 2001; 86:800–803.
- De Stefano V, Martinelli I, Rossi E, Battaglioli T, Za T, Mannucci PM, Leone G. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006; 135:386–391.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995; 173:1869–1873.
- Ensom MH, Stephenson MD. Pharmacokinetics of low molecular weight heparin and unfractionated heparin in pregnancy. J Soc Gynecol Investig 2004; 11:377–383.
- Crowther MA, Berry LR, Monagle PT, Chan AK. Mechanisms responsible for the failure of protamine to inactivate low-molecular-weight heparin. Br J Haematol 2002; 116:178–186.
- Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997; 336:1506–1511.
- Thomas LA, Summers RR, Cardwell MS. Use of Greenfield filters in pregnant women at risk for pulmonary embolism. South Med J 1997; 90:215–217.
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs 2007; 67:1429–1440.
- Information from LactMed: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT, LactMed Record Number: 279. Accessed 11/26/2008.
- Gerhardt A, Scharf RE, Beckmann MW, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 2000; 342:374–380.
- Hirsch DR, Mikkola KM, Marks PW, et al. Pulmonary embolism and deep venous thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J 1996; 131:1145–1148.
- Dizon-Townson DS, Nelson LM, Jang H, Varner MW, Ward K. The incidence of the factor V Leiden mutation in an obstetric population and its relationship to deep vein thrombosis. Am J Obstet Gynecol 1997; 176:883–886.
- McColl MD, Ramsay JE, Tait RC, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 1997; 78:1183–1188.
- Kupferminc MJ, Fait G, Many A, Gordon D, Eldor A, Lessing JB. Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 2000; 96:45–49.
- Kupferminc MJ, Eldor A, Steinman N, et al. Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 1999; 340:9–13.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006; 132:171–196.
- Brenner B, Hoffman R, Blumenfeld Z, Weiner Z, Younis JS. Gestational outcome in thrombophilic women with recurrent pregnancy loss treated by enoxaparin. Thromb Haemost 2000; 83:693–697.
- Carp H, Dolitzky M, Inbal A. Thromboprophylaxis improves the live birth rate in women with consecutive recurrent miscarriages and hereditary thrombophilia. J Thromb Haemost 2003; 1:433–438.
- Gris JC, Mercier E, Quere I, et al. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder. Blood 2004; 103:3695–3699.
- Salazar E, Izaguirre R, Verdejo J, Mutchinick O. Failure of adjusted doses of subcutaneous heparin to prevent thromboembolic phenomena in pregnant patients with mechanical cardiac valve prostheses. J Am Coll Cardiol 1996; 27:1698–1703.
- Iturbe-Alessio I, Fonseca MC, Mutchinik O, Santos MA, Zajarias A, Salazar E. Risks of anticoagulant therapy in pregnant women with artificial heart valves. N Engl J Med 1986; 315:1390–1393.
- Rowan JA, McCowan LM, Raudkivi PJ, North RA. Enoxaparin treatment in women with mechanical heart valves during pregnancy. Am J Obstet Gynecol 2001; 185:633–637.
- Oran B, Lee-Parritz A, Ansell J. Low molecular weight heparin for the prophylaxis of thromboembolism in women with prosthetic mechanical heart valves during pregnancy. Thromb Haemost 2004; 92:747–751.
Anticoagulation is essential in a wide variety of conditions in women of child-bearing age. Some, such as venous thromboembolism, occur more often during pregnancy. Others, such as recurrent fetal loss in the setting of antiphospholipid antibodies, are specific to pregnancy.
While anticoagulants are useful in many circumstances, their use during pregnancy increases the risk of hemorrhage and other adverse effects on the mother and the fetus. Treatment with anticoagulants during pregnancy must therefore be carefully considered, with judicious selection of the agent, and with reflection on the physiologic changes of pregnancy to ensure appropriate dosing. In this article, we review these issues.
WHY IS THROMBOTIC RISK HIGHER DURING PREGNANCY?
Venous thromboembolism is among the leading causes of maternal death in developed countries.1–3 Modern care has dramatically reduced the risk of maternal death from hemorrhage, infection, and hypertension, but rates of morbidity and death from thrombosis have remained stable or increased in recent years.4
- Much higher levels of fibrinogen and factors VII, VIII, IX, and X
- Lower levels of protein S and increased resistance to activated protein C
- Impaired fibrinolysis, due to inhibitors derived from the placenta.
Acquired antithrombin deficiency may also occur in high-proteinuric states such as nephrotic syndrome or preeclampsia, further increasing thrombotic risk. Pooling of venous blood, caused by progesterone-mediated venous dilation and compounded by compression of the inferior vena cava by the uterus in later pregnancy, also increases thrombotic risk. And endothelial disruption of the pelvic vessels may occur during delivery, particularly during cesarean section.
Additional factors that increase thrombotic risk include immobilization, such as bed rest for pregnancy complications; surgery, including cesarean section; ovarian hyperstimulation during gonadotropin use for in vitro fertilization; trauma; malignancy; and hereditary or acquired hypercoagulable states.6 These hypercoagulable states include deficiencies of antithrombin or the intrinsic anticoagulant proteins C or S; resistance to activated protein C, usually due to the factor V Leiden mutation; the PT20210A mutation of the prothrombin gene; hyperhomocystinemia due to mutation of the methyltetrahydrofolate reductase (MTHFR) gene; and the sustained presence of antiphospholipid antibodies, including lupus anticoagulant antibodies, sometimes also with moderately high titers of anticardiolipin or beta-2-glycoprotein I antibodies.
Other conditions that increase thrombotic risk include hyperemesis gravidarum, obesity, inflammatory bowel disease, infection, smoking, and indwelling intravenous catheters.6 Given the multitude of risk factors, pregnant women have a risk of thrombotic complications three to five times higher than nonpregnant women.7
HEPARIN USE DURING PREGNANCY
Low-molecular-weight heparins (LMWHs)8 and unfractionated heparin bind to anti-thrombin and thus change the shape of the antithrombin molecule, dramatically increasing its interaction with the clotting factors Xa and prothrombin (factor II). The enhanced clearance of these procoagulant proteins leads to the anticoagulant effect. Unfractionated heparin has roughly equivalent interaction with factors Xa and II and prolongs the activated partial thromboplastin time (aPTT), which is therefore used to monitor the intensity of anticoagulation.
LMWHs, on the other hand, interact relatively little with factor II and do not predictably prolong the aPTT. Monitoring their effect is therefore more difficult and requires direct measurement of anti-factor-Xa activity. This test is widely available, but it is time-consuming (it takes several hours and results may not be available within 24 hours if the test is requested “after hours”), and therefore it is of limited use in the acute clinical setting. While weight-based dosing of LMWHs is reliable and safe in nonpregnant patients, it has not yet been validated for pregnant women.
Unfractionated heparin has been used for decades for many indications during pregnancy. It is a large molecule, so it does not cross the placenta and thus, in contrast to the coumarin derivatives, does not cause teratogenesis or toxic fetal effects. Its main limitations in pregnancy are its inconvenient dosing (at least twice daily when given subcutaneously) and its potential maternal adverse effects (mainly osteoporosis and heparin-induced thrombocytopenia).
Over the last 10 years LMWHs have become the preferred anticoagulants for treating and preventing thromboembolism in all patients. They are equivalent or superior to unfractionated heparin in efficacy and safety in the initial treatment of acute deep venous thrombosis9,10 and pulmonary embolism11,12 outside of pregnancy. While comparative data are much less robust in pregnant patients, several series have confirmed the safety and efficacy of LMWHs in pregnancy.13–15 LMWHs do not cross the placenta15–17 and thus have a fetal safety profile equivalent to that of unfractionated heparin.
Pregnancy alters metabolism of LMWHs
The physiologic changes of pregnancy alter the metabolism of LMWH, resulting in lower peak levels and a higher rate of clearance,18,19 and so a pregnant woman may need higher doses or more frequent dosing.
Recent evidence suggests that thromboprophylaxis can be done with lower, fixed, once-daily doses of LMWH throughout pregnancy,20 although some clinicians still prefer twice-daily dosing (particularly during the latter half of pregnancy).
Pending more research on weight-based dosing of LMWH in pregnancy, anti-factor- Xa activity levels should be measured after treatment is started and every 1 to 3 months thereafter during pregnancy.21 Doses should be adjusted to keep the peak anti-Xa level (ie, 4 hours after the dose) at 0.5 to 1.2 U/mL.22
Heparin-induced thrombocytopenia
Type-2 heparin-induced thrombocytopenia is an uncommon but serious adverse effect of unfractionated heparin therapy (and, less commonly of LMWH), caused by heparin-dependent immunoglobulin G (IgG) antibodies that activate platelets via their Fc receptors, potentially precipitating life-threatening arterial or venous thrombosis.
In a trial in nonpregnant orthopedic patients,23 clinical heparin-induced thrombocytopenia occurred in 2.7% of patients receiving unfractionated heparin vs 0% of those receiving LMWH; heparin-dependent IgG was present in 7.8% vs 2.2%, respectively.
Fortunately, heparin-induced thrombocytopenia seems to be very rare in pregnancy: two recent prospective series evaluating prolonged LMWH use in pregnancy13,15 revealed no episodes of this disease. Nonetheless, it is reasonable to measure the platelet count once or twice weekly during the first few weeks of LMWH use and less often thereafter, unless symptoms of heparin-induced thrombocytopenia develop. In pregnant women with heparin-induced thrombocytopenia or heparin-related skin reactions, other anticoagulants must be considered24 (see discussion later).
Heparin-induced osteoporosis
Heparin-induced osteoporosis, a potential effect of prolonged heparin therapy, is of concern, given the prolonged duration and high doses of unfractionated heparin often needed to treat venous thromboembolism during pregnancy. Several studies found significant loss of bone mineral density in the proximal femur25 and lumbar spine26 during extended use of unfractionated heparin in pregnancy.
Fortunately, LMWH appears to be much safer with respect to bone loss. Three recent studies27–30 evaluated the use of LMWH for extended periods during pregnancy, and none found any greater loss of bone mineral density than that seen in normal pregnant controls. Giving supplemental calcium (1,000–1,500 mg/day) and vitamin D (400–1,000 IU/day) concomitantly with unfractionated heparin or LMWH in pregnancy is advisable to further reduce the risk.
Interrupt heparin to permit regional anesthesia
Heparin therapy should be temporarily stopped during the immediate peripartum interval to minimize the risk of hemorrhage and to permit regional anesthesia. Because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, many anesthetists will not perform neuraxial regional anesthesia in women who have recently received heparin.
Since unfractionated heparin has a relatively short duration of action, the American Society of Regional Anesthesia states that subcutaneous unfractionated heparin prophylaxis is not a contraindication to neuraxial regional anesthesia.31 However, LMWHs should be stopped for at least 12 to 24 hours before regional anesthesia can be considered safe. This issue is discussed in more detail in the section on peripartum and postpartum management of anticoagulation, below.
In summary, LMWH during pregnancy offers a number of advantages over unfractionated heparin: equivalent efficacy, once- or twice-daily dosing, lower risk of heparin-induced thrombocytopenia and osteoporosis, and less-intensive monitoring. Unfractionated heparin can be offered to women who cannot afford LMWH (which costs four to five times more), and it may be used peripartum to reduce hemorrhagic risk and to permit regional anesthesia.
COUMARINS
Coumarins are the mainstay of anticoagulant therapy in most nonpregnant women beyond the immediate thrombotic period.
Warfarin (Coumadin) is the most widely used coumarin because it has a predictable onset and duration of action and excellent bioavailability.32 Others, such as acenocoumarol (Sintrom) and phenprocoumon (Marcoumar), are used more outside the United States but can be ordered or brought into the United States.
Coumarins interfere with vitamin K metabolism, inhibiting the generation of vitamin-K-dependent procoagulant proteins (factors II, VII, IX, and X) and thereby preventing clotting. They also inhibit the formation of the vitamin-K-dependent intrinsic anticoagulant proteins C and S.
Major bleeding is the most significant side effect of coumarin therapy, occurring at a rate of 4% to 6% over 3 months when the prothrombin time is maintained at an international normalized ratio (INR) of 2 to 3,33 and more often if the INR is higher.
Other issues with warfarin are the effect of variations in dietary vitamin K intake on anticoagulation and potential drug interactions that may alter the anticoagulant effect. Thus, the INR needs to be monitored closely.
Risks to the fetus and the mother
Unlike the heparins, coumarins freely cross the placenta and thus pose a risk of teratogenicity. A cluster of fetal malformations including “warfarin embryopathy” (nasal bone hypoplasia and chondrodysplasia punctata) can occur when the drug is used between 6 and 12 weeks of gestation. Warfarin embryopathy may be avoided by stopping warfarin prior to 6 weeks from the onset of the last menstrual period (ie, 6-week “menstrual age” or 4-week gestational age34).
Later in pregnancy, warfarin is associated with potential fetal bleeding complications leading to central nervous system abnormalities, increased rates of intrauterine fetal death, and pregnancy loss. In pregnant women with mechanical cardiac valve prostheses who received oral anticoagulants throughout pregnancy, the incidence of congenital anomalies was 6.4% to 10.2%.35 Fetal demise (spontaneous abortion, stillbirth, neonatal death) was also very common (29.7% to 33.6% of pregnancies) in coumarin-treated women.
Severe maternal hemorrhage may also occur in pregnant women on oral anticoagulants, particularly those who remain fully anticoagulated around the time of labor and delivery.
General caveats to warfarin in pregnancy
Because of the many maternal and fetal concerns, oral anticoagulant use in pregnancy is largely restricted to women with older-generation prosthetic heart valves in whom the very high maternal thrombotic risk may outweigh the risk of maternal and fetal side effects.
While there are limited data on warfarin use in pregnant women with antiphospholipid syndrome,36 warfarin use in such patients should be considered only for those at highest risk and with careful informed consent. These issues are discussed further below in the section on mechanical heart valve prostheses.
ANTIPLATELET DRUGS
Aspirin is an antiplatelet agent rather than an anticoagulant. Although considered inadequate for preventing venous thrombosis in high-risk groups when used alone, aspirin can moderately reduce the risk of deep venous thrombosis and pulmonary embolism in nonpregnant patients.37 It also has a well-accepted role in preventing arterial thrombotic events, ie, coronary artery disease and stroke.38
Low-dose aspirin (≤ 100 mg/day) has been extensively evaluated during pregnancy39–41 and has been shown to be safe and effective in reducing the risk of preeclampsia in high-risk women39 and in treating women with antiphospholipid antibodies and recurrent pregnancy loss42 (in conjunction with prophylactic doses of heparin). Although higher doses of aspirin and other nonsteroidal anti-inflammatory drugs can be toxic to the fetus, low doses have been shown to be safe throughout pregnancy.43
Dipyridamole (Persantine) has been studied extensively in pregnancy, and while it appears to be safe, it has not found a well-defined therapeutic role.
Other antiplatelet drugs have been only rarely used, and data on their safety and efficacy during pregnancy are limited to case reports, for example, on ticlopidine44 (Ticlid) and clopidogrel45,46 (Plavix) given during pregnancy in women with cardiac disease. These drugs do not appear to be major teratogens or to cause specific fetal harm. Their use may be reasonable in some high-risk situations, such as recurrent thrombotic stroke despite aspirin therapy. They may be used alone or with other anticoagulants in women with a coronary or other vascular stent if fetal safety is uncertain or if there is an increased risk of maternal bleeding.
NEWER ANTICOAGULANTS
Danaparoid
The heparinoid danaparoid (Orgaran) is an LMWH, a combination of heparan, dermatan, and chondroitin sulfate. Since it is derived from heparin, in theory it can cross-react with antiheparin antibodies, but this is generally not a problem. Danaparoid inhibits factor Xa, and monitoring is via measurement of anti-factor-Xa activity levels. It has been shown to be safe and effective in nonpregnant patients with heparin-induced thrombocytopenia.51
Although no controlled study has been published on danaparoid in pregnancy, at least 51 pregnancies in 49 patients treated with danaparoid have been reported.52 Thirty-two of the patients received danaparoid because of heparin-induced thrombocytopenia and 19 because of heparin-induced skin intolerance. These reports suggest that danaparoid does not cross the placenta53 and that it may be effective and safe during pregnancy.54 For this reason, it is probably the preferred anticoagulant in pregnant patients with heparin-induced thrombocytopenia or other serious reactions to heparin.
Unfortunately, danaparoid has two major disadvantages. First, it has a prolonged half-life and no effective reversing agent, which makes its use problematic close to the time of delivery. Second, and perhaps more relevant to this discussion, it is not readily available in the United States; it was removed from the market by its manufacturer in April 2002 for business reasons rather than because of concerns over toxicity. It is still available in Canada and Europe, and it can be obtained in special circumstances in the United States via the US Food and Drug Administration (FDA); this may be worthwhile in pregnant patients who require a nonurgent alternative to heparin.
Direct thrombin inhibitors
Lepirudin (Refludan), bivalirudin (Angiomax), and argatroban are direct thrombin inhibitors and exert their anticoagulant effect independently of antithrombin. They are given by continuous intravenous infusion, and they have a very short half-life.
Lepirudin and argatroban are typically monitored via the aPTT. Bivalirudin can be monitored with the activated clotting time, partial thromboplastin time, or INR, depending on the circumstances. None of these agents generates or cross-reacts with antibodies generated in heparin-induced thrombocytopenia. None has an antidote, but the short half-life usually obviates the need for one.
Unfortunately, pregnancy data are very sparse for all three of these new agents. Argatroban has a low molecular weight and likely crosses the placenta. Also, because these agents are given intravenously, they are not practical for long-term use in pregnancy.
Fondaparinux
Fondaparinux (Arixtra), a direct factor Xa inhibitor, binds to antithrombin, causing an irreversible conformational change that increases antithrombin’s ability to inactivate factor Xa (as do the heparins). It has no effect on factor IIa (thrombin) and does not predictably affect the aPTT. Its half-life is 17 hours, and no agent is known to reverse its anticoagulant effect, although some experts would recommend a trial of high-dose recombinant factor VIIa (Novo-Seven) in uncontrolled hemorrhage.
While not FDA-approved for treating heparin-induced thrombocytopenia, it has been used for this in some patients.55–58 Animal studies and in vitro human placental perfusion studies suggest that fondaparinux does not cross the placenta in significant amounts.49 Since danaparoid is not available in the United States, fondaparinux would likely be the first choice among the newer anticoagulants when treating heparin-induced thrombocytopenia in pregnancy.
INDICATIONS FOR ANTICOAGULANTS DURING PREGNANCY
Acute deep venous thrombosis and pulmonary embolism
Anticoagulant therapy should begin as full doses of either LMWH or intravenous unfractionated heparin. We prefer starting with LMWH, as it can be started rapidly with less need for nursing care (eg, no need to start and maintain an intravenous line and monitor the aPTT) and has excellent safety. If LMWH is selected, initial dosing should be based on the current weight (Table 2). Subsequent monitoring of the peak anti-factor-Xa activity levels (ie, 4 hours after the dose) is recommended, with the first level drawn in the first few days of treatment, and repeat levels every 1 to 3 months for the rest of treatment. As mentioned earlier, weight-based dosing has not been systematically evaluated in pregnancy.
If unfractionated heparin is the initial agent, it should be given as a bolus followed by a continuous infusion, ideally utilizing a weight-based nomogram to estimate required doses, with adjustment of the infusion rate to maintain the aPTT at 1.5 to 2.5 times the baseline value (obtained during pregnancy). After several days, the heparin may be switched to LMWH in therapeutic doses (Table 2).
Alternatively, in women approaching term or who cannot afford LMWH, anticoagulation may be continued as adjusted-dose subcutaneous unfractionated heparin, ie, two or three large daily doses of subcutaneous heparin to provide therapeutic levels of anticoagulation. The starting dose can be calculated as the total units of heparin required to maintain full anticoagulation intravenously over 24 hours, given as two or three divided doses (Table 2). The aPTT at the mid-dosing interval (eg, 6 hours after the subcutaneous dose during every-12-hour dosing) should be monitored and the dose adjusted to maintain the aPTT at 1.5 to 2.5 times the baseline value.
A therapeutic level of anticoagulation should be maintained for at least 3 months after an acute thrombotic event during pregnancy, though many physicians prefer to continue full anticoagulation for a total of 6 months. Beyond this interval, if the woman is still pregnant, the anticoagulation may be reduced in intensity, perhaps even to a prophylactic level for the duration of the pregnancy (see discussion below on prior venous thromboembolic events) (Table 2). Peripartum and postpartum anticoagulation are discussed further below.
PRIOR VENOUS THROMBOEMBOLIC EVENT
While all pregnant women are at higher risk of venous thrombosis, the overall incidence of thromboembolism is only about one event per 1,000 pregnancies. Routine thromboprophylaxis in all pregnant women is therefore not justified. However, women who have previously had a venous thromboembolic event are at a substantially higher risk of recurrent thrombosis and should be considered for thromboprophylaxis in all subsequent high-risk situations, including pregnancy.
For women on indefinite therapeutic anticoagulation (ie, because of recurrent thrombosis), full therapeutic anticoagulation with LMWH or adjusted-dose unfractionated heparin should be maintained throughout pregnancy, as described above.
Which other women should receive prophylactic anticoagulation is a topic of ongoing debate and controversy.
How great is the risk of recurrent thromboembolism?
A small observational study59 examined the risk of recurrent venous thromboembolism during subsequent pregnancies in women with a prior thrombotic event. Anticoagulation was withheld during the antepartum period and restarted briefly after delivery. Among the 125 women enrolled, recurrent venous thromboembolism occurred in 4.8%, with half of the events occurring during the antepartum period. Among those with underlying thrombophilia, the rate of recurrent venous thromboembolism was 13% (95% confidence interval [CI] 1.7%–40.5%) to 20% (95% CI 2.5%–56.5%), and those with a prior idiopathic clot without thrombophilia had an event rate of 7.7% (95% CI 0.01%–25.1%). The subgroup with a prior reversible risk factor (at the time of their initial venous thromboembolic event) and without detectable thrombophilia had no recurrent events.
This study suggests that women with prior venous thromboembolism and thrombophilia or a prior idiopathic thrombotic event are at a substantial risk of recurrent thrombotic events during pregnancy. And other data confirm the high risk of recurrent venous thromboembolism in thrombophilic pregnant women.60 These women should all be offered active antepartum and postpartum thromboprophylaxis with LMWH or unfractionated heparin (Tables 2 and 4). Women without thrombophilia but with a history of venous thromboembolism related to pregnancy or oral contraceptive use also have a substantial risk of recurrent venous thrombosis and should be offered antepartum and postpartum thromboprophylaxis.61 In contrast, women with a prior “secondary” clot, no thrombophilia, and no additional current risk factors (Table 1) appear to be at low risk of recurrent venous thromboembolism.
The risks should be discussed with these women, with an option for close clinical surveillance during pregnancy (Table 4), but with a low threshold to investigate any worrisome symptoms. Such women may also elect to take LMWH or unfractionated heparin during pregnancy.
Which heparin to use?
Prophylactic anticoagulation during pregnancy can be with either LMWH or unfractionated heparin. For most women this involves “prophylactic” dosing with the goal of maintaining a mid-interval anti-factor-Xa activity level of approximately 0.05 to 0.2 U/mL. Thromboprophylaxis with LMWH can be with lower, fixed, once-daily doses throughout pregnancy20 (Table 2), although some clinicians still prefer twice-daily dosing. The heparin should be started as soon as pregnancy is confirmed, as the pregnancy-associated increase in thrombotic risk begins by the middle of the first trimester.
To maintain effective prophylactic levels, the dose of unfractionated heparin should be increased sequentially over the trimesters62,63: approximately 5,000 units subcutaneously twice daily in the first trimester, then 7,500 units twice daily in the second trimester, and 10,000 units twice daily in the third trimester for a woman of average size.
When to add low-dose aspirin
Women with antiphospholipid antibodies, particularly those with prior recurrent pregnancy loss or fetal demise, should receive aspirin 81 mg/day in addition to heparin.39 The aspirin may be started prior to conception or when pregnancy is confirmed.
Other measures
Women on anticoagulant therapy who are at risk of recurrent venous thromboembolism should be encouraged to wear elastic compression stockings. Intermittent pneumatic compression of the legs via automated devices may be considered for women hospitalized for any reason or on bedrest.
Whichever measures are used, a high index of suspicion and a low threshold for investigating for recurrent thrombosis should be maintained throughout pregnancy and the puerperium.
PERIPARTUM AND POSTPARTUM MANAGEMENT OF ANTICOAGULATION
Heparin therapy must be interrupted temporarily during the immediate peripartum interval to minimize the risk of hemorrhage and to allow for the option of regional anesthesia. As mentioned earlier, because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, the American Society of Regional Anesthesia guidelines advise waiting to insert the needle at least 10 to 12 hours after the last prophylactic dose of LMWH, and at least 24 hours after the last therapeutic dose.31
The guidelines state that neuraxial anesthesia is not contraindicated in patients on prophylactic unfractionated heparin.31
To facilitate use of regional anesthesia in these women, therefore, options include:
- Electively stopping LMWH 24 hours before planned induction of labor
- Electively stopping prophylactic-dose LMWH or unfractionated heparin at about 38 weeks of gestation, to await spontaneous labor, or
- Switching therapeutic or prophylactic LMWH to unfractionated heparin at about 36 weeks of gestation, with instructions to discontinue the injections in the earliest stages of spontaneous labor. This aims to shorten the heparin-free period required before neuraxial anesthesia while minimizing maternal thrombotic risk.
Additional advantages to using unfractionated heparin peripartum include the option of obtaining a rapid aPTT measurement to confirm the absence of a significant ongoing heparin effect prior to regional anesthesia or delivery, and the ability to completely reverse the heparin effect with protamine sulfate if major bleeding occurs. LMWHs are only partially reversible.64
Interrupting anticoagulation after an initial thrombotic event
If therapeutic anticoagulation must be interrupted for labor within 1 month of the initial thrombotic event, the risk of recurrent thrombotic complications is high65; these women must be observed very carefully and may benefit from intravenous heparin before and after delivery. They may even merit placement of a temporary vena cava filter (particularly if less than 2 weeks have elapsed since the venous thromboembolic event and in women with a large deep venous clot burden), a procedure that has been used safely but little studied in pregnant women.66
Fluoroscopic guidance may be needed for filter placement. This exposes the fetus to radiation, but the low-level exposure at this late gestational age is unlikely to pose a significant risk. The filter may be removed within 1 to 2 weeks postpartum, assuming there are no ongoing contraindications to anticoagulation.
In the rare woman with antithrombin deficiency and a recent or prior thrombotic event, giving antithrombin concentrate during the peripartum (heparin-free) interval has been described and may be considered under the guidance of a hematologist.67
Ongoing anticoagulation is essential postpartum, as the puerperium is the period of highest day-to-day risk of thromboembolic events: about one-third of pregnancy-associated events occur during these 6 to 12 weeks.2 Heparin should be resumed 6 to 12 hours after delivery, once hemostasis is confirmed.
Options for women requiring ongoing therapeutic anticoagulation include intravenous heparin started without a bolus, to minimize bleeding risk, with aPTT measured 12 hours later, or an initial prophylactic dose of LMWH 6 to 12 hours postpartum, with therapeutic dosing resumed on postpartum day 1. If prophylactic dosing is desired, unfractionated heparin or LMWH may be given subcutaneously starting at about 6 hours postpartum.
Warfarin in the puerperium
Women may subsequently be maintained on either LMWH or unfractionated heparin, or switched to an oral anticoagulant such as warfarin. Although warfarin may appear in minute amounts in breast milk, it has not been associated with adverse events in newborns and is considered compatible with breastfeeding.68 Heparin should be continued during the initial days of warfarin therapy, until the INR is at a therapeutic level for 24 hours. Some physicians prefer to delay warfarin for several days, giving LMWH alone in the immediate postpartum period, to allow wound-healing and to reduce bleeding risk.
Postpartum, anticoagulation should be continued for at least 6 to 12 weeks, at which point the physiologic changes in the coagulation system related to pregnancy will have returned to normal.
THROMBOPHILIA WITHOUT A PREVIOUS THROMBOEMBOLIC EVENT
Over the last 5 to 10 years, practitioners have been seeing many more young women with genetic or acquired thrombophilias who have never had a venous thromboembolic event. Physicians must advise these women about their risk of thromboembolic events during pregnancy and about the appropriateness of anticoagulant use.
Thrombophilias are often detected in women who develop venous thrombosis during pregnancy,69–71 but they are also very common in the general population (around 15%). While women with thrombophilia are at above-average risk of venous thromboembolism during pregnancy, the magnitude of risk in an individual patient is often difficult to estimate.
Data suggest that some types of thrombophilia confer greater thrombotic risk than others. McColl et al72 derived risk estimates for a primary event in women with several of the disorders: 0.23% in women heterozygous for the factor V Leiden mutation, 0.88% in women with protein C deficiency, and 2.4% to 35.7% in women with antithrombin deficiency. A case-control study70 found that all thrombophilic states were more common in women with pregnancy-associated venous thromboembolism than in healthy pregnant controls, except those with the MTHFR mutation and protein S deficiency. The estimated risk during pregnancy was 0.03% in women with no defect, 0.1% in women with protein C deficiency, 0.25% in women with the factor V Leiden mutation, 0.4% in those with antithrombin deficiency, 0.5% in those with the prothrombin gene mutation, and 4.6% in those with both factor V Leiden and prothrombin gene mutations.
Routine anticoagulation not advised in pregnant thrombophilic women
Because the risk of a primary venous thromboembolic event is less than 1% for most thrombophilic women, routine anticoagulant therapy does not seem prudent for this indication. Given the low absolute risk of venous thromboembolism, the cost and potential side effects of anticoagulant use are difficult to justify.
The women who seem at higher risk and in whom anticoagulation should be considered include those with antithrombin deficiency; those with high-titer anticardiolipin antibodies or a lupus anticoagulant antibody (treat with heparin and low-dose aspirin); those with combined thrombophilic defects or who are homozygotes for the factor V Leiden or prothrombin gene mutations; and those with multiple other current risk factors for venous thromboembolism (Table 1).
Since anticoagulants for primary prevention of adverse pregnancy outcomes in thrombophilic women have not yet been shown to have a definitive benefit, they are not recommended for this purpose.
ADVERSE PREGNANCY OUTCOMES IN WOMEN WITH THROMBOPHILIAS
Women with antiphospholipid antibodies and a previous poor obstetric outcome are clearly at increased risk of recurrent adverse pregnancy outcomes such as recurrent spontaneous abortion, unexplained fetal death, placental insufficiency, and early or severe preeclampsia. In such women who have both antiphospholipid antibodies and a history of venous thromboembolism or adverse pregnancy outcome, treatment during subsequent pregnancy with low-dose aspirin and prophylactic-dose LMWH or unfractionated heparin improves pregnancy outcomes.36–42 Women with antiphospholipid antibodies without previous thrombosis or pregnancy complications may also be at increased risk, but it is unclear whether thromboprophylaxis improves their outcomes.
Recent epidemiologic data reveal that women with other thrombophilic conditions also are at increased risk of early, severe preeclampsia73 as well as other pregnancy complications, including recurrent pregnancy loss, placental abruption, fetal growth restriction, and stillbirth.74 A recent meta-analysis75 looked at individual thrombophilias and found that factor V Leiden and prothrombin gene mutations were associated with recurrent fetal loss, stillbirth, and preeclampsia; that protein S deficiency was associated with recurrent fetal loss and stillbirth; that antiphospholipid antibodies were associated with recurrent pregnancy loss, preeclampsia, and intrauterine growth restriction; that the MTHFR mutation (homozygous) was associated with preeclampsia; and that protein C and antithrombin deficiencies were not significantly associated with adverse pregnancy outcomes. Data were scant for some of the rarer thrombophilias.75
Several recent small studies76–78 suggest that anticoagulants may improve pregnancy outcomes in women with genetic thrombophilias and recurrent pregnancy loss. These findings have not yet been confirmed in high-quality clinical trials, but such trials are under way. It is still unclear whether anticoagulants also reduce the risk of other adverse pregnancy outcomes associated with thrombophilias.
The current American College of Chest Physicians guidelines recommend testing of women with adverse pregnancy outcomes (recurrent pregnancy loss, prior severe or recurrent preeclampsia, abruptions, or otherwise unexplained intrauterine death) for congenital thrombophilias and antiphospholipid antibodies, and offering treatment to such women, if thrombophilic, with low-dose aspirin plus prophylactic heparin (unfractionated or LMWH).22 The authors of the guidelines admit that the evidence for this recommendation is weak, but they argue that the heparin will also serve as thromboprophylaxis in this high-risk group. Hopefully, the randomized clinical trials currently under way will provide clearer guidance regarding the most appropriate therapy in this difficult clinical situation.
MECHANICAL HEART VALVES
Internists may occasionally encounter a woman with a mechanical heart valve prosthesis who is either pregnant or is planning a pregnancy and therefore needs advice regarding optimal anticoagulant management. This should generally be undertaken in a multi-disciplinary fashion, with input from cardiology, hematology, and maternal-fetal medicine. The substantial maternal and fetal risks and the lack of definitive data on which to base treatment decisions make it a treacherous and stressful undertaking. Nonetheless, all internists should have a basic understanding of the complex issues regarding this management.
Outside of pregnancy, oral anticoagulants are the mainstay of therapy for patients with mechanical heart valves. Unfortunately, as discussed above, the use of these agents during pregnancy carries a risk of teratogenicity and toxic fetal effects and increases the risk of pregnancy loss and maternal hemorrhage. Heparins have been used in this setting for many years, but data on their efficacy and safety are very limited, and there are numerous reports of catastrophic maternal thrombotic complications.79,80
A systematic review of anticoagulation in pregnant women with prosthetic heart valves34 found very limited data on heparin use throughout pregnancy. Women maintained on warfarin vs heparin between pregnancy weeks 6 and 12 had higher rates of congenital anomalies (6.4% with warfarin vs 3.4% with heparin) and total fetal wastage (33.6% vs 26.5%). The warfarin group had fewer maternal thromboembolic complications (3.9% vs 9.2%), however, and a slightly lower rate of maternal death (1.8% vs 4.2%). Most of the women had higher-risk older-generation valves in the mitral position.
Recent data on LMWH consist mainly of case reports and case series,81 with a likely bias to publication of worse outcomes. Controlled trials in this area will be difficult to conduct. Still, aggressive anticoagulation with LMWH or unfractionated heparin, with close monitoring of the intensity of anticoagulation, may be safe and effective for pregnant women with newer-generation mechanical heart valves.82 A recent consensus statement22 suggested several regimens for pregnant women with mechanical heart valves:
- Twice-daily LMWH throughout pregnancy, with the dose adjusted either by weight, or to keep the 4-hour postinjection anti-factor-Xa activity level around 1.0 to 1.2 U/mL
- Aggressive adjusted-dose unfractionated heparin throughout pregnancy, given subcutaneously every 12 hours and adjusted to keep the mid-interval aPTT at least twice the control value or to attain a mid-interval anti-factor-Xa activity level of 0.35 to 0.70 U/mL
- Unfractionated heparin or LMWH (as above) until gestation week 13, then warfarin until the middle of the third trimester, and then heparin again.22
The authors also recommended adding low-dose aspirin (75–162 mg/day) in high-risk women.22
These options all seem reasonable, given our current knowledge, though warfarin use during pregnancy should be restricted to very-high-risk situations, such as women with older-generation mitral prostheses. LM-WHs may become the preferred therapy for this indication once further controlled data regarding their efficacy and safety become available.
Anticoagulation is essential in a wide variety of conditions in women of child-bearing age. Some, such as venous thromboembolism, occur more often during pregnancy. Others, such as recurrent fetal loss in the setting of antiphospholipid antibodies, are specific to pregnancy.
While anticoagulants are useful in many circumstances, their use during pregnancy increases the risk of hemorrhage and other adverse effects on the mother and the fetus. Treatment with anticoagulants during pregnancy must therefore be carefully considered, with judicious selection of the agent, and with reflection on the physiologic changes of pregnancy to ensure appropriate dosing. In this article, we review these issues.
WHY IS THROMBOTIC RISK HIGHER DURING PREGNANCY?
Venous thromboembolism is among the leading causes of maternal death in developed countries.1–3 Modern care has dramatically reduced the risk of maternal death from hemorrhage, infection, and hypertension, but rates of morbidity and death from thrombosis have remained stable or increased in recent years.4
- Much higher levels of fibrinogen and factors VII, VIII, IX, and X
- Lower levels of protein S and increased resistance to activated protein C
- Impaired fibrinolysis, due to inhibitors derived from the placenta.
Acquired antithrombin deficiency may also occur in high-proteinuric states such as nephrotic syndrome or preeclampsia, further increasing thrombotic risk. Pooling of venous blood, caused by progesterone-mediated venous dilation and compounded by compression of the inferior vena cava by the uterus in later pregnancy, also increases thrombotic risk. And endothelial disruption of the pelvic vessels may occur during delivery, particularly during cesarean section.
Additional factors that increase thrombotic risk include immobilization, such as bed rest for pregnancy complications; surgery, including cesarean section; ovarian hyperstimulation during gonadotropin use for in vitro fertilization; trauma; malignancy; and hereditary or acquired hypercoagulable states.6 These hypercoagulable states include deficiencies of antithrombin or the intrinsic anticoagulant proteins C or S; resistance to activated protein C, usually due to the factor V Leiden mutation; the PT20210A mutation of the prothrombin gene; hyperhomocystinemia due to mutation of the methyltetrahydrofolate reductase (MTHFR) gene; and the sustained presence of antiphospholipid antibodies, including lupus anticoagulant antibodies, sometimes also with moderately high titers of anticardiolipin or beta-2-glycoprotein I antibodies.
Other conditions that increase thrombotic risk include hyperemesis gravidarum, obesity, inflammatory bowel disease, infection, smoking, and indwelling intravenous catheters.6 Given the multitude of risk factors, pregnant women have a risk of thrombotic complications three to five times higher than nonpregnant women.7
HEPARIN USE DURING PREGNANCY
Low-molecular-weight heparins (LMWHs)8 and unfractionated heparin bind to anti-thrombin and thus change the shape of the antithrombin molecule, dramatically increasing its interaction with the clotting factors Xa and prothrombin (factor II). The enhanced clearance of these procoagulant proteins leads to the anticoagulant effect. Unfractionated heparin has roughly equivalent interaction with factors Xa and II and prolongs the activated partial thromboplastin time (aPTT), which is therefore used to monitor the intensity of anticoagulation.
LMWHs, on the other hand, interact relatively little with factor II and do not predictably prolong the aPTT. Monitoring their effect is therefore more difficult and requires direct measurement of anti-factor-Xa activity. This test is widely available, but it is time-consuming (it takes several hours and results may not be available within 24 hours if the test is requested “after hours”), and therefore it is of limited use in the acute clinical setting. While weight-based dosing of LMWHs is reliable and safe in nonpregnant patients, it has not yet been validated for pregnant women.
Unfractionated heparin has been used for decades for many indications during pregnancy. It is a large molecule, so it does not cross the placenta and thus, in contrast to the coumarin derivatives, does not cause teratogenesis or toxic fetal effects. Its main limitations in pregnancy are its inconvenient dosing (at least twice daily when given subcutaneously) and its potential maternal adverse effects (mainly osteoporosis and heparin-induced thrombocytopenia).
Over the last 10 years LMWHs have become the preferred anticoagulants for treating and preventing thromboembolism in all patients. They are equivalent or superior to unfractionated heparin in efficacy and safety in the initial treatment of acute deep venous thrombosis9,10 and pulmonary embolism11,12 outside of pregnancy. While comparative data are much less robust in pregnant patients, several series have confirmed the safety and efficacy of LMWHs in pregnancy.13–15 LMWHs do not cross the placenta15–17 and thus have a fetal safety profile equivalent to that of unfractionated heparin.
Pregnancy alters metabolism of LMWHs
The physiologic changes of pregnancy alter the metabolism of LMWH, resulting in lower peak levels and a higher rate of clearance,18,19 and so a pregnant woman may need higher doses or more frequent dosing.
Recent evidence suggests that thromboprophylaxis can be done with lower, fixed, once-daily doses of LMWH throughout pregnancy,20 although some clinicians still prefer twice-daily dosing (particularly during the latter half of pregnancy).
Pending more research on weight-based dosing of LMWH in pregnancy, anti-factor- Xa activity levels should be measured after treatment is started and every 1 to 3 months thereafter during pregnancy.21 Doses should be adjusted to keep the peak anti-Xa level (ie, 4 hours after the dose) at 0.5 to 1.2 U/mL.22
Heparin-induced thrombocytopenia
Type-2 heparin-induced thrombocytopenia is an uncommon but serious adverse effect of unfractionated heparin therapy (and, less commonly of LMWH), caused by heparin-dependent immunoglobulin G (IgG) antibodies that activate platelets via their Fc receptors, potentially precipitating life-threatening arterial or venous thrombosis.
In a trial in nonpregnant orthopedic patients,23 clinical heparin-induced thrombocytopenia occurred in 2.7% of patients receiving unfractionated heparin vs 0% of those receiving LMWH; heparin-dependent IgG was present in 7.8% vs 2.2%, respectively.
Fortunately, heparin-induced thrombocytopenia seems to be very rare in pregnancy: two recent prospective series evaluating prolonged LMWH use in pregnancy13,15 revealed no episodes of this disease. Nonetheless, it is reasonable to measure the platelet count once or twice weekly during the first few weeks of LMWH use and less often thereafter, unless symptoms of heparin-induced thrombocytopenia develop. In pregnant women with heparin-induced thrombocytopenia or heparin-related skin reactions, other anticoagulants must be considered24 (see discussion later).
Heparin-induced osteoporosis
Heparin-induced osteoporosis, a potential effect of prolonged heparin therapy, is of concern, given the prolonged duration and high doses of unfractionated heparin often needed to treat venous thromboembolism during pregnancy. Several studies found significant loss of bone mineral density in the proximal femur25 and lumbar spine26 during extended use of unfractionated heparin in pregnancy.
Fortunately, LMWH appears to be much safer with respect to bone loss. Three recent studies27–30 evaluated the use of LMWH for extended periods during pregnancy, and none found any greater loss of bone mineral density than that seen in normal pregnant controls. Giving supplemental calcium (1,000–1,500 mg/day) and vitamin D (400–1,000 IU/day) concomitantly with unfractionated heparin or LMWH in pregnancy is advisable to further reduce the risk.
Interrupt heparin to permit regional anesthesia
Heparin therapy should be temporarily stopped during the immediate peripartum interval to minimize the risk of hemorrhage and to permit regional anesthesia. Because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, many anesthetists will not perform neuraxial regional anesthesia in women who have recently received heparin.
Since unfractionated heparin has a relatively short duration of action, the American Society of Regional Anesthesia states that subcutaneous unfractionated heparin prophylaxis is not a contraindication to neuraxial regional anesthesia.31 However, LMWHs should be stopped for at least 12 to 24 hours before regional anesthesia can be considered safe. This issue is discussed in more detail in the section on peripartum and postpartum management of anticoagulation, below.
In summary, LMWH during pregnancy offers a number of advantages over unfractionated heparin: equivalent efficacy, once- or twice-daily dosing, lower risk of heparin-induced thrombocytopenia and osteoporosis, and less-intensive monitoring. Unfractionated heparin can be offered to women who cannot afford LMWH (which costs four to five times more), and it may be used peripartum to reduce hemorrhagic risk and to permit regional anesthesia.
COUMARINS
Coumarins are the mainstay of anticoagulant therapy in most nonpregnant women beyond the immediate thrombotic period.
Warfarin (Coumadin) is the most widely used coumarin because it has a predictable onset and duration of action and excellent bioavailability.32 Others, such as acenocoumarol (Sintrom) and phenprocoumon (Marcoumar), are used more outside the United States but can be ordered or brought into the United States.
Coumarins interfere with vitamin K metabolism, inhibiting the generation of vitamin-K-dependent procoagulant proteins (factors II, VII, IX, and X) and thereby preventing clotting. They also inhibit the formation of the vitamin-K-dependent intrinsic anticoagulant proteins C and S.
Major bleeding is the most significant side effect of coumarin therapy, occurring at a rate of 4% to 6% over 3 months when the prothrombin time is maintained at an international normalized ratio (INR) of 2 to 3,33 and more often if the INR is higher.
Other issues with warfarin are the effect of variations in dietary vitamin K intake on anticoagulation and potential drug interactions that may alter the anticoagulant effect. Thus, the INR needs to be monitored closely.
Risks to the fetus and the mother
Unlike the heparins, coumarins freely cross the placenta and thus pose a risk of teratogenicity. A cluster of fetal malformations including “warfarin embryopathy” (nasal bone hypoplasia and chondrodysplasia punctata) can occur when the drug is used between 6 and 12 weeks of gestation. Warfarin embryopathy may be avoided by stopping warfarin prior to 6 weeks from the onset of the last menstrual period (ie, 6-week “menstrual age” or 4-week gestational age34).
Later in pregnancy, warfarin is associated with potential fetal bleeding complications leading to central nervous system abnormalities, increased rates of intrauterine fetal death, and pregnancy loss. In pregnant women with mechanical cardiac valve prostheses who received oral anticoagulants throughout pregnancy, the incidence of congenital anomalies was 6.4% to 10.2%.35 Fetal demise (spontaneous abortion, stillbirth, neonatal death) was also very common (29.7% to 33.6% of pregnancies) in coumarin-treated women.
Severe maternal hemorrhage may also occur in pregnant women on oral anticoagulants, particularly those who remain fully anticoagulated around the time of labor and delivery.
General caveats to warfarin in pregnancy
Because of the many maternal and fetal concerns, oral anticoagulant use in pregnancy is largely restricted to women with older-generation prosthetic heart valves in whom the very high maternal thrombotic risk may outweigh the risk of maternal and fetal side effects.
While there are limited data on warfarin use in pregnant women with antiphospholipid syndrome,36 warfarin use in such patients should be considered only for those at highest risk and with careful informed consent. These issues are discussed further below in the section on mechanical heart valve prostheses.
ANTIPLATELET DRUGS
Aspirin is an antiplatelet agent rather than an anticoagulant. Although considered inadequate for preventing venous thrombosis in high-risk groups when used alone, aspirin can moderately reduce the risk of deep venous thrombosis and pulmonary embolism in nonpregnant patients.37 It also has a well-accepted role in preventing arterial thrombotic events, ie, coronary artery disease and stroke.38
Low-dose aspirin (≤ 100 mg/day) has been extensively evaluated during pregnancy39–41 and has been shown to be safe and effective in reducing the risk of preeclampsia in high-risk women39 and in treating women with antiphospholipid antibodies and recurrent pregnancy loss42 (in conjunction with prophylactic doses of heparin). Although higher doses of aspirin and other nonsteroidal anti-inflammatory drugs can be toxic to the fetus, low doses have been shown to be safe throughout pregnancy.43
Dipyridamole (Persantine) has been studied extensively in pregnancy, and while it appears to be safe, it has not found a well-defined therapeutic role.
Other antiplatelet drugs have been only rarely used, and data on their safety and efficacy during pregnancy are limited to case reports, for example, on ticlopidine44 (Ticlid) and clopidogrel45,46 (Plavix) given during pregnancy in women with cardiac disease. These drugs do not appear to be major teratogens or to cause specific fetal harm. Their use may be reasonable in some high-risk situations, such as recurrent thrombotic stroke despite aspirin therapy. They may be used alone or with other anticoagulants in women with a coronary or other vascular stent if fetal safety is uncertain or if there is an increased risk of maternal bleeding.
NEWER ANTICOAGULANTS
Danaparoid
The heparinoid danaparoid (Orgaran) is an LMWH, a combination of heparan, dermatan, and chondroitin sulfate. Since it is derived from heparin, in theory it can cross-react with antiheparin antibodies, but this is generally not a problem. Danaparoid inhibits factor Xa, and monitoring is via measurement of anti-factor-Xa activity levels. It has been shown to be safe and effective in nonpregnant patients with heparin-induced thrombocytopenia.51
Although no controlled study has been published on danaparoid in pregnancy, at least 51 pregnancies in 49 patients treated with danaparoid have been reported.52 Thirty-two of the patients received danaparoid because of heparin-induced thrombocytopenia and 19 because of heparin-induced skin intolerance. These reports suggest that danaparoid does not cross the placenta53 and that it may be effective and safe during pregnancy.54 For this reason, it is probably the preferred anticoagulant in pregnant patients with heparin-induced thrombocytopenia or other serious reactions to heparin.
Unfortunately, danaparoid has two major disadvantages. First, it has a prolonged half-life and no effective reversing agent, which makes its use problematic close to the time of delivery. Second, and perhaps more relevant to this discussion, it is not readily available in the United States; it was removed from the market by its manufacturer in April 2002 for business reasons rather than because of concerns over toxicity. It is still available in Canada and Europe, and it can be obtained in special circumstances in the United States via the US Food and Drug Administration (FDA); this may be worthwhile in pregnant patients who require a nonurgent alternative to heparin.
Direct thrombin inhibitors
Lepirudin (Refludan), bivalirudin (Angiomax), and argatroban are direct thrombin inhibitors and exert their anticoagulant effect independently of antithrombin. They are given by continuous intravenous infusion, and they have a very short half-life.
Lepirudin and argatroban are typically monitored via the aPTT. Bivalirudin can be monitored with the activated clotting time, partial thromboplastin time, or INR, depending on the circumstances. None of these agents generates or cross-reacts with antibodies generated in heparin-induced thrombocytopenia. None has an antidote, but the short half-life usually obviates the need for one.
Unfortunately, pregnancy data are very sparse for all three of these new agents. Argatroban has a low molecular weight and likely crosses the placenta. Also, because these agents are given intravenously, they are not practical for long-term use in pregnancy.
Fondaparinux
Fondaparinux (Arixtra), a direct factor Xa inhibitor, binds to antithrombin, causing an irreversible conformational change that increases antithrombin’s ability to inactivate factor Xa (as do the heparins). It has no effect on factor IIa (thrombin) and does not predictably affect the aPTT. Its half-life is 17 hours, and no agent is known to reverse its anticoagulant effect, although some experts would recommend a trial of high-dose recombinant factor VIIa (Novo-Seven) in uncontrolled hemorrhage.
While not FDA-approved for treating heparin-induced thrombocytopenia, it has been used for this in some patients.55–58 Animal studies and in vitro human placental perfusion studies suggest that fondaparinux does not cross the placenta in significant amounts.49 Since danaparoid is not available in the United States, fondaparinux would likely be the first choice among the newer anticoagulants when treating heparin-induced thrombocytopenia in pregnancy.
INDICATIONS FOR ANTICOAGULANTS DURING PREGNANCY
Acute deep venous thrombosis and pulmonary embolism
Anticoagulant therapy should begin as full doses of either LMWH or intravenous unfractionated heparin. We prefer starting with LMWH, as it can be started rapidly with less need for nursing care (eg, no need to start and maintain an intravenous line and monitor the aPTT) and has excellent safety. If LMWH is selected, initial dosing should be based on the current weight (Table 2). Subsequent monitoring of the peak anti-factor-Xa activity levels (ie, 4 hours after the dose) is recommended, with the first level drawn in the first few days of treatment, and repeat levels every 1 to 3 months for the rest of treatment. As mentioned earlier, weight-based dosing has not been systematically evaluated in pregnancy.
If unfractionated heparin is the initial agent, it should be given as a bolus followed by a continuous infusion, ideally utilizing a weight-based nomogram to estimate required doses, with adjustment of the infusion rate to maintain the aPTT at 1.5 to 2.5 times the baseline value (obtained during pregnancy). After several days, the heparin may be switched to LMWH in therapeutic doses (Table 2).
Alternatively, in women approaching term or who cannot afford LMWH, anticoagulation may be continued as adjusted-dose subcutaneous unfractionated heparin, ie, two or three large daily doses of subcutaneous heparin to provide therapeutic levels of anticoagulation. The starting dose can be calculated as the total units of heparin required to maintain full anticoagulation intravenously over 24 hours, given as two or three divided doses (Table 2). The aPTT at the mid-dosing interval (eg, 6 hours after the subcutaneous dose during every-12-hour dosing) should be monitored and the dose adjusted to maintain the aPTT at 1.5 to 2.5 times the baseline value.
A therapeutic level of anticoagulation should be maintained for at least 3 months after an acute thrombotic event during pregnancy, though many physicians prefer to continue full anticoagulation for a total of 6 months. Beyond this interval, if the woman is still pregnant, the anticoagulation may be reduced in intensity, perhaps even to a prophylactic level for the duration of the pregnancy (see discussion below on prior venous thromboembolic events) (Table 2). Peripartum and postpartum anticoagulation are discussed further below.
PRIOR VENOUS THROMBOEMBOLIC EVENT
While all pregnant women are at higher risk of venous thrombosis, the overall incidence of thromboembolism is only about one event per 1,000 pregnancies. Routine thromboprophylaxis in all pregnant women is therefore not justified. However, women who have previously had a venous thromboembolic event are at a substantially higher risk of recurrent thrombosis and should be considered for thromboprophylaxis in all subsequent high-risk situations, including pregnancy.
For women on indefinite therapeutic anticoagulation (ie, because of recurrent thrombosis), full therapeutic anticoagulation with LMWH or adjusted-dose unfractionated heparin should be maintained throughout pregnancy, as described above.
Which other women should receive prophylactic anticoagulation is a topic of ongoing debate and controversy.
How great is the risk of recurrent thromboembolism?
A small observational study59 examined the risk of recurrent venous thromboembolism during subsequent pregnancies in women with a prior thrombotic event. Anticoagulation was withheld during the antepartum period and restarted briefly after delivery. Among the 125 women enrolled, recurrent venous thromboembolism occurred in 4.8%, with half of the events occurring during the antepartum period. Among those with underlying thrombophilia, the rate of recurrent venous thromboembolism was 13% (95% confidence interval [CI] 1.7%–40.5%) to 20% (95% CI 2.5%–56.5%), and those with a prior idiopathic clot without thrombophilia had an event rate of 7.7% (95% CI 0.01%–25.1%). The subgroup with a prior reversible risk factor (at the time of their initial venous thromboembolic event) and without detectable thrombophilia had no recurrent events.
This study suggests that women with prior venous thromboembolism and thrombophilia or a prior idiopathic thrombotic event are at a substantial risk of recurrent thrombotic events during pregnancy. And other data confirm the high risk of recurrent venous thromboembolism in thrombophilic pregnant women.60 These women should all be offered active antepartum and postpartum thromboprophylaxis with LMWH or unfractionated heparin (Tables 2 and 4). Women without thrombophilia but with a history of venous thromboembolism related to pregnancy or oral contraceptive use also have a substantial risk of recurrent venous thrombosis and should be offered antepartum and postpartum thromboprophylaxis.61 In contrast, women with a prior “secondary” clot, no thrombophilia, and no additional current risk factors (Table 1) appear to be at low risk of recurrent venous thromboembolism.
The risks should be discussed with these women, with an option for close clinical surveillance during pregnancy (Table 4), but with a low threshold to investigate any worrisome symptoms. Such women may also elect to take LMWH or unfractionated heparin during pregnancy.
Which heparin to use?
Prophylactic anticoagulation during pregnancy can be with either LMWH or unfractionated heparin. For most women this involves “prophylactic” dosing with the goal of maintaining a mid-interval anti-factor-Xa activity level of approximately 0.05 to 0.2 U/mL. Thromboprophylaxis with LMWH can be with lower, fixed, once-daily doses throughout pregnancy20 (Table 2), although some clinicians still prefer twice-daily dosing. The heparin should be started as soon as pregnancy is confirmed, as the pregnancy-associated increase in thrombotic risk begins by the middle of the first trimester.
To maintain effective prophylactic levels, the dose of unfractionated heparin should be increased sequentially over the trimesters62,63: approximately 5,000 units subcutaneously twice daily in the first trimester, then 7,500 units twice daily in the second trimester, and 10,000 units twice daily in the third trimester for a woman of average size.
When to add low-dose aspirin
Women with antiphospholipid antibodies, particularly those with prior recurrent pregnancy loss or fetal demise, should receive aspirin 81 mg/day in addition to heparin.39 The aspirin may be started prior to conception or when pregnancy is confirmed.
Other measures
Women on anticoagulant therapy who are at risk of recurrent venous thromboembolism should be encouraged to wear elastic compression stockings. Intermittent pneumatic compression of the legs via automated devices may be considered for women hospitalized for any reason or on bedrest.
Whichever measures are used, a high index of suspicion and a low threshold for investigating for recurrent thrombosis should be maintained throughout pregnancy and the puerperium.
PERIPARTUM AND POSTPARTUM MANAGEMENT OF ANTICOAGULATION
Heparin therapy must be interrupted temporarily during the immediate peripartum interval to minimize the risk of hemorrhage and to allow for the option of regional anesthesia. As mentioned earlier, because of the theoretical risk of paraspinal hemorrhage in women receiving heparin who undergo epidural or spinal anesthesia, the American Society of Regional Anesthesia guidelines advise waiting to insert the needle at least 10 to 12 hours after the last prophylactic dose of LMWH, and at least 24 hours after the last therapeutic dose.31
The guidelines state that neuraxial anesthesia is not contraindicated in patients on prophylactic unfractionated heparin.31
To facilitate use of regional anesthesia in these women, therefore, options include:
- Electively stopping LMWH 24 hours before planned induction of labor
- Electively stopping prophylactic-dose LMWH or unfractionated heparin at about 38 weeks of gestation, to await spontaneous labor, or
- Switching therapeutic or prophylactic LMWH to unfractionated heparin at about 36 weeks of gestation, with instructions to discontinue the injections in the earliest stages of spontaneous labor. This aims to shorten the heparin-free period required before neuraxial anesthesia while minimizing maternal thrombotic risk.
Additional advantages to using unfractionated heparin peripartum include the option of obtaining a rapid aPTT measurement to confirm the absence of a significant ongoing heparin effect prior to regional anesthesia or delivery, and the ability to completely reverse the heparin effect with protamine sulfate if major bleeding occurs. LMWHs are only partially reversible.64
Interrupting anticoagulation after an initial thrombotic event
If therapeutic anticoagulation must be interrupted for labor within 1 month of the initial thrombotic event, the risk of recurrent thrombotic complications is high65; these women must be observed very carefully and may benefit from intravenous heparin before and after delivery. They may even merit placement of a temporary vena cava filter (particularly if less than 2 weeks have elapsed since the venous thromboembolic event and in women with a large deep venous clot burden), a procedure that has been used safely but little studied in pregnant women.66
Fluoroscopic guidance may be needed for filter placement. This exposes the fetus to radiation, but the low-level exposure at this late gestational age is unlikely to pose a significant risk. The filter may be removed within 1 to 2 weeks postpartum, assuming there are no ongoing contraindications to anticoagulation.
In the rare woman with antithrombin deficiency and a recent or prior thrombotic event, giving antithrombin concentrate during the peripartum (heparin-free) interval has been described and may be considered under the guidance of a hematologist.67
Ongoing anticoagulation is essential postpartum, as the puerperium is the period of highest day-to-day risk of thromboembolic events: about one-third of pregnancy-associated events occur during these 6 to 12 weeks.2 Heparin should be resumed 6 to 12 hours after delivery, once hemostasis is confirmed.
Options for women requiring ongoing therapeutic anticoagulation include intravenous heparin started without a bolus, to minimize bleeding risk, with aPTT measured 12 hours later, or an initial prophylactic dose of LMWH 6 to 12 hours postpartum, with therapeutic dosing resumed on postpartum day 1. If prophylactic dosing is desired, unfractionated heparin or LMWH may be given subcutaneously starting at about 6 hours postpartum.
Warfarin in the puerperium
Women may subsequently be maintained on either LMWH or unfractionated heparin, or switched to an oral anticoagulant such as warfarin. Although warfarin may appear in minute amounts in breast milk, it has not been associated with adverse events in newborns and is considered compatible with breastfeeding.68 Heparin should be continued during the initial days of warfarin therapy, until the INR is at a therapeutic level for 24 hours. Some physicians prefer to delay warfarin for several days, giving LMWH alone in the immediate postpartum period, to allow wound-healing and to reduce bleeding risk.
Postpartum, anticoagulation should be continued for at least 6 to 12 weeks, at which point the physiologic changes in the coagulation system related to pregnancy will have returned to normal.
THROMBOPHILIA WITHOUT A PREVIOUS THROMBOEMBOLIC EVENT
Over the last 5 to 10 years, practitioners have been seeing many more young women with genetic or acquired thrombophilias who have never had a venous thromboembolic event. Physicians must advise these women about their risk of thromboembolic events during pregnancy and about the appropriateness of anticoagulant use.
Thrombophilias are often detected in women who develop venous thrombosis during pregnancy,69–71 but they are also very common in the general population (around 15%). While women with thrombophilia are at above-average risk of venous thromboembolism during pregnancy, the magnitude of risk in an individual patient is often difficult to estimate.
Data suggest that some types of thrombophilia confer greater thrombotic risk than others. McColl et al72 derived risk estimates for a primary event in women with several of the disorders: 0.23% in women heterozygous for the factor V Leiden mutation, 0.88% in women with protein C deficiency, and 2.4% to 35.7% in women with antithrombin deficiency. A case-control study70 found that all thrombophilic states were more common in women with pregnancy-associated venous thromboembolism than in healthy pregnant controls, except those with the MTHFR mutation and protein S deficiency. The estimated risk during pregnancy was 0.03% in women with no defect, 0.1% in women with protein C deficiency, 0.25% in women with the factor V Leiden mutation, 0.4% in those with antithrombin deficiency, 0.5% in those with the prothrombin gene mutation, and 4.6% in those with both factor V Leiden and prothrombin gene mutations.
Routine anticoagulation not advised in pregnant thrombophilic women
Because the risk of a primary venous thromboembolic event is less than 1% for most thrombophilic women, routine anticoagulant therapy does not seem prudent for this indication. Given the low absolute risk of venous thromboembolism, the cost and potential side effects of anticoagulant use are difficult to justify.
The women who seem at higher risk and in whom anticoagulation should be considered include those with antithrombin deficiency; those with high-titer anticardiolipin antibodies or a lupus anticoagulant antibody (treat with heparin and low-dose aspirin); those with combined thrombophilic defects or who are homozygotes for the factor V Leiden or prothrombin gene mutations; and those with multiple other current risk factors for venous thromboembolism (Table 1).
Since anticoagulants for primary prevention of adverse pregnancy outcomes in thrombophilic women have not yet been shown to have a definitive benefit, they are not recommended for this purpose.
ADVERSE PREGNANCY OUTCOMES IN WOMEN WITH THROMBOPHILIAS
Women with antiphospholipid antibodies and a previous poor obstetric outcome are clearly at increased risk of recurrent adverse pregnancy outcomes such as recurrent spontaneous abortion, unexplained fetal death, placental insufficiency, and early or severe preeclampsia. In such women who have both antiphospholipid antibodies and a history of venous thromboembolism or adverse pregnancy outcome, treatment during subsequent pregnancy with low-dose aspirin and prophylactic-dose LMWH or unfractionated heparin improves pregnancy outcomes.36–42 Women with antiphospholipid antibodies without previous thrombosis or pregnancy complications may also be at increased risk, but it is unclear whether thromboprophylaxis improves their outcomes.
Recent epidemiologic data reveal that women with other thrombophilic conditions also are at increased risk of early, severe preeclampsia73 as well as other pregnancy complications, including recurrent pregnancy loss, placental abruption, fetal growth restriction, and stillbirth.74 A recent meta-analysis75 looked at individual thrombophilias and found that factor V Leiden and prothrombin gene mutations were associated with recurrent fetal loss, stillbirth, and preeclampsia; that protein S deficiency was associated with recurrent fetal loss and stillbirth; that antiphospholipid antibodies were associated with recurrent pregnancy loss, preeclampsia, and intrauterine growth restriction; that the MTHFR mutation (homozygous) was associated with preeclampsia; and that protein C and antithrombin deficiencies were not significantly associated with adverse pregnancy outcomes. Data were scant for some of the rarer thrombophilias.75
Several recent small studies76–78 suggest that anticoagulants may improve pregnancy outcomes in women with genetic thrombophilias and recurrent pregnancy loss. These findings have not yet been confirmed in high-quality clinical trials, but such trials are under way. It is still unclear whether anticoagulants also reduce the risk of other adverse pregnancy outcomes associated with thrombophilias.
The current American College of Chest Physicians guidelines recommend testing of women with adverse pregnancy outcomes (recurrent pregnancy loss, prior severe or recurrent preeclampsia, abruptions, or otherwise unexplained intrauterine death) for congenital thrombophilias and antiphospholipid antibodies, and offering treatment to such women, if thrombophilic, with low-dose aspirin plus prophylactic heparin (unfractionated or LMWH).22 The authors of the guidelines admit that the evidence for this recommendation is weak, but they argue that the heparin will also serve as thromboprophylaxis in this high-risk group. Hopefully, the randomized clinical trials currently under way will provide clearer guidance regarding the most appropriate therapy in this difficult clinical situation.
MECHANICAL HEART VALVES
Internists may occasionally encounter a woman with a mechanical heart valve prosthesis who is either pregnant or is planning a pregnancy and therefore needs advice regarding optimal anticoagulant management. This should generally be undertaken in a multi-disciplinary fashion, with input from cardiology, hematology, and maternal-fetal medicine. The substantial maternal and fetal risks and the lack of definitive data on which to base treatment decisions make it a treacherous and stressful undertaking. Nonetheless, all internists should have a basic understanding of the complex issues regarding this management.
Outside of pregnancy, oral anticoagulants are the mainstay of therapy for patients with mechanical heart valves. Unfortunately, as discussed above, the use of these agents during pregnancy carries a risk of teratogenicity and toxic fetal effects and increases the risk of pregnancy loss and maternal hemorrhage. Heparins have been used in this setting for many years, but data on their efficacy and safety are very limited, and there are numerous reports of catastrophic maternal thrombotic complications.79,80
A systematic review of anticoagulation in pregnant women with prosthetic heart valves34 found very limited data on heparin use throughout pregnancy. Women maintained on warfarin vs heparin between pregnancy weeks 6 and 12 had higher rates of congenital anomalies (6.4% with warfarin vs 3.4% with heparin) and total fetal wastage (33.6% vs 26.5%). The warfarin group had fewer maternal thromboembolic complications (3.9% vs 9.2%), however, and a slightly lower rate of maternal death (1.8% vs 4.2%). Most of the women had higher-risk older-generation valves in the mitral position.
Recent data on LMWH consist mainly of case reports and case series,81 with a likely bias to publication of worse outcomes. Controlled trials in this area will be difficult to conduct. Still, aggressive anticoagulation with LMWH or unfractionated heparin, with close monitoring of the intensity of anticoagulation, may be safe and effective for pregnant women with newer-generation mechanical heart valves.82 A recent consensus statement22 suggested several regimens for pregnant women with mechanical heart valves:
- Twice-daily LMWH throughout pregnancy, with the dose adjusted either by weight, or to keep the 4-hour postinjection anti-factor-Xa activity level around 1.0 to 1.2 U/mL
- Aggressive adjusted-dose unfractionated heparin throughout pregnancy, given subcutaneously every 12 hours and adjusted to keep the mid-interval aPTT at least twice the control value or to attain a mid-interval anti-factor-Xa activity level of 0.35 to 0.70 U/mL
- Unfractionated heparin or LMWH (as above) until gestation week 13, then warfarin until the middle of the third trimester, and then heparin again.22
The authors also recommended adding low-dose aspirin (75–162 mg/day) in high-risk women.22
These options all seem reasonable, given our current knowledge, though warfarin use during pregnancy should be restricted to very-high-risk situations, such as women with older-generation mitral prostheses. LM-WHs may become the preferred therapy for this indication once further controlled data regarding their efficacy and safety become available.
- Chang J, Elam-Evans LD, Berg CJ, et al. Pregnancy-related mortality surveillance-United States, 1991–1999. MMWR Surveill Summ 2003; 52:1–8.
- Lewis G, Drife JO, Clutton-Brock T, et al. Why Mothers Die, 2000–2002. The Sixth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. London: RCOG Press, 2004.
- Health Canada. Special Report on Maternal Mortality and Severe Morbidity in Canada—Enhanced Surveillance: The Path to Prevention. Ottawa: Minister of Public Works and Government Services Canada, 2004. www.phac-aspc.gc.ca/rhs-ssg/srmm-rsmm/page1-eng.php. Accessed 11/26/2008.
- Stein PD, Hull RD, Kayali F, et al. Venous thromboembolism in pregnancy: 21-year trends. Am J Med 2004; 117:121–125.
- Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet 1999; 353:1258–1265.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353:1167–1173.
- Gherman RB, Goodwin TM, Leung B, et al. Incidence, clinical characteristics, and timing of objectively diagnosed venous thromboembolism during pregnancy. Obstet Gynecol 1999; 94:730–734.
- Weitz JI. Low-molecular-weight heparins. N Engl J Med 1997; 337:688–698.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Simonneau G, Sors H, Charbonnier B, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N Engl J Med 1997; 337:663–669.
- Hull RD, Raskob GE, Brant RF, et al. Low-molecular-weight heparin vs heparin in the treatment of patients with pulmonary embolism. American-Canadian Thrombosis Study Group. Arch Intern Med 2000; 160:229–236.
- Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost 1999; 81:668–672.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005; 106:401–407.
- Melissari E, Parker CJ, Wilson NV, et al. Use of low molecular weight heparin in pregnancy. Thromb Haemost 1992; 68:652–656.
- Forestier F, Daffos F, Capella-Pavlovsky M. Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 1984; 34:557–560.
- Forestier F, Daffos F, Rainaut M, Toulemonde F. Low molecular weight heparin (CY 216) does not cross the placenta during the third trimester of pregnancy. Thromb Haemost 1987; 57:234.
- Barbour LA, Oja JL, Schultz LK. A prospective trial that demonstrates that dalteparin requirements increase in pregnancy to maintain therapeutic levels of anticoagulation. Am J Obstet Gynecol 2004; 191:1024–1029.
- Smith MP, Norris LA, Steer PJ, Savidge GF, Bonnar J. Tinzaparin sodium for thrombosis treatment and prevention during pregnancy. Am J Obstet Gynecol 2004; 190:495–501.
- Ellison J, Walker ID, Greer IA. Antenatal use of enoxaparin for prevention and treatment of thromboembolism in pregnancy. BJOG 2000; 107:1116–1121.
- Sarig G, Brenner B. Monitoring of low molecular weight heparin (LMWH) in pregnancy. Thromb Res 2005; 115 suppl 1:84–86.
- Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:627S–644S.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995; 332:1330–1335.
- Hassell K. The management of patients with heparin-induced thrombocytopenia who require anticoagulant therapy. Chest 2005; 127 suppl 2:1S–8S.
- Barbour LA, Kick SD, Steiner JF, et al. A prospective study of heparin-induced osteoporosis in pregnancy using bone densitometry. Am J Obst Gynecol 1994; 170:862–869.
- Douketis JD, Ginsberg JS, Burrows RF, Duku EK, Webber CE, Brill-Edwards P. The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 1996; 75:254–257.
- Pettila V, Leinonen P, Markkola A, Hiilesmaa V, Kaaja R. Postpartum bone mineral density in women treated for thromboprophylaxis with unfractionated heparin or LMW heparin. Thromb Haemost 2002; 87:182–186.
- Carlin AJ, Farquharson RG, Quenby SM, Topping J, Fraser WD. Prospective observational study of bone mineral density during pregnancy: low molecular weight heparin versus control. Hum Reprod 2004; 19:1211–1214.
- Casele HL, Laifer SA. Prospective evaluation of bone density in pregnant women receiving the low molecular weight heparin enoxaparin sodium. J Matern Fetal Med 2000; 9:122–125.
- Casele H, Haney EI, James A, Rosene-Montella K, Carson M. Bone density changes in women who receive thromboprophylaxis in pregnancy. Am J Obstet Gynecol 2006; 195:1109–1113.
- Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the risks (the second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med 2003; 28:172–197.
- Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 suppl 1:8S–21S.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:287S–310S.
- Holmes LB. Teratogen-induced limb defects. Am J Med Genet 2002; 112:297–303.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Pauzner R, Dulitzki M, Langevitz P, Livneh A, Kenett R, Many A. Low molecular weight heparin and warfarin in the treatment of patients with antiphospholipid syndrome during pregnancy. Thromb Haemost 2001; 86:1379–1384.
- Pulmonary Embolism Prevention (PEP) Trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000; 355:1295–1302.
- Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:234S–264S.
- Duley L, Henderson-Smart DJ, Knight M, King JF. Antiplatelet agents for preventing preeclampsia and its complications. Cochrane Database Syst Rev. 2004; ( 1):CD004659.
- Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol 2003; 101:1319–1332.
- Caritis SN, Sibai BM, Hauth J, et al, and the National Institute of Child Health and Human Development Network of Maternal Fetal Medicine Units. Low-dose aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338:701–705.
- Rai R, Cohen H, Dave M, Regan L. Randomised controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage associated with phospholipid antibodies (or antiphospholipid antibodies). BMJ 1997; 314:253–257.
- Kozer E, Nikfar S, Costei A, Boskovic R, Nulman I, Koren G. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol 2002; 187:1623–1630.
- Sebastian C, Scherlag M, Kugelmass A, Schechter E. Primary stent implantation for acute myocardial infarction during pregnancy: use of abciximab, ticlopidine, and aspirin. Cathet Cardiovasc Diagn 1998; 45:275–249.
- Wilson AM, Boyle AJ, Fox P. Management of ischaemic heart disease in women of child-bearing age. Intern Med J 2004; 34:694–697.
- Klinzing P, Markert UR, Liesaus K, Peiker G. Case report: successful pregnancy and delivery after myocardial infarction and essential thrombocythemia treated with clopidogrel. Clin Exp Obstet Gynecol 2001; 28:215–216.
- Danhof M, de Boer A, Magnani HN, Stiekema JC. Pharmacokinetic considerations on Orgaran (Org 10172) therapy. Haemostasis 1992; 22:73–84.
- Tardy-Poncet B, Tardy B, Reynaud J, et al. Efficacy and safety of danaparoid sodium (ORG 10172) in critically ill patients with heparin-associated thrombocytopenia. Chest 1999; 115:1616–1620.
- Lagrange F, Vergnes C, Brun JL, et al. Absence of placental transfer of pentasaccharide (fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 2002; 87:831–835.
- Dempfle CE. Minor transplacental passge of fondapinux in vivo. N Engl J Med 2004; 350:1914.
- Magnani HN. Heparin-induced thrombocytopenia (HIT): an overview of 230 patients treated with orgaran (Org 10172). Thromb Haemost 1993; 70:554–561.
- Lindhoff-Last E, Kreutzenbeck HJ, Magnani HN. Treatment of 51 pregnancies with danaparoid because of heparin intolerance. Thromb Haemost 2005; 93:63–69.
- Greinacher A, Eckhardt T, Mussmann J, Mueller-Eckhardt C. Pregnancy complicated by heparin associated thrombocytopenia: management by a prospectively in vitro selected heparinoid (Org 10172). Thromb Res 1993; 71:123–126.
- Schindewolf M, Mosch G, Bauersachs RM, Lindhoff-Last E. Safe anticoagulation with danaparoid in pregnancy and lactation. Thromb Haemost 2004; 92:211.
- Harenberg J. Treatment of a woman with lupus and thromboembolism and cutaneous intolerance to heparins using fondaparinux during pregnancy. Thromb Res 2007; 119:385–388.
- Wijesiriwardana A, Lees DA, Lush C. Fondaparinux as anticoagulant in a pregnant woman with heparin allergy. Blood Coagul Fibrinolysis 2006; 17:147–149.
- Mazzolai L, Hohlfeld P, Spertini F, Hayoz D, Schapira M, Duchosal MA. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood 2006; 108:1569–1570.
- Hawkins D, Evans J. Minimizing the risk of heparin-induced osteoporosis during pregnancy. Expert Opin Drug Saf 2005; 4:583–590.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of clot in this pregnancy study group. N Engl J Med 2000; 343:1439–1444.
- Martinelli I, Legnani C, Bucciarelli P, Grandone E, De Stefano V, Mannucci PM. Risk of pregnancy-related venous thrombosis in carriers of severe inherited thrombophilia. Thromb Haemost 2001; 86:800–803.
- De Stefano V, Martinelli I, Rossi E, Battaglioli T, Za T, Mannucci PM, Leone G. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006; 135:386–391.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995; 173:1869–1873.
- Ensom MH, Stephenson MD. Pharmacokinetics of low molecular weight heparin and unfractionated heparin in pregnancy. J Soc Gynecol Investig 2004; 11:377–383.
- Crowther MA, Berry LR, Monagle PT, Chan AK. Mechanisms responsible for the failure of protamine to inactivate low-molecular-weight heparin. Br J Haematol 2002; 116:178–186.
- Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997; 336:1506–1511.
- Thomas LA, Summers RR, Cardwell MS. Use of Greenfield filters in pregnant women at risk for pulmonary embolism. South Med J 1997; 90:215–217.
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs 2007; 67:1429–1440.
- Information from LactMed: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT, LactMed Record Number: 279. Accessed 11/26/2008.
- Gerhardt A, Scharf RE, Beckmann MW, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 2000; 342:374–380.
- Hirsch DR, Mikkola KM, Marks PW, et al. Pulmonary embolism and deep venous thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J 1996; 131:1145–1148.
- Dizon-Townson DS, Nelson LM, Jang H, Varner MW, Ward K. The incidence of the factor V Leiden mutation in an obstetric population and its relationship to deep vein thrombosis. Am J Obstet Gynecol 1997; 176:883–886.
- McColl MD, Ramsay JE, Tait RC, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 1997; 78:1183–1188.
- Kupferminc MJ, Fait G, Many A, Gordon D, Eldor A, Lessing JB. Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 2000; 96:45–49.
- Kupferminc MJ, Eldor A, Steinman N, et al. Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 1999; 340:9–13.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006; 132:171–196.
- Brenner B, Hoffman R, Blumenfeld Z, Weiner Z, Younis JS. Gestational outcome in thrombophilic women with recurrent pregnancy loss treated by enoxaparin. Thromb Haemost 2000; 83:693–697.
- Carp H, Dolitzky M, Inbal A. Thromboprophylaxis improves the live birth rate in women with consecutive recurrent miscarriages and hereditary thrombophilia. J Thromb Haemost 2003; 1:433–438.
- Gris JC, Mercier E, Quere I, et al. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder. Blood 2004; 103:3695–3699.
- Salazar E, Izaguirre R, Verdejo J, Mutchinick O. Failure of adjusted doses of subcutaneous heparin to prevent thromboembolic phenomena in pregnant patients with mechanical cardiac valve prostheses. J Am Coll Cardiol 1996; 27:1698–1703.
- Iturbe-Alessio I, Fonseca MC, Mutchinik O, Santos MA, Zajarias A, Salazar E. Risks of anticoagulant therapy in pregnant women with artificial heart valves. N Engl J Med 1986; 315:1390–1393.
- Rowan JA, McCowan LM, Raudkivi PJ, North RA. Enoxaparin treatment in women with mechanical heart valves during pregnancy. Am J Obstet Gynecol 2001; 185:633–637.
- Oran B, Lee-Parritz A, Ansell J. Low molecular weight heparin for the prophylaxis of thromboembolism in women with prosthetic mechanical heart valves during pregnancy. Thromb Haemost 2004; 92:747–751.
- Chang J, Elam-Evans LD, Berg CJ, et al. Pregnancy-related mortality surveillance-United States, 1991–1999. MMWR Surveill Summ 2003; 52:1–8.
- Lewis G, Drife JO, Clutton-Brock T, et al. Why Mothers Die, 2000–2002. The Sixth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. London: RCOG Press, 2004.
- Health Canada. Special Report on Maternal Mortality and Severe Morbidity in Canada—Enhanced Surveillance: The Path to Prevention. Ottawa: Minister of Public Works and Government Services Canada, 2004. www.phac-aspc.gc.ca/rhs-ssg/srmm-rsmm/page1-eng.php. Accessed 11/26/2008.
- Stein PD, Hull RD, Kayali F, et al. Venous thromboembolism in pregnancy: 21-year trends. Am J Med 2004; 117:121–125.
- Greer IA. Thrombosis in pregnancy: maternal and fetal issues. Lancet 1999; 353:1258–1265.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353:1167–1173.
- Gherman RB, Goodwin TM, Leung B, et al. Incidence, clinical characteristics, and timing of objectively diagnosed venous thromboembolism during pregnancy. Obstet Gynecol 1999; 94:730–734.
- Weitz JI. Low-molecular-weight heparins. N Engl J Med 1997; 337:688–698.
- Levine M, Gent M, Hirsh J, et al. A comparison of low-molecular-weight heparin administered primarily at home with unfractionated heparin administered in the hospital for proximal deep-vein thrombosis. N Engl J Med 1996; 334:677–681.
- Koopman MM, Prandoni P, Piovella F, et al. Treatment of venous thrombosis with intravenous unfractionated heparin administered in the hospital as compared with subcutaneous low-molecular-weight heparin administered at home. The Tasman Study Group. N Engl J Med 1996; 334:682–687.
- Simonneau G, Sors H, Charbonnier B, et al. A comparison of low-molecular-weight heparin with unfractionated heparin for acute pulmonary embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N Engl J Med 1997; 337:663–669.
- Hull RD, Raskob GE, Brant RF, et al. Low-molecular-weight heparin vs heparin in the treatment of patients with pulmonary embolism. American-Canadian Thrombosis Study Group. Arch Intern Med 2000; 160:229–236.
- Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost 1999; 81:668–672.
- Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood 2005; 106:401–407.
- Melissari E, Parker CJ, Wilson NV, et al. Use of low molecular weight heparin in pregnancy. Thromb Haemost 1992; 68:652–656.
- Forestier F, Daffos F, Capella-Pavlovsky M. Low molecular weight heparin (PK 10169) does not cross the placenta during the second trimester of pregnancy study by direct fetal blood sampling under ultrasound. Thromb Res 1984; 34:557–560.
- Forestier F, Daffos F, Rainaut M, Toulemonde F. Low molecular weight heparin (CY 216) does not cross the placenta during the third trimester of pregnancy. Thromb Haemost 1987; 57:234.
- Barbour LA, Oja JL, Schultz LK. A prospective trial that demonstrates that dalteparin requirements increase in pregnancy to maintain therapeutic levels of anticoagulation. Am J Obstet Gynecol 2004; 191:1024–1029.
- Smith MP, Norris LA, Steer PJ, Savidge GF, Bonnar J. Tinzaparin sodium for thrombosis treatment and prevention during pregnancy. Am J Obstet Gynecol 2004; 190:495–501.
- Ellison J, Walker ID, Greer IA. Antenatal use of enoxaparin for prevention and treatment of thromboembolism in pregnancy. BJOG 2000; 107:1116–1121.
- Sarig G, Brenner B. Monitoring of low molecular weight heparin (LMWH) in pregnancy. Thromb Res 2005; 115 suppl 1:84–86.
- Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of antithrombotic agents during pregnancy: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:627S–644S.
- Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med 1995; 332:1330–1335.
- Hassell K. The management of patients with heparin-induced thrombocytopenia who require anticoagulant therapy. Chest 2005; 127 suppl 2:1S–8S.
- Barbour LA, Kick SD, Steiner JF, et al. A prospective study of heparin-induced osteoporosis in pregnancy using bone densitometry. Am J Obst Gynecol 1994; 170:862–869.
- Douketis JD, Ginsberg JS, Burrows RF, Duku EK, Webber CE, Brill-Edwards P. The effects of long-term heparin therapy during pregnancy on bone density. A prospective matched cohort study. Thromb Haemost 1996; 75:254–257.
- Pettila V, Leinonen P, Markkola A, Hiilesmaa V, Kaaja R. Postpartum bone mineral density in women treated for thromboprophylaxis with unfractionated heparin or LMW heparin. Thromb Haemost 2002; 87:182–186.
- Carlin AJ, Farquharson RG, Quenby SM, Topping J, Fraser WD. Prospective observational study of bone mineral density during pregnancy: low molecular weight heparin versus control. Hum Reprod 2004; 19:1211–1214.
- Casele HL, Laifer SA. Prospective evaluation of bone density in pregnant women receiving the low molecular weight heparin enoxaparin sodium. J Matern Fetal Med 2000; 9:122–125.
- Casele H, Haney EI, James A, Rosene-Montella K, Carson M. Bone density changes in women who receive thromboprophylaxis in pregnancy. Am J Obstet Gynecol 2006; 195:1109–1113.
- Horlocker TT, Wedel DJ, Benzon H, et al. Regional anesthesia in the anticoagulated patient: defining the risks (the second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg Anesth Pain Med 2003; 28:172–197.
- Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 suppl 1:8S–21S.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:287S–310S.
- Holmes LB. Teratogen-induced limb defects. Am J Med Genet 2002; 112:297–303.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Pauzner R, Dulitzki M, Langevitz P, Livneh A, Kenett R, Many A. Low molecular weight heparin and warfarin in the treatment of patients with antiphospholipid syndrome during pregnancy. Thromb Haemost 2001; 86:1379–1384.
- Pulmonary Embolism Prevention (PEP) Trial Collaborative Group. Prevention of pulmonary embolism and deep vein thrombosis with low dose aspirin: Pulmonary Embolism Prevention (PEP) trial. Lancet 2000; 355:1295–1302.
- Patrono C, Coller B, FitzGerald GA, Hirsh J, Roth G. Platelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl 3:234S–264S.
- Duley L, Henderson-Smart DJ, Knight M, King JF. Antiplatelet agents for preventing preeclampsia and its complications. Cochrane Database Syst Rev. 2004; ( 1):CD004659.
- Coomarasamy A, Honest H, Papaioannou S, Gee H, Khan KS. Aspirin for prevention of preeclampsia in women with historical risk factors: a systematic review. Obstet Gynecol 2003; 101:1319–1332.
- Caritis SN, Sibai BM, Hauth J, et al, and the National Institute of Child Health and Human Development Network of Maternal Fetal Medicine Units. Low-dose aspirin to prevent preeclampsia in women at high risk. N Engl J Med 1998; 338:701–705.
- Rai R, Cohen H, Dave M, Regan L. Randomised controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriage associated with phospholipid antibodies (or antiphospholipid antibodies). BMJ 1997; 314:253–257.
- Kozer E, Nikfar S, Costei A, Boskovic R, Nulman I, Koren G. Aspirin consumption during the first trimester of pregnancy and congenital anomalies: a meta-analysis. Am J Obstet Gynecol 2002; 187:1623–1630.
- Sebastian C, Scherlag M, Kugelmass A, Schechter E. Primary stent implantation for acute myocardial infarction during pregnancy: use of abciximab, ticlopidine, and aspirin. Cathet Cardiovasc Diagn 1998; 45:275–249.
- Wilson AM, Boyle AJ, Fox P. Management of ischaemic heart disease in women of child-bearing age. Intern Med J 2004; 34:694–697.
- Klinzing P, Markert UR, Liesaus K, Peiker G. Case report: successful pregnancy and delivery after myocardial infarction and essential thrombocythemia treated with clopidogrel. Clin Exp Obstet Gynecol 2001; 28:215–216.
- Danhof M, de Boer A, Magnani HN, Stiekema JC. Pharmacokinetic considerations on Orgaran (Org 10172) therapy. Haemostasis 1992; 22:73–84.
- Tardy-Poncet B, Tardy B, Reynaud J, et al. Efficacy and safety of danaparoid sodium (ORG 10172) in critically ill patients with heparin-associated thrombocytopenia. Chest 1999; 115:1616–1620.
- Lagrange F, Vergnes C, Brun JL, et al. Absence of placental transfer of pentasaccharide (fondaparinux, Arixtra) in the dually perfused human cotyledon in vitro. Thromb Haemost 2002; 87:831–835.
- Dempfle CE. Minor transplacental passge of fondapinux in vivo. N Engl J Med 2004; 350:1914.
- Magnani HN. Heparin-induced thrombocytopenia (HIT): an overview of 230 patients treated with orgaran (Org 10172). Thromb Haemost 1993; 70:554–561.
- Lindhoff-Last E, Kreutzenbeck HJ, Magnani HN. Treatment of 51 pregnancies with danaparoid because of heparin intolerance. Thromb Haemost 2005; 93:63–69.
- Greinacher A, Eckhardt T, Mussmann J, Mueller-Eckhardt C. Pregnancy complicated by heparin associated thrombocytopenia: management by a prospectively in vitro selected heparinoid (Org 10172). Thromb Res 1993; 71:123–126.
- Schindewolf M, Mosch G, Bauersachs RM, Lindhoff-Last E. Safe anticoagulation with danaparoid in pregnancy and lactation. Thromb Haemost 2004; 92:211.
- Harenberg J. Treatment of a woman with lupus and thromboembolism and cutaneous intolerance to heparins using fondaparinux during pregnancy. Thromb Res 2007; 119:385–388.
- Wijesiriwardana A, Lees DA, Lush C. Fondaparinux as anticoagulant in a pregnant woman with heparin allergy. Blood Coagul Fibrinolysis 2006; 17:147–149.
- Mazzolai L, Hohlfeld P, Spertini F, Hayoz D, Schapira M, Duchosal MA. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood 2006; 108:1569–1570.
- Hawkins D, Evans J. Minimizing the risk of heparin-induced osteoporosis during pregnancy. Expert Opin Drug Saf 2005; 4:583–590.
- Brill-Edwards P, Ginsberg JS, Gent M, et al. Safety of withholding heparin in pregnant women with a history of venous thromboembolism. Recurrence of clot in this pregnancy study group. N Engl J Med 2000; 343:1439–1444.
- Martinelli I, Legnani C, Bucciarelli P, Grandone E, De Stefano V, Mannucci PM. Risk of pregnancy-related venous thrombosis in carriers of severe inherited thrombophilia. Thromb Haemost 2001; 86:800–803.
- De Stefano V, Martinelli I, Rossi E, Battaglioli T, Za T, Mannucci PM, Leone G. The risk of recurrent venous thromboembolism in pregnancy and puerperium without antithrombotic prophylaxis. Br J Haematol 2006; 135:386–391.
- Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol 1995; 173:1869–1873.
- Ensom MH, Stephenson MD. Pharmacokinetics of low molecular weight heparin and unfractionated heparin in pregnancy. J Soc Gynecol Investig 2004; 11:377–383.
- Crowther MA, Berry LR, Monagle PT, Chan AK. Mechanisms responsible for the failure of protamine to inactivate low-molecular-weight heparin. Br J Haematol 2002; 116:178–186.
- Kearon C, Hirsh J. Management of anticoagulation before and after elective surgery. N Engl J Med 1997; 336:1506–1511.
- Thomas LA, Summers RR, Cardwell MS. Use of Greenfield filters in pregnant women at risk for pulmonary embolism. South Med J 1997; 90:215–217.
- Maclean PS, Tait RC. Hereditary and acquired antithrombin deficiency: epidemiology, pathogenesis and treatment options. Drugs 2007; 67:1429–1440.
- Information from LactMed: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?LACT, LactMed Record Number: 279. Accessed 11/26/2008.
- Gerhardt A, Scharf RE, Beckmann MW, et al. Prothrombin and factor V mutations in women with a history of thrombosis during pregnancy and the puerperium. N Engl J Med 2000; 342:374–380.
- Hirsch DR, Mikkola KM, Marks PW, et al. Pulmonary embolism and deep venous thrombosis during pregnancy or oral contraceptive use: prevalence of factor V Leiden. Am Heart J 1996; 131:1145–1148.
- Dizon-Townson DS, Nelson LM, Jang H, Varner MW, Ward K. The incidence of the factor V Leiden mutation in an obstetric population and its relationship to deep vein thrombosis. Am J Obstet Gynecol 1997; 176:883–886.
- McColl MD, Ramsay JE, Tait RC, et al. Risk factors for pregnancy associated venous thromboembolism. Thromb Haemost 1997; 78:1183–1188.
- Kupferminc MJ, Fait G, Many A, Gordon D, Eldor A, Lessing JB. Severe preeclampsia and high frequency of genetic thrombophilic mutations. Obstet Gynecol 2000; 96:45–49.
- Kupferminc MJ, Eldor A, Steinman N, et al. Increased frequency of genetic thrombophilia in women with complications of pregnancy. N Engl J Med 1999; 340:9–13.
- Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol 2006; 132:171–196.
- Brenner B, Hoffman R, Blumenfeld Z, Weiner Z, Younis JS. Gestational outcome in thrombophilic women with recurrent pregnancy loss treated by enoxaparin. Thromb Haemost 2000; 83:693–697.
- Carp H, Dolitzky M, Inbal A. Thromboprophylaxis improves the live birth rate in women with consecutive recurrent miscarriages and hereditary thrombophilia. J Thromb Haemost 2003; 1:433–438.
- Gris JC, Mercier E, Quere I, et al. Low-molecular-weight heparin versus low-dose aspirin in women with one fetal loss and a constitutional thrombophilic disorder. Blood 2004; 103:3695–3699.
- Salazar E, Izaguirre R, Verdejo J, Mutchinick O. Failure of adjusted doses of subcutaneous heparin to prevent thromboembolic phenomena in pregnant patients with mechanical cardiac valve prostheses. J Am Coll Cardiol 1996; 27:1698–1703.
- Iturbe-Alessio I, Fonseca MC, Mutchinik O, Santos MA, Zajarias A, Salazar E. Risks of anticoagulant therapy in pregnant women with artificial heart valves. N Engl J Med 1986; 315:1390–1393.
- Rowan JA, McCowan LM, Raudkivi PJ, North RA. Enoxaparin treatment in women with mechanical heart valves during pregnancy. Am J Obstet Gynecol 2001; 185:633–637.
- Oran B, Lee-Parritz A, Ansell J. Low molecular weight heparin for the prophylaxis of thromboembolism in women with prosthetic mechanical heart valves during pregnancy. Thromb Haemost 2004; 92:747–751.
KEY POINTS
- Pregnancy is a hypercoagulable state. Thrombotic risk in an individual pregnancy depends on many maternal and situational factors.
- When indicated, careful anticoagulation can proceed with minimal risk to the mother and fetus.
- Heparins, especially LMWHs, are the main anticoagulants used in pregnancy. Dosing depends on the clinical indications and on the agent selected.
- If anticoagulation is absolutely necessary and LMWH is contraindicated, a newer, alternative anticoagulant should be considered.
- Warfarin should not be used in pregnancy in any but the highest-risk situations.
Which patients benefit from carotid stenting? What recent trials show
Whether carotid stenting has any advantage over carotid surgery (endarterectomy)—and for which patients—is still a topic of study and debate.
Treatment of carotid atherosclerosis and stenosis is important in preventing stroke and its comorbidities. Today, three main treatments exist: medical management (lipid-lowering, antihypertensive, and antiplatelet therapy), surgery, and, more recently, carotid angioplasty and stenting. The rationale for these treatments is to decrease the risk of cerebral infarction by stabilizing or removing plaque and improving blood flow.
Surgery has proven beneficial in patients with symptomatic carotid stenosis greater than 50% or asymptomatic stenosis greater than 60%, but it is risky in some patients. Stenting has evolved in part from the success of surgery and the need for alternative treatments for patients who are at unacceptable risk of perioperative complications. However, it does not have a clear advantage over surgery in patients at average risk. Further, its use in patients with asymptomatic stenosis of any severity is still controversial.
In this paper we review the major trials of carotid endarterectomy and stenting and summarize what we know today about who should undergo these therapies.
NOT ALL STROKES ARE DUE TO CAROTID ATHEROSCLEROSIS
Depending on the institution’s referral pattern and population served, between 80% and 90% of strokes are ischemic (the rest being hemorrhagic).1 Atherosclerosis of large arteries (typically defined as more than 50% stenosis of a major brain artery or branch cortical artery2) is just one cause of ischemic stroke, but it is an important one. Other identifiable causes of ischemic stroke include cardioembolism and small-artery occlusion (lacunar stroke), and some cases are idiopathic.
Large-artery atherosclerotic disease can damage the brain gradually, with carotid stenosis resulting in hypoperfusion and subsequent cerebral infarction. More commonly, however, the carotid plaque often seen in large-artery atherosclerotic disease can ulcerate and occlude the vessel acutely or generate platelet aggregates that may embolize, resulting in cerebral infarction or transient ischemic attack.
In the Lausanne Stroke Registry,3 the rate of ischemic stroke in patients with a greater than 50% large-artery stenosis ranged from 27% in 1979 to 17% in 2003, the decline likely being due to therapeutic advances.
SURGERY BEATS MEDICAL THERAPY FOR CAROTID ATHEROSCLEROSIS
Four landmark trials provided substantial evidence that carotid endarterectomy is better than medical management in patients with symptomatic or asymptomatic high-grade stenosis. These trials indirectly paved the way for carotid stenting.
The North American Symptomatic Carotid Endarterectomy Trial (NASCET)
Patients at 50 clinical centers who had had a hemispheric or retinal transient ischemic attack or a nondisabling stroke were randomized to undergo surgery (carotid endarterectomy) or no surgery. All patients received maximal medical management consisting of blood pressure control, lipid management if indicated, and antiplatelet therapy with aspirin. At baseline, 37% of patients were taking 650 mg or more of aspirin per day, and 11% were taking less than 325 mg per day. The patients were stratified into two prespecified groups on the basis of the severity of carotid stenosis: those with narrowing of 30% to 69% and those with narrowing of 70% to 99%.
Results in high-grade stenosis. In August 1991, the investigators published their results in patients with symptomatic high-grade (70%–99%) stenosis.4 Surgical treatment was more beneficial than medical management alone: the cumulative risk of any ipsilateral stroke at 2 years was 26% in the medical group and 9% in the surgical group, an absolute risk reduction of 17%. The benefit of endarterectomy was still apparent at 8 years of follow-up.5
Results in moderate stenosis. In 1998, the investigators published their results in patients with symptomatic moderate (< 70%) stenosis.5 Surgery was more beneficial than medical therapy in this subgroup as well: at 5 years, the rate of any ipsilateral stroke in patients with 50% to 69% stenosis was 15.7% in those treated surgically and 22.2% in those treated medically (P = .045). In patients with less than 50% stenosis, the 5-year stroke rate was not significantly lower with endarterectomy than with medical therapy.
The European Carotid Surgery Trial (ECST)
The ECST,6 published in 1998, corroborated the NASCET findings. This multicenter, randomized, controlled trial enrolled 3,024 patients with symptoms of at least one transient ischemic attack in the distribution of one or both carotid arteries.
Results. In patients with stenosis of greater than 80% (60% by the NASCET criteria for calculating angiographic stenosis), the frequency of major stroke or death at 3 years was 26.5% in the control group and 14.9% in the surgery group, an absolute difference of 11.6%.
The Endarterectomy for Asymptomatic Carotid Artery Stenosis (ACAS) trial
The NASCET and ECST studies made it clear that select groups of patients with symptomatic carotid stenosis benefit from carotid endarterectomy. But what about patients with stenosis but no prior stroke?
The ACAS trial aimed to find out.7 In this pivotal study, 1,662 patients with asymptomatic carotid artery stenosis greater than 60% were randomized to receive either medical therapy alone or medical plus surgical therapy.
Results were published in 2004. After a median follow-up of 2.7 years, the aggregate 5-year risk of ipsilateral stroke, any perioperative stroke, or death was estimated to be 5.1% in the surgical group and 11.0% in the medical group, a relative risk reduction of 53%. However, for surgery to be beneficial, the rate of perioperative death and other serious complications had to be less than 3%, and the expected patient survival had to be at least 5 years.
Of note, the benefit of carotid endarterectomy in this study was predominantly in men, with less of a benefit for women and diabetic patients. Furthermore, even though endarterectomy was beneficial in this asymptomatic cohort, the overall benefit in terms of stroke risk reduction was small compared with that in NASCET and ECST, in which patients had symptomatic disease.
The Asymptomatic Carotid Surgery Trial (ACST)
In this European version of ACAS, published in 2004, 3,120 patients with asymptomatic carotid narrowing on ultrasonography were randomized to undergo surgery or medical therapy.
Results. The risk of stroke or death within 30 days of carotid endarterectomy was 3.1%. In patients younger than 75 years who had carotid narrowing of 70% or more, immediate surgery decreased the net 5-year stroke risk from 12% to 6%.8
WHO SHOULD NOT UNDERGO CAROTID ENDARTERECTOMY?
From these studies, we can conclude that patients with symptomatic carotid stenosis of 50% or greater and patients with asymptomatic stenosis of 60% or greater benefit from carotid endarterectomy, but only if the perioperative rate of death and other serious complications is less than 3%.7
What are the risk factors for complications during this surgery? In 2006, Cremonesi et al,9 in a consensus paper, defined patients as being at high risk if they had any of the following:
- Contralateral laryngeal nerve palsy
- Radiation therapy to the neck
- Previous carotid endarterectomy with recurrent stenosis
- Lesions high in the cervical internal carotid artery or below the clavicle in the common carotid artery
- Severe tandem lesions
- Age greater than 80 years
- Severe pulmonary disease
- Congestive heart failure (New York Heart
- Association class 3 or 4) or known severe left ventricular dysfunction
- Open heart surgery needed within 6 weeks
- Myocardial infarction within the past 4 weeks
- Unstable angina
- Contralateral carotid occlusion.
Could endovascular treatment be the answer for these patients at high risk who should not undergo carotid endarterectomy? Indeed, the procedure is being studied extensively and performed more frequently. We summarize the major studies below.
STUDIES OF CAROTID STENTING VS ENDARTERECTOMY
The Carotid and Vertebral Artery Transluminal Angioplasty Study (CAVATAS)
This study, published in 2001,10 was the first randomized, multicenter trial to compare the risks and benefits of endovascular treatment (angioplasty with or without stenting) of carotid and vertebral artery stenosis with those of conventional surgery.
To be included, patients had to have carotid artery stenosis (symptomatic or asymptomatic) that was suitable for either carotid endarterectomy or endovascular treatment. Patients were not grouped on the basis of the severity of their stenosis, but the mean stenosis in randomized patients was 86%.
A total of 504 patients were enrolled, of whom 251 were randomized to undergo endovascular treatment. Most patients in this group underwent angioplasty alone, but 26% also received stents because of suboptimal vessel dilatation or at the discretion of the intervening physician.
The primary end point was any disabling stroke or death. Secondary end points were any ipsilateral stroke lasting longer than 7 days and the combination of death or disabling ipsilateral stroke.
The results showed no significant difference between endovascular treatment and surgery in any of these end points at 3 years. However, the overall rates of procedural stroke and death were nearly double those seen in NASCET and ECST. The investigators could not determine the reason for this higher risk, but they hypothesized that CAVATAS included patients at higher risk.
The restenosis rate was higher in the endovascular therapy group (14%) than in the surgery group (4%; P < .001). On the other hand, the surgery group had a higher rate of minor complications, including cranial nerve palsies and neck hematomas.
Carotid Revascularization With Endarterectomy or Stenting Systems (CARESS)
This prospective, multicenter, phase 2 trial, published in 2003, compared the outcomes of standard carotid endarterectomy vs carotid artery stenting using distal embolic protection devices.11 All the patients in this study had at least 50% symptomatic stenosis or 75% asymptomatic stenosis.
Results. At 30 days, 7 (2.4%) of 254 patients in the endarterectomy group had had strokes, and one of the 7 patients with stroke died, so the combined rate of stroke or death (the primary end point) was 2.4%. In the stenting group, 3 (2.1%) of 143 patients had strokes and no patients died. Overall, there was no significant difference in the composite of death, stroke, or myocardial infarction (the secondary end point): 3% for carotid endarterectomy and 2% for stenting patients.
The Stenting and Angioplasty With Protection in Patients at High Risk for Endarterectomy (SAPPHIRE) trial
In this trial,12 published in 2004, patients had to have either symptomatic carotid disease with 50% stenosis or greater or asymptomatic stenosis of 80% or greater, determined by ultrasonography. Further, all patients had to have at least one comorbid condition that increased their perioperative risk. Up until this point, no trial had strictly defined patients at increased risk for complications after carotid endarterectomy and assessed subsequent outcomes. The risk factors included severe cardiac or pulmonary disease, age greater than 80, postendarterectomy carotid stenosis, previous neck surgery, previous neck radiation, contralateral recurrent laryngeal nerve palsy, and contralateral carotid occlusion.
Patients were randomized to undergo carotid artery stenting with distal protection or carotid endarterectomy.
The primary end points of this study were the cumulative incidence of major cardiovascular events at 1 year; death, stroke, or myocardial infarction within 30 days of intervention; and ipsilateral stroke between 31 days and 1 year. Secondary outcomes measured were the rates of target-vessel recanalization at 1 year, cranial nerve palsy, and surgical site complications.
Results. The rate of stroke or death was similar in both groups. The stenting group had fewer adverse cardiac events (mainly non-Q-wave myocardial infarction) than the surgery group. At 1 year the rate of major ipsilateral stroke was 3.3% in the endarterectomy group vs 0% in the stenting group (the difference was not significant), and the cardiovascular event rates continued to be higher in the endarterectomy group.
The investigators noted that myocardial infarction was included as a primary end point because patients with atherosclerotic vascular disease who undergo either stenting or endarterectomy are at a substantial risk of myocardial infarction, and a Q-wave or a non-Q-wave myocardial infarction in the perioperative period increases the risk of future complications and death. A perioperative non-Q-wave infarction increases the risk of death by a factor of 6 and increases the risk of myocardial infarction by a factor of 27 in the subsequent 6 months.
Overall, this study presents evidence that stenting, using distal embolic protection devices, is not inferior to endarterectomy and has fewer cardiovascular complications in patients who have at least one risk factor.
The Endarterectomy Versus Stenting in Patients With Symptomatic Severe Carotid Stenosis (EVA-3S) study
This recent multicenter, randomized study13 was designed to determine if stenting is as good as (not inferior to) carotid endarterectomy in patients with symptomatic carotid stenosis of at least 60%. The primary end point was to be the incidence of stroke or death within 30 days after treatment. However, the trial was stopped early after the inclusion of 527 patients for reasons of safety and futility.
Results. The 30-day incidence of any stroke or death was higher in the stenting group (9.6% vs 3.9%). The relative risk of any stroke or death after stenting as compared with endarterectomy was 2.5. The 30-day incidence of disabling stroke or death was also higher in the stenting group (3.4% vs 1.5%; relative risk 2.2). At 6 months, the incidence of any stroke or death was 6.1% after endarterectomy and 11.7% after stenting (P = .02). There was a trend toward more major local complications after stenting and systemic complications after endarterectomy. Cranial-nerve injury was more common after endarterectomy than after stenting (as expected). Overall, death and stroke rates were lower at 1 month and 6 months with endarterectomy than with stenting.
The Stent-Protected Angioplasty Versus Carotid Endarterectomy (SPACE) trial
This randomized, multicenter study,14 published in 2006, was also designed to compare the safety and efficacy of carotid stenting and endarterectomy. Some 1,200 patients with symptomatic carotid artery stenosis confirmed by ultrasonography were randomly assigned within 180 days of a transient ischemic attack or moderate stroke to undergo carotid artery stenting (n = 605) or carotid endarterectomy (n = 595). The primary end point was ipsilateral ischemic stroke or death 30 days after the procedure. A total of 1,183 patients were included in the analysis.
Results. The rate of the primary end point was 6.84% with stenting and 6.34% with endarterectomy. The study failed to prove the noninferiority of carotid artery stenting compared with carotid endarterectomy for the periprocedural complication rate. Results at 6 to 24 months are awaited.
The Carotid Revascularization Endarterectomy Versus Stenting (CREST) trial
Perhaps the most anxiously awaited results are those of the CREST trial,15 funded by the National Institutes of Health. This is a prospective, randomized, parallel, two-arm, multicenter clinical trial with blinded end point evaluation. Anticipated enrollment will include 2,500 patients. Patients are eligible for enrollment if they have symptoms of carotid stenosis within 180 days of a stroke or transient ischemic attack with ipsilateral carotid stenosis of at least 50% by angiography (70% by ultrasonography), or if they have asymptomatic carotid stenosis of at least 60% by angiography (70% by ultrasonography).
Patients are being randomized to undergo either carotid artery stenting or carotid endarterectomy. All receive aspirin as anti-platelet therapy, treatment for hypertension, and management of other stroke risk factors. Follow-up will last 4 years, with clinic visits at 1, 6, 12, 18, 24, 30, 36, 42, and 48 months. Primary outcome measures will be rates of death, stroke, or myocardial infarction at 30 days postoperatively, and ipsilateral stroke at 30 days postoperatively.
As of February 2007, 1,506 patients had been enrolled and 1,453 had been randomized at 94 sites in North America.
MEDICAID AND MEDICARE NOW PAY FOR THESE THERAPIES
An important practical consideration for patients and physicians is whether Medicaid and Medicare will pay for these therapies.
In July 2001, Medicare began to cover percutaneous transluminal angioplasty of the carotid artery with concurrent stent placement, when furnished in accordance with US Food and Drug Administration (FDA) protocols governing Category B (nonexperimental) investigational device exemption clinical trials.16 Angioplasty of the carotid artery, when provided solely for the purpose of carotid artery dilation concurrent with carotid stent placement, is considered to be a reasonable and necessary service when provided in the context of clinical trials.
In March 2005, Medicare began to provide coverage for percutaneous transluminal angioplasty of the carotid artery concurrent with the placement of an FDA-approved carotid stent with embolic protection for the following groups of patients:
- Those who would be at high risk during carotid endarterectomy and who also have symptomatic carotid artery stenosis of 70% or greater. Coverage is limited to procedures performed using FDA-approved carotid artery stenting systems and embolic protection devices.
- Those who would be at high risk during endarterectomy and who have symptomatic carotid artery stenosis of 50% to 70%, in accordance with the Category B Investigational Device Exemption clinical trials regulation, as a routine cost under the clinical trials policy, or in accordance with the national coverage determination on carotid artery stenting post-approval.
- Those who would be at high risk during carotid endarterectomy and have asymptomatic carotid artery stenosis greater than 80%, in accordance with the Category B Investigational Device Exemption clinical trials regulation, as a routine cost under the clinical trials policy, or in accordance with the national coverage determination on carotid artery stenting postapproval studies.
As noted above, Medicare and Medicaid will only cover carotid stenting if the stent system is FDA-approved, with concomitant use of a distal embolic protection device. However, in view of conflicting data from stenting trials to date, including EVA-3S13 and SPACE,14 it remains to be seen if emboli protection devices significantly reduce periprocedural stroke rates. The FDA recommends that if it is not technically possible to use one of these devices, then the procedure should be aborted due to safety issues.
These coverage decisions are an important practical aspect of carotid stenting and they should be familiar to physicians when they see and refer patients with carotid disease.
WHAT CAN WE SAY AT THIS POINT?
Given the multiple recent and ongoing trials of stenting vs endarterectomy in carotid stenosis, debate continues as to what the role of stenting will be in the future. What can we say at this point?
In patients with asymptomatic carotid stenosis of greater than 60% or symptomatic carotid stenosis of greater than 50%, carotid endarterectomy has been proven to be superior to medical therapy alone.
The efficacy and safety of carotid stenting compared with carotid endarterectomy is still uncertain. In the trials reviewed above, carotid stenting did not appear to have a clear advantage over endarterectomy in patients at average surgical risk. Stenting may be most advantageous when used in patients with symptomatic carotid stenosis who would be at high operative risk, as indicated by the SAPPHIRE trial.
In patients with severe but asymptomatic carotid stenosis who are at high operative risk, the addition of carotid angioplasty and stenting to maximum medical therapy remains controversial. The periprocedural complication rate in these patients may actually exceed the rate of stroke in asymptomatic patients with greater than 60% stenosis who do not undergo stenting or surgery. In addition, subgroup analyses of patients with 70% to 99% symptomatic stenosis in various trials show that surgical benefit is greater in men than in women, and it remains to be seen whether there is any benefit in women with moderate stenoses, asymptomatic lesions, or both.17
Further experience and study are needed, and the results of the Carotid Stenting vs Surgery of Severe Carotid Artery Disease and Stroke Prevention in Asymptomatic Patients (ACT I) study (comparing stenting and surgery in asymptomatic carotid stenosis), and the ongoing CREST trial (comparing stenting and surgery in symptomatic and asymptomatic carotid stenosis) are eagerly awaited. Until then, clinicians should continue to weigh individual patient risks and benefits when referring patients for surgical treatment of carotid athero-sclerotic disease. Regardless of whether surgery is undertaken, maximal medical therapy with the use of antiplatelet agents, blood pressure control, and statin therapy remains the mainstay of treatment.
- Incidence and Prevalence 2006 Chart Book on Cardiovascular and Lung Diseases Bethesda, MD: National Heart, Lung, and Blood Institute; 2006.
- Adams HP, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993; 24:35–41.
- Carrera E, Maeder-Ingvar M, Rossetti AO, Devuyst G, Bogousslavsky JLausanne Stroke Registry. Trends in risk factors, patterns and causes in hospitalized strokes over 25 years: The Lausanne Stroke Registry. Cerebrovasc Dis. 2007; 24:97–103.
- North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. 1991; 325:445–453.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North America Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med. 1998; 339:1415–1425.
- European Carotid Surgery Trialists’ Collaborative Group. Randomized trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet. 1998; 351:1379–1387.
- Halliday A, Mansfield A, Marro J, et al., MRC Asymptomatic Carotid Surgery Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomized controlled trial. Lancet. 2004; 363:1491–1502.
- Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. Endarterectomy for asymptomatic carotid artery stenosis. JAMA. 1995; 273:1421–1428.
- Cremonesi A, Setacci C, Bignamini A, et al. Carotid artery stenting: first consensus document of the ICCS-SPREAD Joint Committee. Stroke. 2006; 37:2400–2409.
- CAVATAS Investigators. Endovascular versus surgical treatment in patients with carotid stenosis in the Carotid and Vertebral Artery Transluminal Angioplasty Study (CA-VATAS): a randomized trial. Lancet. 2001; 357:1729–1737.
- CARESS Steering Committee. Carotid revascularization using endarterectomy or stenting systems (CARESS): phase I clinical trial: J Endovasc Ther 2003; 10:1021–1030.
- Yadav JS, Wholey MD, Kuntz RE, et al; Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy Investigators. Protected carotidartery stenting versus endarterectomy in high-risk patients, N Engl J Med 2004; 351:1493–1501.
- Mas JL, Chatellier G, Beyssen B, et al., EVA-3S Investigators. Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med. 2006; 355:1660–1671.
- Ringleb PA, Allenberg J, Bruckmann H, et al., SPACE Collaborative Group. 30 day results from the SPACE trial of stent-protected angioplasty versus carotid endarterectomy in symptomatic patients: a randomised non-inferiority trial. Lancet. 2006; 368:1239–1247.
- CREST. Carotid Revascularization Endarterectomy vs Stent Trial. The Internet Stroke Center. www.strokecenter.org/trials/trialDetail.aspx?tid=80&search_string=crest.
- Center for Medicare and Medicaid Services. Expansion of coverage for percutaneous transluminal angioplasty (PTA). www.cms.hhs.gov/ContractorLearningResources/downloads/JA3811.pdf.
- Rothwell PM, Goldstein LB. Carotid endarterectomy for asymptomatic carotid stenosis: asymptomatic carotid surgery trial. Stroke. 2004; 35:2425–2427.
Whether carotid stenting has any advantage over carotid surgery (endarterectomy)—and for which patients—is still a topic of study and debate.
Treatment of carotid atherosclerosis and stenosis is important in preventing stroke and its comorbidities. Today, three main treatments exist: medical management (lipid-lowering, antihypertensive, and antiplatelet therapy), surgery, and, more recently, carotid angioplasty and stenting. The rationale for these treatments is to decrease the risk of cerebral infarction by stabilizing or removing plaque and improving blood flow.
Surgery has proven beneficial in patients with symptomatic carotid stenosis greater than 50% or asymptomatic stenosis greater than 60%, but it is risky in some patients. Stenting has evolved in part from the success of surgery and the need for alternative treatments for patients who are at unacceptable risk of perioperative complications. However, it does not have a clear advantage over surgery in patients at average risk. Further, its use in patients with asymptomatic stenosis of any severity is still controversial.
In this paper we review the major trials of carotid endarterectomy and stenting and summarize what we know today about who should undergo these therapies.
NOT ALL STROKES ARE DUE TO CAROTID ATHEROSCLEROSIS
Depending on the institution’s referral pattern and population served, between 80% and 90% of strokes are ischemic (the rest being hemorrhagic).1 Atherosclerosis of large arteries (typically defined as more than 50% stenosis of a major brain artery or branch cortical artery2) is just one cause of ischemic stroke, but it is an important one. Other identifiable causes of ischemic stroke include cardioembolism and small-artery occlusion (lacunar stroke), and some cases are idiopathic.
Large-artery atherosclerotic disease can damage the brain gradually, with carotid stenosis resulting in hypoperfusion and subsequent cerebral infarction. More commonly, however, the carotid plaque often seen in large-artery atherosclerotic disease can ulcerate and occlude the vessel acutely or generate platelet aggregates that may embolize, resulting in cerebral infarction or transient ischemic attack.
In the Lausanne Stroke Registry,3 the rate of ischemic stroke in patients with a greater than 50% large-artery stenosis ranged from 27% in 1979 to 17% in 2003, the decline likely being due to therapeutic advances.
SURGERY BEATS MEDICAL THERAPY FOR CAROTID ATHEROSCLEROSIS
Four landmark trials provided substantial evidence that carotid endarterectomy is better than medical management in patients with symptomatic or asymptomatic high-grade stenosis. These trials indirectly paved the way for carotid stenting.
The North American Symptomatic Carotid Endarterectomy Trial (NASCET)
Patients at 50 clinical centers who had had a hemispheric or retinal transient ischemic attack or a nondisabling stroke were randomized to undergo surgery (carotid endarterectomy) or no surgery. All patients received maximal medical management consisting of blood pressure control, lipid management if indicated, and antiplatelet therapy with aspirin. At baseline, 37% of patients were taking 650 mg or more of aspirin per day, and 11% were taking less than 325 mg per day. The patients were stratified into two prespecified groups on the basis of the severity of carotid stenosis: those with narrowing of 30% to 69% and those with narrowing of 70% to 99%.
Results in high-grade stenosis. In August 1991, the investigators published their results in patients with symptomatic high-grade (70%–99%) stenosis.4 Surgical treatment was more beneficial than medical management alone: the cumulative risk of any ipsilateral stroke at 2 years was 26% in the medical group and 9% in the surgical group, an absolute risk reduction of 17%. The benefit of endarterectomy was still apparent at 8 years of follow-up.5
Results in moderate stenosis. In 1998, the investigators published their results in patients with symptomatic moderate (< 70%) stenosis.5 Surgery was more beneficial than medical therapy in this subgroup as well: at 5 years, the rate of any ipsilateral stroke in patients with 50% to 69% stenosis was 15.7% in those treated surgically and 22.2% in those treated medically (P = .045). In patients with less than 50% stenosis, the 5-year stroke rate was not significantly lower with endarterectomy than with medical therapy.
The European Carotid Surgery Trial (ECST)
The ECST,6 published in 1998, corroborated the NASCET findings. This multicenter, randomized, controlled trial enrolled 3,024 patients with symptoms of at least one transient ischemic attack in the distribution of one or both carotid arteries.
Results. In patients with stenosis of greater than 80% (60% by the NASCET criteria for calculating angiographic stenosis), the frequency of major stroke or death at 3 years was 26.5% in the control group and 14.9% in the surgery group, an absolute difference of 11.6%.
The Endarterectomy for Asymptomatic Carotid Artery Stenosis (ACAS) trial
The NASCET and ECST studies made it clear that select groups of patients with symptomatic carotid stenosis benefit from carotid endarterectomy. But what about patients with stenosis but no prior stroke?
The ACAS trial aimed to find out.7 In this pivotal study, 1,662 patients with asymptomatic carotid artery stenosis greater than 60% were randomized to receive either medical therapy alone or medical plus surgical therapy.
Results were published in 2004. After a median follow-up of 2.7 years, the aggregate 5-year risk of ipsilateral stroke, any perioperative stroke, or death was estimated to be 5.1% in the surgical group and 11.0% in the medical group, a relative risk reduction of 53%. However, for surgery to be beneficial, the rate of perioperative death and other serious complications had to be less than 3%, and the expected patient survival had to be at least 5 years.
Of note, the benefit of carotid endarterectomy in this study was predominantly in men, with less of a benefit for women and diabetic patients. Furthermore, even though endarterectomy was beneficial in this asymptomatic cohort, the overall benefit in terms of stroke risk reduction was small compared with that in NASCET and ECST, in which patients had symptomatic disease.
The Asymptomatic Carotid Surgery Trial (ACST)
In this European version of ACAS, published in 2004, 3,120 patients with asymptomatic carotid narrowing on ultrasonography were randomized to undergo surgery or medical therapy.
Results. The risk of stroke or death within 30 days of carotid endarterectomy was 3.1%. In patients younger than 75 years who had carotid narrowing of 70% or more, immediate surgery decreased the net 5-year stroke risk from 12% to 6%.8
WHO SHOULD NOT UNDERGO CAROTID ENDARTERECTOMY?
From these studies, we can conclude that patients with symptomatic carotid stenosis of 50% or greater and patients with asymptomatic stenosis of 60% or greater benefit from carotid endarterectomy, but only if the perioperative rate of death and other serious complications is less than 3%.7
What are the risk factors for complications during this surgery? In 2006, Cremonesi et al,9 in a consensus paper, defined patients as being at high risk if they had any of the following:
- Contralateral laryngeal nerve palsy
- Radiation therapy to the neck
- Previous carotid endarterectomy with recurrent stenosis
- Lesions high in the cervical internal carotid artery or below the clavicle in the common carotid artery
- Severe tandem lesions
- Age greater than 80 years
- Severe pulmonary disease
- Congestive heart failure (New York Heart
- Association class 3 or 4) or known severe left ventricular dysfunction
- Open heart surgery needed within 6 weeks
- Myocardial infarction within the past 4 weeks
- Unstable angina
- Contralateral carotid occlusion.
Could endovascular treatment be the answer for these patients at high risk who should not undergo carotid endarterectomy? Indeed, the procedure is being studied extensively and performed more frequently. We summarize the major studies below.
STUDIES OF CAROTID STENTING VS ENDARTERECTOMY
The Carotid and Vertebral Artery Transluminal Angioplasty Study (CAVATAS)
This study, published in 2001,10 was the first randomized, multicenter trial to compare the risks and benefits of endovascular treatment (angioplasty with or without stenting) of carotid and vertebral artery stenosis with those of conventional surgery.
To be included, patients had to have carotid artery stenosis (symptomatic or asymptomatic) that was suitable for either carotid endarterectomy or endovascular treatment. Patients were not grouped on the basis of the severity of their stenosis, but the mean stenosis in randomized patients was 86%.
A total of 504 patients were enrolled, of whom 251 were randomized to undergo endovascular treatment. Most patients in this group underwent angioplasty alone, but 26% also received stents because of suboptimal vessel dilatation or at the discretion of the intervening physician.
The primary end point was any disabling stroke or death. Secondary end points were any ipsilateral stroke lasting longer than 7 days and the combination of death or disabling ipsilateral stroke.
The results showed no significant difference between endovascular treatment and surgery in any of these end points at 3 years. However, the overall rates of procedural stroke and death were nearly double those seen in NASCET and ECST. The investigators could not determine the reason for this higher risk, but they hypothesized that CAVATAS included patients at higher risk.
The restenosis rate was higher in the endovascular therapy group (14%) than in the surgery group (4%; P < .001). On the other hand, the surgery group had a higher rate of minor complications, including cranial nerve palsies and neck hematomas.
Carotid Revascularization With Endarterectomy or Stenting Systems (CARESS)
This prospective, multicenter, phase 2 trial, published in 2003, compared the outcomes of standard carotid endarterectomy vs carotid artery stenting using distal embolic protection devices.11 All the patients in this study had at least 50% symptomatic stenosis or 75% asymptomatic stenosis.
Results. At 30 days, 7 (2.4%) of 254 patients in the endarterectomy group had had strokes, and one of the 7 patients with stroke died, so the combined rate of stroke or death (the primary end point) was 2.4%. In the stenting group, 3 (2.1%) of 143 patients had strokes and no patients died. Overall, there was no significant difference in the composite of death, stroke, or myocardial infarction (the secondary end point): 3% for carotid endarterectomy and 2% for stenting patients.
The Stenting and Angioplasty With Protection in Patients at High Risk for Endarterectomy (SAPPHIRE) trial
In this trial,12 published in 2004, patients had to have either symptomatic carotid disease with 50% stenosis or greater or asymptomatic stenosis of 80% or greater, determined by ultrasonography. Further, all patients had to have at least one comorbid condition that increased their perioperative risk. Up until this point, no trial had strictly defined patients at increased risk for complications after carotid endarterectomy and assessed subsequent outcomes. The risk factors included severe cardiac or pulmonary disease, age greater than 80, postendarterectomy carotid stenosis, previous neck surgery, previous neck radiation, contralateral recurrent laryngeal nerve palsy, and contralateral carotid occlusion.
Patients were randomized to undergo carotid artery stenting with distal protection or carotid endarterectomy.
The primary end points of this study were the cumulative incidence of major cardiovascular events at 1 year; death, stroke, or myocardial infarction within 30 days of intervention; and ipsilateral stroke between 31 days and 1 year. Secondary outcomes measured were the rates of target-vessel recanalization at 1 year, cranial nerve palsy, and surgical site complications.
Results. The rate of stroke or death was similar in both groups. The stenting group had fewer adverse cardiac events (mainly non-Q-wave myocardial infarction) than the surgery group. At 1 year the rate of major ipsilateral stroke was 3.3% in the endarterectomy group vs 0% in the stenting group (the difference was not significant), and the cardiovascular event rates continued to be higher in the endarterectomy group.
The investigators noted that myocardial infarction was included as a primary end point because patients with atherosclerotic vascular disease who undergo either stenting or endarterectomy are at a substantial risk of myocardial infarction, and a Q-wave or a non-Q-wave myocardial infarction in the perioperative period increases the risk of future complications and death. A perioperative non-Q-wave infarction increases the risk of death by a factor of 6 and increases the risk of myocardial infarction by a factor of 27 in the subsequent 6 months.
Overall, this study presents evidence that stenting, using distal embolic protection devices, is not inferior to endarterectomy and has fewer cardiovascular complications in patients who have at least one risk factor.
The Endarterectomy Versus Stenting in Patients With Symptomatic Severe Carotid Stenosis (EVA-3S) study
This recent multicenter, randomized study13 was designed to determine if stenting is as good as (not inferior to) carotid endarterectomy in patients with symptomatic carotid stenosis of at least 60%. The primary end point was to be the incidence of stroke or death within 30 days after treatment. However, the trial was stopped early after the inclusion of 527 patients for reasons of safety and futility.
Results. The 30-day incidence of any stroke or death was higher in the stenting group (9.6% vs 3.9%). The relative risk of any stroke or death after stenting as compared with endarterectomy was 2.5. The 30-day incidence of disabling stroke or death was also higher in the stenting group (3.4% vs 1.5%; relative risk 2.2). At 6 months, the incidence of any stroke or death was 6.1% after endarterectomy and 11.7% after stenting (P = .02). There was a trend toward more major local complications after stenting and systemic complications after endarterectomy. Cranial-nerve injury was more common after endarterectomy than after stenting (as expected). Overall, death and stroke rates were lower at 1 month and 6 months with endarterectomy than with stenting.
The Stent-Protected Angioplasty Versus Carotid Endarterectomy (SPACE) trial
This randomized, multicenter study,14 published in 2006, was also designed to compare the safety and efficacy of carotid stenting and endarterectomy. Some 1,200 patients with symptomatic carotid artery stenosis confirmed by ultrasonography were randomly assigned within 180 days of a transient ischemic attack or moderate stroke to undergo carotid artery stenting (n = 605) or carotid endarterectomy (n = 595). The primary end point was ipsilateral ischemic stroke or death 30 days after the procedure. A total of 1,183 patients were included in the analysis.
Results. The rate of the primary end point was 6.84% with stenting and 6.34% with endarterectomy. The study failed to prove the noninferiority of carotid artery stenting compared with carotid endarterectomy for the periprocedural complication rate. Results at 6 to 24 months are awaited.
The Carotid Revascularization Endarterectomy Versus Stenting (CREST) trial
Perhaps the most anxiously awaited results are those of the CREST trial,15 funded by the National Institutes of Health. This is a prospective, randomized, parallel, two-arm, multicenter clinical trial with blinded end point evaluation. Anticipated enrollment will include 2,500 patients. Patients are eligible for enrollment if they have symptoms of carotid stenosis within 180 days of a stroke or transient ischemic attack with ipsilateral carotid stenosis of at least 50% by angiography (70% by ultrasonography), or if they have asymptomatic carotid stenosis of at least 60% by angiography (70% by ultrasonography).
Patients are being randomized to undergo either carotid artery stenting or carotid endarterectomy. All receive aspirin as anti-platelet therapy, treatment for hypertension, and management of other stroke risk factors. Follow-up will last 4 years, with clinic visits at 1, 6, 12, 18, 24, 30, 36, 42, and 48 months. Primary outcome measures will be rates of death, stroke, or myocardial infarction at 30 days postoperatively, and ipsilateral stroke at 30 days postoperatively.
As of February 2007, 1,506 patients had been enrolled and 1,453 had been randomized at 94 sites in North America.
MEDICAID AND MEDICARE NOW PAY FOR THESE THERAPIES
An important practical consideration for patients and physicians is whether Medicaid and Medicare will pay for these therapies.
In July 2001, Medicare began to cover percutaneous transluminal angioplasty of the carotid artery with concurrent stent placement, when furnished in accordance with US Food and Drug Administration (FDA) protocols governing Category B (nonexperimental) investigational device exemption clinical trials.16 Angioplasty of the carotid artery, when provided solely for the purpose of carotid artery dilation concurrent with carotid stent placement, is considered to be a reasonable and necessary service when provided in the context of clinical trials.
In March 2005, Medicare began to provide coverage for percutaneous transluminal angioplasty of the carotid artery concurrent with the placement of an FDA-approved carotid stent with embolic protection for the following groups of patients:
- Those who would be at high risk during carotid endarterectomy and who also have symptomatic carotid artery stenosis of 70% or greater. Coverage is limited to procedures performed using FDA-approved carotid artery stenting systems and embolic protection devices.
- Those who would be at high risk during endarterectomy and who have symptomatic carotid artery stenosis of 50% to 70%, in accordance with the Category B Investigational Device Exemption clinical trials regulation, as a routine cost under the clinical trials policy, or in accordance with the national coverage determination on carotid artery stenting post-approval.
- Those who would be at high risk during carotid endarterectomy and have asymptomatic carotid artery stenosis greater than 80%, in accordance with the Category B Investigational Device Exemption clinical trials regulation, as a routine cost under the clinical trials policy, or in accordance with the national coverage determination on carotid artery stenting postapproval studies.
As noted above, Medicare and Medicaid will only cover carotid stenting if the stent system is FDA-approved, with concomitant use of a distal embolic protection device. However, in view of conflicting data from stenting trials to date, including EVA-3S13 and SPACE,14 it remains to be seen if emboli protection devices significantly reduce periprocedural stroke rates. The FDA recommends that if it is not technically possible to use one of these devices, then the procedure should be aborted due to safety issues.
These coverage decisions are an important practical aspect of carotid stenting and they should be familiar to physicians when they see and refer patients with carotid disease.
WHAT CAN WE SAY AT THIS POINT?
Given the multiple recent and ongoing trials of stenting vs endarterectomy in carotid stenosis, debate continues as to what the role of stenting will be in the future. What can we say at this point?
In patients with asymptomatic carotid stenosis of greater than 60% or symptomatic carotid stenosis of greater than 50%, carotid endarterectomy has been proven to be superior to medical therapy alone.
The efficacy and safety of carotid stenting compared with carotid endarterectomy is still uncertain. In the trials reviewed above, carotid stenting did not appear to have a clear advantage over endarterectomy in patients at average surgical risk. Stenting may be most advantageous when used in patients with symptomatic carotid stenosis who would be at high operative risk, as indicated by the SAPPHIRE trial.
In patients with severe but asymptomatic carotid stenosis who are at high operative risk, the addition of carotid angioplasty and stenting to maximum medical therapy remains controversial. The periprocedural complication rate in these patients may actually exceed the rate of stroke in asymptomatic patients with greater than 60% stenosis who do not undergo stenting or surgery. In addition, subgroup analyses of patients with 70% to 99% symptomatic stenosis in various trials show that surgical benefit is greater in men than in women, and it remains to be seen whether there is any benefit in women with moderate stenoses, asymptomatic lesions, or both.17
Further experience and study are needed, and the results of the Carotid Stenting vs Surgery of Severe Carotid Artery Disease and Stroke Prevention in Asymptomatic Patients (ACT I) study (comparing stenting and surgery in asymptomatic carotid stenosis), and the ongoing CREST trial (comparing stenting and surgery in symptomatic and asymptomatic carotid stenosis) are eagerly awaited. Until then, clinicians should continue to weigh individual patient risks and benefits when referring patients for surgical treatment of carotid athero-sclerotic disease. Regardless of whether surgery is undertaken, maximal medical therapy with the use of antiplatelet agents, blood pressure control, and statin therapy remains the mainstay of treatment.
Whether carotid stenting has any advantage over carotid surgery (endarterectomy)—and for which patients—is still a topic of study and debate.
Treatment of carotid atherosclerosis and stenosis is important in preventing stroke and its comorbidities. Today, three main treatments exist: medical management (lipid-lowering, antihypertensive, and antiplatelet therapy), surgery, and, more recently, carotid angioplasty and stenting. The rationale for these treatments is to decrease the risk of cerebral infarction by stabilizing or removing plaque and improving blood flow.
Surgery has proven beneficial in patients with symptomatic carotid stenosis greater than 50% or asymptomatic stenosis greater than 60%, but it is risky in some patients. Stenting has evolved in part from the success of surgery and the need for alternative treatments for patients who are at unacceptable risk of perioperative complications. However, it does not have a clear advantage over surgery in patients at average risk. Further, its use in patients with asymptomatic stenosis of any severity is still controversial.
In this paper we review the major trials of carotid endarterectomy and stenting and summarize what we know today about who should undergo these therapies.
NOT ALL STROKES ARE DUE TO CAROTID ATHEROSCLEROSIS
Depending on the institution’s referral pattern and population served, between 80% and 90% of strokes are ischemic (the rest being hemorrhagic).1 Atherosclerosis of large arteries (typically defined as more than 50% stenosis of a major brain artery or branch cortical artery2) is just one cause of ischemic stroke, but it is an important one. Other identifiable causes of ischemic stroke include cardioembolism and small-artery occlusion (lacunar stroke), and some cases are idiopathic.
Large-artery atherosclerotic disease can damage the brain gradually, with carotid stenosis resulting in hypoperfusion and subsequent cerebral infarction. More commonly, however, the carotid plaque often seen in large-artery atherosclerotic disease can ulcerate and occlude the vessel acutely or generate platelet aggregates that may embolize, resulting in cerebral infarction or transient ischemic attack.
In the Lausanne Stroke Registry,3 the rate of ischemic stroke in patients with a greater than 50% large-artery stenosis ranged from 27% in 1979 to 17% in 2003, the decline likely being due to therapeutic advances.
SURGERY BEATS MEDICAL THERAPY FOR CAROTID ATHEROSCLEROSIS
Four landmark trials provided substantial evidence that carotid endarterectomy is better than medical management in patients with symptomatic or asymptomatic high-grade stenosis. These trials indirectly paved the way for carotid stenting.
The North American Symptomatic Carotid Endarterectomy Trial (NASCET)
Patients at 50 clinical centers who had had a hemispheric or retinal transient ischemic attack or a nondisabling stroke were randomized to undergo surgery (carotid endarterectomy) or no surgery. All patients received maximal medical management consisting of blood pressure control, lipid management if indicated, and antiplatelet therapy with aspirin. At baseline, 37% of patients were taking 650 mg or more of aspirin per day, and 11% were taking less than 325 mg per day. The patients were stratified into two prespecified groups on the basis of the severity of carotid stenosis: those with narrowing of 30% to 69% and those with narrowing of 70% to 99%.
Results in high-grade stenosis. In August 1991, the investigators published their results in patients with symptomatic high-grade (70%–99%) stenosis.4 Surgical treatment was more beneficial than medical management alone: the cumulative risk of any ipsilateral stroke at 2 years was 26% in the medical group and 9% in the surgical group, an absolute risk reduction of 17%. The benefit of endarterectomy was still apparent at 8 years of follow-up.5
Results in moderate stenosis. In 1998, the investigators published their results in patients with symptomatic moderate (< 70%) stenosis.5 Surgery was more beneficial than medical therapy in this subgroup as well: at 5 years, the rate of any ipsilateral stroke in patients with 50% to 69% stenosis was 15.7% in those treated surgically and 22.2% in those treated medically (P = .045). In patients with less than 50% stenosis, the 5-year stroke rate was not significantly lower with endarterectomy than with medical therapy.
The European Carotid Surgery Trial (ECST)
The ECST,6 published in 1998, corroborated the NASCET findings. This multicenter, randomized, controlled trial enrolled 3,024 patients with symptoms of at least one transient ischemic attack in the distribution of one or both carotid arteries.
Results. In patients with stenosis of greater than 80% (60% by the NASCET criteria for calculating angiographic stenosis), the frequency of major stroke or death at 3 years was 26.5% in the control group and 14.9% in the surgery group, an absolute difference of 11.6%.
The Endarterectomy for Asymptomatic Carotid Artery Stenosis (ACAS) trial
The NASCET and ECST studies made it clear that select groups of patients with symptomatic carotid stenosis benefit from carotid endarterectomy. But what about patients with stenosis but no prior stroke?
The ACAS trial aimed to find out.7 In this pivotal study, 1,662 patients with asymptomatic carotid artery stenosis greater than 60% were randomized to receive either medical therapy alone or medical plus surgical therapy.
Results were published in 2004. After a median follow-up of 2.7 years, the aggregate 5-year risk of ipsilateral stroke, any perioperative stroke, or death was estimated to be 5.1% in the surgical group and 11.0% in the medical group, a relative risk reduction of 53%. However, for surgery to be beneficial, the rate of perioperative death and other serious complications had to be less than 3%, and the expected patient survival had to be at least 5 years.
Of note, the benefit of carotid endarterectomy in this study was predominantly in men, with less of a benefit for women and diabetic patients. Furthermore, even though endarterectomy was beneficial in this asymptomatic cohort, the overall benefit in terms of stroke risk reduction was small compared with that in NASCET and ECST, in which patients had symptomatic disease.
The Asymptomatic Carotid Surgery Trial (ACST)
In this European version of ACAS, published in 2004, 3,120 patients with asymptomatic carotid narrowing on ultrasonography were randomized to undergo surgery or medical therapy.
Results. The risk of stroke or death within 30 days of carotid endarterectomy was 3.1%. In patients younger than 75 years who had carotid narrowing of 70% or more, immediate surgery decreased the net 5-year stroke risk from 12% to 6%.8
WHO SHOULD NOT UNDERGO CAROTID ENDARTERECTOMY?
From these studies, we can conclude that patients with symptomatic carotid stenosis of 50% or greater and patients with asymptomatic stenosis of 60% or greater benefit from carotid endarterectomy, but only if the perioperative rate of death and other serious complications is less than 3%.7
What are the risk factors for complications during this surgery? In 2006, Cremonesi et al,9 in a consensus paper, defined patients as being at high risk if they had any of the following:
- Contralateral laryngeal nerve palsy
- Radiation therapy to the neck
- Previous carotid endarterectomy with recurrent stenosis
- Lesions high in the cervical internal carotid artery or below the clavicle in the common carotid artery
- Severe tandem lesions
- Age greater than 80 years
- Severe pulmonary disease
- Congestive heart failure (New York Heart
- Association class 3 or 4) or known severe left ventricular dysfunction
- Open heart surgery needed within 6 weeks
- Myocardial infarction within the past 4 weeks
- Unstable angina
- Contralateral carotid occlusion.
Could endovascular treatment be the answer for these patients at high risk who should not undergo carotid endarterectomy? Indeed, the procedure is being studied extensively and performed more frequently. We summarize the major studies below.
STUDIES OF CAROTID STENTING VS ENDARTERECTOMY
The Carotid and Vertebral Artery Transluminal Angioplasty Study (CAVATAS)
This study, published in 2001,10 was the first randomized, multicenter trial to compare the risks and benefits of endovascular treatment (angioplasty with or without stenting) of carotid and vertebral artery stenosis with those of conventional surgery.
To be included, patients had to have carotid artery stenosis (symptomatic or asymptomatic) that was suitable for either carotid endarterectomy or endovascular treatment. Patients were not grouped on the basis of the severity of their stenosis, but the mean stenosis in randomized patients was 86%.
A total of 504 patients were enrolled, of whom 251 were randomized to undergo endovascular treatment. Most patients in this group underwent angioplasty alone, but 26% also received stents because of suboptimal vessel dilatation or at the discretion of the intervening physician.
The primary end point was any disabling stroke or death. Secondary end points were any ipsilateral stroke lasting longer than 7 days and the combination of death or disabling ipsilateral stroke.
The results showed no significant difference between endovascular treatment and surgery in any of these end points at 3 years. However, the overall rates of procedural stroke and death were nearly double those seen in NASCET and ECST. The investigators could not determine the reason for this higher risk, but they hypothesized that CAVATAS included patients at higher risk.
The restenosis rate was higher in the endovascular therapy group (14%) than in the surgery group (4%; P < .001). On the other hand, the surgery group had a higher rate of minor complications, including cranial nerve palsies and neck hematomas.
Carotid Revascularization With Endarterectomy or Stenting Systems (CARESS)
This prospective, multicenter, phase 2 trial, published in 2003, compared the outcomes of standard carotid endarterectomy vs carotid artery stenting using distal embolic protection devices.11 All the patients in this study had at least 50% symptomatic stenosis or 75% asymptomatic stenosis.
Results. At 30 days, 7 (2.4%) of 254 patients in the endarterectomy group had had strokes, and one of the 7 patients with stroke died, so the combined rate of stroke or death (the primary end point) was 2.4%. In the stenting group, 3 (2.1%) of 143 patients had strokes and no patients died. Overall, there was no significant difference in the composite of death, stroke, or myocardial infarction (the secondary end point): 3% for carotid endarterectomy and 2% for stenting patients.
The Stenting and Angioplasty With Protection in Patients at High Risk for Endarterectomy (SAPPHIRE) trial
In this trial,12 published in 2004, patients had to have either symptomatic carotid disease with 50% stenosis or greater or asymptomatic stenosis of 80% or greater, determined by ultrasonography. Further, all patients had to have at least one comorbid condition that increased their perioperative risk. Up until this point, no trial had strictly defined patients at increased risk for complications after carotid endarterectomy and assessed subsequent outcomes. The risk factors included severe cardiac or pulmonary disease, age greater than 80, postendarterectomy carotid stenosis, previous neck surgery, previous neck radiation, contralateral recurrent laryngeal nerve palsy, and contralateral carotid occlusion.
Patients were randomized to undergo carotid artery stenting with distal protection or carotid endarterectomy.
The primary end points of this study were the cumulative incidence of major cardiovascular events at 1 year; death, stroke, or myocardial infarction within 30 days of intervention; and ipsilateral stroke between 31 days and 1 year. Secondary outcomes measured were the rates of target-vessel recanalization at 1 year, cranial nerve palsy, and surgical site complications.
Results. The rate of stroke or death was similar in both groups. The stenting group had fewer adverse cardiac events (mainly non-Q-wave myocardial infarction) than the surgery group. At 1 year the rate of major ipsilateral stroke was 3.3% in the endarterectomy group vs 0% in the stenting group (the difference was not significant), and the cardiovascular event rates continued to be higher in the endarterectomy group.
The investigators noted that myocardial infarction was included as a primary end point because patients with atherosclerotic vascular disease who undergo either stenting or endarterectomy are at a substantial risk of myocardial infarction, and a Q-wave or a non-Q-wave myocardial infarction in the perioperative period increases the risk of future complications and death. A perioperative non-Q-wave infarction increases the risk of death by a factor of 6 and increases the risk of myocardial infarction by a factor of 27 in the subsequent 6 months.
Overall, this study presents evidence that stenting, using distal embolic protection devices, is not inferior to endarterectomy and has fewer cardiovascular complications in patients who have at least one risk factor.
The Endarterectomy Versus Stenting in Patients With Symptomatic Severe Carotid Stenosis (EVA-3S) study
This recent multicenter, randomized study13 was designed to determine if stenting is as good as (not inferior to) carotid endarterectomy in patients with symptomatic carotid stenosis of at least 60%. The primary end point was to be the incidence of stroke or death within 30 days after treatment. However, the trial was stopped early after the inclusion of 527 patients for reasons of safety and futility.
Results. The 30-day incidence of any stroke or death was higher in the stenting group (9.6% vs 3.9%). The relative risk of any stroke or death after stenting as compared with endarterectomy was 2.5. The 30-day incidence of disabling stroke or death was also higher in the stenting group (3.4% vs 1.5%; relative risk 2.2). At 6 months, the incidence of any stroke or death was 6.1% after endarterectomy and 11.7% after stenting (P = .02). There was a trend toward more major local complications after stenting and systemic complications after endarterectomy. Cranial-nerve injury was more common after endarterectomy than after stenting (as expected). Overall, death and stroke rates were lower at 1 month and 6 months with endarterectomy than with stenting.
The Stent-Protected Angioplasty Versus Carotid Endarterectomy (SPACE) trial
This randomized, multicenter study,14 published in 2006, was also designed to compare the safety and efficacy of carotid stenting and endarterectomy. Some 1,200 patients with symptomatic carotid artery stenosis confirmed by ultrasonography were randomly assigned within 180 days of a transient ischemic attack or moderate stroke to undergo carotid artery stenting (n = 605) or carotid endarterectomy (n = 595). The primary end point was ipsilateral ischemic stroke or death 30 days after the procedure. A total of 1,183 patients were included in the analysis.
Results. The rate of the primary end point was 6.84% with stenting and 6.34% with endarterectomy. The study failed to prove the noninferiority of carotid artery stenting compared with carotid endarterectomy for the periprocedural complication rate. Results at 6 to 24 months are awaited.
The Carotid Revascularization Endarterectomy Versus Stenting (CREST) trial
Perhaps the most anxiously awaited results are those of the CREST trial,15 funded by the National Institutes of Health. This is a prospective, randomized, parallel, two-arm, multicenter clinical trial with blinded end point evaluation. Anticipated enrollment will include 2,500 patients. Patients are eligible for enrollment if they have symptoms of carotid stenosis within 180 days of a stroke or transient ischemic attack with ipsilateral carotid stenosis of at least 50% by angiography (70% by ultrasonography), or if they have asymptomatic carotid stenosis of at least 60% by angiography (70% by ultrasonography).
Patients are being randomized to undergo either carotid artery stenting or carotid endarterectomy. All receive aspirin as anti-platelet therapy, treatment for hypertension, and management of other stroke risk factors. Follow-up will last 4 years, with clinic visits at 1, 6, 12, 18, 24, 30, 36, 42, and 48 months. Primary outcome measures will be rates of death, stroke, or myocardial infarction at 30 days postoperatively, and ipsilateral stroke at 30 days postoperatively.
As of February 2007, 1,506 patients had been enrolled and 1,453 had been randomized at 94 sites in North America.
MEDICAID AND MEDICARE NOW PAY FOR THESE THERAPIES
An important practical consideration for patients and physicians is whether Medicaid and Medicare will pay for these therapies.
In July 2001, Medicare began to cover percutaneous transluminal angioplasty of the carotid artery with concurrent stent placement, when furnished in accordance with US Food and Drug Administration (FDA) protocols governing Category B (nonexperimental) investigational device exemption clinical trials.16 Angioplasty of the carotid artery, when provided solely for the purpose of carotid artery dilation concurrent with carotid stent placement, is considered to be a reasonable and necessary service when provided in the context of clinical trials.
In March 2005, Medicare began to provide coverage for percutaneous transluminal angioplasty of the carotid artery concurrent with the placement of an FDA-approved carotid stent with embolic protection for the following groups of patients:
- Those who would be at high risk during carotid endarterectomy and who also have symptomatic carotid artery stenosis of 70% or greater. Coverage is limited to procedures performed using FDA-approved carotid artery stenting systems and embolic protection devices.
- Those who would be at high risk during endarterectomy and who have symptomatic carotid artery stenosis of 50% to 70%, in accordance with the Category B Investigational Device Exemption clinical trials regulation, as a routine cost under the clinical trials policy, or in accordance with the national coverage determination on carotid artery stenting post-approval.
- Those who would be at high risk during carotid endarterectomy and have asymptomatic carotid artery stenosis greater than 80%, in accordance with the Category B Investigational Device Exemption clinical trials regulation, as a routine cost under the clinical trials policy, or in accordance with the national coverage determination on carotid artery stenting postapproval studies.
As noted above, Medicare and Medicaid will only cover carotid stenting if the stent system is FDA-approved, with concomitant use of a distal embolic protection device. However, in view of conflicting data from stenting trials to date, including EVA-3S13 and SPACE,14 it remains to be seen if emboli protection devices significantly reduce periprocedural stroke rates. The FDA recommends that if it is not technically possible to use one of these devices, then the procedure should be aborted due to safety issues.
These coverage decisions are an important practical aspect of carotid stenting and they should be familiar to physicians when they see and refer patients with carotid disease.
WHAT CAN WE SAY AT THIS POINT?
Given the multiple recent and ongoing trials of stenting vs endarterectomy in carotid stenosis, debate continues as to what the role of stenting will be in the future. What can we say at this point?
In patients with asymptomatic carotid stenosis of greater than 60% or symptomatic carotid stenosis of greater than 50%, carotid endarterectomy has been proven to be superior to medical therapy alone.
The efficacy and safety of carotid stenting compared with carotid endarterectomy is still uncertain. In the trials reviewed above, carotid stenting did not appear to have a clear advantage over endarterectomy in patients at average surgical risk. Stenting may be most advantageous when used in patients with symptomatic carotid stenosis who would be at high operative risk, as indicated by the SAPPHIRE trial.
In patients with severe but asymptomatic carotid stenosis who are at high operative risk, the addition of carotid angioplasty and stenting to maximum medical therapy remains controversial. The periprocedural complication rate in these patients may actually exceed the rate of stroke in asymptomatic patients with greater than 60% stenosis who do not undergo stenting or surgery. In addition, subgroup analyses of patients with 70% to 99% symptomatic stenosis in various trials show that surgical benefit is greater in men than in women, and it remains to be seen whether there is any benefit in women with moderate stenoses, asymptomatic lesions, or both.17
Further experience and study are needed, and the results of the Carotid Stenting vs Surgery of Severe Carotid Artery Disease and Stroke Prevention in Asymptomatic Patients (ACT I) study (comparing stenting and surgery in asymptomatic carotid stenosis), and the ongoing CREST trial (comparing stenting and surgery in symptomatic and asymptomatic carotid stenosis) are eagerly awaited. Until then, clinicians should continue to weigh individual patient risks and benefits when referring patients for surgical treatment of carotid athero-sclerotic disease. Regardless of whether surgery is undertaken, maximal medical therapy with the use of antiplatelet agents, blood pressure control, and statin therapy remains the mainstay of treatment.
- Incidence and Prevalence 2006 Chart Book on Cardiovascular and Lung Diseases Bethesda, MD: National Heart, Lung, and Blood Institute; 2006.
- Adams HP, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993; 24:35–41.
- Carrera E, Maeder-Ingvar M, Rossetti AO, Devuyst G, Bogousslavsky JLausanne Stroke Registry. Trends in risk factors, patterns and causes in hospitalized strokes over 25 years: The Lausanne Stroke Registry. Cerebrovasc Dis. 2007; 24:97–103.
- North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. 1991; 325:445–453.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North America Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med. 1998; 339:1415–1425.
- European Carotid Surgery Trialists’ Collaborative Group. Randomized trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet. 1998; 351:1379–1387.
- Halliday A, Mansfield A, Marro J, et al., MRC Asymptomatic Carotid Surgery Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomized controlled trial. Lancet. 2004; 363:1491–1502.
- Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. Endarterectomy for asymptomatic carotid artery stenosis. JAMA. 1995; 273:1421–1428.
- Cremonesi A, Setacci C, Bignamini A, et al. Carotid artery stenting: first consensus document of the ICCS-SPREAD Joint Committee. Stroke. 2006; 37:2400–2409.
- CAVATAS Investigators. Endovascular versus surgical treatment in patients with carotid stenosis in the Carotid and Vertebral Artery Transluminal Angioplasty Study (CA-VATAS): a randomized trial. Lancet. 2001; 357:1729–1737.
- CARESS Steering Committee. Carotid revascularization using endarterectomy or stenting systems (CARESS): phase I clinical trial: J Endovasc Ther 2003; 10:1021–1030.
- Yadav JS, Wholey MD, Kuntz RE, et al; Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy Investigators. Protected carotidartery stenting versus endarterectomy in high-risk patients, N Engl J Med 2004; 351:1493–1501.
- Mas JL, Chatellier G, Beyssen B, et al., EVA-3S Investigators. Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med. 2006; 355:1660–1671.
- Ringleb PA, Allenberg J, Bruckmann H, et al., SPACE Collaborative Group. 30 day results from the SPACE trial of stent-protected angioplasty versus carotid endarterectomy in symptomatic patients: a randomised non-inferiority trial. Lancet. 2006; 368:1239–1247.
- CREST. Carotid Revascularization Endarterectomy vs Stent Trial. The Internet Stroke Center. www.strokecenter.org/trials/trialDetail.aspx?tid=80&search_string=crest.
- Center for Medicare and Medicaid Services. Expansion of coverage for percutaneous transluminal angioplasty (PTA). www.cms.hhs.gov/ContractorLearningResources/downloads/JA3811.pdf.
- Rothwell PM, Goldstein LB. Carotid endarterectomy for asymptomatic carotid stenosis: asymptomatic carotid surgery trial. Stroke. 2004; 35:2425–2427.
- Incidence and Prevalence 2006 Chart Book on Cardiovascular and Lung Diseases Bethesda, MD: National Heart, Lung, and Blood Institute; 2006.
- Adams HP, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke. 1993; 24:35–41.
- Carrera E, Maeder-Ingvar M, Rossetti AO, Devuyst G, Bogousslavsky JLausanne Stroke Registry. Trends in risk factors, patterns and causes in hospitalized strokes over 25 years: The Lausanne Stroke Registry. Cerebrovasc Dis. 2007; 24:97–103.
- North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. 1991; 325:445–453.
- Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North America Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med. 1998; 339:1415–1425.
- European Carotid Surgery Trialists’ Collaborative Group. Randomized trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet. 1998; 351:1379–1387.
- Halliday A, Mansfield A, Marro J, et al., MRC Asymptomatic Carotid Surgery Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomized controlled trial. Lancet. 2004; 363:1491–1502.
- Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. Endarterectomy for asymptomatic carotid artery stenosis. JAMA. 1995; 273:1421–1428.
- Cremonesi A, Setacci C, Bignamini A, et al. Carotid artery stenting: first consensus document of the ICCS-SPREAD Joint Committee. Stroke. 2006; 37:2400–2409.
- CAVATAS Investigators. Endovascular versus surgical treatment in patients with carotid stenosis in the Carotid and Vertebral Artery Transluminal Angioplasty Study (CA-VATAS): a randomized trial. Lancet. 2001; 357:1729–1737.
- CARESS Steering Committee. Carotid revascularization using endarterectomy or stenting systems (CARESS): phase I clinical trial: J Endovasc Ther 2003; 10:1021–1030.
- Yadav JS, Wholey MD, Kuntz RE, et al; Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy Investigators. Protected carotidartery stenting versus endarterectomy in high-risk patients, N Engl J Med 2004; 351:1493–1501.
- Mas JL, Chatellier G, Beyssen B, et al., EVA-3S Investigators. Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med. 2006; 355:1660–1671.
- Ringleb PA, Allenberg J, Bruckmann H, et al., SPACE Collaborative Group. 30 day results from the SPACE trial of stent-protected angioplasty versus carotid endarterectomy in symptomatic patients: a randomised non-inferiority trial. Lancet. 2006; 368:1239–1247.
- CREST. Carotid Revascularization Endarterectomy vs Stent Trial. The Internet Stroke Center. www.strokecenter.org/trials/trialDetail.aspx?tid=80&search_string=crest.
- Center for Medicare and Medicaid Services. Expansion of coverage for percutaneous transluminal angioplasty (PTA). www.cms.hhs.gov/ContractorLearningResources/downloads/JA3811.pdf.
- Rothwell PM, Goldstein LB. Carotid endarterectomy for asymptomatic carotid stenosis: asymptomatic carotid surgery trial. Stroke. 2004; 35:2425–2427.
KEY POINTS
- In patients with asymptomatic carotid stenosis greater than 60% or symptomatic carotid stenosis greater than 50%, carotid endarterectomy has been proven to be superior to medical therapy alone.
- In clinical trials, carotid stenting did not appear to have a clear advantage over endarterectomy in patients at average surgical risk.
- Stenting may be most advantageous when used in patients with symptomatic carotid stenosis who would be at high risk of perioperative complications if they were to undergo carotid endarterectomy.