Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Figure 1. Temporal arteritis with intense inflammatory infiltrate within the arterial wall causing intimal thickening with nearly complete occlusion of the arterial lumen (hematoxylin and eosin, × 90).Typical findings in an inflamed temporal artery (Figure 1) include a lymphocytic infiltrate with activated macrophages and multinucleated giant cells (in 50% of cases). Typical panarteritis is not always seen, and infiltrates limited to the adventitia may be the only histologic finding in some patients.18
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
Figure 2. Digital subtraction angiography shows occlusion of the left subclavian artery and the left common carotid artery (black arrow), brachiocephalic dilatation, and post-dilatation stenosis (red arrow).Magnetic resonance angiography and computed angiography. These two noninvasive imaging tests have been used in the diagnosis and serial monitoring of patients with large-vessel involvement from giant cell arteritis (Figure 2). In addition to measuring lumen dimensions, magnetic resonance angiography (edema-weighted images) may also give information on vessel-wall signal intensity that may reflect inflammation. This information may be helpful in serial monitoring of patients with established large-vessel involvement, but it should be interpreted with great caution as it does not always correlate with active inflammation or with new structural changes in the vessel.24,25
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
References
Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med1995; 123:192–194.
Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum1994; 37:1007–1012.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med2003; 349:160–169.
Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum2005; 53:293–297.
Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol1998; 25:99–104.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore)2005; 84:269–276.
Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum1998; 41:1221–1226.
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med2002; 347:261–271.
Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum1995; 24:422–431.
Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med1995; 122:502–507.
Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl1973; 237(suppl 237):1–59.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol1999; 128:211–215.
González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis2001; 60:367–371.
Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med1997; 102:331–336.
Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better?Ann Rheum Dis2006; 65:826–828.
Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol2009; 27(1 suppl 52):S10–S13.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med2003; 139:505–515.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum2001; 45:140–145.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med1997; 337:1336–1342.
Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med2005; 142:359–369.
Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol2010; Epub ahead of print.
Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis2008; 67:1030–1033.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum2002; 46:1634–1642.
Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc1957; 163:821–827.
Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol2002; 86:530–532.
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol2001; 85:1061–1064.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med1975; 82:613–618.
Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum2002; 46:1309–1318.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum2006; 54:3310–3318.
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol2001; 19:495–501.
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med2001; 134:106–114.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum2007; 56:2789–2797.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med2007; 146:621–630.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum2002; 46:457–466.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum2004; 50:1332–1337.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum2006; 54:3306–3309.
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Figure 1. Temporal arteritis with intense inflammatory infiltrate within the arterial wall causing intimal thickening with nearly complete occlusion of the arterial lumen (hematoxylin and eosin, × 90).Typical findings in an inflamed temporal artery (Figure 1) include a lymphocytic infiltrate with activated macrophages and multinucleated giant cells (in 50% of cases). Typical panarteritis is not always seen, and infiltrates limited to the adventitia may be the only histologic finding in some patients.18
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
Figure 2. Digital subtraction angiography shows occlusion of the left subclavian artery and the left common carotid artery (black arrow), brachiocephalic dilatation, and post-dilatation stenosis (red arrow).Magnetic resonance angiography and computed angiography. These two noninvasive imaging tests have been used in the diagnosis and serial monitoring of patients with large-vessel involvement from giant cell arteritis (Figure 2). In addition to measuring lumen dimensions, magnetic resonance angiography (edema-weighted images) may also give information on vessel-wall signal intensity that may reflect inflammation. This information may be helpful in serial monitoring of patients with established large-vessel involvement, but it should be interpreted with great caution as it does not always correlate with active inflammation or with new structural changes in the vessel.24,25
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Figure 1. Temporal arteritis with intense inflammatory infiltrate within the arterial wall causing intimal thickening with nearly complete occlusion of the arterial lumen (hematoxylin and eosin, × 90).Typical findings in an inflamed temporal artery (Figure 1) include a lymphocytic infiltrate with activated macrophages and multinucleated giant cells (in 50% of cases). Typical panarteritis is not always seen, and infiltrates limited to the adventitia may be the only histologic finding in some patients.18
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
Figure 2. Digital subtraction angiography shows occlusion of the left subclavian artery and the left common carotid artery (black arrow), brachiocephalic dilatation, and post-dilatation stenosis (red arrow).Magnetic resonance angiography and computed angiography. These two noninvasive imaging tests have been used in the diagnosis and serial monitoring of patients with large-vessel involvement from giant cell arteritis (Figure 2). In addition to measuring lumen dimensions, magnetic resonance angiography (edema-weighted images) may also give information on vessel-wall signal intensity that may reflect inflammation. This information may be helpful in serial monitoring of patients with established large-vessel involvement, but it should be interpreted with great caution as it does not always correlate with active inflammation or with new structural changes in the vessel.24,25
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
References
Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med1995; 123:192–194.
Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum1994; 37:1007–1012.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med2003; 349:160–169.
Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum2005; 53:293–297.
Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol1998; 25:99–104.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore)2005; 84:269–276.
Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum1998; 41:1221–1226.
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med2002; 347:261–271.
Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum1995; 24:422–431.
Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med1995; 122:502–507.
Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl1973; 237(suppl 237):1–59.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol1999; 128:211–215.
González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis2001; 60:367–371.
Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med1997; 102:331–336.
Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better?Ann Rheum Dis2006; 65:826–828.
Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol2009; 27(1 suppl 52):S10–S13.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med2003; 139:505–515.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum2001; 45:140–145.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med1997; 337:1336–1342.
Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med2005; 142:359–369.
Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol2010; Epub ahead of print.
Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis2008; 67:1030–1033.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum2002; 46:1634–1642.
Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc1957; 163:821–827.
Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol2002; 86:530–532.
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol2001; 85:1061–1064.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med1975; 82:613–618.
Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum2002; 46:1309–1318.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum2006; 54:3310–3318.
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol2001; 19:495–501.
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med2001; 134:106–114.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum2007; 56:2789–2797.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med2007; 146:621–630.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum2002; 46:457–466.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum2004; 50:1332–1337.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum2006; 54:3306–3309.
References
Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med1995; 123:192–194.
Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum1994; 37:1007–1012.
Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med2003; 349:160–169.
Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum2005; 53:293–297.
Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol1998; 25:99–104.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore)2005; 84:269–276.
Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum1998; 41:1221–1226.
Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med2002; 347:261–271.
Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum1995; 24:422–431.
Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med1995; 122:502–507.
Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl1973; 237(suppl 237):1–59.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol1999; 128:211–215.
González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis2001; 60:367–371.
Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med1997; 102:331–336.
Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better?Ann Rheum Dis2006; 65:826–828.
Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol2009; 27(1 suppl 52):S10–S13.
Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med2003; 139:505–515.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum2001; 45:140–145.
Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med1997; 337:1336–1342.
Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med2005; 142:359–369.
Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol2010; Epub ahead of print.
Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis2008; 67:1030–1033.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum2002; 46:1634–1642.
Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc1957; 163:821–827.
Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol2002; 86:530–532.
Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol2001; 85:1061–1064.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med1975; 82:613–618.
Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum2002; 46:1309–1318.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum2006; 54:3310–3318.
Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol2001; 19:495–501.
Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med2001; 134:106–114.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum2007; 56:2789–2797.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med2007; 146:621–630.
Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum2002; 46:457–466.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum2004; 50:1332–1337.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum2006; 54:3306–3309.
Giant cell arteritis is often associated with an intense acute-phase response and cranial symptoms.
Large-vessel involvement is commonly present and may result in serious complications such as visual loss, stroke, limb claudication, and aortic aneurysm.
The diagnosis is usually confirmed by an abnormal temporal artery biopsy.
Symptoms of giant cell arteritis usually respond rapidly and completely to glucocorticoid therapy, still the mainstay of treatment. Most patients need prolonged therapy.
Several studies have evaluated alternative drugs in an attempt to avoid toxicity from long-term use of glucocorticoids. Results have been mixed, and further study is needed.
Editor’s Note: The views expressed in this article are solely those of the authors and do not reflect the official policy or position of the Department of State or the United States Government. This version of the article was peer-reviewed.
Venous thromboembolism (VTE) associated with travel has emerged as an important public health concern over the past decade. Numerous epidemiologic and case control studies have reported air travel as a risk factor for the development of VTE and have attempted to determine who is at risk and which precautions need to be taken to prevent this potentially fatal event.1–7 Often referred to as “traveler’s thrombosis” or “flight-related deep vein thrombosis,” VTE can also develop after long trips by automobile, bus, or train.8,9 Although the absolute risk is very low, this threat appears to be about three times higher in travelers and increases with longer trips.3
This article focuses on defining VTE and recognizing its clinical features, as well as providing recommendations and guidelines to prevent, diagnose, and treat this complication in people who travel.
WHAT IS VENOUS THROMBOEMBOLISM?
Deep vein thrombosis and pulmonary embolism represent different manifestations of the same clinical entity, ie, VTE. VTE is a common, lethal disease that affects hospitalized and nonhospitalized patients, frequently recurs, is often overlooked, may be asymptomatic, and may result in long-term complications that include pulmonary hypertension and the postthrombotic syndrome.
Figure 1.The leg veins are the most common site of deep vein thrombosis, accounting for nearly 90% of all cases; other locations include the arm and pelvic veins (Figure 1). Deep vein thrombosis in a proximal lower extremity (ie, involving the popliteal, femoral, common femoral, or external iliac vein) has an estimated 50% risk of migrating and leading to an acute pulmonary embolism if not treated, while approximately 25% of deep vein thromboses in the calf veins will, if not treated, propagate to involve the aforementioned veins.
Deep vein thrombosis of the upper extremities is generally related to an indwelling venous catheter or a central line being used for long-term administration of antibiotics, chemotherapy, or nutrition. A condition known as Paget-Schroetter syndrome or “effort thrombosis” may be seen in younger or athletic people who have a history of strenuous or unusual arm exercise.
RISK FACTORS FOR VTE
Most patients who develop VTE have one or more risk factors for it (Table 1), the presence of which is often referred to as a hypercoagulable state or thrombophilia. These risk factors are generally classified as either genetic (inherited) or acquired (environmental). Most VTE events are in fact associated with a combination of genetic and acquired risk factors.
Common inherited risk factors include:
Factor V Leiden mutation
Prothrombin gene mutation G20210A
Hyperhomocysteinemia
Deficiency of the natural anticoagulant proteins C, S, or antithrombin
Elevated levels of factor VIII (may be inherited or acquired).
Acquired risk factors include:
Older age
Immobilization or stasis (such as sitting for long periods of time while traveling)
Surgery (most notably orthopedic procedures including hip and knee replacement and repair of a hip fracture)
In addition, emerging risk factors more recently recognized include male sex, persistence of elevated factor VIII levels, and the continued presence of an elevated D-dimer level or deep vein thrombosis on duplex ultrasonography once anticoagulation treatment is completed. There is also evidence of an association between VTE and risk factors for atherosclerotic arterial disease such as smoking, hypertension, hyperlipidemia, and diabetes.
CLINICAL MANIFESTATIONS OF VTE
Patients with deep vein thrombosis may complain of pain, swelling, or both in the leg or arm. Physical examination may reveal increased warmth, tenderness, erythema, edema, or dilated (collateral) veins, most notable on the upper thigh or calf (for deep vein thrombosis in the lower extremity) or the chest wall (for upper-extremity deep vein thrombosis). The examiner may also observe a tender, palpable cord, which represents a superficial vein thrombosis involving the great and small saphenous veins (Figure 1). In extreme situations, the limb may be cyanotic or gangrenous.
A recommended clinical decision algorithm that can help assess a patient’s risk for an acute deep vein thrombosis prior to testing is depicted in Table 2.10 Patients with acute pulmonary embolism are likely to complain of the sudden onset of shortness of breath, pleuritic chest pain (especially with breathing), syncope, cough, or hemoptysis. A clinical decision algorithm that can help assess the risk of acute pulmonary embolism prior to testing is depicted in Table 3.11
DIAGNOSIS OF VTE
Clinical examination alone is generally insufficient to confirm a diagnosis of deep vein thrombosis or pulmonary embolism. Venous duplex ultrasonography is the most dependable investigation for deep vein thrombosis, but other tests include D-dimer and imaging studies such as computed tomographic venography or magnetic resonance venography of the lower extremities. A more invasive approach is venography; formerly considered the gold standard, it is now generally used only when the diagnosis is in doubt after noninvasive testing. The diagnosis of acute pulmonary embolism is best made by spiral computed tomography.
Other studies that may prove helpful include a ventilation-perfusion lung scan for patients who cannot undergo computed tomography due to a contrast allergy or renal insufficiency. Pulmonary angiography, while the gold standard, is less commonly used today, given the specificity and sensitivity of computed tomography.
Echocardiography at the bedside may be useful for patients too sick to move, although the study may not be diagnostic unless thrombi are seen in the heart or pulmonary arteries.
TREATMENT OF VTE
Treatments for VTE are summarized in Table 4. The length of treatment for acute VTE is generally 3 to 6 months. Patients with a known precipitating cause such as recent surgery or oral contraceptive use normally require 3 months of therapy, while those who had an unprovoked (idiopathic) event require longer therapy, sometimes continuing indefinitely.
For acute deep venous thrombosis
Acute deep vein thrombosis is now treated on an outpatient basis under most circumstances.
Unfractionated heparin is given intravenously for patients who need to be hospitalized, or subcutaneously in full dose for inpatient or outpatient treatment.
Low-molecular-weight heparins are available in subcutaneous preparations and can be given on an outpatient basis.
Fondaparinux (Arixtra), a factor Xa inhibitor, can also be given subcutaneously on an outpatient basis. Equivalent products are available outside the United States.
Warfarin (Coumadin), an oral vitamin K inhibitor, is the agent of choice for long-term management of deep vein thrombosis.
Other oral agents are available outside the United States.
For pulmonary embolism
Outpatient treatment of pulmonary embolism is not yet advised: an initial hospitalization is necessary. The same anticoagulants used for deep vein thrombosis are also used for acute pulmonary embolism.
Empiric treatment in underdeveloped countries
VTE may be an even greater concern on an outbound trip to a remote area, where medical care capabilities may be less than ideal and diagnostic and treatment options may be limited.
If there is a high pretest probability of acute VTE (Table 2, Table 3) and no diagnostic methods are available, empiric treatment with any of the parenteral anticoagulant agents listed in Table 4 is an option until the diagnosis can be confirmed. Caveats:
Care must be taken to be certain there is not a strong contraindication to the use of anticoagulation, such as bleeding or a drug allergy.
Neither unfractionated heparin nor any of the low-molecular-weight heparins should be given to a patient who has a history of heparin-induced thrombocytopenia.
In patients who have chronic kidney disease (creatinine clearance less than 30 mL/minute), the dosage of low-molecular-weight heparins must be adjusted and factor Xa inhibitors avoided. Both of these types of anticoagulants should be avoided in patients on hemodialysis.
More aggressive therapy
Under select circumstances a more aggressive approach to the treatment of VTE may be necessary. These options are usually indicated for a patient with a massive deep vein thrombosis of a lower extremity and for certain patients with an upper extremity deep vein thrombosis. Treatments include catheter-directed thrombolytic therapy and endovenous or surgical thrombectomy.
Thrombolytic therapy is recommended for a patient with an acute pulmonary embolism who is clinically unstable (systolic blood pressure lower than 90 mm Hg), if there is no contraindication to its use (bleeding risk or recent stroke or surgery). Thrombolytic therapy is also an option for those at low risk of bleeding with an acute pulmonary embolism who have signs and symptoms of right heart failure proven by echocardiography.
Surgical pulmonary embolectomy for acute massive pulmonary embolism and mechanical thrombectomy for extensive deep vein thrombosis are generally available only at highly sophisticated tertiary care centers.
An inferior vena cava filter is advised in patients with acute deep vein thrombosis or pulmonary embolism who cannot be fully anticoagulated, to prevent the clot from migrating from the lower extremities to the lungs. These filters are available as either permanent or temporary implants. Some temporary versions can remain in place for up to 150 days after insertion.
PREVENTION OF VTE
Prevention is the standard of care for all patients admitted to the hospital and in select individuals as outpatients who are at high risk of VTE.
A number of anticoagulant drugs are available in the United States for prophylaxis, including unfractionated heparin, low-molecular-weight heparin preparations, and fondaparinux (all of these given subcutaneously) and warfarin. In Europe and Canada, additional low-molecular-weight heparin preparations, factor Xa inhibitors, and direct thrombin inhibitors are available that have proven to be equally effective (Table 5).
Mechanical compression (graduated compression stockings, intermittent pneumatic compression devices) has proven effective in reducing the incidence of deep vein thrombosis and pulmonary embolism postoperatively in patients who cannot take anticoagulants. One study has demonstrated that compression stockings may also be effective in preventing VTE during travel.12
ABSOLUTE RISK IS LOW
Over the past decade, special attention has been paid to travel as a risk factor for developing VTE.13 Traveler’s thrombosis has become an important public health concern. Numerous publications and epidemiologic studies have targeted air travel in an attempt to determine who is at risk and what precautions are necessary to prevent this complication.1–7,9
The incidence of VTE following air travel is reported to be 3.2 per 1,000 person-years.4 While this incidence is relatively low, it is still 3.2 times higher than in the healthy population that is not flying.
The more serious complication of VTE, ie, acute pulmonary embolism, occurs less often. In three studies, the reported incidence ranged from 1.65 per million patients in flights longer than 8 hours to a high of 4.8 per million patients in flights longer than 12 hours or distances exceeding 10,000 km (6,200 miles).5,14,15 For the 400 passengers on the average long-haul flight of 12 hours, there is at most a 0.2% chance that somebody on the plane will have a symptomatic VTE).
RISK FACTORS IN LONG-DISTANCE TRAVELERS
The risk of traveler’s thrombosis has recently attracted the attention of passengers and the airline industry. Airlines are now openly discussing the risk and providing reminders such as exercises that should be undertaken in-flight (see the patient information page that accompanies this article). Some airlines are recommending that all patients consult their doctor to assess their personal risk of deep vein thrombosis before flying.
The most common risk factors for VTE in travelers are well established and are additive(Table 1). The extent of the additive risk, however, is not entirely clear.
What is clear is that when VTE occurs it is a life-altering and life-threatening event. If it occurs on an outbound trip, the local resources and capabilities available at the destination may not be adequate for optimal treatment. If a traveler experiences a VTE event on an outbound trip, an emergency return trip to the continental United States or a regional center of expertise may be required. There is an additive risk with this subsequent travel event if the patient is not given immediate treatment first (Table 4). Hence, treatment prior to evacuation should be strongly considered.
The traveler must also be aware that VTE can be recognized up to 2 months after a long-haul flight, though it is especially a concern within the first 2 weeks after travel.2,4,16,17
RECOMMENDATIONS FOR LONG-DISTANCE AIR TRAVELERS
Each person should be evaluated on a case-by-case basis for his or her need for VTE prophylaxis. Medical guidelines for airline passengers have been published by the Aerospace Medical Association and the American College of Chest Physicians (ACCP).18,19 In general, travelers should:
Exercise the legs by flexing and extending the ankles at regular intervals while seated (see the patient information material that accompanies this article) and frequently contracting the calf muscles.
Walk about the cabin periodically, 5 minutes for every hour on longer-duration flights (over 4 hours) and when flight conditions permit.
Drink adequate amounts of water and fruit juices to maintain good hydration.17
Avoid alcohol and caffeinated beverages, which are dehydrating.
Be careful about eating too much during the flight.
Request an aisle seat if you are at risk
Do not place baggage underneath the seat in front of you, because that reduces the ability to move the legs.
Do not sleep in a cramped position, and avoid the use of any type of sleep aid.
Avoid wearing constrictive clothing around the lower extremities or waist.
If a patient has risk factors in addition to more than 8 to 10 hours of flying (Table 1, Table 6), the physician should consider additional preventive measures including compression stockings or an anticoagulant drug as mentioned above, or both.
We recommend that all airplane passengers take the steps listed above to reduce venous stasis and avoid dehydration, even though these measures have not been proven effective in clinical trials.19
The ACCP further advises that decisions about pharmacologic prophylaxis of VTE for airplane passengers at high risk should be made on an individual basis, considering that there are potential adverse effects of prophylaxis and that these may outweigh the benefits. For long-distance travelers with additional risk factors for VTE, we suggest the following:
Use of properly fitted, below-the-knee graduated compression stockings providing 15 to 30 mm Hg of pressure at the ankle (particularly when large varicosities or leg edema is present)
For people at very high risk, a single prophylactic dose of a low-molecular-weight heparin or a factor Xa inhibitor injected just before departure (Table 5)
Aspirin is not recommended as it is not effective for the prevention of VTE.20
SUMMARY FOR THE AIR TRAVELER
All travelers on long flights should perform standard VTE prophylaxis exercises (see the patient information pages accompanying this article). Although VTE is uncommon, people with additional risk factors who travel frequently either on multiple flights in a short period of time or on very long flights should be evaluated on a case-by-case basis for a more aggressive approach to prevention (compression support hose or prophylactic administration of a low-molecular-weight heparin or a factor Xa inhibitor).
Should a VTE event occur during travel, the patient should seek medical care immediately. The standard evaluation of a patient with a suspected VTE should include an estimation of the pretest probability of disease (Table 2, Table 3), followed by duplex ultrasonography of the upper or lower extremity to detect a deep vein thrombosis. If symptoms dictate, then spiral computed tomography, ventilation-perfusion lung scan, or pulmonary angiography (where available) should be ordered to diagnose acute pulmonary embolism. A positive D-dimer blood test alone is not diagnostic and may not be available in more remote locations. A negative D-dimer test result is most helpful to exclude VTE.
Standard therapy for VTE is immediate treatment with one of the anticoagulants listed in Table 4, unless the patient has a contraindication to treatment, such as bleeding or allergy. Immediate evacuation is recommended if the patient has a life-threatening pulmonary embolism, defined as hemodynamic instability (hypotension with a blood pressure under 90 mm Hg systolic or signs of right heart failure) that cannot be treated at a local facility. An air ambulance should be used to transport these patients. If the patient has an iliofemoral deep vein thrombosis, it is also advisable that he or she be considered for evacuation if severe symptoms are present, such as pain, swelling, or cyanosis. Unless contraindicated, all patients should be given either full-dose intravenous or full-dose subcutaneous heparin or subcutaneous injection of a readily available low-molecular-weight heparin preparations or factor Xa inhibitor at once.21
References
Brenner B. Interventions to prevent venous thrombosis after air travel, are they necessary? Yes. J Thromb Haemost2006; 4:2302–2305.
Cannegieter SC, Doggen CJM, van Houwellingen HC, et al. Travel-related venous thrombosis: results from a large population-based case control study (MEGA Study). PLoS Med2006; 3:1258–1265.
Chandra D, Parisini E, Mozaffarian D. Meta-analysis: travel and risk for venous thromboembolism. Ann Intern Med2009; 151:180–190.
Kuipers S, Cannegieter SC, Middeldorp S, et al. The absolute risk of venous thrombosis after air travel: a cohort study of 8,755 employees of international organizations. PLoS Med2007; 4:1508–1514.
Kuipers S, Schreijer AJM, Cannegieter SC, et al. Travel and venous thrombosis: a systematic review. J Intern Med2007; 262:615–634.
Lehmann R, Suess C, Leus M, et al. Incidence, clinical characteristics, and long-term prognosis of travel-associated pulmonary embolism. Eur Heart J2009; 30:233–241.
Philbrick JT, Shumate R, Siadaty MS, et al. Air travel and venous thromboembolism: a systematic review. J Gen Intern Med2007; 22:107–114.
Cruickshank JM, Gorlin R, Jennett B. Air travel and thrombotic episodes: the economy class syndrome. Lancet1988; 2:497–498.
Bagshaw M. Traveler’s thrombosis: a review of deep vein thrombosis associated with travel. Air Transport Medicine Committee, Aerospace Medical Association. Aviat Space Environ Med2001; 72:848–851.
Wells PS, Owens C, Doucette S, et al. Does this patient have deep vein thrombosis?JAMA2006; 295:199–207.
Arnason T, Wells PS, Forester AJ. Appropriateness of diagnostic strategies for evaluating suspected venous thromboembolism. Thromb Haemost2007; 97:195–201.
Clarke M, Hopewell S, Juszcak E, Eisinga A, Kjeldstrøm M. Compression stockings in preventing deep vein thrombosis in airline passengers. Cochrane Database of Syst Rev2006; Apr19( 2):CD004002. DOI: 10.1002/14651858.
Kuipers S, Cannegieter SC, Middeldorp S, et al. Use of preventive measures for travel-related venous thrombosis in professionals who attend medical conferences. J Thromb Haemost2006; 4:2373–2376.
Perez-Rodriguez E, Jimenez D, Diaz G, et al. Incidence of air travel-related pulmonary embolism in the Madrid-Barajas Airport. Arch Intern Med2003; 163:2766–2770.
Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med2001; 345:779–783.
Kelman CW, Kortt MA, Becker NG, et al. Deep vein thrombosis and air travel: record linkage study. BMJ2003; 327:1072–1076.
Eklof B, Kistner RL, Masuda EM, et al. Venous thromboembolism in association with prolonged air travel. Dermatol Surg1996; 22:637–641.
Moyle J. Medical guidelines for airline travel. Aviat Space Environ Med2003: 74:1009.
Geerts WH, Bergqvist B, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines. Chest2008; 133:381S–453S.
Rosendaal FR. Interventions to prevent venous thrombosis after air travel: are they necessary? No. J Thromb Haemost2006; 4:2306–2307.
Kearon C, Ginsberg JS, Julian JA, et al; Fixed-Dose Heparin (FIDO) Investigators. Comparison of fixed-dose weight-adjusted unfractionated heparin and low-molecular-weight heparin for acute treatment of venous thromboembolism. JAMA2006; 296:935–942.
John R. Bartholomew, MD Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Head, Section of Vascular Medicine, Departments of Cardiovascular Medicine and Hematology/Oncology, Heart and Vascular Institute, Cleveland Clinic
Jonathan L. Schaffer, MD, MBA Managing Director, eClevelandClinic, Information Technology Division, Cleveland Clinic
Georges F. McCormick, MD Office of Medical Services, US Department of State, Washington, DC
Address: John R. Bartholomew, MD, Heart and Vascular Institute, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
John R. Bartholomew, MD Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Head, Section of Vascular Medicine, Departments of Cardiovascular Medicine and Hematology/Oncology, Heart and Vascular Institute, Cleveland Clinic
Jonathan L. Schaffer, MD, MBA Managing Director, eClevelandClinic, Information Technology Division, Cleveland Clinic
Georges F. McCormick, MD Office of Medical Services, US Department of State, Washington, DC
Address: John R. Bartholomew, MD, Heart and Vascular Institute, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Author and Disclosure Information
John R. Bartholomew, MD Professor of Medicine, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH; Head, Section of Vascular Medicine, Departments of Cardiovascular Medicine and Hematology/Oncology, Heart and Vascular Institute, Cleveland Clinic
Jonathan L. Schaffer, MD, MBA Managing Director, eClevelandClinic, Information Technology Division, Cleveland Clinic
Georges F. McCormick, MD Office of Medical Services, US Department of State, Washington, DC
Address: John R. Bartholomew, MD, Heart and Vascular Institute, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Editor’s Note: The views expressed in this article are solely those of the authors and do not reflect the official policy or position of the Department of State or the United States Government. This version of the article was peer-reviewed.
Venous thromboembolism (VTE) associated with travel has emerged as an important public health concern over the past decade. Numerous epidemiologic and case control studies have reported air travel as a risk factor for the development of VTE and have attempted to determine who is at risk and which precautions need to be taken to prevent this potentially fatal event.1–7 Often referred to as “traveler’s thrombosis” or “flight-related deep vein thrombosis,” VTE can also develop after long trips by automobile, bus, or train.8,9 Although the absolute risk is very low, this threat appears to be about three times higher in travelers and increases with longer trips.3
This article focuses on defining VTE and recognizing its clinical features, as well as providing recommendations and guidelines to prevent, diagnose, and treat this complication in people who travel.
WHAT IS VENOUS THROMBOEMBOLISM?
Deep vein thrombosis and pulmonary embolism represent different manifestations of the same clinical entity, ie, VTE. VTE is a common, lethal disease that affects hospitalized and nonhospitalized patients, frequently recurs, is often overlooked, may be asymptomatic, and may result in long-term complications that include pulmonary hypertension and the postthrombotic syndrome.
Figure 1.The leg veins are the most common site of deep vein thrombosis, accounting for nearly 90% of all cases; other locations include the arm and pelvic veins (Figure 1). Deep vein thrombosis in a proximal lower extremity (ie, involving the popliteal, femoral, common femoral, or external iliac vein) has an estimated 50% risk of migrating and leading to an acute pulmonary embolism if not treated, while approximately 25% of deep vein thromboses in the calf veins will, if not treated, propagate to involve the aforementioned veins.
Deep vein thrombosis of the upper extremities is generally related to an indwelling venous catheter or a central line being used for long-term administration of antibiotics, chemotherapy, or nutrition. A condition known as Paget-Schroetter syndrome or “effort thrombosis” may be seen in younger or athletic people who have a history of strenuous or unusual arm exercise.
RISK FACTORS FOR VTE
Most patients who develop VTE have one or more risk factors for it (Table 1), the presence of which is often referred to as a hypercoagulable state or thrombophilia. These risk factors are generally classified as either genetic (inherited) or acquired (environmental). Most VTE events are in fact associated with a combination of genetic and acquired risk factors.
Common inherited risk factors include:
Factor V Leiden mutation
Prothrombin gene mutation G20210A
Hyperhomocysteinemia
Deficiency of the natural anticoagulant proteins C, S, or antithrombin
Elevated levels of factor VIII (may be inherited or acquired).
Acquired risk factors include:
Older age
Immobilization or stasis (such as sitting for long periods of time while traveling)
Surgery (most notably orthopedic procedures including hip and knee replacement and repair of a hip fracture)
In addition, emerging risk factors more recently recognized include male sex, persistence of elevated factor VIII levels, and the continued presence of an elevated D-dimer level or deep vein thrombosis on duplex ultrasonography once anticoagulation treatment is completed. There is also evidence of an association between VTE and risk factors for atherosclerotic arterial disease such as smoking, hypertension, hyperlipidemia, and diabetes.
CLINICAL MANIFESTATIONS OF VTE
Patients with deep vein thrombosis may complain of pain, swelling, or both in the leg or arm. Physical examination may reveal increased warmth, tenderness, erythema, edema, or dilated (collateral) veins, most notable on the upper thigh or calf (for deep vein thrombosis in the lower extremity) or the chest wall (for upper-extremity deep vein thrombosis). The examiner may also observe a tender, palpable cord, which represents a superficial vein thrombosis involving the great and small saphenous veins (Figure 1). In extreme situations, the limb may be cyanotic or gangrenous.
A recommended clinical decision algorithm that can help assess a patient’s risk for an acute deep vein thrombosis prior to testing is depicted in Table 2.10 Patients with acute pulmonary embolism are likely to complain of the sudden onset of shortness of breath, pleuritic chest pain (especially with breathing), syncope, cough, or hemoptysis. A clinical decision algorithm that can help assess the risk of acute pulmonary embolism prior to testing is depicted in Table 3.11
DIAGNOSIS OF VTE
Clinical examination alone is generally insufficient to confirm a diagnosis of deep vein thrombosis or pulmonary embolism. Venous duplex ultrasonography is the most dependable investigation for deep vein thrombosis, but other tests include D-dimer and imaging studies such as computed tomographic venography or magnetic resonance venography of the lower extremities. A more invasive approach is venography; formerly considered the gold standard, it is now generally used only when the diagnosis is in doubt after noninvasive testing. The diagnosis of acute pulmonary embolism is best made by spiral computed tomography.
Other studies that may prove helpful include a ventilation-perfusion lung scan for patients who cannot undergo computed tomography due to a contrast allergy or renal insufficiency. Pulmonary angiography, while the gold standard, is less commonly used today, given the specificity and sensitivity of computed tomography.
Echocardiography at the bedside may be useful for patients too sick to move, although the study may not be diagnostic unless thrombi are seen in the heart or pulmonary arteries.
TREATMENT OF VTE
Treatments for VTE are summarized in Table 4. The length of treatment for acute VTE is generally 3 to 6 months. Patients with a known precipitating cause such as recent surgery or oral contraceptive use normally require 3 months of therapy, while those who had an unprovoked (idiopathic) event require longer therapy, sometimes continuing indefinitely.
For acute deep venous thrombosis
Acute deep vein thrombosis is now treated on an outpatient basis under most circumstances.
Unfractionated heparin is given intravenously for patients who need to be hospitalized, or subcutaneously in full dose for inpatient or outpatient treatment.
Low-molecular-weight heparins are available in subcutaneous preparations and can be given on an outpatient basis.
Fondaparinux (Arixtra), a factor Xa inhibitor, can also be given subcutaneously on an outpatient basis. Equivalent products are available outside the United States.
Warfarin (Coumadin), an oral vitamin K inhibitor, is the agent of choice for long-term management of deep vein thrombosis.
Other oral agents are available outside the United States.
For pulmonary embolism
Outpatient treatment of pulmonary embolism is not yet advised: an initial hospitalization is necessary. The same anticoagulants used for deep vein thrombosis are also used for acute pulmonary embolism.
Empiric treatment in underdeveloped countries
VTE may be an even greater concern on an outbound trip to a remote area, where medical care capabilities may be less than ideal and diagnostic and treatment options may be limited.
If there is a high pretest probability of acute VTE (Table 2, Table 3) and no diagnostic methods are available, empiric treatment with any of the parenteral anticoagulant agents listed in Table 4 is an option until the diagnosis can be confirmed. Caveats:
Care must be taken to be certain there is not a strong contraindication to the use of anticoagulation, such as bleeding or a drug allergy.
Neither unfractionated heparin nor any of the low-molecular-weight heparins should be given to a patient who has a history of heparin-induced thrombocytopenia.
In patients who have chronic kidney disease (creatinine clearance less than 30 mL/minute), the dosage of low-molecular-weight heparins must be adjusted and factor Xa inhibitors avoided. Both of these types of anticoagulants should be avoided in patients on hemodialysis.
More aggressive therapy
Under select circumstances a more aggressive approach to the treatment of VTE may be necessary. These options are usually indicated for a patient with a massive deep vein thrombosis of a lower extremity and for certain patients with an upper extremity deep vein thrombosis. Treatments include catheter-directed thrombolytic therapy and endovenous or surgical thrombectomy.
Thrombolytic therapy is recommended for a patient with an acute pulmonary embolism who is clinically unstable (systolic blood pressure lower than 90 mm Hg), if there is no contraindication to its use (bleeding risk or recent stroke or surgery). Thrombolytic therapy is also an option for those at low risk of bleeding with an acute pulmonary embolism who have signs and symptoms of right heart failure proven by echocardiography.
Surgical pulmonary embolectomy for acute massive pulmonary embolism and mechanical thrombectomy for extensive deep vein thrombosis are generally available only at highly sophisticated tertiary care centers.
An inferior vena cava filter is advised in patients with acute deep vein thrombosis or pulmonary embolism who cannot be fully anticoagulated, to prevent the clot from migrating from the lower extremities to the lungs. These filters are available as either permanent or temporary implants. Some temporary versions can remain in place for up to 150 days after insertion.
PREVENTION OF VTE
Prevention is the standard of care for all patients admitted to the hospital and in select individuals as outpatients who are at high risk of VTE.
A number of anticoagulant drugs are available in the United States for prophylaxis, including unfractionated heparin, low-molecular-weight heparin preparations, and fondaparinux (all of these given subcutaneously) and warfarin. In Europe and Canada, additional low-molecular-weight heparin preparations, factor Xa inhibitors, and direct thrombin inhibitors are available that have proven to be equally effective (Table 5).
Mechanical compression (graduated compression stockings, intermittent pneumatic compression devices) has proven effective in reducing the incidence of deep vein thrombosis and pulmonary embolism postoperatively in patients who cannot take anticoagulants. One study has demonstrated that compression stockings may also be effective in preventing VTE during travel.12
ABSOLUTE RISK IS LOW
Over the past decade, special attention has been paid to travel as a risk factor for developing VTE.13 Traveler’s thrombosis has become an important public health concern. Numerous publications and epidemiologic studies have targeted air travel in an attempt to determine who is at risk and what precautions are necessary to prevent this complication.1–7,9
The incidence of VTE following air travel is reported to be 3.2 per 1,000 person-years.4 While this incidence is relatively low, it is still 3.2 times higher than in the healthy population that is not flying.
The more serious complication of VTE, ie, acute pulmonary embolism, occurs less often. In three studies, the reported incidence ranged from 1.65 per million patients in flights longer than 8 hours to a high of 4.8 per million patients in flights longer than 12 hours or distances exceeding 10,000 km (6,200 miles).5,14,15 For the 400 passengers on the average long-haul flight of 12 hours, there is at most a 0.2% chance that somebody on the plane will have a symptomatic VTE).
RISK FACTORS IN LONG-DISTANCE TRAVELERS
The risk of traveler’s thrombosis has recently attracted the attention of passengers and the airline industry. Airlines are now openly discussing the risk and providing reminders such as exercises that should be undertaken in-flight (see the patient information page that accompanies this article). Some airlines are recommending that all patients consult their doctor to assess their personal risk of deep vein thrombosis before flying.
The most common risk factors for VTE in travelers are well established and are additive(Table 1). The extent of the additive risk, however, is not entirely clear.
What is clear is that when VTE occurs it is a life-altering and life-threatening event. If it occurs on an outbound trip, the local resources and capabilities available at the destination may not be adequate for optimal treatment. If a traveler experiences a VTE event on an outbound trip, an emergency return trip to the continental United States or a regional center of expertise may be required. There is an additive risk with this subsequent travel event if the patient is not given immediate treatment first (Table 4). Hence, treatment prior to evacuation should be strongly considered.
The traveler must also be aware that VTE can be recognized up to 2 months after a long-haul flight, though it is especially a concern within the first 2 weeks after travel.2,4,16,17
RECOMMENDATIONS FOR LONG-DISTANCE AIR TRAVELERS
Each person should be evaluated on a case-by-case basis for his or her need for VTE prophylaxis. Medical guidelines for airline passengers have been published by the Aerospace Medical Association and the American College of Chest Physicians (ACCP).18,19 In general, travelers should:
Exercise the legs by flexing and extending the ankles at regular intervals while seated (see the patient information material that accompanies this article) and frequently contracting the calf muscles.
Walk about the cabin periodically, 5 minutes for every hour on longer-duration flights (over 4 hours) and when flight conditions permit.
Drink adequate amounts of water and fruit juices to maintain good hydration.17
Avoid alcohol and caffeinated beverages, which are dehydrating.
Be careful about eating too much during the flight.
Request an aisle seat if you are at risk
Do not place baggage underneath the seat in front of you, because that reduces the ability to move the legs.
Do not sleep in a cramped position, and avoid the use of any type of sleep aid.
Avoid wearing constrictive clothing around the lower extremities or waist.
If a patient has risk factors in addition to more than 8 to 10 hours of flying (Table 1, Table 6), the physician should consider additional preventive measures including compression stockings or an anticoagulant drug as mentioned above, or both.
We recommend that all airplane passengers take the steps listed above to reduce venous stasis and avoid dehydration, even though these measures have not been proven effective in clinical trials.19
The ACCP further advises that decisions about pharmacologic prophylaxis of VTE for airplane passengers at high risk should be made on an individual basis, considering that there are potential adverse effects of prophylaxis and that these may outweigh the benefits. For long-distance travelers with additional risk factors for VTE, we suggest the following:
Use of properly fitted, below-the-knee graduated compression stockings providing 15 to 30 mm Hg of pressure at the ankle (particularly when large varicosities or leg edema is present)
For people at very high risk, a single prophylactic dose of a low-molecular-weight heparin or a factor Xa inhibitor injected just before departure (Table 5)
Aspirin is not recommended as it is not effective for the prevention of VTE.20
SUMMARY FOR THE AIR TRAVELER
All travelers on long flights should perform standard VTE prophylaxis exercises (see the patient information pages accompanying this article). Although VTE is uncommon, people with additional risk factors who travel frequently either on multiple flights in a short period of time or on very long flights should be evaluated on a case-by-case basis for a more aggressive approach to prevention (compression support hose or prophylactic administration of a low-molecular-weight heparin or a factor Xa inhibitor).
Should a VTE event occur during travel, the patient should seek medical care immediately. The standard evaluation of a patient with a suspected VTE should include an estimation of the pretest probability of disease (Table 2, Table 3), followed by duplex ultrasonography of the upper or lower extremity to detect a deep vein thrombosis. If symptoms dictate, then spiral computed tomography, ventilation-perfusion lung scan, or pulmonary angiography (where available) should be ordered to diagnose acute pulmonary embolism. A positive D-dimer blood test alone is not diagnostic and may not be available in more remote locations. A negative D-dimer test result is most helpful to exclude VTE.
Standard therapy for VTE is immediate treatment with one of the anticoagulants listed in Table 4, unless the patient has a contraindication to treatment, such as bleeding or allergy. Immediate evacuation is recommended if the patient has a life-threatening pulmonary embolism, defined as hemodynamic instability (hypotension with a blood pressure under 90 mm Hg systolic or signs of right heart failure) that cannot be treated at a local facility. An air ambulance should be used to transport these patients. If the patient has an iliofemoral deep vein thrombosis, it is also advisable that he or she be considered for evacuation if severe symptoms are present, such as pain, swelling, or cyanosis. Unless contraindicated, all patients should be given either full-dose intravenous or full-dose subcutaneous heparin or subcutaneous injection of a readily available low-molecular-weight heparin preparations or factor Xa inhibitor at once.21
Editor’s Note: The views expressed in this article are solely those of the authors and do not reflect the official policy or position of the Department of State or the United States Government. This version of the article was peer-reviewed.
Venous thromboembolism (VTE) associated with travel has emerged as an important public health concern over the past decade. Numerous epidemiologic and case control studies have reported air travel as a risk factor for the development of VTE and have attempted to determine who is at risk and which precautions need to be taken to prevent this potentially fatal event.1–7 Often referred to as “traveler’s thrombosis” or “flight-related deep vein thrombosis,” VTE can also develop after long trips by automobile, bus, or train.8,9 Although the absolute risk is very low, this threat appears to be about three times higher in travelers and increases with longer trips.3
This article focuses on defining VTE and recognizing its clinical features, as well as providing recommendations and guidelines to prevent, diagnose, and treat this complication in people who travel.
WHAT IS VENOUS THROMBOEMBOLISM?
Deep vein thrombosis and pulmonary embolism represent different manifestations of the same clinical entity, ie, VTE. VTE is a common, lethal disease that affects hospitalized and nonhospitalized patients, frequently recurs, is often overlooked, may be asymptomatic, and may result in long-term complications that include pulmonary hypertension and the postthrombotic syndrome.
Figure 1.The leg veins are the most common site of deep vein thrombosis, accounting for nearly 90% of all cases; other locations include the arm and pelvic veins (Figure 1). Deep vein thrombosis in a proximal lower extremity (ie, involving the popliteal, femoral, common femoral, or external iliac vein) has an estimated 50% risk of migrating and leading to an acute pulmonary embolism if not treated, while approximately 25% of deep vein thromboses in the calf veins will, if not treated, propagate to involve the aforementioned veins.
Deep vein thrombosis of the upper extremities is generally related to an indwelling venous catheter or a central line being used for long-term administration of antibiotics, chemotherapy, or nutrition. A condition known as Paget-Schroetter syndrome or “effort thrombosis” may be seen in younger or athletic people who have a history of strenuous or unusual arm exercise.
RISK FACTORS FOR VTE
Most patients who develop VTE have one or more risk factors for it (Table 1), the presence of which is often referred to as a hypercoagulable state or thrombophilia. These risk factors are generally classified as either genetic (inherited) or acquired (environmental). Most VTE events are in fact associated with a combination of genetic and acquired risk factors.
Common inherited risk factors include:
Factor V Leiden mutation
Prothrombin gene mutation G20210A
Hyperhomocysteinemia
Deficiency of the natural anticoagulant proteins C, S, or antithrombin
Elevated levels of factor VIII (may be inherited or acquired).
Acquired risk factors include:
Older age
Immobilization or stasis (such as sitting for long periods of time while traveling)
Surgery (most notably orthopedic procedures including hip and knee replacement and repair of a hip fracture)
In addition, emerging risk factors more recently recognized include male sex, persistence of elevated factor VIII levels, and the continued presence of an elevated D-dimer level or deep vein thrombosis on duplex ultrasonography once anticoagulation treatment is completed. There is also evidence of an association between VTE and risk factors for atherosclerotic arterial disease such as smoking, hypertension, hyperlipidemia, and diabetes.
CLINICAL MANIFESTATIONS OF VTE
Patients with deep vein thrombosis may complain of pain, swelling, or both in the leg or arm. Physical examination may reveal increased warmth, tenderness, erythema, edema, or dilated (collateral) veins, most notable on the upper thigh or calf (for deep vein thrombosis in the lower extremity) or the chest wall (for upper-extremity deep vein thrombosis). The examiner may also observe a tender, palpable cord, which represents a superficial vein thrombosis involving the great and small saphenous veins (Figure 1). In extreme situations, the limb may be cyanotic or gangrenous.
A recommended clinical decision algorithm that can help assess a patient’s risk for an acute deep vein thrombosis prior to testing is depicted in Table 2.10 Patients with acute pulmonary embolism are likely to complain of the sudden onset of shortness of breath, pleuritic chest pain (especially with breathing), syncope, cough, or hemoptysis. A clinical decision algorithm that can help assess the risk of acute pulmonary embolism prior to testing is depicted in Table 3.11
DIAGNOSIS OF VTE
Clinical examination alone is generally insufficient to confirm a diagnosis of deep vein thrombosis or pulmonary embolism. Venous duplex ultrasonography is the most dependable investigation for deep vein thrombosis, but other tests include D-dimer and imaging studies such as computed tomographic venography or magnetic resonance venography of the lower extremities. A more invasive approach is venography; formerly considered the gold standard, it is now generally used only when the diagnosis is in doubt after noninvasive testing. The diagnosis of acute pulmonary embolism is best made by spiral computed tomography.
Other studies that may prove helpful include a ventilation-perfusion lung scan for patients who cannot undergo computed tomography due to a contrast allergy or renal insufficiency. Pulmonary angiography, while the gold standard, is less commonly used today, given the specificity and sensitivity of computed tomography.
Echocardiography at the bedside may be useful for patients too sick to move, although the study may not be diagnostic unless thrombi are seen in the heart or pulmonary arteries.
TREATMENT OF VTE
Treatments for VTE are summarized in Table 4. The length of treatment for acute VTE is generally 3 to 6 months. Patients with a known precipitating cause such as recent surgery or oral contraceptive use normally require 3 months of therapy, while those who had an unprovoked (idiopathic) event require longer therapy, sometimes continuing indefinitely.
For acute deep venous thrombosis
Acute deep vein thrombosis is now treated on an outpatient basis under most circumstances.
Unfractionated heparin is given intravenously for patients who need to be hospitalized, or subcutaneously in full dose for inpatient or outpatient treatment.
Low-molecular-weight heparins are available in subcutaneous preparations and can be given on an outpatient basis.
Fondaparinux (Arixtra), a factor Xa inhibitor, can also be given subcutaneously on an outpatient basis. Equivalent products are available outside the United States.
Warfarin (Coumadin), an oral vitamin K inhibitor, is the agent of choice for long-term management of deep vein thrombosis.
Other oral agents are available outside the United States.
For pulmonary embolism
Outpatient treatment of pulmonary embolism is not yet advised: an initial hospitalization is necessary. The same anticoagulants used for deep vein thrombosis are also used for acute pulmonary embolism.
Empiric treatment in underdeveloped countries
VTE may be an even greater concern on an outbound trip to a remote area, where medical care capabilities may be less than ideal and diagnostic and treatment options may be limited.
If there is a high pretest probability of acute VTE (Table 2, Table 3) and no diagnostic methods are available, empiric treatment with any of the parenteral anticoagulant agents listed in Table 4 is an option until the diagnosis can be confirmed. Caveats:
Care must be taken to be certain there is not a strong contraindication to the use of anticoagulation, such as bleeding or a drug allergy.
Neither unfractionated heparin nor any of the low-molecular-weight heparins should be given to a patient who has a history of heparin-induced thrombocytopenia.
In patients who have chronic kidney disease (creatinine clearance less than 30 mL/minute), the dosage of low-molecular-weight heparins must be adjusted and factor Xa inhibitors avoided. Both of these types of anticoagulants should be avoided in patients on hemodialysis.
More aggressive therapy
Under select circumstances a more aggressive approach to the treatment of VTE may be necessary. These options are usually indicated for a patient with a massive deep vein thrombosis of a lower extremity and for certain patients with an upper extremity deep vein thrombosis. Treatments include catheter-directed thrombolytic therapy and endovenous or surgical thrombectomy.
Thrombolytic therapy is recommended for a patient with an acute pulmonary embolism who is clinically unstable (systolic blood pressure lower than 90 mm Hg), if there is no contraindication to its use (bleeding risk or recent stroke or surgery). Thrombolytic therapy is also an option for those at low risk of bleeding with an acute pulmonary embolism who have signs and symptoms of right heart failure proven by echocardiography.
Surgical pulmonary embolectomy for acute massive pulmonary embolism and mechanical thrombectomy for extensive deep vein thrombosis are generally available only at highly sophisticated tertiary care centers.
An inferior vena cava filter is advised in patients with acute deep vein thrombosis or pulmonary embolism who cannot be fully anticoagulated, to prevent the clot from migrating from the lower extremities to the lungs. These filters are available as either permanent or temporary implants. Some temporary versions can remain in place for up to 150 days after insertion.
PREVENTION OF VTE
Prevention is the standard of care for all patients admitted to the hospital and in select individuals as outpatients who are at high risk of VTE.
A number of anticoagulant drugs are available in the United States for prophylaxis, including unfractionated heparin, low-molecular-weight heparin preparations, and fondaparinux (all of these given subcutaneously) and warfarin. In Europe and Canada, additional low-molecular-weight heparin preparations, factor Xa inhibitors, and direct thrombin inhibitors are available that have proven to be equally effective (Table 5).
Mechanical compression (graduated compression stockings, intermittent pneumatic compression devices) has proven effective in reducing the incidence of deep vein thrombosis and pulmonary embolism postoperatively in patients who cannot take anticoagulants. One study has demonstrated that compression stockings may also be effective in preventing VTE during travel.12
ABSOLUTE RISK IS LOW
Over the past decade, special attention has been paid to travel as a risk factor for developing VTE.13 Traveler’s thrombosis has become an important public health concern. Numerous publications and epidemiologic studies have targeted air travel in an attempt to determine who is at risk and what precautions are necessary to prevent this complication.1–7,9
The incidence of VTE following air travel is reported to be 3.2 per 1,000 person-years.4 While this incidence is relatively low, it is still 3.2 times higher than in the healthy population that is not flying.
The more serious complication of VTE, ie, acute pulmonary embolism, occurs less often. In three studies, the reported incidence ranged from 1.65 per million patients in flights longer than 8 hours to a high of 4.8 per million patients in flights longer than 12 hours or distances exceeding 10,000 km (6,200 miles).5,14,15 For the 400 passengers on the average long-haul flight of 12 hours, there is at most a 0.2% chance that somebody on the plane will have a symptomatic VTE).
RISK FACTORS IN LONG-DISTANCE TRAVELERS
The risk of traveler’s thrombosis has recently attracted the attention of passengers and the airline industry. Airlines are now openly discussing the risk and providing reminders such as exercises that should be undertaken in-flight (see the patient information page that accompanies this article). Some airlines are recommending that all patients consult their doctor to assess their personal risk of deep vein thrombosis before flying.
The most common risk factors for VTE in travelers are well established and are additive(Table 1). The extent of the additive risk, however, is not entirely clear.
What is clear is that when VTE occurs it is a life-altering and life-threatening event. If it occurs on an outbound trip, the local resources and capabilities available at the destination may not be adequate for optimal treatment. If a traveler experiences a VTE event on an outbound trip, an emergency return trip to the continental United States or a regional center of expertise may be required. There is an additive risk with this subsequent travel event if the patient is not given immediate treatment first (Table 4). Hence, treatment prior to evacuation should be strongly considered.
The traveler must also be aware that VTE can be recognized up to 2 months after a long-haul flight, though it is especially a concern within the first 2 weeks after travel.2,4,16,17
RECOMMENDATIONS FOR LONG-DISTANCE AIR TRAVELERS
Each person should be evaluated on a case-by-case basis for his or her need for VTE prophylaxis. Medical guidelines for airline passengers have been published by the Aerospace Medical Association and the American College of Chest Physicians (ACCP).18,19 In general, travelers should:
Exercise the legs by flexing and extending the ankles at regular intervals while seated (see the patient information material that accompanies this article) and frequently contracting the calf muscles.
Walk about the cabin periodically, 5 minutes for every hour on longer-duration flights (over 4 hours) and when flight conditions permit.
Drink adequate amounts of water and fruit juices to maintain good hydration.17
Avoid alcohol and caffeinated beverages, which are dehydrating.
Be careful about eating too much during the flight.
Request an aisle seat if you are at risk
Do not place baggage underneath the seat in front of you, because that reduces the ability to move the legs.
Do not sleep in a cramped position, and avoid the use of any type of sleep aid.
Avoid wearing constrictive clothing around the lower extremities or waist.
If a patient has risk factors in addition to more than 8 to 10 hours of flying (Table 1, Table 6), the physician should consider additional preventive measures including compression stockings or an anticoagulant drug as mentioned above, or both.
We recommend that all airplane passengers take the steps listed above to reduce venous stasis and avoid dehydration, even though these measures have not been proven effective in clinical trials.19
The ACCP further advises that decisions about pharmacologic prophylaxis of VTE for airplane passengers at high risk should be made on an individual basis, considering that there are potential adverse effects of prophylaxis and that these may outweigh the benefits. For long-distance travelers with additional risk factors for VTE, we suggest the following:
Use of properly fitted, below-the-knee graduated compression stockings providing 15 to 30 mm Hg of pressure at the ankle (particularly when large varicosities or leg edema is present)
For people at very high risk, a single prophylactic dose of a low-molecular-weight heparin or a factor Xa inhibitor injected just before departure (Table 5)
Aspirin is not recommended as it is not effective for the prevention of VTE.20
SUMMARY FOR THE AIR TRAVELER
All travelers on long flights should perform standard VTE prophylaxis exercises (see the patient information pages accompanying this article). Although VTE is uncommon, people with additional risk factors who travel frequently either on multiple flights in a short period of time or on very long flights should be evaluated on a case-by-case basis for a more aggressive approach to prevention (compression support hose or prophylactic administration of a low-molecular-weight heparin or a factor Xa inhibitor).
Should a VTE event occur during travel, the patient should seek medical care immediately. The standard evaluation of a patient with a suspected VTE should include an estimation of the pretest probability of disease (Table 2, Table 3), followed by duplex ultrasonography of the upper or lower extremity to detect a deep vein thrombosis. If symptoms dictate, then spiral computed tomography, ventilation-perfusion lung scan, or pulmonary angiography (where available) should be ordered to diagnose acute pulmonary embolism. A positive D-dimer blood test alone is not diagnostic and may not be available in more remote locations. A negative D-dimer test result is most helpful to exclude VTE.
Standard therapy for VTE is immediate treatment with one of the anticoagulants listed in Table 4, unless the patient has a contraindication to treatment, such as bleeding or allergy. Immediate evacuation is recommended if the patient has a life-threatening pulmonary embolism, defined as hemodynamic instability (hypotension with a blood pressure under 90 mm Hg systolic or signs of right heart failure) that cannot be treated at a local facility. An air ambulance should be used to transport these patients. If the patient has an iliofemoral deep vein thrombosis, it is also advisable that he or she be considered for evacuation if severe symptoms are present, such as pain, swelling, or cyanosis. Unless contraindicated, all patients should be given either full-dose intravenous or full-dose subcutaneous heparin or subcutaneous injection of a readily available low-molecular-weight heparin preparations or factor Xa inhibitor at once.21
References
Brenner B. Interventions to prevent venous thrombosis after air travel, are they necessary? Yes. J Thromb Haemost2006; 4:2302–2305.
Cannegieter SC, Doggen CJM, van Houwellingen HC, et al. Travel-related venous thrombosis: results from a large population-based case control study (MEGA Study). PLoS Med2006; 3:1258–1265.
Chandra D, Parisini E, Mozaffarian D. Meta-analysis: travel and risk for venous thromboembolism. Ann Intern Med2009; 151:180–190.
Kuipers S, Cannegieter SC, Middeldorp S, et al. The absolute risk of venous thrombosis after air travel: a cohort study of 8,755 employees of international organizations. PLoS Med2007; 4:1508–1514.
Kuipers S, Schreijer AJM, Cannegieter SC, et al. Travel and venous thrombosis: a systematic review. J Intern Med2007; 262:615–634.
Lehmann R, Suess C, Leus M, et al. Incidence, clinical characteristics, and long-term prognosis of travel-associated pulmonary embolism. Eur Heart J2009; 30:233–241.
Philbrick JT, Shumate R, Siadaty MS, et al. Air travel and venous thromboembolism: a systematic review. J Gen Intern Med2007; 22:107–114.
Cruickshank JM, Gorlin R, Jennett B. Air travel and thrombotic episodes: the economy class syndrome. Lancet1988; 2:497–498.
Bagshaw M. Traveler’s thrombosis: a review of deep vein thrombosis associated with travel. Air Transport Medicine Committee, Aerospace Medical Association. Aviat Space Environ Med2001; 72:848–851.
Wells PS, Owens C, Doucette S, et al. Does this patient have deep vein thrombosis?JAMA2006; 295:199–207.
Arnason T, Wells PS, Forester AJ. Appropriateness of diagnostic strategies for evaluating suspected venous thromboembolism. Thromb Haemost2007; 97:195–201.
Clarke M, Hopewell S, Juszcak E, Eisinga A, Kjeldstrøm M. Compression stockings in preventing deep vein thrombosis in airline passengers. Cochrane Database of Syst Rev2006; Apr19( 2):CD004002. DOI: 10.1002/14651858.
Kuipers S, Cannegieter SC, Middeldorp S, et al. Use of preventive measures for travel-related venous thrombosis in professionals who attend medical conferences. J Thromb Haemost2006; 4:2373–2376.
Perez-Rodriguez E, Jimenez D, Diaz G, et al. Incidence of air travel-related pulmonary embolism in the Madrid-Barajas Airport. Arch Intern Med2003; 163:2766–2770.
Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med2001; 345:779–783.
Kelman CW, Kortt MA, Becker NG, et al. Deep vein thrombosis and air travel: record linkage study. BMJ2003; 327:1072–1076.
Eklof B, Kistner RL, Masuda EM, et al. Venous thromboembolism in association with prolonged air travel. Dermatol Surg1996; 22:637–641.
Moyle J. Medical guidelines for airline travel. Aviat Space Environ Med2003: 74:1009.
Geerts WH, Bergqvist B, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines. Chest2008; 133:381S–453S.
Rosendaal FR. Interventions to prevent venous thrombosis after air travel: are they necessary? No. J Thromb Haemost2006; 4:2306–2307.
Kearon C, Ginsberg JS, Julian JA, et al; Fixed-Dose Heparin (FIDO) Investigators. Comparison of fixed-dose weight-adjusted unfractionated heparin and low-molecular-weight heparin for acute treatment of venous thromboembolism. JAMA2006; 296:935–942.
References
Brenner B. Interventions to prevent venous thrombosis after air travel, are they necessary? Yes. J Thromb Haemost2006; 4:2302–2305.
Cannegieter SC, Doggen CJM, van Houwellingen HC, et al. Travel-related venous thrombosis: results from a large population-based case control study (MEGA Study). PLoS Med2006; 3:1258–1265.
Chandra D, Parisini E, Mozaffarian D. Meta-analysis: travel and risk for venous thromboembolism. Ann Intern Med2009; 151:180–190.
Kuipers S, Cannegieter SC, Middeldorp S, et al. The absolute risk of venous thrombosis after air travel: a cohort study of 8,755 employees of international organizations. PLoS Med2007; 4:1508–1514.
Kuipers S, Schreijer AJM, Cannegieter SC, et al. Travel and venous thrombosis: a systematic review. J Intern Med2007; 262:615–634.
Lehmann R, Suess C, Leus M, et al. Incidence, clinical characteristics, and long-term prognosis of travel-associated pulmonary embolism. Eur Heart J2009; 30:233–241.
Philbrick JT, Shumate R, Siadaty MS, et al. Air travel and venous thromboembolism: a systematic review. J Gen Intern Med2007; 22:107–114.
Cruickshank JM, Gorlin R, Jennett B. Air travel and thrombotic episodes: the economy class syndrome. Lancet1988; 2:497–498.
Bagshaw M. Traveler’s thrombosis: a review of deep vein thrombosis associated with travel. Air Transport Medicine Committee, Aerospace Medical Association. Aviat Space Environ Med2001; 72:848–851.
Wells PS, Owens C, Doucette S, et al. Does this patient have deep vein thrombosis?JAMA2006; 295:199–207.
Arnason T, Wells PS, Forester AJ. Appropriateness of diagnostic strategies for evaluating suspected venous thromboembolism. Thromb Haemost2007; 97:195–201.
Clarke M, Hopewell S, Juszcak E, Eisinga A, Kjeldstrøm M. Compression stockings in preventing deep vein thrombosis in airline passengers. Cochrane Database of Syst Rev2006; Apr19( 2):CD004002. DOI: 10.1002/14651858.
Kuipers S, Cannegieter SC, Middeldorp S, et al. Use of preventive measures for travel-related venous thrombosis in professionals who attend medical conferences. J Thromb Haemost2006; 4:2373–2376.
Perez-Rodriguez E, Jimenez D, Diaz G, et al. Incidence of air travel-related pulmonary embolism in the Madrid-Barajas Airport. Arch Intern Med2003; 163:2766–2770.
Lapostolle F, Surget V, Borron SW, et al. Severe pulmonary embolism associated with air travel. N Engl J Med2001; 345:779–783.
Kelman CW, Kortt MA, Becker NG, et al. Deep vein thrombosis and air travel: record linkage study. BMJ2003; 327:1072–1076.
Eklof B, Kistner RL, Masuda EM, et al. Venous thromboembolism in association with prolonged air travel. Dermatol Surg1996; 22:637–641.
Moyle J. Medical guidelines for airline travel. Aviat Space Environ Med2003: 74:1009.
Geerts WH, Bergqvist B, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines. Chest2008; 133:381S–453S.
Rosendaal FR. Interventions to prevent venous thrombosis after air travel: are they necessary? No. J Thromb Haemost2006; 4:2306–2307.
Kearon C, Ginsberg JS, Julian JA, et al; Fixed-Dose Heparin (FIDO) Investigators. Comparison of fixed-dose weight-adjusted unfractionated heparin and low-molecular-weight heparin for acute treatment of venous thromboembolism. JAMA2006; 296:935–942.
The risk of VTE is about three times higher in passengers on long-distance flights than in the general population, although the absolute risk is still low.
All long-distance air passengers should perform stretching exercises once an hour while in flight to prevent VTE. They should also stay hydrated.
For patients at higher risk due to hypercoagulable conditions, physicians can consider prescribing compression stockings or an anticoagulant drug (a low-molecular-weight heparin or a factor Xa inhibitor) to be taken before the flight, or both.
The evaluation of a patient with suspected VTE should include an estimation of the pretest probability of disease. If symptoms dictate, duplex ultrasonography of the upper or lower extremity to detect deep vein thrombosis or spiral computed tomography, ventilation-perfusion lung scan, or pulmonary angiography (where available) to diagnose an acute pulmonary embolism should be ordered.
Patients often ask primary care physicians and cardiologists about the measurement of biomarkers for cardiovascular disease and about the efficacy of preventive measures.
Although studies have shown that elevated homocysteine is a risk factor for cardiovascular and peripheral arterial disease1–3 and that supplementation with folic acid, vitamin B6, and vitamin B12 lowers homocysteine levels,4,5 it is unclear whether such supplementation prevents cardiovascular events. As a result, there is no consensus about whose homocysteine levels should be measured and who, if anyone, should receive homocysteine-lowering therapies.
The aim of this paper is to examine whether the evidence is sufficient to recommend homocysteine testing to guide the prevention and treatment of cardiovascular disease, or to recommend using folic acid, vitamin B6, and vitamin B12 for primary or secondary prevention of cardiovascular disease.
HISTORY OF HOMOCYSTEINE AS A RISK MARKER
Homocysteine is an amino acid formed from the metabolism of methionine, an essential amino acid derived from dietary protein. Although homocysteine was first isolated by Butz and du Vigneaud in 1932,6 it was not until 1964 that Gibson et al7 reported that patients with homocystinuria (more about this below) had vascular anomalies and arterial thrombosis. In 1969, McCully8 made the connection between elevated homocysteine levels and the risk of atherosclerosis.
Several possible mechanisms for the association between homocysteine and atherosclerosis have been demonstrated in experimental models. These include stimulation of smooth muscle growth, reduction in endothelial cell growth, impaired endothelial cell relaxation, decreased synthesis of high-density lipoprotein, promotion of autoimmune response, and accumulation of inflammatory monocytes in atherosclerotic plaques.3,9,10
In view of these findings, researchers have been evaluating whether homocysteine-lowering therapies decrease the risk of cardiovascular disease.
CAUSES OF ELEVATED PLASMA HOMOCYSTEINE
An elevated plasma homocysteine level can result from many different factors, including vitamin deficiencies, renal impairment, and inborn errors of homocysteine metabolism (Table 1).9,11,12
Vitamin deficiencies. Vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), and folic acid are cofactors required for homocysteine metabolism, and deficiency in any or all of these leads to disruption of the relevant metabolic pathways.
Renal impairment. A low glomerular filtration rate has also been correlated with an elevated plasma homocysteine concentration. This makes sense, since the kidneys perform up to 70% of the clearance of homocysteine, although a cause-and-effect relationship is unclear.13
Inborn errors of homocysteine metabolism.Homocystinuria, ie, an abnormal elevation of homocysteine in the urine, is caused by several autosomal recessive disorders. People with these genetic variations have extremely high homocysteine levels.
A deficiency in the enzyme cystathionine beta-synthase is quite rare (the incidence in newborn babies has been found to be 1 in 344,000 worldwide and 1 in 65,000 in Ireland and Australia14), but leads to homocysteine levels greater than 100 μmol/L and often causes cardiovascular disease by the age of 30 years.15
A deficiency in the enzyme methylene tetrahydrofolate reductase (MTHFR) is a more common cause of mildly to moderately elevated plasma homocysteine levels.16 The MTHFR deficiency involves a variation at position 677 in the MTHFR gene in which cytosine is replaced by thymidine (thus called C677T or 677C>T).17 Ten percent of the population are homozygous for this variant (TT), 43% are heterozygous (CT), and 47% are unaffected (CC). Heterozygotes have slightly higher homocysteine levels than unaffected people, while people with the TT genotype have approximately 20% higher homocysteine levels.17
ELEVATED HOMOCYSTEINE IS COMMON
In a study of a population in Norway from 1992 to 1993, 8.5% had mild elevations in homocysteine (plasma levels 15–29.99 μmol/L), 0.8% had moderate elevations (30–99.99 μmol/L), and 0.02% had severe elevations (≥ 100 μmol/L).13,18 The prevalence of hyperhomocysteinemia in the United States is probably much lower, given that supplementation of white flour and cereal grains with folic acid has been mandatory since 1998, but this is not well described in the literature.
HOW GREAT IS THE RISK?
Studies over the past 10 to 20 years have shown that elevated homocysteine is a marker of risk of cardiovascular disease. The association was first noted in patients with cystathionine beta-synthase deficiency, who tend to have premature cardiovascular disease.
However, studies of patients with MTHFR 677C>T have yielded mixed results. Although several meta-analyses found up to a 42% higher rate of ischemic heart disease and stroke in patients homozygous for MTHFR 677C>T (the TT genotype) than in those with the CC genotype,17,19,20 two other large meta-analyses did not find an association between this variant and vascular risk.21,22
Nonetheless, in a meta-analysis of the association between homocysteine and cardiovascular disease, Wald et al17 found that for every 5-μmol/L increase in serum homocysteine concentration, the risk of ischemic heart disease increased 20% to 30%.
TRIALS OF HOMOCYSTEINE-LOWERING THERAPY HAVE HAD MIXED RESULTS
Primary prevention of cardiovascular disease
Given the finding that treatment with folic acid lowers homocysteine—initially noted in patients with homocystinuria—researchers hypothesized that treatment with folic acid, vitamin B6, and vitamin B12 would decrease the risk of cardiovascular disease.
Little evidence currently exists to guide recommendations for homocysteine-lowering therapy to prevent first attacks of cardiovascular disease. The few studies published to date have been observational studies of dietary intake (not vitamin supplementation), and many were performed before folic acid fortification was mandated for flour and cereal (Table 2).23–27 Although the studies suggest that higher B-vitamin intake correlates with less vascular disease and its sequelae, there is uncertainty as to whether it is folic acid, vitamin B6, or vitamin B12 that is responsible, and also whether supplements would provide the same protective benefit as the presence of these nutrients in a varied diet.
Thus, in its recent evaluation of novel risk markers of cardiovascular disease, the United States Preventive Services Task Force 28,29 does not recommend measuring the plasma homocysteine level in the evaluation of either low-risk or intermediate-risk populations, finding no evidence that it adds any useful information in predicting major coronary events beyond what one could get from calculating the Framingham Risk Score. The task force also found no evidence that treating people who have elevated homocysteine levels decreases their risk of subsequent cardiovascular events.
In addition, a recent Cochrane Database review of eight randomized controlled trials in patients at low risk did not find a lower risk of myocardial infarction (fatal or nonfatal), stroke, or death from any cause in patients receiving B-complex vitamins.30
Secondary prevention of cardiovascular disease
Results have been mixed with regard to the ability of B vitamins to prevent cardiovascular events in patients with known cardiovascular disease (Table 3).4,5,11,12,31–46
Bazzano et al,47 in a meta-analysis published in 2006, evaluated 12 randomized controlled trials of folic acid supplementation in patients with known cardiovascular disease and did not find that treated patients had better cardiovascular outcomes. The mean homocysteine level was elevated (> 15 μmol/L) at baseline in only 4 of the 12 trials. However, in 1 of these 4 trials, there was no difference in outcomes comparing those with and without elevated homocysteine.31
Albert et al4 more recently evaluated the effect of a combination pill containing folic acid, vitamin B6, and vitamin B12 on cardiovascular events in women at high risk, ie, those with a history of cardiovascular disease or having three or more coronary risk factors. Treatment did not decrease the rate of the composite outcome of cardiovascular disease mortality, stroke, myocardial infarction, or coronary revascularization, although the homocysteine level decreased by a mean of 30% in the treated group. However, only 27.7% of the participants had an elevated homocysteine level. One might not expect patients to benefit from such treatment if they had normal homocysteine levels to begin with.
Ebbing et al,5 in a trial published in 2008, investigated the effect of folic acid, vitamin B12, and vitamin B6 supplements on the risks of death from any cause and of cardiovascular events in patients undergoing coronary angiography. Outcomes were no better in the treatment group than in the control group, despite a mean decrease in homocysteine level of 19%. However, over 90% of the participants had a normal homocysteine level.
Mager et al,32 in a study published in 2009, looked specifically at whether patients with coronary artery disease and elevated homocysteine levels (> 15 μmol/L) would benefit from folate-based vitamin therapy. In this subset, the incidence of death from any cause was lower in the treated group than in the control group (4% vs 32%, P < .001), an association that was not present in patients with normal homocysteine levels.
The SEARCH trial (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),33 recently published, was a double-blind, randomized controlled trial of vitamin B12 and folic acid treatment in 12,064 patients who had survived a myocardial infarction. Although those who received the vitamin therapy had a 28% reduction in homocysteine level, no clinical benefit was demonstrated. Of note, 66% of the patients had a homocysteine level lower than 14 μmol/L at baseline.
Restenosis after angioplasty
Results are also mixed regarding whether folic acid supplements modify the risk of restenosis after coronary angioplasty.
Namazi et al48 evaluated the effect of folic acid supplementation on in-stent restenosis in 200 patients and found no difference between the treatment and placebo groups in the rates of either restenosis or target-vessel revascularization.
Schnyder et al49 evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on the rate of coronary restenosis (in cases of balloon angioplasty) or in-stent restenosis (if a stent was used). Patients receiving treatment had lower rates of restenosis or instent restenosis (40% vs 48%, P = .01) and of need for target-vessel revascularization (11% vs 22%, P = .047). The mean homocysteine level was not elevated in this study either, and the researchers did not analyze the outcomes according to whether patients had high or normal homocysteine levels.
Lange et al35 also evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on coronary in-stent restenosis. Paradoxically, the rate was higher with treatment in the overall group (mean homocysteine level 12.2 μmol/L), leading to a higher incidence of target-vessel revascularization. Patients who had a baseline elevation in homocysteine level had a nonsignificant trend toward a lower rate of in-stent restenosis.
Cerebrovascular and peripheral arterial disease
The evidence is also mixed for using folic acid and other B vitamins to prevent cerebrovascular disease and peripheral vascular disease. Although a 2007 meta-analysis found that folic acid supplementation decreased the risk of a first stroke by 18% (P = .045),50 a later meta-analysis contradicts this finding.51
A 2009 meta-analysis found that patients with peripheral arterial disease had higher homocysteine levels than controls, but it did not find any benefit from supplementation, owing to heterogeneity of the clinical end points used.52 Indeed, a 2009 Cochrane Database Systematic Review found that there were no adequate trials of the treatment of patients with peripheral vascular disease who have elevated plasma homocysteine.53
However, immediately after the Cochrane review was published, Khandanpour et al36 published the results of a trial of the effect of folic acid and 5-methyltetrahydrofolate (an active form of folic acid) supplementation on the ankle-brachial pressure index and the pulse-wave velocity in patients with peripheral arterial disease. These measures improved with 16 weeks of treatment. For the ankle-brachial pressure index, the P value was less than .01 for folic acid and .009 for 5-methyltetrahydrofolate; for the pulse-wave velocity, the P value was .051 for folic acid and .011 for 5-methyltetrahydrofolate.
Kidney disease
One could postulate that patients with end-stage renal disease or chronic kidney disease might benefit the most from folic acid supplementation, given the correlation of elevations in homocysteine levels with decline in glomerular filtration rate.
However, only one study found a lower rate of cardiovascular events with folic acid supplementation in dialysis patients, and the difference was not statistically significant (25% vs 36%, P < .08).31 Further, several studies found no benefit of folic acid supplementation in patients with chronic kidney disease.11,12,37
FUTURE DIRECTIONS AND RECOMMENDATIONS
Many experts have suggested that the existing evidence indicates that the homocysteine-lowering therapies folic acid, vitamin B6, and vitamin B12 do not lower the risk of cardiovascular disease.38,54–59 Indeed, the American Heart Association guidelines for cardiovascular disease prevention in women do not recommend folic acid supplementation to prevent cardiovascular disease.60 (Recommendations for men are the same as for women.) However, most of the clinical trials have not selected and treated patients with elevated homocysteine levels, but have instead included all patients regardless of homocysteine level.
At least two large ongoing trials are currently evaluating B-vitamin therapy for secondary prevention, but neither trial is looking specifically at patients with elevated homocysteine levels.61,62
Thus, instead of concluding that no patients could benefit from homocysteine-lowering treatment, future studies need to clarify:
Whether patients with elevated homocysteine would benefit from such treatment
At what level it would be appropriate to start treatment
The appropriate target homocysteine level with treatment.
Particularly given the recent finding that folic acid supplementation may increase cancer risk,63 these questions need closer scrutiny.
References
Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc2008; 83:1203–1212.
Boers GH. The case for mild hyperhomocysteinaemia as a risk factor. J Inherit Metab Dis1997; 20:301–306.
Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ2004; 11(suppl 1):S56–S64.
Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA2008; 299:2027–2036.
Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA2008; 300:795–804.
Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decompensation of methionine with sulphuric acid. J Biol Chem1932; 99:135–142.
McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol1969; 56:111–128.
Zhang D, Jiang X, Fang P, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation2009; 120:1893–1902.
Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol1999; 34:2002–2006.
Righetti M, Ferrario GM, Milani S, et al. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med Sci Monit2003; 9:PI19–PI24.
Zoungas S, McGrath BP, Branley P, et al. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol2006; 47:1108–1116.
Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press, 2001.
Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol2001; 21:2080–2085.
McKusick V. 236200 Homocystinuria. In:McKusick V, editor. Mendelian Inheritance in Man. 10th ed. Baltimore, MD: The Johns Hopkins University Press, 1992:1444–1446.
McKusick V. 236250 Homocystinuria due to deficiency of N(5,10)-methylenetetrahydrofolate reductase activity. In:McKusick V, editor. Mendelian Inheritance in Man. Baltimore, MD: The Johns Hopkins University Press, 1992:1447–1448.
Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ2002; 325:1202.
Nygård O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA1995; 274:1526–1533.
Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG; MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA2002; 288:2023–2031.
Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology2002; 59:529–536.
Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate?BMJ2005; 331:1053.
Brattström L, Wilcken DE, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation1998; 98:2520–2526.
Cui R, Iso H, Date C, Kikuchi S, Tamakoshi A; Japan Collaborative Cohort Study Group. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan Collaborative Cohort Study. Stroke2010; 41:1285–1289.
Liu S, Stampfer MJ, Hu FB, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses’ Health Study. Am J Clin Nutr1999; 70:412–419.
Liu S, Manson JE, Stampfer MJ, et al. Whole grain consumption and risk of ischemic stroke in women: a prospective study. JAMA2000; 284:1534–1540.
Merchant AT, Hu FB, Spiegelman D, Willett WC, Rimm EB, Ascherio A. The use of B vitamin supplements and peripheral arterial disease risk in men are inversely related. J Nutr2003; 133:2863–2867.
Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA1998; 279:359–364.
US Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med2009; 151:474–482.
Helfand M, Buckley DI, Freeman M, et al. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the US Preventive Services Task Force. Ann Intern Med2009; 151:496–507.
Martí-Carvajal AJ, Solà I, Lathyris D, Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev2009; ( 4):CD006612.
Righetti M, Serbelloni P, Milani S, Ferrario G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif2006; 24:379–386.
Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol2009; 104:745–749.
Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA2010; 303:2486–2494.
Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and percutaneous coronary intervention: the Swiss Heart Study: a randomized controlled trial. JAMA2002; 288:973–979.
Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med2004; 350:2673–2781.
Khandanpour N, Armon MP, Jennings B, et al. Randomized clinical trial of folate supplementation in patients with peripheral arterial disease. Br J Surg2009; 96:990–998.
Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol2004; 15:420–426.
Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med2006; 354:1629–1632.
Bønaa KH, Njølstad I, Ueland PM, et al; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med2006; 354:1578–1588.
Carrero JJ, López-Huertas E, Salmerón LM, Baró L, Ros E. Daily supplementation with (n-3) PUFAs, oleic acid, folic acid, and vitamins B-6 and E increases pain-free walking distance and improves risk factors in men with peripheral vascular disease. J Nutr2005; 135:1393–1399.
Liem AH, van Boven AJ, Veeger NJ, et al; Folic Acid on Risk Diminishment After Acute Myocarial Infarction Study Group. Efficacy of folic acid when added to statin therapy in patients with hypercholesterolemia following acute myocardial infarction: a randomised pilot trial. Int J Cardiol2004; 93:175–179.
Liem A, Reynierse-Buitenwerf GH, Zwinderman AH, Jukema JW, van Veldhuisen DJ. Secondary prevention with folic acid: results of the Goes extension study. Heart2005; 91:1213–1214.
Lonn E, Yusuf S, Arnold MJ, et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med2006; 354:1567–1577.
Sydow K, Schwedhelm E, Arakawa N, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res2003; 57:244–252.
Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA2004; 291:565–575.
Jamison RL, Hartigan P, Kaufman JS, et al; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA2007; 2989:1163–1170. Erratum in JAMA 2008;300:170.
Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA2006; 296:2720–2726.
Namazi MH, Motamedi MR, Safi M, Vakili H, Saadat H, Nazari N. Efficacy of folic acid therapy for prevention of in-stent restenosis: a randomized clinical trial. Arch Iran Med2006; 9:108–110.
Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med2001; 345:1593–1600.
Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet2007; 369:1876–1882.
Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke2010; 41:1205–1212.
Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg2009; 38:316–322.
Hansrani M, Stansby G. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev2002; ( 3):CD003285.
Mosca L. Novel cardiovascular risk factors: do they add value to your practice?Am Fam Physician2003; 67:264,266.
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol2007; 50:e1–e157.
Lonn E. Homocysteine-lowering B vitamin therapy in cardiovascular prevention—wrong again?JAMA2008; 299:2086–2087.
Milani RV, Lavie CJ. Homocysteine: the Rubik’s cube of cardiovascular risk factors. Mayo Clin Proc2008; 83:1200–1202.
Bazzano LA. Folic acid supplementation and cardiovascular disease: the state of the art. Am J Med Sci2009; 338:48–49.
Ntaios G, Savopoulos C, Grekas D, Hatzitolios A. The controversial role of B-vitamins in cardiovascular risk: an update. Arch Cardiovasc Dis2009; 102:847–854.
Mosca L, Banka CL, Benjamin EJ; Expert Panel/Writing Group. Evidence-based guidelines for cardiovascular disease prevention in women: 2007 update. Circulation2007; 115:1481–1501.
Bassuk SS, Albert CM, Cook NR, et al. The Women’s Antioxidant Cardiovascular Study: design and baseline characteristics of participants. J Womens Health (Larchmt)2004; 13:99–117.
SEARCH Study Collaborative Group; Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am Heart J2007; 154:815–823,823.e1–e6.
Ebbing M, Bønaa KH, Nygård O, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA2009; 302:2119–2126.
Patients often ask primary care physicians and cardiologists about the measurement of biomarkers for cardiovascular disease and about the efficacy of preventive measures.
Although studies have shown that elevated homocysteine is a risk factor for cardiovascular and peripheral arterial disease1–3 and that supplementation with folic acid, vitamin B6, and vitamin B12 lowers homocysteine levels,4,5 it is unclear whether such supplementation prevents cardiovascular events. As a result, there is no consensus about whose homocysteine levels should be measured and who, if anyone, should receive homocysteine-lowering therapies.
The aim of this paper is to examine whether the evidence is sufficient to recommend homocysteine testing to guide the prevention and treatment of cardiovascular disease, or to recommend using folic acid, vitamin B6, and vitamin B12 for primary or secondary prevention of cardiovascular disease.
HISTORY OF HOMOCYSTEINE AS A RISK MARKER
Homocysteine is an amino acid formed from the metabolism of methionine, an essential amino acid derived from dietary protein. Although homocysteine was first isolated by Butz and du Vigneaud in 1932,6 it was not until 1964 that Gibson et al7 reported that patients with homocystinuria (more about this below) had vascular anomalies and arterial thrombosis. In 1969, McCully8 made the connection between elevated homocysteine levels and the risk of atherosclerosis.
Several possible mechanisms for the association between homocysteine and atherosclerosis have been demonstrated in experimental models. These include stimulation of smooth muscle growth, reduction in endothelial cell growth, impaired endothelial cell relaxation, decreased synthesis of high-density lipoprotein, promotion of autoimmune response, and accumulation of inflammatory monocytes in atherosclerotic plaques.3,9,10
In view of these findings, researchers have been evaluating whether homocysteine-lowering therapies decrease the risk of cardiovascular disease.
CAUSES OF ELEVATED PLASMA HOMOCYSTEINE
An elevated plasma homocysteine level can result from many different factors, including vitamin deficiencies, renal impairment, and inborn errors of homocysteine metabolism (Table 1).9,11,12
Vitamin deficiencies. Vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), and folic acid are cofactors required for homocysteine metabolism, and deficiency in any or all of these leads to disruption of the relevant metabolic pathways.
Renal impairment. A low glomerular filtration rate has also been correlated with an elevated plasma homocysteine concentration. This makes sense, since the kidneys perform up to 70% of the clearance of homocysteine, although a cause-and-effect relationship is unclear.13
Inborn errors of homocysteine metabolism.Homocystinuria, ie, an abnormal elevation of homocysteine in the urine, is caused by several autosomal recessive disorders. People with these genetic variations have extremely high homocysteine levels.
A deficiency in the enzyme cystathionine beta-synthase is quite rare (the incidence in newborn babies has been found to be 1 in 344,000 worldwide and 1 in 65,000 in Ireland and Australia14), but leads to homocysteine levels greater than 100 μmol/L and often causes cardiovascular disease by the age of 30 years.15
A deficiency in the enzyme methylene tetrahydrofolate reductase (MTHFR) is a more common cause of mildly to moderately elevated plasma homocysteine levels.16 The MTHFR deficiency involves a variation at position 677 in the MTHFR gene in which cytosine is replaced by thymidine (thus called C677T or 677C>T).17 Ten percent of the population are homozygous for this variant (TT), 43% are heterozygous (CT), and 47% are unaffected (CC). Heterozygotes have slightly higher homocysteine levels than unaffected people, while people with the TT genotype have approximately 20% higher homocysteine levels.17
ELEVATED HOMOCYSTEINE IS COMMON
In a study of a population in Norway from 1992 to 1993, 8.5% had mild elevations in homocysteine (plasma levels 15–29.99 μmol/L), 0.8% had moderate elevations (30–99.99 μmol/L), and 0.02% had severe elevations (≥ 100 μmol/L).13,18 The prevalence of hyperhomocysteinemia in the United States is probably much lower, given that supplementation of white flour and cereal grains with folic acid has been mandatory since 1998, but this is not well described in the literature.
HOW GREAT IS THE RISK?
Studies over the past 10 to 20 years have shown that elevated homocysteine is a marker of risk of cardiovascular disease. The association was first noted in patients with cystathionine beta-synthase deficiency, who tend to have premature cardiovascular disease.
However, studies of patients with MTHFR 677C>T have yielded mixed results. Although several meta-analyses found up to a 42% higher rate of ischemic heart disease and stroke in patients homozygous for MTHFR 677C>T (the TT genotype) than in those with the CC genotype,17,19,20 two other large meta-analyses did not find an association between this variant and vascular risk.21,22
Nonetheless, in a meta-analysis of the association between homocysteine and cardiovascular disease, Wald et al17 found that for every 5-μmol/L increase in serum homocysteine concentration, the risk of ischemic heart disease increased 20% to 30%.
TRIALS OF HOMOCYSTEINE-LOWERING THERAPY HAVE HAD MIXED RESULTS
Primary prevention of cardiovascular disease
Given the finding that treatment with folic acid lowers homocysteine—initially noted in patients with homocystinuria—researchers hypothesized that treatment with folic acid, vitamin B6, and vitamin B12 would decrease the risk of cardiovascular disease.
Little evidence currently exists to guide recommendations for homocysteine-lowering therapy to prevent first attacks of cardiovascular disease. The few studies published to date have been observational studies of dietary intake (not vitamin supplementation), and many were performed before folic acid fortification was mandated for flour and cereal (Table 2).23–27 Although the studies suggest that higher B-vitamin intake correlates with less vascular disease and its sequelae, there is uncertainty as to whether it is folic acid, vitamin B6, or vitamin B12 that is responsible, and also whether supplements would provide the same protective benefit as the presence of these nutrients in a varied diet.
Thus, in its recent evaluation of novel risk markers of cardiovascular disease, the United States Preventive Services Task Force 28,29 does not recommend measuring the plasma homocysteine level in the evaluation of either low-risk or intermediate-risk populations, finding no evidence that it adds any useful information in predicting major coronary events beyond what one could get from calculating the Framingham Risk Score. The task force also found no evidence that treating people who have elevated homocysteine levels decreases their risk of subsequent cardiovascular events.
In addition, a recent Cochrane Database review of eight randomized controlled trials in patients at low risk did not find a lower risk of myocardial infarction (fatal or nonfatal), stroke, or death from any cause in patients receiving B-complex vitamins.30
Secondary prevention of cardiovascular disease
Results have been mixed with regard to the ability of B vitamins to prevent cardiovascular events in patients with known cardiovascular disease (Table 3).4,5,11,12,31–46
Bazzano et al,47 in a meta-analysis published in 2006, evaluated 12 randomized controlled trials of folic acid supplementation in patients with known cardiovascular disease and did not find that treated patients had better cardiovascular outcomes. The mean homocysteine level was elevated (> 15 μmol/L) at baseline in only 4 of the 12 trials. However, in 1 of these 4 trials, there was no difference in outcomes comparing those with and without elevated homocysteine.31
Albert et al4 more recently evaluated the effect of a combination pill containing folic acid, vitamin B6, and vitamin B12 on cardiovascular events in women at high risk, ie, those with a history of cardiovascular disease or having three or more coronary risk factors. Treatment did not decrease the rate of the composite outcome of cardiovascular disease mortality, stroke, myocardial infarction, or coronary revascularization, although the homocysteine level decreased by a mean of 30% in the treated group. However, only 27.7% of the participants had an elevated homocysteine level. One might not expect patients to benefit from such treatment if they had normal homocysteine levels to begin with.
Ebbing et al,5 in a trial published in 2008, investigated the effect of folic acid, vitamin B12, and vitamin B6 supplements on the risks of death from any cause and of cardiovascular events in patients undergoing coronary angiography. Outcomes were no better in the treatment group than in the control group, despite a mean decrease in homocysteine level of 19%. However, over 90% of the participants had a normal homocysteine level.
Mager et al,32 in a study published in 2009, looked specifically at whether patients with coronary artery disease and elevated homocysteine levels (> 15 μmol/L) would benefit from folate-based vitamin therapy. In this subset, the incidence of death from any cause was lower in the treated group than in the control group (4% vs 32%, P < .001), an association that was not present in patients with normal homocysteine levels.
The SEARCH trial (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),33 recently published, was a double-blind, randomized controlled trial of vitamin B12 and folic acid treatment in 12,064 patients who had survived a myocardial infarction. Although those who received the vitamin therapy had a 28% reduction in homocysteine level, no clinical benefit was demonstrated. Of note, 66% of the patients had a homocysteine level lower than 14 μmol/L at baseline.
Restenosis after angioplasty
Results are also mixed regarding whether folic acid supplements modify the risk of restenosis after coronary angioplasty.
Namazi et al48 evaluated the effect of folic acid supplementation on in-stent restenosis in 200 patients and found no difference between the treatment and placebo groups in the rates of either restenosis or target-vessel revascularization.
Schnyder et al49 evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on the rate of coronary restenosis (in cases of balloon angioplasty) or in-stent restenosis (if a stent was used). Patients receiving treatment had lower rates of restenosis or instent restenosis (40% vs 48%, P = .01) and of need for target-vessel revascularization (11% vs 22%, P = .047). The mean homocysteine level was not elevated in this study either, and the researchers did not analyze the outcomes according to whether patients had high or normal homocysteine levels.
Lange et al35 also evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on coronary in-stent restenosis. Paradoxically, the rate was higher with treatment in the overall group (mean homocysteine level 12.2 μmol/L), leading to a higher incidence of target-vessel revascularization. Patients who had a baseline elevation in homocysteine level had a nonsignificant trend toward a lower rate of in-stent restenosis.
Cerebrovascular and peripheral arterial disease
The evidence is also mixed for using folic acid and other B vitamins to prevent cerebrovascular disease and peripheral vascular disease. Although a 2007 meta-analysis found that folic acid supplementation decreased the risk of a first stroke by 18% (P = .045),50 a later meta-analysis contradicts this finding.51
A 2009 meta-analysis found that patients with peripheral arterial disease had higher homocysteine levels than controls, but it did not find any benefit from supplementation, owing to heterogeneity of the clinical end points used.52 Indeed, a 2009 Cochrane Database Systematic Review found that there were no adequate trials of the treatment of patients with peripheral vascular disease who have elevated plasma homocysteine.53
However, immediately after the Cochrane review was published, Khandanpour et al36 published the results of a trial of the effect of folic acid and 5-methyltetrahydrofolate (an active form of folic acid) supplementation on the ankle-brachial pressure index and the pulse-wave velocity in patients with peripheral arterial disease. These measures improved with 16 weeks of treatment. For the ankle-brachial pressure index, the P value was less than .01 for folic acid and .009 for 5-methyltetrahydrofolate; for the pulse-wave velocity, the P value was .051 for folic acid and .011 for 5-methyltetrahydrofolate.
Kidney disease
One could postulate that patients with end-stage renal disease or chronic kidney disease might benefit the most from folic acid supplementation, given the correlation of elevations in homocysteine levels with decline in glomerular filtration rate.
However, only one study found a lower rate of cardiovascular events with folic acid supplementation in dialysis patients, and the difference was not statistically significant (25% vs 36%, P < .08).31 Further, several studies found no benefit of folic acid supplementation in patients with chronic kidney disease.11,12,37
FUTURE DIRECTIONS AND RECOMMENDATIONS
Many experts have suggested that the existing evidence indicates that the homocysteine-lowering therapies folic acid, vitamin B6, and vitamin B12 do not lower the risk of cardiovascular disease.38,54–59 Indeed, the American Heart Association guidelines for cardiovascular disease prevention in women do not recommend folic acid supplementation to prevent cardiovascular disease.60 (Recommendations for men are the same as for women.) However, most of the clinical trials have not selected and treated patients with elevated homocysteine levels, but have instead included all patients regardless of homocysteine level.
At least two large ongoing trials are currently evaluating B-vitamin therapy for secondary prevention, but neither trial is looking specifically at patients with elevated homocysteine levels.61,62
Thus, instead of concluding that no patients could benefit from homocysteine-lowering treatment, future studies need to clarify:
Whether patients with elevated homocysteine would benefit from such treatment
At what level it would be appropriate to start treatment
The appropriate target homocysteine level with treatment.
Particularly given the recent finding that folic acid supplementation may increase cancer risk,63 these questions need closer scrutiny.
Patients often ask primary care physicians and cardiologists about the measurement of biomarkers for cardiovascular disease and about the efficacy of preventive measures.
Although studies have shown that elevated homocysteine is a risk factor for cardiovascular and peripheral arterial disease1–3 and that supplementation with folic acid, vitamin B6, and vitamin B12 lowers homocysteine levels,4,5 it is unclear whether such supplementation prevents cardiovascular events. As a result, there is no consensus about whose homocysteine levels should be measured and who, if anyone, should receive homocysteine-lowering therapies.
The aim of this paper is to examine whether the evidence is sufficient to recommend homocysteine testing to guide the prevention and treatment of cardiovascular disease, or to recommend using folic acid, vitamin B6, and vitamin B12 for primary or secondary prevention of cardiovascular disease.
HISTORY OF HOMOCYSTEINE AS A RISK MARKER
Homocysteine is an amino acid formed from the metabolism of methionine, an essential amino acid derived from dietary protein. Although homocysteine was first isolated by Butz and du Vigneaud in 1932,6 it was not until 1964 that Gibson et al7 reported that patients with homocystinuria (more about this below) had vascular anomalies and arterial thrombosis. In 1969, McCully8 made the connection between elevated homocysteine levels and the risk of atherosclerosis.
Several possible mechanisms for the association between homocysteine and atherosclerosis have been demonstrated in experimental models. These include stimulation of smooth muscle growth, reduction in endothelial cell growth, impaired endothelial cell relaxation, decreased synthesis of high-density lipoprotein, promotion of autoimmune response, and accumulation of inflammatory monocytes in atherosclerotic plaques.3,9,10
In view of these findings, researchers have been evaluating whether homocysteine-lowering therapies decrease the risk of cardiovascular disease.
CAUSES OF ELEVATED PLASMA HOMOCYSTEINE
An elevated plasma homocysteine level can result from many different factors, including vitamin deficiencies, renal impairment, and inborn errors of homocysteine metabolism (Table 1).9,11,12
Vitamin deficiencies. Vitamin B6 (pyridoxine), vitamin B12 (cyanocobalamin), and folic acid are cofactors required for homocysteine metabolism, and deficiency in any or all of these leads to disruption of the relevant metabolic pathways.
Renal impairment. A low glomerular filtration rate has also been correlated with an elevated plasma homocysteine concentration. This makes sense, since the kidneys perform up to 70% of the clearance of homocysteine, although a cause-and-effect relationship is unclear.13
Inborn errors of homocysteine metabolism.Homocystinuria, ie, an abnormal elevation of homocysteine in the urine, is caused by several autosomal recessive disorders. People with these genetic variations have extremely high homocysteine levels.
A deficiency in the enzyme cystathionine beta-synthase is quite rare (the incidence in newborn babies has been found to be 1 in 344,000 worldwide and 1 in 65,000 in Ireland and Australia14), but leads to homocysteine levels greater than 100 μmol/L and often causes cardiovascular disease by the age of 30 years.15
A deficiency in the enzyme methylene tetrahydrofolate reductase (MTHFR) is a more common cause of mildly to moderately elevated plasma homocysteine levels.16 The MTHFR deficiency involves a variation at position 677 in the MTHFR gene in which cytosine is replaced by thymidine (thus called C677T or 677C>T).17 Ten percent of the population are homozygous for this variant (TT), 43% are heterozygous (CT), and 47% are unaffected (CC). Heterozygotes have slightly higher homocysteine levels than unaffected people, while people with the TT genotype have approximately 20% higher homocysteine levels.17
ELEVATED HOMOCYSTEINE IS COMMON
In a study of a population in Norway from 1992 to 1993, 8.5% had mild elevations in homocysteine (plasma levels 15–29.99 μmol/L), 0.8% had moderate elevations (30–99.99 μmol/L), and 0.02% had severe elevations (≥ 100 μmol/L).13,18 The prevalence of hyperhomocysteinemia in the United States is probably much lower, given that supplementation of white flour and cereal grains with folic acid has been mandatory since 1998, but this is not well described in the literature.
HOW GREAT IS THE RISK?
Studies over the past 10 to 20 years have shown that elevated homocysteine is a marker of risk of cardiovascular disease. The association was first noted in patients with cystathionine beta-synthase deficiency, who tend to have premature cardiovascular disease.
However, studies of patients with MTHFR 677C>T have yielded mixed results. Although several meta-analyses found up to a 42% higher rate of ischemic heart disease and stroke in patients homozygous for MTHFR 677C>T (the TT genotype) than in those with the CC genotype,17,19,20 two other large meta-analyses did not find an association between this variant and vascular risk.21,22
Nonetheless, in a meta-analysis of the association between homocysteine and cardiovascular disease, Wald et al17 found that for every 5-μmol/L increase in serum homocysteine concentration, the risk of ischemic heart disease increased 20% to 30%.
TRIALS OF HOMOCYSTEINE-LOWERING THERAPY HAVE HAD MIXED RESULTS
Primary prevention of cardiovascular disease
Given the finding that treatment with folic acid lowers homocysteine—initially noted in patients with homocystinuria—researchers hypothesized that treatment with folic acid, vitamin B6, and vitamin B12 would decrease the risk of cardiovascular disease.
Little evidence currently exists to guide recommendations for homocysteine-lowering therapy to prevent first attacks of cardiovascular disease. The few studies published to date have been observational studies of dietary intake (not vitamin supplementation), and many were performed before folic acid fortification was mandated for flour and cereal (Table 2).23–27 Although the studies suggest that higher B-vitamin intake correlates with less vascular disease and its sequelae, there is uncertainty as to whether it is folic acid, vitamin B6, or vitamin B12 that is responsible, and also whether supplements would provide the same protective benefit as the presence of these nutrients in a varied diet.
Thus, in its recent evaluation of novel risk markers of cardiovascular disease, the United States Preventive Services Task Force 28,29 does not recommend measuring the plasma homocysteine level in the evaluation of either low-risk or intermediate-risk populations, finding no evidence that it adds any useful information in predicting major coronary events beyond what one could get from calculating the Framingham Risk Score. The task force also found no evidence that treating people who have elevated homocysteine levels decreases their risk of subsequent cardiovascular events.
In addition, a recent Cochrane Database review of eight randomized controlled trials in patients at low risk did not find a lower risk of myocardial infarction (fatal or nonfatal), stroke, or death from any cause in patients receiving B-complex vitamins.30
Secondary prevention of cardiovascular disease
Results have been mixed with regard to the ability of B vitamins to prevent cardiovascular events in patients with known cardiovascular disease (Table 3).4,5,11,12,31–46
Bazzano et al,47 in a meta-analysis published in 2006, evaluated 12 randomized controlled trials of folic acid supplementation in patients with known cardiovascular disease and did not find that treated patients had better cardiovascular outcomes. The mean homocysteine level was elevated (> 15 μmol/L) at baseline in only 4 of the 12 trials. However, in 1 of these 4 trials, there was no difference in outcomes comparing those with and without elevated homocysteine.31
Albert et al4 more recently evaluated the effect of a combination pill containing folic acid, vitamin B6, and vitamin B12 on cardiovascular events in women at high risk, ie, those with a history of cardiovascular disease or having three or more coronary risk factors. Treatment did not decrease the rate of the composite outcome of cardiovascular disease mortality, stroke, myocardial infarction, or coronary revascularization, although the homocysteine level decreased by a mean of 30% in the treated group. However, only 27.7% of the participants had an elevated homocysteine level. One might not expect patients to benefit from such treatment if they had normal homocysteine levels to begin with.
Ebbing et al,5 in a trial published in 2008, investigated the effect of folic acid, vitamin B12, and vitamin B6 supplements on the risks of death from any cause and of cardiovascular events in patients undergoing coronary angiography. Outcomes were no better in the treatment group than in the control group, despite a mean decrease in homocysteine level of 19%. However, over 90% of the participants had a normal homocysteine level.
Mager et al,32 in a study published in 2009, looked specifically at whether patients with coronary artery disease and elevated homocysteine levels (> 15 μmol/L) would benefit from folate-based vitamin therapy. In this subset, the incidence of death from any cause was lower in the treated group than in the control group (4% vs 32%, P < .001), an association that was not present in patients with normal homocysteine levels.
The SEARCH trial (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),33 recently published, was a double-blind, randomized controlled trial of vitamin B12 and folic acid treatment in 12,064 patients who had survived a myocardial infarction. Although those who received the vitamin therapy had a 28% reduction in homocysteine level, no clinical benefit was demonstrated. Of note, 66% of the patients had a homocysteine level lower than 14 μmol/L at baseline.
Restenosis after angioplasty
Results are also mixed regarding whether folic acid supplements modify the risk of restenosis after coronary angioplasty.
Namazi et al48 evaluated the effect of folic acid supplementation on in-stent restenosis in 200 patients and found no difference between the treatment and placebo groups in the rates of either restenosis or target-vessel revascularization.
Schnyder et al49 evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on the rate of coronary restenosis (in cases of balloon angioplasty) or in-stent restenosis (if a stent was used). Patients receiving treatment had lower rates of restenosis or instent restenosis (40% vs 48%, P = .01) and of need for target-vessel revascularization (11% vs 22%, P = .047). The mean homocysteine level was not elevated in this study either, and the researchers did not analyze the outcomes according to whether patients had high or normal homocysteine levels.
Lange et al35 also evaluated the effect of folic acid, vitamin B6, and vitamin B12 treatment on coronary in-stent restenosis. Paradoxically, the rate was higher with treatment in the overall group (mean homocysteine level 12.2 μmol/L), leading to a higher incidence of target-vessel revascularization. Patients who had a baseline elevation in homocysteine level had a nonsignificant trend toward a lower rate of in-stent restenosis.
Cerebrovascular and peripheral arterial disease
The evidence is also mixed for using folic acid and other B vitamins to prevent cerebrovascular disease and peripheral vascular disease. Although a 2007 meta-analysis found that folic acid supplementation decreased the risk of a first stroke by 18% (P = .045),50 a later meta-analysis contradicts this finding.51
A 2009 meta-analysis found that patients with peripheral arterial disease had higher homocysteine levels than controls, but it did not find any benefit from supplementation, owing to heterogeneity of the clinical end points used.52 Indeed, a 2009 Cochrane Database Systematic Review found that there were no adequate trials of the treatment of patients with peripheral vascular disease who have elevated plasma homocysteine.53
However, immediately after the Cochrane review was published, Khandanpour et al36 published the results of a trial of the effect of folic acid and 5-methyltetrahydrofolate (an active form of folic acid) supplementation on the ankle-brachial pressure index and the pulse-wave velocity in patients with peripheral arterial disease. These measures improved with 16 weeks of treatment. For the ankle-brachial pressure index, the P value was less than .01 for folic acid and .009 for 5-methyltetrahydrofolate; for the pulse-wave velocity, the P value was .051 for folic acid and .011 for 5-methyltetrahydrofolate.
Kidney disease
One could postulate that patients with end-stage renal disease or chronic kidney disease might benefit the most from folic acid supplementation, given the correlation of elevations in homocysteine levels with decline in glomerular filtration rate.
However, only one study found a lower rate of cardiovascular events with folic acid supplementation in dialysis patients, and the difference was not statistically significant (25% vs 36%, P < .08).31 Further, several studies found no benefit of folic acid supplementation in patients with chronic kidney disease.11,12,37
FUTURE DIRECTIONS AND RECOMMENDATIONS
Many experts have suggested that the existing evidence indicates that the homocysteine-lowering therapies folic acid, vitamin B6, and vitamin B12 do not lower the risk of cardiovascular disease.38,54–59 Indeed, the American Heart Association guidelines for cardiovascular disease prevention in women do not recommend folic acid supplementation to prevent cardiovascular disease.60 (Recommendations for men are the same as for women.) However, most of the clinical trials have not selected and treated patients with elevated homocysteine levels, but have instead included all patients regardless of homocysteine level.
At least two large ongoing trials are currently evaluating B-vitamin therapy for secondary prevention, but neither trial is looking specifically at patients with elevated homocysteine levels.61,62
Thus, instead of concluding that no patients could benefit from homocysteine-lowering treatment, future studies need to clarify:
Whether patients with elevated homocysteine would benefit from such treatment
At what level it would be appropriate to start treatment
The appropriate target homocysteine level with treatment.
Particularly given the recent finding that folic acid supplementation may increase cancer risk,63 these questions need closer scrutiny.
References
Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc2008; 83:1203–1212.
Boers GH. The case for mild hyperhomocysteinaemia as a risk factor. J Inherit Metab Dis1997; 20:301–306.
Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ2004; 11(suppl 1):S56–S64.
Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA2008; 299:2027–2036.
Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA2008; 300:795–804.
Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decompensation of methionine with sulphuric acid. J Biol Chem1932; 99:135–142.
McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol1969; 56:111–128.
Zhang D, Jiang X, Fang P, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation2009; 120:1893–1902.
Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol1999; 34:2002–2006.
Righetti M, Ferrario GM, Milani S, et al. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med Sci Monit2003; 9:PI19–PI24.
Zoungas S, McGrath BP, Branley P, et al. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol2006; 47:1108–1116.
Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press, 2001.
Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol2001; 21:2080–2085.
McKusick V. 236200 Homocystinuria. In:McKusick V, editor. Mendelian Inheritance in Man. 10th ed. Baltimore, MD: The Johns Hopkins University Press, 1992:1444–1446.
McKusick V. 236250 Homocystinuria due to deficiency of N(5,10)-methylenetetrahydrofolate reductase activity. In:McKusick V, editor. Mendelian Inheritance in Man. Baltimore, MD: The Johns Hopkins University Press, 1992:1447–1448.
Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ2002; 325:1202.
Nygård O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA1995; 274:1526–1533.
Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG; MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA2002; 288:2023–2031.
Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology2002; 59:529–536.
Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate?BMJ2005; 331:1053.
Brattström L, Wilcken DE, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation1998; 98:2520–2526.
Cui R, Iso H, Date C, Kikuchi S, Tamakoshi A; Japan Collaborative Cohort Study Group. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan Collaborative Cohort Study. Stroke2010; 41:1285–1289.
Liu S, Stampfer MJ, Hu FB, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses’ Health Study. Am J Clin Nutr1999; 70:412–419.
Liu S, Manson JE, Stampfer MJ, et al. Whole grain consumption and risk of ischemic stroke in women: a prospective study. JAMA2000; 284:1534–1540.
Merchant AT, Hu FB, Spiegelman D, Willett WC, Rimm EB, Ascherio A. The use of B vitamin supplements and peripheral arterial disease risk in men are inversely related. J Nutr2003; 133:2863–2867.
Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA1998; 279:359–364.
US Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med2009; 151:474–482.
Helfand M, Buckley DI, Freeman M, et al. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the US Preventive Services Task Force. Ann Intern Med2009; 151:496–507.
Martí-Carvajal AJ, Solà I, Lathyris D, Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev2009; ( 4):CD006612.
Righetti M, Serbelloni P, Milani S, Ferrario G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif2006; 24:379–386.
Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol2009; 104:745–749.
Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA2010; 303:2486–2494.
Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and percutaneous coronary intervention: the Swiss Heart Study: a randomized controlled trial. JAMA2002; 288:973–979.
Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med2004; 350:2673–2781.
Khandanpour N, Armon MP, Jennings B, et al. Randomized clinical trial of folate supplementation in patients with peripheral arterial disease. Br J Surg2009; 96:990–998.
Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol2004; 15:420–426.
Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med2006; 354:1629–1632.
Bønaa KH, Njølstad I, Ueland PM, et al; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med2006; 354:1578–1588.
Carrero JJ, López-Huertas E, Salmerón LM, Baró L, Ros E. Daily supplementation with (n-3) PUFAs, oleic acid, folic acid, and vitamins B-6 and E increases pain-free walking distance and improves risk factors in men with peripheral vascular disease. J Nutr2005; 135:1393–1399.
Liem AH, van Boven AJ, Veeger NJ, et al; Folic Acid on Risk Diminishment After Acute Myocarial Infarction Study Group. Efficacy of folic acid when added to statin therapy in patients with hypercholesterolemia following acute myocardial infarction: a randomised pilot trial. Int J Cardiol2004; 93:175–179.
Liem A, Reynierse-Buitenwerf GH, Zwinderman AH, Jukema JW, van Veldhuisen DJ. Secondary prevention with folic acid: results of the Goes extension study. Heart2005; 91:1213–1214.
Lonn E, Yusuf S, Arnold MJ, et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med2006; 354:1567–1577.
Sydow K, Schwedhelm E, Arakawa N, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res2003; 57:244–252.
Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA2004; 291:565–575.
Jamison RL, Hartigan P, Kaufman JS, et al; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA2007; 2989:1163–1170. Erratum in JAMA 2008;300:170.
Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA2006; 296:2720–2726.
Namazi MH, Motamedi MR, Safi M, Vakili H, Saadat H, Nazari N. Efficacy of folic acid therapy for prevention of in-stent restenosis: a randomized clinical trial. Arch Iran Med2006; 9:108–110.
Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med2001; 345:1593–1600.
Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet2007; 369:1876–1882.
Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke2010; 41:1205–1212.
Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg2009; 38:316–322.
Hansrani M, Stansby G. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev2002; ( 3):CD003285.
Mosca L. Novel cardiovascular risk factors: do they add value to your practice?Am Fam Physician2003; 67:264,266.
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol2007; 50:e1–e157.
Lonn E. Homocysteine-lowering B vitamin therapy in cardiovascular prevention—wrong again?JAMA2008; 299:2086–2087.
Milani RV, Lavie CJ. Homocysteine: the Rubik’s cube of cardiovascular risk factors. Mayo Clin Proc2008; 83:1200–1202.
Bazzano LA. Folic acid supplementation and cardiovascular disease: the state of the art. Am J Med Sci2009; 338:48–49.
Ntaios G, Savopoulos C, Grekas D, Hatzitolios A. The controversial role of B-vitamins in cardiovascular risk: an update. Arch Cardiovasc Dis2009; 102:847–854.
Mosca L, Banka CL, Benjamin EJ; Expert Panel/Writing Group. Evidence-based guidelines for cardiovascular disease prevention in women: 2007 update. Circulation2007; 115:1481–1501.
Bassuk SS, Albert CM, Cook NR, et al. The Women’s Antioxidant Cardiovascular Study: design and baseline characteristics of participants. J Womens Health (Larchmt)2004; 13:99–117.
SEARCH Study Collaborative Group; Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am Heart J2007; 154:815–823,823.e1–e6.
Ebbing M, Bønaa KH, Nygård O, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA2009; 302:2119–2126.
References
Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clin Proc2008; 83:1203–1212.
Boers GH. The case for mild hyperhomocysteinaemia as a risk factor. J Inherit Metab Dis1997; 20:301–306.
Austin RC, Lentz SR, Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ2004; 11(suppl 1):S56–S64.
Albert CM, Cook NR, Gaziano JM, et al. Effect of folic acid and B vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: a randomized trial. JAMA2008; 299:2027–2036.
Ebbing M, Bleie Ø, Ueland PM, et al. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: a randomized controlled trial. JAMA2008; 300:795–804.
Butz LW, du Vigneaud V. The formation of a homologue of cystine by the decompensation of methionine with sulphuric acid. J Biol Chem1932; 99:135–142.
McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol1969; 56:111–128.
Zhang D, Jiang X, Fang P, et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation2009; 120:1893–1902.
Woo KS, Chook P, Lolin YI, Sanderson JE, Metreweli C, Celermajer DS. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J Am Coll Cardiol1999; 34:2002–2006.
Righetti M, Ferrario GM, Milani S, et al. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med Sci Monit2003; 9:PI19–PI24.
Zoungas S, McGrath BP, Branley P, et al. Cardiovascular morbidity and mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in chronic renal failure: a multicenter, randomized, controlled trial. J Am Coll Cardiol2006; 47:1108–1116.
Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press, 2001.
Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocystinuria due to cystathionine beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol2001; 21:2080–2085.
McKusick V. 236200 Homocystinuria. In:McKusick V, editor. Mendelian Inheritance in Man. 10th ed. Baltimore, MD: The Johns Hopkins University Press, 1992:1444–1446.
McKusick V. 236250 Homocystinuria due to deficiency of N(5,10)-methylenetetrahydrofolate reductase activity. In:McKusick V, editor. Mendelian Inheritance in Man. Baltimore, MD: The Johns Hopkins University Press, 1992:1447–1448.
Wald DS, Law M, Morris JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ2002; 325:1202.
Nygård O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile. The Hordaland Homocysteine Study. JAMA1995; 274:1526–1533.
Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG; MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA2002; 288:2023–2031.
Kelly PJ, Rosand J, Kistler JP, et al. Homocysteine, MTHFR 677C-->T polymorphism, and risk of ischemic stroke: results of a meta-analysis. Neurology2002; 59:529–536.
Lewis SJ, Ebrahim S, Davey Smith G. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: does totality of evidence support causal role for homocysteine and preventive potential of folate?BMJ2005; 331:1053.
Brattström L, Wilcken DE, Ohrvik J, Brudin L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation1998; 98:2520–2526.
Cui R, Iso H, Date C, Kikuchi S, Tamakoshi A; Japan Collaborative Cohort Study Group. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan Collaborative Cohort Study. Stroke2010; 41:1285–1289.
Liu S, Stampfer MJ, Hu FB, et al. Whole-grain consumption and risk of coronary heart disease: results from the Nurses’ Health Study. Am J Clin Nutr1999; 70:412–419.
Liu S, Manson JE, Stampfer MJ, et al. Whole grain consumption and risk of ischemic stroke in women: a prospective study. JAMA2000; 284:1534–1540.
Merchant AT, Hu FB, Spiegelman D, Willett WC, Rimm EB, Ascherio A. The use of B vitamin supplements and peripheral arterial disease risk in men are inversely related. J Nutr2003; 133:2863–2867.
Rimm EB, Willett WC, Hu FB, et al. Folate and vitamin B6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA1998; 279:359–364.
US Preventive Services Task Force. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med2009; 151:474–482.
Helfand M, Buckley DI, Freeman M, et al. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the US Preventive Services Task Force. Ann Intern Med2009; 151:496–507.
Martí-Carvajal AJ, Solà I, Lathyris D, Salanti G. Homocysteine lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev2009; ( 4):CD006612.
Righetti M, Serbelloni P, Milani S, Ferrario G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif2006; 24:379–386.
Mager A, Orvin K, Koren-Morag N, et al. Impact of homocysteine-lowering vitamin therapy on long-term outcome of patients with coronary artery disease. Am J Cardiol2009; 104:745–749.
Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group; Armitage JM, Bowman L, Clarke RJ, et al. Effects of homocysteine-lowering with folic acid plus vitamin B12 vs placebo on mortality and major morbidity in myocardial infarction survivors: a randomized trial. JAMA2010; 303:2486–2494.
Schnyder G, Roffi M, Flammer Y, Pin R, Hess OM. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and percutaneous coronary intervention: the Swiss Heart Study: a randomized controlled trial. JAMA2002; 288:973–979.
Lange H, Suryapranata H, De Luca G, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med2004; 350:2673–2781.
Khandanpour N, Armon MP, Jennings B, et al. Randomized clinical trial of folate supplementation in patients with peripheral arterial disease. Br J Surg2009; 96:990–998.
Wrone EM, Hornberger JM, Zehnder JL, McCann LM, Coplon NS, Fortmann SP. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J Am Soc Nephrol2004; 15:420–426.
Loscalzo J. Homocysteine trials—clear outcomes for complex reasons. N Engl J Med2006; 354:1629–1632.
Bønaa KH, Njølstad I, Ueland PM, et al; NORVIT Trial Investigators. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N Engl J Med2006; 354:1578–1588.
Carrero JJ, López-Huertas E, Salmerón LM, Baró L, Ros E. Daily supplementation with (n-3) PUFAs, oleic acid, folic acid, and vitamins B-6 and E increases pain-free walking distance and improves risk factors in men with peripheral vascular disease. J Nutr2005; 135:1393–1399.
Liem AH, van Boven AJ, Veeger NJ, et al; Folic Acid on Risk Diminishment After Acute Myocarial Infarction Study Group. Efficacy of folic acid when added to statin therapy in patients with hypercholesterolemia following acute myocardial infarction: a randomised pilot trial. Int J Cardiol2004; 93:175–179.
Liem A, Reynierse-Buitenwerf GH, Zwinderman AH, Jukema JW, van Veldhuisen DJ. Secondary prevention with folic acid: results of the Goes extension study. Heart2005; 91:1213–1214.
Lonn E, Yusuf S, Arnold MJ, et al; Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N Engl J Med2006; 354:1567–1577.
Sydow K, Schwedhelm E, Arakawa N, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res2003; 57:244–252.
Toole JF, Malinow MR, Chambless LE, et al. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA2004; 291:565–575.
Jamison RL, Hartigan P, Kaufman JS, et al; Veterans Affairs Site Investigators. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA2007; 2989:1163–1170. Erratum in JAMA 2008;300:170.
Bazzano LA, Reynolds K, Holder KN, He J. Effect of folic acid supplementation on risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. JAMA2006; 296:2720–2726.
Namazi MH, Motamedi MR, Safi M, Vakili H, Saadat H, Nazari N. Efficacy of folic acid therapy for prevention of in-stent restenosis: a randomized clinical trial. Arch Iran Med2006; 9:108–110.
Schnyder G, Roffi M, Pin R, et al. Decreased rate of coronary restenosis after lowering of plasma homocysteine levels. N Engl J Med2001; 345:1593–1600.
Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet2007; 369:1876–1882.
Lee M, Hong KS, Chang SC, Saver JL. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: a meta-analysis. Stroke2010; 41:1205–1212.
Khandanpour N, Loke YK, Meyer FJ, Jennings B, Armon MP. Homocysteine and peripheral arterial disease: systematic review and meta-analysis. Eur J Vasc Endovasc Surg2009; 38:316–322.
Hansrani M, Stansby G. Homocysteine lowering interventions for peripheral arterial disease and bypass grafts. Cochrane Database Syst Rev2002; ( 3):CD003285.
Mosca L. Novel cardiovascular risk factors: do they add value to your practice?Am Fam Physician2003; 67:264,266.
Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol2007; 50:e1–e157.
Lonn E. Homocysteine-lowering B vitamin therapy in cardiovascular prevention—wrong again?JAMA2008; 299:2086–2087.
Milani RV, Lavie CJ. Homocysteine: the Rubik’s cube of cardiovascular risk factors. Mayo Clin Proc2008; 83:1200–1202.
Bazzano LA. Folic acid supplementation and cardiovascular disease: the state of the art. Am J Med Sci2009; 338:48–49.
Ntaios G, Savopoulos C, Grekas D, Hatzitolios A. The controversial role of B-vitamins in cardiovascular risk: an update. Arch Cardiovasc Dis2009; 102:847–854.
Mosca L, Banka CL, Benjamin EJ; Expert Panel/Writing Group. Evidence-based guidelines for cardiovascular disease prevention in women: 2007 update. Circulation2007; 115:1481–1501.
Bassuk SS, Albert CM, Cook NR, et al. The Women’s Antioxidant Cardiovascular Study: design and baseline characteristics of participants. J Womens Health (Larchmt)2004; 13:99–117.
SEARCH Study Collaborative Group; Bowman L, Armitage J, Bulbulia R, Parish S, Collins R. Study of the effectiveness of additional reductions in cholesterol and homocysteine (SEARCH): characteristics of a randomized trial among 12064 myocardial infarction survivors. Am Heart J2007; 154:815–823,823.e1–e6.
Ebbing M, Bønaa KH, Nygård O, et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA2009; 302:2119–2126.
Factors that can cause the plasma homocysteine concentration to be high include deficiencies of vitamin B6, vitamin B12, and folic acid; renal insufficiency; and genetic variants in enzymes responsible for homocysteine metabolism.
Higher plasma homocysteine levels are associated with a higher risk of cardiovascular, cerebrovascular, and peripheral arterial disease.
Supplementation of B vitamins and folic acid can lower plasma homocysteine levels.
Randomized controlled trials of supplementation to prevent cardiovascular events and other adverse outcomes have had mostly negative results. However, most patients in these trials had normal baseline plasma homocysteine levels.
Needed are randomized trials to see if supplementation improves outcomes in patients with high homocysteine levels.
For patients with carotid artery stenosis, percutaneous intervention with stenting is as good as surgery (carotid endarterectomy). This was the major finding of the recently completed Carotid Revascularization Endarterectomy Versus Stenting Trial (CREST)1—with some qualifications.
CREST is the latest in a series of clinical trials of treatment of carotid stenosis that have generated reams of numbers and much debate. The topic of surgery vs percutaneous intervention is a moving target, as techniques evolve and improve. We believe the CREST results are valuable and should help inform decisions about treatment in the “real world.”
In this article, we offer a critical review of CREST, with a careful evaluation of its methods, results, and conclusions.
AN EVOLVING FIELD
Despite improvements in diagnosis and management, stroke remains one of the leading causes of morbidity and death in the United States, with an annual incidence of 780,000 cases and 270,000 deaths.2,3
Figure 1. Carotid endarterectomy has long been an established treatment in selected patients with symptomatic carotid artery stenosis of 50% or greater or asymptomatic stenosis of 60% or greater. However, percutaneous carotid artery angioplasty with stenting and placement of an embolic protection device is gaining ground as a reasonable, safe, less invasive alternative.From 10% to 30% of ischemic strokes are due to emboli from the carotid arteries.4–6 Carotid endarterectomy is an established treatment in selected patients with symptomatic carotid stenosis of 50% or greater or asymptomatic stenosis of 60% or greater.7,8 However, percutaneous techniques such as carotid artery angioplasty with stenting have improved, making them a viable, less invasive option (Figure 1).
Randomized trials of stenting have had mixed results, leading the Centers for Medicare and Medicaid Services (CMS) to adopt strict reimbursement policies. Currently, CMS reimburses for stenting only in symptomatic cases with at least 50% carotid artery stenosis. It also reimburses for stenting in asymptomatic cases in patients at high risk with 80% or greater stenosis, but only if the patients are enrolled in ongoing clinical trials or registries.
CREST compared stenting with endarterectomy and provided important insights into each approach.1
BEFORE CREST
Endarterectomy is superior to medical therapy for symptomatic stenosis
First described in 1953, carotid endarterectomy became the most widely used invasive treatment for significant carotid stenosis.9 Several studies have described patient subsets that benefit from this procedure.
NASCET (the North American Symptomatic Carotid Endarterectomy Trial)10 assigned 2,226 patients with symptomatic stenosis (transient ischemic attack or stroke within the past 180 days) to medical management or endarterectomy.
Surgery was associated with a 65% lower rate of ipsilateral cerebral events in patients with 70% or greater stenosis.10 Surgery was also found to be superior in patients with moderate disease (50% to 69% stenosis), but the difference only approached statistical significance. In patients with stenosis of less than 50%, the outcomes were similar with endarterectomy and medical management.11
ECST (the European Carotid Surgery Trial)12 included a similar population of 3,024 patients. Those with high-grade disease (stenosis ≥ 80%) had significantly better outcomes with endarterectomy, but in those with stenosis less than 70%, surgery was no better than drug therapy.
Comment. NASCET and ECST taught us that endarterectomy is clearly superior to medical therapy in patients with severe symptomatic carotid disease. However, both trials excluded patients at high surgical risk, eg, those with severe coronary artery disease, kidney disease, or heart failure. Additionally, medical management was not aggressive by today’s standards in terms of control of blood pressure and hyperlipidemia, and this could have skewed the results in favor of carotid endarterectomy.
The case for carotid endarterectomy for asymptomatic stenosis
Endarterectomy has also been compared with drug therapy for asymp tomatic carotid artery stenosis in several trials.13–15
ACAS (the Asymptomatic Carotid Atherosclerosis Study)15 assigned 1,662 patients who had no symptoms and had at least 60% carotid artery stenosis to endarterectomy or to medical management, and found a relative risk reduction of 53% in favor of surgery.15
The Veterans Affairs Cooperative Study Group14 corroborated these results in 444 patients with asymptomatic stenosis of greater than 50%. Endarterectomy was associated with a 61% lower risk of transient ischemic attack, transient monocular blindness, or stroke compared with medical therapy. However, there was no statistically significant difference in rates of stroke or death at 30 days.14
ACST (the Asymptomatic Carotid Surgery Trial),13 the largest study to compare carotid endarterectomy with drug therapy for asymptomatic stenosis, randomized 3,120 patients to surgery or drug therapy. The net 5-year risk of stroke was 6.4% with endarterectomy vs 11.8% with drug therapy (P < .0001). The rate of fatal stroke was also lower with endarterectomy: 2.1% vs 4.2% (P = .006).13
Comment. The results of these and other studies of endarterectomy vs medical therapy may not be applicable to current practice, since medical therapy has evolved and the risks with current drug therapy are likely much lower than seen in these trials, some of which began 2 decades ago. Another problem with interpreting these trials is that they excluded surgically “high-risk” patients, which limits the generalizability of the findings to this particular patient population.
The American Heart Association and the American Stroke Association have, on the basis of these trials, recommended carotid endarterectomy in patients with7,8,16:
Ipsilateral, symptomatic carotid artery stenosis of 70% to 99% (class I, level of evidence A)
Symptomatic stenosis of 50% to 69%, depending on patient-specific factors such as age, sex, and comorbidities
High-grade asymptomatic carotid stenosis, if the patients are carefully selected and the surgery is performed by surgeons with procedural morbidity and mortality rates of less than 3% (class I, level of evidence A).
In all cases, treatment should be individualized according to the patient’s comorbid conditions and preferences, with a thorough discussion of risks and benefits (Table 1).7,8,16
The case for percutaneous intervention
While carotid endarterectomy is proven to be more efficacious than medical management in certain patient subsets, studies favoring surgery over medical therapy have been criticized because they excluded patients with significant comorbidities. In addition, surgery has been associated with significant cardiovascular events, wound complications, and cranial nerve damage, and it requires general anesthesia in most cases.12,17–19 These and other factors spurred the development of less invasive, percutaneous approaches for patients with substantial comorbidities.
So far, several trials have investigated carotid angioplasty with or without stents and with or without devices to capture distal emboli. This interest set the stage for CREST.20,21
Initial attempts at angioplasty without distal protection were not very successful. A meta-analysis of nonrandomized trials that included 714 patients from the initial 13 studies of angioplasty (with or without stenting) and 6,970 patients from 20 studies of carotid endarterectomy found angioplasty to be possibly associated with higher rates of stroke within 30 days of the procedure.20
With improvements in technology, routine use of embolic protection devices, more experience, and better selection of patients, the outcome of carotid stenting has improved. In fact, a meta-analysis comparing stenting without an embolic protection device (26 trials with 2,357 patients) vs stenting with an embolic protection device (11 trials with 839 patients) showed that embolic protection led to significantly better outcomes with fewer strokes—outcomes arguably similar to those of carotid endarterectomy.21
SAPPHIRE (the Stenting and Angioplasty With Protection in Patients at High Risk for Endarterectomy trial)22 was the only completed US trial until CREST that compared carotid artery stenting with distal protection against surgery. It included 334 high-risk patients with either symptomatic stenosis of 50% or greater or asymptomatic stenosis of 80% or greater.
The results suggested that the outcomes with stenting with embolic protection were in fact similar to those of endarterectomy, with possibly fewer complications.23 The benefit persisted up to 2 years.22
The US Food and Drug Administration (FDA), on the basis of these data, approved the use of stenting with distal protection for high-risk patients, and the CMS reimburses for symptomatic stenosis of 50% or greater and for asymptomatic stenosis of 80% or greater as long as the patient is enrolled in a registry.
SPACE (the Stent-Protected Angioplasty Versus Carotid Endarterectomy in Symptomatic Patients trial),24 conducted in Germany, included 1,214 patients with symptomatic stenosis of at least 50%. Results were similar in terms of the combined primary end point of stroke or death at 30 days. However, the results were not similar enough to prove that stenting is not inferior to surgery, according to preset study criteria.
EVA-3S (the Endarterectomy Versus Stenting in Patients With Symptomatic Severe Carotid Stenosis trial),25 in France, evaluated 527 patients with symptomatic carotid disease (stenosis ≥ 60%), but was terminated early due to significantly higher rates of death or stroke at 30 days in the stenting group.
Comment. SPACE and EVA-3S have been widely criticized for not mandating the use of an embolic protection device (used in 27% of cases in SPACE and in 91.9% of cases in EVA-3S). Questions were also raised about the experience level of the operators who performed the carotid stenting: up to 39% of the primary operators involved in stent placement were trainees.26 Also, myocardial infarction (MI), an important complication of carotid endarterectomy, was not included in the primary end point.
ICSS (the International Carotid Stenting Study)27 compared stenting with endarterectomy in 1,713 patients with symptomatic carotid stenosis of greater than 50%. The primary end point was the rate of fatal or disabling stroke at 3 years.
An interim safety analysis at 120 days of follow-up showed the primary end point had occurred in 4.0% of stenting cases vs 3.2% of endarterectomy cases, a difference that was not statistically significant (hazard ratio [HR] 1.28, 95% confidence interval [CI] 0.77–2.11). However, the risk of any stroke was higher with stenting, with a rate of 7.7% vs 4.1% in the surgical group—a statistically significant difference (HR 1.92, 95% CI 1.27–2.89).
In a substudy of ICSS,28 the investigators corroborated these findings, using magnetic resonance imaging to evaluate for new ischemic brain lesions periprocedurally. They found more new ischemic brain lesions in patients who underwent stenting than in patients who underwent surgery—a statistically significant finding.
Comment. ICSS had limitations: eg, it included only patients with symptoms, and the training for the stenting procedure was not standardized. Furthermore, the use of embolic protection devices was not mandated in stenting procedures.
Because of the controversial and incongruous findings of the above trials, there has been much anticipation for further large, appropriately conducted, randomized controlled trials such as CREST.
CREST STUDY DESIGN
CREST was a prospective, multicenter randomized controlled trial with blinded end point adjudication. Assignment to stenting or surgery occurred in a one-to-one fashion, and patients were stratified by medical center and symptomatic status.
Conducted at 108 sites in the United States and nine sites in Canada, CREST was supported by a grant from the National Institutes of Health and by the manufacturer of the catheter and stent delivery and embolic protection systems. The manufacturer’s representative held a nonvoting position on the executive committee and reviewed the manuscript of the results before submission.
CREST included patients with or without symptoms
CREST was initially designed to compare carotid artery stenting vs carotid endarterectomy in patients with symptoms, but enrollment was later extended to patients without symptoms.
Patients with symptoms were included if they had stenosis of at least 50% on angiography, at least 70% on ultrasonography, or at least 70% on computed tomographic angiography or magnetic resonance angiography if stenosis on ultrasonography was 50% to 69%. Carotid artery stenosis was considered symptomatic if the patient had a transient ischemic attack, amaurosis fugax, or minor disabling stroke in the hemisphere supplied by the target vessel within 180 days of randomization.
Patients without symptoms were eligible if they had at least 60% stenosis on angiography, at least 70% stenosis on ultrasonography, or at least 80% stenosis on computed tomographic angiography or magnetic resonance angiography if the stenosis was 50% to 69% on ultrasonography.
Other eligibility criteria included favorable anatomy and clinical stability for both stenting and surgical procedures.
Exclusion criteria were evolving stroke, history of major stroke, chronic or paroxysmal atrial fibrillation on anticoagulation therapy, MI within the previous 30 days, and unstable angina.
Patients received antiplatelet agents
Patients undergoing stenting received aspirin and clopidogrel (Plavix) before and up to 30 days after the procedure. Continuation of antiplatelet therapy was recommended beyond 1 month.
Patients undergoing endarterectomy received aspirin before surgery and continued to receive aspirin for at least 1 year.
Alternatives to aspirin in both groups were ticlopidine (Ticlid), clopidogrel, or aspirin with extended-release dipyridamole (Aggrenox).
End points: Stroke, MI, death
The primary end point was a composite of periprocedural clinical stroke (any type), MI, or death, and of ipsilateral stroke up to 4 years after the procedure. Secondary analyses were also planned for evaluation of treatment modification by age, symptom status, and sex.
Stroke was defined as any acute neurologic ischemic event lasting at least 24 hours with focal signs and symptoms.
Two separate definitions were applied to distinguish major stroke from nonmajor stroke. Major stroke was defined as a National Institutes of Health Stroke Scale (NIHSS) score greater than 9 or records suggesting that the event was a disabling stroke if admitted to another facility. Nonmajor stroke included an event that did not fit these criteria. The stroke review process was initiated with a significant neurologic event, a positive transient ischemia attack or stroke questionnaire, or a two-point or greater increase in the NIHSS score.
MI was defined as a combination of an elevation of cardiac enzymes to at least twice the laboratory upper limit of normal, as well as clinical signs suggesting MI or electrocardiographic evidence of ischemia.29
Stroke was adjudicated by two independent neurologists, and MI was adjudicated by two independent cardiologists blinded to treatment group assignment.
The Rankin scale, the transient ischemic attack and stroke questionnaire, and the Medical Outcomes Survey were also used to assess for disability and quality of life in long-term follow-up.
Intention-to-treat analysis
Intention-to-treat survival analysis was used along with time-to-event statistical modeling with adjustment for major baseline covariates. Differences in outcomes were assessed, and a noninferiority analysis was performed. Kaplan-Meier estimates were constructed of the proportion of patients remaining free of the composite end point at 30 days, 6 months, 1 year, and annually thereafter, and of the associated confidence intervals. The hazard ratios between groups were estimated after adjustment for important covariates.
Most patients enrolled were available for analysis
From December 2000 to July 2008, 2,522 patients were enrolled; 1,271 were assigned to stenting, and 1,251 were assigned to surgery. After randomization, 2.8% of the patients assigned to stenting withdrew consent, 5.7% underwent surgery, and 2.6% were lost to follow-up. Of those assigned to surgery, 5.1% withdrew consent, 1.0% underwent stenting, and 3.8% were lost to follow-up.
A ‘conventional-risk’ patient population
The trial sought to include a “conventional-risk” patient population to make the study more applicable to real-world practice. The mean age was 69 years in both groups. Of the 2,522 patients enrolled:
35% were women
47% had asymptomatic carotid disease
86% had carotid stenosis of 70% or greater
86% had hypertension
30% had diabetes mellitus
83% had hyperlipidemia
26% were current smokers
42% had a history of cardiovascular disease
21% had undergone coronary artery bypass grafting surgery.
The only statistically significant difference in measured baseline variables between the two treatment groups was a slightly higher rate of dyslipidemia in the group undergoing surgery.
The interventionalists and surgeons were highly experienced
Operators performing stenting underwent a lead-in phase of training, with close supervision and scrutiny before eligibility. Of patients undergoing stenting, 96.1% also received an embolic protection device. Antiplatelet therapy was continued in 99% of the patients.
The surgeons performing endarterectomy were experienced and had documented low complication rates. General anesthesia was used in 90% of surgical patients. Shunts were used during surgery in 57%, and patches were used in 62%. After endarterectomy, 91% of the patients received antiplatelet therapy.
CREST STUDY RESULTS: STENTING WAS AS GOOD AS SURGERY
Periprocedural outcomes
Stroke, MI, or death: 5.2% with stenting vs 4.5% with surgery, HR 1.18, 95% CI 0.82–1.68, P = .38
Stroke: 4.1% vs 2.3%, HR 1.79, 95% CI 1.14–2.82, P = .01
Major ipsilateral stroke: 0.9% vs 0.3%, HR 2.67, 95% CI 0.85–8.40, P = .09.
MI: 1.1% vs 2.3%, HR 0.50, 95% CI 0.26–0.94, P = .03
Cranial nerve palsy: 0.3% vs 4.8%, HR 0.07, 95% CI 0.02–0.18, P < .0001 (Table 2).
Outcomes at 4 years
Brott TG, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23. Copyright 2010, Massachusetts Medical Society. All rights reserved.
Figure 2. Kaplan-Meier analysis of the primary outcome (stroke, myocardial infarction, or death during the periprocedural period or any ipsilateral stroke within 4 years after randomization) for patients undergoing carotid artery stenting or carotid endarterectomy.The primary end point (periprocedural stroke, MI, or death, or ipsilateral stroke within 4 years after the procedure): 7.2% with stenting vs 6.8% with surgery, HR 1.11, 95% CI 0.81–1.51, P = .51. A Kaplan-Meier analysis showed similar findings with statistically similar outcomes (Figure 2).
Ipsilateral stroke: 2.0% vs 2.4%, HR 0.94, 95% CI 0.50–1.76, P = .85.
The primary outcome was analyzed for interactions of baseline variables, and no effect was detected for symptomatic status or sex. There was a suggestion of an interaction with age, with older patients (over age 70) benefiting more from endarterectomy.
Quality-of-life indices showed that both major and minor strokes were likely to produce long-term physical limitations, with minor stroke associated with worse mental and physical health at 1 year. The effect of periprocedural MI on long-term physical and mental health was less certain. The increased incidence of cranial nerve palsy noted with endarterectomy has been found before and has had no effect on quality of life.
WHAT DO THE CREST FINDINGS MEAN?
CREST is the largest trial to date to compare stenting and surgery. It is an important addition to the literature, not only because of its size, but also because it focused on a real-world patient population. For this reason, its results are more applicable to patients seen in primary care clinics, ie, with peripheral vascular disease, coronary artery disease, diabetes mellitus, hypertension, and smoking.
As noted, previous studies of endarterectomy had strict inclusion and exclusion criteria, which selected against patients at high surgical risk. Therefore, the CREST findings are of greater relevance when comparing stenting and endarterectomy.
Periprocedural and long-term neurologic outcomes
CREST showed similar findings for the composite end point of periprocedural stroke, death, or MI (ie, within 30 days of the procedure) and long-term stroke, establishing similar outcomes in patients undergoing stenting and surgery.
However, an analysis of the individual components of the composite end point showed significant differences between the two treatments. The risk of ipsilateral periprocedural stroke was higher with stenting; these events were defined as nonmajor by NIHSS criteria. The risk of contralateral stroke was similar and low with each treatment.
While the increased risk of periprocedural ipsilateral stroke was not synonymous with an increased risk of major stroke, post hoc analysis showed that any stroke was associated with decreased physical and mental health at 1 year. Therefore, patients who had even a minor stroke did worse from a physical and mental standpoint, a finding that argues for the superiority of surgery in selected patients at risk of periprocedural stroke.
If periprocedural stroke is excluded, the risk of long-term ipsilateral stroke was similar for each treatment, and extremely low (2% for stenting, 2.4% for surgery). Despite this, given the importance of periprocedural minor and major stroke, better predictive models are needed to identify patients at risk of procedural neurologic events. These prediction models will allow better patient selection.
The CREST data and medical therapy
The rates of stroke in this trial were similar to those observed with current medical treatment (approximately 1% per year), especially for patients with asymptomatic disease. Such findings introduce fresh controversy in the necessity of performing either procedure for this patient subset and may lead to further studies evaluating current medical therapy vs intervention.
Periprocedural myocardial infarction
Vascular surgery has long been associated with high cardiovascular risk, especially an increased risk of periprocedural MI.30 Findings from CREST provide further evidence of the risk of MI with endarterectomy in a real-world patient population. Given the evidence of a strong correlation between periprocedural cardiac enzyme elevations and adverse outcomes, the increased incidence of periprocedural MI is worrisome.31 As with risk assessment for periprocedural stroke, better predictive models are needed for patients at risk of cardiovascular events during endarterectomy.
Procedural complications
Carotid endarterectomy entails incisions in the neck with disruption of tissue planes, as opposed to catheter entry site wounds with stenting. The more invasive nature of endarterectomy thus carries a higher risk of wound complications. In fact, in the NASCET trial, the risk of wound complications was 9.3%.10,19 In CREST, surgery carried a higher risk of wound complications compared with stenting (42 vs 0 cases), although stenting involved more periprocedural transfusions, presumably due to retroperitoneal bleeding in four patients.
Use of general anesthesia is also associated with adverse outcomes.17,18 In CREST, 90% of endarterectomy procedures required general anesthesia, whereas none of the stenting procedures required this.
Cranial nerve palsy is an often overlooked but real complication after these procedures. Cranial nerve palsies can lead to vocal, swallowing, and sensory problems that can have a transient or permanent impact on quality of life. In CREST, as in EVA-3S, SAPPHIRE, and ICSS, this risk was substantially higher with surgery,23,25,27 although the long-term consequences of these palsies were not found to affect quality of life at 1 year of follow-up.
HOW CREST FINDINGS COMPARE WITH PREVIOUS STUDIES
Patients in CREST enjoyed overall better outcomes than in previous studies. In earlier trials of surgery vs medical therapy, the rates of adverse outcomes were higher than in CREST. In NASCET, the risk of ipsilateral stroke was 9% with surgery, with 2.5% being fatal or disabling strokes.10 In the ECST, rates of major stroke or death with endarterectomy were 7.0% within 30 days of surgery and 37.0% at a mean follow-up of 6.1 years.12
In earlier studies of surgery vs stenting, outcomes at 30 days were also substantially worse than those in CREST. In the EVA-3S trial, the 30-day incidence of stroke or death was 3.9% after surgery and 9.6% after stenting. These findings were similar at 6 months in EVA-3S, with a 6.1% rate of adverse events after surgery and 11.7% after stenting.25 In the SAPPHIRE trial, the cumulative incidence of stroke and death at 1 year was 21.4% for surgery and 13.6% for stenting.23
Overall, the CREST results show better outcomes than in previous trials. This may be due to improvements in technical aspects of the interventions and to more aggressive drug therapy. Also, because of the high number of patients enrolled in CREST, surgeons and interventionalists were required to meet eligibility criteria, which could have contributed to the improved outcomes.32
CREST was also unique in that stenting was done with an embolic protection device whenever possible, and this also likely had an impact on outcomes.
The CREST data suggest that interventions for carotid artery stenosis should only be performed by rigorously trained, experienced personnel at high-volume centers, as this provided lower event rates compared with previous studies. Additional data should also help identify those at risk of periprocedural stroke and MI, thereby helping to match the patient to the most appropriate procedure. The pros and cons of surgery and stenting are shown in Table 3.1,10,23,25,27
CREST vs ICSS
CREST and ICSS, published within a few months of each other, seem to have arrived at entirely different conclusions. As both studies are well-designed randomized controlled trials, these distinct results have yielded much controversy. However, closer scrutiny sheds light as to why the results may be different.
While ICSS focused only on patients with symptoms, CREST also included those without symptoms. The difference in patient populations is itself enough to account for the different outcomes.
Also, the interim analysis of ICSS was at 120 days, which makes periprocedural events a more dominant factor in outcomes, whereas these events likely do not last into the long term, as was the case in CREST. Analysis of the ICSS data at a later follow-up date may show results more similar to those of CREST.
The design of ICSS was also different than CREST. In ICSS, the use of an embolic protection device in stenting was not mandated, and the study lacked a lead-in phase of intensive training for those performing stenting. Furthermore, MI was adjudicated only when clinically recognized, which is different than the more rigorous method used in CREST.
Yet despite these differences, CREST and ICSS shed light on a controversial area of carotid stenosis management, and both studies boasted low rates of periprocedural complications. Clinicians should keep in mind the inclusion criteria and the technical specificities of these trials in order to explain to patients the risks and benefits of stenting and surgery, and to arrive at a decision together.
Limitations
The results of CREST should also be reviewed carefully due to a number of limitations. The study began in 2000 with symptomatic patients only, and began enrolling asymptomatic patients in 2005, so that the methodology of the study was changed midway. However, the investigators performed a subgroup analysis to distinguish between outcomes of the symptomatic and the asymptomatic groups and found no statistical interaction for the primary end point based on symptom status.
Despite careful patient selection, many of the predictors of adverse outcomes with stenting, such as lesion length, level of calcification, and lesion location, were not accounted for in the earlier days of enrollment. This may have had an impact on the incidence of stroke in patients enrolled in the early years of the trial. We await the analysis of predictors of perioperative stroke from CREST.
TAKE-HOME POINTS AND FUTURE DIRECTIONS
The CREST findings show that outcomes with stenting are similar to those with surgery in both the short term and the long term, and that the choice of management should be individualized. Each patient’s risk of MI and stroke should be considered based on a variety of factors, including the severity of coronary artery disease, the length of the carotid lesion, the level of calcification, the location of the lesion, and aortic atheroma. The treatment should be selected after also taking into account the patient’s preference and the available expertise, and only after a comprehensive discussion with the patient.
References
Brott TG, Hobson RW, Howard G, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med2010; 363:11–23.
Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation2006; 113:e85–e151.
Rosamond WD, Folsom AR, Chambless LE, et al. Stroke incidence and survival among middle-aged adults: 9-year follow-up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke1999; 30:736–743.
Chaturvedi S, Bruno A, Feasby T, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Carotid endarterectomy—an evidence-based review: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology2005; 65:794–801.
Howell GM, Makaroun MS, Chaer RA. Current management of extracranial carotid occlusive disease. J Am Coll Surg2009; 208:442–453.
Barnett HJ, Gunton RW, Eliasziw M, et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA2000; 283:1429–1436.
Biller J, Feinberg WM, Castaldo JE, et al. Guidelines for carotid endarterectomy: a statement for healthcare professionals from a Special Writing Group of the Stroke Council, American Heart Association. Circulation1998; 97:501–509.
Goldstein LB, Adams R, Alberts MJ, et al; American Heart Association; American Stroke Association Stroke Council. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation2006; 113:e873–e923.
Strully KJ, Hurwitt ES, Blankenberg HW. Thrombo-endarterectomy for thrombosis of the internal carotid artery in the neck. J Neurosurg1953; 10:474–482.
Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med1991; 325:445–453.
Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med1998; 339:1415–1425.
Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet1998; 351:1379–1387.
Halliday A, Mansfield A, Marro J, et al; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomised controlled trial. Lancet2004; 363:1491–1502.
Hobson RW, Weiss DG, Fields WS, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med1993; 328:221–227.
Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. JAMA1995; 273:1421–1428.
Sacco RL, Adams R, Albers G, et al; American Heart Association/American Stroke Association Council on Stroke; Council on Cardiovascular Radiology and Intervention; American Academy of Neurology. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Circulation2006; 113:e409–e449.
Watts K, Lin PH, Bush RL, et al. The impact of anesthetic modality on the outcome of carotid endarterectomy. Am J Surg2004; 188:741–747.
Weber CF, Friedl H, Hueppe M, et al. Impact of general versus local anesthesia on early postoperative cognitive dysfunction following carotid endarterectomy: GALA Study Subgroup Analysis. World J Surg2009; 33:1526–1532.
Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke1999; 30:1751–1758.
Golledge J, Mitchell A, Greenhalgh RM, Davies AH. Systematic comparison of the early outcome of angioplasty and endarterectomy for symptomatic carotid artery disease. Stroke2000; 31:1439–1443.
Kastrup A, Gröschel K, Krapf H, Brehm BR, Dichgans J, Schulz JB. Early outcome of carotid angioplasty and stenting with and without cerebral protection devices: a systematic review of the literature. Stroke2003; 34:813–819.
Gurm HS, Yadav JS, Fayad P, et al; SAPPHIRE Investigators. Long-term results of carotid stenting versus endarterectomy in high-risk patients. N Engl J Med2008; 358:1572–1579.
Yadav JS, Wholey MH, Kuntz RE, et al; Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy Investigators. Protected carotid-artery stenting versus endarterectomy in high-risk patients. N Engl J Med2004; 351:1493–1501.
Eckstein HH, Ringleb P, Allenberg JR, et al. Results of the Stent-Protected Angioplasty versus Carotid Endarterectomy (SPACE) study to treat symptomatic stenoses at 2 years: a multinational, prospective, randomised trial. Lancet Neurol2008; 7:893–902.
Mas JL, Chatellier G, Beyssen B, et al; EVA-3S Investigators. Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med2006; 355:1660–1771.
Roffi M, Sievert H, Gray WA, et al. Carotid artery stenting versus surgery: adequate comparisons?Lancet Neurol2010; 9:339–341.
International Carotid Stenting Study Investigators; Ederle J, Dobson J, Featherstone RL, et al. Carotid artery stenting compared with endarterectomy in patients with symptomatic carotid stenosis (International Carotid Stenting Study): an interim analysis of a randomised controlled trial. Lancet2010; 375:985–997.
Bonati LH, Jongen LM, Haller S, et al; ICSS-MRI study group. New ischaemic brain lesions on MRI after stenting or endarterectomy for symptomatic carotid stenosis: a sub-study of the International Carotid Stenting Study (ICSS). Lancet Neurol2010; 9:353–362.
Sheffet AJ, Roubin G, Howard G, et al. Design of the Carotid Revascularization Endarterectomy vs. Stenting Trial (CREST). Int J Stroke2010; 5:40–46.
Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery. J Am Coll Cardiol2007; 50:e159–e241.
Bhatt DL, Topol EJ. Does creatinine kinase-MB elevation after percutaneous coronary intervention predict outcomes in 2005? Periprocedural cardiac enzyme elevation predicts adverse outcomes. Circulation2005; 112:906–915.
Hobson RW, Howard VJ, Roubin GS, et al; CREST. Credentialing of surgeons as interventionalists for carotid artery stenting: experience from the lead-in phase of CREST. J Vasc Surg2004; 40:952–957.
Olcay Aksoy, MD Department of Cardiovascular Medicine, Cleveland Clinic
Samir R. Kapadia, MD Department of Cardiovascular Medicine, Cleveland Clinic
Christopher Bajzer, MD Department of Cardiovascular Medicine, Cleveland Clinic
Wayne M. Clark, MD Department of Neurology, Oregon Health & Science University, Portland; Investigator, Carotid Revascularization Endarterectomy Versus Stenting Trial (CREST)
Mehdi H. Shishehbor, DO, MPH, PhD Department of Cardiovascular Medicine, Cleveland Clinic
Address: Mehdi H. Shishehbor, DO, MPH, Heart & Vascular Institute, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Shishehbor has disclosed teaching and speaking for Abbott Vascular.
Olcay Aksoy, MD Department of Cardiovascular Medicine, Cleveland Clinic
Samir R. Kapadia, MD Department of Cardiovascular Medicine, Cleveland Clinic
Christopher Bajzer, MD Department of Cardiovascular Medicine, Cleveland Clinic
Wayne M. Clark, MD Department of Neurology, Oregon Health & Science University, Portland; Investigator, Carotid Revascularization Endarterectomy Versus Stenting Trial (CREST)
Mehdi H. Shishehbor, DO, MPH, PhD Department of Cardiovascular Medicine, Cleveland Clinic
Address: Mehdi H. Shishehbor, DO, MPH, Heart & Vascular Institute, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Shishehbor has disclosed teaching and speaking for Abbott Vascular.
Author and Disclosure Information
Olcay Aksoy, MD Department of Cardiovascular Medicine, Cleveland Clinic
Samir R. Kapadia, MD Department of Cardiovascular Medicine, Cleveland Clinic
Christopher Bajzer, MD Department of Cardiovascular Medicine, Cleveland Clinic
Wayne M. Clark, MD Department of Neurology, Oregon Health & Science University, Portland; Investigator, Carotid Revascularization Endarterectomy Versus Stenting Trial (CREST)
Mehdi H. Shishehbor, DO, MPH, PhD Department of Cardiovascular Medicine, Cleveland Clinic
Address: Mehdi H. Shishehbor, DO, MPH, Heart & Vascular Institute, J3-5, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; e-mail [email protected]
Dr. Shishehbor has disclosed teaching and speaking for Abbott Vascular.
For patients with carotid artery stenosis, percutaneous intervention with stenting is as good as surgery (carotid endarterectomy). This was the major finding of the recently completed Carotid Revascularization Endarterectomy Versus Stenting Trial (CREST)1—with some qualifications.
CREST is the latest in a series of clinical trials of treatment of carotid stenosis that have generated reams of numbers and much debate. The topic of surgery vs percutaneous intervention is a moving target, as techniques evolve and improve. We believe the CREST results are valuable and should help inform decisions about treatment in the “real world.”
In this article, we offer a critical review of CREST, with a careful evaluation of its methods, results, and conclusions.
AN EVOLVING FIELD
Despite improvements in diagnosis and management, stroke remains one of the leading causes of morbidity and death in the United States, with an annual incidence of 780,000 cases and 270,000 deaths.2,3
Figure 1. Carotid endarterectomy has long been an established treatment in selected patients with symptomatic carotid artery stenosis of 50% or greater or asymptomatic stenosis of 60% or greater. However, percutaneous carotid artery angioplasty with stenting and placement of an embolic protection device is gaining ground as a reasonable, safe, less invasive alternative.From 10% to 30% of ischemic strokes are due to emboli from the carotid arteries.4–6 Carotid endarterectomy is an established treatment in selected patients with symptomatic carotid stenosis of 50% or greater or asymptomatic stenosis of 60% or greater.7,8 However, percutaneous techniques such as carotid artery angioplasty with stenting have improved, making them a viable, less invasive option (Figure 1).
Randomized trials of stenting have had mixed results, leading the Centers for Medicare and Medicaid Services (CMS) to adopt strict reimbursement policies. Currently, CMS reimburses for stenting only in symptomatic cases with at least 50% carotid artery stenosis. It also reimburses for stenting in asymptomatic cases in patients at high risk with 80% or greater stenosis, but only if the patients are enrolled in ongoing clinical trials or registries.
CREST compared stenting with endarterectomy and provided important insights into each approach.1
BEFORE CREST
Endarterectomy is superior to medical therapy for symptomatic stenosis
First described in 1953, carotid endarterectomy became the most widely used invasive treatment for significant carotid stenosis.9 Several studies have described patient subsets that benefit from this procedure.
NASCET (the North American Symptomatic Carotid Endarterectomy Trial)10 assigned 2,226 patients with symptomatic stenosis (transient ischemic attack or stroke within the past 180 days) to medical management or endarterectomy.
Surgery was associated with a 65% lower rate of ipsilateral cerebral events in patients with 70% or greater stenosis.10 Surgery was also found to be superior in patients with moderate disease (50% to 69% stenosis), but the difference only approached statistical significance. In patients with stenosis of less than 50%, the outcomes were similar with endarterectomy and medical management.11
ECST (the European Carotid Surgery Trial)12 included a similar population of 3,024 patients. Those with high-grade disease (stenosis ≥ 80%) had significantly better outcomes with endarterectomy, but in those with stenosis less than 70%, surgery was no better than drug therapy.
Comment. NASCET and ECST taught us that endarterectomy is clearly superior to medical therapy in patients with severe symptomatic carotid disease. However, both trials excluded patients at high surgical risk, eg, those with severe coronary artery disease, kidney disease, or heart failure. Additionally, medical management was not aggressive by today’s standards in terms of control of blood pressure and hyperlipidemia, and this could have skewed the results in favor of carotid endarterectomy.
The case for carotid endarterectomy for asymptomatic stenosis
Endarterectomy has also been compared with drug therapy for asymp tomatic carotid artery stenosis in several trials.13–15
ACAS (the Asymptomatic Carotid Atherosclerosis Study)15 assigned 1,662 patients who had no symptoms and had at least 60% carotid artery stenosis to endarterectomy or to medical management, and found a relative risk reduction of 53% in favor of surgery.15
The Veterans Affairs Cooperative Study Group14 corroborated these results in 444 patients with asymptomatic stenosis of greater than 50%. Endarterectomy was associated with a 61% lower risk of transient ischemic attack, transient monocular blindness, or stroke compared with medical therapy. However, there was no statistically significant difference in rates of stroke or death at 30 days.14
ACST (the Asymptomatic Carotid Surgery Trial),13 the largest study to compare carotid endarterectomy with drug therapy for asymptomatic stenosis, randomized 3,120 patients to surgery or drug therapy. The net 5-year risk of stroke was 6.4% with endarterectomy vs 11.8% with drug therapy (P < .0001). The rate of fatal stroke was also lower with endarterectomy: 2.1% vs 4.2% (P = .006).13
Comment. The results of these and other studies of endarterectomy vs medical therapy may not be applicable to current practice, since medical therapy has evolved and the risks with current drug therapy are likely much lower than seen in these trials, some of which began 2 decades ago. Another problem with interpreting these trials is that they excluded surgically “high-risk” patients, which limits the generalizability of the findings to this particular patient population.
The American Heart Association and the American Stroke Association have, on the basis of these trials, recommended carotid endarterectomy in patients with7,8,16:
Ipsilateral, symptomatic carotid artery stenosis of 70% to 99% (class I, level of evidence A)
Symptomatic stenosis of 50% to 69%, depending on patient-specific factors such as age, sex, and comorbidities
High-grade asymptomatic carotid stenosis, if the patients are carefully selected and the surgery is performed by surgeons with procedural morbidity and mortality rates of less than 3% (class I, level of evidence A).
In all cases, treatment should be individualized according to the patient’s comorbid conditions and preferences, with a thorough discussion of risks and benefits (Table 1).7,8,16
The case for percutaneous intervention
While carotid endarterectomy is proven to be more efficacious than medical management in certain patient subsets, studies favoring surgery over medical therapy have been criticized because they excluded patients with significant comorbidities. In addition, surgery has been associated with significant cardiovascular events, wound complications, and cranial nerve damage, and it requires general anesthesia in most cases.12,17–19 These and other factors spurred the development of less invasive, percutaneous approaches for patients with substantial comorbidities.
So far, several trials have investigated carotid angioplasty with or without stents and with or without devices to capture distal emboli. This interest set the stage for CREST.20,21
Initial attempts at angioplasty without distal protection were not very successful. A meta-analysis of nonrandomized trials that included 714 patients from the initial 13 studies of angioplasty (with or without stenting) and 6,970 patients from 20 studies of carotid endarterectomy found angioplasty to be possibly associated with higher rates of stroke within 30 days of the procedure.20
With improvements in technology, routine use of embolic protection devices, more experience, and better selection of patients, the outcome of carotid stenting has improved. In fact, a meta-analysis comparing stenting without an embolic protection device (26 trials with 2,357 patients) vs stenting with an embolic protection device (11 trials with 839 patients) showed that embolic protection led to significantly better outcomes with fewer strokes—outcomes arguably similar to those of carotid endarterectomy.21
SAPPHIRE (the Stenting and Angioplasty With Protection in Patients at High Risk for Endarterectomy trial)22 was the only completed US trial until CREST that compared carotid artery stenting with distal protection against surgery. It included 334 high-risk patients with either symptomatic stenosis of 50% or greater or asymptomatic stenosis of 80% or greater.
The results suggested that the outcomes with stenting with embolic protection were in fact similar to those of endarterectomy, with possibly fewer complications.23 The benefit persisted up to 2 years.22
The US Food and Drug Administration (FDA), on the basis of these data, approved the use of stenting with distal protection for high-risk patients, and the CMS reimburses for symptomatic stenosis of 50% or greater and for asymptomatic stenosis of 80% or greater as long as the patient is enrolled in a registry.
SPACE (the Stent-Protected Angioplasty Versus Carotid Endarterectomy in Symptomatic Patients trial),24 conducted in Germany, included 1,214 patients with symptomatic stenosis of at least 50%. Results were similar in terms of the combined primary end point of stroke or death at 30 days. However, the results were not similar enough to prove that stenting is not inferior to surgery, according to preset study criteria.
EVA-3S (the Endarterectomy Versus Stenting in Patients With Symptomatic Severe Carotid Stenosis trial),25 in France, evaluated 527 patients with symptomatic carotid disease (stenosis ≥ 60%), but was terminated early due to significantly higher rates of death or stroke at 30 days in the stenting group.
Comment. SPACE and EVA-3S have been widely criticized for not mandating the use of an embolic protection device (used in 27% of cases in SPACE and in 91.9% of cases in EVA-3S). Questions were also raised about the experience level of the operators who performed the carotid stenting: up to 39% of the primary operators involved in stent placement were trainees.26 Also, myocardial infarction (MI), an important complication of carotid endarterectomy, was not included in the primary end point.
ICSS (the International Carotid Stenting Study)27 compared stenting with endarterectomy in 1,713 patients with symptomatic carotid stenosis of greater than 50%. The primary end point was the rate of fatal or disabling stroke at 3 years.
An interim safety analysis at 120 days of follow-up showed the primary end point had occurred in 4.0% of stenting cases vs 3.2% of endarterectomy cases, a difference that was not statistically significant (hazard ratio [HR] 1.28, 95% confidence interval [CI] 0.77–2.11). However, the risk of any stroke was higher with stenting, with a rate of 7.7% vs 4.1% in the surgical group—a statistically significant difference (HR 1.92, 95% CI 1.27–2.89).
In a substudy of ICSS,28 the investigators corroborated these findings, using magnetic resonance imaging to evaluate for new ischemic brain lesions periprocedurally. They found more new ischemic brain lesions in patients who underwent stenting than in patients who underwent surgery—a statistically significant finding.
Comment. ICSS had limitations: eg, it included only patients with symptoms, and the training for the stenting procedure was not standardized. Furthermore, the use of embolic protection devices was not mandated in stenting procedures.
Because of the controversial and incongruous findings of the above trials, there has been much anticipation for further large, appropriately conducted, randomized controlled trials such as CREST.
CREST STUDY DESIGN
CREST was a prospective, multicenter randomized controlled trial with blinded end point adjudication. Assignment to stenting or surgery occurred in a one-to-one fashion, and patients were stratified by medical center and symptomatic status.
Conducted at 108 sites in the United States and nine sites in Canada, CREST was supported by a grant from the National Institutes of Health and by the manufacturer of the catheter and stent delivery and embolic protection systems. The manufacturer’s representative held a nonvoting position on the executive committee and reviewed the manuscript of the results before submission.
CREST included patients with or without symptoms
CREST was initially designed to compare carotid artery stenting vs carotid endarterectomy in patients with symptoms, but enrollment was later extended to patients without symptoms.
Patients with symptoms were included if they had stenosis of at least 50% on angiography, at least 70% on ultrasonography, or at least 70% on computed tomographic angiography or magnetic resonance angiography if stenosis on ultrasonography was 50% to 69%. Carotid artery stenosis was considered symptomatic if the patient had a transient ischemic attack, amaurosis fugax, or minor disabling stroke in the hemisphere supplied by the target vessel within 180 days of randomization.
Patients without symptoms were eligible if they had at least 60% stenosis on angiography, at least 70% stenosis on ultrasonography, or at least 80% stenosis on computed tomographic angiography or magnetic resonance angiography if the stenosis was 50% to 69% on ultrasonography.
Other eligibility criteria included favorable anatomy and clinical stability for both stenting and surgical procedures.
Exclusion criteria were evolving stroke, history of major stroke, chronic or paroxysmal atrial fibrillation on anticoagulation therapy, MI within the previous 30 days, and unstable angina.
Patients received antiplatelet agents
Patients undergoing stenting received aspirin and clopidogrel (Plavix) before and up to 30 days after the procedure. Continuation of antiplatelet therapy was recommended beyond 1 month.
Patients undergoing endarterectomy received aspirin before surgery and continued to receive aspirin for at least 1 year.
Alternatives to aspirin in both groups were ticlopidine (Ticlid), clopidogrel, or aspirin with extended-release dipyridamole (Aggrenox).
End points: Stroke, MI, death
The primary end point was a composite of periprocedural clinical stroke (any type), MI, or death, and of ipsilateral stroke up to 4 years after the procedure. Secondary analyses were also planned for evaluation of treatment modification by age, symptom status, and sex.
Stroke was defined as any acute neurologic ischemic event lasting at least 24 hours with focal signs and symptoms.
Two separate definitions were applied to distinguish major stroke from nonmajor stroke. Major stroke was defined as a National Institutes of Health Stroke Scale (NIHSS) score greater than 9 or records suggesting that the event was a disabling stroke if admitted to another facility. Nonmajor stroke included an event that did not fit these criteria. The stroke review process was initiated with a significant neurologic event, a positive transient ischemia attack or stroke questionnaire, or a two-point or greater increase in the NIHSS score.
MI was defined as a combination of an elevation of cardiac enzymes to at least twice the laboratory upper limit of normal, as well as clinical signs suggesting MI or electrocardiographic evidence of ischemia.29
Stroke was adjudicated by two independent neurologists, and MI was adjudicated by two independent cardiologists blinded to treatment group assignment.
The Rankin scale, the transient ischemic attack and stroke questionnaire, and the Medical Outcomes Survey were also used to assess for disability and quality of life in long-term follow-up.
Intention-to-treat analysis
Intention-to-treat survival analysis was used along with time-to-event statistical modeling with adjustment for major baseline covariates. Differences in outcomes were assessed, and a noninferiority analysis was performed. Kaplan-Meier estimates were constructed of the proportion of patients remaining free of the composite end point at 30 days, 6 months, 1 year, and annually thereafter, and of the associated confidence intervals. The hazard ratios between groups were estimated after adjustment for important covariates.
Most patients enrolled were available for analysis
From December 2000 to July 2008, 2,522 patients were enrolled; 1,271 were assigned to stenting, and 1,251 were assigned to surgery. After randomization, 2.8% of the patients assigned to stenting withdrew consent, 5.7% underwent surgery, and 2.6% were lost to follow-up. Of those assigned to surgery, 5.1% withdrew consent, 1.0% underwent stenting, and 3.8% were lost to follow-up.
A ‘conventional-risk’ patient population
The trial sought to include a “conventional-risk” patient population to make the study more applicable to real-world practice. The mean age was 69 years in both groups. Of the 2,522 patients enrolled:
35% were women
47% had asymptomatic carotid disease
86% had carotid stenosis of 70% or greater
86% had hypertension
30% had diabetes mellitus
83% had hyperlipidemia
26% were current smokers
42% had a history of cardiovascular disease
21% had undergone coronary artery bypass grafting surgery.
The only statistically significant difference in measured baseline variables between the two treatment groups was a slightly higher rate of dyslipidemia in the group undergoing surgery.
The interventionalists and surgeons were highly experienced
Operators performing stenting underwent a lead-in phase of training, with close supervision and scrutiny before eligibility. Of patients undergoing stenting, 96.1% also received an embolic protection device. Antiplatelet therapy was continued in 99% of the patients.
The surgeons performing endarterectomy were experienced and had documented low complication rates. General anesthesia was used in 90% of surgical patients. Shunts were used during surgery in 57%, and patches were used in 62%. After endarterectomy, 91% of the patients received antiplatelet therapy.
CREST STUDY RESULTS: STENTING WAS AS GOOD AS SURGERY
Periprocedural outcomes
Stroke, MI, or death: 5.2% with stenting vs 4.5% with surgery, HR 1.18, 95% CI 0.82–1.68, P = .38
Stroke: 4.1% vs 2.3%, HR 1.79, 95% CI 1.14–2.82, P = .01
Major ipsilateral stroke: 0.9% vs 0.3%, HR 2.67, 95% CI 0.85–8.40, P = .09.
MI: 1.1% vs 2.3%, HR 0.50, 95% CI 0.26–0.94, P = .03
Cranial nerve palsy: 0.3% vs 4.8%, HR 0.07, 95% CI 0.02–0.18, P < .0001 (Table 2).
Outcomes at 4 years
Brott TG, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23. Copyright 2010, Massachusetts Medical Society. All rights reserved.
Figure 2. Kaplan-Meier analysis of the primary outcome (stroke, myocardial infarction, or death during the periprocedural period or any ipsilateral stroke within 4 years after randomization) for patients undergoing carotid artery stenting or carotid endarterectomy.The primary end point (periprocedural stroke, MI, or death, or ipsilateral stroke within 4 years after the procedure): 7.2% with stenting vs 6.8% with surgery, HR 1.11, 95% CI 0.81–1.51, P = .51. A Kaplan-Meier analysis showed similar findings with statistically similar outcomes (Figure 2).
Ipsilateral stroke: 2.0% vs 2.4%, HR 0.94, 95% CI 0.50–1.76, P = .85.
The primary outcome was analyzed for interactions of baseline variables, and no effect was detected for symptomatic status or sex. There was a suggestion of an interaction with age, with older patients (over age 70) benefiting more from endarterectomy.
Quality-of-life indices showed that both major and minor strokes were likely to produce long-term physical limitations, with minor stroke associated with worse mental and physical health at 1 year. The effect of periprocedural MI on long-term physical and mental health was less certain. The increased incidence of cranial nerve palsy noted with endarterectomy has been found before and has had no effect on quality of life.
WHAT DO THE CREST FINDINGS MEAN?
CREST is the largest trial to date to compare stenting and surgery. It is an important addition to the literature, not only because of its size, but also because it focused on a real-world patient population. For this reason, its results are more applicable to patients seen in primary care clinics, ie, with peripheral vascular disease, coronary artery disease, diabetes mellitus, hypertension, and smoking.
As noted, previous studies of endarterectomy had strict inclusion and exclusion criteria, which selected against patients at high surgical risk. Therefore, the CREST findings are of greater relevance when comparing stenting and endarterectomy.
Periprocedural and long-term neurologic outcomes
CREST showed similar findings for the composite end point of periprocedural stroke, death, or MI (ie, within 30 days of the procedure) and long-term stroke, establishing similar outcomes in patients undergoing stenting and surgery.
However, an analysis of the individual components of the composite end point showed significant differences between the two treatments. The risk of ipsilateral periprocedural stroke was higher with stenting; these events were defined as nonmajor by NIHSS criteria. The risk of contralateral stroke was similar and low with each treatment.
While the increased risk of periprocedural ipsilateral stroke was not synonymous with an increased risk of major stroke, post hoc analysis showed that any stroke was associated with decreased physical and mental health at 1 year. Therefore, patients who had even a minor stroke did worse from a physical and mental standpoint, a finding that argues for the superiority of surgery in selected patients at risk of periprocedural stroke.
If periprocedural stroke is excluded, the risk of long-term ipsilateral stroke was similar for each treatment, and extremely low (2% for stenting, 2.4% for surgery). Despite this, given the importance of periprocedural minor and major stroke, better predictive models are needed to identify patients at risk of procedural neurologic events. These prediction models will allow better patient selection.
The CREST data and medical therapy
The rates of stroke in this trial were similar to those observed with current medical treatment (approximately 1% per year), especially for patients with asymptomatic disease. Such findings introduce fresh controversy in the necessity of performing either procedure for this patient subset and may lead to further studies evaluating current medical therapy vs intervention.
Periprocedural myocardial infarction
Vascular surgery has long been associated with high cardiovascular risk, especially an increased risk of periprocedural MI.30 Findings from CREST provide further evidence of the risk of MI with endarterectomy in a real-world patient population. Given the evidence of a strong correlation between periprocedural cardiac enzyme elevations and adverse outcomes, the increased incidence of periprocedural MI is worrisome.31 As with risk assessment for periprocedural stroke, better predictive models are needed for patients at risk of cardiovascular events during endarterectomy.
Procedural complications
Carotid endarterectomy entails incisions in the neck with disruption of tissue planes, as opposed to catheter entry site wounds with stenting. The more invasive nature of endarterectomy thus carries a higher risk of wound complications. In fact, in the NASCET trial, the risk of wound complications was 9.3%.10,19 In CREST, surgery carried a higher risk of wound complications compared with stenting (42 vs 0 cases), although stenting involved more periprocedural transfusions, presumably due to retroperitoneal bleeding in four patients.
Use of general anesthesia is also associated with adverse outcomes.17,18 In CREST, 90% of endarterectomy procedures required general anesthesia, whereas none of the stenting procedures required this.
Cranial nerve palsy is an often overlooked but real complication after these procedures. Cranial nerve palsies can lead to vocal, swallowing, and sensory problems that can have a transient or permanent impact on quality of life. In CREST, as in EVA-3S, SAPPHIRE, and ICSS, this risk was substantially higher with surgery,23,25,27 although the long-term consequences of these palsies were not found to affect quality of life at 1 year of follow-up.
HOW CREST FINDINGS COMPARE WITH PREVIOUS STUDIES
Patients in CREST enjoyed overall better outcomes than in previous studies. In earlier trials of surgery vs medical therapy, the rates of adverse outcomes were higher than in CREST. In NASCET, the risk of ipsilateral stroke was 9% with surgery, with 2.5% being fatal or disabling strokes.10 In the ECST, rates of major stroke or death with endarterectomy were 7.0% within 30 days of surgery and 37.0% at a mean follow-up of 6.1 years.12
In earlier studies of surgery vs stenting, outcomes at 30 days were also substantially worse than those in CREST. In the EVA-3S trial, the 30-day incidence of stroke or death was 3.9% after surgery and 9.6% after stenting. These findings were similar at 6 months in EVA-3S, with a 6.1% rate of adverse events after surgery and 11.7% after stenting.25 In the SAPPHIRE trial, the cumulative incidence of stroke and death at 1 year was 21.4% for surgery and 13.6% for stenting.23
Overall, the CREST results show better outcomes than in previous trials. This may be due to improvements in technical aspects of the interventions and to more aggressive drug therapy. Also, because of the high number of patients enrolled in CREST, surgeons and interventionalists were required to meet eligibility criteria, which could have contributed to the improved outcomes.32
CREST was also unique in that stenting was done with an embolic protection device whenever possible, and this also likely had an impact on outcomes.
The CREST data suggest that interventions for carotid artery stenosis should only be performed by rigorously trained, experienced personnel at high-volume centers, as this provided lower event rates compared with previous studies. Additional data should also help identify those at risk of periprocedural stroke and MI, thereby helping to match the patient to the most appropriate procedure. The pros and cons of surgery and stenting are shown in Table 3.1,10,23,25,27
CREST vs ICSS
CREST and ICSS, published within a few months of each other, seem to have arrived at entirely different conclusions. As both studies are well-designed randomized controlled trials, these distinct results have yielded much controversy. However, closer scrutiny sheds light as to why the results may be different.
While ICSS focused only on patients with symptoms, CREST also included those without symptoms. The difference in patient populations is itself enough to account for the different outcomes.
Also, the interim analysis of ICSS was at 120 days, which makes periprocedural events a more dominant factor in outcomes, whereas these events likely do not last into the long term, as was the case in CREST. Analysis of the ICSS data at a later follow-up date may show results more similar to those of CREST.
The design of ICSS was also different than CREST. In ICSS, the use of an embolic protection device in stenting was not mandated, and the study lacked a lead-in phase of intensive training for those performing stenting. Furthermore, MI was adjudicated only when clinically recognized, which is different than the more rigorous method used in CREST.
Yet despite these differences, CREST and ICSS shed light on a controversial area of carotid stenosis management, and both studies boasted low rates of periprocedural complications. Clinicians should keep in mind the inclusion criteria and the technical specificities of these trials in order to explain to patients the risks and benefits of stenting and surgery, and to arrive at a decision together.
Limitations
The results of CREST should also be reviewed carefully due to a number of limitations. The study began in 2000 with symptomatic patients only, and began enrolling asymptomatic patients in 2005, so that the methodology of the study was changed midway. However, the investigators performed a subgroup analysis to distinguish between outcomes of the symptomatic and the asymptomatic groups and found no statistical interaction for the primary end point based on symptom status.
Despite careful patient selection, many of the predictors of adverse outcomes with stenting, such as lesion length, level of calcification, and lesion location, were not accounted for in the earlier days of enrollment. This may have had an impact on the incidence of stroke in patients enrolled in the early years of the trial. We await the analysis of predictors of perioperative stroke from CREST.
TAKE-HOME POINTS AND FUTURE DIRECTIONS
The CREST findings show that outcomes with stenting are similar to those with surgery in both the short term and the long term, and that the choice of management should be individualized. Each patient’s risk of MI and stroke should be considered based on a variety of factors, including the severity of coronary artery disease, the length of the carotid lesion, the level of calcification, the location of the lesion, and aortic atheroma. The treatment should be selected after also taking into account the patient’s preference and the available expertise, and only after a comprehensive discussion with the patient.
For patients with carotid artery stenosis, percutaneous intervention with stenting is as good as surgery (carotid endarterectomy). This was the major finding of the recently completed Carotid Revascularization Endarterectomy Versus Stenting Trial (CREST)1—with some qualifications.
CREST is the latest in a series of clinical trials of treatment of carotid stenosis that have generated reams of numbers and much debate. The topic of surgery vs percutaneous intervention is a moving target, as techniques evolve and improve. We believe the CREST results are valuable and should help inform decisions about treatment in the “real world.”
In this article, we offer a critical review of CREST, with a careful evaluation of its methods, results, and conclusions.
AN EVOLVING FIELD
Despite improvements in diagnosis and management, stroke remains one of the leading causes of morbidity and death in the United States, with an annual incidence of 780,000 cases and 270,000 deaths.2,3
Figure 1. Carotid endarterectomy has long been an established treatment in selected patients with symptomatic carotid artery stenosis of 50% or greater or asymptomatic stenosis of 60% or greater. However, percutaneous carotid artery angioplasty with stenting and placement of an embolic protection device is gaining ground as a reasonable, safe, less invasive alternative.From 10% to 30% of ischemic strokes are due to emboli from the carotid arteries.4–6 Carotid endarterectomy is an established treatment in selected patients with symptomatic carotid stenosis of 50% or greater or asymptomatic stenosis of 60% or greater.7,8 However, percutaneous techniques such as carotid artery angioplasty with stenting have improved, making them a viable, less invasive option (Figure 1).
Randomized trials of stenting have had mixed results, leading the Centers for Medicare and Medicaid Services (CMS) to adopt strict reimbursement policies. Currently, CMS reimburses for stenting only in symptomatic cases with at least 50% carotid artery stenosis. It also reimburses for stenting in asymptomatic cases in patients at high risk with 80% or greater stenosis, but only if the patients are enrolled in ongoing clinical trials or registries.
CREST compared stenting with endarterectomy and provided important insights into each approach.1
BEFORE CREST
Endarterectomy is superior to medical therapy for symptomatic stenosis
First described in 1953, carotid endarterectomy became the most widely used invasive treatment for significant carotid stenosis.9 Several studies have described patient subsets that benefit from this procedure.
NASCET (the North American Symptomatic Carotid Endarterectomy Trial)10 assigned 2,226 patients with symptomatic stenosis (transient ischemic attack or stroke within the past 180 days) to medical management or endarterectomy.
Surgery was associated with a 65% lower rate of ipsilateral cerebral events in patients with 70% or greater stenosis.10 Surgery was also found to be superior in patients with moderate disease (50% to 69% stenosis), but the difference only approached statistical significance. In patients with stenosis of less than 50%, the outcomes were similar with endarterectomy and medical management.11
ECST (the European Carotid Surgery Trial)12 included a similar population of 3,024 patients. Those with high-grade disease (stenosis ≥ 80%) had significantly better outcomes with endarterectomy, but in those with stenosis less than 70%, surgery was no better than drug therapy.
Comment. NASCET and ECST taught us that endarterectomy is clearly superior to medical therapy in patients with severe symptomatic carotid disease. However, both trials excluded patients at high surgical risk, eg, those with severe coronary artery disease, kidney disease, or heart failure. Additionally, medical management was not aggressive by today’s standards in terms of control of blood pressure and hyperlipidemia, and this could have skewed the results in favor of carotid endarterectomy.
The case for carotid endarterectomy for asymptomatic stenosis
Endarterectomy has also been compared with drug therapy for asymp tomatic carotid artery stenosis in several trials.13–15
ACAS (the Asymptomatic Carotid Atherosclerosis Study)15 assigned 1,662 patients who had no symptoms and had at least 60% carotid artery stenosis to endarterectomy or to medical management, and found a relative risk reduction of 53% in favor of surgery.15
The Veterans Affairs Cooperative Study Group14 corroborated these results in 444 patients with asymptomatic stenosis of greater than 50%. Endarterectomy was associated with a 61% lower risk of transient ischemic attack, transient monocular blindness, or stroke compared with medical therapy. However, there was no statistically significant difference in rates of stroke or death at 30 days.14
ACST (the Asymptomatic Carotid Surgery Trial),13 the largest study to compare carotid endarterectomy with drug therapy for asymptomatic stenosis, randomized 3,120 patients to surgery or drug therapy. The net 5-year risk of stroke was 6.4% with endarterectomy vs 11.8% with drug therapy (P < .0001). The rate of fatal stroke was also lower with endarterectomy: 2.1% vs 4.2% (P = .006).13
Comment. The results of these and other studies of endarterectomy vs medical therapy may not be applicable to current practice, since medical therapy has evolved and the risks with current drug therapy are likely much lower than seen in these trials, some of which began 2 decades ago. Another problem with interpreting these trials is that they excluded surgically “high-risk” patients, which limits the generalizability of the findings to this particular patient population.
The American Heart Association and the American Stroke Association have, on the basis of these trials, recommended carotid endarterectomy in patients with7,8,16:
Ipsilateral, symptomatic carotid artery stenosis of 70% to 99% (class I, level of evidence A)
Symptomatic stenosis of 50% to 69%, depending on patient-specific factors such as age, sex, and comorbidities
High-grade asymptomatic carotid stenosis, if the patients are carefully selected and the surgery is performed by surgeons with procedural morbidity and mortality rates of less than 3% (class I, level of evidence A).
In all cases, treatment should be individualized according to the patient’s comorbid conditions and preferences, with a thorough discussion of risks and benefits (Table 1).7,8,16
The case for percutaneous intervention
While carotid endarterectomy is proven to be more efficacious than medical management in certain patient subsets, studies favoring surgery over medical therapy have been criticized because they excluded patients with significant comorbidities. In addition, surgery has been associated with significant cardiovascular events, wound complications, and cranial nerve damage, and it requires general anesthesia in most cases.12,17–19 These and other factors spurred the development of less invasive, percutaneous approaches for patients with substantial comorbidities.
So far, several trials have investigated carotid angioplasty with or without stents and with or without devices to capture distal emboli. This interest set the stage for CREST.20,21
Initial attempts at angioplasty without distal protection were not very successful. A meta-analysis of nonrandomized trials that included 714 patients from the initial 13 studies of angioplasty (with or without stenting) and 6,970 patients from 20 studies of carotid endarterectomy found angioplasty to be possibly associated with higher rates of stroke within 30 days of the procedure.20
With improvements in technology, routine use of embolic protection devices, more experience, and better selection of patients, the outcome of carotid stenting has improved. In fact, a meta-analysis comparing stenting without an embolic protection device (26 trials with 2,357 patients) vs stenting with an embolic protection device (11 trials with 839 patients) showed that embolic protection led to significantly better outcomes with fewer strokes—outcomes arguably similar to those of carotid endarterectomy.21
SAPPHIRE (the Stenting and Angioplasty With Protection in Patients at High Risk for Endarterectomy trial)22 was the only completed US trial until CREST that compared carotid artery stenting with distal protection against surgery. It included 334 high-risk patients with either symptomatic stenosis of 50% or greater or asymptomatic stenosis of 80% or greater.
The results suggested that the outcomes with stenting with embolic protection were in fact similar to those of endarterectomy, with possibly fewer complications.23 The benefit persisted up to 2 years.22
The US Food and Drug Administration (FDA), on the basis of these data, approved the use of stenting with distal protection for high-risk patients, and the CMS reimburses for symptomatic stenosis of 50% or greater and for asymptomatic stenosis of 80% or greater as long as the patient is enrolled in a registry.
SPACE (the Stent-Protected Angioplasty Versus Carotid Endarterectomy in Symptomatic Patients trial),24 conducted in Germany, included 1,214 patients with symptomatic stenosis of at least 50%. Results were similar in terms of the combined primary end point of stroke or death at 30 days. However, the results were not similar enough to prove that stenting is not inferior to surgery, according to preset study criteria.
EVA-3S (the Endarterectomy Versus Stenting in Patients With Symptomatic Severe Carotid Stenosis trial),25 in France, evaluated 527 patients with symptomatic carotid disease (stenosis ≥ 60%), but was terminated early due to significantly higher rates of death or stroke at 30 days in the stenting group.
Comment. SPACE and EVA-3S have been widely criticized for not mandating the use of an embolic protection device (used in 27% of cases in SPACE and in 91.9% of cases in EVA-3S). Questions were also raised about the experience level of the operators who performed the carotid stenting: up to 39% of the primary operators involved in stent placement were trainees.26 Also, myocardial infarction (MI), an important complication of carotid endarterectomy, was not included in the primary end point.
ICSS (the International Carotid Stenting Study)27 compared stenting with endarterectomy in 1,713 patients with symptomatic carotid stenosis of greater than 50%. The primary end point was the rate of fatal or disabling stroke at 3 years.
An interim safety analysis at 120 days of follow-up showed the primary end point had occurred in 4.0% of stenting cases vs 3.2% of endarterectomy cases, a difference that was not statistically significant (hazard ratio [HR] 1.28, 95% confidence interval [CI] 0.77–2.11). However, the risk of any stroke was higher with stenting, with a rate of 7.7% vs 4.1% in the surgical group—a statistically significant difference (HR 1.92, 95% CI 1.27–2.89).
In a substudy of ICSS,28 the investigators corroborated these findings, using magnetic resonance imaging to evaluate for new ischemic brain lesions periprocedurally. They found more new ischemic brain lesions in patients who underwent stenting than in patients who underwent surgery—a statistically significant finding.
Comment. ICSS had limitations: eg, it included only patients with symptoms, and the training for the stenting procedure was not standardized. Furthermore, the use of embolic protection devices was not mandated in stenting procedures.
Because of the controversial and incongruous findings of the above trials, there has been much anticipation for further large, appropriately conducted, randomized controlled trials such as CREST.
CREST STUDY DESIGN
CREST was a prospective, multicenter randomized controlled trial with blinded end point adjudication. Assignment to stenting or surgery occurred in a one-to-one fashion, and patients were stratified by medical center and symptomatic status.
Conducted at 108 sites in the United States and nine sites in Canada, CREST was supported by a grant from the National Institutes of Health and by the manufacturer of the catheter and stent delivery and embolic protection systems. The manufacturer’s representative held a nonvoting position on the executive committee and reviewed the manuscript of the results before submission.
CREST included patients with or without symptoms
CREST was initially designed to compare carotid artery stenting vs carotid endarterectomy in patients with symptoms, but enrollment was later extended to patients without symptoms.
Patients with symptoms were included if they had stenosis of at least 50% on angiography, at least 70% on ultrasonography, or at least 70% on computed tomographic angiography or magnetic resonance angiography if stenosis on ultrasonography was 50% to 69%. Carotid artery stenosis was considered symptomatic if the patient had a transient ischemic attack, amaurosis fugax, or minor disabling stroke in the hemisphere supplied by the target vessel within 180 days of randomization.
Patients without symptoms were eligible if they had at least 60% stenosis on angiography, at least 70% stenosis on ultrasonography, or at least 80% stenosis on computed tomographic angiography or magnetic resonance angiography if the stenosis was 50% to 69% on ultrasonography.
Other eligibility criteria included favorable anatomy and clinical stability for both stenting and surgical procedures.
Exclusion criteria were evolving stroke, history of major stroke, chronic or paroxysmal atrial fibrillation on anticoagulation therapy, MI within the previous 30 days, and unstable angina.
Patients received antiplatelet agents
Patients undergoing stenting received aspirin and clopidogrel (Plavix) before and up to 30 days after the procedure. Continuation of antiplatelet therapy was recommended beyond 1 month.
Patients undergoing endarterectomy received aspirin before surgery and continued to receive aspirin for at least 1 year.
Alternatives to aspirin in both groups were ticlopidine (Ticlid), clopidogrel, or aspirin with extended-release dipyridamole (Aggrenox).
End points: Stroke, MI, death
The primary end point was a composite of periprocedural clinical stroke (any type), MI, or death, and of ipsilateral stroke up to 4 years after the procedure. Secondary analyses were also planned for evaluation of treatment modification by age, symptom status, and sex.
Stroke was defined as any acute neurologic ischemic event lasting at least 24 hours with focal signs and symptoms.
Two separate definitions were applied to distinguish major stroke from nonmajor stroke. Major stroke was defined as a National Institutes of Health Stroke Scale (NIHSS) score greater than 9 or records suggesting that the event was a disabling stroke if admitted to another facility. Nonmajor stroke included an event that did not fit these criteria. The stroke review process was initiated with a significant neurologic event, a positive transient ischemia attack or stroke questionnaire, or a two-point or greater increase in the NIHSS score.
MI was defined as a combination of an elevation of cardiac enzymes to at least twice the laboratory upper limit of normal, as well as clinical signs suggesting MI or electrocardiographic evidence of ischemia.29
Stroke was adjudicated by two independent neurologists, and MI was adjudicated by two independent cardiologists blinded to treatment group assignment.
The Rankin scale, the transient ischemic attack and stroke questionnaire, and the Medical Outcomes Survey were also used to assess for disability and quality of life in long-term follow-up.
Intention-to-treat analysis
Intention-to-treat survival analysis was used along with time-to-event statistical modeling with adjustment for major baseline covariates. Differences in outcomes were assessed, and a noninferiority analysis was performed. Kaplan-Meier estimates were constructed of the proportion of patients remaining free of the composite end point at 30 days, 6 months, 1 year, and annually thereafter, and of the associated confidence intervals. The hazard ratios between groups were estimated after adjustment for important covariates.
Most patients enrolled were available for analysis
From December 2000 to July 2008, 2,522 patients were enrolled; 1,271 were assigned to stenting, and 1,251 were assigned to surgery. After randomization, 2.8% of the patients assigned to stenting withdrew consent, 5.7% underwent surgery, and 2.6% were lost to follow-up. Of those assigned to surgery, 5.1% withdrew consent, 1.0% underwent stenting, and 3.8% were lost to follow-up.
A ‘conventional-risk’ patient population
The trial sought to include a “conventional-risk” patient population to make the study more applicable to real-world practice. The mean age was 69 years in both groups. Of the 2,522 patients enrolled:
35% were women
47% had asymptomatic carotid disease
86% had carotid stenosis of 70% or greater
86% had hypertension
30% had diabetes mellitus
83% had hyperlipidemia
26% were current smokers
42% had a history of cardiovascular disease
21% had undergone coronary artery bypass grafting surgery.
The only statistically significant difference in measured baseline variables between the two treatment groups was a slightly higher rate of dyslipidemia in the group undergoing surgery.
The interventionalists and surgeons were highly experienced
Operators performing stenting underwent a lead-in phase of training, with close supervision and scrutiny before eligibility. Of patients undergoing stenting, 96.1% also received an embolic protection device. Antiplatelet therapy was continued in 99% of the patients.
The surgeons performing endarterectomy were experienced and had documented low complication rates. General anesthesia was used in 90% of surgical patients. Shunts were used during surgery in 57%, and patches were used in 62%. After endarterectomy, 91% of the patients received antiplatelet therapy.
CREST STUDY RESULTS: STENTING WAS AS GOOD AS SURGERY
Periprocedural outcomes
Stroke, MI, or death: 5.2% with stenting vs 4.5% with surgery, HR 1.18, 95% CI 0.82–1.68, P = .38
Stroke: 4.1% vs 2.3%, HR 1.79, 95% CI 1.14–2.82, P = .01
Major ipsilateral stroke: 0.9% vs 0.3%, HR 2.67, 95% CI 0.85–8.40, P = .09.
MI: 1.1% vs 2.3%, HR 0.50, 95% CI 0.26–0.94, P = .03
Cranial nerve palsy: 0.3% vs 4.8%, HR 0.07, 95% CI 0.02–0.18, P < .0001 (Table 2).
Outcomes at 4 years
Brott TG, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23. Copyright 2010, Massachusetts Medical Society. All rights reserved.
Figure 2. Kaplan-Meier analysis of the primary outcome (stroke, myocardial infarction, or death during the periprocedural period or any ipsilateral stroke within 4 years after randomization) for patients undergoing carotid artery stenting or carotid endarterectomy.The primary end point (periprocedural stroke, MI, or death, or ipsilateral stroke within 4 years after the procedure): 7.2% with stenting vs 6.8% with surgery, HR 1.11, 95% CI 0.81–1.51, P = .51. A Kaplan-Meier analysis showed similar findings with statistically similar outcomes (Figure 2).
Ipsilateral stroke: 2.0% vs 2.4%, HR 0.94, 95% CI 0.50–1.76, P = .85.
The primary outcome was analyzed for interactions of baseline variables, and no effect was detected for symptomatic status or sex. There was a suggestion of an interaction with age, with older patients (over age 70) benefiting more from endarterectomy.
Quality-of-life indices showed that both major and minor strokes were likely to produce long-term physical limitations, with minor stroke associated with worse mental and physical health at 1 year. The effect of periprocedural MI on long-term physical and mental health was less certain. The increased incidence of cranial nerve palsy noted with endarterectomy has been found before and has had no effect on quality of life.
WHAT DO THE CREST FINDINGS MEAN?
CREST is the largest trial to date to compare stenting and surgery. It is an important addition to the literature, not only because of its size, but also because it focused on a real-world patient population. For this reason, its results are more applicable to patients seen in primary care clinics, ie, with peripheral vascular disease, coronary artery disease, diabetes mellitus, hypertension, and smoking.
As noted, previous studies of endarterectomy had strict inclusion and exclusion criteria, which selected against patients at high surgical risk. Therefore, the CREST findings are of greater relevance when comparing stenting and endarterectomy.
Periprocedural and long-term neurologic outcomes
CREST showed similar findings for the composite end point of periprocedural stroke, death, or MI (ie, within 30 days of the procedure) and long-term stroke, establishing similar outcomes in patients undergoing stenting and surgery.
However, an analysis of the individual components of the composite end point showed significant differences between the two treatments. The risk of ipsilateral periprocedural stroke was higher with stenting; these events were defined as nonmajor by NIHSS criteria. The risk of contralateral stroke was similar and low with each treatment.
While the increased risk of periprocedural ipsilateral stroke was not synonymous with an increased risk of major stroke, post hoc analysis showed that any stroke was associated with decreased physical and mental health at 1 year. Therefore, patients who had even a minor stroke did worse from a physical and mental standpoint, a finding that argues for the superiority of surgery in selected patients at risk of periprocedural stroke.
If periprocedural stroke is excluded, the risk of long-term ipsilateral stroke was similar for each treatment, and extremely low (2% for stenting, 2.4% for surgery). Despite this, given the importance of periprocedural minor and major stroke, better predictive models are needed to identify patients at risk of procedural neurologic events. These prediction models will allow better patient selection.
The CREST data and medical therapy
The rates of stroke in this trial were similar to those observed with current medical treatment (approximately 1% per year), especially for patients with asymptomatic disease. Such findings introduce fresh controversy in the necessity of performing either procedure for this patient subset and may lead to further studies evaluating current medical therapy vs intervention.
Periprocedural myocardial infarction
Vascular surgery has long been associated with high cardiovascular risk, especially an increased risk of periprocedural MI.30 Findings from CREST provide further evidence of the risk of MI with endarterectomy in a real-world patient population. Given the evidence of a strong correlation between periprocedural cardiac enzyme elevations and adverse outcomes, the increased incidence of periprocedural MI is worrisome.31 As with risk assessment for periprocedural stroke, better predictive models are needed for patients at risk of cardiovascular events during endarterectomy.
Procedural complications
Carotid endarterectomy entails incisions in the neck with disruption of tissue planes, as opposed to catheter entry site wounds with stenting. The more invasive nature of endarterectomy thus carries a higher risk of wound complications. In fact, in the NASCET trial, the risk of wound complications was 9.3%.10,19 In CREST, surgery carried a higher risk of wound complications compared with stenting (42 vs 0 cases), although stenting involved more periprocedural transfusions, presumably due to retroperitoneal bleeding in four patients.
Use of general anesthesia is also associated with adverse outcomes.17,18 In CREST, 90% of endarterectomy procedures required general anesthesia, whereas none of the stenting procedures required this.
Cranial nerve palsy is an often overlooked but real complication after these procedures. Cranial nerve palsies can lead to vocal, swallowing, and sensory problems that can have a transient or permanent impact on quality of life. In CREST, as in EVA-3S, SAPPHIRE, and ICSS, this risk was substantially higher with surgery,23,25,27 although the long-term consequences of these palsies were not found to affect quality of life at 1 year of follow-up.
HOW CREST FINDINGS COMPARE WITH PREVIOUS STUDIES
Patients in CREST enjoyed overall better outcomes than in previous studies. In earlier trials of surgery vs medical therapy, the rates of adverse outcomes were higher than in CREST. In NASCET, the risk of ipsilateral stroke was 9% with surgery, with 2.5% being fatal or disabling strokes.10 In the ECST, rates of major stroke or death with endarterectomy were 7.0% within 30 days of surgery and 37.0% at a mean follow-up of 6.1 years.12
In earlier studies of surgery vs stenting, outcomes at 30 days were also substantially worse than those in CREST. In the EVA-3S trial, the 30-day incidence of stroke or death was 3.9% after surgery and 9.6% after stenting. These findings were similar at 6 months in EVA-3S, with a 6.1% rate of adverse events after surgery and 11.7% after stenting.25 In the SAPPHIRE trial, the cumulative incidence of stroke and death at 1 year was 21.4% for surgery and 13.6% for stenting.23
Overall, the CREST results show better outcomes than in previous trials. This may be due to improvements in technical aspects of the interventions and to more aggressive drug therapy. Also, because of the high number of patients enrolled in CREST, surgeons and interventionalists were required to meet eligibility criteria, which could have contributed to the improved outcomes.32
CREST was also unique in that stenting was done with an embolic protection device whenever possible, and this also likely had an impact on outcomes.
The CREST data suggest that interventions for carotid artery stenosis should only be performed by rigorously trained, experienced personnel at high-volume centers, as this provided lower event rates compared with previous studies. Additional data should also help identify those at risk of periprocedural stroke and MI, thereby helping to match the patient to the most appropriate procedure. The pros and cons of surgery and stenting are shown in Table 3.1,10,23,25,27
CREST vs ICSS
CREST and ICSS, published within a few months of each other, seem to have arrived at entirely different conclusions. As both studies are well-designed randomized controlled trials, these distinct results have yielded much controversy. However, closer scrutiny sheds light as to why the results may be different.
While ICSS focused only on patients with symptoms, CREST also included those without symptoms. The difference in patient populations is itself enough to account for the different outcomes.
Also, the interim analysis of ICSS was at 120 days, which makes periprocedural events a more dominant factor in outcomes, whereas these events likely do not last into the long term, as was the case in CREST. Analysis of the ICSS data at a later follow-up date may show results more similar to those of CREST.
The design of ICSS was also different than CREST. In ICSS, the use of an embolic protection device in stenting was not mandated, and the study lacked a lead-in phase of intensive training for those performing stenting. Furthermore, MI was adjudicated only when clinically recognized, which is different than the more rigorous method used in CREST.
Yet despite these differences, CREST and ICSS shed light on a controversial area of carotid stenosis management, and both studies boasted low rates of periprocedural complications. Clinicians should keep in mind the inclusion criteria and the technical specificities of these trials in order to explain to patients the risks and benefits of stenting and surgery, and to arrive at a decision together.
Limitations
The results of CREST should also be reviewed carefully due to a number of limitations. The study began in 2000 with symptomatic patients only, and began enrolling asymptomatic patients in 2005, so that the methodology of the study was changed midway. However, the investigators performed a subgroup analysis to distinguish between outcomes of the symptomatic and the asymptomatic groups and found no statistical interaction for the primary end point based on symptom status.
Despite careful patient selection, many of the predictors of adverse outcomes with stenting, such as lesion length, level of calcification, and lesion location, were not accounted for in the earlier days of enrollment. This may have had an impact on the incidence of stroke in patients enrolled in the early years of the trial. We await the analysis of predictors of perioperative stroke from CREST.
TAKE-HOME POINTS AND FUTURE DIRECTIONS
The CREST findings show that outcomes with stenting are similar to those with surgery in both the short term and the long term, and that the choice of management should be individualized. Each patient’s risk of MI and stroke should be considered based on a variety of factors, including the severity of coronary artery disease, the length of the carotid lesion, the level of calcification, the location of the lesion, and aortic atheroma. The treatment should be selected after also taking into account the patient’s preference and the available expertise, and only after a comprehensive discussion with the patient.
References
Brott TG, Hobson RW, Howard G, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med2010; 363:11–23.
Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation2006; 113:e85–e151.
Rosamond WD, Folsom AR, Chambless LE, et al. Stroke incidence and survival among middle-aged adults: 9-year follow-up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke1999; 30:736–743.
Chaturvedi S, Bruno A, Feasby T, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Carotid endarterectomy—an evidence-based review: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology2005; 65:794–801.
Howell GM, Makaroun MS, Chaer RA. Current management of extracranial carotid occlusive disease. J Am Coll Surg2009; 208:442–453.
Barnett HJ, Gunton RW, Eliasziw M, et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA2000; 283:1429–1436.
Biller J, Feinberg WM, Castaldo JE, et al. Guidelines for carotid endarterectomy: a statement for healthcare professionals from a Special Writing Group of the Stroke Council, American Heart Association. Circulation1998; 97:501–509.
Goldstein LB, Adams R, Alberts MJ, et al; American Heart Association; American Stroke Association Stroke Council. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation2006; 113:e873–e923.
Strully KJ, Hurwitt ES, Blankenberg HW. Thrombo-endarterectomy for thrombosis of the internal carotid artery in the neck. J Neurosurg1953; 10:474–482.
Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med1991; 325:445–453.
Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med1998; 339:1415–1425.
Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet1998; 351:1379–1387.
Halliday A, Mansfield A, Marro J, et al; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomised controlled trial. Lancet2004; 363:1491–1502.
Hobson RW, Weiss DG, Fields WS, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med1993; 328:221–227.
Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. JAMA1995; 273:1421–1428.
Sacco RL, Adams R, Albers G, et al; American Heart Association/American Stroke Association Council on Stroke; Council on Cardiovascular Radiology and Intervention; American Academy of Neurology. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Circulation2006; 113:e409–e449.
Watts K, Lin PH, Bush RL, et al. The impact of anesthetic modality on the outcome of carotid endarterectomy. Am J Surg2004; 188:741–747.
Weber CF, Friedl H, Hueppe M, et al. Impact of general versus local anesthesia on early postoperative cognitive dysfunction following carotid endarterectomy: GALA Study Subgroup Analysis. World J Surg2009; 33:1526–1532.
Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke1999; 30:1751–1758.
Golledge J, Mitchell A, Greenhalgh RM, Davies AH. Systematic comparison of the early outcome of angioplasty and endarterectomy for symptomatic carotid artery disease. Stroke2000; 31:1439–1443.
Kastrup A, Gröschel K, Krapf H, Brehm BR, Dichgans J, Schulz JB. Early outcome of carotid angioplasty and stenting with and without cerebral protection devices: a systematic review of the literature. Stroke2003; 34:813–819.
Gurm HS, Yadav JS, Fayad P, et al; SAPPHIRE Investigators. Long-term results of carotid stenting versus endarterectomy in high-risk patients. N Engl J Med2008; 358:1572–1579.
Yadav JS, Wholey MH, Kuntz RE, et al; Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy Investigators. Protected carotid-artery stenting versus endarterectomy in high-risk patients. N Engl J Med2004; 351:1493–1501.
Eckstein HH, Ringleb P, Allenberg JR, et al. Results of the Stent-Protected Angioplasty versus Carotid Endarterectomy (SPACE) study to treat symptomatic stenoses at 2 years: a multinational, prospective, randomised trial. Lancet Neurol2008; 7:893–902.
Mas JL, Chatellier G, Beyssen B, et al; EVA-3S Investigators. Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med2006; 355:1660–1771.
Roffi M, Sievert H, Gray WA, et al. Carotid artery stenting versus surgery: adequate comparisons?Lancet Neurol2010; 9:339–341.
International Carotid Stenting Study Investigators; Ederle J, Dobson J, Featherstone RL, et al. Carotid artery stenting compared with endarterectomy in patients with symptomatic carotid stenosis (International Carotid Stenting Study): an interim analysis of a randomised controlled trial. Lancet2010; 375:985–997.
Bonati LH, Jongen LM, Haller S, et al; ICSS-MRI study group. New ischaemic brain lesions on MRI after stenting or endarterectomy for symptomatic carotid stenosis: a sub-study of the International Carotid Stenting Study (ICSS). Lancet Neurol2010; 9:353–362.
Sheffet AJ, Roubin G, Howard G, et al. Design of the Carotid Revascularization Endarterectomy vs. Stenting Trial (CREST). Int J Stroke2010; 5:40–46.
Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery. J Am Coll Cardiol2007; 50:e159–e241.
Bhatt DL, Topol EJ. Does creatinine kinase-MB elevation after percutaneous coronary intervention predict outcomes in 2005? Periprocedural cardiac enzyme elevation predicts adverse outcomes. Circulation2005; 112:906–915.
Hobson RW, Howard VJ, Roubin GS, et al; CREST. Credentialing of surgeons as interventionalists for carotid artery stenting: experience from the lead-in phase of CREST. J Vasc Surg2004; 40:952–957.
References
Brott TG, Hobson RW, Howard G, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med2010; 363:11–23.
Thom T, Haase N, Rosamond W, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation2006; 113:e85–e151.
Rosamond WD, Folsom AR, Chambless LE, et al. Stroke incidence and survival among middle-aged adults: 9-year follow-up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke1999; 30:736–743.
Chaturvedi S, Bruno A, Feasby T, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Carotid endarterectomy—an evidence-based review: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology2005; 65:794–801.
Howell GM, Makaroun MS, Chaer RA. Current management of extracranial carotid occlusive disease. J Am Coll Surg2009; 208:442–453.
Barnett HJ, Gunton RW, Eliasziw M, et al. Causes and severity of ischemic stroke in patients with internal carotid artery stenosis. JAMA2000; 283:1429–1436.
Biller J, Feinberg WM, Castaldo JE, et al. Guidelines for carotid endarterectomy: a statement for healthcare professionals from a Special Writing Group of the Stroke Council, American Heart Association. Circulation1998; 97:501–509.
Goldstein LB, Adams R, Alberts MJ, et al; American Heart Association; American Stroke Association Stroke Council. Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation2006; 113:e873–e923.
Strully KJ, Hurwitt ES, Blankenberg HW. Thrombo-endarterectomy for thrombosis of the internal carotid artery in the neck. J Neurosurg1953; 10:474–482.
Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med1991; 325:445–453.
Barnett HJ, Taylor DW, Eliasziw M, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med1998; 339:1415–1425.
Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet1998; 351:1379–1387.
Halliday A, Mansfield A, Marro J, et al; MRC Asymptomatic Carotid Surgery Trial (ACST) Collaborative Group. Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: randomised controlled trial. Lancet2004; 363:1491–1502.
Hobson RW, Weiss DG, Fields WS, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med1993; 328:221–227.
Endarterectomy for asymptomatic carotid artery stenosis. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. JAMA1995; 273:1421–1428.
Sacco RL, Adams R, Albers G, et al; American Heart Association/American Stroke Association Council on Stroke; Council on Cardiovascular Radiology and Intervention; American Academy of Neurology. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: a statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: co-sponsored by the Council on Cardiovascular Radiology and Intervention: the American Academy of Neurology affirms the value of this guideline. Circulation2006; 113:e409–e449.
Watts K, Lin PH, Bush RL, et al. The impact of anesthetic modality on the outcome of carotid endarterectomy. Am J Surg2004; 188:741–747.
Weber CF, Friedl H, Hueppe M, et al. Impact of general versus local anesthesia on early postoperative cognitive dysfunction following carotid endarterectomy: GALA Study Subgroup Analysis. World J Surg2009; 33:1526–1532.
Ferguson GG, Eliasziw M, Barr HW, et al. The North American Symptomatic Carotid Endarterectomy Trial: surgical results in 1415 patients. Stroke1999; 30:1751–1758.
Golledge J, Mitchell A, Greenhalgh RM, Davies AH. Systematic comparison of the early outcome of angioplasty and endarterectomy for symptomatic carotid artery disease. Stroke2000; 31:1439–1443.
Kastrup A, Gröschel K, Krapf H, Brehm BR, Dichgans J, Schulz JB. Early outcome of carotid angioplasty and stenting with and without cerebral protection devices: a systematic review of the literature. Stroke2003; 34:813–819.
Gurm HS, Yadav JS, Fayad P, et al; SAPPHIRE Investigators. Long-term results of carotid stenting versus endarterectomy in high-risk patients. N Engl J Med2008; 358:1572–1579.
Yadav JS, Wholey MH, Kuntz RE, et al; Stenting and Angioplasty with Protection in Patients at High Risk for Endarterectomy Investigators. Protected carotid-artery stenting versus endarterectomy in high-risk patients. N Engl J Med2004; 351:1493–1501.
Eckstein HH, Ringleb P, Allenberg JR, et al. Results of the Stent-Protected Angioplasty versus Carotid Endarterectomy (SPACE) study to treat symptomatic stenoses at 2 years: a multinational, prospective, randomised trial. Lancet Neurol2008; 7:893–902.
Mas JL, Chatellier G, Beyssen B, et al; EVA-3S Investigators. Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med2006; 355:1660–1771.
Roffi M, Sievert H, Gray WA, et al. Carotid artery stenting versus surgery: adequate comparisons?Lancet Neurol2010; 9:339–341.
International Carotid Stenting Study Investigators; Ederle J, Dobson J, Featherstone RL, et al. Carotid artery stenting compared with endarterectomy in patients with symptomatic carotid stenosis (International Carotid Stenting Study): an interim analysis of a randomised controlled trial. Lancet2010; 375:985–997.
Bonati LH, Jongen LM, Haller S, et al; ICSS-MRI study group. New ischaemic brain lesions on MRI after stenting or endarterectomy for symptomatic carotid stenosis: a sub-study of the International Carotid Stenting Study (ICSS). Lancet Neurol2010; 9:353–362.
Sheffet AJ, Roubin G, Howard G, et al. Design of the Carotid Revascularization Endarterectomy vs. Stenting Trial (CREST). Int J Stroke2010; 5:40–46.
Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, and Society for Vascular Surgery. J Am Coll Cardiol2007; 50:e159–e241.
Bhatt DL, Topol EJ. Does creatinine kinase-MB elevation after percutaneous coronary intervention predict outcomes in 2005? Periprocedural cardiac enzyme elevation predicts adverse outcomes. Circulation2005; 112:906–915.
Hobson RW, Howard VJ, Roubin GS, et al; CREST. Credentialing of surgeons as interventionalists for carotid artery stenting: experience from the lead-in phase of CREST. J Vasc Surg2004; 40:952–957.
In CREST, stenting and surgery had similar combined rates of stroke, MI, and death when performed by highly qualified interventionalists and surgeons in carefully selected patients.
The risk of periprocedural stroke was higher with stenting; most of those strokes were nonmajor. Both major and nonmajor strokes were associated with decreased quality of life in long-term follow-up.
Endarterectomy was associated with higher rates of periprocedural MI than stenting.
Endarterectomy carried a significantly higher rate of cranial nerve damage than stenting.
Although we often approach anticoagulation therapy with a confidence born of familiarity, it is not for the faint of heart. We start chronic anticoagulation in several clinical settings, such as to prevent a recurrence after a thromboembolic event. But this decision requires weighing the increased risk of bleeding from the anticoagulant therapy against the risk of another thromboembolic event.
Along with massive pulmonary embolism, the most feared thromboembolic event is the clot that migrates to the brain, resulting in life-altering stroke. We assess this risk in a semiquantitative manner in patients with atrial fibrillation using the CHADS2 score, hoping to maximize the benefits of anticoagulation while reducing the risks. We recognize that patients at the greatest risk of stroke in this setting are those with a history of a prior stroke. Also, patients bedridden with a recent cerebrovascular accident (CVA) seem to be hypercoagulable, potentially adding risk to recent injury. Thus, we try to start anticoagulation as soon as feasible after the diagnosis of a possible thrombotic event.
But the decision to start or resume anticoagulation is especially agonizing in a patient who has suffered an intracerebral hemorrhage. In this issue of the Journal, Drs. Joshua Goldstein and Steven Greenberg and Dr. Franklin Michota provide a thoughtful discussion of the issues we need to consider in these patients.
While not contributing to the prevention of additional CVAs or other arterial thrombotic events, a modality often underused in the prevention of thrombotic disease is the application (not just the ordering) of compressive leg stockings to bedridden hospitalized patients who cannot, for any reason, be provided pharmacologic anticoagulation therapy. I just completed a stint of hospital consultation, and I was pleased to see the widespread integration of prophylactic anticoagulation therapy, but somewhat dismayed by the number of compressive stockings I watched pumping with vigor, but to no one’s benefit, as they were draped over a bed rail.
As we struggle with complex clinical decisions, we need to also be attentive to the simple and the seemingly mundane: using the foam dispenser at the door, offering the verbal greeting and patient touch at the bedside, and rewrapping the pneumatic stockings that have somehow migrated between mattress and footboard.
Although we often approach anticoagulation therapy with a confidence born of familiarity, it is not for the faint of heart. We start chronic anticoagulation in several clinical settings, such as to prevent a recurrence after a thromboembolic event. But this decision requires weighing the increased risk of bleeding from the anticoagulant therapy against the risk of another thromboembolic event.
Along with massive pulmonary embolism, the most feared thromboembolic event is the clot that migrates to the brain, resulting in life-altering stroke. We assess this risk in a semiquantitative manner in patients with atrial fibrillation using the CHADS2 score, hoping to maximize the benefits of anticoagulation while reducing the risks. We recognize that patients at the greatest risk of stroke in this setting are those with a history of a prior stroke. Also, patients bedridden with a recent cerebrovascular accident (CVA) seem to be hypercoagulable, potentially adding risk to recent injury. Thus, we try to start anticoagulation as soon as feasible after the diagnosis of a possible thrombotic event.
But the decision to start or resume anticoagulation is especially agonizing in a patient who has suffered an intracerebral hemorrhage. In this issue of the Journal, Drs. Joshua Goldstein and Steven Greenberg and Dr. Franklin Michota provide a thoughtful discussion of the issues we need to consider in these patients.
While not contributing to the prevention of additional CVAs or other arterial thrombotic events, a modality often underused in the prevention of thrombotic disease is the application (not just the ordering) of compressive leg stockings to bedridden hospitalized patients who cannot, for any reason, be provided pharmacologic anticoagulation therapy. I just completed a stint of hospital consultation, and I was pleased to see the widespread integration of prophylactic anticoagulation therapy, but somewhat dismayed by the number of compressive stockings I watched pumping with vigor, but to no one’s benefit, as they were draped over a bed rail.
As we struggle with complex clinical decisions, we need to also be attentive to the simple and the seemingly mundane: using the foam dispenser at the door, offering the verbal greeting and patient touch at the bedside, and rewrapping the pneumatic stockings that have somehow migrated between mattress and footboard.
Although we often approach anticoagulation therapy with a confidence born of familiarity, it is not for the faint of heart. We start chronic anticoagulation in several clinical settings, such as to prevent a recurrence after a thromboembolic event. But this decision requires weighing the increased risk of bleeding from the anticoagulant therapy against the risk of another thromboembolic event.
Along with massive pulmonary embolism, the most feared thromboembolic event is the clot that migrates to the brain, resulting in life-altering stroke. We assess this risk in a semiquantitative manner in patients with atrial fibrillation using the CHADS2 score, hoping to maximize the benefits of anticoagulation while reducing the risks. We recognize that patients at the greatest risk of stroke in this setting are those with a history of a prior stroke. Also, patients bedridden with a recent cerebrovascular accident (CVA) seem to be hypercoagulable, potentially adding risk to recent injury. Thus, we try to start anticoagulation as soon as feasible after the diagnosis of a possible thrombotic event.
But the decision to start or resume anticoagulation is especially agonizing in a patient who has suffered an intracerebral hemorrhage. In this issue of the Journal, Drs. Joshua Goldstein and Steven Greenberg and Dr. Franklin Michota provide a thoughtful discussion of the issues we need to consider in these patients.
While not contributing to the prevention of additional CVAs or other arterial thrombotic events, a modality often underused in the prevention of thrombotic disease is the application (not just the ordering) of compressive leg stockings to bedridden hospitalized patients who cannot, for any reason, be provided pharmacologic anticoagulation therapy. I just completed a stint of hospital consultation, and I was pleased to see the widespread integration of prophylactic anticoagulation therapy, but somewhat dismayed by the number of compressive stockings I watched pumping with vigor, but to no one’s benefit, as they were draped over a bed rail.
As we struggle with complex clinical decisions, we need to also be attentive to the simple and the seemingly mundane: using the foam dispenser at the door, offering the verbal greeting and patient touch at the bedside, and rewrapping the pneumatic stockings that have somehow migrated between mattress and footboard.
The reality of blood pressure measurement is that human beings do not do it very well. The time has come to delegate this job to machines that can do it better.
Several automatic devices are available. Used in physicians’ offices and in patients’ homes, they can eliminate some types of observer error as well as the “white-coat effect,” ie, the tendency of some patients to have higher blood pressure when medical personnel are present than in their natural environment. Since a difference of only a few millimeters of mercury can affect the physician’s decision to start or to modify treatment, measurements that more accurately reflect a person’s average blood pressure are to be desired.
In the following pages, we review the problems that plague blood pressure measurement by human observers, and we describe the advantages of automatic devices.
SHORTCOMINGS OF OFFICE BLOOD PRESSURE MEASUREMENT
For decades, large surveys have provided invaluable information on the prevalence of hypertension, its relationship to cardiovascular disease, and the benefits of treating it.1–3 Unfortunately, the percentage of hypertensive patients whose blood pressure is under control has remained low despite our increased knowledge about hypertension’s diagnosis and therapy.4
In these surveys, blood pressure was measured by auscultation by human observers using mercury or aneroid sphygmomanometers, and most physicians still use this method in clinical practice. But in spite of multiple guidelines for accurate measurement of blood pressure in the office, the overall accuracy and reproducibility of office blood pressure measurements remain poor.5–7
The accuracy of blood pressure measurement with aneroid and mercury manometers is affected by a number of observer errors and patient factors.8,9
Observer errors
Failure to prepare the patient. National guidelines5 state that before having their blood pressure taken, patients should be allowed to sit quietly for at least 5 minutes, which often does not happen. Another error is that clinicians rarely discourage patients from smoking cigarettes or drinking coffee in the 30 minutes prior to measurement.
Equipment and layout problems. Equipment should be properly calibrated and validated. 5 However, even if the sphygmomanometer is periodically calibrated, too often it is mounted on the wall adjacent to the examination table in the examination room, making it difficult to provide a comfortable seat with back and arm support during the reading. The measurement should be done with the patient sitting in a chair (not on an examination table), with feet on the floor and the arm supported at the level of the heart. If the forearm is not supported in the horizontal position and with the cuff at heart level, the blood pressure and heart rate tend to be higher.10 Further, the diastolic blood pressure and heart rate may be misleadingly low with the patient supine rather than seated,11,12 so readings should be taken with the patient sitting.
Miscuffing, ie, the use of a blood pressure cuff that is too large or, more often, too small for the patient’s arm, is a common source of error. The cuff bladder should encircle at least 80% of the arm.5 However, some offices do not have a large blood pressure cuff for overweight patients or a pediatric cuff for children or adults with arms of small circumference. It is recommended that a large blood pressure cuff be used routinely in adults, since a smaller cuff gives falsely high readings in people with large upper arms (circumference > 29 cm).13,14
Digit preference. Many physicians round off the blood pressure to the nearest 5 or 10 mm Hg. This problem may go along with:
Deflating the cuff too rapidly.
Talking to the patient while taking the blood pressure can contribute to higher readings.9
Not taking enough readings. Ideally, at the initial visit, blood pressure should be measured in both arms with the patient seated, and another reading should be taken with the patient standing. The arm with the higher pressure should be used for subsequent readings. Physicians should not make any treatment decisions based on blood pressure during an initial clinic visit, and at least two readings should be taken even on subsequent visits. However, owing to time constraints in busy clinical practices, treatment decisions are often based on single readings or on multiple readings on a single visit.
Discrepancies between observers. The blood pressure readings obtained by the nurse or medical assistant may differ significantly from those obtained by the physician. These differences can be large enough to affect treatment decisions,15,16 and they can be partially corrected by adequate training of all medical personnel who take blood pressure, doctors as well as nurses.
Given that time is tight in busy clinical practices and a trained blood pressure nurse or technician is usually not available, we will probably not see any significant improvement in the accuracy of blood pressure measurement using older technology and current physician practices.
The white-coat effect
Most patients have a higher level of anxiety, and therefore higher blood pressure, in the physician’s office or clinic than in their normal environment (as revealed by ambulatory monitoring or home blood pressure measurements), a phenomenon commonly called the white-coat effect.
Several factors can increase this effect, such as observer-patient interaction during the measurement. The effect tends to be greatest in the initial measurement, but can persist through multiple readings by the doctor or nurse during the same visit.
Whether the white-coat effect is due purely to patient anxiety about an office visit or to a conditioned response has been a point of interest in clinical studies. Regardless, it may result in the misdiagnosis of hypertension or in overestimation of the severity of hypertension and may lead to overly aggressive therapy. Antihypertensive treatment may be unnecessary in the absence of concurrent cardiovascular risk factors.17
“White-coat hypertension” or “isolated office hypertension” is the condition in which a patient who is not on antihypertensive drug therapy has persistently elevated blood pressure in the clinic or office (> 140/90 mm Hg) but normal daytime ambulatory blood pressure (< 135/85 mm Hg).18 Since patients may have an elevated reading when seen for a first office visit, at least several visits are required to establish the diagnosis. Multiple studies have suggested that white-coat hypertension may account for 20% to 25% of the hypertensive population, particularly in older patients, mainly women.19,20
Both white-coat hypertension and the white-coat effect can be avoided by using an automatic and programmable device that can take multiple readings after the clinician leaves the examination room (more about this below).21
MEASURING BLOOD PRESSURE OUTSIDE THE OFFICE
Recent studies have reported that the information obtained by 24-hour ambulatory blood pressure monitoring and by self-measurement of blood pressure in the home more accurately reflects the patient’s risk of future cardiovascular events than do conventional blood pressure measurements taken in the physician’s office. 22–24 Current national guidelines recognize this pattern and the frequent measurement inaccuracies observed in clinical practice, and they are recommending including out-of-office measurements in the diagnosis of hypertension. 25,26
Ambulatory monitoring provides the most accurate measurement of out-of-office blood pressure. With ambulatory monitoring, the normal mean daytime pressure is considered to be lower than 135/85 mm Hg, in contrast to the 140/90 mm Hg cutoff used in the physician’s office with standard aneroid or mercury devices.
Self-monitoring of blood pressure at home has now become widely available with single-measurement oscillometric devices. (Oscillometric means that these devices measure the blood pressure by sensing the oscillations in pressure in the cuff induced by the pulsation of the brachial artery, as opposed to auscultating the Korotkoff sounds.) Blood pressures lower than 135/85 mm Hg outside the clinician’s office are considered normal with these devices.
However, despite its proven value, ambulatory monitoring is neither widely available nor cost-effective for the long-term management of hypertension. Furthermore, few physicians recommend that patients take their blood pressure at home, although the information obtained can be of significant value in the patient’s long-term management.
AUTOMATED MEASUREMENT IN THE OFFICE
In recent years, several automated oscillometric sphygmomanometers have been developed for measuring blood pressure in the office, and more are on the way. These devices can be programmed to take multiple readings without a clinician observer in the examination room, thus reducing the white-coat response.
Omron (Kyoto, Japan) makes several devices, including the HEM-907 and the HEM-705, that have been used in the clinical setting. 21,27–29 They can be programmed to take two or three readings at intervals of 1 to 2 minutes, with up to 5 minutes before the first reading. Unfortunately, data were not recorded with the patient alone in the room in many studies of the Omron devices, even though the devices meet national and international standards for accuracy.
The Microlife Watch BP Office (Microlife, Widnau, Switzerland) is currently undergoing development.30
The BpTRU (BpTRU Medical Devices, Coquitlam, BC, Canada) has enjoyed greater clinical acceptance, since it can take up to five blood pressure readings at intervals of 1 to 5 minutes, and calculates the mean of all five readings, taken with the patient resting comfortably in a quiet room without a clinician present.
The accuracy and durability of the device has been well established. Since the BpTRU self-calibrates between every blood pressure measurement, periodic calibration has not been required. The device can be placed on a table, mounted on the wall, or mounted on a cart if used in several locations in the office.
At Cleveland Clinic, several departments are using the BpTRU on a daily basis. Soon, we will be able to transfer data directly from the BpTRU to our electronic medical record system.
Studies of the BpTRU device
To date, most of the studies of automated office blood pressure measurement have used the BpTRU with the recording interval set at 1 to 2 minutes.
Myers31 used the BpTRU device in 50 hypertensive patients. The physician took the patient’s blood pressure with a mercury sphygmomanometer while the BpTRU device made the first reading, and then he left the room. The next five readings were taken at 2-minute intervals with the patient alone in the room. The mean initial reading by the machine was 162/85 mm Hg; the reading by the physician was 163/86 mm Hg. The third automatic reading was the lowest (averaging 140/84 mm Hg), and the mean of the five automated readings was 142/80 mm Hg, which was significantly lower than the initial reading obtained by the physician (P < .001).
In another study, Myers et al32 compared the measurements obtained by 24-hour ambulatory monitoring and by the BpTRU device (the mean of five readings obtained at 1-minute or 2-minute intervals) in 309 hypertensive patients. The mean blood pressure with the Bp-TRU was 132/75 mm Hg, which correlated well with the mean awake ambulatory blood pressure (134/77 mm Hg; r = 0.62 for the systolic pressure and 0.72 for the diastolic pressure).
We recently reviewed the records of 278 patients seen in our preventive medicine clinic (D.G. Vidt, MD, unpublished data, November 2009). The group included patients with and without established hypertension, and among the hypertensive group, both treated and untreated individuals. We had initially set the device to take readings at 3-minute intervals following the initial nurse-initiated reading. But in view of the recent data on the Bp-TRU using shorter intervals, we also obtained readings in 51 patients with the device set to record at 2-minute intervals, and then in 72 additional patients at 1-minute intervals. In all three groups, blood pressure had stabilized by the third reading after the clinician had left the room. These observations support those reported by Myers et al.31,32 Of particular importance is the observation that the white-coat effect dissipates within 2 to 3 minutes after the clinician leaves the room.33
The shorter measurement intervals can add up in a busy office practice, in which the time relegated to taking blood pressure is often limited.
In fact, waiting 5 minutes between measurements may allow the patient to become too relaxed and the blood pressure to drop too low vis-a-vis the gold standard, ambulatory monitoring. Culleton and colleagues34 compared the blood pressure in 107 hypertensive patients as measured four ways: by the referring physician, by a nurse who was trained to adhere to the protocol of the Canadian Hypertension Education Program, by 24-hour ambulatory monitoring, and by the BpTRU (the mean of five readings obtained at 5-minute intervals). The mean measured values were:
150/90 mm Hg by the referring physician
139/86 mm Hg by the nurse
142/85 mm Hg by ambulatory monitoring
132/82 mm Hg by the BpTRU device.
Although the BpTRU reduced the white-coat effect and white-coat hypertension, it underestimated the blood pressure, leading to misclassification of hypertension. Using 140/90 mm Hg as the cutoff for whether the patient was hypertensive and using ambulatory monitoring as the gold standard, the BpTRU misclassified more than half of the patients, agreeing with the classification of hypertensive or not hypertensive by ambulatory monitoring in only 48%. The authors recommended that the BpTRU not be set at 5-minute measurement intervals.34
WHAT ROLE FOR AUTOMATED READINGS IN THE OFFICE?
Although automatic devices, by enabling the physician to leave the room, can eliminate the white-coat effect and white-coat hypertension, physicians must continue to take care to avoid the other potential errors of office blood pressure measurement addressed earlier in this review, for example, by positioning the patient correctly and using a cuff that is large enough. These issues can take on more importance as the clinician leaves the patient alone for brief periods during measurements.
In view of its perennial inaccuracies, some experts have suggested that we abandon routine office measurement of blood pressure.35,36 In its place, ambulatory monitoring would be used for diagnosis and for periodic follow-up. In addition, patients would regularly take their pressure at home with approved, single-measurement oscillometric devices. Unfortunately, in our health care system, periodic ambulatory monitoring for hypertension management would impose a significant financial burden on patients at this time.37
Of particular importance is the observation that the mean of five readings with the BpTRU device, obtained at 1- or 2-minute intervals, closely approximates the mean awake blood pressure obtained in the same patient with an ambulatory monitor.32,38 The ability to obtain readings that correlate exceptionally well with mean daytime ambulatory pressure suggests that this device could well reduce the need for ambulatory monitoring, with its associated cost. The ability to negate the white-coat effect with the use of the BpTRU in the office setting also has particular importance, not only for patient office readings, but for the diagnosis and subsequent treatment of hypertension in individual patients.
Most clinical decisions about the treatment of hypertension are still made on the basis of office determinations of blood pressure. Most office practices still rely on the aneroid manometer or, decreasingly, mercury sphygmomanometers. As noted earlier, although auscultatory blood pressure measurement appears to be simple, it is fraught with a host of observer- or patient-induced errors that not only lead to inaccurate diagnoses, but may also result in the mismanagement of hypertension. Even single-measurement oscillometric devices, now used in a minority of clinical practices, are associated with many of the same measurement issues that lead to overestimation of blood pressure.
We believe the time has come for broader use of oscillometric devices in the outpatient setting. While many available oscillometric devices for use in the home could also be used in the physician’s office, they carry the similar disadvantage of providing only a single measurement. The major disadvantage of all single-measurement devices is the continued presence of the clinician during the reading and the associated white-coat effect observed in most patients.
It is highly likely that the next Joint National Committee Report on Hypertension will further emphasize the role of automated blood pressure devices in the outpatient setting.
Acknowledgment: The authors wish to acknowledge the contributions of Deborah McCoy, RN, and Maria Eckhouse, RN.
References
Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension1995; 25:305–313.
Neaton JD, Wentworth D. Serum cholesterol, blood pressure, cigarette smoking, and death from coronary heart disease. Overall findings and differences by age for 316,099 white men. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med1992; 152:56–64.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet2002; 360:1903–1913.
Lloyd-Jones D, Adams R, Carnethon M, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation2009; 119:e21–e181.
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Grim CM, Grim CE. A curriculum for the training and certification of blood pressure measurement for health care providers. Can J Cardiol1995; 11(suppl H):38H–42H.
Pickering TG, Hall JE, Appel LJ, et al; Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Hypertension2005; 45:142–161.
Langlois S. Measuring blood pressure: how competent are we?Perspect Cardiol1999; 15:29–39.
Le Pailleur C, Helft G, Landais P, et al. The effects of talking, reading, and silence on the “white coat” phenomenon in hypertensive patients. Am J Hypertens1998; 11:203–207.
Webster J, Newnham D, Petrie JC, Lovell HG. Influence of arm position on measurement of blood pressure. Br Med J (Clin Res Ed)1984; 288:1574–1575.
Netea RT, Smits P, Lenders JW, Thien T. Does it matter whether blood pressure measurements are taken with subjects sitting or supine?J Hypertens1998; 16:263–268.
Silverberg DS, Shemesh E, Iaina A. The unsupported arm: a cause of falsely raised blood pressure readings. Br Med J1977; 2:1331.
Iyriboz Y, Hearon CM, Edwards K. Agreement between large and small cuffs in sphygmomanometry: a quantitative assessment. J Clin Monit1994; 10:127–133.
Scherwitz LW, Evans LA, Hennrikus DJ, Vallbona C. Procedures and discrepancies of blood pressure measurements in two community health centers. Med Care1982; 20:727–738.
La Batide-Alanore A, Chatellier G, Bobrie G, Fofol I, Plouin PF. Comparison of nurse- and physician-determined clinic blood pressure levels in patients referred to a hypertension clinic: implications for subsequent management. J Hypertens2000; 18:391–398.
Verdecchia P. Prognostic value of ambulatory blood pressure: current evidence and clinical implications. Hypertension2000; 35:844–851.
Ogedegbe G, Pickering TG, Clemow L, et al. The misdiagnosis of hypertension: the role of patient anxiety. Arch Intern Med2008; 168:2459–2465.
Pickering TG. Stress, white coat hypertension and masked hypertension. In:Izzo JL, Sica DA, Black HR, editors. Hypertension Primer: The Essentials of High Blood Pressure: Basic Science, Population Science, and Clinical Management. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:289–291.
Pickering TG, Coats A, Mallion JM, Mancia G, Verdecchia P. Blood Pressure Monitoring. Task force V: White-coat hypertension. Blood Press Monit1999; 4:333–341.
Myers MG, Valdivieso MA. Use of an automated blood pressure recording device, the BpTRU, to reduce the “white coat effect” in routine practice. Am J Hypertens2003; 16:494–497.
Redon J, Campos C, Narciso ML, Rodicio JL, Pascual JM, Ruilope LM. Prognostic value of ambulatory blood pressure monitoring in refractory hypertension: a prospective study. Hypertension1998; 31:712–718.
Ohkubo T, Imai Y, Tsuji I, et al. Prediction of mortality by ambulatory blood pressure monitoring versus screening blood pressure measurements: a pilot study in Ohasama. J Hypertens1997; 15:357–364.
Verdecchia P, Reboldi G, Porcellati C, et al. Risk of cardiovascular disease in relation to achieved office and ambulatory blood pressure control in treated hypertensive subjects. J Am Coll Cardiol2002; 39:878–885.
Hemmelgarn BR, McAllister FA, Myers MG, et al; Canadian Hypertension Education Program. The 2005 Canadian Hypertension Education Program recommendations for the management of hypertension: part 1 - blood pressure measurement, diagnosis and assessment of risk. Can J Cardiol2005; 21:645–656.
White WB, Anwar YA. Evaluation of the overall efficacy of the Omron office digital blood pressure HEM-907 monitor in adults. Blood Press Monit2001; 6:107–110.
Myers MG, Meglis G, Polemidiotis G. The impact of physician vs automated blood pressure readings on office-induced hypertension. J Hum Hypertens1997; 11:491–493.
Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Measurement of blood pressure in the office: recognizing the problem and proposing the solution. Hypertension2010; 55:195–200.
Stergiou GS, Tzamouranis D, Protogerou A, Nasothimiou E, Kapralos C. Validation of the Microlife Watch BP Office professional device for office blood pressure measurement according to the International protocol. Blood Press Monit2008; 13:299–303.
Myers MG, Valdivieso M, Kiss A. Optimum frequency of office blood pressure measurement using an automated sphygmomanometer. Blood Press Monit2008; 13:333–338.
Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce the white coat response. J Hypertens2009; 27:280–286.
Culleton BF, McKay DW, Campbell NR. Performance of the automated BpTRU measurement device in the assessment of white-coat hypertension and white-coat effect. Blood Press Monit2006; 11:37–42.
Pickering TG, Miller NH, Ogedegbe G, Krakoff LR, Artinian NT, Goff D; American Heart Association. Call to action on use and reimbursement for home blood pressure monitoring: executive summary: a joint scientific statement from the American Heart Association, American Society Of Hypertension, and Preventive Cardiovascular Nurses Association. Hypertension2008; 52:1–9.
Parati G, Stergiou GS, Asmar R, et al; ESH Working Group on Blood Pressure Monitoring. European Society of Hypertension guidelines for blood pressure monitoring at home: a summary report of the Second International Consensus Conference on Home Blood Pressure Monitoring. J Hypertens2008; 26:1505–1526.
O’Brien E. Ambulatory blood pressure measurement: the case for implementation in primary care. Hypertension2008; 51:1435–1441.
Beckett L, Godwin M. The BpTRU automatic blood pressure monitor compared to 24 hour ambulatory blood pressure monitoring in the assessment of blood pressure in patients with hypertension. BMC Cardiovasc Disord2005; 5:18.
Donald G. Vidt, MD Chairman Emeritus and Consultant, Department of Nephrology and Hypertension, Cleveland Clinic; Member, Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure
Richard S. Lang, MD, MPH Chair, Department of Preventive Medicine; Vice Chair, Wellness Institute, Cleveland Clinic
Raul J. Seballos, MD Vice Chair, Department of Preventive Medicine, Cleveland Clinic
Anita Misra-Hebert, MD Department of Preventive Medicine, Cleveland Clinic
John Campbell, MD Department of Preventive Medicine, Cleveland Clinic
James F. Bena, MS Department of Quantitative Health Sciences, Cleveland Clinic
Address: Donald G. Vidt, MD, Emeritus Office AC334, Cleveland Clinic, 3050 Science Park Drive, Beachwood, OH 44122; e-mail [email protected]
Dr. Vidt has disclosed that he has received consulting fees from Astra-Zeneca Pharmaceuticals.
Donald G. Vidt, MD Chairman Emeritus and Consultant, Department of Nephrology and Hypertension, Cleveland Clinic; Member, Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure
Richard S. Lang, MD, MPH Chair, Department of Preventive Medicine; Vice Chair, Wellness Institute, Cleveland Clinic
Raul J. Seballos, MD Vice Chair, Department of Preventive Medicine, Cleveland Clinic
Anita Misra-Hebert, MD Department of Preventive Medicine, Cleveland Clinic
John Campbell, MD Department of Preventive Medicine, Cleveland Clinic
James F. Bena, MS Department of Quantitative Health Sciences, Cleveland Clinic
Address: Donald G. Vidt, MD, Emeritus Office AC334, Cleveland Clinic, 3050 Science Park Drive, Beachwood, OH 44122; e-mail [email protected]
Dr. Vidt has disclosed that he has received consulting fees from Astra-Zeneca Pharmaceuticals.
Author and Disclosure Information
Donald G. Vidt, MD Chairman Emeritus and Consultant, Department of Nephrology and Hypertension, Cleveland Clinic; Member, Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure
Richard S. Lang, MD, MPH Chair, Department of Preventive Medicine; Vice Chair, Wellness Institute, Cleveland Clinic
Raul J. Seballos, MD Vice Chair, Department of Preventive Medicine, Cleveland Clinic
Anita Misra-Hebert, MD Department of Preventive Medicine, Cleveland Clinic
John Campbell, MD Department of Preventive Medicine, Cleveland Clinic
James F. Bena, MS Department of Quantitative Health Sciences, Cleveland Clinic
Address: Donald G. Vidt, MD, Emeritus Office AC334, Cleveland Clinic, 3050 Science Park Drive, Beachwood, OH 44122; e-mail [email protected]
Dr. Vidt has disclosed that he has received consulting fees from Astra-Zeneca Pharmaceuticals.
The reality of blood pressure measurement is that human beings do not do it very well. The time has come to delegate this job to machines that can do it better.
Several automatic devices are available. Used in physicians’ offices and in patients’ homes, they can eliminate some types of observer error as well as the “white-coat effect,” ie, the tendency of some patients to have higher blood pressure when medical personnel are present than in their natural environment. Since a difference of only a few millimeters of mercury can affect the physician’s decision to start or to modify treatment, measurements that more accurately reflect a person’s average blood pressure are to be desired.
In the following pages, we review the problems that plague blood pressure measurement by human observers, and we describe the advantages of automatic devices.
SHORTCOMINGS OF OFFICE BLOOD PRESSURE MEASUREMENT
For decades, large surveys have provided invaluable information on the prevalence of hypertension, its relationship to cardiovascular disease, and the benefits of treating it.1–3 Unfortunately, the percentage of hypertensive patients whose blood pressure is under control has remained low despite our increased knowledge about hypertension’s diagnosis and therapy.4
In these surveys, blood pressure was measured by auscultation by human observers using mercury or aneroid sphygmomanometers, and most physicians still use this method in clinical practice. But in spite of multiple guidelines for accurate measurement of blood pressure in the office, the overall accuracy and reproducibility of office blood pressure measurements remain poor.5–7
The accuracy of blood pressure measurement with aneroid and mercury manometers is affected by a number of observer errors and patient factors.8,9
Observer errors
Failure to prepare the patient. National guidelines5 state that before having their blood pressure taken, patients should be allowed to sit quietly for at least 5 minutes, which often does not happen. Another error is that clinicians rarely discourage patients from smoking cigarettes or drinking coffee in the 30 minutes prior to measurement.
Equipment and layout problems. Equipment should be properly calibrated and validated. 5 However, even if the sphygmomanometer is periodically calibrated, too often it is mounted on the wall adjacent to the examination table in the examination room, making it difficult to provide a comfortable seat with back and arm support during the reading. The measurement should be done with the patient sitting in a chair (not on an examination table), with feet on the floor and the arm supported at the level of the heart. If the forearm is not supported in the horizontal position and with the cuff at heart level, the blood pressure and heart rate tend to be higher.10 Further, the diastolic blood pressure and heart rate may be misleadingly low with the patient supine rather than seated,11,12 so readings should be taken with the patient sitting.
Miscuffing, ie, the use of a blood pressure cuff that is too large or, more often, too small for the patient’s arm, is a common source of error. The cuff bladder should encircle at least 80% of the arm.5 However, some offices do not have a large blood pressure cuff for overweight patients or a pediatric cuff for children or adults with arms of small circumference. It is recommended that a large blood pressure cuff be used routinely in adults, since a smaller cuff gives falsely high readings in people with large upper arms (circumference > 29 cm).13,14
Digit preference. Many physicians round off the blood pressure to the nearest 5 or 10 mm Hg. This problem may go along with:
Deflating the cuff too rapidly.
Talking to the patient while taking the blood pressure can contribute to higher readings.9
Not taking enough readings. Ideally, at the initial visit, blood pressure should be measured in both arms with the patient seated, and another reading should be taken with the patient standing. The arm with the higher pressure should be used for subsequent readings. Physicians should not make any treatment decisions based on blood pressure during an initial clinic visit, and at least two readings should be taken even on subsequent visits. However, owing to time constraints in busy clinical practices, treatment decisions are often based on single readings or on multiple readings on a single visit.
Discrepancies between observers. The blood pressure readings obtained by the nurse or medical assistant may differ significantly from those obtained by the physician. These differences can be large enough to affect treatment decisions,15,16 and they can be partially corrected by adequate training of all medical personnel who take blood pressure, doctors as well as nurses.
Given that time is tight in busy clinical practices and a trained blood pressure nurse or technician is usually not available, we will probably not see any significant improvement in the accuracy of blood pressure measurement using older technology and current physician practices.
The white-coat effect
Most patients have a higher level of anxiety, and therefore higher blood pressure, in the physician’s office or clinic than in their normal environment (as revealed by ambulatory monitoring or home blood pressure measurements), a phenomenon commonly called the white-coat effect.
Several factors can increase this effect, such as observer-patient interaction during the measurement. The effect tends to be greatest in the initial measurement, but can persist through multiple readings by the doctor or nurse during the same visit.
Whether the white-coat effect is due purely to patient anxiety about an office visit or to a conditioned response has been a point of interest in clinical studies. Regardless, it may result in the misdiagnosis of hypertension or in overestimation of the severity of hypertension and may lead to overly aggressive therapy. Antihypertensive treatment may be unnecessary in the absence of concurrent cardiovascular risk factors.17
“White-coat hypertension” or “isolated office hypertension” is the condition in which a patient who is not on antihypertensive drug therapy has persistently elevated blood pressure in the clinic or office (> 140/90 mm Hg) but normal daytime ambulatory blood pressure (< 135/85 mm Hg).18 Since patients may have an elevated reading when seen for a first office visit, at least several visits are required to establish the diagnosis. Multiple studies have suggested that white-coat hypertension may account for 20% to 25% of the hypertensive population, particularly in older patients, mainly women.19,20
Both white-coat hypertension and the white-coat effect can be avoided by using an automatic and programmable device that can take multiple readings after the clinician leaves the examination room (more about this below).21
MEASURING BLOOD PRESSURE OUTSIDE THE OFFICE
Recent studies have reported that the information obtained by 24-hour ambulatory blood pressure monitoring and by self-measurement of blood pressure in the home more accurately reflects the patient’s risk of future cardiovascular events than do conventional blood pressure measurements taken in the physician’s office. 22–24 Current national guidelines recognize this pattern and the frequent measurement inaccuracies observed in clinical practice, and they are recommending including out-of-office measurements in the diagnosis of hypertension. 25,26
Ambulatory monitoring provides the most accurate measurement of out-of-office blood pressure. With ambulatory monitoring, the normal mean daytime pressure is considered to be lower than 135/85 mm Hg, in contrast to the 140/90 mm Hg cutoff used in the physician’s office with standard aneroid or mercury devices.
Self-monitoring of blood pressure at home has now become widely available with single-measurement oscillometric devices. (Oscillometric means that these devices measure the blood pressure by sensing the oscillations in pressure in the cuff induced by the pulsation of the brachial artery, as opposed to auscultating the Korotkoff sounds.) Blood pressures lower than 135/85 mm Hg outside the clinician’s office are considered normal with these devices.
However, despite its proven value, ambulatory monitoring is neither widely available nor cost-effective for the long-term management of hypertension. Furthermore, few physicians recommend that patients take their blood pressure at home, although the information obtained can be of significant value in the patient’s long-term management.
AUTOMATED MEASUREMENT IN THE OFFICE
In recent years, several automated oscillometric sphygmomanometers have been developed for measuring blood pressure in the office, and more are on the way. These devices can be programmed to take multiple readings without a clinician observer in the examination room, thus reducing the white-coat response.
Omron (Kyoto, Japan) makes several devices, including the HEM-907 and the HEM-705, that have been used in the clinical setting. 21,27–29 They can be programmed to take two or three readings at intervals of 1 to 2 minutes, with up to 5 minutes before the first reading. Unfortunately, data were not recorded with the patient alone in the room in many studies of the Omron devices, even though the devices meet national and international standards for accuracy.
The Microlife Watch BP Office (Microlife, Widnau, Switzerland) is currently undergoing development.30
The BpTRU (BpTRU Medical Devices, Coquitlam, BC, Canada) has enjoyed greater clinical acceptance, since it can take up to five blood pressure readings at intervals of 1 to 5 minutes, and calculates the mean of all five readings, taken with the patient resting comfortably in a quiet room without a clinician present.
The accuracy and durability of the device has been well established. Since the BpTRU self-calibrates between every blood pressure measurement, periodic calibration has not been required. The device can be placed on a table, mounted on the wall, or mounted on a cart if used in several locations in the office.
At Cleveland Clinic, several departments are using the BpTRU on a daily basis. Soon, we will be able to transfer data directly from the BpTRU to our electronic medical record system.
Studies of the BpTRU device
To date, most of the studies of automated office blood pressure measurement have used the BpTRU with the recording interval set at 1 to 2 minutes.
Myers31 used the BpTRU device in 50 hypertensive patients. The physician took the patient’s blood pressure with a mercury sphygmomanometer while the BpTRU device made the first reading, and then he left the room. The next five readings were taken at 2-minute intervals with the patient alone in the room. The mean initial reading by the machine was 162/85 mm Hg; the reading by the physician was 163/86 mm Hg. The third automatic reading was the lowest (averaging 140/84 mm Hg), and the mean of the five automated readings was 142/80 mm Hg, which was significantly lower than the initial reading obtained by the physician (P < .001).
In another study, Myers et al32 compared the measurements obtained by 24-hour ambulatory monitoring and by the BpTRU device (the mean of five readings obtained at 1-minute or 2-minute intervals) in 309 hypertensive patients. The mean blood pressure with the Bp-TRU was 132/75 mm Hg, which correlated well with the mean awake ambulatory blood pressure (134/77 mm Hg; r = 0.62 for the systolic pressure and 0.72 for the diastolic pressure).
We recently reviewed the records of 278 patients seen in our preventive medicine clinic (D.G. Vidt, MD, unpublished data, November 2009). The group included patients with and without established hypertension, and among the hypertensive group, both treated and untreated individuals. We had initially set the device to take readings at 3-minute intervals following the initial nurse-initiated reading. But in view of the recent data on the Bp-TRU using shorter intervals, we also obtained readings in 51 patients with the device set to record at 2-minute intervals, and then in 72 additional patients at 1-minute intervals. In all three groups, blood pressure had stabilized by the third reading after the clinician had left the room. These observations support those reported by Myers et al.31,32 Of particular importance is the observation that the white-coat effect dissipates within 2 to 3 minutes after the clinician leaves the room.33
The shorter measurement intervals can add up in a busy office practice, in which the time relegated to taking blood pressure is often limited.
In fact, waiting 5 minutes between measurements may allow the patient to become too relaxed and the blood pressure to drop too low vis-a-vis the gold standard, ambulatory monitoring. Culleton and colleagues34 compared the blood pressure in 107 hypertensive patients as measured four ways: by the referring physician, by a nurse who was trained to adhere to the protocol of the Canadian Hypertension Education Program, by 24-hour ambulatory monitoring, and by the BpTRU (the mean of five readings obtained at 5-minute intervals). The mean measured values were:
150/90 mm Hg by the referring physician
139/86 mm Hg by the nurse
142/85 mm Hg by ambulatory monitoring
132/82 mm Hg by the BpTRU device.
Although the BpTRU reduced the white-coat effect and white-coat hypertension, it underestimated the blood pressure, leading to misclassification of hypertension. Using 140/90 mm Hg as the cutoff for whether the patient was hypertensive and using ambulatory monitoring as the gold standard, the BpTRU misclassified more than half of the patients, agreeing with the classification of hypertensive or not hypertensive by ambulatory monitoring in only 48%. The authors recommended that the BpTRU not be set at 5-minute measurement intervals.34
WHAT ROLE FOR AUTOMATED READINGS IN THE OFFICE?
Although automatic devices, by enabling the physician to leave the room, can eliminate the white-coat effect and white-coat hypertension, physicians must continue to take care to avoid the other potential errors of office blood pressure measurement addressed earlier in this review, for example, by positioning the patient correctly and using a cuff that is large enough. These issues can take on more importance as the clinician leaves the patient alone for brief periods during measurements.
In view of its perennial inaccuracies, some experts have suggested that we abandon routine office measurement of blood pressure.35,36 In its place, ambulatory monitoring would be used for diagnosis and for periodic follow-up. In addition, patients would regularly take their pressure at home with approved, single-measurement oscillometric devices. Unfortunately, in our health care system, periodic ambulatory monitoring for hypertension management would impose a significant financial burden on patients at this time.37
Of particular importance is the observation that the mean of five readings with the BpTRU device, obtained at 1- or 2-minute intervals, closely approximates the mean awake blood pressure obtained in the same patient with an ambulatory monitor.32,38 The ability to obtain readings that correlate exceptionally well with mean daytime ambulatory pressure suggests that this device could well reduce the need for ambulatory monitoring, with its associated cost. The ability to negate the white-coat effect with the use of the BpTRU in the office setting also has particular importance, not only for patient office readings, but for the diagnosis and subsequent treatment of hypertension in individual patients.
Most clinical decisions about the treatment of hypertension are still made on the basis of office determinations of blood pressure. Most office practices still rely on the aneroid manometer or, decreasingly, mercury sphygmomanometers. As noted earlier, although auscultatory blood pressure measurement appears to be simple, it is fraught with a host of observer- or patient-induced errors that not only lead to inaccurate diagnoses, but may also result in the mismanagement of hypertension. Even single-measurement oscillometric devices, now used in a minority of clinical practices, are associated with many of the same measurement issues that lead to overestimation of blood pressure.
We believe the time has come for broader use of oscillometric devices in the outpatient setting. While many available oscillometric devices for use in the home could also be used in the physician’s office, they carry the similar disadvantage of providing only a single measurement. The major disadvantage of all single-measurement devices is the continued presence of the clinician during the reading and the associated white-coat effect observed in most patients.
It is highly likely that the next Joint National Committee Report on Hypertension will further emphasize the role of automated blood pressure devices in the outpatient setting.
Acknowledgment: The authors wish to acknowledge the contributions of Deborah McCoy, RN, and Maria Eckhouse, RN.
The reality of blood pressure measurement is that human beings do not do it very well. The time has come to delegate this job to machines that can do it better.
Several automatic devices are available. Used in physicians’ offices and in patients’ homes, they can eliminate some types of observer error as well as the “white-coat effect,” ie, the tendency of some patients to have higher blood pressure when medical personnel are present than in their natural environment. Since a difference of only a few millimeters of mercury can affect the physician’s decision to start or to modify treatment, measurements that more accurately reflect a person’s average blood pressure are to be desired.
In the following pages, we review the problems that plague blood pressure measurement by human observers, and we describe the advantages of automatic devices.
SHORTCOMINGS OF OFFICE BLOOD PRESSURE MEASUREMENT
For decades, large surveys have provided invaluable information on the prevalence of hypertension, its relationship to cardiovascular disease, and the benefits of treating it.1–3 Unfortunately, the percentage of hypertensive patients whose blood pressure is under control has remained low despite our increased knowledge about hypertension’s diagnosis and therapy.4
In these surveys, blood pressure was measured by auscultation by human observers using mercury or aneroid sphygmomanometers, and most physicians still use this method in clinical practice. But in spite of multiple guidelines for accurate measurement of blood pressure in the office, the overall accuracy and reproducibility of office blood pressure measurements remain poor.5–7
The accuracy of blood pressure measurement with aneroid and mercury manometers is affected by a number of observer errors and patient factors.8,9
Observer errors
Failure to prepare the patient. National guidelines5 state that before having their blood pressure taken, patients should be allowed to sit quietly for at least 5 minutes, which often does not happen. Another error is that clinicians rarely discourage patients from smoking cigarettes or drinking coffee in the 30 minutes prior to measurement.
Equipment and layout problems. Equipment should be properly calibrated and validated. 5 However, even if the sphygmomanometer is periodically calibrated, too often it is mounted on the wall adjacent to the examination table in the examination room, making it difficult to provide a comfortable seat with back and arm support during the reading. The measurement should be done with the patient sitting in a chair (not on an examination table), with feet on the floor and the arm supported at the level of the heart. If the forearm is not supported in the horizontal position and with the cuff at heart level, the blood pressure and heart rate tend to be higher.10 Further, the diastolic blood pressure and heart rate may be misleadingly low with the patient supine rather than seated,11,12 so readings should be taken with the patient sitting.
Miscuffing, ie, the use of a blood pressure cuff that is too large or, more often, too small for the patient’s arm, is a common source of error. The cuff bladder should encircle at least 80% of the arm.5 However, some offices do not have a large blood pressure cuff for overweight patients or a pediatric cuff for children or adults with arms of small circumference. It is recommended that a large blood pressure cuff be used routinely in adults, since a smaller cuff gives falsely high readings in people with large upper arms (circumference > 29 cm).13,14
Digit preference. Many physicians round off the blood pressure to the nearest 5 or 10 mm Hg. This problem may go along with:
Deflating the cuff too rapidly.
Talking to the patient while taking the blood pressure can contribute to higher readings.9
Not taking enough readings. Ideally, at the initial visit, blood pressure should be measured in both arms with the patient seated, and another reading should be taken with the patient standing. The arm with the higher pressure should be used for subsequent readings. Physicians should not make any treatment decisions based on blood pressure during an initial clinic visit, and at least two readings should be taken even on subsequent visits. However, owing to time constraints in busy clinical practices, treatment decisions are often based on single readings or on multiple readings on a single visit.
Discrepancies between observers. The blood pressure readings obtained by the nurse or medical assistant may differ significantly from those obtained by the physician. These differences can be large enough to affect treatment decisions,15,16 and they can be partially corrected by adequate training of all medical personnel who take blood pressure, doctors as well as nurses.
Given that time is tight in busy clinical practices and a trained blood pressure nurse or technician is usually not available, we will probably not see any significant improvement in the accuracy of blood pressure measurement using older technology and current physician practices.
The white-coat effect
Most patients have a higher level of anxiety, and therefore higher blood pressure, in the physician’s office or clinic than in their normal environment (as revealed by ambulatory monitoring or home blood pressure measurements), a phenomenon commonly called the white-coat effect.
Several factors can increase this effect, such as observer-patient interaction during the measurement. The effect tends to be greatest in the initial measurement, but can persist through multiple readings by the doctor or nurse during the same visit.
Whether the white-coat effect is due purely to patient anxiety about an office visit or to a conditioned response has been a point of interest in clinical studies. Regardless, it may result in the misdiagnosis of hypertension or in overestimation of the severity of hypertension and may lead to overly aggressive therapy. Antihypertensive treatment may be unnecessary in the absence of concurrent cardiovascular risk factors.17
“White-coat hypertension” or “isolated office hypertension” is the condition in which a patient who is not on antihypertensive drug therapy has persistently elevated blood pressure in the clinic or office (> 140/90 mm Hg) but normal daytime ambulatory blood pressure (< 135/85 mm Hg).18 Since patients may have an elevated reading when seen for a first office visit, at least several visits are required to establish the diagnosis. Multiple studies have suggested that white-coat hypertension may account for 20% to 25% of the hypertensive population, particularly in older patients, mainly women.19,20
Both white-coat hypertension and the white-coat effect can be avoided by using an automatic and programmable device that can take multiple readings after the clinician leaves the examination room (more about this below).21
MEASURING BLOOD PRESSURE OUTSIDE THE OFFICE
Recent studies have reported that the information obtained by 24-hour ambulatory blood pressure monitoring and by self-measurement of blood pressure in the home more accurately reflects the patient’s risk of future cardiovascular events than do conventional blood pressure measurements taken in the physician’s office. 22–24 Current national guidelines recognize this pattern and the frequent measurement inaccuracies observed in clinical practice, and they are recommending including out-of-office measurements in the diagnosis of hypertension. 25,26
Ambulatory monitoring provides the most accurate measurement of out-of-office blood pressure. With ambulatory monitoring, the normal mean daytime pressure is considered to be lower than 135/85 mm Hg, in contrast to the 140/90 mm Hg cutoff used in the physician’s office with standard aneroid or mercury devices.
Self-monitoring of blood pressure at home has now become widely available with single-measurement oscillometric devices. (Oscillometric means that these devices measure the blood pressure by sensing the oscillations in pressure in the cuff induced by the pulsation of the brachial artery, as opposed to auscultating the Korotkoff sounds.) Blood pressures lower than 135/85 mm Hg outside the clinician’s office are considered normal with these devices.
However, despite its proven value, ambulatory monitoring is neither widely available nor cost-effective for the long-term management of hypertension. Furthermore, few physicians recommend that patients take their blood pressure at home, although the information obtained can be of significant value in the patient’s long-term management.
AUTOMATED MEASUREMENT IN THE OFFICE
In recent years, several automated oscillometric sphygmomanometers have been developed for measuring blood pressure in the office, and more are on the way. These devices can be programmed to take multiple readings without a clinician observer in the examination room, thus reducing the white-coat response.
Omron (Kyoto, Japan) makes several devices, including the HEM-907 and the HEM-705, that have been used in the clinical setting. 21,27–29 They can be programmed to take two or three readings at intervals of 1 to 2 minutes, with up to 5 minutes before the first reading. Unfortunately, data were not recorded with the patient alone in the room in many studies of the Omron devices, even though the devices meet national and international standards for accuracy.
The Microlife Watch BP Office (Microlife, Widnau, Switzerland) is currently undergoing development.30
The BpTRU (BpTRU Medical Devices, Coquitlam, BC, Canada) has enjoyed greater clinical acceptance, since it can take up to five blood pressure readings at intervals of 1 to 5 minutes, and calculates the mean of all five readings, taken with the patient resting comfortably in a quiet room without a clinician present.
The accuracy and durability of the device has been well established. Since the BpTRU self-calibrates between every blood pressure measurement, periodic calibration has not been required. The device can be placed on a table, mounted on the wall, or mounted on a cart if used in several locations in the office.
At Cleveland Clinic, several departments are using the BpTRU on a daily basis. Soon, we will be able to transfer data directly from the BpTRU to our electronic medical record system.
Studies of the BpTRU device
To date, most of the studies of automated office blood pressure measurement have used the BpTRU with the recording interval set at 1 to 2 minutes.
Myers31 used the BpTRU device in 50 hypertensive patients. The physician took the patient’s blood pressure with a mercury sphygmomanometer while the BpTRU device made the first reading, and then he left the room. The next five readings were taken at 2-minute intervals with the patient alone in the room. The mean initial reading by the machine was 162/85 mm Hg; the reading by the physician was 163/86 mm Hg. The third automatic reading was the lowest (averaging 140/84 mm Hg), and the mean of the five automated readings was 142/80 mm Hg, which was significantly lower than the initial reading obtained by the physician (P < .001).
In another study, Myers et al32 compared the measurements obtained by 24-hour ambulatory monitoring and by the BpTRU device (the mean of five readings obtained at 1-minute or 2-minute intervals) in 309 hypertensive patients. The mean blood pressure with the Bp-TRU was 132/75 mm Hg, which correlated well with the mean awake ambulatory blood pressure (134/77 mm Hg; r = 0.62 for the systolic pressure and 0.72 for the diastolic pressure).
We recently reviewed the records of 278 patients seen in our preventive medicine clinic (D.G. Vidt, MD, unpublished data, November 2009). The group included patients with and without established hypertension, and among the hypertensive group, both treated and untreated individuals. We had initially set the device to take readings at 3-minute intervals following the initial nurse-initiated reading. But in view of the recent data on the Bp-TRU using shorter intervals, we also obtained readings in 51 patients with the device set to record at 2-minute intervals, and then in 72 additional patients at 1-minute intervals. In all three groups, blood pressure had stabilized by the third reading after the clinician had left the room. These observations support those reported by Myers et al.31,32 Of particular importance is the observation that the white-coat effect dissipates within 2 to 3 minutes after the clinician leaves the room.33
The shorter measurement intervals can add up in a busy office practice, in which the time relegated to taking blood pressure is often limited.
In fact, waiting 5 minutes between measurements may allow the patient to become too relaxed and the blood pressure to drop too low vis-a-vis the gold standard, ambulatory monitoring. Culleton and colleagues34 compared the blood pressure in 107 hypertensive patients as measured four ways: by the referring physician, by a nurse who was trained to adhere to the protocol of the Canadian Hypertension Education Program, by 24-hour ambulatory monitoring, and by the BpTRU (the mean of five readings obtained at 5-minute intervals). The mean measured values were:
150/90 mm Hg by the referring physician
139/86 mm Hg by the nurse
142/85 mm Hg by ambulatory monitoring
132/82 mm Hg by the BpTRU device.
Although the BpTRU reduced the white-coat effect and white-coat hypertension, it underestimated the blood pressure, leading to misclassification of hypertension. Using 140/90 mm Hg as the cutoff for whether the patient was hypertensive and using ambulatory monitoring as the gold standard, the BpTRU misclassified more than half of the patients, agreeing with the classification of hypertensive or not hypertensive by ambulatory monitoring in only 48%. The authors recommended that the BpTRU not be set at 5-minute measurement intervals.34
WHAT ROLE FOR AUTOMATED READINGS IN THE OFFICE?
Although automatic devices, by enabling the physician to leave the room, can eliminate the white-coat effect and white-coat hypertension, physicians must continue to take care to avoid the other potential errors of office blood pressure measurement addressed earlier in this review, for example, by positioning the patient correctly and using a cuff that is large enough. These issues can take on more importance as the clinician leaves the patient alone for brief periods during measurements.
In view of its perennial inaccuracies, some experts have suggested that we abandon routine office measurement of blood pressure.35,36 In its place, ambulatory monitoring would be used for diagnosis and for periodic follow-up. In addition, patients would regularly take their pressure at home with approved, single-measurement oscillometric devices. Unfortunately, in our health care system, periodic ambulatory monitoring for hypertension management would impose a significant financial burden on patients at this time.37
Of particular importance is the observation that the mean of five readings with the BpTRU device, obtained at 1- or 2-minute intervals, closely approximates the mean awake blood pressure obtained in the same patient with an ambulatory monitor.32,38 The ability to obtain readings that correlate exceptionally well with mean daytime ambulatory pressure suggests that this device could well reduce the need for ambulatory monitoring, with its associated cost. The ability to negate the white-coat effect with the use of the BpTRU in the office setting also has particular importance, not only for patient office readings, but for the diagnosis and subsequent treatment of hypertension in individual patients.
Most clinical decisions about the treatment of hypertension are still made on the basis of office determinations of blood pressure. Most office practices still rely on the aneroid manometer or, decreasingly, mercury sphygmomanometers. As noted earlier, although auscultatory blood pressure measurement appears to be simple, it is fraught with a host of observer- or patient-induced errors that not only lead to inaccurate diagnoses, but may also result in the mismanagement of hypertension. Even single-measurement oscillometric devices, now used in a minority of clinical practices, are associated with many of the same measurement issues that lead to overestimation of blood pressure.
We believe the time has come for broader use of oscillometric devices in the outpatient setting. While many available oscillometric devices for use in the home could also be used in the physician’s office, they carry the similar disadvantage of providing only a single measurement. The major disadvantage of all single-measurement devices is the continued presence of the clinician during the reading and the associated white-coat effect observed in most patients.
It is highly likely that the next Joint National Committee Report on Hypertension will further emphasize the role of automated blood pressure devices in the outpatient setting.
Acknowledgment: The authors wish to acknowledge the contributions of Deborah McCoy, RN, and Maria Eckhouse, RN.
References
Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension1995; 25:305–313.
Neaton JD, Wentworth D. Serum cholesterol, blood pressure, cigarette smoking, and death from coronary heart disease. Overall findings and differences by age for 316,099 white men. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med1992; 152:56–64.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet2002; 360:1903–1913.
Lloyd-Jones D, Adams R, Carnethon M, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation2009; 119:e21–e181.
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Grim CM, Grim CE. A curriculum for the training and certification of blood pressure measurement for health care providers. Can J Cardiol1995; 11(suppl H):38H–42H.
Pickering TG, Hall JE, Appel LJ, et al; Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Hypertension2005; 45:142–161.
Langlois S. Measuring blood pressure: how competent are we?Perspect Cardiol1999; 15:29–39.
Le Pailleur C, Helft G, Landais P, et al. The effects of talking, reading, and silence on the “white coat” phenomenon in hypertensive patients. Am J Hypertens1998; 11:203–207.
Webster J, Newnham D, Petrie JC, Lovell HG. Influence of arm position on measurement of blood pressure. Br Med J (Clin Res Ed)1984; 288:1574–1575.
Netea RT, Smits P, Lenders JW, Thien T. Does it matter whether blood pressure measurements are taken with subjects sitting or supine?J Hypertens1998; 16:263–268.
Silverberg DS, Shemesh E, Iaina A. The unsupported arm: a cause of falsely raised blood pressure readings. Br Med J1977; 2:1331.
Iyriboz Y, Hearon CM, Edwards K. Agreement between large and small cuffs in sphygmomanometry: a quantitative assessment. J Clin Monit1994; 10:127–133.
Scherwitz LW, Evans LA, Hennrikus DJ, Vallbona C. Procedures and discrepancies of blood pressure measurements in two community health centers. Med Care1982; 20:727–738.
La Batide-Alanore A, Chatellier G, Bobrie G, Fofol I, Plouin PF. Comparison of nurse- and physician-determined clinic blood pressure levels in patients referred to a hypertension clinic: implications for subsequent management. J Hypertens2000; 18:391–398.
Verdecchia P. Prognostic value of ambulatory blood pressure: current evidence and clinical implications. Hypertension2000; 35:844–851.
Ogedegbe G, Pickering TG, Clemow L, et al. The misdiagnosis of hypertension: the role of patient anxiety. Arch Intern Med2008; 168:2459–2465.
Pickering TG. Stress, white coat hypertension and masked hypertension. In:Izzo JL, Sica DA, Black HR, editors. Hypertension Primer: The Essentials of High Blood Pressure: Basic Science, Population Science, and Clinical Management. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:289–291.
Pickering TG, Coats A, Mallion JM, Mancia G, Verdecchia P. Blood Pressure Monitoring. Task force V: White-coat hypertension. Blood Press Monit1999; 4:333–341.
Myers MG, Valdivieso MA. Use of an automated blood pressure recording device, the BpTRU, to reduce the “white coat effect” in routine practice. Am J Hypertens2003; 16:494–497.
Redon J, Campos C, Narciso ML, Rodicio JL, Pascual JM, Ruilope LM. Prognostic value of ambulatory blood pressure monitoring in refractory hypertension: a prospective study. Hypertension1998; 31:712–718.
Ohkubo T, Imai Y, Tsuji I, et al. Prediction of mortality by ambulatory blood pressure monitoring versus screening blood pressure measurements: a pilot study in Ohasama. J Hypertens1997; 15:357–364.
Verdecchia P, Reboldi G, Porcellati C, et al. Risk of cardiovascular disease in relation to achieved office and ambulatory blood pressure control in treated hypertensive subjects. J Am Coll Cardiol2002; 39:878–885.
Hemmelgarn BR, McAllister FA, Myers MG, et al; Canadian Hypertension Education Program. The 2005 Canadian Hypertension Education Program recommendations for the management of hypertension: part 1 - blood pressure measurement, diagnosis and assessment of risk. Can J Cardiol2005; 21:645–656.
White WB, Anwar YA. Evaluation of the overall efficacy of the Omron office digital blood pressure HEM-907 monitor in adults. Blood Press Monit2001; 6:107–110.
Myers MG, Meglis G, Polemidiotis G. The impact of physician vs automated blood pressure readings on office-induced hypertension. J Hum Hypertens1997; 11:491–493.
Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Measurement of blood pressure in the office: recognizing the problem and proposing the solution. Hypertension2010; 55:195–200.
Stergiou GS, Tzamouranis D, Protogerou A, Nasothimiou E, Kapralos C. Validation of the Microlife Watch BP Office professional device for office blood pressure measurement according to the International protocol. Blood Press Monit2008; 13:299–303.
Myers MG, Valdivieso M, Kiss A. Optimum frequency of office blood pressure measurement using an automated sphygmomanometer. Blood Press Monit2008; 13:333–338.
Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce the white coat response. J Hypertens2009; 27:280–286.
Culleton BF, McKay DW, Campbell NR. Performance of the automated BpTRU measurement device in the assessment of white-coat hypertension and white-coat effect. Blood Press Monit2006; 11:37–42.
Pickering TG, Miller NH, Ogedegbe G, Krakoff LR, Artinian NT, Goff D; American Heart Association. Call to action on use and reimbursement for home blood pressure monitoring: executive summary: a joint scientific statement from the American Heart Association, American Society Of Hypertension, and Preventive Cardiovascular Nurses Association. Hypertension2008; 52:1–9.
Parati G, Stergiou GS, Asmar R, et al; ESH Working Group on Blood Pressure Monitoring. European Society of Hypertension guidelines for blood pressure monitoring at home: a summary report of the Second International Consensus Conference on Home Blood Pressure Monitoring. J Hypertens2008; 26:1505–1526.
O’Brien E. Ambulatory blood pressure measurement: the case for implementation in primary care. Hypertension2008; 51:1435–1441.
Beckett L, Godwin M. The BpTRU automatic blood pressure monitor compared to 24 hour ambulatory blood pressure monitoring in the assessment of blood pressure in patients with hypertension. BMC Cardiovasc Disord2005; 5:18.
References
Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension1995; 25:305–313.
Neaton JD, Wentworth D. Serum cholesterol, blood pressure, cigarette smoking, and death from coronary heart disease. Overall findings and differences by age for 316,099 white men. Multiple Risk Factor Intervention Trial Research Group. Arch Intern Med1992; 152:56–64.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet2002; 360:1903–1913.
Lloyd-Jones D, Adams R, Carnethon M, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation2009; 119:e21–e181.
Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA2003; 289:2560–2572.
Grim CM, Grim CE. A curriculum for the training and certification of blood pressure measurement for health care providers. Can J Cardiol1995; 11(suppl H):38H–42H.
Pickering TG, Hall JE, Appel LJ, et al; Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Hypertension2005; 45:142–161.
Langlois S. Measuring blood pressure: how competent are we?Perspect Cardiol1999; 15:29–39.
Le Pailleur C, Helft G, Landais P, et al. The effects of talking, reading, and silence on the “white coat” phenomenon in hypertensive patients. Am J Hypertens1998; 11:203–207.
Webster J, Newnham D, Petrie JC, Lovell HG. Influence of arm position on measurement of blood pressure. Br Med J (Clin Res Ed)1984; 288:1574–1575.
Netea RT, Smits P, Lenders JW, Thien T. Does it matter whether blood pressure measurements are taken with subjects sitting or supine?J Hypertens1998; 16:263–268.
Silverberg DS, Shemesh E, Iaina A. The unsupported arm: a cause of falsely raised blood pressure readings. Br Med J1977; 2:1331.
Iyriboz Y, Hearon CM, Edwards K. Agreement between large and small cuffs in sphygmomanometry: a quantitative assessment. J Clin Monit1994; 10:127–133.
Scherwitz LW, Evans LA, Hennrikus DJ, Vallbona C. Procedures and discrepancies of blood pressure measurements in two community health centers. Med Care1982; 20:727–738.
La Batide-Alanore A, Chatellier G, Bobrie G, Fofol I, Plouin PF. Comparison of nurse- and physician-determined clinic blood pressure levels in patients referred to a hypertension clinic: implications for subsequent management. J Hypertens2000; 18:391–398.
Verdecchia P. Prognostic value of ambulatory blood pressure: current evidence and clinical implications. Hypertension2000; 35:844–851.
Ogedegbe G, Pickering TG, Clemow L, et al. The misdiagnosis of hypertension: the role of patient anxiety. Arch Intern Med2008; 168:2459–2465.
Pickering TG. Stress, white coat hypertension and masked hypertension. In:Izzo JL, Sica DA, Black HR, editors. Hypertension Primer: The Essentials of High Blood Pressure: Basic Science, Population Science, and Clinical Management. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:289–291.
Pickering TG, Coats A, Mallion JM, Mancia G, Verdecchia P. Blood Pressure Monitoring. Task force V: White-coat hypertension. Blood Press Monit1999; 4:333–341.
Myers MG, Valdivieso MA. Use of an automated blood pressure recording device, the BpTRU, to reduce the “white coat effect” in routine practice. Am J Hypertens2003; 16:494–497.
Redon J, Campos C, Narciso ML, Rodicio JL, Pascual JM, Ruilope LM. Prognostic value of ambulatory blood pressure monitoring in refractory hypertension: a prospective study. Hypertension1998; 31:712–718.
Ohkubo T, Imai Y, Tsuji I, et al. Prediction of mortality by ambulatory blood pressure monitoring versus screening blood pressure measurements: a pilot study in Ohasama. J Hypertens1997; 15:357–364.
Verdecchia P, Reboldi G, Porcellati C, et al. Risk of cardiovascular disease in relation to achieved office and ambulatory blood pressure control in treated hypertensive subjects. J Am Coll Cardiol2002; 39:878–885.
Hemmelgarn BR, McAllister FA, Myers MG, et al; Canadian Hypertension Education Program. The 2005 Canadian Hypertension Education Program recommendations for the management of hypertension: part 1 - blood pressure measurement, diagnosis and assessment of risk. Can J Cardiol2005; 21:645–656.
White WB, Anwar YA. Evaluation of the overall efficacy of the Omron office digital blood pressure HEM-907 monitor in adults. Blood Press Monit2001; 6:107–110.
Myers MG, Meglis G, Polemidiotis G. The impact of physician vs automated blood pressure readings on office-induced hypertension. J Hum Hypertens1997; 11:491–493.
Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Measurement of blood pressure in the office: recognizing the problem and proposing the solution. Hypertension2010; 55:195–200.
Stergiou GS, Tzamouranis D, Protogerou A, Nasothimiou E, Kapralos C. Validation of the Microlife Watch BP Office professional device for office blood pressure measurement according to the International protocol. Blood Press Monit2008; 13:299–303.
Myers MG, Valdivieso M, Kiss A. Optimum frequency of office blood pressure measurement using an automated sphygmomanometer. Blood Press Monit2008; 13:333–338.
Myers MG, Valdivieso M, Kiss A. Use of automated office blood pressure measurement to reduce the white coat response. J Hypertens2009; 27:280–286.
Culleton BF, McKay DW, Campbell NR. Performance of the automated BpTRU measurement device in the assessment of white-coat hypertension and white-coat effect. Blood Press Monit2006; 11:37–42.
Pickering TG, Miller NH, Ogedegbe G, Krakoff LR, Artinian NT, Goff D; American Heart Association. Call to action on use and reimbursement for home blood pressure monitoring: executive summary: a joint scientific statement from the American Heart Association, American Society Of Hypertension, and Preventive Cardiovascular Nurses Association. Hypertension2008; 52:1–9.
Parati G, Stergiou GS, Asmar R, et al; ESH Working Group on Blood Pressure Monitoring. European Society of Hypertension guidelines for blood pressure monitoring at home: a summary report of the Second International Consensus Conference on Home Blood Pressure Monitoring. J Hypertens2008; 26:1505–1526.
O’Brien E. Ambulatory blood pressure measurement: the case for implementation in primary care. Hypertension2008; 51:1435–1441.
Beckett L, Godwin M. The BpTRU automatic blood pressure monitor compared to 24 hour ambulatory blood pressure monitoring in the assessment of blood pressure in patients with hypertension. BMC Cardiovasc Disord2005; 5:18.
The white-coat effect, ie, the tendency of many patients to have higher blood pressure in the presence of medical personnel than in their own environment, can lead to inappropriate diagnosis of hypertension and unnecessary treatment.
Out-of-office blood pressure correlates better with cardiovascular risk than does the blood pressure in the physician’s office, but ambulatory monitoring is costly and not widely available, and few physicians recommend self-measurement at home.
Several available devices can take a series of blood pressure measurements at preset intervals while the patient sits alone in the examination room, eliminating the white-coat effect.
The mean of five automatic readings taken at intervals of 1 or 2 minutes correlates well with the mean value while awake on ambulatory monitoring.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
The amyloid model is based on early-onset disease, a distinct syndrome with autosomal dominant inheritance. The onset of symptoms usually occurs in the patient’s 40s, but it can occur as early as the 20s or as late as the 50s. The disease is aggressive and almost always follows a 5- to 8-year course to death. In terms of its onset, course, and comorbidities, early-onset Alzheimer disease is very different than the much more common sporadic or late-onset disease found in older adults (Table 1).
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
References
Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol2006; 111:503–509.
Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree?Pract Neurol2006( 4):14–15.
Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie?Neurobiol Aging2002; 23:1101–1105.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol2009; 66:1210–1215.
Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia1996; 39:1392–1397.
Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol2006; 63:1551–1555.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol2006; 5:64–74.
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes2004; 53:474–481.
Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses2004; 63:8–20.
de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci2002; 977:196–215.
Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci2002; 203–204:29–34.
Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci2007; 27:1973–1980.
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA1997; 277:813–817.
McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology1996; 47:425–432.
Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology1997; 48:626–632.
Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol2004; 3:169–178.
Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed2006; 8:25.
Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association2006(2 suppl July):S148.
Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord2010; 30:131–146.
Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA1998; 279:688–695.
Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA2002; 288:872–881.
LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA2001; 285:1489–1499.
Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience2000; 101:485–512.
Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA2004; 291:2947–2958.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol2009; 66:1210–1215.
Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med2010; 170:186–193.
Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil2004; 85:1694–1704.
David S. Geldmacher, MD Harrison Distinguished Teaching Associate Professor, Department of Neurology, University of Virginia Health System, Charlottesville
Adddress: David S. Geldmacher, MD, Department of Neurology, University of Virginia Health Systems, Charlottesville, VA; e-mail [email protected]
Medical Grand Rounds articles are based on edited transcripts from Education Institute Department of Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
David S. Geldmacher, MD Harrison Distinguished Teaching Associate Professor, Department of Neurology, University of Virginia Health System, Charlottesville
Adddress: David S. Geldmacher, MD, Department of Neurology, University of Virginia Health Systems, Charlottesville, VA; e-mail [email protected]
Medical Grand Rounds articles are based on edited transcripts from Education Institute Department of Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Author and Disclosure Information
David S. Geldmacher, MD Harrison Distinguished Teaching Associate Professor, Department of Neurology, University of Virginia Health System, Charlottesville
Adddress: David S. Geldmacher, MD, Department of Neurology, University of Virginia Health Systems, Charlottesville, VA; e-mail [email protected]
Medical Grand Rounds articles are based on edited transcripts from Education Institute Department of Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the author but are not peer-reviewed.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
The amyloid model is based on early-onset disease, a distinct syndrome with autosomal dominant inheritance. The onset of symptoms usually occurs in the patient’s 40s, but it can occur as early as the 20s or as late as the 50s. The disease is aggressive and almost always follows a 5- to 8-year course to death. In terms of its onset, course, and comorbidities, early-onset Alzheimer disease is very different than the much more common sporadic or late-onset disease found in older adults (Table 1).
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
Efforts to modify the relentless course of Alzheimer disease have until now been based on altering the production or clearance of beta-amyloid, the protein found in plaques in the brains of patients with the disease. Results have been disappointing, possibly because our models of the disease—mostly based on the rare, inherited form—may not be applicable to the much more common sporadic form.
Ely Lilly’s recent announcement that it is halting research into semagacestat, a drug designed to reduce amyloid production, only cast further doubt on viability of the amyloid hypothesis as a framework for effective treatments for Alzheimer disease.
Because of the close association of sporadic Alzheimer disease with vascular disease and type 2 diabetes mellitus, increased efforts to treat and prevent these conditions may be the best approach to reducing the incidence of Alzheimer disease.
This article will discuss current thinking of the pathophysiology of Alzheimer disease, with special attention to potential prevention and treatment strategies.
THE CANONICAL VIEW: AMYLOID IS THE CAUSE
The canonical view is that the toxic effects of beta-amyloid are the cause of neuronal dysfunction and loss in Alzheimer disease.
Beta-amyloid is a small peptide, 38 to 42 amino acids long, that accumulates in the extracellular plaque that characterizes Alzheimer pathology. Small amounts of extracellular beta-amyloid can be detected in the brains of elderly people who die of other causes, but the brains of people who die with severe Alzheimer disease show extensive accumulation of plaques.
The amyloid precursor protein is cleaved by normal constitutive enzymes, leaving beta-amyloid as a fragment. The beta-amyloid forms into fibrillar aggregations, which further clump into the extracellular plaque. Plaques can occur in the normal aging process in relatively low amounts. However, in Alzheimer disease, through some unknown trigger, the immune system appears to become activated in reference to the plaque. Microglial cells—the brain’s macrophages—invade the plaque and trigger a cycle of inflammation. The inflammation and its by-products cause local neuronal damage, which seems to propagate the inflammatory cycle to an even greater extent through a feed-forward loop. The damage leads to metabolic stress in the neuron and collapse of the cytoskeleton into a neurofibrillary tangle. Once the neurofibrillary tangle is forming, the neuron is probably on the path to certain death.
This pathway might be interrupted at several points, and in fact, much of the drug development world is working on possible ways to do so.
GENETIC VS SPORADIC DISEASE: WHAT ARE THE KEY DIFFERENCES?
The amyloid model is based on early-onset disease, a distinct syndrome with autosomal dominant inheritance. The onset of symptoms usually occurs in the patient’s 40s, but it can occur as early as the 20s or as late as the 50s. The disease is aggressive and almost always follows a 5- to 8-year course to death. In terms of its onset, course, and comorbidities, early-onset Alzheimer disease is very different than the much more common sporadic or late-onset disease found in older adults (Table 1).
Although the autosomal dominant form of the disease accounts for probably only 1% or 2% of all cases of Alzheimer disease, most animal models and hence much of the basic research and drug testing in Alzheimer disease are based on those dominant mutations. The pathology—the plaques and tangles—in Alzheimer disease in older adults is identical to that in younger adults, but the origins of the disease may not be the same. Therefore, the experimental model for one may not be relevant to the other.
In the last several years, some have questioned whether the amyloid hypothesis applies to all Alzheimer disease.1,2 Arguments go back to at least 2002, when Bishop and Robinson in an article entitled “The amyloid hypothesis: Let sleeping dogmas lie?”3 criticized the hypothesis and suggested that the beta-amyloid peptide appeared to be neuroprotective, not neurotoxic, in most situations. They suggested we await the outcome of antiamyloid therapeutic trials to determine whether the amyloid hypothesis truly explains the disorder.
The antiamyloid trials have now been under way for some time, and we have no definitive answer. Data from the phase II study of the monoclonal antibody agent bapineuzumab suggests there might be some small clinical impact of removing amyloid from the brain through immunotherapy mechanisms, but the benefits thus far are not robust.
COULD AMYLOID BE NEUROPROTECTIVE?
A pivotal question might be, “What if sick neurons made amyloid, instead of amyloid making neurons sick?” A corollary question is, “What if the effect were bidirectional?”
It is possible that in certain concentrations amyloid is neurotoxic, but in other concentrations, it actually facilitates neuronal repair, healing, and connection.
REDUCING METABOLIC STRESS: THE KEY TO PREVENTION?
If our current models of drug therapy are not effective against sporadic Alzheimer disease, perhaps focusing on prevention would be more fruitful.
Consider diabetes mellitus as an analogy. Its manifestations include polydipsia, polyuria, fatigue, and elevated glucose and hemoglobin A1c. Its complications are cardiovascular disease, nephropathy, and retinopathy. Yet diabetes mellitus encompasses two different diseases—type 1 and type 2—with different underlying pathophysiology. We do not treat them the same way. We may be moving toward a similar view of Alzheimer disease.
Links have been hypothesized between vascular risks and dementia. Diabetes, hypertension, dyslipidemia, and obesity might lead to dementia in a process abetted by oxidative stress, endothelial dysfunction, insulin resistance, inflammation, adiposity, and subcortical vascular disease. All of these could be targets of intervention to prevent and treat dementia.4
Instead of a beta-amyloid trigger, let us hypothesize that metabolic stress is the initiating element of the Alzheimer cascade, which then triggers beta-amyloid overproduction or underclearance, and the immune activation damages neurons. By lessening metabolic stress or by preventing immune activation, it may, in theory, be possible to prevent neurons from entering into the terminal pathway of tangle formation and cell death.
LINKS BETWEEN ALZHEIMER DISEASE AND DIABETES
Rates of dementia of all causes are higher in people with diabetes. The strongest effect has been noted in vascular dementia, but Alzheimer disease was also found to be associated with diabetes.5 The Framingham Heart Study6 found the association between dementia and diabetes was significant only when other risk factors for Alzheimer disease were minimal: in an otherwise healthy population, diabetes alone appears to trigger the risk for dementia. But in a population with a lot of vascular comorbidities, the association between diabetes and dementia is not as clear. Perhaps the magnitude of the risk is overwhelmed by greater cerebrovascular and cardiovascular morbidity.
A systematic review7 supported the notion that the risk of dementia is higher in people with diabetes, and even raised the issue of whether we should consider Alzheimer disease “type 3 diabetes.”
Testing of the reverse hypothesis—diabetes is more common in people with Alzheimer disease—also is supportive: diabetes mellitus and even impaired fasting glucose are approximately twice as common in people with Alzheimer disease than in those without.8 Fasting blood glucose levels increase steadily with age, but after age 65, they are higher in people with Alzheimer disease than in those without.
Glucose has some direct effects on brain metabolism that might explain the higher risk. Chronic hyperglycemia is associated with excessive production of free radicals, which leads to reactive oxygen species. These are toxic to neuronal membranes as well as to mitochondria, where many of the reactive oxygen species are generated. Free radicals also facilitate the inflammatory response.
We also see greater neuronal and mitochondrial calcium influx in the presence of hyperglycemia. The excess calcium interferes with mitochondrial metabolism and may trigger the cascade of apoptosis when it reaches critical levels in neurons.
Chronic hyperglycemia is also associated with increased advanced glycation end-products. These are toxic molecules produced by the persistent exposure of proteins to high sugar levels and may be facilitated by the presence of reactive oxygen species that catalyze the reactions between the sugars and the peptides. Glycation end-products are commonly recognized as the same as those occurring during browning of meat (the Maillard reaction).
Hyperglycemia also potentiates neuronal damage from ischemia. Animal experiments show that brain infarction in the presence of hyperglycemia results in worse damage than the same degree of ischemia in the absence of hyperglycemia. Hyperglycemia may exaggerate other blows to neuronal function such as those from small strokes or microvascular ischemia.
AN ALTERNATIVE TO THE AMYLOID HYPOTHESIS: THE ‘MITOCHONDRIAL CASCADE HYPOTHESIS’
Swerdlow and Khan9 have proposed an alternative to the amyloid hypothesis as the cause of Alzheimer disease, known as the “mitochondrial cascade hypothesis.” According to this model, as we age we accumulate more wear-and-tear from oxidative mitochondrial damage, especially the accumulation of toxins leading to reduced cell metabolic activity. This triggers the “3-R response”:
Reset. When toxins alter cell metabolism, neurons try to repair themselves by manufacturing beta-amyloid, which is a “repair-and-reset” synaptic signaling molecule that reduces energy production. Under the lower energy state, beta-pleated sheets develop from beta-amyloid, which aggregate and form amyloid plaque.
Remove. Many cells undergo programmed death when faced with oxidative stress. The first step in neuronal loss is reduced synaptic connections and, hence, losses in neuronal communication. This results in impaired cognition.
Replace. Some cells that are faced with metabolic stress re-enter the cell cycle by undergoing cell division. Neurons, however, are terminally postmitotic and die if they try to divide: by synthesizing cell division proteins, duplicating chromosomes, and reorganizing the complex internal structure, the cell cannot work properly and cell division fails. In the mitochondrial cascade hypothesis, neurofibrillary tangles result from this attempted remodeling of the cytoskeletal filaments, furthering neuronal dysfunction.
ALZHEIMER DISEASE AND STROKE: MORE ALIKE THAN WE THOUGHT?
Although historically clinicians and researchers have tried to distinguish between Alzheimer disease and vascular dementia, growing evidence indicates that the two disorders overlap significantly and that the pathologies may be synergistic.
Alzheimer disease has been hypothesized as being a vascular disorder.10 It shares many of the risk factors of vascular disease, and preclinical detection of Alzheimer disease is possible from measurements of regional cerebral perfusion. Cerebrovascular and neurodegenerative pathology are parallel in Alzheimer disease and vascular disease.
Pure Alzheimer disease and vascular disease are two ends of a pathologic continuum.11 At one end is “pure” Alzheimer disease, in which patients die only with histologic findings of plaques and neurofibrillary tangles. This form may occur only in patients with the autosomal dominant early-onset form. At the other end of the spectrum are people who have serious vascular disease, multiple strokes, and microvascular ischemia and who die demented but with no evidence of the plaques and tangles of Alzheimer disease.
Between these poles is a spectrum of overlapping pathology that is either Alzheimer disease-dominant or vascular disease-dominant, with varying degrees of amyloid plaque and evidence of microvascular infarcts. Cerebral amyloid angiopathy (the accumulation of beta-amyloid in the wall of arteries in the brain) bridges the syndromes.12 In some drug studies that attempted removing amyloid from the brain, vascular permeability was altered, resulting in brain edema.
Along the same lines as Kalaria’s model,11 Snowden et al13 found at autopsy of aged Catholic nuns that for some the accumulation of Alzheimer pathology alone was insufficient to cause dementia, but dementia was nearly universal in nuns with the same burden of Alzheimer pathology commingled with vascular pathology.
DOES INFLAMMATION PLAY A ROLE?
The inflammatory state is a recognized risk factor for Alzheimer disease, but the clinical data are mixed. Epidemiologic evidence is strong: patients who regularly take nonsteroidal anti-inflammatory drugs (NSAIDs) or steroids for chronic, systemic inflammatory diseases (eg, arthritis) have a 45% to 60% reduced risk for Alzheimer disease.14,15
However, multiple clinical trials in patients with Alzheimer disease have failed to show a benefit of taking anti-inflammatory drugs. One preliminary report suggested that indomethacin (Indocin) might offer benefit, but because of gastrointestinal side effects its usefulness in an elderly population is limited.
Diabetes and inflammation are also closely linked: hyperinsulinemia is proinflammatory, promoting the formation of reactive oxygen species, inhibiting the degradation of oxidized proteins, and increasing the risk for lipid per-oxidation. Insulin acts synergistically with endotoxins to raise inflammatory markers, eg, proinflammatory cytokines and C-reactive protein.16
It is possible that anti-inflammatory drugs may not work in Alzheimer disease because inflammation in the brain is mediated more by microglial cells than by prostaglandin pathways. In Alzheimer disease, inflammation is mediated by activated microglial cells, which invade plaques with their processes; these are not evident in the diffuse beta-amyloid-rich plaques seen in typical aging. The trigger for their activation is unclear, but the activated microglial cells and the invasion of plaques are seen in transgenic mouse models of Alzheimer disease, and activation is seen when beta-amyloid is injected into the brain of a healthy mouse.17
Activated microglial cells enlarge and their metabolic rate increases, with a surge in the production of proteins and other protein-mediated inflammatory markers such as alpha-antichymotrypsin, alpha-antitrypsin, serum amyloid P, C-reactive protein, nitric oxide, and proinflammatory cytokines. It is unlikely that it is healthy for cells to be exposed to these inflammatory products. Some of the cytokines are now targets of drug development for Alzheimer disease, and agents targeting these pathways have already been developed for connective tissue diseases.
In a controversial pilot study, Tobinick et al18 studied the use of etanercept (Enbrel), an inhibitor of tumor necrosis factor-alpha (an inflammatory cytokine). They injected etanercept weekly into the spinal canal in 15 patients with mild to severe Alzheimer disease, for 6 months. Patients improved in the Mini-Mental State Examination by more than two points during the study. Patent issues surrounding use of this drug in Alzheimer disease may delay further trials.
The reactive microglial phenotype can be prevented in cell culture by peroxisome proliferator-activated receptor (PPAR) gamma agonists. These include the antidiabetic thiazolidinediones such as pioglitazone (Actos), troglitazone (Rezulin), and rosiglitazone (Avandia), and indomethacin and other NSAIDs.
Using a Veterans Administration database of more than 142,000 patients, Miller et al19 retrospectively found that patients who took a thiazolidinedione for diabetes had a 20% lower risk of developing Alzheimer disease compared with users of insulin or metformin (Glucophage).
However, rosiglitazone showed no benefit against Alzheimer disease in a large clinical trial,20 but this may be because it is rapidly cleared from the brain. Pioglitazone is not actively exported from the brain, so it may be a better candidate, but pharmaceutical industry interest in this agent is low because its patent will soon expire.
Fish oil is another PPAR-gamma agonist, and some studies indicate that eating fish may protect against developing Alzheimer disease; it may also be therapeutic if the disease is present. Double-blind controlled studies have not been carried out and likely will not because of patent issues: the costs of such studies are high, and the potential payback is low.
ESTROGEN: PROTECTIVE OR NOT?
Whether taking estrogen is a risk factor or is protective has not yet been determined. Estrogen directly affects neurons. It increases the number of dendritic spines, which are associated with improved memory. Meta-analyses suggest that hormone replacement therapy reduces the risk of dementia by about one-third. 21,22 Both positive and negative prospective studies exist, but all are complicated by serious methodologic flaws.23,24
Combined analysis of about 7,500 women from two double-blind, randomized, placebo-controlled trials of the Women’s Health Initiative Memory Study found that the risks of dementia and mild cognitive impairment were increased by hormone replacement therapy. The hazard ratio for dementia was found to be 1.76 (P < .005), amounting to 23 new cases of dementia per 10,000 prescriptions annually.25
Patient selection may account for the conflicting results in different studies. Epidemiologic studies consisted mostly of newly postmenopausal women and those who were being treated for symptoms of vasomotor instability. In contrast, the Women’s Health Initiative enrolled only women older than 65 and excluded women with vasomotor instability. Other studies indicate that the greatest cognitive improvements with hormone therapies are seen in women with vasomotor symptoms.
WHICH RISK FACTORS CAN WE CONTROL?
In summary, some of the risk factors for Alzheimer disease can be modified if we do the following.
Aggressively manage diabetes and cardiovascular disease. Vascular risk factors significantly increase dementia risk, providing good targets for prevention: clinicians should aggressively help their patients control diabetes, hypertension, and hyperlipidemia.26 However, aggressive control of hypertension in a patient with already-existing dementia may exacerbate the condition, so caution is warranted.
Optimize diet. Dietary measures include high intake of antioxidants (which are especially high in brightly colored and tart-flavored fruits and vegetables) and polyunsaturated fats.26 Eating a Mediterranean-type diet that includes a high intake of cold-water ocean fish is recommended. Fish should not be fried: the high temperatures may destroy the omega-3 fatty acids, and the high fat content may inhibit their absorption.
Weigh the risks and benefits of estrogen. Although estrogen replacement therapy for postmenopausal women has had mixed results for controlling dementia, it appears to be clinically indicated to control vasomotor symptoms and likely does not increase the risk of dementia for newly menopausal women. Risks and benefits should be carefully weighed for each patient.
Optimize exercise. People who are physically active in midlife have a lower risk of Alzheimer disease.27 Those who adopt new physical activity late in life may also gain some protective or restorative benefit.28
Many measures, such as taking anti-inflammatory or antihypertensive drugs, probably have a very small incremental benefit over time, so it is difficult to measure significant effects during the course of a typical clinical trial.
Clinicians are already recommending actions to reduce the risk of dementia by focusing on lowering cardiovascular risk. Hopefully, as these actions become more commonly practiced as lifelong habits in those reaching the age of risk for Alzheimer disease, we will see a reduced incidence of that devastating and much-feared illness.
References
Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol2006; 111:503–509.
Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree?Pract Neurol2006( 4):14–15.
Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie?Neurobiol Aging2002; 23:1101–1105.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol2009; 66:1210–1215.
Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia1996; 39:1392–1397.
Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol2006; 63:1551–1555.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol2006; 5:64–74.
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes2004; 53:474–481.
Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses2004; 63:8–20.
de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci2002; 977:196–215.
Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci2002; 203–204:29–34.
Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci2007; 27:1973–1980.
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA1997; 277:813–817.
McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology1996; 47:425–432.
Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology1997; 48:626–632.
Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol2004; 3:169–178.
Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed2006; 8:25.
Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association2006(2 suppl July):S148.
Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord2010; 30:131–146.
Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA1998; 279:688–695.
Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA2002; 288:872–881.
LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA2001; 285:1489–1499.
Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience2000; 101:485–512.
Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA2004; 291:2947–2958.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol2009; 66:1210–1215.
Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med2010; 170:186–193.
Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil2004; 85:1694–1704.
References
Castellani RJ, Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Neuropathology of Alzheimer disease: pathognomic but not pathogenic. Acta Neuropathol2006; 111:503–509.
Geldmacher DS. Alzheimer’s pathogenesis: are we barking up the wrong tree?Pract Neurol2006( 4):14–15.
Bishop GM, Robinson SR. The amyloid hypothesis: let sleeping dogmas lie?Neurobiol Aging2002; 23:1101–1105.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol2009; 66:1210–1215.
Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia1996; 39:1392–1397.
Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risks of developing Alzheimer disease: results from the Framingham Study. Arch Neurol2006; 63:1551–1555.
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol2006; 5:64–74.
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes2004; 53:474–481.
Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses2004; 63:8–20.
de la Torre JC. Vascular basis of Alzheimer’s pathogenesis. Ann NY Acad Sci2002; 977:196–215.
Kalaria R. Similarities between Alzheimer’s disease and vascular dementia. J Neurol Sci2002; 203–204:29–34.
Prada CM, Garcia-Alloza M, Betensky RA, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci2007; 27:1973–1980.
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA1997; 277:813–817.
McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology1996; 47:425–432.
Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer’s disease and duration of NSAID use. Neurology1997; 48:626–632.
Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol2004; 3:169–178.
Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed2006; 8:25.
Miller DR, Fincke BG, Davidson JE, Weil JG. Thiazolidinedione use may forestall progression of Alzheimer’s disease in diabetes patients. Alzheimer’s & Dementia: Journal of the Alzheimer’s Association2006(2 suppl July):S148.
Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord2010; 30:131–146.
Yaffe K, Sawaya G, Lieberburg I, Grady D. Estrogen therapy in postmenopausal women: effects on cognitive function and dementia. JAMA1998; 279:688–695.
Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy: scientific review. JAMA2002; 288:872–881.
LeBlanc ES, Janowsky J, Chan BK, Nelson HD. Hormone replacement therapy and cognition: systematic review and meta-analysis. JAMA2001; 285:1489–1499.
Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience2000; 101:485–512.
Shumaker SA, Legault C, Kuller L, et al; Women’s Health Initiative Memory Study. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA2004; 291:2947–2958.
Middleton LE, Yaffe K. Promising strategies for the prevention of dementia. Arch Neurol2009; 66:1210–1215.
Etgen T, Sander D, Huntgeburth U, Poppert H, Förstl H, Bickel H. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch Intern Med2010; 170:186–193.
Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil2004; 85:1694–1704.
Vascular risk factors clearly increase the risk of Alzheimer disease and can be addressed. However, controlled trials in patients with hypertension or with dyslipidemia have had negative results.
Risk is lower with a diet high in antioxidants and polyunsaturated fatty acids.
Estrogen therapy has had mixed results in observational studies, mostly hinting at lower risk. However, a randomized trial of hormone replacement therapy in late life indicated a higher risk of dementia with estrogen.
Physical activity in midlife and in late life was associated with a lower risk of Alzheimer disease in observational studies. Controlled trials were not so positive, but the benefits of exercise may be slowly cumulative.
Reperfusion therapy decreases morbidity and mortality rates in patients with ST-segment elevation myocardial infarction (MI). Primary percutaneous coronary intervention (PCI) is preferred over fibrinolytic therapy as a reperfusion strategy when the delay in the time to treatment is short and the patient presents to a high-volume, well-equipped center with expert interventional cardiologists.
Compared with fibrinolytic therapy in randomized clinical trials, primary PCI produces higher rates of infarct artery patency, complete reperfusion (grade 3 by the criteria of the Thrombolysis in Myocardial Infarction [TIMI] study), and access-site bleeding. It also produces lower rates of recurrent ischemia, reinfarction, emergency repeat revascularization procedures, intracranial hemorrhage, and death.1 If performed early and successfully, primary PCI also greatly decreases the rates of complications of ST-elevation MI that result from longer ischemic times or unsuccessful fibrinolytic therapy, allowing earlier hospital discharge and resumption of daily activities. Primary PCI is also the best reperfusion option in patients who present late after the onset of symptoms and in patients with cardiogenic shock, and it is the only option in patients who have contraindications to fibrinolytic therapy because of bleeding risk.
However, most hospitals do not have PCI capability. Two options at these hospitals are to transfer the patient to a PCI center quickly for primary PCI or to keep the patient on site and give fibrinolytic therapy, with its limitations. Earlier trials suggested that the transfer strategy was superior, but they had limitations: most patients received streptokinase, an inferior fibrinolytic agent, and door-to-door-to-balloon times were rapid, averaging only 95 minutes because of excellent logistical protocols and careful patient selection.2 Most importantly, rescue PCI and routine PCI were seldom performed in patients receiving fibrinolytics, so fibrinolytic therapy was being tested as monotherapy.
In the real world, however, treatment delays are much longer, and fibrinolytic therapy has evolved into a strategy that includes crossover to rescue PCI or routine PCI. Therefore, the initial trials of transfer for primary PCI do not reflect current practice. In fact, recent registry data suggest that prehospital fibrinolytic therapy followed by early angiography is superior to primary PCI because most patients can be treated within 2 hours of symptom onset; they also suggest that on-site fibrinolytic therapy followed by early angiography is equal in efficacy to primary PCI as long as rescue PCI and routine PCI can be performed.3,4
The most important modifiable predictor of outcome in ST-elevation MI is the time to treatment, a biological truth that continues to be supported by clinical evidence despite ideologic arguments by some interventional cardiology enthusiasts who claim that primary PCI is always superior to the fibrinolytic strategy, regardless of delays.
SURPRISINGLY, OUTCOMES WERE WORSE WITH FACILITATED PCI
It made sense, then, to conclude that the perfect strategy for hospitals without PCI capability would be a combined strategy of immediate fibrinolytic therapy to decrease the time delay associated with organizing PCI, and rapid transfer for immediate PCI to improve the limited reperfusion rates associated with fibrinolytic therapy.
Surprisingly, though, randomized trials found worse outcomes with this “facilitated PCI” strategy.5
Again, limitations in trial design might explain the lack of benefit in the trials. Inadequate anticoagulant and antiplatelet therapy were given to the fibrinolytic patients, and primary PCI patients had relatively short treatment delays, with many patients enrolled at hospitals with PCI capability.
PROGRESS IN REPERFUSION THERAPY
Great strides have been made in reperfusion therapy in recent years. Adjunctive therapy with clopidogrel (Plavix) and enoxaparin (Lovenox) has been shown to improve outcomes with fibrinolytic therapy. Bivalirudin (Angiomax) and stents have improved primary PCI’s performance. Reducing bleeding complications has become a clinical priority, with increasing emphasis on adjusting some drug doses according to renal function and using the radial artery for cardiac catheterization.
The American College of Cardiology initiative, “Door-to-Balloon (D2B): An Alliance for Quality,” focused much attention on organizing in-hospital systems of care for primary PCI, thus increasing the national rate of achieving a door-to-balloon time within 90 minutes from 50% to over 75% in patients who presented to hospitals with PCI capability.6
The American Heart Association has launched “Mission: Lifeline,” a national campaign to organize prehospital systems of care with their program,7 working within communities to address their unique needs, resources, and barriers to implementing systems of care for ST-elevation MI. The key aspect of this effort is to help geographic regions develop local solutions, an explicit recognition that there is no one-size-fits-all solution. Early triage by emergency medical services, rapid diagnosis with prehospital electrocardiography, destination and interhospital transfer protocols, and prehospital activation of the cardiac catheterization laboratory can greatly streamline emergency care and decrease treatment delays for primary PCI.
FOR OUTLYING HOSPITALS, A PHARMACOINVASIVE STRATEGY
So what about hospitals without PCI capability that cannot routinely transfer patients to a hospital with PCI capability within 90 minutes?
Lessons learned from the experiences with immediate PCI, rescue PCI, and facilitated PCI have evolved into the “pharmacoinvasive strategy.” Patients with ST-elevation MI are treated as rapidly as possible with a bolus of a fibrinolytic drug, eg, tenecteplase (TNKase) or reteplase (Retavase), and are also given aspirin, clopidogrel, and enoxaparin. Then, they are rapidly transferred to a PCI-capable hospital so that emergency PCI can be performed if reperfusion is not clinically apparent or if the patient develops pulmonary edema or cardiogenic shock. If the clinical signs suggest that reperfusion has been achieved (relief of chest pain, rapid resolution of ST-segment elevation, bursts of accelerated idioventricular rhythm), coronary angiography (and PCI, if indicated) can be performed within 3 to 24 hours of fibrinolytic therapy. This time frame allows the initial fibrinolytic effect to dissipate, while the antiplatelet and anticoagulant drugs achieve therapeutic levels.
Today, the goal is to treat every patient with the best reperfusion strategy available, given the limitations in resources and the geographic location of some centers, and to maximize the possibility of sustained patency of the infarct-related artery by implanting a stent, even if it takes several hours and transfer to another hospital to perform PCI.8 The pharmacoinvasive strategy of rapid administration of fibrinolytic therapy followed by PCI within 24 hours would be practical in most hospitals without PCI capability where treatment delays prohibit performance of primary PCI within 90 minutes of first medical contact.9
THE ‘STREAM’ TRIAL IS UNDER WAY
As proof of concept, the Strategic Reperfusion Early After Myocardial Infarction (STREAM) trial is enrolling 2,000 patients with ST-elevation MI presenting within 3 hours of symptom onset if primary PCI is not feasible within 60 minutes of first medical contact.10 Patients will be randomized to either of the following:
Receive prehospital therapy with tenecteplase, aspirin, clopidogrel, and enoxaparin and undergo cardiac catheterization in 6 to 24 hours (or rescue PCI if reperfusion fails within 90 minutes of fibrinolysis)
Undergo primary PCI performed according to local guidelines.
The primary measure of efficacy will be the composite rate of death, cardiogenic shock, heart failure, and reinfarction at 30 days. Measures of safety include the rates of ischemic stroke, intracranial hemorrhage, and major nonintracranial bleeding.
References
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet2003; 361:13–20.
Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation2003; 108:1809–1814.
Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FASTMI). Circulation2008; 118:268–276.
Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA2010; 303:2148–2155.
Keeley EC, Boura JA, Grines CL. Comparison of primary and facilitated percutaneous coronary interventions for ST-elevation myocardial infarction: quantitative review of randomised trials. Lancet2006; 367:579–588.
Krumholz HM, Bradley EH, Nallamothu BK, et al. A campaign to improve the timeliness of primary percutaneous coronary intervention: Door-to-Balloon: An Alliance for Quality. JACC Cardiovasc Interv. 2008; 1:97–104.
Jacobs AK, Antman EM, Faxon DP, Gregory T, Solis P. Development of systems of care for ST-elevation myocardial infarction patients: executive summary. Circulation2007; 116:217–230.
Kushner FG, Hand M, Smith SC, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation2009; 120:2271–2306.
Bates ER, Nallamothu BK. Commentary: the role of percutaneous coronary intervention in ST-segment-elevation myocardial infarction. Circulation2008; 118:567–573.
Armstrong PW, Gershlick A, Goldstein P, et al; STREAM Steering Committee. The Strategic Reperfusion Early After Myocardial Infarction (STREAM) study. Am Heart J2010; 160:30–35.
Reperfusion therapy decreases morbidity and mortality rates in patients with ST-segment elevation myocardial infarction (MI). Primary percutaneous coronary intervention (PCI) is preferred over fibrinolytic therapy as a reperfusion strategy when the delay in the time to treatment is short and the patient presents to a high-volume, well-equipped center with expert interventional cardiologists.
Compared with fibrinolytic therapy in randomized clinical trials, primary PCI produces higher rates of infarct artery patency, complete reperfusion (grade 3 by the criteria of the Thrombolysis in Myocardial Infarction [TIMI] study), and access-site bleeding. It also produces lower rates of recurrent ischemia, reinfarction, emergency repeat revascularization procedures, intracranial hemorrhage, and death.1 If performed early and successfully, primary PCI also greatly decreases the rates of complications of ST-elevation MI that result from longer ischemic times or unsuccessful fibrinolytic therapy, allowing earlier hospital discharge and resumption of daily activities. Primary PCI is also the best reperfusion option in patients who present late after the onset of symptoms and in patients with cardiogenic shock, and it is the only option in patients who have contraindications to fibrinolytic therapy because of bleeding risk.
However, most hospitals do not have PCI capability. Two options at these hospitals are to transfer the patient to a PCI center quickly for primary PCI or to keep the patient on site and give fibrinolytic therapy, with its limitations. Earlier trials suggested that the transfer strategy was superior, but they had limitations: most patients received streptokinase, an inferior fibrinolytic agent, and door-to-door-to-balloon times were rapid, averaging only 95 minutes because of excellent logistical protocols and careful patient selection.2 Most importantly, rescue PCI and routine PCI were seldom performed in patients receiving fibrinolytics, so fibrinolytic therapy was being tested as monotherapy.
In the real world, however, treatment delays are much longer, and fibrinolytic therapy has evolved into a strategy that includes crossover to rescue PCI or routine PCI. Therefore, the initial trials of transfer for primary PCI do not reflect current practice. In fact, recent registry data suggest that prehospital fibrinolytic therapy followed by early angiography is superior to primary PCI because most patients can be treated within 2 hours of symptom onset; they also suggest that on-site fibrinolytic therapy followed by early angiography is equal in efficacy to primary PCI as long as rescue PCI and routine PCI can be performed.3,4
The most important modifiable predictor of outcome in ST-elevation MI is the time to treatment, a biological truth that continues to be supported by clinical evidence despite ideologic arguments by some interventional cardiology enthusiasts who claim that primary PCI is always superior to the fibrinolytic strategy, regardless of delays.
SURPRISINGLY, OUTCOMES WERE WORSE WITH FACILITATED PCI
It made sense, then, to conclude that the perfect strategy for hospitals without PCI capability would be a combined strategy of immediate fibrinolytic therapy to decrease the time delay associated with organizing PCI, and rapid transfer for immediate PCI to improve the limited reperfusion rates associated with fibrinolytic therapy.
Surprisingly, though, randomized trials found worse outcomes with this “facilitated PCI” strategy.5
Again, limitations in trial design might explain the lack of benefit in the trials. Inadequate anticoagulant and antiplatelet therapy were given to the fibrinolytic patients, and primary PCI patients had relatively short treatment delays, with many patients enrolled at hospitals with PCI capability.
PROGRESS IN REPERFUSION THERAPY
Great strides have been made in reperfusion therapy in recent years. Adjunctive therapy with clopidogrel (Plavix) and enoxaparin (Lovenox) has been shown to improve outcomes with fibrinolytic therapy. Bivalirudin (Angiomax) and stents have improved primary PCI’s performance. Reducing bleeding complications has become a clinical priority, with increasing emphasis on adjusting some drug doses according to renal function and using the radial artery for cardiac catheterization.
The American College of Cardiology initiative, “Door-to-Balloon (D2B): An Alliance for Quality,” focused much attention on organizing in-hospital systems of care for primary PCI, thus increasing the national rate of achieving a door-to-balloon time within 90 minutes from 50% to over 75% in patients who presented to hospitals with PCI capability.6
The American Heart Association has launched “Mission: Lifeline,” a national campaign to organize prehospital systems of care with their program,7 working within communities to address their unique needs, resources, and barriers to implementing systems of care for ST-elevation MI. The key aspect of this effort is to help geographic regions develop local solutions, an explicit recognition that there is no one-size-fits-all solution. Early triage by emergency medical services, rapid diagnosis with prehospital electrocardiography, destination and interhospital transfer protocols, and prehospital activation of the cardiac catheterization laboratory can greatly streamline emergency care and decrease treatment delays for primary PCI.
FOR OUTLYING HOSPITALS, A PHARMACOINVASIVE STRATEGY
So what about hospitals without PCI capability that cannot routinely transfer patients to a hospital with PCI capability within 90 minutes?
Lessons learned from the experiences with immediate PCI, rescue PCI, and facilitated PCI have evolved into the “pharmacoinvasive strategy.” Patients with ST-elevation MI are treated as rapidly as possible with a bolus of a fibrinolytic drug, eg, tenecteplase (TNKase) or reteplase (Retavase), and are also given aspirin, clopidogrel, and enoxaparin. Then, they are rapidly transferred to a PCI-capable hospital so that emergency PCI can be performed if reperfusion is not clinically apparent or if the patient develops pulmonary edema or cardiogenic shock. If the clinical signs suggest that reperfusion has been achieved (relief of chest pain, rapid resolution of ST-segment elevation, bursts of accelerated idioventricular rhythm), coronary angiography (and PCI, if indicated) can be performed within 3 to 24 hours of fibrinolytic therapy. This time frame allows the initial fibrinolytic effect to dissipate, while the antiplatelet and anticoagulant drugs achieve therapeutic levels.
Today, the goal is to treat every patient with the best reperfusion strategy available, given the limitations in resources and the geographic location of some centers, and to maximize the possibility of sustained patency of the infarct-related artery by implanting a stent, even if it takes several hours and transfer to another hospital to perform PCI.8 The pharmacoinvasive strategy of rapid administration of fibrinolytic therapy followed by PCI within 24 hours would be practical in most hospitals without PCI capability where treatment delays prohibit performance of primary PCI within 90 minutes of first medical contact.9
THE ‘STREAM’ TRIAL IS UNDER WAY
As proof of concept, the Strategic Reperfusion Early After Myocardial Infarction (STREAM) trial is enrolling 2,000 patients with ST-elevation MI presenting within 3 hours of symptom onset if primary PCI is not feasible within 60 minutes of first medical contact.10 Patients will be randomized to either of the following:
Receive prehospital therapy with tenecteplase, aspirin, clopidogrel, and enoxaparin and undergo cardiac catheterization in 6 to 24 hours (or rescue PCI if reperfusion fails within 90 minutes of fibrinolysis)
Undergo primary PCI performed according to local guidelines.
The primary measure of efficacy will be the composite rate of death, cardiogenic shock, heart failure, and reinfarction at 30 days. Measures of safety include the rates of ischemic stroke, intracranial hemorrhage, and major nonintracranial bleeding.
Reperfusion therapy decreases morbidity and mortality rates in patients with ST-segment elevation myocardial infarction (MI). Primary percutaneous coronary intervention (PCI) is preferred over fibrinolytic therapy as a reperfusion strategy when the delay in the time to treatment is short and the patient presents to a high-volume, well-equipped center with expert interventional cardiologists.
Compared with fibrinolytic therapy in randomized clinical trials, primary PCI produces higher rates of infarct artery patency, complete reperfusion (grade 3 by the criteria of the Thrombolysis in Myocardial Infarction [TIMI] study), and access-site bleeding. It also produces lower rates of recurrent ischemia, reinfarction, emergency repeat revascularization procedures, intracranial hemorrhage, and death.1 If performed early and successfully, primary PCI also greatly decreases the rates of complications of ST-elevation MI that result from longer ischemic times or unsuccessful fibrinolytic therapy, allowing earlier hospital discharge and resumption of daily activities. Primary PCI is also the best reperfusion option in patients who present late after the onset of symptoms and in patients with cardiogenic shock, and it is the only option in patients who have contraindications to fibrinolytic therapy because of bleeding risk.
However, most hospitals do not have PCI capability. Two options at these hospitals are to transfer the patient to a PCI center quickly for primary PCI or to keep the patient on site and give fibrinolytic therapy, with its limitations. Earlier trials suggested that the transfer strategy was superior, but they had limitations: most patients received streptokinase, an inferior fibrinolytic agent, and door-to-door-to-balloon times were rapid, averaging only 95 minutes because of excellent logistical protocols and careful patient selection.2 Most importantly, rescue PCI and routine PCI were seldom performed in patients receiving fibrinolytics, so fibrinolytic therapy was being tested as monotherapy.
In the real world, however, treatment delays are much longer, and fibrinolytic therapy has evolved into a strategy that includes crossover to rescue PCI or routine PCI. Therefore, the initial trials of transfer for primary PCI do not reflect current practice. In fact, recent registry data suggest that prehospital fibrinolytic therapy followed by early angiography is superior to primary PCI because most patients can be treated within 2 hours of symptom onset; they also suggest that on-site fibrinolytic therapy followed by early angiography is equal in efficacy to primary PCI as long as rescue PCI and routine PCI can be performed.3,4
The most important modifiable predictor of outcome in ST-elevation MI is the time to treatment, a biological truth that continues to be supported by clinical evidence despite ideologic arguments by some interventional cardiology enthusiasts who claim that primary PCI is always superior to the fibrinolytic strategy, regardless of delays.
SURPRISINGLY, OUTCOMES WERE WORSE WITH FACILITATED PCI
It made sense, then, to conclude that the perfect strategy for hospitals without PCI capability would be a combined strategy of immediate fibrinolytic therapy to decrease the time delay associated with organizing PCI, and rapid transfer for immediate PCI to improve the limited reperfusion rates associated with fibrinolytic therapy.
Surprisingly, though, randomized trials found worse outcomes with this “facilitated PCI” strategy.5
Again, limitations in trial design might explain the lack of benefit in the trials. Inadequate anticoagulant and antiplatelet therapy were given to the fibrinolytic patients, and primary PCI patients had relatively short treatment delays, with many patients enrolled at hospitals with PCI capability.
PROGRESS IN REPERFUSION THERAPY
Great strides have been made in reperfusion therapy in recent years. Adjunctive therapy with clopidogrel (Plavix) and enoxaparin (Lovenox) has been shown to improve outcomes with fibrinolytic therapy. Bivalirudin (Angiomax) and stents have improved primary PCI’s performance. Reducing bleeding complications has become a clinical priority, with increasing emphasis on adjusting some drug doses according to renal function and using the radial artery for cardiac catheterization.
The American College of Cardiology initiative, “Door-to-Balloon (D2B): An Alliance for Quality,” focused much attention on organizing in-hospital systems of care for primary PCI, thus increasing the national rate of achieving a door-to-balloon time within 90 minutes from 50% to over 75% in patients who presented to hospitals with PCI capability.6
The American Heart Association has launched “Mission: Lifeline,” a national campaign to organize prehospital systems of care with their program,7 working within communities to address their unique needs, resources, and barriers to implementing systems of care for ST-elevation MI. The key aspect of this effort is to help geographic regions develop local solutions, an explicit recognition that there is no one-size-fits-all solution. Early triage by emergency medical services, rapid diagnosis with prehospital electrocardiography, destination and interhospital transfer protocols, and prehospital activation of the cardiac catheterization laboratory can greatly streamline emergency care and decrease treatment delays for primary PCI.
FOR OUTLYING HOSPITALS, A PHARMACOINVASIVE STRATEGY
So what about hospitals without PCI capability that cannot routinely transfer patients to a hospital with PCI capability within 90 minutes?
Lessons learned from the experiences with immediate PCI, rescue PCI, and facilitated PCI have evolved into the “pharmacoinvasive strategy.” Patients with ST-elevation MI are treated as rapidly as possible with a bolus of a fibrinolytic drug, eg, tenecteplase (TNKase) or reteplase (Retavase), and are also given aspirin, clopidogrel, and enoxaparin. Then, they are rapidly transferred to a PCI-capable hospital so that emergency PCI can be performed if reperfusion is not clinically apparent or if the patient develops pulmonary edema or cardiogenic shock. If the clinical signs suggest that reperfusion has been achieved (relief of chest pain, rapid resolution of ST-segment elevation, bursts of accelerated idioventricular rhythm), coronary angiography (and PCI, if indicated) can be performed within 3 to 24 hours of fibrinolytic therapy. This time frame allows the initial fibrinolytic effect to dissipate, while the antiplatelet and anticoagulant drugs achieve therapeutic levels.
Today, the goal is to treat every patient with the best reperfusion strategy available, given the limitations in resources and the geographic location of some centers, and to maximize the possibility of sustained patency of the infarct-related artery by implanting a stent, even if it takes several hours and transfer to another hospital to perform PCI.8 The pharmacoinvasive strategy of rapid administration of fibrinolytic therapy followed by PCI within 24 hours would be practical in most hospitals without PCI capability where treatment delays prohibit performance of primary PCI within 90 minutes of first medical contact.9
THE ‘STREAM’ TRIAL IS UNDER WAY
As proof of concept, the Strategic Reperfusion Early After Myocardial Infarction (STREAM) trial is enrolling 2,000 patients with ST-elevation MI presenting within 3 hours of symptom onset if primary PCI is not feasible within 60 minutes of first medical contact.10 Patients will be randomized to either of the following:
Receive prehospital therapy with tenecteplase, aspirin, clopidogrel, and enoxaparin and undergo cardiac catheterization in 6 to 24 hours (or rescue PCI if reperfusion fails within 90 minutes of fibrinolysis)
Undergo primary PCI performed according to local guidelines.
The primary measure of efficacy will be the composite rate of death, cardiogenic shock, heart failure, and reinfarction at 30 days. Measures of safety include the rates of ischemic stroke, intracranial hemorrhage, and major nonintracranial bleeding.
References
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet2003; 361:13–20.
Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation2003; 108:1809–1814.
Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FASTMI). Circulation2008; 118:268–276.
Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA2010; 303:2148–2155.
Keeley EC, Boura JA, Grines CL. Comparison of primary and facilitated percutaneous coronary interventions for ST-elevation myocardial infarction: quantitative review of randomised trials. Lancet2006; 367:579–588.
Krumholz HM, Bradley EH, Nallamothu BK, et al. A campaign to improve the timeliness of primary percutaneous coronary intervention: Door-to-Balloon: An Alliance for Quality. JACC Cardiovasc Interv. 2008; 1:97–104.
Jacobs AK, Antman EM, Faxon DP, Gregory T, Solis P. Development of systems of care for ST-elevation myocardial infarction patients: executive summary. Circulation2007; 116:217–230.
Kushner FG, Hand M, Smith SC, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation2009; 120:2271–2306.
Bates ER, Nallamothu BK. Commentary: the role of percutaneous coronary intervention in ST-segment-elevation myocardial infarction. Circulation2008; 118:567–573.
Armstrong PW, Gershlick A, Goldstein P, et al; STREAM Steering Committee. The Strategic Reperfusion Early After Myocardial Infarction (STREAM) study. Am Heart J2010; 160:30–35.
References
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet2003; 361:13–20.
Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation2003; 108:1809–1814.
Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FASTMI). Circulation2008; 118:268–276.
Lambert L, Brown K, Segal E, Brophy J, Rodes-Cabau J, Bogaty P. Association between timeliness of reperfusion therapy and clinical outcomes in ST-elevation myocardial infarction. JAMA2010; 303:2148–2155.
Keeley EC, Boura JA, Grines CL. Comparison of primary and facilitated percutaneous coronary interventions for ST-elevation myocardial infarction: quantitative review of randomised trials. Lancet2006; 367:579–588.
Krumholz HM, Bradley EH, Nallamothu BK, et al. A campaign to improve the timeliness of primary percutaneous coronary intervention: Door-to-Balloon: An Alliance for Quality. JACC Cardiovasc Interv. 2008; 1:97–104.
Jacobs AK, Antman EM, Faxon DP, Gregory T, Solis P. Development of systems of care for ST-elevation myocardial infarction patients: executive summary. Circulation2007; 116:217–230.
Kushner FG, Hand M, Smith SC, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation2009; 120:2271–2306.
Bates ER, Nallamothu BK. Commentary: the role of percutaneous coronary intervention in ST-segment-elevation myocardial infarction. Circulation2008; 118:567–573.
Armstrong PW, Gershlick A, Goldstein P, et al; STREAM Steering Committee. The Strategic Reperfusion Early After Myocardial Infarction (STREAM) study. Am Heart J2010; 160:30–35.
Effective and rapid reperfusion is crucial in patients with acute ST-segment elevation myocardial infarction (MI). The preferred strategy for reperfusion—when it can be performed in a timely fashion at an experienced facility—is primary percutaneous coronary intervention (PCI), which produces outcomes superior to those of pharmacologic thrombolysis.1
Unfortunately, in the United States about half of patients present to hospitals that do not have PCI capability,2 and in one analysis, 91% of transferred patients had a door-to-balloon time greater than the recommended 90 minutes, with a mean of 152 minutes.3 (In this case, the door-to-balloon time was the time that elapsed between entry into the first hospital and inflation of the PCI balloon at the second hospital.)
In situations such as these, a combined approach may be appropriate, with thrombolysis delivered by paramedics or at a local facility, followed by transfer to a PCI facility and performance of PCI within a few hours. However, this is feasible only if standardized community-based or regional protocols for prompt transfer and reperfusion are in place.
In this paper we discuss the rationale and the clinical data behind several approaches to combined reperfusion, as well as experiences with community-based care protocols.
WITHIN 3 HOURS OF SYMPTOM ONSET, THROMBOLYSIS IS AS GOOD AS PCI
The PRAGUE-2 Trial
In the randomized PRAGUE-2 trial,4 patients with ST-elevation MI who presented to a non-PCI facility had better outcomes if they were transferred promptly for PCI (median door-to-balloon time 97 minutes), as opposed to receiving local therapy with streptokinase. However, for patients presenting within 3 hours of symptom onset, the mortality rates were comparable with either strategy.4
See the glossary of clinical trial names below
The CAPTIM trial
In the CAPTIM trial,5 patients who presented within 2 hours of symptom onset and who were randomized to receive prehospital thrombolysis had outcomes similar to those of patients treated with primary PCI, despite a short door-to-balloon time (82 minutes).
The Vienna STEMI Registry
In the Vienna STEMI Registry,6 the mortality rates with primary PCI and with thrombolysis were similar when patients presented within 2 hours of symptom onset. However, as the time from symptom onset increased, primary PCI appeared to offer an increasing survival benefit compared with thrombolysis.
Comments: Thrombolysis is effective mostly in the first 2 to 3 hours, with some benefit up to 12 hours
Previous studies have shown that the sooner thrombolysis is given after symptom onset, the more effective it is. If it is given within an hour of symptom onset, the relative reduction in the mortality rate is 50% and the absolute reduction is 6.5% compared with no reperfusion therapy. If it is started in the second hour, the absolute reduction in the mortality rate drops to 4%, and a lesser benefit extends to patients presenting up to 12 hours after symptom onset.7 This time-dependent benefit is due to the fact that very early reperfusion of the occluded coronary artery may lead to full recovery of ischemic tissue and thus prevent necrosis. In addition, thrombolysis in the first 2 hours is highly efficacious in lysing a fresh thrombus.
These data support the current guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA), which state no preference for either thrombolytic therapy or PCI in ST-elevation MI if the presentation is less than 3 hours after symptom onset.8
Of note, in the CAPTIM trial and in the Vienna STEMI Registry, rescue PCI was available and was in fact used after thrombolysis in about 25% of patients, which might have contributed to the benefit of early thrombolysis.
PRIMARY PCI MAY NOT BE SUPERIOR IF TRANSFER TIME IS LONG
Another time-related factor to consider is the PCI-related delay, ie, the theoretical difference between the expected time from first medical contact to balloon inflation (if the patient undergoes primary PCI) and the time from first medical contact to the start of thrombolytic therapy (if the patient undergoes primary thrombolysis).
A meta-analysis of 13 trials comparing PCI and thrombolysis showed that a PCI-related delay of more than 60 minutes might negate the potential advantage of primary PCI over immediate thrombolysis in terms of deaths.9
This observation has been further refined by data from the National Registry of Myocardial Infarction.10 In this analysis, patient factors, including age, duration of symptoms, and infarct location, significantly affected the point at which the PCI-related delay negated the survival advantage of primary PCI. The survival advantage of primary PCI was lost more rapidly—with a PCI-related delay as short as 40 minutes—in patients who presented sooner, were younger, or had anterior MI. Primary PCI maintained its survival advantage even with a PCI-related delay longer than 100 minutes in older patients or patients with nonanterior MI presenting more than 3 hours after symptom onset. Given that median door-to-balloon times in the United States may exceed 150 minutes when transfer is involved, 3 primary PCI may be no better than primary thrombolysis in transferred patients who present early or who have large infarcts.
Although these results were derived from a post hoc analysis of a registry and the delay times reported were sometimes inaccurate, they suggest that both the PCI-related delay time and patient characteristics should be considered when selecting a reperfusion strategy. Thrombolytic therapy before and in conjunction with primary PCI was considered a potential solution to these concerns.
In addition, while the benefit of any reperfusion strategy depends on the time of presentation, the loss in benefit by later presentation is less pronounced with primary PCI than with thrombolysis, making thrombolysis less attractive in later presentations (> 3 hours).11
Also, while thrombolytic therapy in patients older than 75 years was associated with a lower mortality rate compared with no therapy in a large Swedish registry,12 this benefit was less striking than in younger patients. A meta-analysis of thrombolysis trials failed to show a similar benefit in patients over age 75 vs younger patients,13 whereas primary PCI remained effective and superior to thrombolysis in the elderly, with more absolute reduction in mortality rates in the elderly subgroup than with younger patients. 14 This makes thrombolysis less attractive in the elderly, either as a stand-alone therapy or in conjunction with PCI. Studies of combined thrombolysis and PCI included very few patients over age 75.15–17
THREE COMBINATION REPERFUSION STRATEGIES
Figure 1. The timing of percutaneous coronary intervention (PCI) in relation to thrombolysis in the pharmacoinvasive strategy, rescue PCI strategy, and facilitated PCI strategy, with the respective clinical trials that addressed and defined these strategies. (See the glossary above for complete names of studies.)Three different combination reperfusion strategies for ST-elevation MI have been studied (Figure 1)15,16,18–20:
Facilitated PCI is a strategy of thrombolysis immediately followed by PCI, with a planned door-to-balloon time of 90 to 120 minutes.
Pharmacoinvasive therapy means giving thrombolysis at a non-PCI facility and then promptly and systematically transferring the patient to a PCI facility, where PCI is performed 2 to 24 hours after the start of thrombolytic therapy, regardless of whether thrombolysis results in successful reperfusion. 15 Thus, the time to PCI is longer than with facilitated PCI. Facilitated PCI addresses the value of pretreatment with thrombolytics or glycoprotein IIb/IIIa inhibitors in patients otherwise eligible for primary PCI, whereas pharmacoinvasive therapy addresses the value of routine early PCI after thrombolysis in patients who are not eligible for primary PCI.16
Rescue PCI refers to PCI that is performed urgently if thrombolysis fails, failure being defined as persistent hemodynamic or electrical instability, persistent ischemic symptoms, or failure to achieve at least a 50% to 70% resolution of the maximal ST-segment elevation 90 minutes after the infusion is started.
FACILITATED PCI: NEGATIVE RESULTS IN CLINICAL TRIALS
ASSENT-4 PCI trial
In the ASSENT-4 PCI trial,18 patients receiving full thrombolytic therapy before PCI had a higher rate of in-hospital death, bleeding, and cardiovascular events at 90 days than patients treated with primary PCI.
This trial recruited patients arriving at hospitals with or without PCI capability. The door-to-balloon time was about 110 minutes in both groups, which might not have been prolonged enough to show a benefit from a timely addition of thrombolysis. In addition, antiplatelet therapy was limited in these patients: glycoprotein IIb/IIIa inhibitors were not given, and clopidogrel (Plavix) was not appropriately preloaded, and this might have offset the potential benefit of early PCI. In fact, data suggest that platelet activation and aggregation are heightened after thrombolysis, 21–23 and that glycoprotein IIb/IIIa antagonists can inhibit these effects.23
The FINESSE trial
In the FINESSE trial,19 patients were randomized to undergo primary PCI, to undergo PCI facilitated (ie, preceded) by abciximab (Reo-Pro), or to undergo PCI facilitated by half-dose reteplase (Retavase) and full-dose abciximab. Despite a median door-to-balloon time of 132 minutes, the three strategies were associated with similar rates of death, heart failure, or ischemic outcome at 90 days. Even though the dosage of heparin was weight-adjusted, more major bleeding events occurred with the facilitated strategies.
Comments: Some subgroups may still benefit from facilitated PCI
The results of ASSENT-4 PCI and FINESSE led to the conclusion that PCI facilitated by full-dose thrombolysis should be avoided, and called into question the value of PCI facilitation using glycoprotein IIb/IIIa inhibitors with or without half-dose thrombolytic therapy.
However, subgroup analyses of these trials identified some subgroups that may benefit from a facilitated strategy. In ASSENT-4 PCI, 45% of patients were enrolled at PCI hospitals with a minimal PCI-related delay time. These patients had the worst outcome with the facilitated strategy. In contrast, patients who had a short time from pain onset to thrombolysis (2 to 3 hours) and who were given prehospital thrombolysis had a trend toward better outcomes with facilitated PCI.24 And in FINESSE, 60% of patients were enrolled at centers with PCI capability. Analysis of a small subgroup of patients with a Thrombolysis in Myocardial Infarction study (TIMI) risk score of 3 or greater presenting to non-PCI hospitals within 4 hours of symptom onset suggested a potential reduction of ischemic events with the facilitated strategy in these patients.25
Thus, for patients seen in the first 2 to 3 hours after symptom onset, immediate thrombolysis is recommended if PCI will likely be delayed, with or without plans for subsequent early PCI. “Time is muscle,” especially during the first 3 hours.
PHARMACOINVASIVE STRATEGY: GOOD RESULTS IN HIGH-RISK PATIENTS
A number of randomized studies during the last 10 years have examined the value of a pharmacoinvasive strategy.15,16,26–29
The TRANSFER-AMI trial
The TRANSFER-AMI trial15 randomized 1,059 patients with high-risk ST-elevation MI (ie, anterior or high-risk inferior) at non-PCI centers to undergo either pharmacoinvasive care, ie, full-dose tenecteplase (TNKase) with immediate transfer for PCI or standard care, ie, tenecteplase with transfer for rescue PCI if the patient had persistent ST-segment elevation, chest pain, or hemodynamic instability.15 The goal was to perform PCI within 6 hours of thrombolysis, and the median time to PCI was 3.9 hours (range 2–6 hours). In the standard-care group, 35% of patients needed to be transferred for rescue PCI. Unlike in the ASSENT-4 trial, over 80% of patients received aggressive antiplatelet therapy with both 300 mg of clopidogrel and glycoprotein IIb/IIIa inhibitors.
The rate of cardiovascular events at 30 days was significantly lower with pharmacoinvasive therapy than with standard care and rescue PCI (11% vs 17%, P = .004). This difference was driven by lower rates of recurrent ischemia, reinfarction, and heart failure.
The CARESS-in-AMI study
The CARESS-in-AMI study16 found a similar improvement in ischemic outcomes in 600 patients with high-risk ST-elevation MI arriving at non-PCI centers if they had received pharmacoinvasive therapy. Patients received half-dose reteplase and abciximab and were randomized either to be immediately transferred for PCI (median time to PCI 2.25 hours) or to be transferred only if they had persistent ST-segment elevation or clinical deterioration.16 The event rate was low with pharmacoinvasive therapy, comparable to that achieved in primary PCI trials.
Interestingly, no significant increase was seen in the risk of major and minor bleeding in these two trials despite the use of a femoral approach for PCI in over 80% of the cases; this is probably due to the delays between thrombolytic administration and PCI and to the use of a highly fibrin-specific thrombolytic agent and adjusted-dose heparin.
Meta-analysis of pharmacoinvasive trials
A meta-analysis29 of studies of systematic early PCI (mainly with stenting) within 24 hours of thrombolysis showed a reduction in the rates of mortality and reinfarction with this strategy, without an increase in the risk of major or intracranial bleeding.30 In contrast to the results of the trials of facilitated PCI, a pharmacoinvasive strategy improved outcomes in these trials because the delay between thrombolysis and PCI was more than 2 hours, ie, long enough to prevent bleeding complications, and because most patients randomized in these trials presented within 2 to 3 hours of symptom onset, when the time to reperfusion is critical. After 3 hours, the PCI-mediated myocardial salvage is less time-dependent. Moreover, trials of pharmacoinvasive strategy used aggressive antiplatelet therapy with clopidogrel and glycoprotein IIb/IIIa inhibitors.
Comment: Pharmacoinvasive strategy in the guidelines
These results and those of the subgroup analysis from the FINESSE trial suggest that patients with high-risk ST-elevation MI treated at non-PCI hospitals have better outcomes without an increase in major bleeding events when given thrombolysis and then immediately transferred for routine PCI, rather than being transferred only if reperfusion fails.
Hence, the 2009 update of the ACC/AHA guidelines31 gives a class IIa recommendation for transferring patients with anterior ST-elevation MI or high-risk inferior ST-elevation MI treated with thrombolysis to a PCI-capable facility where PCI is performed as part of a pharmacoinvasive or rescue strategy soon after thrombolysis.
This strategy has been particularly studied in patients younger than 75 years presenting with high-risk types of ST-elevation MI early (< 3 hours) after symptom onset. If not at high risk, the patient may be transferred to a PCI facility after receiving thrombolysis or observed in the initial facility (class IIb recommendation). Consideration should be given to starting anticoagulant and antiplatelet therapy before and during transfer—ie, 300 mg of clopidogrel before transfer for PCI and glycoprotein IIb/IIIa inhibitor therapy during PCI.
The European Society of Cardiology (ESC) guidelines32 recommend early routine angiography 3 to 24 hours after successful thrombolysis. This time window was selected to avoid PCI during the prothrombotic period in the first few hours after thrombolysis and to minimize the risk of reocclusion with PCI delays of more than 24 hours (class IIa recommendation).
Larger randomized trials are still needed to establish whether the pharmacoinvasive strategy confers a survival benefit, to determine its usefulness in low-risk inferior or lateral ST-elevation MI, and to further refine the time window when PCI is both safe and beneficial after thrombolysis.33
RESCUE PCI REDUCES MORTALITY RATES
Rescue PCI is the most accepted form of thrombolysis-PCI combination.
The REACT trial
The REACT trial20 showed that rescue PCI performed at a mean of 4.5 hours after failed thrombolysis reduces the rate of adverse cardiovascular events by more than 50% at 6 to 12 months and reduces the 5-year mortality rate by more than 50% compared with conservative management.20 As in the pharmacoinvasive strategy, aggressive antiplatelet regimens were used in the REACT trial.
A meta-analysis of rescue PCI trials
A meta-analysis of rescue PCI trials34 confirmed these results, showing a reduction in heart failure and reinfarction and a trend toward a lower mortality rate with rescue PCI.34 After thrombolysis, 40% of patients do not achieve grade 3 TIMI flow, which explains why in modern clinical trials 30% of patients treated with thrombolysis require rescue PCI.5,15,16,35
For patients with high-risk ST-elevation MI, current ACC/AHA guidelines assign a class IIa recommendation to rescue PCI.31
WHEN PATIENTS WITH ST-ELEVATION MI PRESENT TO A NON-PCI HOSPITAL
Transfer for primary PCI vs thrombolysis at the non-PCI hospital
The DANAMI-2 trial36 found that immediate transfer for PCI was superior to onsite thrombolytic therapy, as measured by a reduction in the rate of ischemic events (composite of death, myocardial infarction, or stroke at 30 days): 8.5% vs 14.2% (P < .001). There were no deaths during transfer.3
The PRAGUE-2 trial4 showed similar results for patients presenting 3 to 12 hours after symptom onset (30-day mortality rate 6% with immediate transfer vs 15.3% with on-site thrombolysis, P < .002), whereas patients presenting within 3 hours of symptom onset had a similar mortality rate with either therapy.4
Comment. These trials showed that transfer for primary PCI is superior to thrombolytic therapy when performed in a timely fashion. However, they were done in countries with established transfer networks and short distances between community hospitals and PCI centers, with a PCI-related delay of only 44 minutes and a door-to-balloon time of 90 minutes despite transfer. The large-scale application of this prompt transfer policy is not practical in most regions in the United States. Thus, a strategy of local thrombolysis followed by routine early transfer for routine or rescue PCI seems warranted when the door-to-balloon time or the PCI-related delay time is expected to be too long.
Experiences with community-based systems of care and prehospital thrombolysis
In Minnesota, Henry et al37 developed a PCI-based treatment system and an integrated transfer program for ST-elevation MI involving 30 hospitals within 210 miles of the Minneapolis Heart Institute. Participating hospitals were divided into two zones: zone 1 hospitals were within 60 miles, and zone 2 facilities were between 60 and 210 miles from the Heart Institute. Zone 2 patients received half-dose tenecteplase (if thrombolytic therapy was not contraindicated) in anticipation of a lengthy transfer time.
The median door-to-balloon time for zone 1 patients was 95 minutes (interquartile range 82 and 116 minutes) and for zone 2 patients 120 minutes (interquartile range 100 and 145 minutes). The diagnosis of ST-elevation MI was made by the emergency department physician, who activated the system with a phone call. The patient was then directly transferred to the catheterization laboratory, most often by helicopter.
The in-hospital death rate for patients who presented to the PCI center and for patients in zones 1 and 2 was similarly low (about 5%).37
In France, the FAST-MI registry,17 which collected outcome data for different reperfusion strategies, found that thrombolysis yielded in-hospital and midterm results that were comparable to those of primary PCI. Of note, thrombolysis was started early after symptom onset (about 2 hours), and was started in the ambulance in two-thirds of cases. Nearly all patients underwent a pharmacoinvasive strategy that combined thrombolysis with coronary angiography and PCI within 24 hours of symptom onset. These findings suggest that timely thrombolysis followed by semiurgent transfer for PCI is an alternative to primary PCI for patients presenting to hospitals with no PCI capability, and that this alternative offers similar benefit to that of primary PCI.
Five centers in the United States have reported their experience with half-dose thrombolysis in the prehospital setting (in the field or during transfer) or at a non-PCI hospital, followed by prompt transfer to a PCI facility. In this registry of almost 3,000 patients,38 patients treated with thrombolysis had better outcomes than patients directly transferred for primary PCI, with a significantly lower 30-day mortality rate (3.8% vs from 6.4%), and no increase in bleeding.38,39 The mean door-to-balloon time was long (168 minutes in the primary PCI group and 196 minutes in the thrombolysis-PCI group), which might explain the benefit achieved with prompt thrombolysis.
CARDIOGENIC SHOCK
Patients presenting with left ventricular cardiogenic shock derive a large mortality benefit from revascularization, whether they are transferred or directly admitted to a PCI center. 40 Moreover, in the SHOCK registry, patients with predominant right ventricular cardiogenic shock had an in-hospital mortality rate similar to that of patients with predominant left ventricular cardiogenic shock, and revascularization (PCI or surgical revascularization) was associated with a strikingly lower mortality rate in both groups.41
Thus, all patients with left or right cardiogenic shock should be revascularized on an emergency basis, either surgically or percutaneously.
While trials of pharmacoinvasive therapy excluded patients with cardiogenic shock,15,16 thrombolytic therapy was associated with improved outcomes in the drug-therapy group of the SHOCK trial and in hypotensive patients randomized in the early thrombolysis trials.13 Thus, the ACC/AHA guidelines recommend thrombolytic therapy before transfer if a patient presents in shock within 3 to 6 hours of onset of the MI and delays in transport and intervention are anticipated.8
PUTTING IT ALL TOGETHER: MANAGEMENT STRATEGIES
Figure 2. Selecting the appropriate reperfusion strategy in ST-elevation myocardial infarction (MI). Routine early PCI is particularly indicated in high-risk MI, ie, either anterior MI, or inferior MI with one of the following: systolic blood pressure of less than 100 mm Hg, heart rate of more than 100 beats per minute, Killip class II or III, ST-segment depression of 2 mm or more in the anterior leads, or ST-segment elevation of 1 mm or more in the right-sided lead V4, which is indicative of right ventricular involvement. Dual antiplatelet therapy with aspirin and clopidogrel (Plavix) 300 mg should be started as soon as possible in all patients, and consideration should be given to glycoprotein IIb/IIIa inhibition for most patients during PCI (as in the TRANSFER-AMI15 and CARESS-in-AMI16 trials).Taking into account the importance of time to presentation, the PCI-related delay time, and patient and MI characteristics, as well as whether a regional transfer system is in place (as in Minnesota), we suggest an algorithmic approach to the management of ST-elevation MI at a non-PCI facility (Figure 2).
If an effective transfer system is in place, primary PCI not preceded by thrombolytic therapy or glycoprotein IIb/IIIa inhibitor therapy is the preferred approach, according to ACC/AHA and ESC guidelines.31,32 Giving thrombolytics immediately before PCI is harmful and thus should be avoided when the expected door-to-balloon time is 90 minutes or less.
All hospitals (whether or not they offer PCI) and regional emergency medical services should participate in a community-based system of care for ST-elevation MI, with protocols for expeditious transfer as defined and coordinated by the American Heart Association initiative “Mission: Lifeline.” In addition, a system of field triage and direct transport to the catheterization laboratory of a PCI facility after field activation significantly reduces door-to-balloon times and improves outcomes.42
If such a system is not in place, then a pharmacoinvasive strategy seems best: ie, local full-dose thrombolysis (if not contraindicated) followed by transfer to a PCI facility and routine performance of PCI 2 to 6 hours after thrombolysis—in conjunction with aggressive early dual oral antiplatelet therapy and “downstream” glycoprotein IIb/IIIa inhibition. This approach is associated with outcomes similar to those of primary PCI.15–17,37
Prehospital thrombolysis delivered by paramedics and followed by early transfer to a PCI facility has been associated with further reduction in mortality rates compared with in-hospital thrombolysis (as in the Swedish registry43), and a reduction in death rate comparable to that of primary PCI in patients presenting early. This is an adequate strategy in regions where such a system can be established.5,17,38,43,44
Patients presenting more than 3 to 4 hours after symptom onset, older patients, and patients with lower-risk MI or a higher risk of bleeding may still be suited for primary PCI even when the door-to-balloon time is 90 to 120 minutes, as stated by the European guidelines,32 or when the PCI-related delay time is as long as 100 minutes. 10 On the other hand, while the ACC/AHA guidelines recognize that in these patients the mortality advantage of primary PCI vs thrombolytic therapy is maintained with more prolonged door-to-balloon times, they nevertheless state that the focus should be on developing systems of care to increase the number of patients with access to primary PCI in less than 90 minutes rather than extending the acceptable window for door-to-balloon time.
In conclusion, for patients presenting with ST-elevation MI who cannot undergo timely primary PCI, the best approach seems to be prehospital thrombolysis delivered by paramedics or local thrombolysis at the non-PCI hospital followed by transferring the patient and performing PCI within a few hours. This is especially important in patients with high-risk ST-elevation MI who present early after symptom onset, when the extent of myocardial necrosis associated with delayed primary PCI is largest.
In addition, every community should develop a coordinated transfer strategy between non-PCI and PCI hospitals.
References
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet2003; 361:13–20.
Waters RE, Singh KP, Roe MT, et al. Rationale and strategies for implementing community-based transfer protocols for primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. J Am Coll Cardiol2004; 43:2153–2159.
Chakrabarti A, Krumholz HM, Wang Y, Rumsfeld JS, Nallamothu BK; National Cardiovascular Data Registry. Time-to-reperfusion in patients undergoing interhospital transfer for primary percutaneous coronary intervention in the U.S: an analysis of 2005 and 2006 data from the National Cardiovascular Data Registry. J Am Coll Cardiol2008; 51:2442–2443.
Widimský P, Budesínský T, Vorác D, et al; ‘PRAGUE’ Study Group Investigators. Long distance transport for primary angioplasty vs immediate thrombolysis in acute myocardial infarction. Final results of the randomized national multicentre trial—PRAGUE-2. Eur Heart J2003; 24:94–104.
Steg PG, Bonnefoy E, Chabaud S, et al; Comparison of Angioplasty and Prehospital Thrombolysis in Acute Myocardial infarction (CAPTIM) Investigators. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation2003; 108:2851–2856.
Kalla K, Christ G, Karnik R, et al; Vienna STEMI Registry Group. Implementation of guidelines improves the standard of care: the Viennese registry on reperfusion strategies in ST-elevation myocardial infarction (Vienna STEMI registry). Circulation2006; 113:2398–2405.
Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet1996; 348:771–775.
Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation2004; 110:e82–e292.
Nallamothu BK, Bates ER. Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction: is timing (almost) everything?Am J Cardiol2003; 92:824–826.
Pinto DS, Kirtane AJ, Nallamothu BK, et al. Hospital delays in reperfusion for ST-elevation myocardial infarction: implications when selecting a reperfusion strategy. Circulation2006; 114:2019–2025.
Boersma E; Primary Coronary Angioplasty vs Thrombolysis Group. Does time matter? A pooled analysis of randomized clinical trials comparing primary percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J2006; 27:779–788.
Stenestrand U, Wallentin L; Register of Information and Knowledge About Swedish Heart Intensive Care Admissions (RIKS-HIA). Fibrinolytic therapy in patients 75 years and older with ST-segment-elevation myocardial infarction: one-year follow-up of a large prospective cohort. Arch Intern Med2003; 163:965–971.
Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet1994; 343:311–322.
Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med1993; 328:673–679.
Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med2009; 360:2705–2718.
Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab RE-teplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet2008; 371:559–568.
Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FAST-MI). Circulation2008; 118:268–276.
Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet2006; 367:569–578.
Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med2008; 358:2205–2217.
Carver A, Rafelt S, Gershlick AH, Fairbrother KL, Hughes S, Wilcox R; REACT Investigators. Longer-term follow-up of patients recruited to the REACT (Rescue Angioplasty Versus Conservative Treatment or Repeat Thrombolysis) trial. J Am Coll Cardiol2009; 54:118–126.
Rasmanis G, Vesterqvist O, Gréen K, Edhag O, Henriksson P. Evidence of increased platelet activation after thrombolysis in patients with acute myocardial infarction. Br Heart J1992; 68:374–376.
Gurbel PA, Serebruany VL, Shustov AR, et al. Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. GUSTO-III Investigators. Global Use of Strategies to Open Occluded Coronary Arteries. J Am Coll Cardiol1998; 31:1466–1473.
Coulter SA, Cannon CP, Ault KA, et al. High levels of platelet inhibition with abciximab despite heightened platelet activation and aggregation during thrombolysis for acute myocardial infarction: results from TIMI (thrombolysis in myocardial infarction) 14. Circulation2000; 101:2690–2695.
Ross AM, Huber K, Zeymer U, et al. The impact of place of enrollment and delay to reperfusion on 90-day post-infarction mortality in the ASSENT-4 PCI trial: assessment of the safety and efficacy of a new treatment strategy with percutaneous coronary intervention. JACC Cardiovasc Interv2009; 2:925–930.
Herrmann HC, Lu J, Brodie BR, et al; FINESSE Investigators. Benefit of facilitated percutaneous coronary intervention in high-risk ST-segment elevation myocardial infarction patients presenting to nonpercutaneous coronary intervention hospitals. JACC Cardiovasc Interv2009; 2:917–924.
Scheller B, Hennen B, Hammer B, et al; SIAM III Study Group. Beneficial effects of immediate stenting after thrombolysis in acute myocardial infarction. J Am Coll Cardiol2003; 42:634–641.
Fernandez-Avilés F, Alonso JJ, Castro-Beiras A, et al; GRACIA (Grupo de Análisis de la Cardiopatía Isquémica Aguda) Group. Routine invasive strategy within 24 hours of thrombolysis versus ischaemiaguided conservative approach for acute myocardial infarction with ST-segment elevation (GRACIA-1): a randomised controlled trial. Lancet2004; 364:1045–1053.
Le May MR, Wells GA, Labinaz M, et al. Combined angioplasty and pharmacological intervention versus thrombolysis alone in acute myocardial infarction (CAPITAL AMI study). J Am Coll Cardiol2005; 46:417–424.
Bøhmer E, Hoffmann P, Abdelnoor M, Arnesen H, Halvorsen S. Efficacy and safety of immediate angioplasty versus ischemia-guided management after thrombolysis in acute myocardial infarction in areas with very long transfer distances results of the NORDISTEMI (NORwegian study on DIstrict treatment of ST-elevation myocardial infarction). J Am Coll Cardiol2010; 55:102–110.
Wijeysundera HC, You JJ, Nallamothu BK, Krumholz HM, Cantor WJ, Ko DT. An early invasive strategy versus ischemia-guided management after fibrinolytic therapy for ST-segment elevation myocardial infarction: a meta-analysis of contemporary randomized controlled trials. Am Heart J2008; 156:564–572,572.e1–e2.
Kushner FG, Hand M, Smith SC, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol2009; 54:2205–2241.
Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J2008; 29:2909–2945.
Mukherjee D, Moliterno DJ. The timely coupling of mechanical revascularization following thrombolysis for myocardial infarction. Cardiology2007; 107:337–339.
Wijeysundera HC, Vijayaraghavan R, Nallamothu BK, et al. Rescue angioplasty or repeat fibrinolysis after failed fibrinolytic therapy for ST-segment myocardial infarction: a meta-analysis of randomized trials. J Am Coll Cardiol2007; 49:422–430.
The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med1993; 329:1615–1622.
Andersen HR, Nielsen TT, Rasmussen K, et al; DANAMI-2 Investigators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N Engl J Med2003; 349:733–742.
Henry TD, Sharkey SW, Burke MN, et al. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation2007; 116:721–728.
Denktas AE, Athar H, Henry TD, et al. Reduced-dose fibrinolytic acceleration of ST-segment elevation myocardial infarction treatment coupled with urgent percutaneous coronary intervention compared to primary percutaneous coronary intervention alone results of the AMICO (Alliance for Myocardial Infarction Care Optimization) Registry. JACC Cardiovasc Interv2008; 1:504–510.
Smalling RW. Ischemic time: the new gold standard for ST-segment elevation myocardial infarction care. J Am Coll Cardiol2009; 54:2154–2156.
Hochman JS, Sleeper LA, White HD, et al; SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. One-year survival following early revascularization for cardiogenic shock. JAMA2001; 285:190–192.
Jacobs AK, Leopold JA, Bates E, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol2003; 41:1273–1279.
Pedersen SH, Galatius S, Hansen PR, et al. Field triage reduces treatment delay and improves long-term clinical outcome in patients with acute ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol2009; 54:2296–2302.
Björklund E, Stenestrand U, Lindbäck J, Svensson L, Wallentin L, Lindahl B. Pre-hospital thrombolysis delivered by paramedics is associated with reduced time delay and mortality in ambulance-transported real-life patients with ST-elevation myocardial infarction. Eur Heart J2006; 27:1146–1152.
The European Myocardial Infarction Project Group. Prehospital thrombolytic therapy in patients with suspected acute myocardial infarction. N Engl J Med1993; 329:383–389.
Elias B. Hanna, MD Currently in the Department of Medicine, Cardiovascular Section, Louisiana State University, New Orleans; this article written while in the Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Thomas A. Hennebry, MD Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Mazen S. Abu-Fadel, MD Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Address: Elias B. Hanna, MD, Department of Medicine, Section of Cardiology, Louisiana State University, Room 323, Box T4M-2, 1542 Tulane Avenue, New Orleans, LA 70112; e-mail [email protected]
Elias B. Hanna, MD Currently in the Department of Medicine, Cardiovascular Section, Louisiana State University, New Orleans; this article written while in the Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Thomas A. Hennebry, MD Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Mazen S. Abu-Fadel, MD Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Address: Elias B. Hanna, MD, Department of Medicine, Section of Cardiology, Louisiana State University, Room 323, Box T4M-2, 1542 Tulane Avenue, New Orleans, LA 70112; e-mail [email protected]
Author and Disclosure Information
Elias B. Hanna, MD Currently in the Department of Medicine, Cardiovascular Section, Louisiana State University, New Orleans; this article written while in the Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Thomas A. Hennebry, MD Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Mazen S. Abu-Fadel, MD Department of Medicine, Cardiovascular Section, University of Oklahoma Health Sciences Center, Oklahoma City
Address: Elias B. Hanna, MD, Department of Medicine, Section of Cardiology, Louisiana State University, Room 323, Box T4M-2, 1542 Tulane Avenue, New Orleans, LA 70112; e-mail [email protected]
Effective and rapid reperfusion is crucial in patients with acute ST-segment elevation myocardial infarction (MI). The preferred strategy for reperfusion—when it can be performed in a timely fashion at an experienced facility—is primary percutaneous coronary intervention (PCI), which produces outcomes superior to those of pharmacologic thrombolysis.1
Unfortunately, in the United States about half of patients present to hospitals that do not have PCI capability,2 and in one analysis, 91% of transferred patients had a door-to-balloon time greater than the recommended 90 minutes, with a mean of 152 minutes.3 (In this case, the door-to-balloon time was the time that elapsed between entry into the first hospital and inflation of the PCI balloon at the second hospital.)
In situations such as these, a combined approach may be appropriate, with thrombolysis delivered by paramedics or at a local facility, followed by transfer to a PCI facility and performance of PCI within a few hours. However, this is feasible only if standardized community-based or regional protocols for prompt transfer and reperfusion are in place.
In this paper we discuss the rationale and the clinical data behind several approaches to combined reperfusion, as well as experiences with community-based care protocols.
WITHIN 3 HOURS OF SYMPTOM ONSET, THROMBOLYSIS IS AS GOOD AS PCI
The PRAGUE-2 Trial
In the randomized PRAGUE-2 trial,4 patients with ST-elevation MI who presented to a non-PCI facility had better outcomes if they were transferred promptly for PCI (median door-to-balloon time 97 minutes), as opposed to receiving local therapy with streptokinase. However, for patients presenting within 3 hours of symptom onset, the mortality rates were comparable with either strategy.4
See the glossary of clinical trial names below
The CAPTIM trial
In the CAPTIM trial,5 patients who presented within 2 hours of symptom onset and who were randomized to receive prehospital thrombolysis had outcomes similar to those of patients treated with primary PCI, despite a short door-to-balloon time (82 minutes).
The Vienna STEMI Registry
In the Vienna STEMI Registry,6 the mortality rates with primary PCI and with thrombolysis were similar when patients presented within 2 hours of symptom onset. However, as the time from symptom onset increased, primary PCI appeared to offer an increasing survival benefit compared with thrombolysis.
Comments: Thrombolysis is effective mostly in the first 2 to 3 hours, with some benefit up to 12 hours
Previous studies have shown that the sooner thrombolysis is given after symptom onset, the more effective it is. If it is given within an hour of symptom onset, the relative reduction in the mortality rate is 50% and the absolute reduction is 6.5% compared with no reperfusion therapy. If it is started in the second hour, the absolute reduction in the mortality rate drops to 4%, and a lesser benefit extends to patients presenting up to 12 hours after symptom onset.7 This time-dependent benefit is due to the fact that very early reperfusion of the occluded coronary artery may lead to full recovery of ischemic tissue and thus prevent necrosis. In addition, thrombolysis in the first 2 hours is highly efficacious in lysing a fresh thrombus.
These data support the current guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA), which state no preference for either thrombolytic therapy or PCI in ST-elevation MI if the presentation is less than 3 hours after symptom onset.8
Of note, in the CAPTIM trial and in the Vienna STEMI Registry, rescue PCI was available and was in fact used after thrombolysis in about 25% of patients, which might have contributed to the benefit of early thrombolysis.
PRIMARY PCI MAY NOT BE SUPERIOR IF TRANSFER TIME IS LONG
Another time-related factor to consider is the PCI-related delay, ie, the theoretical difference between the expected time from first medical contact to balloon inflation (if the patient undergoes primary PCI) and the time from first medical contact to the start of thrombolytic therapy (if the patient undergoes primary thrombolysis).
A meta-analysis of 13 trials comparing PCI and thrombolysis showed that a PCI-related delay of more than 60 minutes might negate the potential advantage of primary PCI over immediate thrombolysis in terms of deaths.9
This observation has been further refined by data from the National Registry of Myocardial Infarction.10 In this analysis, patient factors, including age, duration of symptoms, and infarct location, significantly affected the point at which the PCI-related delay negated the survival advantage of primary PCI. The survival advantage of primary PCI was lost more rapidly—with a PCI-related delay as short as 40 minutes—in patients who presented sooner, were younger, or had anterior MI. Primary PCI maintained its survival advantage even with a PCI-related delay longer than 100 minutes in older patients or patients with nonanterior MI presenting more than 3 hours after symptom onset. Given that median door-to-balloon times in the United States may exceed 150 minutes when transfer is involved, 3 primary PCI may be no better than primary thrombolysis in transferred patients who present early or who have large infarcts.
Although these results were derived from a post hoc analysis of a registry and the delay times reported were sometimes inaccurate, they suggest that both the PCI-related delay time and patient characteristics should be considered when selecting a reperfusion strategy. Thrombolytic therapy before and in conjunction with primary PCI was considered a potential solution to these concerns.
In addition, while the benefit of any reperfusion strategy depends on the time of presentation, the loss in benefit by later presentation is less pronounced with primary PCI than with thrombolysis, making thrombolysis less attractive in later presentations (> 3 hours).11
Also, while thrombolytic therapy in patients older than 75 years was associated with a lower mortality rate compared with no therapy in a large Swedish registry,12 this benefit was less striking than in younger patients. A meta-analysis of thrombolysis trials failed to show a similar benefit in patients over age 75 vs younger patients,13 whereas primary PCI remained effective and superior to thrombolysis in the elderly, with more absolute reduction in mortality rates in the elderly subgroup than with younger patients. 14 This makes thrombolysis less attractive in the elderly, either as a stand-alone therapy or in conjunction with PCI. Studies of combined thrombolysis and PCI included very few patients over age 75.15–17
THREE COMBINATION REPERFUSION STRATEGIES
Figure 1. The timing of percutaneous coronary intervention (PCI) in relation to thrombolysis in the pharmacoinvasive strategy, rescue PCI strategy, and facilitated PCI strategy, with the respective clinical trials that addressed and defined these strategies. (See the glossary above for complete names of studies.)Three different combination reperfusion strategies for ST-elevation MI have been studied (Figure 1)15,16,18–20:
Facilitated PCI is a strategy of thrombolysis immediately followed by PCI, with a planned door-to-balloon time of 90 to 120 minutes.
Pharmacoinvasive therapy means giving thrombolysis at a non-PCI facility and then promptly and systematically transferring the patient to a PCI facility, where PCI is performed 2 to 24 hours after the start of thrombolytic therapy, regardless of whether thrombolysis results in successful reperfusion. 15 Thus, the time to PCI is longer than with facilitated PCI. Facilitated PCI addresses the value of pretreatment with thrombolytics or glycoprotein IIb/IIIa inhibitors in patients otherwise eligible for primary PCI, whereas pharmacoinvasive therapy addresses the value of routine early PCI after thrombolysis in patients who are not eligible for primary PCI.16
Rescue PCI refers to PCI that is performed urgently if thrombolysis fails, failure being defined as persistent hemodynamic or electrical instability, persistent ischemic symptoms, or failure to achieve at least a 50% to 70% resolution of the maximal ST-segment elevation 90 minutes after the infusion is started.
FACILITATED PCI: NEGATIVE RESULTS IN CLINICAL TRIALS
ASSENT-4 PCI trial
In the ASSENT-4 PCI trial,18 patients receiving full thrombolytic therapy before PCI had a higher rate of in-hospital death, bleeding, and cardiovascular events at 90 days than patients treated with primary PCI.
This trial recruited patients arriving at hospitals with or without PCI capability. The door-to-balloon time was about 110 minutes in both groups, which might not have been prolonged enough to show a benefit from a timely addition of thrombolysis. In addition, antiplatelet therapy was limited in these patients: glycoprotein IIb/IIIa inhibitors were not given, and clopidogrel (Plavix) was not appropriately preloaded, and this might have offset the potential benefit of early PCI. In fact, data suggest that platelet activation and aggregation are heightened after thrombolysis, 21–23 and that glycoprotein IIb/IIIa antagonists can inhibit these effects.23
The FINESSE trial
In the FINESSE trial,19 patients were randomized to undergo primary PCI, to undergo PCI facilitated (ie, preceded) by abciximab (Reo-Pro), or to undergo PCI facilitated by half-dose reteplase (Retavase) and full-dose abciximab. Despite a median door-to-balloon time of 132 minutes, the three strategies were associated with similar rates of death, heart failure, or ischemic outcome at 90 days. Even though the dosage of heparin was weight-adjusted, more major bleeding events occurred with the facilitated strategies.
Comments: Some subgroups may still benefit from facilitated PCI
The results of ASSENT-4 PCI and FINESSE led to the conclusion that PCI facilitated by full-dose thrombolysis should be avoided, and called into question the value of PCI facilitation using glycoprotein IIb/IIIa inhibitors with or without half-dose thrombolytic therapy.
However, subgroup analyses of these trials identified some subgroups that may benefit from a facilitated strategy. In ASSENT-4 PCI, 45% of patients were enrolled at PCI hospitals with a minimal PCI-related delay time. These patients had the worst outcome with the facilitated strategy. In contrast, patients who had a short time from pain onset to thrombolysis (2 to 3 hours) and who were given prehospital thrombolysis had a trend toward better outcomes with facilitated PCI.24 And in FINESSE, 60% of patients were enrolled at centers with PCI capability. Analysis of a small subgroup of patients with a Thrombolysis in Myocardial Infarction study (TIMI) risk score of 3 or greater presenting to non-PCI hospitals within 4 hours of symptom onset suggested a potential reduction of ischemic events with the facilitated strategy in these patients.25
Thus, for patients seen in the first 2 to 3 hours after symptom onset, immediate thrombolysis is recommended if PCI will likely be delayed, with or without plans for subsequent early PCI. “Time is muscle,” especially during the first 3 hours.
PHARMACOINVASIVE STRATEGY: GOOD RESULTS IN HIGH-RISK PATIENTS
A number of randomized studies during the last 10 years have examined the value of a pharmacoinvasive strategy.15,16,26–29
The TRANSFER-AMI trial
The TRANSFER-AMI trial15 randomized 1,059 patients with high-risk ST-elevation MI (ie, anterior or high-risk inferior) at non-PCI centers to undergo either pharmacoinvasive care, ie, full-dose tenecteplase (TNKase) with immediate transfer for PCI or standard care, ie, tenecteplase with transfer for rescue PCI if the patient had persistent ST-segment elevation, chest pain, or hemodynamic instability.15 The goal was to perform PCI within 6 hours of thrombolysis, and the median time to PCI was 3.9 hours (range 2–6 hours). In the standard-care group, 35% of patients needed to be transferred for rescue PCI. Unlike in the ASSENT-4 trial, over 80% of patients received aggressive antiplatelet therapy with both 300 mg of clopidogrel and glycoprotein IIb/IIIa inhibitors.
The rate of cardiovascular events at 30 days was significantly lower with pharmacoinvasive therapy than with standard care and rescue PCI (11% vs 17%, P = .004). This difference was driven by lower rates of recurrent ischemia, reinfarction, and heart failure.
The CARESS-in-AMI study
The CARESS-in-AMI study16 found a similar improvement in ischemic outcomes in 600 patients with high-risk ST-elevation MI arriving at non-PCI centers if they had received pharmacoinvasive therapy. Patients received half-dose reteplase and abciximab and were randomized either to be immediately transferred for PCI (median time to PCI 2.25 hours) or to be transferred only if they had persistent ST-segment elevation or clinical deterioration.16 The event rate was low with pharmacoinvasive therapy, comparable to that achieved in primary PCI trials.
Interestingly, no significant increase was seen in the risk of major and minor bleeding in these two trials despite the use of a femoral approach for PCI in over 80% of the cases; this is probably due to the delays between thrombolytic administration and PCI and to the use of a highly fibrin-specific thrombolytic agent and adjusted-dose heparin.
Meta-analysis of pharmacoinvasive trials
A meta-analysis29 of studies of systematic early PCI (mainly with stenting) within 24 hours of thrombolysis showed a reduction in the rates of mortality and reinfarction with this strategy, without an increase in the risk of major or intracranial bleeding.30 In contrast to the results of the trials of facilitated PCI, a pharmacoinvasive strategy improved outcomes in these trials because the delay between thrombolysis and PCI was more than 2 hours, ie, long enough to prevent bleeding complications, and because most patients randomized in these trials presented within 2 to 3 hours of symptom onset, when the time to reperfusion is critical. After 3 hours, the PCI-mediated myocardial salvage is less time-dependent. Moreover, trials of pharmacoinvasive strategy used aggressive antiplatelet therapy with clopidogrel and glycoprotein IIb/IIIa inhibitors.
Comment: Pharmacoinvasive strategy in the guidelines
These results and those of the subgroup analysis from the FINESSE trial suggest that patients with high-risk ST-elevation MI treated at non-PCI hospitals have better outcomes without an increase in major bleeding events when given thrombolysis and then immediately transferred for routine PCI, rather than being transferred only if reperfusion fails.
Hence, the 2009 update of the ACC/AHA guidelines31 gives a class IIa recommendation for transferring patients with anterior ST-elevation MI or high-risk inferior ST-elevation MI treated with thrombolysis to a PCI-capable facility where PCI is performed as part of a pharmacoinvasive or rescue strategy soon after thrombolysis.
This strategy has been particularly studied in patients younger than 75 years presenting with high-risk types of ST-elevation MI early (< 3 hours) after symptom onset. If not at high risk, the patient may be transferred to a PCI facility after receiving thrombolysis or observed in the initial facility (class IIb recommendation). Consideration should be given to starting anticoagulant and antiplatelet therapy before and during transfer—ie, 300 mg of clopidogrel before transfer for PCI and glycoprotein IIb/IIIa inhibitor therapy during PCI.
The European Society of Cardiology (ESC) guidelines32 recommend early routine angiography 3 to 24 hours after successful thrombolysis. This time window was selected to avoid PCI during the prothrombotic period in the first few hours after thrombolysis and to minimize the risk of reocclusion with PCI delays of more than 24 hours (class IIa recommendation).
Larger randomized trials are still needed to establish whether the pharmacoinvasive strategy confers a survival benefit, to determine its usefulness in low-risk inferior or lateral ST-elevation MI, and to further refine the time window when PCI is both safe and beneficial after thrombolysis.33
RESCUE PCI REDUCES MORTALITY RATES
Rescue PCI is the most accepted form of thrombolysis-PCI combination.
The REACT trial
The REACT trial20 showed that rescue PCI performed at a mean of 4.5 hours after failed thrombolysis reduces the rate of adverse cardiovascular events by more than 50% at 6 to 12 months and reduces the 5-year mortality rate by more than 50% compared with conservative management.20 As in the pharmacoinvasive strategy, aggressive antiplatelet regimens were used in the REACT trial.
A meta-analysis of rescue PCI trials
A meta-analysis of rescue PCI trials34 confirmed these results, showing a reduction in heart failure and reinfarction and a trend toward a lower mortality rate with rescue PCI.34 After thrombolysis, 40% of patients do not achieve grade 3 TIMI flow, which explains why in modern clinical trials 30% of patients treated with thrombolysis require rescue PCI.5,15,16,35
For patients with high-risk ST-elevation MI, current ACC/AHA guidelines assign a class IIa recommendation to rescue PCI.31
WHEN PATIENTS WITH ST-ELEVATION MI PRESENT TO A NON-PCI HOSPITAL
Transfer for primary PCI vs thrombolysis at the non-PCI hospital
The DANAMI-2 trial36 found that immediate transfer for PCI was superior to onsite thrombolytic therapy, as measured by a reduction in the rate of ischemic events (composite of death, myocardial infarction, or stroke at 30 days): 8.5% vs 14.2% (P < .001). There were no deaths during transfer.3
The PRAGUE-2 trial4 showed similar results for patients presenting 3 to 12 hours after symptom onset (30-day mortality rate 6% with immediate transfer vs 15.3% with on-site thrombolysis, P < .002), whereas patients presenting within 3 hours of symptom onset had a similar mortality rate with either therapy.4
Comment. These trials showed that transfer for primary PCI is superior to thrombolytic therapy when performed in a timely fashion. However, they were done in countries with established transfer networks and short distances between community hospitals and PCI centers, with a PCI-related delay of only 44 minutes and a door-to-balloon time of 90 minutes despite transfer. The large-scale application of this prompt transfer policy is not practical in most regions in the United States. Thus, a strategy of local thrombolysis followed by routine early transfer for routine or rescue PCI seems warranted when the door-to-balloon time or the PCI-related delay time is expected to be too long.
Experiences with community-based systems of care and prehospital thrombolysis
In Minnesota, Henry et al37 developed a PCI-based treatment system and an integrated transfer program for ST-elevation MI involving 30 hospitals within 210 miles of the Minneapolis Heart Institute. Participating hospitals were divided into two zones: zone 1 hospitals were within 60 miles, and zone 2 facilities were between 60 and 210 miles from the Heart Institute. Zone 2 patients received half-dose tenecteplase (if thrombolytic therapy was not contraindicated) in anticipation of a lengthy transfer time.
The median door-to-balloon time for zone 1 patients was 95 minutes (interquartile range 82 and 116 minutes) and for zone 2 patients 120 minutes (interquartile range 100 and 145 minutes). The diagnosis of ST-elevation MI was made by the emergency department physician, who activated the system with a phone call. The patient was then directly transferred to the catheterization laboratory, most often by helicopter.
The in-hospital death rate for patients who presented to the PCI center and for patients in zones 1 and 2 was similarly low (about 5%).37
In France, the FAST-MI registry,17 which collected outcome data for different reperfusion strategies, found that thrombolysis yielded in-hospital and midterm results that were comparable to those of primary PCI. Of note, thrombolysis was started early after symptom onset (about 2 hours), and was started in the ambulance in two-thirds of cases. Nearly all patients underwent a pharmacoinvasive strategy that combined thrombolysis with coronary angiography and PCI within 24 hours of symptom onset. These findings suggest that timely thrombolysis followed by semiurgent transfer for PCI is an alternative to primary PCI for patients presenting to hospitals with no PCI capability, and that this alternative offers similar benefit to that of primary PCI.
Five centers in the United States have reported their experience with half-dose thrombolysis in the prehospital setting (in the field or during transfer) or at a non-PCI hospital, followed by prompt transfer to a PCI facility. In this registry of almost 3,000 patients,38 patients treated with thrombolysis had better outcomes than patients directly transferred for primary PCI, with a significantly lower 30-day mortality rate (3.8% vs from 6.4%), and no increase in bleeding.38,39 The mean door-to-balloon time was long (168 minutes in the primary PCI group and 196 minutes in the thrombolysis-PCI group), which might explain the benefit achieved with prompt thrombolysis.
CARDIOGENIC SHOCK
Patients presenting with left ventricular cardiogenic shock derive a large mortality benefit from revascularization, whether they are transferred or directly admitted to a PCI center. 40 Moreover, in the SHOCK registry, patients with predominant right ventricular cardiogenic shock had an in-hospital mortality rate similar to that of patients with predominant left ventricular cardiogenic shock, and revascularization (PCI or surgical revascularization) was associated with a strikingly lower mortality rate in both groups.41
Thus, all patients with left or right cardiogenic shock should be revascularized on an emergency basis, either surgically or percutaneously.
While trials of pharmacoinvasive therapy excluded patients with cardiogenic shock,15,16 thrombolytic therapy was associated with improved outcomes in the drug-therapy group of the SHOCK trial and in hypotensive patients randomized in the early thrombolysis trials.13 Thus, the ACC/AHA guidelines recommend thrombolytic therapy before transfer if a patient presents in shock within 3 to 6 hours of onset of the MI and delays in transport and intervention are anticipated.8
PUTTING IT ALL TOGETHER: MANAGEMENT STRATEGIES
Figure 2. Selecting the appropriate reperfusion strategy in ST-elevation myocardial infarction (MI). Routine early PCI is particularly indicated in high-risk MI, ie, either anterior MI, or inferior MI with one of the following: systolic blood pressure of less than 100 mm Hg, heart rate of more than 100 beats per minute, Killip class II or III, ST-segment depression of 2 mm or more in the anterior leads, or ST-segment elevation of 1 mm or more in the right-sided lead V4, which is indicative of right ventricular involvement. Dual antiplatelet therapy with aspirin and clopidogrel (Plavix) 300 mg should be started as soon as possible in all patients, and consideration should be given to glycoprotein IIb/IIIa inhibition for most patients during PCI (as in the TRANSFER-AMI15 and CARESS-in-AMI16 trials).Taking into account the importance of time to presentation, the PCI-related delay time, and patient and MI characteristics, as well as whether a regional transfer system is in place (as in Minnesota), we suggest an algorithmic approach to the management of ST-elevation MI at a non-PCI facility (Figure 2).
If an effective transfer system is in place, primary PCI not preceded by thrombolytic therapy or glycoprotein IIb/IIIa inhibitor therapy is the preferred approach, according to ACC/AHA and ESC guidelines.31,32 Giving thrombolytics immediately before PCI is harmful and thus should be avoided when the expected door-to-balloon time is 90 minutes or less.
All hospitals (whether or not they offer PCI) and regional emergency medical services should participate in a community-based system of care for ST-elevation MI, with protocols for expeditious transfer as defined and coordinated by the American Heart Association initiative “Mission: Lifeline.” In addition, a system of field triage and direct transport to the catheterization laboratory of a PCI facility after field activation significantly reduces door-to-balloon times and improves outcomes.42
If such a system is not in place, then a pharmacoinvasive strategy seems best: ie, local full-dose thrombolysis (if not contraindicated) followed by transfer to a PCI facility and routine performance of PCI 2 to 6 hours after thrombolysis—in conjunction with aggressive early dual oral antiplatelet therapy and “downstream” glycoprotein IIb/IIIa inhibition. This approach is associated with outcomes similar to those of primary PCI.15–17,37
Prehospital thrombolysis delivered by paramedics and followed by early transfer to a PCI facility has been associated with further reduction in mortality rates compared with in-hospital thrombolysis (as in the Swedish registry43), and a reduction in death rate comparable to that of primary PCI in patients presenting early. This is an adequate strategy in regions where such a system can be established.5,17,38,43,44
Patients presenting more than 3 to 4 hours after symptom onset, older patients, and patients with lower-risk MI or a higher risk of bleeding may still be suited for primary PCI even when the door-to-balloon time is 90 to 120 minutes, as stated by the European guidelines,32 or when the PCI-related delay time is as long as 100 minutes. 10 On the other hand, while the ACC/AHA guidelines recognize that in these patients the mortality advantage of primary PCI vs thrombolytic therapy is maintained with more prolonged door-to-balloon times, they nevertheless state that the focus should be on developing systems of care to increase the number of patients with access to primary PCI in less than 90 minutes rather than extending the acceptable window for door-to-balloon time.
In conclusion, for patients presenting with ST-elevation MI who cannot undergo timely primary PCI, the best approach seems to be prehospital thrombolysis delivered by paramedics or local thrombolysis at the non-PCI hospital followed by transferring the patient and performing PCI within a few hours. This is especially important in patients with high-risk ST-elevation MI who present early after symptom onset, when the extent of myocardial necrosis associated with delayed primary PCI is largest.
In addition, every community should develop a coordinated transfer strategy between non-PCI and PCI hospitals.
Effective and rapid reperfusion is crucial in patients with acute ST-segment elevation myocardial infarction (MI). The preferred strategy for reperfusion—when it can be performed in a timely fashion at an experienced facility—is primary percutaneous coronary intervention (PCI), which produces outcomes superior to those of pharmacologic thrombolysis.1
Unfortunately, in the United States about half of patients present to hospitals that do not have PCI capability,2 and in one analysis, 91% of transferred patients had a door-to-balloon time greater than the recommended 90 minutes, with a mean of 152 minutes.3 (In this case, the door-to-balloon time was the time that elapsed between entry into the first hospital and inflation of the PCI balloon at the second hospital.)
In situations such as these, a combined approach may be appropriate, with thrombolysis delivered by paramedics or at a local facility, followed by transfer to a PCI facility and performance of PCI within a few hours. However, this is feasible only if standardized community-based or regional protocols for prompt transfer and reperfusion are in place.
In this paper we discuss the rationale and the clinical data behind several approaches to combined reperfusion, as well as experiences with community-based care protocols.
WITHIN 3 HOURS OF SYMPTOM ONSET, THROMBOLYSIS IS AS GOOD AS PCI
The PRAGUE-2 Trial
In the randomized PRAGUE-2 trial,4 patients with ST-elevation MI who presented to a non-PCI facility had better outcomes if they were transferred promptly for PCI (median door-to-balloon time 97 minutes), as opposed to receiving local therapy with streptokinase. However, for patients presenting within 3 hours of symptom onset, the mortality rates were comparable with either strategy.4
See the glossary of clinical trial names below
The CAPTIM trial
In the CAPTIM trial,5 patients who presented within 2 hours of symptom onset and who were randomized to receive prehospital thrombolysis had outcomes similar to those of patients treated with primary PCI, despite a short door-to-balloon time (82 minutes).
The Vienna STEMI Registry
In the Vienna STEMI Registry,6 the mortality rates with primary PCI and with thrombolysis were similar when patients presented within 2 hours of symptom onset. However, as the time from symptom onset increased, primary PCI appeared to offer an increasing survival benefit compared with thrombolysis.
Comments: Thrombolysis is effective mostly in the first 2 to 3 hours, with some benefit up to 12 hours
Previous studies have shown that the sooner thrombolysis is given after symptom onset, the more effective it is. If it is given within an hour of symptom onset, the relative reduction in the mortality rate is 50% and the absolute reduction is 6.5% compared with no reperfusion therapy. If it is started in the second hour, the absolute reduction in the mortality rate drops to 4%, and a lesser benefit extends to patients presenting up to 12 hours after symptom onset.7 This time-dependent benefit is due to the fact that very early reperfusion of the occluded coronary artery may lead to full recovery of ischemic tissue and thus prevent necrosis. In addition, thrombolysis in the first 2 hours is highly efficacious in lysing a fresh thrombus.
These data support the current guidelines of the American College of Cardiology (ACC) and the American Heart Association (AHA), which state no preference for either thrombolytic therapy or PCI in ST-elevation MI if the presentation is less than 3 hours after symptom onset.8
Of note, in the CAPTIM trial and in the Vienna STEMI Registry, rescue PCI was available and was in fact used after thrombolysis in about 25% of patients, which might have contributed to the benefit of early thrombolysis.
PRIMARY PCI MAY NOT BE SUPERIOR IF TRANSFER TIME IS LONG
Another time-related factor to consider is the PCI-related delay, ie, the theoretical difference between the expected time from first medical contact to balloon inflation (if the patient undergoes primary PCI) and the time from first medical contact to the start of thrombolytic therapy (if the patient undergoes primary thrombolysis).
A meta-analysis of 13 trials comparing PCI and thrombolysis showed that a PCI-related delay of more than 60 minutes might negate the potential advantage of primary PCI over immediate thrombolysis in terms of deaths.9
This observation has been further refined by data from the National Registry of Myocardial Infarction.10 In this analysis, patient factors, including age, duration of symptoms, and infarct location, significantly affected the point at which the PCI-related delay negated the survival advantage of primary PCI. The survival advantage of primary PCI was lost more rapidly—with a PCI-related delay as short as 40 minutes—in patients who presented sooner, were younger, or had anterior MI. Primary PCI maintained its survival advantage even with a PCI-related delay longer than 100 minutes in older patients or patients with nonanterior MI presenting more than 3 hours after symptom onset. Given that median door-to-balloon times in the United States may exceed 150 minutes when transfer is involved, 3 primary PCI may be no better than primary thrombolysis in transferred patients who present early or who have large infarcts.
Although these results were derived from a post hoc analysis of a registry and the delay times reported were sometimes inaccurate, they suggest that both the PCI-related delay time and patient characteristics should be considered when selecting a reperfusion strategy. Thrombolytic therapy before and in conjunction with primary PCI was considered a potential solution to these concerns.
In addition, while the benefit of any reperfusion strategy depends on the time of presentation, the loss in benefit by later presentation is less pronounced with primary PCI than with thrombolysis, making thrombolysis less attractive in later presentations (> 3 hours).11
Also, while thrombolytic therapy in patients older than 75 years was associated with a lower mortality rate compared with no therapy in a large Swedish registry,12 this benefit was less striking than in younger patients. A meta-analysis of thrombolysis trials failed to show a similar benefit in patients over age 75 vs younger patients,13 whereas primary PCI remained effective and superior to thrombolysis in the elderly, with more absolute reduction in mortality rates in the elderly subgroup than with younger patients. 14 This makes thrombolysis less attractive in the elderly, either as a stand-alone therapy or in conjunction with PCI. Studies of combined thrombolysis and PCI included very few patients over age 75.15–17
THREE COMBINATION REPERFUSION STRATEGIES
Figure 1. The timing of percutaneous coronary intervention (PCI) in relation to thrombolysis in the pharmacoinvasive strategy, rescue PCI strategy, and facilitated PCI strategy, with the respective clinical trials that addressed and defined these strategies. (See the glossary above for complete names of studies.)Three different combination reperfusion strategies for ST-elevation MI have been studied (Figure 1)15,16,18–20:
Facilitated PCI is a strategy of thrombolysis immediately followed by PCI, with a planned door-to-balloon time of 90 to 120 minutes.
Pharmacoinvasive therapy means giving thrombolysis at a non-PCI facility and then promptly and systematically transferring the patient to a PCI facility, where PCI is performed 2 to 24 hours after the start of thrombolytic therapy, regardless of whether thrombolysis results in successful reperfusion. 15 Thus, the time to PCI is longer than with facilitated PCI. Facilitated PCI addresses the value of pretreatment with thrombolytics or glycoprotein IIb/IIIa inhibitors in patients otherwise eligible for primary PCI, whereas pharmacoinvasive therapy addresses the value of routine early PCI after thrombolysis in patients who are not eligible for primary PCI.16
Rescue PCI refers to PCI that is performed urgently if thrombolysis fails, failure being defined as persistent hemodynamic or electrical instability, persistent ischemic symptoms, or failure to achieve at least a 50% to 70% resolution of the maximal ST-segment elevation 90 minutes after the infusion is started.
FACILITATED PCI: NEGATIVE RESULTS IN CLINICAL TRIALS
ASSENT-4 PCI trial
In the ASSENT-4 PCI trial,18 patients receiving full thrombolytic therapy before PCI had a higher rate of in-hospital death, bleeding, and cardiovascular events at 90 days than patients treated with primary PCI.
This trial recruited patients arriving at hospitals with or without PCI capability. The door-to-balloon time was about 110 minutes in both groups, which might not have been prolonged enough to show a benefit from a timely addition of thrombolysis. In addition, antiplatelet therapy was limited in these patients: glycoprotein IIb/IIIa inhibitors were not given, and clopidogrel (Plavix) was not appropriately preloaded, and this might have offset the potential benefit of early PCI. In fact, data suggest that platelet activation and aggregation are heightened after thrombolysis, 21–23 and that glycoprotein IIb/IIIa antagonists can inhibit these effects.23
The FINESSE trial
In the FINESSE trial,19 patients were randomized to undergo primary PCI, to undergo PCI facilitated (ie, preceded) by abciximab (Reo-Pro), or to undergo PCI facilitated by half-dose reteplase (Retavase) and full-dose abciximab. Despite a median door-to-balloon time of 132 minutes, the three strategies were associated with similar rates of death, heart failure, or ischemic outcome at 90 days. Even though the dosage of heparin was weight-adjusted, more major bleeding events occurred with the facilitated strategies.
Comments: Some subgroups may still benefit from facilitated PCI
The results of ASSENT-4 PCI and FINESSE led to the conclusion that PCI facilitated by full-dose thrombolysis should be avoided, and called into question the value of PCI facilitation using glycoprotein IIb/IIIa inhibitors with or without half-dose thrombolytic therapy.
However, subgroup analyses of these trials identified some subgroups that may benefit from a facilitated strategy. In ASSENT-4 PCI, 45% of patients were enrolled at PCI hospitals with a minimal PCI-related delay time. These patients had the worst outcome with the facilitated strategy. In contrast, patients who had a short time from pain onset to thrombolysis (2 to 3 hours) and who were given prehospital thrombolysis had a trend toward better outcomes with facilitated PCI.24 And in FINESSE, 60% of patients were enrolled at centers with PCI capability. Analysis of a small subgroup of patients with a Thrombolysis in Myocardial Infarction study (TIMI) risk score of 3 or greater presenting to non-PCI hospitals within 4 hours of symptom onset suggested a potential reduction of ischemic events with the facilitated strategy in these patients.25
Thus, for patients seen in the first 2 to 3 hours after symptom onset, immediate thrombolysis is recommended if PCI will likely be delayed, with or without plans for subsequent early PCI. “Time is muscle,” especially during the first 3 hours.
PHARMACOINVASIVE STRATEGY: GOOD RESULTS IN HIGH-RISK PATIENTS
A number of randomized studies during the last 10 years have examined the value of a pharmacoinvasive strategy.15,16,26–29
The TRANSFER-AMI trial
The TRANSFER-AMI trial15 randomized 1,059 patients with high-risk ST-elevation MI (ie, anterior or high-risk inferior) at non-PCI centers to undergo either pharmacoinvasive care, ie, full-dose tenecteplase (TNKase) with immediate transfer for PCI or standard care, ie, tenecteplase with transfer for rescue PCI if the patient had persistent ST-segment elevation, chest pain, or hemodynamic instability.15 The goal was to perform PCI within 6 hours of thrombolysis, and the median time to PCI was 3.9 hours (range 2–6 hours). In the standard-care group, 35% of patients needed to be transferred for rescue PCI. Unlike in the ASSENT-4 trial, over 80% of patients received aggressive antiplatelet therapy with both 300 mg of clopidogrel and glycoprotein IIb/IIIa inhibitors.
The rate of cardiovascular events at 30 days was significantly lower with pharmacoinvasive therapy than with standard care and rescue PCI (11% vs 17%, P = .004). This difference was driven by lower rates of recurrent ischemia, reinfarction, and heart failure.
The CARESS-in-AMI study
The CARESS-in-AMI study16 found a similar improvement in ischemic outcomes in 600 patients with high-risk ST-elevation MI arriving at non-PCI centers if they had received pharmacoinvasive therapy. Patients received half-dose reteplase and abciximab and were randomized either to be immediately transferred for PCI (median time to PCI 2.25 hours) or to be transferred only if they had persistent ST-segment elevation or clinical deterioration.16 The event rate was low with pharmacoinvasive therapy, comparable to that achieved in primary PCI trials.
Interestingly, no significant increase was seen in the risk of major and minor bleeding in these two trials despite the use of a femoral approach for PCI in over 80% of the cases; this is probably due to the delays between thrombolytic administration and PCI and to the use of a highly fibrin-specific thrombolytic agent and adjusted-dose heparin.
Meta-analysis of pharmacoinvasive trials
A meta-analysis29 of studies of systematic early PCI (mainly with stenting) within 24 hours of thrombolysis showed a reduction in the rates of mortality and reinfarction with this strategy, without an increase in the risk of major or intracranial bleeding.30 In contrast to the results of the trials of facilitated PCI, a pharmacoinvasive strategy improved outcomes in these trials because the delay between thrombolysis and PCI was more than 2 hours, ie, long enough to prevent bleeding complications, and because most patients randomized in these trials presented within 2 to 3 hours of symptom onset, when the time to reperfusion is critical. After 3 hours, the PCI-mediated myocardial salvage is less time-dependent. Moreover, trials of pharmacoinvasive strategy used aggressive antiplatelet therapy with clopidogrel and glycoprotein IIb/IIIa inhibitors.
Comment: Pharmacoinvasive strategy in the guidelines
These results and those of the subgroup analysis from the FINESSE trial suggest that patients with high-risk ST-elevation MI treated at non-PCI hospitals have better outcomes without an increase in major bleeding events when given thrombolysis and then immediately transferred for routine PCI, rather than being transferred only if reperfusion fails.
Hence, the 2009 update of the ACC/AHA guidelines31 gives a class IIa recommendation for transferring patients with anterior ST-elevation MI or high-risk inferior ST-elevation MI treated with thrombolysis to a PCI-capable facility where PCI is performed as part of a pharmacoinvasive or rescue strategy soon after thrombolysis.
This strategy has been particularly studied in patients younger than 75 years presenting with high-risk types of ST-elevation MI early (< 3 hours) after symptom onset. If not at high risk, the patient may be transferred to a PCI facility after receiving thrombolysis or observed in the initial facility (class IIb recommendation). Consideration should be given to starting anticoagulant and antiplatelet therapy before and during transfer—ie, 300 mg of clopidogrel before transfer for PCI and glycoprotein IIb/IIIa inhibitor therapy during PCI.
The European Society of Cardiology (ESC) guidelines32 recommend early routine angiography 3 to 24 hours after successful thrombolysis. This time window was selected to avoid PCI during the prothrombotic period in the first few hours after thrombolysis and to minimize the risk of reocclusion with PCI delays of more than 24 hours (class IIa recommendation).
Larger randomized trials are still needed to establish whether the pharmacoinvasive strategy confers a survival benefit, to determine its usefulness in low-risk inferior or lateral ST-elevation MI, and to further refine the time window when PCI is both safe and beneficial after thrombolysis.33
RESCUE PCI REDUCES MORTALITY RATES
Rescue PCI is the most accepted form of thrombolysis-PCI combination.
The REACT trial
The REACT trial20 showed that rescue PCI performed at a mean of 4.5 hours after failed thrombolysis reduces the rate of adverse cardiovascular events by more than 50% at 6 to 12 months and reduces the 5-year mortality rate by more than 50% compared with conservative management.20 As in the pharmacoinvasive strategy, aggressive antiplatelet regimens were used in the REACT trial.
A meta-analysis of rescue PCI trials
A meta-analysis of rescue PCI trials34 confirmed these results, showing a reduction in heart failure and reinfarction and a trend toward a lower mortality rate with rescue PCI.34 After thrombolysis, 40% of patients do not achieve grade 3 TIMI flow, which explains why in modern clinical trials 30% of patients treated with thrombolysis require rescue PCI.5,15,16,35
For patients with high-risk ST-elevation MI, current ACC/AHA guidelines assign a class IIa recommendation to rescue PCI.31
WHEN PATIENTS WITH ST-ELEVATION MI PRESENT TO A NON-PCI HOSPITAL
Transfer for primary PCI vs thrombolysis at the non-PCI hospital
The DANAMI-2 trial36 found that immediate transfer for PCI was superior to onsite thrombolytic therapy, as measured by a reduction in the rate of ischemic events (composite of death, myocardial infarction, or stroke at 30 days): 8.5% vs 14.2% (P < .001). There were no deaths during transfer.3
The PRAGUE-2 trial4 showed similar results for patients presenting 3 to 12 hours after symptom onset (30-day mortality rate 6% with immediate transfer vs 15.3% with on-site thrombolysis, P < .002), whereas patients presenting within 3 hours of symptom onset had a similar mortality rate with either therapy.4
Comment. These trials showed that transfer for primary PCI is superior to thrombolytic therapy when performed in a timely fashion. However, they were done in countries with established transfer networks and short distances between community hospitals and PCI centers, with a PCI-related delay of only 44 minutes and a door-to-balloon time of 90 minutes despite transfer. The large-scale application of this prompt transfer policy is not practical in most regions in the United States. Thus, a strategy of local thrombolysis followed by routine early transfer for routine or rescue PCI seems warranted when the door-to-balloon time or the PCI-related delay time is expected to be too long.
Experiences with community-based systems of care and prehospital thrombolysis
In Minnesota, Henry et al37 developed a PCI-based treatment system and an integrated transfer program for ST-elevation MI involving 30 hospitals within 210 miles of the Minneapolis Heart Institute. Participating hospitals were divided into two zones: zone 1 hospitals were within 60 miles, and zone 2 facilities were between 60 and 210 miles from the Heart Institute. Zone 2 patients received half-dose tenecteplase (if thrombolytic therapy was not contraindicated) in anticipation of a lengthy transfer time.
The median door-to-balloon time for zone 1 patients was 95 minutes (interquartile range 82 and 116 minutes) and for zone 2 patients 120 minutes (interquartile range 100 and 145 minutes). The diagnosis of ST-elevation MI was made by the emergency department physician, who activated the system with a phone call. The patient was then directly transferred to the catheterization laboratory, most often by helicopter.
The in-hospital death rate for patients who presented to the PCI center and for patients in zones 1 and 2 was similarly low (about 5%).37
In France, the FAST-MI registry,17 which collected outcome data for different reperfusion strategies, found that thrombolysis yielded in-hospital and midterm results that were comparable to those of primary PCI. Of note, thrombolysis was started early after symptom onset (about 2 hours), and was started in the ambulance in two-thirds of cases. Nearly all patients underwent a pharmacoinvasive strategy that combined thrombolysis with coronary angiography and PCI within 24 hours of symptom onset. These findings suggest that timely thrombolysis followed by semiurgent transfer for PCI is an alternative to primary PCI for patients presenting to hospitals with no PCI capability, and that this alternative offers similar benefit to that of primary PCI.
Five centers in the United States have reported their experience with half-dose thrombolysis in the prehospital setting (in the field or during transfer) or at a non-PCI hospital, followed by prompt transfer to a PCI facility. In this registry of almost 3,000 patients,38 patients treated with thrombolysis had better outcomes than patients directly transferred for primary PCI, with a significantly lower 30-day mortality rate (3.8% vs from 6.4%), and no increase in bleeding.38,39 The mean door-to-balloon time was long (168 minutes in the primary PCI group and 196 minutes in the thrombolysis-PCI group), which might explain the benefit achieved with prompt thrombolysis.
CARDIOGENIC SHOCK
Patients presenting with left ventricular cardiogenic shock derive a large mortality benefit from revascularization, whether they are transferred or directly admitted to a PCI center. 40 Moreover, in the SHOCK registry, patients with predominant right ventricular cardiogenic shock had an in-hospital mortality rate similar to that of patients with predominant left ventricular cardiogenic shock, and revascularization (PCI or surgical revascularization) was associated with a strikingly lower mortality rate in both groups.41
Thus, all patients with left or right cardiogenic shock should be revascularized on an emergency basis, either surgically or percutaneously.
While trials of pharmacoinvasive therapy excluded patients with cardiogenic shock,15,16 thrombolytic therapy was associated with improved outcomes in the drug-therapy group of the SHOCK trial and in hypotensive patients randomized in the early thrombolysis trials.13 Thus, the ACC/AHA guidelines recommend thrombolytic therapy before transfer if a patient presents in shock within 3 to 6 hours of onset of the MI and delays in transport and intervention are anticipated.8
PUTTING IT ALL TOGETHER: MANAGEMENT STRATEGIES
Figure 2. Selecting the appropriate reperfusion strategy in ST-elevation myocardial infarction (MI). Routine early PCI is particularly indicated in high-risk MI, ie, either anterior MI, or inferior MI with one of the following: systolic blood pressure of less than 100 mm Hg, heart rate of more than 100 beats per minute, Killip class II or III, ST-segment depression of 2 mm or more in the anterior leads, or ST-segment elevation of 1 mm or more in the right-sided lead V4, which is indicative of right ventricular involvement. Dual antiplatelet therapy with aspirin and clopidogrel (Plavix) 300 mg should be started as soon as possible in all patients, and consideration should be given to glycoprotein IIb/IIIa inhibition for most patients during PCI (as in the TRANSFER-AMI15 and CARESS-in-AMI16 trials).Taking into account the importance of time to presentation, the PCI-related delay time, and patient and MI characteristics, as well as whether a regional transfer system is in place (as in Minnesota), we suggest an algorithmic approach to the management of ST-elevation MI at a non-PCI facility (Figure 2).
If an effective transfer system is in place, primary PCI not preceded by thrombolytic therapy or glycoprotein IIb/IIIa inhibitor therapy is the preferred approach, according to ACC/AHA and ESC guidelines.31,32 Giving thrombolytics immediately before PCI is harmful and thus should be avoided when the expected door-to-balloon time is 90 minutes or less.
All hospitals (whether or not they offer PCI) and regional emergency medical services should participate in a community-based system of care for ST-elevation MI, with protocols for expeditious transfer as defined and coordinated by the American Heart Association initiative “Mission: Lifeline.” In addition, a system of field triage and direct transport to the catheterization laboratory of a PCI facility after field activation significantly reduces door-to-balloon times and improves outcomes.42
If such a system is not in place, then a pharmacoinvasive strategy seems best: ie, local full-dose thrombolysis (if not contraindicated) followed by transfer to a PCI facility and routine performance of PCI 2 to 6 hours after thrombolysis—in conjunction with aggressive early dual oral antiplatelet therapy and “downstream” glycoprotein IIb/IIIa inhibition. This approach is associated with outcomes similar to those of primary PCI.15–17,37
Prehospital thrombolysis delivered by paramedics and followed by early transfer to a PCI facility has been associated with further reduction in mortality rates compared with in-hospital thrombolysis (as in the Swedish registry43), and a reduction in death rate comparable to that of primary PCI in patients presenting early. This is an adequate strategy in regions where such a system can be established.5,17,38,43,44
Patients presenting more than 3 to 4 hours after symptom onset, older patients, and patients with lower-risk MI or a higher risk of bleeding may still be suited for primary PCI even when the door-to-balloon time is 90 to 120 minutes, as stated by the European guidelines,32 or when the PCI-related delay time is as long as 100 minutes. 10 On the other hand, while the ACC/AHA guidelines recognize that in these patients the mortality advantage of primary PCI vs thrombolytic therapy is maintained with more prolonged door-to-balloon times, they nevertheless state that the focus should be on developing systems of care to increase the number of patients with access to primary PCI in less than 90 minutes rather than extending the acceptable window for door-to-balloon time.
In conclusion, for patients presenting with ST-elevation MI who cannot undergo timely primary PCI, the best approach seems to be prehospital thrombolysis delivered by paramedics or local thrombolysis at the non-PCI hospital followed by transferring the patient and performing PCI within a few hours. This is especially important in patients with high-risk ST-elevation MI who present early after symptom onset, when the extent of myocardial necrosis associated with delayed primary PCI is largest.
In addition, every community should develop a coordinated transfer strategy between non-PCI and PCI hospitals.
References
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet2003; 361:13–20.
Waters RE, Singh KP, Roe MT, et al. Rationale and strategies for implementing community-based transfer protocols for primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. J Am Coll Cardiol2004; 43:2153–2159.
Chakrabarti A, Krumholz HM, Wang Y, Rumsfeld JS, Nallamothu BK; National Cardiovascular Data Registry. Time-to-reperfusion in patients undergoing interhospital transfer for primary percutaneous coronary intervention in the U.S: an analysis of 2005 and 2006 data from the National Cardiovascular Data Registry. J Am Coll Cardiol2008; 51:2442–2443.
Widimský P, Budesínský T, Vorác D, et al; ‘PRAGUE’ Study Group Investigators. Long distance transport for primary angioplasty vs immediate thrombolysis in acute myocardial infarction. Final results of the randomized national multicentre trial—PRAGUE-2. Eur Heart J2003; 24:94–104.
Steg PG, Bonnefoy E, Chabaud S, et al; Comparison of Angioplasty and Prehospital Thrombolysis in Acute Myocardial infarction (CAPTIM) Investigators. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation2003; 108:2851–2856.
Kalla K, Christ G, Karnik R, et al; Vienna STEMI Registry Group. Implementation of guidelines improves the standard of care: the Viennese registry on reperfusion strategies in ST-elevation myocardial infarction (Vienna STEMI registry). Circulation2006; 113:2398–2405.
Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet1996; 348:771–775.
Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation2004; 110:e82–e292.
Nallamothu BK, Bates ER. Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction: is timing (almost) everything?Am J Cardiol2003; 92:824–826.
Pinto DS, Kirtane AJ, Nallamothu BK, et al. Hospital delays in reperfusion for ST-elevation myocardial infarction: implications when selecting a reperfusion strategy. Circulation2006; 114:2019–2025.
Boersma E; Primary Coronary Angioplasty vs Thrombolysis Group. Does time matter? A pooled analysis of randomized clinical trials comparing primary percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J2006; 27:779–788.
Stenestrand U, Wallentin L; Register of Information and Knowledge About Swedish Heart Intensive Care Admissions (RIKS-HIA). Fibrinolytic therapy in patients 75 years and older with ST-segment-elevation myocardial infarction: one-year follow-up of a large prospective cohort. Arch Intern Med2003; 163:965–971.
Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet1994; 343:311–322.
Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med1993; 328:673–679.
Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med2009; 360:2705–2718.
Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab RE-teplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet2008; 371:559–568.
Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FAST-MI). Circulation2008; 118:268–276.
Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet2006; 367:569–578.
Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med2008; 358:2205–2217.
Carver A, Rafelt S, Gershlick AH, Fairbrother KL, Hughes S, Wilcox R; REACT Investigators. Longer-term follow-up of patients recruited to the REACT (Rescue Angioplasty Versus Conservative Treatment or Repeat Thrombolysis) trial. J Am Coll Cardiol2009; 54:118–126.
Rasmanis G, Vesterqvist O, Gréen K, Edhag O, Henriksson P. Evidence of increased platelet activation after thrombolysis in patients with acute myocardial infarction. Br Heart J1992; 68:374–376.
Gurbel PA, Serebruany VL, Shustov AR, et al. Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. GUSTO-III Investigators. Global Use of Strategies to Open Occluded Coronary Arteries. J Am Coll Cardiol1998; 31:1466–1473.
Coulter SA, Cannon CP, Ault KA, et al. High levels of platelet inhibition with abciximab despite heightened platelet activation and aggregation during thrombolysis for acute myocardial infarction: results from TIMI (thrombolysis in myocardial infarction) 14. Circulation2000; 101:2690–2695.
Ross AM, Huber K, Zeymer U, et al. The impact of place of enrollment and delay to reperfusion on 90-day post-infarction mortality in the ASSENT-4 PCI trial: assessment of the safety and efficacy of a new treatment strategy with percutaneous coronary intervention. JACC Cardiovasc Interv2009; 2:925–930.
Herrmann HC, Lu J, Brodie BR, et al; FINESSE Investigators. Benefit of facilitated percutaneous coronary intervention in high-risk ST-segment elevation myocardial infarction patients presenting to nonpercutaneous coronary intervention hospitals. JACC Cardiovasc Interv2009; 2:917–924.
Scheller B, Hennen B, Hammer B, et al; SIAM III Study Group. Beneficial effects of immediate stenting after thrombolysis in acute myocardial infarction. J Am Coll Cardiol2003; 42:634–641.
Fernandez-Avilés F, Alonso JJ, Castro-Beiras A, et al; GRACIA (Grupo de Análisis de la Cardiopatía Isquémica Aguda) Group. Routine invasive strategy within 24 hours of thrombolysis versus ischaemiaguided conservative approach for acute myocardial infarction with ST-segment elevation (GRACIA-1): a randomised controlled trial. Lancet2004; 364:1045–1053.
Le May MR, Wells GA, Labinaz M, et al. Combined angioplasty and pharmacological intervention versus thrombolysis alone in acute myocardial infarction (CAPITAL AMI study). J Am Coll Cardiol2005; 46:417–424.
Bøhmer E, Hoffmann P, Abdelnoor M, Arnesen H, Halvorsen S. Efficacy and safety of immediate angioplasty versus ischemia-guided management after thrombolysis in acute myocardial infarction in areas with very long transfer distances results of the NORDISTEMI (NORwegian study on DIstrict treatment of ST-elevation myocardial infarction). J Am Coll Cardiol2010; 55:102–110.
Wijeysundera HC, You JJ, Nallamothu BK, Krumholz HM, Cantor WJ, Ko DT. An early invasive strategy versus ischemia-guided management after fibrinolytic therapy for ST-segment elevation myocardial infarction: a meta-analysis of contemporary randomized controlled trials. Am Heart J2008; 156:564–572,572.e1–e2.
Kushner FG, Hand M, Smith SC, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol2009; 54:2205–2241.
Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J2008; 29:2909–2945.
Mukherjee D, Moliterno DJ. The timely coupling of mechanical revascularization following thrombolysis for myocardial infarction. Cardiology2007; 107:337–339.
Wijeysundera HC, Vijayaraghavan R, Nallamothu BK, et al. Rescue angioplasty or repeat fibrinolysis after failed fibrinolytic therapy for ST-segment myocardial infarction: a meta-analysis of randomized trials. J Am Coll Cardiol2007; 49:422–430.
The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med1993; 329:1615–1622.
Andersen HR, Nielsen TT, Rasmussen K, et al; DANAMI-2 Investigators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N Engl J Med2003; 349:733–742.
Henry TD, Sharkey SW, Burke MN, et al. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation2007; 116:721–728.
Denktas AE, Athar H, Henry TD, et al. Reduced-dose fibrinolytic acceleration of ST-segment elevation myocardial infarction treatment coupled with urgent percutaneous coronary intervention compared to primary percutaneous coronary intervention alone results of the AMICO (Alliance for Myocardial Infarction Care Optimization) Registry. JACC Cardiovasc Interv2008; 1:504–510.
Smalling RW. Ischemic time: the new gold standard for ST-segment elevation myocardial infarction care. J Am Coll Cardiol2009; 54:2154–2156.
Hochman JS, Sleeper LA, White HD, et al; SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. One-year survival following early revascularization for cardiogenic shock. JAMA2001; 285:190–192.
Jacobs AK, Leopold JA, Bates E, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol2003; 41:1273–1279.
Pedersen SH, Galatius S, Hansen PR, et al. Field triage reduces treatment delay and improves long-term clinical outcome in patients with acute ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol2009; 54:2296–2302.
Björklund E, Stenestrand U, Lindbäck J, Svensson L, Wallentin L, Lindahl B. Pre-hospital thrombolysis delivered by paramedics is associated with reduced time delay and mortality in ambulance-transported real-life patients with ST-elevation myocardial infarction. Eur Heart J2006; 27:1146–1152.
The European Myocardial Infarction Project Group. Prehospital thrombolytic therapy in patients with suspected acute myocardial infarction. N Engl J Med1993; 329:383–389.
References
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet2003; 361:13–20.
Waters RE, Singh KP, Roe MT, et al. Rationale and strategies for implementing community-based transfer protocols for primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. J Am Coll Cardiol2004; 43:2153–2159.
Chakrabarti A, Krumholz HM, Wang Y, Rumsfeld JS, Nallamothu BK; National Cardiovascular Data Registry. Time-to-reperfusion in patients undergoing interhospital transfer for primary percutaneous coronary intervention in the U.S: an analysis of 2005 and 2006 data from the National Cardiovascular Data Registry. J Am Coll Cardiol2008; 51:2442–2443.
Widimský P, Budesínský T, Vorác D, et al; ‘PRAGUE’ Study Group Investigators. Long distance transport for primary angioplasty vs immediate thrombolysis in acute myocardial infarction. Final results of the randomized national multicentre trial—PRAGUE-2. Eur Heart J2003; 24:94–104.
Steg PG, Bonnefoy E, Chabaud S, et al; Comparison of Angioplasty and Prehospital Thrombolysis in Acute Myocardial infarction (CAPTIM) Investigators. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation2003; 108:2851–2856.
Kalla K, Christ G, Karnik R, et al; Vienna STEMI Registry Group. Implementation of guidelines improves the standard of care: the Viennese registry on reperfusion strategies in ST-elevation myocardial infarction (Vienna STEMI registry). Circulation2006; 113:2398–2405.
Boersma E, Maas AC, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet1996; 348:771–775.
Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation2004; 110:e82–e292.
Nallamothu BK, Bates ER. Percutaneous coronary intervention versus fibrinolytic therapy in acute myocardial infarction: is timing (almost) everything?Am J Cardiol2003; 92:824–826.
Pinto DS, Kirtane AJ, Nallamothu BK, et al. Hospital delays in reperfusion for ST-elevation myocardial infarction: implications when selecting a reperfusion strategy. Circulation2006; 114:2019–2025.
Boersma E; Primary Coronary Angioplasty vs Thrombolysis Group. Does time matter? A pooled analysis of randomized clinical trials comparing primary percutaneous coronary intervention and in-hospital fibrinolysis in acute myocardial infarction patients. Eur Heart J2006; 27:779–788.
Stenestrand U, Wallentin L; Register of Information and Knowledge About Swedish Heart Intensive Care Admissions (RIKS-HIA). Fibrinolytic therapy in patients 75 years and older with ST-segment-elevation myocardial infarction: one-year follow-up of a large prospective cohort. Arch Intern Med2003; 163:965–971.
Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet1994; 343:311–322.
Grines CL, Browne KF, Marco J, et al. A comparison of immediate angioplasty with thrombolytic therapy for acute myocardial infarction. The Primary Angioplasty in Myocardial Infarction Study Group. N Engl J Med1993; 328:673–679.
Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med2009; 360:2705–2718.
Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab RE-teplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet2008; 371:559–568.
Danchin N, Coste P, Ferrières J, et al; FAST-MI Investigators. Comparison of thrombolysis followed by broad use of percutaneous coronary intervention with primary percutaneous coronary intervention for ST-segment-elevation acute myocardial infarction: data from the French registry on Acute ST-elevation Myocardial Infarction (FAST-MI). Circulation2008; 118:268–276.
Assessment of the Safety and Efficacy of a New Treatment Strategy with Percutaneous Coronary Intervention (ASSENT-4 PCI) investigators. Primary versus tenecteplase-facilitated percutaneous coronary intervention in patients with ST-segment elevation acute myocardial infarction (ASSENT-4 PCI): randomised trial. Lancet2006; 367:569–578.
Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med2008; 358:2205–2217.
Carver A, Rafelt S, Gershlick AH, Fairbrother KL, Hughes S, Wilcox R; REACT Investigators. Longer-term follow-up of patients recruited to the REACT (Rescue Angioplasty Versus Conservative Treatment or Repeat Thrombolysis) trial. J Am Coll Cardiol2009; 54:118–126.
Rasmanis G, Vesterqvist O, Gréen K, Edhag O, Henriksson P. Evidence of increased platelet activation after thrombolysis in patients with acute myocardial infarction. Br Heart J1992; 68:374–376.
Gurbel PA, Serebruany VL, Shustov AR, et al. Effects of reteplase and alteplase on platelet aggregation and major receptor expression during the first 24 hours of acute myocardial infarction treatment. GUSTO-III Investigators. Global Use of Strategies to Open Occluded Coronary Arteries. J Am Coll Cardiol1998; 31:1466–1473.
Coulter SA, Cannon CP, Ault KA, et al. High levels of platelet inhibition with abciximab despite heightened platelet activation and aggregation during thrombolysis for acute myocardial infarction: results from TIMI (thrombolysis in myocardial infarction) 14. Circulation2000; 101:2690–2695.
Ross AM, Huber K, Zeymer U, et al. The impact of place of enrollment and delay to reperfusion on 90-day post-infarction mortality in the ASSENT-4 PCI trial: assessment of the safety and efficacy of a new treatment strategy with percutaneous coronary intervention. JACC Cardiovasc Interv2009; 2:925–930.
Herrmann HC, Lu J, Brodie BR, et al; FINESSE Investigators. Benefit of facilitated percutaneous coronary intervention in high-risk ST-segment elevation myocardial infarction patients presenting to nonpercutaneous coronary intervention hospitals. JACC Cardiovasc Interv2009; 2:917–924.
Scheller B, Hennen B, Hammer B, et al; SIAM III Study Group. Beneficial effects of immediate stenting after thrombolysis in acute myocardial infarction. J Am Coll Cardiol2003; 42:634–641.
Fernandez-Avilés F, Alonso JJ, Castro-Beiras A, et al; GRACIA (Grupo de Análisis de la Cardiopatía Isquémica Aguda) Group. Routine invasive strategy within 24 hours of thrombolysis versus ischaemiaguided conservative approach for acute myocardial infarction with ST-segment elevation (GRACIA-1): a randomised controlled trial. Lancet2004; 364:1045–1053.
Le May MR, Wells GA, Labinaz M, et al. Combined angioplasty and pharmacological intervention versus thrombolysis alone in acute myocardial infarction (CAPITAL AMI study). J Am Coll Cardiol2005; 46:417–424.
Bøhmer E, Hoffmann P, Abdelnoor M, Arnesen H, Halvorsen S. Efficacy and safety of immediate angioplasty versus ischemia-guided management after thrombolysis in acute myocardial infarction in areas with very long transfer distances results of the NORDISTEMI (NORwegian study on DIstrict treatment of ST-elevation myocardial infarction). J Am Coll Cardiol2010; 55:102–110.
Wijeysundera HC, You JJ, Nallamothu BK, Krumholz HM, Cantor WJ, Ko DT. An early invasive strategy versus ischemia-guided management after fibrinolytic therapy for ST-segment elevation myocardial infarction: a meta-analysis of contemporary randomized controlled trials. Am Heart J2008; 156:564–572,572.e1–e2.
Kushner FG, Hand M, Smith SC, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update) a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol2009; 54:2205–2241.
Van de Werf F, Bax J, Betriu A, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J2008; 29:2909–2945.
Mukherjee D, Moliterno DJ. The timely coupling of mechanical revascularization following thrombolysis for myocardial infarction. Cardiology2007; 107:337–339.
Wijeysundera HC, Vijayaraghavan R, Nallamothu BK, et al. Rescue angioplasty or repeat fibrinolysis after failed fibrinolytic therapy for ST-segment myocardial infarction: a meta-analysis of randomized trials. J Am Coll Cardiol2007; 49:422–430.
The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med1993; 329:1615–1622.
Andersen HR, Nielsen TT, Rasmussen K, et al; DANAMI-2 Investigators. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N Engl J Med2003; 349:733–742.
Henry TD, Sharkey SW, Burke MN, et al. A regional system to provide timely access to percutaneous coronary intervention for ST-elevation myocardial infarction. Circulation2007; 116:721–728.
Denktas AE, Athar H, Henry TD, et al. Reduced-dose fibrinolytic acceleration of ST-segment elevation myocardial infarction treatment coupled with urgent percutaneous coronary intervention compared to primary percutaneous coronary intervention alone results of the AMICO (Alliance for Myocardial Infarction Care Optimization) Registry. JACC Cardiovasc Interv2008; 1:504–510.
Smalling RW. Ischemic time: the new gold standard for ST-segment elevation myocardial infarction care. J Am Coll Cardiol2009; 54:2154–2156.
Hochman JS, Sleeper LA, White HD, et al; SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock. One-year survival following early revascularization for cardiogenic shock. JAMA2001; 285:190–192.
Jacobs AK, Leopold JA, Bates E, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol2003; 41:1273–1279.
Pedersen SH, Galatius S, Hansen PR, et al. Field triage reduces treatment delay and improves long-term clinical outcome in patients with acute ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol2009; 54:2296–2302.
Björklund E, Stenestrand U, Lindbäck J, Svensson L, Wallentin L, Lindahl B. Pre-hospital thrombolysis delivered by paramedics is associated with reduced time delay and mortality in ambulance-transported real-life patients with ST-elevation myocardial infarction. Eur Heart J2006; 27:1146–1152.
The European Myocardial Infarction Project Group. Prehospital thrombolytic therapy in patients with suspected acute myocardial infarction. N Engl J Med1993; 329:383–389.
When the expected door-to-balloon time is less than 90 minutes and the door-to-balloon time minus the door-to-needle time is less than 60 minutes, the preferred approach is PCI not preceded by thrombolysis.
Evidence suggests that routine early (but not immediate) PCI—ie, 2 to 6 hours after thrombolysis—is beneficial, particularly in patients with high-risk ST-elevation MI.
Hospitals and emergency services should participate in community-based and regional systems of care, with standardized protocols to ensure expeditious transfer and prompt reperfusion.
Prehospital thrombolysis followed by early transfer to a PCI facility as part of a community-based system of care may further improve outcomes of patients with very long transfer times.
Disallow All Ads
Alternative CME
Consolidated Pubs: Do Not Show Source Publication Logo
Yes. Treatment with beta-adrenergic receptor blockers decreases the mortality rate in patients with coronary artery disease or heart failure, as well as during the perioperative period in selected patients (eg, those with a history of myocardial infarction, a positive stress test, or current chest pain due to myocardial ischemia). The current evidence supports giving beta-blockers to patients with coronary artery disease and chronic obstructive pulmonary disease (COPD) or asthma, which lowers the 1-year mortality rate to a degree similar to that in patients without COPD or asthma, and without worsening respiratory function.1 However, many clinicians still hesitate to start patients with COPD or asthma on a beta-blocker due to the fear of bronchoconstriction.2
THE RISKS
In patients with reversible airway disease, beta-blockers may increase airway reactivity and bronchospasm, as well as decrease the response to inhaled or oral beta-receptor agonists.3 Even topical ophthalmic nonselective beta-blockers for glaucoma can cause a worsening of pulmonary function.4 However, these data are from small trials in the 1970s and 1980s.
On the other hand, not giving beta-blockers can pose a risk of death. In a retrospective study of more than 200,000 patients with myocardial infarction, Gottlieb et al5 found that beta-blockers were associated with a 40% reduction in mortality rates in patients with conditions often considered a contraindication to beta-blocker therapy, such as congestive heart failure, pulmonary disease, and older age.5
CARDIOSELECTIVE BETA-BLOCKERS
Cardioselective beta-blockers with an affinity for the beta-1 receptor theoretically result in fewer adverse effects on the lungs. They competitively block the response to beta-adrenergic stimulation and selectively block beta-1 receptors with little or no effect on beta-2 receptors, except perhaps at high doses. However, this possible high-dose effect requires further study.
The effect of cardioselective beta-blockers on respiratory function was evaluated in two meta-analyses,6,7 one in patients with mild to moderate reactive airway disease, the other in patients with mild to severe COPD. Patients with reactive airway disease who received a single dose of a beta-blocker had a 7.46% reduction in forced expiratory volume in the first second of expiration (FEV1), an effect that was completely reversed by treatment with a beta-agonist inhaler. The FEV1 increased by a statistically significantly greater amount in response to beta-agonists in patients who received beta-blockers (a single dose or continuous therapy) than in those who did not receive beta-blockers. Patients who received continuous cardioselective beta-blockers experienced no significant drop in FEV1, and no new symptoms developed. These results led the authors to conclude that cardioselective beta-blockers do not cause a significant reduction in pulmonary function in patients with mild to moderate reactive airway disease and COPD and are therefore safe to use. A single dose of a cardioselective beta-blocker may produce a small decrease in FEV1, especially in patients with reactive airway disease, but as therapy is continued over days to weeks, there is no significant change in symptoms or FEV1 and no increase in the need for beta-agonist inhalers.
A major limitation of the two meta-analyses was that the patients were younger than most patients who require beta-blockers: the average age was 40 in patients with reactive airway disease, and 54 in patients with COPD. Also important to consider is that only patients with mild to moderate reactive airway disease were included. Patients with severe asthma, especially those with active bronchospasm, may react differently to even cardioselective beta-blockers.
NONSELECTIVE BETA-BLOCKERS
Recent studies suggest that nonselective beta-blockers can affect respiratory function in patients with COPD, but they have failed to show any harm. For example, propranolol (Inderal) was shown to worsen pulmonary function and to decrease the sensitivity of the airway to the effects of long-acting beta-2-agonists, but the 15 patients included in this study had no increase in respiratory symptoms.8
It has also been suggested that combined nonselective beta- and alpha-receptor blockade—eg, with labetalol (Trandate) or carvedilol (Coreg)—might be better tolerated than nonselective beta-blockers in patients with COPD.9 However, from limited data, Kotlyar et al10 suggested that carvedilol may be less well tolerated in patients with asthma than with COPD. All current evidence on combined nonselective beta-and alpha-blockade is observational, and it is not yet clear whether this class of beta-blockers is better tolerated due to alpha-blockade or merely because nonselective beta-blockers themselves are well tolerated.
OUR RECOMMENDATIONS
Beta-blockers improve survival rates in patients with chronic systolic heart failure and after myocardial infarction, including in those patients with coexisting COPD and reactive airway disease. But not all beta-blockers are the same (Table 1). Cardioselective beta-blockers (ie, those that block predominantly beta-1 receptors) are our beta-blockers of choice based on stronger evidence from clinical studies. Nonselective agents that include alpha-adrenergic blockade can be considered, although less is known about their effect on respiratory function. However, the use of even beta-1-selective drugs merits caution and close follow-up in patients with severe asthma (for which clinical study data are limited).
References
Chen J, Radford MJ, Wang Y, Marciniak TA, Krumholz HM. Effectiveness of beta-blocker therapy after acute myocardial infarction in elderly patients with chronic obstructive pulmonary disease or asthma. J Am Coll Cardiol2001; 37:1950–1956.
The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med1997; 157:2413–2446.
Benson MK, Berrill WT, Cruickshank JM, Sterling GS. A comparison of four beta-adrenoceptor antagonists in patients with asthma. Br J Clin Pharmacol1978; 5:415–419.
Fraunfelder FT, Barker AF. Respiratory effects of timolol. N Engl J Med1984; 311:1441.
Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med1998; 339:489–497.
Salpeter SR, Ormiston TM, Salpeter EE, Poole PJ, Cates CJ. Cardioselective beta-blockers for chronic obstructive pulmonary disease: a meta-analysis. Respir Med2003; 97:1094–1101.
Salpeter SR, Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with reactive airway disease: a meta-analysis. Ann Intern Med2002; 137:715–725.
van der Woude HJ, Zaagsma J, Postma DS, Winter TH, van Hulst M, Aalbers R. Detrimental effects of beta-blockers in COPD: a concern for nonselective beta-blockers. Chest2005; 127:818–824.
Sirak TE, Jelic S, Le Jemtel TH. Therapeutic update: non-selective beta- and alpha-adrenergic blockade in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol2004; 44:497–502.
Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. J Heart Lung Transplant2002; 21:1290–1295.
Yes. Treatment with beta-adrenergic receptor blockers decreases the mortality rate in patients with coronary artery disease or heart failure, as well as during the perioperative period in selected patients (eg, those with a history of myocardial infarction, a positive stress test, or current chest pain due to myocardial ischemia). The current evidence supports giving beta-blockers to patients with coronary artery disease and chronic obstructive pulmonary disease (COPD) or asthma, which lowers the 1-year mortality rate to a degree similar to that in patients without COPD or asthma, and without worsening respiratory function.1 However, many clinicians still hesitate to start patients with COPD or asthma on a beta-blocker due to the fear of bronchoconstriction.2
THE RISKS
In patients with reversible airway disease, beta-blockers may increase airway reactivity and bronchospasm, as well as decrease the response to inhaled or oral beta-receptor agonists.3 Even topical ophthalmic nonselective beta-blockers for glaucoma can cause a worsening of pulmonary function.4 However, these data are from small trials in the 1970s and 1980s.
On the other hand, not giving beta-blockers can pose a risk of death. In a retrospective study of more than 200,000 patients with myocardial infarction, Gottlieb et al5 found that beta-blockers were associated with a 40% reduction in mortality rates in patients with conditions often considered a contraindication to beta-blocker therapy, such as congestive heart failure, pulmonary disease, and older age.5
CARDIOSELECTIVE BETA-BLOCKERS
Cardioselective beta-blockers with an affinity for the beta-1 receptor theoretically result in fewer adverse effects on the lungs. They competitively block the response to beta-adrenergic stimulation and selectively block beta-1 receptors with little or no effect on beta-2 receptors, except perhaps at high doses. However, this possible high-dose effect requires further study.
The effect of cardioselective beta-blockers on respiratory function was evaluated in two meta-analyses,6,7 one in patients with mild to moderate reactive airway disease, the other in patients with mild to severe COPD. Patients with reactive airway disease who received a single dose of a beta-blocker had a 7.46% reduction in forced expiratory volume in the first second of expiration (FEV1), an effect that was completely reversed by treatment with a beta-agonist inhaler. The FEV1 increased by a statistically significantly greater amount in response to beta-agonists in patients who received beta-blockers (a single dose or continuous therapy) than in those who did not receive beta-blockers. Patients who received continuous cardioselective beta-blockers experienced no significant drop in FEV1, and no new symptoms developed. These results led the authors to conclude that cardioselective beta-blockers do not cause a significant reduction in pulmonary function in patients with mild to moderate reactive airway disease and COPD and are therefore safe to use. A single dose of a cardioselective beta-blocker may produce a small decrease in FEV1, especially in patients with reactive airway disease, but as therapy is continued over days to weeks, there is no significant change in symptoms or FEV1 and no increase in the need for beta-agonist inhalers.
A major limitation of the two meta-analyses was that the patients were younger than most patients who require beta-blockers: the average age was 40 in patients with reactive airway disease, and 54 in patients with COPD. Also important to consider is that only patients with mild to moderate reactive airway disease were included. Patients with severe asthma, especially those with active bronchospasm, may react differently to even cardioselective beta-blockers.
NONSELECTIVE BETA-BLOCKERS
Recent studies suggest that nonselective beta-blockers can affect respiratory function in patients with COPD, but they have failed to show any harm. For example, propranolol (Inderal) was shown to worsen pulmonary function and to decrease the sensitivity of the airway to the effects of long-acting beta-2-agonists, but the 15 patients included in this study had no increase in respiratory symptoms.8
It has also been suggested that combined nonselective beta- and alpha-receptor blockade—eg, with labetalol (Trandate) or carvedilol (Coreg)—might be better tolerated than nonselective beta-blockers in patients with COPD.9 However, from limited data, Kotlyar et al10 suggested that carvedilol may be less well tolerated in patients with asthma than with COPD. All current evidence on combined nonselective beta-and alpha-blockade is observational, and it is not yet clear whether this class of beta-blockers is better tolerated due to alpha-blockade or merely because nonselective beta-blockers themselves are well tolerated.
OUR RECOMMENDATIONS
Beta-blockers improve survival rates in patients with chronic systolic heart failure and after myocardial infarction, including in those patients with coexisting COPD and reactive airway disease. But not all beta-blockers are the same (Table 1). Cardioselective beta-blockers (ie, those that block predominantly beta-1 receptors) are our beta-blockers of choice based on stronger evidence from clinical studies. Nonselective agents that include alpha-adrenergic blockade can be considered, although less is known about their effect on respiratory function. However, the use of even beta-1-selective drugs merits caution and close follow-up in patients with severe asthma (for which clinical study data are limited).
Yes. Treatment with beta-adrenergic receptor blockers decreases the mortality rate in patients with coronary artery disease or heart failure, as well as during the perioperative period in selected patients (eg, those with a history of myocardial infarction, a positive stress test, or current chest pain due to myocardial ischemia). The current evidence supports giving beta-blockers to patients with coronary artery disease and chronic obstructive pulmonary disease (COPD) or asthma, which lowers the 1-year mortality rate to a degree similar to that in patients without COPD or asthma, and without worsening respiratory function.1 However, many clinicians still hesitate to start patients with COPD or asthma on a beta-blocker due to the fear of bronchoconstriction.2
THE RISKS
In patients with reversible airway disease, beta-blockers may increase airway reactivity and bronchospasm, as well as decrease the response to inhaled or oral beta-receptor agonists.3 Even topical ophthalmic nonselective beta-blockers for glaucoma can cause a worsening of pulmonary function.4 However, these data are from small trials in the 1970s and 1980s.
On the other hand, not giving beta-blockers can pose a risk of death. In a retrospective study of more than 200,000 patients with myocardial infarction, Gottlieb et al5 found that beta-blockers were associated with a 40% reduction in mortality rates in patients with conditions often considered a contraindication to beta-blocker therapy, such as congestive heart failure, pulmonary disease, and older age.5
CARDIOSELECTIVE BETA-BLOCKERS
Cardioselective beta-blockers with an affinity for the beta-1 receptor theoretically result in fewer adverse effects on the lungs. They competitively block the response to beta-adrenergic stimulation and selectively block beta-1 receptors with little or no effect on beta-2 receptors, except perhaps at high doses. However, this possible high-dose effect requires further study.
The effect of cardioselective beta-blockers on respiratory function was evaluated in two meta-analyses,6,7 one in patients with mild to moderate reactive airway disease, the other in patients with mild to severe COPD. Patients with reactive airway disease who received a single dose of a beta-blocker had a 7.46% reduction in forced expiratory volume in the first second of expiration (FEV1), an effect that was completely reversed by treatment with a beta-agonist inhaler. The FEV1 increased by a statistically significantly greater amount in response to beta-agonists in patients who received beta-blockers (a single dose or continuous therapy) than in those who did not receive beta-blockers. Patients who received continuous cardioselective beta-blockers experienced no significant drop in FEV1, and no new symptoms developed. These results led the authors to conclude that cardioselective beta-blockers do not cause a significant reduction in pulmonary function in patients with mild to moderate reactive airway disease and COPD and are therefore safe to use. A single dose of a cardioselective beta-blocker may produce a small decrease in FEV1, especially in patients with reactive airway disease, but as therapy is continued over days to weeks, there is no significant change in symptoms or FEV1 and no increase in the need for beta-agonist inhalers.
A major limitation of the two meta-analyses was that the patients were younger than most patients who require beta-blockers: the average age was 40 in patients with reactive airway disease, and 54 in patients with COPD. Also important to consider is that only patients with mild to moderate reactive airway disease were included. Patients with severe asthma, especially those with active bronchospasm, may react differently to even cardioselective beta-blockers.
NONSELECTIVE BETA-BLOCKERS
Recent studies suggest that nonselective beta-blockers can affect respiratory function in patients with COPD, but they have failed to show any harm. For example, propranolol (Inderal) was shown to worsen pulmonary function and to decrease the sensitivity of the airway to the effects of long-acting beta-2-agonists, but the 15 patients included in this study had no increase in respiratory symptoms.8
It has also been suggested that combined nonselective beta- and alpha-receptor blockade—eg, with labetalol (Trandate) or carvedilol (Coreg)—might be better tolerated than nonselective beta-blockers in patients with COPD.9 However, from limited data, Kotlyar et al10 suggested that carvedilol may be less well tolerated in patients with asthma than with COPD. All current evidence on combined nonselective beta-and alpha-blockade is observational, and it is not yet clear whether this class of beta-blockers is better tolerated due to alpha-blockade or merely because nonselective beta-blockers themselves are well tolerated.
OUR RECOMMENDATIONS
Beta-blockers improve survival rates in patients with chronic systolic heart failure and after myocardial infarction, including in those patients with coexisting COPD and reactive airway disease. But not all beta-blockers are the same (Table 1). Cardioselective beta-blockers (ie, those that block predominantly beta-1 receptors) are our beta-blockers of choice based on stronger evidence from clinical studies. Nonselective agents that include alpha-adrenergic blockade can be considered, although less is known about their effect on respiratory function. However, the use of even beta-1-selective drugs merits caution and close follow-up in patients with severe asthma (for which clinical study data are limited).
References
Chen J, Radford MJ, Wang Y, Marciniak TA, Krumholz HM. Effectiveness of beta-blocker therapy after acute myocardial infarction in elderly patients with chronic obstructive pulmonary disease or asthma. J Am Coll Cardiol2001; 37:1950–1956.
The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med1997; 157:2413–2446.
Benson MK, Berrill WT, Cruickshank JM, Sterling GS. A comparison of four beta-adrenoceptor antagonists in patients with asthma. Br J Clin Pharmacol1978; 5:415–419.
Fraunfelder FT, Barker AF. Respiratory effects of timolol. N Engl J Med1984; 311:1441.
Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med1998; 339:489–497.
Salpeter SR, Ormiston TM, Salpeter EE, Poole PJ, Cates CJ. Cardioselective beta-blockers for chronic obstructive pulmonary disease: a meta-analysis. Respir Med2003; 97:1094–1101.
Salpeter SR, Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with reactive airway disease: a meta-analysis. Ann Intern Med2002; 137:715–725.
van der Woude HJ, Zaagsma J, Postma DS, Winter TH, van Hulst M, Aalbers R. Detrimental effects of beta-blockers in COPD: a concern for nonselective beta-blockers. Chest2005; 127:818–824.
Sirak TE, Jelic S, Le Jemtel TH. Therapeutic update: non-selective beta- and alpha-adrenergic blockade in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol2004; 44:497–502.
Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. J Heart Lung Transplant2002; 21:1290–1295.
References
Chen J, Radford MJ, Wang Y, Marciniak TA, Krumholz HM. Effectiveness of beta-blocker therapy after acute myocardial infarction in elderly patients with chronic obstructive pulmonary disease or asthma. J Am Coll Cardiol2001; 37:1950–1956.
The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med1997; 157:2413–2446.
Benson MK, Berrill WT, Cruickshank JM, Sterling GS. A comparison of four beta-adrenoceptor antagonists in patients with asthma. Br J Clin Pharmacol1978; 5:415–419.
Fraunfelder FT, Barker AF. Respiratory effects of timolol. N Engl J Med1984; 311:1441.
Gottlieb SS, McCarter RJ, Vogel RA. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med1998; 339:489–497.
Salpeter SR, Ormiston TM, Salpeter EE, Poole PJ, Cates CJ. Cardioselective beta-blockers for chronic obstructive pulmonary disease: a meta-analysis. Respir Med2003; 97:1094–1101.
Salpeter SR, Ormiston TM, Salpeter EE. Cardioselective beta-blockers in patients with reactive airway disease: a meta-analysis. Ann Intern Med2002; 137:715–725.
van der Woude HJ, Zaagsma J, Postma DS, Winter TH, van Hulst M, Aalbers R. Detrimental effects of beta-blockers in COPD: a concern for nonselective beta-blockers. Chest2005; 127:818–824.
Sirak TE, Jelic S, Le Jemtel TH. Therapeutic update: non-selective beta- and alpha-adrenergic blockade in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol2004; 44:497–502.
Kotlyar E, Keogh AM, Macdonald PS, Arnold RH, McCaffrey DJ, Glanville AR. Tolerability of carvedilol in patients with heart failure and concomitant chronic obstructive pulmonary disease or asthma. J Heart Lung Transplant2002; 21:1290–1295.
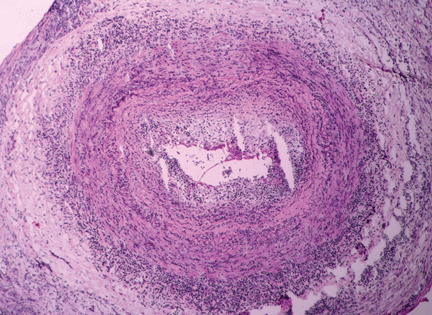
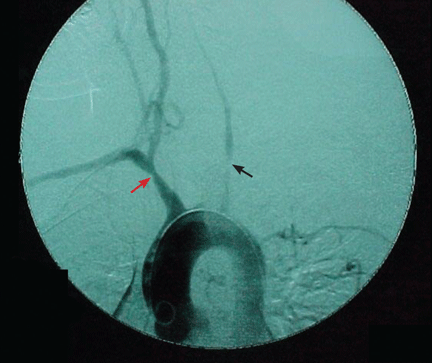
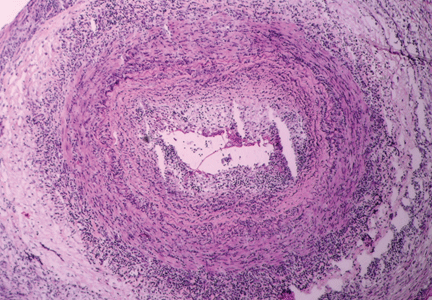
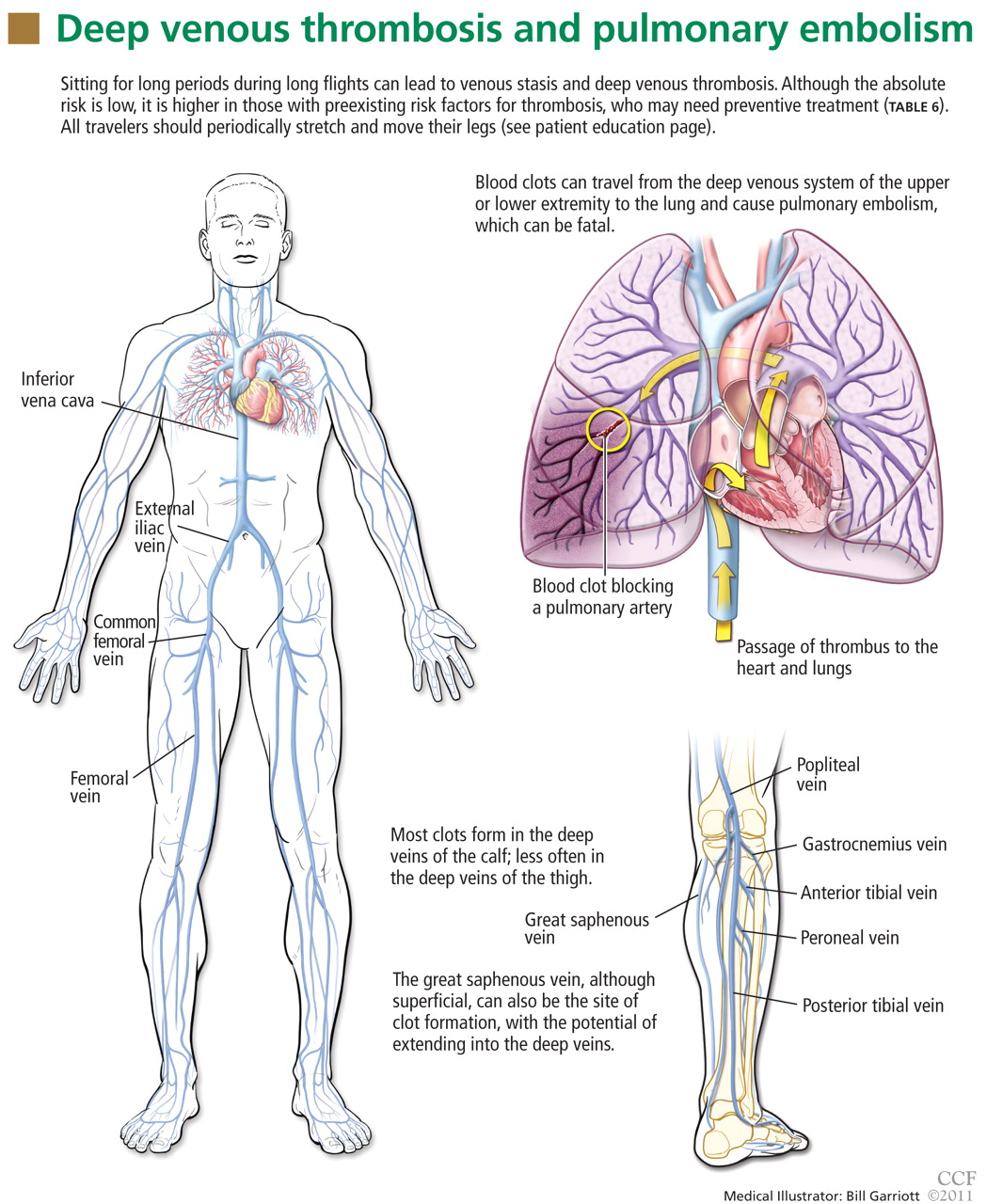






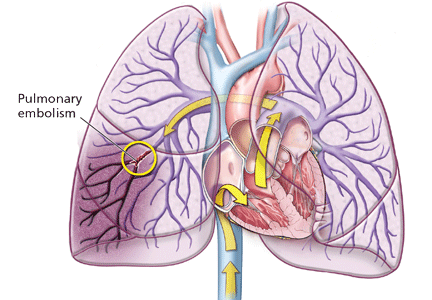

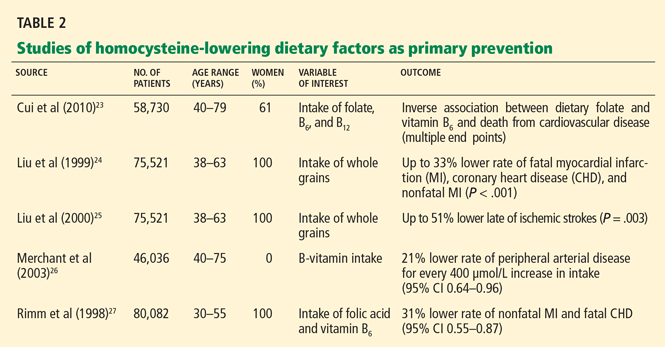
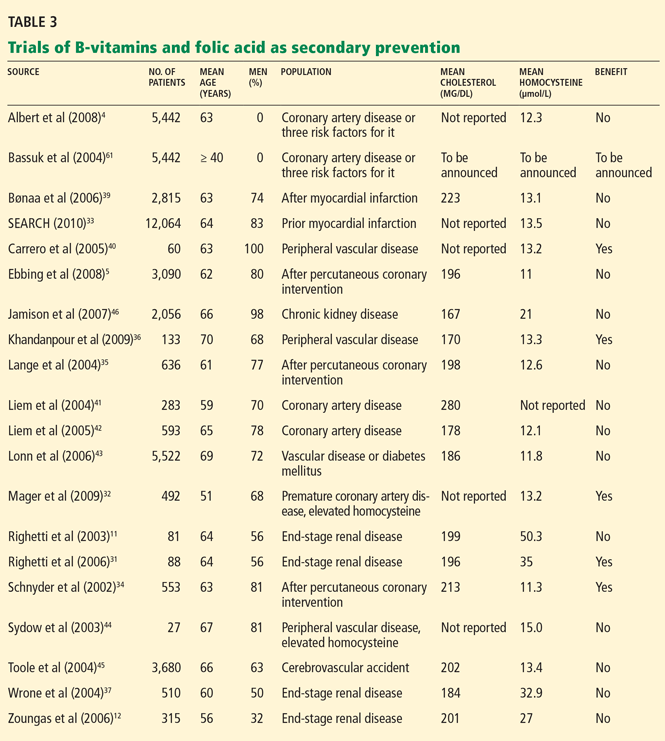
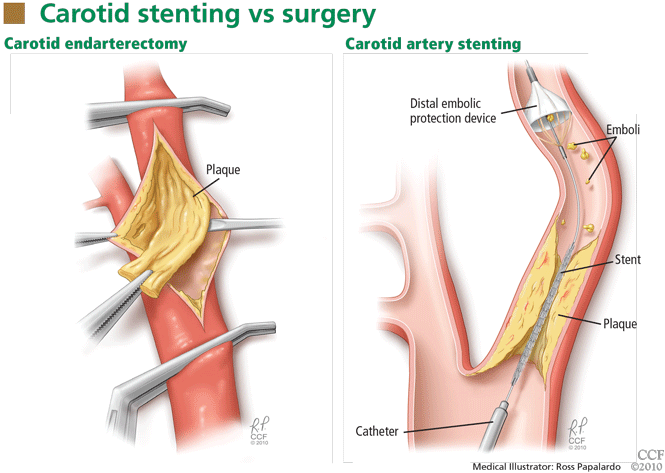
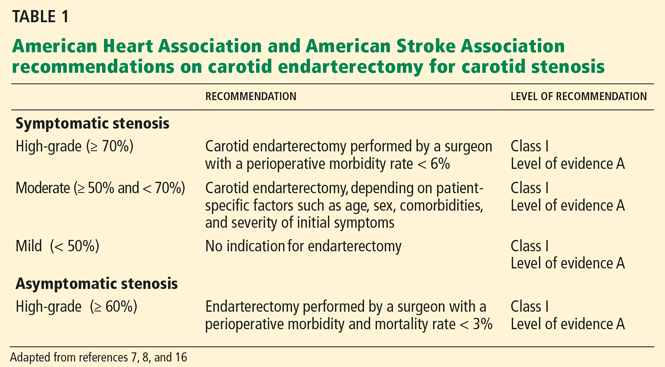
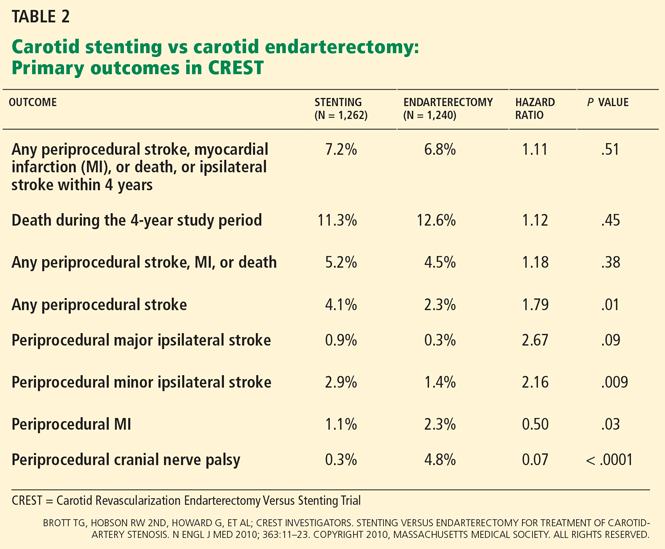
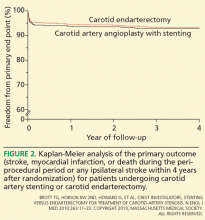 Brott TG, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23. Copyright 2010, Massachusetts Medical Society. All rights reserved.
Brott TG, et al; CREST Investigators. Stenting versus endarterectomy for treatment of carotid-artery stenosis. N Engl J Med 2010; 363:11–23. Copyright 2010, Massachusetts Medical Society. All rights reserved.