User login
Antibiotic Beads Perform Well to Prevent Surgical Infection
NATIONAL HARBOR, MD – Local delivery of an antibiotic using impregnated bone-cement beads enabled sterile cultures to be achieved in the majority of infected surgical sites before final repair or graft replacement, according to a small, retrospective study reported by Dr. Dennis F. Bandyk.
This type of nonbiodegradable antibiotic implant is especially useful in cases of infection related to a groin incision, he said at the annual meeting of the Eastern Vascular Society.
"You can make these drug-delivery beads in the operating room in about 10 to 15 minutes.
"After EVAR [endovascular aneurysm repair], we have about a 5% incidence of surgical site infection. Arterial revascularization in the literature has a 10% to 30% infection rate, [and] it’s 18% in lengthy lower limb revascularization at our institution in Tampa. Major amputations generally range from a 15% to 25% infection rate, with 22% for the last 3 years at Tampa. The problem likely exists because of colonization with staph, particularly MRSA [methicillin-resistant Staphylococcus aureus], of injured and ischemic tissue, especially with involvement of the groin," Dr. Bandyk said.
"We have no decent methods of preventing these sorts of surgical site infections," he noted. The approach that his group takes to treat these infections is to use sequential in situ antibiotic treatment.
"I believe that many of the SSIs [surgical site infections] we have follow this theme of a biofilm-mediated infection," he said. Specific pathogenic strains colonize the area; they produce an extracellular matrix that then creates selective antibiotic resistance, since many of the antibiotics don’t penetrate biofilms.
The reported study comprised a 7-year case audit of 78 patients (55% male) who had complex SSI following peripheral arterial repair, treatment of an infected hip, or above- or below-knee lower limb amputation (12 infected stumps).
Antibiotic delivery directly to the wounds was mediated via the use of polymethyl methacrylate (PMMA) bone-cement beads. For gram-positive infection, which occurred in 70% of patients, the beads were impregnated with vancomycin (2 g/40 g PMMA) in the early part of the case series. Daptomycin (1.5 g/40 g PMMA) was found superior to vancomycin during in vitro testing, and became the antibiotic of choice for patients seen later in the case series. Tobramycin (2 g/40 g PMMA) was used for gram-negative infection, seen in 30% of patients. This was coupled to culture-specific parenteral antibiotics for 3-6 weeks. MRSA accounted for at least half of all early and late infections, and thus MRSA must be taken into account when comparing therapy options.
Infected surgical sites were explored and cultured, and based on a Gram stain of pus or a prior culture result, PMMA antibiotic-impregnated beads were implanted into the wound after soft tissue debridement, including the adjunct use of wound irrigants such as the "brown volcano" – a mixture of salt, peroxide, and Betadine – which disrupts biofilms.
Arterial infections underwent an average of 2.3 debridements. Surgical wounds were primarily closed with a planned bead exchange 3-5 days later (often repeated one to three times) to confirm sterilization prior to graft preservation or in situ graft replacement. The main outcomes were rates of wound sterilization (negative culture) based on wound type, procedures for persistent infection, and freedom from arterial repair infection.
In terms of outcomes, there were no cases of limb loss, higher-level amputation, or death at 30 days. The rate of recurrent infection was 7% over a mean follow-up period of 3 years. Sterile wound cultures were achieved in 91% of cases after 1-3 bead exchanges. Daptomycin beads appeared to work the most rapidly, Dr. Bandyk added.
"So you can sterilize the wound with the prosthetic in place," he noted.
Because of this strategy’s success, his group has transitioned to treating almost 58% of their SSIs in this way, said Dr. Bandyk, professor of surgery at the University of South Florida, Tampa.
"You can make these drug-delivery beads in the operating room in about 10 to 15 minutes. Vancomycin is bacteriostatic and does not penetrate biofilms; daptomycin is a bacteriocidal antibiotic that does penetrate biofilms," he explained.
In addition, because the use of antibiotic beads in general "isn’t anything new, you get paid for putting it in and paid for pulling it out."
Discussion after the presentation focused on the expense of daptomycin versus vancomycin and the difficulty of obtaining it in many institutions due to cost and issues of antibiotic stewardship. Dr. Bandyk responded, "I thought we were surgeons. Most surgeons believe that we should use a bactericidal agent that can get to the tissue with a chance of killing the bacteria that are there. I didn’t realize that we were in this sort of price war with the hospital. If you look at what a biofilm infection is, you will understand why vancomycin doesn’t work."
In response to a question as to whether the type of incision was an issue regarding SSI, Dr. Bandyk said: "It’s the patient that’s producing the infection, and certain patient characteristics, so it isn’t necessarily how we’re closing, which way we are putting the incision ... So don’t blame yourself for every surgical site infection. It’s the patient factors that are operative in many of these cases. Of almost all of our infections that occurred, two-thirds came from the MRSA-colonized patient," he said.
Dr. Bandyk reported being on the speakers bureau and having received funding from Cubist Pharmaceuticals, the manufacturer of daptomycin (Cubicin).
Postoperative infection has long been the bane of bypass surgery, especially when prosthetic conduits are used. While excision of the graft and extra-anatomic bypass is feasible, there are many downsides to this approach. More recently, in situ treatment has been promulgated, with methods such as wound vacuum, in situ replacement with antibiotic-bonded or -soaked grafts, and now antibiotic beads. Dr. Bandyk’s method allows for more rapid closure of the wound, instead of allowing healing by secondary intent, but it does require several trips to the OR for bead exchanges. His outcomes have certainly been excellent, with only 7% of these complex patients developing recurrent infections over a 3-year period and just one to three bead exchanges per patient.
Dr. Linda Harris is vice chair, faculty development, department of surgery, Millard Fillmore Gates Hospital-Kaleida, Buffalo, N.Y.She has no relevant disclosures.
Postoperative infection has long been the bane of bypass surgery, especially when prosthetic conduits are used. While excision of the graft and extra-anatomic bypass is feasible, there are many downsides to this approach. More recently, in situ treatment has been promulgated, with methods such as wound vacuum, in situ replacement with antibiotic-bonded or -soaked grafts, and now antibiotic beads. Dr. Bandyk’s method allows for more rapid closure of the wound, instead of allowing healing by secondary intent, but it does require several trips to the OR for bead exchanges. His outcomes have certainly been excellent, with only 7% of these complex patients developing recurrent infections over a 3-year period and just one to three bead exchanges per patient.
Dr. Linda Harris is vice chair, faculty development, department of surgery, Millard Fillmore Gates Hospital-Kaleida, Buffalo, N.Y.She has no relevant disclosures.
Postoperative infection has long been the bane of bypass surgery, especially when prosthetic conduits are used. While excision of the graft and extra-anatomic bypass is feasible, there are many downsides to this approach. More recently, in situ treatment has been promulgated, with methods such as wound vacuum, in situ replacement with antibiotic-bonded or -soaked grafts, and now antibiotic beads. Dr. Bandyk’s method allows for more rapid closure of the wound, instead of allowing healing by secondary intent, but it does require several trips to the OR for bead exchanges. His outcomes have certainly been excellent, with only 7% of these complex patients developing recurrent infections over a 3-year period and just one to three bead exchanges per patient.
Dr. Linda Harris is vice chair, faculty development, department of surgery, Millard Fillmore Gates Hospital-Kaleida, Buffalo, N.Y.She has no relevant disclosures.
NATIONAL HARBOR, MD – Local delivery of an antibiotic using impregnated bone-cement beads enabled sterile cultures to be achieved in the majority of infected surgical sites before final repair or graft replacement, according to a small, retrospective study reported by Dr. Dennis F. Bandyk.
This type of nonbiodegradable antibiotic implant is especially useful in cases of infection related to a groin incision, he said at the annual meeting of the Eastern Vascular Society.
"You can make these drug-delivery beads in the operating room in about 10 to 15 minutes.
"After EVAR [endovascular aneurysm repair], we have about a 5% incidence of surgical site infection. Arterial revascularization in the literature has a 10% to 30% infection rate, [and] it’s 18% in lengthy lower limb revascularization at our institution in Tampa. Major amputations generally range from a 15% to 25% infection rate, with 22% for the last 3 years at Tampa. The problem likely exists because of colonization with staph, particularly MRSA [methicillin-resistant Staphylococcus aureus], of injured and ischemic tissue, especially with involvement of the groin," Dr. Bandyk said.
"We have no decent methods of preventing these sorts of surgical site infections," he noted. The approach that his group takes to treat these infections is to use sequential in situ antibiotic treatment.
"I believe that many of the SSIs [surgical site infections] we have follow this theme of a biofilm-mediated infection," he said. Specific pathogenic strains colonize the area; they produce an extracellular matrix that then creates selective antibiotic resistance, since many of the antibiotics don’t penetrate biofilms.
The reported study comprised a 7-year case audit of 78 patients (55% male) who had complex SSI following peripheral arterial repair, treatment of an infected hip, or above- or below-knee lower limb amputation (12 infected stumps).
Antibiotic delivery directly to the wounds was mediated via the use of polymethyl methacrylate (PMMA) bone-cement beads. For gram-positive infection, which occurred in 70% of patients, the beads were impregnated with vancomycin (2 g/40 g PMMA) in the early part of the case series. Daptomycin (1.5 g/40 g PMMA) was found superior to vancomycin during in vitro testing, and became the antibiotic of choice for patients seen later in the case series. Tobramycin (2 g/40 g PMMA) was used for gram-negative infection, seen in 30% of patients. This was coupled to culture-specific parenteral antibiotics for 3-6 weeks. MRSA accounted for at least half of all early and late infections, and thus MRSA must be taken into account when comparing therapy options.
Infected surgical sites were explored and cultured, and based on a Gram stain of pus or a prior culture result, PMMA antibiotic-impregnated beads were implanted into the wound after soft tissue debridement, including the adjunct use of wound irrigants such as the "brown volcano" – a mixture of salt, peroxide, and Betadine – which disrupts biofilms.
Arterial infections underwent an average of 2.3 debridements. Surgical wounds were primarily closed with a planned bead exchange 3-5 days later (often repeated one to three times) to confirm sterilization prior to graft preservation or in situ graft replacement. The main outcomes were rates of wound sterilization (negative culture) based on wound type, procedures for persistent infection, and freedom from arterial repair infection.
In terms of outcomes, there were no cases of limb loss, higher-level amputation, or death at 30 days. The rate of recurrent infection was 7% over a mean follow-up period of 3 years. Sterile wound cultures were achieved in 91% of cases after 1-3 bead exchanges. Daptomycin beads appeared to work the most rapidly, Dr. Bandyk added.
"So you can sterilize the wound with the prosthetic in place," he noted.
Because of this strategy’s success, his group has transitioned to treating almost 58% of their SSIs in this way, said Dr. Bandyk, professor of surgery at the University of South Florida, Tampa.
"You can make these drug-delivery beads in the operating room in about 10 to 15 minutes. Vancomycin is bacteriostatic and does not penetrate biofilms; daptomycin is a bacteriocidal antibiotic that does penetrate biofilms," he explained.
In addition, because the use of antibiotic beads in general "isn’t anything new, you get paid for putting it in and paid for pulling it out."
Discussion after the presentation focused on the expense of daptomycin versus vancomycin and the difficulty of obtaining it in many institutions due to cost and issues of antibiotic stewardship. Dr. Bandyk responded, "I thought we were surgeons. Most surgeons believe that we should use a bactericidal agent that can get to the tissue with a chance of killing the bacteria that are there. I didn’t realize that we were in this sort of price war with the hospital. If you look at what a biofilm infection is, you will understand why vancomycin doesn’t work."
In response to a question as to whether the type of incision was an issue regarding SSI, Dr. Bandyk said: "It’s the patient that’s producing the infection, and certain patient characteristics, so it isn’t necessarily how we’re closing, which way we are putting the incision ... So don’t blame yourself for every surgical site infection. It’s the patient factors that are operative in many of these cases. Of almost all of our infections that occurred, two-thirds came from the MRSA-colonized patient," he said.
Dr. Bandyk reported being on the speakers bureau and having received funding from Cubist Pharmaceuticals, the manufacturer of daptomycin (Cubicin).
NATIONAL HARBOR, MD – Local delivery of an antibiotic using impregnated bone-cement beads enabled sterile cultures to be achieved in the majority of infected surgical sites before final repair or graft replacement, according to a small, retrospective study reported by Dr. Dennis F. Bandyk.
This type of nonbiodegradable antibiotic implant is especially useful in cases of infection related to a groin incision, he said at the annual meeting of the Eastern Vascular Society.
"You can make these drug-delivery beads in the operating room in about 10 to 15 minutes.
"After EVAR [endovascular aneurysm repair], we have about a 5% incidence of surgical site infection. Arterial revascularization in the literature has a 10% to 30% infection rate, [and] it’s 18% in lengthy lower limb revascularization at our institution in Tampa. Major amputations generally range from a 15% to 25% infection rate, with 22% for the last 3 years at Tampa. The problem likely exists because of colonization with staph, particularly MRSA [methicillin-resistant Staphylococcus aureus], of injured and ischemic tissue, especially with involvement of the groin," Dr. Bandyk said.
"We have no decent methods of preventing these sorts of surgical site infections," he noted. The approach that his group takes to treat these infections is to use sequential in situ antibiotic treatment.
"I believe that many of the SSIs [surgical site infections] we have follow this theme of a biofilm-mediated infection," he said. Specific pathogenic strains colonize the area; they produce an extracellular matrix that then creates selective antibiotic resistance, since many of the antibiotics don’t penetrate biofilms.
The reported study comprised a 7-year case audit of 78 patients (55% male) who had complex SSI following peripheral arterial repair, treatment of an infected hip, or above- or below-knee lower limb amputation (12 infected stumps).
Antibiotic delivery directly to the wounds was mediated via the use of polymethyl methacrylate (PMMA) bone-cement beads. For gram-positive infection, which occurred in 70% of patients, the beads were impregnated with vancomycin (2 g/40 g PMMA) in the early part of the case series. Daptomycin (1.5 g/40 g PMMA) was found superior to vancomycin during in vitro testing, and became the antibiotic of choice for patients seen later in the case series. Tobramycin (2 g/40 g PMMA) was used for gram-negative infection, seen in 30% of patients. This was coupled to culture-specific parenteral antibiotics for 3-6 weeks. MRSA accounted for at least half of all early and late infections, and thus MRSA must be taken into account when comparing therapy options.
Infected surgical sites were explored and cultured, and based on a Gram stain of pus or a prior culture result, PMMA antibiotic-impregnated beads were implanted into the wound after soft tissue debridement, including the adjunct use of wound irrigants such as the "brown volcano" – a mixture of salt, peroxide, and Betadine – which disrupts biofilms.
Arterial infections underwent an average of 2.3 debridements. Surgical wounds were primarily closed with a planned bead exchange 3-5 days later (often repeated one to three times) to confirm sterilization prior to graft preservation or in situ graft replacement. The main outcomes were rates of wound sterilization (negative culture) based on wound type, procedures for persistent infection, and freedom from arterial repair infection.
In terms of outcomes, there were no cases of limb loss, higher-level amputation, or death at 30 days. The rate of recurrent infection was 7% over a mean follow-up period of 3 years. Sterile wound cultures were achieved in 91% of cases after 1-3 bead exchanges. Daptomycin beads appeared to work the most rapidly, Dr. Bandyk added.
"So you can sterilize the wound with the prosthetic in place," he noted.
Because of this strategy’s success, his group has transitioned to treating almost 58% of their SSIs in this way, said Dr. Bandyk, professor of surgery at the University of South Florida, Tampa.
"You can make these drug-delivery beads in the operating room in about 10 to 15 minutes. Vancomycin is bacteriostatic and does not penetrate biofilms; daptomycin is a bacteriocidal antibiotic that does penetrate biofilms," he explained.
In addition, because the use of antibiotic beads in general "isn’t anything new, you get paid for putting it in and paid for pulling it out."
Discussion after the presentation focused on the expense of daptomycin versus vancomycin and the difficulty of obtaining it in many institutions due to cost and issues of antibiotic stewardship. Dr. Bandyk responded, "I thought we were surgeons. Most surgeons believe that we should use a bactericidal agent that can get to the tissue with a chance of killing the bacteria that are there. I didn’t realize that we were in this sort of price war with the hospital. If you look at what a biofilm infection is, you will understand why vancomycin doesn’t work."
In response to a question as to whether the type of incision was an issue regarding SSI, Dr. Bandyk said: "It’s the patient that’s producing the infection, and certain patient characteristics, so it isn’t necessarily how we’re closing, which way we are putting the incision ... So don’t blame yourself for every surgical site infection. It’s the patient factors that are operative in many of these cases. Of almost all of our infections that occurred, two-thirds came from the MRSA-colonized patient," he said.
Dr. Bandyk reported being on the speakers bureau and having received funding from Cubist Pharmaceuticals, the manufacturer of daptomycin (Cubicin).
FROM THE ANNUAL MEETING OF THE EASTERN VASCULAR SOCIETY
Dabigatran: Will it change clinical practice?
Dabigatran etexilate (Pradaxa) is a new oral anticoagulant that has distinct advantages over warfarin (Coumadin) in terms of its ease of administration, efficacy, and safety.
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY trial),1 in patients with nonvalvular atrial fibrillation, dabigatran 110 mg twice a day was found to be as good as warfarin in preventing systemic embolization and stroke (the primary outcome of the study), and at 150 mg twice a day it was superior.1 It has also shown efficacy in treating acute deep vein thrombosis and pulmonary embolism and in preventing these complications in orthopedic surgical patients.2–4
Dabigatran has been approved in 75 countries. It carries the trade name Pradaxa in Europe and the United States and Pradax in Canada. In October 2010, the US Food and Drug Administration (FDA) Cardiovascular and Renal Drugs Advisory Committee endorsed two twice-daily doses (75 mg and 150 mg) of dabigatran for the prevention of systemic embolization and stroke in patients with nonvalvular atrial fibrillation.
However, dabigatran is relatively expensive, and its current high cost might be a barrier to its wider use.
MANY PATIENTS NEED ANTICOAGULATION
Anticoagulation plays a vital role in the primary and secondary prevention of stroke in patients with atrial fibrillation and of pulmonary embolism in patients with venous thromboembolism. It is also used during cardiothoracic and vascular surgery, endovascular procedures, and dialysis and in patients with mechanical heart valves and hypercoagulable conditions.
Atrial fibrillation affects 3.03 million people in the United States (2005 figures), and this number is predicted to be as high as 7.56 million by 2050.5 More than 10% of people over the age of 80 years have it, and the lifetime risk of developing it is approximately 25%.6,7 Its most serious complication is ischemic stroke (the risk of which increases with age) and systemic embolization.5,8
Until the recent introduction of dabigatran, the only oral anticoagulant available in the United States for treating patients with atrial fibrillation was warfarin. Although warfarin has a number of disadvantages (see below), it is actually very effective for preventing ischemic stroke, reducing the incidence by as much as 65%.9,10
Venous thromboembolism is the third most common cardiovascular disorder after myocardial infarction and stroke.11 Although its exact incidence is unknown, nearly 1 million cases of it (incident or recurrent, fatal and nonfatal events) occur in the United States each year.12 Many patients with venous thromboembolism need oral anticoagulation long-term, and currently warfarin remains the only option for them as well.
NEEDED: A BETTER ANTICOAGULANT
Warfarin has been the most commonly prescribed oral anticoagulant in the United States for more than 60 years. As of 2004, more than 30 million outpatient prescriptions for it were filled annually in this country alone.13 However, warfarin has several important limitations.
Warfarin has a narrow therapeutic index. Patients taking it require monitoring of their international normalized ratio (INR) and frequent dose adjustments, and this is time-consuming and inconvenient. The target INR for patients with venous thromboembolism and atrial fibrillation is 2.0 to 3.0, whereas patients with a mechanical heart valve need a higher INR (2.5 to 3.5). If the INR is below these ranges, warfarin is less effective, with a risk of new thrombosis. On the other hand, if the INR is too high, there is a risk of bleeding.14 In fact, the most important side effect of warfarin is the risk of major and minor bleeding.13 However, even in well-designed clinical trials in which patients are closely managed, only 55% to 60% of patients regularly achieve their therapeutic target INR.1,2,14,15
Warfarin also interacts with many drugs and with some foods. Compliance is difficult. It has a slow onset of action. Genetic variations require dose adjustments. When switching from a parenteral anticoagulant, overlapping is required. Skin necrosis is a possible side effect. And warfarin is teratogenic.
Despite these limitations, the American College of Chest Physicians endorses warfarin to prevent or treat venous thromboembolism, and to prevent stroke in patients with atrial fibrillation.16

DABIGATRAN, A THROMBIN INHIBITOR

A prodrug, dabigatran is rapidly absorbed and converted to its active form. Its plasma concentration reaches a peak 1.5 to 3 hours after an oral dose, and it has an elimination half-life of 12 to 14 hours. About 80% of its excretion is by the kidneys and the remaining 20% is through bile.
Dabigatran is not metabolized by cytochrome P450 isoenzymes, and therefore it has few major interactions with other drugs. An exception is rifampin, a P-glycoprotein inducer that blocks dabigatran’s absorption in the gut, so this combination should be avoided. Another is quinidine, a strong P-glycoprotein inhibitor that is contraindicated for use with dabigatran. Also, amiodarone (Cordarone), another P-glycoprotein inhibitor, increases blood levels of dabigatran, and therefore a lower dose of dabigatran is recommended if these drugs are given together.18–20
DOES DABIGATRAN NEED MONITORING? CAN IT EVEN BE MONITORED?
Dabigatran has a predictable pharmacodynamic effect, and current data indicate it does not need regular monitoring.18–20 However, one may need to be able to measure the drug’s activity in certain situations, such as suspected overdose, bleeding, need for emergency surgery, impaired renal function, pregnancy, and obesity, and in children.20
Dabigatran has little effect on the prothrombin time or the INR, even at therapeutic concentrations.19 Further, its effect on the activated partial thromboplastin time (aPTT) is neither linear nor dose-dependent, and the aPTT reaches a plateau and becomes less sensitive at very high concentrations. Therefore, the aPTT does not appear to be an appropriate test to monitor dabigatran’s therapeutic anticoagulant effect, although it does provide a qualitative indication of anticoagulant activity.18,19
The thrombin time is a very sensitive method for determining if dabigatran is present, but the test lacks standardization; the ecarin clotting time provides better evidence of the dose but is not readily available at most institutions.18,19,21
EVALUATED IN CLINICAL TRIALS
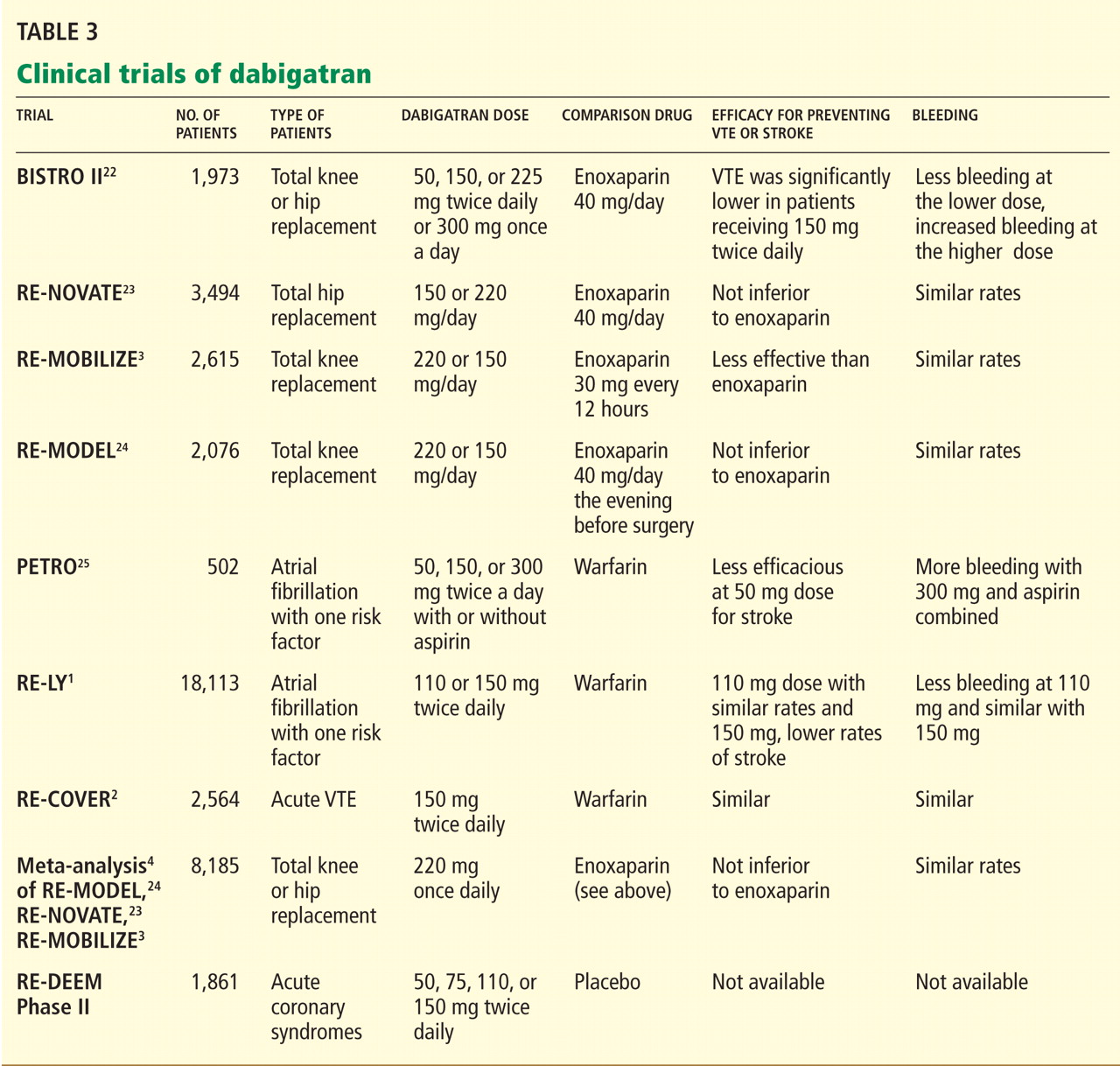
DABIGATRAN IS EXPENSIVE BUT MAY BE COST-EFFECTIVE
The estimated price of dabigatran 150 mg twice a day in the United States is about $6.75 to $8.00 per day.26,27
Warfarin, in contrast, costs as little as $50 per year.28 However, this low price does not include the cost of monitoring the INR (office visits and laboratory testing), and these combined expenses are much higher than the price of the warfarin itself.29 In addition, warfarin requires time-consuming management when bridging to a parenteral anticoagulant (for reversal of its anticoagulant action) before routine health maintenance procedures such as dental work and colonoscopy and interventional procedures and surgery. Any bleeding complication will also add to its cost and will be associated with a decrease in the patient’s perceived health and quality of life, but this is true for both drugs.30
In today’s health care environment, controlling costs is a universal priority, but it may be unfair to compare the cost of dabigatran with that of warfarin alone. The expense and morbidity associated with stroke and intracranial bleeding are high, and if patients on dabigatran have fewer strokes (as seen in the RE-LY trial with dabigatran 150 mg twice a day) and no added expense of monitoring, then dabigatran may be cost-effective.
Freeman et al31 analyzed the cost-effectiveness of dabigatran, using an estimated cost of $13.70 per day and data from the RE-LY trial. They concluded that dabigatran may be a cost-effective alternative to warfarin in preventing ischemic stroke in patients considered at higher risk for ischemic stroke or intracranial hemorrhage, ie, those with a CHADS2 score of 1 or higher or equivalent. (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack.)
As more new-generation oral anticoagulants become available (see below), the price of dabigatran will undoubtedly decrease. Until then, warfarin will remain a cost-effective and cost-saving drug that cannot yet be considered obsolete.
WHO SHOULD RECEIVE DABIGATRAN?
The ideal patient for dabigatran treatment is not yet defined. The decision to convert a patient’s treatment from warfarin to dabigatran will likely depend on several factors, including the patient’s response to warfarin and the physician’s comfort with this new drug.
Many patients do extremely well with warfarin, requiring infrequent monitoring to maintain a therapeutic INR and having no bleeding complications. For them, it would be more practical to continue warfarin. Another reason for staying with warfarin would be if twice-a-day dosing would pose a problem.
Dabigatran would be a reasonable choice for a patient whose INR is erratic, who requires more frequent monitoring, for whom cost is not an issue, and for whom there is concern about dietary or drug interactions.
Another consideration is whether the patient has access to a health care facility for warfarin monitoring: this is difficult for those who cannot drive, who depend on others for transportation, and who live in rural areas.
Additionally, dabigatran may be a cost-effective alternative to warfarin for a patient with a high CHADS2 score who is considered at a higher risk for stroke.31
In all cases, the option should be considered only after an open discussion with the patient about the risks and benefits of this new drug.
WHO SHOULD NOT RECEIVE IT?
Dabigatran is a twice-daily drug with a short half-life. No patient with a history of poor compliance will be a good candidate for dabigatran. Since there are no practical laboratory tests for monitoring compliance, one will have to reinforce at every visit the importance of taking this medication according to instructions.
Patients with underlying kidney disease will need close monitoring of their creatinine clearance, with dose adjustment if renal function deteriorates.
Additionally, one should use caution when prescribing dabigatran to obese patients, pregnant women, or children until more is known about its use in these populations.
ADVANTAGES AND DISADVANTAGES OF DABIGATRAN

A reason may be that patients with atrial fibrillation and poor INR control have higher rates of death, stroke, myocardial infarction, and major bleeding.14 In most clinical trials, only 55% to 60% of patients achieve a therapeutic INR on warfarin, leaving them at risk of thrombosis or, conversely, bleeding.1,2,15,32 Dabigatran has predictable pharmacokinetics, and its twice-daily dosing allows for less variability in its anticoagulant effect, making it more consistently therapeutic with less potential for bleeding or thrombosis.1
The Canadian Cardiovascular Society included dabigatran in its 2010 guidelines on atrial fibrillation, recommending it or warfarin.33 The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society now give dabigatran a class I B recommendation (benefit greater than risk, but limited populations studied) in secondary stroke prevention.34
On the other hand, major concerns are the lack of an antidote for dabigatran and a lack of experience in treating bleeding complications. Since dabigatran is not monitored, physicians may be uncertain if we are overdosing or undertreating. As we gain experience, we will learn how to treat bleeding complications. Until then, it will be important to anticipate this problem and to develop an algorithm based on the best available evidence in managing this complication.
Although the overall rates of bleeding in the RE-LY trial were lower with dabigatran than with warfarin, there were more gastrointestinal bleeding events with the 150-mg dose of dabigatran, which was not readily explained.
Further, the rate of dyspepsia was almost twice as high with dabigatran than with warfarin, regardless of the dose of dabigatran. There were also more dropouts in the 2nd year of follow-up in the dabigatran groups, with gastrointestinal intolerance being one of the major reasons. Therefore, dyspepsia may cause intolerance and noncompliance.1
Dabigatran must be taken twice a day and has a relatively short half-life. For a noncompliant patient, missing one or two doses will cause a reversal of its anticoagulation effect, leaving the patient susceptible to thrombosis. In comparison, warfarin has a longer half-life and is taken once a day, so missing a dose is less likely to result in a similar reversal of its anticoagulant effect.
SPECIAL CONDITIONS
Switching from other anticoagulants to dabigatran
When making the transition from a subcutaneously administered anticoagulant, ie, a low-molecular-weight heparin or the anti-Xa inhibitor fondaparinux (Arixtra), dabigatran should be started 0 to 2 hours before the next subcutaneous dose of the parenteral anticoagulant was to be given.21,35
When switching from unfractionated heparin given by continuous intravenous infusion, the first dose of dabigatran should be given at the time the infusion is stopped.
When switching from warfarin, dabigatran should be started once the patient’s INR is less than 2.0.
Switching from dabigatran to a parenteral anticoagulant
When switching from dabigatran back to a parenteral anticoagulant, allow 12 to 24 hours after the last dabigatran dose before starting the parenteral agent.21,35
Elective surgery or invasive procedures
The manufacturer recommends stopping dabigatran 1 to 2 days before elective surgery for patients who have normal renal function and a low risk of bleeding, or 3 to 5 days before surgery for patients who have a creatinine clearance of 50 mL/min or less. Before major surgery or placement of a spinal or epidural catheter, the manufacturer recommends that dabigatran be held even longer.35
If emergency surgery is needed
If emergency surgery is needed, the clinician must use his or her judgment as to the risks of bleeding vs those of postponing the surgery.21,35
Overdose or bleeding
No antidote for dabigatran is currently available. It has a short half-life (12–14 hours), and the treatment for overdose or bleeding is to discontinue it immediately, maintain adequate diuresis, and transfuse fresh-frozen plasma or red blood cells as indicated.
The role of activated charcoal given orally to reduce absorption is under evaluation, but the charcoal must be given within 1 to 2 hours after the overdose is taken.21
Dabigatran does not bind very much to plasma proteins and hence is dialyzable—an approach that may be necessary in cases of persistent or life-threatening bleeding.
Recombinant activated factor VII or prothrombin complex concentrates may be additional options in cases of severe bleeding.18,21
TOPICS OF FUTURE RESEARCH
A limitation of the dabigatran trials was that they did not enroll patients who had renal or liver impairment, cancer, or other comorbidities; pregnant women; or children. Other topics of future research include its use in patients weighing less than 48 kg or more than 110 kg, its efficacy in patients with thrombophilia, in patients with mechanical heart valves, and in long-term follow-up and the use of thrombolytics in patients with acute stroke who are on dabigatran.
WILL DABIGATRAN CHANGE CLINICAL PRACTICE?
Despite some of the challenges listed above, we believe that dabigatran is likely to change medical practice in patients requiring anticoagulation.
Dabigatran’s biggest use will most likely be in patients with atrial fibrillation, mainly because this is the largest group of people receiving anticoagulation. In addition, the incidence of atrial fibrillation rises with age, the US population is living longer, and patients generally require life-long anticoagulation once this condition develops.
Dabigatran may be approved for additional indications in the near future. It has already shown efficacy in primary and secondary prevention of venous thromboembolism. Other important areas to be studied include its use in patients with mechanical heart valves and thrombophilia.
Whether dabigatran will be a worthy substitute for the parenteral anticoagulants (heparin, low-molecular-weight heparins, or factor Xa inhibitors) is not yet known, but it will have an enormous impact on anticoagulation management if proved efficacious.
If dabigatran becomes a major substitute for warfarin, it will affect the anticoagulation clinics, with their well-trained staff, that are currently monitoring millions of patients in the United States. These clinics would no longer be needed, and laboratory and technical costs could be saved. A downside is that patients on dabigatran will not be as closely supervised and reminded to take their medication as patients on warfarin are now at these clinics. Instead, they will likely be supervised by their own physician (or assistants), who will need to become familiar with this anticoagulant. This may affect compliance with dabigatran.
OTHER NEW ORAL ANTICOAGULANTS ARE ON THE WAY
Other oral anticoagulants, including rivaroxaban (Xarelto) and apixaban (Eliquis), have been under study and show promise in preventing both thrombotic stroke and venous thromboembolism. They will likely compete with dabigatran once they are approved.
Rivaroxaban, an oral direct factor Xa inhibitor, is being investigated for stroke prevention in patients with atrial fibrillation. It has also been shown to be not inferior to (and to be less expensive than) enoxaparin in treating and preventing venous thromboembolism in patients undergoing hip or knee arthroplasty.32,36,37 Rivaroxaban has recently been approved by the FDA for this indication.
Apixaban, another direct factor Xa inhibitor, is also being studied for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. To date, there are no head-to-head trials comparing dabigatran with either of these new oral anticoagulants.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- RE-MOBILIZE Writing Committee; Ginsberg JS, Davidson BL, Comp PC, et al. Oral thrombin inhibitor dabigatran etexilate vs North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty 2009; 24:1–9.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 1995; 98:476–484.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 1991; 22:983–988.
- Go AS, Hylek EM, Chang Y, et al. Anticoagulation therapy for stroke prevention in atrial fibrillation: how well do randomized trials translate into clinical practice? JAMA 2003; 290:2685–2692.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol 1992; 19:246–247.
- Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008; 28:370–372.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med 2007; 167:1414–1419.
- White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med 2007; 167:239–245.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial Fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 suppl):381S–453S.
- Mungall D. BIBR-1048 Boehringer Ingelheim. Curr Opin Investig Drugs 2002; 3:905–907.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Eisert WG, Hauel N, Stangier J, Wienen W, Clemens A, van Ryn J. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol 2010; 30:1885–1889.
- Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against. J Thromb Haemost 2010; 8:627–630.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 2005; 3:103–111.
- Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100:1419–1426.
- Burger L. Bayer rival Boehringer prices blood pill at $6.75. Reuters, October 26, 2010. Available at http://www.reuters.com. Accessed September 12, 2011.
- Drugstore.com. Pradaxa. http://www.drugstore.com/pradaxa/bottle-60-150mg-capsules/qxn00597013554. Accessed September 10, 2011.
- Wal-Mart Stores, Inc. Retail Prescription Program Drug List. http://i.walmartimages.com/i/if/hmp/fusion/customer_list.pdf. Accessed September 10, 2011.
- Teachey DT. Dabigatran versus warfarin for venous thromboembolism (letter). N Engl J Med 2010; 362:1050; author reply1050–1051.
- Lancaster TR, Singer DE, Sheehan MA, et al. The impact of long-term warfarin therapy on quality of life. Evidence from a randomized trial. Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. Arch Intern Med 1991; 151:1944–1949.
- Freeman JV, Zhu RP, Owens DK, et al. Cost-effectiveness of dabigatran compared with warfarin for stroke prevention in atrial fibrillation. Ann Intern Med 2011; 154:1–11.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- Cairns JA, Connolly S, McMurtry S, Stephenson M, Talajic M; CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society atrial fibrillation guidelines 2010: prevention of stroke and systemic embolization in atrial fibrillation and flutter. Can J Cardiol 2011; 27:74–90.
- Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Boehringer Ingelheim. Pradaxa prescribing information. http://www.pradaxa.com. Accessed September 8, 2011.
- Huisman MV, Quinlan DJ, Dahl OE, Schulman S. Enoxaparin versus dabigatran or rivaroxaban for thromboprophylaxis after hip or knee arthroplasty: results of separate pooled analyses of phase III multicenter randomized trials. Circ Cardiovasc Qual Outcomes 2010; 3:652–660.
- McCullagh L, Tilson L, Walsh C, Barry M. A cost-effectiveness model comparing rivaroxaban and dabigatran etexilate with enoxaparin sodium as thromboprophylaxis after total hip and total knee replacement in the Irish healthcare setting. Pharmacoeconomics 2009; 27:829–846.
Dabigatran etexilate (Pradaxa) is a new oral anticoagulant that has distinct advantages over warfarin (Coumadin) in terms of its ease of administration, efficacy, and safety.
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY trial),1 in patients with nonvalvular atrial fibrillation, dabigatran 110 mg twice a day was found to be as good as warfarin in preventing systemic embolization and stroke (the primary outcome of the study), and at 150 mg twice a day it was superior.1 It has also shown efficacy in treating acute deep vein thrombosis and pulmonary embolism and in preventing these complications in orthopedic surgical patients.2–4
Dabigatran has been approved in 75 countries. It carries the trade name Pradaxa in Europe and the United States and Pradax in Canada. In October 2010, the US Food and Drug Administration (FDA) Cardiovascular and Renal Drugs Advisory Committee endorsed two twice-daily doses (75 mg and 150 mg) of dabigatran for the prevention of systemic embolization and stroke in patients with nonvalvular atrial fibrillation.
However, dabigatran is relatively expensive, and its current high cost might be a barrier to its wider use.
MANY PATIENTS NEED ANTICOAGULATION
Anticoagulation plays a vital role in the primary and secondary prevention of stroke in patients with atrial fibrillation and of pulmonary embolism in patients with venous thromboembolism. It is also used during cardiothoracic and vascular surgery, endovascular procedures, and dialysis and in patients with mechanical heart valves and hypercoagulable conditions.
Atrial fibrillation affects 3.03 million people in the United States (2005 figures), and this number is predicted to be as high as 7.56 million by 2050.5 More than 10% of people over the age of 80 years have it, and the lifetime risk of developing it is approximately 25%.6,7 Its most serious complication is ischemic stroke (the risk of which increases with age) and systemic embolization.5,8
Until the recent introduction of dabigatran, the only oral anticoagulant available in the United States for treating patients with atrial fibrillation was warfarin. Although warfarin has a number of disadvantages (see below), it is actually very effective for preventing ischemic stroke, reducing the incidence by as much as 65%.9,10
Venous thromboembolism is the third most common cardiovascular disorder after myocardial infarction and stroke.11 Although its exact incidence is unknown, nearly 1 million cases of it (incident or recurrent, fatal and nonfatal events) occur in the United States each year.12 Many patients with venous thromboembolism need oral anticoagulation long-term, and currently warfarin remains the only option for them as well.
NEEDED: A BETTER ANTICOAGULANT
Warfarin has been the most commonly prescribed oral anticoagulant in the United States for more than 60 years. As of 2004, more than 30 million outpatient prescriptions for it were filled annually in this country alone.13 However, warfarin has several important limitations.
Warfarin has a narrow therapeutic index. Patients taking it require monitoring of their international normalized ratio (INR) and frequent dose adjustments, and this is time-consuming and inconvenient. The target INR for patients with venous thromboembolism and atrial fibrillation is 2.0 to 3.0, whereas patients with a mechanical heart valve need a higher INR (2.5 to 3.5). If the INR is below these ranges, warfarin is less effective, with a risk of new thrombosis. On the other hand, if the INR is too high, there is a risk of bleeding.14 In fact, the most important side effect of warfarin is the risk of major and minor bleeding.13 However, even in well-designed clinical trials in which patients are closely managed, only 55% to 60% of patients regularly achieve their therapeutic target INR.1,2,14,15
Warfarin also interacts with many drugs and with some foods. Compliance is difficult. It has a slow onset of action. Genetic variations require dose adjustments. When switching from a parenteral anticoagulant, overlapping is required. Skin necrosis is a possible side effect. And warfarin is teratogenic.
Despite these limitations, the American College of Chest Physicians endorses warfarin to prevent or treat venous thromboembolism, and to prevent stroke in patients with atrial fibrillation.16
DABIGATRAN, A THROMBIN INHIBITOR
A prodrug, dabigatran is rapidly absorbed and converted to its active form. Its plasma concentration reaches a peak 1.5 to 3 hours after an oral dose, and it has an elimination half-life of 12 to 14 hours. About 80% of its excretion is by the kidneys and the remaining 20% is through bile.
Dabigatran is not metabolized by cytochrome P450 isoenzymes, and therefore it has few major interactions with other drugs. An exception is rifampin, a P-glycoprotein inducer that blocks dabigatran’s absorption in the gut, so this combination should be avoided. Another is quinidine, a strong P-glycoprotein inhibitor that is contraindicated for use with dabigatran. Also, amiodarone (Cordarone), another P-glycoprotein inhibitor, increases blood levels of dabigatran, and therefore a lower dose of dabigatran is recommended if these drugs are given together.18–20
DOES DABIGATRAN NEED MONITORING? CAN IT EVEN BE MONITORED?
Dabigatran has a predictable pharmacodynamic effect, and current data indicate it does not need regular monitoring.18–20 However, one may need to be able to measure the drug’s activity in certain situations, such as suspected overdose, bleeding, need for emergency surgery, impaired renal function, pregnancy, and obesity, and in children.20
Dabigatran has little effect on the prothrombin time or the INR, even at therapeutic concentrations.19 Further, its effect on the activated partial thromboplastin time (aPTT) is neither linear nor dose-dependent, and the aPTT reaches a plateau and becomes less sensitive at very high concentrations. Therefore, the aPTT does not appear to be an appropriate test to monitor dabigatran’s therapeutic anticoagulant effect, although it does provide a qualitative indication of anticoagulant activity.18,19
The thrombin time is a very sensitive method for determining if dabigatran is present, but the test lacks standardization; the ecarin clotting time provides better evidence of the dose but is not readily available at most institutions.18,19,21
EVALUATED IN CLINICAL TRIALS
DABIGATRAN IS EXPENSIVE BUT MAY BE COST-EFFECTIVE
The estimated price of dabigatran 150 mg twice a day in the United States is about $6.75 to $8.00 per day.26,27
Warfarin, in contrast, costs as little as $50 per year.28 However, this low price does not include the cost of monitoring the INR (office visits and laboratory testing), and these combined expenses are much higher than the price of the warfarin itself.29 In addition, warfarin requires time-consuming management when bridging to a parenteral anticoagulant (for reversal of its anticoagulant action) before routine health maintenance procedures such as dental work and colonoscopy and interventional procedures and surgery. Any bleeding complication will also add to its cost and will be associated with a decrease in the patient’s perceived health and quality of life, but this is true for both drugs.30
In today’s health care environment, controlling costs is a universal priority, but it may be unfair to compare the cost of dabigatran with that of warfarin alone. The expense and morbidity associated with stroke and intracranial bleeding are high, and if patients on dabigatran have fewer strokes (as seen in the RE-LY trial with dabigatran 150 mg twice a day) and no added expense of monitoring, then dabigatran may be cost-effective.
Freeman et al31 analyzed the cost-effectiveness of dabigatran, using an estimated cost of $13.70 per day and data from the RE-LY trial. They concluded that dabigatran may be a cost-effective alternative to warfarin in preventing ischemic stroke in patients considered at higher risk for ischemic stroke or intracranial hemorrhage, ie, those with a CHADS2 score of 1 or higher or equivalent. (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack.)
As more new-generation oral anticoagulants become available (see below), the price of dabigatran will undoubtedly decrease. Until then, warfarin will remain a cost-effective and cost-saving drug that cannot yet be considered obsolete.
WHO SHOULD RECEIVE DABIGATRAN?
The ideal patient for dabigatran treatment is not yet defined. The decision to convert a patient’s treatment from warfarin to dabigatran will likely depend on several factors, including the patient’s response to warfarin and the physician’s comfort with this new drug.
Many patients do extremely well with warfarin, requiring infrequent monitoring to maintain a therapeutic INR and having no bleeding complications. For them, it would be more practical to continue warfarin. Another reason for staying with warfarin would be if twice-a-day dosing would pose a problem.
Dabigatran would be a reasonable choice for a patient whose INR is erratic, who requires more frequent monitoring, for whom cost is not an issue, and for whom there is concern about dietary or drug interactions.
Another consideration is whether the patient has access to a health care facility for warfarin monitoring: this is difficult for those who cannot drive, who depend on others for transportation, and who live in rural areas.
Additionally, dabigatran may be a cost-effective alternative to warfarin for a patient with a high CHADS2 score who is considered at a higher risk for stroke.31
In all cases, the option should be considered only after an open discussion with the patient about the risks and benefits of this new drug.
WHO SHOULD NOT RECEIVE IT?
Dabigatran is a twice-daily drug with a short half-life. No patient with a history of poor compliance will be a good candidate for dabigatran. Since there are no practical laboratory tests for monitoring compliance, one will have to reinforce at every visit the importance of taking this medication according to instructions.
Patients with underlying kidney disease will need close monitoring of their creatinine clearance, with dose adjustment if renal function deteriorates.
Additionally, one should use caution when prescribing dabigatran to obese patients, pregnant women, or children until more is known about its use in these populations.
ADVANTAGES AND DISADVANTAGES OF DABIGATRAN
A reason may be that patients with atrial fibrillation and poor INR control have higher rates of death, stroke, myocardial infarction, and major bleeding.14 In most clinical trials, only 55% to 60% of patients achieve a therapeutic INR on warfarin, leaving them at risk of thrombosis or, conversely, bleeding.1,2,15,32 Dabigatran has predictable pharmacokinetics, and its twice-daily dosing allows for less variability in its anticoagulant effect, making it more consistently therapeutic with less potential for bleeding or thrombosis.1
The Canadian Cardiovascular Society included dabigatran in its 2010 guidelines on atrial fibrillation, recommending it or warfarin.33 The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society now give dabigatran a class I B recommendation (benefit greater than risk, but limited populations studied) in secondary stroke prevention.34
On the other hand, major concerns are the lack of an antidote for dabigatran and a lack of experience in treating bleeding complications. Since dabigatran is not monitored, physicians may be uncertain if we are overdosing or undertreating. As we gain experience, we will learn how to treat bleeding complications. Until then, it will be important to anticipate this problem and to develop an algorithm based on the best available evidence in managing this complication.
Although the overall rates of bleeding in the RE-LY trial were lower with dabigatran than with warfarin, there were more gastrointestinal bleeding events with the 150-mg dose of dabigatran, which was not readily explained.
Further, the rate of dyspepsia was almost twice as high with dabigatran than with warfarin, regardless of the dose of dabigatran. There were also more dropouts in the 2nd year of follow-up in the dabigatran groups, with gastrointestinal intolerance being one of the major reasons. Therefore, dyspepsia may cause intolerance and noncompliance.1
Dabigatran must be taken twice a day and has a relatively short half-life. For a noncompliant patient, missing one or two doses will cause a reversal of its anticoagulation effect, leaving the patient susceptible to thrombosis. In comparison, warfarin has a longer half-life and is taken once a day, so missing a dose is less likely to result in a similar reversal of its anticoagulant effect.
SPECIAL CONDITIONS
Switching from other anticoagulants to dabigatran
When making the transition from a subcutaneously administered anticoagulant, ie, a low-molecular-weight heparin or the anti-Xa inhibitor fondaparinux (Arixtra), dabigatran should be started 0 to 2 hours before the next subcutaneous dose of the parenteral anticoagulant was to be given.21,35
When switching from unfractionated heparin given by continuous intravenous infusion, the first dose of dabigatran should be given at the time the infusion is stopped.
When switching from warfarin, dabigatran should be started once the patient’s INR is less than 2.0.
Switching from dabigatran to a parenteral anticoagulant
When switching from dabigatran back to a parenteral anticoagulant, allow 12 to 24 hours after the last dabigatran dose before starting the parenteral agent.21,35
Elective surgery or invasive procedures
The manufacturer recommends stopping dabigatran 1 to 2 days before elective surgery for patients who have normal renal function and a low risk of bleeding, or 3 to 5 days before surgery for patients who have a creatinine clearance of 50 mL/min or less. Before major surgery or placement of a spinal or epidural catheter, the manufacturer recommends that dabigatran be held even longer.35
If emergency surgery is needed
If emergency surgery is needed, the clinician must use his or her judgment as to the risks of bleeding vs those of postponing the surgery.21,35
Overdose or bleeding
No antidote for dabigatran is currently available. It has a short half-life (12–14 hours), and the treatment for overdose or bleeding is to discontinue it immediately, maintain adequate diuresis, and transfuse fresh-frozen plasma or red blood cells as indicated.
The role of activated charcoal given orally to reduce absorption is under evaluation, but the charcoal must be given within 1 to 2 hours after the overdose is taken.21
Dabigatran does not bind very much to plasma proteins and hence is dialyzable—an approach that may be necessary in cases of persistent or life-threatening bleeding.
Recombinant activated factor VII or prothrombin complex concentrates may be additional options in cases of severe bleeding.18,21
TOPICS OF FUTURE RESEARCH
A limitation of the dabigatran trials was that they did not enroll patients who had renal or liver impairment, cancer, or other comorbidities; pregnant women; or children. Other topics of future research include its use in patients weighing less than 48 kg or more than 110 kg, its efficacy in patients with thrombophilia, in patients with mechanical heart valves, and in long-term follow-up and the use of thrombolytics in patients with acute stroke who are on dabigatran.
WILL DABIGATRAN CHANGE CLINICAL PRACTICE?
Despite some of the challenges listed above, we believe that dabigatran is likely to change medical practice in patients requiring anticoagulation.
Dabigatran’s biggest use will most likely be in patients with atrial fibrillation, mainly because this is the largest group of people receiving anticoagulation. In addition, the incidence of atrial fibrillation rises with age, the US population is living longer, and patients generally require life-long anticoagulation once this condition develops.
Dabigatran may be approved for additional indications in the near future. It has already shown efficacy in primary and secondary prevention of venous thromboembolism. Other important areas to be studied include its use in patients with mechanical heart valves and thrombophilia.
Whether dabigatran will be a worthy substitute for the parenteral anticoagulants (heparin, low-molecular-weight heparins, or factor Xa inhibitors) is not yet known, but it will have an enormous impact on anticoagulation management if proved efficacious.
If dabigatran becomes a major substitute for warfarin, it will affect the anticoagulation clinics, with their well-trained staff, that are currently monitoring millions of patients in the United States. These clinics would no longer be needed, and laboratory and technical costs could be saved. A downside is that patients on dabigatran will not be as closely supervised and reminded to take their medication as patients on warfarin are now at these clinics. Instead, they will likely be supervised by their own physician (or assistants), who will need to become familiar with this anticoagulant. This may affect compliance with dabigatran.
OTHER NEW ORAL ANTICOAGULANTS ARE ON THE WAY
Other oral anticoagulants, including rivaroxaban (Xarelto) and apixaban (Eliquis), have been under study and show promise in preventing both thrombotic stroke and venous thromboembolism. They will likely compete with dabigatran once they are approved.
Rivaroxaban, an oral direct factor Xa inhibitor, is being investigated for stroke prevention in patients with atrial fibrillation. It has also been shown to be not inferior to (and to be less expensive than) enoxaparin in treating and preventing venous thromboembolism in patients undergoing hip or knee arthroplasty.32,36,37 Rivaroxaban has recently been approved by the FDA for this indication.
Apixaban, another direct factor Xa inhibitor, is also being studied for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. To date, there are no head-to-head trials comparing dabigatran with either of these new oral anticoagulants.
Dabigatran etexilate (Pradaxa) is a new oral anticoagulant that has distinct advantages over warfarin (Coumadin) in terms of its ease of administration, efficacy, and safety.
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY trial),1 in patients with nonvalvular atrial fibrillation, dabigatran 110 mg twice a day was found to be as good as warfarin in preventing systemic embolization and stroke (the primary outcome of the study), and at 150 mg twice a day it was superior.1 It has also shown efficacy in treating acute deep vein thrombosis and pulmonary embolism and in preventing these complications in orthopedic surgical patients.2–4
Dabigatran has been approved in 75 countries. It carries the trade name Pradaxa in Europe and the United States and Pradax in Canada. In October 2010, the US Food and Drug Administration (FDA) Cardiovascular and Renal Drugs Advisory Committee endorsed two twice-daily doses (75 mg and 150 mg) of dabigatran for the prevention of systemic embolization and stroke in patients with nonvalvular atrial fibrillation.
However, dabigatran is relatively expensive, and its current high cost might be a barrier to its wider use.
MANY PATIENTS NEED ANTICOAGULATION
Anticoagulation plays a vital role in the primary and secondary prevention of stroke in patients with atrial fibrillation and of pulmonary embolism in patients with venous thromboembolism. It is also used during cardiothoracic and vascular surgery, endovascular procedures, and dialysis and in patients with mechanical heart valves and hypercoagulable conditions.
Atrial fibrillation affects 3.03 million people in the United States (2005 figures), and this number is predicted to be as high as 7.56 million by 2050.5 More than 10% of people over the age of 80 years have it, and the lifetime risk of developing it is approximately 25%.6,7 Its most serious complication is ischemic stroke (the risk of which increases with age) and systemic embolization.5,8
Until the recent introduction of dabigatran, the only oral anticoagulant available in the United States for treating patients with atrial fibrillation was warfarin. Although warfarin has a number of disadvantages (see below), it is actually very effective for preventing ischemic stroke, reducing the incidence by as much as 65%.9,10
Venous thromboembolism is the third most common cardiovascular disorder after myocardial infarction and stroke.11 Although its exact incidence is unknown, nearly 1 million cases of it (incident or recurrent, fatal and nonfatal events) occur in the United States each year.12 Many patients with venous thromboembolism need oral anticoagulation long-term, and currently warfarin remains the only option for them as well.
NEEDED: A BETTER ANTICOAGULANT
Warfarin has been the most commonly prescribed oral anticoagulant in the United States for more than 60 years. As of 2004, more than 30 million outpatient prescriptions for it were filled annually in this country alone.13 However, warfarin has several important limitations.
Warfarin has a narrow therapeutic index. Patients taking it require monitoring of their international normalized ratio (INR) and frequent dose adjustments, and this is time-consuming and inconvenient. The target INR for patients with venous thromboembolism and atrial fibrillation is 2.0 to 3.0, whereas patients with a mechanical heart valve need a higher INR (2.5 to 3.5). If the INR is below these ranges, warfarin is less effective, with a risk of new thrombosis. On the other hand, if the INR is too high, there is a risk of bleeding.14 In fact, the most important side effect of warfarin is the risk of major and minor bleeding.13 However, even in well-designed clinical trials in which patients are closely managed, only 55% to 60% of patients regularly achieve their therapeutic target INR.1,2,14,15
Warfarin also interacts with many drugs and with some foods. Compliance is difficult. It has a slow onset of action. Genetic variations require dose adjustments. When switching from a parenteral anticoagulant, overlapping is required. Skin necrosis is a possible side effect. And warfarin is teratogenic.
Despite these limitations, the American College of Chest Physicians endorses warfarin to prevent or treat venous thromboembolism, and to prevent stroke in patients with atrial fibrillation.16
DABIGATRAN, A THROMBIN INHIBITOR
A prodrug, dabigatran is rapidly absorbed and converted to its active form. Its plasma concentration reaches a peak 1.5 to 3 hours after an oral dose, and it has an elimination half-life of 12 to 14 hours. About 80% of its excretion is by the kidneys and the remaining 20% is through bile.
Dabigatran is not metabolized by cytochrome P450 isoenzymes, and therefore it has few major interactions with other drugs. An exception is rifampin, a P-glycoprotein inducer that blocks dabigatran’s absorption in the gut, so this combination should be avoided. Another is quinidine, a strong P-glycoprotein inhibitor that is contraindicated for use with dabigatran. Also, amiodarone (Cordarone), another P-glycoprotein inhibitor, increases blood levels of dabigatran, and therefore a lower dose of dabigatran is recommended if these drugs are given together.18–20
DOES DABIGATRAN NEED MONITORING? CAN IT EVEN BE MONITORED?
Dabigatran has a predictable pharmacodynamic effect, and current data indicate it does not need regular monitoring.18–20 However, one may need to be able to measure the drug’s activity in certain situations, such as suspected overdose, bleeding, need for emergency surgery, impaired renal function, pregnancy, and obesity, and in children.20
Dabigatran has little effect on the prothrombin time or the INR, even at therapeutic concentrations.19 Further, its effect on the activated partial thromboplastin time (aPTT) is neither linear nor dose-dependent, and the aPTT reaches a plateau and becomes less sensitive at very high concentrations. Therefore, the aPTT does not appear to be an appropriate test to monitor dabigatran’s therapeutic anticoagulant effect, although it does provide a qualitative indication of anticoagulant activity.18,19
The thrombin time is a very sensitive method for determining if dabigatran is present, but the test lacks standardization; the ecarin clotting time provides better evidence of the dose but is not readily available at most institutions.18,19,21
EVALUATED IN CLINICAL TRIALS
DABIGATRAN IS EXPENSIVE BUT MAY BE COST-EFFECTIVE
The estimated price of dabigatran 150 mg twice a day in the United States is about $6.75 to $8.00 per day.26,27
Warfarin, in contrast, costs as little as $50 per year.28 However, this low price does not include the cost of monitoring the INR (office visits and laboratory testing), and these combined expenses are much higher than the price of the warfarin itself.29 In addition, warfarin requires time-consuming management when bridging to a parenteral anticoagulant (for reversal of its anticoagulant action) before routine health maintenance procedures such as dental work and colonoscopy and interventional procedures and surgery. Any bleeding complication will also add to its cost and will be associated with a decrease in the patient’s perceived health and quality of life, but this is true for both drugs.30
In today’s health care environment, controlling costs is a universal priority, but it may be unfair to compare the cost of dabigatran with that of warfarin alone. The expense and morbidity associated with stroke and intracranial bleeding are high, and if patients on dabigatran have fewer strokes (as seen in the RE-LY trial with dabigatran 150 mg twice a day) and no added expense of monitoring, then dabigatran may be cost-effective.
Freeman et al31 analyzed the cost-effectiveness of dabigatran, using an estimated cost of $13.70 per day and data from the RE-LY trial. They concluded that dabigatran may be a cost-effective alternative to warfarin in preventing ischemic stroke in patients considered at higher risk for ischemic stroke or intracranial hemorrhage, ie, those with a CHADS2 score of 1 or higher or equivalent. (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack.)
As more new-generation oral anticoagulants become available (see below), the price of dabigatran will undoubtedly decrease. Until then, warfarin will remain a cost-effective and cost-saving drug that cannot yet be considered obsolete.
WHO SHOULD RECEIVE DABIGATRAN?
The ideal patient for dabigatran treatment is not yet defined. The decision to convert a patient’s treatment from warfarin to dabigatran will likely depend on several factors, including the patient’s response to warfarin and the physician’s comfort with this new drug.
Many patients do extremely well with warfarin, requiring infrequent monitoring to maintain a therapeutic INR and having no bleeding complications. For them, it would be more practical to continue warfarin. Another reason for staying with warfarin would be if twice-a-day dosing would pose a problem.
Dabigatran would be a reasonable choice for a patient whose INR is erratic, who requires more frequent monitoring, for whom cost is not an issue, and for whom there is concern about dietary or drug interactions.
Another consideration is whether the patient has access to a health care facility for warfarin monitoring: this is difficult for those who cannot drive, who depend on others for transportation, and who live in rural areas.
Additionally, dabigatran may be a cost-effective alternative to warfarin for a patient with a high CHADS2 score who is considered at a higher risk for stroke.31
In all cases, the option should be considered only after an open discussion with the patient about the risks and benefits of this new drug.
WHO SHOULD NOT RECEIVE IT?
Dabigatran is a twice-daily drug with a short half-life. No patient with a history of poor compliance will be a good candidate for dabigatran. Since there are no practical laboratory tests for monitoring compliance, one will have to reinforce at every visit the importance of taking this medication according to instructions.
Patients with underlying kidney disease will need close monitoring of their creatinine clearance, with dose adjustment if renal function deteriorates.
Additionally, one should use caution when prescribing dabigatran to obese patients, pregnant women, or children until more is known about its use in these populations.
ADVANTAGES AND DISADVANTAGES OF DABIGATRAN
A reason may be that patients with atrial fibrillation and poor INR control have higher rates of death, stroke, myocardial infarction, and major bleeding.14 In most clinical trials, only 55% to 60% of patients achieve a therapeutic INR on warfarin, leaving them at risk of thrombosis or, conversely, bleeding.1,2,15,32 Dabigatran has predictable pharmacokinetics, and its twice-daily dosing allows for less variability in its anticoagulant effect, making it more consistently therapeutic with less potential for bleeding or thrombosis.1
The Canadian Cardiovascular Society included dabigatran in its 2010 guidelines on atrial fibrillation, recommending it or warfarin.33 The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society now give dabigatran a class I B recommendation (benefit greater than risk, but limited populations studied) in secondary stroke prevention.34
On the other hand, major concerns are the lack of an antidote for dabigatran and a lack of experience in treating bleeding complications. Since dabigatran is not monitored, physicians may be uncertain if we are overdosing or undertreating. As we gain experience, we will learn how to treat bleeding complications. Until then, it will be important to anticipate this problem and to develop an algorithm based on the best available evidence in managing this complication.
Although the overall rates of bleeding in the RE-LY trial were lower with dabigatran than with warfarin, there were more gastrointestinal bleeding events with the 150-mg dose of dabigatran, which was not readily explained.
Further, the rate of dyspepsia was almost twice as high with dabigatran than with warfarin, regardless of the dose of dabigatran. There were also more dropouts in the 2nd year of follow-up in the dabigatran groups, with gastrointestinal intolerance being one of the major reasons. Therefore, dyspepsia may cause intolerance and noncompliance.1
Dabigatran must be taken twice a day and has a relatively short half-life. For a noncompliant patient, missing one or two doses will cause a reversal of its anticoagulation effect, leaving the patient susceptible to thrombosis. In comparison, warfarin has a longer half-life and is taken once a day, so missing a dose is less likely to result in a similar reversal of its anticoagulant effect.
SPECIAL CONDITIONS
Switching from other anticoagulants to dabigatran
When making the transition from a subcutaneously administered anticoagulant, ie, a low-molecular-weight heparin or the anti-Xa inhibitor fondaparinux (Arixtra), dabigatran should be started 0 to 2 hours before the next subcutaneous dose of the parenteral anticoagulant was to be given.21,35
When switching from unfractionated heparin given by continuous intravenous infusion, the first dose of dabigatran should be given at the time the infusion is stopped.
When switching from warfarin, dabigatran should be started once the patient’s INR is less than 2.0.
Switching from dabigatran to a parenteral anticoagulant
When switching from dabigatran back to a parenteral anticoagulant, allow 12 to 24 hours after the last dabigatran dose before starting the parenteral agent.21,35
Elective surgery or invasive procedures
The manufacturer recommends stopping dabigatran 1 to 2 days before elective surgery for patients who have normal renal function and a low risk of bleeding, or 3 to 5 days before surgery for patients who have a creatinine clearance of 50 mL/min or less. Before major surgery or placement of a spinal or epidural catheter, the manufacturer recommends that dabigatran be held even longer.35
If emergency surgery is needed
If emergency surgery is needed, the clinician must use his or her judgment as to the risks of bleeding vs those of postponing the surgery.21,35
Overdose or bleeding
No antidote for dabigatran is currently available. It has a short half-life (12–14 hours), and the treatment for overdose or bleeding is to discontinue it immediately, maintain adequate diuresis, and transfuse fresh-frozen plasma or red blood cells as indicated.
The role of activated charcoal given orally to reduce absorption is under evaluation, but the charcoal must be given within 1 to 2 hours after the overdose is taken.21
Dabigatran does not bind very much to plasma proteins and hence is dialyzable—an approach that may be necessary in cases of persistent or life-threatening bleeding.
Recombinant activated factor VII or prothrombin complex concentrates may be additional options in cases of severe bleeding.18,21
TOPICS OF FUTURE RESEARCH
A limitation of the dabigatran trials was that they did not enroll patients who had renal or liver impairment, cancer, or other comorbidities; pregnant women; or children. Other topics of future research include its use in patients weighing less than 48 kg or more than 110 kg, its efficacy in patients with thrombophilia, in patients with mechanical heart valves, and in long-term follow-up and the use of thrombolytics in patients with acute stroke who are on dabigatran.
WILL DABIGATRAN CHANGE CLINICAL PRACTICE?
Despite some of the challenges listed above, we believe that dabigatran is likely to change medical practice in patients requiring anticoagulation.
Dabigatran’s biggest use will most likely be in patients with atrial fibrillation, mainly because this is the largest group of people receiving anticoagulation. In addition, the incidence of atrial fibrillation rises with age, the US population is living longer, and patients generally require life-long anticoagulation once this condition develops.
Dabigatran may be approved for additional indications in the near future. It has already shown efficacy in primary and secondary prevention of venous thromboembolism. Other important areas to be studied include its use in patients with mechanical heart valves and thrombophilia.
Whether dabigatran will be a worthy substitute for the parenteral anticoagulants (heparin, low-molecular-weight heparins, or factor Xa inhibitors) is not yet known, but it will have an enormous impact on anticoagulation management if proved efficacious.
If dabigatran becomes a major substitute for warfarin, it will affect the anticoagulation clinics, with their well-trained staff, that are currently monitoring millions of patients in the United States. These clinics would no longer be needed, and laboratory and technical costs could be saved. A downside is that patients on dabigatran will not be as closely supervised and reminded to take their medication as patients on warfarin are now at these clinics. Instead, they will likely be supervised by their own physician (or assistants), who will need to become familiar with this anticoagulant. This may affect compliance with dabigatran.
OTHER NEW ORAL ANTICOAGULANTS ARE ON THE WAY
Other oral anticoagulants, including rivaroxaban (Xarelto) and apixaban (Eliquis), have been under study and show promise in preventing both thrombotic stroke and venous thromboembolism. They will likely compete with dabigatran once they are approved.
Rivaroxaban, an oral direct factor Xa inhibitor, is being investigated for stroke prevention in patients with atrial fibrillation. It has also been shown to be not inferior to (and to be less expensive than) enoxaparin in treating and preventing venous thromboembolism in patients undergoing hip or knee arthroplasty.32,36,37 Rivaroxaban has recently been approved by the FDA for this indication.
Apixaban, another direct factor Xa inhibitor, is also being studied for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. To date, there are no head-to-head trials comparing dabigatran with either of these new oral anticoagulants.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- RE-MOBILIZE Writing Committee; Ginsberg JS, Davidson BL, Comp PC, et al. Oral thrombin inhibitor dabigatran etexilate vs North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty 2009; 24:1–9.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 1995; 98:476–484.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 1991; 22:983–988.
- Go AS, Hylek EM, Chang Y, et al. Anticoagulation therapy for stroke prevention in atrial fibrillation: how well do randomized trials translate into clinical practice? JAMA 2003; 290:2685–2692.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol 1992; 19:246–247.
- Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008; 28:370–372.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med 2007; 167:1414–1419.
- White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med 2007; 167:239–245.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial Fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 suppl):381S–453S.
- Mungall D. BIBR-1048 Boehringer Ingelheim. Curr Opin Investig Drugs 2002; 3:905–907.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Eisert WG, Hauel N, Stangier J, Wienen W, Clemens A, van Ryn J. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol 2010; 30:1885–1889.
- Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against. J Thromb Haemost 2010; 8:627–630.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 2005; 3:103–111.
- Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100:1419–1426.
- Burger L. Bayer rival Boehringer prices blood pill at $6.75. Reuters, October 26, 2010. Available at http://www.reuters.com. Accessed September 12, 2011.
- Drugstore.com. Pradaxa. http://www.drugstore.com/pradaxa/bottle-60-150mg-capsules/qxn00597013554. Accessed September 10, 2011.
- Wal-Mart Stores, Inc. Retail Prescription Program Drug List. http://i.walmartimages.com/i/if/hmp/fusion/customer_list.pdf. Accessed September 10, 2011.
- Teachey DT. Dabigatran versus warfarin for venous thromboembolism (letter). N Engl J Med 2010; 362:1050; author reply1050–1051.
- Lancaster TR, Singer DE, Sheehan MA, et al. The impact of long-term warfarin therapy on quality of life. Evidence from a randomized trial. Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. Arch Intern Med 1991; 151:1944–1949.
- Freeman JV, Zhu RP, Owens DK, et al. Cost-effectiveness of dabigatran compared with warfarin for stroke prevention in atrial fibrillation. Ann Intern Med 2011; 154:1–11.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- Cairns JA, Connolly S, McMurtry S, Stephenson M, Talajic M; CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society atrial fibrillation guidelines 2010: prevention of stroke and systemic embolization in atrial fibrillation and flutter. Can J Cardiol 2011; 27:74–90.
- Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Boehringer Ingelheim. Pradaxa prescribing information. http://www.pradaxa.com. Accessed September 8, 2011.
- Huisman MV, Quinlan DJ, Dahl OE, Schulman S. Enoxaparin versus dabigatran or rivaroxaban for thromboprophylaxis after hip or knee arthroplasty: results of separate pooled analyses of phase III multicenter randomized trials. Circ Cardiovasc Qual Outcomes 2010; 3:652–660.
- McCullagh L, Tilson L, Walsh C, Barry M. A cost-effectiveness model comparing rivaroxaban and dabigatran etexilate with enoxaparin sodium as thromboprophylaxis after total hip and total knee replacement in the Irish healthcare setting. Pharmacoeconomics 2009; 27:829–846.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- RE-MOBILIZE Writing Committee; Ginsberg JS, Davidson BL, Comp PC, et al. Oral thrombin inhibitor dabigatran etexilate vs North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty 2009; 24:1–9.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 1995; 98:476–484.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 1991; 22:983–988.
- Go AS, Hylek EM, Chang Y, et al. Anticoagulation therapy for stroke prevention in atrial fibrillation: how well do randomized trials translate into clinical practice? JAMA 2003; 290:2685–2692.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol 1992; 19:246–247.
- Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008; 28:370–372.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med 2007; 167:1414–1419.
- White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med 2007; 167:239–245.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial Fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 suppl):381S–453S.
- Mungall D. BIBR-1048 Boehringer Ingelheim. Curr Opin Investig Drugs 2002; 3:905–907.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Eisert WG, Hauel N, Stangier J, Wienen W, Clemens A, van Ryn J. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol 2010; 30:1885–1889.
- Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against. J Thromb Haemost 2010; 8:627–630.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 2005; 3:103–111.
- Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100:1419–1426.
- Burger L. Bayer rival Boehringer prices blood pill at $6.75. Reuters, October 26, 2010. Available at http://www.reuters.com. Accessed September 12, 2011.
- Drugstore.com. Pradaxa. http://www.drugstore.com/pradaxa/bottle-60-150mg-capsules/qxn00597013554. Accessed September 10, 2011.
- Wal-Mart Stores, Inc. Retail Prescription Program Drug List. http://i.walmartimages.com/i/if/hmp/fusion/customer_list.pdf. Accessed September 10, 2011.
- Teachey DT. Dabigatran versus warfarin for venous thromboembolism (letter). N Engl J Med 2010; 362:1050; author reply1050–1051.
- Lancaster TR, Singer DE, Sheehan MA, et al. The impact of long-term warfarin therapy on quality of life. Evidence from a randomized trial. Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. Arch Intern Med 1991; 151:1944–1949.
- Freeman JV, Zhu RP, Owens DK, et al. Cost-effectiveness of dabigatran compared with warfarin for stroke prevention in atrial fibrillation. Ann Intern Med 2011; 154:1–11.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- Cairns JA, Connolly S, McMurtry S, Stephenson M, Talajic M; CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society atrial fibrillation guidelines 2010: prevention of stroke and systemic embolization in atrial fibrillation and flutter. Can J Cardiol 2011; 27:74–90.
- Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Boehringer Ingelheim. Pradaxa prescribing information. http://www.pradaxa.com. Accessed September 8, 2011.
- Huisman MV, Quinlan DJ, Dahl OE, Schulman S. Enoxaparin versus dabigatran or rivaroxaban for thromboprophylaxis after hip or knee arthroplasty: results of separate pooled analyses of phase III multicenter randomized trials. Circ Cardiovasc Qual Outcomes 2010; 3:652–660.
- McCullagh L, Tilson L, Walsh C, Barry M. A cost-effectiveness model comparing rivaroxaban and dabigatran etexilate with enoxaparin sodium as thromboprophylaxis after total hip and total knee replacement in the Irish healthcare setting. Pharmacoeconomics 2009; 27:829–846.
KEY POINTS
- Dabigatran is a potent, reversible, direct thrombin inhibitor. Available only in oral form, it has a rapid onset of action, a predictable anticoagulant response, and few major interactions.
- Dabigatran does not require dose adjustments (except for renal insufficiency) or monitoring of its effect during treatment.
- In trials in patients with nonvalvular atrial fibrillation, two different doses of dabigatran were compared with warfarin. Less bleeding occurred with the lower dose than with warfarin, while the higher dose was more effective than warfarin in preventing stroke and systemic embolization.
- The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society have given dabigatran a class I B recommendation for secondary stroke prevention in patients with nonvalvular atrial fibrillation.
A discussion of dissection

Dr. Alan C. Braverman, in this issue of the Journal, discusses thoracic aortic dissection. To most of us who do not routinely treat aortic disease, it may not seem that much has changed since that Thanksgiving in Philadelphia. Atherosclerosis is still a common risk, surgery is the treatment for ascending dissection, beta-blockers are useful for chronic descending dissections, and the mortality rate is enormously high when dissections bleed.
As internists, we consider the possibility of genetic disorders in patients with a family history of dissection or aneurysm, but we don’t really expect to find many, and most of us don’t often track advances in the understanding of these disorders at the molecular level. At the time I was working in that emergency room, Marfan syndrome was viewed as a connective tissue disorder, with a structurally weak aortic wall and variable other morphologic features. When the molecular defect was defined as fibrillin-1 deficiency, I didn’t think much more than that the weak link of the aorta’s fibrous belt was identified.
But it turns out that fibrillin is not just an aortic girdle; fibrillin lowers the concentration of the cytokine transforming growth factor (TGF)-beta in the aorta (and other organs) by promoting its sequestration in the extracellular matrix. Absence of fibrillin enhances TGF-beta activity, and excess TGF-beta can produce Marfan syndrome in young mice. In maybe the most striking consequence of this line of research, Dietz and colleagues1 have demonstrated that the specific antagonism of the angiotensin II type 1 receptor by the drug losartan (Cozaar) also blocks the effects of TGF-beta and consequently blocks the development of murine Marfan syndrome. And in a preliminary study, it slowed aneurysm progression in a small group of children with Marfan syndrome.
This does not imply that the same pathophysiology is at play in all aortic aneurysms. But at a time of new guidelines for screening for abdominal aneurysm, these observations offer a novel paradigm for developing drug therapies as an alternative to the mad rush for the vascular operating suite.
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358:2787–2795.
Dr. Alan C. Braverman, in this issue of the Journal, discusses thoracic aortic dissection. To most of us who do not routinely treat aortic disease, it may not seem that much has changed since that Thanksgiving in Philadelphia. Atherosclerosis is still a common risk, surgery is the treatment for ascending dissection, beta-blockers are useful for chronic descending dissections, and the mortality rate is enormously high when dissections bleed.
As internists, we consider the possibility of genetic disorders in patients with a family history of dissection or aneurysm, but we don’t really expect to find many, and most of us don’t often track advances in the understanding of these disorders at the molecular level. At the time I was working in that emergency room, Marfan syndrome was viewed as a connective tissue disorder, with a structurally weak aortic wall and variable other morphologic features. When the molecular defect was defined as fibrillin-1 deficiency, I didn’t think much more than that the weak link of the aorta’s fibrous belt was identified.
But it turns out that fibrillin is not just an aortic girdle; fibrillin lowers the concentration of the cytokine transforming growth factor (TGF)-beta in the aorta (and other organs) by promoting its sequestration in the extracellular matrix. Absence of fibrillin enhances TGF-beta activity, and excess TGF-beta can produce Marfan syndrome in young mice. In maybe the most striking consequence of this line of research, Dietz and colleagues1 have demonstrated that the specific antagonism of the angiotensin II type 1 receptor by the drug losartan (Cozaar) also blocks the effects of TGF-beta and consequently blocks the development of murine Marfan syndrome. And in a preliminary study, it slowed aneurysm progression in a small group of children with Marfan syndrome.
This does not imply that the same pathophysiology is at play in all aortic aneurysms. But at a time of new guidelines for screening for abdominal aneurysm, these observations offer a novel paradigm for developing drug therapies as an alternative to the mad rush for the vascular operating suite.
Dr. Alan C. Braverman, in this issue of the Journal, discusses thoracic aortic dissection. To most of us who do not routinely treat aortic disease, it may not seem that much has changed since that Thanksgiving in Philadelphia. Atherosclerosis is still a common risk, surgery is the treatment for ascending dissection, beta-blockers are useful for chronic descending dissections, and the mortality rate is enormously high when dissections bleed.
As internists, we consider the possibility of genetic disorders in patients with a family history of dissection or aneurysm, but we don’t really expect to find many, and most of us don’t often track advances in the understanding of these disorders at the molecular level. At the time I was working in that emergency room, Marfan syndrome was viewed as a connective tissue disorder, with a structurally weak aortic wall and variable other morphologic features. When the molecular defect was defined as fibrillin-1 deficiency, I didn’t think much more than that the weak link of the aorta’s fibrous belt was identified.
But it turns out that fibrillin is not just an aortic girdle; fibrillin lowers the concentration of the cytokine transforming growth factor (TGF)-beta in the aorta (and other organs) by promoting its sequestration in the extracellular matrix. Absence of fibrillin enhances TGF-beta activity, and excess TGF-beta can produce Marfan syndrome in young mice. In maybe the most striking consequence of this line of research, Dietz and colleagues1 have demonstrated that the specific antagonism of the angiotensin II type 1 receptor by the drug losartan (Cozaar) also blocks the effects of TGF-beta and consequently blocks the development of murine Marfan syndrome. And in a preliminary study, it slowed aneurysm progression in a small group of children with Marfan syndrome.
This does not imply that the same pathophysiology is at play in all aortic aneurysms. But at a time of new guidelines for screening for abdominal aneurysm, these observations offer a novel paradigm for developing drug therapies as an alternative to the mad rush for the vascular operating suite.
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358:2787–2795.
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358:2787–2795.
Aortic dissection: Prompt diagnosis and emergency treatment are critical
A 50-year-old man developed severe chest pain and collapsed to the floor. The pain was sudden in onset, was burning in quality, and was located in the center of his chest. Emergency medical services arrived a few minutes later and found the patient diaphoretic and cyanotic, with an initial blood pressure of 74/54 mm Hg and a heart rate of 125 beats per minute. He was rushed to the hospital.
His medical history was unremarkable. He smoked one pack of cigarettes per day for 20 years. His father died of a “heart attack” at age 52.
In the emergency department he underwent echocardiography with a portable handheld unit, which showed a pericardial effusion and cardiac tamponade. He was sent for emergency computed tomography of the chest, which revealed an aneurysm of the aortic root and acute type A (Stanford classification) aortic dissection with hemopericardium.
He underwent emergency cardiac surgery. At the time of surgery, he was in cardiogenic shock from aortic dissection complicated by severe aortic regurgitation and cardiac tamponade with hemopericardium. The aortic valve was trileaflet. A 27-mm St. Jude composite valve graft root replacement was performed.
The patient did well and was discharged home 7 days after surgery. Pathologic study of the aorta revealed cystic medial degeneration. He did not have any features of Marfan syndrome or Loeys-Dietz syndrome. His three children underwent evaluation, and each had a normal physical examination and echocardiographic test results.
A HIGH INDEX OF SUSPICION IS CRITICAL
Acute aortic dissection is the most common aortic catastrophe, with an incidence estimated at 5 to 30 per 1 million people per year, amounting to nearly 10,000 cases per year in the United States.1–4
The diagnosis of acute aortic dissection has many potential pitfalls.2,3 Aortic dissection may mimic other more common conditions, such as coronary ischemia, pleurisy, heart failure, stroke, and acute abdominal illness. Because acute aortic dissection may be rapidly fatal, one must maintain a high index of suspicion.2,3 Prompt diagnosis and emergency treatment are critical.
WHAT CAUSES AORTIC DISSECTION?
One hypothesis is that acute aortic dissection is caused by a primary tear in the aortic intima, with blood from the aortic lumen penetrating into the diseased media leading to dissection and creating a true and false lumen.2 Another is that rupture of the vasa vasorum leads to hemorrhage in the aortic wall with subsequent intimal disruption, creating the intimal tear and aortic dissection.
Once a dissection starts, pulsatile flow of blood within the aortic wall causes it to extend. The dissection flap may be localized, but it often spirals the entire length of the aorta. Distention of the false lumen with blood may cause the intimal flap to compress the true lumen and potentially lead to malperfusion syndromes.
CLASSIFIED ACCORDING TO LOCATION
It is important to recognize the location of the dissection, as the prognosis and treatment depend on whether the ascending aorta is involved.2,3 For classification purposes, the ascending aorta is the portion proximal to the brachiocephalic artery, while the descending aorta is the portion distal to the left subclavian artery.3
The DeBakey classification defines a type I aortic dissection as one that begins in the ascending aorta and extends at least to the aortic arch or beyond. Type II dissections involve the ascending aorta only, while type III dissections begin in the descending aorta, most often just distal to the left subclavian artery.
The Stanford classification scheme divides dissections into type A and type B. Type A dissections involve the ascending aorta, while type B dissections do not involve the ascending aorta.
Which classification scheme is used is not important. However, identifying patients with dissection of the ascending aorta (DeBakey type I or type II or Stanford type A) is critical, as emergency cardiac surgery is recommended for this type of dissection.2,3 For the purposes of this paper, the Stanford classification scheme will be used.
Dissection that involves the ascending aorta most commonly occurs in people ages 50 to 60, whereas acute dissection of the descending aorta typically occurs in people 10 years older.1,2
An acute aortic dissection is one that has occurred within 2 weeks of symptom onset. A chronic dissection is one that occurred more than 2 weeks after symptoms began.
DISEASES AND CONDITIONS ASSOCIATED WITH AORTIC DISSECTION

Hypertension and disorders leading to disruption of the normal structure and function of the aortic wall. About 75% of patients with acute aortic dissection have underlying hypertension.1–3
Cystic medial degeneration is a common pathologic feature in many cases of aortic dissection.

Cocaine use and intense weight-lifting increase the shear stresses on the aorta.2,3
Inflammatory aortic diseases such as giant cell arteritis.
Pregnancy can be complicated by aortic dissection, usually in the setting of an underlying aortopathy.5
Iatrogenic aortic dissection accounts for about 4% of cases, as a result of cardiac surgery, catheterization, stenting, or use of an intra-aortic balloon pump.1
Aortic aneurysm. Patients with thoracic aortic aneurysm are at higher risk of aortic dissection, and the larger the aortic diameter, the higher the risk.2,3,6 In the International Registry of Acute Aortic Dissection (IRAD), the average size of the aorta was about 5.3 cm at the time of acute dissection. Importantly, about 40% of acute dissections of the ascending aorta occur in patients with ascending aortic diameters less than 5.0 cm.7,8
Thus, many factors are associated with acute dissection, and specific reasons leading to an individual’s susceptibility to sudden dissection are poorly understood.
CLINICAL FEATURES OF ACUTE AORTIC DISSECTION
Because the symptoms of acute dissection may mimic other, more common conditions, one of the most important factors in the diagnosis of aortic dissection is a high clinical suspicion.1–3
What is the pretest risk of disease?
Recently, the American College of Cardiology (ACC) and the American Heart Association (AHA) released joint guidelines on thoracic aortic disease.3 These guidelines provide an approach to patients who have complaints that may represent acute thoracic aortic dissection, the intent being to establish a pretest risk of disease to be used to guide decision-making.3
The focused evaluation includes specific questions about underlying conditions, symptoms, and findings on examination that may greatly increase the likelihood of acute dissection. These include:
- High-risk conditions and historical features associated with aortic dissection, such as Marfan syndrome and other genetic disorders (Table 2), bicuspid aortic valve, family history of thoracic aortic aneurysm or dissection, known thoracic aortic aneurysm, and recent aortic manipulation
- Pain in the chest, back, or abdomen with high-risk features (eg, abrupt onset, severe intensity, or a ripping or tearing quality)
- High-risk findings on examination (eg, pulse deficits, new aortic regurgitation, hypotension, shock, or systolic blood pressure differences).
Using this information, expedited aortic imaging and treatment algorithms have been devised to improve the diagnosis.3
Using the IRAD database of more than 2,500 acute dissections, the diagnostic algorithm proposed in the ACC/AHA guidelines was shown to be highly sensitive (about 95%) for detecting acute aortic dissection.4 In addition, using this score may expedite evaluation by classifying certain patients as being at high risk of acute dissection.3,4
Important to recognize is that almost two-thirds of patients who suffered dissection in this large database did not have one of the “high-risk conditions” associated with dissection.4 Additionally, the specificity of the ACC/AHA algorithm is unknown, and further testing is necessary.4
Acute onset of severe pain
More than 90% of acute dissections present with acute pain in the chest or the back, or both.1–3 The pain is usually severe, of sudden onset, and often described as sharp or, occasionally, tearing, ripping, or stabbing. The pain usually differs from that of coronary ischemia, being most severe at its onset as opposed to the less intense, crescendo-like pain of angina or myocardial infarction. The pain may migrate as the dissection progresses along the length of the aorta or to branch vessels. It may abate, leading to a false sense of security in the patient and the physician.3 “Painless” dissection occurs in a minority, usually in those with syncope, neurologic symptoms, or heart failure.1–3
The patient with acute dissection may be anxious and may feel a sense of doom.
Acute heart failure, related to severe aortic regurgitation, may be a predominant symptom in dissection of the ascending aorta.
Syncope may occur as a result of aortic rupture, hemopericardium with cardiac tamponade, or acute neurologic complications.
Vascular insufficiency may occur in any branch vessel, leading to clinical syndromes that include acute myocardial infarction, stroke, paraplegia, paraparesis, mesenteric ischemia, and limb ischemia.
PHYSICAL FINDINGS CAN VARY WIDELY
Findings on physical examination in acute aortic dissection vary widely depending on underlying conditions and on the specific complications of the dissection.
Although the classic presentation is acute, severe pain in the chest or back in a severely hypertensive patient with aortic regurgitation and pulse deficits, most patients do not have all these characteristics.4 Most patients with type B dissection are hypertensive on presentation, but many with type A dissection present with normal blood pressure or hypotension.1 Pulse deficits (unequal or absent pulses) are reported in 10% to 30% of acute dissections and may be intermittent as the dynamic movement of the dissection flap interferes with branch vessel perfusion.1–3
- Aortic leaflet prolapse or distortion of the leaflet alignment
- Malcoaptation of the aortic leaflets from dilation of the aortic root and annulus
- Prolapse of the intimal flap across the aortic valve, interfering with valve function
- Preexisting aortic regurgitation from underlying aortic root aneurysm or primary aortic valve disease (such as a bicuspid aortic valve).
Neurologic manifestations are most common in dissection of the ascending aorta and are particularly important to recognize, as they may dominate the clinical presentation and lead to delay in the diagnosis of dissection.2,3 Neurologic syndromes include:
- Persistent or transient ischemic stroke
- Spinal cord ischemia
- Ischemic neuropathy
- Hypoxic encephalopathy.
These manifestations are related to malperfusion to branches supplying the brain, spinal cord, or peripheral nerves.9
Syncope is relatively common in aortic dissection and may be related to acute hypotension caused by cardiac tamponade or aortic rupture, cerebral vessel obstruction, or activation of cerebral baroreceptors.2,9 It is important to consider aortic dissection in the differential diagnosis in cases of unexplained syncope.3
Aortic dissections may extend into the abdominal aorta, leading to vascular complications involving one or more branch vessels.10 The renal artery is involved in at least 5% to 10% of cases and may lead to renal ischemia, infarction, renal insufficiency, or refractory hypertension.2Mesenteric ischemia or infarction occurs in about 5% of dissections, may be difficult to diagnose, and is particularly dangerous.2,8 Aortic dissection may extend into the iliac arteries and may cause acute lower extremity ischemia.
Acute myocardial infarction due to involvement of the dissection flap causing malperfusion of a coronary artery occurs in 1% to 7% of acute type A aortic dissections.1–3 The right coronary artery (Figure 2) is most commonly involved, leading to acute inferior myocardial infarction. Acute myocardial ischemia and infarction in the setting of dissection may lead to a delay in the diagnosis of dissection and to bleeding complications from antiplatelet and anticoagulant drugs given to treat the acute coronary syndrome.
Cardiac tamponade, occurring in about 10% of acute type A dissections, portends a higher risk of death.2,3
Additional clinical features of aortic dissection include a left-sided pleural effusion, usually related to an inflammatory response. An acute hemothorax may occur from rupture or leaking of a descending aortic dissection.
FINDINGS ON RADIOGRAPHY AND ELECTROCARDIOGRAPHY
Electrocardiography usually has normal or nonspecific findings, unless acute myocardial infarction complicates the dissection.
D-DIMER LEVELS
Biomarkers for the diagnosis of acute aortic dissection are of great interest.
D-dimer levels rise in acute aortic dissection as they do in pulmonary embolism.11 A D-dimer level greater than 1,600 ng/mL within the first 6 hours has a very high positive likelihood ratio for dissection, so this test may be useful in identifying patients with a high probability for dissection. In the first 24 hours after symptom onset, a D-dimer level of less than 500 ng/mL has a negative predictive value of 95%. Thus, elevations in D-dimer may help decide which imaging to perform in a patient presenting with chest pain or suspicion of dissection.11
However, D-dimer levels may not be elevated in dissection variants, such as aortic intramural hematoma or penetrating aortic ulcer. Additionally, once 24 hours have elapsed since the dissection started, D-dimer levels may no longer be elevated. The current ACC/AHA guidelines on thoracic aortic disease concluded that the D-dimer level cannot be used to rule out aortic dissection in high-risk individuals.3
Additional studies may clarify the appropriate role of the D-dimer assay in diagnosing aortic dissection.
DEFINITIVE IMAGING STUDIES: CT, MRI, TEE
Contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), and transesophageal echocardiography (TEE) all have very high sensitivity and specificity for the diagnosis of aortic dissection.2,3 The choice of imaging study often depends on the availability of these studies, with CT and TEE being the most commonly performed initial studies.
MRI is outstanding for detecting and following aortic dissection, but it is usually not the initial study performed because of the time required for image acquisition and because it is generally not available on an emergency basis.
While transthoracic echocardiography (TTE) can detect aortic dissection, its sensitivity is much lower than that of other imaging tests.2,3 Therefore, negative findings on TTE do not exclude aortic dissection.
MANAGEMENT OF AORTIC DISSECTION
When acute aortic dissection is diagnosed, multidisciplinary evaluation and treatment are necessary. Time is of the essence, as the death rate in acute dissection may be as high as 1% per hour during the first 24 hours.1–3 All patients with acute aortic dissection, whether type A or type B, should be transferred to a tertiary care center with a staff experienced in managing aortic dissection and its complications.3 Emergency surgery is recommended for type A aortic dissection, whereas type B dissection is generally treated medically unless complications occur.2,3
The cornerstone of drug therapy is the prompt reduction in blood pressure with a beta-blocker to reduce shear stresses on the aorta. Intravenous agents such as esmolol (Brevibloc) or labetalol (Normodyne) are usually chosen. Sodium nitroprusside may be added to beta-blocker therapy for rapid blood pressure control in appropriate patients. The patient may require multiple antihypertensive medications. If hypertension is refractory, one must consider renal artery hypertension due to the dissection causing renal malperfusion.2 Acute pain may also worsen hypertension, and appropriate analgesia should be used.
Definitive therapy in acute dissection
Patients with acute type A dissection require emergency surgery,2,3 as they are at risk for life-threatening complications including cardiac tamponade from hemopericardium, aortic rupture, stroke, visceral ischemia, and heart failure due to severe aortic regurgitation. When aortic regurgitation complicates acute type A dissection, some patients are adequately treated by resuspension of the aortic valve leaflets, while others require valve-sparing root replacement or prosthetic aortic valve replacement.
Surgical therapy is associated with a survival benefit compared with medical therapy in acute type A dissection.1 The 14-day mortality rate for acute type A dissection treated surgically is about 25%.1 Patients with high-risk features such as heart failure, shock, tamponade, and mesenteric ischemia have a worse prognosis compared with those without these features.2,12,13
Acute type B aortic dissection carries a lower rate of death than type A dissection.1–3 In the IRAD cohort, the early mortality rate in those with type B dissection treated medically was about 10%.1 However, when complications such as malperfusion, shock, or requirement for surgery occur in type B dissection, the mortality rate is much higher,2,14 with rates of 25% to 50% reported.2
Thus, initial medical therapy is the preferred approach to acute type B dissection, and surgery or endovascular therapy is reserved for patients with acute complications.2,3 Typical indications for surgery or endovascular therapy in type B dissection include visceral or limb ischemia, aortic rupture, refractory pain, and aneurysmal dilation (Table 3).2
Endovascular therapy in aortic dissection
The high mortality rate with open surgery in acute type B dissection has spurred tremendous interest in endovascular treatments for complications involving the descending aorta and branch vessels.2
Fenestration of the aorta and stenting of branch vessels were the earliest techniques used in complicated type B dissection. By fenestrating (ie, opening) the intimal flap, blood can flow from the false lumen into the true lumen, decompressing the distended false lumen.
Endovascular stenting is used for acute aortic rupture, for malperfusion syndromes, and for rapidly enlarging false lumens. Endovascular grafts may cover the area of a primary intimal tear and thus eliminate the flow into the false channel and promote false-lumen thrombosis. Many patients with complicated type B dissection are treated with a hybrid approach, in which one segment of the aorta, such as the aortic arch, is treated surgically, while the descending aorta receives an endovascular graft.2
Patients with a type B dissection treated medically are at risk for late complications, including aneurysmal enlargement and subsequent aortic rupture. The Investigation of Stent Grafts in Aortic Dissection (INSTEAD) trial included 140 patients with uncomplicated type B dissection and compared drug therapy with endovascular stent grafting.15 After 2 years of follow-up, there was no difference in the rate of death between the two treatment groups. Patients receiving endovascular grafts had a higher rate of false-lumen thrombosis.
More studies are under way to examine the role of endovascular therapy in uncomplicated type B dissection.
AORTIC DISSECTION VARIANTS
Aortic intramural hematoma
Aortic intramural hematoma is a form of acute aortic syndrome in which a hematoma develops in the aortic media and no intimal flap is visualized either by imaging or at surgery.2,3,16 It is important to recognize this clinical entity in a patient presenting with acute chest or back pain, as sometimes it is mistaken for a “thrombus in a nonaneurysmal aorta.” Intramural hematoma accounts for 5% to 25% of acute aortic syndromes, depending on the study population (it is more common in Asian studies).2,3,17 It may present with symptoms similar to classic aortic dissection and is classified as type A or type B, depending on whether the ascending aorta is involved.
Patients with an intramural hematoma may progress to having complications such as hemopericardium, classic aortic dissection, aortic rupture, or aneurysmal dilation.2,3 However, many cases of type B aortic intramural hematoma result in complete resorption of the hematoma over time. In general, like classic aortic dissection, type A intramural hematoma is treated with emergency surgery and type B with initial medical therapy.2,3
There are reports from Southeast Asia of successful initial medical therapy for type A intramural hematoma, with surgery used for acute complications.18 In the Western literature, improved outcomes are reported with initial surgical therapy.17 Given the unpredictable nature of type A intramural hematoma, most experts recommend surgical therapy for appropriate candidates with acute type A intramural hematoma.2,3,19
Penetrating atherosclerotic ulcer of the aorta
Penetrating atherosclerotic ulcer of the aorta, another acute aortic syndrome, results from acute penetration of an atherosclerotic aortic lesion through the internal elastic lamina into the media.2,3,20 It is often associated with bleeding into the media, or intramural hematoma. While the ulcer may be found incidentally on imaging studies, especially in patients with severe aortic atherosclerosis, the typical presentation is acute, severe chest or back pain. It occurs most often in the descending aorta and the abdominal aorta.
Penetrating atherosclerotic ulcer may lead to pseudoaneurysm formation, focal aortic dissection, aortic rupture, or late aortic aneurysm.2
LONG-TERM MANAGEMENT AFTER AORTIC DISSECTION
After hospital discharge, patients with aortic dissection require lifelong management. This includes blood pressure control, lifestyle modification, serial imaging of the aorta with CT or MRI, patient education about the condition, and, when appropriate, screening of family members for aortic disease.5,21
Reported survival rates after hospitalization for type A dissection are 52% to 94% at 1 year and 45% to 88% at 5 years.2,22 The 10-year actuarial survival rate for those with acute dissection who survive the acute hospitalization is reported as 30% to 60%. Long-term survival rates after acute type B dissection have been reported at 56% to 92% at 1 year and 48% to 82% at 5 years.23 Survival rates depend on many factors, including the underlying condition, the age of the patient, and comorbidities.
It is important to treat hypertension after aortic dissection, with a goal blood pressure of 120/80 mm Hg or less for most patients. Older studies found higher mortality rates with poorly controlled hypertension. Beta-blockers are the drugs of first choice. Even in the absence of hypertension, long-term beta-blocker therapy should be used to lessen the aortic stress and force of ventricular contraction.
Genetic evaluation
Genetically triggered causes of aortic dissection should be considered. In many circumstances, referral to a medical geneticist or other practitioner knowledgeable in these conditions is important when these disorders are being evaluated (Table 2).
Many of these disorders have an autosomal dominant inheritance, and the patient should be asked about a family history of aortic disease, aortic dissection, or unexplained sudden death. Features of Marfan syndrome, Loeys-Dietz syndrome, and familial thoracic aortic aneurysm syndromes should be sought. Through comprehensive family studies, it is now recognized that up to 20% of patients with thoracic aortic disease (such as aneurysm or dissection) have another first-degree relative with thoracic aortic disease.2,3,24 Thus, first-degree relatives of patients with aortic aneurysm or dissection should be screened for thoracic aortic aneurysm disease.
Research into molecular genetics is providing a better understanding of the genetics of aortic dissection.3 New mutations associated with aortic dissection are being discovered in signaling pathways as well as elements critical for the integrity of the vascular wall.2,3 However, at present, most patients with aortic dissection will not have a specific identifiable genetic defect.
Not only does genetic testing enable the accurate diagnosis of the affected individual, but also treatments are often based on this diagnosis.3 Importantly, the identification of a specific gene mutation (ie, in TGFBR1 or 2, FBN1, ACTA2, MYH11, and COL3A1) in an affected individual has the potential to identify other family members at risk.3
Follow-up imaging
It is important to continue to image the aorta after aortic dissection. Patients may develop progressive dilation or aneurysm formation of the dissected aorta, pseudoaneurysm formation after repair, or recurrent dissection. Many patients require additional surgery on the aorta because of late aneurysm formation.
CT or MRI is usually performed at least every 6 months in the first 2 years after dissection and at least annually thereafter. More centers are choosing MRI for long-term follow-up to avoid the repeated radiation exposure with serial CT.
Patient education
Besides receiving medical therapy and undergoing imaging, patients with aortic dissection should be educated about this condition.5,21 The patient should be aware of symptoms suggesting dissection and should be instructed to seek attention for any concerning symptoms.
Lifestyle modifications are also important. The patient should be educated about safe activity levels and to avoid heavy isometric exercise, such as weight-lifting. Some patients will have to cease their current occupation because of activity restrictions.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. International Registry of Acute Aortic Dissection (IRAD): new insights from an old disease. JAMA 2000; 283:897–903.
- Braverman AC, Thompson R, Sanchez L. Diseases of the aorta. In:Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease, 9th Edition. Elsevier, Philadelphia, 2011.
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. Guidelines for the management of patients with thoracic aortic disease. Circulation 2010; 121:e266–e369.
- Rogers AM, Herman LK, Booher AM, et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation. Results from the International Registry of Acute Aortic Dissection. Circulation 2011; 123:2213–2228.
- Braverman AC. Acute aortic dissection: clinician update. Circulation 2010; 122:184–188.
- Davies RR, Gallo A, Coady MA, et al. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg 2006; 81:169–177.
- Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >5.5 cm is not a good predictor of type A aortic dissection. Observations from the International Registry of Acute Aortic Dissection. Circulation 2007; 116:1120–1127.
- Parish LM, Gorman JH, Kahn S, et al. Aortic size in acute type A dissection: implications for preventative ascending aortic replacement. Eur J Cardiothorac Surg 2009; 35:941–945.
- Gaul C, Dietrich W, Erbguth FJ. Neurological symptoms in acute aortic dissection: a challenge for neurologists. Cerebrovasc Dis 2008; 26:1–8.
- Upchurch GR, Nienaber C, Fattori R, et al Acute aortic dissection presenting with primarily abdominal pain: a rare manifestation of a deadly disease. Ann Vasc Surg 2005; 19:367–373.
- Suzuki T, Distante A, Zizza A, et al. Diagnosis of acute aortic dissection by D-dimer: the International Registry of Acute Aortic Dissection substudy on biomarkers (IRAD-bio) experience. Circulation 2009; 119:2702–2707.
- Tsai TT, Trimarchi S, Neinaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg 2009; 37:149–159.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg 2005; 129:112–122.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Role and results of surgery in acute type B aortic dissection. Insights from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2006; 114(suppl 1):I-357–I-364.
- Nienaber CA, Rousseau H, Eggbrecht H, et al. Randomized comparison of strategies for type B aortic dissection. The Investigation of STEnt grafts in Aortic Dissection (INSTEAD) Trial. Circulation 2009; 120:2519–2528.
- Evangelista A, Mukherjee D, Mehta RH, et al. Acute intramural hematoma of the aorta. Circulation 2005; 111:1063–1070.
- Pelzel JM, Braverman AC, Hirsch AT, Harris KM. International heterogeneity in diagnostic frequency and clinical outcomes of ascending aortic intramural hematoma. J Am Soc Echo 2007; 20:1260–1268.
- Song JK, Yim JH, Ahn JM, et al. Outcomes of patients with acute type A aortic intramural hematoma. Circulation 2009; 120:2046–2052.
- Harris KM, Pelzel JM, Braverman AC. Letter regarding article, “Outcomes of patients with acute type A intramural hematoma.” Circulation 2010; 121:e456.
- Sundt TM. Intramural hematoma and penetrating atherosclerotic ulcer of the aorta. Ann Thorac Surg 2007; 83:S835–S841.
- Juang D, Braverman A, Eagle K. Aortic dissection. Circulation 2008; 118:e507–e510.
- Tsai TT, Evangelista A, Nienaber CA, et al. Long-term survival in patients presenting with type A acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114(suppl I):I-350–I-356.
- Tsai TT, Fattori R, Trimarchi S, et al. Long-term survival in patients presenting with type B acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114:2226–2231.
- Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections: incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82:1400–1405.
A 50-year-old man developed severe chest pain and collapsed to the floor. The pain was sudden in onset, was burning in quality, and was located in the center of his chest. Emergency medical services arrived a few minutes later and found the patient diaphoretic and cyanotic, with an initial blood pressure of 74/54 mm Hg and a heart rate of 125 beats per minute. He was rushed to the hospital.
His medical history was unremarkable. He smoked one pack of cigarettes per day for 20 years. His father died of a “heart attack” at age 52.
In the emergency department he underwent echocardiography with a portable handheld unit, which showed a pericardial effusion and cardiac tamponade. He was sent for emergency computed tomography of the chest, which revealed an aneurysm of the aortic root and acute type A (Stanford classification) aortic dissection with hemopericardium.
He underwent emergency cardiac surgery. At the time of surgery, he was in cardiogenic shock from aortic dissection complicated by severe aortic regurgitation and cardiac tamponade with hemopericardium. The aortic valve was trileaflet. A 27-mm St. Jude composite valve graft root replacement was performed.
The patient did well and was discharged home 7 days after surgery. Pathologic study of the aorta revealed cystic medial degeneration. He did not have any features of Marfan syndrome or Loeys-Dietz syndrome. His three children underwent evaluation, and each had a normal physical examination and echocardiographic test results.
A HIGH INDEX OF SUSPICION IS CRITICAL
Acute aortic dissection is the most common aortic catastrophe, with an incidence estimated at 5 to 30 per 1 million people per year, amounting to nearly 10,000 cases per year in the United States.1–4
The diagnosis of acute aortic dissection has many potential pitfalls.2,3 Aortic dissection may mimic other more common conditions, such as coronary ischemia, pleurisy, heart failure, stroke, and acute abdominal illness. Because acute aortic dissection may be rapidly fatal, one must maintain a high index of suspicion.2,3 Prompt diagnosis and emergency treatment are critical.
WHAT CAUSES AORTIC DISSECTION?
One hypothesis is that acute aortic dissection is caused by a primary tear in the aortic intima, with blood from the aortic lumen penetrating into the diseased media leading to dissection and creating a true and false lumen.2 Another is that rupture of the vasa vasorum leads to hemorrhage in the aortic wall with subsequent intimal disruption, creating the intimal tear and aortic dissection.
Once a dissection starts, pulsatile flow of blood within the aortic wall causes it to extend. The dissection flap may be localized, but it often spirals the entire length of the aorta. Distention of the false lumen with blood may cause the intimal flap to compress the true lumen and potentially lead to malperfusion syndromes.
CLASSIFIED ACCORDING TO LOCATION
It is important to recognize the location of the dissection, as the prognosis and treatment depend on whether the ascending aorta is involved.2,3 For classification purposes, the ascending aorta is the portion proximal to the brachiocephalic artery, while the descending aorta is the portion distal to the left subclavian artery.3
The DeBakey classification defines a type I aortic dissection as one that begins in the ascending aorta and extends at least to the aortic arch or beyond. Type II dissections involve the ascending aorta only, while type III dissections begin in the descending aorta, most often just distal to the left subclavian artery.
The Stanford classification scheme divides dissections into type A and type B. Type A dissections involve the ascending aorta, while type B dissections do not involve the ascending aorta.
Which classification scheme is used is not important. However, identifying patients with dissection of the ascending aorta (DeBakey type I or type II or Stanford type A) is critical, as emergency cardiac surgery is recommended for this type of dissection.2,3 For the purposes of this paper, the Stanford classification scheme will be used.
Dissection that involves the ascending aorta most commonly occurs in people ages 50 to 60, whereas acute dissection of the descending aorta typically occurs in people 10 years older.1,2
An acute aortic dissection is one that has occurred within 2 weeks of symptom onset. A chronic dissection is one that occurred more than 2 weeks after symptoms began.
DISEASES AND CONDITIONS ASSOCIATED WITH AORTIC DISSECTION
Hypertension and disorders leading to disruption of the normal structure and function of the aortic wall. About 75% of patients with acute aortic dissection have underlying hypertension.1–3
Cystic medial degeneration is a common pathologic feature in many cases of aortic dissection.
Cocaine use and intense weight-lifting increase the shear stresses on the aorta.2,3
Inflammatory aortic diseases such as giant cell arteritis.
Pregnancy can be complicated by aortic dissection, usually in the setting of an underlying aortopathy.5
Iatrogenic aortic dissection accounts for about 4% of cases, as a result of cardiac surgery, catheterization, stenting, or use of an intra-aortic balloon pump.1
Aortic aneurysm. Patients with thoracic aortic aneurysm are at higher risk of aortic dissection, and the larger the aortic diameter, the higher the risk.2,3,6 In the International Registry of Acute Aortic Dissection (IRAD), the average size of the aorta was about 5.3 cm at the time of acute dissection. Importantly, about 40% of acute dissections of the ascending aorta occur in patients with ascending aortic diameters less than 5.0 cm.7,8
Thus, many factors are associated with acute dissection, and specific reasons leading to an individual’s susceptibility to sudden dissection are poorly understood.
CLINICAL FEATURES OF ACUTE AORTIC DISSECTION
Because the symptoms of acute dissection may mimic other, more common conditions, one of the most important factors in the diagnosis of aortic dissection is a high clinical suspicion.1–3
What is the pretest risk of disease?
Recently, the American College of Cardiology (ACC) and the American Heart Association (AHA) released joint guidelines on thoracic aortic disease.3 These guidelines provide an approach to patients who have complaints that may represent acute thoracic aortic dissection, the intent being to establish a pretest risk of disease to be used to guide decision-making.3
The focused evaluation includes specific questions about underlying conditions, symptoms, and findings on examination that may greatly increase the likelihood of acute dissection. These include:
- High-risk conditions and historical features associated with aortic dissection, such as Marfan syndrome and other genetic disorders (Table 2), bicuspid aortic valve, family history of thoracic aortic aneurysm or dissection, known thoracic aortic aneurysm, and recent aortic manipulation
- Pain in the chest, back, or abdomen with high-risk features (eg, abrupt onset, severe intensity, or a ripping or tearing quality)
- High-risk findings on examination (eg, pulse deficits, new aortic regurgitation, hypotension, shock, or systolic blood pressure differences).
Using this information, expedited aortic imaging and treatment algorithms have been devised to improve the diagnosis.3
Using the IRAD database of more than 2,500 acute dissections, the diagnostic algorithm proposed in the ACC/AHA guidelines was shown to be highly sensitive (about 95%) for detecting acute aortic dissection.4 In addition, using this score may expedite evaluation by classifying certain patients as being at high risk of acute dissection.3,4
Important to recognize is that almost two-thirds of patients who suffered dissection in this large database did not have one of the “high-risk conditions” associated with dissection.4 Additionally, the specificity of the ACC/AHA algorithm is unknown, and further testing is necessary.4
Acute onset of severe pain
More than 90% of acute dissections present with acute pain in the chest or the back, or both.1–3 The pain is usually severe, of sudden onset, and often described as sharp or, occasionally, tearing, ripping, or stabbing. The pain usually differs from that of coronary ischemia, being most severe at its onset as opposed to the less intense, crescendo-like pain of angina or myocardial infarction. The pain may migrate as the dissection progresses along the length of the aorta or to branch vessels. It may abate, leading to a false sense of security in the patient and the physician.3 “Painless” dissection occurs in a minority, usually in those with syncope, neurologic symptoms, or heart failure.1–3
The patient with acute dissection may be anxious and may feel a sense of doom.
Acute heart failure, related to severe aortic regurgitation, may be a predominant symptom in dissection of the ascending aorta.
Syncope may occur as a result of aortic rupture, hemopericardium with cardiac tamponade, or acute neurologic complications.
Vascular insufficiency may occur in any branch vessel, leading to clinical syndromes that include acute myocardial infarction, stroke, paraplegia, paraparesis, mesenteric ischemia, and limb ischemia.
PHYSICAL FINDINGS CAN VARY WIDELY
Findings on physical examination in acute aortic dissection vary widely depending on underlying conditions and on the specific complications of the dissection.
Although the classic presentation is acute, severe pain in the chest or back in a severely hypertensive patient with aortic regurgitation and pulse deficits, most patients do not have all these characteristics.4 Most patients with type B dissection are hypertensive on presentation, but many with type A dissection present with normal blood pressure or hypotension.1 Pulse deficits (unequal or absent pulses) are reported in 10% to 30% of acute dissections and may be intermittent as the dynamic movement of the dissection flap interferes with branch vessel perfusion.1–3
- Aortic leaflet prolapse or distortion of the leaflet alignment
- Malcoaptation of the aortic leaflets from dilation of the aortic root and annulus
- Prolapse of the intimal flap across the aortic valve, interfering with valve function
- Preexisting aortic regurgitation from underlying aortic root aneurysm or primary aortic valve disease (such as a bicuspid aortic valve).
Neurologic manifestations are most common in dissection of the ascending aorta and are particularly important to recognize, as they may dominate the clinical presentation and lead to delay in the diagnosis of dissection.2,3 Neurologic syndromes include:
- Persistent or transient ischemic stroke
- Spinal cord ischemia
- Ischemic neuropathy
- Hypoxic encephalopathy.
These manifestations are related to malperfusion to branches supplying the brain, spinal cord, or peripheral nerves.9
Syncope is relatively common in aortic dissection and may be related to acute hypotension caused by cardiac tamponade or aortic rupture, cerebral vessel obstruction, or activation of cerebral baroreceptors.2,9 It is important to consider aortic dissection in the differential diagnosis in cases of unexplained syncope.3
Aortic dissections may extend into the abdominal aorta, leading to vascular complications involving one or more branch vessels.10 The renal artery is involved in at least 5% to 10% of cases and may lead to renal ischemia, infarction, renal insufficiency, or refractory hypertension.2Mesenteric ischemia or infarction occurs in about 5% of dissections, may be difficult to diagnose, and is particularly dangerous.2,8 Aortic dissection may extend into the iliac arteries and may cause acute lower extremity ischemia.
Acute myocardial infarction due to involvement of the dissection flap causing malperfusion of a coronary artery occurs in 1% to 7% of acute type A aortic dissections.1–3 The right coronary artery (Figure 2) is most commonly involved, leading to acute inferior myocardial infarction. Acute myocardial ischemia and infarction in the setting of dissection may lead to a delay in the diagnosis of dissection and to bleeding complications from antiplatelet and anticoagulant drugs given to treat the acute coronary syndrome.
Cardiac tamponade, occurring in about 10% of acute type A dissections, portends a higher risk of death.2,3
Additional clinical features of aortic dissection include a left-sided pleural effusion, usually related to an inflammatory response. An acute hemothorax may occur from rupture or leaking of a descending aortic dissection.
FINDINGS ON RADIOGRAPHY AND ELECTROCARDIOGRAPHY
Electrocardiography usually has normal or nonspecific findings, unless acute myocardial infarction complicates the dissection.
D-DIMER LEVELS
Biomarkers for the diagnosis of acute aortic dissection are of great interest.
D-dimer levels rise in acute aortic dissection as they do in pulmonary embolism.11 A D-dimer level greater than 1,600 ng/mL within the first 6 hours has a very high positive likelihood ratio for dissection, so this test may be useful in identifying patients with a high probability for dissection. In the first 24 hours after symptom onset, a D-dimer level of less than 500 ng/mL has a negative predictive value of 95%. Thus, elevations in D-dimer may help decide which imaging to perform in a patient presenting with chest pain or suspicion of dissection.11
However, D-dimer levels may not be elevated in dissection variants, such as aortic intramural hematoma or penetrating aortic ulcer. Additionally, once 24 hours have elapsed since the dissection started, D-dimer levels may no longer be elevated. The current ACC/AHA guidelines on thoracic aortic disease concluded that the D-dimer level cannot be used to rule out aortic dissection in high-risk individuals.3
Additional studies may clarify the appropriate role of the D-dimer assay in diagnosing aortic dissection.
DEFINITIVE IMAGING STUDIES: CT, MRI, TEE
Contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), and transesophageal echocardiography (TEE) all have very high sensitivity and specificity for the diagnosis of aortic dissection.2,3 The choice of imaging study often depends on the availability of these studies, with CT and TEE being the most commonly performed initial studies.
MRI is outstanding for detecting and following aortic dissection, but it is usually not the initial study performed because of the time required for image acquisition and because it is generally not available on an emergency basis.
While transthoracic echocardiography (TTE) can detect aortic dissection, its sensitivity is much lower than that of other imaging tests.2,3 Therefore, negative findings on TTE do not exclude aortic dissection.
MANAGEMENT OF AORTIC DISSECTION
When acute aortic dissection is diagnosed, multidisciplinary evaluation and treatment are necessary. Time is of the essence, as the death rate in acute dissection may be as high as 1% per hour during the first 24 hours.1–3 All patients with acute aortic dissection, whether type A or type B, should be transferred to a tertiary care center with a staff experienced in managing aortic dissection and its complications.3 Emergency surgery is recommended for type A aortic dissection, whereas type B dissection is generally treated medically unless complications occur.2,3
The cornerstone of drug therapy is the prompt reduction in blood pressure with a beta-blocker to reduce shear stresses on the aorta. Intravenous agents such as esmolol (Brevibloc) or labetalol (Normodyne) are usually chosen. Sodium nitroprusside may be added to beta-blocker therapy for rapid blood pressure control in appropriate patients. The patient may require multiple antihypertensive medications. If hypertension is refractory, one must consider renal artery hypertension due to the dissection causing renal malperfusion.2 Acute pain may also worsen hypertension, and appropriate analgesia should be used.
Definitive therapy in acute dissection
Patients with acute type A dissection require emergency surgery,2,3 as they are at risk for life-threatening complications including cardiac tamponade from hemopericardium, aortic rupture, stroke, visceral ischemia, and heart failure due to severe aortic regurgitation. When aortic regurgitation complicates acute type A dissection, some patients are adequately treated by resuspension of the aortic valve leaflets, while others require valve-sparing root replacement or prosthetic aortic valve replacement.
Surgical therapy is associated with a survival benefit compared with medical therapy in acute type A dissection.1 The 14-day mortality rate for acute type A dissection treated surgically is about 25%.1 Patients with high-risk features such as heart failure, shock, tamponade, and mesenteric ischemia have a worse prognosis compared with those without these features.2,12,13
Acute type B aortic dissection carries a lower rate of death than type A dissection.1–3 In the IRAD cohort, the early mortality rate in those with type B dissection treated medically was about 10%.1 However, when complications such as malperfusion, shock, or requirement for surgery occur in type B dissection, the mortality rate is much higher,2,14 with rates of 25% to 50% reported.2
Thus, initial medical therapy is the preferred approach to acute type B dissection, and surgery or endovascular therapy is reserved for patients with acute complications.2,3 Typical indications for surgery or endovascular therapy in type B dissection include visceral or limb ischemia, aortic rupture, refractory pain, and aneurysmal dilation (Table 3).2
Endovascular therapy in aortic dissection
The high mortality rate with open surgery in acute type B dissection has spurred tremendous interest in endovascular treatments for complications involving the descending aorta and branch vessels.2
Fenestration of the aorta and stenting of branch vessels were the earliest techniques used in complicated type B dissection. By fenestrating (ie, opening) the intimal flap, blood can flow from the false lumen into the true lumen, decompressing the distended false lumen.
Endovascular stenting is used for acute aortic rupture, for malperfusion syndromes, and for rapidly enlarging false lumens. Endovascular grafts may cover the area of a primary intimal tear and thus eliminate the flow into the false channel and promote false-lumen thrombosis. Many patients with complicated type B dissection are treated with a hybrid approach, in which one segment of the aorta, such as the aortic arch, is treated surgically, while the descending aorta receives an endovascular graft.2
Patients with a type B dissection treated medically are at risk for late complications, including aneurysmal enlargement and subsequent aortic rupture. The Investigation of Stent Grafts in Aortic Dissection (INSTEAD) trial included 140 patients with uncomplicated type B dissection and compared drug therapy with endovascular stent grafting.15 After 2 years of follow-up, there was no difference in the rate of death between the two treatment groups. Patients receiving endovascular grafts had a higher rate of false-lumen thrombosis.
More studies are under way to examine the role of endovascular therapy in uncomplicated type B dissection.
AORTIC DISSECTION VARIANTS
Aortic intramural hematoma
Aortic intramural hematoma is a form of acute aortic syndrome in which a hematoma develops in the aortic media and no intimal flap is visualized either by imaging or at surgery.2,3,16 It is important to recognize this clinical entity in a patient presenting with acute chest or back pain, as sometimes it is mistaken for a “thrombus in a nonaneurysmal aorta.” Intramural hematoma accounts for 5% to 25% of acute aortic syndromes, depending on the study population (it is more common in Asian studies).2,3,17 It may present with symptoms similar to classic aortic dissection and is classified as type A or type B, depending on whether the ascending aorta is involved.
Patients with an intramural hematoma may progress to having complications such as hemopericardium, classic aortic dissection, aortic rupture, or aneurysmal dilation.2,3 However, many cases of type B aortic intramural hematoma result in complete resorption of the hematoma over time. In general, like classic aortic dissection, type A intramural hematoma is treated with emergency surgery and type B with initial medical therapy.2,3
There are reports from Southeast Asia of successful initial medical therapy for type A intramural hematoma, with surgery used for acute complications.18 In the Western literature, improved outcomes are reported with initial surgical therapy.17 Given the unpredictable nature of type A intramural hematoma, most experts recommend surgical therapy for appropriate candidates with acute type A intramural hematoma.2,3,19
Penetrating atherosclerotic ulcer of the aorta
Penetrating atherosclerotic ulcer of the aorta, another acute aortic syndrome, results from acute penetration of an atherosclerotic aortic lesion through the internal elastic lamina into the media.2,3,20 It is often associated with bleeding into the media, or intramural hematoma. While the ulcer may be found incidentally on imaging studies, especially in patients with severe aortic atherosclerosis, the typical presentation is acute, severe chest or back pain. It occurs most often in the descending aorta and the abdominal aorta.
Penetrating atherosclerotic ulcer may lead to pseudoaneurysm formation, focal aortic dissection, aortic rupture, or late aortic aneurysm.2
LONG-TERM MANAGEMENT AFTER AORTIC DISSECTION
After hospital discharge, patients with aortic dissection require lifelong management. This includes blood pressure control, lifestyle modification, serial imaging of the aorta with CT or MRI, patient education about the condition, and, when appropriate, screening of family members for aortic disease.5,21
Reported survival rates after hospitalization for type A dissection are 52% to 94% at 1 year and 45% to 88% at 5 years.2,22 The 10-year actuarial survival rate for those with acute dissection who survive the acute hospitalization is reported as 30% to 60%. Long-term survival rates after acute type B dissection have been reported at 56% to 92% at 1 year and 48% to 82% at 5 years.23 Survival rates depend on many factors, including the underlying condition, the age of the patient, and comorbidities.
It is important to treat hypertension after aortic dissection, with a goal blood pressure of 120/80 mm Hg or less for most patients. Older studies found higher mortality rates with poorly controlled hypertension. Beta-blockers are the drugs of first choice. Even in the absence of hypertension, long-term beta-blocker therapy should be used to lessen the aortic stress and force of ventricular contraction.
Genetic evaluation
Genetically triggered causes of aortic dissection should be considered. In many circumstances, referral to a medical geneticist or other practitioner knowledgeable in these conditions is important when these disorders are being evaluated (Table 2).
Many of these disorders have an autosomal dominant inheritance, and the patient should be asked about a family history of aortic disease, aortic dissection, or unexplained sudden death. Features of Marfan syndrome, Loeys-Dietz syndrome, and familial thoracic aortic aneurysm syndromes should be sought. Through comprehensive family studies, it is now recognized that up to 20% of patients with thoracic aortic disease (such as aneurysm or dissection) have another first-degree relative with thoracic aortic disease.2,3,24 Thus, first-degree relatives of patients with aortic aneurysm or dissection should be screened for thoracic aortic aneurysm disease.
Research into molecular genetics is providing a better understanding of the genetics of aortic dissection.3 New mutations associated with aortic dissection are being discovered in signaling pathways as well as elements critical for the integrity of the vascular wall.2,3 However, at present, most patients with aortic dissection will not have a specific identifiable genetic defect.
Not only does genetic testing enable the accurate diagnosis of the affected individual, but also treatments are often based on this diagnosis.3 Importantly, the identification of a specific gene mutation (ie, in TGFBR1 or 2, FBN1, ACTA2, MYH11, and COL3A1) in an affected individual has the potential to identify other family members at risk.3
Follow-up imaging
It is important to continue to image the aorta after aortic dissection. Patients may develop progressive dilation or aneurysm formation of the dissected aorta, pseudoaneurysm formation after repair, or recurrent dissection. Many patients require additional surgery on the aorta because of late aneurysm formation.
CT or MRI is usually performed at least every 6 months in the first 2 years after dissection and at least annually thereafter. More centers are choosing MRI for long-term follow-up to avoid the repeated radiation exposure with serial CT.
Patient education
Besides receiving medical therapy and undergoing imaging, patients with aortic dissection should be educated about this condition.5,21 The patient should be aware of symptoms suggesting dissection and should be instructed to seek attention for any concerning symptoms.
Lifestyle modifications are also important. The patient should be educated about safe activity levels and to avoid heavy isometric exercise, such as weight-lifting. Some patients will have to cease their current occupation because of activity restrictions.
A 50-year-old man developed severe chest pain and collapsed to the floor. The pain was sudden in onset, was burning in quality, and was located in the center of his chest. Emergency medical services arrived a few minutes later and found the patient diaphoretic and cyanotic, with an initial blood pressure of 74/54 mm Hg and a heart rate of 125 beats per minute. He was rushed to the hospital.
His medical history was unremarkable. He smoked one pack of cigarettes per day for 20 years. His father died of a “heart attack” at age 52.
In the emergency department he underwent echocardiography with a portable handheld unit, which showed a pericardial effusion and cardiac tamponade. He was sent for emergency computed tomography of the chest, which revealed an aneurysm of the aortic root and acute type A (Stanford classification) aortic dissection with hemopericardium.
He underwent emergency cardiac surgery. At the time of surgery, he was in cardiogenic shock from aortic dissection complicated by severe aortic regurgitation and cardiac tamponade with hemopericardium. The aortic valve was trileaflet. A 27-mm St. Jude composite valve graft root replacement was performed.
The patient did well and was discharged home 7 days after surgery. Pathologic study of the aorta revealed cystic medial degeneration. He did not have any features of Marfan syndrome or Loeys-Dietz syndrome. His three children underwent evaluation, and each had a normal physical examination and echocardiographic test results.
A HIGH INDEX OF SUSPICION IS CRITICAL
Acute aortic dissection is the most common aortic catastrophe, with an incidence estimated at 5 to 30 per 1 million people per year, amounting to nearly 10,000 cases per year in the United States.1–4
The diagnosis of acute aortic dissection has many potential pitfalls.2,3 Aortic dissection may mimic other more common conditions, such as coronary ischemia, pleurisy, heart failure, stroke, and acute abdominal illness. Because acute aortic dissection may be rapidly fatal, one must maintain a high index of suspicion.2,3 Prompt diagnosis and emergency treatment are critical.
WHAT CAUSES AORTIC DISSECTION?
One hypothesis is that acute aortic dissection is caused by a primary tear in the aortic intima, with blood from the aortic lumen penetrating into the diseased media leading to dissection and creating a true and false lumen.2 Another is that rupture of the vasa vasorum leads to hemorrhage in the aortic wall with subsequent intimal disruption, creating the intimal tear and aortic dissection.
Once a dissection starts, pulsatile flow of blood within the aortic wall causes it to extend. The dissection flap may be localized, but it often spirals the entire length of the aorta. Distention of the false lumen with blood may cause the intimal flap to compress the true lumen and potentially lead to malperfusion syndromes.
CLASSIFIED ACCORDING TO LOCATION
It is important to recognize the location of the dissection, as the prognosis and treatment depend on whether the ascending aorta is involved.2,3 For classification purposes, the ascending aorta is the portion proximal to the brachiocephalic artery, while the descending aorta is the portion distal to the left subclavian artery.3
The DeBakey classification defines a type I aortic dissection as one that begins in the ascending aorta and extends at least to the aortic arch or beyond. Type II dissections involve the ascending aorta only, while type III dissections begin in the descending aorta, most often just distal to the left subclavian artery.
The Stanford classification scheme divides dissections into type A and type B. Type A dissections involve the ascending aorta, while type B dissections do not involve the ascending aorta.
Which classification scheme is used is not important. However, identifying patients with dissection of the ascending aorta (DeBakey type I or type II or Stanford type A) is critical, as emergency cardiac surgery is recommended for this type of dissection.2,3 For the purposes of this paper, the Stanford classification scheme will be used.
Dissection that involves the ascending aorta most commonly occurs in people ages 50 to 60, whereas acute dissection of the descending aorta typically occurs in people 10 years older.1,2
An acute aortic dissection is one that has occurred within 2 weeks of symptom onset. A chronic dissection is one that occurred more than 2 weeks after symptoms began.
DISEASES AND CONDITIONS ASSOCIATED WITH AORTIC DISSECTION
Hypertension and disorders leading to disruption of the normal structure and function of the aortic wall. About 75% of patients with acute aortic dissection have underlying hypertension.1–3
Cystic medial degeneration is a common pathologic feature in many cases of aortic dissection.
Cocaine use and intense weight-lifting increase the shear stresses on the aorta.2,3
Inflammatory aortic diseases such as giant cell arteritis.
Pregnancy can be complicated by aortic dissection, usually in the setting of an underlying aortopathy.5
Iatrogenic aortic dissection accounts for about 4% of cases, as a result of cardiac surgery, catheterization, stenting, or use of an intra-aortic balloon pump.1
Aortic aneurysm. Patients with thoracic aortic aneurysm are at higher risk of aortic dissection, and the larger the aortic diameter, the higher the risk.2,3,6 In the International Registry of Acute Aortic Dissection (IRAD), the average size of the aorta was about 5.3 cm at the time of acute dissection. Importantly, about 40% of acute dissections of the ascending aorta occur in patients with ascending aortic diameters less than 5.0 cm.7,8
Thus, many factors are associated with acute dissection, and specific reasons leading to an individual’s susceptibility to sudden dissection are poorly understood.
CLINICAL FEATURES OF ACUTE AORTIC DISSECTION
Because the symptoms of acute dissection may mimic other, more common conditions, one of the most important factors in the diagnosis of aortic dissection is a high clinical suspicion.1–3
What is the pretest risk of disease?
Recently, the American College of Cardiology (ACC) and the American Heart Association (AHA) released joint guidelines on thoracic aortic disease.3 These guidelines provide an approach to patients who have complaints that may represent acute thoracic aortic dissection, the intent being to establish a pretest risk of disease to be used to guide decision-making.3
The focused evaluation includes specific questions about underlying conditions, symptoms, and findings on examination that may greatly increase the likelihood of acute dissection. These include:
- High-risk conditions and historical features associated with aortic dissection, such as Marfan syndrome and other genetic disorders (Table 2), bicuspid aortic valve, family history of thoracic aortic aneurysm or dissection, known thoracic aortic aneurysm, and recent aortic manipulation
- Pain in the chest, back, or abdomen with high-risk features (eg, abrupt onset, severe intensity, or a ripping or tearing quality)
- High-risk findings on examination (eg, pulse deficits, new aortic regurgitation, hypotension, shock, or systolic blood pressure differences).
Using this information, expedited aortic imaging and treatment algorithms have been devised to improve the diagnosis.3
Using the IRAD database of more than 2,500 acute dissections, the diagnostic algorithm proposed in the ACC/AHA guidelines was shown to be highly sensitive (about 95%) for detecting acute aortic dissection.4 In addition, using this score may expedite evaluation by classifying certain patients as being at high risk of acute dissection.3,4
Important to recognize is that almost two-thirds of patients who suffered dissection in this large database did not have one of the “high-risk conditions” associated with dissection.4 Additionally, the specificity of the ACC/AHA algorithm is unknown, and further testing is necessary.4
Acute onset of severe pain
More than 90% of acute dissections present with acute pain in the chest or the back, or both.1–3 The pain is usually severe, of sudden onset, and often described as sharp or, occasionally, tearing, ripping, or stabbing. The pain usually differs from that of coronary ischemia, being most severe at its onset as opposed to the less intense, crescendo-like pain of angina or myocardial infarction. The pain may migrate as the dissection progresses along the length of the aorta or to branch vessels. It may abate, leading to a false sense of security in the patient and the physician.3 “Painless” dissection occurs in a minority, usually in those with syncope, neurologic symptoms, or heart failure.1–3
The patient with acute dissection may be anxious and may feel a sense of doom.
Acute heart failure, related to severe aortic regurgitation, may be a predominant symptom in dissection of the ascending aorta.
Syncope may occur as a result of aortic rupture, hemopericardium with cardiac tamponade, or acute neurologic complications.
Vascular insufficiency may occur in any branch vessel, leading to clinical syndromes that include acute myocardial infarction, stroke, paraplegia, paraparesis, mesenteric ischemia, and limb ischemia.
PHYSICAL FINDINGS CAN VARY WIDELY
Findings on physical examination in acute aortic dissection vary widely depending on underlying conditions and on the specific complications of the dissection.
Although the classic presentation is acute, severe pain in the chest or back in a severely hypertensive patient with aortic regurgitation and pulse deficits, most patients do not have all these characteristics.4 Most patients with type B dissection are hypertensive on presentation, but many with type A dissection present with normal blood pressure or hypotension.1 Pulse deficits (unequal or absent pulses) are reported in 10% to 30% of acute dissections and may be intermittent as the dynamic movement of the dissection flap interferes with branch vessel perfusion.1–3
- Aortic leaflet prolapse or distortion of the leaflet alignment
- Malcoaptation of the aortic leaflets from dilation of the aortic root and annulus
- Prolapse of the intimal flap across the aortic valve, interfering with valve function
- Preexisting aortic regurgitation from underlying aortic root aneurysm or primary aortic valve disease (such as a bicuspid aortic valve).
Neurologic manifestations are most common in dissection of the ascending aorta and are particularly important to recognize, as they may dominate the clinical presentation and lead to delay in the diagnosis of dissection.2,3 Neurologic syndromes include:
- Persistent or transient ischemic stroke
- Spinal cord ischemia
- Ischemic neuropathy
- Hypoxic encephalopathy.
These manifestations are related to malperfusion to branches supplying the brain, spinal cord, or peripheral nerves.9
Syncope is relatively common in aortic dissection and may be related to acute hypotension caused by cardiac tamponade or aortic rupture, cerebral vessel obstruction, or activation of cerebral baroreceptors.2,9 It is important to consider aortic dissection in the differential diagnosis in cases of unexplained syncope.3
Aortic dissections may extend into the abdominal aorta, leading to vascular complications involving one or more branch vessels.10 The renal artery is involved in at least 5% to 10% of cases and may lead to renal ischemia, infarction, renal insufficiency, or refractory hypertension.2Mesenteric ischemia or infarction occurs in about 5% of dissections, may be difficult to diagnose, and is particularly dangerous.2,8 Aortic dissection may extend into the iliac arteries and may cause acute lower extremity ischemia.
Acute myocardial infarction due to involvement of the dissection flap causing malperfusion of a coronary artery occurs in 1% to 7% of acute type A aortic dissections.1–3 The right coronary artery (Figure 2) is most commonly involved, leading to acute inferior myocardial infarction. Acute myocardial ischemia and infarction in the setting of dissection may lead to a delay in the diagnosis of dissection and to bleeding complications from antiplatelet and anticoagulant drugs given to treat the acute coronary syndrome.
Cardiac tamponade, occurring in about 10% of acute type A dissections, portends a higher risk of death.2,3
Additional clinical features of aortic dissection include a left-sided pleural effusion, usually related to an inflammatory response. An acute hemothorax may occur from rupture or leaking of a descending aortic dissection.
FINDINGS ON RADIOGRAPHY AND ELECTROCARDIOGRAPHY
Electrocardiography usually has normal or nonspecific findings, unless acute myocardial infarction complicates the dissection.
D-DIMER LEVELS
Biomarkers for the diagnosis of acute aortic dissection are of great interest.
D-dimer levels rise in acute aortic dissection as they do in pulmonary embolism.11 A D-dimer level greater than 1,600 ng/mL within the first 6 hours has a very high positive likelihood ratio for dissection, so this test may be useful in identifying patients with a high probability for dissection. In the first 24 hours after symptom onset, a D-dimer level of less than 500 ng/mL has a negative predictive value of 95%. Thus, elevations in D-dimer may help decide which imaging to perform in a patient presenting with chest pain or suspicion of dissection.11
However, D-dimer levels may not be elevated in dissection variants, such as aortic intramural hematoma or penetrating aortic ulcer. Additionally, once 24 hours have elapsed since the dissection started, D-dimer levels may no longer be elevated. The current ACC/AHA guidelines on thoracic aortic disease concluded that the D-dimer level cannot be used to rule out aortic dissection in high-risk individuals.3
Additional studies may clarify the appropriate role of the D-dimer assay in diagnosing aortic dissection.
DEFINITIVE IMAGING STUDIES: CT, MRI, TEE
Contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), and transesophageal echocardiography (TEE) all have very high sensitivity and specificity for the diagnosis of aortic dissection.2,3 The choice of imaging study often depends on the availability of these studies, with CT and TEE being the most commonly performed initial studies.
MRI is outstanding for detecting and following aortic dissection, but it is usually not the initial study performed because of the time required for image acquisition and because it is generally not available on an emergency basis.
While transthoracic echocardiography (TTE) can detect aortic dissection, its sensitivity is much lower than that of other imaging tests.2,3 Therefore, negative findings on TTE do not exclude aortic dissection.
MANAGEMENT OF AORTIC DISSECTION
When acute aortic dissection is diagnosed, multidisciplinary evaluation and treatment are necessary. Time is of the essence, as the death rate in acute dissection may be as high as 1% per hour during the first 24 hours.1–3 All patients with acute aortic dissection, whether type A or type B, should be transferred to a tertiary care center with a staff experienced in managing aortic dissection and its complications.3 Emergency surgery is recommended for type A aortic dissection, whereas type B dissection is generally treated medically unless complications occur.2,3
The cornerstone of drug therapy is the prompt reduction in blood pressure with a beta-blocker to reduce shear stresses on the aorta. Intravenous agents such as esmolol (Brevibloc) or labetalol (Normodyne) are usually chosen. Sodium nitroprusside may be added to beta-blocker therapy for rapid blood pressure control in appropriate patients. The patient may require multiple antihypertensive medications. If hypertension is refractory, one must consider renal artery hypertension due to the dissection causing renal malperfusion.2 Acute pain may also worsen hypertension, and appropriate analgesia should be used.
Definitive therapy in acute dissection
Patients with acute type A dissection require emergency surgery,2,3 as they are at risk for life-threatening complications including cardiac tamponade from hemopericardium, aortic rupture, stroke, visceral ischemia, and heart failure due to severe aortic regurgitation. When aortic regurgitation complicates acute type A dissection, some patients are adequately treated by resuspension of the aortic valve leaflets, while others require valve-sparing root replacement or prosthetic aortic valve replacement.
Surgical therapy is associated with a survival benefit compared with medical therapy in acute type A dissection.1 The 14-day mortality rate for acute type A dissection treated surgically is about 25%.1 Patients with high-risk features such as heart failure, shock, tamponade, and mesenteric ischemia have a worse prognosis compared with those without these features.2,12,13
Acute type B aortic dissection carries a lower rate of death than type A dissection.1–3 In the IRAD cohort, the early mortality rate in those with type B dissection treated medically was about 10%.1 However, when complications such as malperfusion, shock, or requirement for surgery occur in type B dissection, the mortality rate is much higher,2,14 with rates of 25% to 50% reported.2
Thus, initial medical therapy is the preferred approach to acute type B dissection, and surgery or endovascular therapy is reserved for patients with acute complications.2,3 Typical indications for surgery or endovascular therapy in type B dissection include visceral or limb ischemia, aortic rupture, refractory pain, and aneurysmal dilation (Table 3).2
Endovascular therapy in aortic dissection
The high mortality rate with open surgery in acute type B dissection has spurred tremendous interest in endovascular treatments for complications involving the descending aorta and branch vessels.2
Fenestration of the aorta and stenting of branch vessels were the earliest techniques used in complicated type B dissection. By fenestrating (ie, opening) the intimal flap, blood can flow from the false lumen into the true lumen, decompressing the distended false lumen.
Endovascular stenting is used for acute aortic rupture, for malperfusion syndromes, and for rapidly enlarging false lumens. Endovascular grafts may cover the area of a primary intimal tear and thus eliminate the flow into the false channel and promote false-lumen thrombosis. Many patients with complicated type B dissection are treated with a hybrid approach, in which one segment of the aorta, such as the aortic arch, is treated surgically, while the descending aorta receives an endovascular graft.2
Patients with a type B dissection treated medically are at risk for late complications, including aneurysmal enlargement and subsequent aortic rupture. The Investigation of Stent Grafts in Aortic Dissection (INSTEAD) trial included 140 patients with uncomplicated type B dissection and compared drug therapy with endovascular stent grafting.15 After 2 years of follow-up, there was no difference in the rate of death between the two treatment groups. Patients receiving endovascular grafts had a higher rate of false-lumen thrombosis.
More studies are under way to examine the role of endovascular therapy in uncomplicated type B dissection.
AORTIC DISSECTION VARIANTS
Aortic intramural hematoma
Aortic intramural hematoma is a form of acute aortic syndrome in which a hematoma develops in the aortic media and no intimal flap is visualized either by imaging or at surgery.2,3,16 It is important to recognize this clinical entity in a patient presenting with acute chest or back pain, as sometimes it is mistaken for a “thrombus in a nonaneurysmal aorta.” Intramural hematoma accounts for 5% to 25% of acute aortic syndromes, depending on the study population (it is more common in Asian studies).2,3,17 It may present with symptoms similar to classic aortic dissection and is classified as type A or type B, depending on whether the ascending aorta is involved.
Patients with an intramural hematoma may progress to having complications such as hemopericardium, classic aortic dissection, aortic rupture, or aneurysmal dilation.2,3 However, many cases of type B aortic intramural hematoma result in complete resorption of the hematoma over time. In general, like classic aortic dissection, type A intramural hematoma is treated with emergency surgery and type B with initial medical therapy.2,3
There are reports from Southeast Asia of successful initial medical therapy for type A intramural hematoma, with surgery used for acute complications.18 In the Western literature, improved outcomes are reported with initial surgical therapy.17 Given the unpredictable nature of type A intramural hematoma, most experts recommend surgical therapy for appropriate candidates with acute type A intramural hematoma.2,3,19
Penetrating atherosclerotic ulcer of the aorta
Penetrating atherosclerotic ulcer of the aorta, another acute aortic syndrome, results from acute penetration of an atherosclerotic aortic lesion through the internal elastic lamina into the media.2,3,20 It is often associated with bleeding into the media, or intramural hematoma. While the ulcer may be found incidentally on imaging studies, especially in patients with severe aortic atherosclerosis, the typical presentation is acute, severe chest or back pain. It occurs most often in the descending aorta and the abdominal aorta.
Penetrating atherosclerotic ulcer may lead to pseudoaneurysm formation, focal aortic dissection, aortic rupture, or late aortic aneurysm.2
LONG-TERM MANAGEMENT AFTER AORTIC DISSECTION
After hospital discharge, patients with aortic dissection require lifelong management. This includes blood pressure control, lifestyle modification, serial imaging of the aorta with CT or MRI, patient education about the condition, and, when appropriate, screening of family members for aortic disease.5,21
Reported survival rates after hospitalization for type A dissection are 52% to 94% at 1 year and 45% to 88% at 5 years.2,22 The 10-year actuarial survival rate for those with acute dissection who survive the acute hospitalization is reported as 30% to 60%. Long-term survival rates after acute type B dissection have been reported at 56% to 92% at 1 year and 48% to 82% at 5 years.23 Survival rates depend on many factors, including the underlying condition, the age of the patient, and comorbidities.
It is important to treat hypertension after aortic dissection, with a goal blood pressure of 120/80 mm Hg or less for most patients. Older studies found higher mortality rates with poorly controlled hypertension. Beta-blockers are the drugs of first choice. Even in the absence of hypertension, long-term beta-blocker therapy should be used to lessen the aortic stress and force of ventricular contraction.
Genetic evaluation
Genetically triggered causes of aortic dissection should be considered. In many circumstances, referral to a medical geneticist or other practitioner knowledgeable in these conditions is important when these disorders are being evaluated (Table 2).
Many of these disorders have an autosomal dominant inheritance, and the patient should be asked about a family history of aortic disease, aortic dissection, or unexplained sudden death. Features of Marfan syndrome, Loeys-Dietz syndrome, and familial thoracic aortic aneurysm syndromes should be sought. Through comprehensive family studies, it is now recognized that up to 20% of patients with thoracic aortic disease (such as aneurysm or dissection) have another first-degree relative with thoracic aortic disease.2,3,24 Thus, first-degree relatives of patients with aortic aneurysm or dissection should be screened for thoracic aortic aneurysm disease.
Research into molecular genetics is providing a better understanding of the genetics of aortic dissection.3 New mutations associated with aortic dissection are being discovered in signaling pathways as well as elements critical for the integrity of the vascular wall.2,3 However, at present, most patients with aortic dissection will not have a specific identifiable genetic defect.
Not only does genetic testing enable the accurate diagnosis of the affected individual, but also treatments are often based on this diagnosis.3 Importantly, the identification of a specific gene mutation (ie, in TGFBR1 or 2, FBN1, ACTA2, MYH11, and COL3A1) in an affected individual has the potential to identify other family members at risk.3
Follow-up imaging
It is important to continue to image the aorta after aortic dissection. Patients may develop progressive dilation or aneurysm formation of the dissected aorta, pseudoaneurysm formation after repair, or recurrent dissection. Many patients require additional surgery on the aorta because of late aneurysm formation.
CT or MRI is usually performed at least every 6 months in the first 2 years after dissection and at least annually thereafter. More centers are choosing MRI for long-term follow-up to avoid the repeated radiation exposure with serial CT.
Patient education
Besides receiving medical therapy and undergoing imaging, patients with aortic dissection should be educated about this condition.5,21 The patient should be aware of symptoms suggesting dissection and should be instructed to seek attention for any concerning symptoms.
Lifestyle modifications are also important. The patient should be educated about safe activity levels and to avoid heavy isometric exercise, such as weight-lifting. Some patients will have to cease their current occupation because of activity restrictions.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. International Registry of Acute Aortic Dissection (IRAD): new insights from an old disease. JAMA 2000; 283:897–903.
- Braverman AC, Thompson R, Sanchez L. Diseases of the aorta. In:Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease, 9th Edition. Elsevier, Philadelphia, 2011.
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. Guidelines for the management of patients with thoracic aortic disease. Circulation 2010; 121:e266–e369.
- Rogers AM, Herman LK, Booher AM, et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation. Results from the International Registry of Acute Aortic Dissection. Circulation 2011; 123:2213–2228.
- Braverman AC. Acute aortic dissection: clinician update. Circulation 2010; 122:184–188.
- Davies RR, Gallo A, Coady MA, et al. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg 2006; 81:169–177.
- Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >5.5 cm is not a good predictor of type A aortic dissection. Observations from the International Registry of Acute Aortic Dissection. Circulation 2007; 116:1120–1127.
- Parish LM, Gorman JH, Kahn S, et al. Aortic size in acute type A dissection: implications for preventative ascending aortic replacement. Eur J Cardiothorac Surg 2009; 35:941–945.
- Gaul C, Dietrich W, Erbguth FJ. Neurological symptoms in acute aortic dissection: a challenge for neurologists. Cerebrovasc Dis 2008; 26:1–8.
- Upchurch GR, Nienaber C, Fattori R, et al Acute aortic dissection presenting with primarily abdominal pain: a rare manifestation of a deadly disease. Ann Vasc Surg 2005; 19:367–373.
- Suzuki T, Distante A, Zizza A, et al. Diagnosis of acute aortic dissection by D-dimer: the International Registry of Acute Aortic Dissection substudy on biomarkers (IRAD-bio) experience. Circulation 2009; 119:2702–2707.
- Tsai TT, Trimarchi S, Neinaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg 2009; 37:149–159.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg 2005; 129:112–122.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Role and results of surgery in acute type B aortic dissection. Insights from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2006; 114(suppl 1):I-357–I-364.
- Nienaber CA, Rousseau H, Eggbrecht H, et al. Randomized comparison of strategies for type B aortic dissection. The Investigation of STEnt grafts in Aortic Dissection (INSTEAD) Trial. Circulation 2009; 120:2519–2528.
- Evangelista A, Mukherjee D, Mehta RH, et al. Acute intramural hematoma of the aorta. Circulation 2005; 111:1063–1070.
- Pelzel JM, Braverman AC, Hirsch AT, Harris KM. International heterogeneity in diagnostic frequency and clinical outcomes of ascending aortic intramural hematoma. J Am Soc Echo 2007; 20:1260–1268.
- Song JK, Yim JH, Ahn JM, et al. Outcomes of patients with acute type A aortic intramural hematoma. Circulation 2009; 120:2046–2052.
- Harris KM, Pelzel JM, Braverman AC. Letter regarding article, “Outcomes of patients with acute type A intramural hematoma.” Circulation 2010; 121:e456.
- Sundt TM. Intramural hematoma and penetrating atherosclerotic ulcer of the aorta. Ann Thorac Surg 2007; 83:S835–S841.
- Juang D, Braverman A, Eagle K. Aortic dissection. Circulation 2008; 118:e507–e510.
- Tsai TT, Evangelista A, Nienaber CA, et al. Long-term survival in patients presenting with type A acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114(suppl I):I-350–I-356.
- Tsai TT, Fattori R, Trimarchi S, et al. Long-term survival in patients presenting with type B acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114:2226–2231.
- Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections: incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82:1400–1405.
- Hagan PG, Nienaber CA, Isselbacher EM, et al. International Registry of Acute Aortic Dissection (IRAD): new insights from an old disease. JAMA 2000; 283:897–903.
- Braverman AC, Thompson R, Sanchez L. Diseases of the aorta. In:Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease, 9th Edition. Elsevier, Philadelphia, 2011.
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. Guidelines for the management of patients with thoracic aortic disease. Circulation 2010; 121:e266–e369.
- Rogers AM, Herman LK, Booher AM, et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation. Results from the International Registry of Acute Aortic Dissection. Circulation 2011; 123:2213–2228.
- Braverman AC. Acute aortic dissection: clinician update. Circulation 2010; 122:184–188.
- Davies RR, Gallo A, Coady MA, et al. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg 2006; 81:169–177.
- Pape LA, Tsai TT, Isselbacher EM, et al. Aortic diameter >5.5 cm is not a good predictor of type A aortic dissection. Observations from the International Registry of Acute Aortic Dissection. Circulation 2007; 116:1120–1127.
- Parish LM, Gorman JH, Kahn S, et al. Aortic size in acute type A dissection: implications for preventative ascending aortic replacement. Eur J Cardiothorac Surg 2009; 35:941–945.
- Gaul C, Dietrich W, Erbguth FJ. Neurological symptoms in acute aortic dissection: a challenge for neurologists. Cerebrovasc Dis 2008; 26:1–8.
- Upchurch GR, Nienaber C, Fattori R, et al Acute aortic dissection presenting with primarily abdominal pain: a rare manifestation of a deadly disease. Ann Vasc Surg 2005; 19:367–373.
- Suzuki T, Distante A, Zizza A, et al. Diagnosis of acute aortic dissection by D-dimer: the International Registry of Acute Aortic Dissection substudy on biomarkers (IRAD-bio) experience. Circulation 2009; 119:2702–2707.
- Tsai TT, Trimarchi S, Neinaber CA. Acute aortic dissection: perspectives from the International Registry of Acute Aortic Dissection (IRAD). Eur J Vasc Endovasc Surg 2009; 37:149–159.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Contemporary results of surgery in acute type A aortic dissection: the International Registry of Acute Aortic Dissection experience. J Thorac Cardiovasc Surg 2005; 129:112–122.
- Trimarchi S, Nienaber CA, Rampoldi V, et al. Role and results of surgery in acute type B aortic dissection. Insights from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2006; 114(suppl 1):I-357–I-364.
- Nienaber CA, Rousseau H, Eggbrecht H, et al. Randomized comparison of strategies for type B aortic dissection. The Investigation of STEnt grafts in Aortic Dissection (INSTEAD) Trial. Circulation 2009; 120:2519–2528.
- Evangelista A, Mukherjee D, Mehta RH, et al. Acute intramural hematoma of the aorta. Circulation 2005; 111:1063–1070.
- Pelzel JM, Braverman AC, Hirsch AT, Harris KM. International heterogeneity in diagnostic frequency and clinical outcomes of ascending aortic intramural hematoma. J Am Soc Echo 2007; 20:1260–1268.
- Song JK, Yim JH, Ahn JM, et al. Outcomes of patients with acute type A aortic intramural hematoma. Circulation 2009; 120:2046–2052.
- Harris KM, Pelzel JM, Braverman AC. Letter regarding article, “Outcomes of patients with acute type A intramural hematoma.” Circulation 2010; 121:e456.
- Sundt TM. Intramural hematoma and penetrating atherosclerotic ulcer of the aorta. Ann Thorac Surg 2007; 83:S835–S841.
- Juang D, Braverman A, Eagle K. Aortic dissection. Circulation 2008; 118:e507–e510.
- Tsai TT, Evangelista A, Nienaber CA, et al. Long-term survival in patients presenting with type A acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114(suppl I):I-350–I-356.
- Tsai TT, Fattori R, Trimarchi S, et al. Long-term survival in patients presenting with type B acute aortic dissection. Insights from the international registry of acute aortic dissection. Circulation 2006; 114:2226–2231.
- Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections: incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82:1400–1405.
KEY POINTS
- Aortic surgery is the treatment of choice for dissection of the ascending aorta, whereas dissection of the descending aorta is initially managed medically.
- Look for an underlying genetic predisposition to aortic disease and, in many instances, screen first-degree relatives for aortic disease.
- Long-term management requires serial imaging of the aorta, blood pressure control, and, for many, future aortic procedures.
- Measuring the D-dimer levels may help in decision-making for appropriate imaging in patients presenting with chest pain, as an elevated level raises the suspicion of dissection. However, more study of this and other biomarkers is needed.
- Advances in molecular genetics and the biology of the aortic wall promise to improve the diagnosis and prognosis of aortic disease.
Vitamin D Deficiency Boosts CV Events After Vascular Surgery
PARIS – Patients with severe vitamin D deficiency who underwent vascular surgery faced a significant, 56% increased risk for developing a cardiovascular event during the 30 days following surgery compared with patients who had a sufficient vitamin D level, in a review of 269 patients treated at a single Dutch center.
The study findings did not address whether patients who were vitamin D deficient and then received a vitamin D supplement had a reduced number of postsurgical events, Dr. Koen M. van de Luijtgaarden and his associates from Erasmus University Medical Center in Rotterdam, The Netherlands reported in a poster at the meeting.
The study also documented the frequency of vitamin D deficiency or insufficiency in patients scheduled for vascular surgery. Of the 269 patients included in the review who underwent vascular surgery during 2008-2010 at Erasmus Medical Center, 78 (29%) had a sufficient plasma level of vitamin D at baseline, defined as a 25-hydroxyvitamin D level of greater than 75 nmol/L. In all 38 of the patients (14%) had severe vitamin D deficiency at baseline, defined as 25 nmol/L or less. A total of 76 (28%) had moderate deficiency, defined as more than 25 nmol/L to 50 nmol/L, and 77 (29%) had vitamin D insufficiency, defined as more than 50 nmol/L to 75 nmol/L.
About two-thirds of the surgery patients were men, and the average age of all patients in the review was 66.
The primary endpoint of the study was the rate of cardiovascular events during the first 30 days after surgery, a composite tally that included the rates of cardiovascular deaths, nonfatal myocardial damage, or stroke. The incidence of these events was 38% in the severely deficient patients, 24% in patients with moderate deficiency, 32% in patients with vitamin D insufficiency, and 13% in those with a sufficient level of the vitamin.
In a multivariate analysis that controlled for baseline differences in demographics, medical history, and medications, patients in the severely deficient group had a statistically significant, 56% increased incidence of cardiovascular events, compared with patients with a sufficient vitamin D level. Patients with moderate deficiency or insufficiency both showed trends toward more events compared with the sufficient group, but in neither case was the difference statistically significant.
The findings are consistent with prior reports that vitamin D deficiency links with an increased risk for atherosclerosis and cardiovascular events, said Dr. van de Luijtgaarden, a researcher in the department of vascular surgery at Erasmus, and his associates.
Dr. van de Luijtgaarden said that he and his associates had no disclosures.
PARIS – Patients with severe vitamin D deficiency who underwent vascular surgery faced a significant, 56% increased risk for developing a cardiovascular event during the 30 days following surgery compared with patients who had a sufficient vitamin D level, in a review of 269 patients treated at a single Dutch center.
The study findings did not address whether patients who were vitamin D deficient and then received a vitamin D supplement had a reduced number of postsurgical events, Dr. Koen M. van de Luijtgaarden and his associates from Erasmus University Medical Center in Rotterdam, The Netherlands reported in a poster at the meeting.
The study also documented the frequency of vitamin D deficiency or insufficiency in patients scheduled for vascular surgery. Of the 269 patients included in the review who underwent vascular surgery during 2008-2010 at Erasmus Medical Center, 78 (29%) had a sufficient plasma level of vitamin D at baseline, defined as a 25-hydroxyvitamin D level of greater than 75 nmol/L. In all 38 of the patients (14%) had severe vitamin D deficiency at baseline, defined as 25 nmol/L or less. A total of 76 (28%) had moderate deficiency, defined as more than 25 nmol/L to 50 nmol/L, and 77 (29%) had vitamin D insufficiency, defined as more than 50 nmol/L to 75 nmol/L.
About two-thirds of the surgery patients were men, and the average age of all patients in the review was 66.
The primary endpoint of the study was the rate of cardiovascular events during the first 30 days after surgery, a composite tally that included the rates of cardiovascular deaths, nonfatal myocardial damage, or stroke. The incidence of these events was 38% in the severely deficient patients, 24% in patients with moderate deficiency, 32% in patients with vitamin D insufficiency, and 13% in those with a sufficient level of the vitamin.
In a multivariate analysis that controlled for baseline differences in demographics, medical history, and medications, patients in the severely deficient group had a statistically significant, 56% increased incidence of cardiovascular events, compared with patients with a sufficient vitamin D level. Patients with moderate deficiency or insufficiency both showed trends toward more events compared with the sufficient group, but in neither case was the difference statistically significant.
The findings are consistent with prior reports that vitamin D deficiency links with an increased risk for atherosclerosis and cardiovascular events, said Dr. van de Luijtgaarden, a researcher in the department of vascular surgery at Erasmus, and his associates.
Dr. van de Luijtgaarden said that he and his associates had no disclosures.
PARIS – Patients with severe vitamin D deficiency who underwent vascular surgery faced a significant, 56% increased risk for developing a cardiovascular event during the 30 days following surgery compared with patients who had a sufficient vitamin D level, in a review of 269 patients treated at a single Dutch center.
The study findings did not address whether patients who were vitamin D deficient and then received a vitamin D supplement had a reduced number of postsurgical events, Dr. Koen M. van de Luijtgaarden and his associates from Erasmus University Medical Center in Rotterdam, The Netherlands reported in a poster at the meeting.
The study also documented the frequency of vitamin D deficiency or insufficiency in patients scheduled for vascular surgery. Of the 269 patients included in the review who underwent vascular surgery during 2008-2010 at Erasmus Medical Center, 78 (29%) had a sufficient plasma level of vitamin D at baseline, defined as a 25-hydroxyvitamin D level of greater than 75 nmol/L. In all 38 of the patients (14%) had severe vitamin D deficiency at baseline, defined as 25 nmol/L or less. A total of 76 (28%) had moderate deficiency, defined as more than 25 nmol/L to 50 nmol/L, and 77 (29%) had vitamin D insufficiency, defined as more than 50 nmol/L to 75 nmol/L.
About two-thirds of the surgery patients were men, and the average age of all patients in the review was 66.
The primary endpoint of the study was the rate of cardiovascular events during the first 30 days after surgery, a composite tally that included the rates of cardiovascular deaths, nonfatal myocardial damage, or stroke. The incidence of these events was 38% in the severely deficient patients, 24% in patients with moderate deficiency, 32% in patients with vitamin D insufficiency, and 13% in those with a sufficient level of the vitamin.
In a multivariate analysis that controlled for baseline differences in demographics, medical history, and medications, patients in the severely deficient group had a statistically significant, 56% increased incidence of cardiovascular events, compared with patients with a sufficient vitamin D level. Patients with moderate deficiency or insufficiency both showed trends toward more events compared with the sufficient group, but in neither case was the difference statistically significant.
The findings are consistent with prior reports that vitamin D deficiency links with an increased risk for atherosclerosis and cardiovascular events, said Dr. van de Luijtgaarden, a researcher in the department of vascular surgery at Erasmus, and his associates.
Dr. van de Luijtgaarden said that he and his associates had no disclosures.
FROM THE ANNUAL CONGRESS OF THE EUROPEAN SOCIETY OF CARDIOLOGY
Major Finding: During the 30 days following vascular surgery, patients with severe vitamin D deficiency at baseline had a statistically significant, 56% increased rate of cardiovascular events, compared with patients with sufficient plasma levels of vitamin D in a multivariate analysis that controlled for baseline differences in demographics, medical history, and medication.
Data Source: Review of 269 patients who underwent vascular surgery at Erasmus University Medical Center during 2008-2010.
Disclosures: Dr. van de Luijtgaarden said that he and his associates had no disclosures.
Venous thromboembolism: What to do after anticoagulation is started
Deep vein thrombosis and pulmonary embolism are collectively referred to as venous thromboembolic (VTE) disease. They affect approximately 100,000 to 300,000 patients per year in the United States.1 Although patients with deep vein thrombosis can be treated as outpatients, many are admitted for the initiation of anticoagulation. Initial anticoagulation usually requires the overlap of a parenteral anticoagulant (unfractionated heparin, low-molecular-weight heparin [LMWH] or fondaparinux) with warfarin for a minimum of 5 days and until the international normalized ratio (INR) of the prothrombin time is above 2.0 for at least 24 hours.2
Three clinical issues need to be addressed after the initiation of anticoagulation for VTE:
- Determination of the length of anticoagulation with the correct anticoagulant
- Prevention of postthrombotic syndrome
- Appropriate screening for occult malignancy.
HOW LONG SHOULD VTE BE TREATED?
The duration of anticoagulation has been a matter of debate.
The risk of recurrent VTE appears related to clinical risk factors that a patient has at the time of the initial thrombotic event. An epidemiologic study3 found that patients with VTE treated for approximately 6 months had a low rate of recurrence (0% at 2 years of follow-up) if surgery was the risk factor. The risk climbed to 9% if the risk factor was nonsurgical and to 19% if there were no provoking risk factors.
The likelihood of VTE recurrence and therefore the recommended duration of treatment depend on whether the VTE event was provoked, cancer-related, recurrent, thrombophilia-related, or idiopathic. We address each of these scenarios below.
HOW LONG TO TREAT PROVOKED VTE
A VTE event is considered provoked if the patient had a clear inciting risk factor. As defined in various clinical trials, these risk factors include:
- Hospitalization with confinement to bed for 3 or more consecutive days in the last 3 months
- Surgery or general anesthesia in the last 3 months
- Immobilization for more than 7 days, regardless of the cause
- Trauma in the last 3 months
- Pregnancy
- Use of an oral contraceptive, regardless of which estrogen or progesterone analogue it contains
- Travel for more than 4 hours
- Recent childbirth.
However, the trials that tested different lengths of anticoagulation have varied markedly in how they defined provoked deep vein thrombosis.4–7
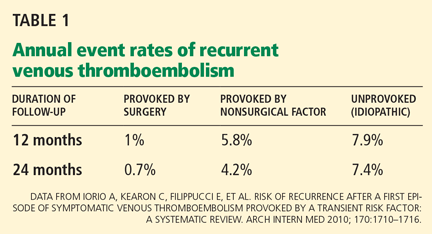
Recommendation: Warfarin or equivalent for 3 months
The American College of Chest Physicians (ACCP) recommends 3 months of anticoagulation with warfarin or another vitamin K antagonist for patients with VTE secondary to a transient (reversible) risk factor,2 and we agree.
HOW LONG TO TREAT CANCER-RELATED VTE
Patients with cancer are at higher risk of developing VTE. Furthermore, in one study,9 compared with other patients with VTE, patients with cancer were three times more likely to have another episode of VTE, with a cumulative rate of recurrence at 1 year of 21% vs 7%. Cancer patients were also twice as likely to suffer major bleeding complications while on anticoagulation.9
Warfarin is a difficult drug to manage because it has many interactions with foods, diseases, and other drugs. These difficulties are amplified in many cancer patients during chemotherapy.
Warfarin was compared with a LMWH in four randomized trials in cancer patients, and a meta-analysis10 found a 50% relative reduction in the rates of recurrent deep vein thrombosis and pulmonary embolism with LMWH treatment. These results were driven primarily by the CLOT trial (Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer),11 which showed an 8% absolute risk reduction (number needed to treat 13) without an increase in major bleeding when cancer-related VTE was treated with an LMWH—ie, dalteparin (Fragmin)—for 6 months compared with warfarin.
Current thinking suggests that VTE should be treated until the cancer is resolved. However, this hypothesis has not been adequately tested, and consequently, the ACCP gives it only a level 1C recommendation.2 The largest of the four trials comparing warfarin and an LMWH lasted only 6 months. The safety of extending LMWH treatment beyond 6 months is currently unknown but is under investigation (clinicaltrials.gov identifier NCT00942968).
Recommendation: LMWH therapy for at least 6 months
The ACCP guidelines recommend LMWH therapy for 3 to 6 months, followed by warfarin or another vitamin K antagonist or continued LMWH treatment until the cancer is resolved.2
The National Comprehensive Cancer Network guidelines recommend an LMWH for 6 months as monotherapy and indefinite anticoagulation if the cancer is still active.12
The American Society of Clinical Oncology guidelines recommend an LMWH for at least 6 months and indefinite anticoagulant therapy for selected patients with active cancer.13
We agree that patients with active cancer should receive an LMWH for at least 6 months and indefinite anticoagulation until the cancer is resolved.
In our experience, many patients are reluctant to give themselves the daily injections that LMWH therapy requires, and so they need to be well-informed about the marked decrease in VTE recurrence with this less-convenient and more-expensive therapy. Many patients face insurance barriers to cover the cost of LMWH therapy; however, careful attention to preauthorization can usually overcome this obstacle.
HOW LONG TO TREAT RECURRENT VTE
It makes clinical sense that patients who have a second VTE event should be treated indefinitely. This theory was tested in a randomized clinical trial14 in which patients with provoked or unprovoked VTE were randomized after their second event to receive anticoagulation for 6 months vs indefinitely.
After 4 years of follow-up, the recurrence rate was 21% in patients assigned to 6 months of treatment and only 3% in patients who continued anticoagulation throughout the trial. On the other hand, major hemorrhage occurred in 3% of patients treated for 6 months and in 9% in patients who continued anticoagulation indefinitely.
Of note, most of the patients in this trial had unprovoked (idiopathic) VTE, so the results should not be extrapolated to patients with provoked VTE, who accounted for only 20% of the study population.14
Recommendation: Long-term anticoagulation
We agree with the ACCP recommendation2 that patients who have had a second episode of unprovoked VTE should receive long-term anticoagulation. Because of a lack of data, the duration of therapy for patients with a second episode of provoked VTE should be individualized.
HOW LONG TO TREAT THROMBOPHILIA-RELATED VTE
Inherited thrombophilias
Patients with VTE that is not related to a clear provoking risk factor or cancer frequently have testing to evaluate for a hypercoagulable state. This workup traditionally includes the most common inherited thrombophilias for gene mutations for factor V and prothrombin as well as for deficiencies in protein C, protein S, antithrombin and the acquired antiphospholipid syndrome.
The key questions that should be asked prior to embarking on this workup are:
- Will the results change the length of therapy for the patient?
- Will testing the patient help with genetic counseling and possible testing of family members?
- Will the results change the targeted INR range for warfarin or other vitamin K antagonist therapy?
Patients with inherited thrombophilia have a greater risk of developing an initial VTE event; however, these tests do not help predict the recurrence of VTE in patients with established disease more than clinical risk factors do. A prospective study demonstrated this by looking at the effect of thrombophilia and clinical factors on the recurrence of venous thrombosis and found that inherited prothrombotic abnormalities do not appear to play an important role in the risk of a recurrent event.15 On the other hand, clinical factors, such as whether the first event was idiopathic or provoked, appear more important in determining the duration of anticoagulation therapy.15 A systematic review of the common inherited thrombophilias showed the VTE recurrence rate of patients with factor V Leiden was higher than in patients without the mutation; however, the absolute rates of recurrence were not much different than what would be expected in patients with idiopathic VTE.16
A retrospective study involving a large cohort of families of patients who already had experienced a first episode of either idiopathic or provoked VTE showed high annual risks of recurrent VTE associated with hereditary deficiencies of protein S (8.4%), protein C (6.0%), and antithrombin (10%).17 However, for the more commonly occurring genetic thrombophilias, the factor V Leiden and prothrombin G20210A mutations, family members with either gene abnormality had low rates of VTE, suggesting that testing of relatives of probands is not clinically useful.16
Antiphospholipid syndrome
Antiphospholipid syndrome is an acquired thrombophilia. A patient has thrombotic antiphospholipid syndrome when there is a history of vascular thrombosis in the presence of persistently positive tests (at least 12 weeks apart) for lupus anticoagulants, anticardiolipin antibodies, or anti-beta-2 glycoprotein I. A prospective study of 412 patients with a first episode of VTE found that 15% were positive for anticardiolipin antibody at the end of 6 months of anticoagulation. The risk of recurrent VTE after 4 years was 29% in patients with antibodies and 14% in those without antibodies (relative risk 2.1; 95% confidence interval [CI] 1.3–3.3; P =.0013).18
Recent reviews advise indefinite warfarin anticoagulation in patients with VTE and persistence of antiphospholipid antibodies.19 However, the optimal duration of anticoagulation is uncertain. Until well-designed clinical trials are done, the current general consensus is to anticoagulate these patients indefinitely.20,21 Retrospective studies had suggested that patients with antiphospholipid antibodies required a higher therapeutic INR range; however, this observation was tested in two trials that found no difference in thromboembolic rates when patients were randomized to an INR of 2.0–3.0 vs 3.1–4.0,22 or 2.0–3.0 vs 3.0–4.5.23
No formal recommendations
In the absence of strong evidence, the ACCP guidelines do not include a recommendation on the duration of anticoagulation treatment specific to inherited thrombophilias. We believe that clinical factors are more important than inherited thrombophilias for deciding the duration of anticoagulation, and that testing is almost never indicated or useful. However, patients with antiphospholipid syndrome are at high risk of recurrence, and it is our practice to anticoagulate these patients indefinitely.
HOW LONG TO TREAT UNPROVOKED (IDIOPATHIC) VTE
A VTE event is thought to be idiopathic if it occurs without a clearly identified provoking factor.
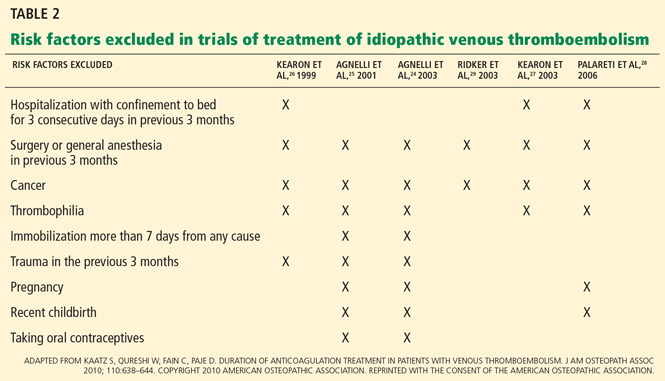
In an observational study,3 patients with oral contraceptive use, transient illness, immobilization, or a history of travel had an 8.8% risk of recurrence vs 19.4% in patients with unprovoked VTE. The meta-analysis discussed above (Table 1)8 also shows that patients with these nonsurgical risk factors have a lower rate of recurrence than patients with idiopathic VTE.
The high rate of recurrence of idiopathic VTE (4% to 27% after 3 months of anticoagulation24–26) suggests that a longer duration of treatment is reasonable. However, increasing the length of therapy from 3 to 12 months delays but does not prevent recurrence, the risk of which begins to accumulate once anticoagulation is stopped.24,25
Three promising strategies to identify subgroups of patients with idiopathic VTE who are at highest risk of recurrence and who would benefit the most from prolonged anticoagulation are d-dimer testing, evaluation for residual vein thrombosis in patients who present with a deep vein thrombosis, and clinical prediction rules.
d-dimer testing
d-dimer is a degradation product of fibrin and is an indirect marker of residual thrombosis.30
In a systematic review of patients with a first episode of unprovoked VTE,31 a normal d-dimer concentration at the end of at least 3 months of anticoagulation was associated with a 3.5% annual risk of recurrence, whereas an elevated d-dimer level at that time was associated with an annual risk of 8.9%. These results were confirmed in a systematic review of individual patient data.32
In a randomized trial,28 patients with an idiopathic VTE event who received anticoagulation for at least 3 months had their d-dimer level measured 1 month after cessation of treatment. Those with an elevated level were randomized to either resume anticoagulation or not. Patients who resumed anticoagulation had an annual recurrence rate of 2%; however, those who were allocated not to restart anticoagulation had a recurrence rate of 10.9% per year. There was no difference in the rate of major bleeding between the two groups. Patients in this clinical trial who had a normal d-dimer level did not restart anticoagulation and had an annual recurrence rate of 4.4%.
Evaluation for residual thrombosis

A randomized trial34 in patients with both provoked and idiopathic deep vein thrombosis showed a reduction in recurrence when those who had residual vein thrombosis were given extended anticoagulation. In the subset of patients whose deep vein thrombosis was idiopathic, the recurrence rate was 17% per year when treatment lasted only 3 months and 10% when it was extended for up to 1 year.
Another trial35 randomized patients with provoked and idiopathic deep vein thrombosis to receive anticoagulation for the usual duration or to continue treatment until recanalization of the residual thrombus was demonstrated on follow-up Doppler ultrasonography. Patients who received this ultrasonography-tailored treatment had a lower rate of recurrence of VTE; however, the absolute reductions in recurrence rates cannot be calculated from this report for patients with idiopathic deep vein thrombosis.
A prospective observational study36 of the predictive value of d-dimer status and residual vein thrombus found that only d-dimer was an independent risk factor for recurrent VTE after vitamin K antagonist withdrawal.
A clinical prediction rule: ‘Men and HERDOO2’
A promising tool for predicting if a patient is at low risk of recurrent VTE after the first episode of proximal deep vein thrombosis or pulmonary embolism is known by the mnemonic device “Men and HERDOO2.” It is based on data prospectively derived by Rodger et al37 to identify patients with less than a 3% annual risk of recurrent VTE after their first event of idiopathic proximal deep vein thrombosis or pulmonary embolism. Risk factors for recurrent VTE were male sex (the “men” of “Men and HERDOO2”), signs of postthrombotic syndrome, including hyperpigmentation of the lower extremities, edema or redness of either leg, a d-dimer level > 250 μg/L, obesity (body mass index > 30 kg/m2, and older age (> 65 years).
Overall, one-fourth of the population were women with no risk factors or one risk factor, and their risk of recurrence was 1.6% per year. Men and women who had two or more risk factors for postthrombotic syndrome (hyperpigmentation, edema, or redness), elevated d-dimer, obesity, or older age were predicted to be at higher risk of recurrent VTE. Patients such as this should be considered for indefinite anticoagulation.
Ideally, clinical prediction rules should be validated in a separate group of patients before they are used routinely in practice,38 and this clinical prediction rule is currently being tested in the REVERSE II study. If the results are consistent, this will be an easy-to-use tool to help identify patients who likely can safely stop anticoagulation therapy after 3 to 6 months (clinicaltrials.gov Identifier: NCT00967304).
The location of the thrombosis also influences the likelihood of recurrence. Patients with isolated distal (calf) deep vein thrombosis are less likely to suffer recurrent VTE than those who present with proximal deep vein thrombosis. However, trials focusing specifically on the precise subset of idiopathic isolated distal deep vein thrombosis are lacking. In a randomized trial39 comparing 6 vs 12 weeks of anticoagulation for isolated distal deep vein thrombosis and 12 vs 24 weeks for proximal deep vein thrombosis, the annual rates of recurrence after 12 weeks of treatment were approximately 3.4% for isolated distal and 8.1% for proximal deep vein thrombosis.39
Recommendation: At least 3 months of warfarin or equivalent
We agree with the ACCP recommendation2 that patients with unprovoked VTE should receive at least 3 months of anticoagulation with a vitamin K antagonist.
If the patient has no risk factors for bleeding and good anticoagulant monitoring is achievable, we agree with long-term anticoagulation for proximal unprovoked deep vein thrombosis or pulmonary embolism, and 3 months of therapy for isolated distal unprovoked deep vein thrombosis.
Patient preferences and the risk of recurrence vs the risk of bleeding should be discussed with patients when contemplating indefinite anticoagulation.
If testing is being considered to assist in the decision to prescribe indefinite anticoagulation, we prefer using d-dimer levels rather than ultrasonography to detect residual venous thrombosis because of its ease of use and the strength of the current evidence.
PREVENTING POSTTHROMBOTIC SYNDROME
The postthrombotic (postphlebitic) syndrome is a chronic and burdensome consequence of deep vein thrombosis that occurs despite anticoagulation therapy. It is estimated to affect 23% to 60% of patients and typically manifests in the first 2 years.40 It is not only costly in clinical terms, with decreased quality of life for the patient, but health care expenditures have been estimated to range from $400 per year in a Brazilian study to $7,000 per year in a US study.40
Typical symptoms include leg pain, heaviness, swelling, and cramping. In severe cases, chronic venous ulcers can occur and are difficult to treat.41
The definition of postthrombotic syndrome has been unclear over the years, and six different scales that measure signs and symptoms have been reported.42
The Villalta scale has been proposed by the International Society of Thrombosis and Hemostasis as a diagnostic standard to define postthrombotic syndrome.42 This validated scale is based on five clinical symptoms, six clinical signs, and the presence or absence of venous ulcers. Each clinical symptom and sign is scored as mild (1 point), moderate (2 points), or severe (3 points). Symptoms include pain, cramps, heaviness, paresthesia, and pruritus; the six clinical signs are pretibial edema, skin induration, hyperpigmentation, redness, venous ectasia, and pain on calf compression.
According to the International Society of Thrombosis and Hemostasis, postthrombotic syndrome is present if the Villalta score is 5 or greater or if a venous ulcer is present in a leg with previous deep vein thrombosis. Further, using the Villalta scale, postthrombotic syndrome can be categorized as mild (score 5–9), moderate (10–14), or severe (≥ 15).
A limitation of the Villalta scale is that the presence or absence of a venous ulcer has not been assigned a score. Since a venous ulcer requires more aggressive measures, the society defines postthrombotic syndrome as severe if venous ulcers are present.42
Acute symptoms of deep vein thrombosis may take months to resolve and, indeed, acute symptoms may transition to chronic symptoms without a symptom-free interval. It is recommended that postthrombotic syndrome not be diagnosed before 3 months to avoid inappropriately attributing acute symptoms and signs of deep vein thrombosis to the postthrombotic syndrome.42
Studies of stockings
A systematic review of three randomized trials44 concluded that elastic compression stockings reduce the risk of postthrombotic syndrome (any severity) from 43% to 20% and severe postthrombotic syndrome from 15% to 7%.43
The first of these trials44 randomized patients soon after the diagnosis of deep vein thrombosis to receive made-to-order compression stockings that were rated at 30 to 40 mm Hg or to be in a control group that did not receive stockings. The second trial45 randomized patients 1 year after the index event of deep vein thrombosis to receive 20- to 30-mm Hg stockings or stockings that were two sizes too large (the control group). The third study46 randomly allocated patients to receive “off-the-shelf” stockings (30–40 mm Hg) or no stockings. Each study used its own definition of postthrombotic syndrome.
Although these studies strongly suggest compression stockings prevent postthrombotic syndrome, several methodologic issues remain:
- A standard definition of postthrombotic syndrome was not used
- The amount of compression varied between studies
- The studies were not blinded.
Lack of blinding becomes most significant when an outcome is based on subjective findings, like the symptoms that make up a large part of the diagnosis of postthrombotic syndrome.
The SOX trial, currently under way, is designed to address these methodologic issues and should be completed in 2012 (clinicaltrials.gov Identifier: NCT00143598).
Recommendation: Stockings for at least 2 years
We agree with the ACCP recommendation that a patient who has had a symptomatic proximal deep vein thrombosis should wear an elastic compression stocking with an ankle pressure gradient of 30 to 40 mm Hg as soon as possible after starting anticoagulant therapy and continuing for a minimum of 2 years.2
SCREENING FOR OCCULT MALIGNANCY
VTE can be the first manifestation of cancer.
French physician Armand Trousseau, in the 1860s, was the first to describe disseminated intravascular coagulation closely associated with adenocarcinoma. Ironically, several years later, after suffering for weeks from abdominal pain, he declared to one of his students that he had developed thrombosis, and he died of gastric cancer shortly thereafter.47
Since cancer is a well-known risk factor for VTE, it is logical to screen for cancer as an explanation for an idiopathic VTE event.48 To make an informed decision, one needs to understand the rate of occult cancer at the time VTE is diagnosed, the risk of future development of cancer, and the utility of extensive cancer screening.
The clinical efficacy, side effects, and cost-effectiveness of cancer screening in patients with idiopathic VTE are unknown. However, a systematic review47 of 34 studies found that, in patients with idiopathic VTE, cancer was diagnosed within 1 month in 6.1%, within 6 months in 8.6%, and within 1 year in 10.0% (95% CI 8.6–11.3).
A subset of studies compared two strategies for screening soon after the diagnosis of idiopathic VTE: a strategy limited to the history, physical examination, basic blood work, and chest radiography vs an extensive screening strategy that also included serum tumor markers or abdominal ultrasonography or computed tomography. Limited screening detected 49% of the prevalent cancers; extensive screening increased this rate to 70%. Stated another way, the detection rate for prevalent cancers was 5% with limited screening and 7% with extensive screening soon after the diagnosis of idiopathic VTE.47
Patients with idiopathic VTE had higher rates of cancer within 1 month of diagnosis than patients with provoked VTE (6.1% vs 1.9%), and this difference persisted at 1 year (10.0% vs 2.6%).47
Recommendation: Individualized cancer screening
Patients with idiopathic VTE have a significant risk of occult cancer within the first year after diagnosis, and cancer screening should be considered. Our practice for patients with idiopathic VTE is to perform a history and physical examination and ensure that the patient is up to date on age- and sex-specific cancer screening.
The use of additional imaging or biomarkers should be discussed with patients so they can balance the risks (radiation and potential false-positive results with their downstream consequences), costs, and potential benefits, given the lack of proven survival benefit or cost-effectiveness.
ORAL ANTICOAGULANT MANAGEMENT
Warfarin’s multiple interactions, along with the need for INR monitoring, make it a difficult medication to manage.
The Joint Commission, the US organization for health service accreditation and certification, has defined National Patient Safety Goals and quality measures for the management of anticoagulation.49 Organized anticoagulation management services, dosing algorithms, and patient self-testing using capillary INR meters or patient self-management of warfarin were recommended as tools to improve the time patients spend in the therapeutic INR range.50
Two new oral anticoagulants
The limitations of warfarin have stimulated the search for newer oral anticoagulants that do not require laboratory monitoring or have as many diet and drug interactions.
Two trials have been published with experimental oral anticoagulants that had similar efficacy and safety as warfarin in the treatment of VTE.
The study of dabigatran (Pradaxa) vs warfarin in the treatment of acute VTE (the RECOVER trial)51 randomized 2,539 patients with acute VTE to receive the oral direct thrombin inhibitor dabigatran or warfarin for approximately 6 months. Of note, each treatment group received a median of 6 days of heparin, LMWH, or fondaparinux at the beginning of blinded therapy. The rates of recurrent VTE and major bleeding were similar between the treatment arms, and overall bleeding was less with dabigatran. Dabigatran was approved in the United States in October 2010 for stroke prevention in atrial fibrillation but has yet to be approved for the treatment of VTE pending further study (clinicaltrials.gov Identifier: NCT00680186).
A study of oral rivaroxaban (Xarelto) for symptomatic VTE (the EINSTEIN-DVT trial) 52 randomized 3,449 patients with acute deep vein thrombosis to rivaroxaban or enoxaparin (Lovenox) overlapped with warfarin or another vitamin K antagonist in the usual manner. No difference was noted between the treatments in the rate of recurrence of VTE or of major bleeding. Of note, patients randomized to rivaroxaban received 15 mg twice a day for the first 3 weeks of treatment and then 20 mg per day for the remainder of their therapy and did not require parenteral anticoagulant overlap.
The long-awaited promise of easier-to-use oral anticoagulants for the treatment of VTE is drawing near and has the potential to revolutionize the treatment of this common disorder. In the meantime, close monitoring of warfarin and careful patient education regarding its use are essential. And even with the development of new drugs in the future, it is still imperative that patients with acute VTE receive the correct length of anticoagulation treatment, are prescribed stockings to prevent postthrombotic syndrome, and are updated on routine cancer screening.
- Spencer FA, Emery C, Lessard D, et al. The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 2006; 21:722–727.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523–526.
- Schulman S, Lockner D, Juhlin-Dannfelt A. The duration of oral anticoagulation after deep vein thrombosis. A randomized study. Acta Med Scand 1985; 217:547–552.
- Optimum duration of anticoagulation for deep-vein thrombosis and pulmonary embolism. Research Committee of the British Thoracic Society. Lancet 1992; 340:873–876.
- Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. Duration of Anticoagulation Trial Study Group. N Engl J Med 1995; 332:1661–1665.
- Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743–749.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med 2010; 170:1710–1716.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hull RD, Pineo GF, Brant RF, et al; LITE Trial Investigators. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology, Venous Thromboembolic Disease. http://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed August 3, 2011.
- Lyman GH, Khorana AA, Falanga A, et al; American Society of Clinical Oncology. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Schulman S, Granqvist S, Holmström M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–2485.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost 2009; 101:93–99.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med 1998; 104:332–338.
- Derksen RH, de Groot PG. Towards evidence-based treatment of thrombotic antiphospholipid syndrome. Lupus 2010; 19:470–474.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Fonseca AG, D’Cruz DP. Controversies in the antiphospholipid syndrome: can we ever stop warfarin? J Autoimmune Dis 2008; 5:6.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3:848–853.
- Agnelli G, Prandoni P, Becattini C, et al; Warfarin Optimal Duration Italian Trial Investigators. Extended oral anticoagulant therapy after a first episode of pulmonary embolism. Ann Intern Med 2003; 139:19–25.
- Agnelli G, Prandoni P, Santamaria MG, et al; Warfarin Optimal Duration Italian Trial Investigators. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Kearon C, Ginsberg JS, Kovacs MJ, et al; Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–639.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Ridker PM, Goldhaber SZ, Glynn RJ. Low-intensity versus conventional-intensity warfarin for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:2164–2167.
- Bockenstedt P. D-dimer in venous thromboembolism. N Engl J Med 2003; 349:1203–1204.
- Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008; 149:481–490,W94.
- Douketis J, Tosetto A, Marcucci M, et al. Patient-level metaanalysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med 2010; 153:523–531.
- Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med 2002; 137:955–960.
- Siragusa S, Malato A, Anastasio R, et al. Residual vein thrombosis to establish duration of anticoagulation after a first episode of deep vein thrombosis: the Duration of Anticoagulation based on Compression UltraSonography (DACUS) study. Blood 2008; 112:511–515.
- Prandoni P, Prins MH, Lensing AW, et al; AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep venous thrombosis: a randomized trial. Ann Intern Med 2009; 150:577–585.
- Cosmi B, Legnani C, Cini M, Guazzaloca G, Palareti G. D-dimer levels in combination with residual venous obstruction and the risk of recurrence after anticoagulation withdrawal for a first idiopathic deep vein thrombosis. Thromb Haemost 2005; 94:969–974.
- Rodger MA, Kahn SR, Wells PS, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ 2008; 179:417–426.
- McGinn TG, Guyatt GH, Wyer PC, Naylor CD, Stiell IG, Richardson WS. Users’ guides to the medical literature: XXII: how to use articles about clinical decision rules. Evidence-Based Medicine Working Group. JAMA 2000; 284:79–84.
- Pinede L, Ninet J, Duhaut P, et al; Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103:2453–2460.
- Ashrani AA, Heit JA. Incidence and cost burden of postthrombotic syndrome. J Thromb Thrombolysis 2009; 28:465–476.
- Kahn SR, Shrier I, Julian JA, et al. Determinants and time course of the postthrombotic syndrome after acute deep venous thrombosis. Ann Intern Med 2008; 149:698–707.
- Kahn SR, Partsch H, Vedantham S, Prandoni P, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of post-thrombotic syndrome of the leg for use in clinical investigations: a recommendation for standardization. J Thromb Haemost 2009; 7:879–883.
- Kolbach DN, Sandbrink MW, Hamulyak K, Neumann HA, Prins MH. Non-pharmaceutical measures for prevention of post-thrombotic syndrome. Cochrane Database Syst Rev 2004;CD004174.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet 1997; 349:759–762.
- Ginsberg JS, Hirsh J, Julian J, et al. Prevention and treatment of postphlebitic syndrome: results of a 3-part study. Arch Intern Med 2001; 161:2105–2109.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the post-thrombotic syndrome: a randomized, controlled trial. Ann Intern Med 2004; 141:249–256.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008; 149:323–333.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Kaatz S. Impact on patient care: patient case through the continuum of care. J Thromb Thrombolysis 2010; 29:167–170.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Schulman S, Kearon C, Kakkar AK, et al; for the RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2452.
- The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363;2499–2510.
Deep vein thrombosis and pulmonary embolism are collectively referred to as venous thromboembolic (VTE) disease. They affect approximately 100,000 to 300,000 patients per year in the United States.1 Although patients with deep vein thrombosis can be treated as outpatients, many are admitted for the initiation of anticoagulation. Initial anticoagulation usually requires the overlap of a parenteral anticoagulant (unfractionated heparin, low-molecular-weight heparin [LMWH] or fondaparinux) with warfarin for a minimum of 5 days and until the international normalized ratio (INR) of the prothrombin time is above 2.0 for at least 24 hours.2
Three clinical issues need to be addressed after the initiation of anticoagulation for VTE:
- Determination of the length of anticoagulation with the correct anticoagulant
- Prevention of postthrombotic syndrome
- Appropriate screening for occult malignancy.
HOW LONG SHOULD VTE BE TREATED?
The duration of anticoagulation has been a matter of debate.
The risk of recurrent VTE appears related to clinical risk factors that a patient has at the time of the initial thrombotic event. An epidemiologic study3 found that patients with VTE treated for approximately 6 months had a low rate of recurrence (0% at 2 years of follow-up) if surgery was the risk factor. The risk climbed to 9% if the risk factor was nonsurgical and to 19% if there were no provoking risk factors.
The likelihood of VTE recurrence and therefore the recommended duration of treatment depend on whether the VTE event was provoked, cancer-related, recurrent, thrombophilia-related, or idiopathic. We address each of these scenarios below.
HOW LONG TO TREAT PROVOKED VTE
A VTE event is considered provoked if the patient had a clear inciting risk factor. As defined in various clinical trials, these risk factors include:
- Hospitalization with confinement to bed for 3 or more consecutive days in the last 3 months
- Surgery or general anesthesia in the last 3 months
- Immobilization for more than 7 days, regardless of the cause
- Trauma in the last 3 months
- Pregnancy
- Use of an oral contraceptive, regardless of which estrogen or progesterone analogue it contains
- Travel for more than 4 hours
- Recent childbirth.
However, the trials that tested different lengths of anticoagulation have varied markedly in how they defined provoked deep vein thrombosis.4–7
Recommendation: Warfarin or equivalent for 3 months
The American College of Chest Physicians (ACCP) recommends 3 months of anticoagulation with warfarin or another vitamin K antagonist for patients with VTE secondary to a transient (reversible) risk factor,2 and we agree.
HOW LONG TO TREAT CANCER-RELATED VTE
Patients with cancer are at higher risk of developing VTE. Furthermore, in one study,9 compared with other patients with VTE, patients with cancer were three times more likely to have another episode of VTE, with a cumulative rate of recurrence at 1 year of 21% vs 7%. Cancer patients were also twice as likely to suffer major bleeding complications while on anticoagulation.9
Warfarin is a difficult drug to manage because it has many interactions with foods, diseases, and other drugs. These difficulties are amplified in many cancer patients during chemotherapy.
Warfarin was compared with a LMWH in four randomized trials in cancer patients, and a meta-analysis10 found a 50% relative reduction in the rates of recurrent deep vein thrombosis and pulmonary embolism with LMWH treatment. These results were driven primarily by the CLOT trial (Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer),11 which showed an 8% absolute risk reduction (number needed to treat 13) without an increase in major bleeding when cancer-related VTE was treated with an LMWH—ie, dalteparin (Fragmin)—for 6 months compared with warfarin.
Current thinking suggests that VTE should be treated until the cancer is resolved. However, this hypothesis has not been adequately tested, and consequently, the ACCP gives it only a level 1C recommendation.2 The largest of the four trials comparing warfarin and an LMWH lasted only 6 months. The safety of extending LMWH treatment beyond 6 months is currently unknown but is under investigation (clinicaltrials.gov identifier NCT00942968).
Recommendation: LMWH therapy for at least 6 months
The ACCP guidelines recommend LMWH therapy for 3 to 6 months, followed by warfarin or another vitamin K antagonist or continued LMWH treatment until the cancer is resolved.2
The National Comprehensive Cancer Network guidelines recommend an LMWH for 6 months as monotherapy and indefinite anticoagulation if the cancer is still active.12
The American Society of Clinical Oncology guidelines recommend an LMWH for at least 6 months and indefinite anticoagulant therapy for selected patients with active cancer.13
We agree that patients with active cancer should receive an LMWH for at least 6 months and indefinite anticoagulation until the cancer is resolved.
In our experience, many patients are reluctant to give themselves the daily injections that LMWH therapy requires, and so they need to be well-informed about the marked decrease in VTE recurrence with this less-convenient and more-expensive therapy. Many patients face insurance barriers to cover the cost of LMWH therapy; however, careful attention to preauthorization can usually overcome this obstacle.
HOW LONG TO TREAT RECURRENT VTE
It makes clinical sense that patients who have a second VTE event should be treated indefinitely. This theory was tested in a randomized clinical trial14 in which patients with provoked or unprovoked VTE were randomized after their second event to receive anticoagulation for 6 months vs indefinitely.
After 4 years of follow-up, the recurrence rate was 21% in patients assigned to 6 months of treatment and only 3% in patients who continued anticoagulation throughout the trial. On the other hand, major hemorrhage occurred in 3% of patients treated for 6 months and in 9% in patients who continued anticoagulation indefinitely.
Of note, most of the patients in this trial had unprovoked (idiopathic) VTE, so the results should not be extrapolated to patients with provoked VTE, who accounted for only 20% of the study population.14
Recommendation: Long-term anticoagulation
We agree with the ACCP recommendation2 that patients who have had a second episode of unprovoked VTE should receive long-term anticoagulation. Because of a lack of data, the duration of therapy for patients with a second episode of provoked VTE should be individualized.
HOW LONG TO TREAT THROMBOPHILIA-RELATED VTE
Inherited thrombophilias
Patients with VTE that is not related to a clear provoking risk factor or cancer frequently have testing to evaluate for a hypercoagulable state. This workup traditionally includes the most common inherited thrombophilias for gene mutations for factor V and prothrombin as well as for deficiencies in protein C, protein S, antithrombin and the acquired antiphospholipid syndrome.
The key questions that should be asked prior to embarking on this workup are:
- Will the results change the length of therapy for the patient?
- Will testing the patient help with genetic counseling and possible testing of family members?
- Will the results change the targeted INR range for warfarin or other vitamin K antagonist therapy?
Patients with inherited thrombophilia have a greater risk of developing an initial VTE event; however, these tests do not help predict the recurrence of VTE in patients with established disease more than clinical risk factors do. A prospective study demonstrated this by looking at the effect of thrombophilia and clinical factors on the recurrence of venous thrombosis and found that inherited prothrombotic abnormalities do not appear to play an important role in the risk of a recurrent event.15 On the other hand, clinical factors, such as whether the first event was idiopathic or provoked, appear more important in determining the duration of anticoagulation therapy.15 A systematic review of the common inherited thrombophilias showed the VTE recurrence rate of patients with factor V Leiden was higher than in patients without the mutation; however, the absolute rates of recurrence were not much different than what would be expected in patients with idiopathic VTE.16
A retrospective study involving a large cohort of families of patients who already had experienced a first episode of either idiopathic or provoked VTE showed high annual risks of recurrent VTE associated with hereditary deficiencies of protein S (8.4%), protein C (6.0%), and antithrombin (10%).17 However, for the more commonly occurring genetic thrombophilias, the factor V Leiden and prothrombin G20210A mutations, family members with either gene abnormality had low rates of VTE, suggesting that testing of relatives of probands is not clinically useful.16
Antiphospholipid syndrome
Antiphospholipid syndrome is an acquired thrombophilia. A patient has thrombotic antiphospholipid syndrome when there is a history of vascular thrombosis in the presence of persistently positive tests (at least 12 weeks apart) for lupus anticoagulants, anticardiolipin antibodies, or anti-beta-2 glycoprotein I. A prospective study of 412 patients with a first episode of VTE found that 15% were positive for anticardiolipin antibody at the end of 6 months of anticoagulation. The risk of recurrent VTE after 4 years was 29% in patients with antibodies and 14% in those without antibodies (relative risk 2.1; 95% confidence interval [CI] 1.3–3.3; P =.0013).18
Recent reviews advise indefinite warfarin anticoagulation in patients with VTE and persistence of antiphospholipid antibodies.19 However, the optimal duration of anticoagulation is uncertain. Until well-designed clinical trials are done, the current general consensus is to anticoagulate these patients indefinitely.20,21 Retrospective studies had suggested that patients with antiphospholipid antibodies required a higher therapeutic INR range; however, this observation was tested in two trials that found no difference in thromboembolic rates when patients were randomized to an INR of 2.0–3.0 vs 3.1–4.0,22 or 2.0–3.0 vs 3.0–4.5.23
No formal recommendations
In the absence of strong evidence, the ACCP guidelines do not include a recommendation on the duration of anticoagulation treatment specific to inherited thrombophilias. We believe that clinical factors are more important than inherited thrombophilias for deciding the duration of anticoagulation, and that testing is almost never indicated or useful. However, patients with antiphospholipid syndrome are at high risk of recurrence, and it is our practice to anticoagulate these patients indefinitely.
HOW LONG TO TREAT UNPROVOKED (IDIOPATHIC) VTE
A VTE event is thought to be idiopathic if it occurs without a clearly identified provoking factor.
In an observational study,3 patients with oral contraceptive use, transient illness, immobilization, or a history of travel had an 8.8% risk of recurrence vs 19.4% in patients with unprovoked VTE. The meta-analysis discussed above (Table 1)8 also shows that patients with these nonsurgical risk factors have a lower rate of recurrence than patients with idiopathic VTE.
The high rate of recurrence of idiopathic VTE (4% to 27% after 3 months of anticoagulation24–26) suggests that a longer duration of treatment is reasonable. However, increasing the length of therapy from 3 to 12 months delays but does not prevent recurrence, the risk of which begins to accumulate once anticoagulation is stopped.24,25
Three promising strategies to identify subgroups of patients with idiopathic VTE who are at highest risk of recurrence and who would benefit the most from prolonged anticoagulation are d-dimer testing, evaluation for residual vein thrombosis in patients who present with a deep vein thrombosis, and clinical prediction rules.
d-dimer testing
d-dimer is a degradation product of fibrin and is an indirect marker of residual thrombosis.30
In a systematic review of patients with a first episode of unprovoked VTE,31 a normal d-dimer concentration at the end of at least 3 months of anticoagulation was associated with a 3.5% annual risk of recurrence, whereas an elevated d-dimer level at that time was associated with an annual risk of 8.9%. These results were confirmed in a systematic review of individual patient data.32
In a randomized trial,28 patients with an idiopathic VTE event who received anticoagulation for at least 3 months had their d-dimer level measured 1 month after cessation of treatment. Those with an elevated level were randomized to either resume anticoagulation or not. Patients who resumed anticoagulation had an annual recurrence rate of 2%; however, those who were allocated not to restart anticoagulation had a recurrence rate of 10.9% per year. There was no difference in the rate of major bleeding between the two groups. Patients in this clinical trial who had a normal d-dimer level did not restart anticoagulation and had an annual recurrence rate of 4.4%.
Evaluation for residual thrombosis
A randomized trial34 in patients with both provoked and idiopathic deep vein thrombosis showed a reduction in recurrence when those who had residual vein thrombosis were given extended anticoagulation. In the subset of patients whose deep vein thrombosis was idiopathic, the recurrence rate was 17% per year when treatment lasted only 3 months and 10% when it was extended for up to 1 year.
Another trial35 randomized patients with provoked and idiopathic deep vein thrombosis to receive anticoagulation for the usual duration or to continue treatment until recanalization of the residual thrombus was demonstrated on follow-up Doppler ultrasonography. Patients who received this ultrasonography-tailored treatment had a lower rate of recurrence of VTE; however, the absolute reductions in recurrence rates cannot be calculated from this report for patients with idiopathic deep vein thrombosis.
A prospective observational study36 of the predictive value of d-dimer status and residual vein thrombus found that only d-dimer was an independent risk factor for recurrent VTE after vitamin K antagonist withdrawal.
A clinical prediction rule: ‘Men and HERDOO2’
A promising tool for predicting if a patient is at low risk of recurrent VTE after the first episode of proximal deep vein thrombosis or pulmonary embolism is known by the mnemonic device “Men and HERDOO2.” It is based on data prospectively derived by Rodger et al37 to identify patients with less than a 3% annual risk of recurrent VTE after their first event of idiopathic proximal deep vein thrombosis or pulmonary embolism. Risk factors for recurrent VTE were male sex (the “men” of “Men and HERDOO2”), signs of postthrombotic syndrome, including hyperpigmentation of the lower extremities, edema or redness of either leg, a d-dimer level > 250 μg/L, obesity (body mass index > 30 kg/m2, and older age (> 65 years).
Overall, one-fourth of the population were women with no risk factors or one risk factor, and their risk of recurrence was 1.6% per year. Men and women who had two or more risk factors for postthrombotic syndrome (hyperpigmentation, edema, or redness), elevated d-dimer, obesity, or older age were predicted to be at higher risk of recurrent VTE. Patients such as this should be considered for indefinite anticoagulation.
Ideally, clinical prediction rules should be validated in a separate group of patients before they are used routinely in practice,38 and this clinical prediction rule is currently being tested in the REVERSE II study. If the results are consistent, this will be an easy-to-use tool to help identify patients who likely can safely stop anticoagulation therapy after 3 to 6 months (clinicaltrials.gov Identifier: NCT00967304).
The location of the thrombosis also influences the likelihood of recurrence. Patients with isolated distal (calf) deep vein thrombosis are less likely to suffer recurrent VTE than those who present with proximal deep vein thrombosis. However, trials focusing specifically on the precise subset of idiopathic isolated distal deep vein thrombosis are lacking. In a randomized trial39 comparing 6 vs 12 weeks of anticoagulation for isolated distal deep vein thrombosis and 12 vs 24 weeks for proximal deep vein thrombosis, the annual rates of recurrence after 12 weeks of treatment were approximately 3.4% for isolated distal and 8.1% for proximal deep vein thrombosis.39
Recommendation: At least 3 months of warfarin or equivalent
We agree with the ACCP recommendation2 that patients with unprovoked VTE should receive at least 3 months of anticoagulation with a vitamin K antagonist.
If the patient has no risk factors for bleeding and good anticoagulant monitoring is achievable, we agree with long-term anticoagulation for proximal unprovoked deep vein thrombosis or pulmonary embolism, and 3 months of therapy for isolated distal unprovoked deep vein thrombosis.
Patient preferences and the risk of recurrence vs the risk of bleeding should be discussed with patients when contemplating indefinite anticoagulation.
If testing is being considered to assist in the decision to prescribe indefinite anticoagulation, we prefer using d-dimer levels rather than ultrasonography to detect residual venous thrombosis because of its ease of use and the strength of the current evidence.
PREVENTING POSTTHROMBOTIC SYNDROME
The postthrombotic (postphlebitic) syndrome is a chronic and burdensome consequence of deep vein thrombosis that occurs despite anticoagulation therapy. It is estimated to affect 23% to 60% of patients and typically manifests in the first 2 years.40 It is not only costly in clinical terms, with decreased quality of life for the patient, but health care expenditures have been estimated to range from $400 per year in a Brazilian study to $7,000 per year in a US study.40
Typical symptoms include leg pain, heaviness, swelling, and cramping. In severe cases, chronic venous ulcers can occur and are difficult to treat.41
The definition of postthrombotic syndrome has been unclear over the years, and six different scales that measure signs and symptoms have been reported.42
The Villalta scale has been proposed by the International Society of Thrombosis and Hemostasis as a diagnostic standard to define postthrombotic syndrome.42 This validated scale is based on five clinical symptoms, six clinical signs, and the presence or absence of venous ulcers. Each clinical symptom and sign is scored as mild (1 point), moderate (2 points), or severe (3 points). Symptoms include pain, cramps, heaviness, paresthesia, and pruritus; the six clinical signs are pretibial edema, skin induration, hyperpigmentation, redness, venous ectasia, and pain on calf compression.
According to the International Society of Thrombosis and Hemostasis, postthrombotic syndrome is present if the Villalta score is 5 or greater or if a venous ulcer is present in a leg with previous deep vein thrombosis. Further, using the Villalta scale, postthrombotic syndrome can be categorized as mild (score 5–9), moderate (10–14), or severe (≥ 15).
A limitation of the Villalta scale is that the presence or absence of a venous ulcer has not been assigned a score. Since a venous ulcer requires more aggressive measures, the society defines postthrombotic syndrome as severe if venous ulcers are present.42
Acute symptoms of deep vein thrombosis may take months to resolve and, indeed, acute symptoms may transition to chronic symptoms without a symptom-free interval. It is recommended that postthrombotic syndrome not be diagnosed before 3 months to avoid inappropriately attributing acute symptoms and signs of deep vein thrombosis to the postthrombotic syndrome.42
Studies of stockings
A systematic review of three randomized trials44 concluded that elastic compression stockings reduce the risk of postthrombotic syndrome (any severity) from 43% to 20% and severe postthrombotic syndrome from 15% to 7%.43
The first of these trials44 randomized patients soon after the diagnosis of deep vein thrombosis to receive made-to-order compression stockings that were rated at 30 to 40 mm Hg or to be in a control group that did not receive stockings. The second trial45 randomized patients 1 year after the index event of deep vein thrombosis to receive 20- to 30-mm Hg stockings or stockings that were two sizes too large (the control group). The third study46 randomly allocated patients to receive “off-the-shelf” stockings (30–40 mm Hg) or no stockings. Each study used its own definition of postthrombotic syndrome.
Although these studies strongly suggest compression stockings prevent postthrombotic syndrome, several methodologic issues remain:
- A standard definition of postthrombotic syndrome was not used
- The amount of compression varied between studies
- The studies were not blinded.
Lack of blinding becomes most significant when an outcome is based on subjective findings, like the symptoms that make up a large part of the diagnosis of postthrombotic syndrome.
The SOX trial, currently under way, is designed to address these methodologic issues and should be completed in 2012 (clinicaltrials.gov Identifier: NCT00143598).
Recommendation: Stockings for at least 2 years
We agree with the ACCP recommendation that a patient who has had a symptomatic proximal deep vein thrombosis should wear an elastic compression stocking with an ankle pressure gradient of 30 to 40 mm Hg as soon as possible after starting anticoagulant therapy and continuing for a minimum of 2 years.2
SCREENING FOR OCCULT MALIGNANCY
VTE can be the first manifestation of cancer.
French physician Armand Trousseau, in the 1860s, was the first to describe disseminated intravascular coagulation closely associated with adenocarcinoma. Ironically, several years later, after suffering for weeks from abdominal pain, he declared to one of his students that he had developed thrombosis, and he died of gastric cancer shortly thereafter.47
Since cancer is a well-known risk factor for VTE, it is logical to screen for cancer as an explanation for an idiopathic VTE event.48 To make an informed decision, one needs to understand the rate of occult cancer at the time VTE is diagnosed, the risk of future development of cancer, and the utility of extensive cancer screening.
The clinical efficacy, side effects, and cost-effectiveness of cancer screening in patients with idiopathic VTE are unknown. However, a systematic review47 of 34 studies found that, in patients with idiopathic VTE, cancer was diagnosed within 1 month in 6.1%, within 6 months in 8.6%, and within 1 year in 10.0% (95% CI 8.6–11.3).
A subset of studies compared two strategies for screening soon after the diagnosis of idiopathic VTE: a strategy limited to the history, physical examination, basic blood work, and chest radiography vs an extensive screening strategy that also included serum tumor markers or abdominal ultrasonography or computed tomography. Limited screening detected 49% of the prevalent cancers; extensive screening increased this rate to 70%. Stated another way, the detection rate for prevalent cancers was 5% with limited screening and 7% with extensive screening soon after the diagnosis of idiopathic VTE.47
Patients with idiopathic VTE had higher rates of cancer within 1 month of diagnosis than patients with provoked VTE (6.1% vs 1.9%), and this difference persisted at 1 year (10.0% vs 2.6%).47
Recommendation: Individualized cancer screening
Patients with idiopathic VTE have a significant risk of occult cancer within the first year after diagnosis, and cancer screening should be considered. Our practice for patients with idiopathic VTE is to perform a history and physical examination and ensure that the patient is up to date on age- and sex-specific cancer screening.
The use of additional imaging or biomarkers should be discussed with patients so they can balance the risks (radiation and potential false-positive results with their downstream consequences), costs, and potential benefits, given the lack of proven survival benefit or cost-effectiveness.
ORAL ANTICOAGULANT MANAGEMENT
Warfarin’s multiple interactions, along with the need for INR monitoring, make it a difficult medication to manage.
The Joint Commission, the US organization for health service accreditation and certification, has defined National Patient Safety Goals and quality measures for the management of anticoagulation.49 Organized anticoagulation management services, dosing algorithms, and patient self-testing using capillary INR meters or patient self-management of warfarin were recommended as tools to improve the time patients spend in the therapeutic INR range.50
Two new oral anticoagulants
The limitations of warfarin have stimulated the search for newer oral anticoagulants that do not require laboratory monitoring or have as many diet and drug interactions.
Two trials have been published with experimental oral anticoagulants that had similar efficacy and safety as warfarin in the treatment of VTE.
The study of dabigatran (Pradaxa) vs warfarin in the treatment of acute VTE (the RECOVER trial)51 randomized 2,539 patients with acute VTE to receive the oral direct thrombin inhibitor dabigatran or warfarin for approximately 6 months. Of note, each treatment group received a median of 6 days of heparin, LMWH, or fondaparinux at the beginning of blinded therapy. The rates of recurrent VTE and major bleeding were similar between the treatment arms, and overall bleeding was less with dabigatran. Dabigatran was approved in the United States in October 2010 for stroke prevention in atrial fibrillation but has yet to be approved for the treatment of VTE pending further study (clinicaltrials.gov Identifier: NCT00680186).
A study of oral rivaroxaban (Xarelto) for symptomatic VTE (the EINSTEIN-DVT trial) 52 randomized 3,449 patients with acute deep vein thrombosis to rivaroxaban or enoxaparin (Lovenox) overlapped with warfarin or another vitamin K antagonist in the usual manner. No difference was noted between the treatments in the rate of recurrence of VTE or of major bleeding. Of note, patients randomized to rivaroxaban received 15 mg twice a day for the first 3 weeks of treatment and then 20 mg per day for the remainder of their therapy and did not require parenteral anticoagulant overlap.
The long-awaited promise of easier-to-use oral anticoagulants for the treatment of VTE is drawing near and has the potential to revolutionize the treatment of this common disorder. In the meantime, close monitoring of warfarin and careful patient education regarding its use are essential. And even with the development of new drugs in the future, it is still imperative that patients with acute VTE receive the correct length of anticoagulation treatment, are prescribed stockings to prevent postthrombotic syndrome, and are updated on routine cancer screening.
Deep vein thrombosis and pulmonary embolism are collectively referred to as venous thromboembolic (VTE) disease. They affect approximately 100,000 to 300,000 patients per year in the United States.1 Although patients with deep vein thrombosis can be treated as outpatients, many are admitted for the initiation of anticoagulation. Initial anticoagulation usually requires the overlap of a parenteral anticoagulant (unfractionated heparin, low-molecular-weight heparin [LMWH] or fondaparinux) with warfarin for a minimum of 5 days and until the international normalized ratio (INR) of the prothrombin time is above 2.0 for at least 24 hours.2
Three clinical issues need to be addressed after the initiation of anticoagulation for VTE:
- Determination of the length of anticoagulation with the correct anticoagulant
- Prevention of postthrombotic syndrome
- Appropriate screening for occult malignancy.
HOW LONG SHOULD VTE BE TREATED?
The duration of anticoagulation has been a matter of debate.
The risk of recurrent VTE appears related to clinical risk factors that a patient has at the time of the initial thrombotic event. An epidemiologic study3 found that patients with VTE treated for approximately 6 months had a low rate of recurrence (0% at 2 years of follow-up) if surgery was the risk factor. The risk climbed to 9% if the risk factor was nonsurgical and to 19% if there were no provoking risk factors.
The likelihood of VTE recurrence and therefore the recommended duration of treatment depend on whether the VTE event was provoked, cancer-related, recurrent, thrombophilia-related, or idiopathic. We address each of these scenarios below.
HOW LONG TO TREAT PROVOKED VTE
A VTE event is considered provoked if the patient had a clear inciting risk factor. As defined in various clinical trials, these risk factors include:
- Hospitalization with confinement to bed for 3 or more consecutive days in the last 3 months
- Surgery or general anesthesia in the last 3 months
- Immobilization for more than 7 days, regardless of the cause
- Trauma in the last 3 months
- Pregnancy
- Use of an oral contraceptive, regardless of which estrogen or progesterone analogue it contains
- Travel for more than 4 hours
- Recent childbirth.
However, the trials that tested different lengths of anticoagulation have varied markedly in how they defined provoked deep vein thrombosis.4–7
Recommendation: Warfarin or equivalent for 3 months
The American College of Chest Physicians (ACCP) recommends 3 months of anticoagulation with warfarin or another vitamin K antagonist for patients with VTE secondary to a transient (reversible) risk factor,2 and we agree.
HOW LONG TO TREAT CANCER-RELATED VTE
Patients with cancer are at higher risk of developing VTE. Furthermore, in one study,9 compared with other patients with VTE, patients with cancer were three times more likely to have another episode of VTE, with a cumulative rate of recurrence at 1 year of 21% vs 7%. Cancer patients were also twice as likely to suffer major bleeding complications while on anticoagulation.9
Warfarin is a difficult drug to manage because it has many interactions with foods, diseases, and other drugs. These difficulties are amplified in many cancer patients during chemotherapy.
Warfarin was compared with a LMWH in four randomized trials in cancer patients, and a meta-analysis10 found a 50% relative reduction in the rates of recurrent deep vein thrombosis and pulmonary embolism with LMWH treatment. These results were driven primarily by the CLOT trial (Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer),11 which showed an 8% absolute risk reduction (number needed to treat 13) without an increase in major bleeding when cancer-related VTE was treated with an LMWH—ie, dalteparin (Fragmin)—for 6 months compared with warfarin.
Current thinking suggests that VTE should be treated until the cancer is resolved. However, this hypothesis has not been adequately tested, and consequently, the ACCP gives it only a level 1C recommendation.2 The largest of the four trials comparing warfarin and an LMWH lasted only 6 months. The safety of extending LMWH treatment beyond 6 months is currently unknown but is under investigation (clinicaltrials.gov identifier NCT00942968).
Recommendation: LMWH therapy for at least 6 months
The ACCP guidelines recommend LMWH therapy for 3 to 6 months, followed by warfarin or another vitamin K antagonist or continued LMWH treatment until the cancer is resolved.2
The National Comprehensive Cancer Network guidelines recommend an LMWH for 6 months as monotherapy and indefinite anticoagulation if the cancer is still active.12
The American Society of Clinical Oncology guidelines recommend an LMWH for at least 6 months and indefinite anticoagulant therapy for selected patients with active cancer.13
We agree that patients with active cancer should receive an LMWH for at least 6 months and indefinite anticoagulation until the cancer is resolved.
In our experience, many patients are reluctant to give themselves the daily injections that LMWH therapy requires, and so they need to be well-informed about the marked decrease in VTE recurrence with this less-convenient and more-expensive therapy. Many patients face insurance barriers to cover the cost of LMWH therapy; however, careful attention to preauthorization can usually overcome this obstacle.
HOW LONG TO TREAT RECURRENT VTE
It makes clinical sense that patients who have a second VTE event should be treated indefinitely. This theory was tested in a randomized clinical trial14 in which patients with provoked or unprovoked VTE were randomized after their second event to receive anticoagulation for 6 months vs indefinitely.
After 4 years of follow-up, the recurrence rate was 21% in patients assigned to 6 months of treatment and only 3% in patients who continued anticoagulation throughout the trial. On the other hand, major hemorrhage occurred in 3% of patients treated for 6 months and in 9% in patients who continued anticoagulation indefinitely.
Of note, most of the patients in this trial had unprovoked (idiopathic) VTE, so the results should not be extrapolated to patients with provoked VTE, who accounted for only 20% of the study population.14
Recommendation: Long-term anticoagulation
We agree with the ACCP recommendation2 that patients who have had a second episode of unprovoked VTE should receive long-term anticoagulation. Because of a lack of data, the duration of therapy for patients with a second episode of provoked VTE should be individualized.
HOW LONG TO TREAT THROMBOPHILIA-RELATED VTE
Inherited thrombophilias
Patients with VTE that is not related to a clear provoking risk factor or cancer frequently have testing to evaluate for a hypercoagulable state. This workup traditionally includes the most common inherited thrombophilias for gene mutations for factor V and prothrombin as well as for deficiencies in protein C, protein S, antithrombin and the acquired antiphospholipid syndrome.
The key questions that should be asked prior to embarking on this workup are:
- Will the results change the length of therapy for the patient?
- Will testing the patient help with genetic counseling and possible testing of family members?
- Will the results change the targeted INR range for warfarin or other vitamin K antagonist therapy?
Patients with inherited thrombophilia have a greater risk of developing an initial VTE event; however, these tests do not help predict the recurrence of VTE in patients with established disease more than clinical risk factors do. A prospective study demonstrated this by looking at the effect of thrombophilia and clinical factors on the recurrence of venous thrombosis and found that inherited prothrombotic abnormalities do not appear to play an important role in the risk of a recurrent event.15 On the other hand, clinical factors, such as whether the first event was idiopathic or provoked, appear more important in determining the duration of anticoagulation therapy.15 A systematic review of the common inherited thrombophilias showed the VTE recurrence rate of patients with factor V Leiden was higher than in patients without the mutation; however, the absolute rates of recurrence were not much different than what would be expected in patients with idiopathic VTE.16
A retrospective study involving a large cohort of families of patients who already had experienced a first episode of either idiopathic or provoked VTE showed high annual risks of recurrent VTE associated with hereditary deficiencies of protein S (8.4%), protein C (6.0%), and antithrombin (10%).17 However, for the more commonly occurring genetic thrombophilias, the factor V Leiden and prothrombin G20210A mutations, family members with either gene abnormality had low rates of VTE, suggesting that testing of relatives of probands is not clinically useful.16
Antiphospholipid syndrome
Antiphospholipid syndrome is an acquired thrombophilia. A patient has thrombotic antiphospholipid syndrome when there is a history of vascular thrombosis in the presence of persistently positive tests (at least 12 weeks apart) for lupus anticoagulants, anticardiolipin antibodies, or anti-beta-2 glycoprotein I. A prospective study of 412 patients with a first episode of VTE found that 15% were positive for anticardiolipin antibody at the end of 6 months of anticoagulation. The risk of recurrent VTE after 4 years was 29% in patients with antibodies and 14% in those without antibodies (relative risk 2.1; 95% confidence interval [CI] 1.3–3.3; P =.0013).18
Recent reviews advise indefinite warfarin anticoagulation in patients with VTE and persistence of antiphospholipid antibodies.19 However, the optimal duration of anticoagulation is uncertain. Until well-designed clinical trials are done, the current general consensus is to anticoagulate these patients indefinitely.20,21 Retrospective studies had suggested that patients with antiphospholipid antibodies required a higher therapeutic INR range; however, this observation was tested in two trials that found no difference in thromboembolic rates when patients were randomized to an INR of 2.0–3.0 vs 3.1–4.0,22 or 2.0–3.0 vs 3.0–4.5.23
No formal recommendations
In the absence of strong evidence, the ACCP guidelines do not include a recommendation on the duration of anticoagulation treatment specific to inherited thrombophilias. We believe that clinical factors are more important than inherited thrombophilias for deciding the duration of anticoagulation, and that testing is almost never indicated or useful. However, patients with antiphospholipid syndrome are at high risk of recurrence, and it is our practice to anticoagulate these patients indefinitely.
HOW LONG TO TREAT UNPROVOKED (IDIOPATHIC) VTE
A VTE event is thought to be idiopathic if it occurs without a clearly identified provoking factor.
In an observational study,3 patients with oral contraceptive use, transient illness, immobilization, or a history of travel had an 8.8% risk of recurrence vs 19.4% in patients with unprovoked VTE. The meta-analysis discussed above (Table 1)8 also shows that patients with these nonsurgical risk factors have a lower rate of recurrence than patients with idiopathic VTE.
The high rate of recurrence of idiopathic VTE (4% to 27% after 3 months of anticoagulation24–26) suggests that a longer duration of treatment is reasonable. However, increasing the length of therapy from 3 to 12 months delays but does not prevent recurrence, the risk of which begins to accumulate once anticoagulation is stopped.24,25
Three promising strategies to identify subgroups of patients with idiopathic VTE who are at highest risk of recurrence and who would benefit the most from prolonged anticoagulation are d-dimer testing, evaluation for residual vein thrombosis in patients who present with a deep vein thrombosis, and clinical prediction rules.
d-dimer testing
d-dimer is a degradation product of fibrin and is an indirect marker of residual thrombosis.30
In a systematic review of patients with a first episode of unprovoked VTE,31 a normal d-dimer concentration at the end of at least 3 months of anticoagulation was associated with a 3.5% annual risk of recurrence, whereas an elevated d-dimer level at that time was associated with an annual risk of 8.9%. These results were confirmed in a systematic review of individual patient data.32
In a randomized trial,28 patients with an idiopathic VTE event who received anticoagulation for at least 3 months had their d-dimer level measured 1 month after cessation of treatment. Those with an elevated level were randomized to either resume anticoagulation or not. Patients who resumed anticoagulation had an annual recurrence rate of 2%; however, those who were allocated not to restart anticoagulation had a recurrence rate of 10.9% per year. There was no difference in the rate of major bleeding between the two groups. Patients in this clinical trial who had a normal d-dimer level did not restart anticoagulation and had an annual recurrence rate of 4.4%.
Evaluation for residual thrombosis
A randomized trial34 in patients with both provoked and idiopathic deep vein thrombosis showed a reduction in recurrence when those who had residual vein thrombosis were given extended anticoagulation. In the subset of patients whose deep vein thrombosis was idiopathic, the recurrence rate was 17% per year when treatment lasted only 3 months and 10% when it was extended for up to 1 year.
Another trial35 randomized patients with provoked and idiopathic deep vein thrombosis to receive anticoagulation for the usual duration or to continue treatment until recanalization of the residual thrombus was demonstrated on follow-up Doppler ultrasonography. Patients who received this ultrasonography-tailored treatment had a lower rate of recurrence of VTE; however, the absolute reductions in recurrence rates cannot be calculated from this report for patients with idiopathic deep vein thrombosis.
A prospective observational study36 of the predictive value of d-dimer status and residual vein thrombus found that only d-dimer was an independent risk factor for recurrent VTE after vitamin K antagonist withdrawal.
A clinical prediction rule: ‘Men and HERDOO2’
A promising tool for predicting if a patient is at low risk of recurrent VTE after the first episode of proximal deep vein thrombosis or pulmonary embolism is known by the mnemonic device “Men and HERDOO2.” It is based on data prospectively derived by Rodger et al37 to identify patients with less than a 3% annual risk of recurrent VTE after their first event of idiopathic proximal deep vein thrombosis or pulmonary embolism. Risk factors for recurrent VTE were male sex (the “men” of “Men and HERDOO2”), signs of postthrombotic syndrome, including hyperpigmentation of the lower extremities, edema or redness of either leg, a d-dimer level > 250 μg/L, obesity (body mass index > 30 kg/m2, and older age (> 65 years).
Overall, one-fourth of the population were women with no risk factors or one risk factor, and their risk of recurrence was 1.6% per year. Men and women who had two or more risk factors for postthrombotic syndrome (hyperpigmentation, edema, or redness), elevated d-dimer, obesity, or older age were predicted to be at higher risk of recurrent VTE. Patients such as this should be considered for indefinite anticoagulation.
Ideally, clinical prediction rules should be validated in a separate group of patients before they are used routinely in practice,38 and this clinical prediction rule is currently being tested in the REVERSE II study. If the results are consistent, this will be an easy-to-use tool to help identify patients who likely can safely stop anticoagulation therapy after 3 to 6 months (clinicaltrials.gov Identifier: NCT00967304).
The location of the thrombosis also influences the likelihood of recurrence. Patients with isolated distal (calf) deep vein thrombosis are less likely to suffer recurrent VTE than those who present with proximal deep vein thrombosis. However, trials focusing specifically on the precise subset of idiopathic isolated distal deep vein thrombosis are lacking. In a randomized trial39 comparing 6 vs 12 weeks of anticoagulation for isolated distal deep vein thrombosis and 12 vs 24 weeks for proximal deep vein thrombosis, the annual rates of recurrence after 12 weeks of treatment were approximately 3.4% for isolated distal and 8.1% for proximal deep vein thrombosis.39
Recommendation: At least 3 months of warfarin or equivalent
We agree with the ACCP recommendation2 that patients with unprovoked VTE should receive at least 3 months of anticoagulation with a vitamin K antagonist.
If the patient has no risk factors for bleeding and good anticoagulant monitoring is achievable, we agree with long-term anticoagulation for proximal unprovoked deep vein thrombosis or pulmonary embolism, and 3 months of therapy for isolated distal unprovoked deep vein thrombosis.
Patient preferences and the risk of recurrence vs the risk of bleeding should be discussed with patients when contemplating indefinite anticoagulation.
If testing is being considered to assist in the decision to prescribe indefinite anticoagulation, we prefer using d-dimer levels rather than ultrasonography to detect residual venous thrombosis because of its ease of use and the strength of the current evidence.
PREVENTING POSTTHROMBOTIC SYNDROME
The postthrombotic (postphlebitic) syndrome is a chronic and burdensome consequence of deep vein thrombosis that occurs despite anticoagulation therapy. It is estimated to affect 23% to 60% of patients and typically manifests in the first 2 years.40 It is not only costly in clinical terms, with decreased quality of life for the patient, but health care expenditures have been estimated to range from $400 per year in a Brazilian study to $7,000 per year in a US study.40
Typical symptoms include leg pain, heaviness, swelling, and cramping. In severe cases, chronic venous ulcers can occur and are difficult to treat.41
The definition of postthrombotic syndrome has been unclear over the years, and six different scales that measure signs and symptoms have been reported.42
The Villalta scale has been proposed by the International Society of Thrombosis and Hemostasis as a diagnostic standard to define postthrombotic syndrome.42 This validated scale is based on five clinical symptoms, six clinical signs, and the presence or absence of venous ulcers. Each clinical symptom and sign is scored as mild (1 point), moderate (2 points), or severe (3 points). Symptoms include pain, cramps, heaviness, paresthesia, and pruritus; the six clinical signs are pretibial edema, skin induration, hyperpigmentation, redness, venous ectasia, and pain on calf compression.
According to the International Society of Thrombosis and Hemostasis, postthrombotic syndrome is present if the Villalta score is 5 or greater or if a venous ulcer is present in a leg with previous deep vein thrombosis. Further, using the Villalta scale, postthrombotic syndrome can be categorized as mild (score 5–9), moderate (10–14), or severe (≥ 15).
A limitation of the Villalta scale is that the presence or absence of a venous ulcer has not been assigned a score. Since a venous ulcer requires more aggressive measures, the society defines postthrombotic syndrome as severe if venous ulcers are present.42
Acute symptoms of deep vein thrombosis may take months to resolve and, indeed, acute symptoms may transition to chronic symptoms without a symptom-free interval. It is recommended that postthrombotic syndrome not be diagnosed before 3 months to avoid inappropriately attributing acute symptoms and signs of deep vein thrombosis to the postthrombotic syndrome.42
Studies of stockings
A systematic review of three randomized trials44 concluded that elastic compression stockings reduce the risk of postthrombotic syndrome (any severity) from 43% to 20% and severe postthrombotic syndrome from 15% to 7%.43
The first of these trials44 randomized patients soon after the diagnosis of deep vein thrombosis to receive made-to-order compression stockings that were rated at 30 to 40 mm Hg or to be in a control group that did not receive stockings. The second trial45 randomized patients 1 year after the index event of deep vein thrombosis to receive 20- to 30-mm Hg stockings or stockings that were two sizes too large (the control group). The third study46 randomly allocated patients to receive “off-the-shelf” stockings (30–40 mm Hg) or no stockings. Each study used its own definition of postthrombotic syndrome.
Although these studies strongly suggest compression stockings prevent postthrombotic syndrome, several methodologic issues remain:
- A standard definition of postthrombotic syndrome was not used
- The amount of compression varied between studies
- The studies were not blinded.
Lack of blinding becomes most significant when an outcome is based on subjective findings, like the symptoms that make up a large part of the diagnosis of postthrombotic syndrome.
The SOX trial, currently under way, is designed to address these methodologic issues and should be completed in 2012 (clinicaltrials.gov Identifier: NCT00143598).
Recommendation: Stockings for at least 2 years
We agree with the ACCP recommendation that a patient who has had a symptomatic proximal deep vein thrombosis should wear an elastic compression stocking with an ankle pressure gradient of 30 to 40 mm Hg as soon as possible after starting anticoagulant therapy and continuing for a minimum of 2 years.2
SCREENING FOR OCCULT MALIGNANCY
VTE can be the first manifestation of cancer.
French physician Armand Trousseau, in the 1860s, was the first to describe disseminated intravascular coagulation closely associated with adenocarcinoma. Ironically, several years later, after suffering for weeks from abdominal pain, he declared to one of his students that he had developed thrombosis, and he died of gastric cancer shortly thereafter.47
Since cancer is a well-known risk factor for VTE, it is logical to screen for cancer as an explanation for an idiopathic VTE event.48 To make an informed decision, one needs to understand the rate of occult cancer at the time VTE is diagnosed, the risk of future development of cancer, and the utility of extensive cancer screening.
The clinical efficacy, side effects, and cost-effectiveness of cancer screening in patients with idiopathic VTE are unknown. However, a systematic review47 of 34 studies found that, in patients with idiopathic VTE, cancer was diagnosed within 1 month in 6.1%, within 6 months in 8.6%, and within 1 year in 10.0% (95% CI 8.6–11.3).
A subset of studies compared two strategies for screening soon after the diagnosis of idiopathic VTE: a strategy limited to the history, physical examination, basic blood work, and chest radiography vs an extensive screening strategy that also included serum tumor markers or abdominal ultrasonography or computed tomography. Limited screening detected 49% of the prevalent cancers; extensive screening increased this rate to 70%. Stated another way, the detection rate for prevalent cancers was 5% with limited screening and 7% with extensive screening soon after the diagnosis of idiopathic VTE.47
Patients with idiopathic VTE had higher rates of cancer within 1 month of diagnosis than patients with provoked VTE (6.1% vs 1.9%), and this difference persisted at 1 year (10.0% vs 2.6%).47
Recommendation: Individualized cancer screening
Patients with idiopathic VTE have a significant risk of occult cancer within the first year after diagnosis, and cancer screening should be considered. Our practice for patients with idiopathic VTE is to perform a history and physical examination and ensure that the patient is up to date on age- and sex-specific cancer screening.
The use of additional imaging or biomarkers should be discussed with patients so they can balance the risks (radiation and potential false-positive results with their downstream consequences), costs, and potential benefits, given the lack of proven survival benefit or cost-effectiveness.
ORAL ANTICOAGULANT MANAGEMENT
Warfarin’s multiple interactions, along with the need for INR monitoring, make it a difficult medication to manage.
The Joint Commission, the US organization for health service accreditation and certification, has defined National Patient Safety Goals and quality measures for the management of anticoagulation.49 Organized anticoagulation management services, dosing algorithms, and patient self-testing using capillary INR meters or patient self-management of warfarin were recommended as tools to improve the time patients spend in the therapeutic INR range.50
Two new oral anticoagulants
The limitations of warfarin have stimulated the search for newer oral anticoagulants that do not require laboratory monitoring or have as many diet and drug interactions.
Two trials have been published with experimental oral anticoagulants that had similar efficacy and safety as warfarin in the treatment of VTE.
The study of dabigatran (Pradaxa) vs warfarin in the treatment of acute VTE (the RECOVER trial)51 randomized 2,539 patients with acute VTE to receive the oral direct thrombin inhibitor dabigatran or warfarin for approximately 6 months. Of note, each treatment group received a median of 6 days of heparin, LMWH, or fondaparinux at the beginning of blinded therapy. The rates of recurrent VTE and major bleeding were similar between the treatment arms, and overall bleeding was less with dabigatran. Dabigatran was approved in the United States in October 2010 for stroke prevention in atrial fibrillation but has yet to be approved for the treatment of VTE pending further study (clinicaltrials.gov Identifier: NCT00680186).
A study of oral rivaroxaban (Xarelto) for symptomatic VTE (the EINSTEIN-DVT trial) 52 randomized 3,449 patients with acute deep vein thrombosis to rivaroxaban or enoxaparin (Lovenox) overlapped with warfarin or another vitamin K antagonist in the usual manner. No difference was noted between the treatments in the rate of recurrence of VTE or of major bleeding. Of note, patients randomized to rivaroxaban received 15 mg twice a day for the first 3 weeks of treatment and then 20 mg per day for the remainder of their therapy and did not require parenteral anticoagulant overlap.
The long-awaited promise of easier-to-use oral anticoagulants for the treatment of VTE is drawing near and has the potential to revolutionize the treatment of this common disorder. In the meantime, close monitoring of warfarin and careful patient education regarding its use are essential. And even with the development of new drugs in the future, it is still imperative that patients with acute VTE receive the correct length of anticoagulation treatment, are prescribed stockings to prevent postthrombotic syndrome, and are updated on routine cancer screening.
- Spencer FA, Emery C, Lessard D, et al. The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 2006; 21:722–727.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523–526.
- Schulman S, Lockner D, Juhlin-Dannfelt A. The duration of oral anticoagulation after deep vein thrombosis. A randomized study. Acta Med Scand 1985; 217:547–552.
- Optimum duration of anticoagulation for deep-vein thrombosis and pulmonary embolism. Research Committee of the British Thoracic Society. Lancet 1992; 340:873–876.
- Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. Duration of Anticoagulation Trial Study Group. N Engl J Med 1995; 332:1661–1665.
- Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743–749.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med 2010; 170:1710–1716.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hull RD, Pineo GF, Brant RF, et al; LITE Trial Investigators. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology, Venous Thromboembolic Disease. http://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed August 3, 2011.
- Lyman GH, Khorana AA, Falanga A, et al; American Society of Clinical Oncology. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Schulman S, Granqvist S, Holmström M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–2485.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost 2009; 101:93–99.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med 1998; 104:332–338.
- Derksen RH, de Groot PG. Towards evidence-based treatment of thrombotic antiphospholipid syndrome. Lupus 2010; 19:470–474.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Fonseca AG, D’Cruz DP. Controversies in the antiphospholipid syndrome: can we ever stop warfarin? J Autoimmune Dis 2008; 5:6.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3:848–853.
- Agnelli G, Prandoni P, Becattini C, et al; Warfarin Optimal Duration Italian Trial Investigators. Extended oral anticoagulant therapy after a first episode of pulmonary embolism. Ann Intern Med 2003; 139:19–25.
- Agnelli G, Prandoni P, Santamaria MG, et al; Warfarin Optimal Duration Italian Trial Investigators. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Kearon C, Ginsberg JS, Kovacs MJ, et al; Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–639.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Ridker PM, Goldhaber SZ, Glynn RJ. Low-intensity versus conventional-intensity warfarin for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:2164–2167.
- Bockenstedt P. D-dimer in venous thromboembolism. N Engl J Med 2003; 349:1203–1204.
- Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008; 149:481–490,W94.
- Douketis J, Tosetto A, Marcucci M, et al. Patient-level metaanalysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med 2010; 153:523–531.
- Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med 2002; 137:955–960.
- Siragusa S, Malato A, Anastasio R, et al. Residual vein thrombosis to establish duration of anticoagulation after a first episode of deep vein thrombosis: the Duration of Anticoagulation based on Compression UltraSonography (DACUS) study. Blood 2008; 112:511–515.
- Prandoni P, Prins MH, Lensing AW, et al; AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep venous thrombosis: a randomized trial. Ann Intern Med 2009; 150:577–585.
- Cosmi B, Legnani C, Cini M, Guazzaloca G, Palareti G. D-dimer levels in combination with residual venous obstruction and the risk of recurrence after anticoagulation withdrawal for a first idiopathic deep vein thrombosis. Thromb Haemost 2005; 94:969–974.
- Rodger MA, Kahn SR, Wells PS, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ 2008; 179:417–426.
- McGinn TG, Guyatt GH, Wyer PC, Naylor CD, Stiell IG, Richardson WS. Users’ guides to the medical literature: XXII: how to use articles about clinical decision rules. Evidence-Based Medicine Working Group. JAMA 2000; 284:79–84.
- Pinede L, Ninet J, Duhaut P, et al; Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103:2453–2460.
- Ashrani AA, Heit JA. Incidence and cost burden of postthrombotic syndrome. J Thromb Thrombolysis 2009; 28:465–476.
- Kahn SR, Shrier I, Julian JA, et al. Determinants and time course of the postthrombotic syndrome after acute deep venous thrombosis. Ann Intern Med 2008; 149:698–707.
- Kahn SR, Partsch H, Vedantham S, Prandoni P, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of post-thrombotic syndrome of the leg for use in clinical investigations: a recommendation for standardization. J Thromb Haemost 2009; 7:879–883.
- Kolbach DN, Sandbrink MW, Hamulyak K, Neumann HA, Prins MH. Non-pharmaceutical measures for prevention of post-thrombotic syndrome. Cochrane Database Syst Rev 2004;CD004174.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet 1997; 349:759–762.
- Ginsberg JS, Hirsh J, Julian J, et al. Prevention and treatment of postphlebitic syndrome: results of a 3-part study. Arch Intern Med 2001; 161:2105–2109.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the post-thrombotic syndrome: a randomized, controlled trial. Ann Intern Med 2004; 141:249–256.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008; 149:323–333.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Kaatz S. Impact on patient care: patient case through the continuum of care. J Thromb Thrombolysis 2010; 29:167–170.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Schulman S, Kearon C, Kakkar AK, et al; for the RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2452.
- The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363;2499–2510.
- Spencer FA, Emery C, Lessard D, et al. The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 2006; 21:722–727.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523–526.
- Schulman S, Lockner D, Juhlin-Dannfelt A. The duration of oral anticoagulation after deep vein thrombosis. A randomized study. Acta Med Scand 1985; 217:547–552.
- Optimum duration of anticoagulation for deep-vein thrombosis and pulmonary embolism. Research Committee of the British Thoracic Society. Lancet 1992; 340:873–876.
- Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. Duration of Anticoagulation Trial Study Group. N Engl J Med 1995; 332:1661–1665.
- Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743–749.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med 2010; 170:1710–1716.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hull RD, Pineo GF, Brant RF, et al; LITE Trial Investigators. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology, Venous Thromboembolic Disease. http://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed August 3, 2011.
- Lyman GH, Khorana AA, Falanga A, et al; American Society of Clinical Oncology. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Schulman S, Granqvist S, Holmström M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–2485.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost 2009; 101:93–99.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med 1998; 104:332–338.
- Derksen RH, de Groot PG. Towards evidence-based treatment of thrombotic antiphospholipid syndrome. Lupus 2010; 19:470–474.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Fonseca AG, D’Cruz DP. Controversies in the antiphospholipid syndrome: can we ever stop warfarin? J Autoimmune Dis 2008; 5:6.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3:848–853.
- Agnelli G, Prandoni P, Becattini C, et al; Warfarin Optimal Duration Italian Trial Investigators. Extended oral anticoagulant therapy after a first episode of pulmonary embolism. Ann Intern Med 2003; 139:19–25.
- Agnelli G, Prandoni P, Santamaria MG, et al; Warfarin Optimal Duration Italian Trial Investigators. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Kearon C, Ginsberg JS, Kovacs MJ, et al; Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–639.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Ridker PM, Goldhaber SZ, Glynn RJ. Low-intensity versus conventional-intensity warfarin for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:2164–2167.
- Bockenstedt P. D-dimer in venous thromboembolism. N Engl J Med 2003; 349:1203–1204.
- Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008; 149:481–490,W94.
- Douketis J, Tosetto A, Marcucci M, et al. Patient-level metaanalysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med 2010; 153:523–531.
- Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med 2002; 137:955–960.
- Siragusa S, Malato A, Anastasio R, et al. Residual vein thrombosis to establish duration of anticoagulation after a first episode of deep vein thrombosis: the Duration of Anticoagulation based on Compression UltraSonography (DACUS) study. Blood 2008; 112:511–515.
- Prandoni P, Prins MH, Lensing AW, et al; AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep venous thrombosis: a randomized trial. Ann Intern Med 2009; 150:577–585.
- Cosmi B, Legnani C, Cini M, Guazzaloca G, Palareti G. D-dimer levels in combination with residual venous obstruction and the risk of recurrence after anticoagulation withdrawal for a first idiopathic deep vein thrombosis. Thromb Haemost 2005; 94:969–974.
- Rodger MA, Kahn SR, Wells PS, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ 2008; 179:417–426.
- McGinn TG, Guyatt GH, Wyer PC, Naylor CD, Stiell IG, Richardson WS. Users’ guides to the medical literature: XXII: how to use articles about clinical decision rules. Evidence-Based Medicine Working Group. JAMA 2000; 284:79–84.
- Pinede L, Ninet J, Duhaut P, et al; Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103:2453–2460.
- Ashrani AA, Heit JA. Incidence and cost burden of postthrombotic syndrome. J Thromb Thrombolysis 2009; 28:465–476.
- Kahn SR, Shrier I, Julian JA, et al. Determinants and time course of the postthrombotic syndrome after acute deep venous thrombosis. Ann Intern Med 2008; 149:698–707.
- Kahn SR, Partsch H, Vedantham S, Prandoni P, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of post-thrombotic syndrome of the leg for use in clinical investigations: a recommendation for standardization. J Thromb Haemost 2009; 7:879–883.
- Kolbach DN, Sandbrink MW, Hamulyak K, Neumann HA, Prins MH. Non-pharmaceutical measures for prevention of post-thrombotic syndrome. Cochrane Database Syst Rev 2004;CD004174.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet 1997; 349:759–762.
- Ginsberg JS, Hirsh J, Julian J, et al. Prevention and treatment of postphlebitic syndrome: results of a 3-part study. Arch Intern Med 2001; 161:2105–2109.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the post-thrombotic syndrome: a randomized, controlled trial. Ann Intern Med 2004; 141:249–256.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008; 149:323–333.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Kaatz S. Impact on patient care: patient case through the continuum of care. J Thromb Thrombolysis 2010; 29:167–170.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Schulman S, Kearon C, Kakkar AK, et al; for the RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2452.
- The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363;2499–2510.
KEY POINTS
- A low-molecular-weight heparin for at least 6 months is the treatment of choice for cancer-related VTE.
- We recommend 3 months of anticoagulation for VTE caused by a reversible risk factor and indefinite treatment for idiopathic VTE in patients without risk factors for bleeding who can get anticoagulation monitoring.
- Clinical factors are more important in deciding the duration of anticoagulation therapy than evidence of an inherited thrombophilic state.
- Elastic compression stockings reduce the risk of postthrombotic syndrome substantially.
- Patients with idiopathic VTE should have a basic screening for malignancy.
In reply: Giant cell arteritis
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
Giant cell arteritis
To the Editor: As a practicing internist, I found Dr. Alexandra Villa-Forte’s review of giant-cell arteritis (Cleve Clin J Med 2011; 78:265–270) both interesting and useful, as usual for the Cleveland Clinic Journal of Medicine. However, she did not mention the recommendation by some experts that patients who have had temporal arteritis should receive annual chest x-rays, for a decade or longer, to screen for the development of thoracic aortic aneurysm. Does she agree with this precaution? Is it advisable, in addition, to screen for abdominal aortic aneurysm by means of abdominal ultrasonography? If so, at what time intervals should this be done?
To the Editor: As a practicing internist, I found Dr. Alexandra Villa-Forte’s review of giant-cell arteritis (Cleve Clin J Med 2011; 78:265–270) both interesting and useful, as usual for the Cleveland Clinic Journal of Medicine. However, she did not mention the recommendation by some experts that patients who have had temporal arteritis should receive annual chest x-rays, for a decade or longer, to screen for the development of thoracic aortic aneurysm. Does she agree with this precaution? Is it advisable, in addition, to screen for abdominal aortic aneurysm by means of abdominal ultrasonography? If so, at what time intervals should this be done?
To the Editor: As a practicing internist, I found Dr. Alexandra Villa-Forte’s review of giant-cell arteritis (Cleve Clin J Med 2011; 78:265–270) both interesting and useful, as usual for the Cleveland Clinic Journal of Medicine. However, she did not mention the recommendation by some experts that patients who have had temporal arteritis should receive annual chest x-rays, for a decade or longer, to screen for the development of thoracic aortic aneurysm. Does she agree with this precaution? Is it advisable, in addition, to screen for abdominal aortic aneurysm by means of abdominal ultrasonography? If so, at what time intervals should this be done?
Immune thrombocytopenia: No longer ‘idiopathic’
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.

FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
Once regarded as idiopathic, immune thrombocytopenia (ITP) is now understood to have a complex pathogenesis, involving the evolution of antibodies against multiple platelet antigens leading to reduced platelet survival as well as impaired platelet production. For this reason, multiple therapies with different mechanisms of action are available to treat ITP, though not all of them are effective for individual patients.
In this article, I discuss the pathogenesis, demographics, manifestations, diagnosis, and management of ITP.
THE NAME AND THE CUTOFF HAVE CHANGED
The term ITP formerly was used to refer to “idiopathic” or “immune” thrombocytopenic purpura. However, although not all aspects of the pathogenesis of ITP are understood, the disease can no longer be considered idiopathic. In addition, many patients do not have purpura at the time of diagnosis. Though the abbreviation “ITP” remains the same, it now refers to immune thrombocytopenia, which can be either primary or secondary.1
ITP is defined as a platelet count of less than 100 × 109/L (100,000/μL) with no evidence of leukopenia or anemia. This cutoff point is new: in the past, ITP was defined as a platelet count of less than 150 × 109/L, which is the threshold for a normal platelet count in most laboratories.
The platelet threshold of 100 × 109/L was based on a study by Stasi et al,2 who followed 217 otherwise healthy people who had an incidental finding of mild thrombocytopenia (platelet count 100–150 × 109/L). Within 6 months, the platelet count rose to more than 150 × 109/L in 23, while three had either worsening thrombocytopenia or were diagnosed with other conditions. During long-term follow-up (median 64 months), 109 of the remaining 191 individuals remained stable, 13 developed counts greater than 150 × 109/L, 12 developed ITP, 13 developed an autoimmune disorder, 18 developed other disorders, and 26 were lost to follow-up. The 10-year probability of developing ITP, defined as a platelet count persistently below 100 × 109/L, was only 6.9%, indicating that the chances are small that a person with an isolated finding of mild, stable thrombocytopenia will develop ITP.
Categories of ITP
An international working group designated to standardize terminology has divided ITP into two major diagnostic categories.1 The proportion of patients within each is not well established and varies by region and demographic characteristics.
Primary ITP accounts for the majority of cases in most studies; other conditions associated with thrombocytopenia are absent.
Secondary ITP can be due to infection with a number of agents, including hepatitis C virus (HCV), human immunodeficiency virus (HIV), and Helicobacter pylori. Other causes include underlying autoimmune and lymphoproliferative disorders such as systemic lupus erythematosus, Wiskott-Aldrich syndrome, chronic lymphocytic leukemia, antiphospholipid syndrome, and common variable immunodeficiency, as well as drugs such as quinine and trimethoprim-sulfamethoxazole.
Categories of ITP have also been established to facilitate management decisions, as follows:
Newly diagnosed ITP refers to ITP diagnosed within the preceding 3 months.
Persistent ITP refers to ITP diagnosed 3 to 12 months previously, and includes ITP in patients not reaching spontaneous remission and in those not maintaining a complete response off therapy. (When ITP spontaneously remits in adults, it usually does so within the first 12 months after the condition is diagnosed.)
Chronic ITP: Lasting for more than 12 months.
Severe ITP is defined by bleeding at presentation sufficient to mandate treatment, or new bleeding symptoms requiring additional therapeutic intervention with a different platelet-enhancing agent or an increased dosage of a current agent.
ITP IS COMMON IN OLDER ADULTS
We previously believed that ITP was a disorder that primarily affected women in their third and fourth decades. However, this was not borne out in recent epidemiologic studies, which have demonstrated that the highest age-specific incidence of ITP occurs in the elderly. This may potentially reflect the development of immune dysregulation as a consequence of aging. There is a female preponderance in the incidence of ITP throughout adulthood until around age 60, after which the overall incidence increases in both sexes, and the ratio of affected women to men is about equal.3,4 Thus, even though thrombocytopenia in the elderly may reflect myelodysplasia in some individuals, ITP is much more common than previously appreciated.
Previous guidelines from the American Society of Hematology suggested that a bone marrow examination be strongly considered in patients over age 60 with suspected ITP. With the realization that ITP occurs more commonly in the elderly, it is apparent that bone marrow examination is not necessary in this group if there are no other cytopenias present and the physical examination and blood smear are consistent with ITP.
In children, ITP has a peak incidence between ages 5 and 6, and behaves differently from the adult syndrome. ITP in children usually follows an apparent viral infection and tends to be self-limited, with approximately 80% of cases resolving spontaneously within 6 months. In contrast, adult ITP usually develops into a chronic disease.
BLEEDING MAY NOT BE PRESENT AT DIAGNOSIS
ITP is now recognized as a diverse syndrome with a constellation of signs and symptoms.
Petechiae are pinpoint microvascular hemorrhages that do not blanch with pressure. This distinguishes them from small hemangiomas, which look similar but blanch transiently with pressure. Petechiae tend to occur on dependent areas, particularly the hands and feet, when the platelet count drops below approximately 15 × 109/L.
Ecchymoses (dry purpura) appear as large bruises.
Mucosal bleeding (wet purpura) involves the oral mucosa. Particularly in children, wet purpura tends to be associated with systemic bleeding complications, involving the gastrointestinal tract for example. The incidence of intracranial hemorrhage, though very low, may also be increased in patients with wet purpura.
Other bleeding manifestations may include heavy menstrual bleeding, oral bleeding, and epistaxis.
Bleeding is generally but not entirely proportional to the platelet count. In a study of adults with newly diagnosed ITP and a platelet count of less than 50 × 109/L,4 the presenting symptom was hemorrhage in 12% and purpura in 58%.4 Remarkably, 28% of cases were asymptomatic, with some patients remaining free of symptoms for years despite very low platelet counts. More than half of patients with a platelet count of 30 to 50 × 109/L have no symptoms.3,4
A PARADOXICAL RISK OF THROMBOSIS
Although ITP is primarily a bleeding disorder, it is paradoxically also associated with thrombosis. Sarpatwari et al,5 in a study in the United Kingdom, found that the 4-year incidence of thromboembolic events was about 1.3 times higher in patients with ITP than in matched controls.
The reason for the increased risk of thrombosis is not clear. It is possible that in some patients, antiphospholipid antibodies may contribute to the development of thrombosis, although this has not been confirmed in all studies.
A DIAGNOSIS OF EXCLUSION
The evaluation of any patient suspected of having ITP should include the following:
- Personal history, with special attention to drugs and to medical conditions that could cause thrombocytopenia.
- Family history. ITP may occasionally be mistaken for an inherited cause of thrombocytopenia. The presence of the latter can often be confirmed by review of the peripheral blood film of the patient as well as other family members with thrombocytopenia. ITP is generally not considered to be an inherited disorder, although some HLA alleles may be more prevalent in ITP patients.
- Physical examination, with special attention to lymphadenopathy or splenomegaly, which may suggest an underlying malignancy such as a lymphoproliferative disorder. In general, patients with ITP have a normal physical examination, except for signs of bleeding or bruising in some.
- Laboratory tests, including a complete blood cell count, blood smear, reticulocyte count, Rh typing, and direct antiglobulin (Coombs) test.
In ITP, the peripheral blood smear should appear normal except for the presence of thrombocytopenia, although platelets may be mildly enlarged in some individuals. Red cell and leukocyte morphology is normal. It is important to exclude the presence of schistocytes (red cell fragments) and nucleated red blood cells, which often indicate a microangiopathic hemolytic anemia caused by disorders such as thrombotic thrombocytopenic purpura.
International guidelines suggest that testing for reduced immunoglobulin levels (as seen in common variable hypogammaglobulinemia) and HIV, HCV, and H pylori infections should also be considered. Coincident HCV infection is particularly high in some regions. Other cytopenias or abnormalities in the history or physical examination may prompt bone marrow examination. Testing for antiphospholipid antibodies, antinuclear antibodies, parvovirus, and cytomegalovirus may also be indicated in specific individuals. Testing for antiplatelet antibodies is not commonly performed in the current era because of its relatively low sensitivity and specificity.
Ultimately, the diagnosis of ITP is clinical, however, and cannot be established by any specific laboratory assay. Perhaps the best diagnostic study is assessment of the patient’s response to ITP therapy.
ITP INVOLVES ACCELERATED PLATELET DESTRUCTION
In 1951, William Harrington, a fellow at Washington University, infused blood from a patient with ITP into himself and, subsequently, into normal volunteers.6 The majority of recipients demonstrated significant reductions in the platelet count, sometimes severe. This fascinating and bold experiment provided the first demonstration that ITP was caused by a factor that circulates in blood. What is often not emphasized, however, is that some recipients did not develop thrombocytopenia, suggesting an alternative mechanism.
Later, Luiken et al7 and Hirschman and Shulman8 demonstrated that the transmissible agent in the blood was immunoglobulin, primarily immunoglobulin G (IgG). We now understand that much of the pathogenesis of ITP is caused by antibodies against platelet glycoproteins, most commonly platelet glycoprotein IIb/IIIa, the platelet fibrinogen receptor. Most patients, especially those with chronic ITP, also have antibodies against other platelet glycoproteins, including glycoprotein Ib/IX (the receptor for von Willebrand factor), and glycoprotein Ia/IIa, a collagen receptor. It is commonly believed that ITP may begin with antibodies against a single glycoprotein, which leads to accelerated clearance of antibody-coated platelets in the spleen. Degradation of cleared platelets by splenic macrophages leads to the release and subsequent presentation of antigenic peptides from proteolyzed platelet components, including glycoproteins, on the macrophage or dendritic cell. This may lead to recruitment and activation of specific T cells that in turn interact with and stimulate B cells to produce new antibodies against the platelet-derived peptides. This phenomenon, known as epitope spreading, may be responsible for the fact that most patients with long-standing, chronic ITP develop autoantibodies against multiple platelet glycoprotein targets.9
Several agents used in the treatment of ITP may work by impairing clearance of antibody-coated platelets by the reticuloendothelial system. One of many potential mechanisms underlying the therapeutic efficacy of intravenous immunoglobulin (IVIG) may be its ability to interact with a specific type of Fc gamma receptor, Fc gamma RIIb. IVIG therapy stimulates increased expression of this receptor, which in turn may impair the function of other “activating” Fc gamma receptors responsible for platelet clearance.10,11
ITP associated with infection may arise due to molecular mimicry. HCV, HIV, and H pylori contain amino acid sequences that may have structural similarity to regions within platelet glycoproteins. Thus, antibodies directed against the pathogen may cross-react with the glycoprotein, leading to thrombocytopenia.12–15
HCV has been found in up to one-third of cases of ITP in some centers.16–20H pylori-associated ITP is very common in some regions, particularly in Japan, and may often resolve after eradication of the infection. However, in the United States, eradication of H pylori generally does not improve the course of ITP. This may reflect antigen mimicry, in particular the fact that different cagA proteins are expressed by different strains of H pylori in certain regions of the world.
Our understanding of the immunologic basis of ITP has greatly expanded over the last decade. Although it has long been known that B cells produce autoantibodies, T cells have more recently been shown to play a critical role in regulating B-cell-mediated autoantibody production in ITP. In some situations, T cells may directly lyse platelets, or suppress megakaryopoiesis. This may explain why some patients who do not respond to standard B-cell-targeted therapy may respond to cyclosporine or other T-cell-directed agents.
ANOTHER MECHANISM OF ITP: REDUCED PLATELET PRODUCTION
In addition to accelerated platelet destruction, ITP is also associated with decreased platelet production by megakaryocytes in the bone marrow.21–25
Increased platelet destruction and reduced platelet production are likely two ends of a spectrum of ITP, and most patients likely have some degree of both processes. This concept helps explain why different drug strategies are more effective in some patients than in others.
ARE THE RISKS OF THERAPY JUSTIFIED?
It is important to understand the natural history of ITP to determine whether the risks of therapy are justified.
A 2001 study from the Netherlands26 followed 134 patients with primary ITP for 10 years: 90% had taken prednisone or had had a splenectomy. Within 2 years, 85% of these patients had platelet counts above 30 × 109/L off therapy. Although this group likely experienced more bleeding and bruising than the general population, the mortality rate was not increased. Another 6% also achieved a platelet count above 30 × 109/L, but required chronic maintenance therapy (usually steroids) to do so. This group led a nearly normal life but had more hospitalizations. The remaining 9% of patients had refractory ITP, with platelet counts remaining below 30 × 109/L despite therapy. This group had a death rate 4.2 times that of age-matched controls. About half died of bleeding and the others died of opportunistic infections to which they were susceptible because of long-term steroid therapy.
This study was influential in the general opinion that 30 × 109/L is a reasonable cutoff for treating ITP. An international consensus report states that treatment is rarely indicated in patients with platelet counts above 50 × 109/L in the absence of bleeding due to platelet dysfunction or other hemostatic defect, trauma, or surgery.27 Although this number is not supported by evidence-based data, it is a reasonable threshold endorsed by an international working group.27 Individual factors must be weighed heavily: for example, an athlete involved in contact sports requires a higher platelet count in order to play safely.
FIRST-LINE THERAPIES
First-line therapies for ITP include corticosteroids, IVIG, and anti-Rho(D) immune globulin (WinRho).27
Corticosteroids are standard therapy
Corticosteroids can be given in one of two ways:
Standard prednisone therapy, ie, 1 to 2 mg/kg per day, is given until a response is seen, and then tapered. Some maintain therapy for an additional week before tapering. There are no guidelines on how to taper: some decrease the dosage by 50% per week, although many recommend going more slowly, particularly at the lower range of dosing.
Up to 85% of patients achieve a clinical response, usually within 7 to 10 days, with platelet counts peaking in 2 to 4 weeks. Unfortunately, only about 15% of patients maintain the response over the subsequent 6 to 12 months. Restarting prednisone often initiates a vicious circle and makes patients vulnerable to steroid toxicities.
“Pulse” dexamethasone therapy consists of 40 mg per day for 4 days for one to three cycles. (Dexamethasone 1 mg is equivalent to about 10 mg of prednisone.)
Pulse dexamethasone therapy as an initial approach to ITP has been developed during the past decade and has been used primarily in research studies. This regimen evolved from studies of patients with multiple myelomas and has the potential to induce more durable remissions in some patients with newly diagnosed ITP.29 However, high-dose corticosteroids may be associated with increased toxicity, at least in the short term, and should be used cautiously. A study to address the role of high-dose vs standard-dose steroid therapy has recently been opened under the guidance of the Transfusion Medicine–Hemostasis Clinical Trials Network of the National Heart, Lung, and Blood Institute.
Immunoglobulin is useful for very low platelet counts and bleeding
Another primary therapy for ITP is IVIG 0.5 to 2.0 g/kg over 2 to 5 days. Its efficacy is similar to that of prednisone: about 65% of patients achieve a platelet count above 100 × 109/L, and 85% achieve 50 × 109/L. However, most responses are transient, and a significant minority of cases become refractory to IVIG after repeated infusions.
IVIG is associated with numerous adverse effects, including thrombosis, renal insufficiency, headache, and anaphylaxis in IgA-deficient patients. It also converts the direct antiglobulin test to positive. IVIG is expensive, is inconvenient to administer, and may require lengthy infusions depending on the formulation.
Although IVIG is not a good long-term therapy, it can help raise the platelet count relatively quickly in patients who present with severe thrombocytopenia accompanied by bleeding. Such patients should be treated with high-dose steroids, IVIG, and platelet transfusions. IVIG may also be useful to increase platelet counts prior to interventional procedures.
Intravenous anti-Rho(D)
Anti-Rho(D) is an alternative to IVIG in patients who are Rho(D)-positive and have an intact spleen. Anti-Rho(D) binds to Rh-positive red blood cells, causing them to be cleared in the reticuloendothelial system and blocking the clearance of antibody-coated platelets. In effect, red cells are sacrificed to save platelets, but because there are many more red cells than platelets, the benefits usually outweigh the risks.
The initial dose is 50 μg/kg given intravenously over 2 to 5 minutes. Anti-Rho(D) should not be given to patients whose hemoglobin level is less than 10 g/dL or who have compromised bone marrow function. It is ineffective in Rh-negative patients or those who have undergone splenectomy.
Accelerated hemolysis is a rare but severe possible adverse event associated with this therapy, occurring in slightly more than 1 in 1,000 infusions. About 1 out of every 20,000 patients develops disseminated intravascular coagulation.30 Its cause is poorly understood, and it is probably an accelerated extravascular rather than an intravascular event. The US Food and Drug Administration has recently issued a black-box warning cautioning that patients who receive anti-Rho(D) should remain in a health care setting for 8 hours after treatment, although most cases of accelerated hemolysis occur within 4 hours. Moreover, it is possible that many of these cases can be avoided by appropriate patient selection.
SECOND-LINE THERAPIES
Second-line therapies, as designated by the international working group, include azathioprine (Imuran), cyclosporine A, cyclophosphamide (Cytoxan), danazol (Danocrine), dapsone, mycophenolate mofetil (CellCept), rituximab (Rituxan), splenectomy, thrombopoietin receptor agonists, and vinca alkaloids.27 Only the most commonly used therapies will be briefly discussed below.
The evidence for efficacy of the cytotoxic agents, ie, cyclophosphamide, the vinca alkaloids, and azathioprine, comes from small, nonrandomized studies.31 Although these agents are useful in some patients, they may be associated with significant toxicities, and they are used less commonly than in the past.
Splenectomy has a high success rate
Splenectomy probably offers the best response of any treatment for ITP. About 80% of patients with ITP respond rapidly—often within 1 week. Of those, 15% relapse within the first year, and after 10 years, two-thirds remain in remission.32,33
Because there is no well-accepted predictor of a short- or long-term response to splenectomy, and because more medical options are currently available, the use of splenectomy has declined over the past 10 years. Nevertheless, splenectomy remains a useful option for therapy of ITP.
Whether and which second-line drugs should be tried before splenectomy is still controversial and should be determined on a case-by-case basis. Some patients are poor candidates for splenectomy because of comorbidities. If possible, splenectomy should be delayed until at least a year after diagnosis to allow an opportunity for spontaneous remission.
Splenectomy increases the risk of subsequent infection by encapsulated organisms, and patients should be immunized with pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines, preferably at least 3 weeks before the spleen is removed.
Splenectomy is associated with pulmonary hypertension and thrombosis, primarily in patients who have had their spleens removed because of accelerated red cell destruction. Whether these risks are applicable to patients with ITP is unknown, but if so they are probably much lower than in patients with red cell disorders.
Rituximab
Rituximab, a humanized monoclonal antibody against the CD20 antigen on B lymphocytes, was developed for treating lymphoma. However, it has been found to have significant activity in a number of immunohematologic disorders. Although many studies of rituximab for ITP have been published,34–38 it has never been tested in a randomized controlled study. The response rate is generally around 50%, and it is effective in patients with or without a spleen.
In one study,39 44 (32%) of 137 patients with chronic ITP who were given rituximab achieved a complete remission that was sustained 1 year. After more than 5 years, 63% of this group (ie, approximately 20% of the original group) were still in remission.
Potential drawbacks of rituximab include its expense as well as the risk of first-infusion reactions, which may be severe or, rarely, fatal. Rituxan has also been associated with rare cases of progressive multifocal leukoencephalopathy, usually in patients heavily treated with other immunosuppressive agents; however, very rare cases of progressive multifocal leukoencephalopathy have been reported in patients with ITP who received rituximab.
Thrombopoietin receptor agonists increase platelet production
Thrombopoietin receptor agonists are approved for patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. Rather than inhibit platelet destruction, as do all the other ITP therapies, they enhance platelet production.
Diseases involving bone marrow failure that also involve a low platelet count tend to be associated with very high levels of serum thrombopoietin, which is produced constitutively by the liver. In ITP, thrombopoeitin levels tend to be close to normal and not significantly elevated, most likely because of accelerated thrombopoietin clearance when bound to antibody-coated platelets.40 This provides a rationale for the use of thrombopoietic agents in the treatment of ITP.
Earlier-generation thrombopoietic drugs had significant amino acid homology with natural thrombopoietin, and some patients who were treated with these drugs developed antibodies against them that cross-reacted with endogenous thrombopoietin. In some cases, this led to severe, refractory thrombocytopenia. Because the newer thrombopoietic agents have no sequence homology to natural thrombopoietin, antibody production has not been a significant problem.
Two drugs in this class are currently available for treating ITP:
Romiplostim (Nplate) is a peptibody (comprising an IgG Fc region and four peptidometic regions that interact with the thrombopoietin receptor, c-mpl) that is given subcutaneously once a week.
Romiplostim performed well in several phase I clinical trials.41 In a 24-week phase III trial that compared romiplostim against placebo in patients with ITP that had been refractory to other primary treatments, 79% of splenectomized patients and 88% of nonsplenectomized patients had an overall response (defined as a platelet count > 50 × 109/L for 4 weeks during the study period), and 38% of splenectomized patients and 61% of nonsplenectomized patients had a durable response (platelet count > 50 × 109/L for 6 of the last 8 weeks of the study).42
In an ongoing long-term extension study of romiplostim that allows dose adjustments to maintain a platelet count between 50 and 200 × 109/L, romiplostim dosage and efficacy have remained stable over 5 years.42,43
Eltrombopag (Promacta) is a nonpeptide small-molecule c-mpl agonist that is taken orally once daily. A recent randomized, placebo-controlled study in patients with ITP refractory to other primary treatments found that eltrombopag was highly effective in raising platelet counts over the 6 months of the study.44 Like romiplostim, it was effective in both splenectomized and nonsplenectomized patients.
Although eltrombopag has not been studied for as long as romiplostim, data over 3 years indicate that increased platelet counts are maintained without the emergence of drug resistance or cumulative toxicity.45
Several other drugs in this class are currently in development.
Adverse effects of thrombopoietic agents
Thrombopoietic agents have several associated toxicities:
Rebound thrombocytopenia occurs in up to 10% of patients following treatment with either romiplostim or eltrombopag. Rebound thrombocytopenia is defined as a fall in the platelet count that occurs following discontinuation of a thrombopoietic agent that may result in more severe thrombocytopenia, transiently, than before the drug was initiated. Thus, the platelet count must be closely monitored after treatment with these drugs is discontinued.
Bone marrow fibrosis, which consists primarily of increased marrow reticulin content, occurs in less than 10% of treated patients, and all patients on therapy must be monitored for this potential complication by close examination of the peripheral blood film on a frequent basis. Appearance of abnormalities such as teardrop cells or nucleated red blood cells in the peripheral blood smear should prompt at least temporary discontinuation of the drug and consideration of bone marrow examination. There have been no cases of actual irreversible myelofibrosis in which thrombopoietic agents have been clearly implicated in causation. Interestingly, some reports suggest that increased reticulin is a common finding in marrow from ITP patients who have not been treated with thrombopoietic agents.46
Thrombosis must be considered a risk of treatment with thrombopoietic agents, which increase the platelet count in a disease that may already be thrombogenic. However, in the placebo-controlled studies, a significantly increased incidence of thrombosis was not observed in the treatment arms vs placebo. Moreover, even in treated patients who developed thrombosis, there was no clear association with the degree of elevation in the platelet count. Nevertheless, thrombopoietic agents should be used according to the manufacturer’s recommendations, to increase the platelet count to a range of 50 to 200 × 109/L, but not to exceed that.
Progression of hematologic malignancies. Thrombopoietin receptor agonists act not only on megakaryocytes but also on stem cells and other hematopoieic precursors. Although trials for treating patients with hematologic malignancies and bone marrow failure with thrombopoietic agents are ongoing, there is concern that they could worsen certain hematologic malignancies, though there are no controlled data to either support or refute this concern at present. At this time, these drugs are approved only for ITP and should not be used for other conditions.
Hepatotoxicity has been seen with eltrombopag, but it is usually reversible and may resolve with continued therapy. Nevertheless, close monitoring for this potential complication is indicated.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Stasi R, Amadori S, Osborn J, Newland AC, Provan D. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006; 3:e24.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytoenic purpura among adults: a population-based study and literature review. Eur Haematol 2009; 83:83–89.
- Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR; Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol 2003; 122:966–974.
- Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica 2010; 95:1167–1175.
- Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J Lab Clin Med 1951; 38:1–10.
- Luiken GA, McMillan R, Lightsey AL, et al. Platelet-associated IgG in immune thrombocytopenic purpura. Blood 1977; 50:317–325.
- Hirschman RJ, Schulman NR. Utilization of the platelet release reaction to measure ITP factor and platelet antibodies. Trans Assoc Am Physicians 1972; 85:325–334.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Karpatkin S. Autoimmune (idiopathic) thrombocytopenic purpura. Lancet 1997; 349:1531–1536.
- Psaila B, Bussel JB. Fc receptors in immune thrombocytopenias: a target for immunomodulation? J Clin Invest 2008; 118:2677–2681.
- Aster RH. Molecular mimicry and immune thrombocytopenia (comment). Blood 2009; 113:3887–3888.
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol 2004; 124:91–96.
- Nardi MA, Liu LX, Karpatkin S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc Natl Acad Sci U S A 1997; 94:7589–7594.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Pivetti S, Novarino A, Merico F, et al. High prevalence of autoimmune phenomena in hepatitis C virus antibody positive patients with lymphoproliferative and connective tissue disorders. Br J Haematol 1996; 95:204–211.
- Pawlotsky JM, Bouvier M, Fromont P, et al. Hepatitis C virus infection and autoimmune thrombocytopenic purpura. J Hepatol 1995; 23:635–639.
- Sakuraya M, Murakami H, Uchiumi H, et al. Steroid-refractory chronic idiopathic thrombocytopenic purpura associated with hepatitis C virus infection. Eur J Haematol 2002; 68:49–53.
- García-Suárez J, Burgaleta C, Hernanz N, Albarran F, Tobaruela P, Alvarez-Mon M. HCV-associated thrombocytopenia: clinical characteristics and platelet response after recombinant alpha2b-interferon therapy. Br J Haematol 2000; 110:98–103.
- Rajan SK, Espina BM, Liebman HA. Hepatitis C virus-related thrombocytopenia: clinical and laboratory characteristics compared with chronic immune thrombocytopenic purpura. Br J Haematol 2005; 129:818–824.
- Harker LA. Thrombokinetics in idiopathic thrombocytopenic purpura. Br J Haematol 1970; 19:95–104.
- Branehög I, Kutti J, Weinfeld A. Platelet survival and platelet production in idiopathic thrombocytopenic purpura (ITP). Br J Haematol 1974; 27:127–143.
- Stoll D, Cines DB, Aster RH, Murphy S. Platelet kinetics in patients with idiopathic thrombocytopenic purpura and moderate thrombocytopenia. Blood 1985; 65:584–588.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- British Committee for Standards in Haematology General Haematology Task Force. Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120:574–596.
- Mazzucconi MG, Fazi P, Bernasconi S, et al; Gruppo Italiano Malattie Ematoligiche dell’Adulto (GIMEMA) Thrombocytopenia Working Party. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007; 109:1401–1407.
- Gaines AR. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following RH9O)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005; 106:1532–1537.
- George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000; 37:290–298.
- Schwartz J, Leber MD, Gillis S, Giunta A, Eldor A, Bussel JB. Long term follow-up after splenectomy performed for immune thrombocytopenic purpura (ITP). Am J Hematol 2003; 72:94–98.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Stasi R, Provan D. Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 2004; 79:504–522.
- Giagounidis AA, Anhuf J, Schneider P, et al. Treatment of relapsed idiopathic thrombocytopenic purpura with the anti-CD20 monoclonal antibody rituximab: a pilot study. Eur J Haematol 2002; 69:95–100.
- Stasi R, Stipa E, Forte V, Meo P, Amadori S. Variable patterns of response to rituximab treatment in adults with chronic idiopathic thrombocytopenic purpura (letter). Blood 2002; 99:3872–3873.
- Cooper N, Stasi R, Cunningham-Rundles S, et al. The efficacy and safetyof B-cell depletion with anti-CD20 monocloncal antibody in adults with chronic immune thrombocytopenic purpura. Br J Haematol 2004; 125:232–239.
- Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytoopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003; 78:1340–1346.
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB. Long term follow-up of patients with immune thrombocytopenic purpura (ITP) whose initial response to rituximab lasted a minimum of 1 year (abstract). Blood (ASH Annual Meeting Abstracts): 2006;108:Abstract 479.
- Mukai HY, Kojima H, Todokoro K, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76:675–678.
- Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006; 355:1672–1681. (Published correction in N Engl J Med 2006; 355:2054.)
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010; 8:1372–1382.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2010 Aug 23(Epub ahead of print).
- Saleh MN, Bussel JB, Cheng G, et al. Long-term treatment of chronic immune thrombocytopenic purpura with oral eltrombopag. Abstract #682 presented at the 51st American Society of Hematology Annual Meeting and Exposition, New Orleans, LA, December 5–8, 2009; http://ash.confex.com/ash/2009/webprogram/Paper24081.html. Accessed April 26, 2011.
- Kuter DJ, Mufti GJ, Bain BJ, Hasserjian RP, Davis W, Rutstein M. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood 2009; 114:3748–3756.
KEY POINTS
- ITP is defined as an isolated platelet count of less than 100 × 109/L (100,000/μL) and usually presents without symptoms.
- Patients without symptoms who have a platelet count above 30 × 109/L should generally not be treated unless they have an increased risk of bleeding.
- Recent studies suggest that viruses and other pathogens play an important role in secondary ITP.
- Initially, corticosteroids are usually given as prednisone (1–2 mg/kg/day, then tapered), though recent studies suggest that dexamethasone pulses (40 mg/day for 4 days) may provide more durable responses when used in this setting.
- Thrombopoietic agents are important new treatments, although their place in the overall therapy of ITP has not been established.
Correction: Giant cell arteritis
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.
There was an error in the caption for Figure 2 in: Villa-Forte A. Giant cell arteritis: Suspect it, treat it promptly. Cleve Clin J Med 2011; 78:265–270. The image was of digital subtraction angiography, not magnetic resonance angiography. The caption has been corrected in the online version of the article.