User login
Sober today, but lethargic and confused
CASE Confused and weak
Mr. W, age 26, is brought to the emergency department (ED) by his parents for intermittent confusion, weakness, and increasing lethargy over the past 4 days. He is jaundiced with mild abdominal pain, nausea, and vomiting.
Mr. W has a history of alcohol use disorder, drinking as much as 1 L of vodka a day. Six months ago, he was hospitalized for alcoholic hepatitis and severe hyponatremia.
In the ED, Mr. W is awake, alert, and oriented to person, place, and time. Vital signs are: pulse 89 beats per minute; blood pressure, 117/50 mm Hg; respirations, 15 breaths per minute; and temperature, 98.5ºF. Physical examination is notable for scleral icterus, jaundice, tender hepatomegaly, and asterixis.
Mr. W is not taking any medications. He reports that his most recent drink was the day before; however, his current alcohol intake is unknown.
Laboratory tests reveal altered hepatic function, including elevated aspartate aminotransferase (251 U/L), alanine aminotransferase (56 U/L), alkaline phosphatase (179 U/L), total bilirubin (15.4 mg/dL), and ammonia (143 U/L), impaired coagulation (international normalized ratio 2.39), and decreased albumin (2.7 g/dL). Other metabolic disturbances include: sodium, 104 mEq/L; chloride, <60 mEq/L; potassium, 2.2 mEq/L; and CO2, 44.5 mEq/L.
What is your differential diagnosis for Mr. W’s altered mental status?
a) hepatic encephalopathy
b) Wernicke’s encephalopathy
c) hyponatremia
d) drug intoxication
e) head trauma
The authors’ observations
Hyponatremia is defined as a serum sodium concentration <136 mEq/L. Mr. W is considered to have severe hyponatremia because his serum sodium concentration is <125 mEq/L. Although commonly caused by an inability to suppress antidiuretic hormone, hyponatremia has several possible causes (Figure 1).1 Symptoms are nonspecific and are more visible when there is a large or rapid decrease in the serum sodium concentration. Most patients with a serum sodium concentration >125 mEq/L are asymptomatic. Mr. W, who had a serum sodium of 104 mEq/L, presented with several symptoms, including confusion, lethargy, nausea, vomiting, and weakness. Headache, muscle spasms, depressed reflexes, restlessness, and disorientation also might be observed.2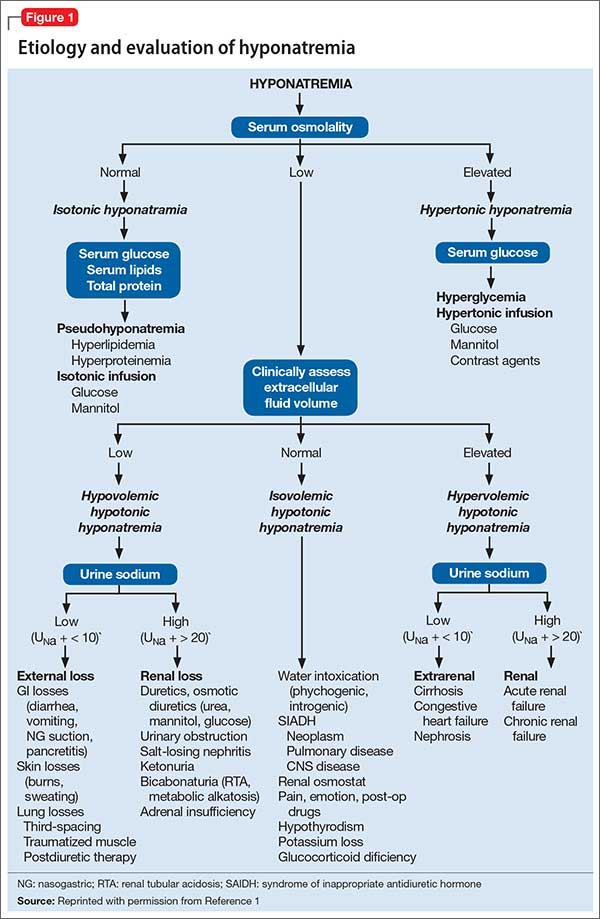
Because of Mr. W’s impaired hepatic function, elevated ammonia, and asterixis, hepatic encephalopathy could be contributing to his altered mental status. Suspect Wernicke’s encephalopathy in a patient with neurologic symptoms and a history of chronic alcohol abuse. In its classic form, Wernicke’s encephalopathy has acute onset, characterized by the triad of ataxia, global confusion, and ocular abnormalities. However, this triad is not consistently or frequently encountered.3
Which tests would you order next?
a) blood ethanol level
b) urine drug screen
c) serum osmolality
d) CT of the head
EVALUATION Sober, yet sick
To rule out intoxication as the cause of Mr. W’s altered mental status, blood ethanol level and urine drug screens are obtained and found to be negative. CT of the head is negative for acute intracranial pathology.
Mr. W is admitted to the medical intensive care unit (MICU) for severe hyponatremia and altered mental status. Serum osmolality is 220 mOsm/kg (normal range 281 to 304 mOsm/kg). To further classify his hypotonic hyponatremia, volume status is assessed, and Mr. W is determined to be euvolemic. Thyroid-stimulating hormone and cortisol are within normal limits, eliminating hypothyroidism and adrenal insufficiency as causes of his euvolemic hypotonic hyponatremia. Mr. W is treated for hyponatremia likely secondary to syndrome of inappropriate antidiuretic hormone (SIADH). SIADH is a diagnosis of exclusion that first requires ruling out hypothyroidism and glucocorticoid insufficiency (Figure 1).1
The authors’ observations
Because hypokalemia is an independent predictive factor for development of hyponatremia, it is necessary to evaluate the potassium level in all hyponatremic patients. Mr. W’s potassium level was 2.2 mEq/L on admission. Serum sodium concentration is related to total exchangeable sodium, total body water, and total exchangeable potassium. Potassium depletion causes a shift of sodium into cells with a comparable exit of potassium from cells into extracellular fluid. The reverse process occurs during potassium repletion, leading to an increase in serum sodium concentration and making hypokalemia a risk factor for developing osmotic demyelination syndrome (ODS).4
Treating hyponatremia
Hyponatremia treatment depends on its severity, presence or absence of symptoms, and whether the hyponatremia is acute (<24 hours) or chronic (>48 hours).5
Because of Mr. W’s extremely low serum sodium concentration, predisposition to hyponatremia secondary to alcoholism, and history of severe hyponatremia, it is likely he is chronically hyponatremic.
In patients with chronic hyponatremia, neurological sequelae are associated with the need for a more rapid rate of correction of serum sodium. For most patients with chronic hyponatremia, a correction rate of ≤10 to 12 mEq/L during the first 24 hours and <18 mEq/L over 48 hours is recommended to avoid ODS.6
Evidence suggests, however, that this 1-day limit might be too high for some patients. Alcoholism, hypokalemia, malnutrition, and liver disease are present in a high percentage of patients who develop
ODS after correcting hyponatremia (Table 1).6 Therefore, for patients such as Mr. W who are at high risk of ODS, experts recommend a goal of 4 to 6 mEq/L/d with a correction rate of ≤8 mEq/L in any 24-hour period (Table 2).6


TREATMENT Sodium normalizes
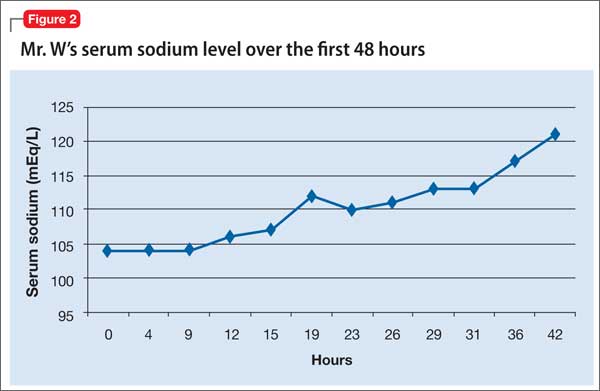
Mr. W receives 1 L of normal saline in the ED before admission to the MICU. Once in the MICU, despite likely chronic hyponatremia, he receives hypertonic (3%) saline, followed by normal saline. Initially, he responds when the serum sodium concentration improves. Because of his likely SIADH, Mr. W is fluid-restricted for 4 days. Serum sodium returns to normal over 7 hospital days (Figure 2). To address the profound hypokalemia, Mr. S receives 30 mEq of potassium chloride in the ED, and potassium is repeated daily throughout his stay in the MICU.
Mr. W remains lethargic, with intermittent periods of confusion throughout the hospital stay. His altered mental status is attributed to hepatic encephalopathy secondary to alcoholic hepatitis. The Maddrey discriminant function is a calculation that stratifies patients with alcoholic hepatitis for risk of mortality and the use of steroids. Because Mr. W shows a Maddrey discriminant function ≥32, he receives methylprednisolone, followed by pentoxifylline, and liver function tests trend down. He also receives lactulose throughout hospitalization.
By discharge on hospital day 9, Mr. W’s serum sodium is 138 mEq/L; serum potassium, 4.1 mEq/L. Total bilirubin and prothrombin remain elevated. Mr. W is discharged on lactulose, thiamine, folic acid, and a 1-month course of pentoxifylline, 400 mg, 3 times a day.
READMISSION Unsteady gait, nausea
Three days after discharge, Mr. W returns to the ED after experiencing a 20-second episode of total body rigidity. He has an unsteady gait and worsening nausea and vomiting.
When Mr. W arrives in the ED, he confirms he is taking his discharge medications as prescribed. His parents report that he has consumed alcohol and Cannabis since discharge and has been taking his sibling’s prescription medications, including quetiapine.
In the ED, Mr. W is awake, alert, and oriented to person, place, and time. Vital signs are: pulse, 118 beats per minute; blood pressure, 128/73 mm Hg; respirations, 16 breaths per minute; and temperature, 98.5ºF. Physical examination, again, is notable for scleral icterus, jaundice, and asterixis. No focal neurologic deficits are noted.
Consistent with Mr. W’s previous admission, laboratory values reveal altered hepatic function and impaired coagulation. The serum sodium level remains within normal limits at 136 mEq/L. However, again, metabolic disturbances include decreased chloride (97 mEq/L), potassium (2.9 mEq/L), and CO2 (18.2 mEq/L). CT on readmission is unchanged from the earlier hospitalization.
What is your differential diagnosis for Mr. W’s total body rigidity?
a) seizure
b) ODS
c) drug intoxication
d) neuroleptic malignant syndrome
EVALUATION Shaking and weakness
Once admitted to the hospital, Mr. W reports an episode of right upper-extremity “shaking,” followed by weakness. He remembers the entire event and denies tongue biting or incontinence. He is evaluated for possible seizure, given his multiple risk factors, including drug and alcohol use, ingestion of quetiapine, and history of hyponatremia. Routine EEG is negative but prolactin level is elevated.
Mr. W’s mental status continues to wax and wane, prompting a neurology consult and MRI for further evaluation. MRI of the brain without contrast reveals restricted diffusion in the pons centrally, with extension bilaterally to the midbrain and thalami—findings consistent with central pontine myelinolysis. A neurology consultation reveals quadriparesis, paraparesis, dysarthria, and diplopia on examination, all symptoms associated with central pontine myelinolysis.
The authors’ observations
ODS, including central and extrapontine myelinolysis, is a demyelinating condition that occurs because of severe osmotic stress, most commonly secondary to the overly rapid correction of hyponatremia in patients with conditions leading to nutritional or electrolyte stress.7 Mr. W is considered at high risk of developing ODS because he fulfills the 5 criteria listed in Table 1.
Several psychiatric illnesses and neuropsychiatric medications could lead to hyponatremia. Many studies8-10 have documented hyponatremia and resulting ODS in patients with alcoholism, schizophrenia, anorexia, primary psychogenic polydipsia, and MDMA (3,4-methylenedioxymethamphetamine) abuse. Hyponatremia is a side effect of several neuropsychiatric medications, including serotonin reuptake inhibitors, lithium, tricyclic antidepressants, opioids, carbamazepine, oxcarbazepine, and antipsychotic polypharmacy. Other commonly used medications associated with hyponatremia include salt-losing diuretics, nonsteroidal anti-inflammatory drugs, and acetaminophen.7
Disease severity varies from asymptomatic to coma or death. Symptoms, although some could reverse completely, typically are a combination of neuropsychiatric (ie, emotional lability, disinhibition, and other bizarre behaviors) and neurologic. Neurologic symptoms include confusion, impaired cognition, dysarthria, dysphagia, gait instability, weakness or paralysis, and generalized seizures. Severely affected patients could experience “locked-in syndrome,” in which they are awake but unable to move or communicate. Also consistent with Mr. W’s case, ODS often presents initially with delirium, seizures, or encephalopathy, followed by a lucid interval before symptoms develop.7
Diagnosis is based on the appearance of demyelinating brain lesions on CT or MRI. MRI is more sensitive than CT; however, even an MRI scan can appear normal for as long as 4 weeks after symptoms appear.7 Therefore, an initial negative radiologic study in a high-risk patient who develops neurologic symptoms does not exclude ODS. Earlier detection is possible with diffusion-weighted MRI, which is most sensitive and can detect lesions within 24 hours of developing symptoms.11 The severity of the lesion does not correlate with severity of symptoms.
Studies reveal a considerable range in prognosis of patients with clinically symptomatic ODS. A study of 44 patients with central pontine myelinolysis, of which 42 had chronic alcoholism, reported that 34% had no significant functional deficits at follow-up, 34% had minor neurologic deficits, and 31% became dependent on personal help. Outcome did not depend on the extent or severity of neurologic symptoms or the severity of concomitant systemic complications.12
Because of its poor prognosis, prevention of ODS is important. Because ODS commonly is caused by overly rapid correction of hyponatremia, it is necessary to adhere to guidelines for treating chronic hyponatremia (Table 2). If overcorrection occurs, therapeutic re-lowering of serum sodium can be considered, but has not been validated in controlled trials. Based mainly on case reports that suggest benefit from early re-lowering serum sodium in patients with ODS symptoms, experts recommend the following:
• administer desmopressin, 2 to 4 μg, every 8 hours parenterally
• replace water orally or as 5% dextrose in water intravenously (3 mL/kg/hr)
• check serum sodium hourly until serum is reduced to goal.6
Bottom Line
Hyponatremia is the most common electrolyte disorder encountered in practice. Osmotic demyelination syndrome often is preventable, with considerable morbidity and mortality. Psychiatrists should be aware of this condition because it could be an adverse effect of many psychiatric medications and there are some psychiatric illnesses in which hyponatremia is a potential risk. In hyponatremic patients with persistent nonspecific neurologic or neuropsychiatric symptoms and negative CT imaging, additional imaging, such as MRI, is warranted.
Related Resources
- Braun MM, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
- Vaidya C, Ho W, Freda BJ. Management of hyponatremia: providing treatment and avoiding harm. Cleve Clin J Med. 2010;77(10):715-726.
Drug Brand Names
Carbamazepine • Tegretol
Oxcarbazepine • Trileptal
Desmopressin • Stimate, DDAVP
Lithium • Eskalith, Lithobid
Pentoxifylline • Trental, Pentoxil
Methylprednisolone • Medrol
Quetiapine • Seroquel
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Elhassen EA, Schrier RW. Disorders of sodium and water balance. In: McKean SC, Ross JJ, Dressler DD, et al, eds. Principles and practice of hospital medicine. New York, NY: McGraw-Hill; 2012:2084-2093.
2. Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581-1589.
3. Reuler JB, Girard DE, Cooney TG. Current concepts. Wernicke’s encephalopathy. N Engl J Med. 1985;312(16):1035-1039.
4. Edelman IS, Leibman J, O’Meara MP, et al. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest. 1958;37(9):1236-1256.
5. Reynolds RM, Seckl JR. Hyponatraemia for the clinical endocrinologist. Clin Endocrinol (Oxf). 2005;63(4):366-374.
6. Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 suppl 1):S1-S42.
7. Hurley RA, Filley CM, Taber KH. Central pontine myelinolysis: a metabolic disorder of myelin. J Neuropsychiatry Clin Neurosci. 2011;23(4):369-374.
8. Goldman MB. The assessment and treatment of water imbalance in patients with psychosis. Clin Schizophr Related Psychoses. 2010;4(2):115-123.
9. Patel AS, Matthews L, Bruce-Jones W. Central pontine myelinolysis as a complication of refeeding syndrome in a patient with anorexia nervosa. J Neuropsychiatry Clin Neurosci. 2008;20(3):371-373.
10. Bhuvaneswar CG, Baldessarini RJ, Harsh VL, et al. Adverse endocrine and metabolic effects of psychotropic drugs: selective clinical review. CNS Drugs. 2009;23(12):1003-1021.
11. Ruzek KA, Campeau NG, Miller GM. Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. AJNR Am J Neuroradiol. 2004;25(2):210-213.
12. Menger H, Jörg J. Outcome of central pontine and extrapontine myelinolysis (n = 44). J Neurol. 1999;246(8):700-705.
sodium concentration, Wernicke’s
encephalopathy, osmotic demyelination syndrome, electrolyte disorder
CASE Confused and weak
Mr. W, age 26, is brought to the emergency department (ED) by his parents for intermittent confusion, weakness, and increasing lethargy over the past 4 days. He is jaundiced with mild abdominal pain, nausea, and vomiting.
Mr. W has a history of alcohol use disorder, drinking as much as 1 L of vodka a day. Six months ago, he was hospitalized for alcoholic hepatitis and severe hyponatremia.
In the ED, Mr. W is awake, alert, and oriented to person, place, and time. Vital signs are: pulse 89 beats per minute; blood pressure, 117/50 mm Hg; respirations, 15 breaths per minute; and temperature, 98.5ºF. Physical examination is notable for scleral icterus, jaundice, tender hepatomegaly, and asterixis.
Mr. W is not taking any medications. He reports that his most recent drink was the day before; however, his current alcohol intake is unknown.
Laboratory tests reveal altered hepatic function, including elevated aspartate aminotransferase (251 U/L), alanine aminotransferase (56 U/L), alkaline phosphatase (179 U/L), total bilirubin (15.4 mg/dL), and ammonia (143 U/L), impaired coagulation (international normalized ratio 2.39), and decreased albumin (2.7 g/dL). Other metabolic disturbances include: sodium, 104 mEq/L; chloride, <60 mEq/L; potassium, 2.2 mEq/L; and CO2, 44.5 mEq/L.
What is your differential diagnosis for Mr. W’s altered mental status?
a) hepatic encephalopathy
b) Wernicke’s encephalopathy
c) hyponatremia
d) drug intoxication
e) head trauma
The authors’ observations
Hyponatremia is defined as a serum sodium concentration <136 mEq/L. Mr. W is considered to have severe hyponatremia because his serum sodium concentration is <125 mEq/L. Although commonly caused by an inability to suppress antidiuretic hormone, hyponatremia has several possible causes (Figure 1).1 Symptoms are nonspecific and are more visible when there is a large or rapid decrease in the serum sodium concentration. Most patients with a serum sodium concentration >125 mEq/L are asymptomatic. Mr. W, who had a serum sodium of 104 mEq/L, presented with several symptoms, including confusion, lethargy, nausea, vomiting, and weakness. Headache, muscle spasms, depressed reflexes, restlessness, and disorientation also might be observed.2
Because of Mr. W’s impaired hepatic function, elevated ammonia, and asterixis, hepatic encephalopathy could be contributing to his altered mental status. Suspect Wernicke’s encephalopathy in a patient with neurologic symptoms and a history of chronic alcohol abuse. In its classic form, Wernicke’s encephalopathy has acute onset, characterized by the triad of ataxia, global confusion, and ocular abnormalities. However, this triad is not consistently or frequently encountered.3
Which tests would you order next?
a) blood ethanol level
b) urine drug screen
c) serum osmolality
d) CT of the head
EVALUATION Sober, yet sick
To rule out intoxication as the cause of Mr. W’s altered mental status, blood ethanol level and urine drug screens are obtained and found to be negative. CT of the head is negative for acute intracranial pathology.
Mr. W is admitted to the medical intensive care unit (MICU) for severe hyponatremia and altered mental status. Serum osmolality is 220 mOsm/kg (normal range 281 to 304 mOsm/kg). To further classify his hypotonic hyponatremia, volume status is assessed, and Mr. W is determined to be euvolemic. Thyroid-stimulating hormone and cortisol are within normal limits, eliminating hypothyroidism and adrenal insufficiency as causes of his euvolemic hypotonic hyponatremia. Mr. W is treated for hyponatremia likely secondary to syndrome of inappropriate antidiuretic hormone (SIADH). SIADH is a diagnosis of exclusion that first requires ruling out hypothyroidism and glucocorticoid insufficiency (Figure 1).1
The authors’ observations
Because hypokalemia is an independent predictive factor for development of hyponatremia, it is necessary to evaluate the potassium level in all hyponatremic patients. Mr. W’s potassium level was 2.2 mEq/L on admission. Serum sodium concentration is related to total exchangeable sodium, total body water, and total exchangeable potassium. Potassium depletion causes a shift of sodium into cells with a comparable exit of potassium from cells into extracellular fluid. The reverse process occurs during potassium repletion, leading to an increase in serum sodium concentration and making hypokalemia a risk factor for developing osmotic demyelination syndrome (ODS).4
Treating hyponatremia
Hyponatremia treatment depends on its severity, presence or absence of symptoms, and whether the hyponatremia is acute (<24 hours) or chronic (>48 hours).5
Because of Mr. W’s extremely low serum sodium concentration, predisposition to hyponatremia secondary to alcoholism, and history of severe hyponatremia, it is likely he is chronically hyponatremic.
In patients with chronic hyponatremia, neurological sequelae are associated with the need for a more rapid rate of correction of serum sodium. For most patients with chronic hyponatremia, a correction rate of ≤10 to 12 mEq/L during the first 24 hours and <18 mEq/L over 48 hours is recommended to avoid ODS.6
Evidence suggests, however, that this 1-day limit might be too high for some patients. Alcoholism, hypokalemia, malnutrition, and liver disease are present in a high percentage of patients who develop
ODS after correcting hyponatremia (Table 1).6 Therefore, for patients such as Mr. W who are at high risk of ODS, experts recommend a goal of 4 to 6 mEq/L/d with a correction rate of ≤8 mEq/L in any 24-hour period (Table 2).6
TREATMENT Sodium normalizes
Mr. W receives 1 L of normal saline in the ED before admission to the MICU. Once in the MICU, despite likely chronic hyponatremia, he receives hypertonic (3%) saline, followed by normal saline. Initially, he responds when the serum sodium concentration improves. Because of his likely SIADH, Mr. W is fluid-restricted for 4 days. Serum sodium returns to normal over 7 hospital days (Figure 2). To address the profound hypokalemia, Mr. S receives 30 mEq of potassium chloride in the ED, and potassium is repeated daily throughout his stay in the MICU.
Mr. W remains lethargic, with intermittent periods of confusion throughout the hospital stay. His altered mental status is attributed to hepatic encephalopathy secondary to alcoholic hepatitis. The Maddrey discriminant function is a calculation that stratifies patients with alcoholic hepatitis for risk of mortality and the use of steroids. Because Mr. W shows a Maddrey discriminant function ≥32, he receives methylprednisolone, followed by pentoxifylline, and liver function tests trend down. He also receives lactulose throughout hospitalization.
By discharge on hospital day 9, Mr. W’s serum sodium is 138 mEq/L; serum potassium, 4.1 mEq/L. Total bilirubin and prothrombin remain elevated. Mr. W is discharged on lactulose, thiamine, folic acid, and a 1-month course of pentoxifylline, 400 mg, 3 times a day.
READMISSION Unsteady gait, nausea
Three days after discharge, Mr. W returns to the ED after experiencing a 20-second episode of total body rigidity. He has an unsteady gait and worsening nausea and vomiting.
When Mr. W arrives in the ED, he confirms he is taking his discharge medications as prescribed. His parents report that he has consumed alcohol and Cannabis since discharge and has been taking his sibling’s prescription medications, including quetiapine.
In the ED, Mr. W is awake, alert, and oriented to person, place, and time. Vital signs are: pulse, 118 beats per minute; blood pressure, 128/73 mm Hg; respirations, 16 breaths per minute; and temperature, 98.5ºF. Physical examination, again, is notable for scleral icterus, jaundice, and asterixis. No focal neurologic deficits are noted.
Consistent with Mr. W’s previous admission, laboratory values reveal altered hepatic function and impaired coagulation. The serum sodium level remains within normal limits at 136 mEq/L. However, again, metabolic disturbances include decreased chloride (97 mEq/L), potassium (2.9 mEq/L), and CO2 (18.2 mEq/L). CT on readmission is unchanged from the earlier hospitalization.
What is your differential diagnosis for Mr. W’s total body rigidity?
a) seizure
b) ODS
c) drug intoxication
d) neuroleptic malignant syndrome
EVALUATION Shaking and weakness
Once admitted to the hospital, Mr. W reports an episode of right upper-extremity “shaking,” followed by weakness. He remembers the entire event and denies tongue biting or incontinence. He is evaluated for possible seizure, given his multiple risk factors, including drug and alcohol use, ingestion of quetiapine, and history of hyponatremia. Routine EEG is negative but prolactin level is elevated.
Mr. W’s mental status continues to wax and wane, prompting a neurology consult and MRI for further evaluation. MRI of the brain without contrast reveals restricted diffusion in the pons centrally, with extension bilaterally to the midbrain and thalami—findings consistent with central pontine myelinolysis. A neurology consultation reveals quadriparesis, paraparesis, dysarthria, and diplopia on examination, all symptoms associated with central pontine myelinolysis.
The authors’ observations
ODS, including central and extrapontine myelinolysis, is a demyelinating condition that occurs because of severe osmotic stress, most commonly secondary to the overly rapid correction of hyponatremia in patients with conditions leading to nutritional or electrolyte stress.7 Mr. W is considered at high risk of developing ODS because he fulfills the 5 criteria listed in Table 1.
Several psychiatric illnesses and neuropsychiatric medications could lead to hyponatremia. Many studies8-10 have documented hyponatremia and resulting ODS in patients with alcoholism, schizophrenia, anorexia, primary psychogenic polydipsia, and MDMA (3,4-methylenedioxymethamphetamine) abuse. Hyponatremia is a side effect of several neuropsychiatric medications, including serotonin reuptake inhibitors, lithium, tricyclic antidepressants, opioids, carbamazepine, oxcarbazepine, and antipsychotic polypharmacy. Other commonly used medications associated with hyponatremia include salt-losing diuretics, nonsteroidal anti-inflammatory drugs, and acetaminophen.7
Disease severity varies from asymptomatic to coma or death. Symptoms, although some could reverse completely, typically are a combination of neuropsychiatric (ie, emotional lability, disinhibition, and other bizarre behaviors) and neurologic. Neurologic symptoms include confusion, impaired cognition, dysarthria, dysphagia, gait instability, weakness or paralysis, and generalized seizures. Severely affected patients could experience “locked-in syndrome,” in which they are awake but unable to move or communicate. Also consistent with Mr. W’s case, ODS often presents initially with delirium, seizures, or encephalopathy, followed by a lucid interval before symptoms develop.7
Diagnosis is based on the appearance of demyelinating brain lesions on CT or MRI. MRI is more sensitive than CT; however, even an MRI scan can appear normal for as long as 4 weeks after symptoms appear.7 Therefore, an initial negative radiologic study in a high-risk patient who develops neurologic symptoms does not exclude ODS. Earlier detection is possible with diffusion-weighted MRI, which is most sensitive and can detect lesions within 24 hours of developing symptoms.11 The severity of the lesion does not correlate with severity of symptoms.
Studies reveal a considerable range in prognosis of patients with clinically symptomatic ODS. A study of 44 patients with central pontine myelinolysis, of which 42 had chronic alcoholism, reported that 34% had no significant functional deficits at follow-up, 34% had minor neurologic deficits, and 31% became dependent on personal help. Outcome did not depend on the extent or severity of neurologic symptoms or the severity of concomitant systemic complications.12
Because of its poor prognosis, prevention of ODS is important. Because ODS commonly is caused by overly rapid correction of hyponatremia, it is necessary to adhere to guidelines for treating chronic hyponatremia (Table 2). If overcorrection occurs, therapeutic re-lowering of serum sodium can be considered, but has not been validated in controlled trials. Based mainly on case reports that suggest benefit from early re-lowering serum sodium in patients with ODS symptoms, experts recommend the following:
• administer desmopressin, 2 to 4 μg, every 8 hours parenterally
• replace water orally or as 5% dextrose in water intravenously (3 mL/kg/hr)
• check serum sodium hourly until serum is reduced to goal.6
Bottom Line
Hyponatremia is the most common electrolyte disorder encountered in practice. Osmotic demyelination syndrome often is preventable, with considerable morbidity and mortality. Psychiatrists should be aware of this condition because it could be an adverse effect of many psychiatric medications and there are some psychiatric illnesses in which hyponatremia is a potential risk. In hyponatremic patients with persistent nonspecific neurologic or neuropsychiatric symptoms and negative CT imaging, additional imaging, such as MRI, is warranted.
Related Resources
- Braun MM, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
- Vaidya C, Ho W, Freda BJ. Management of hyponatremia: providing treatment and avoiding harm. Cleve Clin J Med. 2010;77(10):715-726.
Drug Brand Names
Carbamazepine • Tegretol
Oxcarbazepine • Trileptal
Desmopressin • Stimate, DDAVP
Lithium • Eskalith, Lithobid
Pentoxifylline • Trental, Pentoxil
Methylprednisolone • Medrol
Quetiapine • Seroquel
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Confused and weak
Mr. W, age 26, is brought to the emergency department (ED) by his parents for intermittent confusion, weakness, and increasing lethargy over the past 4 days. He is jaundiced with mild abdominal pain, nausea, and vomiting.
Mr. W has a history of alcohol use disorder, drinking as much as 1 L of vodka a day. Six months ago, he was hospitalized for alcoholic hepatitis and severe hyponatremia.
In the ED, Mr. W is awake, alert, and oriented to person, place, and time. Vital signs are: pulse 89 beats per minute; blood pressure, 117/50 mm Hg; respirations, 15 breaths per minute; and temperature, 98.5ºF. Physical examination is notable for scleral icterus, jaundice, tender hepatomegaly, and asterixis.
Mr. W is not taking any medications. He reports that his most recent drink was the day before; however, his current alcohol intake is unknown.
Laboratory tests reveal altered hepatic function, including elevated aspartate aminotransferase (251 U/L), alanine aminotransferase (56 U/L), alkaline phosphatase (179 U/L), total bilirubin (15.4 mg/dL), and ammonia (143 U/L), impaired coagulation (international normalized ratio 2.39), and decreased albumin (2.7 g/dL). Other metabolic disturbances include: sodium, 104 mEq/L; chloride, <60 mEq/L; potassium, 2.2 mEq/L; and CO2, 44.5 mEq/L.
What is your differential diagnosis for Mr. W’s altered mental status?
a) hepatic encephalopathy
b) Wernicke’s encephalopathy
c) hyponatremia
d) drug intoxication
e) head trauma
The authors’ observations
Hyponatremia is defined as a serum sodium concentration <136 mEq/L. Mr. W is considered to have severe hyponatremia because his serum sodium concentration is <125 mEq/L. Although commonly caused by an inability to suppress antidiuretic hormone, hyponatremia has several possible causes (Figure 1).1 Symptoms are nonspecific and are more visible when there is a large or rapid decrease in the serum sodium concentration. Most patients with a serum sodium concentration >125 mEq/L are asymptomatic. Mr. W, who had a serum sodium of 104 mEq/L, presented with several symptoms, including confusion, lethargy, nausea, vomiting, and weakness. Headache, muscle spasms, depressed reflexes, restlessness, and disorientation also might be observed.2
Because of Mr. W’s impaired hepatic function, elevated ammonia, and asterixis, hepatic encephalopathy could be contributing to his altered mental status. Suspect Wernicke’s encephalopathy in a patient with neurologic symptoms and a history of chronic alcohol abuse. In its classic form, Wernicke’s encephalopathy has acute onset, characterized by the triad of ataxia, global confusion, and ocular abnormalities. However, this triad is not consistently or frequently encountered.3
Which tests would you order next?
a) blood ethanol level
b) urine drug screen
c) serum osmolality
d) CT of the head
EVALUATION Sober, yet sick
To rule out intoxication as the cause of Mr. W’s altered mental status, blood ethanol level and urine drug screens are obtained and found to be negative. CT of the head is negative for acute intracranial pathology.
Mr. W is admitted to the medical intensive care unit (MICU) for severe hyponatremia and altered mental status. Serum osmolality is 220 mOsm/kg (normal range 281 to 304 mOsm/kg). To further classify his hypotonic hyponatremia, volume status is assessed, and Mr. W is determined to be euvolemic. Thyroid-stimulating hormone and cortisol are within normal limits, eliminating hypothyroidism and adrenal insufficiency as causes of his euvolemic hypotonic hyponatremia. Mr. W is treated for hyponatremia likely secondary to syndrome of inappropriate antidiuretic hormone (SIADH). SIADH is a diagnosis of exclusion that first requires ruling out hypothyroidism and glucocorticoid insufficiency (Figure 1).1
The authors’ observations
Because hypokalemia is an independent predictive factor for development of hyponatremia, it is necessary to evaluate the potassium level in all hyponatremic patients. Mr. W’s potassium level was 2.2 mEq/L on admission. Serum sodium concentration is related to total exchangeable sodium, total body water, and total exchangeable potassium. Potassium depletion causes a shift of sodium into cells with a comparable exit of potassium from cells into extracellular fluid. The reverse process occurs during potassium repletion, leading to an increase in serum sodium concentration and making hypokalemia a risk factor for developing osmotic demyelination syndrome (ODS).4
Treating hyponatremia
Hyponatremia treatment depends on its severity, presence or absence of symptoms, and whether the hyponatremia is acute (<24 hours) or chronic (>48 hours).5
Because of Mr. W’s extremely low serum sodium concentration, predisposition to hyponatremia secondary to alcoholism, and history of severe hyponatremia, it is likely he is chronically hyponatremic.
In patients with chronic hyponatremia, neurological sequelae are associated with the need for a more rapid rate of correction of serum sodium. For most patients with chronic hyponatremia, a correction rate of ≤10 to 12 mEq/L during the first 24 hours and <18 mEq/L over 48 hours is recommended to avoid ODS.6
Evidence suggests, however, that this 1-day limit might be too high for some patients. Alcoholism, hypokalemia, malnutrition, and liver disease are present in a high percentage of patients who develop
ODS after correcting hyponatremia (Table 1).6 Therefore, for patients such as Mr. W who are at high risk of ODS, experts recommend a goal of 4 to 6 mEq/L/d with a correction rate of ≤8 mEq/L in any 24-hour period (Table 2).6
TREATMENT Sodium normalizes
Mr. W receives 1 L of normal saline in the ED before admission to the MICU. Once in the MICU, despite likely chronic hyponatremia, he receives hypertonic (3%) saline, followed by normal saline. Initially, he responds when the serum sodium concentration improves. Because of his likely SIADH, Mr. W is fluid-restricted for 4 days. Serum sodium returns to normal over 7 hospital days (Figure 2). To address the profound hypokalemia, Mr. S receives 30 mEq of potassium chloride in the ED, and potassium is repeated daily throughout his stay in the MICU.
Mr. W remains lethargic, with intermittent periods of confusion throughout the hospital stay. His altered mental status is attributed to hepatic encephalopathy secondary to alcoholic hepatitis. The Maddrey discriminant function is a calculation that stratifies patients with alcoholic hepatitis for risk of mortality and the use of steroids. Because Mr. W shows a Maddrey discriminant function ≥32, he receives methylprednisolone, followed by pentoxifylline, and liver function tests trend down. He also receives lactulose throughout hospitalization.
By discharge on hospital day 9, Mr. W’s serum sodium is 138 mEq/L; serum potassium, 4.1 mEq/L. Total bilirubin and prothrombin remain elevated. Mr. W is discharged on lactulose, thiamine, folic acid, and a 1-month course of pentoxifylline, 400 mg, 3 times a day.
READMISSION Unsteady gait, nausea
Three days after discharge, Mr. W returns to the ED after experiencing a 20-second episode of total body rigidity. He has an unsteady gait and worsening nausea and vomiting.
When Mr. W arrives in the ED, he confirms he is taking his discharge medications as prescribed. His parents report that he has consumed alcohol and Cannabis since discharge and has been taking his sibling’s prescription medications, including quetiapine.
In the ED, Mr. W is awake, alert, and oriented to person, place, and time. Vital signs are: pulse, 118 beats per minute; blood pressure, 128/73 mm Hg; respirations, 16 breaths per minute; and temperature, 98.5ºF. Physical examination, again, is notable for scleral icterus, jaundice, and asterixis. No focal neurologic deficits are noted.
Consistent with Mr. W’s previous admission, laboratory values reveal altered hepatic function and impaired coagulation. The serum sodium level remains within normal limits at 136 mEq/L. However, again, metabolic disturbances include decreased chloride (97 mEq/L), potassium (2.9 mEq/L), and CO2 (18.2 mEq/L). CT on readmission is unchanged from the earlier hospitalization.
What is your differential diagnosis for Mr. W’s total body rigidity?
a) seizure
b) ODS
c) drug intoxication
d) neuroleptic malignant syndrome
EVALUATION Shaking and weakness
Once admitted to the hospital, Mr. W reports an episode of right upper-extremity “shaking,” followed by weakness. He remembers the entire event and denies tongue biting or incontinence. He is evaluated for possible seizure, given his multiple risk factors, including drug and alcohol use, ingestion of quetiapine, and history of hyponatremia. Routine EEG is negative but prolactin level is elevated.
Mr. W’s mental status continues to wax and wane, prompting a neurology consult and MRI for further evaluation. MRI of the brain without contrast reveals restricted diffusion in the pons centrally, with extension bilaterally to the midbrain and thalami—findings consistent with central pontine myelinolysis. A neurology consultation reveals quadriparesis, paraparesis, dysarthria, and diplopia on examination, all symptoms associated with central pontine myelinolysis.
The authors’ observations
ODS, including central and extrapontine myelinolysis, is a demyelinating condition that occurs because of severe osmotic stress, most commonly secondary to the overly rapid correction of hyponatremia in patients with conditions leading to nutritional or electrolyte stress.7 Mr. W is considered at high risk of developing ODS because he fulfills the 5 criteria listed in Table 1.
Several psychiatric illnesses and neuropsychiatric medications could lead to hyponatremia. Many studies8-10 have documented hyponatremia and resulting ODS in patients with alcoholism, schizophrenia, anorexia, primary psychogenic polydipsia, and MDMA (3,4-methylenedioxymethamphetamine) abuse. Hyponatremia is a side effect of several neuropsychiatric medications, including serotonin reuptake inhibitors, lithium, tricyclic antidepressants, opioids, carbamazepine, oxcarbazepine, and antipsychotic polypharmacy. Other commonly used medications associated with hyponatremia include salt-losing diuretics, nonsteroidal anti-inflammatory drugs, and acetaminophen.7
Disease severity varies from asymptomatic to coma or death. Symptoms, although some could reverse completely, typically are a combination of neuropsychiatric (ie, emotional lability, disinhibition, and other bizarre behaviors) and neurologic. Neurologic symptoms include confusion, impaired cognition, dysarthria, dysphagia, gait instability, weakness or paralysis, and generalized seizures. Severely affected patients could experience “locked-in syndrome,” in which they are awake but unable to move or communicate. Also consistent with Mr. W’s case, ODS often presents initially with delirium, seizures, or encephalopathy, followed by a lucid interval before symptoms develop.7
Diagnosis is based on the appearance of demyelinating brain lesions on CT or MRI. MRI is more sensitive than CT; however, even an MRI scan can appear normal for as long as 4 weeks after symptoms appear.7 Therefore, an initial negative radiologic study in a high-risk patient who develops neurologic symptoms does not exclude ODS. Earlier detection is possible with diffusion-weighted MRI, which is most sensitive and can detect lesions within 24 hours of developing symptoms.11 The severity of the lesion does not correlate with severity of symptoms.
Studies reveal a considerable range in prognosis of patients with clinically symptomatic ODS. A study of 44 patients with central pontine myelinolysis, of which 42 had chronic alcoholism, reported that 34% had no significant functional deficits at follow-up, 34% had minor neurologic deficits, and 31% became dependent on personal help. Outcome did not depend on the extent or severity of neurologic symptoms or the severity of concomitant systemic complications.12
Because of its poor prognosis, prevention of ODS is important. Because ODS commonly is caused by overly rapid correction of hyponatremia, it is necessary to adhere to guidelines for treating chronic hyponatremia (Table 2). If overcorrection occurs, therapeutic re-lowering of serum sodium can be considered, but has not been validated in controlled trials. Based mainly on case reports that suggest benefit from early re-lowering serum sodium in patients with ODS symptoms, experts recommend the following:
• administer desmopressin, 2 to 4 μg, every 8 hours parenterally
• replace water orally or as 5% dextrose in water intravenously (3 mL/kg/hr)
• check serum sodium hourly until serum is reduced to goal.6
Bottom Line
Hyponatremia is the most common electrolyte disorder encountered in practice. Osmotic demyelination syndrome often is preventable, with considerable morbidity and mortality. Psychiatrists should be aware of this condition because it could be an adverse effect of many psychiatric medications and there are some psychiatric illnesses in which hyponatremia is a potential risk. In hyponatremic patients with persistent nonspecific neurologic or neuropsychiatric symptoms and negative CT imaging, additional imaging, such as MRI, is warranted.
Related Resources
- Braun MM, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
- Vaidya C, Ho W, Freda BJ. Management of hyponatremia: providing treatment and avoiding harm. Cleve Clin J Med. 2010;77(10):715-726.
Drug Brand Names
Carbamazepine • Tegretol
Oxcarbazepine • Trileptal
Desmopressin • Stimate, DDAVP
Lithium • Eskalith, Lithobid
Pentoxifylline • Trental, Pentoxil
Methylprednisolone • Medrol
Quetiapine • Seroquel
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Elhassen EA, Schrier RW. Disorders of sodium and water balance. In: McKean SC, Ross JJ, Dressler DD, et al, eds. Principles and practice of hospital medicine. New York, NY: McGraw-Hill; 2012:2084-2093.
2. Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581-1589.
3. Reuler JB, Girard DE, Cooney TG. Current concepts. Wernicke’s encephalopathy. N Engl J Med. 1985;312(16):1035-1039.
4. Edelman IS, Leibman J, O’Meara MP, et al. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest. 1958;37(9):1236-1256.
5. Reynolds RM, Seckl JR. Hyponatraemia for the clinical endocrinologist. Clin Endocrinol (Oxf). 2005;63(4):366-374.
6. Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 suppl 1):S1-S42.
7. Hurley RA, Filley CM, Taber KH. Central pontine myelinolysis: a metabolic disorder of myelin. J Neuropsychiatry Clin Neurosci. 2011;23(4):369-374.
8. Goldman MB. The assessment and treatment of water imbalance in patients with psychosis. Clin Schizophr Related Psychoses. 2010;4(2):115-123.
9. Patel AS, Matthews L, Bruce-Jones W. Central pontine myelinolysis as a complication of refeeding syndrome in a patient with anorexia nervosa. J Neuropsychiatry Clin Neurosci. 2008;20(3):371-373.
10. Bhuvaneswar CG, Baldessarini RJ, Harsh VL, et al. Adverse endocrine and metabolic effects of psychotropic drugs: selective clinical review. CNS Drugs. 2009;23(12):1003-1021.
11. Ruzek KA, Campeau NG, Miller GM. Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. AJNR Am J Neuroradiol. 2004;25(2):210-213.
12. Menger H, Jörg J. Outcome of central pontine and extrapontine myelinolysis (n = 44). J Neurol. 1999;246(8):700-705.
1. Elhassen EA, Schrier RW. Disorders of sodium and water balance. In: McKean SC, Ross JJ, Dressler DD, et al, eds. Principles and practice of hospital medicine. New York, NY: McGraw-Hill; 2012:2084-2093.
2. Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581-1589.
3. Reuler JB, Girard DE, Cooney TG. Current concepts. Wernicke’s encephalopathy. N Engl J Med. 1985;312(16):1035-1039.
4. Edelman IS, Leibman J, O’Meara MP, et al. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest. 1958;37(9):1236-1256.
5. Reynolds RM, Seckl JR. Hyponatraemia for the clinical endocrinologist. Clin Endocrinol (Oxf). 2005;63(4):366-374.
6. Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 suppl 1):S1-S42.
7. Hurley RA, Filley CM, Taber KH. Central pontine myelinolysis: a metabolic disorder of myelin. J Neuropsychiatry Clin Neurosci. 2011;23(4):369-374.
8. Goldman MB. The assessment and treatment of water imbalance in patients with psychosis. Clin Schizophr Related Psychoses. 2010;4(2):115-123.
9. Patel AS, Matthews L, Bruce-Jones W. Central pontine myelinolysis as a complication of refeeding syndrome in a patient with anorexia nervosa. J Neuropsychiatry Clin Neurosci. 2008;20(3):371-373.
10. Bhuvaneswar CG, Baldessarini RJ, Harsh VL, et al. Adverse endocrine and metabolic effects of psychotropic drugs: selective clinical review. CNS Drugs. 2009;23(12):1003-1021.
11. Ruzek KA, Campeau NG, Miller GM. Early diagnosis of central pontine myelinolysis with diffusion-weighted imaging. AJNR Am J Neuroradiol. 2004;25(2):210-213.
12. Menger H, Jörg J. Outcome of central pontine and extrapontine myelinolysis (n = 44). J Neurol. 1999;246(8):700-705.
sodium concentration, Wernicke’s
encephalopathy, osmotic demyelination syndrome, electrolyte disorder
sodium concentration, Wernicke’s
encephalopathy, osmotic demyelination syndrome, electrolyte disorder
Psychoneurogastroenterology: The abdominal brain, the microbiome, and psychiatry
This nervous system is located inside the wall of the GI tract, extending from the esophagus to the rectum. Technically, it is known as the enteric nervous system, or ENS, but it has been given other labels, too: “second brain,”2 “abdominal brain,” “other brain,” and “back-up brain.” Its neurologic disorders include abdominal epilepsy, abdominal migraine, and autism with intestinal symptoms, such as chronic enterocolitis.3
Impressive brain-like features
The ENS includes 100 million neurons (same as the spinal cord) with glia-like support cells. It contains >30 neurotransmitters, including several closely linked to psychopathology (serotonin, dopamine, γ-aminobutyric acid, and acetylcholine). The ENS is not part of the autonomic nervous system. It communicates with the brain via the vagus nerve.
A vast system of gut bacteria
The ENS maintains close links with, and is influenced by, the microbiome, an extensive universe of commensal (that is, symbiotic) bacteria in the gut that play a vital role in immune health, brain function, and signaling systems within the CNS. The role of the microbiome in neuropsychiatric disorders has become a sizzling area of research.
The numbers of the microbiome are astonishing, including approximately 1,000 species of bacteria; 100 trillion total bacterial organisms (outnumbering cells of the body by 100-fold); 4 million bacterial genes (compared with 26,000 genes in the host human genome); and a density as high as 1 trillion bacteria in a cubic milliliter—higher than any known microbial system.4
Significant GI−brain connections
It is of great relevance to psychiatry that 90% of the body’s serotonin and 50% of dopamine are found in the GI brain. Selective serotonin reuptake inhibitors often are associated with GI symptoms, such as nausea and diarrhea; antipsychotics, which are dopamine antagonists, are known for antiemetic effects. Clozapine’s potent anticholinergic effects can cause serious ileus.
Things get more interesting when one considers the association of GI disorders and psychiatric symptoms:
Irritable bowel syndrome is associated with panic disorder, generalized anxiety disorder, social phobia, dysthymia, and major depression.
Inflammatory bowel disease (IBD)— such as Crohn’s disease and ulcerative colitis (prevalence ranging from 6% in Canada to 14% in the United States to 46% in Mexico5)—is commonly associated with mood and anxiety disorders and personality changes. The psychiatric manifestations of IBD are so common that the authors of a recent article in World Journal of Gastroenterology urged gastroenterologists to collaborate with psychiatrists when managing IBD.6
Celiac disease has been repeatedly associated with several neuropsychiatric disorders, including ataxia, epilepsy, peripheral neuropathy, headache, anxiety, attention-deficit/hyperactivity disorder, autism spectrum disorder, and schizophrenia.
New, exciting challenges for medical science
There potentially are important implications for possible exploitation of the ENS and the microbiome in the diagnosis and treatment of neuropsychiatric disorders. For example, consider these speculative challenges:
• Can intestinal biopsy reveal neurotransmitter pathology in schizophrenia?
• Can early dopamine deficiency predict Parkinson’s disease, enabling early intervention?
• Can β-amyloid deposits, the degenerative neurologic stigmata of Alzheimer’s disease, be detected in abdominal neurons years before onset of symptoms to allow early intervention?
• Can the ENS become a therapeutic pathway by targeting the various neurotransmitters found there or by engaging the enormous human microbiome to manipulate its beneficial properties?
• Can foods or probiotic supplements be prescribed as microbiomal adjuncts to improve the mood and anxiety spectrum?
One recommendation I came across is that ingesting 10 to 100 million beneficial bacteria, such as Lactobacillus plantarum and Bifidobacterium infantis, might be helpful. Such prescriptions obviously are speculative but also are reasonably testable hypotheses of ways to exploit the “other brain” and the microbiome.
We must summon the guts to seize this opportunity
An independent second brain and a remarkable microbiome appear to be significant evolutionary adaptations and advantages for humans. For too long, neuropsychiatric researchers have ignored the ENS and the microbiome; now, they must focus on how to exploit these entities to yield innovative diagnostic and therapeutic advances. Integrating the ENS and the microbiome and enmeshing them into neuropsychiatric research and clinical applications hold great promise.
The field of psychoneurogastroenterology is in its infancy, but its growth and relevance will be momentous for neuropsychiatry. A major intellectual peristalsis is underway.
1. Robinson B. The abdominal and pelvic brain. Hammond, IN: Frank S. Betz; 1907.
2. Gershon M. The second brain: a groundbreaking new understanding of nervous disorders of the stomach and intestine. New York, NY: HarperCollins Publishers; 1998.
3. McMillin DL, Richards DG, Mein EA, et al. The abdominal brain and enteric nervous system. J Altern Complement Med. 1999;5(6):575-586.
4. Hill JM, Bhattacharjee S, Pogue AI, et al. The gastrointestinal tract microbiome and potential link to Alzheimer’s disease. Front Neurol. 2014;5:43.
5. Olden KW, Lydiard RB. Gastrointestinal disorders. In: Rundell JR, Wise MG. Textbook of consultation-liaison psychiatry. Washington, DC: American Psychiatric Association; 1994.
6. Filipovic BR, Filipovic BF. World J Gastroenterol. 2014;20(13):3552-3563.
This nervous system is located inside the wall of the GI tract, extending from the esophagus to the rectum. Technically, it is known as the enteric nervous system, or ENS, but it has been given other labels, too: “second brain,”2 “abdominal brain,” “other brain,” and “back-up brain.” Its neurologic disorders include abdominal epilepsy, abdominal migraine, and autism with intestinal symptoms, such as chronic enterocolitis.3
Impressive brain-like features
The ENS includes 100 million neurons (same as the spinal cord) with glia-like support cells. It contains >30 neurotransmitters, including several closely linked to psychopathology (serotonin, dopamine, γ-aminobutyric acid, and acetylcholine). The ENS is not part of the autonomic nervous system. It communicates with the brain via the vagus nerve.
A vast system of gut bacteria
The ENS maintains close links with, and is influenced by, the microbiome, an extensive universe of commensal (that is, symbiotic) bacteria in the gut that play a vital role in immune health, brain function, and signaling systems within the CNS. The role of the microbiome in neuropsychiatric disorders has become a sizzling area of research.
The numbers of the microbiome are astonishing, including approximately 1,000 species of bacteria; 100 trillion total bacterial organisms (outnumbering cells of the body by 100-fold); 4 million bacterial genes (compared with 26,000 genes in the host human genome); and a density as high as 1 trillion bacteria in a cubic milliliter—higher than any known microbial system.4
Significant GI−brain connections
It is of great relevance to psychiatry that 90% of the body’s serotonin and 50% of dopamine are found in the GI brain. Selective serotonin reuptake inhibitors often are associated with GI symptoms, such as nausea and diarrhea; antipsychotics, which are dopamine antagonists, are known for antiemetic effects. Clozapine’s potent anticholinergic effects can cause serious ileus.
Things get more interesting when one considers the association of GI disorders and psychiatric symptoms:
Irritable bowel syndrome is associated with panic disorder, generalized anxiety disorder, social phobia, dysthymia, and major depression.
Inflammatory bowel disease (IBD)— such as Crohn’s disease and ulcerative colitis (prevalence ranging from 6% in Canada to 14% in the United States to 46% in Mexico5)—is commonly associated with mood and anxiety disorders and personality changes. The psychiatric manifestations of IBD are so common that the authors of a recent article in World Journal of Gastroenterology urged gastroenterologists to collaborate with psychiatrists when managing IBD.6
Celiac disease has been repeatedly associated with several neuropsychiatric disorders, including ataxia, epilepsy, peripheral neuropathy, headache, anxiety, attention-deficit/hyperactivity disorder, autism spectrum disorder, and schizophrenia.
New, exciting challenges for medical science
There potentially are important implications for possible exploitation of the ENS and the microbiome in the diagnosis and treatment of neuropsychiatric disorders. For example, consider these speculative challenges:
• Can intestinal biopsy reveal neurotransmitter pathology in schizophrenia?
• Can early dopamine deficiency predict Parkinson’s disease, enabling early intervention?
• Can β-amyloid deposits, the degenerative neurologic stigmata of Alzheimer’s disease, be detected in abdominal neurons years before onset of symptoms to allow early intervention?
• Can the ENS become a therapeutic pathway by targeting the various neurotransmitters found there or by engaging the enormous human microbiome to manipulate its beneficial properties?
• Can foods or probiotic supplements be prescribed as microbiomal adjuncts to improve the mood and anxiety spectrum?
One recommendation I came across is that ingesting 10 to 100 million beneficial bacteria, such as Lactobacillus plantarum and Bifidobacterium infantis, might be helpful. Such prescriptions obviously are speculative but also are reasonably testable hypotheses of ways to exploit the “other brain” and the microbiome.
We must summon the guts to seize this opportunity
An independent second brain and a remarkable microbiome appear to be significant evolutionary adaptations and advantages for humans. For too long, neuropsychiatric researchers have ignored the ENS and the microbiome; now, they must focus on how to exploit these entities to yield innovative diagnostic and therapeutic advances. Integrating the ENS and the microbiome and enmeshing them into neuropsychiatric research and clinical applications hold great promise.
The field of psychoneurogastroenterology is in its infancy, but its growth and relevance will be momentous for neuropsychiatry. A major intellectual peristalsis is underway.
This nervous system is located inside the wall of the GI tract, extending from the esophagus to the rectum. Technically, it is known as the enteric nervous system, or ENS, but it has been given other labels, too: “second brain,”2 “abdominal brain,” “other brain,” and “back-up brain.” Its neurologic disorders include abdominal epilepsy, abdominal migraine, and autism with intestinal symptoms, such as chronic enterocolitis.3
Impressive brain-like features
The ENS includes 100 million neurons (same as the spinal cord) with glia-like support cells. It contains >30 neurotransmitters, including several closely linked to psychopathology (serotonin, dopamine, γ-aminobutyric acid, and acetylcholine). The ENS is not part of the autonomic nervous system. It communicates with the brain via the vagus nerve.
A vast system of gut bacteria
The ENS maintains close links with, and is influenced by, the microbiome, an extensive universe of commensal (that is, symbiotic) bacteria in the gut that play a vital role in immune health, brain function, and signaling systems within the CNS. The role of the microbiome in neuropsychiatric disorders has become a sizzling area of research.
The numbers of the microbiome are astonishing, including approximately 1,000 species of bacteria; 100 trillion total bacterial organisms (outnumbering cells of the body by 100-fold); 4 million bacterial genes (compared with 26,000 genes in the host human genome); and a density as high as 1 trillion bacteria in a cubic milliliter—higher than any known microbial system.4
Significant GI−brain connections
It is of great relevance to psychiatry that 90% of the body’s serotonin and 50% of dopamine are found in the GI brain. Selective serotonin reuptake inhibitors often are associated with GI symptoms, such as nausea and diarrhea; antipsychotics, which are dopamine antagonists, are known for antiemetic effects. Clozapine’s potent anticholinergic effects can cause serious ileus.
Things get more interesting when one considers the association of GI disorders and psychiatric symptoms:
Irritable bowel syndrome is associated with panic disorder, generalized anxiety disorder, social phobia, dysthymia, and major depression.
Inflammatory bowel disease (IBD)— such as Crohn’s disease and ulcerative colitis (prevalence ranging from 6% in Canada to 14% in the United States to 46% in Mexico5)—is commonly associated with mood and anxiety disorders and personality changes. The psychiatric manifestations of IBD are so common that the authors of a recent article in World Journal of Gastroenterology urged gastroenterologists to collaborate with psychiatrists when managing IBD.6
Celiac disease has been repeatedly associated with several neuropsychiatric disorders, including ataxia, epilepsy, peripheral neuropathy, headache, anxiety, attention-deficit/hyperactivity disorder, autism spectrum disorder, and schizophrenia.
New, exciting challenges for medical science
There potentially are important implications for possible exploitation of the ENS and the microbiome in the diagnosis and treatment of neuropsychiatric disorders. For example, consider these speculative challenges:
• Can intestinal biopsy reveal neurotransmitter pathology in schizophrenia?
• Can early dopamine deficiency predict Parkinson’s disease, enabling early intervention?
• Can β-amyloid deposits, the degenerative neurologic stigmata of Alzheimer’s disease, be detected in abdominal neurons years before onset of symptoms to allow early intervention?
• Can the ENS become a therapeutic pathway by targeting the various neurotransmitters found there or by engaging the enormous human microbiome to manipulate its beneficial properties?
• Can foods or probiotic supplements be prescribed as microbiomal adjuncts to improve the mood and anxiety spectrum?
One recommendation I came across is that ingesting 10 to 100 million beneficial bacteria, such as Lactobacillus plantarum and Bifidobacterium infantis, might be helpful. Such prescriptions obviously are speculative but also are reasonably testable hypotheses of ways to exploit the “other brain” and the microbiome.
We must summon the guts to seize this opportunity
An independent second brain and a remarkable microbiome appear to be significant evolutionary adaptations and advantages for humans. For too long, neuropsychiatric researchers have ignored the ENS and the microbiome; now, they must focus on how to exploit these entities to yield innovative diagnostic and therapeutic advances. Integrating the ENS and the microbiome and enmeshing them into neuropsychiatric research and clinical applications hold great promise.
The field of psychoneurogastroenterology is in its infancy, but its growth and relevance will be momentous for neuropsychiatry. A major intellectual peristalsis is underway.
1. Robinson B. The abdominal and pelvic brain. Hammond, IN: Frank S. Betz; 1907.
2. Gershon M. The second brain: a groundbreaking new understanding of nervous disorders of the stomach and intestine. New York, NY: HarperCollins Publishers; 1998.
3. McMillin DL, Richards DG, Mein EA, et al. The abdominal brain and enteric nervous system. J Altern Complement Med. 1999;5(6):575-586.
4. Hill JM, Bhattacharjee S, Pogue AI, et al. The gastrointestinal tract microbiome and potential link to Alzheimer’s disease. Front Neurol. 2014;5:43.
5. Olden KW, Lydiard RB. Gastrointestinal disorders. In: Rundell JR, Wise MG. Textbook of consultation-liaison psychiatry. Washington, DC: American Psychiatric Association; 1994.
6. Filipovic BR, Filipovic BF. World J Gastroenterol. 2014;20(13):3552-3563.
1. Robinson B. The abdominal and pelvic brain. Hammond, IN: Frank S. Betz; 1907.
2. Gershon M. The second brain: a groundbreaking new understanding of nervous disorders of the stomach and intestine. New York, NY: HarperCollins Publishers; 1998.
3. McMillin DL, Richards DG, Mein EA, et al. The abdominal brain and enteric nervous system. J Altern Complement Med. 1999;5(6):575-586.
4. Hill JM, Bhattacharjee S, Pogue AI, et al. The gastrointestinal tract microbiome and potential link to Alzheimer’s disease. Front Neurol. 2014;5:43.
5. Olden KW, Lydiard RB. Gastrointestinal disorders. In: Rundell JR, Wise MG. Textbook of consultation-liaison psychiatry. Washington, DC: American Psychiatric Association; 1994.
6. Filipovic BR, Filipovic BF. World J Gastroenterol. 2014;20(13):3552-3563.
Young, pregnant, ataxic—and jilted
CASE Difficulty walking
Ms. M, age 15, is a pregnant, Spanish-speaking Guatemalan woman who is brought to obstetrics triage in a large academic medical center at 35 weeks gestational age. She complains of dizziness, tinnitus, left orbital headache, and difficulty walking.
The neurology service finds profound truncal ataxia, astasia-abasia, and buckling of the knees; a normal brain and spine MRI are not consistent with a neurologic etiology. Otolaryngology service evaluates Ms. M to rule out a cholesteatoma and suggests a head CT and endoscopy, which are normal.
Ms. M’s symptoms resolve after 3 days, although the gait disturbances persist. When no clear cause is found for her difficulty walking, the psychiatry service is consulted to evaluate whether an underlying psychiatric disorder is contributing to symptoms.
What could be causing Ms. M’s symptoms?
a) malingering
b) factitious disorder
c) undiagnosed neurologic disorder
d) conversion disorder
The authors’ observations
Women are vulnerable to a variety of psychiatric illnesses during pregnancy1 that have deleterious effects on mother, baby, and family.2-6 Although there is a burgeoning literature on affective and anxiety disorders occurring in pregnancy, there is a dearth of information about somatoform disorders.
HISTORY Abandonment
Ms. M reports that, although her boyfriend deserted her after learning about the unexpected pregnancy, she will welcome the baby and looks forward to motherhood. She seems unaware of the responsibilities of being a mother.
Ms. M acknowledges a history of depression and self-harm a few years earlier, yet says she feels better now and thinks that psychiatric care is unnecessary. Because she does not endorse a history of trauma or symptoms suggesting an affective, anxiety, or psychotic illness, the psychiatrist does not recommend treatment with psychotropic medication.
At age 5, Ms. M’s parents sent her to the United States with her aunt, hoping that she would have a better life than she would have had in Guatemala. Her aunt reports that Ms. M initially had difficulty adjusting to life in the United States without her parents, yet she has made substantial strides over the years and is now quite accustomed to the country. Her aunt describes Ms. M as an independent high school student who earns good grades.
During the interview, the psychiatrist observes that Ms. M exhibits childlike mannerisms, including sleeping with stuffed toys and coloring in Disney books with crayons. She also is indifferent to her gait difficulty, pregnancy, and psychosocial stressors. Her affect is inconsistent with the content of her speech and she is alexithymic.
Ms. M’s aunt reports that her niece is becoming more dependent on her, which is not consistent with her baseline. Her aunt also notes that several years earlier, Ms. M’s nephew was diagnosed with a cholesteatoma after he presented with similar symptoms.
The combination of (1) Ms. M’s clinical presentation, which was causing her significant impairment in her social functioning, (2) the incompatibility of symptoms with any recognized neurologic and medical disease, and (3) prior family experience with cholesteatoma leads the consulting psychiatrist to suspect conversion disorder. Ms. M’s alexithymia, indifference to her symptoms, and recent abandonment by the baby’s father also support a conversion disorder diagnosis.
From a psychodynamic perspective, the ataxia appears to be her way of protecting herself from the abandonment she is experiencing by being left again to “stand alone” by her boyfriend as she had been when her parents sent her to the United States. Her regressive behavior could be her way of securing her aunt’s love and support.
The authors’ observations
This is the first case of psychogenic gait disturbance during pregnancy described in the literature. Authors have reported on pseudotoxemia,7 hyperemesis gravidarum,8 and pesudocyesis,9 yet there is a paucity of information on psychogenic gait disturbance during pregnancy. Ms. M’s case elucidates many of the clinical quandaries that occur when managing psychiatric illness—and, more specifically, conversion disorder— during pregnancy. Many women are hesitant to seek psychiatric treatment during pregnancy because of shame, stigma, and fear of loss of personal or parental rights10,11; it is not surprising that emotionally distressed women communicate their feelings or troubled thoughts through physical symptoms.
Likely diagnosis
Conversion disorder is the presence of neurologic symptoms in the absence of a neurologic diagnosis that fully explains those symptoms. Conversion disorder, previously known as hysteria, is called functional neurologic symptom disorder in DSM-5 (Box).12 Symptoms are not feigned; instead, they represent “conversion” of emotional distress into neurologic symptoms.13,14 Although misdiagnosing conversion disorder in patients with true neurologic disease is uncommon, clinicians often are uncomfortable making the diagnosis until all medical causes have been ruled out.14 It is not always possible to find a psychological explanation for conversion disorder, but a history of childhood abuse, particularly sexual abuse, could play a role.14

Because of the variety of presentations, clinicians in all specialties should be familiar with somatoform disorders; this is especially important in obstetrics and gynecology because women are more likely than men to develop these disorders.15 It is important to consider that Ms. M is a teenager and somatoform disorders can present differently in adults. The diagnostic process should include a diligent somatic workup and a personal and social history to identify the patient’s developmental tasks, stressors, and coping style.15
How would you treat Ms. M?
a) destigmatize psychiatric illness and provide psychoeducation regarding treatment benefits
b) identify and treat any comorbid psychiatric disorders
c) maintain a proactive and multidisciplinary approach that includes assessment of psychosocial stressors and psychodynamic factors, particularly those related to the pregnancy
d) all of the above
TREATMENT Close follow-up
The psychiatrist recommends continued close psychiatric follow-up as well as multidisciplinary involvement, including physical therapy, neurology, and obstetrics.
Ms. M initially is resistant to psychiatric follow-up because she says that “people on the street” told her that, if she saw a psychiatrist, her baby would be taken away. After the psychiatrist explains that it is unlikely her baby would be taken away, Ms. M immediately appears relieved, smiles, and readily agrees to outpatient psychotherapy.
Over the next 24 hours, she continues to work with a physical therapist and her gait significantly improves. She is discharged home 2 days later with a walking aid (Zimmer frame) for assistance.
Four days later, however, Ms. M is readmitted with worsening ataxia. Her aunt reports that, at home, Ms. M’s regressed behaviors are worsening; she is sleeping in bed with her and had several episodes of enuresis at home.
Ms. M continues to deny psychiatric symptoms or anxiety about the delivery. Although she shows some improvement when working with physical therapists, they note that Ms. M is still unable to ambulate or stand on her own. The psychiatrist is increasingly concerned about her regressed behavior and continued ataxia.
A family meeting is held and the psychiatrist and social worker educate Ms. M and her aunt about conversion disorder, including how some emotionally distressed women communicate their feelings or troubled thoughts through physical symptoms and how that may apply to Ms. M. During the meeting, the team also destigmatizes psychiatric illness and treatment and provides psychoeducation regarding its benefits. The psychiatrist and social worker also provide a psychodynamic interpretation that her ataxia could be a way of protecting herself against the abandonment she is experiencing by being left to “stand alone” by her boyfriend— as she had been when her parents sent her to the United States, and that her behavior could be her way of securing her aunt’s love and support.Ms. M and her aunt both readily agree with this interpretation. The aunt notes that her niece is more anxious about motherhood than she acknowledges and is concerned that Ms. M expects her to be the primary caregiver for the baby. Those present note that Ms. M is becoming increasingly dependent on her aunt, and that it is important for her to retain her independence, especially once she becomes a mother.
Ms. M immediately begins to display more affect; she smiles and reports feeling relieved. Similar to the previous admission, her gait significantly improves over the next 2 days and she is discharged home with a walking aid.
The authors’ observations
A broad differential diagnosis and early multidisciplinary involvement might facilitate earlier diagnosis and treatment.16 Assessment of psychosocial stressors in the patient’s personal and family life, including circumstances around the pregnancy and the meaning of motherhood, as well as investigation of what the patient may gain from the sick role, are paramount. In Ms. M’s case, cultural background, separation from her parents at a young age, and recent abandonment by her boyfriend have contributed to her inability to “stand alone,” which manifested as ataxia. Young age, regressed behavior, and her minimization of stressors also point to her difficulty acknowledging and coping with psychosocial stressors.
Successful delivery of the diagnosis is key to treatment success. After building a therapeutic alliance, a multidisciplinary discussion should take place that allows the patient to understand the diagnosis and treatment plan.17,18 The patient and family should be reassured that the fetus is healthy and all organic causes of symptoms have been investigated.17 Although management of conversion disorder during pregnancy is similar to that in non-pregnant women, several additional avenues of investigation should be considered:
• Explore the psychodynamic basis of the disorder and the role of the pregnancy and motherhood.
• Identify any comorbid psychiatric disorders, particularly those specific to pregnancy or the postpartum period.
• Because of the shame and stigma associated with seeking psychiatric treatment during pregnancy,10,11 it is imperative to destigmatize treatment and provide psychoeducation regarding its benefits.
A treatment plan can then be developed that involves psychotherapy, psychoeducation, stress management, and, when appropriate, pharmacotherapy.17
Providing psychoeducation about postpartum depression and other perinatal psychiatric illness could be beneficial. Physical therapy often is culturally acceptable and can help re-establish healthy patterns of motor function.19 Ms. M’s gait showed some improvement with physical therapy as part of the multidisciplinary approach, which also should include a thorough medical workup. Appropriate psychiatric treatment can help patients give up the sick role and return to their previous level of functioning.17
Maintain close communication with the outpatient perinatal care team as they monitor the patient’s parenting capacity. The outpatient perinatal care team also should engage pregnant or postpartum women in prioritizing their emotional well-being and encourage outpatient mental health treatment. Despite a dearth of data on the regressive symptoms and prognosis for future pregnancies, it is important to monitor maternal capacity and discuss the possibility of symptom recurrence.
OUTCOME Healthy baby
Three days later, Ms. M returns in labor with improved gait yet still using a walking aid. She has a normal vaginal delivery of a healthy baby boy at 37 weeks’ gestational age.
After the birth, Ms. M reports feeling well and enjoying motherhood, and denies psychiatric symptoms. She is ambulating without assistance within hours of delivery. This spontaneous resolution of symptoms could have been because of the psychodynamically oriented multidisciplinary approach to her care, which may have helped her realize that she did not have to “stand alone” as she embarked on motherhood.
Before being discharged home, Ms. M and her aunt meet with the inpatient obstetric social worker to assess Ms. M’s ability to care for the baby and discuss the importance of continued emotional support. The social worker does not contact the Department of Children and Families because Ms. M is walking independently and not endorsing or exhibiting regressive behaviors. Ms. M also reports that she will ask her aunt to take care of the baby should ataxia recur. Her aunt reassures the social workers that she will encourage Ms. M to attend outpatient psychotherapy and will contact the social worker if she becomes concerned about Ms. M’s or the baby’s well-being.
During her postpartum obstetric visit, Ms. M is walking independently and does not exhibit or endorse neurologic symptoms. The social worker provides psychoeducation about the importance of outpatient psychotherapy and schedules an initial appointment; Ms. M does not attend outpatient psychotherapy after discharge.
Bottom Line
Consider conversion disorder in obstetric patients who present with ataxia without a neurologic cause. Management involves a proactive and multidisciplinary approach that includes a thorough medical workup and assessment of psychosocial stressors and psychodynamic factors, particularly those related to the pregnancy. Early identification and delivery of the diagnosis, destigmatization, and provision of appropriate psychiatric treatment can facilitate treatment success.
Disclosures
Dr. Byatt has received grant funding/support for this project from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR000160. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Dr. Toor reports no financial relationships with any company whose products are mentioned in this article or manufacturers of competing products.
1. Vesga-Lopez O, Blanco C, Keyes K, et al. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry. 2008;65(7):805-815.
2. Britton HL, Gronwaldt V, Britton JR. Maternal postpartum behaviors and mother-infant relationship during the first year of life. J Pediatr. 2001;138(6):905-909.
3. Deave T, Heron J, Evans J, et al. The impact of maternal depression in pregnancy on early child development. BJOG. 2008;115(8):1043-1051.
4. Paulson JF, Keefe HA, Leiferman JA. Early parental depression and child language development. J Child Psychol Psychiatry. 2009;50(3):254-262.
5. Zuckerman B, Amaro H, Bauchner H, et al. Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol. 1989;160(5 pt 1):1107-1111.
6. Forman DR, O’Hara MW, Stuart S, et al. Effective treatment for postpartum depression is not sufficient to improve the developing mother-child relationship. Dev Psychopathol. 2007;19(2):585-602.
7. Brady WJ Jr, Huff JS. Pseudotoxemia: new onset psychogenic seizure in third trimester pregnancy. J Emerg Med. 1997;15(6):815-820.
8. el-Mallakh RS, Liebowitz NR, Hale MS. Hyperemesis gravidarum as conversion disorder. J Nerv Ment Dis. 1990; 178(10):655-659.
9. Paulman PM, Sadat A. Pseudocyesis. J Fam Pract. 1990;30(5):575-576.
10. Dennis CL, Chung-Lee L. Postpartum depression help-seeking barriers and maternal treatment p: a qualitative systematic review. Birth. 2006;33(4):323-331.
11. Byatt N, Simas TA, Lundquist RS, et al. Strategies for improving perinatal depression treatment in North American outpatient obstetric settings. J Psychosom Obstetr Gynaecol. 2012;33(4):143-161.
12. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
13. Feinstein A. Conversion disorder: advances in our understanding. CMAJ. 2011;183(8):915-920.
14. Nicholson TR, Stone J, Kanaan RA. Conversion disorder: a problematic diagnosis. J Neurol Neurosurg Psychiatry. 2011;82(11):1267-1273.
15. Bitzer J. Somatization disorders in obstetrics and gynecology. Arch Womens Mental health, 2003;6(2):99-107.
16. Smith HE, Rynning RE, Okafor C, et al. Evaluation of neurologic deficit without apparent cause: the importance of a multidisciplinary approach. J Spinal Cord Med. 2007;30(5):509-517.
17. Hinson VK, Haren WB. Psychogenic movement disorders. Lancet Neurol. 2006;5(8):695-700.
18. Oyama O, Paltoo C, Greengold J. Somatoform disorders. Am Fam Physician. 2007;76(9):1333-1338.
19. Ness D. Physical therapy management for conversion disorder: case series. J Neurol Phys Ther. 2007;31(1):30-39.
CASE Difficulty walking
Ms. M, age 15, is a pregnant, Spanish-speaking Guatemalan woman who is brought to obstetrics triage in a large academic medical center at 35 weeks gestational age. She complains of dizziness, tinnitus, left orbital headache, and difficulty walking.
The neurology service finds profound truncal ataxia, astasia-abasia, and buckling of the knees; a normal brain and spine MRI are not consistent with a neurologic etiology. Otolaryngology service evaluates Ms. M to rule out a cholesteatoma and suggests a head CT and endoscopy, which are normal.
Ms. M’s symptoms resolve after 3 days, although the gait disturbances persist. When no clear cause is found for her difficulty walking, the psychiatry service is consulted to evaluate whether an underlying psychiatric disorder is contributing to symptoms.
What could be causing Ms. M’s symptoms?
a) malingering
b) factitious disorder
c) undiagnosed neurologic disorder
d) conversion disorder
The authors’ observations
Women are vulnerable to a variety of psychiatric illnesses during pregnancy1 that have deleterious effects on mother, baby, and family.2-6 Although there is a burgeoning literature on affective and anxiety disorders occurring in pregnancy, there is a dearth of information about somatoform disorders.
HISTORY Abandonment
Ms. M reports that, although her boyfriend deserted her after learning about the unexpected pregnancy, she will welcome the baby and looks forward to motherhood. She seems unaware of the responsibilities of being a mother.
Ms. M acknowledges a history of depression and self-harm a few years earlier, yet says she feels better now and thinks that psychiatric care is unnecessary. Because she does not endorse a history of trauma or symptoms suggesting an affective, anxiety, or psychotic illness, the psychiatrist does not recommend treatment with psychotropic medication.
At age 5, Ms. M’s parents sent her to the United States with her aunt, hoping that she would have a better life than she would have had in Guatemala. Her aunt reports that Ms. M initially had difficulty adjusting to life in the United States without her parents, yet she has made substantial strides over the years and is now quite accustomed to the country. Her aunt describes Ms. M as an independent high school student who earns good grades.
During the interview, the psychiatrist observes that Ms. M exhibits childlike mannerisms, including sleeping with stuffed toys and coloring in Disney books with crayons. She also is indifferent to her gait difficulty, pregnancy, and psychosocial stressors. Her affect is inconsistent with the content of her speech and she is alexithymic.
Ms. M’s aunt reports that her niece is becoming more dependent on her, which is not consistent with her baseline. Her aunt also notes that several years earlier, Ms. M’s nephew was diagnosed with a cholesteatoma after he presented with similar symptoms.
The combination of (1) Ms. M’s clinical presentation, which was causing her significant impairment in her social functioning, (2) the incompatibility of symptoms with any recognized neurologic and medical disease, and (3) prior family experience with cholesteatoma leads the consulting psychiatrist to suspect conversion disorder. Ms. M’s alexithymia, indifference to her symptoms, and recent abandonment by the baby’s father also support a conversion disorder diagnosis.
From a psychodynamic perspective, the ataxia appears to be her way of protecting herself from the abandonment she is experiencing by being left again to “stand alone” by her boyfriend as she had been when her parents sent her to the United States. Her regressive behavior could be her way of securing her aunt’s love and support.
The authors’ observations
This is the first case of psychogenic gait disturbance during pregnancy described in the literature. Authors have reported on pseudotoxemia,7 hyperemesis gravidarum,8 and pesudocyesis,9 yet there is a paucity of information on psychogenic gait disturbance during pregnancy. Ms. M’s case elucidates many of the clinical quandaries that occur when managing psychiatric illness—and, more specifically, conversion disorder— during pregnancy. Many women are hesitant to seek psychiatric treatment during pregnancy because of shame, stigma, and fear of loss of personal or parental rights10,11; it is not surprising that emotionally distressed women communicate their feelings or troubled thoughts through physical symptoms.
Likely diagnosis
Conversion disorder is the presence of neurologic symptoms in the absence of a neurologic diagnosis that fully explains those symptoms. Conversion disorder, previously known as hysteria, is called functional neurologic symptom disorder in DSM-5 (Box).12 Symptoms are not feigned; instead, they represent “conversion” of emotional distress into neurologic symptoms.13,14 Although misdiagnosing conversion disorder in patients with true neurologic disease is uncommon, clinicians often are uncomfortable making the diagnosis until all medical causes have been ruled out.14 It is not always possible to find a psychological explanation for conversion disorder, but a history of childhood abuse, particularly sexual abuse, could play a role.14
Because of the variety of presentations, clinicians in all specialties should be familiar with somatoform disorders; this is especially important in obstetrics and gynecology because women are more likely than men to develop these disorders.15 It is important to consider that Ms. M is a teenager and somatoform disorders can present differently in adults. The diagnostic process should include a diligent somatic workup and a personal and social history to identify the patient’s developmental tasks, stressors, and coping style.15
How would you treat Ms. M?
a) destigmatize psychiatric illness and provide psychoeducation regarding treatment benefits
b) identify and treat any comorbid psychiatric disorders
c) maintain a proactive and multidisciplinary approach that includes assessment of psychosocial stressors and psychodynamic factors, particularly those related to the pregnancy
d) all of the above
TREATMENT Close follow-up
The psychiatrist recommends continued close psychiatric follow-up as well as multidisciplinary involvement, including physical therapy, neurology, and obstetrics.
Ms. M initially is resistant to psychiatric follow-up because she says that “people on the street” told her that, if she saw a psychiatrist, her baby would be taken away. After the psychiatrist explains that it is unlikely her baby would be taken away, Ms. M immediately appears relieved, smiles, and readily agrees to outpatient psychotherapy.
Over the next 24 hours, she continues to work with a physical therapist and her gait significantly improves. She is discharged home 2 days later with a walking aid (Zimmer frame) for assistance.
Four days later, however, Ms. M is readmitted with worsening ataxia. Her aunt reports that, at home, Ms. M’s regressed behaviors are worsening; she is sleeping in bed with her and had several episodes of enuresis at home.
Ms. M continues to deny psychiatric symptoms or anxiety about the delivery. Although she shows some improvement when working with physical therapists, they note that Ms. M is still unable to ambulate or stand on her own. The psychiatrist is increasingly concerned about her regressed behavior and continued ataxia.
A family meeting is held and the psychiatrist and social worker educate Ms. M and her aunt about conversion disorder, including how some emotionally distressed women communicate their feelings or troubled thoughts through physical symptoms and how that may apply to Ms. M. During the meeting, the team also destigmatizes psychiatric illness and treatment and provides psychoeducation regarding its benefits. The psychiatrist and social worker also provide a psychodynamic interpretation that her ataxia could be a way of protecting herself against the abandonment she is experiencing by being left to “stand alone” by her boyfriend— as she had been when her parents sent her to the United States, and that her behavior could be her way of securing her aunt’s love and support.Ms. M and her aunt both readily agree with this interpretation. The aunt notes that her niece is more anxious about motherhood than she acknowledges and is concerned that Ms. M expects her to be the primary caregiver for the baby. Those present note that Ms. M is becoming increasingly dependent on her aunt, and that it is important for her to retain her independence, especially once she becomes a mother.
Ms. M immediately begins to display more affect; she smiles and reports feeling relieved. Similar to the previous admission, her gait significantly improves over the next 2 days and she is discharged home with a walking aid.
The authors’ observations
A broad differential diagnosis and early multidisciplinary involvement might facilitate earlier diagnosis and treatment.16 Assessment of psychosocial stressors in the patient’s personal and family life, including circumstances around the pregnancy and the meaning of motherhood, as well as investigation of what the patient may gain from the sick role, are paramount. In Ms. M’s case, cultural background, separation from her parents at a young age, and recent abandonment by her boyfriend have contributed to her inability to “stand alone,” which manifested as ataxia. Young age, regressed behavior, and her minimization of stressors also point to her difficulty acknowledging and coping with psychosocial stressors.
Successful delivery of the diagnosis is key to treatment success. After building a therapeutic alliance, a multidisciplinary discussion should take place that allows the patient to understand the diagnosis and treatment plan.17,18 The patient and family should be reassured that the fetus is healthy and all organic causes of symptoms have been investigated.17 Although management of conversion disorder during pregnancy is similar to that in non-pregnant women, several additional avenues of investigation should be considered:
• Explore the psychodynamic basis of the disorder and the role of the pregnancy and motherhood.
• Identify any comorbid psychiatric disorders, particularly those specific to pregnancy or the postpartum period.
• Because of the shame and stigma associated with seeking psychiatric treatment during pregnancy,10,11 it is imperative to destigmatize treatment and provide psychoeducation regarding its benefits.
A treatment plan can then be developed that involves psychotherapy, psychoeducation, stress management, and, when appropriate, pharmacotherapy.17
Providing psychoeducation about postpartum depression and other perinatal psychiatric illness could be beneficial. Physical therapy often is culturally acceptable and can help re-establish healthy patterns of motor function.19 Ms. M’s gait showed some improvement with physical therapy as part of the multidisciplinary approach, which also should include a thorough medical workup. Appropriate psychiatric treatment can help patients give up the sick role and return to their previous level of functioning.17
Maintain close communication with the outpatient perinatal care team as they monitor the patient’s parenting capacity. The outpatient perinatal care team also should engage pregnant or postpartum women in prioritizing their emotional well-being and encourage outpatient mental health treatment. Despite a dearth of data on the regressive symptoms and prognosis for future pregnancies, it is important to monitor maternal capacity and discuss the possibility of symptom recurrence.
OUTCOME Healthy baby
Three days later, Ms. M returns in labor with improved gait yet still using a walking aid. She has a normal vaginal delivery of a healthy baby boy at 37 weeks’ gestational age.
After the birth, Ms. M reports feeling well and enjoying motherhood, and denies psychiatric symptoms. She is ambulating without assistance within hours of delivery. This spontaneous resolution of symptoms could have been because of the psychodynamically oriented multidisciplinary approach to her care, which may have helped her realize that she did not have to “stand alone” as she embarked on motherhood.
Before being discharged home, Ms. M and her aunt meet with the inpatient obstetric social worker to assess Ms. M’s ability to care for the baby and discuss the importance of continued emotional support. The social worker does not contact the Department of Children and Families because Ms. M is walking independently and not endorsing or exhibiting regressive behaviors. Ms. M also reports that she will ask her aunt to take care of the baby should ataxia recur. Her aunt reassures the social workers that she will encourage Ms. M to attend outpatient psychotherapy and will contact the social worker if she becomes concerned about Ms. M’s or the baby’s well-being.
During her postpartum obstetric visit, Ms. M is walking independently and does not exhibit or endorse neurologic symptoms. The social worker provides psychoeducation about the importance of outpatient psychotherapy and schedules an initial appointment; Ms. M does not attend outpatient psychotherapy after discharge.
Bottom Line
Consider conversion disorder in obstetric patients who present with ataxia without a neurologic cause. Management involves a proactive and multidisciplinary approach that includes a thorough medical workup and assessment of psychosocial stressors and psychodynamic factors, particularly those related to the pregnancy. Early identification and delivery of the diagnosis, destigmatization, and provision of appropriate psychiatric treatment can facilitate treatment success.
Disclosures
Dr. Byatt has received grant funding/support for this project from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR000160. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Dr. Toor reports no financial relationships with any company whose products are mentioned in this article or manufacturers of competing products.
CASE Difficulty walking
Ms. M, age 15, is a pregnant, Spanish-speaking Guatemalan woman who is brought to obstetrics triage in a large academic medical center at 35 weeks gestational age. She complains of dizziness, tinnitus, left orbital headache, and difficulty walking.
The neurology service finds profound truncal ataxia, astasia-abasia, and buckling of the knees; a normal brain and spine MRI are not consistent with a neurologic etiology. Otolaryngology service evaluates Ms. M to rule out a cholesteatoma and suggests a head CT and endoscopy, which are normal.
Ms. M’s symptoms resolve after 3 days, although the gait disturbances persist. When no clear cause is found for her difficulty walking, the psychiatry service is consulted to evaluate whether an underlying psychiatric disorder is contributing to symptoms.
What could be causing Ms. M’s symptoms?
a) malingering
b) factitious disorder
c) undiagnosed neurologic disorder
d) conversion disorder
The authors’ observations
Women are vulnerable to a variety of psychiatric illnesses during pregnancy1 that have deleterious effects on mother, baby, and family.2-6 Although there is a burgeoning literature on affective and anxiety disorders occurring in pregnancy, there is a dearth of information about somatoform disorders.
HISTORY Abandonment
Ms. M reports that, although her boyfriend deserted her after learning about the unexpected pregnancy, she will welcome the baby and looks forward to motherhood. She seems unaware of the responsibilities of being a mother.
Ms. M acknowledges a history of depression and self-harm a few years earlier, yet says she feels better now and thinks that psychiatric care is unnecessary. Because she does not endorse a history of trauma or symptoms suggesting an affective, anxiety, or psychotic illness, the psychiatrist does not recommend treatment with psychotropic medication.
At age 5, Ms. M’s parents sent her to the United States with her aunt, hoping that she would have a better life than she would have had in Guatemala. Her aunt reports that Ms. M initially had difficulty adjusting to life in the United States without her parents, yet she has made substantial strides over the years and is now quite accustomed to the country. Her aunt describes Ms. M as an independent high school student who earns good grades.
During the interview, the psychiatrist observes that Ms. M exhibits childlike mannerisms, including sleeping with stuffed toys and coloring in Disney books with crayons. She also is indifferent to her gait difficulty, pregnancy, and psychosocial stressors. Her affect is inconsistent with the content of her speech and she is alexithymic.
Ms. M’s aunt reports that her niece is becoming more dependent on her, which is not consistent with her baseline. Her aunt also notes that several years earlier, Ms. M’s nephew was diagnosed with a cholesteatoma after he presented with similar symptoms.
The combination of (1) Ms. M’s clinical presentation, which was causing her significant impairment in her social functioning, (2) the incompatibility of symptoms with any recognized neurologic and medical disease, and (3) prior family experience with cholesteatoma leads the consulting psychiatrist to suspect conversion disorder. Ms. M’s alexithymia, indifference to her symptoms, and recent abandonment by the baby’s father also support a conversion disorder diagnosis.
From a psychodynamic perspective, the ataxia appears to be her way of protecting herself from the abandonment she is experiencing by being left again to “stand alone” by her boyfriend as she had been when her parents sent her to the United States. Her regressive behavior could be her way of securing her aunt’s love and support.
The authors’ observations
This is the first case of psychogenic gait disturbance during pregnancy described in the literature. Authors have reported on pseudotoxemia,7 hyperemesis gravidarum,8 and pesudocyesis,9 yet there is a paucity of information on psychogenic gait disturbance during pregnancy. Ms. M’s case elucidates many of the clinical quandaries that occur when managing psychiatric illness—and, more specifically, conversion disorder— during pregnancy. Many women are hesitant to seek psychiatric treatment during pregnancy because of shame, stigma, and fear of loss of personal or parental rights10,11; it is not surprising that emotionally distressed women communicate their feelings or troubled thoughts through physical symptoms.
Likely diagnosis
Conversion disorder is the presence of neurologic symptoms in the absence of a neurologic diagnosis that fully explains those symptoms. Conversion disorder, previously known as hysteria, is called functional neurologic symptom disorder in DSM-5 (Box).12 Symptoms are not feigned; instead, they represent “conversion” of emotional distress into neurologic symptoms.13,14 Although misdiagnosing conversion disorder in patients with true neurologic disease is uncommon, clinicians often are uncomfortable making the diagnosis until all medical causes have been ruled out.14 It is not always possible to find a psychological explanation for conversion disorder, but a history of childhood abuse, particularly sexual abuse, could play a role.14
Because of the variety of presentations, clinicians in all specialties should be familiar with somatoform disorders; this is especially important in obstetrics and gynecology because women are more likely than men to develop these disorders.15 It is important to consider that Ms. M is a teenager and somatoform disorders can present differently in adults. The diagnostic process should include a diligent somatic workup and a personal and social history to identify the patient’s developmental tasks, stressors, and coping style.15
How would you treat Ms. M?
a) destigmatize psychiatric illness and provide psychoeducation regarding treatment benefits
b) identify and treat any comorbid psychiatric disorders
c) maintain a proactive and multidisciplinary approach that includes assessment of psychosocial stressors and psychodynamic factors, particularly those related to the pregnancy
d) all of the above
TREATMENT Close follow-up
The psychiatrist recommends continued close psychiatric follow-up as well as multidisciplinary involvement, including physical therapy, neurology, and obstetrics.
Ms. M initially is resistant to psychiatric follow-up because she says that “people on the street” told her that, if she saw a psychiatrist, her baby would be taken away. After the psychiatrist explains that it is unlikely her baby would be taken away, Ms. M immediately appears relieved, smiles, and readily agrees to outpatient psychotherapy.
Over the next 24 hours, she continues to work with a physical therapist and her gait significantly improves. She is discharged home 2 days later with a walking aid (Zimmer frame) for assistance.
Four days later, however, Ms. M is readmitted with worsening ataxia. Her aunt reports that, at home, Ms. M’s regressed behaviors are worsening; she is sleeping in bed with her and had several episodes of enuresis at home.
Ms. M continues to deny psychiatric symptoms or anxiety about the delivery. Although she shows some improvement when working with physical therapists, they note that Ms. M is still unable to ambulate or stand on her own. The psychiatrist is increasingly concerned about her regressed behavior and continued ataxia.
A family meeting is held and the psychiatrist and social worker educate Ms. M and her aunt about conversion disorder, including how some emotionally distressed women communicate their feelings or troubled thoughts through physical symptoms and how that may apply to Ms. M. During the meeting, the team also destigmatizes psychiatric illness and treatment and provides psychoeducation regarding its benefits. The psychiatrist and social worker also provide a psychodynamic interpretation that her ataxia could be a way of protecting herself against the abandonment she is experiencing by being left to “stand alone” by her boyfriend— as she had been when her parents sent her to the United States, and that her behavior could be her way of securing her aunt’s love and support.Ms. M and her aunt both readily agree with this interpretation. The aunt notes that her niece is more anxious about motherhood than she acknowledges and is concerned that Ms. M expects her to be the primary caregiver for the baby. Those present note that Ms. M is becoming increasingly dependent on her aunt, and that it is important for her to retain her independence, especially once she becomes a mother.
Ms. M immediately begins to display more affect; she smiles and reports feeling relieved. Similar to the previous admission, her gait significantly improves over the next 2 days and she is discharged home with a walking aid.
The authors’ observations
A broad differential diagnosis and early multidisciplinary involvement might facilitate earlier diagnosis and treatment.16 Assessment of psychosocial stressors in the patient’s personal and family life, including circumstances around the pregnancy and the meaning of motherhood, as well as investigation of what the patient may gain from the sick role, are paramount. In Ms. M’s case, cultural background, separation from her parents at a young age, and recent abandonment by her boyfriend have contributed to her inability to “stand alone,” which manifested as ataxia. Young age, regressed behavior, and her minimization of stressors also point to her difficulty acknowledging and coping with psychosocial stressors.
Successful delivery of the diagnosis is key to treatment success. After building a therapeutic alliance, a multidisciplinary discussion should take place that allows the patient to understand the diagnosis and treatment plan.17,18 The patient and family should be reassured that the fetus is healthy and all organic causes of symptoms have been investigated.17 Although management of conversion disorder during pregnancy is similar to that in non-pregnant women, several additional avenues of investigation should be considered:
• Explore the psychodynamic basis of the disorder and the role of the pregnancy and motherhood.
• Identify any comorbid psychiatric disorders, particularly those specific to pregnancy or the postpartum period.
• Because of the shame and stigma associated with seeking psychiatric treatment during pregnancy,10,11 it is imperative to destigmatize treatment and provide psychoeducation regarding its benefits.
A treatment plan can then be developed that involves psychotherapy, psychoeducation, stress management, and, when appropriate, pharmacotherapy.17
Providing psychoeducation about postpartum depression and other perinatal psychiatric illness could be beneficial. Physical therapy often is culturally acceptable and can help re-establish healthy patterns of motor function.19 Ms. M’s gait showed some improvement with physical therapy as part of the multidisciplinary approach, which also should include a thorough medical workup. Appropriate psychiatric treatment can help patients give up the sick role and return to their previous level of functioning.17
Maintain close communication with the outpatient perinatal care team as they monitor the patient’s parenting capacity. The outpatient perinatal care team also should engage pregnant or postpartum women in prioritizing their emotional well-being and encourage outpatient mental health treatment. Despite a dearth of data on the regressive symptoms and prognosis for future pregnancies, it is important to monitor maternal capacity and discuss the possibility of symptom recurrence.
OUTCOME Healthy baby
Three days later, Ms. M returns in labor with improved gait yet still using a walking aid. She has a normal vaginal delivery of a healthy baby boy at 37 weeks’ gestational age.
After the birth, Ms. M reports feeling well and enjoying motherhood, and denies psychiatric symptoms. She is ambulating without assistance within hours of delivery. This spontaneous resolution of symptoms could have been because of the psychodynamically oriented multidisciplinary approach to her care, which may have helped her realize that she did not have to “stand alone” as she embarked on motherhood.
Before being discharged home, Ms. M and her aunt meet with the inpatient obstetric social worker to assess Ms. M’s ability to care for the baby and discuss the importance of continued emotional support. The social worker does not contact the Department of Children and Families because Ms. M is walking independently and not endorsing or exhibiting regressive behaviors. Ms. M also reports that she will ask her aunt to take care of the baby should ataxia recur. Her aunt reassures the social workers that she will encourage Ms. M to attend outpatient psychotherapy and will contact the social worker if she becomes concerned about Ms. M’s or the baby’s well-being.
During her postpartum obstetric visit, Ms. M is walking independently and does not exhibit or endorse neurologic symptoms. The social worker provides psychoeducation about the importance of outpatient psychotherapy and schedules an initial appointment; Ms. M does not attend outpatient psychotherapy after discharge.
Bottom Line
Consider conversion disorder in obstetric patients who present with ataxia without a neurologic cause. Management involves a proactive and multidisciplinary approach that includes a thorough medical workup and assessment of psychosocial stressors and psychodynamic factors, particularly those related to the pregnancy. Early identification and delivery of the diagnosis, destigmatization, and provision of appropriate psychiatric treatment can facilitate treatment success.
Disclosures
Dr. Byatt has received grant funding/support for this project from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR000160. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Dr. Toor reports no financial relationships with any company whose products are mentioned in this article or manufacturers of competing products.
1. Vesga-Lopez O, Blanco C, Keyes K, et al. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry. 2008;65(7):805-815.
2. Britton HL, Gronwaldt V, Britton JR. Maternal postpartum behaviors and mother-infant relationship during the first year of life. J Pediatr. 2001;138(6):905-909.
3. Deave T, Heron J, Evans J, et al. The impact of maternal depression in pregnancy on early child development. BJOG. 2008;115(8):1043-1051.
4. Paulson JF, Keefe HA, Leiferman JA. Early parental depression and child language development. J Child Psychol Psychiatry. 2009;50(3):254-262.
5. Zuckerman B, Amaro H, Bauchner H, et al. Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol. 1989;160(5 pt 1):1107-1111.
6. Forman DR, O’Hara MW, Stuart S, et al. Effective treatment for postpartum depression is not sufficient to improve the developing mother-child relationship. Dev Psychopathol. 2007;19(2):585-602.
7. Brady WJ Jr, Huff JS. Pseudotoxemia: new onset psychogenic seizure in third trimester pregnancy. J Emerg Med. 1997;15(6):815-820.
8. el-Mallakh RS, Liebowitz NR, Hale MS. Hyperemesis gravidarum as conversion disorder. J Nerv Ment Dis. 1990; 178(10):655-659.
9. Paulman PM, Sadat A. Pseudocyesis. J Fam Pract. 1990;30(5):575-576.
10. Dennis CL, Chung-Lee L. Postpartum depression help-seeking barriers and maternal treatment p: a qualitative systematic review. Birth. 2006;33(4):323-331.
11. Byatt N, Simas TA, Lundquist RS, et al. Strategies for improving perinatal depression treatment in North American outpatient obstetric settings. J Psychosom Obstetr Gynaecol. 2012;33(4):143-161.
12. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
13. Feinstein A. Conversion disorder: advances in our understanding. CMAJ. 2011;183(8):915-920.
14. Nicholson TR, Stone J, Kanaan RA. Conversion disorder: a problematic diagnosis. J Neurol Neurosurg Psychiatry. 2011;82(11):1267-1273.
15. Bitzer J. Somatization disorders in obstetrics and gynecology. Arch Womens Mental health, 2003;6(2):99-107.
16. Smith HE, Rynning RE, Okafor C, et al. Evaluation of neurologic deficit without apparent cause: the importance of a multidisciplinary approach. J Spinal Cord Med. 2007;30(5):509-517.
17. Hinson VK, Haren WB. Psychogenic movement disorders. Lancet Neurol. 2006;5(8):695-700.
18. Oyama O, Paltoo C, Greengold J. Somatoform disorders. Am Fam Physician. 2007;76(9):1333-1338.
19. Ness D. Physical therapy management for conversion disorder: case series. J Neurol Phys Ther. 2007;31(1):30-39.
1. Vesga-Lopez O, Blanco C, Keyes K, et al. Psychiatric disorders in pregnant and postpartum women in the United States. Arch Gen Psychiatry. 2008;65(7):805-815.
2. Britton HL, Gronwaldt V, Britton JR. Maternal postpartum behaviors and mother-infant relationship during the first year of life. J Pediatr. 2001;138(6):905-909.
3. Deave T, Heron J, Evans J, et al. The impact of maternal depression in pregnancy on early child development. BJOG. 2008;115(8):1043-1051.
4. Paulson JF, Keefe HA, Leiferman JA. Early parental depression and child language development. J Child Psychol Psychiatry. 2009;50(3):254-262.
5. Zuckerman B, Amaro H, Bauchner H, et al. Depressive symptoms during pregnancy: relationship to poor health behaviors. Am J Obstet Gynecol. 1989;160(5 pt 1):1107-1111.
6. Forman DR, O’Hara MW, Stuart S, et al. Effective treatment for postpartum depression is not sufficient to improve the developing mother-child relationship. Dev Psychopathol. 2007;19(2):585-602.
7. Brady WJ Jr, Huff JS. Pseudotoxemia: new onset psychogenic seizure in third trimester pregnancy. J Emerg Med. 1997;15(6):815-820.
8. el-Mallakh RS, Liebowitz NR, Hale MS. Hyperemesis gravidarum as conversion disorder. J Nerv Ment Dis. 1990; 178(10):655-659.
9. Paulman PM, Sadat A. Pseudocyesis. J Fam Pract. 1990;30(5):575-576.
10. Dennis CL, Chung-Lee L. Postpartum depression help-seeking barriers and maternal treatment p: a qualitative systematic review. Birth. 2006;33(4):323-331.
11. Byatt N, Simas TA, Lundquist RS, et al. Strategies for improving perinatal depression treatment in North American outpatient obstetric settings. J Psychosom Obstetr Gynaecol. 2012;33(4):143-161.
12. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
13. Feinstein A. Conversion disorder: advances in our understanding. CMAJ. 2011;183(8):915-920.
14. Nicholson TR, Stone J, Kanaan RA. Conversion disorder: a problematic diagnosis. J Neurol Neurosurg Psychiatry. 2011;82(11):1267-1273.
15. Bitzer J. Somatization disorders in obstetrics and gynecology. Arch Womens Mental health, 2003;6(2):99-107.
16. Smith HE, Rynning RE, Okafor C, et al. Evaluation of neurologic deficit without apparent cause: the importance of a multidisciplinary approach. J Spinal Cord Med. 2007;30(5):509-517.
17. Hinson VK, Haren WB. Psychogenic movement disorders. Lancet Neurol. 2006;5(8):695-700.
18. Oyama O, Paltoo C, Greengold J. Somatoform disorders. Am Fam Physician. 2007;76(9):1333-1338.
19. Ness D. Physical therapy management for conversion disorder: case series. J Neurol Phys Ther. 2007;31(1):30-39.
Have you RULED O2uT medical illness in the presumptive psychiatric patient?
What a practitioner might identify and report as “psychiatric” symptoms or signs cannot always be explained in terms of psychological stress or a psychiatric disorder. In fact, a range of medical1 and neurologic2 illnesses can manifest in ways that appear psychiatric in nature. Common examples are sleep and thyroid disorders; deficiencies of vitamin D, folate, and B12; Parkinson’s disease; and anti-N-methyl-d-aspartate receptor autoimmune encephalitis.
People who have a medical illness with what appear to be psychiatric manifestations often elude identification and diagnosis because they do not visit a health care provider for any of several reasons, including difficulty obtaining health insurance. Instead, they might seek care in an emergency room (ER).
When such patients present for evaluation, it is easy—especially in a fast-paced ER—to miss the underlying cause of their illness. Some are then treated on the assumption that their diagnosis is psychiatric, while their medical illness goes unidentified.3
We propose a mnemonic, RULED O2uT, as a reminder in the ER (and any other setting) of the need to rule out physical illness before treating a patient for a psychiatric disorder. To demonstrate how the work-up of a patient whose medical illness was obscured by psychiatric signs and symptoms could benefit from applying RULED O2uT, we also present a case.
CASE REPORT
A man with a medical illness who presented with psychiatric symptoms
Mr. Z, in his late 40s, is brought to the ER by his sister for evaluation of depressed mood of 6 to 8 months’ duration. He has no psychiatric history.
On evaluation, Mr. Z does not remember an event or stressors that could have triggered depression. He describes complete loss of motivation for activities of daily living, such as personal grooming. He has stopped leaving the house and meeting friends and family members.
Mr. Z’s sister is concerned for his well-being because he has been living without heat and electricity, which were disconnected for nonpayment. Mr. Z reveals that he has not seen his primary care physician “for 20 or 25 years,” although he recently sought care in the ER of another hospital because of mild gait instability for several months.
Mr. Z has a blunted affect, with linear and goal-directed thought processes. He denies suicidal ideation. Laboratory testing, including a comprehensive metabolic panel, complete blood count, and urine toxicology and urinalysis, are negative.
A non-contrast CT scan of the head reveals foci of low attenuation in the left frontal corona radiata. Follow-up MRI of the brain, with and without contrast, shows extensive supratentorial and infratentorial demyelinating lesions consistent with multiple sclerosis (MS). Several cerebral lesions in the white matter are consistent with active demyelination.
Mr. Z is admitted to the neurology service and started on methylprednisolone for MS. The psychiatry consultation-liaison team prescribes sertraline, 50 mg titrated to 100 mg, for depression.
Detailed history means better overall evaluation
Mr. Z presented to the busy ER with psychiatric symptoms. It was easy to make a diagnosis of depression and refer him to the outpatient psychiatrist. However, a detailed history provided pertinent information about Mr. Z such as no regular medical check-ups, no family history of mental illness, and gait disturbance in absence of physical injury. This enabled the physicians to conduct a thorough evaluation including a neurologic examination, laboratory tests, and imaging of the brain.
The 8 components of RULED O2uT
Rx interactions. Review medications that the patient is taking or recently stopped taking, to rule out drug−drug interactions and adverse effects.
Unusual presentation. Be mindful of any unusual presentation. For example, sudden onset of psychiatric symptoms with seizures or hypersensitivity to the sun with depression or psychosis.
Labs. Obtain appropriate blood work, including:
• comprehensive metabolic panel
• complete blood count
• thyroid-stimulating hormone (myxedema, thyrotoxicosis)
• delta-aminolevulinic acid and porphobilinogen (acute intermittent porphyria)
• antinuclear antibody (systemic lupus erythematosus)
• B12 level
• fluorescent treponemal antibody absorption test (neurosyphilis)
• serum ceruloplasmin and copper (Wilson’s disease).
Examination. Perform a thorough examination, including a proper neurological exam. This is especially important when you see signs, or the patient reports symptoms, that cannot be explained by depression alone. An abnormality or change in gait, for example, might be a consequence of injury or a manifestation of MS, stroke, or Parkinson’s disease. Additional testing, such as CT of the head or lumbar puncture, might be appropriate to supplement or clarify findings of the exam. Mr. Z’s neurologic exam revealed weakness in his left leg with variability in reflexes.
Drugs. Ensure that a patient presenting with new-onset psychosis is not taking dopaminergic medications or steroids and, based on results of a toxicology screen, is not under the influence of stimulants or hallucinogens.
Onset and Office. Determine:
• the time since onset of symptoms; this is crucial to differentiate psychiatric disorders and ruling out a nonpsychiatric cause of the patient’s presentation
• if the patient gets a regular medical check-up with her (his) primary care physician.
Thorough history. Obtain a thorough history so that you have a clear picture of the patient’s current situation; this includes medical history and family history of neurologic and psychiatric disorders and substance abuse.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rahul R, Pieters T. An unusual psychiatric presentation of polycythaemia ‘Difficulties lie in our habits of thought rather than in the nature of things’ Andre Tardieu. BMJ Case Rep. 2013. pii: bcr2012008215. doi: 10.1136/bcr-2012-008215.
2. Butler C, Zeman AZ. Neurological syndromes which can be mistaken for psychiatric conditions. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i31-i38.
3. Roie EV, Labarque V, Renard M, et al. Obsessive-compulsive behavior as presenting symptom of primary antiphospholipid syndrome. Psychosom Med. 2013;75(3):326-330.
What a practitioner might identify and report as “psychiatric” symptoms or signs cannot always be explained in terms of psychological stress or a psychiatric disorder. In fact, a range of medical1 and neurologic2 illnesses can manifest in ways that appear psychiatric in nature. Common examples are sleep and thyroid disorders; deficiencies of vitamin D, folate, and B12; Parkinson’s disease; and anti-N-methyl-d-aspartate receptor autoimmune encephalitis.
People who have a medical illness with what appear to be psychiatric manifestations often elude identification and diagnosis because they do not visit a health care provider for any of several reasons, including difficulty obtaining health insurance. Instead, they might seek care in an emergency room (ER).
When such patients present for evaluation, it is easy—especially in a fast-paced ER—to miss the underlying cause of their illness. Some are then treated on the assumption that their diagnosis is psychiatric, while their medical illness goes unidentified.3
We propose a mnemonic, RULED O2uT, as a reminder in the ER (and any other setting) of the need to rule out physical illness before treating a patient for a psychiatric disorder. To demonstrate how the work-up of a patient whose medical illness was obscured by psychiatric signs and symptoms could benefit from applying RULED O2uT, we also present a case.
CASE REPORT
A man with a medical illness who presented with psychiatric symptoms
Mr. Z, in his late 40s, is brought to the ER by his sister for evaluation of depressed mood of 6 to 8 months’ duration. He has no psychiatric history.
On evaluation, Mr. Z does not remember an event or stressors that could have triggered depression. He describes complete loss of motivation for activities of daily living, such as personal grooming. He has stopped leaving the house and meeting friends and family members.
Mr. Z’s sister is concerned for his well-being because he has been living without heat and electricity, which were disconnected for nonpayment. Mr. Z reveals that he has not seen his primary care physician “for 20 or 25 years,” although he recently sought care in the ER of another hospital because of mild gait instability for several months.
Mr. Z has a blunted affect, with linear and goal-directed thought processes. He denies suicidal ideation. Laboratory testing, including a comprehensive metabolic panel, complete blood count, and urine toxicology and urinalysis, are negative.
A non-contrast CT scan of the head reveals foci of low attenuation in the left frontal corona radiata. Follow-up MRI of the brain, with and without contrast, shows extensive supratentorial and infratentorial demyelinating lesions consistent with multiple sclerosis (MS). Several cerebral lesions in the white matter are consistent with active demyelination.
Mr. Z is admitted to the neurology service and started on methylprednisolone for MS. The psychiatry consultation-liaison team prescribes sertraline, 50 mg titrated to 100 mg, for depression.
Detailed history means better overall evaluation
Mr. Z presented to the busy ER with psychiatric symptoms. It was easy to make a diagnosis of depression and refer him to the outpatient psychiatrist. However, a detailed history provided pertinent information about Mr. Z such as no regular medical check-ups, no family history of mental illness, and gait disturbance in absence of physical injury. This enabled the physicians to conduct a thorough evaluation including a neurologic examination, laboratory tests, and imaging of the brain.
The 8 components of RULED O2uT
Rx interactions. Review medications that the patient is taking or recently stopped taking, to rule out drug−drug interactions and adverse effects.
Unusual presentation. Be mindful of any unusual presentation. For example, sudden onset of psychiatric symptoms with seizures or hypersensitivity to the sun with depression or psychosis.
Labs. Obtain appropriate blood work, including:
• comprehensive metabolic panel
• complete blood count
• thyroid-stimulating hormone (myxedema, thyrotoxicosis)
• delta-aminolevulinic acid and porphobilinogen (acute intermittent porphyria)
• antinuclear antibody (systemic lupus erythematosus)
• B12 level
• fluorescent treponemal antibody absorption test (neurosyphilis)
• serum ceruloplasmin and copper (Wilson’s disease).
Examination. Perform a thorough examination, including a proper neurological exam. This is especially important when you see signs, or the patient reports symptoms, that cannot be explained by depression alone. An abnormality or change in gait, for example, might be a consequence of injury or a manifestation of MS, stroke, or Parkinson’s disease. Additional testing, such as CT of the head or lumbar puncture, might be appropriate to supplement or clarify findings of the exam. Mr. Z’s neurologic exam revealed weakness in his left leg with variability in reflexes.
Drugs. Ensure that a patient presenting with new-onset psychosis is not taking dopaminergic medications or steroids and, based on results of a toxicology screen, is not under the influence of stimulants or hallucinogens.
Onset and Office. Determine:
• the time since onset of symptoms; this is crucial to differentiate psychiatric disorders and ruling out a nonpsychiatric cause of the patient’s presentation
• if the patient gets a regular medical check-up with her (his) primary care physician.
Thorough history. Obtain a thorough history so that you have a clear picture of the patient’s current situation; this includes medical history and family history of neurologic and psychiatric disorders and substance abuse.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
What a practitioner might identify and report as “psychiatric” symptoms or signs cannot always be explained in terms of psychological stress or a psychiatric disorder. In fact, a range of medical1 and neurologic2 illnesses can manifest in ways that appear psychiatric in nature. Common examples are sleep and thyroid disorders; deficiencies of vitamin D, folate, and B12; Parkinson’s disease; and anti-N-methyl-d-aspartate receptor autoimmune encephalitis.
People who have a medical illness with what appear to be psychiatric manifestations often elude identification and diagnosis because they do not visit a health care provider for any of several reasons, including difficulty obtaining health insurance. Instead, they might seek care in an emergency room (ER).
When such patients present for evaluation, it is easy—especially in a fast-paced ER—to miss the underlying cause of their illness. Some are then treated on the assumption that their diagnosis is psychiatric, while their medical illness goes unidentified.3
We propose a mnemonic, RULED O2uT, as a reminder in the ER (and any other setting) of the need to rule out physical illness before treating a patient for a psychiatric disorder. To demonstrate how the work-up of a patient whose medical illness was obscured by psychiatric signs and symptoms could benefit from applying RULED O2uT, we also present a case.
CASE REPORT
A man with a medical illness who presented with psychiatric symptoms
Mr. Z, in his late 40s, is brought to the ER by his sister for evaluation of depressed mood of 6 to 8 months’ duration. He has no psychiatric history.
On evaluation, Mr. Z does not remember an event or stressors that could have triggered depression. He describes complete loss of motivation for activities of daily living, such as personal grooming. He has stopped leaving the house and meeting friends and family members.
Mr. Z’s sister is concerned for his well-being because he has been living without heat and electricity, which were disconnected for nonpayment. Mr. Z reveals that he has not seen his primary care physician “for 20 or 25 years,” although he recently sought care in the ER of another hospital because of mild gait instability for several months.
Mr. Z has a blunted affect, with linear and goal-directed thought processes. He denies suicidal ideation. Laboratory testing, including a comprehensive metabolic panel, complete blood count, and urine toxicology and urinalysis, are negative.
A non-contrast CT scan of the head reveals foci of low attenuation in the left frontal corona radiata. Follow-up MRI of the brain, with and without contrast, shows extensive supratentorial and infratentorial demyelinating lesions consistent with multiple sclerosis (MS). Several cerebral lesions in the white matter are consistent with active demyelination.
Mr. Z is admitted to the neurology service and started on methylprednisolone for MS. The psychiatry consultation-liaison team prescribes sertraline, 50 mg titrated to 100 mg, for depression.
Detailed history means better overall evaluation
Mr. Z presented to the busy ER with psychiatric symptoms. It was easy to make a diagnosis of depression and refer him to the outpatient psychiatrist. However, a detailed history provided pertinent information about Mr. Z such as no regular medical check-ups, no family history of mental illness, and gait disturbance in absence of physical injury. This enabled the physicians to conduct a thorough evaluation including a neurologic examination, laboratory tests, and imaging of the brain.
The 8 components of RULED O2uT
Rx interactions. Review medications that the patient is taking or recently stopped taking, to rule out drug−drug interactions and adverse effects.
Unusual presentation. Be mindful of any unusual presentation. For example, sudden onset of psychiatric symptoms with seizures or hypersensitivity to the sun with depression or psychosis.
Labs. Obtain appropriate blood work, including:
• comprehensive metabolic panel
• complete blood count
• thyroid-stimulating hormone (myxedema, thyrotoxicosis)
• delta-aminolevulinic acid and porphobilinogen (acute intermittent porphyria)
• antinuclear antibody (systemic lupus erythematosus)
• B12 level
• fluorescent treponemal antibody absorption test (neurosyphilis)
• serum ceruloplasmin and copper (Wilson’s disease).
Examination. Perform a thorough examination, including a proper neurological exam. This is especially important when you see signs, or the patient reports symptoms, that cannot be explained by depression alone. An abnormality or change in gait, for example, might be a consequence of injury or a manifestation of MS, stroke, or Parkinson’s disease. Additional testing, such as CT of the head or lumbar puncture, might be appropriate to supplement or clarify findings of the exam. Mr. Z’s neurologic exam revealed weakness in his left leg with variability in reflexes.
Drugs. Ensure that a patient presenting with new-onset psychosis is not taking dopaminergic medications or steroids and, based on results of a toxicology screen, is not under the influence of stimulants or hallucinogens.
Onset and Office. Determine:
• the time since onset of symptoms; this is crucial to differentiate psychiatric disorders and ruling out a nonpsychiatric cause of the patient’s presentation
• if the patient gets a regular medical check-up with her (his) primary care physician.
Thorough history. Obtain a thorough history so that you have a clear picture of the patient’s current situation; this includes medical history and family history of neurologic and psychiatric disorders and substance abuse.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rahul R, Pieters T. An unusual psychiatric presentation of polycythaemia ‘Difficulties lie in our habits of thought rather than in the nature of things’ Andre Tardieu. BMJ Case Rep. 2013. pii: bcr2012008215. doi: 10.1136/bcr-2012-008215.
2. Butler C, Zeman AZ. Neurological syndromes which can be mistaken for psychiatric conditions. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i31-i38.
3. Roie EV, Labarque V, Renard M, et al. Obsessive-compulsive behavior as presenting symptom of primary antiphospholipid syndrome. Psychosom Med. 2013;75(3):326-330.
1. Rahul R, Pieters T. An unusual psychiatric presentation of polycythaemia ‘Difficulties lie in our habits of thought rather than in the nature of things’ Andre Tardieu. BMJ Case Rep. 2013. pii: bcr2012008215. doi: 10.1136/bcr-2012-008215.
2. Butler C, Zeman AZ. Neurological syndromes which can be mistaken for psychiatric conditions. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i31-i38.
3. Roie EV, Labarque V, Renard M, et al. Obsessive-compulsive behavior as presenting symptom of primary antiphospholipid syndrome. Psychosom Med. 2013;75(3):326-330.
Assessing tremor to rule out psychogenic origin: It’s tricky
Tremors are a rhythmic and oscillatory movement of a body part with a relatively constant frequency.1 Several subtypes of tremors are classified on the basis of whether they occur during static or kinetic body positioning. Assessing tremors to rule out psychogenic origin is one of the trickiest tasks for a psychiatrist (Table 12). Non-organic movement disorders are not rare, and all common organic movement disorders can be mimicked by non-organic presentations.

Diagnostic approach
Start by categorizing the tremor based on its activation condition (at rest, kinetic or intentional, postural or isometric), topographic distribution, and frequency. Observe the patient sitting in a chair with his hands on his lap for resting tremor. Postural or kinetic tremors can be assessed by stretching the arms and performing a finger-to-nose test. A resting tremor can indicate parkinsonism; intention tremor may indicate a cerebellar lesion. A psychogenic tremor can occur at rest or during postural or active movement, and often will occur in all 3 situations (Table 2).1-3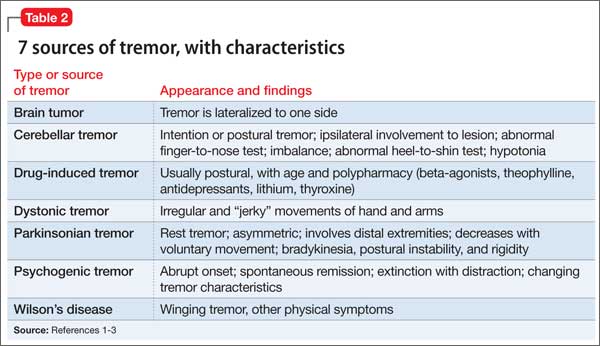
Some of the maneuvers listed in Table 3 are helpful to distinguish a psychogenic from an organic cause. The key is to look for variability in direction, amplitude, and frequency. Psychogenic tremor often increases when the limb is examined and reduces upon distraction, and also might be exacerbated with movement of other limbs. Patients with psychogenic tremor often have other “non-organic” neurologic signs, such as give-way weakness, deliberate slowness carrying out requested voluntary movement, and sensory signs that contradict neuroanatomical principles.

Investigation
Proceed as follows:
1. Perform laboratory testing: thyroid function panel and serum copper and ceruloplasmin levels.2
2. Perform surface electromyography to differentiate Parkinson’s disease and benign tremor disorders.2
3. Obtain a MRI to assess atypical tremor; findings might reveal Wilson’s disease (basal ganglia and brainstem involvement) or fragile X-associated tremor/ataxia syndrome (pontocerebellar hypoplasia or cerebral white matter involvement).3
4. Consider dopaminergic functional imaging scanning. When positive, the scan can reveal symptoms of parkinsonism; negative findings can help consolidate a diagnosis of psychogenic tremor.3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 suppl 4):S7.
2. Alty JE, Kempster PA. A practical guide to the differential diagnosis of tremor. Postgrad Med J. 2011;87(1031):623-629.
3. Crawford P, Zimmerman EE. Differentiation and diagnosis of tremor. Am Fam Physician. 2011;83(6):697-702.
Tremors are a rhythmic and oscillatory movement of a body part with a relatively constant frequency.1 Several subtypes of tremors are classified on the basis of whether they occur during static or kinetic body positioning. Assessing tremors to rule out psychogenic origin is one of the trickiest tasks for a psychiatrist (Table 12). Non-organic movement disorders are not rare, and all common organic movement disorders can be mimicked by non-organic presentations.
Diagnostic approach
Start by categorizing the tremor based on its activation condition (at rest, kinetic or intentional, postural or isometric), topographic distribution, and frequency. Observe the patient sitting in a chair with his hands on his lap for resting tremor. Postural or kinetic tremors can be assessed by stretching the arms and performing a finger-to-nose test. A resting tremor can indicate parkinsonism; intention tremor may indicate a cerebellar lesion. A psychogenic tremor can occur at rest or during postural or active movement, and often will occur in all 3 situations (Table 2).1-3
Some of the maneuvers listed in Table 3 are helpful to distinguish a psychogenic from an organic cause. The key is to look for variability in direction, amplitude, and frequency. Psychogenic tremor often increases when the limb is examined and reduces upon distraction, and also might be exacerbated with movement of other limbs. Patients with psychogenic tremor often have other “non-organic” neurologic signs, such as give-way weakness, deliberate slowness carrying out requested voluntary movement, and sensory signs that contradict neuroanatomical principles.
Investigation
Proceed as follows:
1. Perform laboratory testing: thyroid function panel and serum copper and ceruloplasmin levels.2
2. Perform surface electromyography to differentiate Parkinson’s disease and benign tremor disorders.2
3. Obtain a MRI to assess atypical tremor; findings might reveal Wilson’s disease (basal ganglia and brainstem involvement) or fragile X-associated tremor/ataxia syndrome (pontocerebellar hypoplasia or cerebral white matter involvement).3
4. Consider dopaminergic functional imaging scanning. When positive, the scan can reveal symptoms of parkinsonism; negative findings can help consolidate a diagnosis of psychogenic tremor.3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Tremors are a rhythmic and oscillatory movement of a body part with a relatively constant frequency.1 Several subtypes of tremors are classified on the basis of whether they occur during static or kinetic body positioning. Assessing tremors to rule out psychogenic origin is one of the trickiest tasks for a psychiatrist (Table 12). Non-organic movement disorders are not rare, and all common organic movement disorders can be mimicked by non-organic presentations.
Diagnostic approach
Start by categorizing the tremor based on its activation condition (at rest, kinetic or intentional, postural or isometric), topographic distribution, and frequency. Observe the patient sitting in a chair with his hands on his lap for resting tremor. Postural or kinetic tremors can be assessed by stretching the arms and performing a finger-to-nose test. A resting tremor can indicate parkinsonism; intention tremor may indicate a cerebellar lesion. A psychogenic tremor can occur at rest or during postural or active movement, and often will occur in all 3 situations (Table 2).1-3
Some of the maneuvers listed in Table 3 are helpful to distinguish a psychogenic from an organic cause. The key is to look for variability in direction, amplitude, and frequency. Psychogenic tremor often increases when the limb is examined and reduces upon distraction, and also might be exacerbated with movement of other limbs. Patients with psychogenic tremor often have other “non-organic” neurologic signs, such as give-way weakness, deliberate slowness carrying out requested voluntary movement, and sensory signs that contradict neuroanatomical principles.
Investigation
Proceed as follows:
1. Perform laboratory testing: thyroid function panel and serum copper and ceruloplasmin levels.2
2. Perform surface electromyography to differentiate Parkinson’s disease and benign tremor disorders.2
3. Obtain a MRI to assess atypical tremor; findings might reveal Wilson’s disease (basal ganglia and brainstem involvement) or fragile X-associated tremor/ataxia syndrome (pontocerebellar hypoplasia or cerebral white matter involvement).3
4. Consider dopaminergic functional imaging scanning. When positive, the scan can reveal symptoms of parkinsonism; negative findings can help consolidate a diagnosis of psychogenic tremor.3
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 suppl 4):S7.
2. Alty JE, Kempster PA. A practical guide to the differential diagnosis of tremor. Postgrad Med J. 2011;87(1031):623-629.
3. Crawford P, Zimmerman EE. Differentiation and diagnosis of tremor. Am Fam Physician. 2011;83(6):697-702.
1. Bain P, Brin M, Deuschl G, et al. Criteria for the diagnosis of essential tremor. Neurology. 2000;54(11 suppl 4):S7.
2. Alty JE, Kempster PA. A practical guide to the differential diagnosis of tremor. Postgrad Med J. 2011;87(1031):623-629.
3. Crawford P, Zimmerman EE. Differentiation and diagnosis of tremor. Am Fam Physician. 2011;83(6):697-702.
Akathisia: Is restlessness a primary condition or an adverse drug effect?
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112). 
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
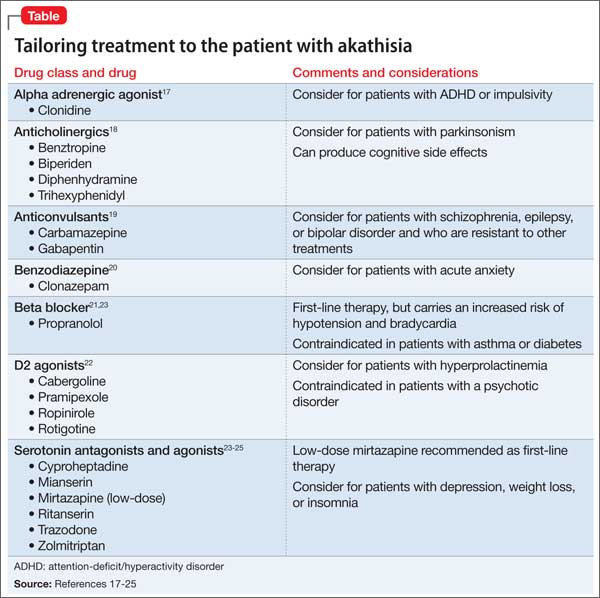
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.

VIDEO: Psychogenic seizure patients probably okay to drive
SEATTLE – To be safe, neurologists usually tell patients with psychogenic nonepileptic seizures to limit their driving as much as patients with confirmed epilepsy.
That might be an unnecessary restriction on their quality of life, however, according to a study from the Phoenix branch of the Mayo Clinic.
Epileptologist Dr. Kristine Ziemba, who conducted the study while a fellow at the Mayo Clinic, and is now a neurologist in St. Petersburg, Fla., explained why in a video interview at the annual meeting of the American Epilepsy Society.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SEATTLE – To be safe, neurologists usually tell patients with psychogenic nonepileptic seizures to limit their driving as much as patients with confirmed epilepsy.
That might be an unnecessary restriction on their quality of life, however, according to a study from the Phoenix branch of the Mayo Clinic.
Epileptologist Dr. Kristine Ziemba, who conducted the study while a fellow at the Mayo Clinic, and is now a neurologist in St. Petersburg, Fla., explained why in a video interview at the annual meeting of the American Epilepsy Society.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SEATTLE – To be safe, neurologists usually tell patients with psychogenic nonepileptic seizures to limit their driving as much as patients with confirmed epilepsy.
That might be an unnecessary restriction on their quality of life, however, according to a study from the Phoenix branch of the Mayo Clinic.
Epileptologist Dr. Kristine Ziemba, who conducted the study while a fellow at the Mayo Clinic, and is now a neurologist in St. Petersburg, Fla., explained why in a video interview at the annual meeting of the American Epilepsy Society.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AES 2014

How to modify psychotropic therapy for patients who have liver dysfunction
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3

Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
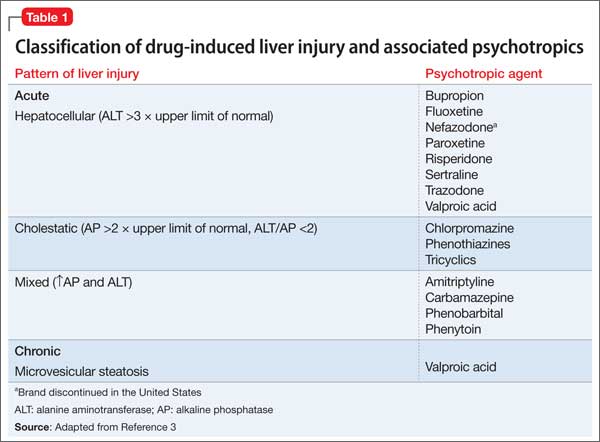
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5

When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3
Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5
When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3
Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5
When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
Brain changes identified in chronic fatigue syndrome
Imaging to assess brain microstructure identified increased fractional anisotropy in the anterior right arcuate fasciculus of patients with chronic fatigue syndrome, which was not present in age- and sex-matched control subjects.
The right-sided abnormality was particularly marked in right-handed chronic fatigue syndrome (CFS) patients, and the degree of increase correlated with the severity of their disorder. A finding of increased fractional anisotropy in this area of the brain may eventually serve as a biomarker for CFS, said Dr. Michael M. Zeineh of the department of radiology at Stanford (Calif.) University, and his associates (Radiology 2014 Oct. 29 [doi:10.1148/radiol.14141079]).
In what they described as the first study to examine brain microstructure in CFS, the investigators used volumetric MRI with diffusion-tensor imaging (DTI) to detect microstructural abnormalities in the patients – who had been evaluated at the university’s CFS Clinic during the preceding 5 years – and in healthy volunteers. The researchers also performed gray- and white-matter volumetric studies to identify differences between the two study groups in gross brain structure, as well as pseudocontinuous arterial spin labeling to assess global alterations in brain perfusion.
The mean age of the study participants was 46 years, and the mean duration of CFS symptoms was 12 years.
In the DTI analysis, the only significant difference between the two study groups was that the anterior right arcuate fasciculus – identified in 13 of 15 CFS patients and in all 14 control subjects in the study – showed significantly higher fractional anisotropy in CFS patients than in control subjects. This was confirmed in a second dataset from the same study subjects. The difference between CFS patients and control subjects was most pronounced in the subgroup of right-handed participants, “suggesting that hemispheric differences with handedness and language are an additional source of variance,” Dr. Zeineh and his associates said.
In addition, “two cortical regions connected via the arcuate fasciculus exhibited increased thickness: the right middle temporal and precentral gyri,” they noted.
The volumetric studies showed significantly lower total supratentorial white-matter volume in CFS, even after the data were adjusted to account for subject age, total intracranial volume, and handedness. However, total cortical gray-matter volume was equivalent between the two study groups, as were prefrontal cortex volume and global cortical thickness.
There were no differences between patients and controls in any perfusion measure, including perfusion to the cortex, supratentorial white matter, basal ganglia, thalami, or hippocampi. This finding is consistent with the results of a previous perfusion study involving monozygotic twins. Taken together, the findings suggest that brain perfusion is not affected in CFS.
Given the small number of subjects in this study, the findings must be verified in larger studies. It also would be valuable to assess possible changes in brain microstructure over time in a longitudinal study, and to determine whether treatment interventions exert any beneficial effects, the investigators added.
Imaging to assess brain microstructure identified increased fractional anisotropy in the anterior right arcuate fasciculus of patients with chronic fatigue syndrome, which was not present in age- and sex-matched control subjects.
The right-sided abnormality was particularly marked in right-handed chronic fatigue syndrome (CFS) patients, and the degree of increase correlated with the severity of their disorder. A finding of increased fractional anisotropy in this area of the brain may eventually serve as a biomarker for CFS, said Dr. Michael M. Zeineh of the department of radiology at Stanford (Calif.) University, and his associates (Radiology 2014 Oct. 29 [doi:10.1148/radiol.14141079]).
In what they described as the first study to examine brain microstructure in CFS, the investigators used volumetric MRI with diffusion-tensor imaging (DTI) to detect microstructural abnormalities in the patients – who had been evaluated at the university’s CFS Clinic during the preceding 5 years – and in healthy volunteers. The researchers also performed gray- and white-matter volumetric studies to identify differences between the two study groups in gross brain structure, as well as pseudocontinuous arterial spin labeling to assess global alterations in brain perfusion.
The mean age of the study participants was 46 years, and the mean duration of CFS symptoms was 12 years.
In the DTI analysis, the only significant difference between the two study groups was that the anterior right arcuate fasciculus – identified in 13 of 15 CFS patients and in all 14 control subjects in the study – showed significantly higher fractional anisotropy in CFS patients than in control subjects. This was confirmed in a second dataset from the same study subjects. The difference between CFS patients and control subjects was most pronounced in the subgroup of right-handed participants, “suggesting that hemispheric differences with handedness and language are an additional source of variance,” Dr. Zeineh and his associates said.
In addition, “two cortical regions connected via the arcuate fasciculus exhibited increased thickness: the right middle temporal and precentral gyri,” they noted.
The volumetric studies showed significantly lower total supratentorial white-matter volume in CFS, even after the data were adjusted to account for subject age, total intracranial volume, and handedness. However, total cortical gray-matter volume was equivalent between the two study groups, as were prefrontal cortex volume and global cortical thickness.
There were no differences between patients and controls in any perfusion measure, including perfusion to the cortex, supratentorial white matter, basal ganglia, thalami, or hippocampi. This finding is consistent with the results of a previous perfusion study involving monozygotic twins. Taken together, the findings suggest that brain perfusion is not affected in CFS.
Given the small number of subjects in this study, the findings must be verified in larger studies. It also would be valuable to assess possible changes in brain microstructure over time in a longitudinal study, and to determine whether treatment interventions exert any beneficial effects, the investigators added.
Imaging to assess brain microstructure identified increased fractional anisotropy in the anterior right arcuate fasciculus of patients with chronic fatigue syndrome, which was not present in age- and sex-matched control subjects.
The right-sided abnormality was particularly marked in right-handed chronic fatigue syndrome (CFS) patients, and the degree of increase correlated with the severity of their disorder. A finding of increased fractional anisotropy in this area of the brain may eventually serve as a biomarker for CFS, said Dr. Michael M. Zeineh of the department of radiology at Stanford (Calif.) University, and his associates (Radiology 2014 Oct. 29 [doi:10.1148/radiol.14141079]).
In what they described as the first study to examine brain microstructure in CFS, the investigators used volumetric MRI with diffusion-tensor imaging (DTI) to detect microstructural abnormalities in the patients – who had been evaluated at the university’s CFS Clinic during the preceding 5 years – and in healthy volunteers. The researchers also performed gray- and white-matter volumetric studies to identify differences between the two study groups in gross brain structure, as well as pseudocontinuous arterial spin labeling to assess global alterations in brain perfusion.
The mean age of the study participants was 46 years, and the mean duration of CFS symptoms was 12 years.
In the DTI analysis, the only significant difference between the two study groups was that the anterior right arcuate fasciculus – identified in 13 of 15 CFS patients and in all 14 control subjects in the study – showed significantly higher fractional anisotropy in CFS patients than in control subjects. This was confirmed in a second dataset from the same study subjects. The difference between CFS patients and control subjects was most pronounced in the subgroup of right-handed participants, “suggesting that hemispheric differences with handedness and language are an additional source of variance,” Dr. Zeineh and his associates said.
In addition, “two cortical regions connected via the arcuate fasciculus exhibited increased thickness: the right middle temporal and precentral gyri,” they noted.
The volumetric studies showed significantly lower total supratentorial white-matter volume in CFS, even after the data were adjusted to account for subject age, total intracranial volume, and handedness. However, total cortical gray-matter volume was equivalent between the two study groups, as were prefrontal cortex volume and global cortical thickness.
There were no differences between patients and controls in any perfusion measure, including perfusion to the cortex, supratentorial white matter, basal ganglia, thalami, or hippocampi. This finding is consistent with the results of a previous perfusion study involving monozygotic twins. Taken together, the findings suggest that brain perfusion is not affected in CFS.
Given the small number of subjects in this study, the findings must be verified in larger studies. It also would be valuable to assess possible changes in brain microstructure over time in a longitudinal study, and to determine whether treatment interventions exert any beneficial effects, the investigators added.
FROM RADIOLOGY
Key clinical point: Increased fractional anisotropy in the anterior right arcuate fasciculus may eventually serve as a biomarker for CFS if larger studies confirm the finding.
Major finding: The anterior right arcuate fasciculus showed significantly higher fractional anisotropy in CFS patients than in control subjects
Data source: A case-control study in which brain imaging was compared between 15 patients with chronic fatigue syndrome and 14 age- and sex-matched control subjects.
Disclosures: This study was supported by Stanford’s division of infectious disease CFS Fund. Dr. Zeineh reported receiving support from GE Healthcare unrelated to this study. He and his associates reported having no other financial disclosures.