User login
JIA arthritis and uveitis flares ‘often run parallel’
Children with juvenile idiopathic arthritis–associated uveitis (JIA-U) are significantly more likely to experience a flare in their eye disease if their arthritis is also worsening, a team of U.S.-based researchers has found.
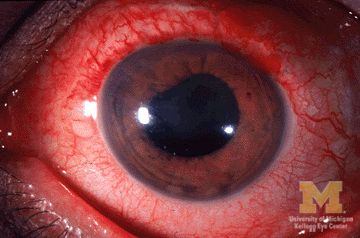
In a longitudinal cohort study, children with active arthritis at the time of a routine rheumatology assessment had an almost 2.5-fold increased risk of also having active uveitis 45 days before or after the assessment than did children whose arthritis was not flaring at the rheumatology assessment.
“We demonstrate that the two diseases often run parallel courses,” corresponding author Emily J. Liebling, MD, of the Children’s Hospital of Philadelphia and associates state in Arthritis Care & Research, noting that the magnitude of the association is striking.
“Although there are known risk factors associated with uveitis development in children with JIA, less data are available about factors associated with uveitis flare or activity,” said Sheila T. Angeles-Han, MD, MSc, of the departments of pediatrics and ophthalmology at Cincinnati Children’s Hospital Medical Center who commented on the study in an interview.
“If proven, this knowledge has the potential to impact practice patterns and current guidelines wherein a pediatric rheumatologist who evaluates a child with JIA-associated uveitis and finds active arthritis would request an expedited ophthalmic examination,” Dr. Angeles-Han suggested.
Dr. Angeles-Han led the development of the first American College of Rheumatology/Arthritis Foundation Guideline for the Screening, Monitoring, and Treatment of JIA-Associated Uveitis, which recommends regular screening for uveitis in all children with JIA. Children found to have uveitis should then be screened at least every 3 months, and more frequently if they are taking glucocorticoids and treatment is being tapered.

JIA-associated uveitis accounts for around 20%-40% of all cases of noninfectious childhood eye inflammation, and it can run an insidious and chronic course.
“Children with acute anterior uveitis are symptomatic and tend to have a painful red eye, thus prompting an ophthalmic evaluation,” Dr. Angeles-Han explained. “This is different from children with chronic anterior uveitis who tend not to have any symptoms, thus a screening examination is critical to detect ocular inflammation.”
While the ACR/AF guideline distinguishes between acute and chronic uveitis, Dr. Liebling and colleagues explain that they did not because their experience shows that “even patients with chronic anterior uveitis, typically thought to have silent disease, may exhibit symptoms of eye pain, redness, vision changes, and photophobia.”
Conversely, they say “the JIA subtypes usually associated with acute anterior uveitis may instead manifest as asymptomatic eye disease.”
For their study, Dr. Liebling and coinvestigators examined the records of children seen at the Children’s Hospital of Philadelphia over a 6.5-year period. For inclusion, children had to have a physician diagnosis of JIA of any subtype and a history of uveitis.
A total of 98 children were included in the retrospective evaluation; the median age at diagnosis of JIA was 3.3 years, and the median age at first uveitis diagnosis was 5.1 years. The majority (82%) were female, 69% were antinuclear antibody (ANA) positive, and 60% had oligoarthritis – all of which have been associated with having a higher risk for developing uveitis.
However, independent of these and several other factors, the probability of having active uveitis within 45 days of a rheumatology assessment was 65% in those with active arthritis versus 42% for those with no active joints.
Their data are based on 1,229 rheumatology visits that occurred between 2013 and 2019, with a median of 13 visits per patient. Overall, arthritis was defined as being active in 17% of visits, and active uveitis was observed in 18% of rheumatology visits.
Concordance between arthritis and uveitis activity was observed 73% of the time, the researchers reported. A sensitivity analysis that excluded children with the enthesitis-related arthritis subtype of JIA, who may not undergo frequent eye exams, did not change their findings.
Decreased odds of active uveitis at any time point were seen with the use of combination biologic and nonbiologic disease-modifying antirheumatic drugs. Years from uveitis diagnosis was also associated with lower odds of active uveitis over time.
Other factors associated with lower odds of uveitis were female sex, HLA-B27 positivity, and having any subtype of JIA other than the oligoarticular subtype.
Dr. Liebling and coinvestigators concluded that, contrary to the historical dogma, arthritis and uveitis do not run distinct and unrelated courses: “In patients with JIA-U, there is a significant temporal association between arthritis and uveitis disease activity.”
The study was sponsored by the Children’s Hospital of Philadelphia Rheumatology Research Fund. The investigators for the study had no financial support from commercial sources or any other potential conflicts of interest. Dr. Angeles-Han had no conflicts of interest to disclose.
SOURCE: Liebling EJ et al. Arthritis Care Res. 2020 Oct 12. doi: 10.1002/acr.24483.
Children with juvenile idiopathic arthritis–associated uveitis (JIA-U) are significantly more likely to experience a flare in their eye disease if their arthritis is also worsening, a team of U.S.-based researchers has found.
In a longitudinal cohort study, children with active arthritis at the time of a routine rheumatology assessment had an almost 2.5-fold increased risk of also having active uveitis 45 days before or after the assessment than did children whose arthritis was not flaring at the rheumatology assessment.
“We demonstrate that the two diseases often run parallel courses,” corresponding author Emily J. Liebling, MD, of the Children’s Hospital of Philadelphia and associates state in Arthritis Care & Research, noting that the magnitude of the association is striking.
“Although there are known risk factors associated with uveitis development in children with JIA, less data are available about factors associated with uveitis flare or activity,” said Sheila T. Angeles-Han, MD, MSc, of the departments of pediatrics and ophthalmology at Cincinnati Children’s Hospital Medical Center who commented on the study in an interview.
“If proven, this knowledge has the potential to impact practice patterns and current guidelines wherein a pediatric rheumatologist who evaluates a child with JIA-associated uveitis and finds active arthritis would request an expedited ophthalmic examination,” Dr. Angeles-Han suggested.
Dr. Angeles-Han led the development of the first American College of Rheumatology/Arthritis Foundation Guideline for the Screening, Monitoring, and Treatment of JIA-Associated Uveitis, which recommends regular screening for uveitis in all children with JIA. Children found to have uveitis should then be screened at least every 3 months, and more frequently if they are taking glucocorticoids and treatment is being tapered.
JIA-associated uveitis accounts for around 20%-40% of all cases of noninfectious childhood eye inflammation, and it can run an insidious and chronic course.
“Children with acute anterior uveitis are symptomatic and tend to have a painful red eye, thus prompting an ophthalmic evaluation,” Dr. Angeles-Han explained. “This is different from children with chronic anterior uveitis who tend not to have any symptoms, thus a screening examination is critical to detect ocular inflammation.”
While the ACR/AF guideline distinguishes between acute and chronic uveitis, Dr. Liebling and colleagues explain that they did not because their experience shows that “even patients with chronic anterior uveitis, typically thought to have silent disease, may exhibit symptoms of eye pain, redness, vision changes, and photophobia.”
Conversely, they say “the JIA subtypes usually associated with acute anterior uveitis may instead manifest as asymptomatic eye disease.”
For their study, Dr. Liebling and coinvestigators examined the records of children seen at the Children’s Hospital of Philadelphia over a 6.5-year period. For inclusion, children had to have a physician diagnosis of JIA of any subtype and a history of uveitis.
A total of 98 children were included in the retrospective evaluation; the median age at diagnosis of JIA was 3.3 years, and the median age at first uveitis diagnosis was 5.1 years. The majority (82%) were female, 69% were antinuclear antibody (ANA) positive, and 60% had oligoarthritis – all of which have been associated with having a higher risk for developing uveitis.
However, independent of these and several other factors, the probability of having active uveitis within 45 days of a rheumatology assessment was 65% in those with active arthritis versus 42% for those with no active joints.
Their data are based on 1,229 rheumatology visits that occurred between 2013 and 2019, with a median of 13 visits per patient. Overall, arthritis was defined as being active in 17% of visits, and active uveitis was observed in 18% of rheumatology visits.
Concordance between arthritis and uveitis activity was observed 73% of the time, the researchers reported. A sensitivity analysis that excluded children with the enthesitis-related arthritis subtype of JIA, who may not undergo frequent eye exams, did not change their findings.
Decreased odds of active uveitis at any time point were seen with the use of combination biologic and nonbiologic disease-modifying antirheumatic drugs. Years from uveitis diagnosis was also associated with lower odds of active uveitis over time.
Other factors associated with lower odds of uveitis were female sex, HLA-B27 positivity, and having any subtype of JIA other than the oligoarticular subtype.
Dr. Liebling and coinvestigators concluded that, contrary to the historical dogma, arthritis and uveitis do not run distinct and unrelated courses: “In patients with JIA-U, there is a significant temporal association between arthritis and uveitis disease activity.”
The study was sponsored by the Children’s Hospital of Philadelphia Rheumatology Research Fund. The investigators for the study had no financial support from commercial sources or any other potential conflicts of interest. Dr. Angeles-Han had no conflicts of interest to disclose.
SOURCE: Liebling EJ et al. Arthritis Care Res. 2020 Oct 12. doi: 10.1002/acr.24483.
Children with juvenile idiopathic arthritis–associated uveitis (JIA-U) are significantly more likely to experience a flare in their eye disease if their arthritis is also worsening, a team of U.S.-based researchers has found.
In a longitudinal cohort study, children with active arthritis at the time of a routine rheumatology assessment had an almost 2.5-fold increased risk of also having active uveitis 45 days before or after the assessment than did children whose arthritis was not flaring at the rheumatology assessment.
“We demonstrate that the two diseases often run parallel courses,” corresponding author Emily J. Liebling, MD, of the Children’s Hospital of Philadelphia and associates state in Arthritis Care & Research, noting that the magnitude of the association is striking.
“Although there are known risk factors associated with uveitis development in children with JIA, less data are available about factors associated with uveitis flare or activity,” said Sheila T. Angeles-Han, MD, MSc, of the departments of pediatrics and ophthalmology at Cincinnati Children’s Hospital Medical Center who commented on the study in an interview.
“If proven, this knowledge has the potential to impact practice patterns and current guidelines wherein a pediatric rheumatologist who evaluates a child with JIA-associated uveitis and finds active arthritis would request an expedited ophthalmic examination,” Dr. Angeles-Han suggested.
Dr. Angeles-Han led the development of the first American College of Rheumatology/Arthritis Foundation Guideline for the Screening, Monitoring, and Treatment of JIA-Associated Uveitis, which recommends regular screening for uveitis in all children with JIA. Children found to have uveitis should then be screened at least every 3 months, and more frequently if they are taking glucocorticoids and treatment is being tapered.
JIA-associated uveitis accounts for around 20%-40% of all cases of noninfectious childhood eye inflammation, and it can run an insidious and chronic course.
“Children with acute anterior uveitis are symptomatic and tend to have a painful red eye, thus prompting an ophthalmic evaluation,” Dr. Angeles-Han explained. “This is different from children with chronic anterior uveitis who tend not to have any symptoms, thus a screening examination is critical to detect ocular inflammation.”
While the ACR/AF guideline distinguishes between acute and chronic uveitis, Dr. Liebling and colleagues explain that they did not because their experience shows that “even patients with chronic anterior uveitis, typically thought to have silent disease, may exhibit symptoms of eye pain, redness, vision changes, and photophobia.”
Conversely, they say “the JIA subtypes usually associated with acute anterior uveitis may instead manifest as asymptomatic eye disease.”
For their study, Dr. Liebling and coinvestigators examined the records of children seen at the Children’s Hospital of Philadelphia over a 6.5-year period. For inclusion, children had to have a physician diagnosis of JIA of any subtype and a history of uveitis.
A total of 98 children were included in the retrospective evaluation; the median age at diagnosis of JIA was 3.3 years, and the median age at first uveitis diagnosis was 5.1 years. The majority (82%) were female, 69% were antinuclear antibody (ANA) positive, and 60% had oligoarthritis – all of which have been associated with having a higher risk for developing uveitis.
However, independent of these and several other factors, the probability of having active uveitis within 45 days of a rheumatology assessment was 65% in those with active arthritis versus 42% for those with no active joints.
Their data are based on 1,229 rheumatology visits that occurred between 2013 and 2019, with a median of 13 visits per patient. Overall, arthritis was defined as being active in 17% of visits, and active uveitis was observed in 18% of rheumatology visits.
Concordance between arthritis and uveitis activity was observed 73% of the time, the researchers reported. A sensitivity analysis that excluded children with the enthesitis-related arthritis subtype of JIA, who may not undergo frequent eye exams, did not change their findings.
Decreased odds of active uveitis at any time point were seen with the use of combination biologic and nonbiologic disease-modifying antirheumatic drugs. Years from uveitis diagnosis was also associated with lower odds of active uveitis over time.
Other factors associated with lower odds of uveitis were female sex, HLA-B27 positivity, and having any subtype of JIA other than the oligoarticular subtype.
Dr. Liebling and coinvestigators concluded that, contrary to the historical dogma, arthritis and uveitis do not run distinct and unrelated courses: “In patients with JIA-U, there is a significant temporal association between arthritis and uveitis disease activity.”
The study was sponsored by the Children’s Hospital of Philadelphia Rheumatology Research Fund. The investigators for the study had no financial support from commercial sources or any other potential conflicts of interest. Dr. Angeles-Han had no conflicts of interest to disclose.
SOURCE: Liebling EJ et al. Arthritis Care Res. 2020 Oct 12. doi: 10.1002/acr.24483.
FROM ARTHRITIS CARE & RESEARCH
Nusinersen provides continued benefits to presymptomatic children with SMA
according to an analysis presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year.
“Children are developing in a manner more consistent with normal development than that expected for children with two and three SMN2 gene copies,” said Russell Chin, MD, a neurologist at New York–Presbyterian Hospital. “These data demonstrate the durability of effect over a median of 3.8 years of follow-up, with children aged 2.8-4.8 years at the last visit.”
Many participants in the study achieved motor milestones within normal time limits, and no participant lost any major motor milestones. The investigators did not identify any new safety concerns during a maximum of 4.7 years of follow-up. They will follow participants until they reach approximately 8 years of age.
An ongoing open-label study
Dr. Chin presented interim results of the ongoing NURTURE study, which is examining the efficacy and safety of intrathecal nusinersen when administered to presymptomatic infants with SMA. The open-label, single-arm, phase 2 study is being conducted in various countries. Eligible participants were 6 weeks old or younger at first dose and had two or three copies of SMN2. The primary end point of NURTURE is time to death or respiratory intervention (i.e., invasive or noninvasive ventilation for 6 or more hours per day continuously for 7 or more days or tracheostomy). The natural history of SMA type 1 indicates that the median age at death or requirement for ventilation support is 13.5 months.
The investigators enrolled 25 infants: 15 with two copies of the gene and 10 with three copies. At the February 2020 interim analysis, participants had been in the study for 3.8 years and were aged 2.8-4.8 years at the last visit. No children had discontinued treatment or withdrawn from the study. All participants are alive, and four participants (all of whom have two copies of SMN2) required respiratory intervention. The latter children initiated respiratory support during an acute reversible illness. No subjects have required permanent ventilation, which the investigators define as ventilation for 16 or more hours per day for more than 21 days in the absence of an acute reversible event, or tracheostomy.
Treatment improved motor development
Approximately 84% of children achieved a maximum score on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scale. The population’s mean CHOP INTEND score increased steadily from baseline and stabilized at approximately the maximum score of 64. The population’s mean change in CHOP INTEND score from baseline to last visit was 13.6 points. The mean score at last visit was 62.0 among patients with two copies of SMN2 and 63.4 among patients with three copies. In addition, the time to first achievement of maximum CHOP INTEND score was shorter in participants with three copies of SMN2, compared with those with two. Four participants with two copies of the gene have not yet achieved a maximum CHOP INTEND score.
Many of the children in the study achieved World Health Organization motor milestones within time frames consistent with normal development. About 84% of participants became able to sit without support within the normal time frame in healthy children. Approximately 60% of children achieved walking with assistance within the normal window, and 64% achieved walking alone within the normal window. Of 25 participants, 24 are walking with assistance, and 22 of 25 (88%) can walk alone. Dr. Chin and colleagues observed that lower levels of phosphorylated neurofilament heavy chain in plasma and cerebrospinal fluid on treatment at day 64 were significantly correlated with higher total score on the Hammersmith Infant Neurological Examination at day 302 and with earlier achievement of the WHO milestone walking alone.
Nusinersen and lumbar puncture were well tolerated. No children discontinued treatment or withdrew from the study because of an adverse event. The investigators did not consider any adverse events or serious adverse events to be related to the study drug. They also did not observe any clinically relevant trends related to nusinersen in hematology, blood chemistry, urinalysis, coagulation, vital signs, or ECGs.
Dr. Chin is an employee of and holds stock in Biogen, which manufactures nusinersen and is sponsoring the study.
SOURCE: Chin R et al. CNS-ICNA 2020, Abstract PL78.
according to an analysis presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year.
“Children are developing in a manner more consistent with normal development than that expected for children with two and three SMN2 gene copies,” said Russell Chin, MD, a neurologist at New York–Presbyterian Hospital. “These data demonstrate the durability of effect over a median of 3.8 years of follow-up, with children aged 2.8-4.8 years at the last visit.”
Many participants in the study achieved motor milestones within normal time limits, and no participant lost any major motor milestones. The investigators did not identify any new safety concerns during a maximum of 4.7 years of follow-up. They will follow participants until they reach approximately 8 years of age.
An ongoing open-label study
Dr. Chin presented interim results of the ongoing NURTURE study, which is examining the efficacy and safety of intrathecal nusinersen when administered to presymptomatic infants with SMA. The open-label, single-arm, phase 2 study is being conducted in various countries. Eligible participants were 6 weeks old or younger at first dose and had two or three copies of SMN2. The primary end point of NURTURE is time to death or respiratory intervention (i.e., invasive or noninvasive ventilation for 6 or more hours per day continuously for 7 or more days or tracheostomy). The natural history of SMA type 1 indicates that the median age at death or requirement for ventilation support is 13.5 months.
The investigators enrolled 25 infants: 15 with two copies of the gene and 10 with three copies. At the February 2020 interim analysis, participants had been in the study for 3.8 years and were aged 2.8-4.8 years at the last visit. No children had discontinued treatment or withdrawn from the study. All participants are alive, and four participants (all of whom have two copies of SMN2) required respiratory intervention. The latter children initiated respiratory support during an acute reversible illness. No subjects have required permanent ventilation, which the investigators define as ventilation for 16 or more hours per day for more than 21 days in the absence of an acute reversible event, or tracheostomy.
Treatment improved motor development
Approximately 84% of children achieved a maximum score on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scale. The population’s mean CHOP INTEND score increased steadily from baseline and stabilized at approximately the maximum score of 64. The population’s mean change in CHOP INTEND score from baseline to last visit was 13.6 points. The mean score at last visit was 62.0 among patients with two copies of SMN2 and 63.4 among patients with three copies. In addition, the time to first achievement of maximum CHOP INTEND score was shorter in participants with three copies of SMN2, compared with those with two. Four participants with two copies of the gene have not yet achieved a maximum CHOP INTEND score.
Many of the children in the study achieved World Health Organization motor milestones within time frames consistent with normal development. About 84% of participants became able to sit without support within the normal time frame in healthy children. Approximately 60% of children achieved walking with assistance within the normal window, and 64% achieved walking alone within the normal window. Of 25 participants, 24 are walking with assistance, and 22 of 25 (88%) can walk alone. Dr. Chin and colleagues observed that lower levels of phosphorylated neurofilament heavy chain in plasma and cerebrospinal fluid on treatment at day 64 were significantly correlated with higher total score on the Hammersmith Infant Neurological Examination at day 302 and with earlier achievement of the WHO milestone walking alone.
Nusinersen and lumbar puncture were well tolerated. No children discontinued treatment or withdrew from the study because of an adverse event. The investigators did not consider any adverse events or serious adverse events to be related to the study drug. They also did not observe any clinically relevant trends related to nusinersen in hematology, blood chemistry, urinalysis, coagulation, vital signs, or ECGs.
Dr. Chin is an employee of and holds stock in Biogen, which manufactures nusinersen and is sponsoring the study.
SOURCE: Chin R et al. CNS-ICNA 2020, Abstract PL78.
according to an analysis presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year.
“Children are developing in a manner more consistent with normal development than that expected for children with two and three SMN2 gene copies,” said Russell Chin, MD, a neurologist at New York–Presbyterian Hospital. “These data demonstrate the durability of effect over a median of 3.8 years of follow-up, with children aged 2.8-4.8 years at the last visit.”
Many participants in the study achieved motor milestones within normal time limits, and no participant lost any major motor milestones. The investigators did not identify any new safety concerns during a maximum of 4.7 years of follow-up. They will follow participants until they reach approximately 8 years of age.
An ongoing open-label study
Dr. Chin presented interim results of the ongoing NURTURE study, which is examining the efficacy and safety of intrathecal nusinersen when administered to presymptomatic infants with SMA. The open-label, single-arm, phase 2 study is being conducted in various countries. Eligible participants were 6 weeks old or younger at first dose and had two or three copies of SMN2. The primary end point of NURTURE is time to death or respiratory intervention (i.e., invasive or noninvasive ventilation for 6 or more hours per day continuously for 7 or more days or tracheostomy). The natural history of SMA type 1 indicates that the median age at death or requirement for ventilation support is 13.5 months.
The investigators enrolled 25 infants: 15 with two copies of the gene and 10 with three copies. At the February 2020 interim analysis, participants had been in the study for 3.8 years and were aged 2.8-4.8 years at the last visit. No children had discontinued treatment or withdrawn from the study. All participants are alive, and four participants (all of whom have two copies of SMN2) required respiratory intervention. The latter children initiated respiratory support during an acute reversible illness. No subjects have required permanent ventilation, which the investigators define as ventilation for 16 or more hours per day for more than 21 days in the absence of an acute reversible event, or tracheostomy.
Treatment improved motor development
Approximately 84% of children achieved a maximum score on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scale. The population’s mean CHOP INTEND score increased steadily from baseline and stabilized at approximately the maximum score of 64. The population’s mean change in CHOP INTEND score from baseline to last visit was 13.6 points. The mean score at last visit was 62.0 among patients with two copies of SMN2 and 63.4 among patients with three copies. In addition, the time to first achievement of maximum CHOP INTEND score was shorter in participants with three copies of SMN2, compared with those with two. Four participants with two copies of the gene have not yet achieved a maximum CHOP INTEND score.
Many of the children in the study achieved World Health Organization motor milestones within time frames consistent with normal development. About 84% of participants became able to sit without support within the normal time frame in healthy children. Approximately 60% of children achieved walking with assistance within the normal window, and 64% achieved walking alone within the normal window. Of 25 participants, 24 are walking with assistance, and 22 of 25 (88%) can walk alone. Dr. Chin and colleagues observed that lower levels of phosphorylated neurofilament heavy chain in plasma and cerebrospinal fluid on treatment at day 64 were significantly correlated with higher total score on the Hammersmith Infant Neurological Examination at day 302 and with earlier achievement of the WHO milestone walking alone.
Nusinersen and lumbar puncture were well tolerated. No children discontinued treatment or withdrew from the study because of an adverse event. The investigators did not consider any adverse events or serious adverse events to be related to the study drug. They also did not observe any clinically relevant trends related to nusinersen in hematology, blood chemistry, urinalysis, coagulation, vital signs, or ECGs.
Dr. Chin is an employee of and holds stock in Biogen, which manufactures nusinersen and is sponsoring the study.
SOURCE: Chin R et al. CNS-ICNA 2020, Abstract PL78.
FROM CNS-ICNA 2020
CBD for LGS: Fewer seizures, but thrombocytopenia risk

At the 2020 CNS-ICNA Conjoint Meeting, held virtually this year, Anul Patel, MD, section chief of Pediatric neurology at Nationwide Children’s and associate professor of clinical pediatrics and neurology at the Ohio State University, both in Columbus, Ohio, reported 156-week results of an open-label extension trial called GWPCARE5 that showed patients with LGS taking Epidiolex had a 60% or greater average reduction in seizures, compared with baseline. Epidiolex, a highly purified form of CBD, was approved by the Food and Drug Administration in 2018 for LGS and Dravet syndrome.
In a separate presentation, Nancy A. McNamara, MD, an assistant professor at the C.S. Mott Children’s Hospital at the University of Michigan, Ann Arbor, said that more than one-third of patients taking both Epidiolex and valproic acid (VPA) developed thrombocytopenia after starting CBD therapy. The single-center chart review she reported on included 83 patients.
Daniel Friedman, MD, an epilepsy specialist at New York University who’s researched CBD in children with autism spectrum disorder, said, “These studies show that, while purified CBD has durable effects on the most disabling seizures in children and adults with LGS, like all treatments, it is not without risks that warrant attention and monitoring.”
Open-label extension study
The open-label extension study included 366 patients who participated in the two previous clinical trials. They were given varying doses of CBD titrated over 2 weeks with 20 mg/kg as the target dose, Dr. Patel said. The most common concurrent therapies they were taking were clobazam, valproate or VPA, lamotrigine, levetiracetam, and rufinamide. At weeks 145-156, 67% of patients had a 50% or greater reduction in seizures, 44% had a 75% or greater reduction, and 9% stopped having seizures altogether, Dr. Patel said.
“CBD treatment had a similar safety profile to what was observed in the completed parent randomized clinical trials,” Dr. Patel said. “Sustained reductions in drop and total seizures were observed up to the 156-week follow-up point. So these results demonstrate the potential long-term benefits of CBD treatment for patients with LGS as it relates to reduction of their seizures.”
Adverse event profiles in this analysis were similar to previous clinical trials, he noted. The three most common adverse events were diarrhea (38%), convulsion (38%) and pyrexia (34%), but high percentages of those adverse events resolved during follow-up: 78%, 80%, and 96%, respectively.
Dr. Patel also noted that 31% of patients had elevated liver enzymes (alanine aminotransferase or aspartate aminotransferase), but most of these patients – 78 of 113, or 69% – were on concomitant VPA. “Importantly, no patient met the standard criteria for severe drug-induced liver injury, known as Hy’s law,” he said.
Retention rates for patients were 81% at 1 year, 69% at 2 years and 65% at 3 years, Dr. Patel said.
“An urgent systemic review”

Dr. McNamara’s research drilled down into the interaction of CBD and VPA. “Over the past several months we have made observations that several patients that had been started on CBD, also known as Epidiolex, had developed thrombocytopenia, some of which were symptomatic,” she said. Symptoms included hematuria, easy bruising, and gingival bleeding.
That prompted what Dr. McNamara called “an urgent systemic review” of all patients on CBD. Of 83 patients started on CBD for LGS from January to August 2019, 9 (11%) developed thrombocytopenia. “All of these patients were on concurrent VPA and no patients started on CBD without VPA developed thrombocytopenia,” she said. In all, 23 patients were taking CBD concurrently with VPA. Four of nine cases were symptomatic.
“The thrombocytopenia was reversible in all patients with reduction of medication and one patient recovered spontaneously without intervention,” Dr. McNamara noted.
“This was an important finding because this was not something that had come out of the clinical trials prior to FDA approval,” Dr. McNamara said. “This requires closer monitoring for patients who are started on CBD who are already on VPA.”
Of the 23 patients taking concurrent VPA, 10 had low platelet counts after starting CBD. In six patients, platelet counts dropped from normal before CBD therapy to low afterward.
The study used a McNemar test to determine if an observed adverse event occurred by chance or was related to starting a drug, which yielded a P value of .125, Dr. McNamara said. “While this did not achieve statistical significance, we suggest that prescribers closely monitor platelet levels after starting CBD, particularly when a patient is also on concurrent VPA,” she said.
Her group obtained a complete blood count at baseline and then at 1, 3, and 6 months after starting the patient on CBD, along with evaluation of alanine aminotransferase and aspartate aminotransferase. “We believe that this is helpful because most of the patients that develop low platelets did so within 3 months of starting cannabidiol,” Dr. McNamara said.
She acknowledged the limits of the single-center study. “Future research will need to be done with larger cohorts with standardized surveillance labs,” she said in an interview.
Dr. Patel disclosed financial relationships with GW Research and Greenwich Biosciences. Dr. McNamara has no relevant disclosures.
At the 2020 CNS-ICNA Conjoint Meeting, held virtually this year, Anul Patel, MD, section chief of Pediatric neurology at Nationwide Children’s and associate professor of clinical pediatrics and neurology at the Ohio State University, both in Columbus, Ohio, reported 156-week results of an open-label extension trial called GWPCARE5 that showed patients with LGS taking Epidiolex had a 60% or greater average reduction in seizures, compared with baseline. Epidiolex, a highly purified form of CBD, was approved by the Food and Drug Administration in 2018 for LGS and Dravet syndrome.
In a separate presentation, Nancy A. McNamara, MD, an assistant professor at the C.S. Mott Children’s Hospital at the University of Michigan, Ann Arbor, said that more than one-third of patients taking both Epidiolex and valproic acid (VPA) developed thrombocytopenia after starting CBD therapy. The single-center chart review she reported on included 83 patients.
Daniel Friedman, MD, an epilepsy specialist at New York University who’s researched CBD in children with autism spectrum disorder, said, “These studies show that, while purified CBD has durable effects on the most disabling seizures in children and adults with LGS, like all treatments, it is not without risks that warrant attention and monitoring.”
Open-label extension study
The open-label extension study included 366 patients who participated in the two previous clinical trials. They were given varying doses of CBD titrated over 2 weeks with 20 mg/kg as the target dose, Dr. Patel said. The most common concurrent therapies they were taking were clobazam, valproate or VPA, lamotrigine, levetiracetam, and rufinamide. At weeks 145-156, 67% of patients had a 50% or greater reduction in seizures, 44% had a 75% or greater reduction, and 9% stopped having seizures altogether, Dr. Patel said.
“CBD treatment had a similar safety profile to what was observed in the completed parent randomized clinical trials,” Dr. Patel said. “Sustained reductions in drop and total seizures were observed up to the 156-week follow-up point. So these results demonstrate the potential long-term benefits of CBD treatment for patients with LGS as it relates to reduction of their seizures.”
Adverse event profiles in this analysis were similar to previous clinical trials, he noted. The three most common adverse events were diarrhea (38%), convulsion (38%) and pyrexia (34%), but high percentages of those adverse events resolved during follow-up: 78%, 80%, and 96%, respectively.
Dr. Patel also noted that 31% of patients had elevated liver enzymes (alanine aminotransferase or aspartate aminotransferase), but most of these patients – 78 of 113, or 69% – were on concomitant VPA. “Importantly, no patient met the standard criteria for severe drug-induced liver injury, known as Hy’s law,” he said.
Retention rates for patients were 81% at 1 year, 69% at 2 years and 65% at 3 years, Dr. Patel said.
“An urgent systemic review”
Dr. McNamara’s research drilled down into the interaction of CBD and VPA. “Over the past several months we have made observations that several patients that had been started on CBD, also known as Epidiolex, had developed thrombocytopenia, some of which were symptomatic,” she said. Symptoms included hematuria, easy bruising, and gingival bleeding.
That prompted what Dr. McNamara called “an urgent systemic review” of all patients on CBD. Of 83 patients started on CBD for LGS from January to August 2019, 9 (11%) developed thrombocytopenia. “All of these patients were on concurrent VPA and no patients started on CBD without VPA developed thrombocytopenia,” she said. In all, 23 patients were taking CBD concurrently with VPA. Four of nine cases were symptomatic.
“The thrombocytopenia was reversible in all patients with reduction of medication and one patient recovered spontaneously without intervention,” Dr. McNamara noted.
“This was an important finding because this was not something that had come out of the clinical trials prior to FDA approval,” Dr. McNamara said. “This requires closer monitoring for patients who are started on CBD who are already on VPA.”
Of the 23 patients taking concurrent VPA, 10 had low platelet counts after starting CBD. In six patients, platelet counts dropped from normal before CBD therapy to low afterward.
The study used a McNemar test to determine if an observed adverse event occurred by chance or was related to starting a drug, which yielded a P value of .125, Dr. McNamara said. “While this did not achieve statistical significance, we suggest that prescribers closely monitor platelet levels after starting CBD, particularly when a patient is also on concurrent VPA,” she said.
Her group obtained a complete blood count at baseline and then at 1, 3, and 6 months after starting the patient on CBD, along with evaluation of alanine aminotransferase and aspartate aminotransferase. “We believe that this is helpful because most of the patients that develop low platelets did so within 3 months of starting cannabidiol,” Dr. McNamara said.
She acknowledged the limits of the single-center study. “Future research will need to be done with larger cohorts with standardized surveillance labs,” she said in an interview.
Dr. Patel disclosed financial relationships with GW Research and Greenwich Biosciences. Dr. McNamara has no relevant disclosures.
At the 2020 CNS-ICNA Conjoint Meeting, held virtually this year, Anul Patel, MD, section chief of Pediatric neurology at Nationwide Children’s and associate professor of clinical pediatrics and neurology at the Ohio State University, both in Columbus, Ohio, reported 156-week results of an open-label extension trial called GWPCARE5 that showed patients with LGS taking Epidiolex had a 60% or greater average reduction in seizures, compared with baseline. Epidiolex, a highly purified form of CBD, was approved by the Food and Drug Administration in 2018 for LGS and Dravet syndrome.
In a separate presentation, Nancy A. McNamara, MD, an assistant professor at the C.S. Mott Children’s Hospital at the University of Michigan, Ann Arbor, said that more than one-third of patients taking both Epidiolex and valproic acid (VPA) developed thrombocytopenia after starting CBD therapy. The single-center chart review she reported on included 83 patients.
Daniel Friedman, MD, an epilepsy specialist at New York University who’s researched CBD in children with autism spectrum disorder, said, “These studies show that, while purified CBD has durable effects on the most disabling seizures in children and adults with LGS, like all treatments, it is not without risks that warrant attention and monitoring.”
Open-label extension study
The open-label extension study included 366 patients who participated in the two previous clinical trials. They were given varying doses of CBD titrated over 2 weeks with 20 mg/kg as the target dose, Dr. Patel said. The most common concurrent therapies they were taking were clobazam, valproate or VPA, lamotrigine, levetiracetam, and rufinamide. At weeks 145-156, 67% of patients had a 50% or greater reduction in seizures, 44% had a 75% or greater reduction, and 9% stopped having seizures altogether, Dr. Patel said.
“CBD treatment had a similar safety profile to what was observed in the completed parent randomized clinical trials,” Dr. Patel said. “Sustained reductions in drop and total seizures were observed up to the 156-week follow-up point. So these results demonstrate the potential long-term benefits of CBD treatment for patients with LGS as it relates to reduction of their seizures.”
Adverse event profiles in this analysis were similar to previous clinical trials, he noted. The three most common adverse events were diarrhea (38%), convulsion (38%) and pyrexia (34%), but high percentages of those adverse events resolved during follow-up: 78%, 80%, and 96%, respectively.
Dr. Patel also noted that 31% of patients had elevated liver enzymes (alanine aminotransferase or aspartate aminotransferase), but most of these patients – 78 of 113, or 69% – were on concomitant VPA. “Importantly, no patient met the standard criteria for severe drug-induced liver injury, known as Hy’s law,” he said.
Retention rates for patients were 81% at 1 year, 69% at 2 years and 65% at 3 years, Dr. Patel said.
“An urgent systemic review”
Dr. McNamara’s research drilled down into the interaction of CBD and VPA. “Over the past several months we have made observations that several patients that had been started on CBD, also known as Epidiolex, had developed thrombocytopenia, some of which were symptomatic,” she said. Symptoms included hematuria, easy bruising, and gingival bleeding.
That prompted what Dr. McNamara called “an urgent systemic review” of all patients on CBD. Of 83 patients started on CBD for LGS from January to August 2019, 9 (11%) developed thrombocytopenia. “All of these patients were on concurrent VPA and no patients started on CBD without VPA developed thrombocytopenia,” she said. In all, 23 patients were taking CBD concurrently with VPA. Four of nine cases were symptomatic.
“The thrombocytopenia was reversible in all patients with reduction of medication and one patient recovered spontaneously without intervention,” Dr. McNamara noted.
“This was an important finding because this was not something that had come out of the clinical trials prior to FDA approval,” Dr. McNamara said. “This requires closer monitoring for patients who are started on CBD who are already on VPA.”
Of the 23 patients taking concurrent VPA, 10 had low platelet counts after starting CBD. In six patients, platelet counts dropped from normal before CBD therapy to low afterward.
The study used a McNemar test to determine if an observed adverse event occurred by chance or was related to starting a drug, which yielded a P value of .125, Dr. McNamara said. “While this did not achieve statistical significance, we suggest that prescribers closely monitor platelet levels after starting CBD, particularly when a patient is also on concurrent VPA,” she said.
Her group obtained a complete blood count at baseline and then at 1, 3, and 6 months after starting the patient on CBD, along with evaluation of alanine aminotransferase and aspartate aminotransferase. “We believe that this is helpful because most of the patients that develop low platelets did so within 3 months of starting cannabidiol,” Dr. McNamara said.
She acknowledged the limits of the single-center study. “Future research will need to be done with larger cohorts with standardized surveillance labs,” she said in an interview.
Dr. Patel disclosed financial relationships with GW Research and Greenwich Biosciences. Dr. McNamara has no relevant disclosures.
FROM CNS-ICNA 2020
NMOSD challenges in children
, but have led to some uncertainty and confusion as well.
At the2020 CNS-ICNA Conjoint Meeting, held virtually this year, presenters discussed some of the challenges of differential diagnosis and treatment choice in pediatric NMOSD, which is easily confused with multiple sclerosis.
NMOSD used to be considered a monophasic disease restricted to the optic nerve and spinal cord, but is now known to affect other regions of the central nervous system and to relapse in some patients.
Diagnosis
The disease is often mediated by antibodies to the aquaporin-4 (AQP-4) water channel, but about 30% of adult patients lack the antibody, and AQP-4 seronegativity is more common in the pediatric population. Another common antibody found in 40%–50% of children with NMOSD targets myelin oligodendrocyte glycoprotein (MOG).
It is important to be aware that false negatives can occur in serology assays, and false positives are common, particularly in ELISA assays, Silvia N. Tenembaum, MD, said during her presentation. For those reasons, serology is not enough for a diagnosis. “Patients should also have compatible symptoms and MRI findings,” said Dr. Tenembaum, director of the pediatric neuroimmunology program at National Pediatric Hospital in Buenos Aires.
According to international consensus criteria, to be diagnosed with NMOSD, AQP-4 seropositive patients should also have at least one core clinical symptom: optic neuritis, acute myelitis, area postrema syndrome, other acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome, or symptomatic cerebral syndrome. AQP-4 seronegative patients or with unknown status should have at least two core symptoms, one of which must be optic neuritis, acute myelitis, or area postrema syndrome. Both conventional MRI and advanced new techniques are important for achieving differential diagnosis.
The most common symptom in children is optic neuritis, which occurs in 50%-70% of patients. Cerebral syndromes with or without encephalopathy and large tumefactive white matter lesions are also common, according to Dr. Tenembaum.
There are many conditions that mimic the spinal cord and optic nerve symptoms of NMOSD, which must be ruled out. One example is optic myelopathy and vision loss from late-onset biotinylase deficiency. It is critical to rule that out because it is treatable with supplements. Optic neuropathy, papillitis, and papilledema can also resemble NMOSD.
It is critical to achieve an early diagnosis of NMOSD in children, because some MS drugs can worsen NMOSD, according to Thaís Armangue, MD, PhD, head of neuroimmunology at SJD Barcelona Children’s Hospital, who also presented at the session. She pointed out that the MOG antibody, while common in children, is also associated with many demyelinating diseases. Some 50%-60% of children with acute disseminated encephalomyelitis (ADEM) have high titers of MOG antibodies. Although early studies suggested that persistent anti-MOG antibodies were associated with risk of developing MS, more recent studies show it predicts a non-MS disease course, particularly at titers greater than 1:1280, according to Dr. Tenembaum. Persistent anti-MOG antibodies are also associated with relapsing disease, but it is associated with other syndromes besides NMOSD. “The probability is that [MOG antibodies are] useful, but they cannot guide chronic immunotherapy, because even monophasic patients can last maybe 12 months before they become MOG negative, and we cannot wait so many months” to determine treatment course, said Dr. Tenembaum.
For monophasic ADEM or NMOSD, there is no need for chronic treatment. But children with MS and recurrent NMOSD require early chronic immunotherapy because specific therapies have been shown to improve prognosis.
Acute treatment
When it comes to acute treatment of NMOSD, the goal is to suppress the inflammatory attack but also to minimize long-term damage and optimize long-term neurological function. “The potential for irreversible injury with an attack is very high, and cumulative disabilities in NMOSD can result directly from attacks,” E. Ann Yeh, MD, director of the Pediatric MS and Neuroinflammatory Disorders Program at the Hospital for Sick Children at the University of Toronto, said during her talk.
IV steroids are generally the first choice, with a preference for methylprednisolone. Pediatric patients that are MOG antibody positive usually respond better and more quickly than do adults, with rapid daily improvements in mobility, vomiting, and eyesight. Dr. Yeh recommends weaning good responders off steroids because AQP-4 positive patients are likely to relapse without a steroid wean, and antibody testing may be unavailable or results may be delayed. The wean can range from 4 weeks to 4-6 months, depending on antibody status, likelihood of AQP-4 positivity, and clinical parameters.
Inadequate responses are usually pretty evident. If there is only light perception by day 4 or 5, or paralyzed patients are nonambulatory and achieve only twitchy movements by that time, second-line therapies should be considered, including therapeutic plasma exchange (TPE) with 5-7 exchanges or intravenous immunoglobulins (IVIg).
Dr. Yeh called for quick treatment. Whatever you do, “please do it sooner rather than later if you think there’s no response [to steroids],” Dr. Yeh said.
TPE is the first choice, according to Dr. Yeh. “There seems to be a fair amount of information that suggests that if you’re having difficulty getting a response to steroids, TPE can make a difference in these patients,” she said. But in some cases TPE may not be available, and IVIg can be attempted first. If it achieves no or only marginal improvement, TPE can be attempted later, but it must be kept in mind that TPE conducted too soon could wash out IVIg. Patients who get much better on IVIg can undergo a steroid wean, and then be evaluated for prophylactic therapy, said Dr. Yeh.
The evidence for IVIg is limited, reflecting the difficulty of studying treatments in rare populations. Still, when TPE is not available and the patient is quite impaired, IVIg makes sense to try. “Absence of evidence does not mean that the therapy doesn’t work, and I don’t think we should throw out the baby with the bath water,” said Dr. Yeh.
Although IVIg treatment is generally well tolerated, there have been a few serious adverse events, such as anaphylactic shock and aseptic meningitis, according to Andrea Savransky, MD, a pediatrician at National Pediatric Hospital in Buenos Aires, who also spoke at the session. “I think it is important to weigh the benefits against the risk,” Dr. Savransky said. She noted that TPE should not be taken lightly. One study showed more complications in pediatric patients than in adult patients, and it must be performed in specialized centers.
Emerging treaments
Tanuja Chitnis, MD, director of the Partners Pediatric MS Center at Massachusetts General Hospital, Boston, discussed some of the emerging treatments for pediatric NMOSD. Rituximab has been associated with success in some retrospective studies, but dosing should be personalized. Dr. Chitnis reported that B cells can return before 6 months, so she monitors B cells beginning 2 months after induction, redosing after 4 or 5 months rather than 6 if B cells return.
Nevertheless, relapses can still occur after rituximab therapy. “There is room for additional therapies to address this gap,” said Dr. Chitnis. Three new antibodies have received approval for treatment of NMOSD in adults. These include the complement inhibitor eculizumab, the IL-6 receptor antibody satralizumab, and the anti-CD19 antibody inebilizumab. Phase 3 clinical trials in children have been conducted for eculizumab and are in the planning stage for inebilizumab, and pediatric patients were included in pivotal trials for satralizumab.
Eculizumab treatment resulted in a 94.2% reduction in relapse risk in AQP4-positive adults. Satralizumab showed a 79% reduction in relapse risk among AQP-4 positive subjects with NMOSD or neuromyelitis optica and a 34% reduction in those who were AQP-4 negative. The pediatric subgroup had similar levels of response to adults, though the numbers were too small for a subgroup analysis.
In AQP-4 positive patients, inebilizumab treatment yielded a 77% reduction in relapse rate. In all patients, there was a 73% reduction.
For MOG antibody-positive patients with AQP-4 negative disease, novel therapies are at earlier stages of development. Typical MS therapies such as interferon beta and glatiramer acetate don’t seem to be effective. Some that have shown signs of efficacy include azathioprine, mycophenylate mofetil, rituximab, and IVIg infusion, but the state of the field is not encouraging. “This is an observation now being studied in larger cohorts, but in general I have not found that there’s a very strong response to any of these therapies, possibly with the exception of IVIg,” said Dr. Chitnis.
Dr. Tenembaum has no relevant financial disclosures. Dr. Armangue has received speaking honoraria from Novartis and travel expenses for scientific meetings from Merck, Biogen, and Roche. Dr. Yeh is on the scientific advisory board of Juno Therapeutics and has received research support from Biogen. Dr. Chitnis advises Biogen-Idec, Novartis, and Alexion, serves on clinical trial advisory boards for Novartis and Sanofi Aventis, and has received research support from Verily, EMD Serono, and Novartis. Dr. Savransky has received honoraria from Genzyme de Argentina SA.
, but have led to some uncertainty and confusion as well.
At the2020 CNS-ICNA Conjoint Meeting, held virtually this year, presenters discussed some of the challenges of differential diagnosis and treatment choice in pediatric NMOSD, which is easily confused with multiple sclerosis.
NMOSD used to be considered a monophasic disease restricted to the optic nerve and spinal cord, but is now known to affect other regions of the central nervous system and to relapse in some patients.
Diagnosis
The disease is often mediated by antibodies to the aquaporin-4 (AQP-4) water channel, but about 30% of adult patients lack the antibody, and AQP-4 seronegativity is more common in the pediatric population. Another common antibody found in 40%–50% of children with NMOSD targets myelin oligodendrocyte glycoprotein (MOG).
It is important to be aware that false negatives can occur in serology assays, and false positives are common, particularly in ELISA assays, Silvia N. Tenembaum, MD, said during her presentation. For those reasons, serology is not enough for a diagnosis. “Patients should also have compatible symptoms and MRI findings,” said Dr. Tenembaum, director of the pediatric neuroimmunology program at National Pediatric Hospital in Buenos Aires.
According to international consensus criteria, to be diagnosed with NMOSD, AQP-4 seropositive patients should also have at least one core clinical symptom: optic neuritis, acute myelitis, area postrema syndrome, other acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome, or symptomatic cerebral syndrome. AQP-4 seronegative patients or with unknown status should have at least two core symptoms, one of which must be optic neuritis, acute myelitis, or area postrema syndrome. Both conventional MRI and advanced new techniques are important for achieving differential diagnosis.
The most common symptom in children is optic neuritis, which occurs in 50%-70% of patients. Cerebral syndromes with or without encephalopathy and large tumefactive white matter lesions are also common, according to Dr. Tenembaum.
There are many conditions that mimic the spinal cord and optic nerve symptoms of NMOSD, which must be ruled out. One example is optic myelopathy and vision loss from late-onset biotinylase deficiency. It is critical to rule that out because it is treatable with supplements. Optic neuropathy, papillitis, and papilledema can also resemble NMOSD.
It is critical to achieve an early diagnosis of NMOSD in children, because some MS drugs can worsen NMOSD, according to Thaís Armangue, MD, PhD, head of neuroimmunology at SJD Barcelona Children’s Hospital, who also presented at the session. She pointed out that the MOG antibody, while common in children, is also associated with many demyelinating diseases. Some 50%-60% of children with acute disseminated encephalomyelitis (ADEM) have high titers of MOG antibodies. Although early studies suggested that persistent anti-MOG antibodies were associated with risk of developing MS, more recent studies show it predicts a non-MS disease course, particularly at titers greater than 1:1280, according to Dr. Tenembaum. Persistent anti-MOG antibodies are also associated with relapsing disease, but it is associated with other syndromes besides NMOSD. “The probability is that [MOG antibodies are] useful, but they cannot guide chronic immunotherapy, because even monophasic patients can last maybe 12 months before they become MOG negative, and we cannot wait so many months” to determine treatment course, said Dr. Tenembaum.
For monophasic ADEM or NMOSD, there is no need for chronic treatment. But children with MS and recurrent NMOSD require early chronic immunotherapy because specific therapies have been shown to improve prognosis.
Acute treatment
When it comes to acute treatment of NMOSD, the goal is to suppress the inflammatory attack but also to minimize long-term damage and optimize long-term neurological function. “The potential for irreversible injury with an attack is very high, and cumulative disabilities in NMOSD can result directly from attacks,” E. Ann Yeh, MD, director of the Pediatric MS and Neuroinflammatory Disorders Program at the Hospital for Sick Children at the University of Toronto, said during her talk.
IV steroids are generally the first choice, with a preference for methylprednisolone. Pediatric patients that are MOG antibody positive usually respond better and more quickly than do adults, with rapid daily improvements in mobility, vomiting, and eyesight. Dr. Yeh recommends weaning good responders off steroids because AQP-4 positive patients are likely to relapse without a steroid wean, and antibody testing may be unavailable or results may be delayed. The wean can range from 4 weeks to 4-6 months, depending on antibody status, likelihood of AQP-4 positivity, and clinical parameters.
Inadequate responses are usually pretty evident. If there is only light perception by day 4 or 5, or paralyzed patients are nonambulatory and achieve only twitchy movements by that time, second-line therapies should be considered, including therapeutic plasma exchange (TPE) with 5-7 exchanges or intravenous immunoglobulins (IVIg).
Dr. Yeh called for quick treatment. Whatever you do, “please do it sooner rather than later if you think there’s no response [to steroids],” Dr. Yeh said.
TPE is the first choice, according to Dr. Yeh. “There seems to be a fair amount of information that suggests that if you’re having difficulty getting a response to steroids, TPE can make a difference in these patients,” she said. But in some cases TPE may not be available, and IVIg can be attempted first. If it achieves no or only marginal improvement, TPE can be attempted later, but it must be kept in mind that TPE conducted too soon could wash out IVIg. Patients who get much better on IVIg can undergo a steroid wean, and then be evaluated for prophylactic therapy, said Dr. Yeh.
The evidence for IVIg is limited, reflecting the difficulty of studying treatments in rare populations. Still, when TPE is not available and the patient is quite impaired, IVIg makes sense to try. “Absence of evidence does not mean that the therapy doesn’t work, and I don’t think we should throw out the baby with the bath water,” said Dr. Yeh.
Although IVIg treatment is generally well tolerated, there have been a few serious adverse events, such as anaphylactic shock and aseptic meningitis, according to Andrea Savransky, MD, a pediatrician at National Pediatric Hospital in Buenos Aires, who also spoke at the session. “I think it is important to weigh the benefits against the risk,” Dr. Savransky said. She noted that TPE should not be taken lightly. One study showed more complications in pediatric patients than in adult patients, and it must be performed in specialized centers.
Emerging treaments
Tanuja Chitnis, MD, director of the Partners Pediatric MS Center at Massachusetts General Hospital, Boston, discussed some of the emerging treatments for pediatric NMOSD. Rituximab has been associated with success in some retrospective studies, but dosing should be personalized. Dr. Chitnis reported that B cells can return before 6 months, so she monitors B cells beginning 2 months after induction, redosing after 4 or 5 months rather than 6 if B cells return.
Nevertheless, relapses can still occur after rituximab therapy. “There is room for additional therapies to address this gap,” said Dr. Chitnis. Three new antibodies have received approval for treatment of NMOSD in adults. These include the complement inhibitor eculizumab, the IL-6 receptor antibody satralizumab, and the anti-CD19 antibody inebilizumab. Phase 3 clinical trials in children have been conducted for eculizumab and are in the planning stage for inebilizumab, and pediatric patients were included in pivotal trials for satralizumab.
Eculizumab treatment resulted in a 94.2% reduction in relapse risk in AQP4-positive adults. Satralizumab showed a 79% reduction in relapse risk among AQP-4 positive subjects with NMOSD or neuromyelitis optica and a 34% reduction in those who were AQP-4 negative. The pediatric subgroup had similar levels of response to adults, though the numbers were too small for a subgroup analysis.
In AQP-4 positive patients, inebilizumab treatment yielded a 77% reduction in relapse rate. In all patients, there was a 73% reduction.
For MOG antibody-positive patients with AQP-4 negative disease, novel therapies are at earlier stages of development. Typical MS therapies such as interferon beta and glatiramer acetate don’t seem to be effective. Some that have shown signs of efficacy include azathioprine, mycophenylate mofetil, rituximab, and IVIg infusion, but the state of the field is not encouraging. “This is an observation now being studied in larger cohorts, but in general I have not found that there’s a very strong response to any of these therapies, possibly with the exception of IVIg,” said Dr. Chitnis.
Dr. Tenembaum has no relevant financial disclosures. Dr. Armangue has received speaking honoraria from Novartis and travel expenses for scientific meetings from Merck, Biogen, and Roche. Dr. Yeh is on the scientific advisory board of Juno Therapeutics and has received research support from Biogen. Dr. Chitnis advises Biogen-Idec, Novartis, and Alexion, serves on clinical trial advisory boards for Novartis and Sanofi Aventis, and has received research support from Verily, EMD Serono, and Novartis. Dr. Savransky has received honoraria from Genzyme de Argentina SA.
, but have led to some uncertainty and confusion as well.
At the2020 CNS-ICNA Conjoint Meeting, held virtually this year, presenters discussed some of the challenges of differential diagnosis and treatment choice in pediatric NMOSD, which is easily confused with multiple sclerosis.
NMOSD used to be considered a monophasic disease restricted to the optic nerve and spinal cord, but is now known to affect other regions of the central nervous system and to relapse in some patients.
Diagnosis
The disease is often mediated by antibodies to the aquaporin-4 (AQP-4) water channel, but about 30% of adult patients lack the antibody, and AQP-4 seronegativity is more common in the pediatric population. Another common antibody found in 40%–50% of children with NMOSD targets myelin oligodendrocyte glycoprotein (MOG).
It is important to be aware that false negatives can occur in serology assays, and false positives are common, particularly in ELISA assays, Silvia N. Tenembaum, MD, said during her presentation. For those reasons, serology is not enough for a diagnosis. “Patients should also have compatible symptoms and MRI findings,” said Dr. Tenembaum, director of the pediatric neuroimmunology program at National Pediatric Hospital in Buenos Aires.
According to international consensus criteria, to be diagnosed with NMOSD, AQP-4 seropositive patients should also have at least one core clinical symptom: optic neuritis, acute myelitis, area postrema syndrome, other acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic clinical syndrome, or symptomatic cerebral syndrome. AQP-4 seronegative patients or with unknown status should have at least two core symptoms, one of which must be optic neuritis, acute myelitis, or area postrema syndrome. Both conventional MRI and advanced new techniques are important for achieving differential diagnosis.
The most common symptom in children is optic neuritis, which occurs in 50%-70% of patients. Cerebral syndromes with or without encephalopathy and large tumefactive white matter lesions are also common, according to Dr. Tenembaum.
There are many conditions that mimic the spinal cord and optic nerve symptoms of NMOSD, which must be ruled out. One example is optic myelopathy and vision loss from late-onset biotinylase deficiency. It is critical to rule that out because it is treatable with supplements. Optic neuropathy, papillitis, and papilledema can also resemble NMOSD.
It is critical to achieve an early diagnosis of NMOSD in children, because some MS drugs can worsen NMOSD, according to Thaís Armangue, MD, PhD, head of neuroimmunology at SJD Barcelona Children’s Hospital, who also presented at the session. She pointed out that the MOG antibody, while common in children, is also associated with many demyelinating diseases. Some 50%-60% of children with acute disseminated encephalomyelitis (ADEM) have high titers of MOG antibodies. Although early studies suggested that persistent anti-MOG antibodies were associated with risk of developing MS, more recent studies show it predicts a non-MS disease course, particularly at titers greater than 1:1280, according to Dr. Tenembaum. Persistent anti-MOG antibodies are also associated with relapsing disease, but it is associated with other syndromes besides NMOSD. “The probability is that [MOG antibodies are] useful, but they cannot guide chronic immunotherapy, because even monophasic patients can last maybe 12 months before they become MOG negative, and we cannot wait so many months” to determine treatment course, said Dr. Tenembaum.
For monophasic ADEM or NMOSD, there is no need for chronic treatment. But children with MS and recurrent NMOSD require early chronic immunotherapy because specific therapies have been shown to improve prognosis.
Acute treatment
When it comes to acute treatment of NMOSD, the goal is to suppress the inflammatory attack but also to minimize long-term damage and optimize long-term neurological function. “The potential for irreversible injury with an attack is very high, and cumulative disabilities in NMOSD can result directly from attacks,” E. Ann Yeh, MD, director of the Pediatric MS and Neuroinflammatory Disorders Program at the Hospital for Sick Children at the University of Toronto, said during her talk.
IV steroids are generally the first choice, with a preference for methylprednisolone. Pediatric patients that are MOG antibody positive usually respond better and more quickly than do adults, with rapid daily improvements in mobility, vomiting, and eyesight. Dr. Yeh recommends weaning good responders off steroids because AQP-4 positive patients are likely to relapse without a steroid wean, and antibody testing may be unavailable or results may be delayed. The wean can range from 4 weeks to 4-6 months, depending on antibody status, likelihood of AQP-4 positivity, and clinical parameters.
Inadequate responses are usually pretty evident. If there is only light perception by day 4 or 5, or paralyzed patients are nonambulatory and achieve only twitchy movements by that time, second-line therapies should be considered, including therapeutic plasma exchange (TPE) with 5-7 exchanges or intravenous immunoglobulins (IVIg).
Dr. Yeh called for quick treatment. Whatever you do, “please do it sooner rather than later if you think there’s no response [to steroids],” Dr. Yeh said.
TPE is the first choice, according to Dr. Yeh. “There seems to be a fair amount of information that suggests that if you’re having difficulty getting a response to steroids, TPE can make a difference in these patients,” she said. But in some cases TPE may not be available, and IVIg can be attempted first. If it achieves no or only marginal improvement, TPE can be attempted later, but it must be kept in mind that TPE conducted too soon could wash out IVIg. Patients who get much better on IVIg can undergo a steroid wean, and then be evaluated for prophylactic therapy, said Dr. Yeh.
The evidence for IVIg is limited, reflecting the difficulty of studying treatments in rare populations. Still, when TPE is not available and the patient is quite impaired, IVIg makes sense to try. “Absence of evidence does not mean that the therapy doesn’t work, and I don’t think we should throw out the baby with the bath water,” said Dr. Yeh.
Although IVIg treatment is generally well tolerated, there have been a few serious adverse events, such as anaphylactic shock and aseptic meningitis, according to Andrea Savransky, MD, a pediatrician at National Pediatric Hospital in Buenos Aires, who also spoke at the session. “I think it is important to weigh the benefits against the risk,” Dr. Savransky said. She noted that TPE should not be taken lightly. One study showed more complications in pediatric patients than in adult patients, and it must be performed in specialized centers.
Emerging treaments
Tanuja Chitnis, MD, director of the Partners Pediatric MS Center at Massachusetts General Hospital, Boston, discussed some of the emerging treatments for pediatric NMOSD. Rituximab has been associated with success in some retrospective studies, but dosing should be personalized. Dr. Chitnis reported that B cells can return before 6 months, so she monitors B cells beginning 2 months after induction, redosing after 4 or 5 months rather than 6 if B cells return.
Nevertheless, relapses can still occur after rituximab therapy. “There is room for additional therapies to address this gap,” said Dr. Chitnis. Three new antibodies have received approval for treatment of NMOSD in adults. These include the complement inhibitor eculizumab, the IL-6 receptor antibody satralizumab, and the anti-CD19 antibody inebilizumab. Phase 3 clinical trials in children have been conducted for eculizumab and are in the planning stage for inebilizumab, and pediatric patients were included in pivotal trials for satralizumab.
Eculizumab treatment resulted in a 94.2% reduction in relapse risk in AQP4-positive adults. Satralizumab showed a 79% reduction in relapse risk among AQP-4 positive subjects with NMOSD or neuromyelitis optica and a 34% reduction in those who were AQP-4 negative. The pediatric subgroup had similar levels of response to adults, though the numbers were too small for a subgroup analysis.
In AQP-4 positive patients, inebilizumab treatment yielded a 77% reduction in relapse rate. In all patients, there was a 73% reduction.
For MOG antibody-positive patients with AQP-4 negative disease, novel therapies are at earlier stages of development. Typical MS therapies such as interferon beta and glatiramer acetate don’t seem to be effective. Some that have shown signs of efficacy include azathioprine, mycophenylate mofetil, rituximab, and IVIg infusion, but the state of the field is not encouraging. “This is an observation now being studied in larger cohorts, but in general I have not found that there’s a very strong response to any of these therapies, possibly with the exception of IVIg,” said Dr. Chitnis.
Dr. Tenembaum has no relevant financial disclosures. Dr. Armangue has received speaking honoraria from Novartis and travel expenses for scientific meetings from Merck, Biogen, and Roche. Dr. Yeh is on the scientific advisory board of Juno Therapeutics and has received research support from Biogen. Dr. Chitnis advises Biogen-Idec, Novartis, and Alexion, serves on clinical trial advisory boards for Novartis and Sanofi Aventis, and has received research support from Verily, EMD Serono, and Novartis. Dr. Savransky has received honoraria from Genzyme de Argentina SA.
FROM CNS-ICNA 2020
Systemic sclerosis patients share their perspectives and needs in treatment trials
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.

The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.

Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.
The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.
Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.
The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.
Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
FROM AN FDA PATIENT-FOCUSED DRUG DEVELOPMENT MEETING
Burosumab is a ‘game changer,’ effective in all subgroups of XLH
A recently approved agent, burosumab (Crysvita), was better than placebo across a range of efficacy outcomes for 14 predefined subgroups of adults with X-linked hypophosphatemia (XLH), new research shows.
The authors analyzed data from the initial 24-week randomized blinded phase of the pivotal phase 3 trial that led to regulatory approval of this drug in the United States in 2018 for XLH, a rare form of rickets characterized by low serum phosphorus levels, skeletal defects, pain, and stiffness.
As in the main analysis, in the subgroups, among patients who received burosumab, serum phosphorus levels were improved, and outcomes were better on the following measures: Western Ontario and McMaster Universities Arthritis Index (WOMAC) stiffness scale, the WOMAC physical function measure, and the Brief Pain Inventory (BPI), which were the main efficacy outcomes. Improvements were seen for many other outcomes as well.
Maria-Luisa Brandi, MD, Careggi University Hospital, Florence, Italy, presented the new subanalysis during the virtual American Society of Bone and Mineral Research (ASBMR) 2020 annual meeting.
The subgroup results were consistent with the overall trial findings, “showing a favorable direction of effect of burosumab relative to placebo” except for results in patients recruited in Asia and non-White patients; those results were considered inconclusive because there were too few participants in those categories, she told Medscape Medical News,.
Lorenz Hofbauer, MD, scientific chair of the ASBMR meeting, said that the take-away message is that the drug “works to reduce pain and disability” in adults with XLH with more severe/less severe symptoms, and “it provides new hope for many patients suffering from this disease,” he told Medscape Medical News.
Burosemab also appears superior to what has previously been considered standard therapy for XLH, phosphate/calcitriol, the experts say.
‘Rare is relative,’ burosumab is a ‘transformative therapy’
“The disease prevalence is 1 to 9 in a million,” Brandi said. “Undiagnosed adults are treated by the doctor that makes the diagnosis, usually a nephrologist or a rheumatologist or a bone doctor; this depends on the prevalent complications in a given patient. The endocrinologist who treats this patient is the one expert in bone disorders.”
Hofbauer noted, however, that “[r]are is relative. If you run a bone clinic, you will see four to five patients with XLH; if you are a regional center, 20 to 30 patients. People with rare disease travel more than 1000 miles to see experts.”
The US Food and Drug Administration approved burosumab for use in children and adults with XLH 2 years ago. The European Medicines Agency (EMA) approved it for use in children.
The drug is expected to be approved by the EMA for adults with XLH some time this year, said Hofbauer, who is from Dresden Technical University, Dresden, Germany.
Burosumab is a “game changer” with respect to previous treatments, he stressed.
This study is one of the top five clinical abstracts of the ASBMR meeting, which are selected on the basis of “scientific content/novelty, making a difference in clinical practice,” Hofbauer explained. He noted that “new drugs that work are always in the top ranks.”
Craig Munns, PhD, who was senior author of a recent review about burosumab, agrees.
“Burosumab is transformative, as it is a paradigm shift in the way we manage XLH,” he told Medscape Medical News.
“Standard therapy for children is with oral phosphate and calcitriol, and many adults do not receive any therapy,” said Munns, from the University of Sydney, Sydney, Australia.
“Phosphate and calcitriol need to be taken multiple times per day, is an incomplete therapy, and has many complications. Burosumab offers a 2-weekly (children) or 4-weekly (adult) dosing regime with superior outcomes compared to no treatment or phosphate/calcitriol,” he emphasized.
Efficacy in 14 predefined subgroups
“Burosumab is an anti-FGF-23 [anti–fibroblast growth factor-23] antibody for a rare genetic disease, XLH, in which the gene for PHEX is defective,” Hofbauer explained.
“PHEX is an enzyme that clears FGF-23; if it does not work, then FGF-23 accumulates in the body and causes phosphate wasting with wide consequences for bone, muscle, and joints. Burosumab is a smart approach, since it blocks these excessive FGF-23 effects.”
Children with XLH have rickets, deformities in the lower skeleton, and short stature, Brandi noted, whereas adults have fractures, pseudofractures, enthesopathy (calcification of joint capsule, tendon insertions, and ligaments), pain, stiffness, and impaired physical function.
However, “treatment with oral phosphate and vitamin D is associated with nephrocalcinosis and hyperparathyroidism,” she said.
In the phase 3 trial, 134 adults (aged 18 to 65 years) with XLH were randomly assigned in a double-blind manner to receive either burosumab or placebo for 24 weeks, followed by 24 weeks of open-label burosumab. The patients’ serum phosphorus levels were <2.5 mg/dL, and they were experiencing measurable bone/joint pain.
Baseline characteristics were similar for the patients who received placebo (66) and those who received burosumab (68). The mean age of the patients was 40 years; 65% were women; and 81% were White.
The current exploratory analysis examined efficacy outcomes in patients grouped according to the following factors and characteristics: sex; age (≤41 years or >41 years); race (non-White, White); region (Asia, North America/Europe); baseline WOMAC pain score; WOMAC total pain; WOMAC stiffness; WOMAC physical function; BPI worst pain; BPI average pain; opioid use; pain medication use; active fractures and pseudofractures; and 6-minute walking test distance.
The efficacy outcomes were as follows: serum phosphorus level (primary outcome), BPI worst pain, WOMAC stiffness, and WOMAC physical function (key secondary outcomes); and WOMAC pain, WOMAC total score, BPI average pain, BPI pain interference, BPI worst fatigue, BPI global score, patient global impression (PGI), and 6-minute walking distance.
In the overall cohort, at 24 weeks, in comparison with patients who received placebo, patients who received burosumab had favorable responses with respect to serum phosphorus level, WOMAC stiffness (P =. 012),WOMAC physical function (P = .048), and BPI worst pain (P = .092, not significant), as well as significant improvements in WOMAC total score and the 6-minute walk test. There were nonsignificant improvements in WOMAC pain and BPI average pain.
In the subgroup analysis, burosumab was superior to placebo for the primary outcome (serum phosphorus) in all subgroups. It was also superior to placebo for the key secondary outcomes (worst pain, stiffness, and physical function) across all subgroups except for patients from Asia (18 patients) and non-White patients (26).
The study was funded by Kyowa Kirin in partnership with Ultragenyx. Brandi receives consultancy and speaker fees as well as research grants from Kyowa Kirin and other pharmaceutical companies. Munns has received research funding from Kyowa Kirin.
This article first appeared on Medscape.com.
A recently approved agent, burosumab (Crysvita), was better than placebo across a range of efficacy outcomes for 14 predefined subgroups of adults with X-linked hypophosphatemia (XLH), new research shows.
The authors analyzed data from the initial 24-week randomized blinded phase of the pivotal phase 3 trial that led to regulatory approval of this drug in the United States in 2018 for XLH, a rare form of rickets characterized by low serum phosphorus levels, skeletal defects, pain, and stiffness.
As in the main analysis, in the subgroups, among patients who received burosumab, serum phosphorus levels were improved, and outcomes were better on the following measures: Western Ontario and McMaster Universities Arthritis Index (WOMAC) stiffness scale, the WOMAC physical function measure, and the Brief Pain Inventory (BPI), which were the main efficacy outcomes. Improvements were seen for many other outcomes as well.
Maria-Luisa Brandi, MD, Careggi University Hospital, Florence, Italy, presented the new subanalysis during the virtual American Society of Bone and Mineral Research (ASBMR) 2020 annual meeting.
The subgroup results were consistent with the overall trial findings, “showing a favorable direction of effect of burosumab relative to placebo” except for results in patients recruited in Asia and non-White patients; those results were considered inconclusive because there were too few participants in those categories, she told Medscape Medical News,.
Lorenz Hofbauer, MD, scientific chair of the ASBMR meeting, said that the take-away message is that the drug “works to reduce pain and disability” in adults with XLH with more severe/less severe symptoms, and “it provides new hope for many patients suffering from this disease,” he told Medscape Medical News.
Burosemab also appears superior to what has previously been considered standard therapy for XLH, phosphate/calcitriol, the experts say.
‘Rare is relative,’ burosumab is a ‘transformative therapy’
“The disease prevalence is 1 to 9 in a million,” Brandi said. “Undiagnosed adults are treated by the doctor that makes the diagnosis, usually a nephrologist or a rheumatologist or a bone doctor; this depends on the prevalent complications in a given patient. The endocrinologist who treats this patient is the one expert in bone disorders.”
Hofbauer noted, however, that “[r]are is relative. If you run a bone clinic, you will see four to five patients with XLH; if you are a regional center, 20 to 30 patients. People with rare disease travel more than 1000 miles to see experts.”
The US Food and Drug Administration approved burosumab for use in children and adults with XLH 2 years ago. The European Medicines Agency (EMA) approved it for use in children.
The drug is expected to be approved by the EMA for adults with XLH some time this year, said Hofbauer, who is from Dresden Technical University, Dresden, Germany.
Burosumab is a “game changer” with respect to previous treatments, he stressed.
This study is one of the top five clinical abstracts of the ASBMR meeting, which are selected on the basis of “scientific content/novelty, making a difference in clinical practice,” Hofbauer explained. He noted that “new drugs that work are always in the top ranks.”
Craig Munns, PhD, who was senior author of a recent review about burosumab, agrees.
“Burosumab is transformative, as it is a paradigm shift in the way we manage XLH,” he told Medscape Medical News.
“Standard therapy for children is with oral phosphate and calcitriol, and many adults do not receive any therapy,” said Munns, from the University of Sydney, Sydney, Australia.
“Phosphate and calcitriol need to be taken multiple times per day, is an incomplete therapy, and has many complications. Burosumab offers a 2-weekly (children) or 4-weekly (adult) dosing regime with superior outcomes compared to no treatment or phosphate/calcitriol,” he emphasized.
Efficacy in 14 predefined subgroups
“Burosumab is an anti-FGF-23 [anti–fibroblast growth factor-23] antibody for a rare genetic disease, XLH, in which the gene for PHEX is defective,” Hofbauer explained.
“PHEX is an enzyme that clears FGF-23; if it does not work, then FGF-23 accumulates in the body and causes phosphate wasting with wide consequences for bone, muscle, and joints. Burosumab is a smart approach, since it blocks these excessive FGF-23 effects.”
Children with XLH have rickets, deformities in the lower skeleton, and short stature, Brandi noted, whereas adults have fractures, pseudofractures, enthesopathy (calcification of joint capsule, tendon insertions, and ligaments), pain, stiffness, and impaired physical function.
However, “treatment with oral phosphate and vitamin D is associated with nephrocalcinosis and hyperparathyroidism,” she said.
In the phase 3 trial, 134 adults (aged 18 to 65 years) with XLH were randomly assigned in a double-blind manner to receive either burosumab or placebo for 24 weeks, followed by 24 weeks of open-label burosumab. The patients’ serum phosphorus levels were <2.5 mg/dL, and they were experiencing measurable bone/joint pain.
Baseline characteristics were similar for the patients who received placebo (66) and those who received burosumab (68). The mean age of the patients was 40 years; 65% were women; and 81% were White.
The current exploratory analysis examined efficacy outcomes in patients grouped according to the following factors and characteristics: sex; age (≤41 years or >41 years); race (non-White, White); region (Asia, North America/Europe); baseline WOMAC pain score; WOMAC total pain; WOMAC stiffness; WOMAC physical function; BPI worst pain; BPI average pain; opioid use; pain medication use; active fractures and pseudofractures; and 6-minute walking test distance.
The efficacy outcomes were as follows: serum phosphorus level (primary outcome), BPI worst pain, WOMAC stiffness, and WOMAC physical function (key secondary outcomes); and WOMAC pain, WOMAC total score, BPI average pain, BPI pain interference, BPI worst fatigue, BPI global score, patient global impression (PGI), and 6-minute walking distance.
In the overall cohort, at 24 weeks, in comparison with patients who received placebo, patients who received burosumab had favorable responses with respect to serum phosphorus level, WOMAC stiffness (P =. 012),WOMAC physical function (P = .048), and BPI worst pain (P = .092, not significant), as well as significant improvements in WOMAC total score and the 6-minute walk test. There were nonsignificant improvements in WOMAC pain and BPI average pain.
In the subgroup analysis, burosumab was superior to placebo for the primary outcome (serum phosphorus) in all subgroups. It was also superior to placebo for the key secondary outcomes (worst pain, stiffness, and physical function) across all subgroups except for patients from Asia (18 patients) and non-White patients (26).
The study was funded by Kyowa Kirin in partnership with Ultragenyx. Brandi receives consultancy and speaker fees as well as research grants from Kyowa Kirin and other pharmaceutical companies. Munns has received research funding from Kyowa Kirin.
This article first appeared on Medscape.com.
A recently approved agent, burosumab (Crysvita), was better than placebo across a range of efficacy outcomes for 14 predefined subgroups of adults with X-linked hypophosphatemia (XLH), new research shows.
The authors analyzed data from the initial 24-week randomized blinded phase of the pivotal phase 3 trial that led to regulatory approval of this drug in the United States in 2018 for XLH, a rare form of rickets characterized by low serum phosphorus levels, skeletal defects, pain, and stiffness.
As in the main analysis, in the subgroups, among patients who received burosumab, serum phosphorus levels were improved, and outcomes were better on the following measures: Western Ontario and McMaster Universities Arthritis Index (WOMAC) stiffness scale, the WOMAC physical function measure, and the Brief Pain Inventory (BPI), which were the main efficacy outcomes. Improvements were seen for many other outcomes as well.
Maria-Luisa Brandi, MD, Careggi University Hospital, Florence, Italy, presented the new subanalysis during the virtual American Society of Bone and Mineral Research (ASBMR) 2020 annual meeting.
The subgroup results were consistent with the overall trial findings, “showing a favorable direction of effect of burosumab relative to placebo” except for results in patients recruited in Asia and non-White patients; those results were considered inconclusive because there were too few participants in those categories, she told Medscape Medical News,.
Lorenz Hofbauer, MD, scientific chair of the ASBMR meeting, said that the take-away message is that the drug “works to reduce pain and disability” in adults with XLH with more severe/less severe symptoms, and “it provides new hope for many patients suffering from this disease,” he told Medscape Medical News.
Burosemab also appears superior to what has previously been considered standard therapy for XLH, phosphate/calcitriol, the experts say.
‘Rare is relative,’ burosumab is a ‘transformative therapy’
“The disease prevalence is 1 to 9 in a million,” Brandi said. “Undiagnosed adults are treated by the doctor that makes the diagnosis, usually a nephrologist or a rheumatologist or a bone doctor; this depends on the prevalent complications in a given patient. The endocrinologist who treats this patient is the one expert in bone disorders.”
Hofbauer noted, however, that “[r]are is relative. If you run a bone clinic, you will see four to five patients with XLH; if you are a regional center, 20 to 30 patients. People with rare disease travel more than 1000 miles to see experts.”
The US Food and Drug Administration approved burosumab for use in children and adults with XLH 2 years ago. The European Medicines Agency (EMA) approved it for use in children.
The drug is expected to be approved by the EMA for adults with XLH some time this year, said Hofbauer, who is from Dresden Technical University, Dresden, Germany.
Burosumab is a “game changer” with respect to previous treatments, he stressed.
This study is one of the top five clinical abstracts of the ASBMR meeting, which are selected on the basis of “scientific content/novelty, making a difference in clinical practice,” Hofbauer explained. He noted that “new drugs that work are always in the top ranks.”
Craig Munns, PhD, who was senior author of a recent review about burosumab, agrees.
“Burosumab is transformative, as it is a paradigm shift in the way we manage XLH,” he told Medscape Medical News.
“Standard therapy for children is with oral phosphate and calcitriol, and many adults do not receive any therapy,” said Munns, from the University of Sydney, Sydney, Australia.
“Phosphate and calcitriol need to be taken multiple times per day, is an incomplete therapy, and has many complications. Burosumab offers a 2-weekly (children) or 4-weekly (adult) dosing regime with superior outcomes compared to no treatment or phosphate/calcitriol,” he emphasized.
Efficacy in 14 predefined subgroups
“Burosumab is an anti-FGF-23 [anti–fibroblast growth factor-23] antibody for a rare genetic disease, XLH, in which the gene for PHEX is defective,” Hofbauer explained.
“PHEX is an enzyme that clears FGF-23; if it does not work, then FGF-23 accumulates in the body and causes phosphate wasting with wide consequences for bone, muscle, and joints. Burosumab is a smart approach, since it blocks these excessive FGF-23 effects.”
Children with XLH have rickets, deformities in the lower skeleton, and short stature, Brandi noted, whereas adults have fractures, pseudofractures, enthesopathy (calcification of joint capsule, tendon insertions, and ligaments), pain, stiffness, and impaired physical function.
However, “treatment with oral phosphate and vitamin D is associated with nephrocalcinosis and hyperparathyroidism,” she said.
In the phase 3 trial, 134 adults (aged 18 to 65 years) with XLH were randomly assigned in a double-blind manner to receive either burosumab or placebo for 24 weeks, followed by 24 weeks of open-label burosumab. The patients’ serum phosphorus levels were <2.5 mg/dL, and they were experiencing measurable bone/joint pain.
Baseline characteristics were similar for the patients who received placebo (66) and those who received burosumab (68). The mean age of the patients was 40 years; 65% were women; and 81% were White.
The current exploratory analysis examined efficacy outcomes in patients grouped according to the following factors and characteristics: sex; age (≤41 years or >41 years); race (non-White, White); region (Asia, North America/Europe); baseline WOMAC pain score; WOMAC total pain; WOMAC stiffness; WOMAC physical function; BPI worst pain; BPI average pain; opioid use; pain medication use; active fractures and pseudofractures; and 6-minute walking test distance.
The efficacy outcomes were as follows: serum phosphorus level (primary outcome), BPI worst pain, WOMAC stiffness, and WOMAC physical function (key secondary outcomes); and WOMAC pain, WOMAC total score, BPI average pain, BPI pain interference, BPI worst fatigue, BPI global score, patient global impression (PGI), and 6-minute walking distance.
In the overall cohort, at 24 weeks, in comparison with patients who received placebo, patients who received burosumab had favorable responses with respect to serum phosphorus level, WOMAC stiffness (P =. 012),WOMAC physical function (P = .048), and BPI worst pain (P = .092, not significant), as well as significant improvements in WOMAC total score and the 6-minute walk test. There were nonsignificant improvements in WOMAC pain and BPI average pain.
In the subgroup analysis, burosumab was superior to placebo for the primary outcome (serum phosphorus) in all subgroups. It was also superior to placebo for the key secondary outcomes (worst pain, stiffness, and physical function) across all subgroups except for patients from Asia (18 patients) and non-White patients (26).
The study was funded by Kyowa Kirin in partnership with Ultragenyx. Brandi receives consultancy and speaker fees as well as research grants from Kyowa Kirin and other pharmaceutical companies. Munns has received research funding from Kyowa Kirin.
This article first appeared on Medscape.com.
Hidradenitis Suppurativa in the Military
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Practice Points
- Hidradenitis suppurativa (HS) can be more difficult to treat in physically active military servicemembers (SMs).
- Patient education and primary care physician awareness of HS is critical to initial diagnosis and long-term management.
- Primary care physician knowledge of HS as well as an understanding of the capabilities at local military medical facilities is important for optimal treatment of HS in military SMs.

CDER chief reflects on advances in rare diseases
, from helping to usher the approval of the first treatments for cystic fibrosis and multiple sclerosis during her tenure as director of the Office of Therapeutics Research and Review, to introducing the concept of risk management in the agency’s analysis of drug safety during her role as acting director of the Center for Drug Evaluation and Research (CDER).
During an online event on Oct. 9, Dr. Woodcock, who became CDER’s director in 2008, will receive a lifetime achievement award from the National Organization for Rare Disorders*. In this interview, she reflects on the CDER’s accomplishments in the field of rare diseases, from which she draws inspiration, and what it’s like to be overseeing the therapeutics component of Operation Warp Speed amid the COVID-19 pandemic.

Q: What does this lifetime achievement award from the National Organization for Rare Disorders mean to you at this stage in your career?
Dr. Woodcock: According to NORD, there are more than 7,000 rare diseases that affect an estimated 25 million Americans. More than half of those affected are children. Many of these diseases are very serious, so there is a great deal of suffering that goes on, sometimes for a lifetime. I’ve always felt that people suffering like this don’t really have a voice. I’ve always tried to push the regulatory science, the science behind evaluation, and all of the efforts we can make to help those who are trying to develop products for people suffering from these rare diseases. The science is really picking up. We’re seeing more drug approvals every year for rare disorders. Hopefully, the lives of people with rare disorders will improve and we will continue to see a trajectory of better outcomes for people.
Q: Who inspired you most early in your career as a physician? What was it about that person (or persons) that made a difference to you?
Dr. Woodcock: During my training I had the privilege to be exposed to a wide range of stellar diagnosticians and people who were good clinicians who cared about their patients. That experience modeled for me what I would like to be as a doctor.
Q: In 2017, the National Consumers League described you as “a passionate advocate for American patients and consumers, an ally to patient advocacy groups, and a fearless leader at the FDA.” In your own words, how do you describe your leadership style?
Dr. Woodcock: People always call me fearless, but I feel like I just state the facts. I care about getting technical input from everyone, but I’m not terribly concerned about people’s disapproval of my actions. I’m a leader who tries to do the right thing, the thing that will benefit patients. I try to keep them at the center of what we’re doing, who we’re regulating for. We work for the American public. As far as CDER, it’s the people who take medicine, people who administer medicine, and people who need treatments.
Q: Since joining CDER as director in 2008, what are some accomplishments you are most proud of as it relates to treatments for patients with rare diseases?
Dr. Woodcock: I undertook a transformation and modernization of the New Drugs Regulatory Program, which created offices that align interrelated disease areas, and divisions with clearer and more focused areas of expertise. These changes will bring efficiency and effectiveness. We also set up an Office of Translational Sciences. All of these actions are important. In developing drugs for rare disorders, we need more flexibility. We have a lot of critics who say, “Rare disease trials are too small.” If you look at a cardiovascular trial of 25,000 people, for example, the investigators might only have .1% of the affected population enrolled. On the other hand, a rare disease trial of 100 people might represent half of the entire population with that disease. We often get criticism because it’s more difficult to define endpoints. The diseases aren’t that well understood, and you’re going to have smaller trials because there aren’t that many people with the disease. We need to figure out how to appropriately exercise that flexibility in regulation and make sure people have access, but have a high probability of getting products that work and have been adequately tested for safely. We also started a Rare Disease Cures Accelerator, which is enrolling people online in natural history studies to see what happens to them so we can better plan studies. We have Patient-Focused Drug Development meetings as a way to gather patients’ perspectives on their conditions and available therapies to treat those conditions. That is eye-opening, because what the doctor thinks about the disease may not be what the patient thinks about the disease. The patients are the ones taking the medicine, so we need to collect their opinions. Such approaches make it easier to study rare diseases and get new treatments.
Q: How do the challenges of drug research and development in the field of rare diseases differ from those associated with more prevalent diseases?
Dr. Woodcock: There is one advantage today for people with rare diseases. That is, when there is a known genetic mutation causing a disease, RNA interference and other gene therapy approaches can be used. There are challenges, though. Patients with rare disorders often don’t have a uniform disease course. They often have a multisystem impact, so they might have things wrong with their GI tract and/or skin, so it’s difficult to know what to measure. We’re trying to remedy this by gathering better natural history information on what happens to people. That is empowering for patients as well.
Q: In what practical ways can physicians become advocates for patients and their families who are navigating life with a rare disease?
Dr. Woodcock: I advise people to get involved in the association or advocacy group for their rare disease. It’s empowering. They can share stories and information with others who have been suffering from the disease. Also, they would get information about what trials might be available. As for physicians themselves, they have a bewildering variety of jobs they’re supposed to do, so it’s hard to be good in any one of them. People with rare disease often suffer terribly because they don’t get diagnosed for 10 years even though they have classic symptoms of a particular disorder. If physicians have never seen it or never heard of it, they may not know how to treat it. It’s a huge problem.
Q: Who inspires you most in your work today?
Dr. Woodcock: The dedication of the staff at the FDA is unbelievable. When you look at responses to the Federal Employee Viewpoint Survey administered by the Office of Personnel Management, FDA workers consistently express a strong sense of mission and dedication. It’s out of the park, really. They have worked night and day during this pandemic. I’m inspired by everyone who works at the FDA and their incredible dedication to their work.
Q: In what ways do you cope with the pressure that comes with your line of work? Do you have a favorite hobby or that activity that helps keep you grounded?
Dr. Woodcock: I’m an avid gardener, so I have a garden with vegetables, fruits, and flowers, including a large orchid collection. I’m also a hiker and a physical fitness buff, so I feel like there isn’t enough time in the day for all of my hobbies. Formal hiking trails near me are very crowded now, so I’ve been hiking around my neighborhood, taking long walks and going up and down hills quickly. Last November, I went hiking in New Zealand with my daughter. We hiked the Milford Track, which is about 33 miles long. It goes from an inland lake, over a mountain pass, and to the Pacific Ocean. It was fun, with unbelievable scenery.
Q: What novel treatment developments in rare disorders are you most excited about in the next 5 years?
Dr. Woodcock: I think gene therapy will come into its own. I think that could be a game-changer for people with genetic mutations causing rare diseases, and even cancer. We’ll see. It takes the technology a long time to mature. There are also gene-directed therapies such as RNA inhibition. We’ve already approved a couple of products like that for rare diseases, including treatments for the cardiomyopathy and neuropathy associated with ATTR amyloidosis. As our knowledge of biology continues to grow, I think more of these diseases will be amenable to interventions.
Q: In May of 2020 you were asked to temporarily step aside from your post as director of CDER to work on Operation Warp Speed. Please describe what your role is in this effort to accelerate COVID-19 treatments.
Dr. Woodcock: I’m the lead on therapeutics. Operation Warp Speed is mainly focused on developing vaccines for COVID-19. In the meantime, people who don’t respond to vaccines are going to need therapeutics, such as the elderly, or those who refuse to take vaccines, or those who are immunosuppressed and can’t mount a response to a vaccine. If we can develop those therapeutics now, that would be good to get that populous vaccinated. The team identified what we thought were the five highest priority agents to work on, and we’re testing them. We have identified many more in a priority list. We have five master protocols running for different times in the disease, such as when you’re an outpatient, when you’re an inpatient, or when you’re in the ICU. The work is stressful, because we need these treatments as soon as possible, but we have a great team working on this. I feel like I’m making a contribution in this role, because I know people in industry and in the National Institutes of Health. I try to bring everyone together and get things done.
*Correction, 10/22/20: An earlier version of this article misstated the name of the National Organization for Rare Disorders.
, from helping to usher the approval of the first treatments for cystic fibrosis and multiple sclerosis during her tenure as director of the Office of Therapeutics Research and Review, to introducing the concept of risk management in the agency’s analysis of drug safety during her role as acting director of the Center for Drug Evaluation and Research (CDER).
During an online event on Oct. 9, Dr. Woodcock, who became CDER’s director in 2008, will receive a lifetime achievement award from the National Organization for Rare Disorders*. In this interview, she reflects on the CDER’s accomplishments in the field of rare diseases, from which she draws inspiration, and what it’s like to be overseeing the therapeutics component of Operation Warp Speed amid the COVID-19 pandemic.
Q: What does this lifetime achievement award from the National Organization for Rare Disorders mean to you at this stage in your career?
Dr. Woodcock: According to NORD, there are more than 7,000 rare diseases that affect an estimated 25 million Americans. More than half of those affected are children. Many of these diseases are very serious, so there is a great deal of suffering that goes on, sometimes for a lifetime. I’ve always felt that people suffering like this don’t really have a voice. I’ve always tried to push the regulatory science, the science behind evaluation, and all of the efforts we can make to help those who are trying to develop products for people suffering from these rare diseases. The science is really picking up. We’re seeing more drug approvals every year for rare disorders. Hopefully, the lives of people with rare disorders will improve and we will continue to see a trajectory of better outcomes for people.
Q: Who inspired you most early in your career as a physician? What was it about that person (or persons) that made a difference to you?
Dr. Woodcock: During my training I had the privilege to be exposed to a wide range of stellar diagnosticians and people who were good clinicians who cared about their patients. That experience modeled for me what I would like to be as a doctor.
Q: In 2017, the National Consumers League described you as “a passionate advocate for American patients and consumers, an ally to patient advocacy groups, and a fearless leader at the FDA.” In your own words, how do you describe your leadership style?
Dr. Woodcock: People always call me fearless, but I feel like I just state the facts. I care about getting technical input from everyone, but I’m not terribly concerned about people’s disapproval of my actions. I’m a leader who tries to do the right thing, the thing that will benefit patients. I try to keep them at the center of what we’re doing, who we’re regulating for. We work for the American public. As far as CDER, it’s the people who take medicine, people who administer medicine, and people who need treatments.
Q: Since joining CDER as director in 2008, what are some accomplishments you are most proud of as it relates to treatments for patients with rare diseases?
Dr. Woodcock: I undertook a transformation and modernization of the New Drugs Regulatory Program, which created offices that align interrelated disease areas, and divisions with clearer and more focused areas of expertise. These changes will bring efficiency and effectiveness. We also set up an Office of Translational Sciences. All of these actions are important. In developing drugs for rare disorders, we need more flexibility. We have a lot of critics who say, “Rare disease trials are too small.” If you look at a cardiovascular trial of 25,000 people, for example, the investigators might only have .1% of the affected population enrolled. On the other hand, a rare disease trial of 100 people might represent half of the entire population with that disease. We often get criticism because it’s more difficult to define endpoints. The diseases aren’t that well understood, and you’re going to have smaller trials because there aren’t that many people with the disease. We need to figure out how to appropriately exercise that flexibility in regulation and make sure people have access, but have a high probability of getting products that work and have been adequately tested for safely. We also started a Rare Disease Cures Accelerator, which is enrolling people online in natural history studies to see what happens to them so we can better plan studies. We have Patient-Focused Drug Development meetings as a way to gather patients’ perspectives on their conditions and available therapies to treat those conditions. That is eye-opening, because what the doctor thinks about the disease may not be what the patient thinks about the disease. The patients are the ones taking the medicine, so we need to collect their opinions. Such approaches make it easier to study rare diseases and get new treatments.
Q: How do the challenges of drug research and development in the field of rare diseases differ from those associated with more prevalent diseases?
Dr. Woodcock: There is one advantage today for people with rare diseases. That is, when there is a known genetic mutation causing a disease, RNA interference and other gene therapy approaches can be used. There are challenges, though. Patients with rare disorders often don’t have a uniform disease course. They often have a multisystem impact, so they might have things wrong with their GI tract and/or skin, so it’s difficult to know what to measure. We’re trying to remedy this by gathering better natural history information on what happens to people. That is empowering for patients as well.
Q: In what practical ways can physicians become advocates for patients and their families who are navigating life with a rare disease?
Dr. Woodcock: I advise people to get involved in the association or advocacy group for their rare disease. It’s empowering. They can share stories and information with others who have been suffering from the disease. Also, they would get information about what trials might be available. As for physicians themselves, they have a bewildering variety of jobs they’re supposed to do, so it’s hard to be good in any one of them. People with rare disease often suffer terribly because they don’t get diagnosed for 10 years even though they have classic symptoms of a particular disorder. If physicians have never seen it or never heard of it, they may not know how to treat it. It’s a huge problem.
Q: Who inspires you most in your work today?
Dr. Woodcock: The dedication of the staff at the FDA is unbelievable. When you look at responses to the Federal Employee Viewpoint Survey administered by the Office of Personnel Management, FDA workers consistently express a strong sense of mission and dedication. It’s out of the park, really. They have worked night and day during this pandemic. I’m inspired by everyone who works at the FDA and their incredible dedication to their work.
Q: In what ways do you cope with the pressure that comes with your line of work? Do you have a favorite hobby or that activity that helps keep you grounded?
Dr. Woodcock: I’m an avid gardener, so I have a garden with vegetables, fruits, and flowers, including a large orchid collection. I’m also a hiker and a physical fitness buff, so I feel like there isn’t enough time in the day for all of my hobbies. Formal hiking trails near me are very crowded now, so I’ve been hiking around my neighborhood, taking long walks and going up and down hills quickly. Last November, I went hiking in New Zealand with my daughter. We hiked the Milford Track, which is about 33 miles long. It goes from an inland lake, over a mountain pass, and to the Pacific Ocean. It was fun, with unbelievable scenery.
Q: What novel treatment developments in rare disorders are you most excited about in the next 5 years?
Dr. Woodcock: I think gene therapy will come into its own. I think that could be a game-changer for people with genetic mutations causing rare diseases, and even cancer. We’ll see. It takes the technology a long time to mature. There are also gene-directed therapies such as RNA inhibition. We’ve already approved a couple of products like that for rare diseases, including treatments for the cardiomyopathy and neuropathy associated with ATTR amyloidosis. As our knowledge of biology continues to grow, I think more of these diseases will be amenable to interventions.
Q: In May of 2020 you were asked to temporarily step aside from your post as director of CDER to work on Operation Warp Speed. Please describe what your role is in this effort to accelerate COVID-19 treatments.
Dr. Woodcock: I’m the lead on therapeutics. Operation Warp Speed is mainly focused on developing vaccines for COVID-19. In the meantime, people who don’t respond to vaccines are going to need therapeutics, such as the elderly, or those who refuse to take vaccines, or those who are immunosuppressed and can’t mount a response to a vaccine. If we can develop those therapeutics now, that would be good to get that populous vaccinated. The team identified what we thought were the five highest priority agents to work on, and we’re testing them. We have identified many more in a priority list. We have five master protocols running for different times in the disease, such as when you’re an outpatient, when you’re an inpatient, or when you’re in the ICU. The work is stressful, because we need these treatments as soon as possible, but we have a great team working on this. I feel like I’m making a contribution in this role, because I know people in industry and in the National Institutes of Health. I try to bring everyone together and get things done.
*Correction, 10/22/20: An earlier version of this article misstated the name of the National Organization for Rare Disorders.
, from helping to usher the approval of the first treatments for cystic fibrosis and multiple sclerosis during her tenure as director of the Office of Therapeutics Research and Review, to introducing the concept of risk management in the agency’s analysis of drug safety during her role as acting director of the Center for Drug Evaluation and Research (CDER).
During an online event on Oct. 9, Dr. Woodcock, who became CDER’s director in 2008, will receive a lifetime achievement award from the National Organization for Rare Disorders*. In this interview, she reflects on the CDER’s accomplishments in the field of rare diseases, from which she draws inspiration, and what it’s like to be overseeing the therapeutics component of Operation Warp Speed amid the COVID-19 pandemic.
Q: What does this lifetime achievement award from the National Organization for Rare Disorders mean to you at this stage in your career?
Dr. Woodcock: According to NORD, there are more than 7,000 rare diseases that affect an estimated 25 million Americans. More than half of those affected are children. Many of these diseases are very serious, so there is a great deal of suffering that goes on, sometimes for a lifetime. I’ve always felt that people suffering like this don’t really have a voice. I’ve always tried to push the regulatory science, the science behind evaluation, and all of the efforts we can make to help those who are trying to develop products for people suffering from these rare diseases. The science is really picking up. We’re seeing more drug approvals every year for rare disorders. Hopefully, the lives of people with rare disorders will improve and we will continue to see a trajectory of better outcomes for people.
Q: Who inspired you most early in your career as a physician? What was it about that person (or persons) that made a difference to you?
Dr. Woodcock: During my training I had the privilege to be exposed to a wide range of stellar diagnosticians and people who were good clinicians who cared about their patients. That experience modeled for me what I would like to be as a doctor.
Q: In 2017, the National Consumers League described you as “a passionate advocate for American patients and consumers, an ally to patient advocacy groups, and a fearless leader at the FDA.” In your own words, how do you describe your leadership style?
Dr. Woodcock: People always call me fearless, but I feel like I just state the facts. I care about getting technical input from everyone, but I’m not terribly concerned about people’s disapproval of my actions. I’m a leader who tries to do the right thing, the thing that will benefit patients. I try to keep them at the center of what we’re doing, who we’re regulating for. We work for the American public. As far as CDER, it’s the people who take medicine, people who administer medicine, and people who need treatments.
Q: Since joining CDER as director in 2008, what are some accomplishments you are most proud of as it relates to treatments for patients with rare diseases?
Dr. Woodcock: I undertook a transformation and modernization of the New Drugs Regulatory Program, which created offices that align interrelated disease areas, and divisions with clearer and more focused areas of expertise. These changes will bring efficiency and effectiveness. We also set up an Office of Translational Sciences. All of these actions are important. In developing drugs for rare disorders, we need more flexibility. We have a lot of critics who say, “Rare disease trials are too small.” If you look at a cardiovascular trial of 25,000 people, for example, the investigators might only have .1% of the affected population enrolled. On the other hand, a rare disease trial of 100 people might represent half of the entire population with that disease. We often get criticism because it’s more difficult to define endpoints. The diseases aren’t that well understood, and you’re going to have smaller trials because there aren’t that many people with the disease. We need to figure out how to appropriately exercise that flexibility in regulation and make sure people have access, but have a high probability of getting products that work and have been adequately tested for safely. We also started a Rare Disease Cures Accelerator, which is enrolling people online in natural history studies to see what happens to them so we can better plan studies. We have Patient-Focused Drug Development meetings as a way to gather patients’ perspectives on their conditions and available therapies to treat those conditions. That is eye-opening, because what the doctor thinks about the disease may not be what the patient thinks about the disease. The patients are the ones taking the medicine, so we need to collect their opinions. Such approaches make it easier to study rare diseases and get new treatments.
Q: How do the challenges of drug research and development in the field of rare diseases differ from those associated with more prevalent diseases?
Dr. Woodcock: There is one advantage today for people with rare diseases. That is, when there is a known genetic mutation causing a disease, RNA interference and other gene therapy approaches can be used. There are challenges, though. Patients with rare disorders often don’t have a uniform disease course. They often have a multisystem impact, so they might have things wrong with their GI tract and/or skin, so it’s difficult to know what to measure. We’re trying to remedy this by gathering better natural history information on what happens to people. That is empowering for patients as well.
Q: In what practical ways can physicians become advocates for patients and their families who are navigating life with a rare disease?
Dr. Woodcock: I advise people to get involved in the association or advocacy group for their rare disease. It’s empowering. They can share stories and information with others who have been suffering from the disease. Also, they would get information about what trials might be available. As for physicians themselves, they have a bewildering variety of jobs they’re supposed to do, so it’s hard to be good in any one of them. People with rare disease often suffer terribly because they don’t get diagnosed for 10 years even though they have classic symptoms of a particular disorder. If physicians have never seen it or never heard of it, they may not know how to treat it. It’s a huge problem.
Q: Who inspires you most in your work today?
Dr. Woodcock: The dedication of the staff at the FDA is unbelievable. When you look at responses to the Federal Employee Viewpoint Survey administered by the Office of Personnel Management, FDA workers consistently express a strong sense of mission and dedication. It’s out of the park, really. They have worked night and day during this pandemic. I’m inspired by everyone who works at the FDA and their incredible dedication to their work.
Q: In what ways do you cope with the pressure that comes with your line of work? Do you have a favorite hobby or that activity that helps keep you grounded?
Dr. Woodcock: I’m an avid gardener, so I have a garden with vegetables, fruits, and flowers, including a large orchid collection. I’m also a hiker and a physical fitness buff, so I feel like there isn’t enough time in the day for all of my hobbies. Formal hiking trails near me are very crowded now, so I’ve been hiking around my neighborhood, taking long walks and going up and down hills quickly. Last November, I went hiking in New Zealand with my daughter. We hiked the Milford Track, which is about 33 miles long. It goes from an inland lake, over a mountain pass, and to the Pacific Ocean. It was fun, with unbelievable scenery.
Q: What novel treatment developments in rare disorders are you most excited about in the next 5 years?
Dr. Woodcock: I think gene therapy will come into its own. I think that could be a game-changer for people with genetic mutations causing rare diseases, and even cancer. We’ll see. It takes the technology a long time to mature. There are also gene-directed therapies such as RNA inhibition. We’ve already approved a couple of products like that for rare diseases, including treatments for the cardiomyopathy and neuropathy associated with ATTR amyloidosis. As our knowledge of biology continues to grow, I think more of these diseases will be amenable to interventions.
Q: In May of 2020 you were asked to temporarily step aside from your post as director of CDER to work on Operation Warp Speed. Please describe what your role is in this effort to accelerate COVID-19 treatments.
Dr. Woodcock: I’m the lead on therapeutics. Operation Warp Speed is mainly focused on developing vaccines for COVID-19. In the meantime, people who don’t respond to vaccines are going to need therapeutics, such as the elderly, or those who refuse to take vaccines, or those who are immunosuppressed and can’t mount a response to a vaccine. If we can develop those therapeutics now, that would be good to get that populous vaccinated. The team identified what we thought were the five highest priority agents to work on, and we’re testing them. We have identified many more in a priority list. We have five master protocols running for different times in the disease, such as when you’re an outpatient, when you’re an inpatient, or when you’re in the ICU. The work is stressful, because we need these treatments as soon as possible, but we have a great team working on this. I feel like I’m making a contribution in this role, because I know people in industry and in the National Institutes of Health. I try to bring everyone together and get things done.
*Correction, 10/22/20: An earlier version of this article misstated the name of the National Organization for Rare Disorders.
Clinical factors and treatment tied to COVID-19 mortality in cancer patients
according to two presentations at the European Society for Medical Oncology Virtual Congress 2020.
Two analyses of data from the COVID-19 and Cancer Consortium (CCC19) were presented at the meeting.
The data suggest that older age, male sex, more comorbidities, poor performance status, progressive cancer or multiple cancers, hematologic malignancy, and recent cancer therapy are all associated with higher mortality among patients with cancer and COVID-19. Anti-CD20 therapy is associated with an especially high mortality rate, according to an investigator.
Among hospitalized patients, increased absolute neutrophil count as well as abnormal D-dimer, high-sensitivity troponin, and C-reactive protein are associated with a higher risk of mortality.
Prior analyses of CCC19 data pointed to several factors associated with higher COVID-19 death rates, according to Petros Grivas, MD, PhD, of University of Washington, Seattle, who presented some CCC19 data at the meeting. However, the prior analyses were limited by weak statistical power and low event rates, Dr. Grivas said.
Clinical and laboratory factors: Abstract LBA72
The aim of Dr. Grivas’s analysis was to validate a priori identified demographic and clinicopathologic factors associated with 30-day all-cause mortality in patients with COVID-19 and cancer. Dr. Grivas and colleagues also explored the potential association between laboratory parameters and 30-day all-cause mortality.
The analysis included 3,899 patients with cancer and COVID-19 from 124 centers. Most centers are in the United States, but 4% are in Canada, and 2% are in Spain. About two-thirds of patients were 60 years of age or younger at baseline, half were men, 79% had solid tumors, and 21% had hematologic malignancies.
Cancer-specific factors associated with an increased risk of 30-day all-cause mortality were having progressive cancer (adjusted odds ratio, 2.9), receiving cancer therapy within 3 months (aOR, 1.2), having a hematologic versus solid tumor (aOR, 1.7), and having multiple malignancies (aOR, 1.5).
Clinical factors associated with an increased risk of 30-day all-cause mortality were Black versus White race (aOR, 1.5), older age (aOR, 1.7 per 10 years), three or more actively treated comorbidities (versus none; aOR, 2.1), and Eastern Cooperative Oncology Group performance status of 2 or more (versus 0; aOR, 4.6).
In hospitalized patients, several laboratory variables were associated with an increased risk of 30-day all-cause mortality. Having an absolute neutrophil count above the upper limit of normal doubled the risk (aOR, 2.0), while abnormal D-dimer, high-sensitivity troponin, and C-reactive protein all more than doubled the risk of mortality (aORs of 2.5, 2.5, and 2.4, respectively).
Further risk modeling with multivariable analysis will be performed after longer follow-up, Dr. Grivas noted.
Treatment-related outcomes: Abstract LBA71
An additional analysis of CCC19 data encompassed 3,654 patients. In this analysis, researchers investigated the correlation between timing of cancer treatment and COVID-19–related complications and 30-day mortality.
Mortality was highest among cancer patients treated 1-3 months prior to COVID-19 diagnosis, with all-cause mortality at 28%, said Trisha M. Wise-Draper, MD, PhD, of University of Cincinnati, when presenting the data at the meeting.
Rates for other complications (hospitalization, oxygen required, ICU admission, and mechanical ventilation) were similar regardless of treatment timing.
The unadjusted 30-day mortality rate was highest for patients treated most recently with chemoimmunotherapy (30%), followed by chemotherapy (18%), chemoradiotherapy (18%), and targeted therapy (17%).
The mortality rate was “particularly high,” at 50%, in patients receiving anti-CD20 therapy 1-3 months prior to COVID-19 diagnosis – the time period for which significant B-cell depletion develops, Dr. Wise-Draper observed.
An analysis of disease status among 1,449 patients treated within 3 months of COVID-19 diagnosis showed mortality risk increasing from 6% among patients in remission or with newly emergent disease, to 22% in patients with any active cancer, to 34% in those with progressing disease, Dr. Wise-Draper said.
Discussant Benjamin Solomon, MD, PhD, of Peter MacCallum Cancer Centre in Melbourne, made note of the high 30-day mortality rate seen in patients receiving anti-CD20 therapy as well as the elevated standardized mortality ratios with recent chemoimmunotherapy and targeted therapy.
“Although there are some limitations of this analysis, it provides the best data we have to date about the effects of treatment on early mortality in patients with COVID-19 and cancer. It points to a modest but heterogeneous effect of treatment on outcome, one which is likely to become clearer with larger cohorts and additional analysis,” Dr. Solomon said.
This research was funded by the American Cancer Society, Hope Foundation for Cancer Research, Jim and Carol O’Hare Fund, National Cancer Institute, National Human Genome Research Institute, Vanderbilt Institute for Clinical and Translational Research, and Fonds de Recherche du Quebec-Sante. Dr. Grivas disclosed relationships with many companies, but none are related to this work. Dr. Wise-Draper disclosed relationships with Merck, Bristol-Myers Squibb, Tesaro, GlaxoSmithKline, AstraZeneca, Shattuck Labs, and Rakuten. Dr. Solomon disclosed relationships with Amgen, AstraZeneca, Merck, Bristol-Myers Squibb, Novartis, Pfizer, and Roche-Genentech.
SOURCES: Grivas P et al. ESMO 2020, Abstract LBA72; Wise-Draper TM et al. ESMO 2020, Abstract LBA71.
according to two presentations at the European Society for Medical Oncology Virtual Congress 2020.
Two analyses of data from the COVID-19 and Cancer Consortium (CCC19) were presented at the meeting.
The data suggest that older age, male sex, more comorbidities, poor performance status, progressive cancer or multiple cancers, hematologic malignancy, and recent cancer therapy are all associated with higher mortality among patients with cancer and COVID-19. Anti-CD20 therapy is associated with an especially high mortality rate, according to an investigator.
Among hospitalized patients, increased absolute neutrophil count as well as abnormal D-dimer, high-sensitivity troponin, and C-reactive protein are associated with a higher risk of mortality.
Prior analyses of CCC19 data pointed to several factors associated with higher COVID-19 death rates, according to Petros Grivas, MD, PhD, of University of Washington, Seattle, who presented some CCC19 data at the meeting. However, the prior analyses were limited by weak statistical power and low event rates, Dr. Grivas said.
Clinical and laboratory factors: Abstract LBA72
The aim of Dr. Grivas’s analysis was to validate a priori identified demographic and clinicopathologic factors associated with 30-day all-cause mortality in patients with COVID-19 and cancer. Dr. Grivas and colleagues also explored the potential association between laboratory parameters and 30-day all-cause mortality.
The analysis included 3,899 patients with cancer and COVID-19 from 124 centers. Most centers are in the United States, but 4% are in Canada, and 2% are in Spain. About two-thirds of patients were 60 years of age or younger at baseline, half were men, 79% had solid tumors, and 21% had hematologic malignancies.
Cancer-specific factors associated with an increased risk of 30-day all-cause mortality were having progressive cancer (adjusted odds ratio, 2.9), receiving cancer therapy within 3 months (aOR, 1.2), having a hematologic versus solid tumor (aOR, 1.7), and having multiple malignancies (aOR, 1.5).
Clinical factors associated with an increased risk of 30-day all-cause mortality were Black versus White race (aOR, 1.5), older age (aOR, 1.7 per 10 years), three or more actively treated comorbidities (versus none; aOR, 2.1), and Eastern Cooperative Oncology Group performance status of 2 or more (versus 0; aOR, 4.6).
In hospitalized patients, several laboratory variables were associated with an increased risk of 30-day all-cause mortality. Having an absolute neutrophil count above the upper limit of normal doubled the risk (aOR, 2.0), while abnormal D-dimer, high-sensitivity troponin, and C-reactive protein all more than doubled the risk of mortality (aORs of 2.5, 2.5, and 2.4, respectively).
Further risk modeling with multivariable analysis will be performed after longer follow-up, Dr. Grivas noted.
Treatment-related outcomes: Abstract LBA71
An additional analysis of CCC19 data encompassed 3,654 patients. In this analysis, researchers investigated the correlation between timing of cancer treatment and COVID-19–related complications and 30-day mortality.
Mortality was highest among cancer patients treated 1-3 months prior to COVID-19 diagnosis, with all-cause mortality at 28%, said Trisha M. Wise-Draper, MD, PhD, of University of Cincinnati, when presenting the data at the meeting.
Rates for other complications (hospitalization, oxygen required, ICU admission, and mechanical ventilation) were similar regardless of treatment timing.
The unadjusted 30-day mortality rate was highest for patients treated most recently with chemoimmunotherapy (30%), followed by chemotherapy (18%), chemoradiotherapy (18%), and targeted therapy (17%).
The mortality rate was “particularly high,” at 50%, in patients receiving anti-CD20 therapy 1-3 months prior to COVID-19 diagnosis – the time period for which significant B-cell depletion develops, Dr. Wise-Draper observed.
An analysis of disease status among 1,449 patients treated within 3 months of COVID-19 diagnosis showed mortality risk increasing from 6% among patients in remission or with newly emergent disease, to 22% in patients with any active cancer, to 34% in those with progressing disease, Dr. Wise-Draper said.
Discussant Benjamin Solomon, MD, PhD, of Peter MacCallum Cancer Centre in Melbourne, made note of the high 30-day mortality rate seen in patients receiving anti-CD20 therapy as well as the elevated standardized mortality ratios with recent chemoimmunotherapy and targeted therapy.
“Although there are some limitations of this analysis, it provides the best data we have to date about the effects of treatment on early mortality in patients with COVID-19 and cancer. It points to a modest but heterogeneous effect of treatment on outcome, one which is likely to become clearer with larger cohorts and additional analysis,” Dr. Solomon said.
This research was funded by the American Cancer Society, Hope Foundation for Cancer Research, Jim and Carol O’Hare Fund, National Cancer Institute, National Human Genome Research Institute, Vanderbilt Institute for Clinical and Translational Research, and Fonds de Recherche du Quebec-Sante. Dr. Grivas disclosed relationships with many companies, but none are related to this work. Dr. Wise-Draper disclosed relationships with Merck, Bristol-Myers Squibb, Tesaro, GlaxoSmithKline, AstraZeneca, Shattuck Labs, and Rakuten. Dr. Solomon disclosed relationships with Amgen, AstraZeneca, Merck, Bristol-Myers Squibb, Novartis, Pfizer, and Roche-Genentech.
SOURCES: Grivas P et al. ESMO 2020, Abstract LBA72; Wise-Draper TM et al. ESMO 2020, Abstract LBA71.
according to two presentations at the European Society for Medical Oncology Virtual Congress 2020.
Two analyses of data from the COVID-19 and Cancer Consortium (CCC19) were presented at the meeting.
The data suggest that older age, male sex, more comorbidities, poor performance status, progressive cancer or multiple cancers, hematologic malignancy, and recent cancer therapy are all associated with higher mortality among patients with cancer and COVID-19. Anti-CD20 therapy is associated with an especially high mortality rate, according to an investigator.
Among hospitalized patients, increased absolute neutrophil count as well as abnormal D-dimer, high-sensitivity troponin, and C-reactive protein are associated with a higher risk of mortality.
Prior analyses of CCC19 data pointed to several factors associated with higher COVID-19 death rates, according to Petros Grivas, MD, PhD, of University of Washington, Seattle, who presented some CCC19 data at the meeting. However, the prior analyses were limited by weak statistical power and low event rates, Dr. Grivas said.
Clinical and laboratory factors: Abstract LBA72
The aim of Dr. Grivas’s analysis was to validate a priori identified demographic and clinicopathologic factors associated with 30-day all-cause mortality in patients with COVID-19 and cancer. Dr. Grivas and colleagues also explored the potential association between laboratory parameters and 30-day all-cause mortality.
The analysis included 3,899 patients with cancer and COVID-19 from 124 centers. Most centers are in the United States, but 4% are in Canada, and 2% are in Spain. About two-thirds of patients were 60 years of age or younger at baseline, half were men, 79% had solid tumors, and 21% had hematologic malignancies.
Cancer-specific factors associated with an increased risk of 30-day all-cause mortality were having progressive cancer (adjusted odds ratio, 2.9), receiving cancer therapy within 3 months (aOR, 1.2), having a hematologic versus solid tumor (aOR, 1.7), and having multiple malignancies (aOR, 1.5).
Clinical factors associated with an increased risk of 30-day all-cause mortality were Black versus White race (aOR, 1.5), older age (aOR, 1.7 per 10 years), three or more actively treated comorbidities (versus none; aOR, 2.1), and Eastern Cooperative Oncology Group performance status of 2 or more (versus 0; aOR, 4.6).
In hospitalized patients, several laboratory variables were associated with an increased risk of 30-day all-cause mortality. Having an absolute neutrophil count above the upper limit of normal doubled the risk (aOR, 2.0), while abnormal D-dimer, high-sensitivity troponin, and C-reactive protein all more than doubled the risk of mortality (aORs of 2.5, 2.5, and 2.4, respectively).
Further risk modeling with multivariable analysis will be performed after longer follow-up, Dr. Grivas noted.
Treatment-related outcomes: Abstract LBA71
An additional analysis of CCC19 data encompassed 3,654 patients. In this analysis, researchers investigated the correlation between timing of cancer treatment and COVID-19–related complications and 30-day mortality.
Mortality was highest among cancer patients treated 1-3 months prior to COVID-19 diagnosis, with all-cause mortality at 28%, said Trisha M. Wise-Draper, MD, PhD, of University of Cincinnati, when presenting the data at the meeting.
Rates for other complications (hospitalization, oxygen required, ICU admission, and mechanical ventilation) were similar regardless of treatment timing.
The unadjusted 30-day mortality rate was highest for patients treated most recently with chemoimmunotherapy (30%), followed by chemotherapy (18%), chemoradiotherapy (18%), and targeted therapy (17%).
The mortality rate was “particularly high,” at 50%, in patients receiving anti-CD20 therapy 1-3 months prior to COVID-19 diagnosis – the time period for which significant B-cell depletion develops, Dr. Wise-Draper observed.
An analysis of disease status among 1,449 patients treated within 3 months of COVID-19 diagnosis showed mortality risk increasing from 6% among patients in remission or with newly emergent disease, to 22% in patients with any active cancer, to 34% in those with progressing disease, Dr. Wise-Draper said.
Discussant Benjamin Solomon, MD, PhD, of Peter MacCallum Cancer Centre in Melbourne, made note of the high 30-day mortality rate seen in patients receiving anti-CD20 therapy as well as the elevated standardized mortality ratios with recent chemoimmunotherapy and targeted therapy.
“Although there are some limitations of this analysis, it provides the best data we have to date about the effects of treatment on early mortality in patients with COVID-19 and cancer. It points to a modest but heterogeneous effect of treatment on outcome, one which is likely to become clearer with larger cohorts and additional analysis,” Dr. Solomon said.
This research was funded by the American Cancer Society, Hope Foundation for Cancer Research, Jim and Carol O’Hare Fund, National Cancer Institute, National Human Genome Research Institute, Vanderbilt Institute for Clinical and Translational Research, and Fonds de Recherche du Quebec-Sante. Dr. Grivas disclosed relationships with many companies, but none are related to this work. Dr. Wise-Draper disclosed relationships with Merck, Bristol-Myers Squibb, Tesaro, GlaxoSmithKline, AstraZeneca, Shattuck Labs, and Rakuten. Dr. Solomon disclosed relationships with Amgen, AstraZeneca, Merck, Bristol-Myers Squibb, Novartis, Pfizer, and Roche-Genentech.
SOURCES: Grivas P et al. ESMO 2020, Abstract LBA72; Wise-Draper TM et al. ESMO 2020, Abstract LBA71.
FROM ESMO 2020
Golimumab approval extended to polyarticular-course JIA and juvenile PsA
after the Food and Drug Administration approved the tumor necrosis factor inhibitor for these indications on Sept. 30, according to an announcement from its manufacturer, Janssen.
Results from the open-label, single-arm, multicenter, phase 3, GO-VIVA clinical trial formed the basis for the agency’s approval of IV golimumab. GO-VIVA was conducted in 127 patients aged 2-17 years with JIA with arthritis in five or more joints (despite receiving treatment with methotrexate for at least 2 months) as part of a postmarketing requirement under the Pediatric Research Equity Act after the intravenous formulation of the biologic was approved for adults with rheumatoid arthritis in 2013. It demonstrated that pediatric patients had a level of pharmacokinetic exposure to golimumab that was similar to what was observed in two pivotal phase 3 trials in adults with moderately to severely active RA and active PsA, as well as efficacy that was generally consistent with responses seen in adult patients with RA, the manufacturer said.
Besides RA, intravenous golimumab was previously approved for adults with PsA and ankylosing spondylitis. As opposed to the IV dosing for adults with RA, PsA, and ankylosing spondylitis at 2 mg/kg infused over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter, dosing for pediatric patients with pJIA and PsA is based on body surface area at 80 mg/m2, also given as an IV infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter.
The adverse reactions observed in GO-VIVA were consistent with the established safety profile of intravenous golimumab in adult patients with RA and PsA, according to Janssen.
The full prescribing information for intravenous golimumab can be found on the FDA website.
after the Food and Drug Administration approved the tumor necrosis factor inhibitor for these indications on Sept. 30, according to an announcement from its manufacturer, Janssen.
Results from the open-label, single-arm, multicenter, phase 3, GO-VIVA clinical trial formed the basis for the agency’s approval of IV golimumab. GO-VIVA was conducted in 127 patients aged 2-17 years with JIA with arthritis in five or more joints (despite receiving treatment with methotrexate for at least 2 months) as part of a postmarketing requirement under the Pediatric Research Equity Act after the intravenous formulation of the biologic was approved for adults with rheumatoid arthritis in 2013. It demonstrated that pediatric patients had a level of pharmacokinetic exposure to golimumab that was similar to what was observed in two pivotal phase 3 trials in adults with moderately to severely active RA and active PsA, as well as efficacy that was generally consistent with responses seen in adult patients with RA, the manufacturer said.
Besides RA, intravenous golimumab was previously approved for adults with PsA and ankylosing spondylitis. As opposed to the IV dosing for adults with RA, PsA, and ankylosing spondylitis at 2 mg/kg infused over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter, dosing for pediatric patients with pJIA and PsA is based on body surface area at 80 mg/m2, also given as an IV infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter.
The adverse reactions observed in GO-VIVA were consistent with the established safety profile of intravenous golimumab in adult patients with RA and PsA, according to Janssen.
The full prescribing information for intravenous golimumab can be found on the FDA website.
after the Food and Drug Administration approved the tumor necrosis factor inhibitor for these indications on Sept. 30, according to an announcement from its manufacturer, Janssen.
Results from the open-label, single-arm, multicenter, phase 3, GO-VIVA clinical trial formed the basis for the agency’s approval of IV golimumab. GO-VIVA was conducted in 127 patients aged 2-17 years with JIA with arthritis in five or more joints (despite receiving treatment with methotrexate for at least 2 months) as part of a postmarketing requirement under the Pediatric Research Equity Act after the intravenous formulation of the biologic was approved for adults with rheumatoid arthritis in 2013. It demonstrated that pediatric patients had a level of pharmacokinetic exposure to golimumab that was similar to what was observed in two pivotal phase 3 trials in adults with moderately to severely active RA and active PsA, as well as efficacy that was generally consistent with responses seen in adult patients with RA, the manufacturer said.
Besides RA, intravenous golimumab was previously approved for adults with PsA and ankylosing spondylitis. As opposed to the IV dosing for adults with RA, PsA, and ankylosing spondylitis at 2 mg/kg infused over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter, dosing for pediatric patients with pJIA and PsA is based on body surface area at 80 mg/m2, also given as an IV infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter.
The adverse reactions observed in GO-VIVA were consistent with the established safety profile of intravenous golimumab in adult patients with RA and PsA, according to Janssen.
The full prescribing information for intravenous golimumab can be found on the FDA website.