User login
GLIMMER of hope for itch in primary biliary cholangitis
Patients with primary biliary cholangitis experienced rapid improvements in itch and quality of life after treatment with linerixibat in a randomized, placebo-controlled trial of the safety, efficacy, and tolerability of the small-molecule drug.
Moderate to severe pruritus “affects patients’ quality of life and is a huge burden for them,” said investigator Cynthia Levy, MD, from the University of Miami Health System.
“Finally having a medication that controls those symptoms is really important,” she said in an interview.
With a twice-daily mid-range dose of the drug for 12 weeks, patients with moderate to severe itch reported significantly less itch and better social and emotional quality of life, Dr. Levy reported at the Liver Meeting, where she presented findings from the phase 2 GLIMMER trial.
After a single-blind 4-week placebo run-in period for patients with itch scores of at least 4 on a 10-point rating scale, those with itch scores of at least 3 were then randomly assigned to one of five treatment regimens – once-daily linerixibat at doses of 20 mg, 90 mg, or 180 mg, or twice-daily doses of 40 mg or 90 mg – or to placebo.
After 12 weeks of treatment, all 147 participants once again received placebo for 4 weeks.
During the trial, participants recorded itch levels twice daily. The worst of these daily scores was averaged every 7 days to determine the mean worst daily itch.
The primary study endpoint was the change in worst daily itch from baseline after 12 weeks of treatment. Participants whose self-rated itch improved by 2 points on the 10-point scale were considered to have had a response to the drug.
Participants also completed the PBC-40, an instrument to measure quality of life in patients with primary biliary cholangitis, answering questions about itch and social and emotional status.
Reductions in worst daily itch from baseline to 12 weeks were steepest in the 40-mg twice-daily group, at 2.86 points, and in the 90-mg twice-daily group, at 2.25 points. In the placebo group, the mean decrease was 1.73 points.
During the subsequent 4 weeks of placebo, after treatment ended, the itch relief faded in all groups.
Scores on the PBC-40 itch domain improved significantly in every group, including placebo. However, only those in the twice-daily 40-mg group saw significant improvements on the social (P = .0016) and emotional (P = .0025) domains.
‘Between incremental and revolutionary’
The results are on a “kind of continuum between incremental and revolutionary,” said Jonathan A. Dranoff, MD, from the University of Arkansas for Medical Sciences, Little Rock, who was not involved in the study. “It doesn’t hit either extreme, but it’s the first new drug for this purpose in forever, which by itself is a good thing.”
The placebo effect suggests that “maybe the actual contribution of the noncognitive brain to pruritus is bigger than we thought, and that’s worth noting,” he added. Nevertheless, “the drug still appears to have effects that are statistically different from placebo.”
The placebo effect in itching studies is always high but tends to wane over time, said Dr. Levy. This trial had a 4-week placebo run-in period to allow that effect to fade somewhat, she explained.
About 10% of the study cohort experienced drug-related diarrhea, which was expected, and about 10% dropped out of the trial because of drug-related adverse events.
Linerixibat is an ileal sodium-dependent bile acid transporter inhibitor, so the gut has to deal with the excess bile acid fallout, but the diarrhea is likely manageable with antidiarrheals, said Dr. Levy.
It is unlikely that diarrhea will deter patients with severe itch from using an effective drug when other drugs have failed them. “These patients are consumed by itch most of the time,” said Dr. Dranoff. “I think for people who don’t regularly treat patients with primary biliary cholangitis, it’s one of the underappreciated aspects of the disease.”
The improvements in social and emotional quality of life seen with linerixibat are not only statistically significant, they are also clinically significant, said Dr. Levy. “We are really expecting this to impact the lives of our patients and are looking forward to phase 3.”
Dr. Levy disclosed support from GlaxoSmithKline. Dr. Dranoff disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Patients with primary biliary cholangitis experienced rapid improvements in itch and quality of life after treatment with linerixibat in a randomized, placebo-controlled trial of the safety, efficacy, and tolerability of the small-molecule drug.
Moderate to severe pruritus “affects patients’ quality of life and is a huge burden for them,” said investigator Cynthia Levy, MD, from the University of Miami Health System.
“Finally having a medication that controls those symptoms is really important,” she said in an interview.
With a twice-daily mid-range dose of the drug for 12 weeks, patients with moderate to severe itch reported significantly less itch and better social and emotional quality of life, Dr. Levy reported at the Liver Meeting, where she presented findings from the phase 2 GLIMMER trial.
After a single-blind 4-week placebo run-in period for patients with itch scores of at least 4 on a 10-point rating scale, those with itch scores of at least 3 were then randomly assigned to one of five treatment regimens – once-daily linerixibat at doses of 20 mg, 90 mg, or 180 mg, or twice-daily doses of 40 mg or 90 mg – or to placebo.
After 12 weeks of treatment, all 147 participants once again received placebo for 4 weeks.
During the trial, participants recorded itch levels twice daily. The worst of these daily scores was averaged every 7 days to determine the mean worst daily itch.
The primary study endpoint was the change in worst daily itch from baseline after 12 weeks of treatment. Participants whose self-rated itch improved by 2 points on the 10-point scale were considered to have had a response to the drug.
Participants also completed the PBC-40, an instrument to measure quality of life in patients with primary biliary cholangitis, answering questions about itch and social and emotional status.
Reductions in worst daily itch from baseline to 12 weeks were steepest in the 40-mg twice-daily group, at 2.86 points, and in the 90-mg twice-daily group, at 2.25 points. In the placebo group, the mean decrease was 1.73 points.
During the subsequent 4 weeks of placebo, after treatment ended, the itch relief faded in all groups.
Scores on the PBC-40 itch domain improved significantly in every group, including placebo. However, only those in the twice-daily 40-mg group saw significant improvements on the social (P = .0016) and emotional (P = .0025) domains.
‘Between incremental and revolutionary’
The results are on a “kind of continuum between incremental and revolutionary,” said Jonathan A. Dranoff, MD, from the University of Arkansas for Medical Sciences, Little Rock, who was not involved in the study. “It doesn’t hit either extreme, but it’s the first new drug for this purpose in forever, which by itself is a good thing.”
The placebo effect suggests that “maybe the actual contribution of the noncognitive brain to pruritus is bigger than we thought, and that’s worth noting,” he added. Nevertheless, “the drug still appears to have effects that are statistically different from placebo.”
The placebo effect in itching studies is always high but tends to wane over time, said Dr. Levy. This trial had a 4-week placebo run-in period to allow that effect to fade somewhat, she explained.
About 10% of the study cohort experienced drug-related diarrhea, which was expected, and about 10% dropped out of the trial because of drug-related adverse events.
Linerixibat is an ileal sodium-dependent bile acid transporter inhibitor, so the gut has to deal with the excess bile acid fallout, but the diarrhea is likely manageable with antidiarrheals, said Dr. Levy.
It is unlikely that diarrhea will deter patients with severe itch from using an effective drug when other drugs have failed them. “These patients are consumed by itch most of the time,” said Dr. Dranoff. “I think for people who don’t regularly treat patients with primary biliary cholangitis, it’s one of the underappreciated aspects of the disease.”
The improvements in social and emotional quality of life seen with linerixibat are not only statistically significant, they are also clinically significant, said Dr. Levy. “We are really expecting this to impact the lives of our patients and are looking forward to phase 3.”
Dr. Levy disclosed support from GlaxoSmithKline. Dr. Dranoff disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Patients with primary biliary cholangitis experienced rapid improvements in itch and quality of life after treatment with linerixibat in a randomized, placebo-controlled trial of the safety, efficacy, and tolerability of the small-molecule drug.
Moderate to severe pruritus “affects patients’ quality of life and is a huge burden for them,” said investigator Cynthia Levy, MD, from the University of Miami Health System.
“Finally having a medication that controls those symptoms is really important,” she said in an interview.
With a twice-daily mid-range dose of the drug for 12 weeks, patients with moderate to severe itch reported significantly less itch and better social and emotional quality of life, Dr. Levy reported at the Liver Meeting, where she presented findings from the phase 2 GLIMMER trial.
After a single-blind 4-week placebo run-in period for patients with itch scores of at least 4 on a 10-point rating scale, those with itch scores of at least 3 were then randomly assigned to one of five treatment regimens – once-daily linerixibat at doses of 20 mg, 90 mg, or 180 mg, or twice-daily doses of 40 mg or 90 mg – or to placebo.
After 12 weeks of treatment, all 147 participants once again received placebo for 4 weeks.
During the trial, participants recorded itch levels twice daily. The worst of these daily scores was averaged every 7 days to determine the mean worst daily itch.
The primary study endpoint was the change in worst daily itch from baseline after 12 weeks of treatment. Participants whose self-rated itch improved by 2 points on the 10-point scale were considered to have had a response to the drug.
Participants also completed the PBC-40, an instrument to measure quality of life in patients with primary biliary cholangitis, answering questions about itch and social and emotional status.
Reductions in worst daily itch from baseline to 12 weeks were steepest in the 40-mg twice-daily group, at 2.86 points, and in the 90-mg twice-daily group, at 2.25 points. In the placebo group, the mean decrease was 1.73 points.
During the subsequent 4 weeks of placebo, after treatment ended, the itch relief faded in all groups.
Scores on the PBC-40 itch domain improved significantly in every group, including placebo. However, only those in the twice-daily 40-mg group saw significant improvements on the social (P = .0016) and emotional (P = .0025) domains.
‘Between incremental and revolutionary’
The results are on a “kind of continuum between incremental and revolutionary,” said Jonathan A. Dranoff, MD, from the University of Arkansas for Medical Sciences, Little Rock, who was not involved in the study. “It doesn’t hit either extreme, but it’s the first new drug for this purpose in forever, which by itself is a good thing.”
The placebo effect suggests that “maybe the actual contribution of the noncognitive brain to pruritus is bigger than we thought, and that’s worth noting,” he added. Nevertheless, “the drug still appears to have effects that are statistically different from placebo.”
The placebo effect in itching studies is always high but tends to wane over time, said Dr. Levy. This trial had a 4-week placebo run-in period to allow that effect to fade somewhat, she explained.
About 10% of the study cohort experienced drug-related diarrhea, which was expected, and about 10% dropped out of the trial because of drug-related adverse events.
Linerixibat is an ileal sodium-dependent bile acid transporter inhibitor, so the gut has to deal with the excess bile acid fallout, but the diarrhea is likely manageable with antidiarrheals, said Dr. Levy.
It is unlikely that diarrhea will deter patients with severe itch from using an effective drug when other drugs have failed them. “These patients are consumed by itch most of the time,” said Dr. Dranoff. “I think for people who don’t regularly treat patients with primary biliary cholangitis, it’s one of the underappreciated aspects of the disease.”
The improvements in social and emotional quality of life seen with linerixibat are not only statistically significant, they are also clinically significant, said Dr. Levy. “We are really expecting this to impact the lives of our patients and are looking forward to phase 3.”
Dr. Levy disclosed support from GlaxoSmithKline. Dr. Dranoff disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Using telehealth to deliver palliative care to cancer patients
Traditional delivery of palliative care to outpatients with cancer is associated with many challenges.

Telehealth can eliminate some of these challenges but comes with issues of its own, according to results of the REACH PC trial.
Jennifer S. Temel, MD, of Massachusetts General Hospital in Boston, discussed the use of telemedicine in palliative care, including results from REACH PC, during an educational session at the ASCO Virtual Quality Care Symposium 2020.
Dr. Temel noted that, for cancer patients, an in-person visit with a palliative care specialist can cost time, induce fatigue, and increase financial burden from transportation and parking expenses.
For caregivers and family, an in-person visit may necessitate absence from family and/or work, require complex scheduling to coordinate with other office visits, and result in additional transportation and/or parking expenses.
For health care systems, to have a dedicated palliative care clinic requires precious space and financial expenditures for office personnel and other resources.
These issues make it attractive to consider whether telehealth could be used for palliative care services.
Scarcity of palliative care specialists
In the United States, there is roughly 1 palliative care physician for every 20,000 older adults with a life-limiting illness, according to research published in Annual Review of Public Health in 2014.
In its 2019 state-by-state report card, the Center to Advance Palliative Care noted that only 72% of U.S. hospitals with 50 or more beds have a palliative care team.
For patients with serious illnesses and those who are socioeconomically or geographically disadvantaged, palliative care is often inaccessible.
Inefficiencies in the current system are an additional impediment. Palliative care specialists frequently see patients during a portion of the patient’s routine visit to subspecialty or primary care clinics. This limits the palliative care specialist’s ability to perform comprehensive assessments and provide patient-centered care efficiently.
Special considerations regarding telehealth for palliative care
As a specialty, palliative care involves interactions that could make the use of telehealth problematic. For example, conveyance of interest, warmth, and touch are challenging or impossible in a video format.
Palliative care specialists engage with patients regarding relatively serious topics such as prognosis and end-of-life preferences. There is uncertainty about how those discussions would be received by patients and their caregivers via video.
Furthermore, there are logistical impediments such as prescribing opioids with video or across state lines.
Despite these concerns, the ENABLE study showed that supplementing usual oncology care with weekly (transitioning to monthly) telephone-based educational palliative care produced higher quality of life and mood than did usual oncology care alone. These results were published in JAMA in 2009.
REACH PC study demonstrates feasibility of telehealth model
Dr. Temel described the ongoing REACH PC trial in which palliative care is delivered via video visits and compared with in-person palliative care for patients with advanced non–small cell lung cancer.
The primary aim of REACH PC is to determine whether telehealth palliative care is equivalent to traditional palliative care in improving quality of life as a supplement to routine oncology care.
Currently, REACH PC has enrolled 581 patients at its 20 sites, spanning a geographically diverse area. Just over half of patients approached about REACH PC agreed to enroll in it. Ultimately, 1,250 enrollees are sought.
Among patients who declined to participate, 7.6% indicated “discomfort with technology” as the reason. Most refusals were due to lack of interest in research (35.1%) and/or palliative care (22.9%).
Older adults were prominent among enrollees. More than 60% were older than 60 years of age, and more than one-third were older than 70 years.
Among patients who began the trial, there were slightly more withdrawals in the telehealth participants, in comparison with in-person participants (13.6% versus 9.1%).
When palliative care clinicians were queried about video visits, 64.3% said there were no challenges. This is comparable to the 65.5% of clinicians who had no challenges with in-person visits.
When problems occurred with video visits, they were most frequently technical (19.1%). Only 1.4% of clinicians reported difficulty addressing topics that felt uncomfortable over video, and 1.5% reported difficulty establishing rapport.
The success rates of video and in-person visits were similar. About 80% of visits accomplished planned goals.
‘Webside’ manner
Strategies such as reflective listening and summarizing what patients say (to verify an accurate understanding of the patient’s perspective) are key to successful palliative care visits, regardless of the setting.
For telehealth visits, Dr. Temel described techniques she defined as “webside manner,” to compensate for the inability of the clinician to touch a patient. These techniques include leaning in toward the camera, nodding, and pausing to be certain the patient has finished speaking before the clinician speaks again.
Is telehealth the future of palliative care?
I include myself among those oncologists who have voiced concern about moving from face-to-face to remote visits for complicated consultations such as those required for palliative care. Nonetheless, from the preliminary results of the REACH PC trial, it appears that telehealth could be a valuable tool.
To minimize differences between in-person and remote delivery of palliative care, practical strategies for ensuring rapport and facilitating a trusting relationship should be defined further and disseminated.
In addition, we need to be vigilant for widening inequities of care from rapid movement to the use of technology (i.e., an equity gap). In their telehealth experience during the COVID-19 pandemic, investigators at Houston Methodist Cancer Center found that patients declining virtual visits tended to be older, lower-income, and less likely to have commercial insurance. These results were recently published in JCO Oncology Practice.
For the foregoing reasons, hybrid systems for palliative care services will probably always be needed.
Going forward, we should heed the advice of Alvin Toffler in his book Future Shock. Mr. Toffler said, “The illiterate of the 21st century will not be those who cannot read and write, but those who cannot learn, unlearn, and relearn.”
The traditional model for delivering palliative care will almost certainly need to be reimagined and relearned.
Dr. Temel disclosed institutional research funding from Pfizer.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Traditional delivery of palliative care to outpatients with cancer is associated with many challenges.
Telehealth can eliminate some of these challenges but comes with issues of its own, according to results of the REACH PC trial.
Jennifer S. Temel, MD, of Massachusetts General Hospital in Boston, discussed the use of telemedicine in palliative care, including results from REACH PC, during an educational session at the ASCO Virtual Quality Care Symposium 2020.
Dr. Temel noted that, for cancer patients, an in-person visit with a palliative care specialist can cost time, induce fatigue, and increase financial burden from transportation and parking expenses.
For caregivers and family, an in-person visit may necessitate absence from family and/or work, require complex scheduling to coordinate with other office visits, and result in additional transportation and/or parking expenses.
For health care systems, to have a dedicated palliative care clinic requires precious space and financial expenditures for office personnel and other resources.
These issues make it attractive to consider whether telehealth could be used for palliative care services.
Scarcity of palliative care specialists
In the United States, there is roughly 1 palliative care physician for every 20,000 older adults with a life-limiting illness, according to research published in Annual Review of Public Health in 2014.
In its 2019 state-by-state report card, the Center to Advance Palliative Care noted that only 72% of U.S. hospitals with 50 or more beds have a palliative care team.
For patients with serious illnesses and those who are socioeconomically or geographically disadvantaged, palliative care is often inaccessible.
Inefficiencies in the current system are an additional impediment. Palliative care specialists frequently see patients during a portion of the patient’s routine visit to subspecialty or primary care clinics. This limits the palliative care specialist’s ability to perform comprehensive assessments and provide patient-centered care efficiently.
Special considerations regarding telehealth for palliative care
As a specialty, palliative care involves interactions that could make the use of telehealth problematic. For example, conveyance of interest, warmth, and touch are challenging or impossible in a video format.
Palliative care specialists engage with patients regarding relatively serious topics such as prognosis and end-of-life preferences. There is uncertainty about how those discussions would be received by patients and their caregivers via video.
Furthermore, there are logistical impediments such as prescribing opioids with video or across state lines.
Despite these concerns, the ENABLE study showed that supplementing usual oncology care with weekly (transitioning to monthly) telephone-based educational palliative care produced higher quality of life and mood than did usual oncology care alone. These results were published in JAMA in 2009.
REACH PC study demonstrates feasibility of telehealth model
Dr. Temel described the ongoing REACH PC trial in which palliative care is delivered via video visits and compared with in-person palliative care for patients with advanced non–small cell lung cancer.
The primary aim of REACH PC is to determine whether telehealth palliative care is equivalent to traditional palliative care in improving quality of life as a supplement to routine oncology care.
Currently, REACH PC has enrolled 581 patients at its 20 sites, spanning a geographically diverse area. Just over half of patients approached about REACH PC agreed to enroll in it. Ultimately, 1,250 enrollees are sought.
Among patients who declined to participate, 7.6% indicated “discomfort with technology” as the reason. Most refusals were due to lack of interest in research (35.1%) and/or palliative care (22.9%).
Older adults were prominent among enrollees. More than 60% were older than 60 years of age, and more than one-third were older than 70 years.
Among patients who began the trial, there were slightly more withdrawals in the telehealth participants, in comparison with in-person participants (13.6% versus 9.1%).
When palliative care clinicians were queried about video visits, 64.3% said there were no challenges. This is comparable to the 65.5% of clinicians who had no challenges with in-person visits.
When problems occurred with video visits, they were most frequently technical (19.1%). Only 1.4% of clinicians reported difficulty addressing topics that felt uncomfortable over video, and 1.5% reported difficulty establishing rapport.
The success rates of video and in-person visits were similar. About 80% of visits accomplished planned goals.
‘Webside’ manner
Strategies such as reflective listening and summarizing what patients say (to verify an accurate understanding of the patient’s perspective) are key to successful palliative care visits, regardless of the setting.
For telehealth visits, Dr. Temel described techniques she defined as “webside manner,” to compensate for the inability of the clinician to touch a patient. These techniques include leaning in toward the camera, nodding, and pausing to be certain the patient has finished speaking before the clinician speaks again.
Is telehealth the future of palliative care?
I include myself among those oncologists who have voiced concern about moving from face-to-face to remote visits for complicated consultations such as those required for palliative care. Nonetheless, from the preliminary results of the REACH PC trial, it appears that telehealth could be a valuable tool.
To minimize differences between in-person and remote delivery of palliative care, practical strategies for ensuring rapport and facilitating a trusting relationship should be defined further and disseminated.
In addition, we need to be vigilant for widening inequities of care from rapid movement to the use of technology (i.e., an equity gap). In their telehealth experience during the COVID-19 pandemic, investigators at Houston Methodist Cancer Center found that patients declining virtual visits tended to be older, lower-income, and less likely to have commercial insurance. These results were recently published in JCO Oncology Practice.
For the foregoing reasons, hybrid systems for palliative care services will probably always be needed.
Going forward, we should heed the advice of Alvin Toffler in his book Future Shock. Mr. Toffler said, “The illiterate of the 21st century will not be those who cannot read and write, but those who cannot learn, unlearn, and relearn.”
The traditional model for delivering palliative care will almost certainly need to be reimagined and relearned.
Dr. Temel disclosed institutional research funding from Pfizer.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Traditional delivery of palliative care to outpatients with cancer is associated with many challenges.
Telehealth can eliminate some of these challenges but comes with issues of its own, according to results of the REACH PC trial.
Jennifer S. Temel, MD, of Massachusetts General Hospital in Boston, discussed the use of telemedicine in palliative care, including results from REACH PC, during an educational session at the ASCO Virtual Quality Care Symposium 2020.
Dr. Temel noted that, for cancer patients, an in-person visit with a palliative care specialist can cost time, induce fatigue, and increase financial burden from transportation and parking expenses.
For caregivers and family, an in-person visit may necessitate absence from family and/or work, require complex scheduling to coordinate with other office visits, and result in additional transportation and/or parking expenses.
For health care systems, to have a dedicated palliative care clinic requires precious space and financial expenditures for office personnel and other resources.
These issues make it attractive to consider whether telehealth could be used for palliative care services.
Scarcity of palliative care specialists
In the United States, there is roughly 1 palliative care physician for every 20,000 older adults with a life-limiting illness, according to research published in Annual Review of Public Health in 2014.
In its 2019 state-by-state report card, the Center to Advance Palliative Care noted that only 72% of U.S. hospitals with 50 or more beds have a palliative care team.
For patients with serious illnesses and those who are socioeconomically or geographically disadvantaged, palliative care is often inaccessible.
Inefficiencies in the current system are an additional impediment. Palliative care specialists frequently see patients during a portion of the patient’s routine visit to subspecialty or primary care clinics. This limits the palliative care specialist’s ability to perform comprehensive assessments and provide patient-centered care efficiently.
Special considerations regarding telehealth for palliative care
As a specialty, palliative care involves interactions that could make the use of telehealth problematic. For example, conveyance of interest, warmth, and touch are challenging or impossible in a video format.
Palliative care specialists engage with patients regarding relatively serious topics such as prognosis and end-of-life preferences. There is uncertainty about how those discussions would be received by patients and their caregivers via video.
Furthermore, there are logistical impediments such as prescribing opioids with video or across state lines.
Despite these concerns, the ENABLE study showed that supplementing usual oncology care with weekly (transitioning to monthly) telephone-based educational palliative care produced higher quality of life and mood than did usual oncology care alone. These results were published in JAMA in 2009.
REACH PC study demonstrates feasibility of telehealth model
Dr. Temel described the ongoing REACH PC trial in which palliative care is delivered via video visits and compared with in-person palliative care for patients with advanced non–small cell lung cancer.
The primary aim of REACH PC is to determine whether telehealth palliative care is equivalent to traditional palliative care in improving quality of life as a supplement to routine oncology care.
Currently, REACH PC has enrolled 581 patients at its 20 sites, spanning a geographically diverse area. Just over half of patients approached about REACH PC agreed to enroll in it. Ultimately, 1,250 enrollees are sought.
Among patients who declined to participate, 7.6% indicated “discomfort with technology” as the reason. Most refusals were due to lack of interest in research (35.1%) and/or palliative care (22.9%).
Older adults were prominent among enrollees. More than 60% were older than 60 years of age, and more than one-third were older than 70 years.
Among patients who began the trial, there were slightly more withdrawals in the telehealth participants, in comparison with in-person participants (13.6% versus 9.1%).
When palliative care clinicians were queried about video visits, 64.3% said there were no challenges. This is comparable to the 65.5% of clinicians who had no challenges with in-person visits.
When problems occurred with video visits, they were most frequently technical (19.1%). Only 1.4% of clinicians reported difficulty addressing topics that felt uncomfortable over video, and 1.5% reported difficulty establishing rapport.
The success rates of video and in-person visits were similar. About 80% of visits accomplished planned goals.
‘Webside’ manner
Strategies such as reflective listening and summarizing what patients say (to verify an accurate understanding of the patient’s perspective) are key to successful palliative care visits, regardless of the setting.
For telehealth visits, Dr. Temel described techniques she defined as “webside manner,” to compensate for the inability of the clinician to touch a patient. These techniques include leaning in toward the camera, nodding, and pausing to be certain the patient has finished speaking before the clinician speaks again.
Is telehealth the future of palliative care?
I include myself among those oncologists who have voiced concern about moving from face-to-face to remote visits for complicated consultations such as those required for palliative care. Nonetheless, from the preliminary results of the REACH PC trial, it appears that telehealth could be a valuable tool.
To minimize differences between in-person and remote delivery of palliative care, practical strategies for ensuring rapport and facilitating a trusting relationship should be defined further and disseminated.
In addition, we need to be vigilant for widening inequities of care from rapid movement to the use of technology (i.e., an equity gap). In their telehealth experience during the COVID-19 pandemic, investigators at Houston Methodist Cancer Center found that patients declining virtual visits tended to be older, lower-income, and less likely to have commercial insurance. These results were recently published in JCO Oncology Practice.
For the foregoing reasons, hybrid systems for palliative care services will probably always be needed.
Going forward, we should heed the advice of Alvin Toffler in his book Future Shock. Mr. Toffler said, “The illiterate of the 21st century will not be those who cannot read and write, but those who cannot learn, unlearn, and relearn.”
The traditional model for delivering palliative care will almost certainly need to be reimagined and relearned.
Dr. Temel disclosed institutional research funding from Pfizer.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
FROM ASCO QUALITY CARE SYMPOSIUM 2020
GLIMMER of hope for itch in primary biliary cholangitis
Patients with primary biliary cholangitis experienced rapid improvements in itch and quality of life after treatment with linerixibat in a randomized, placebo-controlled trial of the safety, efficacy, and tolerability of the small-molecule drug.
Moderate to severe pruritus “affects patients’ quality of life and is a huge burden for them,” said investigator Cynthia Levy, MD, from the University of Miami Health System.
“Finally having a medication that controls those symptoms is really important,” she said in an interview.
With a twice-daily mid-range dose of the drug for 12 weeks, patients with moderate to severe itch reported significantly less itch and better social and emotional quality of life, Dr. Levy reported at the Liver Meeting, where she presented findings from the phase 2 GLIMMER trial.
After a single-blind 4-week placebo run-in period for patients with itch scores of at least 4 on a 10-point rating scale, those with itch scores of at least 3 were then randomly assigned to one of five treatment regimens – once-daily linerixibat at doses of 20 mg, 90 mg, or 180 mg, or twice-daily doses of 40 mg or 90 mg – or to placebo.
After 12 weeks of treatment, all 147 participants once again received placebo for 4 weeks.
During the trial, participants recorded itch levels twice daily. The worst of these daily scores was averaged every 7 days to determine the mean worst daily itch.
The primary study endpoint was the change in worst daily itch from baseline after 12 weeks of treatment. Participants whose self-rated itch improved by 2 points on the 10-point scale were considered to have had a response to the drug.
Participants also completed the PBC-40, an instrument to measure quality of life in patients with primary biliary cholangitis, answering questions about itch and social and emotional status.
Reductions in worst daily itch from baseline to 12 weeks were steepest in the 40-mg twice-daily group, at 2.86 points, and in the 90-mg twice-daily group, at 2.25 points. In the placebo group, the mean decrease was 1.73 points.
During the subsequent 4 weeks of placebo, after treatment ended, the itch relief faded in all groups.
Scores on the PBC-40 itch domain improved significantly in every group, including placebo. However, only those in the twice-daily 40-mg group saw significant improvements on the social (P = .0016) and emotional (P = .0025) domains.
‘Between incremental and revolutionary’
The results are on a “kind of continuum between incremental and revolutionary,” said Jonathan A. Dranoff, MD, from the University of Arkansas for Medical Sciences, Little Rock, who was not involved in the study. “It doesn’t hit either extreme, but it’s the first new drug for this purpose in forever, which by itself is a good thing.”
The placebo effect suggests that “maybe the actual contribution of the noncognitive brain to pruritus is bigger than we thought, and that’s worth noting,” he added. Nevertheless, “the drug still appears to have effects that are statistically different from placebo.”
The placebo effect in itching studies is always high but tends to wane over time, said Dr. Levy. This trial had a 4-week placebo run-in period to allow that effect to fade somewhat, she explained.
About 10% of the study cohort experienced drug-related diarrhea, which was expected, and about 10% dropped out of the trial because of drug-related adverse events.
Linerixibat is an ileal sodium-dependent bile acid transporter inhibitor, so the gut has to deal with the excess bile acid fallout, but the diarrhea is likely manageable with antidiarrheals, said Dr. Levy.
It is unlikely that diarrhea will deter patients with severe itch from using an effective drug when other drugs have failed them. “These patients are consumed by itch most of the time,” said Dr. Dranoff. “I think for people who don’t regularly treat patients with primary biliary cholangitis, it’s one of the underappreciated aspects of the disease.”
The improvements in social and emotional quality of life seen with linerixibat are not only statistically significant, they are also clinically significant, said Dr. Levy. “We are really expecting this to impact the lives of our patients and are looking forward to phase 3.”
Dr. Levy disclosed support from GlaxoSmithKline. Dr. Dranoff disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Patients with primary biliary cholangitis experienced rapid improvements in itch and quality of life after treatment with linerixibat in a randomized, placebo-controlled trial of the safety, efficacy, and tolerability of the small-molecule drug.
Moderate to severe pruritus “affects patients’ quality of life and is a huge burden for them,” said investigator Cynthia Levy, MD, from the University of Miami Health System.
“Finally having a medication that controls those symptoms is really important,” she said in an interview.
With a twice-daily mid-range dose of the drug for 12 weeks, patients with moderate to severe itch reported significantly less itch and better social and emotional quality of life, Dr. Levy reported at the Liver Meeting, where she presented findings from the phase 2 GLIMMER trial.
After a single-blind 4-week placebo run-in period for patients with itch scores of at least 4 on a 10-point rating scale, those with itch scores of at least 3 were then randomly assigned to one of five treatment regimens – once-daily linerixibat at doses of 20 mg, 90 mg, or 180 mg, or twice-daily doses of 40 mg or 90 mg – or to placebo.
After 12 weeks of treatment, all 147 participants once again received placebo for 4 weeks.
During the trial, participants recorded itch levels twice daily. The worst of these daily scores was averaged every 7 days to determine the mean worst daily itch.
The primary study endpoint was the change in worst daily itch from baseline after 12 weeks of treatment. Participants whose self-rated itch improved by 2 points on the 10-point scale were considered to have had a response to the drug.
Participants also completed the PBC-40, an instrument to measure quality of life in patients with primary biliary cholangitis, answering questions about itch and social and emotional status.
Reductions in worst daily itch from baseline to 12 weeks were steepest in the 40-mg twice-daily group, at 2.86 points, and in the 90-mg twice-daily group, at 2.25 points. In the placebo group, the mean decrease was 1.73 points.
During the subsequent 4 weeks of placebo, after treatment ended, the itch relief faded in all groups.
Scores on the PBC-40 itch domain improved significantly in every group, including placebo. However, only those in the twice-daily 40-mg group saw significant improvements on the social (P = .0016) and emotional (P = .0025) domains.
‘Between incremental and revolutionary’
The results are on a “kind of continuum between incremental and revolutionary,” said Jonathan A. Dranoff, MD, from the University of Arkansas for Medical Sciences, Little Rock, who was not involved in the study. “It doesn’t hit either extreme, but it’s the first new drug for this purpose in forever, which by itself is a good thing.”
The placebo effect suggests that “maybe the actual contribution of the noncognitive brain to pruritus is bigger than we thought, and that’s worth noting,” he added. Nevertheless, “the drug still appears to have effects that are statistically different from placebo.”
The placebo effect in itching studies is always high but tends to wane over time, said Dr. Levy. This trial had a 4-week placebo run-in period to allow that effect to fade somewhat, she explained.
About 10% of the study cohort experienced drug-related diarrhea, which was expected, and about 10% dropped out of the trial because of drug-related adverse events.
Linerixibat is an ileal sodium-dependent bile acid transporter inhibitor, so the gut has to deal with the excess bile acid fallout, but the diarrhea is likely manageable with antidiarrheals, said Dr. Levy.
It is unlikely that diarrhea will deter patients with severe itch from using an effective drug when other drugs have failed them. “These patients are consumed by itch most of the time,” said Dr. Dranoff. “I think for people who don’t regularly treat patients with primary biliary cholangitis, it’s one of the underappreciated aspects of the disease.”
The improvements in social and emotional quality of life seen with linerixibat are not only statistically significant, they are also clinically significant, said Dr. Levy. “We are really expecting this to impact the lives of our patients and are looking forward to phase 3.”
Dr. Levy disclosed support from GlaxoSmithKline. Dr. Dranoff disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Patients with primary biliary cholangitis experienced rapid improvements in itch and quality of life after treatment with linerixibat in a randomized, placebo-controlled trial of the safety, efficacy, and tolerability of the small-molecule drug.
Moderate to severe pruritus “affects patients’ quality of life and is a huge burden for them,” said investigator Cynthia Levy, MD, from the University of Miami Health System.
“Finally having a medication that controls those symptoms is really important,” she said in an interview.
With a twice-daily mid-range dose of the drug for 12 weeks, patients with moderate to severe itch reported significantly less itch and better social and emotional quality of life, Dr. Levy reported at the Liver Meeting, where she presented findings from the phase 2 GLIMMER trial.
After a single-blind 4-week placebo run-in period for patients with itch scores of at least 4 on a 10-point rating scale, those with itch scores of at least 3 were then randomly assigned to one of five treatment regimens – once-daily linerixibat at doses of 20 mg, 90 mg, or 180 mg, or twice-daily doses of 40 mg or 90 mg – or to placebo.
After 12 weeks of treatment, all 147 participants once again received placebo for 4 weeks.
During the trial, participants recorded itch levels twice daily. The worst of these daily scores was averaged every 7 days to determine the mean worst daily itch.
The primary study endpoint was the change in worst daily itch from baseline after 12 weeks of treatment. Participants whose self-rated itch improved by 2 points on the 10-point scale were considered to have had a response to the drug.
Participants also completed the PBC-40, an instrument to measure quality of life in patients with primary biliary cholangitis, answering questions about itch and social and emotional status.
Reductions in worst daily itch from baseline to 12 weeks were steepest in the 40-mg twice-daily group, at 2.86 points, and in the 90-mg twice-daily group, at 2.25 points. In the placebo group, the mean decrease was 1.73 points.
During the subsequent 4 weeks of placebo, after treatment ended, the itch relief faded in all groups.
Scores on the PBC-40 itch domain improved significantly in every group, including placebo. However, only those in the twice-daily 40-mg group saw significant improvements on the social (P = .0016) and emotional (P = .0025) domains.
‘Between incremental and revolutionary’
The results are on a “kind of continuum between incremental and revolutionary,” said Jonathan A. Dranoff, MD, from the University of Arkansas for Medical Sciences, Little Rock, who was not involved in the study. “It doesn’t hit either extreme, but it’s the first new drug for this purpose in forever, which by itself is a good thing.”
The placebo effect suggests that “maybe the actual contribution of the noncognitive brain to pruritus is bigger than we thought, and that’s worth noting,” he added. Nevertheless, “the drug still appears to have effects that are statistically different from placebo.”
The placebo effect in itching studies is always high but tends to wane over time, said Dr. Levy. This trial had a 4-week placebo run-in period to allow that effect to fade somewhat, she explained.
About 10% of the study cohort experienced drug-related diarrhea, which was expected, and about 10% dropped out of the trial because of drug-related adverse events.
Linerixibat is an ileal sodium-dependent bile acid transporter inhibitor, so the gut has to deal with the excess bile acid fallout, but the diarrhea is likely manageable with antidiarrheals, said Dr. Levy.
It is unlikely that diarrhea will deter patients with severe itch from using an effective drug when other drugs have failed them. “These patients are consumed by itch most of the time,” said Dr. Dranoff. “I think for people who don’t regularly treat patients with primary biliary cholangitis, it’s one of the underappreciated aspects of the disease.”
The improvements in social and emotional quality of life seen with linerixibat are not only statistically significant, they are also clinically significant, said Dr. Levy. “We are really expecting this to impact the lives of our patients and are looking forward to phase 3.”
Dr. Levy disclosed support from GlaxoSmithKline. Dr. Dranoff disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
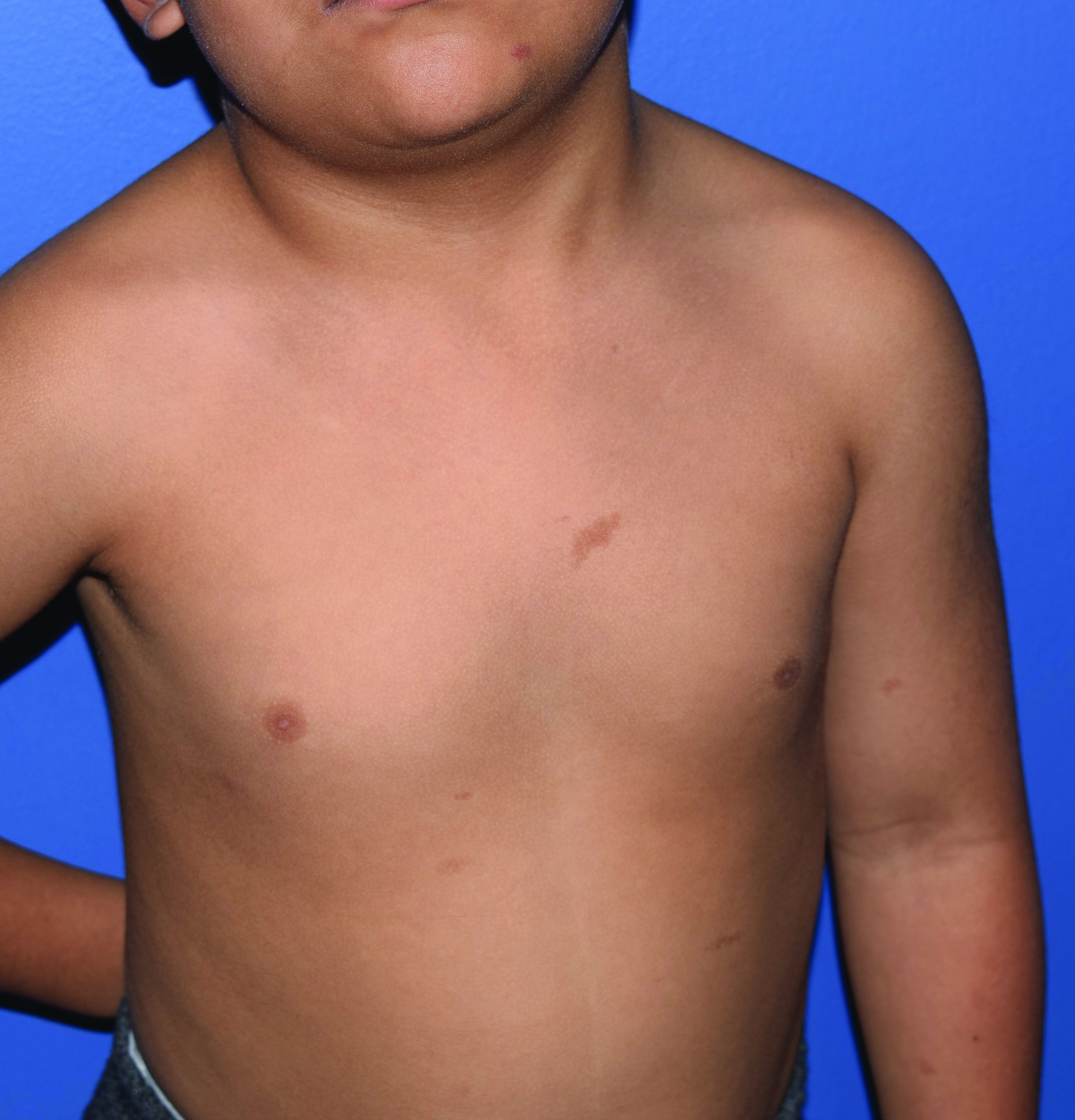
A 4-year-old presented to our pediatric dermatology clinic for evaluation of asymptomatic "brown spots."
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1

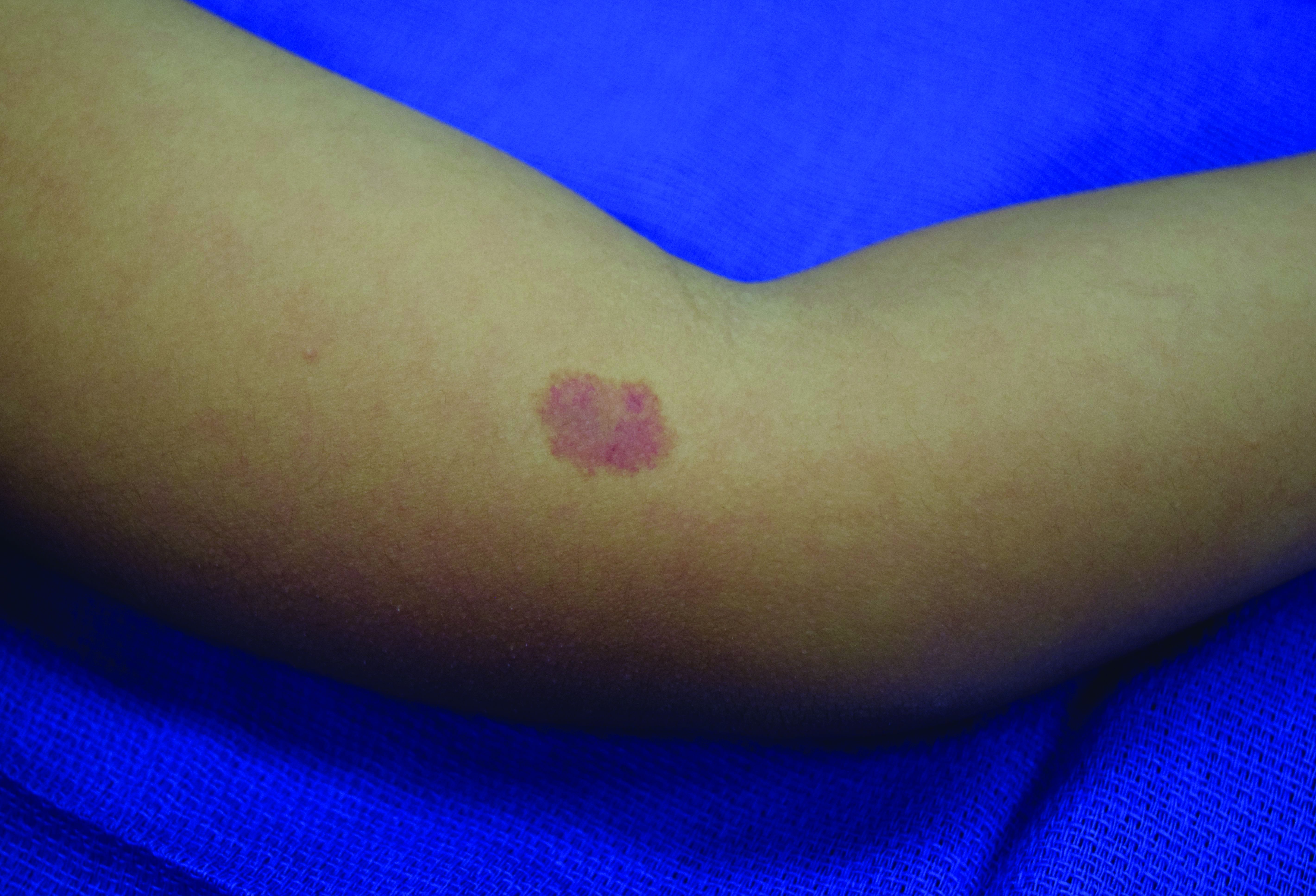
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4

It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.

Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Capillary malformation-arteriovenous malformation syndrome
with or without arteriovenous malformations, as well as arteriovenous fistulas (AVFs). CM-AVM is an autosomal dominant disorder.1 CM-AVM type 1 is caused by mutations in the RASA1 gene, and CM-AVM type 2 is caused by mutations in the EPHB4 gene.2 Approximately 70% of patients with RASA1-associated CM-AVM syndrome and 80% of patients with EPHB4-associated CM-AVM syndrome have an affected parent, while the remainder have de novo variants.1
In patients with CM-AVM syndrome, CMs are often present at birth and more are typically acquired over time. CMs are characteristically 1-3 cm in diameter, round or oval, dull red or red-brown macules and patches with a blanched halo.3 Some CMs may be warm to touch indicating a possible underlying AVM or AVF.4 This can be confirmed by Doppler ultrasound, which would demonstrate increased arterial flow.4 CMs are most commonly located on the face and limbs and may present in isolation, but approximately one-third of patients have associated AVMs and AVFs.1,5 These high-flow vascular malformations may be present in skin, muscle, bone, brain, and/or spine and may be asymptomatic or lead to serious sequelae, including bleeding, congestive heart failure, and neurologic complications, such as migraine headaches, seizures, or even stroke.5 Symptoms from intracranial and spinal high-flow lesions usually present in early childhood and affect approximately 7% of patients.3
The diagnosis of CM-AVM should be suspected in an individual with numerous characteristic CMs and may be supported by the presence of AVMs and AVFs, family history of CM-AVM, and/or identification of RASA1 or EPHB4 mutation by molecular genetic testing.1,3 Although there are no consensus protocols for imaging CM-AVM patients, MRI of the brain and spine is recommended at diagnosis to identify underlying high-flow lesions.1 This may allow for early treatment before the development of symptoms.1 Any lesions identified on screening imaging may require regular surveillance, which is best determined by discussion with the radiologist.1 Although there are no reports of patients with negative results on screening imaging who later develop AVMs or AVFs, there should be a low threshold for repeat imaging in patients who develop new symptoms or physical exam findings.3,4
It has previously been suggested that the CMs in CM-AVM may actually represent early or small AVMs and pulsed-dye laser (PDL) treatment was not recommended because of concern for potential progression of lesions.4 However, a recent study demonstrated good response to PDL in patients with CM-AVM with no evidence of worsening or recurrence of lesions with long-term follow-up.6 Treatment of CMs that cause cosmetic concerns may be considered following discussion of risks and benefits with a dermatologist. Management of AVMs and AVFs requires a multidisciplinary team that, depending on location and symptoms of these features, may require the expertise of specialists such as neurosurgery, surgery, orthopedics, cardiology, and/or interventional radiology.1
Given the suspicion for CM-AVM in our patient, further workup was completed. A skin biopsy was consistent with CM. Genetic testing with the Vascular Malformations Panel, Sequencing and Deletion/Duplication revealed a pathogenic variant in the RASA1 gene and a variant of unknown clinical significance in the TEK gene. Parental genetic testing for the RASA1 mutation was negative, supporting a de novo mutation in the patient. CNS imaging showed a small developmental venous malformation in the brain that neurosurgery did not think was clinically significant. At the most recent follow-up at age 8 years, our patient had developed a few new small CMs but was otherwise well.
Dr. Leszczynska is trained in pediatrics and is the current dermatology research fellow at the University of Texas at Austin. Ms. Croce is a dermatology-trained pediatric nurse practitioner and PhD student at the University of Texas at Austin School of Nursing. Dr. Diaz is chief of pediatric dermatology at Dell Children’s Medical Center, Austin, assistant professor of pediatrics and medicine (dermatology), and dermatology residency associate program director at University of Texas at Austin . The authors have no relevant conflicts of interest to disclose. Donna Bilu Martin, MD, is the editor of this column.
References
1. Bayrak-Toydemir P, Stevenson D. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle: University of Washington, Seattle; February 22, 2011.
2.Yu J et al. Pediatr Dermatol. 2017 Sep;34(5):e227-30.
3. Orme CM et al. Pediatr Dermatol. 2013 Jul-Aug;30(4):409-15.
4. Weitz NA et al. Pediatr Dermatol. 2015 Jan-Feb;32(1):76-84.
5. Revencu N et al. Hum Mutat. 2013 Dec;34(12):1632-41.
6. Iznardo H et al. Pediatr Dermatol. 2020 Mar;37(2):342-44.
Higher cardiovascular risks in Kawasaki disease persist 10-plus years
Risks are highest in first year.
Survivors of Kawasaki disease remain at a higher long-term risk for cardiovascular events into young adulthood, including myocardial infarction, compared to people without the disease, new evidence reveals. The elevated risks emerged in survivors both with and without cardiovascular involvement at the time of initial diagnosis.

Overall risk of cardiovascular events was highest in the first year following Kawasaki disease diagnosis, and about 10 times greater than in healthy children, Cal Robinson, MD, said during a press conference at the virtual annual meeting of the American College of Rheumatology.
“The risk gradually decreased over time. However, even 10 years after diagnosis of their illness, they still had a 39% higher risk,” said study author Dr. Robinson, a PGY4 pediatric nephrology fellow at The Hospital for Sick Children in Toronto.
Dr. Robinson also put the numbers in perspective. “We fully acknowledged these are very rare events in children, especially healthy children, which is why we needed such a large cohort to study this. Interpret the numbers cautiously.”
In terms of patient and family counseling, “I would say children with Kawasaki disease have a higher risk of myocardial infarction, but the absolute risk is still low,” he added. For example, 16 Kawasaki disease survivors experienced a heart attack during follow-up, or 0.4% of the affected study population, compared to a rate of 0.1% among matched controls.
“These families are often very frightened after the initial Kawasaki disease diagnosis,” Dr. Robinson said. “We have to balance some discussion with what we know about Kawasaki disease without overly scaring or terrifying these families, who are already anxious.”
To quantify the incidence and timing of cardiovascular events and cardiac disease following diagnosis, Dr. Robinson and colleagues assessed large databases representing approximately 3 million children. They focused on children hospitalized with a Kawasaki disease diagnosis between 1995 and 2018. These children had a median length of stay of 3 days and 2.5% were admitted to critical care. The investigators matched his population 1:100 to unaffected children in Ontario.
Follow-up was up to 24 years (median, 11 years) in this retrospective, population-based cohort study.
Risks raised over a decade and beyond
Compared to matched controls, Kawasaki disease survivors had a higher risk for a cardiac event in the first year following diagnosis (adjusted hazard ratio, 11.65; 95% confidence interval, 10.34-13.13). The 1- to 5-year risk was lower (aHR, 3.35), a trend that continued between 5 and 10 years (aHR, 1.87) and as well as after more than 10 years (aHR, 1.39).
The risk of major adverse cardiac events (MACE, a composite of myocardial infarction, stroke, or cardiovascular death) was likewise highest in the first year after diagnosis (aHR, 3.27), followed by a 51% greater risk at 1-5 years, a 113% increased risk at 5-10 years, and a 17% elevated risk after 10 years.
The investigators compared the 144 Kawasaki disease survivors who experienced a coronary artery aneurysm (CAA) within 90 days of hospital admission to the 4,453 others who did not have a CAA. The risk for a composite cardiovascular event was elevated at each time point among those with a history of CAA, especially in the first year. The adjusted HR was 33.12 in the CAA group versus 10.44 in the non-CAA group.
“The most interesting finding of this study was that children with Kawasaki syndrome are at higher risk for composite cardiovascular events and major adverse cardiac events even if they were not diagnosed with coronary artery aneurysm,” session comoderator Shervin Assassi, MD, professor of medicine and director of division of rheumatology at the University of Texas Health Science Center at Houston, said when asked to comment.
Dr. Robinson and colleagues also looked at outcomes based on presence or absence of coronary involvement at the time of Kawasaki disease diagnosis. For example, among those with initial coronary involvement, 15% later experienced a cardiovascular event and 10% experienced a major cardiovascular event.
“However, we were specifically interested in looking at children without initial coronary involvement. In this group, we also found these children were at increased risk for cardiovascular events compared to children without Kawasaki disease,” Dr. Robinson said. He said the distinction is important because approximately 95% of children diagnosed with Kawasaki disease do not feature initial coronary involvement.
In terms of clinical care, “our data provides an early signal that Kawasaki disease survivors – including those without initial coronary involvement – may be at higher risk of cardiovascular events into early adulthood.”
A call for closer monitoring
“Based on our results, we find that Kawasaki disease survivors may benefit from additional follow-up and surveillance for cardiovascular disease risk factors, such as obesity, high blood pressure, and high cholesterol,” Dr. Robinson said. Early identification of heightened risk could allow physicians to more closely monitor this subgroup and emphasize potentially beneficial lifestyle modifications, including increasing physical activity, implementing a heart healthy diet, and avoiding smoking.
Mortality was not significantly different between groups. “Despite the risk of cardiac events we found, death was uncommon,” Dr. Robinson said. Among children with Kawasaki disease, 1 in 500 died during follow-up, so “the risk of death was actually lower than for children without Kawasaki disease.”
Similar findings of lower mortality have been reported in research out of Japan, he added during a plenary presentation at ACR 2020. Future research is warranted to evaluate this finding further, Dr. Robinson said.
Future plans
Going forward, the investigators plan to evaluate noncardiovascular outcomes in this patient population. They would also like to examine health care utilization following a diagnosis of Kawasaki disease “to better understand what kind of follow-up is happening now in Ontario,” Dr. Robinson said.
Another unanswered question is whether the cardiovascular events observed in the study stem from atherosclerotic disease or a different mechanism among survivors of Kawasaki disease.
The research was supported by a McMaster University Resident Research Grant, a Hamilton Health Sciences New Investigator Award, and Ontario’s Institute for Clinical Evaluative Sciences. Dr. Robinson had no relevant financial disclosures.
SOURCE: Robinson C et al. Arthritis Rheumatol. 2020;72(suppl 10): Abstract 0937.
Risks are highest in first year.
Risks are highest in first year.
Survivors of Kawasaki disease remain at a higher long-term risk for cardiovascular events into young adulthood, including myocardial infarction, compared to people without the disease, new evidence reveals. The elevated risks emerged in survivors both with and without cardiovascular involvement at the time of initial diagnosis.
Overall risk of cardiovascular events was highest in the first year following Kawasaki disease diagnosis, and about 10 times greater than in healthy children, Cal Robinson, MD, said during a press conference at the virtual annual meeting of the American College of Rheumatology.
“The risk gradually decreased over time. However, even 10 years after diagnosis of their illness, they still had a 39% higher risk,” said study author Dr. Robinson, a PGY4 pediatric nephrology fellow at The Hospital for Sick Children in Toronto.
Dr. Robinson also put the numbers in perspective. “We fully acknowledged these are very rare events in children, especially healthy children, which is why we needed such a large cohort to study this. Interpret the numbers cautiously.”
In terms of patient and family counseling, “I would say children with Kawasaki disease have a higher risk of myocardial infarction, but the absolute risk is still low,” he added. For example, 16 Kawasaki disease survivors experienced a heart attack during follow-up, or 0.4% of the affected study population, compared to a rate of 0.1% among matched controls.
“These families are often very frightened after the initial Kawasaki disease diagnosis,” Dr. Robinson said. “We have to balance some discussion with what we know about Kawasaki disease without overly scaring or terrifying these families, who are already anxious.”
To quantify the incidence and timing of cardiovascular events and cardiac disease following diagnosis, Dr. Robinson and colleagues assessed large databases representing approximately 3 million children. They focused on children hospitalized with a Kawasaki disease diagnosis between 1995 and 2018. These children had a median length of stay of 3 days and 2.5% were admitted to critical care. The investigators matched his population 1:100 to unaffected children in Ontario.
Follow-up was up to 24 years (median, 11 years) in this retrospective, population-based cohort study.
Risks raised over a decade and beyond
Compared to matched controls, Kawasaki disease survivors had a higher risk for a cardiac event in the first year following diagnosis (adjusted hazard ratio, 11.65; 95% confidence interval, 10.34-13.13). The 1- to 5-year risk was lower (aHR, 3.35), a trend that continued between 5 and 10 years (aHR, 1.87) and as well as after more than 10 years (aHR, 1.39).
The risk of major adverse cardiac events (MACE, a composite of myocardial infarction, stroke, or cardiovascular death) was likewise highest in the first year after diagnosis (aHR, 3.27), followed by a 51% greater risk at 1-5 years, a 113% increased risk at 5-10 years, and a 17% elevated risk after 10 years.
The investigators compared the 144 Kawasaki disease survivors who experienced a coronary artery aneurysm (CAA) within 90 days of hospital admission to the 4,453 others who did not have a CAA. The risk for a composite cardiovascular event was elevated at each time point among those with a history of CAA, especially in the first year. The adjusted HR was 33.12 in the CAA group versus 10.44 in the non-CAA group.
“The most interesting finding of this study was that children with Kawasaki syndrome are at higher risk for composite cardiovascular events and major adverse cardiac events even if they were not diagnosed with coronary artery aneurysm,” session comoderator Shervin Assassi, MD, professor of medicine and director of division of rheumatology at the University of Texas Health Science Center at Houston, said when asked to comment.
Dr. Robinson and colleagues also looked at outcomes based on presence or absence of coronary involvement at the time of Kawasaki disease diagnosis. For example, among those with initial coronary involvement, 15% later experienced a cardiovascular event and 10% experienced a major cardiovascular event.
“However, we were specifically interested in looking at children without initial coronary involvement. In this group, we also found these children were at increased risk for cardiovascular events compared to children without Kawasaki disease,” Dr. Robinson said. He said the distinction is important because approximately 95% of children diagnosed with Kawasaki disease do not feature initial coronary involvement.
In terms of clinical care, “our data provides an early signal that Kawasaki disease survivors – including those without initial coronary involvement – may be at higher risk of cardiovascular events into early adulthood.”
A call for closer monitoring
“Based on our results, we find that Kawasaki disease survivors may benefit from additional follow-up and surveillance for cardiovascular disease risk factors, such as obesity, high blood pressure, and high cholesterol,” Dr. Robinson said. Early identification of heightened risk could allow physicians to more closely monitor this subgroup and emphasize potentially beneficial lifestyle modifications, including increasing physical activity, implementing a heart healthy diet, and avoiding smoking.
Mortality was not significantly different between groups. “Despite the risk of cardiac events we found, death was uncommon,” Dr. Robinson said. Among children with Kawasaki disease, 1 in 500 died during follow-up, so “the risk of death was actually lower than for children without Kawasaki disease.”
Similar findings of lower mortality have been reported in research out of Japan, he added during a plenary presentation at ACR 2020. Future research is warranted to evaluate this finding further, Dr. Robinson said.
Future plans
Going forward, the investigators plan to evaluate noncardiovascular outcomes in this patient population. They would also like to examine health care utilization following a diagnosis of Kawasaki disease “to better understand what kind of follow-up is happening now in Ontario,” Dr. Robinson said.
Another unanswered question is whether the cardiovascular events observed in the study stem from atherosclerotic disease or a different mechanism among survivors of Kawasaki disease.
The research was supported by a McMaster University Resident Research Grant, a Hamilton Health Sciences New Investigator Award, and Ontario’s Institute for Clinical Evaluative Sciences. Dr. Robinson had no relevant financial disclosures.
SOURCE: Robinson C et al. Arthritis Rheumatol. 2020;72(suppl 10): Abstract 0937.
Survivors of Kawasaki disease remain at a higher long-term risk for cardiovascular events into young adulthood, including myocardial infarction, compared to people without the disease, new evidence reveals. The elevated risks emerged in survivors both with and without cardiovascular involvement at the time of initial diagnosis.
Overall risk of cardiovascular events was highest in the first year following Kawasaki disease diagnosis, and about 10 times greater than in healthy children, Cal Robinson, MD, said during a press conference at the virtual annual meeting of the American College of Rheumatology.
“The risk gradually decreased over time. However, even 10 years after diagnosis of their illness, they still had a 39% higher risk,” said study author Dr. Robinson, a PGY4 pediatric nephrology fellow at The Hospital for Sick Children in Toronto.
Dr. Robinson also put the numbers in perspective. “We fully acknowledged these are very rare events in children, especially healthy children, which is why we needed such a large cohort to study this. Interpret the numbers cautiously.”
In terms of patient and family counseling, “I would say children with Kawasaki disease have a higher risk of myocardial infarction, but the absolute risk is still low,” he added. For example, 16 Kawasaki disease survivors experienced a heart attack during follow-up, or 0.4% of the affected study population, compared to a rate of 0.1% among matched controls.
“These families are often very frightened after the initial Kawasaki disease diagnosis,” Dr. Robinson said. “We have to balance some discussion with what we know about Kawasaki disease without overly scaring or terrifying these families, who are already anxious.”
To quantify the incidence and timing of cardiovascular events and cardiac disease following diagnosis, Dr. Robinson and colleagues assessed large databases representing approximately 3 million children. They focused on children hospitalized with a Kawasaki disease diagnosis between 1995 and 2018. These children had a median length of stay of 3 days and 2.5% were admitted to critical care. The investigators matched his population 1:100 to unaffected children in Ontario.
Follow-up was up to 24 years (median, 11 years) in this retrospective, population-based cohort study.
Risks raised over a decade and beyond
Compared to matched controls, Kawasaki disease survivors had a higher risk for a cardiac event in the first year following diagnosis (adjusted hazard ratio, 11.65; 95% confidence interval, 10.34-13.13). The 1- to 5-year risk was lower (aHR, 3.35), a trend that continued between 5 and 10 years (aHR, 1.87) and as well as after more than 10 years (aHR, 1.39).
The risk of major adverse cardiac events (MACE, a composite of myocardial infarction, stroke, or cardiovascular death) was likewise highest in the first year after diagnosis (aHR, 3.27), followed by a 51% greater risk at 1-5 years, a 113% increased risk at 5-10 years, and a 17% elevated risk after 10 years.
The investigators compared the 144 Kawasaki disease survivors who experienced a coronary artery aneurysm (CAA) within 90 days of hospital admission to the 4,453 others who did not have a CAA. The risk for a composite cardiovascular event was elevated at each time point among those with a history of CAA, especially in the first year. The adjusted HR was 33.12 in the CAA group versus 10.44 in the non-CAA group.
“The most interesting finding of this study was that children with Kawasaki syndrome are at higher risk for composite cardiovascular events and major adverse cardiac events even if they were not diagnosed with coronary artery aneurysm,” session comoderator Shervin Assassi, MD, professor of medicine and director of division of rheumatology at the University of Texas Health Science Center at Houston, said when asked to comment.
Dr. Robinson and colleagues also looked at outcomes based on presence or absence of coronary involvement at the time of Kawasaki disease diagnosis. For example, among those with initial coronary involvement, 15% later experienced a cardiovascular event and 10% experienced a major cardiovascular event.
“However, we were specifically interested in looking at children without initial coronary involvement. In this group, we also found these children were at increased risk for cardiovascular events compared to children without Kawasaki disease,” Dr. Robinson said. He said the distinction is important because approximately 95% of children diagnosed with Kawasaki disease do not feature initial coronary involvement.
In terms of clinical care, “our data provides an early signal that Kawasaki disease survivors – including those without initial coronary involvement – may be at higher risk of cardiovascular events into early adulthood.”
A call for closer monitoring
“Based on our results, we find that Kawasaki disease survivors may benefit from additional follow-up and surveillance for cardiovascular disease risk factors, such as obesity, high blood pressure, and high cholesterol,” Dr. Robinson said. Early identification of heightened risk could allow physicians to more closely monitor this subgroup and emphasize potentially beneficial lifestyle modifications, including increasing physical activity, implementing a heart healthy diet, and avoiding smoking.
Mortality was not significantly different between groups. “Despite the risk of cardiac events we found, death was uncommon,” Dr. Robinson said. Among children with Kawasaki disease, 1 in 500 died during follow-up, so “the risk of death was actually lower than for children without Kawasaki disease.”
Similar findings of lower mortality have been reported in research out of Japan, he added during a plenary presentation at ACR 2020. Future research is warranted to evaluate this finding further, Dr. Robinson said.
Future plans
Going forward, the investigators plan to evaluate noncardiovascular outcomes in this patient population. They would also like to examine health care utilization following a diagnosis of Kawasaki disease “to better understand what kind of follow-up is happening now in Ontario,” Dr. Robinson said.
Another unanswered question is whether the cardiovascular events observed in the study stem from atherosclerotic disease or a different mechanism among survivors of Kawasaki disease.
The research was supported by a McMaster University Resident Research Grant, a Hamilton Health Sciences New Investigator Award, and Ontario’s Institute for Clinical Evaluative Sciences. Dr. Robinson had no relevant financial disclosures.
SOURCE: Robinson C et al. Arthritis Rheumatol. 2020;72(suppl 10): Abstract 0937.
FROM ACR 2020
Key clinical point: Kawasaki disease survivors remain at elevated long-term risk for cardiovascular events.
Major finding: Overall cardiovascular event risk was 39% higher, even after 10 years.
Study details: A retrospective, population-based cohort study of more than 4,597 Kawasaki disease survivors and 459,700 matched children without Kawasaki disease.
Disclosures: The research was supported by a McMaster University Resident Research Grant, a Hamilton Health Sciences New Investigator Award, and Ontario’s Institute for Clinical Evaluative Sciences. Dr. Robinson had no relevant financial disclosures.
Source: Robinson C et al. Arthritis Rheumatol. 2020;72(suppl 10): Abstract 0937.
Gene-replacement therapy shows promise in X-linked myotubular myopathy
, according to research presented at the 2020 CNS-ICNA Conjoint Meeting, which was held virtually this year. The treatment also appears to improve patients’ motor function significantly and help them to achieve motor milestones.

The results come from a phase 1/2 study of two doses of AT132. Three of 17 patients who received the higher dose had fatal liver dysfunction. The researchers are investigating these cases and will communicate their findings.
X-linked myotubular myopathy is a rare and often fatal neuromuscular disease. Mutations in MTM1, which encodes the myotubularin enzyme that is required for the development and function of skeletal muscle, cause the disease, which affects about one in 50,000 to one in 40,000 newborn boys. The disease is associated with profound muscle weakness and impairment of neuromuscular and respiratory function. Patients with X-linked myotubular myopathy achieve motor milestones much later or not at all, and most require a ventilator or a feeding tube. The mortality by age 18 months is approximately 50%.
The ASPIRO trial
Investigators theorized that muscle tissue would be an appropriate therapeutic target because it does not display dystrophic or inflammatory changes in most patients. They identified adeno-associated virus AAV8 as a potential carrier for gene therapy, since it targets skeletal muscle effectively.
Nancy L. Kuntz, MD, an attending physician at Ann and Robert H. Lurie Children’s Hospital of Chicago, and colleagues conducted the ASPIRO trial to examine AT132 as a potential treatment for X-linked myotubular myopathy. Eligible patients were younger than 5 years or had previously enrolled in a natural history study of the disease, required ventilator support at baseline, and had no clinically significant underlying liver disease. Patients were randomly assigned to 1 × 1014 vg/kg of AAT132, 3 × 1014 vg/kg of AT132, or delayed treatment. Participants assigned to delayed treatment served as the study’s control group.
The study’s primary end points were safety and change in hours of daily ventilator support from baseline to week 24 after dosing. The investigators also examined a respiratory endpoint (i.e., maximal inspiratory pressure [MIP]) and neuromuscular endpoints (i.e., motor milestones, CHOP INTEND score, and muscle biopsy).
Treatment improved respiratory function
As of July 28, Dr. Kuntz and colleagues had enrolled 23 patients in the trial. Six participants received the lower dose of therapy, and 17 received the higher dose. Median age was 1.7 years for the low-dose group and 2.6 years for the high-dose group.
Patients assigned to receive the higher dose of therapy received treatment more recently than the low-dose group, and not all of the former have reached 48 weeks since treatment, said Dr. Kuntz. Fewer efficacy data are thus available for the high-dose group.
Each dose of AT132 was associated with a significantly greater decrease from baseline in least squares mean daily hours of ventilator dependence, compared with the control condition. At week 48, the mean reduction was approximately 19 hours/day for patients receiving 1 × 1014 vg/kg of AAT132 and approximately 13 hours per day for patients receiving 3 × 1014 vg/kg of AT132. The investigators did not perform a statistical comparison of the two doses because of differing protocols for ventilator weaning between groups. All six patients who received the lower dose achieved ventilator independence, as did one patient who received the higher dose.
In addition, all treated patients had significantly greater increases from baseline in least squares mean MIP, compared with controls. The mean increase was 45.7 cmH2O for the low-dose group, 46.1 cmH2O for the high-dose group, and −8.0 cmH2O for controls.
Before treatment, most patients had not achieved any of the motor milestones that investigators assessed. After treatment, five of six patients receiving the low dose achieved independent walking, as did one in 10 patients receiving the high dose. No controls achieved this milestone. Treated patients also had significantly greater increases from baseline in least squares mean CHOP INTEND scores, compared with controls. At least at one time point, five of six patients receiving the low dose, six of 10 patients receiving the high dose, and one control patient achieved the mean score observed in healthy infants.
Patients in both treatment arms had improvements in muscle pathology at weeks 24 and 48, including improvements in organelle localization and fiber size. In addition, patients in both treatment arms had continued detectable vector copies and myotubularin protein expression at both time points.
Deaths under investigation
In the low-dose group, one patient had four serious treatment-emergent adverse events, and in the high-dose group, eight patients had 27 serious treatment-emergent adverse events. The three patients in the high-dose group who developed fatal liver dysfunction were among the older, heavier patients in the study and, consequently, received among the highest total doses of treatment. These patients had evidence of likely preexisting intrahepatic cholestasis.
“This clinical trial is on hold pending discussions between regulatory agencies and the study sponsor regarding additional recruitment and the duration of follow-up,” said Dr. Kuntz.
Audentes Therapeutics, which is developing AT132, funded the trial. Dr. Kuntz had no conflicts of interest.
SOURCE: Bönnemann CG et al. CNS-ICNA 2020, Abstract P.62.
, according to research presented at the 2020 CNS-ICNA Conjoint Meeting, which was held virtually this year. The treatment also appears to improve patients’ motor function significantly and help them to achieve motor milestones.
The results come from a phase 1/2 study of two doses of AT132. Three of 17 patients who received the higher dose had fatal liver dysfunction. The researchers are investigating these cases and will communicate their findings.
X-linked myotubular myopathy is a rare and often fatal neuromuscular disease. Mutations in MTM1, which encodes the myotubularin enzyme that is required for the development and function of skeletal muscle, cause the disease, which affects about one in 50,000 to one in 40,000 newborn boys. The disease is associated with profound muscle weakness and impairment of neuromuscular and respiratory function. Patients with X-linked myotubular myopathy achieve motor milestones much later or not at all, and most require a ventilator or a feeding tube. The mortality by age 18 months is approximately 50%.
The ASPIRO trial
Investigators theorized that muscle tissue would be an appropriate therapeutic target because it does not display dystrophic or inflammatory changes in most patients. They identified adeno-associated virus AAV8 as a potential carrier for gene therapy, since it targets skeletal muscle effectively.
Nancy L. Kuntz, MD, an attending physician at Ann and Robert H. Lurie Children’s Hospital of Chicago, and colleagues conducted the ASPIRO trial to examine AT132 as a potential treatment for X-linked myotubular myopathy. Eligible patients were younger than 5 years or had previously enrolled in a natural history study of the disease, required ventilator support at baseline, and had no clinically significant underlying liver disease. Patients were randomly assigned to 1 × 1014 vg/kg of AAT132, 3 × 1014 vg/kg of AT132, or delayed treatment. Participants assigned to delayed treatment served as the study’s control group.
The study’s primary end points were safety and change in hours of daily ventilator support from baseline to week 24 after dosing. The investigators also examined a respiratory endpoint (i.e., maximal inspiratory pressure [MIP]) and neuromuscular endpoints (i.e., motor milestones, CHOP INTEND score, and muscle biopsy).
Treatment improved respiratory function
As of July 28, Dr. Kuntz and colleagues had enrolled 23 patients in the trial. Six participants received the lower dose of therapy, and 17 received the higher dose. Median age was 1.7 years for the low-dose group and 2.6 years for the high-dose group.
Patients assigned to receive the higher dose of therapy received treatment more recently than the low-dose group, and not all of the former have reached 48 weeks since treatment, said Dr. Kuntz. Fewer efficacy data are thus available for the high-dose group.
Each dose of AT132 was associated with a significantly greater decrease from baseline in least squares mean daily hours of ventilator dependence, compared with the control condition. At week 48, the mean reduction was approximately 19 hours/day for patients receiving 1 × 1014 vg/kg of AAT132 and approximately 13 hours per day for patients receiving 3 × 1014 vg/kg of AT132. The investigators did not perform a statistical comparison of the two doses because of differing protocols for ventilator weaning between groups. All six patients who received the lower dose achieved ventilator independence, as did one patient who received the higher dose.
In addition, all treated patients had significantly greater increases from baseline in least squares mean MIP, compared with controls. The mean increase was 45.7 cmH2O for the low-dose group, 46.1 cmH2O for the high-dose group, and −8.0 cmH2O for controls.
Before treatment, most patients had not achieved any of the motor milestones that investigators assessed. After treatment, five of six patients receiving the low dose achieved independent walking, as did one in 10 patients receiving the high dose. No controls achieved this milestone. Treated patients also had significantly greater increases from baseline in least squares mean CHOP INTEND scores, compared with controls. At least at one time point, five of six patients receiving the low dose, six of 10 patients receiving the high dose, and one control patient achieved the mean score observed in healthy infants.
Patients in both treatment arms had improvements in muscle pathology at weeks 24 and 48, including improvements in organelle localization and fiber size. In addition, patients in both treatment arms had continued detectable vector copies and myotubularin protein expression at both time points.
Deaths under investigation
In the low-dose group, one patient had four serious treatment-emergent adverse events, and in the high-dose group, eight patients had 27 serious treatment-emergent adverse events. The three patients in the high-dose group who developed fatal liver dysfunction were among the older, heavier patients in the study and, consequently, received among the highest total doses of treatment. These patients had evidence of likely preexisting intrahepatic cholestasis.
“This clinical trial is on hold pending discussions between regulatory agencies and the study sponsor regarding additional recruitment and the duration of follow-up,” said Dr. Kuntz.
Audentes Therapeutics, which is developing AT132, funded the trial. Dr. Kuntz had no conflicts of interest.
SOURCE: Bönnemann CG et al. CNS-ICNA 2020, Abstract P.62.
, according to research presented at the 2020 CNS-ICNA Conjoint Meeting, which was held virtually this year. The treatment also appears to improve patients’ motor function significantly and help them to achieve motor milestones.
The results come from a phase 1/2 study of two doses of AT132. Three of 17 patients who received the higher dose had fatal liver dysfunction. The researchers are investigating these cases and will communicate their findings.
X-linked myotubular myopathy is a rare and often fatal neuromuscular disease. Mutations in MTM1, which encodes the myotubularin enzyme that is required for the development and function of skeletal muscle, cause the disease, which affects about one in 50,000 to one in 40,000 newborn boys. The disease is associated with profound muscle weakness and impairment of neuromuscular and respiratory function. Patients with X-linked myotubular myopathy achieve motor milestones much later or not at all, and most require a ventilator or a feeding tube. The mortality by age 18 months is approximately 50%.
The ASPIRO trial
Investigators theorized that muscle tissue would be an appropriate therapeutic target because it does not display dystrophic or inflammatory changes in most patients. They identified adeno-associated virus AAV8 as a potential carrier for gene therapy, since it targets skeletal muscle effectively.
Nancy L. Kuntz, MD, an attending physician at Ann and Robert H. Lurie Children’s Hospital of Chicago, and colleagues conducted the ASPIRO trial to examine AT132 as a potential treatment for X-linked myotubular myopathy. Eligible patients were younger than 5 years or had previously enrolled in a natural history study of the disease, required ventilator support at baseline, and had no clinically significant underlying liver disease. Patients were randomly assigned to 1 × 1014 vg/kg of AAT132, 3 × 1014 vg/kg of AT132, or delayed treatment. Participants assigned to delayed treatment served as the study’s control group.
The study’s primary end points were safety and change in hours of daily ventilator support from baseline to week 24 after dosing. The investigators also examined a respiratory endpoint (i.e., maximal inspiratory pressure [MIP]) and neuromuscular endpoints (i.e., motor milestones, CHOP INTEND score, and muscle biopsy).
Treatment improved respiratory function
As of July 28, Dr. Kuntz and colleagues had enrolled 23 patients in the trial. Six participants received the lower dose of therapy, and 17 received the higher dose. Median age was 1.7 years for the low-dose group and 2.6 years for the high-dose group.
Patients assigned to receive the higher dose of therapy received treatment more recently than the low-dose group, and not all of the former have reached 48 weeks since treatment, said Dr. Kuntz. Fewer efficacy data are thus available for the high-dose group.
Each dose of AT132 was associated with a significantly greater decrease from baseline in least squares mean daily hours of ventilator dependence, compared with the control condition. At week 48, the mean reduction was approximately 19 hours/day for patients receiving 1 × 1014 vg/kg of AAT132 and approximately 13 hours per day for patients receiving 3 × 1014 vg/kg of AT132. The investigators did not perform a statistical comparison of the two doses because of differing protocols for ventilator weaning between groups. All six patients who received the lower dose achieved ventilator independence, as did one patient who received the higher dose.
In addition, all treated patients had significantly greater increases from baseline in least squares mean MIP, compared with controls. The mean increase was 45.7 cmH2O for the low-dose group, 46.1 cmH2O for the high-dose group, and −8.0 cmH2O for controls.
Before treatment, most patients had not achieved any of the motor milestones that investigators assessed. After treatment, five of six patients receiving the low dose achieved independent walking, as did one in 10 patients receiving the high dose. No controls achieved this milestone. Treated patients also had significantly greater increases from baseline in least squares mean CHOP INTEND scores, compared with controls. At least at one time point, five of six patients receiving the low dose, six of 10 patients receiving the high dose, and one control patient achieved the mean score observed in healthy infants.
Patients in both treatment arms had improvements in muscle pathology at weeks 24 and 48, including improvements in organelle localization and fiber size. In addition, patients in both treatment arms had continued detectable vector copies and myotubularin protein expression at both time points.
Deaths under investigation
In the low-dose group, one patient had four serious treatment-emergent adverse events, and in the high-dose group, eight patients had 27 serious treatment-emergent adverse events. The three patients in the high-dose group who developed fatal liver dysfunction were among the older, heavier patients in the study and, consequently, received among the highest total doses of treatment. These patients had evidence of likely preexisting intrahepatic cholestasis.
“This clinical trial is on hold pending discussions between regulatory agencies and the study sponsor regarding additional recruitment and the duration of follow-up,” said Dr. Kuntz.
Audentes Therapeutics, which is developing AT132, funded the trial. Dr. Kuntz had no conflicts of interest.
SOURCE: Bönnemann CG et al. CNS-ICNA 2020, Abstract P.62.
FROM CNS-ICNA 2020
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis Overlap Syndrome in a Patient With Relapsing Polychondritis
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.

Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
Practice Points
- The clinical presentation of relapsing polychondritis (RP) may demonstrate cutaneous manifestations other than the typical inflammation of cartilage-rich structures.
- Approximately 30% of patients with RP will have another autoimmune disease.
Birch bark derivative gel found effective for EB, in phase 3 study
A . The results come from the largest double-blind, randomized trial performed in this patient population.

More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
A . The results come from the largest double-blind, randomized trial performed in this patient population.
More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
A . The results come from the largest double-blind, randomized trial performed in this patient population.
More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
Ataluren delays disease milestones in patients with nonsense mutation DMD
(nmDMD), according to study results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Because so few patients in the study reached one of the negative pulmonary endpoints, longer follow-up will be needed to assess more conclusively the effect of ataluren on pulmonary function, said Francesco Bibbiani, MD, vice president of clinical development at PTC Therapeutics.

DMD is a rare and fatal neuromuscular disorder that causes progressive muscle weakness. Between 10% and 15% of patients with DMD have a nonsense mutation in the DMD gene. This mutation creates a premature stop codon that prevents the translation of a full-length dystrophin protein. Ataluren is designed to promote readthrough of this premature stop codon, thus enabling the production of a full-length dystrophin protein. An oral formulation of the drug has been approved in several European and South American countries.
Comparing treatment and standard of care
Study 019 was a phase 3, multicenter, open-label, long-term safety study of ataluren that enrolled international patients with nmDMD, most of whom had participated previously in a trial of ataluren. Dr. Bibbiani and colleagues conducted a post hoc analysis of Study 019 data to determine whether patients with nmDMD who received ataluren and standard of care for as long as 240 weeks had a different time to loss of ambulation and to decline of pulmonary function, compared with patients who received standard of care alone. Patients who were eligible to participate in Study 019 were male, had nmDMD, and had completed the blinded study drug treatment in a previous PTC-sponsored study. Treatment consisted of two 10-mg/kg doses and one 20-mg/kg dose of ataluren per day.
Dr. Bibbiani and colleagues used participants in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) as a control group. CINRG DNHS was a prospective, longitudinal study of patients with DMD who received standard of care at 20 centers worldwide from 2006 to 2016. Dr. Bibbiani and colleagues used propensity-score matching to pair participants in this study with participants in Study 019. They matched patients with respect to age at onset of first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of use of other corticosteroids. These factors are established predictors of disease progression in DMD.
Patients were eligible for inclusion in the post hoc analysis if they had available data for age, loss of ambulation, and the covariates selected for matching. Of 94 Study 019 participants, 60 were eligible for propensity-score matching with participants in CINRG DNHS. Forty-five nonambulatory patients were eligible for matching in the analysis of age at the decline in pulmonary function because data for age at loss of ambulation and for the three pulmonary endpoints measured were available for them. Thus, comparable population sizes were available for each analysis.
Treatment delayed disease milestones
Kaplan–Meier analysis indicated that the median age at various disease milestones was higher among patients who received ataluren and standard of care, compared with those who received standard of care alone. The median age at loss of ambulation was 15.5 years for Study 019 participants and 13.3 years for CINRG DNHS patients. The median age at predicted forced vital capacity (FVC) of less than 60% was 18.1 years for Study 019 participants and 15.8 years for CINRG DNHS participants. The median age at predicted FVC of less than 50% was 19.1 years for Study 019 participants and 17.9 years for CINRG DNHS participants. Finally, the median age at FVC of less than 1 L was not calculable for Study 019 participants and 23.8 years for CINRG DNHS participants.
The Study 019 and CINRG DNHS study groups are sponsored by PTC Therapeutics, which developed ataluren. Dr. Bibbiani is an employee of PTC Therapeutics.
SOURCE: McDonald C, et al. CNS-ICNA 2020. Abstract PL69.
(nmDMD), according to study results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Because so few patients in the study reached one of the negative pulmonary endpoints, longer follow-up will be needed to assess more conclusively the effect of ataluren on pulmonary function, said Francesco Bibbiani, MD, vice president of clinical development at PTC Therapeutics.
DMD is a rare and fatal neuromuscular disorder that causes progressive muscle weakness. Between 10% and 15% of patients with DMD have a nonsense mutation in the DMD gene. This mutation creates a premature stop codon that prevents the translation of a full-length dystrophin protein. Ataluren is designed to promote readthrough of this premature stop codon, thus enabling the production of a full-length dystrophin protein. An oral formulation of the drug has been approved in several European and South American countries.
Comparing treatment and standard of care
Study 019 was a phase 3, multicenter, open-label, long-term safety study of ataluren that enrolled international patients with nmDMD, most of whom had participated previously in a trial of ataluren. Dr. Bibbiani and colleagues conducted a post hoc analysis of Study 019 data to determine whether patients with nmDMD who received ataluren and standard of care for as long as 240 weeks had a different time to loss of ambulation and to decline of pulmonary function, compared with patients who received standard of care alone. Patients who were eligible to participate in Study 019 were male, had nmDMD, and had completed the blinded study drug treatment in a previous PTC-sponsored study. Treatment consisted of two 10-mg/kg doses and one 20-mg/kg dose of ataluren per day.
Dr. Bibbiani and colleagues used participants in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) as a control group. CINRG DNHS was a prospective, longitudinal study of patients with DMD who received standard of care at 20 centers worldwide from 2006 to 2016. Dr. Bibbiani and colleagues used propensity-score matching to pair participants in this study with participants in Study 019. They matched patients with respect to age at onset of first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of use of other corticosteroids. These factors are established predictors of disease progression in DMD.
Patients were eligible for inclusion in the post hoc analysis if they had available data for age, loss of ambulation, and the covariates selected for matching. Of 94 Study 019 participants, 60 were eligible for propensity-score matching with participants in CINRG DNHS. Forty-five nonambulatory patients were eligible for matching in the analysis of age at the decline in pulmonary function because data for age at loss of ambulation and for the three pulmonary endpoints measured were available for them. Thus, comparable population sizes were available for each analysis.
Treatment delayed disease milestones
Kaplan–Meier analysis indicated that the median age at various disease milestones was higher among patients who received ataluren and standard of care, compared with those who received standard of care alone. The median age at loss of ambulation was 15.5 years for Study 019 participants and 13.3 years for CINRG DNHS patients. The median age at predicted forced vital capacity (FVC) of less than 60% was 18.1 years for Study 019 participants and 15.8 years for CINRG DNHS participants. The median age at predicted FVC of less than 50% was 19.1 years for Study 019 participants and 17.9 years for CINRG DNHS participants. Finally, the median age at FVC of less than 1 L was not calculable for Study 019 participants and 23.8 years for CINRG DNHS participants.
The Study 019 and CINRG DNHS study groups are sponsored by PTC Therapeutics, which developed ataluren. Dr. Bibbiani is an employee of PTC Therapeutics.
SOURCE: McDonald C, et al. CNS-ICNA 2020. Abstract PL69.
(nmDMD), according to study results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Because so few patients in the study reached one of the negative pulmonary endpoints, longer follow-up will be needed to assess more conclusively the effect of ataluren on pulmonary function, said Francesco Bibbiani, MD, vice president of clinical development at PTC Therapeutics.
DMD is a rare and fatal neuromuscular disorder that causes progressive muscle weakness. Between 10% and 15% of patients with DMD have a nonsense mutation in the DMD gene. This mutation creates a premature stop codon that prevents the translation of a full-length dystrophin protein. Ataluren is designed to promote readthrough of this premature stop codon, thus enabling the production of a full-length dystrophin protein. An oral formulation of the drug has been approved in several European and South American countries.
Comparing treatment and standard of care
Study 019 was a phase 3, multicenter, open-label, long-term safety study of ataluren that enrolled international patients with nmDMD, most of whom had participated previously in a trial of ataluren. Dr. Bibbiani and colleagues conducted a post hoc analysis of Study 019 data to determine whether patients with nmDMD who received ataluren and standard of care for as long as 240 weeks had a different time to loss of ambulation and to decline of pulmonary function, compared with patients who received standard of care alone. Patients who were eligible to participate in Study 019 were male, had nmDMD, and had completed the blinded study drug treatment in a previous PTC-sponsored study. Treatment consisted of two 10-mg/kg doses and one 20-mg/kg dose of ataluren per day.
Dr. Bibbiani and colleagues used participants in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) as a control group. CINRG DNHS was a prospective, longitudinal study of patients with DMD who received standard of care at 20 centers worldwide from 2006 to 2016. Dr. Bibbiani and colleagues used propensity-score matching to pair participants in this study with participants in Study 019. They matched patients with respect to age at onset of first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of use of other corticosteroids. These factors are established predictors of disease progression in DMD.
Patients were eligible for inclusion in the post hoc analysis if they had available data for age, loss of ambulation, and the covariates selected for matching. Of 94 Study 019 participants, 60 were eligible for propensity-score matching with participants in CINRG DNHS. Forty-five nonambulatory patients were eligible for matching in the analysis of age at the decline in pulmonary function because data for age at loss of ambulation and for the three pulmonary endpoints measured were available for them. Thus, comparable population sizes were available for each analysis.
Treatment delayed disease milestones
Kaplan–Meier analysis indicated that the median age at various disease milestones was higher among patients who received ataluren and standard of care, compared with those who received standard of care alone. The median age at loss of ambulation was 15.5 years for Study 019 participants and 13.3 years for CINRG DNHS patients. The median age at predicted forced vital capacity (FVC) of less than 60% was 18.1 years for Study 019 participants and 15.8 years for CINRG DNHS participants. The median age at predicted FVC of less than 50% was 19.1 years for Study 019 participants and 17.9 years for CINRG DNHS participants. Finally, the median age at FVC of less than 1 L was not calculable for Study 019 participants and 23.8 years for CINRG DNHS participants.
The Study 019 and CINRG DNHS study groups are sponsored by PTC Therapeutics, which developed ataluren. Dr. Bibbiani is an employee of PTC Therapeutics.
SOURCE: McDonald C, et al. CNS-ICNA 2020. Abstract PL69.
FROM CNS-ICNA 2020

ASSET extension supports abatacept as treatment for diffuse cutaneous systemic sclerosis
“Exploratory outcome measures during the open-label extension, including the composite ACR CRISS [American College of Rheumatology Combined Response Index in diffuse cutaneous Systemic Sclerosis] score, indicate that abatacept [Orencia] might promote overall global improvement in these participants,” Lorinda Chung, MD, of Stanford (Calif.) University, and colleagues wrote in the Lancet Rheumatology.
To continue to determine the safety and efficacy of abatacept as a treatment for dcSSc, the researchers launched the open-label extension of the randomized, double-blind ASSET trial. After patients in ASSET completed 12 months of treatment with either 125 mg weekly of subcutaneous abatacept or placebo, they were invited to join the extension period. ASSET’s primary endpoint was the change in modified Rodnan Skin Score (mRSS) from baseline to 12 months.
Of the 88 patients who began the ASSET trial – half of which were assigned to the abatacept group and the other half to the placebo group – 33 and 34 transitioned to weekly open-label treatment with 125 mg of abatacept, respectively. In the trial’s primary endpoint at 12 months, mean improvement in mRSS with abatacept was –6.6 (standard deviation, 6.4), compared to –3.7 (SD, 7.6) with placebo.
All told, 32 patients in each group completed 18 months of treatment. Patients who received abatacept in both periods had even more improvement in mRSS score (–9.8 [SD, 8.1]), as did patients who received placebo and then abatacept (–6.3 [SD, 9.3]). After 12 months, 68% of patients (23 of 34) in the abatacept group had a 5-unit or greater improvement in mRSS, compared with 50% of patients (19 of 38) in the placebo group. After 18 months, that percentage went up to 72% (23 of 32) among patients who took abatacept in both periods and 65% (20 of 31) for those who received it only in the extension.
Although the median ACR CRISS score was significantly greater at 12 months with abatacept, compared with placebo, both groups improved during the open-label period. After 12 months, an ACR CRISS score of 0.60 or higher was achieved by 55% (18 of 33) of the abatacept group and 36% (13 of 36) of the placebo group. In the extension, those percentages leapt to 66% (19 of 29) and 50% (13 of 26), respectively.
Throughout the double-blind phase, adverse events – including infectious events, serious events, and those that led to study withdrawal – were more frequent with placebo than with abatacept. During the extension period, although adverse events occurred slightly more among 18-month abatacept users, they ultimately occurred in fewer participants than in the double-blind phase.
Time to push forward with additional studies on abatacept
The results fortified by this open-label extension from Dr. Chung and colleagues should lay the groundwork for a phase 3 study on treatment with abatacept, wrote Francesco Del Galdo, MD, PhD, of the University of Leeds (England), in an accompanying editorial.
Along with clinically important improvements in several key measures, Dr. Del Galdo restated the value of abatacept’s “very benign” safety profile over the 18-month study period. However, he also acknowledged the “absence of concurrent immunosuppression” in the ASSET trial, noting that there is not yet a record of combination therapy safety across the board.
Beyond safety, he wondered if perhaps the time has passed for a placebo arm in dcSSc patients. He noted that participants in the initial placebo group were more likely to have “clinically relevant worsening of disease that required escape treatment,” which could make it ethically and analytically difficult to justify placebo.
The authors acknowledged the extension’s limitations, including the possibility that the survivors of the 12-month trial who joined the extension were more responsive to treatment or suffered from less severe disease. In addition, they noted that the study “was not powered for formal statistical comparison of the two treatment arms,” meaning all open-label results are considered exploratory. Finally, the number of participants in the extension period was small and likely contributed to low rates of adverse events, such as infection.
The trial was funded by Bristol-Myers Squibb, which markets abatacept, and the National Institutes of Health. The authors reported numerous potential conflicts of interest, including receiving grants, clinical trial support, and personal fees from various organizations and pharmaceutical companies, as well as serving on the advisory boards for such companies. Dr. Del Galdo reported receiving consultancy fees and research grants from several pharmaceutical companies.
SOURCE: Chung L et al. Lancet Rheumatol. 2020 Oct 19. doi: 10.1016/S2665-9913(20)30237-X.
“Exploratory outcome measures during the open-label extension, including the composite ACR CRISS [American College of Rheumatology Combined Response Index in diffuse cutaneous Systemic Sclerosis] score, indicate that abatacept [Orencia] might promote overall global improvement in these participants,” Lorinda Chung, MD, of Stanford (Calif.) University, and colleagues wrote in the Lancet Rheumatology.
To continue to determine the safety and efficacy of abatacept as a treatment for dcSSc, the researchers launched the open-label extension of the randomized, double-blind ASSET trial. After patients in ASSET completed 12 months of treatment with either 125 mg weekly of subcutaneous abatacept or placebo, they were invited to join the extension period. ASSET’s primary endpoint was the change in modified Rodnan Skin Score (mRSS) from baseline to 12 months.
Of the 88 patients who began the ASSET trial – half of which were assigned to the abatacept group and the other half to the placebo group – 33 and 34 transitioned to weekly open-label treatment with 125 mg of abatacept, respectively. In the trial’s primary endpoint at 12 months, mean improvement in mRSS with abatacept was –6.6 (standard deviation, 6.4), compared to –3.7 (SD, 7.6) with placebo.
All told, 32 patients in each group completed 18 months of treatment. Patients who received abatacept in both periods had even more improvement in mRSS score (–9.8 [SD, 8.1]), as did patients who received placebo and then abatacept (–6.3 [SD, 9.3]). After 12 months, 68% of patients (23 of 34) in the abatacept group had a 5-unit or greater improvement in mRSS, compared with 50% of patients (19 of 38) in the placebo group. After 18 months, that percentage went up to 72% (23 of 32) among patients who took abatacept in both periods and 65% (20 of 31) for those who received it only in the extension.
Although the median ACR CRISS score was significantly greater at 12 months with abatacept, compared with placebo, both groups improved during the open-label period. After 12 months, an ACR CRISS score of 0.60 or higher was achieved by 55% (18 of 33) of the abatacept group and 36% (13 of 36) of the placebo group. In the extension, those percentages leapt to 66% (19 of 29) and 50% (13 of 26), respectively.
Throughout the double-blind phase, adverse events – including infectious events, serious events, and those that led to study withdrawal – were more frequent with placebo than with abatacept. During the extension period, although adverse events occurred slightly more among 18-month abatacept users, they ultimately occurred in fewer participants than in the double-blind phase.
Time to push forward with additional studies on abatacept
The results fortified by this open-label extension from Dr. Chung and colleagues should lay the groundwork for a phase 3 study on treatment with abatacept, wrote Francesco Del Galdo, MD, PhD, of the University of Leeds (England), in an accompanying editorial.
Along with clinically important improvements in several key measures, Dr. Del Galdo restated the value of abatacept’s “very benign” safety profile over the 18-month study period. However, he also acknowledged the “absence of concurrent immunosuppression” in the ASSET trial, noting that there is not yet a record of combination therapy safety across the board.
Beyond safety, he wondered if perhaps the time has passed for a placebo arm in dcSSc patients. He noted that participants in the initial placebo group were more likely to have “clinically relevant worsening of disease that required escape treatment,” which could make it ethically and analytically difficult to justify placebo.
The authors acknowledged the extension’s limitations, including the possibility that the survivors of the 12-month trial who joined the extension were more responsive to treatment or suffered from less severe disease. In addition, they noted that the study “was not powered for formal statistical comparison of the two treatment arms,” meaning all open-label results are considered exploratory. Finally, the number of participants in the extension period was small and likely contributed to low rates of adverse events, such as infection.
The trial was funded by Bristol-Myers Squibb, which markets abatacept, and the National Institutes of Health. The authors reported numerous potential conflicts of interest, including receiving grants, clinical trial support, and personal fees from various organizations and pharmaceutical companies, as well as serving on the advisory boards for such companies. Dr. Del Galdo reported receiving consultancy fees and research grants from several pharmaceutical companies.
SOURCE: Chung L et al. Lancet Rheumatol. 2020 Oct 19. doi: 10.1016/S2665-9913(20)30237-X.
“Exploratory outcome measures during the open-label extension, including the composite ACR CRISS [American College of Rheumatology Combined Response Index in diffuse cutaneous Systemic Sclerosis] score, indicate that abatacept [Orencia] might promote overall global improvement in these participants,” Lorinda Chung, MD, of Stanford (Calif.) University, and colleagues wrote in the Lancet Rheumatology.
To continue to determine the safety and efficacy of abatacept as a treatment for dcSSc, the researchers launched the open-label extension of the randomized, double-blind ASSET trial. After patients in ASSET completed 12 months of treatment with either 125 mg weekly of subcutaneous abatacept or placebo, they were invited to join the extension period. ASSET’s primary endpoint was the change in modified Rodnan Skin Score (mRSS) from baseline to 12 months.
Of the 88 patients who began the ASSET trial – half of which were assigned to the abatacept group and the other half to the placebo group – 33 and 34 transitioned to weekly open-label treatment with 125 mg of abatacept, respectively. In the trial’s primary endpoint at 12 months, mean improvement in mRSS with abatacept was –6.6 (standard deviation, 6.4), compared to –3.7 (SD, 7.6) with placebo.
All told, 32 patients in each group completed 18 months of treatment. Patients who received abatacept in both periods had even more improvement in mRSS score (–9.8 [SD, 8.1]), as did patients who received placebo and then abatacept (–6.3 [SD, 9.3]). After 12 months, 68% of patients (23 of 34) in the abatacept group had a 5-unit or greater improvement in mRSS, compared with 50% of patients (19 of 38) in the placebo group. After 18 months, that percentage went up to 72% (23 of 32) among patients who took abatacept in both periods and 65% (20 of 31) for those who received it only in the extension.
Although the median ACR CRISS score was significantly greater at 12 months with abatacept, compared with placebo, both groups improved during the open-label period. After 12 months, an ACR CRISS score of 0.60 or higher was achieved by 55% (18 of 33) of the abatacept group and 36% (13 of 36) of the placebo group. In the extension, those percentages leapt to 66% (19 of 29) and 50% (13 of 26), respectively.
Throughout the double-blind phase, adverse events – including infectious events, serious events, and those that led to study withdrawal – were more frequent with placebo than with abatacept. During the extension period, although adverse events occurred slightly more among 18-month abatacept users, they ultimately occurred in fewer participants than in the double-blind phase.
Time to push forward with additional studies on abatacept
The results fortified by this open-label extension from Dr. Chung and colleagues should lay the groundwork for a phase 3 study on treatment with abatacept, wrote Francesco Del Galdo, MD, PhD, of the University of Leeds (England), in an accompanying editorial.
Along with clinically important improvements in several key measures, Dr. Del Galdo restated the value of abatacept’s “very benign” safety profile over the 18-month study period. However, he also acknowledged the “absence of concurrent immunosuppression” in the ASSET trial, noting that there is not yet a record of combination therapy safety across the board.
Beyond safety, he wondered if perhaps the time has passed for a placebo arm in dcSSc patients. He noted that participants in the initial placebo group were more likely to have “clinically relevant worsening of disease that required escape treatment,” which could make it ethically and analytically difficult to justify placebo.
The authors acknowledged the extension’s limitations, including the possibility that the survivors of the 12-month trial who joined the extension were more responsive to treatment or suffered from less severe disease. In addition, they noted that the study “was not powered for formal statistical comparison of the two treatment arms,” meaning all open-label results are considered exploratory. Finally, the number of participants in the extension period was small and likely contributed to low rates of adverse events, such as infection.
The trial was funded by Bristol-Myers Squibb, which markets abatacept, and the National Institutes of Health. The authors reported numerous potential conflicts of interest, including receiving grants, clinical trial support, and personal fees from various organizations and pharmaceutical companies, as well as serving on the advisory boards for such companies. Dr. Del Galdo reported receiving consultancy fees and research grants from several pharmaceutical companies.
SOURCE: Chung L et al. Lancet Rheumatol. 2020 Oct 19. doi: 10.1016/S2665-9913(20)30237-X.
FROM THE LANCET RHEUMATOLOGY