User login
Cutaneous Insulin-Derived Amyloidosis Presenting as Hyperkeratotic Nodules
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
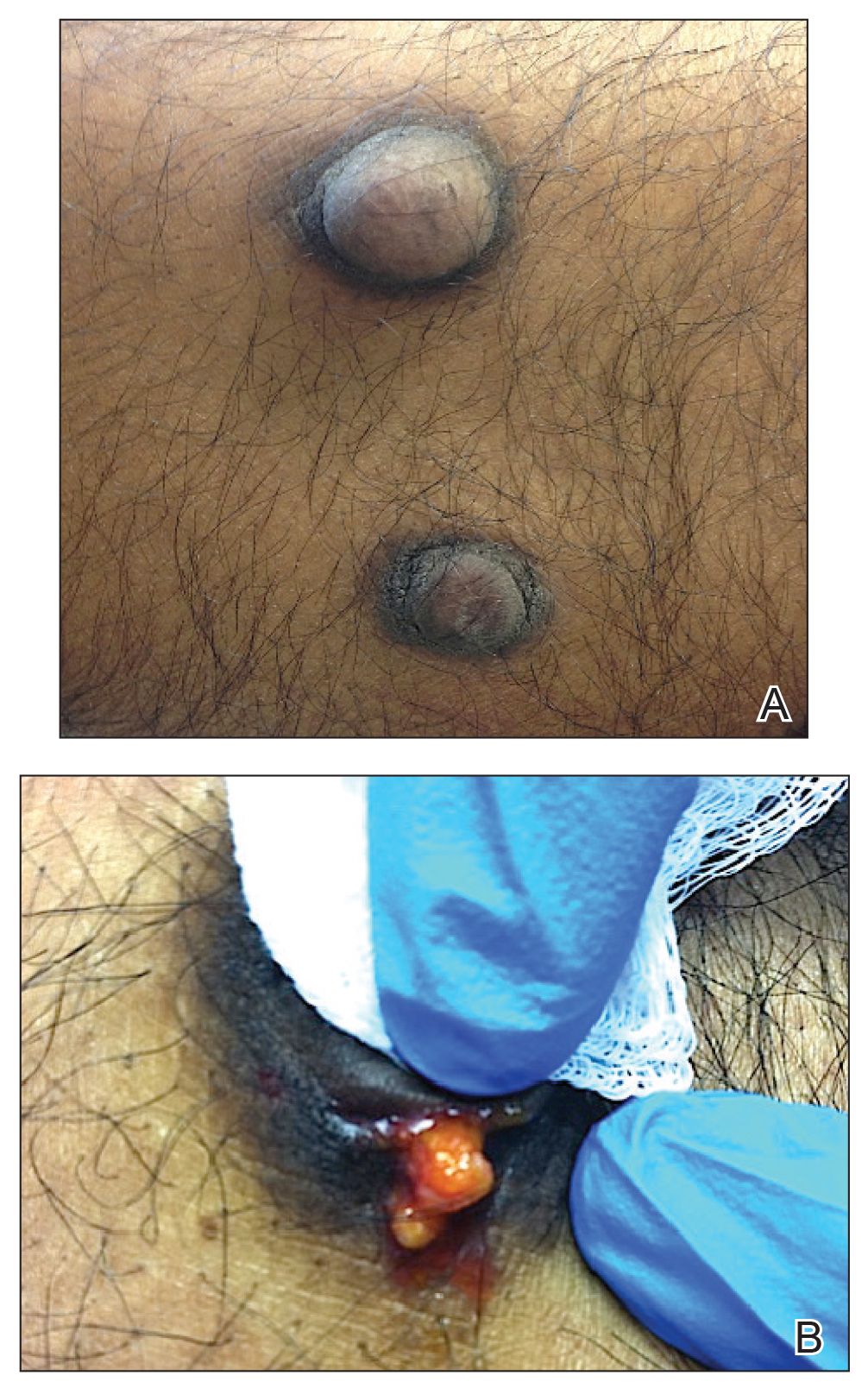
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
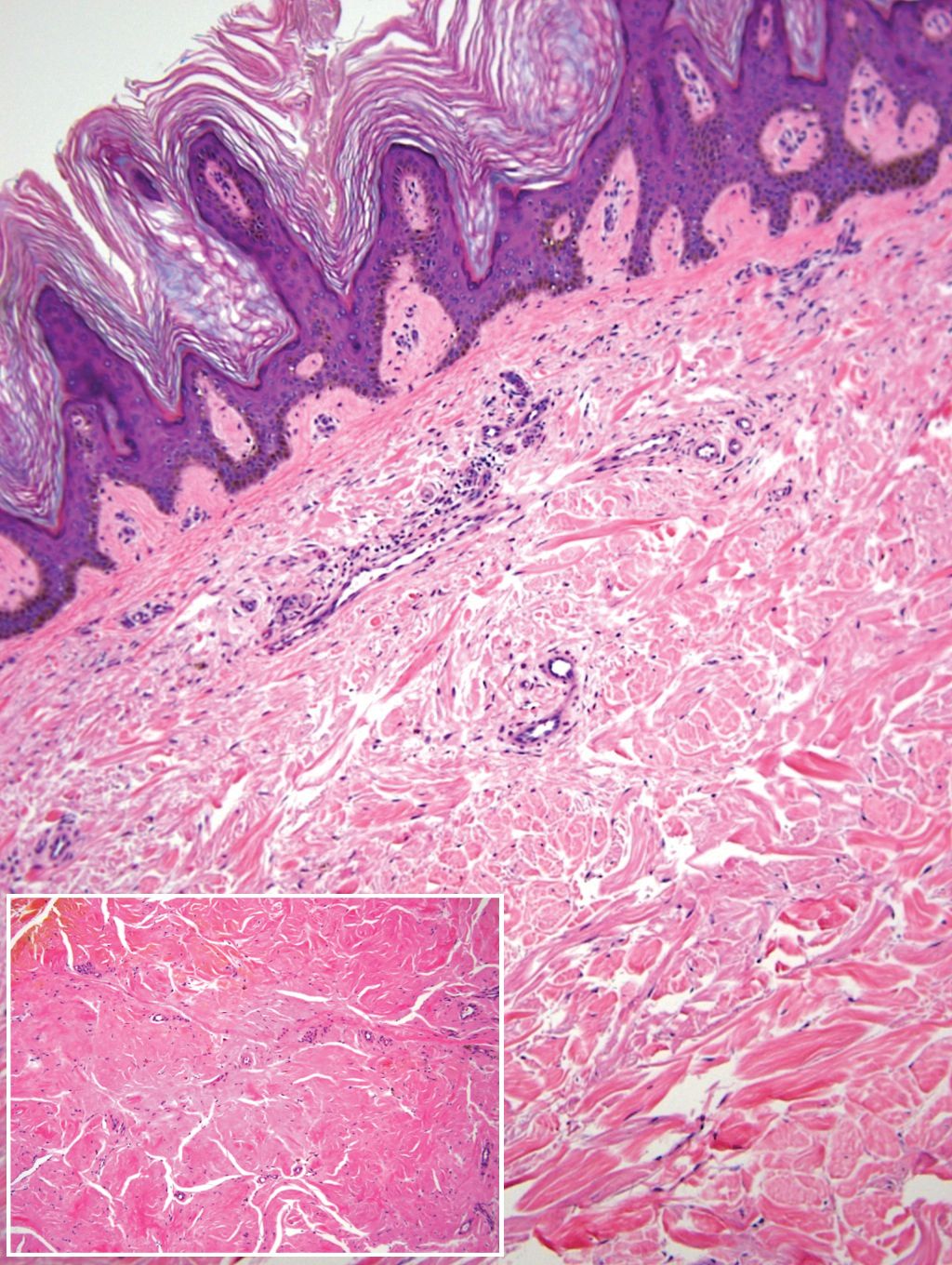
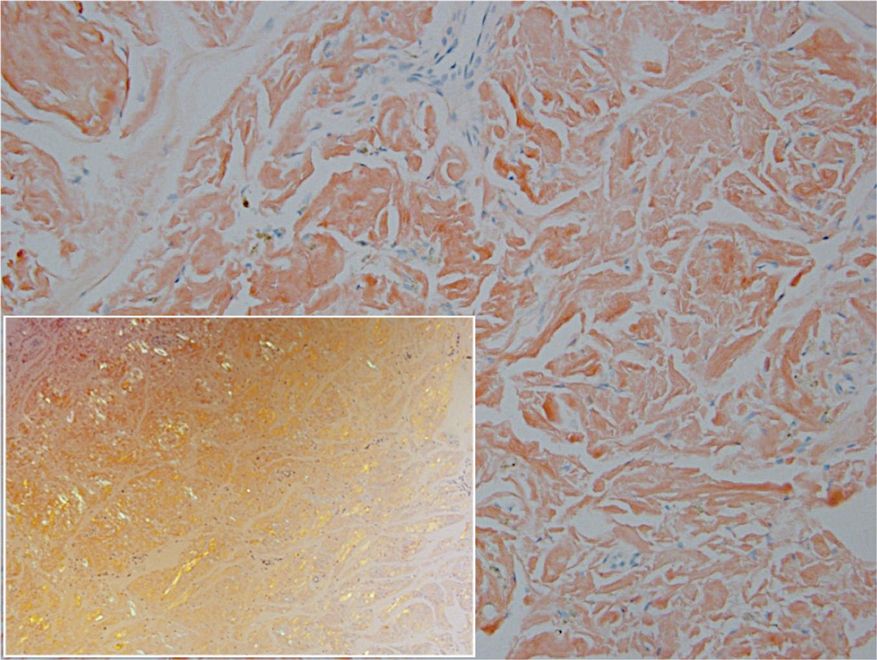
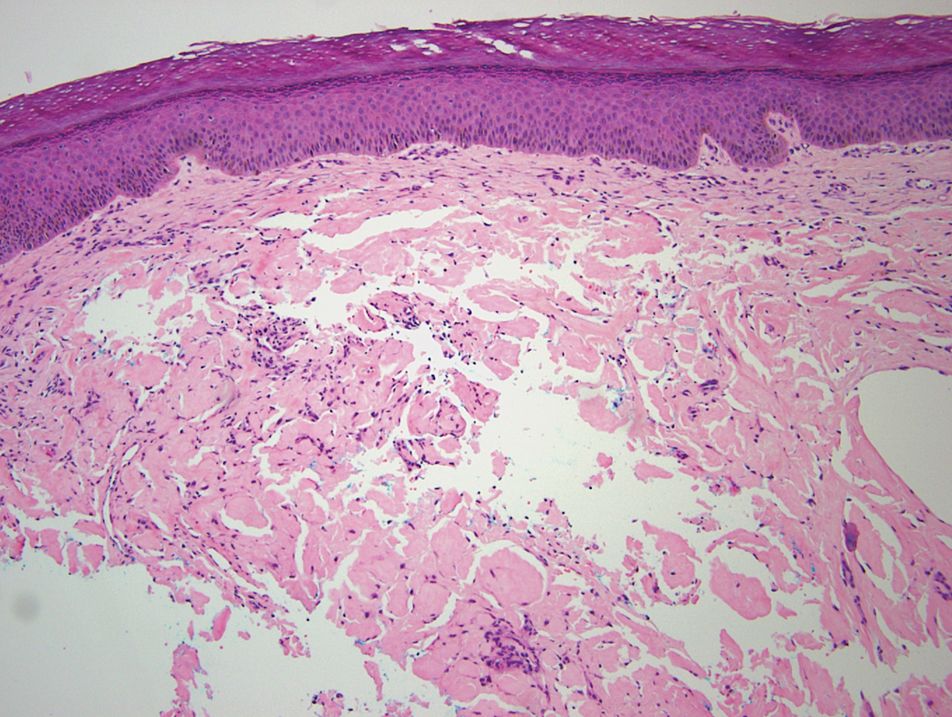
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
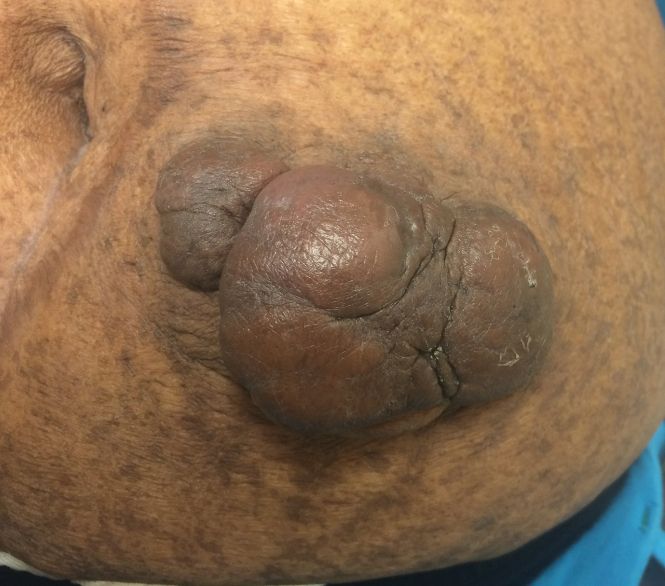
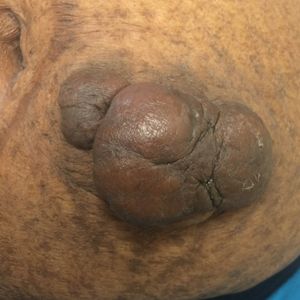
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
Amyloidosis consists of approximately 30 protein-folding disorders sharing the common feature of abnormal extracellular amyloid deposition. In each condition, a specific soluble precursor protein aggregates to form the insoluble fibrils of amyloid, characterized by the beta-pleated sheet structure.1 Amyloidosis occurs as either a systemic or localized process. Insulin-derived (AIns) amyloidosis, a localized process occurring at insulin injection sites, was first reported in 1983.2 There were fewer than 20 reported cases until 2014, when 57 additional cases were reported by just 2 institutions,3,4 indicating that AIns amyloidosis may be more common than previously thought.3,5
Despite the increasing prevalence of diabetes mellitus and insulin use, there is a paucity of published cases of AIns amyloidosis. The lack of awareness of this condition among both dermatologists and general practitioners may be in part due to its variable clinical manifestations. We describe 2 patients with unique presentations of localized amyloidosis at repeated insulin injection sites.
Case Reports
Patient 1
A 39-year-old man with a history of type 1 diabetes mellitus presented with 4 asymptomatic nodules on the lateral thighs in areas of previous insulin injection. He first noticed the lesions 9 months prior to presentation and subsequently switched the injection site to the abdomen without development of new nodules. Despite being compliant with his insulin regimen, he had a long history of irregular glucose control, including frequent hypoglycemic episodes. The patient was using regular and neutral protamine hagedorn insulin.
On physical examination, 2 soft, nontender, exophytic nodules were noted on each upper thigh with surrounding hyperpigmented and hyperkeratotic collarettes (Figure 1). The nodules ranged in size from 2 to 3.5 cm in diameter.
Remarkable laboratory data included a fasting glucose level of 207 mg/dL (reference range, 70–110 mg/dL) and a glycohemoglobin of 8.8% (reference range, <5.7%). Serum protein electrophoresis and immunofixation were normal. Histopathology of the lesions demonstrated diffuse deposition of pink amorphous material associated with prominent papillomatosis, hyperkeratosis, and acanthosis (Figure 2). Congo red staining was positive with green birefringence under polarized light, indicative of amyloid deposits (Figure 3). Liquid chromatography–tandem mass spectrometry of the specimens was consistent with deposition of AIns amyloidosis.
Due to the size and persistent nature of the lesions, the nodules were removed by tangential excision. In addition, the patient was advised to continue rotating injection sites frequently. His blood glucose levels are now well controlled, and he has not developed any new nodules.
Patient 2
A 53-year-old woman with a history of type 2 diabetes mellitus presented with painful subcutaneous nodules on the lower abdomen at sites of previous insulin injections. The nodules developed approximately 1 month after she started treatment with neutral protamine hagedorn insulin and had been slowly enlarging over the past year. She tried switching injection sites after noticing the lesions, but the nodules persisted. The patient had a long history of poor glucose control with chronically elevated glycohemoglobin and blood glucose levels.
On physical examination, 2 hyperpigmented, exophytic, smooth nodules were noted on the right and left lower abdomen, ranging in size from 2.5 to 5.5 cm in diameter (Figure 4).
Relevant laboratory data included a fasting glucose level of 197 mg/dL and a glycohemoglobin of 9.3%. A biopsy of the lesion on the left lower abdomen revealed eosinophilic amorphous deposits with fissuring in the dermis (Figure 5). Congo red stain was positive with green birefringence under polarized light. Liquid chromatography–tandem mass spectrometry of the specimen showed deposition of AIns amyloid. The patient began injecting away from the amyloid nodules without development of any new lesions. The original nodules have persisted, and surgical excision is planned.
Comment
Insulin is the suspected precursor protein in AIns amyloidosis, but the exact pathogenesis is unknown. The protein that is derived from insulin in these tumors is now identified as AIns amyloidosis.5,6 It is hypothesized that insulin accumulates locally and is converted to amyloid by an unknown mechanism.7 Other potential contributory factors include chronic inflammation and foreign body reactions developing around amyloid deposits, as well as repeated trauma from injections into a single site.4,5 It appears that lesions may derive from a wide range of insulin types and occur after variable time periods.
A majority of cases of iatrogenic amyloid have been described as single, firm, subcutaneous masses at an injection site that commonly are misdiagnosed as lipomas or lipohypertrophy.7-11 To our knowledge, none of the reported cases resembled the multiple, discrete, exophytic nodules seen in our patients.3,4 The surrounding hyperkeratosis noted in patient 1 is another uncommon feature of AIns amyloidosis (Figures 1 and 2). Only 3 AIns amyloidosis cases described lesions with acanthosis nigricans–like changes, only 1 of which provided a clinical image.6,7,12The mechanism for the acanthosis nigricans–like changes may have been due to the high levels of insulin at the injection site. It has been suggested that the activation of insulinlike growth factor receptor by insulin leads to the proliferation of keratinocytes and fibroblasts.6 Histologic examination of AIns amyloidosis lesions generally demonstrates deposition of homogenous eosinophilic material consistent with amyloid, as well as positive Congo red staining with green birefringence by polarization. Immunohistologic staining with insulin antibody with or without proteomic analysis of the amyloid deposits can confirm the diagnosis. In both of our patients’ specimens, liquid chromatography–tandem mass spectrometry was performed for proteomic analysis, and results were consistent with AIns amyloidosis.
Reports in the literature have suggested that the deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.4,11,13 Hence, injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis. In their study of 4 patients, Nagase et al4 compared serum insulin levels after insulin injection into amyloid nodules vs insulin levels after injection into normal skin. Insulin absorption at the amyloid sites was 34% of that at normal sites. Given these results, patients should be instructed to inject away from the amyloid deposit once it is identified.6 Glucose levels should be monitored closely when patients first inject away from the amyloid mass, as injection of the same dosage to an area of normal skin can lead to increased insulin absorption and hypoglycemia.4,6 It is possible that the frequent hypoglycemic episodes noted in patient 1 were due to increased insulin sensitivity after switching to injection sites away from amyloid lesions.
Conclusion
Our patients demonstrate unique presentations of localized cutaneous amyloidosis at repeated insulin injection sites. We report these cases to complement the current data of iatrogenic amyloidosis and provide insight into this likely underreported phenomenon.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39:323-345.
- Storkel S, Schneider HM, Muntefering H, et al. Iatrogenic, insulin-dependent, local amyloidosis. Lab Invest. 1983;48:108-111.
- D’souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Nagase T, Iwaya K, Iwaki Y, et al. Insulin-derived amyloidosis and poor glycemic control: a case series. Am J Med. 2014;127:450-454.
- Gupta Y, Singla G, Singla R. Insulin-derived amyloidosis. Indian J Endocrinol Metab. 2015;19:174-177.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Yumlu S, Barany R, Eriksson M, et al. Localized insulin-derived amyloidosis in patients with diabetes mellitus: a case report. Hum Pathol. 2009;40:1655-1660.
- Okamura S, Hayashino Y, Kore-Eda S, et al. Localized amyloidosis at the site of repeated insulin injection in a patient with type 2 diabetes. Diabetes Care. 2013;36:E200.
- Dische FE, Wernstedt C, Westermark GT, et al. Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient. Diabetologia. 1988;31:158-161.
- Swift B, Hawkins PN, Richards C, et al. Examination of insulin injection sites: an unexpected finding of localized amyloidosis. Diabetic Med. 2002;19:881-882.
- Albert SG, Obadiah J, Parseghian SA, et al. Severe insulin resistance associated with subcutaneous amyloid deposition. Diabetes Res Clin Pract. 2007;75:374-376.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Endo JO, Rocken C, Lamb S, et al. Nodular amyloidosis in a diabetic patient with frequent hypoglycemia: sequelae of repeatedly injecting insulin without site rotation. J Am Acad Dermatol. 2010;63:E113-E114.
Practice Points
- Deposition of amyloid at insulin injection sites has the potential to interfere with insulin absorption, leading to poor glucose control.
- Patients with insulin-derived (AIns) amyloidosis may initially present after noticing nodular deposits.
- Insulin injection site rotation is a crucial aspect of treatment and prevention of AIns amyloidosis.
Data call for biologics trials in undertreated juvenile arthritis subtype
Children with enthesitis-related arthritis often have a high burden of disease and could benefit from medications currently approved for adults with spondyloarthritis, according to a review published in Arthritis Care & Research.

“Enthesitis-related arthritis (ERA) was the JIA [juvenile idiopathic arthritis] category applied to children with spondyloarthritis (SpA), recognizing enthesitis as a defining characteristic,” wrote Pamela F. Weiss, MD, of Children’s Hospital of Philadelphia, and colleagues.
The ERA criteria include “arthritis plus enthesitis; or arthritis or enthesitis plus at least two of the following: sacroiliac tenderness or inflammatory back pain, HLA-B27 positivity, first-degree relative with HLA-B27–associated disease, acute anterior uveitis, and arthritis in a male older than 6 years,” the review authors noted.
“None of the [Food and Drug Administration]–approved therapies for peripheral SpA or nonradiographic axial SpA” have been studied or approved for use in children with ERA, but data support biologic similarity to SpA in adults; notably, studies of the HLA-B27 allele have identified it as a risk factor for both SpA and ERA, they said.
Common factors in adult and childhood conditions
“The principal commonalities of children with ERA and axial arthritis, and adults with nonradiographic axial SpA, include enthesitis, arthritis, inflammatory back pain, anterior uveitis, HLA-B27 positivity, and family history of HLA-B27–associated disease,” the review authors wrote.
The first-line treatment for both ERA with axial arthritis and nonradiographic axial SpA is NSAIDs, followed by tumor necrosis factor (TNF) inhibitors if needed, they said. However, conventional disease-modifying antirheumatic drugs (cDMARDs) may be used in cases of peripheral disease affecting five or more joints. Studies of treatment response show similarities between ERA in children and SpA in adults, the authors added, with nearly half of adults with axial disease unable to achieve remission and approximately one-third of children with ERA failing to respond to therapy.
Clinical trials could improve options and outcomes for those with ERA who need advanced therapy and such trials should evaluate response of axial and peripheral disease separately, the review authors emphasized. For example, “Eligibility criteria for children with ERA and axial features could include the presence of some of the following disease features: active inflammatory sacroiliitis based on typical MRI changes according to ASAS/OMERACT [Assessment of SpondyloArthritis international Society/Outcome Measures in Rheumatology Clinical Trials] criteria; elevated CRP [C-reactive protein]; and inadequate response or intolerance to NSAIDs,” they noted. “Considering the similarities between adult spondyloarthritis and ERA in terms of etiology, genetics, pathogenesis, and clinical manifestations, it is evident that medications approved for axial or peripheral SpA should be studied in children with ERA involving axial or peripheral joints, respectively, with the intent to achieve labeling for use in children,” they concluded.
New data highlight ERA disease burden
The need for additional therapies for ERA patients gained more support from a recent study in which a majority of children with ERA or juvenile psoriatic arthritis (jPsA) used biologics, but those with sacroiliitis in particular showed a significant disease burden despite high biologic use.

The International Leagues Against Rheumatism criteria include seven categories of juvenile idiopathic arthritis, of which ERA and jPsA are the most common; however, characteristics of these children have not been well described, wrote Dax G. Rumsey, MD, of the University of Alberta, Edmonton, and colleagues.
“Children with ERA are more likely to have a clinical picture with predominantly peripheral arthritis, typically described as an oligoarthritis involving the lower limbs with high risk of axial disease, relative to the other categories of JIA,” and report more intense pain and worse health status, compared with children in other categories, the researchers wrote.
To more completely characterize children with ERA and jPsA, the researchers assessed 522 children with ERA and 380 with jPsA. The children were enrolled in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) Registry. The findings were published in a brief report in Arthritis Care & Research.
Overall, 69% of the children took at least one biologic, including 72% with ERA and 64% with jPsA. Biologic use was even higher (81%) among the 28% of patients with sacroiliitis (40% of ERA patients and 12% of jPsA patients). Approximately 36% of the patients with sacroiliitis were positive for HLA-B27. In addition, Physician Global Assessment scores and clinical Juvenile Arthritis Disease Activity Score-10 (cJADAS10) scores were significantly higher at the first clinical visit with sacroiliitis, compared with the first visit without, which confirms “the clinical impression that active sacroiliitis significantly impacts children and their families,” the researchers said.
The average age at diagnosis was 10.8 years for ERA and 8.2 years for jPsA, and significantly more ERA patients were male (56% vs. 38%). However, more of the patients with sacroiliitis (54%) were female. More than half of the patients reported polyarticular involvement.
The study findings were limited by several factors, including the classification of ERA or jPsA and the reliance on physician diagnoses, as well as the variation in identifying sacroiliitis, the researchers said. However, the results increase understanding of the pathophysiology of ERA and jPsA to help determine optimal treatment, they concluded.
Data highlight research and treatment gaps
“Recent research demonstrates a large, unmet medical need in the treatment of JIA with 52%-65% of all JIA patients, including those with ERA and jPsA, having been treated with at least one biologic DMARD and 15%-19% having been treated with an FDA-unapproved biologic. In those with ERA or jPsA, 72%-79% of the children had been treated with a biologic DMARD, although no biologic DMARD has ever been FDA approved for these JIA categories,” Daniel J. Lovell, MD, and Hermine I. Brunner, MD, both with Cincinnati Children’s Hospital Medical Center, wrote in an editorial that accompanied the new study. Dr. Lovell and Dr. Brunner also were coauthors of the review article.

The new study supports findings from other recent publications, the editorialists noted. The new results showed “a significant proportion of the JIA population with active sacroiliitis with high disease burden despite very frequent (over 80% of the population) [treatment] with unstudied and unapproved biologic DMARDs,” they said. “These children with sacroiliitis had significantly greater disease burden with higher physician assessment of disease activity, higher parent assessment of disease impact, and higher disease activity as measured by the Juvenile Idiopathic Arthritis Disease Activity Score, compared to the children with ERA or jPsA without sacroiliitis,” they noted.

Previously, “the FDA granted pharmaceutical companies studying new treatments in adult SpA automatic full waivers from doing studies in children for new medications for ‘axial spondyloarthropathies including ankylosing spondylitis’ up until July 2020,” the editorialists said. However, “It is now time now for the pharmaceutical industry to perform FDA-monitored clinical trials of children and adolescents with SpA,” they emphasized. “This will allow for the scientific assessment of proper dosing, efficacy, and safety of the increasing number of new medications that are being licensed by the FDA for the treatment of SpA, such as the anti-TNF, anti–IL[interleukin]-17, and anti–IL-23 biologics, and perhaps JAK [Janus kinase] agents, to address this unmet medical need in these patients with juvenile SpA,” they concluded.
Dr. Weiss disclosed grant support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), and financial relationships with Eli Lilly and Pfizer. Dr. Lovell disclosed relationships with companies including Abbott, AbbVie Amgen, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Hoffmann-La Roche, Janssen, Novartis, Pfizer, Takeda, UCB, and Wyeth, as well as serving on the data and safety monitoring board for Forest Research and NIAMS. Dr. Brunner disclosed relationships with companies including Ablynx, AbbVie, AstraZeneca-MedImmune, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli Lilly, EMD Serono, F. Hoffmann-La Roche, Genzyme, GlaxoSmithKline, Merck, Novartis, R-Pharm, and Sanofi. The study by Dr. Rumsey and colleagues was supported by Amgen. Dr. Rumsey and colleagues had no relevant financial conflicts to disclose.
SOURCES: Weiss PF et al. Arthritis Care Res. 2020 Dec 5. doi: 10.1002/acr.24529; Rumsey DG et al. Arthritis Care Res. 2020 Dec. 16. doi: 10.1002/acr.24537; Lovell DJ and Brunner HI. Arthritis Care Res. 2020 Dec 16. doi: 10.1002/acr.24536.
Children with enthesitis-related arthritis often have a high burden of disease and could benefit from medications currently approved for adults with spondyloarthritis, according to a review published in Arthritis Care & Research.
“Enthesitis-related arthritis (ERA) was the JIA [juvenile idiopathic arthritis] category applied to children with spondyloarthritis (SpA), recognizing enthesitis as a defining characteristic,” wrote Pamela F. Weiss, MD, of Children’s Hospital of Philadelphia, and colleagues.
The ERA criteria include “arthritis plus enthesitis; or arthritis or enthesitis plus at least two of the following: sacroiliac tenderness or inflammatory back pain, HLA-B27 positivity, first-degree relative with HLA-B27–associated disease, acute anterior uveitis, and arthritis in a male older than 6 years,” the review authors noted.
“None of the [Food and Drug Administration]–approved therapies for peripheral SpA or nonradiographic axial SpA” have been studied or approved for use in children with ERA, but data support biologic similarity to SpA in adults; notably, studies of the HLA-B27 allele have identified it as a risk factor for both SpA and ERA, they said.
Common factors in adult and childhood conditions
“The principal commonalities of children with ERA and axial arthritis, and adults with nonradiographic axial SpA, include enthesitis, arthritis, inflammatory back pain, anterior uveitis, HLA-B27 positivity, and family history of HLA-B27–associated disease,” the review authors wrote.
The first-line treatment for both ERA with axial arthritis and nonradiographic axial SpA is NSAIDs, followed by tumor necrosis factor (TNF) inhibitors if needed, they said. However, conventional disease-modifying antirheumatic drugs (cDMARDs) may be used in cases of peripheral disease affecting five or more joints. Studies of treatment response show similarities between ERA in children and SpA in adults, the authors added, with nearly half of adults with axial disease unable to achieve remission and approximately one-third of children with ERA failing to respond to therapy.
Clinical trials could improve options and outcomes for those with ERA who need advanced therapy and such trials should evaluate response of axial and peripheral disease separately, the review authors emphasized. For example, “Eligibility criteria for children with ERA and axial features could include the presence of some of the following disease features: active inflammatory sacroiliitis based on typical MRI changes according to ASAS/OMERACT [Assessment of SpondyloArthritis international Society/Outcome Measures in Rheumatology Clinical Trials] criteria; elevated CRP [C-reactive protein]; and inadequate response or intolerance to NSAIDs,” they noted. “Considering the similarities between adult spondyloarthritis and ERA in terms of etiology, genetics, pathogenesis, and clinical manifestations, it is evident that medications approved for axial or peripheral SpA should be studied in children with ERA involving axial or peripheral joints, respectively, with the intent to achieve labeling for use in children,” they concluded.
New data highlight ERA disease burden
The need for additional therapies for ERA patients gained more support from a recent study in which a majority of children with ERA or juvenile psoriatic arthritis (jPsA) used biologics, but those with sacroiliitis in particular showed a significant disease burden despite high biologic use.
The International Leagues Against Rheumatism criteria include seven categories of juvenile idiopathic arthritis, of which ERA and jPsA are the most common; however, characteristics of these children have not been well described, wrote Dax G. Rumsey, MD, of the University of Alberta, Edmonton, and colleagues.
“Children with ERA are more likely to have a clinical picture with predominantly peripheral arthritis, typically described as an oligoarthritis involving the lower limbs with high risk of axial disease, relative to the other categories of JIA,” and report more intense pain and worse health status, compared with children in other categories, the researchers wrote.
To more completely characterize children with ERA and jPsA, the researchers assessed 522 children with ERA and 380 with jPsA. The children were enrolled in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) Registry. The findings were published in a brief report in Arthritis Care & Research.
Overall, 69% of the children took at least one biologic, including 72% with ERA and 64% with jPsA. Biologic use was even higher (81%) among the 28% of patients with sacroiliitis (40% of ERA patients and 12% of jPsA patients). Approximately 36% of the patients with sacroiliitis were positive for HLA-B27. In addition, Physician Global Assessment scores and clinical Juvenile Arthritis Disease Activity Score-10 (cJADAS10) scores were significantly higher at the first clinical visit with sacroiliitis, compared with the first visit without, which confirms “the clinical impression that active sacroiliitis significantly impacts children and their families,” the researchers said.
The average age at diagnosis was 10.8 years for ERA and 8.2 years for jPsA, and significantly more ERA patients were male (56% vs. 38%). However, more of the patients with sacroiliitis (54%) were female. More than half of the patients reported polyarticular involvement.
The study findings were limited by several factors, including the classification of ERA or jPsA and the reliance on physician diagnoses, as well as the variation in identifying sacroiliitis, the researchers said. However, the results increase understanding of the pathophysiology of ERA and jPsA to help determine optimal treatment, they concluded.
Data highlight research and treatment gaps
“Recent research demonstrates a large, unmet medical need in the treatment of JIA with 52%-65% of all JIA patients, including those with ERA and jPsA, having been treated with at least one biologic DMARD and 15%-19% having been treated with an FDA-unapproved biologic. In those with ERA or jPsA, 72%-79% of the children had been treated with a biologic DMARD, although no biologic DMARD has ever been FDA approved for these JIA categories,” Daniel J. Lovell, MD, and Hermine I. Brunner, MD, both with Cincinnati Children’s Hospital Medical Center, wrote in an editorial that accompanied the new study. Dr. Lovell and Dr. Brunner also were coauthors of the review article.
The new study supports findings from other recent publications, the editorialists noted. The new results showed “a significant proportion of the JIA population with active sacroiliitis with high disease burden despite very frequent (over 80% of the population) [treatment] with unstudied and unapproved biologic DMARDs,” they said. “These children with sacroiliitis had significantly greater disease burden with higher physician assessment of disease activity, higher parent assessment of disease impact, and higher disease activity as measured by the Juvenile Idiopathic Arthritis Disease Activity Score, compared to the children with ERA or jPsA without sacroiliitis,” they noted.
Previously, “the FDA granted pharmaceutical companies studying new treatments in adult SpA automatic full waivers from doing studies in children for new medications for ‘axial spondyloarthropathies including ankylosing spondylitis’ up until July 2020,” the editorialists said. However, “It is now time now for the pharmaceutical industry to perform FDA-monitored clinical trials of children and adolescents with SpA,” they emphasized. “This will allow for the scientific assessment of proper dosing, efficacy, and safety of the increasing number of new medications that are being licensed by the FDA for the treatment of SpA, such as the anti-TNF, anti–IL[interleukin]-17, and anti–IL-23 biologics, and perhaps JAK [Janus kinase] agents, to address this unmet medical need in these patients with juvenile SpA,” they concluded.
Dr. Weiss disclosed grant support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), and financial relationships with Eli Lilly and Pfizer. Dr. Lovell disclosed relationships with companies including Abbott, AbbVie Amgen, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Hoffmann-La Roche, Janssen, Novartis, Pfizer, Takeda, UCB, and Wyeth, as well as serving on the data and safety monitoring board for Forest Research and NIAMS. Dr. Brunner disclosed relationships with companies including Ablynx, AbbVie, AstraZeneca-MedImmune, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli Lilly, EMD Serono, F. Hoffmann-La Roche, Genzyme, GlaxoSmithKline, Merck, Novartis, R-Pharm, and Sanofi. The study by Dr. Rumsey and colleagues was supported by Amgen. Dr. Rumsey and colleagues had no relevant financial conflicts to disclose.
SOURCES: Weiss PF et al. Arthritis Care Res. 2020 Dec 5. doi: 10.1002/acr.24529; Rumsey DG et al. Arthritis Care Res. 2020 Dec. 16. doi: 10.1002/acr.24537; Lovell DJ and Brunner HI. Arthritis Care Res. 2020 Dec 16. doi: 10.1002/acr.24536.
Children with enthesitis-related arthritis often have a high burden of disease and could benefit from medications currently approved for adults with spondyloarthritis, according to a review published in Arthritis Care & Research.
“Enthesitis-related arthritis (ERA) was the JIA [juvenile idiopathic arthritis] category applied to children with spondyloarthritis (SpA), recognizing enthesitis as a defining characteristic,” wrote Pamela F. Weiss, MD, of Children’s Hospital of Philadelphia, and colleagues.
The ERA criteria include “arthritis plus enthesitis; or arthritis or enthesitis plus at least two of the following: sacroiliac tenderness or inflammatory back pain, HLA-B27 positivity, first-degree relative with HLA-B27–associated disease, acute anterior uveitis, and arthritis in a male older than 6 years,” the review authors noted.
“None of the [Food and Drug Administration]–approved therapies for peripheral SpA or nonradiographic axial SpA” have been studied or approved for use in children with ERA, but data support biologic similarity to SpA in adults; notably, studies of the HLA-B27 allele have identified it as a risk factor for both SpA and ERA, they said.
Common factors in adult and childhood conditions
“The principal commonalities of children with ERA and axial arthritis, and adults with nonradiographic axial SpA, include enthesitis, arthritis, inflammatory back pain, anterior uveitis, HLA-B27 positivity, and family history of HLA-B27–associated disease,” the review authors wrote.
The first-line treatment for both ERA with axial arthritis and nonradiographic axial SpA is NSAIDs, followed by tumor necrosis factor (TNF) inhibitors if needed, they said. However, conventional disease-modifying antirheumatic drugs (cDMARDs) may be used in cases of peripheral disease affecting five or more joints. Studies of treatment response show similarities between ERA in children and SpA in adults, the authors added, with nearly half of adults with axial disease unable to achieve remission and approximately one-third of children with ERA failing to respond to therapy.
Clinical trials could improve options and outcomes for those with ERA who need advanced therapy and such trials should evaluate response of axial and peripheral disease separately, the review authors emphasized. For example, “Eligibility criteria for children with ERA and axial features could include the presence of some of the following disease features: active inflammatory sacroiliitis based on typical MRI changes according to ASAS/OMERACT [Assessment of SpondyloArthritis international Society/Outcome Measures in Rheumatology Clinical Trials] criteria; elevated CRP [C-reactive protein]; and inadequate response or intolerance to NSAIDs,” they noted. “Considering the similarities between adult spondyloarthritis and ERA in terms of etiology, genetics, pathogenesis, and clinical manifestations, it is evident that medications approved for axial or peripheral SpA should be studied in children with ERA involving axial or peripheral joints, respectively, with the intent to achieve labeling for use in children,” they concluded.
New data highlight ERA disease burden
The need for additional therapies for ERA patients gained more support from a recent study in which a majority of children with ERA or juvenile psoriatic arthritis (jPsA) used biologics, but those with sacroiliitis in particular showed a significant disease burden despite high biologic use.
The International Leagues Against Rheumatism criteria include seven categories of juvenile idiopathic arthritis, of which ERA and jPsA are the most common; however, characteristics of these children have not been well described, wrote Dax G. Rumsey, MD, of the University of Alberta, Edmonton, and colleagues.
“Children with ERA are more likely to have a clinical picture with predominantly peripheral arthritis, typically described as an oligoarthritis involving the lower limbs with high risk of axial disease, relative to the other categories of JIA,” and report more intense pain and worse health status, compared with children in other categories, the researchers wrote.
To more completely characterize children with ERA and jPsA, the researchers assessed 522 children with ERA and 380 with jPsA. The children were enrolled in the Childhood Arthritis and Rheumatology Research Alliance (CARRA) Registry. The findings were published in a brief report in Arthritis Care & Research.
Overall, 69% of the children took at least one biologic, including 72% with ERA and 64% with jPsA. Biologic use was even higher (81%) among the 28% of patients with sacroiliitis (40% of ERA patients and 12% of jPsA patients). Approximately 36% of the patients with sacroiliitis were positive for HLA-B27. In addition, Physician Global Assessment scores and clinical Juvenile Arthritis Disease Activity Score-10 (cJADAS10) scores were significantly higher at the first clinical visit with sacroiliitis, compared with the first visit without, which confirms “the clinical impression that active sacroiliitis significantly impacts children and their families,” the researchers said.
The average age at diagnosis was 10.8 years for ERA and 8.2 years for jPsA, and significantly more ERA patients were male (56% vs. 38%). However, more of the patients with sacroiliitis (54%) were female. More than half of the patients reported polyarticular involvement.
The study findings were limited by several factors, including the classification of ERA or jPsA and the reliance on physician diagnoses, as well as the variation in identifying sacroiliitis, the researchers said. However, the results increase understanding of the pathophysiology of ERA and jPsA to help determine optimal treatment, they concluded.
Data highlight research and treatment gaps
“Recent research demonstrates a large, unmet medical need in the treatment of JIA with 52%-65% of all JIA patients, including those with ERA and jPsA, having been treated with at least one biologic DMARD and 15%-19% having been treated with an FDA-unapproved biologic. In those with ERA or jPsA, 72%-79% of the children had been treated with a biologic DMARD, although no biologic DMARD has ever been FDA approved for these JIA categories,” Daniel J. Lovell, MD, and Hermine I. Brunner, MD, both with Cincinnati Children’s Hospital Medical Center, wrote in an editorial that accompanied the new study. Dr. Lovell and Dr. Brunner also were coauthors of the review article.
The new study supports findings from other recent publications, the editorialists noted. The new results showed “a significant proportion of the JIA population with active sacroiliitis with high disease burden despite very frequent (over 80% of the population) [treatment] with unstudied and unapproved biologic DMARDs,” they said. “These children with sacroiliitis had significantly greater disease burden with higher physician assessment of disease activity, higher parent assessment of disease impact, and higher disease activity as measured by the Juvenile Idiopathic Arthritis Disease Activity Score, compared to the children with ERA or jPsA without sacroiliitis,” they noted.
Previously, “the FDA granted pharmaceutical companies studying new treatments in adult SpA automatic full waivers from doing studies in children for new medications for ‘axial spondyloarthropathies including ankylosing spondylitis’ up until July 2020,” the editorialists said. However, “It is now time now for the pharmaceutical industry to perform FDA-monitored clinical trials of children and adolescents with SpA,” they emphasized. “This will allow for the scientific assessment of proper dosing, efficacy, and safety of the increasing number of new medications that are being licensed by the FDA for the treatment of SpA, such as the anti-TNF, anti–IL[interleukin]-17, and anti–IL-23 biologics, and perhaps JAK [Janus kinase] agents, to address this unmet medical need in these patients with juvenile SpA,” they concluded.
Dr. Weiss disclosed grant support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), and financial relationships with Eli Lilly and Pfizer. Dr. Lovell disclosed relationships with companies including Abbott, AbbVie Amgen, AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, GlaxoSmithKline, Hoffmann-La Roche, Janssen, Novartis, Pfizer, Takeda, UCB, and Wyeth, as well as serving on the data and safety monitoring board for Forest Research and NIAMS. Dr. Brunner disclosed relationships with companies including Ablynx, AbbVie, AstraZeneca-MedImmune, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli Lilly, EMD Serono, F. Hoffmann-La Roche, Genzyme, GlaxoSmithKline, Merck, Novartis, R-Pharm, and Sanofi. The study by Dr. Rumsey and colleagues was supported by Amgen. Dr. Rumsey and colleagues had no relevant financial conflicts to disclose.
SOURCES: Weiss PF et al. Arthritis Care Res. 2020 Dec 5. doi: 10.1002/acr.24529; Rumsey DG et al. Arthritis Care Res. 2020 Dec. 16. doi: 10.1002/acr.24537; Lovell DJ and Brunner HI. Arthritis Care Res. 2020 Dec 16. doi: 10.1002/acr.24536.
FROM ARTHRITIS CARE & RESEARCH
Long-Term Successful Treatment of Indolent Systemic Mastocytosis With Omalizumab
This case study suggests that omalizumab may help prevent anaphylaxis and reduce disease burden associated with systemic mastocytosis, but further studies and formal clinical trials are needed to confirm these findings.
Mastocytosis is a rare disease that causes allergic and anaphylactic symptoms due to chronic or episodic, excessive mast cell degranulation as well as mast cell infiltration of the skin or other organs.1 Mast cells aid in innate immunity by generation of a vasodilatory and inflammatory response and are significant contributors to allergic reactions. Cutaneous mastocytosis is defined by isolated skin involvement. Systemic mastocytosis (SM) is characterized by mast cell infiltration of extracutaneous organs, most often bone marrow.2

Background
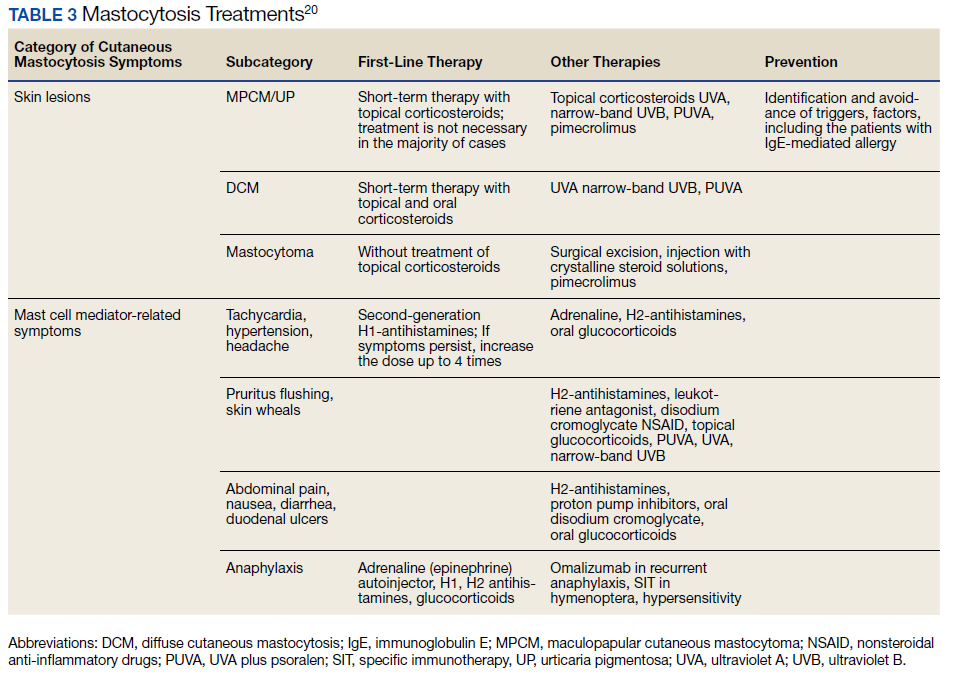
SM is divided into distinct subtypes (Table 1). Nonadvanced SM subtypes include indolent SM and smoldering SM. These are the most common forms and tend to have more slowly progressing courses without evidence of organ tissue dysfunction, a myelodysplastic syndrome, or of a myeloproliferative disorder.3 Advanced SM is less common and is associated with organ tissue dysfunction. It also may be associated with myeloproliferative, myelodysplastic, or lymphoproliferative hematologic neoplasms, and subtypes include aggressive SM, SM with an associated hematologic neoplasm, and mast cell leukemia (Table 2).4

Treatment options approved by the US Food and Drug Administration (FDA) for advanced SM include disease-altering medications, such as tyrosine kinase inhibitors (eg, imatinib), but the approved treatment options for nonadvanced SM are generally aimed at managing only symptoms (Table 3). Although not approved by the FDA for the treatment of SM, omalizumab may aid in the prevention of anaphylaxis, the reduction of disease burden, and the improvement in quality of life for patients with SM.5 Omalizumab is a humanized monoclonal antibody against the Fc portion of immunoglobulin E (IgE). It is approved by the FDA for treatment of asthma as well as chronic idiopathic urticaria.6
Case Presentation
A 32-year-old female initially presented to Womack Army Medical Center at Fort Bragg, North Carolina, for evaluation due to recurrent episodes of anaphylaxis occurring 1 to 2 times per month as well as chronic skin rashes that progressed over the previous 5 years (Figure). She initially was diagnosed with idiopathic anaphylaxis and subsequently had multiple emergency department (ED) and clinic visits for vasovagal syncope, unexplained allergic reactions, dizziness, giddiness, and shortness of breath. More recently, she was diagnosed with idiopathic urticaria.
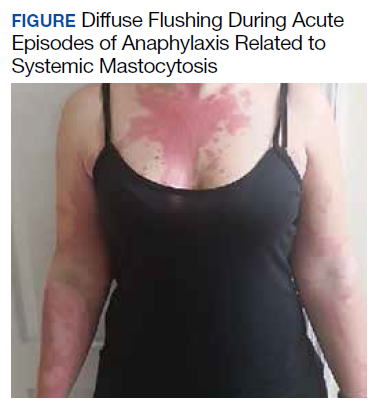
The patient reported at least 12 episodes in the previous year involving facial flushing that proceeded inferiorly, chest tightness, shortness of breath, labored breathing, crampy abdominal pain, and nausea without urticaria or significant pruritus. These bouts often were accompanied by mild facial angioedema, acute sinus pressure, vomiting, tachycardia, and lightheadedness. She reported experiencing brief losses of consciousness with at least 4 of these episodes. Home and ED blood pressure measurements revealed hypotension on several occasions with systolic readings in the 80s. She also developed nonpruritic freckles on her upper chest initially with subsequent increase in number and spread to involve her entire trunk, proximal extremities, and eventually distal extremities.
The patient had received intramuscular epinephrine several times, which led to rapid resolution of her symptoms. Intensive care unit admission for observation overnight was deemed necessary following one of her first episodes, but she did not require intubation or vasopressor support. Eventually, she began treating most episodes at home with diphenhydramine, ranitidine, and occasionally an epinephrine auto-injector, only presenting to the ED for severe dyspnea or loss of consciousness. Some episodes awoke her from sleeping but no triggers were identified (eg, foods, alcohol, supplements, medications, insect stings, latex exposure, exercise, strong emotions, or menstrual cycle).
Examination revealed hyperpigmented macules and papules scattered on the trunk and extremities, with a positive Darier sign. Punch biopsy of one of the macules revealed focal basal cell hyperpigmentation and sheets of benign-appearing mast cells in the superficial dermis, highlighted by CD117 immunohistochemical stain. A serum tryptase level was obtained and found to be significantly elevated (134 mcg/L). The patient was diagnosed with maculopapular cutaneous mastocytosis (urticaria pigmentosa).
A bone marrow biopsy revealed multiple prominent infiltrates of monomorphic, spindled, CD117-positive, CD2-positive, and CD25-positive mast cells arranged interstitially and paratrabecularly, with associated reticulin fibrosis. Indolent SM was diagnosed according to the World Health Organization classification system with multifocal, dense aggregates of mast cells (> 25%) in the bone marrow and with persistently elevated serum tryptase levels (134, 134, 151, and 159 ng/mL) without laboratory evidence of an associated clonal myeloid disorder or findings consistent with infiltrating bone lesions on full body magnetic resonance imaging scan.4
Despite maximal antihistamine and antileukotriene therapy with ranitidine (150 mg twice daily), cetirizine (10 mg twice daily), montelukast (10 mg daily), and cromolyn sodium (200 mg daily), the patient continued to experience recurrent episodes of anaphylaxis requiring subcutaneous epinephrine and systemic corticosteroids. In May 2016, the patient began a trial of off-label therapy with omalizumab injections (300 mg subcutaneous every 4 weeks). She has continued on therapy for more than 4 years and experienced only 1 anaphylactic episode. She also has had significant improvement in cutaneous symptoms.
Discussion
Mast cell overactivation and degranulation in mastocytosis is largely driven by the IgE antibody, which plays a significant role in atopic conditions, immediate hypersensitivity reactions, and anaphylaxis, as well as in the immunologic response to parasitic infections. The severity of atopic disease seems to be associated with serum IgE levels in many patients.7 IgE binding to surface receptors on mast cells and eosinophils prompts the release of toxic mediators, incites inflammation, and induces allergic symptoms.8 Activation of mast cells is classically elicited by IgE binding to the high-affinity Fcε RI receptor, the expression of which correlates with IgE levels.9
The anti-IgE, recombinant, humanized immunoglobulin G monoclonal antibody, omalizumab, decreases mastocytic and eosinophilic symptoms by binding and inhibiting IgE. This diminishes free IgE levels, inhibits IgE binding to the Fcε RI receptor, and affects downregulation of this high-affinity receptor on mast cells and basophils.6 Omalizumab is currently FDA approved only for the treatment of moderate-to-severe, persistent, allergic asthma that is not controlled by inhaled corticosteroids in patients aged ≥ 6 years, and for chronic idiopathic urticaria not controlled by H1 antihistamine therapy in patients aged ≥ 12 years.10 However, it stands to reason that this therapy also should be effective in the treatment of other poorly controlled atopic conditions, especially mastocytosis, the symptoms of which are driven by excessive mast cell degranulation and tissue infiltration.
As early as 2007, preliminary data showed that treatment with omalizumab could decrease the frequency of episodes of anaphylaxis.11 A National Institutes of Health case report followed 2 patients, one for 5 months and the other for 24 months. Both patients experienced a decrease in frequency of anaphylaxis following initiation of omalizumab. In 2010, a second case report described the treatment of an Australian patient with recurrent idiopathic anaphylaxis also diagnosed with SM. After initiation of treatment with omalizumab, she, too, experienced decreased frequency of episodes of anaphylaxis over 14 months.12 A review of patients treated at the Mastocytosis Centre Odense University Hospital in Denmark was published in 2017. Of 13 patients with SM treated with omalizumab, 5 experienced what was considered a complete response to the medication, with 3 each experiencing major and partial responses.5 The median treatment time in these patients was 27 months. Each of these cases showed significant promise in the use of omalizumab to treat SM, informing the decision to attempt this treatment in our patient.
The potential positive effects of omalizumab in reducing symptom severity in patients with SM was further supported by a 2017 meta-analysis. This review included several individual case reports noting that omalizumab could decrease frequency of pulmonary and gastrointestinal manifestations of SM.13 A small randomized control trial of omalizumab for treatment of mild symptoms of SM found improvement in disease severity, although neither primary nor secondary endpoints reached statistical significance.14
This case demonstrates a substantial, long-term, clinical benefit and quality of life improvement with omalizumab therapy in a patient with indolent SM that was not adequately controlled by conventional therapies. This is evidenced by an impressive decline in the frequency of mastocytic anaphylactic episodes as well as diminished patient-endorsed cutaneous symptoms.
This case provides further evidence of the efficacy of this therapy in diminishing disease burden for patients with SM who are otherwise limited to treatments aimed at transient symptomatic relief without significant alteration of the underlying cause of symptoms. At the time this article was written, our patient had now 52 months of continuous treatment without any adverse reactions noted, suggesting the treatment's long-term efficacy. It also adds to a small but growing body of literature that supports the use of anti-IgE therapy as a treatment option for improved management of this distressing, life-altering illness. Even in the time that our patient has been receiving omalizumab for SM, another small case series of 2 patients has been published showing sustained treatment effect at 12 years of therapy.15 This adds further insight that omalizumab can offer long-term, safe treatment for this limiting condition.
Omalizumab therapy is not without risk, but for patients afflicted by unrestrained mastocytic disease, the benefits may outweigh the risks. The most common significant risk with this medication is anaphylaxis, occurring in 1 to 2 per 1,000 patients, usually within 2 hours of an injection.16 This may correlate to the underlying degree of atopy in patients receiving omalizumab, and the risk of anaphylaxis is relatively low compared with that of many other biologic medications.17 Additionally, early data from initial phases of clinical trials indicated a potentially elevated malignancy risk with omalizumab. However, subsequent pooled analysis of larger numbers of patients has decreased suspicion that a causal relationship exists.18
Conclusions
Omalizumab has proven value in the treatment of atopic conditions, such as asthma and idiopathic urticaria, for which it has been approved for use by the FDA. Its effectiveness in significantly decreasing free serum IgE levels, and inhibiting IgE activation of mast cells makes it a possible treatment option for patients with SM who are not sufficiently controlled with conventional therapy. The findings in this case suggest that omalizumab may be effective in the prevention of anaphylaxis and in the reduction of disease burden associated with SM. Further studies and formal clinical trials are needed to confirm these findings. Patients should be counseled appropriately concerning the risks, benefits, and off-label status of this treatment option.
1. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163-172. doi:10.1056/NEJMra1409760
2. Valent P, Sperr WR, Schwartz LB, Horny H-P. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3-11. doi:10.1016/j.jaci.2004.02.045
3. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691-1700. doi:10.1093/annonc/mdu047
4. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
5. Broesby-Olsen S, Vestergaard H, Mortz CG, et al. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. 2018;73(1):230-238. doi:10.1111/all.13237
6. Kaplan AP, Giménez-Arnau AM, Saini SS.Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72(4):519-533. doi:10.1111/all.13083
7. Borish L, Chipps B, Deniz Y, Gujrathi S, Zheng B, Dolan C; TENOR Study Group. Total serum IgE levels in a large cohort of patients with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2005;95(3):247-253. doi:10.1016/S1081-1206(10)61221-5
8. Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402(suppl 6760):18-23. doi:10.1038/35037014
9. MacGlashan D, McKenzie-White J, Chichester K, et al. In vitro regulation of FcRIα expression on human basophils by IgE antibody. Blood. 1998;91(5):1633-1643.
10. XOLAIR [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. Revised 2019. Accessed November 11, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf
11. Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GC, and Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550-1551. doi:10.1016/j.jaci.2007.03.032
12. Douglass JA, Carroll K, Voskamp A, Bourke P, Wei A, O’Hehir RE. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy. 2010; 65(7):926-927. doi:10.1111/j.1398-9995.2009.02259.x
13. Le M, Miedzybrodzki B, Olynych T, Chapdelaine H, Ben-Shoshan M. Natural history and treatment of cutaneous and systemic mastocytosis. Postgrad Med. 2017;129(8):896-901. doi:10.1080/00325481.2017.1364124
14. Distler M, Maul J-T, Steiner T, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA Study) [published online ahead of print January 20, 2020]. Dermatology. doi:10.1159/000504842
15. Constantine G, Bressler P, Petroni D, Metcalfe D, Carter M. Twelve-year follow-up of omalizumab for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract. 2019;7(4)1314-1316. doi:10.1016/j.jaip.2018.07.041
16. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002-1014. doi:10.1056/NEJMra0804579
17. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013;2(10):e26333. doi:10.4161/onci.26333
18. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6. doi:10.1016/j.jaci.2012.01.033.
19. Castells M, Akin C. Mastocytosis (cutaneous and systemic): epidemiology, pathogenesis, and clinical manifestations. Accessed December 8, 2020. Updated June 12, 2018. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-epidemiology-pathogenesis-and-clinical-manifestations
20. Czarny J, Lange M, Lugowska-Umer H, Nowicki R. Cutaneous mastocytosis treatment: strategies, limitations, and perspectives. Postepy Dermatol Alergol. 2018;35(6):541-545. doi:10.5114/ada.2018.77605
This case study suggests that omalizumab may help prevent anaphylaxis and reduce disease burden associated with systemic mastocytosis, but further studies and formal clinical trials are needed to confirm these findings.
This case study suggests that omalizumab may help prevent anaphylaxis and reduce disease burden associated with systemic mastocytosis, but further studies and formal clinical trials are needed to confirm these findings.
Mastocytosis is a rare disease that causes allergic and anaphylactic symptoms due to chronic or episodic, excessive mast cell degranulation as well as mast cell infiltration of the skin or other organs.1 Mast cells aid in innate immunity by generation of a vasodilatory and inflammatory response and are significant contributors to allergic reactions. Cutaneous mastocytosis is defined by isolated skin involvement. Systemic mastocytosis (SM) is characterized by mast cell infiltration of extracutaneous organs, most often bone marrow.2
Background
SM is divided into distinct subtypes (Table 1). Nonadvanced SM subtypes include indolent SM and smoldering SM. These are the most common forms and tend to have more slowly progressing courses without evidence of organ tissue dysfunction, a myelodysplastic syndrome, or of a myeloproliferative disorder.3 Advanced SM is less common and is associated with organ tissue dysfunction. It also may be associated with myeloproliferative, myelodysplastic, or lymphoproliferative hematologic neoplasms, and subtypes include aggressive SM, SM with an associated hematologic neoplasm, and mast cell leukemia (Table 2).4
Treatment options approved by the US Food and Drug Administration (FDA) for advanced SM include disease-altering medications, such as tyrosine kinase inhibitors (eg, imatinib), but the approved treatment options for nonadvanced SM are generally aimed at managing only symptoms (Table 3). Although not approved by the FDA for the treatment of SM, omalizumab may aid in the prevention of anaphylaxis, the reduction of disease burden, and the improvement in quality of life for patients with SM.5 Omalizumab is a humanized monoclonal antibody against the Fc portion of immunoglobulin E (IgE). It is approved by the FDA for treatment of asthma as well as chronic idiopathic urticaria.6
Case Presentation
A 32-year-old female initially presented to Womack Army Medical Center at Fort Bragg, North Carolina, for evaluation due to recurrent episodes of anaphylaxis occurring 1 to 2 times per month as well as chronic skin rashes that progressed over the previous 5 years (Figure). She initially was diagnosed with idiopathic anaphylaxis and subsequently had multiple emergency department (ED) and clinic visits for vasovagal syncope, unexplained allergic reactions, dizziness, giddiness, and shortness of breath. More recently, she was diagnosed with idiopathic urticaria.
The patient reported at least 12 episodes in the previous year involving facial flushing that proceeded inferiorly, chest tightness, shortness of breath, labored breathing, crampy abdominal pain, and nausea without urticaria or significant pruritus. These bouts often were accompanied by mild facial angioedema, acute sinus pressure, vomiting, tachycardia, and lightheadedness. She reported experiencing brief losses of consciousness with at least 4 of these episodes. Home and ED blood pressure measurements revealed hypotension on several occasions with systolic readings in the 80s. She also developed nonpruritic freckles on her upper chest initially with subsequent increase in number and spread to involve her entire trunk, proximal extremities, and eventually distal extremities.
The patient had received intramuscular epinephrine several times, which led to rapid resolution of her symptoms. Intensive care unit admission for observation overnight was deemed necessary following one of her first episodes, but she did not require intubation or vasopressor support. Eventually, she began treating most episodes at home with diphenhydramine, ranitidine, and occasionally an epinephrine auto-injector, only presenting to the ED for severe dyspnea or loss of consciousness. Some episodes awoke her from sleeping but no triggers were identified (eg, foods, alcohol, supplements, medications, insect stings, latex exposure, exercise, strong emotions, or menstrual cycle).
Examination revealed hyperpigmented macules and papules scattered on the trunk and extremities, with a positive Darier sign. Punch biopsy of one of the macules revealed focal basal cell hyperpigmentation and sheets of benign-appearing mast cells in the superficial dermis, highlighted by CD117 immunohistochemical stain. A serum tryptase level was obtained and found to be significantly elevated (134 mcg/L). The patient was diagnosed with maculopapular cutaneous mastocytosis (urticaria pigmentosa).
A bone marrow biopsy revealed multiple prominent infiltrates of monomorphic, spindled, CD117-positive, CD2-positive, and CD25-positive mast cells arranged interstitially and paratrabecularly, with associated reticulin fibrosis. Indolent SM was diagnosed according to the World Health Organization classification system with multifocal, dense aggregates of mast cells (> 25%) in the bone marrow and with persistently elevated serum tryptase levels (134, 134, 151, and 159 ng/mL) without laboratory evidence of an associated clonal myeloid disorder or findings consistent with infiltrating bone lesions on full body magnetic resonance imaging scan.4
Despite maximal antihistamine and antileukotriene therapy with ranitidine (150 mg twice daily), cetirizine (10 mg twice daily), montelukast (10 mg daily), and cromolyn sodium (200 mg daily), the patient continued to experience recurrent episodes of anaphylaxis requiring subcutaneous epinephrine and systemic corticosteroids. In May 2016, the patient began a trial of off-label therapy with omalizumab injections (300 mg subcutaneous every 4 weeks). She has continued on therapy for more than 4 years and experienced only 1 anaphylactic episode. She also has had significant improvement in cutaneous symptoms.
Discussion
Mast cell overactivation and degranulation in mastocytosis is largely driven by the IgE antibody, which plays a significant role in atopic conditions, immediate hypersensitivity reactions, and anaphylaxis, as well as in the immunologic response to parasitic infections. The severity of atopic disease seems to be associated with serum IgE levels in many patients.7 IgE binding to surface receptors on mast cells and eosinophils prompts the release of toxic mediators, incites inflammation, and induces allergic symptoms.8 Activation of mast cells is classically elicited by IgE binding to the high-affinity Fcε RI receptor, the expression of which correlates with IgE levels.9
The anti-IgE, recombinant, humanized immunoglobulin G monoclonal antibody, omalizumab, decreases mastocytic and eosinophilic symptoms by binding and inhibiting IgE. This diminishes free IgE levels, inhibits IgE binding to the Fcε RI receptor, and affects downregulation of this high-affinity receptor on mast cells and basophils.6 Omalizumab is currently FDA approved only for the treatment of moderate-to-severe, persistent, allergic asthma that is not controlled by inhaled corticosteroids in patients aged ≥ 6 years, and for chronic idiopathic urticaria not controlled by H1 antihistamine therapy in patients aged ≥ 12 years.10 However, it stands to reason that this therapy also should be effective in the treatment of other poorly controlled atopic conditions, especially mastocytosis, the symptoms of which are driven by excessive mast cell degranulation and tissue infiltration.
As early as 2007, preliminary data showed that treatment with omalizumab could decrease the frequency of episodes of anaphylaxis.11 A National Institutes of Health case report followed 2 patients, one for 5 months and the other for 24 months. Both patients experienced a decrease in frequency of anaphylaxis following initiation of omalizumab. In 2010, a second case report described the treatment of an Australian patient with recurrent idiopathic anaphylaxis also diagnosed with SM. After initiation of treatment with omalizumab, she, too, experienced decreased frequency of episodes of anaphylaxis over 14 months.12 A review of patients treated at the Mastocytosis Centre Odense University Hospital in Denmark was published in 2017. Of 13 patients with SM treated with omalizumab, 5 experienced what was considered a complete response to the medication, with 3 each experiencing major and partial responses.5 The median treatment time in these patients was 27 months. Each of these cases showed significant promise in the use of omalizumab to treat SM, informing the decision to attempt this treatment in our patient.
The potential positive effects of omalizumab in reducing symptom severity in patients with SM was further supported by a 2017 meta-analysis. This review included several individual case reports noting that omalizumab could decrease frequency of pulmonary and gastrointestinal manifestations of SM.13 A small randomized control trial of omalizumab for treatment of mild symptoms of SM found improvement in disease severity, although neither primary nor secondary endpoints reached statistical significance.14
This case demonstrates a substantial, long-term, clinical benefit and quality of life improvement with omalizumab therapy in a patient with indolent SM that was not adequately controlled by conventional therapies. This is evidenced by an impressive decline in the frequency of mastocytic anaphylactic episodes as well as diminished patient-endorsed cutaneous symptoms.
This case provides further evidence of the efficacy of this therapy in diminishing disease burden for patients with SM who are otherwise limited to treatments aimed at transient symptomatic relief without significant alteration of the underlying cause of symptoms. At the time this article was written, our patient had now 52 months of continuous treatment without any adverse reactions noted, suggesting the treatment's long-term efficacy. It also adds to a small but growing body of literature that supports the use of anti-IgE therapy as a treatment option for improved management of this distressing, life-altering illness. Even in the time that our patient has been receiving omalizumab for SM, another small case series of 2 patients has been published showing sustained treatment effect at 12 years of therapy.15 This adds further insight that omalizumab can offer long-term, safe treatment for this limiting condition.
Omalizumab therapy is not without risk, but for patients afflicted by unrestrained mastocytic disease, the benefits may outweigh the risks. The most common significant risk with this medication is anaphylaxis, occurring in 1 to 2 per 1,000 patients, usually within 2 hours of an injection.16 This may correlate to the underlying degree of atopy in patients receiving omalizumab, and the risk of anaphylaxis is relatively low compared with that of many other biologic medications.17 Additionally, early data from initial phases of clinical trials indicated a potentially elevated malignancy risk with omalizumab. However, subsequent pooled analysis of larger numbers of patients has decreased suspicion that a causal relationship exists.18
Conclusions
Omalizumab has proven value in the treatment of atopic conditions, such as asthma and idiopathic urticaria, for which it has been approved for use by the FDA. Its effectiveness in significantly decreasing free serum IgE levels, and inhibiting IgE activation of mast cells makes it a possible treatment option for patients with SM who are not sufficiently controlled with conventional therapy. The findings in this case suggest that omalizumab may be effective in the prevention of anaphylaxis and in the reduction of disease burden associated with SM. Further studies and formal clinical trials are needed to confirm these findings. Patients should be counseled appropriately concerning the risks, benefits, and off-label status of this treatment option.
Mastocytosis is a rare disease that causes allergic and anaphylactic symptoms due to chronic or episodic, excessive mast cell degranulation as well as mast cell infiltration of the skin or other organs.1 Mast cells aid in innate immunity by generation of a vasodilatory and inflammatory response and are significant contributors to allergic reactions. Cutaneous mastocytosis is defined by isolated skin involvement. Systemic mastocytosis (SM) is characterized by mast cell infiltration of extracutaneous organs, most often bone marrow.2
Background
SM is divided into distinct subtypes (Table 1). Nonadvanced SM subtypes include indolent SM and smoldering SM. These are the most common forms and tend to have more slowly progressing courses without evidence of organ tissue dysfunction, a myelodysplastic syndrome, or of a myeloproliferative disorder.3 Advanced SM is less common and is associated with organ tissue dysfunction. It also may be associated with myeloproliferative, myelodysplastic, or lymphoproliferative hematologic neoplasms, and subtypes include aggressive SM, SM with an associated hematologic neoplasm, and mast cell leukemia (Table 2).4
Treatment options approved by the US Food and Drug Administration (FDA) for advanced SM include disease-altering medications, such as tyrosine kinase inhibitors (eg, imatinib), but the approved treatment options for nonadvanced SM are generally aimed at managing only symptoms (Table 3). Although not approved by the FDA for the treatment of SM, omalizumab may aid in the prevention of anaphylaxis, the reduction of disease burden, and the improvement in quality of life for patients with SM.5 Omalizumab is a humanized monoclonal antibody against the Fc portion of immunoglobulin E (IgE). It is approved by the FDA for treatment of asthma as well as chronic idiopathic urticaria.6
Case Presentation
A 32-year-old female initially presented to Womack Army Medical Center at Fort Bragg, North Carolina, for evaluation due to recurrent episodes of anaphylaxis occurring 1 to 2 times per month as well as chronic skin rashes that progressed over the previous 5 years (Figure). She initially was diagnosed with idiopathic anaphylaxis and subsequently had multiple emergency department (ED) and clinic visits for vasovagal syncope, unexplained allergic reactions, dizziness, giddiness, and shortness of breath. More recently, she was diagnosed with idiopathic urticaria.
The patient reported at least 12 episodes in the previous year involving facial flushing that proceeded inferiorly, chest tightness, shortness of breath, labored breathing, crampy abdominal pain, and nausea without urticaria or significant pruritus. These bouts often were accompanied by mild facial angioedema, acute sinus pressure, vomiting, tachycardia, and lightheadedness. She reported experiencing brief losses of consciousness with at least 4 of these episodes. Home and ED blood pressure measurements revealed hypotension on several occasions with systolic readings in the 80s. She also developed nonpruritic freckles on her upper chest initially with subsequent increase in number and spread to involve her entire trunk, proximal extremities, and eventually distal extremities.
The patient had received intramuscular epinephrine several times, which led to rapid resolution of her symptoms. Intensive care unit admission for observation overnight was deemed necessary following one of her first episodes, but she did not require intubation or vasopressor support. Eventually, she began treating most episodes at home with diphenhydramine, ranitidine, and occasionally an epinephrine auto-injector, only presenting to the ED for severe dyspnea or loss of consciousness. Some episodes awoke her from sleeping but no triggers were identified (eg, foods, alcohol, supplements, medications, insect stings, latex exposure, exercise, strong emotions, or menstrual cycle).
Examination revealed hyperpigmented macules and papules scattered on the trunk and extremities, with a positive Darier sign. Punch biopsy of one of the macules revealed focal basal cell hyperpigmentation and sheets of benign-appearing mast cells in the superficial dermis, highlighted by CD117 immunohistochemical stain. A serum tryptase level was obtained and found to be significantly elevated (134 mcg/L). The patient was diagnosed with maculopapular cutaneous mastocytosis (urticaria pigmentosa).
A bone marrow biopsy revealed multiple prominent infiltrates of monomorphic, spindled, CD117-positive, CD2-positive, and CD25-positive mast cells arranged interstitially and paratrabecularly, with associated reticulin fibrosis. Indolent SM was diagnosed according to the World Health Organization classification system with multifocal, dense aggregates of mast cells (> 25%) in the bone marrow and with persistently elevated serum tryptase levels (134, 134, 151, and 159 ng/mL) without laboratory evidence of an associated clonal myeloid disorder or findings consistent with infiltrating bone lesions on full body magnetic resonance imaging scan.4
Despite maximal antihistamine and antileukotriene therapy with ranitidine (150 mg twice daily), cetirizine (10 mg twice daily), montelukast (10 mg daily), and cromolyn sodium (200 mg daily), the patient continued to experience recurrent episodes of anaphylaxis requiring subcutaneous epinephrine and systemic corticosteroids. In May 2016, the patient began a trial of off-label therapy with omalizumab injections (300 mg subcutaneous every 4 weeks). She has continued on therapy for more than 4 years and experienced only 1 anaphylactic episode. She also has had significant improvement in cutaneous symptoms.
Discussion
Mast cell overactivation and degranulation in mastocytosis is largely driven by the IgE antibody, which plays a significant role in atopic conditions, immediate hypersensitivity reactions, and anaphylaxis, as well as in the immunologic response to parasitic infections. The severity of atopic disease seems to be associated with serum IgE levels in many patients.7 IgE binding to surface receptors on mast cells and eosinophils prompts the release of toxic mediators, incites inflammation, and induces allergic symptoms.8 Activation of mast cells is classically elicited by IgE binding to the high-affinity Fcε RI receptor, the expression of which correlates with IgE levels.9
The anti-IgE, recombinant, humanized immunoglobulin G monoclonal antibody, omalizumab, decreases mastocytic and eosinophilic symptoms by binding and inhibiting IgE. This diminishes free IgE levels, inhibits IgE binding to the Fcε RI receptor, and affects downregulation of this high-affinity receptor on mast cells and basophils.6 Omalizumab is currently FDA approved only for the treatment of moderate-to-severe, persistent, allergic asthma that is not controlled by inhaled corticosteroids in patients aged ≥ 6 years, and for chronic idiopathic urticaria not controlled by H1 antihistamine therapy in patients aged ≥ 12 years.10 However, it stands to reason that this therapy also should be effective in the treatment of other poorly controlled atopic conditions, especially mastocytosis, the symptoms of which are driven by excessive mast cell degranulation and tissue infiltration.
As early as 2007, preliminary data showed that treatment with omalizumab could decrease the frequency of episodes of anaphylaxis.11 A National Institutes of Health case report followed 2 patients, one for 5 months and the other for 24 months. Both patients experienced a decrease in frequency of anaphylaxis following initiation of omalizumab. In 2010, a second case report described the treatment of an Australian patient with recurrent idiopathic anaphylaxis also diagnosed with SM. After initiation of treatment with omalizumab, she, too, experienced decreased frequency of episodes of anaphylaxis over 14 months.12 A review of patients treated at the Mastocytosis Centre Odense University Hospital in Denmark was published in 2017. Of 13 patients with SM treated with omalizumab, 5 experienced what was considered a complete response to the medication, with 3 each experiencing major and partial responses.5 The median treatment time in these patients was 27 months. Each of these cases showed significant promise in the use of omalizumab to treat SM, informing the decision to attempt this treatment in our patient.
The potential positive effects of omalizumab in reducing symptom severity in patients with SM was further supported by a 2017 meta-analysis. This review included several individual case reports noting that omalizumab could decrease frequency of pulmonary and gastrointestinal manifestations of SM.13 A small randomized control trial of omalizumab for treatment of mild symptoms of SM found improvement in disease severity, although neither primary nor secondary endpoints reached statistical significance.14
This case demonstrates a substantial, long-term, clinical benefit and quality of life improvement with omalizumab therapy in a patient with indolent SM that was not adequately controlled by conventional therapies. This is evidenced by an impressive decline in the frequency of mastocytic anaphylactic episodes as well as diminished patient-endorsed cutaneous symptoms.
This case provides further evidence of the efficacy of this therapy in diminishing disease burden for patients with SM who are otherwise limited to treatments aimed at transient symptomatic relief without significant alteration of the underlying cause of symptoms. At the time this article was written, our patient had now 52 months of continuous treatment without any adverse reactions noted, suggesting the treatment's long-term efficacy. It also adds to a small but growing body of literature that supports the use of anti-IgE therapy as a treatment option for improved management of this distressing, life-altering illness. Even in the time that our patient has been receiving omalizumab for SM, another small case series of 2 patients has been published showing sustained treatment effect at 12 years of therapy.15 This adds further insight that omalizumab can offer long-term, safe treatment for this limiting condition.
Omalizumab therapy is not without risk, but for patients afflicted by unrestrained mastocytic disease, the benefits may outweigh the risks. The most common significant risk with this medication is anaphylaxis, occurring in 1 to 2 per 1,000 patients, usually within 2 hours of an injection.16 This may correlate to the underlying degree of atopy in patients receiving omalizumab, and the risk of anaphylaxis is relatively low compared with that of many other biologic medications.17 Additionally, early data from initial phases of clinical trials indicated a potentially elevated malignancy risk with omalizumab. However, subsequent pooled analysis of larger numbers of patients has decreased suspicion that a causal relationship exists.18
Conclusions
Omalizumab has proven value in the treatment of atopic conditions, such as asthma and idiopathic urticaria, for which it has been approved for use by the FDA. Its effectiveness in significantly decreasing free serum IgE levels, and inhibiting IgE activation of mast cells makes it a possible treatment option for patients with SM who are not sufficiently controlled with conventional therapy. The findings in this case suggest that omalizumab may be effective in the prevention of anaphylaxis and in the reduction of disease burden associated with SM. Further studies and formal clinical trials are needed to confirm these findings. Patients should be counseled appropriately concerning the risks, benefits, and off-label status of this treatment option.
1. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163-172. doi:10.1056/NEJMra1409760
2. Valent P, Sperr WR, Schwartz LB, Horny H-P. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3-11. doi:10.1016/j.jaci.2004.02.045
3. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691-1700. doi:10.1093/annonc/mdu047
4. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
5. Broesby-Olsen S, Vestergaard H, Mortz CG, et al. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. 2018;73(1):230-238. doi:10.1111/all.13237
6. Kaplan AP, Giménez-Arnau AM, Saini SS.Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72(4):519-533. doi:10.1111/all.13083
7. Borish L, Chipps B, Deniz Y, Gujrathi S, Zheng B, Dolan C; TENOR Study Group. Total serum IgE levels in a large cohort of patients with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2005;95(3):247-253. doi:10.1016/S1081-1206(10)61221-5
8. Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402(suppl 6760):18-23. doi:10.1038/35037014
9. MacGlashan D, McKenzie-White J, Chichester K, et al. In vitro regulation of FcRIα expression on human basophils by IgE antibody. Blood. 1998;91(5):1633-1643.
10. XOLAIR [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. Revised 2019. Accessed November 11, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf
11. Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GC, and Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550-1551. doi:10.1016/j.jaci.2007.03.032
12. Douglass JA, Carroll K, Voskamp A, Bourke P, Wei A, O’Hehir RE. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy. 2010; 65(7):926-927. doi:10.1111/j.1398-9995.2009.02259.x
13. Le M, Miedzybrodzki B, Olynych T, Chapdelaine H, Ben-Shoshan M. Natural history and treatment of cutaneous and systemic mastocytosis. Postgrad Med. 2017;129(8):896-901. doi:10.1080/00325481.2017.1364124
14. Distler M, Maul J-T, Steiner T, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA Study) [published online ahead of print January 20, 2020]. Dermatology. doi:10.1159/000504842
15. Constantine G, Bressler P, Petroni D, Metcalfe D, Carter M. Twelve-year follow-up of omalizumab for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract. 2019;7(4)1314-1316. doi:10.1016/j.jaip.2018.07.041
16. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002-1014. doi:10.1056/NEJMra0804579
17. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013;2(10):e26333. doi:10.4161/onci.26333
18. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6. doi:10.1016/j.jaci.2012.01.033.
19. Castells M, Akin C. Mastocytosis (cutaneous and systemic): epidemiology, pathogenesis, and clinical manifestations. Accessed December 8, 2020. Updated June 12, 2018. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-epidemiology-pathogenesis-and-clinical-manifestations
20. Czarny J, Lange M, Lugowska-Umer H, Nowicki R. Cutaneous mastocytosis treatment: strategies, limitations, and perspectives. Postepy Dermatol Alergol. 2018;35(6):541-545. doi:10.5114/ada.2018.77605
1. Theoharides TC, Valent P, Akin C. Mast cells, mastocytosis, and related disorders. N Engl J Med. 2015;373(2):163-172. doi:10.1056/NEJMra1409760
2. Valent P, Sperr WR, Schwartz LB, Horny H-P. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114(1):3-11. doi:10.1016/j.jaci.2004.02.045
3. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691-1700. doi:10.1093/annonc/mdu047
4. Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420-1427. doi:10.1182/blood-2016-09-731893
5. Broesby-Olsen S, Vestergaard H, Mortz CG, et al. Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. 2018;73(1):230-238. doi:10.1111/all.13237
6. Kaplan AP, Giménez-Arnau AM, Saini SS.Mechanisms of action that contribute to efficacy of omalizumab in chronic spontaneous urticaria. Allergy. 2017;72(4):519-533. doi:10.1111/all.13083
7. Borish L, Chipps B, Deniz Y, Gujrathi S, Zheng B, Dolan C; TENOR Study Group. Total serum IgE levels in a large cohort of patients with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2005;95(3):247-253. doi:10.1016/S1081-1206(10)61221-5
8. Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999;402(suppl 6760):18-23. doi:10.1038/35037014
9. MacGlashan D, McKenzie-White J, Chichester K, et al. In vitro regulation of FcRIα expression on human basophils by IgE antibody. Blood. 1998;91(5):1633-1643.
10. XOLAIR [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation. Revised 2019. Accessed November 11, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/103976s5234lbl.pdf
11. Carter MC, Robyn JA, Bressler PB, Walker JC, Shapiro GC, and Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550-1551. doi:10.1016/j.jaci.2007.03.032
12. Douglass JA, Carroll K, Voskamp A, Bourke P, Wei A, O’Hehir RE. Omalizumab is effective in treating systemic mastocytosis in a nonatopic patient. Allergy. 2010; 65(7):926-927. doi:10.1111/j.1398-9995.2009.02259.x
13. Le M, Miedzybrodzki B, Olynych T, Chapdelaine H, Ben-Shoshan M. Natural history and treatment of cutaneous and systemic mastocytosis. Postgrad Med. 2017;129(8):896-901. doi:10.1080/00325481.2017.1364124
14. Distler M, Maul J-T, Steiner T, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA Study) [published online ahead of print January 20, 2020]. Dermatology. doi:10.1159/000504842
15. Constantine G, Bressler P, Petroni D, Metcalfe D, Carter M. Twelve-year follow-up of omalizumab for anaphylaxis in 2 patients with systemic mastocytosis. J Allergy Clin Immunol Pract. 2019;7(4)1314-1316. doi:10.1016/j.jaip.2018.07.041
16. Fanta CH. Asthma. N Engl J Med. 2009;360(10):1002-1014. doi:10.1056/NEJMra0804579
17. Baldo BA. Adverse events to monoclonal antibodies used for cancer therapy: focus on hypersensitivity responses. Oncoimmunology. 2013;2(10):e26333. doi:10.4161/onci.26333
18. Busse W, Buhl R, Fernandez Vidaurre C, et al. Omalizumab and the risk of malignancy: results from a pooled analysis. J Allergy Clin Immunol. 2012;129(4):983-989.e6. doi:10.1016/j.jaci.2012.01.033.
19. Castells M, Akin C. Mastocytosis (cutaneous and systemic): epidemiology, pathogenesis, and clinical manifestations. Accessed December 8, 2020. Updated June 12, 2018. https://www.uptodate.com/contents/mastocytosis-cutaneous-and-systemic-epidemiology-pathogenesis-and-clinical-manifestations
20. Czarny J, Lange M, Lugowska-Umer H, Nowicki R. Cutaneous mastocytosis treatment: strategies, limitations, and perspectives. Postepy Dermatol Alergol. 2018;35(6):541-545. doi:10.5114/ada.2018.77605
FDA clears device to remove dead pancreatic tissue
The Food and Drug Administration has approved the EndoRotor System (Interscope, Inc.) for removal of necrotic tissue in patients with walled-off pancreatic necrosis (WOPN).
“This device has shown its potential to provide a minimally invasive way to remove harmful necrotic pancreatic tissue in patients with walled-off pancreatic necrosis,” Charles Viviano, MD, PhD, acting director, Reproductive, Gastro-Renal, Urological, General Hospital Device and Human Factors Office, FDA Center for Devices and Radiological Health, said in a statement.
“Currently, in order to remove dead tissue from a patient’s necrotic pancreatic cavity, health care providers need to perform an invasive surgery or use other endoscopic tools not specifically indicated to treat this condition. With [this] marketing authorization, patients with walled-off pancreatic necrosis now have a new treatment option,” said Dr. Viviano.
WOPN is a potentially deadly condition that occurs in about 15% of patients with severe pancreatitis. Often, the dead tissue must be removed.
The EndoRotor System is made up of a power console, foot control, specimen trap, and single-use catheter.
The device is used to perform endoscopic necrosectomy. In this procedure, a stent is used to create a portal between the stomach and the necrotic cavity in the pancreas to accommodate a standard endoscope through which the EndoRotor cuts and removes necrotized tissue.
The FDA approved the EndoRotor System on the basis of a clinical trial involving 30 patients with WOPN who underwent a total of 63 direct endoscopic necrosectomies with the EndoRotor System (average, 2.1 procedures per patient).
The effectiveness of the EndoRotor System was determined by how well it cleared pancreatic necrotic tissue measured during CT with contrast before and after the procedure, endoscopy, or MRI 14 to 28 days after the last procedure.
Results showed an average 85% reduction in the amount of necrotic tissue, with half of the patients having 98.5% clearance of necrotic tissue, the FDA said.
Three patients suffered procedure-related serious adverse events (10% complication rate). Two patients experienced gastrointestinal bleeding. One patient had a pneumoperitoneum and later died after suffering from sepsis and multiorgan system failure caused by massive collections of infected pancreatic necrotic tissue.
Other serious adverse events, which were thought to be due to the patient’s underlying condition and not related to the device or procedure, included hematemesis, deep vein thrombosis, and pancreatitis.
The EndoRotor System should not be used for patients with known or suspected pancreatic cancer, and the device will carry a boxed warning stating this.
The FDA said it knows of one patient who died from pancreatic cancer 3 months after having necrotic pancreatic tissue removed with the EndoRotor System.
“This patient did not have a diagnosis of pancreatic cancer prior to treatment, although the patient’s outcome is believed to be unrelated to the device or procedure,” the FDA said.
The EndoRotor System should be used only after patients have undergone other procedures to drain the WOPN.
It is also not appropriate for patients with walled-off necrosis who have a documented pseudoaneurysm greater than 1 cm within the cavity or with intervening gastric varices or unavoidable blood vessels within the access tract.
The EndoRotor System was approved under the de novo premarket review pathway for new low- to moderate-risk devices.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the EndoRotor System (Interscope, Inc.) for removal of necrotic tissue in patients with walled-off pancreatic necrosis (WOPN).
“This device has shown its potential to provide a minimally invasive way to remove harmful necrotic pancreatic tissue in patients with walled-off pancreatic necrosis,” Charles Viviano, MD, PhD, acting director, Reproductive, Gastro-Renal, Urological, General Hospital Device and Human Factors Office, FDA Center for Devices and Radiological Health, said in a statement.
“Currently, in order to remove dead tissue from a patient’s necrotic pancreatic cavity, health care providers need to perform an invasive surgery or use other endoscopic tools not specifically indicated to treat this condition. With [this] marketing authorization, patients with walled-off pancreatic necrosis now have a new treatment option,” said Dr. Viviano.
WOPN is a potentially deadly condition that occurs in about 15% of patients with severe pancreatitis. Often, the dead tissue must be removed.
The EndoRotor System is made up of a power console, foot control, specimen trap, and single-use catheter.
The device is used to perform endoscopic necrosectomy. In this procedure, a stent is used to create a portal between the stomach and the necrotic cavity in the pancreas to accommodate a standard endoscope through which the EndoRotor cuts and removes necrotized tissue.
The FDA approved the EndoRotor System on the basis of a clinical trial involving 30 patients with WOPN who underwent a total of 63 direct endoscopic necrosectomies with the EndoRotor System (average, 2.1 procedures per patient).
The effectiveness of the EndoRotor System was determined by how well it cleared pancreatic necrotic tissue measured during CT with contrast before and after the procedure, endoscopy, or MRI 14 to 28 days after the last procedure.
Results showed an average 85% reduction in the amount of necrotic tissue, with half of the patients having 98.5% clearance of necrotic tissue, the FDA said.
Three patients suffered procedure-related serious adverse events (10% complication rate). Two patients experienced gastrointestinal bleeding. One patient had a pneumoperitoneum and later died after suffering from sepsis and multiorgan system failure caused by massive collections of infected pancreatic necrotic tissue.
Other serious adverse events, which were thought to be due to the patient’s underlying condition and not related to the device or procedure, included hematemesis, deep vein thrombosis, and pancreatitis.
The EndoRotor System should not be used for patients with known or suspected pancreatic cancer, and the device will carry a boxed warning stating this.
The FDA said it knows of one patient who died from pancreatic cancer 3 months after having necrotic pancreatic tissue removed with the EndoRotor System.
“This patient did not have a diagnosis of pancreatic cancer prior to treatment, although the patient’s outcome is believed to be unrelated to the device or procedure,” the FDA said.
The EndoRotor System should be used only after patients have undergone other procedures to drain the WOPN.
It is also not appropriate for patients with walled-off necrosis who have a documented pseudoaneurysm greater than 1 cm within the cavity or with intervening gastric varices or unavoidable blood vessels within the access tract.
The EndoRotor System was approved under the de novo premarket review pathway for new low- to moderate-risk devices.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved the EndoRotor System (Interscope, Inc.) for removal of necrotic tissue in patients with walled-off pancreatic necrosis (WOPN).
“This device has shown its potential to provide a minimally invasive way to remove harmful necrotic pancreatic tissue in patients with walled-off pancreatic necrosis,” Charles Viviano, MD, PhD, acting director, Reproductive, Gastro-Renal, Urological, General Hospital Device and Human Factors Office, FDA Center for Devices and Radiological Health, said in a statement.
“Currently, in order to remove dead tissue from a patient’s necrotic pancreatic cavity, health care providers need to perform an invasive surgery or use other endoscopic tools not specifically indicated to treat this condition. With [this] marketing authorization, patients with walled-off pancreatic necrosis now have a new treatment option,” said Dr. Viviano.
WOPN is a potentially deadly condition that occurs in about 15% of patients with severe pancreatitis. Often, the dead tissue must be removed.
The EndoRotor System is made up of a power console, foot control, specimen trap, and single-use catheter.
The device is used to perform endoscopic necrosectomy. In this procedure, a stent is used to create a portal between the stomach and the necrotic cavity in the pancreas to accommodate a standard endoscope through which the EndoRotor cuts and removes necrotized tissue.
The FDA approved the EndoRotor System on the basis of a clinical trial involving 30 patients with WOPN who underwent a total of 63 direct endoscopic necrosectomies with the EndoRotor System (average, 2.1 procedures per patient).
The effectiveness of the EndoRotor System was determined by how well it cleared pancreatic necrotic tissue measured during CT with contrast before and after the procedure, endoscopy, or MRI 14 to 28 days after the last procedure.
Results showed an average 85% reduction in the amount of necrotic tissue, with half of the patients having 98.5% clearance of necrotic tissue, the FDA said.
Three patients suffered procedure-related serious adverse events (10% complication rate). Two patients experienced gastrointestinal bleeding. One patient had a pneumoperitoneum and later died after suffering from sepsis and multiorgan system failure caused by massive collections of infected pancreatic necrotic tissue.
Other serious adverse events, which were thought to be due to the patient’s underlying condition and not related to the device or procedure, included hematemesis, deep vein thrombosis, and pancreatitis.
The EndoRotor System should not be used for patients with known or suspected pancreatic cancer, and the device will carry a boxed warning stating this.
The FDA said it knows of one patient who died from pancreatic cancer 3 months after having necrotic pancreatic tissue removed with the EndoRotor System.
“This patient did not have a diagnosis of pancreatic cancer prior to treatment, although the patient’s outcome is believed to be unrelated to the device or procedure,” the FDA said.
The EndoRotor System should be used only after patients have undergone other procedures to drain the WOPN.
It is also not appropriate for patients with walled-off necrosis who have a documented pseudoaneurysm greater than 1 cm within the cavity or with intervening gastric varices or unavoidable blood vessels within the access tract.
The EndoRotor System was approved under the de novo premarket review pathway for new low- to moderate-risk devices.
A version of this article first appeared on Medscape.com.
Analysis characterizes common wound microbes in epidermolysis bullosa
– in a retrospective analysis of over 700 wound cultures from 158 patients across the United States and Canada.
The findings from the EB Clinical Characterization and Outcomes Database speak to the value of surveillance cultures with routine testing for microbial resistance – including mupirocin resistance – and to the importance of antibiotic stewardship not only for oral antibiotics but for topicals as well, according to Laura E. Levin, MD, and Kimberly D. Morel, MD, of the departments of dermatology and pediatrics, Columbia University Irving Medical Center, New York, the lead and senior authors, respectively, of the paper recently published in Pediatric Dermatology.
Almost all of the 158 patients with at least one wound culture recorded in the database from the period of 2001-2018 had one or more positive culture results. Of 152 patients with positive cultures, 131 (86%) were positive for SA and 56 (37%) and 34 (22%) were positive for PA and GAS, respectively. Other bacteria isolated included Corynebacterium spp and Proteus spp. Nearly half (47%) of patients with SA-positive cultures had methicillin-resistant SA, and 68% had methicillin-susceptible SA. (Some patients grew both MSSA and MRSA at different points in time.)
Mupirocin-susceptibility testing was performed at only some of the 13 participating centers. Of 15 patients whose cultures had recorded SA mupirocin-susceptibility testing, 11 had cultures positive for mupirocin-susceptible SA and 6 (40%) had mupirocin-resistant SA isolates (2 patients grew both). Of these six patients, half had isolates that were also methicillin-resistant.
Mupirocin, a topical antibiotic, has been a cornerstone of decolonization regimens for MSSA and MRSA, but resistance has been demonstrated in other research as well and is not specific to EB, wrote Dr. Levin, Dr. Morel, and coauthors.
“Pediatric dermatologists often rely on topical antimicrobials in the treatment of patients’ open wounds to both prevent and treat infection, depending on the clinical scenario,” and surveillance cultures with routine testing for mupirocin resistance can help guide antibiotic choice and management strategies, Dr. Levin said in an interview.
More broadly, she added, “it’s helpful to know what bacteria are routinely colonizing wounds, not causing infection, versus those that are more likely to be associated with infection, chronic wounds, or the risk of developing skin cancer ... [to know] which wounds need to be treated more aggressively.”
A subset of patients with EB have been known to be at risk for squamous cell carcinoma, and research is implicating certain bacteria “as contributing to wound inflammation,” Dr. Morel said in an interview.
SCC was reported in 23 out of 717 patients in the database – but fewer than half of the patients with SCC had recorded wound cultures. The small numbers precluded the identification of microbes that may confer significant risk.
Correlating particular microbes with clinical features also will take more research. About half (57%) of the patients with recorded wound cultures had wounds with purulent exudate or other features of clinical infection. However, the presence or absence of clinical signs of infection was not temporally correlated with culture results in the database.
The 158 patients with recorded wound cultures had a mean age of 12.8 years and represented a range of EB subtypes.
PA was present in the wounds of patients as young as 1 month old, the authors noted. Investigators are “looking to further study PA and characterize clinical features ... to understand more about this microbe and its impact on patients with EB,” Dr. Morel said.
In the meantime, the analysis reaffirms the importance of antibiotic stewardship. Mupirocin is labeled to be used three times a day for a short period of time, but “tends to be prescribed and used less judiciously than intended,” Dr. Morel said. “It’s important [not to overuse it]. We have seen that patients’ culture results become sensitive to mupirocin again in the future when they avoid it for a period of time.”
The work was supported by the EB Research Partnership and EB Medical Research Foundation, as well as an NIH/NCATS grant. No investigator disclosures were listed.
SOURCE: Pediatr Dermatol. 2020 Nov 28. doi: 10.1111/pde.14444.
– in a retrospective analysis of over 700 wound cultures from 158 patients across the United States and Canada.
The findings from the EB Clinical Characterization and Outcomes Database speak to the value of surveillance cultures with routine testing for microbial resistance – including mupirocin resistance – and to the importance of antibiotic stewardship not only for oral antibiotics but for topicals as well, according to Laura E. Levin, MD, and Kimberly D. Morel, MD, of the departments of dermatology and pediatrics, Columbia University Irving Medical Center, New York, the lead and senior authors, respectively, of the paper recently published in Pediatric Dermatology.
Almost all of the 158 patients with at least one wound culture recorded in the database from the period of 2001-2018 had one or more positive culture results. Of 152 patients with positive cultures, 131 (86%) were positive for SA and 56 (37%) and 34 (22%) were positive for PA and GAS, respectively. Other bacteria isolated included Corynebacterium spp and Proteus spp. Nearly half (47%) of patients with SA-positive cultures had methicillin-resistant SA, and 68% had methicillin-susceptible SA. (Some patients grew both MSSA and MRSA at different points in time.)
Mupirocin-susceptibility testing was performed at only some of the 13 participating centers. Of 15 patients whose cultures had recorded SA mupirocin-susceptibility testing, 11 had cultures positive for mupirocin-susceptible SA and 6 (40%) had mupirocin-resistant SA isolates (2 patients grew both). Of these six patients, half had isolates that were also methicillin-resistant.
Mupirocin, a topical antibiotic, has been a cornerstone of decolonization regimens for MSSA and MRSA, but resistance has been demonstrated in other research as well and is not specific to EB, wrote Dr. Levin, Dr. Morel, and coauthors.
“Pediatric dermatologists often rely on topical antimicrobials in the treatment of patients’ open wounds to both prevent and treat infection, depending on the clinical scenario,” and surveillance cultures with routine testing for mupirocin resistance can help guide antibiotic choice and management strategies, Dr. Levin said in an interview.
More broadly, she added, “it’s helpful to know what bacteria are routinely colonizing wounds, not causing infection, versus those that are more likely to be associated with infection, chronic wounds, or the risk of developing skin cancer ... [to know] which wounds need to be treated more aggressively.”
A subset of patients with EB have been known to be at risk for squamous cell carcinoma, and research is implicating certain bacteria “as contributing to wound inflammation,” Dr. Morel said in an interview.
SCC was reported in 23 out of 717 patients in the database – but fewer than half of the patients with SCC had recorded wound cultures. The small numbers precluded the identification of microbes that may confer significant risk.
Correlating particular microbes with clinical features also will take more research. About half (57%) of the patients with recorded wound cultures had wounds with purulent exudate or other features of clinical infection. However, the presence or absence of clinical signs of infection was not temporally correlated with culture results in the database.
The 158 patients with recorded wound cultures had a mean age of 12.8 years and represented a range of EB subtypes.
PA was present in the wounds of patients as young as 1 month old, the authors noted. Investigators are “looking to further study PA and characterize clinical features ... to understand more about this microbe and its impact on patients with EB,” Dr. Morel said.
In the meantime, the analysis reaffirms the importance of antibiotic stewardship. Mupirocin is labeled to be used three times a day for a short period of time, but “tends to be prescribed and used less judiciously than intended,” Dr. Morel said. “It’s important [not to overuse it]. We have seen that patients’ culture results become sensitive to mupirocin again in the future when they avoid it for a period of time.”
The work was supported by the EB Research Partnership and EB Medical Research Foundation, as well as an NIH/NCATS grant. No investigator disclosures were listed.
SOURCE: Pediatr Dermatol. 2020 Nov 28. doi: 10.1111/pde.14444.
– in a retrospective analysis of over 700 wound cultures from 158 patients across the United States and Canada.
The findings from the EB Clinical Characterization and Outcomes Database speak to the value of surveillance cultures with routine testing for microbial resistance – including mupirocin resistance – and to the importance of antibiotic stewardship not only for oral antibiotics but for topicals as well, according to Laura E. Levin, MD, and Kimberly D. Morel, MD, of the departments of dermatology and pediatrics, Columbia University Irving Medical Center, New York, the lead and senior authors, respectively, of the paper recently published in Pediatric Dermatology.
Almost all of the 158 patients with at least one wound culture recorded in the database from the period of 2001-2018 had one or more positive culture results. Of 152 patients with positive cultures, 131 (86%) were positive for SA and 56 (37%) and 34 (22%) were positive for PA and GAS, respectively. Other bacteria isolated included Corynebacterium spp and Proteus spp. Nearly half (47%) of patients with SA-positive cultures had methicillin-resistant SA, and 68% had methicillin-susceptible SA. (Some patients grew both MSSA and MRSA at different points in time.)
Mupirocin-susceptibility testing was performed at only some of the 13 participating centers. Of 15 patients whose cultures had recorded SA mupirocin-susceptibility testing, 11 had cultures positive for mupirocin-susceptible SA and 6 (40%) had mupirocin-resistant SA isolates (2 patients grew both). Of these six patients, half had isolates that were also methicillin-resistant.
Mupirocin, a topical antibiotic, has been a cornerstone of decolonization regimens for MSSA and MRSA, but resistance has been demonstrated in other research as well and is not specific to EB, wrote Dr. Levin, Dr. Morel, and coauthors.
“Pediatric dermatologists often rely on topical antimicrobials in the treatment of patients’ open wounds to both prevent and treat infection, depending on the clinical scenario,” and surveillance cultures with routine testing for mupirocin resistance can help guide antibiotic choice and management strategies, Dr. Levin said in an interview.
More broadly, she added, “it’s helpful to know what bacteria are routinely colonizing wounds, not causing infection, versus those that are more likely to be associated with infection, chronic wounds, or the risk of developing skin cancer ... [to know] which wounds need to be treated more aggressively.”
A subset of patients with EB have been known to be at risk for squamous cell carcinoma, and research is implicating certain bacteria “as contributing to wound inflammation,” Dr. Morel said in an interview.
SCC was reported in 23 out of 717 patients in the database – but fewer than half of the patients with SCC had recorded wound cultures. The small numbers precluded the identification of microbes that may confer significant risk.
Correlating particular microbes with clinical features also will take more research. About half (57%) of the patients with recorded wound cultures had wounds with purulent exudate or other features of clinical infection. However, the presence or absence of clinical signs of infection was not temporally correlated with culture results in the database.
The 158 patients with recorded wound cultures had a mean age of 12.8 years and represented a range of EB subtypes.
PA was present in the wounds of patients as young as 1 month old, the authors noted. Investigators are “looking to further study PA and characterize clinical features ... to understand more about this microbe and its impact on patients with EB,” Dr. Morel said.
In the meantime, the analysis reaffirms the importance of antibiotic stewardship. Mupirocin is labeled to be used three times a day for a short period of time, but “tends to be prescribed and used less judiciously than intended,” Dr. Morel said. “It’s important [not to overuse it]. We have seen that patients’ culture results become sensitive to mupirocin again in the future when they avoid it for a period of time.”
The work was supported by the EB Research Partnership and EB Medical Research Foundation, as well as an NIH/NCATS grant. No investigator disclosures were listed.
SOURCE: Pediatr Dermatol. 2020 Nov 28. doi: 10.1111/pde.14444.
FROM PEDIATRIC DERMATOLOGY
DART trial hits the target in angiosarcoma
Rare cancers comprise about 20% of all cancers in the United States and Europe, according to recent estimates, but patients with rare cancers are vastly underrepresented in clinical trials.

Recently, there has been a focus on immune checkpoint blockade (ICB) in common cancer types. Since several rare tumor types share similar biologic features with the more common tumors, there is a need to test ICB in rare tumors, particularly because remissions with ICB can be durable.
Enter the DART trial, a phase 2, single-arm study of combinatorial ICB with ipilimumab plus nivolumab in patients with unresectable or metastatic rare cancers.
Results from DART were recently presented at the Society for Immunotherapy of Cancer’s 35th Anniversary Annual Meeting. Michael J. Wagner, MD, of the University of Washington, Seattle, reported results in patients with advanced or unresectable angiosarcoma, one of the rare tumor types included in DART.
About angiosarcomas
Angiosarcomas account for less than 3% of all adult soft-tissue sarcomas, according to a review published in The Lancet Oncology. Angiosarcomas may arise in any part of the body, especially the head and neck (27%), breast (19.7%), and extremities (15.3%). These cancers can be primary or secondary (i.e., associated with prior radiation therapy or chronic lymphedema).
Angiosarcomas are aggressive, difficult to treat, and confer high mortality. The tumors are responsive to chemotherapy, but responses are brief. The estimated 5-year survival rate for all patients with angiosarcoma, including those who present with localized disease, is 30%-40%.
According to Dr. Wagner, a subset of angiosarcomas are characterized by high tumor mutational burden (TMB) and COSMIC signature 7, a DNA mutational signature that is consistent with other cancers caused by ultraviolet light exposure.
The high TMB subset of angiosarcomas is comparable with other cancer types that are responsive to ICB. Indeed, patients with angiosarcoma treated with ICB have shown responses, according to research published in the Journal for Immunotherapy of Cancer. However, no prospective studies of ICB in angiosarcoma have been published.
About DART
The DART trial includes more than 50 cohorts of rare cancer subtypes. Patients receive IV ipilimumab at 1 mg/kg every 6 weeks and IV nivolumab at 240 mg every 2 weeks.
The primary endpoint is objective response rate, as assessed by RECIST v1.1. Secondary endpoints include progression-free survival, overall survival, stable disease at 6 months, and toxicity.
The trial has a two-stage design. Six patients are enrolled in the first stage, and, if at least one patient responds to treatment, an additional 10 patients are enrolled in the second stage.
If at least two responses are seen among the 16 patients enrolled, further study of ICB is considered warranted.
Results in angiosarcoma
Dr. Wagner reported on the 16 angiosarcoma patients enrolled in DART. Nine patients had cutaneous primary tumors, seven had noncutaneous primary tumors, and three patients had radiation-associated angiosarcoma of the breast or chest wall.
Patients had received a median of two (range, zero to five) prior lines of therapy.
Adverse events (AEs) were consistent with prior safety results of the ipilimumab-nivolumab combination. Three-quarters of patients experienced an AE of any grade. The most common AEs were transaminase elevation, anemia, diarrhea, fatigue, hypothyroidism, pneumonitis, pruritus, and rash.
A quarter of patients had a grade 3-4 AE, and 12.5% of AEs led to premature treatment discontinuation. There were no fatal AEs.
The ORR was 25%. Responses occurred in 4 of the 16 patients, including 3 of 5 patients with primary cutaneous tumors of the scalp or face and 1 of 3 patients with radiation-associated breast angiosarcoma.
Two of the four responses and one case of stable disease have persisted for almost a year, and these patients remain on treatment. To put these results into perspective, Dr. Wagner noted that responses to cytotoxic chemotherapy rarely last 6 months.
The 6-month progression-free survival rate was 38%. The median overall survival has not yet been reached.
Dr. Wagner concluded that the combinatorial ICB regimen employed in DART was well tolerated and had an ORR of 25% in angiosarcoma regardless of primary site. Per the criteria of the DART trial, further investigation of ICB in angiosarcoma is warranted.
Molecular insights
Although correlative analyses of tumor tissue and peripheral blood are embedded in the DART trial, those analyses have not yet been performed. Eight of the 16 angiosarcoma patients had diagnostic molecular studies performed at their parent institutions, utilizing a variety of commercial platforms.

All eight patients for whom molecular data were available had at least two deleterious genomic alterations detected, but each had a distinct molecular profile.
Seven patients had TMB analyzed, including two partial responders to ICB. One of the seven patients had a high TMB, and this patient was one of the two responders. The other responder had an intermediate TMB.
Three patients had programmed death–ligand 1 staining on their tumors. Two of the three had high expression of PD-L1, including the responder with an intermediate TMB.
The real impact of DART
The DART trial is a “basket trial,” employing a similar treatment regimen for multiple tumor types. It provides a uniform framework for studying tumors that have been neglected in clinical trials heretofore.
Although the cohort of angiosarcoma patients is small, central pathology review was not required, and the treatment regimen was not compared directly with other potential therapies, the reported results of the ipilimumab-nivolumab regimen justify further study.
The biospecimens collected in DART will provide a rich source of data to identify common themes among responders and nonresponders, among patients who experience durable remissions and those who do not.
Angiosarcoma is not the only rare cancer for which combinatorial ICB has been valuable under the auspices of the DART trial. In Clinical Cancer Research, investigators reported an ORR of 44% among patients with high-grade neuroendocrine cancers, independent of primary site of origin. Progression-free survival at 6 months was 31%.
The DART trial is available at more than 800 sites, providing access to potentially promising treatment in a rigorous, scientifically valuable study for geographically underserved populations, including patients who live in rural areas.
The key message for practicing oncologists and clinical investigators is that clinical trials in rare tumors are feasible and can yield hope for patients who might lack it otherwise.
DART is funded by the National Cancer Institute and Bristol-Myers Squibb. Dr. Wagner disclosed relationships with Deciphera, Adaptimmune, GlaxoSmithKline, Athenex, and Incyte.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
SOURCE: Wagner M et al. SITC 2020, Abstract 795.
Rare cancers comprise about 20% of all cancers in the United States and Europe, according to recent estimates, but patients with rare cancers are vastly underrepresented in clinical trials.
Recently, there has been a focus on immune checkpoint blockade (ICB) in common cancer types. Since several rare tumor types share similar biologic features with the more common tumors, there is a need to test ICB in rare tumors, particularly because remissions with ICB can be durable.
Enter the DART trial, a phase 2, single-arm study of combinatorial ICB with ipilimumab plus nivolumab in patients with unresectable or metastatic rare cancers.
Results from DART were recently presented at the Society for Immunotherapy of Cancer’s 35th Anniversary Annual Meeting. Michael J. Wagner, MD, of the University of Washington, Seattle, reported results in patients with advanced or unresectable angiosarcoma, one of the rare tumor types included in DART.
About angiosarcomas
Angiosarcomas account for less than 3% of all adult soft-tissue sarcomas, according to a review published in The Lancet Oncology. Angiosarcomas may arise in any part of the body, especially the head and neck (27%), breast (19.7%), and extremities (15.3%). These cancers can be primary or secondary (i.e., associated with prior radiation therapy or chronic lymphedema).
Angiosarcomas are aggressive, difficult to treat, and confer high mortality. The tumors are responsive to chemotherapy, but responses are brief. The estimated 5-year survival rate for all patients with angiosarcoma, including those who present with localized disease, is 30%-40%.
According to Dr. Wagner, a subset of angiosarcomas are characterized by high tumor mutational burden (TMB) and COSMIC signature 7, a DNA mutational signature that is consistent with other cancers caused by ultraviolet light exposure.
The high TMB subset of angiosarcomas is comparable with other cancer types that are responsive to ICB. Indeed, patients with angiosarcoma treated with ICB have shown responses, according to research published in the Journal for Immunotherapy of Cancer. However, no prospective studies of ICB in angiosarcoma have been published.
About DART
The DART trial includes more than 50 cohorts of rare cancer subtypes. Patients receive IV ipilimumab at 1 mg/kg every 6 weeks and IV nivolumab at 240 mg every 2 weeks.
The primary endpoint is objective response rate, as assessed by RECIST v1.1. Secondary endpoints include progression-free survival, overall survival, stable disease at 6 months, and toxicity.
The trial has a two-stage design. Six patients are enrolled in the first stage, and, if at least one patient responds to treatment, an additional 10 patients are enrolled in the second stage.
If at least two responses are seen among the 16 patients enrolled, further study of ICB is considered warranted.
Results in angiosarcoma
Dr. Wagner reported on the 16 angiosarcoma patients enrolled in DART. Nine patients had cutaneous primary tumors, seven had noncutaneous primary tumors, and three patients had radiation-associated angiosarcoma of the breast or chest wall.
Patients had received a median of two (range, zero to five) prior lines of therapy.
Adverse events (AEs) were consistent with prior safety results of the ipilimumab-nivolumab combination. Three-quarters of patients experienced an AE of any grade. The most common AEs were transaminase elevation, anemia, diarrhea, fatigue, hypothyroidism, pneumonitis, pruritus, and rash.
A quarter of patients had a grade 3-4 AE, and 12.5% of AEs led to premature treatment discontinuation. There were no fatal AEs.
The ORR was 25%. Responses occurred in 4 of the 16 patients, including 3 of 5 patients with primary cutaneous tumors of the scalp or face and 1 of 3 patients with radiation-associated breast angiosarcoma.
Two of the four responses and one case of stable disease have persisted for almost a year, and these patients remain on treatment. To put these results into perspective, Dr. Wagner noted that responses to cytotoxic chemotherapy rarely last 6 months.
The 6-month progression-free survival rate was 38%. The median overall survival has not yet been reached.
Dr. Wagner concluded that the combinatorial ICB regimen employed in DART was well tolerated and had an ORR of 25% in angiosarcoma regardless of primary site. Per the criteria of the DART trial, further investigation of ICB in angiosarcoma is warranted.
Molecular insights
Although correlative analyses of tumor tissue and peripheral blood are embedded in the DART trial, those analyses have not yet been performed. Eight of the 16 angiosarcoma patients had diagnostic molecular studies performed at their parent institutions, utilizing a variety of commercial platforms.
All eight patients for whom molecular data were available had at least two deleterious genomic alterations detected, but each had a distinct molecular profile.
Seven patients had TMB analyzed, including two partial responders to ICB. One of the seven patients had a high TMB, and this patient was one of the two responders. The other responder had an intermediate TMB.
Three patients had programmed death–ligand 1 staining on their tumors. Two of the three had high expression of PD-L1, including the responder with an intermediate TMB.
The real impact of DART
The DART trial is a “basket trial,” employing a similar treatment regimen for multiple tumor types. It provides a uniform framework for studying tumors that have been neglected in clinical trials heretofore.
Although the cohort of angiosarcoma patients is small, central pathology review was not required, and the treatment regimen was not compared directly with other potential therapies, the reported results of the ipilimumab-nivolumab regimen justify further study.
The biospecimens collected in DART will provide a rich source of data to identify common themes among responders and nonresponders, among patients who experience durable remissions and those who do not.
Angiosarcoma is not the only rare cancer for which combinatorial ICB has been valuable under the auspices of the DART trial. In Clinical Cancer Research, investigators reported an ORR of 44% among patients with high-grade neuroendocrine cancers, independent of primary site of origin. Progression-free survival at 6 months was 31%.
The DART trial is available at more than 800 sites, providing access to potentially promising treatment in a rigorous, scientifically valuable study for geographically underserved populations, including patients who live in rural areas.
The key message for practicing oncologists and clinical investigators is that clinical trials in rare tumors are feasible and can yield hope for patients who might lack it otherwise.
DART is funded by the National Cancer Institute and Bristol-Myers Squibb. Dr. Wagner disclosed relationships with Deciphera, Adaptimmune, GlaxoSmithKline, Athenex, and Incyte.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
SOURCE: Wagner M et al. SITC 2020, Abstract 795.
Rare cancers comprise about 20% of all cancers in the United States and Europe, according to recent estimates, but patients with rare cancers are vastly underrepresented in clinical trials.
Recently, there has been a focus on immune checkpoint blockade (ICB) in common cancer types. Since several rare tumor types share similar biologic features with the more common tumors, there is a need to test ICB in rare tumors, particularly because remissions with ICB can be durable.
Enter the DART trial, a phase 2, single-arm study of combinatorial ICB with ipilimumab plus nivolumab in patients with unresectable or metastatic rare cancers.
Results from DART were recently presented at the Society for Immunotherapy of Cancer’s 35th Anniversary Annual Meeting. Michael J. Wagner, MD, of the University of Washington, Seattle, reported results in patients with advanced or unresectable angiosarcoma, one of the rare tumor types included in DART.
About angiosarcomas
Angiosarcomas account for less than 3% of all adult soft-tissue sarcomas, according to a review published in The Lancet Oncology. Angiosarcomas may arise in any part of the body, especially the head and neck (27%), breast (19.7%), and extremities (15.3%). These cancers can be primary or secondary (i.e., associated with prior radiation therapy or chronic lymphedema).
Angiosarcomas are aggressive, difficult to treat, and confer high mortality. The tumors are responsive to chemotherapy, but responses are brief. The estimated 5-year survival rate for all patients with angiosarcoma, including those who present with localized disease, is 30%-40%.
According to Dr. Wagner, a subset of angiosarcomas are characterized by high tumor mutational burden (TMB) and COSMIC signature 7, a DNA mutational signature that is consistent with other cancers caused by ultraviolet light exposure.
The high TMB subset of angiosarcomas is comparable with other cancer types that are responsive to ICB. Indeed, patients with angiosarcoma treated with ICB have shown responses, according to research published in the Journal for Immunotherapy of Cancer. However, no prospective studies of ICB in angiosarcoma have been published.
About DART
The DART trial includes more than 50 cohorts of rare cancer subtypes. Patients receive IV ipilimumab at 1 mg/kg every 6 weeks and IV nivolumab at 240 mg every 2 weeks.
The primary endpoint is objective response rate, as assessed by RECIST v1.1. Secondary endpoints include progression-free survival, overall survival, stable disease at 6 months, and toxicity.
The trial has a two-stage design. Six patients are enrolled in the first stage, and, if at least one patient responds to treatment, an additional 10 patients are enrolled in the second stage.
If at least two responses are seen among the 16 patients enrolled, further study of ICB is considered warranted.
Results in angiosarcoma
Dr. Wagner reported on the 16 angiosarcoma patients enrolled in DART. Nine patients had cutaneous primary tumors, seven had noncutaneous primary tumors, and three patients had radiation-associated angiosarcoma of the breast or chest wall.
Patients had received a median of two (range, zero to five) prior lines of therapy.
Adverse events (AEs) were consistent with prior safety results of the ipilimumab-nivolumab combination. Three-quarters of patients experienced an AE of any grade. The most common AEs were transaminase elevation, anemia, diarrhea, fatigue, hypothyroidism, pneumonitis, pruritus, and rash.
A quarter of patients had a grade 3-4 AE, and 12.5% of AEs led to premature treatment discontinuation. There were no fatal AEs.
The ORR was 25%. Responses occurred in 4 of the 16 patients, including 3 of 5 patients with primary cutaneous tumors of the scalp or face and 1 of 3 patients with radiation-associated breast angiosarcoma.
Two of the four responses and one case of stable disease have persisted for almost a year, and these patients remain on treatment. To put these results into perspective, Dr. Wagner noted that responses to cytotoxic chemotherapy rarely last 6 months.
The 6-month progression-free survival rate was 38%. The median overall survival has not yet been reached.
Dr. Wagner concluded that the combinatorial ICB regimen employed in DART was well tolerated and had an ORR of 25% in angiosarcoma regardless of primary site. Per the criteria of the DART trial, further investigation of ICB in angiosarcoma is warranted.
Molecular insights
Although correlative analyses of tumor tissue and peripheral blood are embedded in the DART trial, those analyses have not yet been performed. Eight of the 16 angiosarcoma patients had diagnostic molecular studies performed at their parent institutions, utilizing a variety of commercial platforms.
All eight patients for whom molecular data were available had at least two deleterious genomic alterations detected, but each had a distinct molecular profile.
Seven patients had TMB analyzed, including two partial responders to ICB. One of the seven patients had a high TMB, and this patient was one of the two responders. The other responder had an intermediate TMB.
Three patients had programmed death–ligand 1 staining on their tumors. Two of the three had high expression of PD-L1, including the responder with an intermediate TMB.
The real impact of DART
The DART trial is a “basket trial,” employing a similar treatment regimen for multiple tumor types. It provides a uniform framework for studying tumors that have been neglected in clinical trials heretofore.
Although the cohort of angiosarcoma patients is small, central pathology review was not required, and the treatment regimen was not compared directly with other potential therapies, the reported results of the ipilimumab-nivolumab regimen justify further study.
The biospecimens collected in DART will provide a rich source of data to identify common themes among responders and nonresponders, among patients who experience durable remissions and those who do not.
Angiosarcoma is not the only rare cancer for which combinatorial ICB has been valuable under the auspices of the DART trial. In Clinical Cancer Research, investigators reported an ORR of 44% among patients with high-grade neuroendocrine cancers, independent of primary site of origin. Progression-free survival at 6 months was 31%.
The DART trial is available at more than 800 sites, providing access to potentially promising treatment in a rigorous, scientifically valuable study for geographically underserved populations, including patients who live in rural areas.
The key message for practicing oncologists and clinical investigators is that clinical trials in rare tumors are feasible and can yield hope for patients who might lack it otherwise.
DART is funded by the National Cancer Institute and Bristol-Myers Squibb. Dr. Wagner disclosed relationships with Deciphera, Adaptimmune, GlaxoSmithKline, Athenex, and Incyte.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
SOURCE: Wagner M et al. SITC 2020, Abstract 795.
FROM SITC 2020
COVID-19 vaccines and cancer patients: 4 things to know
Earlier this week, Medscape spoke with Nora Disis, MD, about vaccinating cancer patients. Disis is a medical oncologist and director of both the Institute of Translational Health Sciences and the Cancer Vaccine Institute, the University of Washington, Seattle, Washington. As editor-in-chief of JAMA Oncology, she has watched COVID-19 developments in the oncology community over the past year.
Here are a few themes that Disis said oncologists should be aware of as vaccines eventually begin reaching cancer patients.
We should expect cancer patients to respond to vaccines. Historically, some believed that cancer patients would be unable to mount an immune response to vaccines. Data on other viral vaccines have shown otherwise. For example, there has been a long history of studies of flu vaccination in cancer patients, and in general, those vaccines confer protection. Likewise for pneumococcal vaccine, which, generally speaking, cancer patients should receive.
Special cases may include hematologic malignancies in which the immune system has been destroyed and profound immunosuppression occurs. Data on immunization during this immunosuppressed period are scarce, but what data are available suggest that once cancer patients are through this immunosuppressed period, they can be vaccinated successfully.
The type of vaccine will probably be important for cancer patients. Currently, there are 61 coronavirus vaccines in human clinical trials, and 17 have reached the final stages of testing. At least 85 preclinical vaccines are under active investigation in animals.
Both the Pfizer-BioNTech and Moderna COVID vaccines are mRNA type. There are many other types, including protein-based vaccines, viral vector vaccines based on adenoviruses, and inactivated or attenuated coronavirus vaccines.
The latter vaccines, particularly attenuated live virus vaccines, may not be a good choice for cancer patients. Especially in those with rapidly progressing disease or on chemotherapy, attenuated live viruses may cause a low-grade infection.
Incidentally, the technology used in the genetic, or mRNA, vaccines developed by both Pfizer-BioNTech and Moderna was initially developed for fighting cancer, and studies have shown that patients can generate immune responses to cancer-associated proteins with this type of vaccine.
These genetic vaccines could turn out to be the most effective for cancer patients, especially those with solid tumors.
Our understanding is very limited right now. Neither the Pfizer-BioNTech nor the Moderna early data discuss cancer patients. Two of the most important questions for cancer patients are dosing and booster scheduling. Potential defects in lymphocyte function among cancer patients may require unique initial dosing and booster schedules. In terms of timing, it is unclear how active therapy might affect a patient’s immune response to vaccination and whether vaccines should be timed with therapy cycles.
Vaccine access may depend on whether cancer patients are viewed as a vulnerable population. Those at higher risk for severe COVID-19 clearly have a greater need for vaccination. While there are data suggesting that cancer patients are at higher risk, they are a bit murky, in part because cancer patients are a heterogeneous group. For example, there are data suggesting that lung and blood cancer patients fare worse. There is also a suggestion that, like in the general population, COVID risk in cancer patients remains driven by comorbidities.
It is likely, then, that personalized risk factors such as type of cancer therapy, site of disease, and comorbidities will shape individual choices about vaccination among cancer patients.
A version of this article first appeared on Medscape.com.
Earlier this week, Medscape spoke with Nora Disis, MD, about vaccinating cancer patients. Disis is a medical oncologist and director of both the Institute of Translational Health Sciences and the Cancer Vaccine Institute, the University of Washington, Seattle, Washington. As editor-in-chief of JAMA Oncology, she has watched COVID-19 developments in the oncology community over the past year.
Here are a few themes that Disis said oncologists should be aware of as vaccines eventually begin reaching cancer patients.
We should expect cancer patients to respond to vaccines. Historically, some believed that cancer patients would be unable to mount an immune response to vaccines. Data on other viral vaccines have shown otherwise. For example, there has been a long history of studies of flu vaccination in cancer patients, and in general, those vaccines confer protection. Likewise for pneumococcal vaccine, which, generally speaking, cancer patients should receive.
Special cases may include hematologic malignancies in which the immune system has been destroyed and profound immunosuppression occurs. Data on immunization during this immunosuppressed period are scarce, but what data are available suggest that once cancer patients are through this immunosuppressed period, they can be vaccinated successfully.
The type of vaccine will probably be important for cancer patients. Currently, there are 61 coronavirus vaccines in human clinical trials, and 17 have reached the final stages of testing. At least 85 preclinical vaccines are under active investigation in animals.
Both the Pfizer-BioNTech and Moderna COVID vaccines are mRNA type. There are many other types, including protein-based vaccines, viral vector vaccines based on adenoviruses, and inactivated or attenuated coronavirus vaccines.
The latter vaccines, particularly attenuated live virus vaccines, may not be a good choice for cancer patients. Especially in those with rapidly progressing disease or on chemotherapy, attenuated live viruses may cause a low-grade infection.
Incidentally, the technology used in the genetic, or mRNA, vaccines developed by both Pfizer-BioNTech and Moderna was initially developed for fighting cancer, and studies have shown that patients can generate immune responses to cancer-associated proteins with this type of vaccine.
These genetic vaccines could turn out to be the most effective for cancer patients, especially those with solid tumors.
Our understanding is very limited right now. Neither the Pfizer-BioNTech nor the Moderna early data discuss cancer patients. Two of the most important questions for cancer patients are dosing and booster scheduling. Potential defects in lymphocyte function among cancer patients may require unique initial dosing and booster schedules. In terms of timing, it is unclear how active therapy might affect a patient’s immune response to vaccination and whether vaccines should be timed with therapy cycles.
Vaccine access may depend on whether cancer patients are viewed as a vulnerable population. Those at higher risk for severe COVID-19 clearly have a greater need for vaccination. While there are data suggesting that cancer patients are at higher risk, they are a bit murky, in part because cancer patients are a heterogeneous group. For example, there are data suggesting that lung and blood cancer patients fare worse. There is also a suggestion that, like in the general population, COVID risk in cancer patients remains driven by comorbidities.
It is likely, then, that personalized risk factors such as type of cancer therapy, site of disease, and comorbidities will shape individual choices about vaccination among cancer patients.
A version of this article first appeared on Medscape.com.
Earlier this week, Medscape spoke with Nora Disis, MD, about vaccinating cancer patients. Disis is a medical oncologist and director of both the Institute of Translational Health Sciences and the Cancer Vaccine Institute, the University of Washington, Seattle, Washington. As editor-in-chief of JAMA Oncology, she has watched COVID-19 developments in the oncology community over the past year.
Here are a few themes that Disis said oncologists should be aware of as vaccines eventually begin reaching cancer patients.
We should expect cancer patients to respond to vaccines. Historically, some believed that cancer patients would be unable to mount an immune response to vaccines. Data on other viral vaccines have shown otherwise. For example, there has been a long history of studies of flu vaccination in cancer patients, and in general, those vaccines confer protection. Likewise for pneumococcal vaccine, which, generally speaking, cancer patients should receive.
Special cases may include hematologic malignancies in which the immune system has been destroyed and profound immunosuppression occurs. Data on immunization during this immunosuppressed period are scarce, but what data are available suggest that once cancer patients are through this immunosuppressed period, they can be vaccinated successfully.
The type of vaccine will probably be important for cancer patients. Currently, there are 61 coronavirus vaccines in human clinical trials, and 17 have reached the final stages of testing. At least 85 preclinical vaccines are under active investigation in animals.
Both the Pfizer-BioNTech and Moderna COVID vaccines are mRNA type. There are many other types, including protein-based vaccines, viral vector vaccines based on adenoviruses, and inactivated or attenuated coronavirus vaccines.
The latter vaccines, particularly attenuated live virus vaccines, may not be a good choice for cancer patients. Especially in those with rapidly progressing disease or on chemotherapy, attenuated live viruses may cause a low-grade infection.
Incidentally, the technology used in the genetic, or mRNA, vaccines developed by both Pfizer-BioNTech and Moderna was initially developed for fighting cancer, and studies have shown that patients can generate immune responses to cancer-associated proteins with this type of vaccine.
These genetic vaccines could turn out to be the most effective for cancer patients, especially those with solid tumors.
Our understanding is very limited right now. Neither the Pfizer-BioNTech nor the Moderna early data discuss cancer patients. Two of the most important questions for cancer patients are dosing and booster scheduling. Potential defects in lymphocyte function among cancer patients may require unique initial dosing and booster schedules. In terms of timing, it is unclear how active therapy might affect a patient’s immune response to vaccination and whether vaccines should be timed with therapy cycles.
Vaccine access may depend on whether cancer patients are viewed as a vulnerable population. Those at higher risk for severe COVID-19 clearly have a greater need for vaccination. While there are data suggesting that cancer patients are at higher risk, they are a bit murky, in part because cancer patients are a heterogeneous group. For example, there are data suggesting that lung and blood cancer patients fare worse. There is also a suggestion that, like in the general population, COVID risk in cancer patients remains driven by comorbidities.
It is likely, then, that personalized risk factors such as type of cancer therapy, site of disease, and comorbidities will shape individual choices about vaccination among cancer patients.
A version of this article first appeared on Medscape.com.
RARE DISEASES REPORT: Cancers
Rare cancers, though individually rare by definition, impose a tremendous burden on adult and pediatric patient populations, especially when considering hematological cancers. In this Rare Diseases Report: Cancers, we bring you the latest information on new and ongoing developments in the treatment of some of these cancers through interviews with frontline researchers in the field.

- Survey reveals special impact of COVID-19 on patients with rare disorders
- New agents boost survival – and complexity – in chronic lymphocytic leukemia
- Targeted therapies may alter the landscape of MCL treatment
- Will CAR T push beyond lymphoma? There’s no guarantee
Rare cancers, though individually rare by definition, impose a tremendous burden on adult and pediatric patient populations, especially when considering hematological cancers. In this Rare Diseases Report: Cancers, we bring you the latest information on new and ongoing developments in the treatment of some of these cancers through interviews with frontline researchers in the field.
- Survey reveals special impact of COVID-19 on patients with rare disorders
- New agents boost survival – and complexity – in chronic lymphocytic leukemia
- Targeted therapies may alter the landscape of MCL treatment
- Will CAR T push beyond lymphoma? There’s no guarantee
Rare cancers, though individually rare by definition, impose a tremendous burden on adult and pediatric patient populations, especially when considering hematological cancers. In this Rare Diseases Report: Cancers, we bring you the latest information on new and ongoing developments in the treatment of some of these cancers through interviews with frontline researchers in the field.
- Survey reveals special impact of COVID-19 on patients with rare disorders
- New agents boost survival – and complexity – in chronic lymphocytic leukemia
- Targeted therapies may alter the landscape of MCL treatment
- Will CAR T push beyond lymphoma? There’s no guarantee

Childhood Hodgkin survivors have neurocognitive impairment
More than 2 decades on, adult survivors of childhood Hodgkin lymphoma report significantly more neurocognitive impairment than their siblings, but the differences may be related to risk factors in adulthood rather than to treatment in childhood, investigators say.
Among adults with a history of childhood Hodgkin lymphoma and their siblings as controls, the survivors reported significantly worse functioning than their brothers or sisters in four domains of neurocognitive functioning.
In multivariate analysis, however, while sex, race, activity level and smoking status were all significant predictors for worse neurocognitive impairment, there were no significant associations between chemotherapy drugs or chest radiation and neurocognitive impairment, said Annalynn M. Williams, PhD, from St. Jude Children’s Research Hospital in Memphis.
“Hodgkin lymphoma is the most common cancer diagnosed in adolescents, and for many years we’ve had high cure rates, resulting in a growing population of survivors who are now, unfortunately, at an increased risk for cardiovascular, respiratory, endocrine and neurologic late morbidity. The neurocognitive morbidity in this population, however, is unknown,” she said in oral abstract presented at the annual meeting of the American Society of Hematology.
Survivors and sibs
To better characterize the potential late neurocognitive effects of intensive Hodgkin lymphoma therapy in childhood, Dr. Williams and colleagues polled survivors of childhood Hodgkin lymphoma and randomly selected sibling controls who were participants in the Childhood Cancer Survivor Study (CCSS).
Participants were asked to complete questionnaires regarding four domains of neurocognitive impairment: task efficiency, emotional regulation, organization, and memory. The investigators defined impairment in each domain as a score lower than that of the 90th percentile of community controls from the St. Jude Lifetime Cohort.
A total of 1,564 survivors and 725 controls completed the questionnaires and were included in the study.
The median age at follow-up was slightly higher among survivors, at 37 versus 32 years. The median age at diagnosis was 14, and the median time since diagnosis was 23 years.
In all, 10.8% of survivors reported impaired task efficiency, compared with 7.7% of controls. Problems with emotional regulation were reported by 16.6% of survivors versus 11.5% of siblings, and difficulties with organization and memory were reported by 12.1% versus10.3%, and 8.1% versus 5.7%, respectively.
In a model adjusted for age, sex, and race, the relative risks for neurocognitive impairment among survivors versus siblings, were as follows: task efficiency (RR,1.37); emotional regulation (RR, 1.56); organization (RR, 1.32); memory (RR, 1.72) (all significant by confidence interval).
In a model adjusted for sex, race, smoking status, exercise, age, time since diagnosis, and treatment exposures, risk factors for neurocognitive impairment among survivors included female versus male sex (significant for emotional regulation and memory deficits); non-White versus White (significant for task efficiency); former smoker versus never (significant for all domains except organization); current smoker versus never (significant for task efficiency and emotional regulation); and meeting Centers for Disease Control and Prevention exercise criteria versus not (negatively significant for task efficiency and organization); (P < .05 for all above comparisons).
However, in a model adjusted for relapse, second malignancy, treatment exposures, age, sex, race, time since diagnosis, smoking status and physical activity, only relapse or second malignancy – surrogates for additional treatment exposures – were significantly associated with neurocognitive impairment, and then only in the domain of task efficiency.
Chronic conditions significantly associated with risk for impairment included cardiovascular disease (significant across all domains), respiratory comorbidities (significant for task efficiency), endocrine disorders (significant for task efficiency), and neurologic disorders (significant in all domains except organization).
“While these analyses give us a sense of the presence of neurocognitive impairment in a large sample of Hodgkin lymphoma survivors from across the U.S., these analyses are limited by the self-reported nature of the data,” Dr. Williams acknowledged.
“Because survivors self-report impairments, these likely represent overt, symptomatic neurocognitive impairments. Many more survivors may experience more subtle neurocognitive impairments, and additional research with objective measures of both chronic health conditions and neurocognitive functioning are warranted,” she added.
Smoking gun?
In the question-and-answer session following the presentation, session comoderator Pallawi Torka, MD, from Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., who was not involved in the research, commented that the finding regarding a link between current and former smoking as risk factors for neurocognitive impairment was “intriguing.”
“Do you think that smoking is a cause or an effect of having that impairment in childhood survivors of Hodgkin lymphoma?” she asked.
“That’s a great question, and actually one we have spent a great deal of time discussing, and we’re still trying to tease that apart. We’re still not really sure where that association is coming from,” Dr. Williams replied.
She noted that, in a different sample of CCSS participants from whom biospecimens were collected, the investigators plan to see whether smoking drives inflammation and oxidative stress mechanisms that may be contributing to neurocognitive impairment, or whether smoking is a coping mechanism related to anxiety and depression, which have also been seen in survivors.
Kara Kelly, MD, a pediatric oncologist at Roswell Park, commented that some survivors report symptoms of cognitive dysfunction shortly after treatment, and asked whether there might be a relationship to Hodgkin-specific factors such as B symptoms, in which cytokine-mediated inflammation may play a role.
Dr. Williams said that, “unfortunately, in CCSS these survivors had to be at least 5 years from diagnosis, but in many cases were recruited years after their diagnosis and treatment, so we don’t have data on B symptoms.”
The CCSS is funded by the National Cancer Institute. Dr. Williams, Dr. Palawi, and Dr. Kelly all reported no relevant conflicts of interest to disclose.
SOURCE: Williams AM et al. ASH 2020, Abstract 370.
More than 2 decades on, adult survivors of childhood Hodgkin lymphoma report significantly more neurocognitive impairment than their siblings, but the differences may be related to risk factors in adulthood rather than to treatment in childhood, investigators say.
Among adults with a history of childhood Hodgkin lymphoma and their siblings as controls, the survivors reported significantly worse functioning than their brothers or sisters in four domains of neurocognitive functioning.
In multivariate analysis, however, while sex, race, activity level and smoking status were all significant predictors for worse neurocognitive impairment, there were no significant associations between chemotherapy drugs or chest radiation and neurocognitive impairment, said Annalynn M. Williams, PhD, from St. Jude Children’s Research Hospital in Memphis.
“Hodgkin lymphoma is the most common cancer diagnosed in adolescents, and for many years we’ve had high cure rates, resulting in a growing population of survivors who are now, unfortunately, at an increased risk for cardiovascular, respiratory, endocrine and neurologic late morbidity. The neurocognitive morbidity in this population, however, is unknown,” she said in oral abstract presented at the annual meeting of the American Society of Hematology.
Survivors and sibs
To better characterize the potential late neurocognitive effects of intensive Hodgkin lymphoma therapy in childhood, Dr. Williams and colleagues polled survivors of childhood Hodgkin lymphoma and randomly selected sibling controls who were participants in the Childhood Cancer Survivor Study (CCSS).
Participants were asked to complete questionnaires regarding four domains of neurocognitive impairment: task efficiency, emotional regulation, organization, and memory. The investigators defined impairment in each domain as a score lower than that of the 90th percentile of community controls from the St. Jude Lifetime Cohort.
A total of 1,564 survivors and 725 controls completed the questionnaires and were included in the study.
The median age at follow-up was slightly higher among survivors, at 37 versus 32 years. The median age at diagnosis was 14, and the median time since diagnosis was 23 years.
In all, 10.8% of survivors reported impaired task efficiency, compared with 7.7% of controls. Problems with emotional regulation were reported by 16.6% of survivors versus 11.5% of siblings, and difficulties with organization and memory were reported by 12.1% versus10.3%, and 8.1% versus 5.7%, respectively.
In a model adjusted for age, sex, and race, the relative risks for neurocognitive impairment among survivors versus siblings, were as follows: task efficiency (RR,1.37); emotional regulation (RR, 1.56); organization (RR, 1.32); memory (RR, 1.72) (all significant by confidence interval).
In a model adjusted for sex, race, smoking status, exercise, age, time since diagnosis, and treatment exposures, risk factors for neurocognitive impairment among survivors included female versus male sex (significant for emotional regulation and memory deficits); non-White versus White (significant for task efficiency); former smoker versus never (significant for all domains except organization); current smoker versus never (significant for task efficiency and emotional regulation); and meeting Centers for Disease Control and Prevention exercise criteria versus not (negatively significant for task efficiency and organization); (P < .05 for all above comparisons).
However, in a model adjusted for relapse, second malignancy, treatment exposures, age, sex, race, time since diagnosis, smoking status and physical activity, only relapse or second malignancy – surrogates for additional treatment exposures – were significantly associated with neurocognitive impairment, and then only in the domain of task efficiency.
Chronic conditions significantly associated with risk for impairment included cardiovascular disease (significant across all domains), respiratory comorbidities (significant for task efficiency), endocrine disorders (significant for task efficiency), and neurologic disorders (significant in all domains except organization).
“While these analyses give us a sense of the presence of neurocognitive impairment in a large sample of Hodgkin lymphoma survivors from across the U.S., these analyses are limited by the self-reported nature of the data,” Dr. Williams acknowledged.
“Because survivors self-report impairments, these likely represent overt, symptomatic neurocognitive impairments. Many more survivors may experience more subtle neurocognitive impairments, and additional research with objective measures of both chronic health conditions and neurocognitive functioning are warranted,” she added.
Smoking gun?
In the question-and-answer session following the presentation, session comoderator Pallawi Torka, MD, from Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., who was not involved in the research, commented that the finding regarding a link between current and former smoking as risk factors for neurocognitive impairment was “intriguing.”
“Do you think that smoking is a cause or an effect of having that impairment in childhood survivors of Hodgkin lymphoma?” she asked.
“That’s a great question, and actually one we have spent a great deal of time discussing, and we’re still trying to tease that apart. We’re still not really sure where that association is coming from,” Dr. Williams replied.
She noted that, in a different sample of CCSS participants from whom biospecimens were collected, the investigators plan to see whether smoking drives inflammation and oxidative stress mechanisms that may be contributing to neurocognitive impairment, or whether smoking is a coping mechanism related to anxiety and depression, which have also been seen in survivors.
Kara Kelly, MD, a pediatric oncologist at Roswell Park, commented that some survivors report symptoms of cognitive dysfunction shortly after treatment, and asked whether there might be a relationship to Hodgkin-specific factors such as B symptoms, in which cytokine-mediated inflammation may play a role.
Dr. Williams said that, “unfortunately, in CCSS these survivors had to be at least 5 years from diagnosis, but in many cases were recruited years after their diagnosis and treatment, so we don’t have data on B symptoms.”
The CCSS is funded by the National Cancer Institute. Dr. Williams, Dr. Palawi, and Dr. Kelly all reported no relevant conflicts of interest to disclose.
SOURCE: Williams AM et al. ASH 2020, Abstract 370.
More than 2 decades on, adult survivors of childhood Hodgkin lymphoma report significantly more neurocognitive impairment than their siblings, but the differences may be related to risk factors in adulthood rather than to treatment in childhood, investigators say.
Among adults with a history of childhood Hodgkin lymphoma and their siblings as controls, the survivors reported significantly worse functioning than their brothers or sisters in four domains of neurocognitive functioning.
In multivariate analysis, however, while sex, race, activity level and smoking status were all significant predictors for worse neurocognitive impairment, there were no significant associations between chemotherapy drugs or chest radiation and neurocognitive impairment, said Annalynn M. Williams, PhD, from St. Jude Children’s Research Hospital in Memphis.
“Hodgkin lymphoma is the most common cancer diagnosed in adolescents, and for many years we’ve had high cure rates, resulting in a growing population of survivors who are now, unfortunately, at an increased risk for cardiovascular, respiratory, endocrine and neurologic late morbidity. The neurocognitive morbidity in this population, however, is unknown,” she said in oral abstract presented at the annual meeting of the American Society of Hematology.
Survivors and sibs
To better characterize the potential late neurocognitive effects of intensive Hodgkin lymphoma therapy in childhood, Dr. Williams and colleagues polled survivors of childhood Hodgkin lymphoma and randomly selected sibling controls who were participants in the Childhood Cancer Survivor Study (CCSS).
Participants were asked to complete questionnaires regarding four domains of neurocognitive impairment: task efficiency, emotional regulation, organization, and memory. The investigators defined impairment in each domain as a score lower than that of the 90th percentile of community controls from the St. Jude Lifetime Cohort.
A total of 1,564 survivors and 725 controls completed the questionnaires and were included in the study.
The median age at follow-up was slightly higher among survivors, at 37 versus 32 years. The median age at diagnosis was 14, and the median time since diagnosis was 23 years.
In all, 10.8% of survivors reported impaired task efficiency, compared with 7.7% of controls. Problems with emotional regulation were reported by 16.6% of survivors versus 11.5% of siblings, and difficulties with organization and memory were reported by 12.1% versus10.3%, and 8.1% versus 5.7%, respectively.
In a model adjusted for age, sex, and race, the relative risks for neurocognitive impairment among survivors versus siblings, were as follows: task efficiency (RR,1.37); emotional regulation (RR, 1.56); organization (RR, 1.32); memory (RR, 1.72) (all significant by confidence interval).
In a model adjusted for sex, race, smoking status, exercise, age, time since diagnosis, and treatment exposures, risk factors for neurocognitive impairment among survivors included female versus male sex (significant for emotional regulation and memory deficits); non-White versus White (significant for task efficiency); former smoker versus never (significant for all domains except organization); current smoker versus never (significant for task efficiency and emotional regulation); and meeting Centers for Disease Control and Prevention exercise criteria versus not (negatively significant for task efficiency and organization); (P < .05 for all above comparisons).
However, in a model adjusted for relapse, second malignancy, treatment exposures, age, sex, race, time since diagnosis, smoking status and physical activity, only relapse or second malignancy – surrogates for additional treatment exposures – were significantly associated with neurocognitive impairment, and then only in the domain of task efficiency.
Chronic conditions significantly associated with risk for impairment included cardiovascular disease (significant across all domains), respiratory comorbidities (significant for task efficiency), endocrine disorders (significant for task efficiency), and neurologic disorders (significant in all domains except organization).
“While these analyses give us a sense of the presence of neurocognitive impairment in a large sample of Hodgkin lymphoma survivors from across the U.S., these analyses are limited by the self-reported nature of the data,” Dr. Williams acknowledged.
“Because survivors self-report impairments, these likely represent overt, symptomatic neurocognitive impairments. Many more survivors may experience more subtle neurocognitive impairments, and additional research with objective measures of both chronic health conditions and neurocognitive functioning are warranted,” she added.
Smoking gun?
In the question-and-answer session following the presentation, session comoderator Pallawi Torka, MD, from Roswell Park Comprehensive Cancer Center in Buffalo, N.Y., who was not involved in the research, commented that the finding regarding a link between current and former smoking as risk factors for neurocognitive impairment was “intriguing.”
“Do you think that smoking is a cause or an effect of having that impairment in childhood survivors of Hodgkin lymphoma?” she asked.
“That’s a great question, and actually one we have spent a great deal of time discussing, and we’re still trying to tease that apart. We’re still not really sure where that association is coming from,” Dr. Williams replied.
She noted that, in a different sample of CCSS participants from whom biospecimens were collected, the investigators plan to see whether smoking drives inflammation and oxidative stress mechanisms that may be contributing to neurocognitive impairment, or whether smoking is a coping mechanism related to anxiety and depression, which have also been seen in survivors.
Kara Kelly, MD, a pediatric oncologist at Roswell Park, commented that some survivors report symptoms of cognitive dysfunction shortly after treatment, and asked whether there might be a relationship to Hodgkin-specific factors such as B symptoms, in which cytokine-mediated inflammation may play a role.
Dr. Williams said that, “unfortunately, in CCSS these survivors had to be at least 5 years from diagnosis, but in many cases were recruited years after their diagnosis and treatment, so we don’t have data on B symptoms.”
The CCSS is funded by the National Cancer Institute. Dr. Williams, Dr. Palawi, and Dr. Kelly all reported no relevant conflicts of interest to disclose.
SOURCE: Williams AM et al. ASH 2020, Abstract 370.
FROM ASH 2020
FDA clears first drug for rare genetic causes of severe obesity
The Food and Drug Administration has approved setmelanotide (Imcivree, Rhythm Pharmaceuticals) for weight management in adults and children as young as 6 years with obesity because of proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing.
Individuals with these rare genetic causes of severe obesity have a normal weight at birth but develop persistent severe obesity within months because of insatiable hunger (hyperphagia).
Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first FDA-approved therapy for these disorders.
“Many patients and families who live with these diseases face an often-burdensome stigma associated with severe obesity. To manage this obesity and control disruptive food-seeking behavior, caregivers often lock cabinets and refrigerators and significantly limit social activities,” said Jennifer Miller, MD, a pediatric endocrinologist at University of Florida Health, Gainesville, in a press release issued by the company.
“This FDA approval marks an important turning point, providing a much needed therapy and supporting the use of genetic testing to identify and properly diagnose patients with these rare genetic diseases of obesity,” she noted.
David Meeker, MD, chair, president, and CEO of Rhythm Pharmaceuticals, added: “We are advancing a first-in-class, precision medicine that is designed to directly address the underlying cause of obesities driven by genetic deficits in the MC4R pathway.”
Setmelanotide was evaluated in two phase 3 clinical trials. In one trial, 80% of patients with obesity caused by POMC or PCSK1 deficiency achieved greater than 10% weight loss after 1 year of treatment.
In the other trial, 45.5% of patients with obesity caused by LEPR deficiency achieved greater than 10% weight loss with 1 year of treatment.
Results for the two trials were recently published in The Lancet Diabetes & Endocrinology and discussed at the ObesityWeek Interactive 2020 meeting.
Setmelanotide was generally well tolerated in both trials. The most common adverse events were injection-site reactions, skin hyperpigmentation, and nausea.
The drug label notes that disturbances in sexual arousal, depression, and suicidal ideation; skin pigmentation; and darkening of preexisting nevi may occur with setmelanotide treatment.
The drug label also notes a risk for serious adverse reactions because of benzyl alcohol preservative in neonates and low-birth-weight infants. Setmelanotide is not approved for use in neonates or infants.
The company expects the drug to be commercially available in the United States in the first quarter of 2021.
Setmelanotide for the treatment of obesity associated with rare genetic defects had FDA breakthrough therapy designation as well as orphan drug designation.
The company is also evaluating setmelanotide for reduction in hunger and body weight in a pivotal phase 3 trial in people living with Bardet-Biedl or Alström syndrome, and top-line data are due soon.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved setmelanotide (Imcivree, Rhythm Pharmaceuticals) for weight management in adults and children as young as 6 years with obesity because of proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing.
Individuals with these rare genetic causes of severe obesity have a normal weight at birth but develop persistent severe obesity within months because of insatiable hunger (hyperphagia).
Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first FDA-approved therapy for these disorders.
“Many patients and families who live with these diseases face an often-burdensome stigma associated with severe obesity. To manage this obesity and control disruptive food-seeking behavior, caregivers often lock cabinets and refrigerators and significantly limit social activities,” said Jennifer Miller, MD, a pediatric endocrinologist at University of Florida Health, Gainesville, in a press release issued by the company.
“This FDA approval marks an important turning point, providing a much needed therapy and supporting the use of genetic testing to identify and properly diagnose patients with these rare genetic diseases of obesity,” she noted.
David Meeker, MD, chair, president, and CEO of Rhythm Pharmaceuticals, added: “We are advancing a first-in-class, precision medicine that is designed to directly address the underlying cause of obesities driven by genetic deficits in the MC4R pathway.”
Setmelanotide was evaluated in two phase 3 clinical trials. In one trial, 80% of patients with obesity caused by POMC or PCSK1 deficiency achieved greater than 10% weight loss after 1 year of treatment.
In the other trial, 45.5% of patients with obesity caused by LEPR deficiency achieved greater than 10% weight loss with 1 year of treatment.
Results for the two trials were recently published in The Lancet Diabetes & Endocrinology and discussed at the ObesityWeek Interactive 2020 meeting.
Setmelanotide was generally well tolerated in both trials. The most common adverse events were injection-site reactions, skin hyperpigmentation, and nausea.
The drug label notes that disturbances in sexual arousal, depression, and suicidal ideation; skin pigmentation; and darkening of preexisting nevi may occur with setmelanotide treatment.
The drug label also notes a risk for serious adverse reactions because of benzyl alcohol preservative in neonates and low-birth-weight infants. Setmelanotide is not approved for use in neonates or infants.
The company expects the drug to be commercially available in the United States in the first quarter of 2021.
Setmelanotide for the treatment of obesity associated with rare genetic defects had FDA breakthrough therapy designation as well as orphan drug designation.
The company is also evaluating setmelanotide for reduction in hunger and body weight in a pivotal phase 3 trial in people living with Bardet-Biedl or Alström syndrome, and top-line data are due soon.
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved setmelanotide (Imcivree, Rhythm Pharmaceuticals) for weight management in adults and children as young as 6 years with obesity because of proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing.
Individuals with these rare genetic causes of severe obesity have a normal weight at birth but develop persistent severe obesity within months because of insatiable hunger (hyperphagia).
Setmelanotide, a melanocortin-4 receptor (MC4R) agonist, is the first FDA-approved therapy for these disorders.
“Many patients and families who live with these diseases face an often-burdensome stigma associated with severe obesity. To manage this obesity and control disruptive food-seeking behavior, caregivers often lock cabinets and refrigerators and significantly limit social activities,” said Jennifer Miller, MD, a pediatric endocrinologist at University of Florida Health, Gainesville, in a press release issued by the company.
“This FDA approval marks an important turning point, providing a much needed therapy and supporting the use of genetic testing to identify and properly diagnose patients with these rare genetic diseases of obesity,” she noted.
David Meeker, MD, chair, president, and CEO of Rhythm Pharmaceuticals, added: “We are advancing a first-in-class, precision medicine that is designed to directly address the underlying cause of obesities driven by genetic deficits in the MC4R pathway.”
Setmelanotide was evaluated in two phase 3 clinical trials. In one trial, 80% of patients with obesity caused by POMC or PCSK1 deficiency achieved greater than 10% weight loss after 1 year of treatment.
In the other trial, 45.5% of patients with obesity caused by LEPR deficiency achieved greater than 10% weight loss with 1 year of treatment.
Results for the two trials were recently published in The Lancet Diabetes & Endocrinology and discussed at the ObesityWeek Interactive 2020 meeting.
Setmelanotide was generally well tolerated in both trials. The most common adverse events were injection-site reactions, skin hyperpigmentation, and nausea.
The drug label notes that disturbances in sexual arousal, depression, and suicidal ideation; skin pigmentation; and darkening of preexisting nevi may occur with setmelanotide treatment.
The drug label also notes a risk for serious adverse reactions because of benzyl alcohol preservative in neonates and low-birth-weight infants. Setmelanotide is not approved for use in neonates or infants.
The company expects the drug to be commercially available in the United States in the first quarter of 2021.
Setmelanotide for the treatment of obesity associated with rare genetic defects had FDA breakthrough therapy designation as well as orphan drug designation.
The company is also evaluating setmelanotide for reduction in hunger and body weight in a pivotal phase 3 trial in people living with Bardet-Biedl or Alström syndrome, and top-line data are due soon.
A version of this article originally appeared on Medscape.com.