User login
Asymptomatic Hemorrhagic Lesions in an Anemic Woman
The Diagnosis: Bullous Amyloidosis
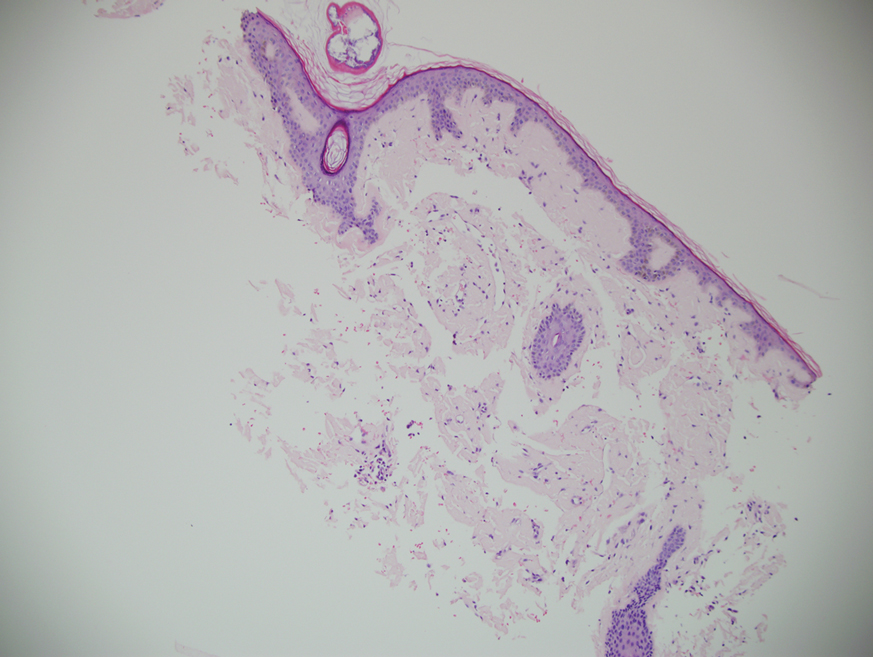
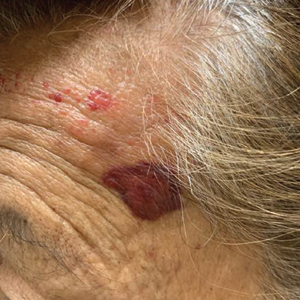
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
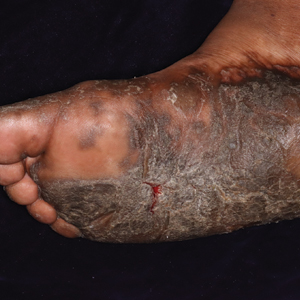
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
The Diagnosis: Bullous Amyloidosis
A punch biopsy from the left temple showed deposits of amorphous eosinophilic material at the tips of dermal papillae and in the papillary dermis with hemorrhage present (Figure 1). A diagnosis of amyloidosis was confirmed on the biopsy of the skin bulla. The low κ/λ light chain ratio and M-spike with notably elevated free λ light chains in both serum and urine were consistent with a λ light chain primary systemic amyloidosis. The patient was seen by hematology and oncology. A bone marrow biopsy demonstrated that 15% to 20% of the clonal-cell population was λ light chain restricted. Eosinophilic extracellular deposits found in the adjacent soft tissue and bone marrow space were confirmed as amyloid with apple green birefringence under polarized light on Congo red stain and metachromatic staining with crystal violet. The patient ultimately was diagnosed with λ light chain multiple myeloma and primary systemic amyloidosis.
Our patient was treated with a combination therapy of bortezomib, cyclophosphamide, and dexamethasone on 21-day cycles, with bortezomib on days 1, 4, 8, and 11. She had received 3 cycles of chemotherapy before developing diarrhea, hypotension, acute on chronic heart failure, and acute renal failure requiring hospitalization. She had several related complications due to amyloid light chain (AL) amyloidosis and subsequently died 16 days after her initial hospitalization from complications of methicillin-resistant Staphylococcus aureus bacteremia and septic shock.
Amyloidosis is the pathologic deposition of abnormal protein in the extracellular space of any tissue. Various soluble precursor proteins can make up amyloid, and these proteins polymerize into insoluble fibrils that damage the surrounding parenchyma. The clinical presentation of amyloidosis varies depending on the affected tissue as well as the constituent protein. The amyloidoses are divided into localized cutaneous, primary systemic, and secondary systemic variants. The initial distinction in amyloidosis is determining whether it is skin limited or systemic. Localized cutaneous amyloidosis comprises 30% to 40% of all amyloidosis cases and is further divided into 3 main subtypes: macular, lichen, and nodular amyloidosis.1 Macular and lichen amyloidosis are composed of keratin derivatives and typically are induced by patients when rubbing or scratching the skin. Histologically, macular and lichen amyloidosis are restricted to the superficial papillary dermis.1 Nodular amyloidosis is composed of λ or κ light chain immunoglobulins, which are produced by cutaneous infiltrates of monoclonal plasma cells. Histologically, nodular amyloidosis is characterized by a diffuse dermal infiltrate of amorphous eosinophilic material.1 Primary systemic amyloidosis is associated with an underlying plasma cell dyscrasia, and unlike secondary keratinocyte-derived amyloid, it can involve internal organs. Similar to nodular amyloidosis, primary systemic amyloidosis is composed of AL proteins, and it is histologically similar to nodular amyloidosis.1
Primary systemic AL amyloidosis commonly affects individuals aged 50 to 60 years. Males and females are equally affected. Macroglossia and periorbital purpura are some of the pathognomonic presentations in AL amyloidosis. The major cause of death in these patients is cardiac and renal involvement. Renal involvement commonly presents as nephrotic syndrome, and cardiac involvement can present as a restrictive cardiomyopathy with dyspnea. Other symptoms include edema, hepatosplenomegaly, bleeding diathesis, and carpal tunnel syndrome.2 An evaluation for AL amyloidosis should include a complete review of systems and physical examination with studies such as complete blood cell count, comprehensive metabolic panel, serum and urine protein electrophoresis and immunofixation, and electrocardiogram.
Cutaneous involvement in AL amyloidosis most commonly includes yellowish waxy papules, nodules, and plaques but also can include purpura and petechiae.2 Bullous amyloidosis, as seen in our patient, is a rare cutaneous presentation of AL amyloidosis that usually is negative for the Nikolsky sign (Figure 2). Bullae form due to weakness in amyloid-laden dermal connective tissue.3 Eighty-eight percent of cases of bullous amyloidosis have systemic involvement.1 Some cases have reported a familial linkage, suggesting there might be a genetic component to the disease.4 A PubMed search of articles indexed for MEDLINE using the terms bullous amyloidosis, bullous, amyloidosis, and amyloid revealed fewer than 35 cases of bullous amyloidosis in the English-language literature.5 Bullae can be located intradermally or subepidermally and commonly are hemorrhagic but also can be translucent, tender, and tense.
A study of electron microscopy in a patient with systemic bullous amyloidosis demonstrated amyloid and keratinocyte protrusions that perforated the dermis through the spaces in the lamina densa. The study concluded that the disintegration of the lamina densa and expansion of the intercellular spaces between keratinocytes were the causes of skin fragility as well as fluid exudation.5 Trauma or friction to the skin are local precipitating factors for blister formation in bullous amyloidosis.
Bullae can become apparent at any stage of AL amyloidosis, but they generally increase in size and number over time and are most common in intertriginous areas. Bullous amyloid lesions, especially those located in intertriginous areas, can have secondary impetiginization.6 In many cases, patients who present with bullous amyloidosis ultimately will be diagnosed with multiple myeloma or another plasma cell dyscrasia. In AL amyloidosis, only 10% to 15% of cases meet criteria for multiple myeloma, whereas 80% or more patients have a monoclonal gammopathy of undetermined significance.7
The prognosis of cutaneous amyloidosis depends on the extent of organ involvement and response to treatment. Treatment is aimed at eliminating clonal plasma cell populations to decrease the production of light chains, thereby decreasing protein burden and amyloid progression. Historically, treatment options included cytotoxic chemotherapy such as oral melphalan and dexamethasone, followed by hematopoietic stem cell transplant. More recent treatment options include bortezomib, thalidomide, pomalidomide, and lenalidomide.8 Our patient received a regimen of bortezomib, cyclophosphamide, and dexamethasone that is used for patients with extensive multiple myeloma.
The differential diagnosis in our patient included bullous drug eruption, which should be considered if the bullae are reoccurring at the same location and in association with the administration of a culprit drug. Bullous pemphigoid is preceded by pruritus, and biopsy demonstrates subepidermal bullae with associated eosinophilic infiltrate. Epidermolysis bullosa acquisita can present with milia and a linear pattern along the basement membrane zone with direct immunofluorescence. Traumatic purpura usually present with the classic shape and hue of an ecchymosis, and the patient will have a history of trauma.
Cutaneous involvement of amyloidosis can be an early clue to the diagnosis of plasma cell dyscrasia. Early diagnosis and treatment can portend a better prognosis and prevent progression to renal or cardiac disease.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
- Heaton J, Steinhoff N, Wanner B, et al. A review of primary cutaneous amyloidosis. J Am Osteopath Coll Dermatol. doi:10.1007/springerreference_42272
- Ventarola DJ, Schuster MW, Cohen JA, et al. JAAD grand rounds quiz. bullae and nodules on the legs of a 57-year-old woman. J Am Acad Dermatol. 2014;71:1035-1037.
- Chang SL, Lai PC, Cheng CJ, et al. Bullous amyloidosis in a hemodialysis patient is myeloma-associated rather than hemodialysis-associated amyloidosis. Amyloid. 2007;14:153-156.
- Suranagi VV, Siddramappa B, Bannur HB, et al. Bullous variant of familial biphasic lichen amyloidosis: a unique combination of three rare presentations. Indian J Dermatol. 2015;60:105.
- Antúnez-Lay A, Jaque A, González S. Hemorrhagic bullous skin lesions. Int J Dermatol. 2017;56:145-147.
- Reddy K, Hoda S, Penstein A, et al. Bullous amyloidosis complicated by cellulitis and sepsis: a case report. Arch Dermatol. 2011;147:126-127.
- Chu CH, Chan JY, Hsieh SW, et al. Diffuse ecchymoses and blisters on a yellowish waxy base: a case of bullous amyloidosis. J Dermatol. 2016;43:713-714.
- Gonzalez-Ramos J, Garrido-Gutiérrez C, González-Silva Y, et al. Relapsing bullous amyloidosis of the oral mucosa and acquired cutis laxa in a patient with multiple myeloma: a rare triple association. Clin Exp Dermatol. 2017;42:410-412.
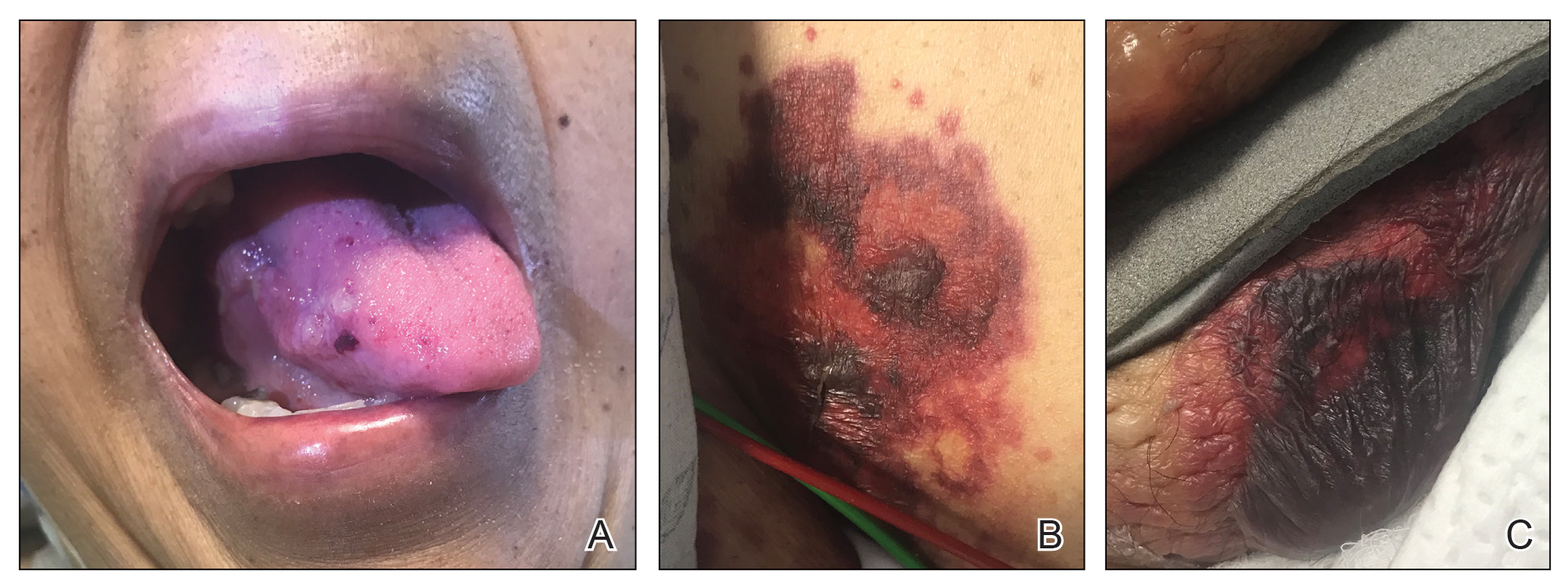
A 67-year-old woman with a medical history of type 2 diabetes mellitus, unspecified leukocytosis, and anemia presented to the dermatology clinic with asymptomatic hemorrhagic bullae on the face, chest, and tongue, as well as a large, tender, tense, hemorrhagic bulla on the groin of 3 to 4 months’ duration. A review of systems was negative for fever, chills, night sweats, malaise, shortness of breath, and dyspnea on exertion. A complete blood cell count showed mild leukocytosis, anemia, and thrombocytopenia. Her creatinine level was slightly elevated. Chest computed tomography showed early pulmonary fibrosis and coronary artery calcification. An echocardiogram showed diastolic dysfunction with moderate left ventricle thickening. A serum and urine electrophoresis demonstrated elevated free λ light chains with an M-spike. A punch biopsy was performed.
Gynecologic and Obstetric Implications of Darier Disease: A Dermatologist’s Perspective
Darier disease (DD)(also known as dyskeratosis follicularis) is a rare, autosomal-dominant genodermatosis characterized by greasy, rough, keratotic papules; typical nail abnormalities; mucosal changes; and characteristic dyskeratotic acantholysis that is called corps ronds and grains on histopathologic analysis. Darier disease is caused by mutations of the ATP2A2 gene on chromosome 12q23-24.1,2
Because of the autosomal-dominant pattern of inheritance in DD, if either parent is affected by DD, approximately 50% of their offspring will have the disorder. Therefore, couples need to be offered genetic counseling at a preconception visit or early in pregnancy. Although penetrance of DD is complete, spontaneous mutations are frequent and expressivity is variable1; prenatal diagnosis, though available since the 1980s, is therefore unreliable in DD, given the considerable variation in phenotypic expressivity. Differing phenotypes underscore the importance of proper counseling by the treating dermatologist or other provider. Females with a mild or nearly undetectable phenotype can give birth to a child with severe disease.
Lack of clear understanding about the variable phenotypic expressivity of DD can cause considerable anger, anxiety, guilt, psychological trauma, and fear in parents, should their child later develop a severe phenotype. They may feel that they were not properly prepared for the outcome. The physician-parent or physician-patient relationship can be negatively impacted if ongoing counseling is inadequate.
Clinically, DD presents in early adolescence (age range, 6–20 years) in most patients, which means that the disease and female reproductive years are contemporaneous. However, gynecologic and obstetric issues and complications of DD rarely have been addressed.3 Oromucosal involvement in DD is reported in 13% to 50% of cases, yet vaginal and cervical mucosal involvement rarely has been described,4,5 likely due to underreporting. Therefore, in this rare disease, it is important to address these aspects so that the patients are provided with appropriate management options.
Implications for Cervical Screening and Papanicolaou Tests
Cytopathologic findings of a Papanicolaou test taken from a patient with DD can lead to erroneous diagnosis of a low-grade squamous intraepithelial lesion due to cervical involvement by the disease process; therefore, correct interpretation of a smear may be inappropriate and erroneous. The cytopathologist needs to be informed of the patient’s diagnosis of DD in advance for appropriate reporting.5,6
Obstetric Implications
Fertility is normal in DD patients, and pregnancy usually has a normal course; however, exacerbation and remission of disease have been reported. de la Rosa Carrillo7 reported a case of vegetating DD during pregnancy. He described it as an exacerbation with concurrent bacterial infection and bilateral external otitis.7 Spouge et al8 reported a case of a 58-year-old woman who was the mother of 4 DD patients. She experienced an exacerbation of DD during all 6 pregnancies but improved immediately postpartum.8 Espy et al9 evaluated 8 cases of women with DD and described spontaneous improvement of the disorder during pregnancy (1 case) or while taking an oral contraceptive (3 cases).
Prenatal Counseling
Women with DD should be encouraged to talk to their dermatologist, obstetrician, or other provider of prenatal care regarding plans for pregnancy, labor, and delivery, as these events might be affected by the disorder. During pregnancy, careful monitoring and self-care remain essential. Simple measures to reduce the impact of irritants on DD during pregnancy include keeping the skin cool, using a soothing moisturizer, applying photoprotection, and using sunscreen. Treatment with systemic retinoids must be avoided if pregnancy is planned.
Warty plaques and papules of DD can involve flexures (groin, vulva, and perineum), with resultant malodor and pruritus10 as well as the potential for (drug resistant) secondary infection (eg, Staphylococcus aureus, group B Streptococcus, viruses [eg, Kaposi varicelliform eruption]). Skin swabs should be taken for culture and susceptibility testing, and infection should be treated at the earliest sign.
Management Concerns During Pregnancy and Delivery
Because the benefits of treating DD might outweigh risk in certain cases, thorough discussion with the patient about options is recommended, including the following concerns:
• Because mucocutaneous elasticity of the birth canal, including the vulva, perineum, and groin, is essential for nontraumatic vaginal delivery, it might be necessary to schedule an elective cesarean delivery in DD patients in whom these regions are involved.11
• In females with lower abdominal lesions, using a Pfannenstiel-Kerr incision for cesarean delivery might be problematic.11
• A single case report has described successful anesthetic management of labor, delivery, and postpartum care in a DD patient.12 Involvement of the skin of the back might preclude safe administration of regional anesthesia; however, because DD lesions are considered noninfectious, the authors operatively administered a subarachnoid block at the L3-L4 interspace through a lesion-free area. Postpartum, the patient was observed in the intensive care unit. She and the baby remained stable; she did not develop infectious complications, including a central nervous system infection.12
•Mucosal involvement is relatively rare in DD and has not been reported to compromise airway management.8
Postnatal Considerations
Breastfeeding might have to be stopped early or withheld altogether if there is widespread involvement of the skin of the breast or the nipple.11 Darier disease has been associated with neuropsychiatric manifestations, including major depression (30%), suicide attempts (13%), suicidal thoughts (31%), cyclothymia, bipolar disorder (4%), and epilepsy (3%).13,14 Therefore, patients should be screened for postpartum psychiatric manifestations at an early follow-up visit.
Final Thoughts
Although the etiology of DD is well known, the gynelogic and obstretric implications of this genodermatosis have rarely been described. This brief commentary is an attempt to provide the important information to a practicing dermatologist for appropriate management of female DD patients.
- Bale SJ, Toro JR. Genetic basis of Darier-White disease: bad pumps cause bumps. J Cutan Med Surg. 2000;4:103-106. doi:10.1177/120347540000400212
- Kansal NK, Hazarika N, Rao S. Familial case of Darier disease with guttate leukoderma: a case series from India. Indian Dermatol Online J. 2018;9:62-63. doi:10.4103/idoj.IDOJ_52_17
- Lynch PJ. Vulvar dermatoses: the eczematous diseases. In: Black M, Ambros-Rudolph CM, Edwards L, Lynch P, eds. Obstetric and Gynecologic Dermatology. 3rd ed. Mosby-Elsevier; 2008:192-194.
- Adam AE. Ectopic Darier’s disease of the cervix: an extraordinary cause of an abnormal smear. Cytopathology. 1996;7:414-421. doi:10.1111/j.1365-2303.1996.tb00547.x
- Suárez-Peñaranda JM, Antúnez JR, Del Rio E, et al. Vaginal involvement in a woman with Darier’s disease: a case report. Acta Cytol. 2005;49:530-532. doi:10.1159/000326200
- Boon ME. Dr. Darier’s lesson: it can be advantageous to the patient to ignore evident cytonuclear changes. Acta Cytol. 2005;49:469-470. doi:10.1159/000326189
- de la Rosa Carrillo D. Vegetating Darier’s disease during pregnancy. Acta Derm Venereol. 2006;86:259-260. doi:10.2340/00015555-0066
- Spouge JD, Trott JR, Chesko G. Darier-White’s disease: a cause of white lesions of the mucosa. report of four cases. Oral Surg Oral Med Oral Pathol. 1966;21:441-457. doi:10.1016/0030-4220(66)90401-4
- Espy PD, Stone S, Jolly HW Jr. Hormonal dependency in Darier disease. Cutis. 1976;17:315-320.
- De D, Kanwar AJ, Saikia UN. Uncommon flexural presentation of Darier disease. J Cutan Med Surg. 2008;12:249-252. doi:10.2310/7750.2008.07035
- Quinlivan JA, O'Halloran LC. Darier’s disease and pregnancy. Dermatol Aspects. 2013;1:1-3. doi:10.7243/2053-5309-1-1
- Sharma R, Singh BP, Das SN. Anesthetic management of cesarean section in a parturient with Darier’s disease. Acta Anaesthesiol Taiwan. 2010;48:158-159. doi:10.1016/S1875-4597(10)60051-3
- Gordon-Smith K, Jones LA, Burge SM, et al. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515-522. doi:10.1111/j.1365-2133.2010.09834.x
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol. 2016;174:562-568. doi:10.1111/bjd.14220
Darier disease (DD)(also known as dyskeratosis follicularis) is a rare, autosomal-dominant genodermatosis characterized by greasy, rough, keratotic papules; typical nail abnormalities; mucosal changes; and characteristic dyskeratotic acantholysis that is called corps ronds and grains on histopathologic analysis. Darier disease is caused by mutations of the ATP2A2 gene on chromosome 12q23-24.1,2
Because of the autosomal-dominant pattern of inheritance in DD, if either parent is affected by DD, approximately 50% of their offspring will have the disorder. Therefore, couples need to be offered genetic counseling at a preconception visit or early in pregnancy. Although penetrance of DD is complete, spontaneous mutations are frequent and expressivity is variable1; prenatal diagnosis, though available since the 1980s, is therefore unreliable in DD, given the considerable variation in phenotypic expressivity. Differing phenotypes underscore the importance of proper counseling by the treating dermatologist or other provider. Females with a mild or nearly undetectable phenotype can give birth to a child with severe disease.
Lack of clear understanding about the variable phenotypic expressivity of DD can cause considerable anger, anxiety, guilt, psychological trauma, and fear in parents, should their child later develop a severe phenotype. They may feel that they were not properly prepared for the outcome. The physician-parent or physician-patient relationship can be negatively impacted if ongoing counseling is inadequate.
Clinically, DD presents in early adolescence (age range, 6–20 years) in most patients, which means that the disease and female reproductive years are contemporaneous. However, gynecologic and obstetric issues and complications of DD rarely have been addressed.3 Oromucosal involvement in DD is reported in 13% to 50% of cases, yet vaginal and cervical mucosal involvement rarely has been described,4,5 likely due to underreporting. Therefore, in this rare disease, it is important to address these aspects so that the patients are provided with appropriate management options.
Implications for Cervical Screening and Papanicolaou Tests
Cytopathologic findings of a Papanicolaou test taken from a patient with DD can lead to erroneous diagnosis of a low-grade squamous intraepithelial lesion due to cervical involvement by the disease process; therefore, correct interpretation of a smear may be inappropriate and erroneous. The cytopathologist needs to be informed of the patient’s diagnosis of DD in advance for appropriate reporting.5,6
Obstetric Implications
Fertility is normal in DD patients, and pregnancy usually has a normal course; however, exacerbation and remission of disease have been reported. de la Rosa Carrillo7 reported a case of vegetating DD during pregnancy. He described it as an exacerbation with concurrent bacterial infection and bilateral external otitis.7 Spouge et al8 reported a case of a 58-year-old woman who was the mother of 4 DD patients. She experienced an exacerbation of DD during all 6 pregnancies but improved immediately postpartum.8 Espy et al9 evaluated 8 cases of women with DD and described spontaneous improvement of the disorder during pregnancy (1 case) or while taking an oral contraceptive (3 cases).
Prenatal Counseling
Women with DD should be encouraged to talk to their dermatologist, obstetrician, or other provider of prenatal care regarding plans for pregnancy, labor, and delivery, as these events might be affected by the disorder. During pregnancy, careful monitoring and self-care remain essential. Simple measures to reduce the impact of irritants on DD during pregnancy include keeping the skin cool, using a soothing moisturizer, applying photoprotection, and using sunscreen. Treatment with systemic retinoids must be avoided if pregnancy is planned.
Warty plaques and papules of DD can involve flexures (groin, vulva, and perineum), with resultant malodor and pruritus10 as well as the potential for (drug resistant) secondary infection (eg, Staphylococcus aureus, group B Streptococcus, viruses [eg, Kaposi varicelliform eruption]). Skin swabs should be taken for culture and susceptibility testing, and infection should be treated at the earliest sign.
Management Concerns During Pregnancy and Delivery
Because the benefits of treating DD might outweigh risk in certain cases, thorough discussion with the patient about options is recommended, including the following concerns:
• Because mucocutaneous elasticity of the birth canal, including the vulva, perineum, and groin, is essential for nontraumatic vaginal delivery, it might be necessary to schedule an elective cesarean delivery in DD patients in whom these regions are involved.11
• In females with lower abdominal lesions, using a Pfannenstiel-Kerr incision for cesarean delivery might be problematic.11
• A single case report has described successful anesthetic management of labor, delivery, and postpartum care in a DD patient.12 Involvement of the skin of the back might preclude safe administration of regional anesthesia; however, because DD lesions are considered noninfectious, the authors operatively administered a subarachnoid block at the L3-L4 interspace through a lesion-free area. Postpartum, the patient was observed in the intensive care unit. She and the baby remained stable; she did not develop infectious complications, including a central nervous system infection.12
•Mucosal involvement is relatively rare in DD and has not been reported to compromise airway management.8
Postnatal Considerations
Breastfeeding might have to be stopped early or withheld altogether if there is widespread involvement of the skin of the breast or the nipple.11 Darier disease has been associated with neuropsychiatric manifestations, including major depression (30%), suicide attempts (13%), suicidal thoughts (31%), cyclothymia, bipolar disorder (4%), and epilepsy (3%).13,14 Therefore, patients should be screened for postpartum psychiatric manifestations at an early follow-up visit.
Final Thoughts
Although the etiology of DD is well known, the gynelogic and obstretric implications of this genodermatosis have rarely been described. This brief commentary is an attempt to provide the important information to a practicing dermatologist for appropriate management of female DD patients.
Darier disease (DD)(also known as dyskeratosis follicularis) is a rare, autosomal-dominant genodermatosis characterized by greasy, rough, keratotic papules; typical nail abnormalities; mucosal changes; and characteristic dyskeratotic acantholysis that is called corps ronds and grains on histopathologic analysis. Darier disease is caused by mutations of the ATP2A2 gene on chromosome 12q23-24.1,2
Because of the autosomal-dominant pattern of inheritance in DD, if either parent is affected by DD, approximately 50% of their offspring will have the disorder. Therefore, couples need to be offered genetic counseling at a preconception visit or early in pregnancy. Although penetrance of DD is complete, spontaneous mutations are frequent and expressivity is variable1; prenatal diagnosis, though available since the 1980s, is therefore unreliable in DD, given the considerable variation in phenotypic expressivity. Differing phenotypes underscore the importance of proper counseling by the treating dermatologist or other provider. Females with a mild or nearly undetectable phenotype can give birth to a child with severe disease.
Lack of clear understanding about the variable phenotypic expressivity of DD can cause considerable anger, anxiety, guilt, psychological trauma, and fear in parents, should their child later develop a severe phenotype. They may feel that they were not properly prepared for the outcome. The physician-parent or physician-patient relationship can be negatively impacted if ongoing counseling is inadequate.
Clinically, DD presents in early adolescence (age range, 6–20 years) in most patients, which means that the disease and female reproductive years are contemporaneous. However, gynecologic and obstetric issues and complications of DD rarely have been addressed.3 Oromucosal involvement in DD is reported in 13% to 50% of cases, yet vaginal and cervical mucosal involvement rarely has been described,4,5 likely due to underreporting. Therefore, in this rare disease, it is important to address these aspects so that the patients are provided with appropriate management options.
Implications for Cervical Screening and Papanicolaou Tests
Cytopathologic findings of a Papanicolaou test taken from a patient with DD can lead to erroneous diagnosis of a low-grade squamous intraepithelial lesion due to cervical involvement by the disease process; therefore, correct interpretation of a smear may be inappropriate and erroneous. The cytopathologist needs to be informed of the patient’s diagnosis of DD in advance for appropriate reporting.5,6
Obstetric Implications
Fertility is normal in DD patients, and pregnancy usually has a normal course; however, exacerbation and remission of disease have been reported. de la Rosa Carrillo7 reported a case of vegetating DD during pregnancy. He described it as an exacerbation with concurrent bacterial infection and bilateral external otitis.7 Spouge et al8 reported a case of a 58-year-old woman who was the mother of 4 DD patients. She experienced an exacerbation of DD during all 6 pregnancies but improved immediately postpartum.8 Espy et al9 evaluated 8 cases of women with DD and described spontaneous improvement of the disorder during pregnancy (1 case) or while taking an oral contraceptive (3 cases).
Prenatal Counseling
Women with DD should be encouraged to talk to their dermatologist, obstetrician, or other provider of prenatal care regarding plans for pregnancy, labor, and delivery, as these events might be affected by the disorder. During pregnancy, careful monitoring and self-care remain essential. Simple measures to reduce the impact of irritants on DD during pregnancy include keeping the skin cool, using a soothing moisturizer, applying photoprotection, and using sunscreen. Treatment with systemic retinoids must be avoided if pregnancy is planned.
Warty plaques and papules of DD can involve flexures (groin, vulva, and perineum), with resultant malodor and pruritus10 as well as the potential for (drug resistant) secondary infection (eg, Staphylococcus aureus, group B Streptococcus, viruses [eg, Kaposi varicelliform eruption]). Skin swabs should be taken for culture and susceptibility testing, and infection should be treated at the earliest sign.
Management Concerns During Pregnancy and Delivery
Because the benefits of treating DD might outweigh risk in certain cases, thorough discussion with the patient about options is recommended, including the following concerns:
• Because mucocutaneous elasticity of the birth canal, including the vulva, perineum, and groin, is essential for nontraumatic vaginal delivery, it might be necessary to schedule an elective cesarean delivery in DD patients in whom these regions are involved.11
• In females with lower abdominal lesions, using a Pfannenstiel-Kerr incision for cesarean delivery might be problematic.11
• A single case report has described successful anesthetic management of labor, delivery, and postpartum care in a DD patient.12 Involvement of the skin of the back might preclude safe administration of regional anesthesia; however, because DD lesions are considered noninfectious, the authors operatively administered a subarachnoid block at the L3-L4 interspace through a lesion-free area. Postpartum, the patient was observed in the intensive care unit. She and the baby remained stable; she did not develop infectious complications, including a central nervous system infection.12
•Mucosal involvement is relatively rare in DD and has not been reported to compromise airway management.8
Postnatal Considerations
Breastfeeding might have to be stopped early or withheld altogether if there is widespread involvement of the skin of the breast or the nipple.11 Darier disease has been associated with neuropsychiatric manifestations, including major depression (30%), suicide attempts (13%), suicidal thoughts (31%), cyclothymia, bipolar disorder (4%), and epilepsy (3%).13,14 Therefore, patients should be screened for postpartum psychiatric manifestations at an early follow-up visit.
Final Thoughts
Although the etiology of DD is well known, the gynelogic and obstretric implications of this genodermatosis have rarely been described. This brief commentary is an attempt to provide the important information to a practicing dermatologist for appropriate management of female DD patients.
- Bale SJ, Toro JR. Genetic basis of Darier-White disease: bad pumps cause bumps. J Cutan Med Surg. 2000;4:103-106. doi:10.1177/120347540000400212
- Kansal NK, Hazarika N, Rao S. Familial case of Darier disease with guttate leukoderma: a case series from India. Indian Dermatol Online J. 2018;9:62-63. doi:10.4103/idoj.IDOJ_52_17
- Lynch PJ. Vulvar dermatoses: the eczematous diseases. In: Black M, Ambros-Rudolph CM, Edwards L, Lynch P, eds. Obstetric and Gynecologic Dermatology. 3rd ed. Mosby-Elsevier; 2008:192-194.
- Adam AE. Ectopic Darier’s disease of the cervix: an extraordinary cause of an abnormal smear. Cytopathology. 1996;7:414-421. doi:10.1111/j.1365-2303.1996.tb00547.x
- Suárez-Peñaranda JM, Antúnez JR, Del Rio E, et al. Vaginal involvement in a woman with Darier’s disease: a case report. Acta Cytol. 2005;49:530-532. doi:10.1159/000326200
- Boon ME. Dr. Darier’s lesson: it can be advantageous to the patient to ignore evident cytonuclear changes. Acta Cytol. 2005;49:469-470. doi:10.1159/000326189
- de la Rosa Carrillo D. Vegetating Darier’s disease during pregnancy. Acta Derm Venereol. 2006;86:259-260. doi:10.2340/00015555-0066
- Spouge JD, Trott JR, Chesko G. Darier-White’s disease: a cause of white lesions of the mucosa. report of four cases. Oral Surg Oral Med Oral Pathol. 1966;21:441-457. doi:10.1016/0030-4220(66)90401-4
- Espy PD, Stone S, Jolly HW Jr. Hormonal dependency in Darier disease. Cutis. 1976;17:315-320.
- De D, Kanwar AJ, Saikia UN. Uncommon flexural presentation of Darier disease. J Cutan Med Surg. 2008;12:249-252. doi:10.2310/7750.2008.07035
- Quinlivan JA, O'Halloran LC. Darier’s disease and pregnancy. Dermatol Aspects. 2013;1:1-3. doi:10.7243/2053-5309-1-1
- Sharma R, Singh BP, Das SN. Anesthetic management of cesarean section in a parturient with Darier’s disease. Acta Anaesthesiol Taiwan. 2010;48:158-159. doi:10.1016/S1875-4597(10)60051-3
- Gordon-Smith K, Jones LA, Burge SM, et al. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515-522. doi:10.1111/j.1365-2133.2010.09834.x
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol. 2016;174:562-568. doi:10.1111/bjd.14220
- Bale SJ, Toro JR. Genetic basis of Darier-White disease: bad pumps cause bumps. J Cutan Med Surg. 2000;4:103-106. doi:10.1177/120347540000400212
- Kansal NK, Hazarika N, Rao S. Familial case of Darier disease with guttate leukoderma: a case series from India. Indian Dermatol Online J. 2018;9:62-63. doi:10.4103/idoj.IDOJ_52_17
- Lynch PJ. Vulvar dermatoses: the eczematous diseases. In: Black M, Ambros-Rudolph CM, Edwards L, Lynch P, eds. Obstetric and Gynecologic Dermatology. 3rd ed. Mosby-Elsevier; 2008:192-194.
- Adam AE. Ectopic Darier’s disease of the cervix: an extraordinary cause of an abnormal smear. Cytopathology. 1996;7:414-421. doi:10.1111/j.1365-2303.1996.tb00547.x
- Suárez-Peñaranda JM, Antúnez JR, Del Rio E, et al. Vaginal involvement in a woman with Darier’s disease: a case report. Acta Cytol. 2005;49:530-532. doi:10.1159/000326200
- Boon ME. Dr. Darier’s lesson: it can be advantageous to the patient to ignore evident cytonuclear changes. Acta Cytol. 2005;49:469-470. doi:10.1159/000326189
- de la Rosa Carrillo D. Vegetating Darier’s disease during pregnancy. Acta Derm Venereol. 2006;86:259-260. doi:10.2340/00015555-0066
- Spouge JD, Trott JR, Chesko G. Darier-White’s disease: a cause of white lesions of the mucosa. report of four cases. Oral Surg Oral Med Oral Pathol. 1966;21:441-457. doi:10.1016/0030-4220(66)90401-4
- Espy PD, Stone S, Jolly HW Jr. Hormonal dependency in Darier disease. Cutis. 1976;17:315-320.
- De D, Kanwar AJ, Saikia UN. Uncommon flexural presentation of Darier disease. J Cutan Med Surg. 2008;12:249-252. doi:10.2310/7750.2008.07035
- Quinlivan JA, O'Halloran LC. Darier’s disease and pregnancy. Dermatol Aspects. 2013;1:1-3. doi:10.7243/2053-5309-1-1
- Sharma R, Singh BP, Das SN. Anesthetic management of cesarean section in a parturient with Darier’s disease. Acta Anaesthesiol Taiwan. 2010;48:158-159. doi:10.1016/S1875-4597(10)60051-3
- Gordon-Smith K, Jones LA, Burge SM, et al. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515-522. doi:10.1111/j.1365-2133.2010.09834.x
- Dodiuk-Gad RP, Cohen-Barak E, Khayat M, et al. Darier disease in Israel: combined evaluation of genetic and neuropsychiatric aspects. Br J Dermatol. 2016;174:562-568. doi:10.1111/bjd.14220
Practice Points
- Because Darier disease (DD) manifests during reproductive years, systemic retinoids should be used carefully in female patients.
- For a Papanicolaou test to be properly interpreted in a patient with DD, the cytopathologist must be informed of the DD diagnosis.
- Darier disease may be exacerbated or relieved during pregnancy.
Despite new ichthyosis treatment recommendations, ‘many questions still exist’
.

According to a consensus statement published in the February issue of Pediatric Dermatology, adequate data exist in the medical literature to demonstrate an improvement in use of systemic retinoids for select genotypes of congenital ichthyosiform erythroderma, epidermolytic ichthyosis, erythrokeratodermia variabilis, harlequin ichthyosis, IFAP syndrome (ichthyosis with confetti, ichthyosis follicularis, atrichia, and photophobia), KID syndrome (keratitis-ichthyosis-deafness), KLICK syndrome (keratosis linearis with ichthyosis congenita and sclerosing keratoderma), lamellar ichthyosis, loricrin keratoderma, neutral lipid storage disease with ichthyosis, recessive X-linked ichthyosis, and Sjögren-Larsson syndrome.
At the same time, limited or no data exist to support the use of systemic retinoids for CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects), CHIME syndrome (colobomas, heart defects, ichthyosiform dermatosis, intellectual disability, and either ear defects or epilepsy), Conradi-Hunermann-Happle syndrome, ichthyosis-hypotrichosis, ichthyosis-hypotrichosis-sclerosis cholangitis, ichthyosis prematurity syndrome, MEDNIK syndrome (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma), peeling skin disease, Refsum syndrome, and trichothiodystrophy, according to the statement.
“In particular, we did note that, with any disorder that was associated with atopy, the retinoids were often counterproductive,” one of the consensus statement cochairs, Andrea L. Zaenglein, MD, said during the Society for Pediatric Dermatology pre-AAD meeting. “In Netherton syndrome, for example, retinoids seemed to make the skin fragility a lot worse, so typically, they would be avoided in those patients.”
The statement, which she assembled with cochair pediatric dermatologist Moise L. Levy, MD, professor of pediatrics, University of Texas at Austin, and 21 other multidisciplinary experts, recommends considering use of topical retinoids to help decrease scaling of the skin,“but [they] are particularly helpful for more localized complications of ichthyosis, such as digital contractures and ectropion,” said Dr. Zaenglein, professor of dermatology and pediatrics at Penn State University, Hershey. “A lot of it has to do with the size and the volume of the tubes and getting enough [product] to be able to apply it over larger areas. We do tend to use them more focally.”
While systemic absorption can occur with widespread use, no specific lab monitoring is required. Dr. Zaenglein and her colleagues also recommend avoiding the use of tazarotene during pregnancy, since it is contraindicated in pregnancy (category X), but monthly pregnancy tests are not recommended.
During an overview of the document at the meeting, she noted that the recommended dosing for both isotretinoin and acitretin is 0.5-1.0 mg/kg per day and the side effects tend to be dose dependent, “except teratogenicity, which can occur with even low doses of systemic retinoid exposure and early on in pregnancy.” The authors also advise patients to consider drug holidays or lower doses “especially during warmer, more humid months, where you might not need the higher doses to achieve cutaneous effects,” she said.
They emphasized the importance of avoiding pregnancy for 3 years after completion of treatment with acitretin. “While the half-life of acitretin is 49 hours, it’s easily converted with any alcohol exposure to etretinate,” Dr. Zaenglein noted. “Then, the half-life is 120 days.”

The statement, which was sponsored by the Pediatric Dermatology Research Alliance (PEDRA), also addresses the clinical considerations and consequences of long-term systemic retinoid use on bone health, such as premature epiphyseal closure in preadolescent children. “In general, this risk is greater with higher doses of therapies – above 1 mg/kg per day – and over prolonged periods of time, typically 4-6 years,” she said. Other potential effects on bone health include calcifications of tendons and ligaments, osteophytes or “bone spurs,” DISH (diffuse idiopathic skeletal hyperostosis), and potential alterations in bone density and growth.
“We also have to worry about concomitant effects of contraception, particularly if you’re using progestin-only formulations that carry a black box warning for osteoporosis,” Dr. Zaenglein said. “It is recommended that you limit their use to 3 years.” Other factors to consider include genetic risk and modifiable factors that affect bone health, such as diet and physical activity, which may impact susceptibility to systemic retinoid bone toxicity and should be discussed with the patient.
Recommended bone monitoring in children starts with a comprehensive family and personal medical history for skeletal toxicity risk factors, followed by an annual growth assessment (height, weight, body mass index, and growth curve), asking regularly about musculoskeletal symptoms, and following up with appropriate imaging. “Inquiring about their diet is recommended as well, so making sure they’re getting sufficient amounts of calcium and vitamin D, and no additional vitamin A sources that may compound the side effects from systemic retinoids,” Dr. Zaenglein said.
The document also advises that a baseline skeletal radiographic survey be performed in patients aged 16-18 years. This may include imaging of the lateral cervical and thoracic spine, lateral view of the calcanei to include Achilles tendon, hips and symptomatic areas, and bone density evaluation.
The statement addressed the psychiatric considerations and consequences of long-term systemic retinoid use. One cross-sectional study of children with ichthyosis found that 30% screened positive for depression and 38% screened positive for anxiety, “but the role of retinoids is unclear,” Dr. Zaenglein said. “It’s a complicated matter, but patients with a personal history of depression, anxiety, and other affective disorders prior to initiation of systemic retinoid treatment should be monitored carefully for exacerbation of symptoms. Comanagement with a mental health provider should be considered.”
As for contraception considerations with long-term systemic retinoid therapy use, the authors recommend that two forms of contraception be used. “Consider long-acting reversible contraception, especially in sexually active adolescents who have a history of noncompliance, or to remove the risk of teratogenicity for them,” she said. “We’re not sure what additive effects progestin/lower estrogen have on long-term cardiovascular health, including lipids and bone density.”
The authors noted that iPLEDGE is not designed for long-term use. “It’s really designed for the on-label use of systemic retinoids in severe acne, where you’re using it for 5-6 months, not for 5-6 years,” Dr. Zaenglein said. “iPLEDGE does impose significant and financial barriers for our patients. More advocacy is needed to adapt that program for our patients.”
She and her coauthors acknowledged practice gaps and unmet needs in patients with disorders of cornification/types of ichthyosis, including the optimal formulation of retinoids based on ichthyosis subtype, whether there is a benefit to intermittent therapy with respect to risk of toxicity and maintenance of efficacy, and how to minimize the bone-related changes that can occur with treatment. “These are some of the things that we can look further into,” she said. “For now, though, retinoids can improve function and quality of life in patients with ichthyosis and disorders of cornification. Many questions still exist, and more data and research are needed.”
Sun Pharmaceuticals and the Foundation for Ichthyosis and Related Skin Types (FIRST) provided an unrestricted grant for development of the recommendations.
Dr. Zaenglein disclosed that she is a consultant for Pfizer. She is also an advisory board member for Dermata, Sol-Gel, Regeneron, Verrica, and Cassiopea, and has conducted contracted research for AbbVie, Incyte, Arcutis, and Pfizer. The other authors disclosed serving as investigators, advisers, consultants, and/or had other relationships with various pharmaceutical companies.
.
According to a consensus statement published in the February issue of Pediatric Dermatology, adequate data exist in the medical literature to demonstrate an improvement in use of systemic retinoids for select genotypes of congenital ichthyosiform erythroderma, epidermolytic ichthyosis, erythrokeratodermia variabilis, harlequin ichthyosis, IFAP syndrome (ichthyosis with confetti, ichthyosis follicularis, atrichia, and photophobia), KID syndrome (keratitis-ichthyosis-deafness), KLICK syndrome (keratosis linearis with ichthyosis congenita and sclerosing keratoderma), lamellar ichthyosis, loricrin keratoderma, neutral lipid storage disease with ichthyosis, recessive X-linked ichthyosis, and Sjögren-Larsson syndrome.
At the same time, limited or no data exist to support the use of systemic retinoids for CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects), CHIME syndrome (colobomas, heart defects, ichthyosiform dermatosis, intellectual disability, and either ear defects or epilepsy), Conradi-Hunermann-Happle syndrome, ichthyosis-hypotrichosis, ichthyosis-hypotrichosis-sclerosis cholangitis, ichthyosis prematurity syndrome, MEDNIK syndrome (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma), peeling skin disease, Refsum syndrome, and trichothiodystrophy, according to the statement.
“In particular, we did note that, with any disorder that was associated with atopy, the retinoids were often counterproductive,” one of the consensus statement cochairs, Andrea L. Zaenglein, MD, said during the Society for Pediatric Dermatology pre-AAD meeting. “In Netherton syndrome, for example, retinoids seemed to make the skin fragility a lot worse, so typically, they would be avoided in those patients.”
The statement, which she assembled with cochair pediatric dermatologist Moise L. Levy, MD, professor of pediatrics, University of Texas at Austin, and 21 other multidisciplinary experts, recommends considering use of topical retinoids to help decrease scaling of the skin,“but [they] are particularly helpful for more localized complications of ichthyosis, such as digital contractures and ectropion,” said Dr. Zaenglein, professor of dermatology and pediatrics at Penn State University, Hershey. “A lot of it has to do with the size and the volume of the tubes and getting enough [product] to be able to apply it over larger areas. We do tend to use them more focally.”
While systemic absorption can occur with widespread use, no specific lab monitoring is required. Dr. Zaenglein and her colleagues also recommend avoiding the use of tazarotene during pregnancy, since it is contraindicated in pregnancy (category X), but monthly pregnancy tests are not recommended.
During an overview of the document at the meeting, she noted that the recommended dosing for both isotretinoin and acitretin is 0.5-1.0 mg/kg per day and the side effects tend to be dose dependent, “except teratogenicity, which can occur with even low doses of systemic retinoid exposure and early on in pregnancy.” The authors also advise patients to consider drug holidays or lower doses “especially during warmer, more humid months, where you might not need the higher doses to achieve cutaneous effects,” she said.
They emphasized the importance of avoiding pregnancy for 3 years after completion of treatment with acitretin. “While the half-life of acitretin is 49 hours, it’s easily converted with any alcohol exposure to etretinate,” Dr. Zaenglein noted. “Then, the half-life is 120 days.”
The statement, which was sponsored by the Pediatric Dermatology Research Alliance (PEDRA), also addresses the clinical considerations and consequences of long-term systemic retinoid use on bone health, such as premature epiphyseal closure in preadolescent children. “In general, this risk is greater with higher doses of therapies – above 1 mg/kg per day – and over prolonged periods of time, typically 4-6 years,” she said. Other potential effects on bone health include calcifications of tendons and ligaments, osteophytes or “bone spurs,” DISH (diffuse idiopathic skeletal hyperostosis), and potential alterations in bone density and growth.
“We also have to worry about concomitant effects of contraception, particularly if you’re using progestin-only formulations that carry a black box warning for osteoporosis,” Dr. Zaenglein said. “It is recommended that you limit their use to 3 years.” Other factors to consider include genetic risk and modifiable factors that affect bone health, such as diet and physical activity, which may impact susceptibility to systemic retinoid bone toxicity and should be discussed with the patient.
Recommended bone monitoring in children starts with a comprehensive family and personal medical history for skeletal toxicity risk factors, followed by an annual growth assessment (height, weight, body mass index, and growth curve), asking regularly about musculoskeletal symptoms, and following up with appropriate imaging. “Inquiring about their diet is recommended as well, so making sure they’re getting sufficient amounts of calcium and vitamin D, and no additional vitamin A sources that may compound the side effects from systemic retinoids,” Dr. Zaenglein said.
The document also advises that a baseline skeletal radiographic survey be performed in patients aged 16-18 years. This may include imaging of the lateral cervical and thoracic spine, lateral view of the calcanei to include Achilles tendon, hips and symptomatic areas, and bone density evaluation.
The statement addressed the psychiatric considerations and consequences of long-term systemic retinoid use. One cross-sectional study of children with ichthyosis found that 30% screened positive for depression and 38% screened positive for anxiety, “but the role of retinoids is unclear,” Dr. Zaenglein said. “It’s a complicated matter, but patients with a personal history of depression, anxiety, and other affective disorders prior to initiation of systemic retinoid treatment should be monitored carefully for exacerbation of symptoms. Comanagement with a mental health provider should be considered.”
As for contraception considerations with long-term systemic retinoid therapy use, the authors recommend that two forms of contraception be used. “Consider long-acting reversible contraception, especially in sexually active adolescents who have a history of noncompliance, or to remove the risk of teratogenicity for them,” she said. “We’re not sure what additive effects progestin/lower estrogen have on long-term cardiovascular health, including lipids and bone density.”
The authors noted that iPLEDGE is not designed for long-term use. “It’s really designed for the on-label use of systemic retinoids in severe acne, where you’re using it for 5-6 months, not for 5-6 years,” Dr. Zaenglein said. “iPLEDGE does impose significant and financial barriers for our patients. More advocacy is needed to adapt that program for our patients.”
She and her coauthors acknowledged practice gaps and unmet needs in patients with disorders of cornification/types of ichthyosis, including the optimal formulation of retinoids based on ichthyosis subtype, whether there is a benefit to intermittent therapy with respect to risk of toxicity and maintenance of efficacy, and how to minimize the bone-related changes that can occur with treatment. “These are some of the things that we can look further into,” she said. “For now, though, retinoids can improve function and quality of life in patients with ichthyosis and disorders of cornification. Many questions still exist, and more data and research are needed.”
Sun Pharmaceuticals and the Foundation for Ichthyosis and Related Skin Types (FIRST) provided an unrestricted grant for development of the recommendations.
Dr. Zaenglein disclosed that she is a consultant for Pfizer. She is also an advisory board member for Dermata, Sol-Gel, Regeneron, Verrica, and Cassiopea, and has conducted contracted research for AbbVie, Incyte, Arcutis, and Pfizer. The other authors disclosed serving as investigators, advisers, consultants, and/or had other relationships with various pharmaceutical companies.
.
According to a consensus statement published in the February issue of Pediatric Dermatology, adequate data exist in the medical literature to demonstrate an improvement in use of systemic retinoids for select genotypes of congenital ichthyosiform erythroderma, epidermolytic ichthyosis, erythrokeratodermia variabilis, harlequin ichthyosis, IFAP syndrome (ichthyosis with confetti, ichthyosis follicularis, atrichia, and photophobia), KID syndrome (keratitis-ichthyosis-deafness), KLICK syndrome (keratosis linearis with ichthyosis congenita and sclerosing keratoderma), lamellar ichthyosis, loricrin keratoderma, neutral lipid storage disease with ichthyosis, recessive X-linked ichthyosis, and Sjögren-Larsson syndrome.
At the same time, limited or no data exist to support the use of systemic retinoids for CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects), CHIME syndrome (colobomas, heart defects, ichthyosiform dermatosis, intellectual disability, and either ear defects or epilepsy), Conradi-Hunermann-Happle syndrome, ichthyosis-hypotrichosis, ichthyosis-hypotrichosis-sclerosis cholangitis, ichthyosis prematurity syndrome, MEDNIK syndrome (mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma), peeling skin disease, Refsum syndrome, and trichothiodystrophy, according to the statement.
“In particular, we did note that, with any disorder that was associated with atopy, the retinoids were often counterproductive,” one of the consensus statement cochairs, Andrea L. Zaenglein, MD, said during the Society for Pediatric Dermatology pre-AAD meeting. “In Netherton syndrome, for example, retinoids seemed to make the skin fragility a lot worse, so typically, they would be avoided in those patients.”
The statement, which she assembled with cochair pediatric dermatologist Moise L. Levy, MD, professor of pediatrics, University of Texas at Austin, and 21 other multidisciplinary experts, recommends considering use of topical retinoids to help decrease scaling of the skin,“but [they] are particularly helpful for more localized complications of ichthyosis, such as digital contractures and ectropion,” said Dr. Zaenglein, professor of dermatology and pediatrics at Penn State University, Hershey. “A lot of it has to do with the size and the volume of the tubes and getting enough [product] to be able to apply it over larger areas. We do tend to use them more focally.”
While systemic absorption can occur with widespread use, no specific lab monitoring is required. Dr. Zaenglein and her colleagues also recommend avoiding the use of tazarotene during pregnancy, since it is contraindicated in pregnancy (category X), but monthly pregnancy tests are not recommended.
During an overview of the document at the meeting, she noted that the recommended dosing for both isotretinoin and acitretin is 0.5-1.0 mg/kg per day and the side effects tend to be dose dependent, “except teratogenicity, which can occur with even low doses of systemic retinoid exposure and early on in pregnancy.” The authors also advise patients to consider drug holidays or lower doses “especially during warmer, more humid months, where you might not need the higher doses to achieve cutaneous effects,” she said.
They emphasized the importance of avoiding pregnancy for 3 years after completion of treatment with acitretin. “While the half-life of acitretin is 49 hours, it’s easily converted with any alcohol exposure to etretinate,” Dr. Zaenglein noted. “Then, the half-life is 120 days.”
The statement, which was sponsored by the Pediatric Dermatology Research Alliance (PEDRA), also addresses the clinical considerations and consequences of long-term systemic retinoid use on bone health, such as premature epiphyseal closure in preadolescent children. “In general, this risk is greater with higher doses of therapies – above 1 mg/kg per day – and over prolonged periods of time, typically 4-6 years,” she said. Other potential effects on bone health include calcifications of tendons and ligaments, osteophytes or “bone spurs,” DISH (diffuse idiopathic skeletal hyperostosis), and potential alterations in bone density and growth.
“We also have to worry about concomitant effects of contraception, particularly if you’re using progestin-only formulations that carry a black box warning for osteoporosis,” Dr. Zaenglein said. “It is recommended that you limit their use to 3 years.” Other factors to consider include genetic risk and modifiable factors that affect bone health, such as diet and physical activity, which may impact susceptibility to systemic retinoid bone toxicity and should be discussed with the patient.
Recommended bone monitoring in children starts with a comprehensive family and personal medical history for skeletal toxicity risk factors, followed by an annual growth assessment (height, weight, body mass index, and growth curve), asking regularly about musculoskeletal symptoms, and following up with appropriate imaging. “Inquiring about their diet is recommended as well, so making sure they’re getting sufficient amounts of calcium and vitamin D, and no additional vitamin A sources that may compound the side effects from systemic retinoids,” Dr. Zaenglein said.
The document also advises that a baseline skeletal radiographic survey be performed in patients aged 16-18 years. This may include imaging of the lateral cervical and thoracic spine, lateral view of the calcanei to include Achilles tendon, hips and symptomatic areas, and bone density evaluation.
The statement addressed the psychiatric considerations and consequences of long-term systemic retinoid use. One cross-sectional study of children with ichthyosis found that 30% screened positive for depression and 38% screened positive for anxiety, “but the role of retinoids is unclear,” Dr. Zaenglein said. “It’s a complicated matter, but patients with a personal history of depression, anxiety, and other affective disorders prior to initiation of systemic retinoid treatment should be monitored carefully for exacerbation of symptoms. Comanagement with a mental health provider should be considered.”
As for contraception considerations with long-term systemic retinoid therapy use, the authors recommend that two forms of contraception be used. “Consider long-acting reversible contraception, especially in sexually active adolescents who have a history of noncompliance, or to remove the risk of teratogenicity for them,” she said. “We’re not sure what additive effects progestin/lower estrogen have on long-term cardiovascular health, including lipids and bone density.”
The authors noted that iPLEDGE is not designed for long-term use. “It’s really designed for the on-label use of systemic retinoids in severe acne, where you’re using it for 5-6 months, not for 5-6 years,” Dr. Zaenglein said. “iPLEDGE does impose significant and financial barriers for our patients. More advocacy is needed to adapt that program for our patients.”
She and her coauthors acknowledged practice gaps and unmet needs in patients with disorders of cornification/types of ichthyosis, including the optimal formulation of retinoids based on ichthyosis subtype, whether there is a benefit to intermittent therapy with respect to risk of toxicity and maintenance of efficacy, and how to minimize the bone-related changes that can occur with treatment. “These are some of the things that we can look further into,” she said. “For now, though, retinoids can improve function and quality of life in patients with ichthyosis and disorders of cornification. Many questions still exist, and more data and research are needed.”
Sun Pharmaceuticals and the Foundation for Ichthyosis and Related Skin Types (FIRST) provided an unrestricted grant for development of the recommendations.
Dr. Zaenglein disclosed that she is a consultant for Pfizer. She is also an advisory board member for Dermata, Sol-Gel, Regeneron, Verrica, and Cassiopea, and has conducted contracted research for AbbVie, Incyte, Arcutis, and Pfizer. The other authors disclosed serving as investigators, advisers, consultants, and/or had other relationships with various pharmaceutical companies.
FROM THE SPD PRE-AAD MEETING
VEXAS: A novel rheumatologic, hematologic syndrome that’s making waves
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
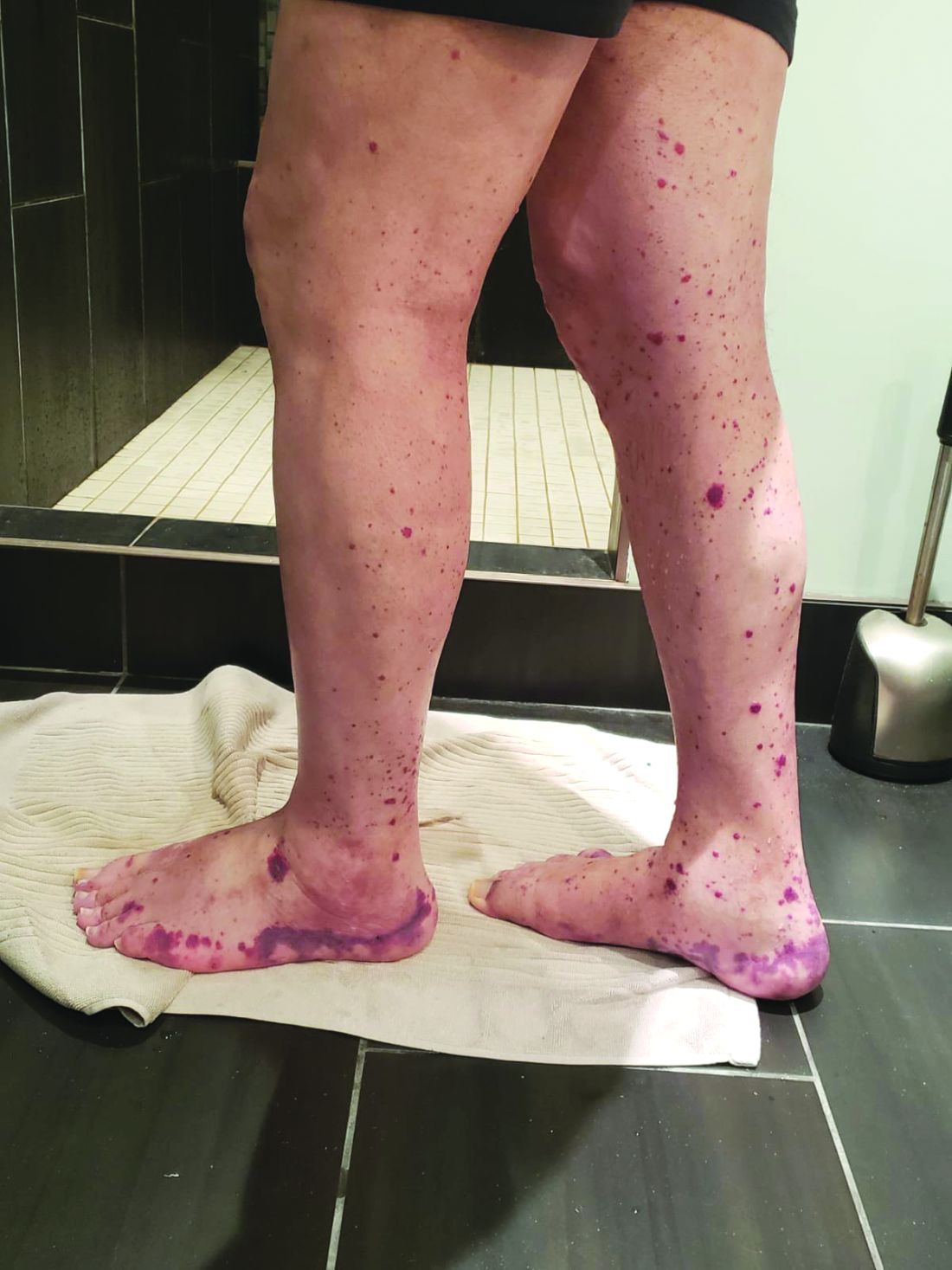
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
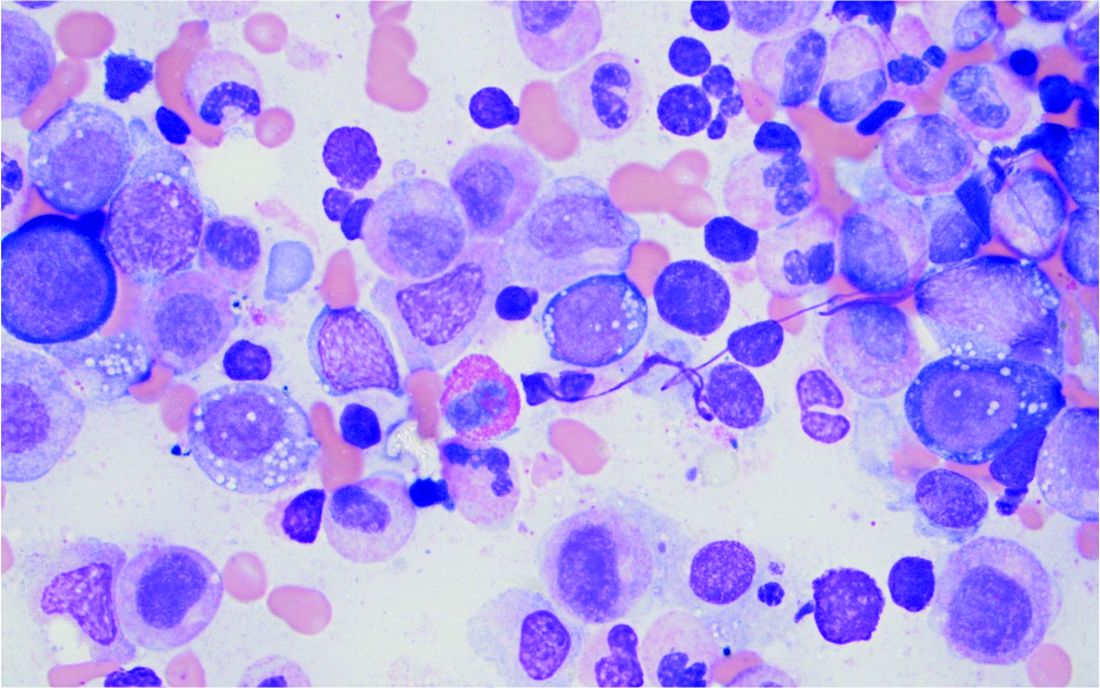
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Hyperphagia, anxiety eased with carbetocin in patients with Prader-Willi syndrome
Children and adolescents with Prader-Willi syndrome (PWS) who received three daily, intranasal doses of carbetocin, an investigational, long-acting oxytocin analogue, had significant improvement in hyperphagia and anxiety during 8 weeks on treatment, compared with placebo in a multicenter, phase 3 trial with 119 patients.

The treatment also appeared safe during up to 56 additional weeks on active treatment, with no serious adverse effects nor “unexpected” events, and once completing the study about 95% of enrolled patients opted to remain on active treatment, Cheri L. Deal, MD, PhD, said at the annual meeting of the Endocrine Society.
Based on “the significant results for the placebo-controlled period, as well as for those finishing the 56-week extension, we may well have a new armament for helping these kids and their families deal with the unrelenting hunger of patients with PWS as well as some of the behavioral symptoms,” Dr. Deal, chief of endocrinology and diabetes at the Sainte-Justine Mother-Child University of Montreal Hospital, said in an interview. No treatment currently has labeling for addressing the hyperphagia or anxiety that is characteristic and often problematic for children and adolescents with PWS, an autosomal dominant genetic disease with an incidence of about 1 in 15,000 births and an estimated U.S. prevalence of about 9,000 cases, or about 1 case for every 37,000 people.
‘Gorgeous’ safety
“The results looked pretty positive, and we’re encouraged by what appears to be a good safety profile, so overall I think the PWS community is very excited by the results and is very interested in getting access to this drug,” commented Theresa V. Strong, PhD, director of research programs for the Foundation for Prader-Willi Research in Walnut, Calif., a group not involved with the study. Currently, “we have no effective treatments for these difficult behaviors” of hyperphagia and anxiety. Surveys and studies run by the foundation have documented that hyperphagia and anxiety “were the two most important symptoms that families would like to see treated,” Dr. Strong added in an interview.
PWS “is complex and affects almost every aspect of the lives of affected people and their families. Any treatment that can chip away at some of the problems these patients have can be a huge benefit to the patients and their families,” said Jennifer L. Miller, MD, a professor of pediatric endocrinology at the University of Florida, Gainesville, and a coinvestigator on the study.
But the finding that carbetocin appeared to address, at least in part, this unmet need while compiling a safety record that Dr. Miller called “gorgeous” and “remarkable,” also came with a few limitations.
Fewer patients than planned, and muddled outcomes
The CARE-PWS trial aimed to enroll 175 patients, but fell short once the COVID-19 pandemic hit. Plus the trial had two prespecified primary endpoints – improvements in a measure of hyperphagia, and in a measure of obsessive and compulsive behaviors – specifically in the 40 patients who received the higher of the two dosages studied, 9.6 mg t.i.d. intranasally. Neither endpoint showed significant improvement among the patients on this dosage, compared with the 40 patients who received placebo, although both outcomes trended in the right direction in the actively treated patients.
The study’s positive results came in a secondary treatment group, 39 patients who received 3.2 mg t.i.d., also intranasally. This subgroup had significant benefit, compared with placebo, for reducing hyperphagia symptoms as measured on the Hyperphagia Questionnaire for Clinical Trials (HQ-CT) Total Score. After the first 8 weeks on treatment, patients on the lower carbetocin dosage had an average reduction in their HQ-CT score of greater than 5 points, more than double the reduction seen among control patients who received placebo.
Those on the 3.2-mg t.i.d. dosage also showed significant improvements, compared with placebo, for anxiety, measured by the PWS Anxiety and Distress Questionnaire Total Score, as well as on measures of clinical global impression of severity, and of clinical global impression of change. Like the higher-dosage patients the lower-dosage subgroup did not show a significant difference compared with placebo for the other primary endpoint, change in obsessive and compulsive behaviors as measured by the Children’s Yale-Brown Obsessive-Compulsive Scale Total Score, although also like the higher dosage the effect from the lower dosage trended toward benefit.
A further limitation was that, at the time of her report, presented in abstract OR16-3 at the meeting, Dr. Deal could only present complete 64-week follow-up for 72 patients, although this reassuringly showed that, as time on the 9.6-mg t.i.d. dosage continued beyond 8 weeks, patients gradually improved their HQ-CT response so that by 64 weeks on treatment their hyperphagia score had improved as much as in the patients who received the lower dosage.
In short, documented benefits occurred on the lower dosage, especially for clinically meaningful symptoms like hyperphagia and anxiety, but the study’s overall results were not fully consistent by statistical criteria.
Benefiting an unmet need?
“While it is regrettable that we did not get to 175 patients because of COVID-19, the dataset is significant enough for me to feel that the FDA [Food and Drug Administration] needs to take a very serious look and consider approval,” Dr. Deal said in an interview. “Once safety is assured, which I think it is, I can only hope that regulatory officials understand their unmet needs of this rare disease community and will allow the drug to move to the next stage.”
“This is a very rare disease, and having families participate in trials is really challenging,” especially while the COVID-19 pandemic continues, Dr. Strong said. For the pediatric and adolescent patients targeted in this study “it will take a while for COVID to go away and for families to feel safe again being in a trial, so a real concern is that a need for more clinical trials is not terribly feasible now. Given that the safety profile looked good and one dose seemed to have good efficacy, as long as the long-term data continue to look good we’d love for the FDA to look at the existing data and see whether there is a path forward.”
Dr. Miller highlighted the limitations of what the CARE-PWS findings show.
“Given that it was only an 8-week trial of drug against placebo, and the fact that the primary outcomes weren’t met for the higher dose, my thought is that potentially we need to study more patients for a longer period at the 3.2-mg dose,” she said. She acknowledged that the metric used in the study to assess obsessive and compulsive behaviors is “very difficult” to apply to patients with PWS because of uncertainties in scoring obsessions in patients “who are not very good at telling you what they’re thinking.” Plus, “it’s absolutely not a problem that we did not see an effect on obsession and compulsions if the treatment potentially improves anxiety and hyperphagia, which are very common.” A treatment that reliably reduces these symptoms “would be amazing,” Dr. Miller added.
“PWS is very rare, so it’s very hard to do trials. Maybe the FDA will approve carbetocin because it was safe and gave a signal of efficacy at the lower dose. But my thought is that additional treatment trials are needed with only the lower dose and with longer duration,” she said.
CARE-PWS enrolled patients with nutritional phase 3 PWS who were aged 7-18 years at any of 24 sites in the United States, Canada, or Australia during 2018-2020. They averaged about 12 years of age, and 56% were girls.
The most common adverse effect from carbetocin was flushing, occurring in 14% of those on the lower dose and 21% on the higher dose, but not in any placebo patient. Other adverse effects more common on the lower dose than in the placebo group included headache in 16%, and diarrhea in 9%.
Carbetocin is not only long-lasting in circulation, it also has better affinity for oxytocin receptors than for vasopressin receptors, reducing the potential for causing hyponatremia. The idea to use it in patients with PWS followed prior studies with oxytocin, which had shown dopamine interactions that reduced anxiety and influenced food ingestion behavior. Brain autopsy studies had shown that patients with Prader-Willi syndrome have substantially fewer neurons than usual producing oxytocin. Treatment with intranasal carbetocin had shown efficacy for improving hyperphagia and behavior in a controlled phase 2 study with 37 patients.
Carbetocin is approved for use in reducing excessive bleeding after childbirth, particularly cesarean, in more than 20 countries outside the United States.
CARE-PWS was sponsored by Levo Therapeutics, the company developing carbetocin. Dr. Deal has been an adviser to Levo Therapeutics. Dr. Strong is an employee of the Foundation for Prader-Willi Research, which has received support from Levo Therapeutics as well as from other drug companies, but which receives most of its funding from individuals. Dr. Miller has received research funding from Levo Therapeutics and also from Harmony Biosciences, Rhythm Pharmaceuticals, and Soleno Therapeutics.
Children and adolescents with Prader-Willi syndrome (PWS) who received three daily, intranasal doses of carbetocin, an investigational, long-acting oxytocin analogue, had significant improvement in hyperphagia and anxiety during 8 weeks on treatment, compared with placebo in a multicenter, phase 3 trial with 119 patients.
The treatment also appeared safe during up to 56 additional weeks on active treatment, with no serious adverse effects nor “unexpected” events, and once completing the study about 95% of enrolled patients opted to remain on active treatment, Cheri L. Deal, MD, PhD, said at the annual meeting of the Endocrine Society.
Based on “the significant results for the placebo-controlled period, as well as for those finishing the 56-week extension, we may well have a new armament for helping these kids and their families deal with the unrelenting hunger of patients with PWS as well as some of the behavioral symptoms,” Dr. Deal, chief of endocrinology and diabetes at the Sainte-Justine Mother-Child University of Montreal Hospital, said in an interview. No treatment currently has labeling for addressing the hyperphagia or anxiety that is characteristic and often problematic for children and adolescents with PWS, an autosomal dominant genetic disease with an incidence of about 1 in 15,000 births and an estimated U.S. prevalence of about 9,000 cases, or about 1 case for every 37,000 people.
‘Gorgeous’ safety
“The results looked pretty positive, and we’re encouraged by what appears to be a good safety profile, so overall I think the PWS community is very excited by the results and is very interested in getting access to this drug,” commented Theresa V. Strong, PhD, director of research programs for the Foundation for Prader-Willi Research in Walnut, Calif., a group not involved with the study. Currently, “we have no effective treatments for these difficult behaviors” of hyperphagia and anxiety. Surveys and studies run by the foundation have documented that hyperphagia and anxiety “were the two most important symptoms that families would like to see treated,” Dr. Strong added in an interview.
PWS “is complex and affects almost every aspect of the lives of affected people and their families. Any treatment that can chip away at some of the problems these patients have can be a huge benefit to the patients and their families,” said Jennifer L. Miller, MD, a professor of pediatric endocrinology at the University of Florida, Gainesville, and a coinvestigator on the study.
But the finding that carbetocin appeared to address, at least in part, this unmet need while compiling a safety record that Dr. Miller called “gorgeous” and “remarkable,” also came with a few limitations.
Fewer patients than planned, and muddled outcomes
The CARE-PWS trial aimed to enroll 175 patients, but fell short once the COVID-19 pandemic hit. Plus the trial had two prespecified primary endpoints – improvements in a measure of hyperphagia, and in a measure of obsessive and compulsive behaviors – specifically in the 40 patients who received the higher of the two dosages studied, 9.6 mg t.i.d. intranasally. Neither endpoint showed significant improvement among the patients on this dosage, compared with the 40 patients who received placebo, although both outcomes trended in the right direction in the actively treated patients.
The study’s positive results came in a secondary treatment group, 39 patients who received 3.2 mg t.i.d., also intranasally. This subgroup had significant benefit, compared with placebo, for reducing hyperphagia symptoms as measured on the Hyperphagia Questionnaire for Clinical Trials (HQ-CT) Total Score. After the first 8 weeks on treatment, patients on the lower carbetocin dosage had an average reduction in their HQ-CT score of greater than 5 points, more than double the reduction seen among control patients who received placebo.
Those on the 3.2-mg t.i.d. dosage also showed significant improvements, compared with placebo, for anxiety, measured by the PWS Anxiety and Distress Questionnaire Total Score, as well as on measures of clinical global impression of severity, and of clinical global impression of change. Like the higher-dosage patients the lower-dosage subgroup did not show a significant difference compared with placebo for the other primary endpoint, change in obsessive and compulsive behaviors as measured by the Children’s Yale-Brown Obsessive-Compulsive Scale Total Score, although also like the higher dosage the effect from the lower dosage trended toward benefit.
A further limitation was that, at the time of her report, presented in abstract OR16-3 at the meeting, Dr. Deal could only present complete 64-week follow-up for 72 patients, although this reassuringly showed that, as time on the 9.6-mg t.i.d. dosage continued beyond 8 weeks, patients gradually improved their HQ-CT response so that by 64 weeks on treatment their hyperphagia score had improved as much as in the patients who received the lower dosage.
In short, documented benefits occurred on the lower dosage, especially for clinically meaningful symptoms like hyperphagia and anxiety, but the study’s overall results were not fully consistent by statistical criteria.
Benefiting an unmet need?
“While it is regrettable that we did not get to 175 patients because of COVID-19, the dataset is significant enough for me to feel that the FDA [Food and Drug Administration] needs to take a very serious look and consider approval,” Dr. Deal said in an interview. “Once safety is assured, which I think it is, I can only hope that regulatory officials understand their unmet needs of this rare disease community and will allow the drug to move to the next stage.”
“This is a very rare disease, and having families participate in trials is really challenging,” especially while the COVID-19 pandemic continues, Dr. Strong said. For the pediatric and adolescent patients targeted in this study “it will take a while for COVID to go away and for families to feel safe again being in a trial, so a real concern is that a need for more clinical trials is not terribly feasible now. Given that the safety profile looked good and one dose seemed to have good efficacy, as long as the long-term data continue to look good we’d love for the FDA to look at the existing data and see whether there is a path forward.”
Dr. Miller highlighted the limitations of what the CARE-PWS findings show.
“Given that it was only an 8-week trial of drug against placebo, and the fact that the primary outcomes weren’t met for the higher dose, my thought is that potentially we need to study more patients for a longer period at the 3.2-mg dose,” she said. She acknowledged that the metric used in the study to assess obsessive and compulsive behaviors is “very difficult” to apply to patients with PWS because of uncertainties in scoring obsessions in patients “who are not very good at telling you what they’re thinking.” Plus, “it’s absolutely not a problem that we did not see an effect on obsession and compulsions if the treatment potentially improves anxiety and hyperphagia, which are very common.” A treatment that reliably reduces these symptoms “would be amazing,” Dr. Miller added.
“PWS is very rare, so it’s very hard to do trials. Maybe the FDA will approve carbetocin because it was safe and gave a signal of efficacy at the lower dose. But my thought is that additional treatment trials are needed with only the lower dose and with longer duration,” she said.
CARE-PWS enrolled patients with nutritional phase 3 PWS who were aged 7-18 years at any of 24 sites in the United States, Canada, or Australia during 2018-2020. They averaged about 12 years of age, and 56% were girls.
The most common adverse effect from carbetocin was flushing, occurring in 14% of those on the lower dose and 21% on the higher dose, but not in any placebo patient. Other adverse effects more common on the lower dose than in the placebo group included headache in 16%, and diarrhea in 9%.
Carbetocin is not only long-lasting in circulation, it also has better affinity for oxytocin receptors than for vasopressin receptors, reducing the potential for causing hyponatremia. The idea to use it in patients with PWS followed prior studies with oxytocin, which had shown dopamine interactions that reduced anxiety and influenced food ingestion behavior. Brain autopsy studies had shown that patients with Prader-Willi syndrome have substantially fewer neurons than usual producing oxytocin. Treatment with intranasal carbetocin had shown efficacy for improving hyperphagia and behavior in a controlled phase 2 study with 37 patients.
Carbetocin is approved for use in reducing excessive bleeding after childbirth, particularly cesarean, in more than 20 countries outside the United States.
CARE-PWS was sponsored by Levo Therapeutics, the company developing carbetocin. Dr. Deal has been an adviser to Levo Therapeutics. Dr. Strong is an employee of the Foundation for Prader-Willi Research, which has received support from Levo Therapeutics as well as from other drug companies, but which receives most of its funding from individuals. Dr. Miller has received research funding from Levo Therapeutics and also from Harmony Biosciences, Rhythm Pharmaceuticals, and Soleno Therapeutics.
Children and adolescents with Prader-Willi syndrome (PWS) who received three daily, intranasal doses of carbetocin, an investigational, long-acting oxytocin analogue, had significant improvement in hyperphagia and anxiety during 8 weeks on treatment, compared with placebo in a multicenter, phase 3 trial with 119 patients.
The treatment also appeared safe during up to 56 additional weeks on active treatment, with no serious adverse effects nor “unexpected” events, and once completing the study about 95% of enrolled patients opted to remain on active treatment, Cheri L. Deal, MD, PhD, said at the annual meeting of the Endocrine Society.
Based on “the significant results for the placebo-controlled period, as well as for those finishing the 56-week extension, we may well have a new armament for helping these kids and their families deal with the unrelenting hunger of patients with PWS as well as some of the behavioral symptoms,” Dr. Deal, chief of endocrinology and diabetes at the Sainte-Justine Mother-Child University of Montreal Hospital, said in an interview. No treatment currently has labeling for addressing the hyperphagia or anxiety that is characteristic and often problematic for children and adolescents with PWS, an autosomal dominant genetic disease with an incidence of about 1 in 15,000 births and an estimated U.S. prevalence of about 9,000 cases, or about 1 case for every 37,000 people.
‘Gorgeous’ safety
“The results looked pretty positive, and we’re encouraged by what appears to be a good safety profile, so overall I think the PWS community is very excited by the results and is very interested in getting access to this drug,” commented Theresa V. Strong, PhD, director of research programs for the Foundation for Prader-Willi Research in Walnut, Calif., a group not involved with the study. Currently, “we have no effective treatments for these difficult behaviors” of hyperphagia and anxiety. Surveys and studies run by the foundation have documented that hyperphagia and anxiety “were the two most important symptoms that families would like to see treated,” Dr. Strong added in an interview.
PWS “is complex and affects almost every aspect of the lives of affected people and their families. Any treatment that can chip away at some of the problems these patients have can be a huge benefit to the patients and their families,” said Jennifer L. Miller, MD, a professor of pediatric endocrinology at the University of Florida, Gainesville, and a coinvestigator on the study.
But the finding that carbetocin appeared to address, at least in part, this unmet need while compiling a safety record that Dr. Miller called “gorgeous” and “remarkable,” also came with a few limitations.
Fewer patients than planned, and muddled outcomes
The CARE-PWS trial aimed to enroll 175 patients, but fell short once the COVID-19 pandemic hit. Plus the trial had two prespecified primary endpoints – improvements in a measure of hyperphagia, and in a measure of obsessive and compulsive behaviors – specifically in the 40 patients who received the higher of the two dosages studied, 9.6 mg t.i.d. intranasally. Neither endpoint showed significant improvement among the patients on this dosage, compared with the 40 patients who received placebo, although both outcomes trended in the right direction in the actively treated patients.
The study’s positive results came in a secondary treatment group, 39 patients who received 3.2 mg t.i.d., also intranasally. This subgroup had significant benefit, compared with placebo, for reducing hyperphagia symptoms as measured on the Hyperphagia Questionnaire for Clinical Trials (HQ-CT) Total Score. After the first 8 weeks on treatment, patients on the lower carbetocin dosage had an average reduction in their HQ-CT score of greater than 5 points, more than double the reduction seen among control patients who received placebo.
Those on the 3.2-mg t.i.d. dosage also showed significant improvements, compared with placebo, for anxiety, measured by the PWS Anxiety and Distress Questionnaire Total Score, as well as on measures of clinical global impression of severity, and of clinical global impression of change. Like the higher-dosage patients the lower-dosage subgroup did not show a significant difference compared with placebo for the other primary endpoint, change in obsessive and compulsive behaviors as measured by the Children’s Yale-Brown Obsessive-Compulsive Scale Total Score, although also like the higher dosage the effect from the lower dosage trended toward benefit.
A further limitation was that, at the time of her report, presented in abstract OR16-3 at the meeting, Dr. Deal could only present complete 64-week follow-up for 72 patients, although this reassuringly showed that, as time on the 9.6-mg t.i.d. dosage continued beyond 8 weeks, patients gradually improved their HQ-CT response so that by 64 weeks on treatment their hyperphagia score had improved as much as in the patients who received the lower dosage.
In short, documented benefits occurred on the lower dosage, especially for clinically meaningful symptoms like hyperphagia and anxiety, but the study’s overall results were not fully consistent by statistical criteria.
Benefiting an unmet need?
“While it is regrettable that we did not get to 175 patients because of COVID-19, the dataset is significant enough for me to feel that the FDA [Food and Drug Administration] needs to take a very serious look and consider approval,” Dr. Deal said in an interview. “Once safety is assured, which I think it is, I can only hope that regulatory officials understand their unmet needs of this rare disease community and will allow the drug to move to the next stage.”
“This is a very rare disease, and having families participate in trials is really challenging,” especially while the COVID-19 pandemic continues, Dr. Strong said. For the pediatric and adolescent patients targeted in this study “it will take a while for COVID to go away and for families to feel safe again being in a trial, so a real concern is that a need for more clinical trials is not terribly feasible now. Given that the safety profile looked good and one dose seemed to have good efficacy, as long as the long-term data continue to look good we’d love for the FDA to look at the existing data and see whether there is a path forward.”
Dr. Miller highlighted the limitations of what the CARE-PWS findings show.
“Given that it was only an 8-week trial of drug against placebo, and the fact that the primary outcomes weren’t met for the higher dose, my thought is that potentially we need to study more patients for a longer period at the 3.2-mg dose,” she said. She acknowledged that the metric used in the study to assess obsessive and compulsive behaviors is “very difficult” to apply to patients with PWS because of uncertainties in scoring obsessions in patients “who are not very good at telling you what they’re thinking.” Plus, “it’s absolutely not a problem that we did not see an effect on obsession and compulsions if the treatment potentially improves anxiety and hyperphagia, which are very common.” A treatment that reliably reduces these symptoms “would be amazing,” Dr. Miller added.
“PWS is very rare, so it’s very hard to do trials. Maybe the FDA will approve carbetocin because it was safe and gave a signal of efficacy at the lower dose. But my thought is that additional treatment trials are needed with only the lower dose and with longer duration,” she said.
CARE-PWS enrolled patients with nutritional phase 3 PWS who were aged 7-18 years at any of 24 sites in the United States, Canada, or Australia during 2018-2020. They averaged about 12 years of age, and 56% were girls.
The most common adverse effect from carbetocin was flushing, occurring in 14% of those on the lower dose and 21% on the higher dose, but not in any placebo patient. Other adverse effects more common on the lower dose than in the placebo group included headache in 16%, and diarrhea in 9%.
Carbetocin is not only long-lasting in circulation, it also has better affinity for oxytocin receptors than for vasopressin receptors, reducing the potential for causing hyponatremia. The idea to use it in patients with PWS followed prior studies with oxytocin, which had shown dopamine interactions that reduced anxiety and influenced food ingestion behavior. Brain autopsy studies had shown that patients with Prader-Willi syndrome have substantially fewer neurons than usual producing oxytocin. Treatment with intranasal carbetocin had shown efficacy for improving hyperphagia and behavior in a controlled phase 2 study with 37 patients.
Carbetocin is approved for use in reducing excessive bleeding after childbirth, particularly cesarean, in more than 20 countries outside the United States.
CARE-PWS was sponsored by Levo Therapeutics, the company developing carbetocin. Dr. Deal has been an adviser to Levo Therapeutics. Dr. Strong is an employee of the Foundation for Prader-Willi Research, which has received support from Levo Therapeutics as well as from other drug companies, but which receives most of its funding from individuals. Dr. Miller has received research funding from Levo Therapeutics and also from Harmony Biosciences, Rhythm Pharmaceuticals, and Soleno Therapeutics.
FROM ENDO 2021
Enfuvirtide-Induced Cutaneous Amyloidosis
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
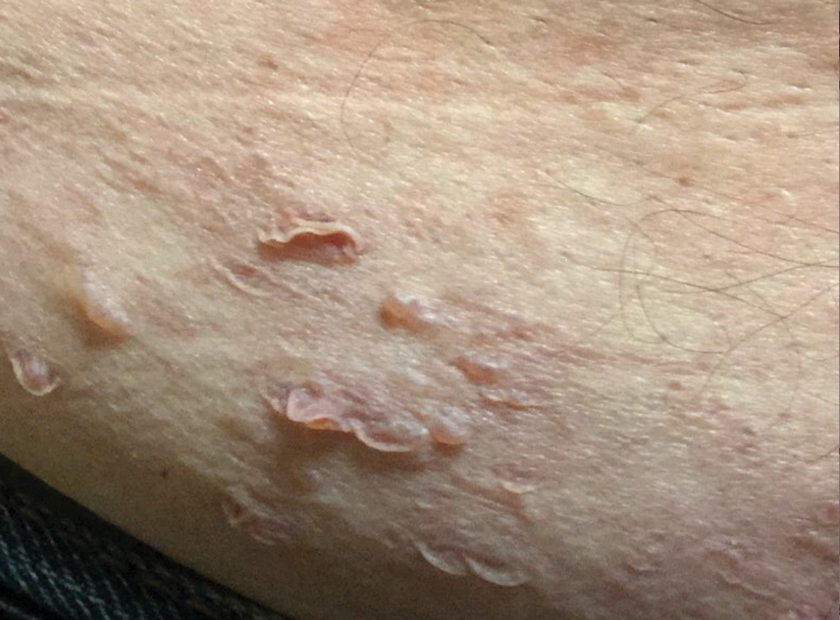
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
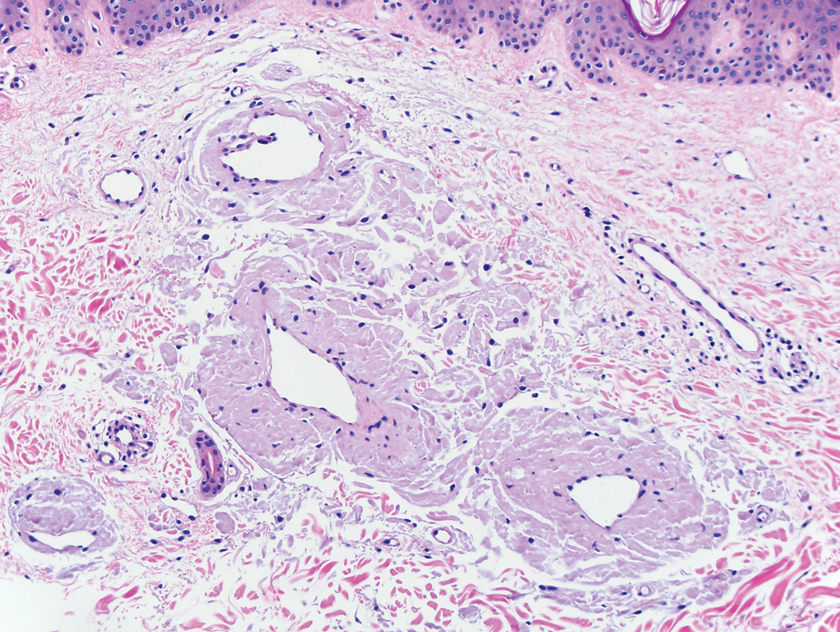
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
Practice Points
- There are multiple types of cutaneous amyloidosis, and proper diagnosis is essential to direct treatment and follow-up care.
- Medication-associated amyloidosis is a rare type of amyloidosis that is not associated with systemic amyloidosis and is treated by switching to alternative medicines.
Crusted Papules on the Bilateral Helices and Lobules
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.

Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
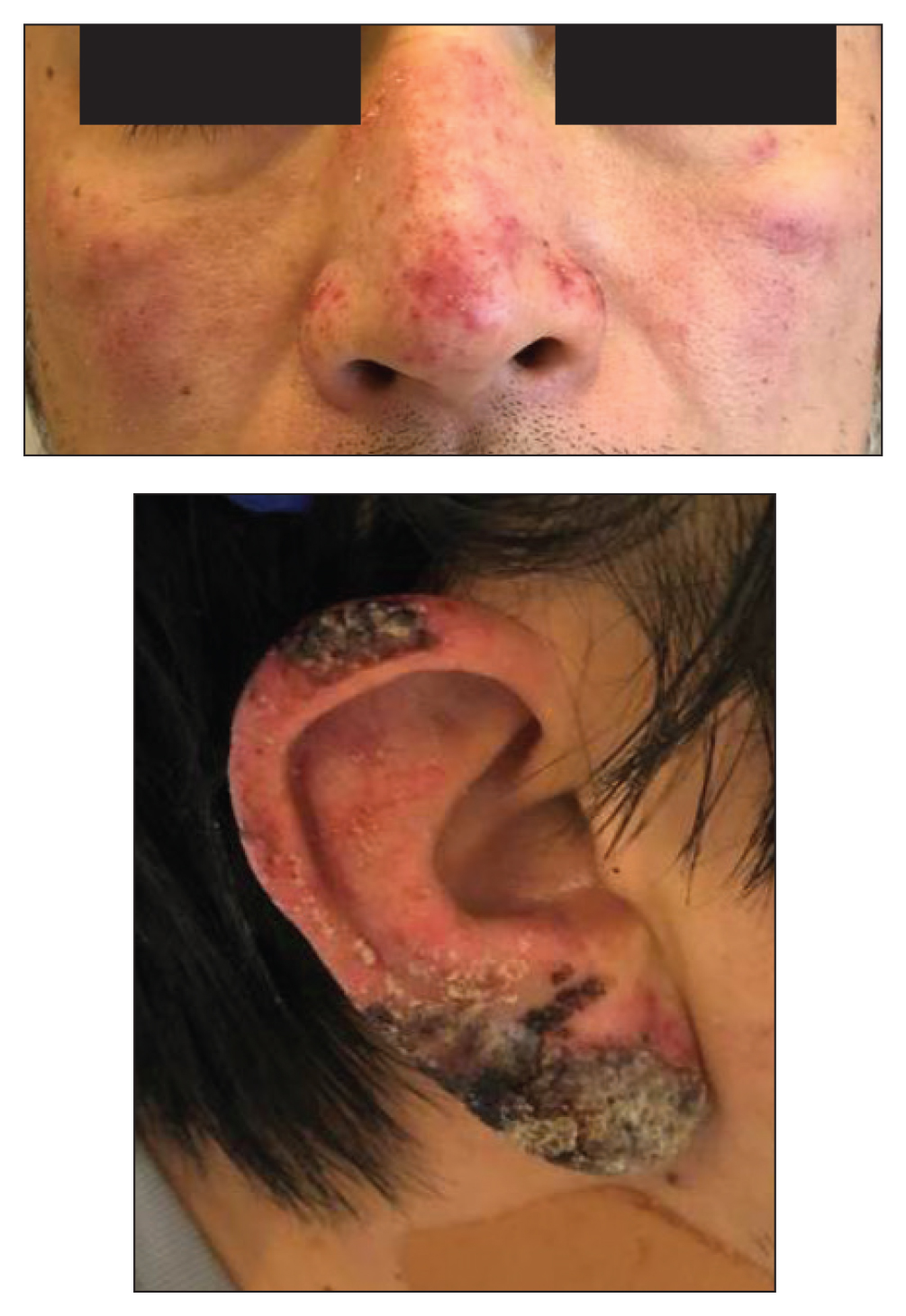
A healthy 42-year-old Japanese man presented with painful lymphadenopathy and fevers of 1 month’s duration as well as a pruritic rash and bilateral ear redness and crusting of 1 week’s duration. He initially was seen at an outside facility and was treated with antibiotics and supportive care for cervical adenitis. During clinical evaluation, he denied joint pain, photosensitivity, and oral lesions. His medical and family history were noncontributory. Although he reported recent travel to multiple countries, he denied exposure to animals, ticks, or sick individuals. Physical examination revealed erythematous blanching papules on the nose and cheeks (top) as well as crusted papules coalescing into plaques on the bilateral helices and lobules (bottom).
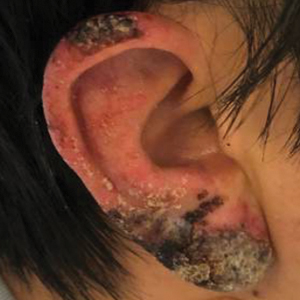
Success in achondroplasia spurs testing vosoritide in more growth disorders
On the basis of the quality of sustained bone growth achieved with vosoritide in dwarfism, studies are underway or being considered for more diseases that impair bone growth, according to discussion that followed the presentation of a phase 3 trial extension study at the annual meeting of the Endocrine Society.
After 1 year on active therapy in the randomized trial and another year in the extension study, patients in the vosoritide group had sustained growth velocity while placebo group patients who crossed over to active therapy caught up, reported Ravi Savarirayan, MD, Murdoch Children’s Research Institute, University of Melbourne, Australia.
Moreover, the quality and type of bone growth, such as the improvement in body segment ratios over the second year of the study, support a durable benefit. Dr. Savarirayan said that improvements in activities of daily living are expected from this improvement in upper-to-lower body segment ratios, as well as the growth seen in the limbs.
Currently there is no approved pharmacologic therapy for achondroplasia in the United States. Growth hormone has been approved in Japan, but Dr. Savarirayan said its effects have been limited. Surgery such as limb lengthening is another option, but this approach is not uniformly effective and carries risks.
The 52-week results from the multinational phase 3 trial with vosoritide, which stimulates bone growth, were published last year in The Lancet. In that trial, 121 patients between the ages of 5 and 18 years with achondroplasia were randomized to vosoritide at a dose of 15 μg/kg once daily or placebo.
Relative to those in the placebo arm, which did not experience any change in growth, the median growth at the end of 52 weeks was 1.75 cm/year greater (6.71 vs. 3.99 cm).
After crossover, placebo patients catch up
In the extension study, the placebo patients were crossed over to the active therapy and both groups were followed for an additional 52 weeks. Over this period, velocity declined modestly in those in the group initially randomized to vosoritide but climbed steeply in the placebo group so that rates after 1 year were nearly identical (5.57 vs. 5.65 cm, respectively).
“The results suggest this medication may well have a durable effect,” said Dr. Savarirayan, who believes that the benefit is derived from stimulation of the growth plates. Based on the very similar efficacy observed in the placebo group once switched to active therapy, the response to vosoritide appears to be predictable.
Of the 60 patients initially randomized to vosoritide, 58 entered the extension. Of the patients who did not remain in the study, two left due to discomfort from injection-site reactions. All 61 patients initially assigned to placebo crossed over.
“We did not see any evidence of tachyphylaxis in the randomized study or in the extension,” Dr. Savarirayan said.
Although two more patients initiated on vosoritide discontinued treatment before the end of 2 years, there were no new adverse events observed. Rather, injection-site pain, which self-resolved in all patients, appears to be the most significant side effect.
“In children, the daily subcutaneous injections can be an issue,” Dr. Savarirayan acknowledged.
Injection site reactions most common adverse event
In a detailed evaluation of safety in a previously published dose-finding phase 2 study, injection-site reactions were also the most common of treatment-related adverse events, but there were no episodes of anaphylaxis or other grade 3 or higher hypersensitivity reactions (N Engl J Med. 2019 Jul 4;381:25-35).
Prior to clinical trials, continuous infusion of endogenous C-type natriuretic peptide demonstrated an ability to stimulate long-bone growth in experimental studies. Vosoritide, a recombinant analogue of C-type natriuretic peptide, appears to provide the same activity but offers a longer half-life.
Based on the benefits observed in achondroplasia, other applications are now being explored.
“When you evaluate the quality of the bone growth associated with vosoritide, it is normal,” said Melita Irving, MD, a consultant in clinical genetics at the Guy’s and St .Thomas’ NHS Trust, London. Dr. Irving has been involved in other research initiatives with this therapy and she cited a variety of evidence that has supported healthy bone development, including favorable changes in markers of bone growth such as type 10 collagen.
As a result, vosoritide, which is now under review by the U.S. Food and Drug Administration for treatment of dwarfism, is being pursued for several other diseases that result in abnormal bone growth, such as hypochondroplasia. Not least, clinical studies in idiopathic short stature have reached “early stages,” Dr. Irving said.
Dr. Savarirayan and Dr. Irving report no relevant conflicts of interest.
On the basis of the quality of sustained bone growth achieved with vosoritide in dwarfism, studies are underway or being considered for more diseases that impair bone growth, according to discussion that followed the presentation of a phase 3 trial extension study at the annual meeting of the Endocrine Society.
After 1 year on active therapy in the randomized trial and another year in the extension study, patients in the vosoritide group had sustained growth velocity while placebo group patients who crossed over to active therapy caught up, reported Ravi Savarirayan, MD, Murdoch Children’s Research Institute, University of Melbourne, Australia.
Moreover, the quality and type of bone growth, such as the improvement in body segment ratios over the second year of the study, support a durable benefit. Dr. Savarirayan said that improvements in activities of daily living are expected from this improvement in upper-to-lower body segment ratios, as well as the growth seen in the limbs.
Currently there is no approved pharmacologic therapy for achondroplasia in the United States. Growth hormone has been approved in Japan, but Dr. Savarirayan said its effects have been limited. Surgery such as limb lengthening is another option, but this approach is not uniformly effective and carries risks.
The 52-week results from the multinational phase 3 trial with vosoritide, which stimulates bone growth, were published last year in The Lancet. In that trial, 121 patients between the ages of 5 and 18 years with achondroplasia were randomized to vosoritide at a dose of 15 μg/kg once daily or placebo.
Relative to those in the placebo arm, which did not experience any change in growth, the median growth at the end of 52 weeks was 1.75 cm/year greater (6.71 vs. 3.99 cm).
After crossover, placebo patients catch up
In the extension study, the placebo patients were crossed over to the active therapy and both groups were followed for an additional 52 weeks. Over this period, velocity declined modestly in those in the group initially randomized to vosoritide but climbed steeply in the placebo group so that rates after 1 year were nearly identical (5.57 vs. 5.65 cm, respectively).
“The results suggest this medication may well have a durable effect,” said Dr. Savarirayan, who believes that the benefit is derived from stimulation of the growth plates. Based on the very similar efficacy observed in the placebo group once switched to active therapy, the response to vosoritide appears to be predictable.
Of the 60 patients initially randomized to vosoritide, 58 entered the extension. Of the patients who did not remain in the study, two left due to discomfort from injection-site reactions. All 61 patients initially assigned to placebo crossed over.
“We did not see any evidence of tachyphylaxis in the randomized study or in the extension,” Dr. Savarirayan said.
Although two more patients initiated on vosoritide discontinued treatment before the end of 2 years, there were no new adverse events observed. Rather, injection-site pain, which self-resolved in all patients, appears to be the most significant side effect.
“In children, the daily subcutaneous injections can be an issue,” Dr. Savarirayan acknowledged.
Injection site reactions most common adverse event
In a detailed evaluation of safety in a previously published dose-finding phase 2 study, injection-site reactions were also the most common of treatment-related adverse events, but there were no episodes of anaphylaxis or other grade 3 or higher hypersensitivity reactions (N Engl J Med. 2019 Jul 4;381:25-35).
Prior to clinical trials, continuous infusion of endogenous C-type natriuretic peptide demonstrated an ability to stimulate long-bone growth in experimental studies. Vosoritide, a recombinant analogue of C-type natriuretic peptide, appears to provide the same activity but offers a longer half-life.
Based on the benefits observed in achondroplasia, other applications are now being explored.
“When you evaluate the quality of the bone growth associated with vosoritide, it is normal,” said Melita Irving, MD, a consultant in clinical genetics at the Guy’s and St .Thomas’ NHS Trust, London. Dr. Irving has been involved in other research initiatives with this therapy and she cited a variety of evidence that has supported healthy bone development, including favorable changes in markers of bone growth such as type 10 collagen.
As a result, vosoritide, which is now under review by the U.S. Food and Drug Administration for treatment of dwarfism, is being pursued for several other diseases that result in abnormal bone growth, such as hypochondroplasia. Not least, clinical studies in idiopathic short stature have reached “early stages,” Dr. Irving said.
Dr. Savarirayan and Dr. Irving report no relevant conflicts of interest.
On the basis of the quality of sustained bone growth achieved with vosoritide in dwarfism, studies are underway or being considered for more diseases that impair bone growth, according to discussion that followed the presentation of a phase 3 trial extension study at the annual meeting of the Endocrine Society.
After 1 year on active therapy in the randomized trial and another year in the extension study, patients in the vosoritide group had sustained growth velocity while placebo group patients who crossed over to active therapy caught up, reported Ravi Savarirayan, MD, Murdoch Children’s Research Institute, University of Melbourne, Australia.
Moreover, the quality and type of bone growth, such as the improvement in body segment ratios over the second year of the study, support a durable benefit. Dr. Savarirayan said that improvements in activities of daily living are expected from this improvement in upper-to-lower body segment ratios, as well as the growth seen in the limbs.
Currently there is no approved pharmacologic therapy for achondroplasia in the United States. Growth hormone has been approved in Japan, but Dr. Savarirayan said its effects have been limited. Surgery such as limb lengthening is another option, but this approach is not uniformly effective and carries risks.
The 52-week results from the multinational phase 3 trial with vosoritide, which stimulates bone growth, were published last year in The Lancet. In that trial, 121 patients between the ages of 5 and 18 years with achondroplasia were randomized to vosoritide at a dose of 15 μg/kg once daily or placebo.
Relative to those in the placebo arm, which did not experience any change in growth, the median growth at the end of 52 weeks was 1.75 cm/year greater (6.71 vs. 3.99 cm).
After crossover, placebo patients catch up
In the extension study, the placebo patients were crossed over to the active therapy and both groups were followed for an additional 52 weeks. Over this period, velocity declined modestly in those in the group initially randomized to vosoritide but climbed steeply in the placebo group so that rates after 1 year were nearly identical (5.57 vs. 5.65 cm, respectively).
“The results suggest this medication may well have a durable effect,” said Dr. Savarirayan, who believes that the benefit is derived from stimulation of the growth plates. Based on the very similar efficacy observed in the placebo group once switched to active therapy, the response to vosoritide appears to be predictable.
Of the 60 patients initially randomized to vosoritide, 58 entered the extension. Of the patients who did not remain in the study, two left due to discomfort from injection-site reactions. All 61 patients initially assigned to placebo crossed over.
“We did not see any evidence of tachyphylaxis in the randomized study or in the extension,” Dr. Savarirayan said.
Although two more patients initiated on vosoritide discontinued treatment before the end of 2 years, there were no new adverse events observed. Rather, injection-site pain, which self-resolved in all patients, appears to be the most significant side effect.
“In children, the daily subcutaneous injections can be an issue,” Dr. Savarirayan acknowledged.
Injection site reactions most common adverse event
In a detailed evaluation of safety in a previously published dose-finding phase 2 study, injection-site reactions were also the most common of treatment-related adverse events, but there were no episodes of anaphylaxis or other grade 3 or higher hypersensitivity reactions (N Engl J Med. 2019 Jul 4;381:25-35).
Prior to clinical trials, continuous infusion of endogenous C-type natriuretic peptide demonstrated an ability to stimulate long-bone growth in experimental studies. Vosoritide, a recombinant analogue of C-type natriuretic peptide, appears to provide the same activity but offers a longer half-life.
Based on the benefits observed in achondroplasia, other applications are now being explored.
“When you evaluate the quality of the bone growth associated with vosoritide, it is normal,” said Melita Irving, MD, a consultant in clinical genetics at the Guy’s and St .Thomas’ NHS Trust, London. Dr. Irving has been involved in other research initiatives with this therapy and she cited a variety of evidence that has supported healthy bone development, including favorable changes in markers of bone growth such as type 10 collagen.
As a result, vosoritide, which is now under review by the U.S. Food and Drug Administration for treatment of dwarfism, is being pursued for several other diseases that result in abnormal bone growth, such as hypochondroplasia. Not least, clinical studies in idiopathic short stature have reached “early stages,” Dr. Irving said.
Dr. Savarirayan and Dr. Irving report no relevant conflicts of interest.
FROM ENDO 2021
This Rash Really Stinks!
ANSWER
The correct diagnosis is Darier disease (choice “d”).
DISCUSSION
Darier disease, also known as Darier-White disease or keratosis follicularis, is an inherited defect transmitted by autosomal dominant mode. The pathophysiologic process is a breakdown of cell adhesion that normally binds keratin filaments to tiny connecting fibers called desmosomes.
Darier disease manifests with a “branny” papulosquamous rash, typically arising in the third decade of life and affecting the chest, scalp, back, and intertriginous areas. The nail and intraoral findings noted in this patient are typical. In the author’s experience, the former is more commonly seen and is essentially pathognomic for the disease.
Darier disease is relatively rare, occurring in 1:30,000 to 1:100,000 population, depending on the geographic area studied. Men and women are equally affected, although it is more common in those with darker skin.
The differential outlined in the answer choices is reasonable, considering the condition’s rarity and how unlikely it is to manifest solely in the inframammary area. One could conclude that, just as with psoriasis (choice “b”) and seborrhea, intertrigo (choice “c”) is not always a primary process. And although yeast infection (choice “a”) can complicate any florid rash in this area, topical and oral anti-yeast treatment had utterly failed to help.
TREATMENT
Isotretinoin is used in cases such as this one, but it only offers temporary relief. For less severe cases, oral antibiotics (eg minocycline) or topical steroids (used with caution given the risk for atrophy in the inframammary area) often suffice. This patient’s prognosis is guarded at best, although control of the worst is certainly possible.
ANSWER
The correct diagnosis is Darier disease (choice “d”).
DISCUSSION
Darier disease, also known as Darier-White disease or keratosis follicularis, is an inherited defect transmitted by autosomal dominant mode. The pathophysiologic process is a breakdown of cell adhesion that normally binds keratin filaments to tiny connecting fibers called desmosomes.
Darier disease manifests with a “branny” papulosquamous rash, typically arising in the third decade of life and affecting the chest, scalp, back, and intertriginous areas. The nail and intraoral findings noted in this patient are typical. In the author’s experience, the former is more commonly seen and is essentially pathognomic for the disease.
Darier disease is relatively rare, occurring in 1:30,000 to 1:100,000 population, depending on the geographic area studied. Men and women are equally affected, although it is more common in those with darker skin.
The differential outlined in the answer choices is reasonable, considering the condition’s rarity and how unlikely it is to manifest solely in the inframammary area. One could conclude that, just as with psoriasis (choice “b”) and seborrhea, intertrigo (choice “c”) is not always a primary process. And although yeast infection (choice “a”) can complicate any florid rash in this area, topical and oral anti-yeast treatment had utterly failed to help.
TREATMENT
Isotretinoin is used in cases such as this one, but it only offers temporary relief. For less severe cases, oral antibiotics (eg minocycline) or topical steroids (used with caution given the risk for atrophy in the inframammary area) often suffice. This patient’s prognosis is guarded at best, although control of the worst is certainly possible.
ANSWER
The correct diagnosis is Darier disease (choice “d”).
DISCUSSION
Darier disease, also known as Darier-White disease or keratosis follicularis, is an inherited defect transmitted by autosomal dominant mode. The pathophysiologic process is a breakdown of cell adhesion that normally binds keratin filaments to tiny connecting fibers called desmosomes.
Darier disease manifests with a “branny” papulosquamous rash, typically arising in the third decade of life and affecting the chest, scalp, back, and intertriginous areas. The nail and intraoral findings noted in this patient are typical. In the author’s experience, the former is more commonly seen and is essentially pathognomic for the disease.
Darier disease is relatively rare, occurring in 1:30,000 to 1:100,000 population, depending on the geographic area studied. Men and women are equally affected, although it is more common in those with darker skin.
The differential outlined in the answer choices is reasonable, considering the condition’s rarity and how unlikely it is to manifest solely in the inframammary area. One could conclude that, just as with psoriasis (choice “b”) and seborrhea, intertrigo (choice “c”) is not always a primary process. And although yeast infection (choice “a”) can complicate any florid rash in this area, topical and oral anti-yeast treatment had utterly failed to help.
TREATMENT
Isotretinoin is used in cases such as this one, but it only offers temporary relief. For less severe cases, oral antibiotics (eg minocycline) or topical steroids (used with caution given the risk for atrophy in the inframammary area) often suffice. This patient’s prognosis is guarded at best, although control of the worst is certainly possible.

A 50-year-old woman is referred to dermatology with a “yeast” infection of several years’ duration. The condition causes considerable discomfort, especially during hot weather when the rash emits a very objectionable odor.
The florid, white, scaly rash under her breasts is a stark contrast to the patient’s type V skin. On both sides, the affected skin perfectly matches the inframammary fold. There are sharp margins and uniform moist scaling.
Looking elsewhere, 7 of 10 fingernails exhibit longitudinal white and red streaks, along with triangular nicks in the edges of several nails. The roof of the patient’s mouth is studded with fleshy nodules measuring 0.6 to 1.0 cm. Several pits are seen on her palms.
The patient is in no distress but is quite agitated by the lack of effective treatment. She reports trying a number of prescription and OTC anti-yeast creams, lotions, and oral medications, none of which resolved the problem.
History-taking reveals a family history of skin problems, although neither the patient nor anyone else in the family has ever been seen by a dermatologist. No one has ever suggested that a biopsy be done.
A punch biopsy is performed on the affected inframammary skin. The pathology report shows acantholysis with focal dyskeratotic keratinocytes. Intraepidermal separation is seen throughout the specimen.

Thick Hyperkeratotic Plaques on the Palms and Soles
The Diagnosis: Keratoderma Climactericum
Keratoderma climactericum was first reported in 1934 by Haxthausen1 as nonpruritic circumscribed hyperkeratosis located mainly on the palms and soles. The initial eruption was described as discrete lesions with an oval or round shape that progressed to less-defined, confluent, hyperkeratotic patches with fissures.1 Keratoderma climactericum also may be referred to as Haxthausen disease and is considered an acquired palmoplantar keratoderma.2
Keratoderma climactericum is a rare dermatologic disorder that presents in women of menopausal age who have no family or personal history of skin disease. Keratoderma climactericum is associated with hypertension and obesity.2 Keratotic lesions usually first occur on the plantar surfaces with eventual development of fissuring and hyperkeratosis that causes painful walking. The keratotic lesions on the plantar surfaces often are nonpruritic and gradually become confluent over time. As the disease progresses, keratotic lesions appear on the central palms, which can lead to confluent hyperkeratosis on the palmar surfaces (Figure 1).2 The exact mechanism of keratoderma climactericum has not been described but is believed to be due to hormonal dysregulation.2
In 1986, Deschamps et al3 presented 10 cases of keratoderma climactericum occurring in menopausal women with an average age of 57 years. The lesions began on the soles at areas of greatest pressure. Histopathology for each patient showed orthokeratotic hyperkeratosis, irregular hyperplasia, interpapillary ridges, and exocytosis of lymphocytes in the epidermis. Seven patients were treated with etretinate, which first led to the removal of palmar lesions, followed by improvement in plantar lesions and pain when walking. There was no association of keratoderma climactericum and sex hormones, as hormone levels were negative or normal for each patient.3
Three cases of keratoderma climactericum following bilateral oophorectomy in young women were reported by Wachtel4 in 1981. Unlike in women of menopausal age, there was no association of keratoderma climactericum with hypertension or obesity. Additionally, the lesions on the palms and soles were more diffusely distributed than in women of menopausal age. Estrogen administration completely reversed each patient's hyperkeratotic palms and soles.4 A definitive pathogenic role of estrogens in the development of keratoderma climactericum has yet to be determined.2
Histopathology is not specific for keratoderma climactericum, making the disease a clinical diagnosis. However, a biopsy may be useful to rule out palmoplantar psoriasis.2 Clinical information such as the age and sex of the patient, distribution of disease, presence of fissuring, and progression of disease from soles to palms should be considered when making a diagnosis of keratoderma climactericum. The differential diagnosis of keratoderma climactericum should include fungal infections, contact dermatitis, irritant dermatitis, psoriasis, atopic dermatitis, underlying malignancy, and pityriasis rubra pilaris.
Treatment options for keratoderma climactericum include salicylic acid, emollients, oral retinoids, urea ointments, estriol cream, and topical steroids.5,6 Our patient was prescribed acitretin 25 mg daily and ammonium lactate to apply topically as needed for dry skin. Five months after the initial presentation, fissures and dry skin on the bilateral soles were still present. Ammonium lactate was discontinued, and the patient was prescribed urea cream 40%. Fifteen months after the initial presentation, the patient reported substantial improvement on the hands and feet and noted that she no longer needed the urea cream. Physical examination revealed no presence of hyperkeratosis or fissuring on the palms (Figure 2), and mild hyperkeratosis was present on the plantar surfaces of the feet (Figure 3). The patient continued to use acitretin to prevent disease relapse.
Keratoderma climactericum is an unusual and debilitating condition that occurs in women of menopausal age. It is diagnosed by its specific clinical presentation. More common diagnoses such as tinea and dermatitis should be ruled out before considering keratoderma climactericum.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol. 1934;46:161-167.
- Patel S, Zirwas M, English JC. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica. 1986;172:258-262.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol. 1981;20:270-271.
- Bristow I. The management of heel fissures using a steroid impregnated tape (Haelan) in a patient with Keratoderma climactericum. Podiatry Now. 2008;11:22-23.
- Mendes-Bastos P. Plantar keratoderma climactericum: successful improvement with a topical estriol cream. J Cosmet Dermatol. 2018;17:811-813.
The Diagnosis: Keratoderma Climactericum
Keratoderma climactericum was first reported in 1934 by Haxthausen1 as nonpruritic circumscribed hyperkeratosis located mainly on the palms and soles. The initial eruption was described as discrete lesions with an oval or round shape that progressed to less-defined, confluent, hyperkeratotic patches with fissures.1 Keratoderma climactericum also may be referred to as Haxthausen disease and is considered an acquired palmoplantar keratoderma.2
Keratoderma climactericum is a rare dermatologic disorder that presents in women of menopausal age who have no family or personal history of skin disease. Keratoderma climactericum is associated with hypertension and obesity.2 Keratotic lesions usually first occur on the plantar surfaces with eventual development of fissuring and hyperkeratosis that causes painful walking. The keratotic lesions on the plantar surfaces often are nonpruritic and gradually become confluent over time. As the disease progresses, keratotic lesions appear on the central palms, which can lead to confluent hyperkeratosis on the palmar surfaces (Figure 1).2 The exact mechanism of keratoderma climactericum has not been described but is believed to be due to hormonal dysregulation.2
In 1986, Deschamps et al3 presented 10 cases of keratoderma climactericum occurring in menopausal women with an average age of 57 years. The lesions began on the soles at areas of greatest pressure. Histopathology for each patient showed orthokeratotic hyperkeratosis, irregular hyperplasia, interpapillary ridges, and exocytosis of lymphocytes in the epidermis. Seven patients were treated with etretinate, which first led to the removal of palmar lesions, followed by improvement in plantar lesions and pain when walking. There was no association of keratoderma climactericum and sex hormones, as hormone levels were negative or normal for each patient.3
Three cases of keratoderma climactericum following bilateral oophorectomy in young women were reported by Wachtel4 in 1981. Unlike in women of menopausal age, there was no association of keratoderma climactericum with hypertension or obesity. Additionally, the lesions on the palms and soles were more diffusely distributed than in women of menopausal age. Estrogen administration completely reversed each patient's hyperkeratotic palms and soles.4 A definitive pathogenic role of estrogens in the development of keratoderma climactericum has yet to be determined.2
Histopathology is not specific for keratoderma climactericum, making the disease a clinical diagnosis. However, a biopsy may be useful to rule out palmoplantar psoriasis.2 Clinical information such as the age and sex of the patient, distribution of disease, presence of fissuring, and progression of disease from soles to palms should be considered when making a diagnosis of keratoderma climactericum. The differential diagnosis of keratoderma climactericum should include fungal infections, contact dermatitis, irritant dermatitis, psoriasis, atopic dermatitis, underlying malignancy, and pityriasis rubra pilaris.
Treatment options for keratoderma climactericum include salicylic acid, emollients, oral retinoids, urea ointments, estriol cream, and topical steroids.5,6 Our patient was prescribed acitretin 25 mg daily and ammonium lactate to apply topically as needed for dry skin. Five months after the initial presentation, fissures and dry skin on the bilateral soles were still present. Ammonium lactate was discontinued, and the patient was prescribed urea cream 40%. Fifteen months after the initial presentation, the patient reported substantial improvement on the hands and feet and noted that she no longer needed the urea cream. Physical examination revealed no presence of hyperkeratosis or fissuring on the palms (Figure 2), and mild hyperkeratosis was present on the plantar surfaces of the feet (Figure 3). The patient continued to use acitretin to prevent disease relapse.
Keratoderma climactericum is an unusual and debilitating condition that occurs in women of menopausal age. It is diagnosed by its specific clinical presentation. More common diagnoses such as tinea and dermatitis should be ruled out before considering keratoderma climactericum.
The Diagnosis: Keratoderma Climactericum
Keratoderma climactericum was first reported in 1934 by Haxthausen1 as nonpruritic circumscribed hyperkeratosis located mainly on the palms and soles. The initial eruption was described as discrete lesions with an oval or round shape that progressed to less-defined, confluent, hyperkeratotic patches with fissures.1 Keratoderma climactericum also may be referred to as Haxthausen disease and is considered an acquired palmoplantar keratoderma.2
Keratoderma climactericum is a rare dermatologic disorder that presents in women of menopausal age who have no family or personal history of skin disease. Keratoderma climactericum is associated with hypertension and obesity.2 Keratotic lesions usually first occur on the plantar surfaces with eventual development of fissuring and hyperkeratosis that causes painful walking. The keratotic lesions on the plantar surfaces often are nonpruritic and gradually become confluent over time. As the disease progresses, keratotic lesions appear on the central palms, which can lead to confluent hyperkeratosis on the palmar surfaces (Figure 1).2 The exact mechanism of keratoderma climactericum has not been described but is believed to be due to hormonal dysregulation.2
In 1986, Deschamps et al3 presented 10 cases of keratoderma climactericum occurring in menopausal women with an average age of 57 years. The lesions began on the soles at areas of greatest pressure. Histopathology for each patient showed orthokeratotic hyperkeratosis, irregular hyperplasia, interpapillary ridges, and exocytosis of lymphocytes in the epidermis. Seven patients were treated with etretinate, which first led to the removal of palmar lesions, followed by improvement in plantar lesions and pain when walking. There was no association of keratoderma climactericum and sex hormones, as hormone levels were negative or normal for each patient.3
Three cases of keratoderma climactericum following bilateral oophorectomy in young women were reported by Wachtel4 in 1981. Unlike in women of menopausal age, there was no association of keratoderma climactericum with hypertension or obesity. Additionally, the lesions on the palms and soles were more diffusely distributed than in women of menopausal age. Estrogen administration completely reversed each patient's hyperkeratotic palms and soles.4 A definitive pathogenic role of estrogens in the development of keratoderma climactericum has yet to be determined.2
Histopathology is not specific for keratoderma climactericum, making the disease a clinical diagnosis. However, a biopsy may be useful to rule out palmoplantar psoriasis.2 Clinical information such as the age and sex of the patient, distribution of disease, presence of fissuring, and progression of disease from soles to palms should be considered when making a diagnosis of keratoderma climactericum. The differential diagnosis of keratoderma climactericum should include fungal infections, contact dermatitis, irritant dermatitis, psoriasis, atopic dermatitis, underlying malignancy, and pityriasis rubra pilaris.
Treatment options for keratoderma climactericum include salicylic acid, emollients, oral retinoids, urea ointments, estriol cream, and topical steroids.5,6 Our patient was prescribed acitretin 25 mg daily and ammonium lactate to apply topically as needed for dry skin. Five months after the initial presentation, fissures and dry skin on the bilateral soles were still present. Ammonium lactate was discontinued, and the patient was prescribed urea cream 40%. Fifteen months after the initial presentation, the patient reported substantial improvement on the hands and feet and noted that she no longer needed the urea cream. Physical examination revealed no presence of hyperkeratosis or fissuring on the palms (Figure 2), and mild hyperkeratosis was present on the plantar surfaces of the feet (Figure 3). The patient continued to use acitretin to prevent disease relapse.
Keratoderma climactericum is an unusual and debilitating condition that occurs in women of menopausal age. It is diagnosed by its specific clinical presentation. More common diagnoses such as tinea and dermatitis should be ruled out before considering keratoderma climactericum.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol. 1934;46:161-167.
- Patel S, Zirwas M, English JC. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica. 1986;172:258-262.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol. 1981;20:270-271.
- Bristow I. The management of heel fissures using a steroid impregnated tape (Haelan) in a patient with Keratoderma climactericum. Podiatry Now. 2008;11:22-23.
- Mendes-Bastos P. Plantar keratoderma climactericum: successful improvement with a topical estriol cream. J Cosmet Dermatol. 2018;17:811-813.
- Haxthausen H. Keratoderma climactericum. Br J Dermatol. 1934;46:161-167.
- Patel S, Zirwas M, English JC. Acquired palmoplantar keratoderma. Am J Clin Dermatol. 2007;8:1-11.
- Deschamps P, Leroy D, Pedailles S, et al. Keratoderma climactericum (Haxthausen's disease): clinical signs, laboratory findings and etretinate treatment in 10 patients. Dermatologica. 1986;172:258-262.
- Wachtel TJ. Plantar and palmar hyperkeratosis in young castrated women. Int J Dermatol. 1981;20:270-271.
- Bristow I. The management of heel fissures using a steroid impregnated tape (Haelan) in a patient with Keratoderma climactericum. Podiatry Now. 2008;11:22-23.
- Mendes-Bastos P. Plantar keratoderma climactericum: successful improvement with a topical estriol cream. J Cosmet Dermatol. 2018;17:811-813.
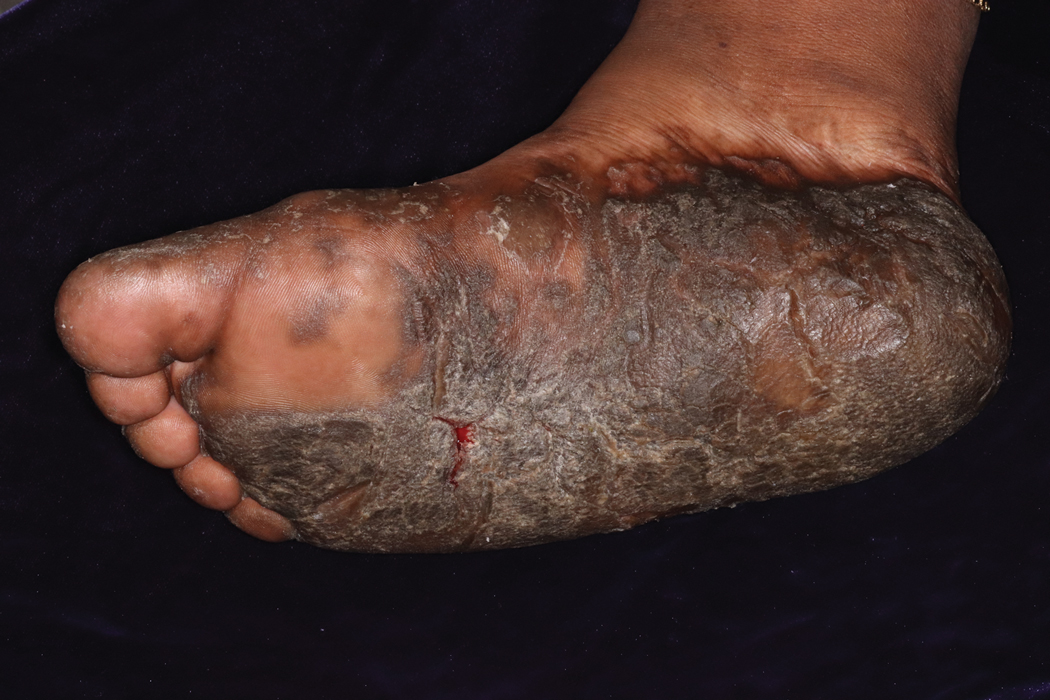
A 52-year-old woman with a history of rheumatoid arthritis presented with a rash on the palms and soles of 7 years' duration that started around the onset of menopause. Physical examination revealed thick hyperkeratotic plaques with multiple deep fissures on the palms and soles. The patient's current medications included methotrexate for rheumatoid arthritis. She previously had been prescribed adalimumab by an outside physician for the rash, which provided no relief, and currently was using urea ointment, which caused a burning sensation on the palms and soles. The patient denied a personal or family history of psoriasis.