User login
FDA OKs first drug for Rett syndrome
Rett syndrome is a rare, genetic neurodevelopmental disorder that affects about 6,000-9,000 people in the United States, mostly females.
Symptoms typically present between 6 and 18 months of age, with patients experiencing a rapid decline with loss of fine motor and communication skills.
Trofinetide is a synthetic analogue of the amino-terminal tripeptide of insulinlike growth factor-1 (IGF-1), which occurs naturally in the brain. The drug is designed to treat the core symptoms of Rett syndrome by potentially reducing neuroinflammation and supporting synaptic function.
The approval of trofinetide was supported by results from the pivotal phase 3 LAVENDER study that tested the efficacy and safety of trofinetide vs. placebo in 187 female patients with Rett syndrome, aged 5-20 years.
A total of 93 participants were randomly assigned to twice-daily oral trofinetide, and 94 received placebo for 12 weeks.
After 12 weeks, trofinetide showed a statistically significant improvement from baseline, compared with placebo, on both the caregiver-assessed Rett Syndrome Behavior Questionnaire (RSBQ) and 7-point Clinical Global Impression-Improvement (CGI-I) scale.
The drug also outperformed placebo at 12 weeks in a key secondary endpoint: the composite score on the Communication and Symbolic Behavior Scales Developmental Profile Infant-Toddler Checklist-Social (CSBS-DP-IT Social), a scale on which caregivers assess nonverbal communication.
The most common adverse events with trofinetide treatment were diarrhea and vomiting. Almost all these events were considered mild or moderate.
‘Historic day’
“This is a historic day for the Rett syndrome community and a meaningful moment for the patients and caregivers who have eagerly awaited the arrival of an approved treatment for this condition,” Melissa Kennedy, MHA, chief executive officer of the International Rett Syndrome Foundation, said in a news release issued by Acadia.
“Rett syndrome is a complicated, devastating disease that affects not only the individual patient, but whole families. With today’s FDA decision, those impacted by Rett have a promising new treatment option that has demonstrated benefit across a variety of Rett symptoms, including those that impact the daily lives of those living with Rett and their loved ones,” Ms. Kennedy said.
Trofinetide is expected to be available in the United States by the end of April.
A version of this article first appeared on Medscape.com.
Rett syndrome is a rare, genetic neurodevelopmental disorder that affects about 6,000-9,000 people in the United States, mostly females.
Symptoms typically present between 6 and 18 months of age, with patients experiencing a rapid decline with loss of fine motor and communication skills.
Trofinetide is a synthetic analogue of the amino-terminal tripeptide of insulinlike growth factor-1 (IGF-1), which occurs naturally in the brain. The drug is designed to treat the core symptoms of Rett syndrome by potentially reducing neuroinflammation and supporting synaptic function.
The approval of trofinetide was supported by results from the pivotal phase 3 LAVENDER study that tested the efficacy and safety of trofinetide vs. placebo in 187 female patients with Rett syndrome, aged 5-20 years.
A total of 93 participants were randomly assigned to twice-daily oral trofinetide, and 94 received placebo for 12 weeks.
After 12 weeks, trofinetide showed a statistically significant improvement from baseline, compared with placebo, on both the caregiver-assessed Rett Syndrome Behavior Questionnaire (RSBQ) and 7-point Clinical Global Impression-Improvement (CGI-I) scale.
The drug also outperformed placebo at 12 weeks in a key secondary endpoint: the composite score on the Communication and Symbolic Behavior Scales Developmental Profile Infant-Toddler Checklist-Social (CSBS-DP-IT Social), a scale on which caregivers assess nonverbal communication.
The most common adverse events with trofinetide treatment were diarrhea and vomiting. Almost all these events were considered mild or moderate.
‘Historic day’
“This is a historic day for the Rett syndrome community and a meaningful moment for the patients and caregivers who have eagerly awaited the arrival of an approved treatment for this condition,” Melissa Kennedy, MHA, chief executive officer of the International Rett Syndrome Foundation, said in a news release issued by Acadia.
“Rett syndrome is a complicated, devastating disease that affects not only the individual patient, but whole families. With today’s FDA decision, those impacted by Rett have a promising new treatment option that has demonstrated benefit across a variety of Rett symptoms, including those that impact the daily lives of those living with Rett and their loved ones,” Ms. Kennedy said.
Trofinetide is expected to be available in the United States by the end of April.
A version of this article first appeared on Medscape.com.
Rett syndrome is a rare, genetic neurodevelopmental disorder that affects about 6,000-9,000 people in the United States, mostly females.
Symptoms typically present between 6 and 18 months of age, with patients experiencing a rapid decline with loss of fine motor and communication skills.
Trofinetide is a synthetic analogue of the amino-terminal tripeptide of insulinlike growth factor-1 (IGF-1), which occurs naturally in the brain. The drug is designed to treat the core symptoms of Rett syndrome by potentially reducing neuroinflammation and supporting synaptic function.
The approval of trofinetide was supported by results from the pivotal phase 3 LAVENDER study that tested the efficacy and safety of trofinetide vs. placebo in 187 female patients with Rett syndrome, aged 5-20 years.
A total of 93 participants were randomly assigned to twice-daily oral trofinetide, and 94 received placebo for 12 weeks.
After 12 weeks, trofinetide showed a statistically significant improvement from baseline, compared with placebo, on both the caregiver-assessed Rett Syndrome Behavior Questionnaire (RSBQ) and 7-point Clinical Global Impression-Improvement (CGI-I) scale.
The drug also outperformed placebo at 12 weeks in a key secondary endpoint: the composite score on the Communication and Symbolic Behavior Scales Developmental Profile Infant-Toddler Checklist-Social (CSBS-DP-IT Social), a scale on which caregivers assess nonverbal communication.
The most common adverse events with trofinetide treatment were diarrhea and vomiting. Almost all these events were considered mild or moderate.
‘Historic day’
“This is a historic day for the Rett syndrome community and a meaningful moment for the patients and caregivers who have eagerly awaited the arrival of an approved treatment for this condition,” Melissa Kennedy, MHA, chief executive officer of the International Rett Syndrome Foundation, said in a news release issued by Acadia.
“Rett syndrome is a complicated, devastating disease that affects not only the individual patient, but whole families. With today’s FDA decision, those impacted by Rett have a promising new treatment option that has demonstrated benefit across a variety of Rett symptoms, including those that impact the daily lives of those living with Rett and their loved ones,” Ms. Kennedy said.
Trofinetide is expected to be available in the United States by the end of April.
A version of this article first appeared on Medscape.com.
Autoantibodies signal reduced cancer risk in dermatomyositis
Adults with the inflammatory autoimmune myopathy dermatomyositis are at increased for concurrent cancers, but new research suggests that certain autoantibodies in patients with a specific dermatomyositis subtype may actually protect against cancer.
A study of cohorts of patients with dermatomyositis, other rheumatic diseases, and those without disease showed that among patients with dermatomyositis positive for antitranscriptional intermediary factor 1 (anti–TIF1-gamma) autoantibodies – a disease subtype associated with increased cancer risk – the presence of autoantibodies directed against cell division cycle and apoptosis regulator 1 (CCAR1) was associated with reduced cancer risk “to a level comparable to that seen in the general population,” Christopher A. Mecoli, MD, MHS, of Johns Hopkins University, Baltimore, and colleagues reported.
“Our prior data suggest that there are autoantigens that, when targeted simultaneously with CCAR1, provide additional cancer protection. Although these autoantigens are less frequently targeted, it is likely that additional, more prevalent ‘autoantigen hubs’ remain undiscovered,” they wrote in Arthritis & Rheumatology.
Identification of other autoantibodies both in the anti–TIF1-gamma–positive and other dermatomyositis subgroups may help with cancer risk stratification in patients with the disease and may ultimately improve cancer screening for adults with dermatomyositis, the investigators said.
Toward precision medicine
“I think this is a step toward precision medicine in patients with rheumatic disease, specifically myositis,” Dr. Mecoli said in an interview.
The study supports earlier work showing that dermatomyositis and related myopathies are heterogeneous, he said, noting that, “if you put 10 myositis patients in the same room, you wouldn’t get that they all have the same disease because they can look so different from one another.”
The association of dermatomyositis with concurrent cancers has been known for decades, but in recent years his team and other investigators have noted that the association holds true for only some patients with dermatomyositis, most notably those patients positive for anti–TIF1-gamma autoantibodies.
“And then, of course, once you really start studying just one gamma-positive dermatomyositis patient, you realize that even among that group it is heterogeneous in terms of their cancer risk, and that was the main focus of this study: to reconcile this clinical observation that I had a lot of patients with TIF1-gamma dermatomyositis who never get diagnosed with cancer,” Dr. Mecoli said.
Study details
Dr. Dr. Mecoli and colleagues previously reported that immune responses to CCAR1 and other autoantigens seen in patients with dermatomyositis were associated with lower probability of cancer occurrence.
In the current study, they focused on the disease specificity, clinical phenotype, and cancer risk for patients with dermatomyositis and anti-CCAR1 autoantibodies.
They looked at all patients aged 18 or older with a probable or definite finding of dermatomyositis, according to 2017 American College of Rheumatology/European Alliance of Associations for Rheumatology Idiopathic Inflammatory Myopathy criteria, who were seen at Stanford (Calif.) University Medical Center from August 2004 to April 2020 (101 patients), or the Johns Hopkins Myositis Center (141 patients) from January 2007 to December 2020.
Controls included 44 patients evaluated at the Johns Hopkins Myositis Center with immune-mediated necrotizing myopathy, 186 patients with anti–TIF1-gamma–negative dermatomyositis (defined as an enzyme-linked immunosorbent assay readout of less than seven units) evaluated at either Stanford or Johns Hopkins, 44 patients with inclusion body myositis evaluated at Johns Hopkins, and 46 patients with systemic lupus erythematosus from the Hopkins Lupus Cohort. The investigators also assayed serum from 32 healthy individuals.
They found that patients with anti–TIF1-gamma–positive dermatomyositis were significantly more likely than those with anti–TIF1-gamma–negative dermatomyositis to have anti-CCAR1 autoantibodies (32% vs. 8%; P < .001). Additionally, they noted that the anti-CCAR1 autoantibodies were not seen in serum from healthy controls and were found at only very low frequencies among patients with other rheumatic diseases.
When they looked at the incidence of cancer from the time of dermatomyositis onset (defined as the first patient-reported symptoms of rash, weakness, myalgia, or dyspnea) they found that the standardized incidence ratio in anti–TIF1-gamma–positive patients in both the Stanford and Hopkins cohorts was higher than expected, with SIRs of 3.49 and 4.54, respectively (P < .001 for each comparison).
However, among those patients who were both anti–TIF1-gamma positive and anti-CCAR1 positive, the SIRs were 1.78 in the Stanford cohort and 1.61 in the Hopkins cohort, and neither SIR was significantly higher than that of the general population.
Risk prediction
Their findings suggest that autoantibody profiles might be used for cancer risk stratification in patients with anti–TIF1-gamma–positive dermatomyositis, Dr. Mecoli said.
“Are we overscreening? What is the cost in terms of patient anxiety, in terms of radiation, and in terms of false positive results?” he asked. “If I had a patient in front of me with anti–TIF1-gamma dermatomyositis, I would probably manage them differently if I knew that they were CCAR1 positive, because the presence of that additional autoantibody attenuates their cancer risk relative to the general population.”
In an editorial accompanying the study, Manabu Fujimoto, MD, of the department of dermatology at Osaka (Japan) University, commented that it “is of clinical importance in that combination of autoantibodies can predict cancer risk with more accuracy. At the same time, this study will give an insight into the pathomechanisms of how antitumor activity may shape autoimmunity in dermatomyositis.”
It will be “intriguing” to discover whether anti-CCAR1 autoantibodies act only against tumors or might also have an impact on dermatomyositis itself, Dr. Fujimoto said.
The research was supported by grants from the National Institutes of Health; Huayi and Siuling Zhang Discovery Fund; Peter Buck, MD; and the Donald B. and Dorothy L. Stabler Foundation. The authors and Dr. Fujimoto reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Adults with the inflammatory autoimmune myopathy dermatomyositis are at increased for concurrent cancers, but new research suggests that certain autoantibodies in patients with a specific dermatomyositis subtype may actually protect against cancer.
A study of cohorts of patients with dermatomyositis, other rheumatic diseases, and those without disease showed that among patients with dermatomyositis positive for antitranscriptional intermediary factor 1 (anti–TIF1-gamma) autoantibodies – a disease subtype associated with increased cancer risk – the presence of autoantibodies directed against cell division cycle and apoptosis regulator 1 (CCAR1) was associated with reduced cancer risk “to a level comparable to that seen in the general population,” Christopher A. Mecoli, MD, MHS, of Johns Hopkins University, Baltimore, and colleagues reported.
“Our prior data suggest that there are autoantigens that, when targeted simultaneously with CCAR1, provide additional cancer protection. Although these autoantigens are less frequently targeted, it is likely that additional, more prevalent ‘autoantigen hubs’ remain undiscovered,” they wrote in Arthritis & Rheumatology.
Identification of other autoantibodies both in the anti–TIF1-gamma–positive and other dermatomyositis subgroups may help with cancer risk stratification in patients with the disease and may ultimately improve cancer screening for adults with dermatomyositis, the investigators said.
Toward precision medicine
“I think this is a step toward precision medicine in patients with rheumatic disease, specifically myositis,” Dr. Mecoli said in an interview.
The study supports earlier work showing that dermatomyositis and related myopathies are heterogeneous, he said, noting that, “if you put 10 myositis patients in the same room, you wouldn’t get that they all have the same disease because they can look so different from one another.”
The association of dermatomyositis with concurrent cancers has been known for decades, but in recent years his team and other investigators have noted that the association holds true for only some patients with dermatomyositis, most notably those patients positive for anti–TIF1-gamma autoantibodies.
“And then, of course, once you really start studying just one gamma-positive dermatomyositis patient, you realize that even among that group it is heterogeneous in terms of their cancer risk, and that was the main focus of this study: to reconcile this clinical observation that I had a lot of patients with TIF1-gamma dermatomyositis who never get diagnosed with cancer,” Dr. Mecoli said.
Study details
Dr. Dr. Mecoli and colleagues previously reported that immune responses to CCAR1 and other autoantigens seen in patients with dermatomyositis were associated with lower probability of cancer occurrence.
In the current study, they focused on the disease specificity, clinical phenotype, and cancer risk for patients with dermatomyositis and anti-CCAR1 autoantibodies.
They looked at all patients aged 18 or older with a probable or definite finding of dermatomyositis, according to 2017 American College of Rheumatology/European Alliance of Associations for Rheumatology Idiopathic Inflammatory Myopathy criteria, who were seen at Stanford (Calif.) University Medical Center from August 2004 to April 2020 (101 patients), or the Johns Hopkins Myositis Center (141 patients) from January 2007 to December 2020.
Controls included 44 patients evaluated at the Johns Hopkins Myositis Center with immune-mediated necrotizing myopathy, 186 patients with anti–TIF1-gamma–negative dermatomyositis (defined as an enzyme-linked immunosorbent assay readout of less than seven units) evaluated at either Stanford or Johns Hopkins, 44 patients with inclusion body myositis evaluated at Johns Hopkins, and 46 patients with systemic lupus erythematosus from the Hopkins Lupus Cohort. The investigators also assayed serum from 32 healthy individuals.
They found that patients with anti–TIF1-gamma–positive dermatomyositis were significantly more likely than those with anti–TIF1-gamma–negative dermatomyositis to have anti-CCAR1 autoantibodies (32% vs. 8%; P < .001). Additionally, they noted that the anti-CCAR1 autoantibodies were not seen in serum from healthy controls and were found at only very low frequencies among patients with other rheumatic diseases.
When they looked at the incidence of cancer from the time of dermatomyositis onset (defined as the first patient-reported symptoms of rash, weakness, myalgia, or dyspnea) they found that the standardized incidence ratio in anti–TIF1-gamma–positive patients in both the Stanford and Hopkins cohorts was higher than expected, with SIRs of 3.49 and 4.54, respectively (P < .001 for each comparison).
However, among those patients who were both anti–TIF1-gamma positive and anti-CCAR1 positive, the SIRs were 1.78 in the Stanford cohort and 1.61 in the Hopkins cohort, and neither SIR was significantly higher than that of the general population.
Risk prediction
Their findings suggest that autoantibody profiles might be used for cancer risk stratification in patients with anti–TIF1-gamma–positive dermatomyositis, Dr. Mecoli said.
“Are we overscreening? What is the cost in terms of patient anxiety, in terms of radiation, and in terms of false positive results?” he asked. “If I had a patient in front of me with anti–TIF1-gamma dermatomyositis, I would probably manage them differently if I knew that they were CCAR1 positive, because the presence of that additional autoantibody attenuates their cancer risk relative to the general population.”
In an editorial accompanying the study, Manabu Fujimoto, MD, of the department of dermatology at Osaka (Japan) University, commented that it “is of clinical importance in that combination of autoantibodies can predict cancer risk with more accuracy. At the same time, this study will give an insight into the pathomechanisms of how antitumor activity may shape autoimmunity in dermatomyositis.”
It will be “intriguing” to discover whether anti-CCAR1 autoantibodies act only against tumors or might also have an impact on dermatomyositis itself, Dr. Fujimoto said.
The research was supported by grants from the National Institutes of Health; Huayi and Siuling Zhang Discovery Fund; Peter Buck, MD; and the Donald B. and Dorothy L. Stabler Foundation. The authors and Dr. Fujimoto reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Adults with the inflammatory autoimmune myopathy dermatomyositis are at increased for concurrent cancers, but new research suggests that certain autoantibodies in patients with a specific dermatomyositis subtype may actually protect against cancer.
A study of cohorts of patients with dermatomyositis, other rheumatic diseases, and those without disease showed that among patients with dermatomyositis positive for antitranscriptional intermediary factor 1 (anti–TIF1-gamma) autoantibodies – a disease subtype associated with increased cancer risk – the presence of autoantibodies directed against cell division cycle and apoptosis regulator 1 (CCAR1) was associated with reduced cancer risk “to a level comparable to that seen in the general population,” Christopher A. Mecoli, MD, MHS, of Johns Hopkins University, Baltimore, and colleagues reported.
“Our prior data suggest that there are autoantigens that, when targeted simultaneously with CCAR1, provide additional cancer protection. Although these autoantigens are less frequently targeted, it is likely that additional, more prevalent ‘autoantigen hubs’ remain undiscovered,” they wrote in Arthritis & Rheumatology.
Identification of other autoantibodies both in the anti–TIF1-gamma–positive and other dermatomyositis subgroups may help with cancer risk stratification in patients with the disease and may ultimately improve cancer screening for adults with dermatomyositis, the investigators said.
Toward precision medicine
“I think this is a step toward precision medicine in patients with rheumatic disease, specifically myositis,” Dr. Mecoli said in an interview.
The study supports earlier work showing that dermatomyositis and related myopathies are heterogeneous, he said, noting that, “if you put 10 myositis patients in the same room, you wouldn’t get that they all have the same disease because they can look so different from one another.”
The association of dermatomyositis with concurrent cancers has been known for decades, but in recent years his team and other investigators have noted that the association holds true for only some patients with dermatomyositis, most notably those patients positive for anti–TIF1-gamma autoantibodies.
“And then, of course, once you really start studying just one gamma-positive dermatomyositis patient, you realize that even among that group it is heterogeneous in terms of their cancer risk, and that was the main focus of this study: to reconcile this clinical observation that I had a lot of patients with TIF1-gamma dermatomyositis who never get diagnosed with cancer,” Dr. Mecoli said.
Study details
Dr. Dr. Mecoli and colleagues previously reported that immune responses to CCAR1 and other autoantigens seen in patients with dermatomyositis were associated with lower probability of cancer occurrence.
In the current study, they focused on the disease specificity, clinical phenotype, and cancer risk for patients with dermatomyositis and anti-CCAR1 autoantibodies.
They looked at all patients aged 18 or older with a probable or definite finding of dermatomyositis, according to 2017 American College of Rheumatology/European Alliance of Associations for Rheumatology Idiopathic Inflammatory Myopathy criteria, who were seen at Stanford (Calif.) University Medical Center from August 2004 to April 2020 (101 patients), or the Johns Hopkins Myositis Center (141 patients) from January 2007 to December 2020.
Controls included 44 patients evaluated at the Johns Hopkins Myositis Center with immune-mediated necrotizing myopathy, 186 patients with anti–TIF1-gamma–negative dermatomyositis (defined as an enzyme-linked immunosorbent assay readout of less than seven units) evaluated at either Stanford or Johns Hopkins, 44 patients with inclusion body myositis evaluated at Johns Hopkins, and 46 patients with systemic lupus erythematosus from the Hopkins Lupus Cohort. The investigators also assayed serum from 32 healthy individuals.
They found that patients with anti–TIF1-gamma–positive dermatomyositis were significantly more likely than those with anti–TIF1-gamma–negative dermatomyositis to have anti-CCAR1 autoantibodies (32% vs. 8%; P < .001). Additionally, they noted that the anti-CCAR1 autoantibodies were not seen in serum from healthy controls and were found at only very low frequencies among patients with other rheumatic diseases.
When they looked at the incidence of cancer from the time of dermatomyositis onset (defined as the first patient-reported symptoms of rash, weakness, myalgia, or dyspnea) they found that the standardized incidence ratio in anti–TIF1-gamma–positive patients in both the Stanford and Hopkins cohorts was higher than expected, with SIRs of 3.49 and 4.54, respectively (P < .001 for each comparison).
However, among those patients who were both anti–TIF1-gamma positive and anti-CCAR1 positive, the SIRs were 1.78 in the Stanford cohort and 1.61 in the Hopkins cohort, and neither SIR was significantly higher than that of the general population.
Risk prediction
Their findings suggest that autoantibody profiles might be used for cancer risk stratification in patients with anti–TIF1-gamma–positive dermatomyositis, Dr. Mecoli said.
“Are we overscreening? What is the cost in terms of patient anxiety, in terms of radiation, and in terms of false positive results?” he asked. “If I had a patient in front of me with anti–TIF1-gamma dermatomyositis, I would probably manage them differently if I knew that they were CCAR1 positive, because the presence of that additional autoantibody attenuates their cancer risk relative to the general population.”
In an editorial accompanying the study, Manabu Fujimoto, MD, of the department of dermatology at Osaka (Japan) University, commented that it “is of clinical importance in that combination of autoantibodies can predict cancer risk with more accuracy. At the same time, this study will give an insight into the pathomechanisms of how antitumor activity may shape autoimmunity in dermatomyositis.”
It will be “intriguing” to discover whether anti-CCAR1 autoantibodies act only against tumors or might also have an impact on dermatomyositis itself, Dr. Fujimoto said.
The research was supported by grants from the National Institutes of Health; Huayi and Siuling Zhang Discovery Fund; Peter Buck, MD; and the Donald B. and Dorothy L. Stabler Foundation. The authors and Dr. Fujimoto reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ARTHRITIS & RHEUMATOLOGY
Maternal infection in pregnancy ups risk for childhood leukemia?
Children born to mothers who had urinary or genital tract infections during pregnancy appear to have an increased risk for childhood leukemia, said researchers reporting a Danish registry analysis that may point to preventive strategies for the disease.
The research was published online in JAMA Network Open.
The team studied more than 2.2 million children born in Denmark over more than 3 decades, linking their records across multiple national registries to examine both later cancer risk and maternal infection rates.
They found that, overall, at least one maternal infection during pregnancy was associated with a 35% increased risk for leukemia in the children, rising to 65% for urinary tract infections, and 142% for genital infections.
“The findings of this large population-based cohort study suggest that maternal urinary and genital tract infections during pregnancy are associated with a higher risk of childhood leukemia in offspring,” said lead author Jian-Rong He, DPhil, division of birth cohort study, Guangzhou (China) Women and Children’s Medical Center.
However, he added, “the associated absolute risk remained small given the rarity” of the disease. In absolute terms, the risk difference between exposed and unexposed children was 1.8 cases per 100,000 person-years for any infection, 3.4 cases per 100,000 person-years for urinary traction infection, and 7.1 cases per 100,000 person-years for genital tract infection.
Maternal infections during pregnancy may be associated with chromosomal and immunologic alterations in the fetus, the authors speculated.
“Given that little is known about the etiology of childhood leukemia,” these results “suggest an important direction for research on the etiology of childhood leukemia as well as development of potential preventive measures,” they wrote.
In many countries, pregnant women are tested for urinary tract infection and bacterial vaginosis, and treated with antibiotics in antenatal care, as these infections are linked to adverse perinatal outcomes, they pointed out.
Study details
The team conducted a large population-based study that included all live births in Denmark between 1978 and 2015.
After exclusions, they gathered information on 2,222,797 children, linking data from several national registries, including the Danish Medical Birth Register, the Danish National Patient Registry, and the Danish National Cancer Registry, to identify cases of childhood cancers and maternal infection during pregnancy.
The results were then validated by comparing them with those in 2.6 million live births in Sweden between 1988 and 2014, for whom similar data were available through linkage with several Swedish registries.
The Danish cohort was followed up for a mean of 12 years per person, yielding a total of 27 million person-years. Just over half (51.3%) were boys.
Cancer was diagnosed in 4,362 children before 15 years of age, of whom 1,307 had leukemia (1,050 had acute lymphocytic leukemia), 1,267 had a brain tumor, 224 had lymphoma, and 1,564 had other cancers.
At least one infection during pregnancy was diagnosed in 81,717 mothers (3.7%). Urinary tract infections were the most common (in 1.7% of women), followed by genital tract infection (in 0.7%), digestive system infection (in 0.5%), and respiratory tract infection (in 0.3%).
Women with any infection during pregnancy were more likely to be younger and primiparous than were women who did not have infections, and they were also more likely to have fewer years of education, higher prepregnancy BMI, diabetes, and to smoke during early pregnancy.
Preterm delivery and low-birth-weight infants were also more common in women with infections during pregnancy.
Cox proportional hazards regression models revealed that, after adjustment for confounders, any maternal infection was associated with a hazard ratio of childhood leukemia of 1.35.
Further analysis revealed that the association was driven by genital tract infection, at a hazard ratio for childhood leukemia of 2.42, and urinary tract infection, at a hazard ratio 1.65.
Moreover, children born to women who had a sexually transmitted infection during pregnancy had a hazard ratio for developing leukemia of 3.13 compared with unexposed children.
There were no associations between other maternal infections and childhood leukemia.
The patterns of association between maternal infections and childhood leukemia were similar when looking at disease subtypes, as well as in the Swedish validation cohort, they added.
When interpreting the results, the researchers caution that, as data on maternal infection were drawn from hospital data, “milder infections and those not diagnosed or treated in specialized health care facilities were not captured.”
“Also, some infections could be captured because the mother sought care for other, more serious conditions, which might bias the association of maternal infections and childhood leukemia.”
The study was supported by grants from the China Scholarship Council–University of Oxford; National Natural Science Foundation of China; Danish Council for Independent Research; Nordic Cancer Union; Novo Nordisk Fonden; and the Swedish Council for Working Life and Social Research. Dr He reported receiving a PhD scholarship from the China Scholarship Council during the conduct of the study. Several other coauthors have disclosures; the full list can be found with the original article.
A version of this article originally appeared on Medscape.com.
Children born to mothers who had urinary or genital tract infections during pregnancy appear to have an increased risk for childhood leukemia, said researchers reporting a Danish registry analysis that may point to preventive strategies for the disease.
The research was published online in JAMA Network Open.
The team studied more than 2.2 million children born in Denmark over more than 3 decades, linking their records across multiple national registries to examine both later cancer risk and maternal infection rates.
They found that, overall, at least one maternal infection during pregnancy was associated with a 35% increased risk for leukemia in the children, rising to 65% for urinary tract infections, and 142% for genital infections.
“The findings of this large population-based cohort study suggest that maternal urinary and genital tract infections during pregnancy are associated with a higher risk of childhood leukemia in offspring,” said lead author Jian-Rong He, DPhil, division of birth cohort study, Guangzhou (China) Women and Children’s Medical Center.
However, he added, “the associated absolute risk remained small given the rarity” of the disease. In absolute terms, the risk difference between exposed and unexposed children was 1.8 cases per 100,000 person-years for any infection, 3.4 cases per 100,000 person-years for urinary traction infection, and 7.1 cases per 100,000 person-years for genital tract infection.
Maternal infections during pregnancy may be associated with chromosomal and immunologic alterations in the fetus, the authors speculated.
“Given that little is known about the etiology of childhood leukemia,” these results “suggest an important direction for research on the etiology of childhood leukemia as well as development of potential preventive measures,” they wrote.
In many countries, pregnant women are tested for urinary tract infection and bacterial vaginosis, and treated with antibiotics in antenatal care, as these infections are linked to adverse perinatal outcomes, they pointed out.
Study details
The team conducted a large population-based study that included all live births in Denmark between 1978 and 2015.
After exclusions, they gathered information on 2,222,797 children, linking data from several national registries, including the Danish Medical Birth Register, the Danish National Patient Registry, and the Danish National Cancer Registry, to identify cases of childhood cancers and maternal infection during pregnancy.
The results were then validated by comparing them with those in 2.6 million live births in Sweden between 1988 and 2014, for whom similar data were available through linkage with several Swedish registries.
The Danish cohort was followed up for a mean of 12 years per person, yielding a total of 27 million person-years. Just over half (51.3%) were boys.
Cancer was diagnosed in 4,362 children before 15 years of age, of whom 1,307 had leukemia (1,050 had acute lymphocytic leukemia), 1,267 had a brain tumor, 224 had lymphoma, and 1,564 had other cancers.
At least one infection during pregnancy was diagnosed in 81,717 mothers (3.7%). Urinary tract infections were the most common (in 1.7% of women), followed by genital tract infection (in 0.7%), digestive system infection (in 0.5%), and respiratory tract infection (in 0.3%).
Women with any infection during pregnancy were more likely to be younger and primiparous than were women who did not have infections, and they were also more likely to have fewer years of education, higher prepregnancy BMI, diabetes, and to smoke during early pregnancy.
Preterm delivery and low-birth-weight infants were also more common in women with infections during pregnancy.
Cox proportional hazards regression models revealed that, after adjustment for confounders, any maternal infection was associated with a hazard ratio of childhood leukemia of 1.35.
Further analysis revealed that the association was driven by genital tract infection, at a hazard ratio for childhood leukemia of 2.42, and urinary tract infection, at a hazard ratio 1.65.
Moreover, children born to women who had a sexually transmitted infection during pregnancy had a hazard ratio for developing leukemia of 3.13 compared with unexposed children.
There were no associations between other maternal infections and childhood leukemia.
The patterns of association between maternal infections and childhood leukemia were similar when looking at disease subtypes, as well as in the Swedish validation cohort, they added.
When interpreting the results, the researchers caution that, as data on maternal infection were drawn from hospital data, “milder infections and those not diagnosed or treated in specialized health care facilities were not captured.”
“Also, some infections could be captured because the mother sought care for other, more serious conditions, which might bias the association of maternal infections and childhood leukemia.”
The study was supported by grants from the China Scholarship Council–University of Oxford; National Natural Science Foundation of China; Danish Council for Independent Research; Nordic Cancer Union; Novo Nordisk Fonden; and the Swedish Council for Working Life and Social Research. Dr He reported receiving a PhD scholarship from the China Scholarship Council during the conduct of the study. Several other coauthors have disclosures; the full list can be found with the original article.
A version of this article originally appeared on Medscape.com.
Children born to mothers who had urinary or genital tract infections during pregnancy appear to have an increased risk for childhood leukemia, said researchers reporting a Danish registry analysis that may point to preventive strategies for the disease.
The research was published online in JAMA Network Open.
The team studied more than 2.2 million children born in Denmark over more than 3 decades, linking their records across multiple national registries to examine both later cancer risk and maternal infection rates.
They found that, overall, at least one maternal infection during pregnancy was associated with a 35% increased risk for leukemia in the children, rising to 65% for urinary tract infections, and 142% for genital infections.
“The findings of this large population-based cohort study suggest that maternal urinary and genital tract infections during pregnancy are associated with a higher risk of childhood leukemia in offspring,” said lead author Jian-Rong He, DPhil, division of birth cohort study, Guangzhou (China) Women and Children’s Medical Center.
However, he added, “the associated absolute risk remained small given the rarity” of the disease. In absolute terms, the risk difference between exposed and unexposed children was 1.8 cases per 100,000 person-years for any infection, 3.4 cases per 100,000 person-years for urinary traction infection, and 7.1 cases per 100,000 person-years for genital tract infection.
Maternal infections during pregnancy may be associated with chromosomal and immunologic alterations in the fetus, the authors speculated.
“Given that little is known about the etiology of childhood leukemia,” these results “suggest an important direction for research on the etiology of childhood leukemia as well as development of potential preventive measures,” they wrote.
In many countries, pregnant women are tested for urinary tract infection and bacterial vaginosis, and treated with antibiotics in antenatal care, as these infections are linked to adverse perinatal outcomes, they pointed out.
Study details
The team conducted a large population-based study that included all live births in Denmark between 1978 and 2015.
After exclusions, they gathered information on 2,222,797 children, linking data from several national registries, including the Danish Medical Birth Register, the Danish National Patient Registry, and the Danish National Cancer Registry, to identify cases of childhood cancers and maternal infection during pregnancy.
The results were then validated by comparing them with those in 2.6 million live births in Sweden between 1988 and 2014, for whom similar data were available through linkage with several Swedish registries.
The Danish cohort was followed up for a mean of 12 years per person, yielding a total of 27 million person-years. Just over half (51.3%) were boys.
Cancer was diagnosed in 4,362 children before 15 years of age, of whom 1,307 had leukemia (1,050 had acute lymphocytic leukemia), 1,267 had a brain tumor, 224 had lymphoma, and 1,564 had other cancers.
At least one infection during pregnancy was diagnosed in 81,717 mothers (3.7%). Urinary tract infections were the most common (in 1.7% of women), followed by genital tract infection (in 0.7%), digestive system infection (in 0.5%), and respiratory tract infection (in 0.3%).
Women with any infection during pregnancy were more likely to be younger and primiparous than were women who did not have infections, and they were also more likely to have fewer years of education, higher prepregnancy BMI, diabetes, and to smoke during early pregnancy.
Preterm delivery and low-birth-weight infants were also more common in women with infections during pregnancy.
Cox proportional hazards regression models revealed that, after adjustment for confounders, any maternal infection was associated with a hazard ratio of childhood leukemia of 1.35.
Further analysis revealed that the association was driven by genital tract infection, at a hazard ratio for childhood leukemia of 2.42, and urinary tract infection, at a hazard ratio 1.65.
Moreover, children born to women who had a sexually transmitted infection during pregnancy had a hazard ratio for developing leukemia of 3.13 compared with unexposed children.
There were no associations between other maternal infections and childhood leukemia.
The patterns of association between maternal infections and childhood leukemia were similar when looking at disease subtypes, as well as in the Swedish validation cohort, they added.
When interpreting the results, the researchers caution that, as data on maternal infection were drawn from hospital data, “milder infections and those not diagnosed or treated in specialized health care facilities were not captured.”
“Also, some infections could be captured because the mother sought care for other, more serious conditions, which might bias the association of maternal infections and childhood leukemia.”
The study was supported by grants from the China Scholarship Council–University of Oxford; National Natural Science Foundation of China; Danish Council for Independent Research; Nordic Cancer Union; Novo Nordisk Fonden; and the Swedish Council for Working Life and Social Research. Dr He reported receiving a PhD scholarship from the China Scholarship Council during the conduct of the study. Several other coauthors have disclosures; the full list can be found with the original article.
A version of this article originally appeared on Medscape.com.
Antibiotics and SJS/TEN: Study provides global prevalence
of SJS/TEN in connection with antibiotics.
“SJS/TEN is considered the most severe form of drug hypersensitivity reaction, and antibiotics are an important risk,” Erika Yue Lee, MD, and associates wrote in JAMA Dermatology.
Their analysis, which involved 38 studies published since 1987 with 2,917 patients from more than 20 countries, showed that 86% of all SJS/TEN cases were associated with a single drug, with the rest involving multiple drug triggers, infections, or other causes. More than a quarter (28%) of those patients had used an antibiotic, and the sulfonamides were the class most often triggering SJS/TEN, said Dr. Lee of the University of Toronto and associates.
Sulfonamides were responsible for 32% of the antibiotic-associated cases, which works out to 11% of all SJS/TEN cases included in the analysis. Penicillins were next with 22% of all antibiotic-associated cases, followed by the cephalosporins (11%), fluoroquinolones (4%), and macrolides (2%), the investigators reported.
A subgroup analysis conducted by age indicated that “there was no difference in the proportion of antibiotics associated with SJS/TEN between adult and pediatric groups,” they noted.
There were differences, however, among the various antibiotic classes. Sulfonamides represented 54% of antibiotic-triggered reactions in children, compared with 25% in adults, but adults were significantly more likely to have cephalosporin (23%) and fluoroquinolone (5%) involvement than were children (2% and 0, respectively). Macrolide-induced SJS/TEN was more common in children (18% vs. 1%), while the penicillin rate was 18% for both age groups, Dr. Lee and associates said.
A second subgroup analysis establishing the proportion of antibiotic-induced SJS/TEN by continent ranked Australia highest with 43%, but that was based on only one study of 42 patients. North America was slightly lower at 37%, but the analysis included 14 studies and 932 patients. Asia’s 16 studies and 1,298 patients were divided into three regions, with the lowest being the southeast at 16%, according to the researchers.
“Global sulfonamide antibiotic use has been decreasing since 2000 despite an ongoing upward trend of use in other antibiotic classes,” they wrote, but “antibiotics remain one of the most common culprit drugs for SJS/TEN in both adults and children worldwide.”
One of Dr. Lee’s associates has received personal fees from Janssen, AstraZeneca, UpToDate, Verve, BioCryst, Regeneron Pharmaceuticals, and Novavax and has served as codirector of IIID Pty Ltd, which holds a patent for HLA-B*57:01 testing and has a patent pending for detection of HLA-A*32:01 in connection with diagnosing drug reaction without any financial remuneration outside this study.
of SJS/TEN in connection with antibiotics.
“SJS/TEN is considered the most severe form of drug hypersensitivity reaction, and antibiotics are an important risk,” Erika Yue Lee, MD, and associates wrote in JAMA Dermatology.
Their analysis, which involved 38 studies published since 1987 with 2,917 patients from more than 20 countries, showed that 86% of all SJS/TEN cases were associated with a single drug, with the rest involving multiple drug triggers, infections, or other causes. More than a quarter (28%) of those patients had used an antibiotic, and the sulfonamides were the class most often triggering SJS/TEN, said Dr. Lee of the University of Toronto and associates.
Sulfonamides were responsible for 32% of the antibiotic-associated cases, which works out to 11% of all SJS/TEN cases included in the analysis. Penicillins were next with 22% of all antibiotic-associated cases, followed by the cephalosporins (11%), fluoroquinolones (4%), and macrolides (2%), the investigators reported.
A subgroup analysis conducted by age indicated that “there was no difference in the proportion of antibiotics associated with SJS/TEN between adult and pediatric groups,” they noted.
There were differences, however, among the various antibiotic classes. Sulfonamides represented 54% of antibiotic-triggered reactions in children, compared with 25% in adults, but adults were significantly more likely to have cephalosporin (23%) and fluoroquinolone (5%) involvement than were children (2% and 0, respectively). Macrolide-induced SJS/TEN was more common in children (18% vs. 1%), while the penicillin rate was 18% for both age groups, Dr. Lee and associates said.
A second subgroup analysis establishing the proportion of antibiotic-induced SJS/TEN by continent ranked Australia highest with 43%, but that was based on only one study of 42 patients. North America was slightly lower at 37%, but the analysis included 14 studies and 932 patients. Asia’s 16 studies and 1,298 patients were divided into three regions, with the lowest being the southeast at 16%, according to the researchers.
“Global sulfonamide antibiotic use has been decreasing since 2000 despite an ongoing upward trend of use in other antibiotic classes,” they wrote, but “antibiotics remain one of the most common culprit drugs for SJS/TEN in both adults and children worldwide.”
One of Dr. Lee’s associates has received personal fees from Janssen, AstraZeneca, UpToDate, Verve, BioCryst, Regeneron Pharmaceuticals, and Novavax and has served as codirector of IIID Pty Ltd, which holds a patent for HLA-B*57:01 testing and has a patent pending for detection of HLA-A*32:01 in connection with diagnosing drug reaction without any financial remuneration outside this study.
of SJS/TEN in connection with antibiotics.
“SJS/TEN is considered the most severe form of drug hypersensitivity reaction, and antibiotics are an important risk,” Erika Yue Lee, MD, and associates wrote in JAMA Dermatology.
Their analysis, which involved 38 studies published since 1987 with 2,917 patients from more than 20 countries, showed that 86% of all SJS/TEN cases were associated with a single drug, with the rest involving multiple drug triggers, infections, or other causes. More than a quarter (28%) of those patients had used an antibiotic, and the sulfonamides were the class most often triggering SJS/TEN, said Dr. Lee of the University of Toronto and associates.
Sulfonamides were responsible for 32% of the antibiotic-associated cases, which works out to 11% of all SJS/TEN cases included in the analysis. Penicillins were next with 22% of all antibiotic-associated cases, followed by the cephalosporins (11%), fluoroquinolones (4%), and macrolides (2%), the investigators reported.
A subgroup analysis conducted by age indicated that “there was no difference in the proportion of antibiotics associated with SJS/TEN between adult and pediatric groups,” they noted.
There were differences, however, among the various antibiotic classes. Sulfonamides represented 54% of antibiotic-triggered reactions in children, compared with 25% in adults, but adults were significantly more likely to have cephalosporin (23%) and fluoroquinolone (5%) involvement than were children (2% and 0, respectively). Macrolide-induced SJS/TEN was more common in children (18% vs. 1%), while the penicillin rate was 18% for both age groups, Dr. Lee and associates said.
A second subgroup analysis establishing the proportion of antibiotic-induced SJS/TEN by continent ranked Australia highest with 43%, but that was based on only one study of 42 patients. North America was slightly lower at 37%, but the analysis included 14 studies and 932 patients. Asia’s 16 studies and 1,298 patients were divided into three regions, with the lowest being the southeast at 16%, according to the researchers.
“Global sulfonamide antibiotic use has been decreasing since 2000 despite an ongoing upward trend of use in other antibiotic classes,” they wrote, but “antibiotics remain one of the most common culprit drugs for SJS/TEN in both adults and children worldwide.”
One of Dr. Lee’s associates has received personal fees from Janssen, AstraZeneca, UpToDate, Verve, BioCryst, Regeneron Pharmaceuticals, and Novavax and has served as codirector of IIID Pty Ltd, which holds a patent for HLA-B*57:01 testing and has a patent pending for detection of HLA-A*32:01 in connection with diagnosing drug reaction without any financial remuneration outside this study.
FROM JAMA DERMATOLOGY
A White male presented with a 1½-year history of a progressive hypoesthetic annular, hyperpigmented plaque on the upper arm
Paucibacillary tuberculoid leprosy is characterized by few anesthetic hypo- or hyperpigmented lesions and can be accompanied by palpable peripheral nerve enlargements.
Tuberculoid leprosy presents histologically with epithelioid histiocytes with lymphocytes and Langhans giant cells. Neurotropic granulomas are also characteristic of tuberculoid leprosy. Fite staining allows for the identification of the acid-fast bacilli of M. leprae, which in some cases are quite few in number. The standard mycobacterium stain, Ziehl-Neelsen, is a good option for M. tuberculosis, but because of the relative weak mycolic acid coat of M. leprae, the Fite stain is more appropriate for identifying M. leprae.
Clinically, other than the presence of fewer than five hypoesthetic lesions that are either hypopigmented or erythematous, tuberculoid leprosy often presents with additional peripheral nerve involvement that manifests as numbness and tingling in hands and feet.1 This patient denied any tingling, weakness, or numbness, outside of the anesthetic lesion on his posterior upper arm.
The patient, born in the United States, had a remote history of military travel to Iraq, Kuwait, and the Philippines, but had not traveled internationally within the last 15 years, apart from a cruise to the Bahamas. He denied any known contact with individuals with similar lesions. He denied a history of contact with armadillos, but acknowledged that they are native to where he resides in central Florida, and that he had seen them in his yard.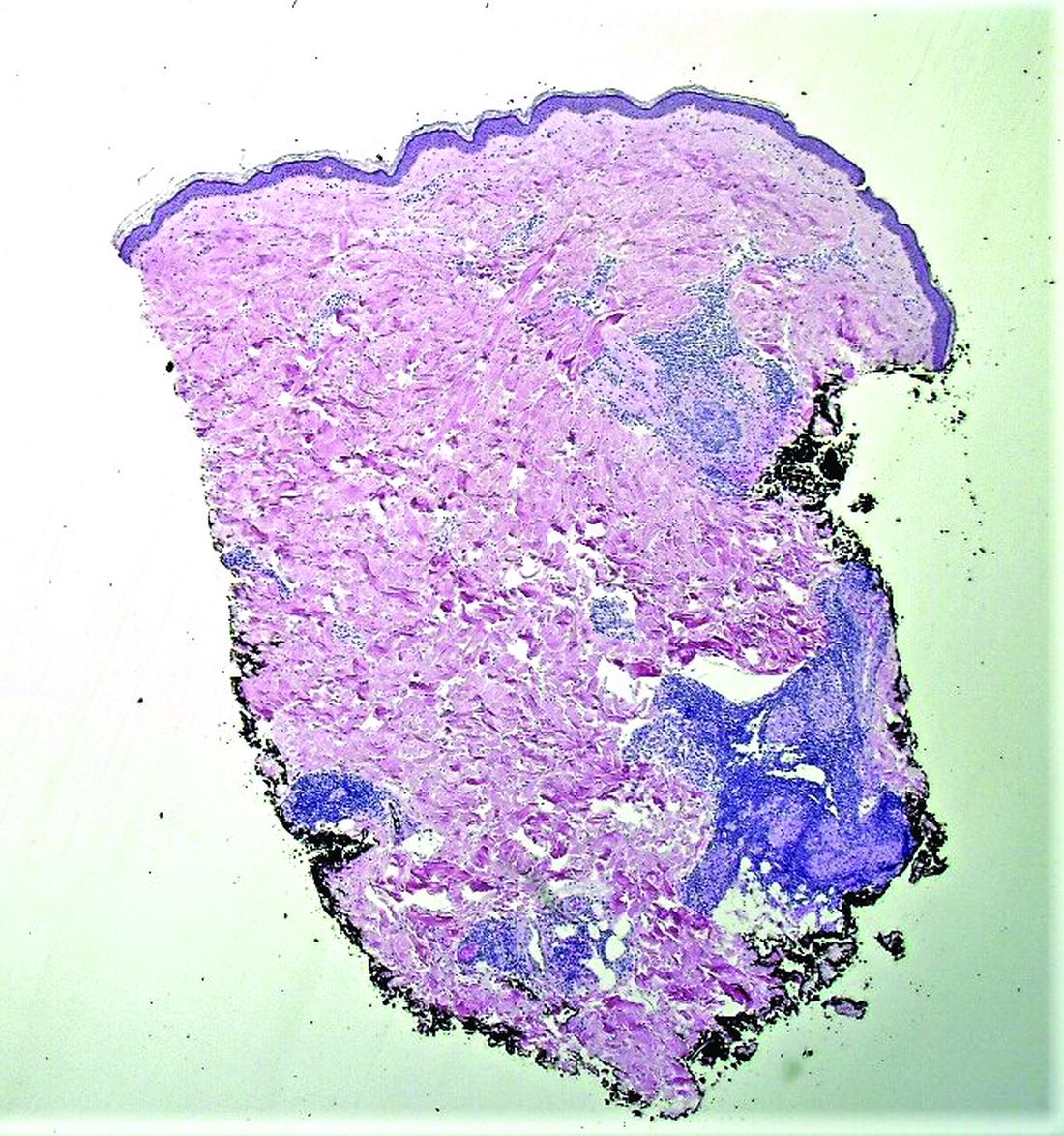
Histopathological examination revealed an unremarkable epidermis with a superficial and deep perivascular, periadnexal, and perineural lymphohistiocytic infiltrate. Fite stain revealed rare rod-shaped organisms (Figure 2). These findings are consistent with a diagnosis of paucibacillary, tuberculoid leprosy.
The patient’s travel history to highly endemic areas (Middle East), as well as possible environmental contact with armadillos – including contact with soil that the armadillos occupied – could explain plausible modes of transmission. Following consultation with our infectious disease department and the National Hansen’s Disease Program, our patient began a planned course of therapy with 18 months of minocycline, rifampin, and moxifloxacin.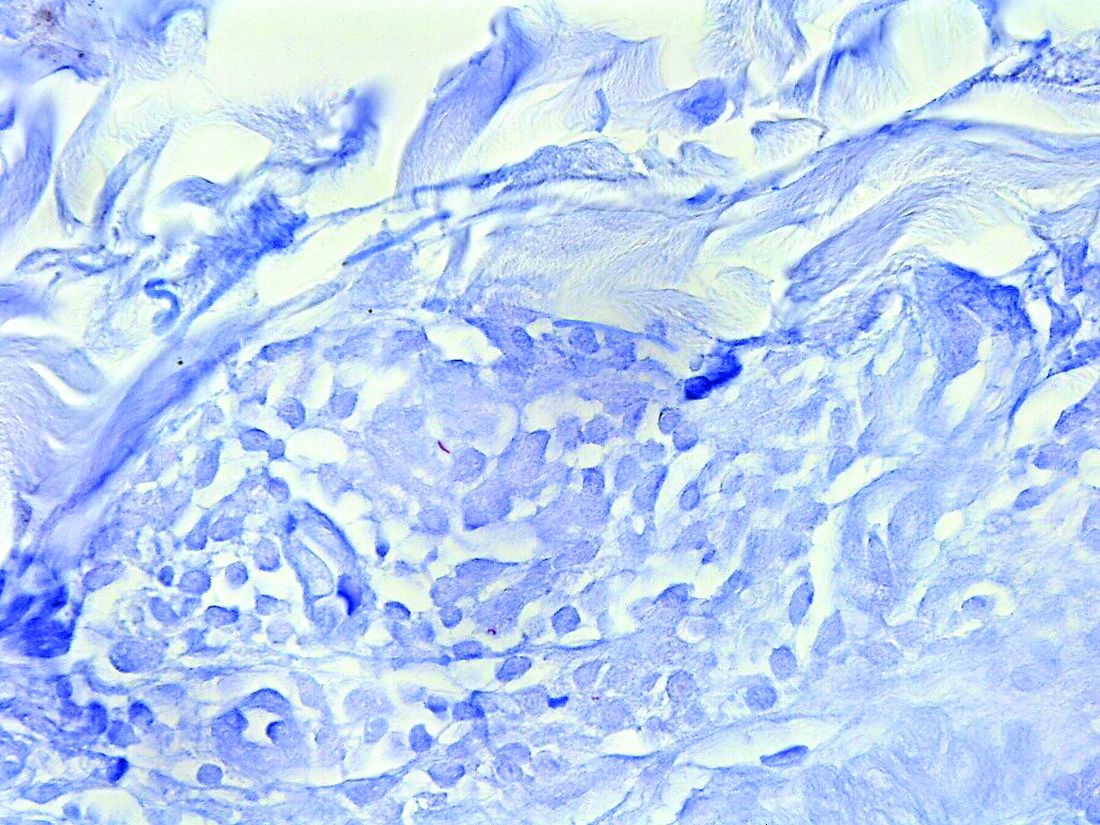
Human-to-human transmission of HD has been well documented; however, zoonotic transmission – specifically via the nine-banded armadillo (Dasypus novemcinctus) – serves as another suggested means of transmission, especially in the Southeastern United States.2-6 Travel to highly-endemic areas increases the risk of contracting HD, which may take up to 20 years following contact with the bacteria to manifest clinically.
While central Florida was previously thought to be a nonendemic area of disease, the incidence of the disease in this region has increased in recent years.7 Human-to-human transmission, which remains a concern with immigration from highly-endemic regions, occurs via long-term contact with nasal droplets of an infected person.8,9

Many patients in regions with very few cases of leprosy deny travel to other endemic regions and contact with infected people. Thus, zoonotic transmission remains a legitimate concern in the Southeastern United States – accounting, at least in part, for many of the non–human-transmitted cases of leprosy.2,10 We encourage clinicians to maintain a high level of clinical suspicion for leprosy when evaluating patients presenting with hypoesthetic cutaneous lesions and to obtain a travel history and to ask about armadillo exposure.
This case and the photos were submitted by Ms. Smith, from the University of South Florida, Tampa; Dr. Hatch and Dr. Sarriera-Lazaro, from the department of dermatology and cutaneous surgery, University of South Florida; and Dr. Turner and Dr. Beachkofsky, from the department of pathology and laboratory medicine at the James A. Haley Veterans’ Hospital, Tampa. Dr. Bilu Martin edited this case. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Leprosy (Hansen’s Disease), in: “Goldman’s Cecil Medicine,” 24th ed. (Philadelphia: W.B. Saunders, 2012: pp. 1950-4.
2. Sharma R et al. Emerg Infect Dis. 2015 Dec;21(12):2127-34.
3. Lane JE et al. J Am Acad Dermatol. 2006 Oct;55(4):714-6.
4. Clark BM et al. Am J Trop Med Hyg. 2008 Jun;78(6):962-7.
5. Bruce S et al. J Am Acad Dermatol. 2000 Aug;43(2 Pt 1):223-8.
6. Loughry WJ et al. J Wildl Dis. 2009 Jan;45(1):144-52.
7. FDo H. Florida charts: Hansen’s Disease (Leprosy). Health FDo. 2019. https://www.flhealthcharts.gov/ChartsReports/rdPage.aspx?rdReport=NonVitalIndNoGrpCounts.DataViewer&cid=174.
8. Maymone MBC et al. J Am Acad Dermatol. 2020 Jul;83(1):1-14.
9. Scollard DM et al. Clin Microbiol Rev. 2006 Apr;19(2):338-81.
10. Domozych R et al. JAAD Case Rep. 2016 May 12;2(3):189-92.
Paucibacillary tuberculoid leprosy is characterized by few anesthetic hypo- or hyperpigmented lesions and can be accompanied by palpable peripheral nerve enlargements.
Tuberculoid leprosy presents histologically with epithelioid histiocytes with lymphocytes and Langhans giant cells. Neurotropic granulomas are also characteristic of tuberculoid leprosy. Fite staining allows for the identification of the acid-fast bacilli of M. leprae, which in some cases are quite few in number. The standard mycobacterium stain, Ziehl-Neelsen, is a good option for M. tuberculosis, but because of the relative weak mycolic acid coat of M. leprae, the Fite stain is more appropriate for identifying M. leprae.
Clinically, other than the presence of fewer than five hypoesthetic lesions that are either hypopigmented or erythematous, tuberculoid leprosy often presents with additional peripheral nerve involvement that manifests as numbness and tingling in hands and feet.1 This patient denied any tingling, weakness, or numbness, outside of the anesthetic lesion on his posterior upper arm.
The patient, born in the United States, had a remote history of military travel to Iraq, Kuwait, and the Philippines, but had not traveled internationally within the last 15 years, apart from a cruise to the Bahamas. He denied any known contact with individuals with similar lesions. He denied a history of contact with armadillos, but acknowledged that they are native to where he resides in central Florida, and that he had seen them in his yard.
Histopathological examination revealed an unremarkable epidermis with a superficial and deep perivascular, periadnexal, and perineural lymphohistiocytic infiltrate. Fite stain revealed rare rod-shaped organisms (Figure 2). These findings are consistent with a diagnosis of paucibacillary, tuberculoid leprosy.
The patient’s travel history to highly endemic areas (Middle East), as well as possible environmental contact with armadillos – including contact with soil that the armadillos occupied – could explain plausible modes of transmission. Following consultation with our infectious disease department and the National Hansen’s Disease Program, our patient began a planned course of therapy with 18 months of minocycline, rifampin, and moxifloxacin.
Human-to-human transmission of HD has been well documented; however, zoonotic transmission – specifically via the nine-banded armadillo (Dasypus novemcinctus) – serves as another suggested means of transmission, especially in the Southeastern United States.2-6 Travel to highly-endemic areas increases the risk of contracting HD, which may take up to 20 years following contact with the bacteria to manifest clinically.
While central Florida was previously thought to be a nonendemic area of disease, the incidence of the disease in this region has increased in recent years.7 Human-to-human transmission, which remains a concern with immigration from highly-endemic regions, occurs via long-term contact with nasal droplets of an infected person.8,9
Many patients in regions with very few cases of leprosy deny travel to other endemic regions and contact with infected people. Thus, zoonotic transmission remains a legitimate concern in the Southeastern United States – accounting, at least in part, for many of the non–human-transmitted cases of leprosy.2,10 We encourage clinicians to maintain a high level of clinical suspicion for leprosy when evaluating patients presenting with hypoesthetic cutaneous lesions and to obtain a travel history and to ask about armadillo exposure.
This case and the photos were submitted by Ms. Smith, from the University of South Florida, Tampa; Dr. Hatch and Dr. Sarriera-Lazaro, from the department of dermatology and cutaneous surgery, University of South Florida; and Dr. Turner and Dr. Beachkofsky, from the department of pathology and laboratory medicine at the James A. Haley Veterans’ Hospital, Tampa. Dr. Bilu Martin edited this case. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Leprosy (Hansen’s Disease), in: “Goldman’s Cecil Medicine,” 24th ed. (Philadelphia: W.B. Saunders, 2012: pp. 1950-4.
2. Sharma R et al. Emerg Infect Dis. 2015 Dec;21(12):2127-34.
3. Lane JE et al. J Am Acad Dermatol. 2006 Oct;55(4):714-6.
4. Clark BM et al. Am J Trop Med Hyg. 2008 Jun;78(6):962-7.
5. Bruce S et al. J Am Acad Dermatol. 2000 Aug;43(2 Pt 1):223-8.
6. Loughry WJ et al. J Wildl Dis. 2009 Jan;45(1):144-52.
7. FDo H. Florida charts: Hansen’s Disease (Leprosy). Health FDo. 2019. https://www.flhealthcharts.gov/ChartsReports/rdPage.aspx?rdReport=NonVitalIndNoGrpCounts.DataViewer&cid=174.
8. Maymone MBC et al. J Am Acad Dermatol. 2020 Jul;83(1):1-14.
9. Scollard DM et al. Clin Microbiol Rev. 2006 Apr;19(2):338-81.
10. Domozych R et al. JAAD Case Rep. 2016 May 12;2(3):189-92.
Paucibacillary tuberculoid leprosy is characterized by few anesthetic hypo- or hyperpigmented lesions and can be accompanied by palpable peripheral nerve enlargements.
Tuberculoid leprosy presents histologically with epithelioid histiocytes with lymphocytes and Langhans giant cells. Neurotropic granulomas are also characteristic of tuberculoid leprosy. Fite staining allows for the identification of the acid-fast bacilli of M. leprae, which in some cases are quite few in number. The standard mycobacterium stain, Ziehl-Neelsen, is a good option for M. tuberculosis, but because of the relative weak mycolic acid coat of M. leprae, the Fite stain is more appropriate for identifying M. leprae.
Clinically, other than the presence of fewer than five hypoesthetic lesions that are either hypopigmented or erythematous, tuberculoid leprosy often presents with additional peripheral nerve involvement that manifests as numbness and tingling in hands and feet.1 This patient denied any tingling, weakness, or numbness, outside of the anesthetic lesion on his posterior upper arm.
The patient, born in the United States, had a remote history of military travel to Iraq, Kuwait, and the Philippines, but had not traveled internationally within the last 15 years, apart from a cruise to the Bahamas. He denied any known contact with individuals with similar lesions. He denied a history of contact with armadillos, but acknowledged that they are native to where he resides in central Florida, and that he had seen them in his yard.
Histopathological examination revealed an unremarkable epidermis with a superficial and deep perivascular, periadnexal, and perineural lymphohistiocytic infiltrate. Fite stain revealed rare rod-shaped organisms (Figure 2). These findings are consistent with a diagnosis of paucibacillary, tuberculoid leprosy.
The patient’s travel history to highly endemic areas (Middle East), as well as possible environmental contact with armadillos – including contact with soil that the armadillos occupied – could explain plausible modes of transmission. Following consultation with our infectious disease department and the National Hansen’s Disease Program, our patient began a planned course of therapy with 18 months of minocycline, rifampin, and moxifloxacin.
Human-to-human transmission of HD has been well documented; however, zoonotic transmission – specifically via the nine-banded armadillo (Dasypus novemcinctus) – serves as another suggested means of transmission, especially in the Southeastern United States.2-6 Travel to highly-endemic areas increases the risk of contracting HD, which may take up to 20 years following contact with the bacteria to manifest clinically.
While central Florida was previously thought to be a nonendemic area of disease, the incidence of the disease in this region has increased in recent years.7 Human-to-human transmission, which remains a concern with immigration from highly-endemic regions, occurs via long-term contact with nasal droplets of an infected person.8,9
Many patients in regions with very few cases of leprosy deny travel to other endemic regions and contact with infected people. Thus, zoonotic transmission remains a legitimate concern in the Southeastern United States – accounting, at least in part, for many of the non–human-transmitted cases of leprosy.2,10 We encourage clinicians to maintain a high level of clinical suspicion for leprosy when evaluating patients presenting with hypoesthetic cutaneous lesions and to obtain a travel history and to ask about armadillo exposure.
This case and the photos were submitted by Ms. Smith, from the University of South Florida, Tampa; Dr. Hatch and Dr. Sarriera-Lazaro, from the department of dermatology and cutaneous surgery, University of South Florida; and Dr. Turner and Dr. Beachkofsky, from the department of pathology and laboratory medicine at the James A. Haley Veterans’ Hospital, Tampa. Dr. Bilu Martin edited this case. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Leprosy (Hansen’s Disease), in: “Goldman’s Cecil Medicine,” 24th ed. (Philadelphia: W.B. Saunders, 2012: pp. 1950-4.
2. Sharma R et al. Emerg Infect Dis. 2015 Dec;21(12):2127-34.
3. Lane JE et al. J Am Acad Dermatol. 2006 Oct;55(4):714-6.
4. Clark BM et al. Am J Trop Med Hyg. 2008 Jun;78(6):962-7.
5. Bruce S et al. J Am Acad Dermatol. 2000 Aug;43(2 Pt 1):223-8.
6. Loughry WJ et al. J Wildl Dis. 2009 Jan;45(1):144-52.
7. FDo H. Florida charts: Hansen’s Disease (Leprosy). Health FDo. 2019. https://www.flhealthcharts.gov/ChartsReports/rdPage.aspx?rdReport=NonVitalIndNoGrpCounts.DataViewer&cid=174.
8. Maymone MBC et al. J Am Acad Dermatol. 2020 Jul;83(1):1-14.
9. Scollard DM et al. Clin Microbiol Rev. 2006 Apr;19(2):338-81.
10. Domozych R et al. JAAD Case Rep. 2016 May 12;2(3):189-92.
A 44-year-old White male presented with a 1½-year history of a progressive hypoesthetic annular, mildly hyperpigmented plaque on the left posterior upper arm.
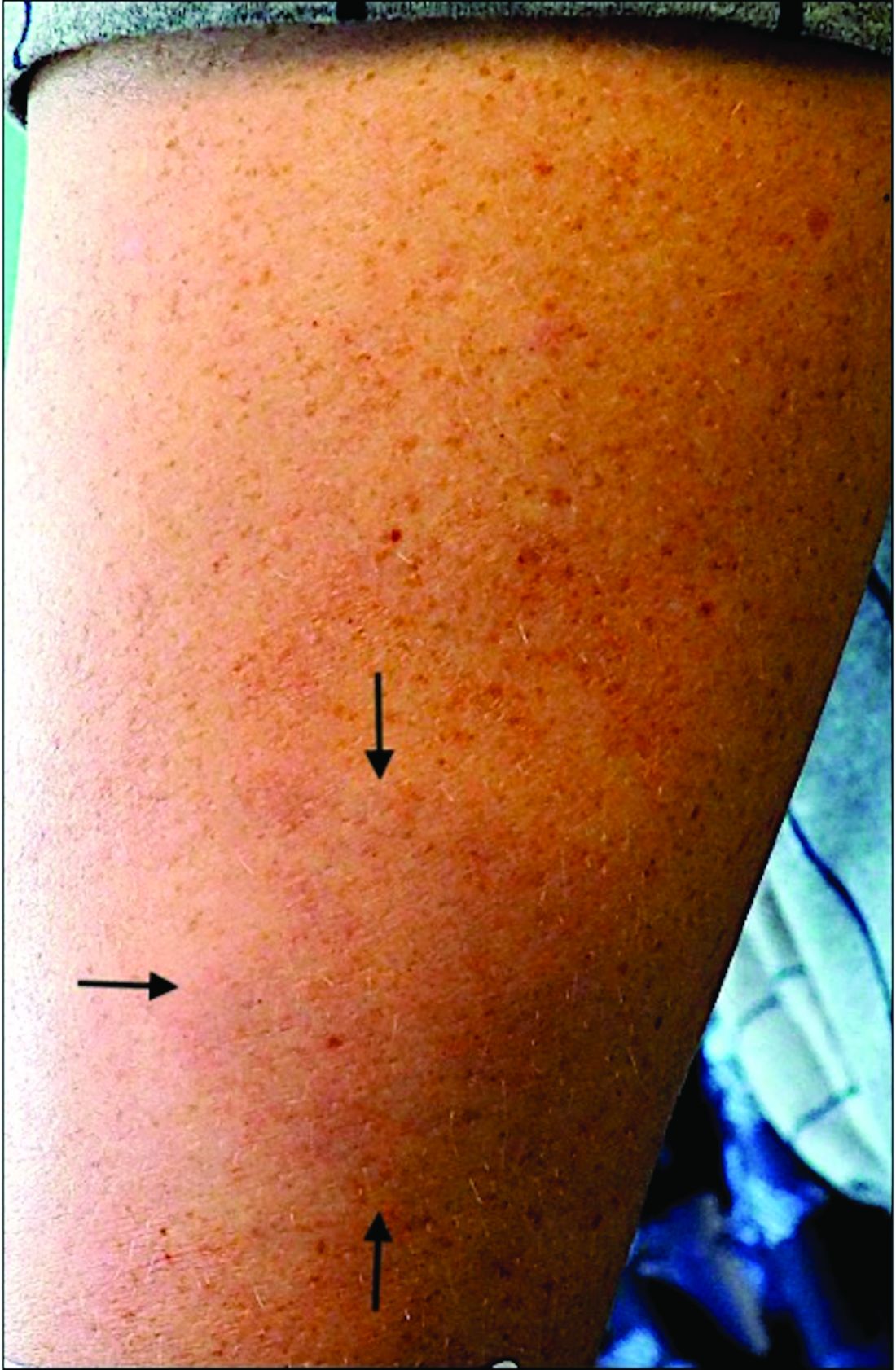
He denied pruritus, pain, or systemic symptoms including weight loss, visual changes, cough, dyspnea, and abdominal pain. He also denied any paresthesia or weakness. On physical examination, there is a subtle, solitary 4-cm annular skin-colored thin plaque on the patient's left posterior upper arm (Figure 1).
Punch biopsy of the lesion was performed, and the histopathological findings are illustrated in Figure 2.
More data back Guillain-Barré risk with Janssen COVID shot
Over 14 months, GBS reporting rates within 21 and 42 days of administration of Janssen’s replication-incompetent adenoviral vector vaccine were approximately 9 to 12 times higher than after administration of the Pfizer-BioNTech (BNT162b2) or the Moderna (mRNA-1273) mRNA COVID vaccines.
Additionally, observed GBS cases after the Janssen shot were 2 to 3 times greater than expected, based on background rates within 21 and 42 days of vaccination.
Conversely, and confirming prior data, there was no increased risk for GBS with the Pfizer or Moderna vaccines and no significant difference between observed and expected numbers of GBS cases after either mRNA COVID-19 vaccine.
The findings were published online in JAMA Network Open.
More precise risk estimates
Winston Abara, MD, with the U.S. Centers for Disease Control and Prevention, and colleagues analyzed GBS reports submitted to the VAERS between December 2020 and January 2022.
Among 487.6 million COVID-19 vaccine doses administered, 3.7% were Janssen’s Ad26.COV2.S vaccine, 54.7% were Pfizer’s BNT162b2 vaccine, and 41.6% were Moderna’s mRNA-1273 vaccine.
There were 295 verified reports of GBS identified after COVID-19 vaccination. Of these, 209 occurred within 21 days of vaccination and 253 within 42 days.
Within 21 days of vaccination, GBS reporting rates per 1 million doses were 3.29 for the Janssen vaccine versus 0.29 and 0.35 for the Pfizer and Moderna vaccines, respectively. Within 42 days of vaccination, reporting rates per 1 million doses were 4.07, 0.34, and 0.44, respectively.
Also within 21 days of vaccination, GBS reporting rates were significantly higher with the Janssen vaccine than the Pfizer vaccine (reporting rate ratio, 11.40) and the Moderna vaccine (RRR, 9.26). Similar findings were observed within 42 days after vaccination.
The observed-to-expected ratios were 3.79 for 21-day and 2.34 for 42-day intervals after receipt of the Janssen vaccine, and less than 1 (not significant) after the Pfizer or Moderna vaccine within both post-vaccination periods.
“Unlike prior studies, our analysis included all U.S. reports of verified GBS cases that met the Brighton Collaboration GBS case definition criteria (Brighton Levels 1, 2, and 3) submitted over a 14-month surveillance period to the to the Vaccine Adverse Event Reporting System,” Dr. Abara said in an interview. “Because we used all U.S. reports, the sample of verified GBS cases in this analysis is larger than other studies. Therefore, it may provide a more precise estimate of the GBS risk within 21 and 42 days after mRNA and Ad26.COV2.S vaccination,” he said.
‘Remarkably low’ use
Nicola Klein, MD, PhD, Kaiser Permanente Vaccine Study Center, Oakland, Calif., noted that this is a “nice confirmatory analysis that supports and further expands what’s been observed before.”
Last year, as reported by this news organization, Dr. Klein and colleagues reported data from the Vaccine Safety Datalink confirming a small but statistically significant increased risk for GBS in the 3 weeks after receipt of the Janssen COVID-19 vaccine but not the Pfizer or Moderna vaccines.
Unlike VAERS, the Vaccine Safety Datalink is not a reporting system. It’s an active surveillance of medical records in the Kaiser Permanente system. The VAERS is a passive system, so it requires individuals to report GBS cases to the VAERS team, Dr. Klein explained.
So although the two studies are slightly different, overall, the VAERS data is “consistent with what we found,” she said.
Also weighing in, C. Buddy Creech, MD, MPH, director of the Vanderbilt Vaccine Research Program and professor of pediatrics at the Vanderbilt University School of Medicine, Nashville, Tenn., said it is “important to realize that GBS had been observed after adenovirus-vectored vaccines earlier in the pandemic, both for the AstraZeneca vaccine and the Janssen vaccine.”
The Advisory Committee on Immunization Practices (ACIP) preferentially recommends that people age 18 years and older receive an mRNA COVID-19 vaccine rather than the Janssen adenoviral vector vaccine when both types of COVID-19 vaccine are available.
“Thus, the use of the Janssen vaccine is remarkably low in the U.S. right now,” Dr. Creech said.
“Nevertheless, we have a firm commitment, both scientifically and ethically, to track potential side effects after vaccination and to make sure that the vaccines in use for COVID, and other important infectious diseases, are safe and effective,” he added.
The study had no commercial funding. Dr. Abara and Dr. Creech have reported no relevant financial relationships. Dr. Klein reported having received grants from Pfizer research support for a COVID vaccine clinical trial, as well as grants from Merck, GlaxoSmithKline, Sanofi Pasteur, and Protein Science (now Sanofi Pasteur).
A version of this article first appeared on Medscape.com.
Over 14 months, GBS reporting rates within 21 and 42 days of administration of Janssen’s replication-incompetent adenoviral vector vaccine were approximately 9 to 12 times higher than after administration of the Pfizer-BioNTech (BNT162b2) or the Moderna (mRNA-1273) mRNA COVID vaccines.
Additionally, observed GBS cases after the Janssen shot were 2 to 3 times greater than expected, based on background rates within 21 and 42 days of vaccination.
Conversely, and confirming prior data, there was no increased risk for GBS with the Pfizer or Moderna vaccines and no significant difference between observed and expected numbers of GBS cases after either mRNA COVID-19 vaccine.
The findings were published online in JAMA Network Open.
More precise risk estimates
Winston Abara, MD, with the U.S. Centers for Disease Control and Prevention, and colleagues analyzed GBS reports submitted to the VAERS between December 2020 and January 2022.
Among 487.6 million COVID-19 vaccine doses administered, 3.7% were Janssen’s Ad26.COV2.S vaccine, 54.7% were Pfizer’s BNT162b2 vaccine, and 41.6% were Moderna’s mRNA-1273 vaccine.
There were 295 verified reports of GBS identified after COVID-19 vaccination. Of these, 209 occurred within 21 days of vaccination and 253 within 42 days.
Within 21 days of vaccination, GBS reporting rates per 1 million doses were 3.29 for the Janssen vaccine versus 0.29 and 0.35 for the Pfizer and Moderna vaccines, respectively. Within 42 days of vaccination, reporting rates per 1 million doses were 4.07, 0.34, and 0.44, respectively.
Also within 21 days of vaccination, GBS reporting rates were significantly higher with the Janssen vaccine than the Pfizer vaccine (reporting rate ratio, 11.40) and the Moderna vaccine (RRR, 9.26). Similar findings were observed within 42 days after vaccination.
The observed-to-expected ratios were 3.79 for 21-day and 2.34 for 42-day intervals after receipt of the Janssen vaccine, and less than 1 (not significant) after the Pfizer or Moderna vaccine within both post-vaccination periods.
“Unlike prior studies, our analysis included all U.S. reports of verified GBS cases that met the Brighton Collaboration GBS case definition criteria (Brighton Levels 1, 2, and 3) submitted over a 14-month surveillance period to the to the Vaccine Adverse Event Reporting System,” Dr. Abara said in an interview. “Because we used all U.S. reports, the sample of verified GBS cases in this analysis is larger than other studies. Therefore, it may provide a more precise estimate of the GBS risk within 21 and 42 days after mRNA and Ad26.COV2.S vaccination,” he said.
‘Remarkably low’ use
Nicola Klein, MD, PhD, Kaiser Permanente Vaccine Study Center, Oakland, Calif., noted that this is a “nice confirmatory analysis that supports and further expands what’s been observed before.”
Last year, as reported by this news organization, Dr. Klein and colleagues reported data from the Vaccine Safety Datalink confirming a small but statistically significant increased risk for GBS in the 3 weeks after receipt of the Janssen COVID-19 vaccine but not the Pfizer or Moderna vaccines.
Unlike VAERS, the Vaccine Safety Datalink is not a reporting system. It’s an active surveillance of medical records in the Kaiser Permanente system. The VAERS is a passive system, so it requires individuals to report GBS cases to the VAERS team, Dr. Klein explained.
So although the two studies are slightly different, overall, the VAERS data is “consistent with what we found,” she said.
Also weighing in, C. Buddy Creech, MD, MPH, director of the Vanderbilt Vaccine Research Program and professor of pediatrics at the Vanderbilt University School of Medicine, Nashville, Tenn., said it is “important to realize that GBS had been observed after adenovirus-vectored vaccines earlier in the pandemic, both for the AstraZeneca vaccine and the Janssen vaccine.”
The Advisory Committee on Immunization Practices (ACIP) preferentially recommends that people age 18 years and older receive an mRNA COVID-19 vaccine rather than the Janssen adenoviral vector vaccine when both types of COVID-19 vaccine are available.
“Thus, the use of the Janssen vaccine is remarkably low in the U.S. right now,” Dr. Creech said.
“Nevertheless, we have a firm commitment, both scientifically and ethically, to track potential side effects after vaccination and to make sure that the vaccines in use for COVID, and other important infectious diseases, are safe and effective,” he added.
The study had no commercial funding. Dr. Abara and Dr. Creech have reported no relevant financial relationships. Dr. Klein reported having received grants from Pfizer research support for a COVID vaccine clinical trial, as well as grants from Merck, GlaxoSmithKline, Sanofi Pasteur, and Protein Science (now Sanofi Pasteur).
A version of this article first appeared on Medscape.com.
Over 14 months, GBS reporting rates within 21 and 42 days of administration of Janssen’s replication-incompetent adenoviral vector vaccine were approximately 9 to 12 times higher than after administration of the Pfizer-BioNTech (BNT162b2) or the Moderna (mRNA-1273) mRNA COVID vaccines.
Additionally, observed GBS cases after the Janssen shot were 2 to 3 times greater than expected, based on background rates within 21 and 42 days of vaccination.
Conversely, and confirming prior data, there was no increased risk for GBS with the Pfizer or Moderna vaccines and no significant difference between observed and expected numbers of GBS cases after either mRNA COVID-19 vaccine.
The findings were published online in JAMA Network Open.
More precise risk estimates
Winston Abara, MD, with the U.S. Centers for Disease Control and Prevention, and colleagues analyzed GBS reports submitted to the VAERS between December 2020 and January 2022.
Among 487.6 million COVID-19 vaccine doses administered, 3.7% were Janssen’s Ad26.COV2.S vaccine, 54.7% were Pfizer’s BNT162b2 vaccine, and 41.6% were Moderna’s mRNA-1273 vaccine.
There were 295 verified reports of GBS identified after COVID-19 vaccination. Of these, 209 occurred within 21 days of vaccination and 253 within 42 days.
Within 21 days of vaccination, GBS reporting rates per 1 million doses were 3.29 for the Janssen vaccine versus 0.29 and 0.35 for the Pfizer and Moderna vaccines, respectively. Within 42 days of vaccination, reporting rates per 1 million doses were 4.07, 0.34, and 0.44, respectively.
Also within 21 days of vaccination, GBS reporting rates were significantly higher with the Janssen vaccine than the Pfizer vaccine (reporting rate ratio, 11.40) and the Moderna vaccine (RRR, 9.26). Similar findings were observed within 42 days after vaccination.
The observed-to-expected ratios were 3.79 for 21-day and 2.34 for 42-day intervals after receipt of the Janssen vaccine, and less than 1 (not significant) after the Pfizer or Moderna vaccine within both post-vaccination periods.
“Unlike prior studies, our analysis included all U.S. reports of verified GBS cases that met the Brighton Collaboration GBS case definition criteria (Brighton Levels 1, 2, and 3) submitted over a 14-month surveillance period to the to the Vaccine Adverse Event Reporting System,” Dr. Abara said in an interview. “Because we used all U.S. reports, the sample of verified GBS cases in this analysis is larger than other studies. Therefore, it may provide a more precise estimate of the GBS risk within 21 and 42 days after mRNA and Ad26.COV2.S vaccination,” he said.
‘Remarkably low’ use
Nicola Klein, MD, PhD, Kaiser Permanente Vaccine Study Center, Oakland, Calif., noted that this is a “nice confirmatory analysis that supports and further expands what’s been observed before.”
Last year, as reported by this news organization, Dr. Klein and colleagues reported data from the Vaccine Safety Datalink confirming a small but statistically significant increased risk for GBS in the 3 weeks after receipt of the Janssen COVID-19 vaccine but not the Pfizer or Moderna vaccines.
Unlike VAERS, the Vaccine Safety Datalink is not a reporting system. It’s an active surveillance of medical records in the Kaiser Permanente system. The VAERS is a passive system, so it requires individuals to report GBS cases to the VAERS team, Dr. Klein explained.
So although the two studies are slightly different, overall, the VAERS data is “consistent with what we found,” she said.
Also weighing in, C. Buddy Creech, MD, MPH, director of the Vanderbilt Vaccine Research Program and professor of pediatrics at the Vanderbilt University School of Medicine, Nashville, Tenn., said it is “important to realize that GBS had been observed after adenovirus-vectored vaccines earlier in the pandemic, both for the AstraZeneca vaccine and the Janssen vaccine.”
The Advisory Committee on Immunization Practices (ACIP) preferentially recommends that people age 18 years and older receive an mRNA COVID-19 vaccine rather than the Janssen adenoviral vector vaccine when both types of COVID-19 vaccine are available.
“Thus, the use of the Janssen vaccine is remarkably low in the U.S. right now,” Dr. Creech said.
“Nevertheless, we have a firm commitment, both scientifically and ethically, to track potential side effects after vaccination and to make sure that the vaccines in use for COVID, and other important infectious diseases, are safe and effective,” he added.
The study had no commercial funding. Dr. Abara and Dr. Creech have reported no relevant financial relationships. Dr. Klein reported having received grants from Pfizer research support for a COVID vaccine clinical trial, as well as grants from Merck, GlaxoSmithKline, Sanofi Pasteur, and Protein Science (now Sanofi Pasteur).
A version of this article first appeared on Medscape.com.
Topical gene therapy heals dystrophic epidermolysis bullosa wounds
.
In a phase 3 study of patients with DEB, “we found that repeated topical application of B-VEC [beremagene geperpavec], an HSV-1–based gene therapy, resulted in a greater likelihood of complete wound healing than the topical application of placebo at up to 6 months,” the authors wrote. The study was published in The New England Journal of Medicine. “Longer and larger trials are warranted to determine the durability of effect and risks of this approach,” the authors noted.
“The results prove that B-VEC, the first topical in vivo gene therapy to reach late-stage development, can heal DEB,” senior author M. Peter Marinkovich, MD, associate professor of dermatology at Stanford University, Redwood City, Calif., said in an interview.

“In the past, DEB was a very specialized disease that only a handful of dermatologists would see but could not do much to treat,” he said. “With gene therapy, many more dermatologists who may not be familiar with DEB will be able to treat these patients in their offices.” It is expected that nurses will be able to administer the treatment to patients at home, he added.
Rare, life-threatening, genetic blistering disease
DEB, a rare disease that affects one to three persons per million in the United States, is caused by mutations in the COL7A1 gene that encodes the alpha-1 chain of collagen type VII (C7) protein. C7 forms the anchoring fibrils that attach the epidermis to the underlying dermal connective tissue.
COL71A mutations that lead to defective, decreased, or absent C7 can make the skin so fragile it tears with the slightest touch. This has led to patients being called “butterfly children.” Epithelial tissues blister and scar, causing esophageal and genitourinary strictures, adhesion of digits, malnutrition, anemia, infection, and bothersome itch and pain. Morbidity and mortality are high. The leading cause of death in adults is chronic wounds leading to aggressive squamous cell cancers.
The first therapy for DEB, under FDA review
B-VEC restores C7 protein by using an engineered replication-defective herpes simplex virus type 1 (HSV-1) vector to deliver the COL7A1 gene directly to skin cells to restore functional C7 protein fibrils that stabilize the skin structure.
On the basis of manufacturing information submitted to the FDA in December 2022, the agency extended the date for a decision on approval by 3 months, to May 19, 2023, according to a statement from Krystal Biotech, the developer of B-VEC and the sponsor of the NEJM study.
Dr. Marinkovich and his colleagues conducted the double-blind, randomized, controlled GEM-3 trial of B-VEC at three sites in the United States. The 31 study participants ranged in age from 1 to 44 years (median age, 16 years) and had genetically confirmed DEB (30 with the recessive form and 1 with the dominant form).
For each participant, a pair of wounds was chosen that were matched in size, region, and appearance. The wounds within each pair were randomly allocated to receive weekly applications of either B-VEC or placebo gel for 26 weeks.
The results of the study included the following:
- Complete healing at 6 months occurred in 67% of the wounds treated with B-VEC (including a wound in the patient with dominant DEB), vs. 22% of those who received placebo (95% confidence interval [CI], 24-68; P = .002).
- Complete healing at 3 months occurred in 71% of the wounds treated with B-VEC, vs. 20% of those who received placebo (95% CI, 29-73; P < .001).
- The mean change from baseline to week 22 in pain severity during wound-dressing changes for patients aged 6 years and older, as determined on the basis of a visual analogue scale, was –0.88 with B-VEC, vs. –0.71 with placebo (adjusted least-squares mean difference, –0.61; 95% CI, –1.10 to –0.13); similar mean changes were seen at weeks 24 and 26.
- Among all patients, 58% had at least one adverse event. Most adverse events were mild or moderate. The most common were pruritus, chills, and squamous cell carcinoma (SCC), which were reported in three patients each (SCC cases occurred at wound sites that had not been exposed to B-VEC or placebo). Serious adverse events, which were unrelated to the treatment, occurred in three patients: diarrhea, anemia, cellulitis, and a positive blood culture related to a hemodialysis catheter.
“With the ability to treat patients with topical gene therapy, dermatology is entering a new age of treatment possibilities,” Dr. Marinkovich said in the interview.
The researchers were surprised that the redosable in vivo gene therapy worked so well, he added. In vivo gene therapy has been plagued by the occurrence of immune reactions against the viral vectors used, Dr. Marinkovich explained. But because the herpes simplex virus has evolved to evade the immune system, his team could use the viral vector every week for 6 months without inflammatory reactions.
“The immune system’s inability to fight off or get rid of the herpes simplex vector makes it bad as a disease, but as a gene therapy vector, it provides a huge advantage,” he added.
Asked to comment on the results, Christen Ebens, MD, MPH, assistant professor in the department of pediatrics at the University of Minnesota, Minneapolis, whose clinical and research interests include EB, called the results exciting for patients, families, and doctors.

“Side effects were minimal, and importantly, use of the replication-incompetent HSV vector means that the payload gene does not integrate into the patient’s DNA,” Dr. Ebens, who was not involved in the study, said in an interview. “B-VEC is not a lifelong cure but potentially an effective maintenance therapy requiring repeated doses,” she added.
Although the researchers found no clinically important immune reactions to B-VEC, Dr. Ebens said she would like to see results from longer studies of the treatment. “We will want to see that patients do not produce neutralizing antibodies against B-VEC or its components, as such antibodies may yield the treatment ineffective or cause significant side effects.”
In an interview, Vanessa R. Holland, MD, associate clinical professor in the division of dermatology at UCLA Health, Burbank, Calif., who was not involved in the study, said that “topical replication-defective HSV-1 is a brilliant vector to deliver the depleted collagen.” She added that “such a vehicle may significantly alter management of these disorders and improve or extend lives by minimizing potentially fatal complications.”
Paras P. Vakharia, MD, PharmD, assistant professor of dermatology at Northwestern University, Chicago, who was not involved in the study, was surprised by the high percentage of healed wounds and wounds that remained healed over time.

In an interview, Dr. Vakharia said that he’d like to know whether patients develop antibodies against HSV and C7 with long-term treatment and whether problems will arise related to drug availability.
B-VEC for treating other conditions
Dr. Marinkovich noted that an ongoing phase 1 clinical trial, also sponsored by Krystal Biotech, is using the HSV-1 vector to deliver a different biologic (KB105) to establish dose and safety in the treatment of ichthyosis. He added that he would like to explore the use of B-VEC to treat DEB at mucosal surfaces, including inside the mouth, the eye, and the esophagus.
Authors of two editorials that accompanied the study also referred to other conditions B-VEC might treat.
This study “highlights potential future investigations,” David V. Schaffer, PhD, professor of chemical and biomolecular engineering, bioengineering, and molecular and cell biology at the University of California, Berkeley, wrotes in one of the editorials.
Important considerations he mentioned include the likelihood of the treatment becoming lifelong; the inability of HSV to penetrate intact skin, making B-VEC unsuitable for preventing the development of new wounds; and the inability of this treatment to treat EB lesions along the digestive tract. “This important trial builds on and extends gene-therapy successes to new targets and vectors, an advance for patients,” he added.
In the second editorial, Aimee S. Payne, MD, PhD, professor of dermatology at the University of Pennsylvania, Philadelphia, raised the question of whether B-VEC’s clinical success for treating DEB can translate to other genetic diseases.
“Formulations for ophthalmic, oral-gastrointestinal, or respiratory delivery would be of great value to address the extracutaneous manifestations of epidermolysis bullosa and other genetic diseases,” she wrote.
Referring to an ongoing trial of a topical gene therapy for cystic fibrosis that is delivered by a nebulizer, Dr. Payne noted, “Ultimately, the completion of clinical trials such as this one will be required to determine whether HSV-1–mediated gene delivery can go more than skin deep.”
Earlier data and more details of the study were presented in a poster at the annual meeting of the Society for Pediatric Dermatology in July 2022.
Dr. Marinkovich has disclosed no relevant financial relationships. Several coauthors are employees of or have other financial relationships with Krystal Biotech, the study’s sponsor and the developer of beremagene geperpavec. Dr. Schaffer and Dr. Payne have financial relationships with the pharmaceutical industry. Dr. Ebens, Dr. Holland, and Dr. Vakharia have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
.
In a phase 3 study of patients with DEB, “we found that repeated topical application of B-VEC [beremagene geperpavec], an HSV-1–based gene therapy, resulted in a greater likelihood of complete wound healing than the topical application of placebo at up to 6 months,” the authors wrote. The study was published in The New England Journal of Medicine. “Longer and larger trials are warranted to determine the durability of effect and risks of this approach,” the authors noted.
“The results prove that B-VEC, the first topical in vivo gene therapy to reach late-stage development, can heal DEB,” senior author M. Peter Marinkovich, MD, associate professor of dermatology at Stanford University, Redwood City, Calif., said in an interview.
“In the past, DEB was a very specialized disease that only a handful of dermatologists would see but could not do much to treat,” he said. “With gene therapy, many more dermatologists who may not be familiar with DEB will be able to treat these patients in their offices.” It is expected that nurses will be able to administer the treatment to patients at home, he added.
Rare, life-threatening, genetic blistering disease
DEB, a rare disease that affects one to three persons per million in the United States, is caused by mutations in the COL7A1 gene that encodes the alpha-1 chain of collagen type VII (C7) protein. C7 forms the anchoring fibrils that attach the epidermis to the underlying dermal connective tissue.
COL71A mutations that lead to defective, decreased, or absent C7 can make the skin so fragile it tears with the slightest touch. This has led to patients being called “butterfly children.” Epithelial tissues blister and scar, causing esophageal and genitourinary strictures, adhesion of digits, malnutrition, anemia, infection, and bothersome itch and pain. Morbidity and mortality are high. The leading cause of death in adults is chronic wounds leading to aggressive squamous cell cancers.
The first therapy for DEB, under FDA review
B-VEC restores C7 protein by using an engineered replication-defective herpes simplex virus type 1 (HSV-1) vector to deliver the COL7A1 gene directly to skin cells to restore functional C7 protein fibrils that stabilize the skin structure.
On the basis of manufacturing information submitted to the FDA in December 2022, the agency extended the date for a decision on approval by 3 months, to May 19, 2023, according to a statement from Krystal Biotech, the developer of B-VEC and the sponsor of the NEJM study.
Dr. Marinkovich and his colleagues conducted the double-blind, randomized, controlled GEM-3 trial of B-VEC at three sites in the United States. The 31 study participants ranged in age from 1 to 44 years (median age, 16 years) and had genetically confirmed DEB (30 with the recessive form and 1 with the dominant form).
For each participant, a pair of wounds was chosen that were matched in size, region, and appearance. The wounds within each pair were randomly allocated to receive weekly applications of either B-VEC or placebo gel for 26 weeks.
The results of the study included the following:
- Complete healing at 6 months occurred in 67% of the wounds treated with B-VEC (including a wound in the patient with dominant DEB), vs. 22% of those who received placebo (95% confidence interval [CI], 24-68; P = .002).
- Complete healing at 3 months occurred in 71% of the wounds treated with B-VEC, vs. 20% of those who received placebo (95% CI, 29-73; P < .001).
- The mean change from baseline to week 22 in pain severity during wound-dressing changes for patients aged 6 years and older, as determined on the basis of a visual analogue scale, was –0.88 with B-VEC, vs. –0.71 with placebo (adjusted least-squares mean difference, –0.61; 95% CI, –1.10 to –0.13); similar mean changes were seen at weeks 24 and 26.
- Among all patients, 58% had at least one adverse event. Most adverse events were mild or moderate. The most common were pruritus, chills, and squamous cell carcinoma (SCC), which were reported in three patients each (SCC cases occurred at wound sites that had not been exposed to B-VEC or placebo). Serious adverse events, which were unrelated to the treatment, occurred in three patients: diarrhea, anemia, cellulitis, and a positive blood culture related to a hemodialysis catheter.
“With the ability to treat patients with topical gene therapy, dermatology is entering a new age of treatment possibilities,” Dr. Marinkovich said in the interview.
The researchers were surprised that the redosable in vivo gene therapy worked so well, he added. In vivo gene therapy has been plagued by the occurrence of immune reactions against the viral vectors used, Dr. Marinkovich explained. But because the herpes simplex virus has evolved to evade the immune system, his team could use the viral vector every week for 6 months without inflammatory reactions.
“The immune system’s inability to fight off or get rid of the herpes simplex vector makes it bad as a disease, but as a gene therapy vector, it provides a huge advantage,” he added.
Asked to comment on the results, Christen Ebens, MD, MPH, assistant professor in the department of pediatrics at the University of Minnesota, Minneapolis, whose clinical and research interests include EB, called the results exciting for patients, families, and doctors.
“Side effects were minimal, and importantly, use of the replication-incompetent HSV vector means that the payload gene does not integrate into the patient’s DNA,” Dr. Ebens, who was not involved in the study, said in an interview. “B-VEC is not a lifelong cure but potentially an effective maintenance therapy requiring repeated doses,” she added.
Although the researchers found no clinically important immune reactions to B-VEC, Dr. Ebens said she would like to see results from longer studies of the treatment. “We will want to see that patients do not produce neutralizing antibodies against B-VEC or its components, as such antibodies may yield the treatment ineffective or cause significant side effects.”
In an interview, Vanessa R. Holland, MD, associate clinical professor in the division of dermatology at UCLA Health, Burbank, Calif., who was not involved in the study, said that “topical replication-defective HSV-1 is a brilliant vector to deliver the depleted collagen.” She added that “such a vehicle may significantly alter management of these disorders and improve or extend lives by minimizing potentially fatal complications.”
Paras P. Vakharia, MD, PharmD, assistant professor of dermatology at Northwestern University, Chicago, who was not involved in the study, was surprised by the high percentage of healed wounds and wounds that remained healed over time.
In an interview, Dr. Vakharia said that he’d like to know whether patients develop antibodies against HSV and C7 with long-term treatment and whether problems will arise related to drug availability.
B-VEC for treating other conditions
Dr. Marinkovich noted that an ongoing phase 1 clinical trial, also sponsored by Krystal Biotech, is using the HSV-1 vector to deliver a different biologic (KB105) to establish dose and safety in the treatment of ichthyosis. He added that he would like to explore the use of B-VEC to treat DEB at mucosal surfaces, including inside the mouth, the eye, and the esophagus.
Authors of two editorials that accompanied the study also referred to other conditions B-VEC might treat.
This study “highlights potential future investigations,” David V. Schaffer, PhD, professor of chemical and biomolecular engineering, bioengineering, and molecular and cell biology at the University of California, Berkeley, wrotes in one of the editorials.
Important considerations he mentioned include the likelihood of the treatment becoming lifelong; the inability of HSV to penetrate intact skin, making B-VEC unsuitable for preventing the development of new wounds; and the inability of this treatment to treat EB lesions along the digestive tract. “This important trial builds on and extends gene-therapy successes to new targets and vectors, an advance for patients,” he added.
In the second editorial, Aimee S. Payne, MD, PhD, professor of dermatology at the University of Pennsylvania, Philadelphia, raised the question of whether B-VEC’s clinical success for treating DEB can translate to other genetic diseases.
“Formulations for ophthalmic, oral-gastrointestinal, or respiratory delivery would be of great value to address the extracutaneous manifestations of epidermolysis bullosa and other genetic diseases,” she wrote.
Referring to an ongoing trial of a topical gene therapy for cystic fibrosis that is delivered by a nebulizer, Dr. Payne noted, “Ultimately, the completion of clinical trials such as this one will be required to determine whether HSV-1–mediated gene delivery can go more than skin deep.”
Earlier data and more details of the study were presented in a poster at the annual meeting of the Society for Pediatric Dermatology in July 2022.
Dr. Marinkovich has disclosed no relevant financial relationships. Several coauthors are employees of or have other financial relationships with Krystal Biotech, the study’s sponsor and the developer of beremagene geperpavec. Dr. Schaffer and Dr. Payne have financial relationships with the pharmaceutical industry. Dr. Ebens, Dr. Holland, and Dr. Vakharia have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
.
In a phase 3 study of patients with DEB, “we found that repeated topical application of B-VEC [beremagene geperpavec], an HSV-1–based gene therapy, resulted in a greater likelihood of complete wound healing than the topical application of placebo at up to 6 months,” the authors wrote. The study was published in The New England Journal of Medicine. “Longer and larger trials are warranted to determine the durability of effect and risks of this approach,” the authors noted.
“The results prove that B-VEC, the first topical in vivo gene therapy to reach late-stage development, can heal DEB,” senior author M. Peter Marinkovich, MD, associate professor of dermatology at Stanford University, Redwood City, Calif., said in an interview.
“In the past, DEB was a very specialized disease that only a handful of dermatologists would see but could not do much to treat,” he said. “With gene therapy, many more dermatologists who may not be familiar with DEB will be able to treat these patients in their offices.” It is expected that nurses will be able to administer the treatment to patients at home, he added.
Rare, life-threatening, genetic blistering disease
DEB, a rare disease that affects one to three persons per million in the United States, is caused by mutations in the COL7A1 gene that encodes the alpha-1 chain of collagen type VII (C7) protein. C7 forms the anchoring fibrils that attach the epidermis to the underlying dermal connective tissue.
COL71A mutations that lead to defective, decreased, or absent C7 can make the skin so fragile it tears with the slightest touch. This has led to patients being called “butterfly children.” Epithelial tissues blister and scar, causing esophageal and genitourinary strictures, adhesion of digits, malnutrition, anemia, infection, and bothersome itch and pain. Morbidity and mortality are high. The leading cause of death in adults is chronic wounds leading to aggressive squamous cell cancers.
The first therapy for DEB, under FDA review
B-VEC restores C7 protein by using an engineered replication-defective herpes simplex virus type 1 (HSV-1) vector to deliver the COL7A1 gene directly to skin cells to restore functional C7 protein fibrils that stabilize the skin structure.
On the basis of manufacturing information submitted to the FDA in December 2022, the agency extended the date for a decision on approval by 3 months, to May 19, 2023, according to a statement from Krystal Biotech, the developer of B-VEC and the sponsor of the NEJM study.
Dr. Marinkovich and his colleagues conducted the double-blind, randomized, controlled GEM-3 trial of B-VEC at three sites in the United States. The 31 study participants ranged in age from 1 to 44 years (median age, 16 years) and had genetically confirmed DEB (30 with the recessive form and 1 with the dominant form).
For each participant, a pair of wounds was chosen that were matched in size, region, and appearance. The wounds within each pair were randomly allocated to receive weekly applications of either B-VEC or placebo gel for 26 weeks.
The results of the study included the following:
- Complete healing at 6 months occurred in 67% of the wounds treated with B-VEC (including a wound in the patient with dominant DEB), vs. 22% of those who received placebo (95% confidence interval [CI], 24-68; P = .002).
- Complete healing at 3 months occurred in 71% of the wounds treated with B-VEC, vs. 20% of those who received placebo (95% CI, 29-73; P < .001).
- The mean change from baseline to week 22 in pain severity during wound-dressing changes for patients aged 6 years and older, as determined on the basis of a visual analogue scale, was –0.88 with B-VEC, vs. –0.71 with placebo (adjusted least-squares mean difference, –0.61; 95% CI, –1.10 to –0.13); similar mean changes were seen at weeks 24 and 26.
- Among all patients, 58% had at least one adverse event. Most adverse events were mild or moderate. The most common were pruritus, chills, and squamous cell carcinoma (SCC), which were reported in three patients each (SCC cases occurred at wound sites that had not been exposed to B-VEC or placebo). Serious adverse events, which were unrelated to the treatment, occurred in three patients: diarrhea, anemia, cellulitis, and a positive blood culture related to a hemodialysis catheter.
“With the ability to treat patients with topical gene therapy, dermatology is entering a new age of treatment possibilities,” Dr. Marinkovich said in the interview.
The researchers were surprised that the redosable in vivo gene therapy worked so well, he added. In vivo gene therapy has been plagued by the occurrence of immune reactions against the viral vectors used, Dr. Marinkovich explained. But because the herpes simplex virus has evolved to evade the immune system, his team could use the viral vector every week for 6 months without inflammatory reactions.
“The immune system’s inability to fight off or get rid of the herpes simplex vector makes it bad as a disease, but as a gene therapy vector, it provides a huge advantage,” he added.
Asked to comment on the results, Christen Ebens, MD, MPH, assistant professor in the department of pediatrics at the University of Minnesota, Minneapolis, whose clinical and research interests include EB, called the results exciting for patients, families, and doctors.
“Side effects were minimal, and importantly, use of the replication-incompetent HSV vector means that the payload gene does not integrate into the patient’s DNA,” Dr. Ebens, who was not involved in the study, said in an interview. “B-VEC is not a lifelong cure but potentially an effective maintenance therapy requiring repeated doses,” she added.
Although the researchers found no clinically important immune reactions to B-VEC, Dr. Ebens said she would like to see results from longer studies of the treatment. “We will want to see that patients do not produce neutralizing antibodies against B-VEC or its components, as such antibodies may yield the treatment ineffective or cause significant side effects.”
In an interview, Vanessa R. Holland, MD, associate clinical professor in the division of dermatology at UCLA Health, Burbank, Calif., who was not involved in the study, said that “topical replication-defective HSV-1 is a brilliant vector to deliver the depleted collagen.” She added that “such a vehicle may significantly alter management of these disorders and improve or extend lives by minimizing potentially fatal complications.”
Paras P. Vakharia, MD, PharmD, assistant professor of dermatology at Northwestern University, Chicago, who was not involved in the study, was surprised by the high percentage of healed wounds and wounds that remained healed over time.
In an interview, Dr. Vakharia said that he’d like to know whether patients develop antibodies against HSV and C7 with long-term treatment and whether problems will arise related to drug availability.
B-VEC for treating other conditions
Dr. Marinkovich noted that an ongoing phase 1 clinical trial, also sponsored by Krystal Biotech, is using the HSV-1 vector to deliver a different biologic (KB105) to establish dose and safety in the treatment of ichthyosis. He added that he would like to explore the use of B-VEC to treat DEB at mucosal surfaces, including inside the mouth, the eye, and the esophagus.
Authors of two editorials that accompanied the study also referred to other conditions B-VEC might treat.
This study “highlights potential future investigations,” David V. Schaffer, PhD, professor of chemical and biomolecular engineering, bioengineering, and molecular and cell biology at the University of California, Berkeley, wrotes in one of the editorials.
Important considerations he mentioned include the likelihood of the treatment becoming lifelong; the inability of HSV to penetrate intact skin, making B-VEC unsuitable for preventing the development of new wounds; and the inability of this treatment to treat EB lesions along the digestive tract. “This important trial builds on and extends gene-therapy successes to new targets and vectors, an advance for patients,” he added.
In the second editorial, Aimee S. Payne, MD, PhD, professor of dermatology at the University of Pennsylvania, Philadelphia, raised the question of whether B-VEC’s clinical success for treating DEB can translate to other genetic diseases.
“Formulations for ophthalmic, oral-gastrointestinal, or respiratory delivery would be of great value to address the extracutaneous manifestations of epidermolysis bullosa and other genetic diseases,” she wrote.
Referring to an ongoing trial of a topical gene therapy for cystic fibrosis that is delivered by a nebulizer, Dr. Payne noted, “Ultimately, the completion of clinical trials such as this one will be required to determine whether HSV-1–mediated gene delivery can go more than skin deep.”
Earlier data and more details of the study were presented in a poster at the annual meeting of the Society for Pediatric Dermatology in July 2022.
Dr. Marinkovich has disclosed no relevant financial relationships. Several coauthors are employees of or have other financial relationships with Krystal Biotech, the study’s sponsor and the developer of beremagene geperpavec. Dr. Schaffer and Dr. Payne have financial relationships with the pharmaceutical industry. Dr. Ebens, Dr. Holland, and Dr. Vakharia have disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Expert gives tips on less-discussed dermatologic diseases
ORLANDO – , according to Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington.
These semi-forsaken diseases are important not to miss and can “also be quite challenging when we think about their management,” he said at the ODAC Dermatology, Aesthetic & Surgical Conference.

Dr. Friedman, also director of the GW dermatology residency program, reviewed several of these diseases – along with tips for management – during a session at the meeting.
. It does not always have the classic ring pattern for which it is best known, he said. And in patients with darker skin tones, it is characterized by more of a brown or black color, rather than the pink-red color.
Dr. Friedman said that despite a kind of “Pavlovian response” linking GA with diabetes, this link might not be as strong as the field has come to believe, since the studies on which this belief was based included a patient population with narrow demographics. “Maybe GA and type 1 diabetes aren’t necessarily connected,” he said.
Dyslipidemia, on the other hand, has a strong connection with GA, he said. The disease is also linked to thyroid disease and is linked with malignancy, especially in older patients with generalized or atypical presentations of GA, he said.
Spontaneous resolution of the disease is seen within 2 years for 50% to 75% of patients, so “no treatment may be the best treatment,” but antimalarials can be effective, Dr. Friedman said. “I use antimalarials frequently in my practice,” he said. “The key is, they take time to work (4-5 months),” which should be explained to patients.
Antibiotics, he said, can be “somewhat effective,” but in the case of doxycycline at least, the disease can resolve within weeks but then may return when treatment is stopped.
There is some evidence to support using biologics and more recently, Janus kinase (JAK) inhibitors, off-label, to treat GA. Efficacy has been seen with the tumor necrosis factor (TNF) blocker infliximab and with the JAK inhibitor tofacitinib, he said.
Lichen planus (LP). This is another common disease that can go off-script with its presentation. The disease is often described with the “six P’s” indicating the following characteristics: pruritic, polygonal, planar or flat-topped, purple papules, and plaques. But LP “didn’t read the textbook,” Dr. Friedman said.
“The clinical presentation of lichen planus can be quite broad,” he said. “The P’s aren’t always followed as there are a variety of colors and configurations which can be witnessed.”
With LP, there is a clear association with dyslipidemia and diabetes, so “asking the right questions is going to be important” when talking to the patient. There is also a higher risk of autoimmune diseases, especially of the thyroid type, associated with LP, he said.
No treatment has been Food and Drug Administration approved for LP, but some are expected in the future, he said.
For now, he emphasized creativity in the management of patients with LP. “I love oral retinoids for this,” he said. Antimalarials and methotrexate are also options.
In one case Dr. Friedman saw, nothing seemed to work: light therapy for a year; metronidazole; isotretinoin; halobetasol/tazarotene lotion; and the TNF-blocker adalimumab either weren’t effective or resulted in complications in the patient.
Knowing the recent implication of the interleukin (IL)-17 pathway in the pathophysiology of LP, he then tried the anti-IL17 antibody secukinumab. “This patient had a pretty robust response to treatment,” Dr. Friedman said. “He was very excited. The problem, as always, is access, especially for off-label therapies.”
Tumid lupus erythematosus. This disease is characterized by erythematous, edematous, nonscarring plaques on sun-exposed sites. For treatment, Dr. Friedman said antimalarials can be up to 90% effective, sometimes with rapid resolution of the lesions.
“You want to dose below that 5 mg per kg of true body weight to limit the small potential for ocular toxicity over time,” he said. And, he emphasized, “always combine treatment with good sun-protective measures.”
Dr. Friedman reported financial relationships with Sanova, Pfizer, Novartis, and other companies.
ORLANDO – , according to Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington.
These semi-forsaken diseases are important not to miss and can “also be quite challenging when we think about their management,” he said at the ODAC Dermatology, Aesthetic & Surgical Conference.
Dr. Friedman, also director of the GW dermatology residency program, reviewed several of these diseases – along with tips for management – during a session at the meeting.
. It does not always have the classic ring pattern for which it is best known, he said. And in patients with darker skin tones, it is characterized by more of a brown or black color, rather than the pink-red color.
Dr. Friedman said that despite a kind of “Pavlovian response” linking GA with diabetes, this link might not be as strong as the field has come to believe, since the studies on which this belief was based included a patient population with narrow demographics. “Maybe GA and type 1 diabetes aren’t necessarily connected,” he said.
Dyslipidemia, on the other hand, has a strong connection with GA, he said. The disease is also linked to thyroid disease and is linked with malignancy, especially in older patients with generalized or atypical presentations of GA, he said.
Spontaneous resolution of the disease is seen within 2 years for 50% to 75% of patients, so “no treatment may be the best treatment,” but antimalarials can be effective, Dr. Friedman said. “I use antimalarials frequently in my practice,” he said. “The key is, they take time to work (4-5 months),” which should be explained to patients.
Antibiotics, he said, can be “somewhat effective,” but in the case of doxycycline at least, the disease can resolve within weeks but then may return when treatment is stopped.
There is some evidence to support using biologics and more recently, Janus kinase (JAK) inhibitors, off-label, to treat GA. Efficacy has been seen with the tumor necrosis factor (TNF) blocker infliximab and with the JAK inhibitor tofacitinib, he said.
Lichen planus (LP). This is another common disease that can go off-script with its presentation. The disease is often described with the “six P’s” indicating the following characteristics: pruritic, polygonal, planar or flat-topped, purple papules, and plaques. But LP “didn’t read the textbook,” Dr. Friedman said.
“The clinical presentation of lichen planus can be quite broad,” he said. “The P’s aren’t always followed as there are a variety of colors and configurations which can be witnessed.”
With LP, there is a clear association with dyslipidemia and diabetes, so “asking the right questions is going to be important” when talking to the patient. There is also a higher risk of autoimmune diseases, especially of the thyroid type, associated with LP, he said.
No treatment has been Food and Drug Administration approved for LP, but some are expected in the future, he said.
For now, he emphasized creativity in the management of patients with LP. “I love oral retinoids for this,” he said. Antimalarials and methotrexate are also options.
In one case Dr. Friedman saw, nothing seemed to work: light therapy for a year; metronidazole; isotretinoin; halobetasol/tazarotene lotion; and the TNF-blocker adalimumab either weren’t effective or resulted in complications in the patient.
Knowing the recent implication of the interleukin (IL)-17 pathway in the pathophysiology of LP, he then tried the anti-IL17 antibody secukinumab. “This patient had a pretty robust response to treatment,” Dr. Friedman said. “He was very excited. The problem, as always, is access, especially for off-label therapies.”
Tumid lupus erythematosus. This disease is characterized by erythematous, edematous, nonscarring plaques on sun-exposed sites. For treatment, Dr. Friedman said antimalarials can be up to 90% effective, sometimes with rapid resolution of the lesions.
“You want to dose below that 5 mg per kg of true body weight to limit the small potential for ocular toxicity over time,” he said. And, he emphasized, “always combine treatment with good sun-protective measures.”
Dr. Friedman reported financial relationships with Sanova, Pfizer, Novartis, and other companies.
ORLANDO – , according to Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington.
These semi-forsaken diseases are important not to miss and can “also be quite challenging when we think about their management,” he said at the ODAC Dermatology, Aesthetic & Surgical Conference.
Dr. Friedman, also director of the GW dermatology residency program, reviewed several of these diseases – along with tips for management – during a session at the meeting.
. It does not always have the classic ring pattern for which it is best known, he said. And in patients with darker skin tones, it is characterized by more of a brown or black color, rather than the pink-red color.
Dr. Friedman said that despite a kind of “Pavlovian response” linking GA with diabetes, this link might not be as strong as the field has come to believe, since the studies on which this belief was based included a patient population with narrow demographics. “Maybe GA and type 1 diabetes aren’t necessarily connected,” he said.
Dyslipidemia, on the other hand, has a strong connection with GA, he said. The disease is also linked to thyroid disease and is linked with malignancy, especially in older patients with generalized or atypical presentations of GA, he said.
Spontaneous resolution of the disease is seen within 2 years for 50% to 75% of patients, so “no treatment may be the best treatment,” but antimalarials can be effective, Dr. Friedman said. “I use antimalarials frequently in my practice,” he said. “The key is, they take time to work (4-5 months),” which should be explained to patients.
Antibiotics, he said, can be “somewhat effective,” but in the case of doxycycline at least, the disease can resolve within weeks but then may return when treatment is stopped.
There is some evidence to support using biologics and more recently, Janus kinase (JAK) inhibitors, off-label, to treat GA. Efficacy has been seen with the tumor necrosis factor (TNF) blocker infliximab and with the JAK inhibitor tofacitinib, he said.
Lichen planus (LP). This is another common disease that can go off-script with its presentation. The disease is often described with the “six P’s” indicating the following characteristics: pruritic, polygonal, planar or flat-topped, purple papules, and plaques. But LP “didn’t read the textbook,” Dr. Friedman said.
“The clinical presentation of lichen planus can be quite broad,” he said. “The P’s aren’t always followed as there are a variety of colors and configurations which can be witnessed.”
With LP, there is a clear association with dyslipidemia and diabetes, so “asking the right questions is going to be important” when talking to the patient. There is also a higher risk of autoimmune diseases, especially of the thyroid type, associated with LP, he said.
No treatment has been Food and Drug Administration approved for LP, but some are expected in the future, he said.
For now, he emphasized creativity in the management of patients with LP. “I love oral retinoids for this,” he said. Antimalarials and methotrexate are also options.
In one case Dr. Friedman saw, nothing seemed to work: light therapy for a year; metronidazole; isotretinoin; halobetasol/tazarotene lotion; and the TNF-blocker adalimumab either weren’t effective or resulted in complications in the patient.
Knowing the recent implication of the interleukin (IL)-17 pathway in the pathophysiology of LP, he then tried the anti-IL17 antibody secukinumab. “This patient had a pretty robust response to treatment,” Dr. Friedman said. “He was very excited. The problem, as always, is access, especially for off-label therapies.”
Tumid lupus erythematosus. This disease is characterized by erythematous, edematous, nonscarring plaques on sun-exposed sites. For treatment, Dr. Friedman said antimalarials can be up to 90% effective, sometimes with rapid resolution of the lesions.
“You want to dose below that 5 mg per kg of true body weight to limit the small potential for ocular toxicity over time,” he said. And, he emphasized, “always combine treatment with good sun-protective measures.”
Dr. Friedman reported financial relationships with Sanova, Pfizer, Novartis, and other companies.
AT ODAC 2023
VEXAS syndrome: More common, variable, and severe than expected
A recently discovered inflammatory disease known as VEXAS syndrome is more common, variable, and dangerous than previously understood, according to results of a retrospective observational study of a large health care system database. The findings, published in JAMA, found that it struck 1 in 4,269 men over the age of 50 in a largely White population and caused a wide variety of symptoms.
“The disease is quite severe,” study lead author David Beck, MD, PhD, of the department of medicine at NYU Langone Health, said in an interview. Patients with the condition “have a variety of clinical symptoms affecting different parts of the body and are being managed by different medical specialties.”
Dr. Beck and colleagues first described VEXAS (vacuoles, E1-ubiquitin-activating enzyme, X-linked, autoinflammatory, somatic) syndrome in 2020. They linked it to mutations in the UBA1 (ubiquitin-like modifier activating enzyme 1) gene. The enzyme initiates a process that identifies misfolded proteins as targets for degradation.
“VEXAS syndrome is characterized by anemia and inflammation in the skin, lungs, cartilage, and joints,” Dr. Beck said. “These symptoms are frequently mistaken for other rheumatic or hematologic diseases. However, this syndrome has a different cause, is treated differently, requires additional monitoring, and can be far more severe.”
According to him, hundreds of people have been diagnosed with the disease in the short time since it was defined. The disease is believed to be fatal in some cases. A previous report found that the median survival was 9 years among patients with a certain variant; that was significantly less than patients with two other variants.
For the new study, researchers searched for UBA1 variants in genetic data from 163,096 subjects (mean age, 52.8 years; 94% White, 61% women) who took part in the Geisinger MyCode Community Health Initiative. The 1996-2022 data comes from patients at 10 Pennsylvania hospitals.
Eleven people (9 males, 2 females) had likely UBA1 variants, and all had anemia. The cases accounted for 1 in 13,591 unrelated people (95% confidence interval, 1:7,775-1:23,758), 1 in 4,269 men older than 50 years (95% CI, 1:2,319-1:7,859), and 1 in 26,238 women older than 50 years (95% CI, 1:7,196-1:147,669).
Other common findings included macrocytosis (91%), skin problems (73%), and pulmonary disease (91%). Ten patients (91%) required transfusions.
Five of the 11 subjects didn’t meet the previously defined criteria for VEXAS syndrome. None had been diagnosed with the condition, which is not surprising considering that it hadn’t been discovered and described until recently.
Just over half of the patients – 55% – had a clinical diagnosis that was previously linked to VEXAS syndrome. “This means that slightly less than half of the patients with VEXAS syndrome had no clear associated clinical diagnosis,” Dr. Beck said. “The lack of associated clinical diagnoses may be due to the variety of nonspecific clinical characteristics that span different subspecialities in VEXAS syndrome. VEXAS syndrome represents an example of a multisystem disease where patients and their symptoms may get lost in the shuffle.”
In the future, “professionals should look out for patients with unexplained inflammation – and some combination of hematologic, rheumatologic, pulmonary, and dermatologic clinical manifestations – that either don’t carry a clinical diagnosis or don’t respond to first-line therapies,” Dr. Beck said. “These patients will also frequently be anemic, have low platelet counts, elevated markers of inflammation in the blood, and be dependent on corticosteroids.”
Diagnosis can be made via genetic testing, but the study authors note that it “is not routinely offered on standard workup for myeloid neoplasms or immune dysregulation diagnostic panels.”
As for treatment, Dr. Beck said the disease “can be partially controlled by multiple different anticytokine therapies or biologics. However, in most cases, patients still need additional steroids and/or disease-modifying antirheumatic agents [DMARDs]. In addition, bone marrow transplantation has shown signs of being a highly effective therapy.”
The study authors say more research is needed to understand the disease’s prevalence in more diverse populations.
In an interview, Matthew J. Koster, MD, a rheumatologist at Mayo Clinic in Rochester, Minn., who’s studied the disease but didn’t take part in this research project, said the findings are valid and “highly important.
“The findings of this study highlight what many academic and quaternary referral centers were wondering: Is VEXAS really more common than we think, with patients hiding in plain sight? The answer is yes,” he said. “Currently, there are less than 400 cases reported in the literature of VEXAS, but large centers are diagnosing this condition with some frequency. For example, at Mayo Clinic in Rochester, we diagnose on average one new patient with VEXAS every 7-14 days and have diagnosed 60 in the past 18 months. A national collaborative group in France has diagnosed approximately 250 patients over that same time frame when pooling patients nationwide.”
The prevalence is high enough, he said, that “clinicians should consider that some of the patients with diseases that are not responding to treatment may in fact have VEXAS rather than ‘refractory’ relapsing polychondritis or ‘recalcitrant’ rheumatoid arthritis, etc.”
The National Institute of Health funded the study. Dr. Beck, the other authors, and Dr. Koster report no disclosures.
A recently discovered inflammatory disease known as VEXAS syndrome is more common, variable, and dangerous than previously understood, according to results of a retrospective observational study of a large health care system database. The findings, published in JAMA, found that it struck 1 in 4,269 men over the age of 50 in a largely White population and caused a wide variety of symptoms.
“The disease is quite severe,” study lead author David Beck, MD, PhD, of the department of medicine at NYU Langone Health, said in an interview. Patients with the condition “have a variety of clinical symptoms affecting different parts of the body and are being managed by different medical specialties.”
Dr. Beck and colleagues first described VEXAS (vacuoles, E1-ubiquitin-activating enzyme, X-linked, autoinflammatory, somatic) syndrome in 2020. They linked it to mutations in the UBA1 (ubiquitin-like modifier activating enzyme 1) gene. The enzyme initiates a process that identifies misfolded proteins as targets for degradation.
“VEXAS syndrome is characterized by anemia and inflammation in the skin, lungs, cartilage, and joints,” Dr. Beck said. “These symptoms are frequently mistaken for other rheumatic or hematologic diseases. However, this syndrome has a different cause, is treated differently, requires additional monitoring, and can be far more severe.”
According to him, hundreds of people have been diagnosed with the disease in the short time since it was defined. The disease is believed to be fatal in some cases. A previous report found that the median survival was 9 years among patients with a certain variant; that was significantly less than patients with two other variants.
For the new study, researchers searched for UBA1 variants in genetic data from 163,096 subjects (mean age, 52.8 years; 94% White, 61% women) who took part in the Geisinger MyCode Community Health Initiative. The 1996-2022 data comes from patients at 10 Pennsylvania hospitals.
Eleven people (9 males, 2 females) had likely UBA1 variants, and all had anemia. The cases accounted for 1 in 13,591 unrelated people (95% confidence interval, 1:7,775-1:23,758), 1 in 4,269 men older than 50 years (95% CI, 1:2,319-1:7,859), and 1 in 26,238 women older than 50 years (95% CI, 1:7,196-1:147,669).
Other common findings included macrocytosis (91%), skin problems (73%), and pulmonary disease (91%). Ten patients (91%) required transfusions.
Five of the 11 subjects didn’t meet the previously defined criteria for VEXAS syndrome. None had been diagnosed with the condition, which is not surprising considering that it hadn’t been discovered and described until recently.
Just over half of the patients – 55% – had a clinical diagnosis that was previously linked to VEXAS syndrome. “This means that slightly less than half of the patients with VEXAS syndrome had no clear associated clinical diagnosis,” Dr. Beck said. “The lack of associated clinical diagnoses may be due to the variety of nonspecific clinical characteristics that span different subspecialities in VEXAS syndrome. VEXAS syndrome represents an example of a multisystem disease where patients and their symptoms may get lost in the shuffle.”
In the future, “professionals should look out for patients with unexplained inflammation – and some combination of hematologic, rheumatologic, pulmonary, and dermatologic clinical manifestations – that either don’t carry a clinical diagnosis or don’t respond to first-line therapies,” Dr. Beck said. “These patients will also frequently be anemic, have low platelet counts, elevated markers of inflammation in the blood, and be dependent on corticosteroids.”
Diagnosis can be made via genetic testing, but the study authors note that it “is not routinely offered on standard workup for myeloid neoplasms or immune dysregulation diagnostic panels.”
As for treatment, Dr. Beck said the disease “can be partially controlled by multiple different anticytokine therapies or biologics. However, in most cases, patients still need additional steroids and/or disease-modifying antirheumatic agents [DMARDs]. In addition, bone marrow transplantation has shown signs of being a highly effective therapy.”
The study authors say more research is needed to understand the disease’s prevalence in more diverse populations.
In an interview, Matthew J. Koster, MD, a rheumatologist at Mayo Clinic in Rochester, Minn., who’s studied the disease but didn’t take part in this research project, said the findings are valid and “highly important.
“The findings of this study highlight what many academic and quaternary referral centers were wondering: Is VEXAS really more common than we think, with patients hiding in plain sight? The answer is yes,” he said. “Currently, there are less than 400 cases reported in the literature of VEXAS, but large centers are diagnosing this condition with some frequency. For example, at Mayo Clinic in Rochester, we diagnose on average one new patient with VEXAS every 7-14 days and have diagnosed 60 in the past 18 months. A national collaborative group in France has diagnosed approximately 250 patients over that same time frame when pooling patients nationwide.”
The prevalence is high enough, he said, that “clinicians should consider that some of the patients with diseases that are not responding to treatment may in fact have VEXAS rather than ‘refractory’ relapsing polychondritis or ‘recalcitrant’ rheumatoid arthritis, etc.”
The National Institute of Health funded the study. Dr. Beck, the other authors, and Dr. Koster report no disclosures.
A recently discovered inflammatory disease known as VEXAS syndrome is more common, variable, and dangerous than previously understood, according to results of a retrospective observational study of a large health care system database. The findings, published in JAMA, found that it struck 1 in 4,269 men over the age of 50 in a largely White population and caused a wide variety of symptoms.
“The disease is quite severe,” study lead author David Beck, MD, PhD, of the department of medicine at NYU Langone Health, said in an interview. Patients with the condition “have a variety of clinical symptoms affecting different parts of the body and are being managed by different medical specialties.”
Dr. Beck and colleagues first described VEXAS (vacuoles, E1-ubiquitin-activating enzyme, X-linked, autoinflammatory, somatic) syndrome in 2020. They linked it to mutations in the UBA1 (ubiquitin-like modifier activating enzyme 1) gene. The enzyme initiates a process that identifies misfolded proteins as targets for degradation.
“VEXAS syndrome is characterized by anemia and inflammation in the skin, lungs, cartilage, and joints,” Dr. Beck said. “These symptoms are frequently mistaken for other rheumatic or hematologic diseases. However, this syndrome has a different cause, is treated differently, requires additional monitoring, and can be far more severe.”
According to him, hundreds of people have been diagnosed with the disease in the short time since it was defined. The disease is believed to be fatal in some cases. A previous report found that the median survival was 9 years among patients with a certain variant; that was significantly less than patients with two other variants.
For the new study, researchers searched for UBA1 variants in genetic data from 163,096 subjects (mean age, 52.8 years; 94% White, 61% women) who took part in the Geisinger MyCode Community Health Initiative. The 1996-2022 data comes from patients at 10 Pennsylvania hospitals.
Eleven people (9 males, 2 females) had likely UBA1 variants, and all had anemia. The cases accounted for 1 in 13,591 unrelated people (95% confidence interval, 1:7,775-1:23,758), 1 in 4,269 men older than 50 years (95% CI, 1:2,319-1:7,859), and 1 in 26,238 women older than 50 years (95% CI, 1:7,196-1:147,669).
Other common findings included macrocytosis (91%), skin problems (73%), and pulmonary disease (91%). Ten patients (91%) required transfusions.
Five of the 11 subjects didn’t meet the previously defined criteria for VEXAS syndrome. None had been diagnosed with the condition, which is not surprising considering that it hadn’t been discovered and described until recently.
Just over half of the patients – 55% – had a clinical diagnosis that was previously linked to VEXAS syndrome. “This means that slightly less than half of the patients with VEXAS syndrome had no clear associated clinical diagnosis,” Dr. Beck said. “The lack of associated clinical diagnoses may be due to the variety of nonspecific clinical characteristics that span different subspecialities in VEXAS syndrome. VEXAS syndrome represents an example of a multisystem disease where patients and their symptoms may get lost in the shuffle.”
In the future, “professionals should look out for patients with unexplained inflammation – and some combination of hematologic, rheumatologic, pulmonary, and dermatologic clinical manifestations – that either don’t carry a clinical diagnosis or don’t respond to first-line therapies,” Dr. Beck said. “These patients will also frequently be anemic, have low platelet counts, elevated markers of inflammation in the blood, and be dependent on corticosteroids.”
Diagnosis can be made via genetic testing, but the study authors note that it “is not routinely offered on standard workup for myeloid neoplasms or immune dysregulation diagnostic panels.”
As for treatment, Dr. Beck said the disease “can be partially controlled by multiple different anticytokine therapies or biologics. However, in most cases, patients still need additional steroids and/or disease-modifying antirheumatic agents [DMARDs]. In addition, bone marrow transplantation has shown signs of being a highly effective therapy.”
The study authors say more research is needed to understand the disease’s prevalence in more diverse populations.
In an interview, Matthew J. Koster, MD, a rheumatologist at Mayo Clinic in Rochester, Minn., who’s studied the disease but didn’t take part in this research project, said the findings are valid and “highly important.
“The findings of this study highlight what many academic and quaternary referral centers were wondering: Is VEXAS really more common than we think, with patients hiding in plain sight? The answer is yes,” he said. “Currently, there are less than 400 cases reported in the literature of VEXAS, but large centers are diagnosing this condition with some frequency. For example, at Mayo Clinic in Rochester, we diagnose on average one new patient with VEXAS every 7-14 days and have diagnosed 60 in the past 18 months. A national collaborative group in France has diagnosed approximately 250 patients over that same time frame when pooling patients nationwide.”
The prevalence is high enough, he said, that “clinicians should consider that some of the patients with diseases that are not responding to treatment may in fact have VEXAS rather than ‘refractory’ relapsing polychondritis or ‘recalcitrant’ rheumatoid arthritis, etc.”
The National Institute of Health funded the study. Dr. Beck, the other authors, and Dr. Koster report no disclosures.
FROM JAMA
Mastocytosis: Rare, underdiagnosed, potentially fatal
Nationwide, approximately 1,000 adults are diagnosed with systemic mastocytosis annually. This rare disease is a myeloid neoplasm with a highly variable phenotypic expression, in which abnormal mast cells proliferate and infiltrate organs and tissues. It swings widely from a nonadvanced form, composed of indolent or smoldering disease, to advanced disease that progresses to leukemia in 6% of cases.
More than 80% of systemic mastocytosis is driven by the KIT D816V mutation. Along with a host of other rare KIT mutations, KIT D816V activates KIT-receptor tyrosine kinase to trigger mast cell proliferation.
Dr. Gotlib could not be contacted for an interview. However, there are many good reasons to identify patients with systemic mastocytosis, according to Attilio Orazi, MD, professor and chair of the department of pathology at Texas Tech University, El Paso. The chief reason is that the patient may be in grave peril.
“The degree of heterogeneity is amazing. ... There’s very indolent [disease], which is really not a big deal. And then you have a disease in which you’re dead in 3 months,” Dr. Orazi said. “So you run the gamut between an indolent, no-problem cutaneous disease to a very nasty systemic, aggressive leukemia-like neoplasm.”
Since 2001, the diagnosis of mastocytosis has been guided by the World Health Organization Classification of Tumours, or “Blue Book.” In 2022, Dr. Orazi along with 137 other senior experts, most of whom were involved in past editions of the Blue Book, published their own version: The International Consensus Classification of Myeloid Neoplasms and Acute Leukemias (the ICC 2022).
In September 2021, this group of specialists held a virtual/in-person advisory committee meeting at the University of Chicago to create the document. One factor in their decision to go it alone, Dr. Orazi said, was that WHO decided to proceed with the fifth edition of the Blue Book using its own internal editorial group without convening an advisory committee, despite repeated requests to do so.
ICC 2022 divides advanced systemic mastocytosis into three subtypes: aggressive systemic mastocytosis (ASM), systemic mastocytosis with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL). Median survival is 3.5 years for patients with ASM, 2 years for those with SM-AHN and as low as 2 months for MCL.
The second key reason to increase awareness of mastocytosis among physicians, said Dr. Orazi, is that patients falling through the net are likely to be ambulatory, and their presentation can be “a little confusing.”
Patients with indolent disease are relatively straightforward to recognize, explained Dr. Orazi. Similarly, very sick patients with SM-AHN or MCL are easily recognized by hem-oncs.
“But if you see a patient in an ambulatory setting, in your clinic or whatever, and you’re suspicious, then you need to decide [how] you’re going to investigate that patient further,” he said, Dr. Orazi noted the next step is not always obvious, especially for primary-practice or internal medicine physicians likely to be unfamiliar with such a rare disease.
A practice survey published in 2022 by other researchers backed up Dr. Orazi’s remarks. The study found that community/solo-practice physicians were less likely to have tested systemic mastocytosis patients for KIT816V mutation than academic/specialty physicians (58% vs. 80%; P = .004; n = 111). Clinicians treating these patients estimated that it took an average of 8.5 months for a “typical” patient to receive the diagnosis from the time of symptom onset.
The research was headed by Ruben Mesa, MD, director of University of Texas Health, San Antonio, and funded by Blueprint Medicines, the manufacturer of avapritinib (Ayvakit), a new drug for the disease.
Dr. Orazi urged clinicians to have a high degree of suspicion for mastocytosis in a patient who walks into the clinic with any combination of the following: urticarial-type skin manifestations, especially if persistent into adulthood; history of undue reaction to an insect sting; a big spleen in a patient with a history of cutaneous flushing or rash; chronic diarrhea, especially if a biopsy has shown “too many mast cells” in the lamina propria of the small bowel; and positivity for KIT816V mutation.
Dr. Orazi stressed that the majority of patients will have indolent disease, but for the few patients for whom immediate treatment is essential, “the distinction between indolent and aggressive [disease] is really very, very important.”
Patients with advanced systemic mastocytosis can now be effectively treated, following the arrival of midostaurin (Rydapt, Tauritmo) and avapritinib.
Midostaurin, a multikinase/KIT inhibitor, was approved by the Food and Drug Administration in 2017 for the treatment of advanced systemic mastocytosis (ASM, SM-AHN, and MCL). Avapritinib, a selective kinase inhibitor of KIT816V and platelet-derived growth factor receptor alpha as well as multiple KIT exon 11, 11/17 and 17 mutants, gained the same indication in June 2021.
As with all rare diseases, it is challenging to obtain accurate numbers on how many patients are affected by systemic mastocytosis. The first population-based study of the disorder, presented at the 2018 annual meeting of the American Society of Hematology, used the Surveillance, Epidemiology, and End Results database from 2000 to 2014 to estimate incidence at 0.046 per 10,000, which translates to 1,050 new adult cases per year. The study data have never been published in full.
How many of these cases are advanced disease? There are no U.S. data but extrapolating from a Danish registry study that found 82% of systemic mastocytosis cases to be indolent disease, the incidence of advanced systemic mastocytosis in the United States could be as low as 200 adults a year.
This information, in turn, suggests that identifying more patients with advanced disease would not only benefit those patients but would also benefit clinical trial investigators who are seeking the proverbial needle in the haystack.
Nationwide, five clinical trials are recruiting individuals with advanced systemic mastocytosis, collectively looking for 352 patients in the United States. Two of the studies focus on mast-cell activation (NCT0544944) and cutaneous mastocytoses (NCT04846348). Two trials in a range of hematological malignancies are testing bispecific antibodies flotetuzumab and MGD024 (both from Macrogenics; NCT04681105, NCT05362773).
Apex, a phase 2 study of tyrosine-kinase inhibitor bezuclastinib (a Cogent hopeful), is specifically focusing on advanced disease. Dr. Gotlib and coinvestigators are aiming for 140 participants.
As a pathologist, Dr. Orazi said he find mastocytosis fascinating because he believes he has “a truly useful role,” contrasting with some other hematological diseases in which the molecular profile rules.
“Pathology plays a major role here,” he explained, “because you have to correlate what you see at the microscope with the full clinical picture, selected laboratory tests such as CBC and serum tryptase, and molecular results. You often need integration through a pathologist to put all the pieces together.
“It’s easier to treat once you know exactly what disease you’re dealing with and whether it is an aggressive or indolent subtype,” Dr. Orazi concluded.
Dr. Orazi disclosed no conflicts of interest. Dr. Gotlib has disclosed ties with Blueprint Medicines, Deciphera, Incyte, and Kartos Therapeutics, and has led committees for Blueprint Medicine’s EXPLORER and PATHFINDER studies, Deciphera’s Study Steering Committee for ripretinib in AdvSM, and the Central Response Review Committee for the phase 2 study of bezuclastinib in AdvSM.
Nationwide, approximately 1,000 adults are diagnosed with systemic mastocytosis annually. This rare disease is a myeloid neoplasm with a highly variable phenotypic expression, in which abnormal mast cells proliferate and infiltrate organs and tissues. It swings widely from a nonadvanced form, composed of indolent or smoldering disease, to advanced disease that progresses to leukemia in 6% of cases.
More than 80% of systemic mastocytosis is driven by the KIT D816V mutation. Along with a host of other rare KIT mutations, KIT D816V activates KIT-receptor tyrosine kinase to trigger mast cell proliferation.
Dr. Gotlib could not be contacted for an interview. However, there are many good reasons to identify patients with systemic mastocytosis, according to Attilio Orazi, MD, professor and chair of the department of pathology at Texas Tech University, El Paso. The chief reason is that the patient may be in grave peril.
“The degree of heterogeneity is amazing. ... There’s very indolent [disease], which is really not a big deal. And then you have a disease in which you’re dead in 3 months,” Dr. Orazi said. “So you run the gamut between an indolent, no-problem cutaneous disease to a very nasty systemic, aggressive leukemia-like neoplasm.”
Since 2001, the diagnosis of mastocytosis has been guided by the World Health Organization Classification of Tumours, or “Blue Book.” In 2022, Dr. Orazi along with 137 other senior experts, most of whom were involved in past editions of the Blue Book, published their own version: The International Consensus Classification of Myeloid Neoplasms and Acute Leukemias (the ICC 2022).
In September 2021, this group of specialists held a virtual/in-person advisory committee meeting at the University of Chicago to create the document. One factor in their decision to go it alone, Dr. Orazi said, was that WHO decided to proceed with the fifth edition of the Blue Book using its own internal editorial group without convening an advisory committee, despite repeated requests to do so.
ICC 2022 divides advanced systemic mastocytosis into three subtypes: aggressive systemic mastocytosis (ASM), systemic mastocytosis with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL). Median survival is 3.5 years for patients with ASM, 2 years for those with SM-AHN and as low as 2 months for MCL.
The second key reason to increase awareness of mastocytosis among physicians, said Dr. Orazi, is that patients falling through the net are likely to be ambulatory, and their presentation can be “a little confusing.”
Patients with indolent disease are relatively straightforward to recognize, explained Dr. Orazi. Similarly, very sick patients with SM-AHN or MCL are easily recognized by hem-oncs.
“But if you see a patient in an ambulatory setting, in your clinic or whatever, and you’re suspicious, then you need to decide [how] you’re going to investigate that patient further,” he said, Dr. Orazi noted the next step is not always obvious, especially for primary-practice or internal medicine physicians likely to be unfamiliar with such a rare disease.
A practice survey published in 2022 by other researchers backed up Dr. Orazi’s remarks. The study found that community/solo-practice physicians were less likely to have tested systemic mastocytosis patients for KIT816V mutation than academic/specialty physicians (58% vs. 80%; P = .004; n = 111). Clinicians treating these patients estimated that it took an average of 8.5 months for a “typical” patient to receive the diagnosis from the time of symptom onset.
The research was headed by Ruben Mesa, MD, director of University of Texas Health, San Antonio, and funded by Blueprint Medicines, the manufacturer of avapritinib (Ayvakit), a new drug for the disease.
Dr. Orazi urged clinicians to have a high degree of suspicion for mastocytosis in a patient who walks into the clinic with any combination of the following: urticarial-type skin manifestations, especially if persistent into adulthood; history of undue reaction to an insect sting; a big spleen in a patient with a history of cutaneous flushing or rash; chronic diarrhea, especially if a biopsy has shown “too many mast cells” in the lamina propria of the small bowel; and positivity for KIT816V mutation.
Dr. Orazi stressed that the majority of patients will have indolent disease, but for the few patients for whom immediate treatment is essential, “the distinction between indolent and aggressive [disease] is really very, very important.”
Patients with advanced systemic mastocytosis can now be effectively treated, following the arrival of midostaurin (Rydapt, Tauritmo) and avapritinib.
Midostaurin, a multikinase/KIT inhibitor, was approved by the Food and Drug Administration in 2017 for the treatment of advanced systemic mastocytosis (ASM, SM-AHN, and MCL). Avapritinib, a selective kinase inhibitor of KIT816V and platelet-derived growth factor receptor alpha as well as multiple KIT exon 11, 11/17 and 17 mutants, gained the same indication in June 2021.
As with all rare diseases, it is challenging to obtain accurate numbers on how many patients are affected by systemic mastocytosis. The first population-based study of the disorder, presented at the 2018 annual meeting of the American Society of Hematology, used the Surveillance, Epidemiology, and End Results database from 2000 to 2014 to estimate incidence at 0.046 per 10,000, which translates to 1,050 new adult cases per year. The study data have never been published in full.
How many of these cases are advanced disease? There are no U.S. data but extrapolating from a Danish registry study that found 82% of systemic mastocytosis cases to be indolent disease, the incidence of advanced systemic mastocytosis in the United States could be as low as 200 adults a year.
This information, in turn, suggests that identifying more patients with advanced disease would not only benefit those patients but would also benefit clinical trial investigators who are seeking the proverbial needle in the haystack.
Nationwide, five clinical trials are recruiting individuals with advanced systemic mastocytosis, collectively looking for 352 patients in the United States. Two of the studies focus on mast-cell activation (NCT0544944) and cutaneous mastocytoses (NCT04846348). Two trials in a range of hematological malignancies are testing bispecific antibodies flotetuzumab and MGD024 (both from Macrogenics; NCT04681105, NCT05362773).
Apex, a phase 2 study of tyrosine-kinase inhibitor bezuclastinib (a Cogent hopeful), is specifically focusing on advanced disease. Dr. Gotlib and coinvestigators are aiming for 140 participants.
As a pathologist, Dr. Orazi said he find mastocytosis fascinating because he believes he has “a truly useful role,” contrasting with some other hematological diseases in which the molecular profile rules.
“Pathology plays a major role here,” he explained, “because you have to correlate what you see at the microscope with the full clinical picture, selected laboratory tests such as CBC and serum tryptase, and molecular results. You often need integration through a pathologist to put all the pieces together.
“It’s easier to treat once you know exactly what disease you’re dealing with and whether it is an aggressive or indolent subtype,” Dr. Orazi concluded.
Dr. Orazi disclosed no conflicts of interest. Dr. Gotlib has disclosed ties with Blueprint Medicines, Deciphera, Incyte, and Kartos Therapeutics, and has led committees for Blueprint Medicine’s EXPLORER and PATHFINDER studies, Deciphera’s Study Steering Committee for ripretinib in AdvSM, and the Central Response Review Committee for the phase 2 study of bezuclastinib in AdvSM.
Nationwide, approximately 1,000 adults are diagnosed with systemic mastocytosis annually. This rare disease is a myeloid neoplasm with a highly variable phenotypic expression, in which abnormal mast cells proliferate and infiltrate organs and tissues. It swings widely from a nonadvanced form, composed of indolent or smoldering disease, to advanced disease that progresses to leukemia in 6% of cases.
More than 80% of systemic mastocytosis is driven by the KIT D816V mutation. Along with a host of other rare KIT mutations, KIT D816V activates KIT-receptor tyrosine kinase to trigger mast cell proliferation.
Dr. Gotlib could not be contacted for an interview. However, there are many good reasons to identify patients with systemic mastocytosis, according to Attilio Orazi, MD, professor and chair of the department of pathology at Texas Tech University, El Paso. The chief reason is that the patient may be in grave peril.
“The degree of heterogeneity is amazing. ... There’s very indolent [disease], which is really not a big deal. And then you have a disease in which you’re dead in 3 months,” Dr. Orazi said. “So you run the gamut between an indolent, no-problem cutaneous disease to a very nasty systemic, aggressive leukemia-like neoplasm.”
Since 2001, the diagnosis of mastocytosis has been guided by the World Health Organization Classification of Tumours, or “Blue Book.” In 2022, Dr. Orazi along with 137 other senior experts, most of whom were involved in past editions of the Blue Book, published their own version: The International Consensus Classification of Myeloid Neoplasms and Acute Leukemias (the ICC 2022).
In September 2021, this group of specialists held a virtual/in-person advisory committee meeting at the University of Chicago to create the document. One factor in their decision to go it alone, Dr. Orazi said, was that WHO decided to proceed with the fifth edition of the Blue Book using its own internal editorial group without convening an advisory committee, despite repeated requests to do so.
ICC 2022 divides advanced systemic mastocytosis into three subtypes: aggressive systemic mastocytosis (ASM), systemic mastocytosis with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL). Median survival is 3.5 years for patients with ASM, 2 years for those with SM-AHN and as low as 2 months for MCL.
The second key reason to increase awareness of mastocytosis among physicians, said Dr. Orazi, is that patients falling through the net are likely to be ambulatory, and their presentation can be “a little confusing.”
Patients with indolent disease are relatively straightforward to recognize, explained Dr. Orazi. Similarly, very sick patients with SM-AHN or MCL are easily recognized by hem-oncs.
“But if you see a patient in an ambulatory setting, in your clinic or whatever, and you’re suspicious, then you need to decide [how] you’re going to investigate that patient further,” he said, Dr. Orazi noted the next step is not always obvious, especially for primary-practice or internal medicine physicians likely to be unfamiliar with such a rare disease.
A practice survey published in 2022 by other researchers backed up Dr. Orazi’s remarks. The study found that community/solo-practice physicians were less likely to have tested systemic mastocytosis patients for KIT816V mutation than academic/specialty physicians (58% vs. 80%; P = .004; n = 111). Clinicians treating these patients estimated that it took an average of 8.5 months for a “typical” patient to receive the diagnosis from the time of symptom onset.
The research was headed by Ruben Mesa, MD, director of University of Texas Health, San Antonio, and funded by Blueprint Medicines, the manufacturer of avapritinib (Ayvakit), a new drug for the disease.
Dr. Orazi urged clinicians to have a high degree of suspicion for mastocytosis in a patient who walks into the clinic with any combination of the following: urticarial-type skin manifestations, especially if persistent into adulthood; history of undue reaction to an insect sting; a big spleen in a patient with a history of cutaneous flushing or rash; chronic diarrhea, especially if a biopsy has shown “too many mast cells” in the lamina propria of the small bowel; and positivity for KIT816V mutation.
Dr. Orazi stressed that the majority of patients will have indolent disease, but for the few patients for whom immediate treatment is essential, “the distinction between indolent and aggressive [disease] is really very, very important.”
Patients with advanced systemic mastocytosis can now be effectively treated, following the arrival of midostaurin (Rydapt, Tauritmo) and avapritinib.
Midostaurin, a multikinase/KIT inhibitor, was approved by the Food and Drug Administration in 2017 for the treatment of advanced systemic mastocytosis (ASM, SM-AHN, and MCL). Avapritinib, a selective kinase inhibitor of KIT816V and platelet-derived growth factor receptor alpha as well as multiple KIT exon 11, 11/17 and 17 mutants, gained the same indication in June 2021.
As with all rare diseases, it is challenging to obtain accurate numbers on how many patients are affected by systemic mastocytosis. The first population-based study of the disorder, presented at the 2018 annual meeting of the American Society of Hematology, used the Surveillance, Epidemiology, and End Results database from 2000 to 2014 to estimate incidence at 0.046 per 10,000, which translates to 1,050 new adult cases per year. The study data have never been published in full.
How many of these cases are advanced disease? There are no U.S. data but extrapolating from a Danish registry study that found 82% of systemic mastocytosis cases to be indolent disease, the incidence of advanced systemic mastocytosis in the United States could be as low as 200 adults a year.
This information, in turn, suggests that identifying more patients with advanced disease would not only benefit those patients but would also benefit clinical trial investigators who are seeking the proverbial needle in the haystack.
Nationwide, five clinical trials are recruiting individuals with advanced systemic mastocytosis, collectively looking for 352 patients in the United States. Two of the studies focus on mast-cell activation (NCT0544944) and cutaneous mastocytoses (NCT04846348). Two trials in a range of hematological malignancies are testing bispecific antibodies flotetuzumab and MGD024 (both from Macrogenics; NCT04681105, NCT05362773).
Apex, a phase 2 study of tyrosine-kinase inhibitor bezuclastinib (a Cogent hopeful), is specifically focusing on advanced disease. Dr. Gotlib and coinvestigators are aiming for 140 participants.
As a pathologist, Dr. Orazi said he find mastocytosis fascinating because he believes he has “a truly useful role,” contrasting with some other hematological diseases in which the molecular profile rules.
“Pathology plays a major role here,” he explained, “because you have to correlate what you see at the microscope with the full clinical picture, selected laboratory tests such as CBC and serum tryptase, and molecular results. You often need integration through a pathologist to put all the pieces together.
“It’s easier to treat once you know exactly what disease you’re dealing with and whether it is an aggressive or indolent subtype,” Dr. Orazi concluded.
Dr. Orazi disclosed no conflicts of interest. Dr. Gotlib has disclosed ties with Blueprint Medicines, Deciphera, Incyte, and Kartos Therapeutics, and has led committees for Blueprint Medicine’s EXPLORER and PATHFINDER studies, Deciphera’s Study Steering Committee for ripretinib in AdvSM, and the Central Response Review Committee for the phase 2 study of bezuclastinib in AdvSM.