User login
VIDEO: Oocyte modification might prevent mitochondrial diseases
Clinical trials using genetically modified oocytes to prevent the transmission of mitochondrial diseases in humans may be soon become a reality. But the potentially promising approach to prevent conditions such as Leigh disease and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes) is not without controversy.
In an interview, Dr. Salvatore DiMauro, the Lucy G. Moses Professor of Neurology at Columbia University Medical Center, outlined the impact that mitochondrial DNA–related diseases have on women’s and children’s lives, and he explained why genetically modified oocytes may offer new hope for those affected by these diseases.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Clinical trials using genetically modified oocytes to prevent the transmission of mitochondrial diseases in humans may be soon become a reality. But the potentially promising approach to prevent conditions such as Leigh disease and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes) is not without controversy.
In an interview, Dr. Salvatore DiMauro, the Lucy G. Moses Professor of Neurology at Columbia University Medical Center, outlined the impact that mitochondrial DNA–related diseases have on women’s and children’s lives, and he explained why genetically modified oocytes may offer new hope for those affected by these diseases.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Clinical trials using genetically modified oocytes to prevent the transmission of mitochondrial diseases in humans may be soon become a reality. But the potentially promising approach to prevent conditions such as Leigh disease and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes) is not without controversy.
In an interview, Dr. Salvatore DiMauro, the Lucy G. Moses Professor of Neurology at Columbia University Medical Center, outlined the impact that mitochondrial DNA–related diseases have on women’s and children’s lives, and he explained why genetically modified oocytes may offer new hope for those affected by these diseases.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AN FDA ADVISORY COMMITTEE MEETING

Pseudobulbar affect: More common than you’d think
ORLANDO – The prevalence of pseudobulbar affect symptoms – that is, uncontrollable, disruptive outbursts of crying and/or laughing – is considerably greater across a range of neurologic disorders than previously appreciated, according to the largest-ever study to screen for this condition.
Pseudobulbar affect (PBA) symptoms were found in the study to be more common among neurology patients under age 65; however, the adverse impact of PBA symptoms upon quality of life was greater in the elderly, Dr. David W. Crumpacker reported at the annual meeting of the American Association for Geriatric Psychiatry.
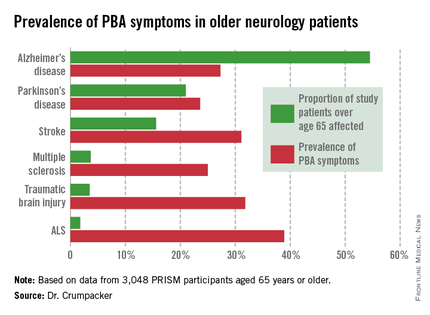
He presented the results of the PRISM (PBA Registry Series) study, which enrolled 5,290 patients on the basis of having any of six neurologic disorders: Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), multiple sclerosis, Parkinson’s disease, stroke, or traumatic brain injury. They were screened for the presence of PBA symptoms using the validated Center for Neurologic Study–Lability Scale (CNS-LS). A score of 13 or more was deemed positive, based upon its demonstrated good predictive value for physician diagnosis of PBA in patients with ALS.
The CNS-LS is a simple test that can be completed quickly by either the patient or caregiver. The test is well-suited for routine use in clinical practice, noted Dr. W. Crumpacker, a psychiatrist at Baylor University Medical Center, Dallas.
The overall prevalence of PBA symptoms among the 3,048 PRISM participants aged 65 years or older was 27.4%, with the highest rate seen in patients having ALS (see chart). In contrast, the prevalence of PBA symptoms among patients under age 65 years was 49.5%, with the highest rate – 56.9% – being seen in traumatic brain injury patients.
Patients or caregivers were asked to rate on a 0-10 scale the impact their primary neurologic disease has had on their quality of life. Patients 65 years and older with PBA symptoms reported a significantly greater negative impact than did those without PBA symptoms, with mean scores on the quality of life impact scale of 6.3 vs. 4.6. The quality of life difference between those with PBA symptoms and those without was significant for patients with each of the neurologic diseases except for ALS.

As another measure of the adverse impact of having PBA symptoms, 56% of affected older patients were on at least one antipsychotic or antidepressant, compared with 35% of older patients without PBA symptoms.
PBA is thought to result from injury to neurologic pathways that regulate emotional expression as a secondary consequence of a variety of neurologic disorders.
In an interview, Dr. Crumpacker said PBA is greatly underdiagnosed and often gets misdiagnosed as depression.
"The symptoms are extremely disturbing to others, and patients are acutely aware of that. I tell my friends in neurology, it’s the psychiatric pathology that causes people problems in their lives. No one gets divorced over neurologic pathology, they get divorced over psychiatric pathology. It’s not, ‘I got a divorce because he had a stroke.’ " "It’s "We got divorced because he had a stroke and it changed his personality; he was a different person and I couldn’t be around him anymore,’ " the psychiatrist said.
PBA became a diagnosable disorder with its own ICD-9 code, albeit a diagnosis that can’t be made in the absence of neurologic pathology, at the behest of the Food and Drug Administration, Dr. Crumpacker explained. The impetus was the discovery of an effective treatment, dextromethorphan HBr and quinidine sulfate (Nuedexta), which received FDA approval for PBA 3 years ago.
Nuedexta’s development as the sole medication indicated for PBA was serendipitous, according to Dr. Crumpacker.
"The drug was being tested in Alzheimer’s disease. The jury is still out on whether it helps. But families of study participants came back saying, ‘You know that stuff dad used to do – the crying, the inappropriate laughter, the anger? He doesn’t do those kinds of things anymore,’ " Dr. Crumpacker recalled.
He reported serving on a scientific advisory board for Avanir Pharmaceuticals, which markets Nuedexta.
ORLANDO – The prevalence of pseudobulbar affect symptoms – that is, uncontrollable, disruptive outbursts of crying and/or laughing – is considerably greater across a range of neurologic disorders than previously appreciated, according to the largest-ever study to screen for this condition.
Pseudobulbar affect (PBA) symptoms were found in the study to be more common among neurology patients under age 65; however, the adverse impact of PBA symptoms upon quality of life was greater in the elderly, Dr. David W. Crumpacker reported at the annual meeting of the American Association for Geriatric Psychiatry.
He presented the results of the PRISM (PBA Registry Series) study, which enrolled 5,290 patients on the basis of having any of six neurologic disorders: Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), multiple sclerosis, Parkinson’s disease, stroke, or traumatic brain injury. They were screened for the presence of PBA symptoms using the validated Center for Neurologic Study–Lability Scale (CNS-LS). A score of 13 or more was deemed positive, based upon its demonstrated good predictive value for physician diagnosis of PBA in patients with ALS.
The CNS-LS is a simple test that can be completed quickly by either the patient or caregiver. The test is well-suited for routine use in clinical practice, noted Dr. W. Crumpacker, a psychiatrist at Baylor University Medical Center, Dallas.
The overall prevalence of PBA symptoms among the 3,048 PRISM participants aged 65 years or older was 27.4%, with the highest rate seen in patients having ALS (see chart). In contrast, the prevalence of PBA symptoms among patients under age 65 years was 49.5%, with the highest rate – 56.9% – being seen in traumatic brain injury patients.
Patients or caregivers were asked to rate on a 0-10 scale the impact their primary neurologic disease has had on their quality of life. Patients 65 years and older with PBA symptoms reported a significantly greater negative impact than did those without PBA symptoms, with mean scores on the quality of life impact scale of 6.3 vs. 4.6. The quality of life difference between those with PBA symptoms and those without was significant for patients with each of the neurologic diseases except for ALS.
As another measure of the adverse impact of having PBA symptoms, 56% of affected older patients were on at least one antipsychotic or antidepressant, compared with 35% of older patients without PBA symptoms.
PBA is thought to result from injury to neurologic pathways that regulate emotional expression as a secondary consequence of a variety of neurologic disorders.
In an interview, Dr. Crumpacker said PBA is greatly underdiagnosed and often gets misdiagnosed as depression.
"The symptoms are extremely disturbing to others, and patients are acutely aware of that. I tell my friends in neurology, it’s the psychiatric pathology that causes people problems in their lives. No one gets divorced over neurologic pathology, they get divorced over psychiatric pathology. It’s not, ‘I got a divorce because he had a stroke.’ " "It’s "We got divorced because he had a stroke and it changed his personality; he was a different person and I couldn’t be around him anymore,’ " the psychiatrist said.
PBA became a diagnosable disorder with its own ICD-9 code, albeit a diagnosis that can’t be made in the absence of neurologic pathology, at the behest of the Food and Drug Administration, Dr. Crumpacker explained. The impetus was the discovery of an effective treatment, dextromethorphan HBr and quinidine sulfate (Nuedexta), which received FDA approval for PBA 3 years ago.
Nuedexta’s development as the sole medication indicated for PBA was serendipitous, according to Dr. Crumpacker.
"The drug was being tested in Alzheimer’s disease. The jury is still out on whether it helps. But families of study participants came back saying, ‘You know that stuff dad used to do – the crying, the inappropriate laughter, the anger? He doesn’t do those kinds of things anymore,’ " Dr. Crumpacker recalled.
He reported serving on a scientific advisory board for Avanir Pharmaceuticals, which markets Nuedexta.
ORLANDO – The prevalence of pseudobulbar affect symptoms – that is, uncontrollable, disruptive outbursts of crying and/or laughing – is considerably greater across a range of neurologic disorders than previously appreciated, according to the largest-ever study to screen for this condition.
Pseudobulbar affect (PBA) symptoms were found in the study to be more common among neurology patients under age 65; however, the adverse impact of PBA symptoms upon quality of life was greater in the elderly, Dr. David W. Crumpacker reported at the annual meeting of the American Association for Geriatric Psychiatry.
He presented the results of the PRISM (PBA Registry Series) study, which enrolled 5,290 patients on the basis of having any of six neurologic disorders: Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), multiple sclerosis, Parkinson’s disease, stroke, or traumatic brain injury. They were screened for the presence of PBA symptoms using the validated Center for Neurologic Study–Lability Scale (CNS-LS). A score of 13 or more was deemed positive, based upon its demonstrated good predictive value for physician diagnosis of PBA in patients with ALS.
The CNS-LS is a simple test that can be completed quickly by either the patient or caregiver. The test is well-suited for routine use in clinical practice, noted Dr. W. Crumpacker, a psychiatrist at Baylor University Medical Center, Dallas.
The overall prevalence of PBA symptoms among the 3,048 PRISM participants aged 65 years or older was 27.4%, with the highest rate seen in patients having ALS (see chart). In contrast, the prevalence of PBA symptoms among patients under age 65 years was 49.5%, with the highest rate – 56.9% – being seen in traumatic brain injury patients.
Patients or caregivers were asked to rate on a 0-10 scale the impact their primary neurologic disease has had on their quality of life. Patients 65 years and older with PBA symptoms reported a significantly greater negative impact than did those without PBA symptoms, with mean scores on the quality of life impact scale of 6.3 vs. 4.6. The quality of life difference between those with PBA symptoms and those without was significant for patients with each of the neurologic diseases except for ALS.
As another measure of the adverse impact of having PBA symptoms, 56% of affected older patients were on at least one antipsychotic or antidepressant, compared with 35% of older patients without PBA symptoms.
PBA is thought to result from injury to neurologic pathways that regulate emotional expression as a secondary consequence of a variety of neurologic disorders.
In an interview, Dr. Crumpacker said PBA is greatly underdiagnosed and often gets misdiagnosed as depression.
"The symptoms are extremely disturbing to others, and patients are acutely aware of that. I tell my friends in neurology, it’s the psychiatric pathology that causes people problems in their lives. No one gets divorced over neurologic pathology, they get divorced over psychiatric pathology. It’s not, ‘I got a divorce because he had a stroke.’ " "It’s "We got divorced because he had a stroke and it changed his personality; he was a different person and I couldn’t be around him anymore,’ " the psychiatrist said.
PBA became a diagnosable disorder with its own ICD-9 code, albeit a diagnosis that can’t be made in the absence of neurologic pathology, at the behest of the Food and Drug Administration, Dr. Crumpacker explained. The impetus was the discovery of an effective treatment, dextromethorphan HBr and quinidine sulfate (Nuedexta), which received FDA approval for PBA 3 years ago.
Nuedexta’s development as the sole medication indicated for PBA was serendipitous, according to Dr. Crumpacker.
"The drug was being tested in Alzheimer’s disease. The jury is still out on whether it helps. But families of study participants came back saying, ‘You know that stuff dad used to do – the crying, the inappropriate laughter, the anger? He doesn’t do those kinds of things anymore,’ " Dr. Crumpacker recalled.
He reported serving on a scientific advisory board for Avanir Pharmaceuticals, which markets Nuedexta.
AT THE AAGP ANNUAL MEETING
Major finding: The prevalence of PBA symptoms among patients over age 65 years with any of six underlying neurologic disorders was 27.4%. That was significantly less than in younger patients with the same disorders, but the adverse effect of having PBA symptoms upon quality of life was markedly greater in the older group.
Data source: The PRISM study included 5,290 patients with Alzheimer’s disease or any of five other less common neurologic disorders, all of whom were screened for the presence of PBA symptoms using a brief validated measure.
Disclosures: The presenter serves on a scientific advisory board for Avanir Pharmaceuticals, which funded the PRISM study.
West Nile virus has cost the United States nearly $800 million
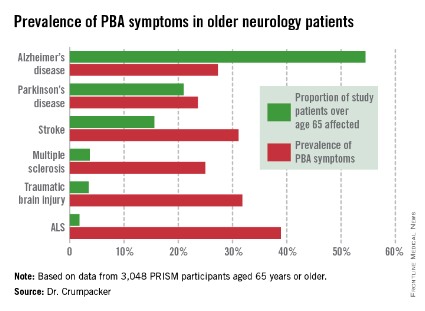
Since the West Nile virus was first detected in New York in 1999, hospitalized patients in the United States have cost an estimated $778.1 million in health care expenditures and lost productivity, according to a study published online Feb. 10 in the American Journal of Tropical Medicine and Hygiene.
Of that $778.1 million, the largest share – $449.5 million, or almost 58% – represents mean lifetime lost productivity from deaths caused by infection. Hospitalization for acute illness was estimated at $252.1 million, long-term medical care cost $27.6 million, long-term lost productivity cost $26.9 million, and short-term lost productivity (survivors only) totaled $22.1 million, investigators from the Centers for Disease Control and Prevention reported.
From 1999 through 2012, there were 37,088 cases of West Nile virus disease reported to the CDC’s ArboNET surveillance system, resulting in more than 18,000 hospitalizations and 1,529 deaths, the investigators said (Am. J. Trop. Med. Hyg. 2014 Feb. 10).
They determined the cost of initial hospitalization for 80 patients in a 2003 West Nile virus outbreak in Colorado, then calculated the cost of additional medical care and missed work for 38 patients who had 5 years of follow-up data available after the initial infection. These costs were then extrapolated to the total number of hospitalized cases in the United States since 1999.

Since the West Nile virus was first detected in New York in 1999, hospitalized patients in the United States have cost an estimated $778.1 million in health care expenditures and lost productivity, according to a study published online Feb. 10 in the American Journal of Tropical Medicine and Hygiene.
Of that $778.1 million, the largest share – $449.5 million, or almost 58% – represents mean lifetime lost productivity from deaths caused by infection. Hospitalization for acute illness was estimated at $252.1 million, long-term medical care cost $27.6 million, long-term lost productivity cost $26.9 million, and short-term lost productivity (survivors only) totaled $22.1 million, investigators from the Centers for Disease Control and Prevention reported.
From 1999 through 2012, there were 37,088 cases of West Nile virus disease reported to the CDC’s ArboNET surveillance system, resulting in more than 18,000 hospitalizations and 1,529 deaths, the investigators said (Am. J. Trop. Med. Hyg. 2014 Feb. 10).
They determined the cost of initial hospitalization for 80 patients in a 2003 West Nile virus outbreak in Colorado, then calculated the cost of additional medical care and missed work for 38 patients who had 5 years of follow-up data available after the initial infection. These costs were then extrapolated to the total number of hospitalized cases in the United States since 1999.
Since the West Nile virus was first detected in New York in 1999, hospitalized patients in the United States have cost an estimated $778.1 million in health care expenditures and lost productivity, according to a study published online Feb. 10 in the American Journal of Tropical Medicine and Hygiene.
Of that $778.1 million, the largest share – $449.5 million, or almost 58% – represents mean lifetime lost productivity from deaths caused by infection. Hospitalization for acute illness was estimated at $252.1 million, long-term medical care cost $27.6 million, long-term lost productivity cost $26.9 million, and short-term lost productivity (survivors only) totaled $22.1 million, investigators from the Centers for Disease Control and Prevention reported.
From 1999 through 2012, there were 37,088 cases of West Nile virus disease reported to the CDC’s ArboNET surveillance system, resulting in more than 18,000 hospitalizations and 1,529 deaths, the investigators said (Am. J. Trop. Med. Hyg. 2014 Feb. 10).
They determined the cost of initial hospitalization for 80 patients in a 2003 West Nile virus outbreak in Colorado, then calculated the cost of additional medical care and missed work for 38 patients who had 5 years of follow-up data available after the initial infection. These costs were then extrapolated to the total number of hospitalized cases in the United States since 1999.
FROM AMERICAN JOURNAL OF TROPICAL MEDICINE AND HYGIENE
Corneal nerve fiber loss may predict diabetic neuropathy
MELBOURNE – Corneal nerve fiber length, measured using corneal confocal microscopy, is significantly reduced in individuals with type 1 diabetes who go on to develop diabetic neuropathy at 3 years, according to data from the longitudinal LANDMark study.
Researchers found that corneal nerve fibre length was significantly lower at baseline in individuals who developed neuropathy than in those who did not over the 3-year follow up (13.3 vs. 17.4 mm/mm2, respectively; P = 0.036).

Corneal nerve fiber length, which is a measure of amount of nerve tissue per unit area in the cornea, may be an useful, noninvasive adjunct to diabetic neuropathy screening, Nicola Pritchard, a researcher for the Institute of Health and Biomedical Innovation at Queensland University of Technology, Brisbane, suggested in her presentation of the results at the World Diabetes Congress.
"In animal models, we know that the dropout of nerves in the cornea actually does precede the dropout of nerves in the foot," Ms. Pritchard said in an interview.
"Our hope is that this technique will be useful to pick up very, very early signs of neuropathy, way before people are getting symptoms and before things develop to a stage where there’s damage," she said.
LANDMark (Longitudinal Assessment of Neuropathy in Diabetes Using Novel Ophthalmic Markers) is a 5-year observational study of 242 individuals with type 1 diabetes.
The 3-year analysis included data from 64 participants without baseline neuropathy, seven (11%) of whom had developed neuropathy by 3 years, as defined by the Toronto criteria.
Study participants undergo annual neuropathy assessments, including measurement of corneal nerve parameters using corneal confocal microscopy and measurements of corneal sensitivity using noncontact corneal esthesiometry.
The study showed that reduced peroneal conduction velocity and cold sensation and increased vibration threshold also were associated with development of diabetic neuropathy.
However, although corneal nerve fiber length was significantly reduced at baseline in individuals who developed neuropathy, compared with those who did not, at the 3-year mark there was no significant difference in corneal nerve fiber length between the two groups.
Ms. Pritchard said that it was unclear why the nerve fibre parameters improved over time, suggesting that perhaps the nerves were growing to fill in the gaps.
Nathan Efron, D.Sc., research leader of the LANDMark study, said corneal confocal microscopy had the potential to be a very simple screening technique for diabetic neuropathy that could be applied at the same time as patients come in for their annual fundus photographs.
"At the very least, it’s a viable alternative technique to the range of techniques neurologists and diabetic specialists already have at their disposal, but the potential advantage of this technique is that it might be a very early marker of diabetic neuropathy," said Dr. Efron, professor in the School of Optometry and Vision Science at Queensland University of Technology.
"We’re not there yet, but down the line, that’s where this all could come to, as long as we can get more firm data and validate it a bit better," he said.
There were no relevant conflicts of interest declared.
MELBOURNE – Corneal nerve fiber length, measured using corneal confocal microscopy, is significantly reduced in individuals with type 1 diabetes who go on to develop diabetic neuropathy at 3 years, according to data from the longitudinal LANDMark study.
Researchers found that corneal nerve fibre length was significantly lower at baseline in individuals who developed neuropathy than in those who did not over the 3-year follow up (13.3 vs. 17.4 mm/mm2, respectively; P = 0.036).
Corneal nerve fiber length, which is a measure of amount of nerve tissue per unit area in the cornea, may be an useful, noninvasive adjunct to diabetic neuropathy screening, Nicola Pritchard, a researcher for the Institute of Health and Biomedical Innovation at Queensland University of Technology, Brisbane, suggested in her presentation of the results at the World Diabetes Congress.
"In animal models, we know that the dropout of nerves in the cornea actually does precede the dropout of nerves in the foot," Ms. Pritchard said in an interview.
"Our hope is that this technique will be useful to pick up very, very early signs of neuropathy, way before people are getting symptoms and before things develop to a stage where there’s damage," she said.
LANDMark (Longitudinal Assessment of Neuropathy in Diabetes Using Novel Ophthalmic Markers) is a 5-year observational study of 242 individuals with type 1 diabetes.
The 3-year analysis included data from 64 participants without baseline neuropathy, seven (11%) of whom had developed neuropathy by 3 years, as defined by the Toronto criteria.
Study participants undergo annual neuropathy assessments, including measurement of corneal nerve parameters using corneal confocal microscopy and measurements of corneal sensitivity using noncontact corneal esthesiometry.
The study showed that reduced peroneal conduction velocity and cold sensation and increased vibration threshold also were associated with development of diabetic neuropathy.
However, although corneal nerve fiber length was significantly reduced at baseline in individuals who developed neuropathy, compared with those who did not, at the 3-year mark there was no significant difference in corneal nerve fiber length between the two groups.
Ms. Pritchard said that it was unclear why the nerve fibre parameters improved over time, suggesting that perhaps the nerves were growing to fill in the gaps.
Nathan Efron, D.Sc., research leader of the LANDMark study, said corneal confocal microscopy had the potential to be a very simple screening technique for diabetic neuropathy that could be applied at the same time as patients come in for their annual fundus photographs.
"At the very least, it’s a viable alternative technique to the range of techniques neurologists and diabetic specialists already have at their disposal, but the potential advantage of this technique is that it might be a very early marker of diabetic neuropathy," said Dr. Efron, professor in the School of Optometry and Vision Science at Queensland University of Technology.
"We’re not there yet, but down the line, that’s where this all could come to, as long as we can get more firm data and validate it a bit better," he said.
There were no relevant conflicts of interest declared.
MELBOURNE – Corneal nerve fiber length, measured using corneal confocal microscopy, is significantly reduced in individuals with type 1 diabetes who go on to develop diabetic neuropathy at 3 years, according to data from the longitudinal LANDMark study.
Researchers found that corneal nerve fibre length was significantly lower at baseline in individuals who developed neuropathy than in those who did not over the 3-year follow up (13.3 vs. 17.4 mm/mm2, respectively; P = 0.036).
Corneal nerve fiber length, which is a measure of amount of nerve tissue per unit area in the cornea, may be an useful, noninvasive adjunct to diabetic neuropathy screening, Nicola Pritchard, a researcher for the Institute of Health and Biomedical Innovation at Queensland University of Technology, Brisbane, suggested in her presentation of the results at the World Diabetes Congress.
"In animal models, we know that the dropout of nerves in the cornea actually does precede the dropout of nerves in the foot," Ms. Pritchard said in an interview.
"Our hope is that this technique will be useful to pick up very, very early signs of neuropathy, way before people are getting symptoms and before things develop to a stage where there’s damage," she said.
LANDMark (Longitudinal Assessment of Neuropathy in Diabetes Using Novel Ophthalmic Markers) is a 5-year observational study of 242 individuals with type 1 diabetes.
The 3-year analysis included data from 64 participants without baseline neuropathy, seven (11%) of whom had developed neuropathy by 3 years, as defined by the Toronto criteria.
Study participants undergo annual neuropathy assessments, including measurement of corneal nerve parameters using corneal confocal microscopy and measurements of corneal sensitivity using noncontact corneal esthesiometry.
The study showed that reduced peroneal conduction velocity and cold sensation and increased vibration threshold also were associated with development of diabetic neuropathy.
However, although corneal nerve fiber length was significantly reduced at baseline in individuals who developed neuropathy, compared with those who did not, at the 3-year mark there was no significant difference in corneal nerve fiber length between the two groups.
Ms. Pritchard said that it was unclear why the nerve fibre parameters improved over time, suggesting that perhaps the nerves were growing to fill in the gaps.
Nathan Efron, D.Sc., research leader of the LANDMark study, said corneal confocal microscopy had the potential to be a very simple screening technique for diabetic neuropathy that could be applied at the same time as patients come in for their annual fundus photographs.
"At the very least, it’s a viable alternative technique to the range of techniques neurologists and diabetic specialists already have at their disposal, but the potential advantage of this technique is that it might be a very early marker of diabetic neuropathy," said Dr. Efron, professor in the School of Optometry and Vision Science at Queensland University of Technology.
"We’re not there yet, but down the line, that’s where this all could come to, as long as we can get more firm data and validate it a bit better," he said.
There were no relevant conflicts of interest declared.
AT THE WORLD DIABETES CONGRESS
Major finding: Corneal nerve fiber length was significantly lower at baseline in individuals who developed neuropathy at 3 years, compared with those who did not develop neuropathy (13.3 vs. 17.4 mm/mm2, respectively; P = 0.036).
Data source: Analysis of baseline and 3-year data from the LANDMark trial looking at novel ophthalmic markers of neuropathy in 242 individuals with type 1 diabetes.
Disclosures: No financial conflicts of interest declared.

Consider small-fiber neuropathies in systemic lupus erythematosus
Small-fiber neuropathy is one of the most common types of peripheral neuropathy affecting patients with systemic lupus erythematosus, but it isn’t even mentioned in the American College of Rheumatology neuropsychiatric case definitions of manifestations of the disorder, according to a retrospective analysis of cohort of 2,097 patients with SLE.
Other types of peripheral neuropathy, such as acute inflammatory demyelinating neuropathies (for example, Guillain-Barré syndrome), plexopathies, and mononeuritis multiplex, are well described in the ACR-NPSLE case definitions but occur much less frequently. This, combined with the fact that small-fiber neuropathies often present as "unorthodox" pain patterns, indicates that they are underdiagnosed, said Dr. Amin Oomatia of the University of Cambridge, England, and his coinvestigators at John Hopkins University, Baltimore.
Small-fiber neuropathies arise through mechanisms that are distinct from those of other neuropathies and require different diagnostic strategies to be properly identified. In particular, small-fiber neuropathies do not always conform to the "stocking-and-glove" pattern of pain that is typical of other neuropathies in SLE, so it is likely that many affected patients "may be regarded in routine clinical care as having a ‘nonorganic’ pain disorder.
"Our findings suggest that rheumatologists and other clinicians who confront SLE patients with seemingly improbable pain patterns should consider the diagnosis of a small-fiber neuropathy," the investigators wrote, especially since it may occur in the face of normal electrodiagnostic studies.
Dr. Oomatia and his colleagues based these conclusions on their retrospective study of one medical center’s 25-year experience treating 2,097 SLE patients – the Johns Hopkins Lupus Cohort. Using details in a database of patients’ electronic medical records, they identified 82 patients who had peripheral neuropathies related to SLE.
Only one patient had peripheral neuropathy attributable to Guillain-Barré syndrome, only one patient had a plexopathy, and only six patients had mononeuritis multiplex, demonstrating that these are very infrequent complications of SLE even though they are included in ACR case definitions.
In contrast, 14 patients (17% of those with peripheral neuropathy) had biopsy-proven small-fiber neuropathies, and most of them presented with "an entirely different and unorthodox pain distribution" characterized as patchy, asymmetric, or proximal.
In particular, nine patients had pain affecting the face, torso, and/or proximal extremities. Three had burning pain over their entire bodies, the investigators said (Arthritis Rheum. 2013 Dec. 10 [doi:10.1002/art.38302]).
In these cases, punch skin biopsy showed abnormalities that disproportionately affected the proximal thigh, "which is considered a surrogate indicator of proximal-most dorsal root ganglia neuronal cell loss," they wrote. In contrast, other patients who had the typical distal pattern of neuropathic pain showed decreased intraepidermal nerve-fiber densities in the distal leg, a surrogate indicator of distal-most axonal degeneration.
Another distinguishing feature of small-fiber neuropathy was its association with a history of herpes zoster virus, opportunistic infections, and osteoporotic fractures, all unrelated to corticosteroid dose, Dr. Oomatia and his associates said.
This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
Small-fiber neuropathy is one of the most common types of peripheral neuropathy affecting patients with systemic lupus erythematosus, but it isn’t even mentioned in the American College of Rheumatology neuropsychiatric case definitions of manifestations of the disorder, according to a retrospective analysis of cohort of 2,097 patients with SLE.
Other types of peripheral neuropathy, such as acute inflammatory demyelinating neuropathies (for example, Guillain-Barré syndrome), plexopathies, and mononeuritis multiplex, are well described in the ACR-NPSLE case definitions but occur much less frequently. This, combined with the fact that small-fiber neuropathies often present as "unorthodox" pain patterns, indicates that they are underdiagnosed, said Dr. Amin Oomatia of the University of Cambridge, England, and his coinvestigators at John Hopkins University, Baltimore.
Small-fiber neuropathies arise through mechanisms that are distinct from those of other neuropathies and require different diagnostic strategies to be properly identified. In particular, small-fiber neuropathies do not always conform to the "stocking-and-glove" pattern of pain that is typical of other neuropathies in SLE, so it is likely that many affected patients "may be regarded in routine clinical care as having a ‘nonorganic’ pain disorder.
"Our findings suggest that rheumatologists and other clinicians who confront SLE patients with seemingly improbable pain patterns should consider the diagnosis of a small-fiber neuropathy," the investigators wrote, especially since it may occur in the face of normal electrodiagnostic studies.
Dr. Oomatia and his colleagues based these conclusions on their retrospective study of one medical center’s 25-year experience treating 2,097 SLE patients – the Johns Hopkins Lupus Cohort. Using details in a database of patients’ electronic medical records, they identified 82 patients who had peripheral neuropathies related to SLE.
Only one patient had peripheral neuropathy attributable to Guillain-Barré syndrome, only one patient had a plexopathy, and only six patients had mononeuritis multiplex, demonstrating that these are very infrequent complications of SLE even though they are included in ACR case definitions.
In contrast, 14 patients (17% of those with peripheral neuropathy) had biopsy-proven small-fiber neuropathies, and most of them presented with "an entirely different and unorthodox pain distribution" characterized as patchy, asymmetric, or proximal.
In particular, nine patients had pain affecting the face, torso, and/or proximal extremities. Three had burning pain over their entire bodies, the investigators said (Arthritis Rheum. 2013 Dec. 10 [doi:10.1002/art.38302]).
In these cases, punch skin biopsy showed abnormalities that disproportionately affected the proximal thigh, "which is considered a surrogate indicator of proximal-most dorsal root ganglia neuronal cell loss," they wrote. In contrast, other patients who had the typical distal pattern of neuropathic pain showed decreased intraepidermal nerve-fiber densities in the distal leg, a surrogate indicator of distal-most axonal degeneration.
Another distinguishing feature of small-fiber neuropathy was its association with a history of herpes zoster virus, opportunistic infections, and osteoporotic fractures, all unrelated to corticosteroid dose, Dr. Oomatia and his associates said.
This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
Small-fiber neuropathy is one of the most common types of peripheral neuropathy affecting patients with systemic lupus erythematosus, but it isn’t even mentioned in the American College of Rheumatology neuropsychiatric case definitions of manifestations of the disorder, according to a retrospective analysis of cohort of 2,097 patients with SLE.
Other types of peripheral neuropathy, such as acute inflammatory demyelinating neuropathies (for example, Guillain-Barré syndrome), plexopathies, and mononeuritis multiplex, are well described in the ACR-NPSLE case definitions but occur much less frequently. This, combined with the fact that small-fiber neuropathies often present as "unorthodox" pain patterns, indicates that they are underdiagnosed, said Dr. Amin Oomatia of the University of Cambridge, England, and his coinvestigators at John Hopkins University, Baltimore.
Small-fiber neuropathies arise through mechanisms that are distinct from those of other neuropathies and require different diagnostic strategies to be properly identified. In particular, small-fiber neuropathies do not always conform to the "stocking-and-glove" pattern of pain that is typical of other neuropathies in SLE, so it is likely that many affected patients "may be regarded in routine clinical care as having a ‘nonorganic’ pain disorder.
"Our findings suggest that rheumatologists and other clinicians who confront SLE patients with seemingly improbable pain patterns should consider the diagnosis of a small-fiber neuropathy," the investigators wrote, especially since it may occur in the face of normal electrodiagnostic studies.
Dr. Oomatia and his colleagues based these conclusions on their retrospective study of one medical center’s 25-year experience treating 2,097 SLE patients – the Johns Hopkins Lupus Cohort. Using details in a database of patients’ electronic medical records, they identified 82 patients who had peripheral neuropathies related to SLE.
Only one patient had peripheral neuropathy attributable to Guillain-Barré syndrome, only one patient had a plexopathy, and only six patients had mononeuritis multiplex, demonstrating that these are very infrequent complications of SLE even though they are included in ACR case definitions.
In contrast, 14 patients (17% of those with peripheral neuropathy) had biopsy-proven small-fiber neuropathies, and most of them presented with "an entirely different and unorthodox pain distribution" characterized as patchy, asymmetric, or proximal.
In particular, nine patients had pain affecting the face, torso, and/or proximal extremities. Three had burning pain over their entire bodies, the investigators said (Arthritis Rheum. 2013 Dec. 10 [doi:10.1002/art.38302]).
In these cases, punch skin biopsy showed abnormalities that disproportionately affected the proximal thigh, "which is considered a surrogate indicator of proximal-most dorsal root ganglia neuronal cell loss," they wrote. In contrast, other patients who had the typical distal pattern of neuropathic pain showed decreased intraepidermal nerve-fiber densities in the distal leg, a surrogate indicator of distal-most axonal degeneration.
Another distinguishing feature of small-fiber neuropathy was its association with a history of herpes zoster virus, opportunistic infections, and osteoporotic fractures, all unrelated to corticosteroid dose, Dr. Oomatia and his associates said.
This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
FROM ARTHRITIS AND RHEUMATISM
Major finding: A total of 14 patients, or 17% of 82 with peripheral neuropathies, had biopsy-proven small-fiber neuropathies and often presented with unorthodox patterns of pain.
Data source: A retrospective analysis of data regarding 2,097 consecutive patients with SLE registered in the Johns Hopkins Lupus Cohort during a 25-year period, including 82 who developed peripheral neuropathies related to the disease.
Disclosures: This study was supported in part by the National Institutes of Health and the National Center for Research Resources. No potential financial conflicts of interest were reported.
Autoantibodies play role in myositis classification, treatment
LAS VEGAS – Autoantibodies and autoantibody subsets can be particularly helpful for classifying and treating patients with myositis, including those with myositis-associated interstitial lung disease, according to Dr. Chester V. Oddis.
Many autoantibody subsets are phenotypically and clinically well defined, and have clinical relevance, said Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh.
Serological classification isn’t always accurate or routinely available for all clinicians, but this may change in the near future as improved techniques for autoantibody detection become available, he said at Perspectives in Rheumatic Diseases 2013.
Anti-MDA-5 and interstitial lung disease
One autoantibody that has gained attention in recent years is anti-MDA-5, also known as anti-CADM-140, which is often seen in patients with amyopathic dermatomyositis (ADM).
Patients with ADM represent a subset of dermatomyositis patients who have cutaneous manifestations of dermatomyositis for 6 months or longer and have no clinical evidence of proximal muscle weakness but may have mild serum muscle enzyme abnormalities. More extensive muscle testing in these patients generally demonstrates no or minimal abnormalities. However, these patients should not be considered to have simply a benign cutaneous form of disease; in fact, they have a frequency of malignancy similar to that of patients with classic dermatomyositis (14% in one series of nearly 300 patients, compared with 15% in classic dermatomyositis).
In addition, ADM patients also have a relatively high frequency of lung disease, Dr. Oddis said. In a published review of the literature of nearly 200 patients with ADM, 10% had interstitial lung disease (ILD) – an important point given that the rash of dermatomyositis may be subtle and missed, he noted.
The Asian population seems to be particularly at risk for this complication. Two studies in recent years have demonstrated that Japanese ADM patients with anti-MDA-5 present with rapidly progressive ILD. A 2011 study showed an increased incidence of acute or subacute interstitial pneumonitis in Chinese patients. Other studies have shown similar findings in Korean and other Asian populations, Dr. Oddis noted.
The presence of anti-MDA-5 represents a novel cutaneous phenotype involving palmar papules and cutaneous ulcerations, severe vasculopathy, and rapidly progressive ILD. The target autoantigen in these cases is MDA-5, which is involved in innate immune defense against viruses, he explained, noting that this supports the possibility that a viral trigger plays a role in the disease.
"I think this complication is filtering into the U.S. population, as we have seen it in our myositis cohort," Dr. Oddis said of the anti-MDA-5 association with ADM and severe ILD. He noted that he recently cared for a 70-year-old white male with "double pneumonia" (a finding that "should always raise suspicion of autoimmune ILD") in June of 2012, a rash of dermatomyositis in September of 2012, and vasculitic skin changes in January of 2013. He presented without muscle weakness.
Anti-synthetase syndrome and ILD
Another autoantibody myositis subset involves the anti-synthetases, including PL-7, PL-12, EJ, and Jo-1.
Patients with anti-synthetase syndrome are generally a clinically homogeneous patient population characterized by fever, myositis, arthritis, Raynaud’s phenomenon, mechanic’s hands, and ILD. About 30%-40% of myositis patients have ILD, which is a significant contributor to morbidity and mortality.
Anti-Jo-1 is found in 50%-75% of these patients, and the coexistence of Ro52 may portend worse prognosis, Dr. Oddis said.
There are certain clinical features of ILD in polymyositis and dermatomyositis, including progressive dyspnea with or without nonproductive cough, lack of digital clubbing (unlike in idiopathic pulmonary fibrosis), and lack of pleuritis and pleural effusion in most cases (unlike in systemic lupus erythematosus). However, presentation can be variable, with about one-third of patients developing ILD before muscle or skin manifestations are apparent. Some patients present with acute disease (acute respiratory distress syndrome), and others present with subacute disease that is chronic and slowly progressing or asymptomatic.
It is important to understand when making a diagnosis of autoimmune ILD that not all patients will present with the classic anti-synthetase syndrome, Dr. Oddis said.
In some cases, patients will have an "incomplete" clinical syndrome with ILD alone or ILD with only subtle connective tissue disease findings, myositis-specific autoantibodies in the absence of myositis, and/or a negative antinuclear antibody (ANA) test, he explained.
The initial symptoms in patients may vary depending on the anti-synthetase autoantibody that is present. In a University of Pittsburgh study, for example, Jo-1 was found in 60% of cases; non-Jo-1 synthetase positive cases more often experienced Raynaud’s as their initial symptom, less often experienced muscle and joint problems as their initial symptom, and had a longer delay in diagnosis, compared with Jo-1 patients. Survival was also decreased, compared with Jo-1 patients.
As for ANA, about half of patients tested positive, whereas 72% demonstrated positive anti-cytoplasmic staining on indirect immunofluorescence.
The diagnosis of autoimmune ILD can be missed when there is a failure to recognize "incomplete" clinical syndromes, when there is a failure to order or detect myositis-specific autoantibodies – even in patients without myositis – and when a negative ANA is considered to be reassuring, Dr. Oddis said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Autoantibodies and autoantibody subsets can be particularly helpful for classifying and treating patients with myositis, including those with myositis-associated interstitial lung disease, according to Dr. Chester V. Oddis.
Many autoantibody subsets are phenotypically and clinically well defined, and have clinical relevance, said Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh.
Serological classification isn’t always accurate or routinely available for all clinicians, but this may change in the near future as improved techniques for autoantibody detection become available, he said at Perspectives in Rheumatic Diseases 2013.
Anti-MDA-5 and interstitial lung disease
One autoantibody that has gained attention in recent years is anti-MDA-5, also known as anti-CADM-140, which is often seen in patients with amyopathic dermatomyositis (ADM).
Patients with ADM represent a subset of dermatomyositis patients who have cutaneous manifestations of dermatomyositis for 6 months or longer and have no clinical evidence of proximal muscle weakness but may have mild serum muscle enzyme abnormalities. More extensive muscle testing in these patients generally demonstrates no or minimal abnormalities. However, these patients should not be considered to have simply a benign cutaneous form of disease; in fact, they have a frequency of malignancy similar to that of patients with classic dermatomyositis (14% in one series of nearly 300 patients, compared with 15% in classic dermatomyositis).
In addition, ADM patients also have a relatively high frequency of lung disease, Dr. Oddis said. In a published review of the literature of nearly 200 patients with ADM, 10% had interstitial lung disease (ILD) – an important point given that the rash of dermatomyositis may be subtle and missed, he noted.
The Asian population seems to be particularly at risk for this complication. Two studies in recent years have demonstrated that Japanese ADM patients with anti-MDA-5 present with rapidly progressive ILD. A 2011 study showed an increased incidence of acute or subacute interstitial pneumonitis in Chinese patients. Other studies have shown similar findings in Korean and other Asian populations, Dr. Oddis noted.
The presence of anti-MDA-5 represents a novel cutaneous phenotype involving palmar papules and cutaneous ulcerations, severe vasculopathy, and rapidly progressive ILD. The target autoantigen in these cases is MDA-5, which is involved in innate immune defense against viruses, he explained, noting that this supports the possibility that a viral trigger plays a role in the disease.
"I think this complication is filtering into the U.S. population, as we have seen it in our myositis cohort," Dr. Oddis said of the anti-MDA-5 association with ADM and severe ILD. He noted that he recently cared for a 70-year-old white male with "double pneumonia" (a finding that "should always raise suspicion of autoimmune ILD") in June of 2012, a rash of dermatomyositis in September of 2012, and vasculitic skin changes in January of 2013. He presented without muscle weakness.
Anti-synthetase syndrome and ILD
Another autoantibody myositis subset involves the anti-synthetases, including PL-7, PL-12, EJ, and Jo-1.
Patients with anti-synthetase syndrome are generally a clinically homogeneous patient population characterized by fever, myositis, arthritis, Raynaud’s phenomenon, mechanic’s hands, and ILD. About 30%-40% of myositis patients have ILD, which is a significant contributor to morbidity and mortality.
Anti-Jo-1 is found in 50%-75% of these patients, and the coexistence of Ro52 may portend worse prognosis, Dr. Oddis said.
There are certain clinical features of ILD in polymyositis and dermatomyositis, including progressive dyspnea with or without nonproductive cough, lack of digital clubbing (unlike in idiopathic pulmonary fibrosis), and lack of pleuritis and pleural effusion in most cases (unlike in systemic lupus erythematosus). However, presentation can be variable, with about one-third of patients developing ILD before muscle or skin manifestations are apparent. Some patients present with acute disease (acute respiratory distress syndrome), and others present with subacute disease that is chronic and slowly progressing or asymptomatic.
It is important to understand when making a diagnosis of autoimmune ILD that not all patients will present with the classic anti-synthetase syndrome, Dr. Oddis said.
In some cases, patients will have an "incomplete" clinical syndrome with ILD alone or ILD with only subtle connective tissue disease findings, myositis-specific autoantibodies in the absence of myositis, and/or a negative antinuclear antibody (ANA) test, he explained.
The initial symptoms in patients may vary depending on the anti-synthetase autoantibody that is present. In a University of Pittsburgh study, for example, Jo-1 was found in 60% of cases; non-Jo-1 synthetase positive cases more often experienced Raynaud’s as their initial symptom, less often experienced muscle and joint problems as their initial symptom, and had a longer delay in diagnosis, compared with Jo-1 patients. Survival was also decreased, compared with Jo-1 patients.
As for ANA, about half of patients tested positive, whereas 72% demonstrated positive anti-cytoplasmic staining on indirect immunofluorescence.
The diagnosis of autoimmune ILD can be missed when there is a failure to recognize "incomplete" clinical syndromes, when there is a failure to order or detect myositis-specific autoantibodies – even in patients without myositis – and when a negative ANA is considered to be reassuring, Dr. Oddis said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Autoantibodies and autoantibody subsets can be particularly helpful for classifying and treating patients with myositis, including those with myositis-associated interstitial lung disease, according to Dr. Chester V. Oddis.
Many autoantibody subsets are phenotypically and clinically well defined, and have clinical relevance, said Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh.
Serological classification isn’t always accurate or routinely available for all clinicians, but this may change in the near future as improved techniques for autoantibody detection become available, he said at Perspectives in Rheumatic Diseases 2013.
Anti-MDA-5 and interstitial lung disease
One autoantibody that has gained attention in recent years is anti-MDA-5, also known as anti-CADM-140, which is often seen in patients with amyopathic dermatomyositis (ADM).
Patients with ADM represent a subset of dermatomyositis patients who have cutaneous manifestations of dermatomyositis for 6 months or longer and have no clinical evidence of proximal muscle weakness but may have mild serum muscle enzyme abnormalities. More extensive muscle testing in these patients generally demonstrates no or minimal abnormalities. However, these patients should not be considered to have simply a benign cutaneous form of disease; in fact, they have a frequency of malignancy similar to that of patients with classic dermatomyositis (14% in one series of nearly 300 patients, compared with 15% in classic dermatomyositis).
In addition, ADM patients also have a relatively high frequency of lung disease, Dr. Oddis said. In a published review of the literature of nearly 200 patients with ADM, 10% had interstitial lung disease (ILD) – an important point given that the rash of dermatomyositis may be subtle and missed, he noted.
The Asian population seems to be particularly at risk for this complication. Two studies in recent years have demonstrated that Japanese ADM patients with anti-MDA-5 present with rapidly progressive ILD. A 2011 study showed an increased incidence of acute or subacute interstitial pneumonitis in Chinese patients. Other studies have shown similar findings in Korean and other Asian populations, Dr. Oddis noted.
The presence of anti-MDA-5 represents a novel cutaneous phenotype involving palmar papules and cutaneous ulcerations, severe vasculopathy, and rapidly progressive ILD. The target autoantigen in these cases is MDA-5, which is involved in innate immune defense against viruses, he explained, noting that this supports the possibility that a viral trigger plays a role in the disease.
"I think this complication is filtering into the U.S. population, as we have seen it in our myositis cohort," Dr. Oddis said of the anti-MDA-5 association with ADM and severe ILD. He noted that he recently cared for a 70-year-old white male with "double pneumonia" (a finding that "should always raise suspicion of autoimmune ILD") in June of 2012, a rash of dermatomyositis in September of 2012, and vasculitic skin changes in January of 2013. He presented without muscle weakness.
Anti-synthetase syndrome and ILD
Another autoantibody myositis subset involves the anti-synthetases, including PL-7, PL-12, EJ, and Jo-1.
Patients with anti-synthetase syndrome are generally a clinically homogeneous patient population characterized by fever, myositis, arthritis, Raynaud’s phenomenon, mechanic’s hands, and ILD. About 30%-40% of myositis patients have ILD, which is a significant contributor to morbidity and mortality.
Anti-Jo-1 is found in 50%-75% of these patients, and the coexistence of Ro52 may portend worse prognosis, Dr. Oddis said.
There are certain clinical features of ILD in polymyositis and dermatomyositis, including progressive dyspnea with or without nonproductive cough, lack of digital clubbing (unlike in idiopathic pulmonary fibrosis), and lack of pleuritis and pleural effusion in most cases (unlike in systemic lupus erythematosus). However, presentation can be variable, with about one-third of patients developing ILD before muscle or skin manifestations are apparent. Some patients present with acute disease (acute respiratory distress syndrome), and others present with subacute disease that is chronic and slowly progressing or asymptomatic.
It is important to understand when making a diagnosis of autoimmune ILD that not all patients will present with the classic anti-synthetase syndrome, Dr. Oddis said.
In some cases, patients will have an "incomplete" clinical syndrome with ILD alone or ILD with only subtle connective tissue disease findings, myositis-specific autoantibodies in the absence of myositis, and/or a negative antinuclear antibody (ANA) test, he explained.
The initial symptoms in patients may vary depending on the anti-synthetase autoantibody that is present. In a University of Pittsburgh study, for example, Jo-1 was found in 60% of cases; non-Jo-1 synthetase positive cases more often experienced Raynaud’s as their initial symptom, less often experienced muscle and joint problems as their initial symptom, and had a longer delay in diagnosis, compared with Jo-1 patients. Survival was also decreased, compared with Jo-1 patients.
As for ANA, about half of patients tested positive, whereas 72% demonstrated positive anti-cytoplasmic staining on indirect immunofluorescence.
The diagnosis of autoimmune ILD can be missed when there is a failure to recognize "incomplete" clinical syndromes, when there is a failure to order or detect myositis-specific autoantibodies – even in patients without myositis – and when a negative ANA is considered to be reassuring, Dr. Oddis said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
AT PERSPECTIVES IN RHEUMATIC DISEASES 2013

Limited data support multiple myositis treatment options
LAS VEGAS – Corticosteroids remain the initial treatment of choice for myositis and myositis-associated interstitial lung disease, but immunosuppressive agents, intravenous immunoglobulin, and biologics can also play a role in the treatment of one or both of these conditions, according to Dr. Chester V. Oddis.
For myositis in general, Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh, recommends an initial divided dose of 30 mg of prednisone twice daily, continued until serum creatine kinase (CK) levels fall to normal. At that time, the total daily prednisone dose can be consolidated into a single morning dose, he said at Perspectives in Rheumatic Diseases 2013.

The prednisone can then be tapered by 25% every 3-4 weeks down to a 5- to 10-mg daily maintenance dose that is continued until active disease is suppressed for 12 months. This is a general guideline that helps prevent disease flares, he noted.
Keep in mind that improvement in strength generally lags behind improvement in CK levels, he added.
Nonsteroidal immunosuppressives
Not all patients will need an additional immunosuppressive agent, but for those who do, methotrexate is a good option, Dr. Oddis said, noting that methotrexate is the drug he is most comfortable using in those cases.
The literature also supports the combined use of methotrexate and azathioprine, which when given together have been shown to be effective in treatment-resistant myositis and in those who failed either of the drugs alone.
"So that’s a regimen you might want to think about," he said.
Another immunosuppressive option is mycophenolate mofetil (MMF), which has been shown in several small studies and case series to be of benefit. In one study, 6 of 10 patients with dermatomyositis successfully tapered corticosteroids with MMF, and 10 of 12 in another study experienced improvement in cutaneous features of the disease.
The use of intravenous immunoglobulin (IVIg) as add-on therapy with MMF was effective in severe refractory patients, including four with polymyositis and three with dermatomyositis. In a retrospective review of 50 patients with juvenile dermatomyositis, MMF for 12 months was well tolerated, improved skin and muscle, and proved to be steroid-sparing, Dr. Oddis said.
Cyclosporine, tacrolimus, and cyclophosphamide are other immunosuppressive options.
While cyclophosphamide is more often used for myositis-associated interstitial lung disease (ILD), it can be of benefit for refractory skin disease, and can be useful in non-ILD myositis cases that involve severe skin disease.
The only available controlled data for IVIg alone are from a study published more than 20 years ago, but that randomized, double-blind, placebo-controlled study showed that treatment was safe, effective, and steroid sparing in 15 patients with dermatomyositis, he said.
Biologics
As for biologics, anti–tumor necrosis factor–alpha (anti-TNF-alpha) therapy and B-cell therapy have both been considered. Anti-TNF-alpha therapy makes sense because TNF-alpha and other proinflammatory cytokines are increased in muscle tissue of myositis patients; TNF-alpha is toxic to myofibers and prevents their regeneration; and TNF-alpha enhances other inflammatory cytokines in both dermatomyositis and polymyositis, but data are lacking on whether targeting TNF-alpha is worthwhile.
B cell therapy, on the other hand, is showing promise. In one open-label pilot study, rituximab was effective in seven patients with refractory dermatomyositis, and in others it was effective in anti-synthetase syndrome. Rituximab also was effective in two studies for refractory myositis and dermatomyositis rash, and it induced longstanding remission in some of the patients. In another study, however, rituximab was not effective for dermatomyositis rash.
The multicenter Rituximab in Myositis (RIM) study, the largest ever done in myositis, evaluated rituximab for the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis patients.
Although the primary and secondary endpoints of the RIM study were not achieved, 83% of refractory adult and juvenile myositis patients met the definition of improvement, there was a significant corticosteroid sparing effect between the baseline dose and the dose at study conclusion, and treatment was generally well tolerated, he said.
Other targets that are being explored include interleukin-6 and type 1 IFN genes. Findings suggest that coordinated dysregulation of type 1 IFN signaling and IL-6 production are contributors to dermatomyositis pathogenesis, he explained.
Treating myositis patients with ILD
The treatment approach to these patients is somewhat similar to those without ILD, with corticosteroids as initial treatment, Dr. Oddis said.
Cyclophosphamide and azathioprine have been used early on, and also in corticosteroid resistant cases, but with variable results. Cyclophosphamide can be given orally or by IV for 3-6 months.
MMF has been used with success in connective tissue disease–associated ILD, and based on small studies it appears to be effective in myositis-associated ILD as well.
Cyclosporine and tacrolimus have been used in both adult and pediatric patients with promising, steroid-sparing results, he said, noting that the use of anti-T-cell therapy in myositis-associated ILD makes sense, because findings from multiple studies have implicated activated CD8-positive T-cells in myositis-associated ILD.
"It’s an exciting time for therapeutic interventions in myositis, but even though we have all these therapeutic options, we have to temper our enthusiasm with what they do long term," he said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Corticosteroids remain the initial treatment of choice for myositis and myositis-associated interstitial lung disease, but immunosuppressive agents, intravenous immunoglobulin, and biologics can also play a role in the treatment of one or both of these conditions, according to Dr. Chester V. Oddis.
For myositis in general, Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh, recommends an initial divided dose of 30 mg of prednisone twice daily, continued until serum creatine kinase (CK) levels fall to normal. At that time, the total daily prednisone dose can be consolidated into a single morning dose, he said at Perspectives in Rheumatic Diseases 2013.
The prednisone can then be tapered by 25% every 3-4 weeks down to a 5- to 10-mg daily maintenance dose that is continued until active disease is suppressed for 12 months. This is a general guideline that helps prevent disease flares, he noted.
Keep in mind that improvement in strength generally lags behind improvement in CK levels, he added.
Nonsteroidal immunosuppressives
Not all patients will need an additional immunosuppressive agent, but for those who do, methotrexate is a good option, Dr. Oddis said, noting that methotrexate is the drug he is most comfortable using in those cases.
The literature also supports the combined use of methotrexate and azathioprine, which when given together have been shown to be effective in treatment-resistant myositis and in those who failed either of the drugs alone.
"So that’s a regimen you might want to think about," he said.
Another immunosuppressive option is mycophenolate mofetil (MMF), which has been shown in several small studies and case series to be of benefit. In one study, 6 of 10 patients with dermatomyositis successfully tapered corticosteroids with MMF, and 10 of 12 in another study experienced improvement in cutaneous features of the disease.
The use of intravenous immunoglobulin (IVIg) as add-on therapy with MMF was effective in severe refractory patients, including four with polymyositis and three with dermatomyositis. In a retrospective review of 50 patients with juvenile dermatomyositis, MMF for 12 months was well tolerated, improved skin and muscle, and proved to be steroid-sparing, Dr. Oddis said.
Cyclosporine, tacrolimus, and cyclophosphamide are other immunosuppressive options.
While cyclophosphamide is more often used for myositis-associated interstitial lung disease (ILD), it can be of benefit for refractory skin disease, and can be useful in non-ILD myositis cases that involve severe skin disease.
The only available controlled data for IVIg alone are from a study published more than 20 years ago, but that randomized, double-blind, placebo-controlled study showed that treatment was safe, effective, and steroid sparing in 15 patients with dermatomyositis, he said.
Biologics
As for biologics, anti–tumor necrosis factor–alpha (anti-TNF-alpha) therapy and B-cell therapy have both been considered. Anti-TNF-alpha therapy makes sense because TNF-alpha and other proinflammatory cytokines are increased in muscle tissue of myositis patients; TNF-alpha is toxic to myofibers and prevents their regeneration; and TNF-alpha enhances other inflammatory cytokines in both dermatomyositis and polymyositis, but data are lacking on whether targeting TNF-alpha is worthwhile.
B cell therapy, on the other hand, is showing promise. In one open-label pilot study, rituximab was effective in seven patients with refractory dermatomyositis, and in others it was effective in anti-synthetase syndrome. Rituximab also was effective in two studies for refractory myositis and dermatomyositis rash, and it induced longstanding remission in some of the patients. In another study, however, rituximab was not effective for dermatomyositis rash.
The multicenter Rituximab in Myositis (RIM) study, the largest ever done in myositis, evaluated rituximab for the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis patients.
Although the primary and secondary endpoints of the RIM study were not achieved, 83% of refractory adult and juvenile myositis patients met the definition of improvement, there was a significant corticosteroid sparing effect between the baseline dose and the dose at study conclusion, and treatment was generally well tolerated, he said.
Other targets that are being explored include interleukin-6 and type 1 IFN genes. Findings suggest that coordinated dysregulation of type 1 IFN signaling and IL-6 production are contributors to dermatomyositis pathogenesis, he explained.
Treating myositis patients with ILD
The treatment approach to these patients is somewhat similar to those without ILD, with corticosteroids as initial treatment, Dr. Oddis said.
Cyclophosphamide and azathioprine have been used early on, and also in corticosteroid resistant cases, but with variable results. Cyclophosphamide can be given orally or by IV for 3-6 months.
MMF has been used with success in connective tissue disease–associated ILD, and based on small studies it appears to be effective in myositis-associated ILD as well.
Cyclosporine and tacrolimus have been used in both adult and pediatric patients with promising, steroid-sparing results, he said, noting that the use of anti-T-cell therapy in myositis-associated ILD makes sense, because findings from multiple studies have implicated activated CD8-positive T-cells in myositis-associated ILD.
"It’s an exciting time for therapeutic interventions in myositis, but even though we have all these therapeutic options, we have to temper our enthusiasm with what they do long term," he said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Corticosteroids remain the initial treatment of choice for myositis and myositis-associated interstitial lung disease, but immunosuppressive agents, intravenous immunoglobulin, and biologics can also play a role in the treatment of one or both of these conditions, according to Dr. Chester V. Oddis.
For myositis in general, Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh, recommends an initial divided dose of 30 mg of prednisone twice daily, continued until serum creatine kinase (CK) levels fall to normal. At that time, the total daily prednisone dose can be consolidated into a single morning dose, he said at Perspectives in Rheumatic Diseases 2013.
The prednisone can then be tapered by 25% every 3-4 weeks down to a 5- to 10-mg daily maintenance dose that is continued until active disease is suppressed for 12 months. This is a general guideline that helps prevent disease flares, he noted.
Keep in mind that improvement in strength generally lags behind improvement in CK levels, he added.
Nonsteroidal immunosuppressives
Not all patients will need an additional immunosuppressive agent, but for those who do, methotrexate is a good option, Dr. Oddis said, noting that methotrexate is the drug he is most comfortable using in those cases.
The literature also supports the combined use of methotrexate and azathioprine, which when given together have been shown to be effective in treatment-resistant myositis and in those who failed either of the drugs alone.
"So that’s a regimen you might want to think about," he said.
Another immunosuppressive option is mycophenolate mofetil (MMF), which has been shown in several small studies and case series to be of benefit. In one study, 6 of 10 patients with dermatomyositis successfully tapered corticosteroids with MMF, and 10 of 12 in another study experienced improvement in cutaneous features of the disease.
The use of intravenous immunoglobulin (IVIg) as add-on therapy with MMF was effective in severe refractory patients, including four with polymyositis and three with dermatomyositis. In a retrospective review of 50 patients with juvenile dermatomyositis, MMF for 12 months was well tolerated, improved skin and muscle, and proved to be steroid-sparing, Dr. Oddis said.
Cyclosporine, tacrolimus, and cyclophosphamide are other immunosuppressive options.
While cyclophosphamide is more often used for myositis-associated interstitial lung disease (ILD), it can be of benefit for refractory skin disease, and can be useful in non-ILD myositis cases that involve severe skin disease.
The only available controlled data for IVIg alone are from a study published more than 20 years ago, but that randomized, double-blind, placebo-controlled study showed that treatment was safe, effective, and steroid sparing in 15 patients with dermatomyositis, he said.
Biologics
As for biologics, anti–tumor necrosis factor–alpha (anti-TNF-alpha) therapy and B-cell therapy have both been considered. Anti-TNF-alpha therapy makes sense because TNF-alpha and other proinflammatory cytokines are increased in muscle tissue of myositis patients; TNF-alpha is toxic to myofibers and prevents their regeneration; and TNF-alpha enhances other inflammatory cytokines in both dermatomyositis and polymyositis, but data are lacking on whether targeting TNF-alpha is worthwhile.
B cell therapy, on the other hand, is showing promise. In one open-label pilot study, rituximab was effective in seven patients with refractory dermatomyositis, and in others it was effective in anti-synthetase syndrome. Rituximab also was effective in two studies for refractory myositis and dermatomyositis rash, and it induced longstanding remission in some of the patients. In another study, however, rituximab was not effective for dermatomyositis rash.
The multicenter Rituximab in Myositis (RIM) study, the largest ever done in myositis, evaluated rituximab for the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis patients.
Although the primary and secondary endpoints of the RIM study were not achieved, 83% of refractory adult and juvenile myositis patients met the definition of improvement, there was a significant corticosteroid sparing effect between the baseline dose and the dose at study conclusion, and treatment was generally well tolerated, he said.
Other targets that are being explored include interleukin-6 and type 1 IFN genes. Findings suggest that coordinated dysregulation of type 1 IFN signaling and IL-6 production are contributors to dermatomyositis pathogenesis, he explained.
Treating myositis patients with ILD
The treatment approach to these patients is somewhat similar to those without ILD, with corticosteroids as initial treatment, Dr. Oddis said.
Cyclophosphamide and azathioprine have been used early on, and also in corticosteroid resistant cases, but with variable results. Cyclophosphamide can be given orally or by IV for 3-6 months.
MMF has been used with success in connective tissue disease–associated ILD, and based on small studies it appears to be effective in myositis-associated ILD as well.
Cyclosporine and tacrolimus have been used in both adult and pediatric patients with promising, steroid-sparing results, he said, noting that the use of anti-T-cell therapy in myositis-associated ILD makes sense, because findings from multiple studies have implicated activated CD8-positive T-cells in myositis-associated ILD.
"It’s an exciting time for therapeutic interventions in myositis, but even though we have all these therapeutic options, we have to temper our enthusiasm with what they do long term," he said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
AT PERSPECTIVES IN RHEUMATIC DISEASES 2013

PML reported in multiple sclerosis patient on fingolimod
The first case of progressive multifocal leukoencephalopathy has occurred in a patient with multiple sclerosis treated with fingolimod, according to the Food and Drug Administration.
The patient was diagnosed with progressive multifocal leukoencephalopathy (PML), the rare, often fatal opportunistic demyelinating infection, after almost 8 months of treatment with fingolimod, the agency reported on Aug. 29.
Fingolimod is a sphingosine 1-phosphate receptor modulator that was approved in 2010 for treating relapsing forms of MS.
The patient had not been treated with natalizumab, the MS drug that is known to increase the risk of the brain infection, but did receive 1 month of treatment with interferon beta-1a and azathioprine prior to beginning treatment with fingolimod. Those drugs were stopped when treatment with fingolimod began. However, the patient was also treated with "multiple courses" of intravenous corticosteroids for several months before and during treatment with fingolimod, which is marketed by Novartis under the name Gilenya.
Treatment with fingolimod was stopped after the diagnosis of PML, made on the basis of clinical symptoms and detection of JC viral DNA in cerebrospinal fluid, according to the FDA statement, which does not say whether the patient survived. The case was reported in Europe.
PML is caused by the JC virus, a common virus that usually does not cause illness, but can cause PML in people who are immunocompromised or are taking medications with immunosuppressive effects.
After several PML cases were reported in patients with MS treated with natalizumab (Tysabri) months after it was approved for treating MS in 2004, it was taken off the market in 2005 and reintroduced in June 2006 with measures to address the risk of PML, including a restricted distribution program.
The FDA is working with Novartis to investigate this case and will make recommendations when the evaluation has been completed, the statement said. The FDA is advising patients not to stop treatment with fingolimod before discussing this with their health care professionals.
The precautions and warnings section of the fingolimod label includes a statement that the treatment causes a dose-dependent reduction in peripheral lymphocyte count, due to reversible sequestration of lymphocytes in lymphoid tissues, and the drug "may therefore increase the risk of infections, some serious in nature," but PML is not mentioned. The label also says that it has not been administered with antineoplastic, immunosuppressive, or immune modulating therapies used to treat MS and that use of fingolimod with any of these treatments "would be expected to increase the risk of immunosuppression."
Fingolimod was approved with a risk evaluation and mitigation strategy, addressing the serious risks associated with treatment, including bradyarrhythmia and atrioventricular block at the start of treatment, infections, macular edema, respiratory effects, hepatic effects, and fetal risk.
According to Novartis, about 71,000 patients worldwide have been treated with fingolimod.
The safety communication is available here. Serious adverse events associated with fingolimod should be reported to the FDA’s MedWatch program at 800-332-1088.
The first case of progressive multifocal leukoencephalopathy has occurred in a patient with multiple sclerosis treated with fingolimod, according to the Food and Drug Administration.
The patient was diagnosed with progressive multifocal leukoencephalopathy (PML), the rare, often fatal opportunistic demyelinating infection, after almost 8 months of treatment with fingolimod, the agency reported on Aug. 29.
Fingolimod is a sphingosine 1-phosphate receptor modulator that was approved in 2010 for treating relapsing forms of MS.
The patient had not been treated with natalizumab, the MS drug that is known to increase the risk of the brain infection, but did receive 1 month of treatment with interferon beta-1a and azathioprine prior to beginning treatment with fingolimod. Those drugs were stopped when treatment with fingolimod began. However, the patient was also treated with "multiple courses" of intravenous corticosteroids for several months before and during treatment with fingolimod, which is marketed by Novartis under the name Gilenya.
Treatment with fingolimod was stopped after the diagnosis of PML, made on the basis of clinical symptoms and detection of JC viral DNA in cerebrospinal fluid, according to the FDA statement, which does not say whether the patient survived. The case was reported in Europe.
PML is caused by the JC virus, a common virus that usually does not cause illness, but can cause PML in people who are immunocompromised or are taking medications with immunosuppressive effects.
After several PML cases were reported in patients with MS treated with natalizumab (Tysabri) months after it was approved for treating MS in 2004, it was taken off the market in 2005 and reintroduced in June 2006 with measures to address the risk of PML, including a restricted distribution program.
The FDA is working with Novartis to investigate this case and will make recommendations when the evaluation has been completed, the statement said. The FDA is advising patients not to stop treatment with fingolimod before discussing this with their health care professionals.
The precautions and warnings section of the fingolimod label includes a statement that the treatment causes a dose-dependent reduction in peripheral lymphocyte count, due to reversible sequestration of lymphocytes in lymphoid tissues, and the drug "may therefore increase the risk of infections, some serious in nature," but PML is not mentioned. The label also says that it has not been administered with antineoplastic, immunosuppressive, or immune modulating therapies used to treat MS and that use of fingolimod with any of these treatments "would be expected to increase the risk of immunosuppression."
Fingolimod was approved with a risk evaluation and mitigation strategy, addressing the serious risks associated with treatment, including bradyarrhythmia and atrioventricular block at the start of treatment, infections, macular edema, respiratory effects, hepatic effects, and fetal risk.
According to Novartis, about 71,000 patients worldwide have been treated with fingolimod.
The safety communication is available here. Serious adverse events associated with fingolimod should be reported to the FDA’s MedWatch program at 800-332-1088.
The first case of progressive multifocal leukoencephalopathy has occurred in a patient with multiple sclerosis treated with fingolimod, according to the Food and Drug Administration.
The patient was diagnosed with progressive multifocal leukoencephalopathy (PML), the rare, often fatal opportunistic demyelinating infection, after almost 8 months of treatment with fingolimod, the agency reported on Aug. 29.
Fingolimod is a sphingosine 1-phosphate receptor modulator that was approved in 2010 for treating relapsing forms of MS.
The patient had not been treated with natalizumab, the MS drug that is known to increase the risk of the brain infection, but did receive 1 month of treatment with interferon beta-1a and azathioprine prior to beginning treatment with fingolimod. Those drugs were stopped when treatment with fingolimod began. However, the patient was also treated with "multiple courses" of intravenous corticosteroids for several months before and during treatment with fingolimod, which is marketed by Novartis under the name Gilenya.
Treatment with fingolimod was stopped after the diagnosis of PML, made on the basis of clinical symptoms and detection of JC viral DNA in cerebrospinal fluid, according to the FDA statement, which does not say whether the patient survived. The case was reported in Europe.
PML is caused by the JC virus, a common virus that usually does not cause illness, but can cause PML in people who are immunocompromised or are taking medications with immunosuppressive effects.
After several PML cases were reported in patients with MS treated with natalizumab (Tysabri) months after it was approved for treating MS in 2004, it was taken off the market in 2005 and reintroduced in June 2006 with measures to address the risk of PML, including a restricted distribution program.
The FDA is working with Novartis to investigate this case and will make recommendations when the evaluation has been completed, the statement said. The FDA is advising patients not to stop treatment with fingolimod before discussing this with their health care professionals.
The precautions and warnings section of the fingolimod label includes a statement that the treatment causes a dose-dependent reduction in peripheral lymphocyte count, due to reversible sequestration of lymphocytes in lymphoid tissues, and the drug "may therefore increase the risk of infections, some serious in nature," but PML is not mentioned. The label also says that it has not been administered with antineoplastic, immunosuppressive, or immune modulating therapies used to treat MS and that use of fingolimod with any of these treatments "would be expected to increase the risk of immunosuppression."
Fingolimod was approved with a risk evaluation and mitigation strategy, addressing the serious risks associated with treatment, including bradyarrhythmia and atrioventricular block at the start of treatment, infections, macular edema, respiratory effects, hepatic effects, and fetal risk.
According to Novartis, about 71,000 patients worldwide have been treated with fingolimod.
The safety communication is available here. Serious adverse events associated with fingolimod should be reported to the FDA’s MedWatch program at 800-332-1088.
FDA requires stronger peripheral neuropathy warning for quinolones
The Food and Drug Administration is requiring a stronger warning about the potential for peripheral neuropathy with fluoroquinolone antibiotics that are taken orally or by injection.
The warning does not apply to topical formulations, which have not been associated with neuropathy. Drug labels and patient medication guides must be updated, said the agency.
The potential for neuropathy was first added to the labels of all drugs in the class in 2004. The new warning was necessary because "the potential rapid onset and risk of permanence were not adequately described" in the 2004 label iteration, the FDA noted.
Before requiring new warnings, the FDA reviewed its Adverse Event Reporting System (FAERS, formerly known as AERS) database. The agency found that cases of fluoroquinolone-associated peripheral neuropathy with an outcome of "disability" had been reported to the AERS database between January 2003 and August 2012, although the FDA did not say how many cases it found. The reports indicated a rapid onset of peripheral neuropathy, often within a few days of starting the quinolone. Some cases reported neuropathy that continued for a year, even though the medication had been stopped.
The database was not able to show whether neuropathy was permanent, however, because it is designed to collect spontaneous reports.
The FDA said it had not been able to identify any risk factors for the development of peripheral neuropathy. But the onset of the condition seemed to have no correlation with the patient’s age or how long they took the antibiotic.
The updated warnings apply to all approved fluoroquinolones: levofloxacin (Levaquin), ciprofloxacin (Cipro), moxifloxacin (Avelox), norfloxacin (Noroxin), ofloxacin (Floxin), and gemifloxacin (Factive).
According to the FDA, 23 million outpatients were prescribed an oral quinolone in 2011. A total of 70% received ciprofloxacin; 28% were prescribed levofloxacin; and 9% were given moxifloxacin. Gemifloxacin, ofloxacin, and norfloxacin each accounted for less than 1% of those patients in 2011.
There were 3.8 million inpatients who received an injectable quinolone in 2011. The most-prescribed was levofloxacin, accounting for 63% of prescriptions, followed by ciprofloxacin (28%) and moxifloxacin (13%).
The agency recommended that patients who develop neuropathy symptoms stop taking the quinolone and be treated with a different antibiotic, unless the benefit outweighs the risk. Patients taking the medications who develop symptoms are urged to tell their physician immediately.
On Twitter @aliciaault
The Food and Drug Administration is requiring a stronger warning about the potential for peripheral neuropathy with fluoroquinolone antibiotics that are taken orally or by injection.
The warning does not apply to topical formulations, which have not been associated with neuropathy. Drug labels and patient medication guides must be updated, said the agency.
The potential for neuropathy was first added to the labels of all drugs in the class in 2004. The new warning was necessary because "the potential rapid onset and risk of permanence were not adequately described" in the 2004 label iteration, the FDA noted.
Before requiring new warnings, the FDA reviewed its Adverse Event Reporting System (FAERS, formerly known as AERS) database. The agency found that cases of fluoroquinolone-associated peripheral neuropathy with an outcome of "disability" had been reported to the AERS database between January 2003 and August 2012, although the FDA did not say how many cases it found. The reports indicated a rapid onset of peripheral neuropathy, often within a few days of starting the quinolone. Some cases reported neuropathy that continued for a year, even though the medication had been stopped.
The database was not able to show whether neuropathy was permanent, however, because it is designed to collect spontaneous reports.
The FDA said it had not been able to identify any risk factors for the development of peripheral neuropathy. But the onset of the condition seemed to have no correlation with the patient’s age or how long they took the antibiotic.
The updated warnings apply to all approved fluoroquinolones: levofloxacin (Levaquin), ciprofloxacin (Cipro), moxifloxacin (Avelox), norfloxacin (Noroxin), ofloxacin (Floxin), and gemifloxacin (Factive).
According to the FDA, 23 million outpatients were prescribed an oral quinolone in 2011. A total of 70% received ciprofloxacin; 28% were prescribed levofloxacin; and 9% were given moxifloxacin. Gemifloxacin, ofloxacin, and norfloxacin each accounted for less than 1% of those patients in 2011.
There were 3.8 million inpatients who received an injectable quinolone in 2011. The most-prescribed was levofloxacin, accounting for 63% of prescriptions, followed by ciprofloxacin (28%) and moxifloxacin (13%).
The agency recommended that patients who develop neuropathy symptoms stop taking the quinolone and be treated with a different antibiotic, unless the benefit outweighs the risk. Patients taking the medications who develop symptoms are urged to tell their physician immediately.
On Twitter @aliciaault
The Food and Drug Administration is requiring a stronger warning about the potential for peripheral neuropathy with fluoroquinolone antibiotics that are taken orally or by injection.
The warning does not apply to topical formulations, which have not been associated with neuropathy. Drug labels and patient medication guides must be updated, said the agency.
The potential for neuropathy was first added to the labels of all drugs in the class in 2004. The new warning was necessary because "the potential rapid onset and risk of permanence were not adequately described" in the 2004 label iteration, the FDA noted.
Before requiring new warnings, the FDA reviewed its Adverse Event Reporting System (FAERS, formerly known as AERS) database. The agency found that cases of fluoroquinolone-associated peripheral neuropathy with an outcome of "disability" had been reported to the AERS database between January 2003 and August 2012, although the FDA did not say how many cases it found. The reports indicated a rapid onset of peripheral neuropathy, often within a few days of starting the quinolone. Some cases reported neuropathy that continued for a year, even though the medication had been stopped.
The database was not able to show whether neuropathy was permanent, however, because it is designed to collect spontaneous reports.
The FDA said it had not been able to identify any risk factors for the development of peripheral neuropathy. But the onset of the condition seemed to have no correlation with the patient’s age or how long they took the antibiotic.
The updated warnings apply to all approved fluoroquinolones: levofloxacin (Levaquin), ciprofloxacin (Cipro), moxifloxacin (Avelox), norfloxacin (Noroxin), ofloxacin (Floxin), and gemifloxacin (Factive).
According to the FDA, 23 million outpatients were prescribed an oral quinolone in 2011. A total of 70% received ciprofloxacin; 28% were prescribed levofloxacin; and 9% were given moxifloxacin. Gemifloxacin, ofloxacin, and norfloxacin each accounted for less than 1% of those patients in 2011.
There were 3.8 million inpatients who received an injectable quinolone in 2011. The most-prescribed was levofloxacin, accounting for 63% of prescriptions, followed by ciprofloxacin (28%) and moxifloxacin (13%).
The agency recommended that patients who develop neuropathy symptoms stop taking the quinolone and be treated with a different antibiotic, unless the benefit outweighs the risk. Patients taking the medications who develop symptoms are urged to tell their physician immediately.
On Twitter @aliciaault

Fingolimod heart effects usually resolve within 6 hours
ORLANDO – The first-dose cardiovascular effects of fingolimod 0.5 mg were transient in most patients with multiple sclerosis and began to resolve within 6 hours after administration, according to analyses of two phase III trials and interim results of a phase IV study sponsored by the drug maker.
The analyses, which were presented in two posters at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers (CMSC) and the Americas Committee for Treatment and Research in Multiple Sclerosis, confirm that the initiation of treatment with fingolimod (Gilenya) is associated with a drop in heart rate (HR) and slowing of atrioventricular conduction, especially in the first 4-6 hours. Previously reported cases of bradycardia and atrioventricular blockhave been mostly transient and self-limited, the authors noted.
Fingolimod’s current prescribing information says that "All patients should be observed and receive hourly pulse and blood pressure measurement for at least 6 hours after first dose and undergo ECG predose and after the 6-hour observation." Its label was revised in May of last year following reports of sudden or unexplained deaths in the United States and Europe after the drug’s 2010 approval for relapsing forms of MS.
In the phase IV EPOC (Evaluate Patient Outcomes, Safety, and Tolerability of Fingolimod) study, 783 patients in the United States and Canada were randomized 3:1 to open-label treatment with once-daily fingolimod 0.5 mg or standard-of-care disease-modifying therapy (DMT) for 6 months.
All patients were fingolimod naive and received at least one dose of the therapy. They had received continual treatment of a single standard-of-care DMT for 6 months or more. The patients’ mean age was 46 years, and most were white (82%) and female (76%). The mean duration of their MS symptoms was 12 years.
Lead investigator Dr. Bruce L. Hughes of Ruan Multiple Sclerosis Center, Des Moines, Iowa, and his colleagues reported that the patients’ mean sitting HR at predose assessment dropped from a mean of 74.1 beats per minute at baseline to a nadir of 65.6 bpm, 5 hours after the therapy’s administration. HR began to recover by the 6th hour.
Most patients (98.6%) were discharged at 6 hours after treatment. Ten required extended observations after the 6th hour; three required a second day of observation and were discharged.
A total of 12 patients (1.5%) had bradycardia during the first-dose observation period. Eight were symptomatic, and four were asymptomatic. None required treatment, the researchers reported. The mean HR in this group decreased to 56.3 plus or minus 8.53 bpm, ranging from 38 to 64 bpm during bradycardia events. The patients recovered to 62.6 plus or minus 9.46 bpm, ranging from 52 to 80 bpm after the symptoms resolved.
Nearly 18% of the patients (137 of 783) had electrocardiography (ECG) performed at 6 hours post dose. In 28, the ECG differed from baseline, and the most common new findings were first-degree atrioventricular block (n = 11), and sinus bradycardia (n = 10). There were no second-degree AV blocks. Other findings included late anterior hemiblock (n = 1), atrial premature complex (n = 2), and biphasic T waves (n = 1).
The two other studies reported at the meeting also analyzed the first-dose cardiovascular effects of fingolimod 0.5 mg. The FIRST trial was a 4-month, open-label, phase IIIb study of 2,417 patients with relapsing MS, and the FREEDOMS II trial was a 2-year, double-blind, placebo-controlled, phase III study of 1,083 patients.
In the FIRST trial, the nadir HR occurred 4-5 hours post dose, and the mean decrease was 7.4 in patients without cardiac factors and 6.5 bpm in those with cardiac factors, the authors reported. The cardiac factors included beta-blocker and/or calcium channel blocker use (n = 120), resting HR of 45-54 bpm, Mobitz type I second-degree atrioventricular block, positive tilt test, or recurrent symptomatic bradycardia.
Dr. Simrat Randhawa and her colleagues at Novartis Pharmaceuticals reported that the mean decrease in HR was 7.2 and 7.3 bpm in patients without and with concomitant beta-blocker and/or calcium channel blocker use, respectively. One patient had a greater than 3-second pause in both screening and post dose ECG results. One patient discontinued the study drug because of second-degree AV block, the authors reported.
In the FREEDOMS II trial, the clinician-observed mean maximal decrease in HR was 8.5 bpm in the fingolimod group. The incidence of Mobitz I second-degree AV block with fingolimod was 3.7%, compared with 2.0% in placebo, while 2:1 AV block occurred in 2% of patients taking fingolimod patients and 0% taking placebo.
Most first-occurrence second-degree AV blocks were observed less than 6 hours after the first dose, the authors reported.
There were no Mobitz II or third-degree AV blocks reported in FIRST and FREEDOMS II.
"This is a good example of ‘I can’t predict who should get fingolimod, but I can say who I should be very careful with and maybe not give [them] fingolimod and give [them] something else,’ " said Dr. Robert P. Lisak, professor of neurology at Wayne State University, Detroit, and president-elect of the CMSC. "But once they’re on it, and if they don’t have other contraindications, they should be OK." Dr. Lisak was not involved in the study.
All the studies were supported by Novartis, which markets fingolimod. Dr. Hughes and another EPOC investigator have served as a speaker and/or advisory board member or received research support from Novartis and other companies involved in MS pharmaceutical research and development. Other investigators involved in the EPOC study are employees of Novartis. Dr. Lisak has received research grants from and has been an adviser for several companies, including, Avanir, Bayer, Novartis, Questcor, and Teva.
On Twitter @NaseemSMiller
ORLANDO – The first-dose cardiovascular effects of fingolimod 0.5 mg were transient in most patients with multiple sclerosis and began to resolve within 6 hours after administration, according to analyses of two phase III trials and interim results of a phase IV study sponsored by the drug maker.
The analyses, which were presented in two posters at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers (CMSC) and the Americas Committee for Treatment and Research in Multiple Sclerosis, confirm that the initiation of treatment with fingolimod (Gilenya) is associated with a drop in heart rate (HR) and slowing of atrioventricular conduction, especially in the first 4-6 hours. Previously reported cases of bradycardia and atrioventricular blockhave been mostly transient and self-limited, the authors noted.
Fingolimod’s current prescribing information says that "All patients should be observed and receive hourly pulse and blood pressure measurement for at least 6 hours after first dose and undergo ECG predose and after the 6-hour observation." Its label was revised in May of last year following reports of sudden or unexplained deaths in the United States and Europe after the drug’s 2010 approval for relapsing forms of MS.
In the phase IV EPOC (Evaluate Patient Outcomes, Safety, and Tolerability of Fingolimod) study, 783 patients in the United States and Canada were randomized 3:1 to open-label treatment with once-daily fingolimod 0.5 mg or standard-of-care disease-modifying therapy (DMT) for 6 months.
All patients were fingolimod naive and received at least one dose of the therapy. They had received continual treatment of a single standard-of-care DMT for 6 months or more. The patients’ mean age was 46 years, and most were white (82%) and female (76%). The mean duration of their MS symptoms was 12 years.
Lead investigator Dr. Bruce L. Hughes of Ruan Multiple Sclerosis Center, Des Moines, Iowa, and his colleagues reported that the patients’ mean sitting HR at predose assessment dropped from a mean of 74.1 beats per minute at baseline to a nadir of 65.6 bpm, 5 hours after the therapy’s administration. HR began to recover by the 6th hour.
Most patients (98.6%) were discharged at 6 hours after treatment. Ten required extended observations after the 6th hour; three required a second day of observation and were discharged.
A total of 12 patients (1.5%) had bradycardia during the first-dose observation period. Eight were symptomatic, and four were asymptomatic. None required treatment, the researchers reported. The mean HR in this group decreased to 56.3 plus or minus 8.53 bpm, ranging from 38 to 64 bpm during bradycardia events. The patients recovered to 62.6 plus or minus 9.46 bpm, ranging from 52 to 80 bpm after the symptoms resolved.
Nearly 18% of the patients (137 of 783) had electrocardiography (ECG) performed at 6 hours post dose. In 28, the ECG differed from baseline, and the most common new findings were first-degree atrioventricular block (n = 11), and sinus bradycardia (n = 10). There were no second-degree AV blocks. Other findings included late anterior hemiblock (n = 1), atrial premature complex (n = 2), and biphasic T waves (n = 1).
The two other studies reported at the meeting also analyzed the first-dose cardiovascular effects of fingolimod 0.5 mg. The FIRST trial was a 4-month, open-label, phase IIIb study of 2,417 patients with relapsing MS, and the FREEDOMS II trial was a 2-year, double-blind, placebo-controlled, phase III study of 1,083 patients.
In the FIRST trial, the nadir HR occurred 4-5 hours post dose, and the mean decrease was 7.4 in patients without cardiac factors and 6.5 bpm in those with cardiac factors, the authors reported. The cardiac factors included beta-blocker and/or calcium channel blocker use (n = 120), resting HR of 45-54 bpm, Mobitz type I second-degree atrioventricular block, positive tilt test, or recurrent symptomatic bradycardia.
Dr. Simrat Randhawa and her colleagues at Novartis Pharmaceuticals reported that the mean decrease in HR was 7.2 and 7.3 bpm in patients without and with concomitant beta-blocker and/or calcium channel blocker use, respectively. One patient had a greater than 3-second pause in both screening and post dose ECG results. One patient discontinued the study drug because of second-degree AV block, the authors reported.
In the FREEDOMS II trial, the clinician-observed mean maximal decrease in HR was 8.5 bpm in the fingolimod group. The incidence of Mobitz I second-degree AV block with fingolimod was 3.7%, compared with 2.0% in placebo, while 2:1 AV block occurred in 2% of patients taking fingolimod patients and 0% taking placebo.
Most first-occurrence second-degree AV blocks were observed less than 6 hours after the first dose, the authors reported.
There were no Mobitz II or third-degree AV blocks reported in FIRST and FREEDOMS II.
"This is a good example of ‘I can’t predict who should get fingolimod, but I can say who I should be very careful with and maybe not give [them] fingolimod and give [them] something else,’ " said Dr. Robert P. Lisak, professor of neurology at Wayne State University, Detroit, and president-elect of the CMSC. "But once they’re on it, and if they don’t have other contraindications, they should be OK." Dr. Lisak was not involved in the study.
All the studies were supported by Novartis, which markets fingolimod. Dr. Hughes and another EPOC investigator have served as a speaker and/or advisory board member or received research support from Novartis and other companies involved in MS pharmaceutical research and development. Other investigators involved in the EPOC study are employees of Novartis. Dr. Lisak has received research grants from and has been an adviser for several companies, including, Avanir, Bayer, Novartis, Questcor, and Teva.
On Twitter @NaseemSMiller
ORLANDO – The first-dose cardiovascular effects of fingolimod 0.5 mg were transient in most patients with multiple sclerosis and began to resolve within 6 hours after administration, according to analyses of two phase III trials and interim results of a phase IV study sponsored by the drug maker.
The analyses, which were presented in two posters at the fifth Cooperative Meeting of the Consortium of Multiple Sclerosis Centers (CMSC) and the Americas Committee for Treatment and Research in Multiple Sclerosis, confirm that the initiation of treatment with fingolimod (Gilenya) is associated with a drop in heart rate (HR) and slowing of atrioventricular conduction, especially in the first 4-6 hours. Previously reported cases of bradycardia and atrioventricular blockhave been mostly transient and self-limited, the authors noted.
Fingolimod’s current prescribing information says that "All patients should be observed and receive hourly pulse and blood pressure measurement for at least 6 hours after first dose and undergo ECG predose and after the 6-hour observation." Its label was revised in May of last year following reports of sudden or unexplained deaths in the United States and Europe after the drug’s 2010 approval for relapsing forms of MS.
In the phase IV EPOC (Evaluate Patient Outcomes, Safety, and Tolerability of Fingolimod) study, 783 patients in the United States and Canada were randomized 3:1 to open-label treatment with once-daily fingolimod 0.5 mg or standard-of-care disease-modifying therapy (DMT) for 6 months.
All patients were fingolimod naive and received at least one dose of the therapy. They had received continual treatment of a single standard-of-care DMT for 6 months or more. The patients’ mean age was 46 years, and most were white (82%) and female (76%). The mean duration of their MS symptoms was 12 years.
Lead investigator Dr. Bruce L. Hughes of Ruan Multiple Sclerosis Center, Des Moines, Iowa, and his colleagues reported that the patients’ mean sitting HR at predose assessment dropped from a mean of 74.1 beats per minute at baseline to a nadir of 65.6 bpm, 5 hours after the therapy’s administration. HR began to recover by the 6th hour.
Most patients (98.6%) were discharged at 6 hours after treatment. Ten required extended observations after the 6th hour; three required a second day of observation and were discharged.
A total of 12 patients (1.5%) had bradycardia during the first-dose observation period. Eight were symptomatic, and four were asymptomatic. None required treatment, the researchers reported. The mean HR in this group decreased to 56.3 plus or minus 8.53 bpm, ranging from 38 to 64 bpm during bradycardia events. The patients recovered to 62.6 plus or minus 9.46 bpm, ranging from 52 to 80 bpm after the symptoms resolved.
Nearly 18% of the patients (137 of 783) had electrocardiography (ECG) performed at 6 hours post dose. In 28, the ECG differed from baseline, and the most common new findings were first-degree atrioventricular block (n = 11), and sinus bradycardia (n = 10). There were no second-degree AV blocks. Other findings included late anterior hemiblock (n = 1), atrial premature complex (n = 2), and biphasic T waves (n = 1).
The two other studies reported at the meeting also analyzed the first-dose cardiovascular effects of fingolimod 0.5 mg. The FIRST trial was a 4-month, open-label, phase IIIb study of 2,417 patients with relapsing MS, and the FREEDOMS II trial was a 2-year, double-blind, placebo-controlled, phase III study of 1,083 patients.
In the FIRST trial, the nadir HR occurred 4-5 hours post dose, and the mean decrease was 7.4 in patients without cardiac factors and 6.5 bpm in those with cardiac factors, the authors reported. The cardiac factors included beta-blocker and/or calcium channel blocker use (n = 120), resting HR of 45-54 bpm, Mobitz type I second-degree atrioventricular block, positive tilt test, or recurrent symptomatic bradycardia.
Dr. Simrat Randhawa and her colleagues at Novartis Pharmaceuticals reported that the mean decrease in HR was 7.2 and 7.3 bpm in patients without and with concomitant beta-blocker and/or calcium channel blocker use, respectively. One patient had a greater than 3-second pause in both screening and post dose ECG results. One patient discontinued the study drug because of second-degree AV block, the authors reported.
In the FREEDOMS II trial, the clinician-observed mean maximal decrease in HR was 8.5 bpm in the fingolimod group. The incidence of Mobitz I second-degree AV block with fingolimod was 3.7%, compared with 2.0% in placebo, while 2:1 AV block occurred in 2% of patients taking fingolimod patients and 0% taking placebo.
Most first-occurrence second-degree AV blocks were observed less than 6 hours after the first dose, the authors reported.
There were no Mobitz II or third-degree AV blocks reported in FIRST and FREEDOMS II.
"This is a good example of ‘I can’t predict who should get fingolimod, but I can say who I should be very careful with and maybe not give [them] fingolimod and give [them] something else,’ " said Dr. Robert P. Lisak, professor of neurology at Wayne State University, Detroit, and president-elect of the CMSC. "But once they’re on it, and if they don’t have other contraindications, they should be OK." Dr. Lisak was not involved in the study.
All the studies were supported by Novartis, which markets fingolimod. Dr. Hughes and another EPOC investigator have served as a speaker and/or advisory board member or received research support from Novartis and other companies involved in MS pharmaceutical research and development. Other investigators involved in the EPOC study are employees of Novartis. Dr. Lisak has received research grants from and has been an adviser for several companies, including, Avanir, Bayer, Novartis, Questcor, and Teva.
On Twitter @NaseemSMiller
AT THE CMSC/ACTRMS ANNUAL MEETING
Major finding: Patients’ mean sitting heart rate at predose assessment dropped from a mean of 74.1 bpm at baseline to a nadir of 65.6 bpm 5 hours after the therapy’s administration. HR began to recover by the 6th hour.
Data source: Analysis of two phase III trials, FIRST and FREEDOMS II, and the interim results of a phase IV study, EPOC.
Disclosures: All the studies were supported by Novartis, which markets fingolimod. Dr. Hughes and another EPOC investigator have served as a speaker and/or advisory board member or received research support from Novartis and other companies involved in MS pharmaceuticals research and development. Other investigators involved in the EPOC study report are employees of Novartis. Dr. Lisak has received research grants from and has been an adviser for several companies, including, Avanir, Bayer, Novartis, Questcor, and Teva.