User login
Consider graded exercise before medications for postural orthostatic tachycardia syndrome
SCOTTSDALE, ARIZ. – Exercise is a cornerstone of treating postural orthostatic tachycardia syndrome, according to Dr. Deborah Tepper.
“Exercise training is really the hot new way to treat this disorder. It’s been found to be very effective,” she said at a symposium sponsored by the American Headache Society.
Postural orthostatic tachycardia syndrome (POTS) is an autonomic disorder that causes symptoms resembling panic attacks, said Dr. Tepper, who is an internist at the Cleveland Clinic Neurological Center for Pain. Patients may report syncope, dizziness, palpitations, rapid or irregular breathing, fatigue, and chest pain, she said. But to be diagnosed with POTS, they must have an increase of at least 30 beats per minute within 10 minutes of standing or with the 60-degree tilt table test. Patients aged 12-19 years must have an increase in heart rate of at least 40 beats per minute. Blood pressure may drop or stay the same, but will not fall to the extent seen in orthostatic hypotension (that is, a systolic drop of 20 mm Hg or a diastolic fall of at least 10 mm Hg), Dr. Tepper noted.

Deconditioning, chronic fatigue, anxiety, dehydration, and various medications increase the frequency and severity of POTS-related symptoms, Dr. Tepper said. Patients sometimes severely restrict exercise in an effort to control symptoms, but lying on the couch or going to bed “is the worst thing you can do with POTS,” she said. Instead, patients should start a graded exercise program by swimming or exercising in recumbent or seated positions. Exercise training increases the aldosterone-to-renin ratio, can reduce migraine, improves overall health and stamina, supports independent functioning, and can help restore the sleep-wake cycle.
Increasing salt and fluid intake and wearing compression socks also can improve symptoms – often to the extent that patients will not need new medications. But this conservative approach is inadequate in patients with risky occupations and is less effective when patients have frequent episodes of syncope, Dr. Tepper said.
“Beta-blockers remain an option if salts, fluids, and patient education are not enough,” she added.
Beta-blockers inhibit epinephrine release and therefore can improve the migraine and anxiety symptoms that can occur in patients with POTS, although they should not be used in patients with asthma, Dr. Tepper emphasized.
Medications such as fludrocortisone and midodrine should be reserved for patients with recurrent or resistant symptoms, according to Dr. Tepper. “Midodrine can be helpful in some people, although the evidence overall rates it as low to moderate,” she said. Midodrine is a direct vasoconstrictor and alpha-adrenergic agonist that increases vasomotor tone, but also can exacerbate headaches and may cause supine hypertension, urinary retention, and insomnia, she noted. Fludrocortisone increases plasma volume, but worsens migraine, increases fluid retention, and cannot be used in diabetic patients, she said.
Clinicians should refer patients for a cardiology evaluation if they do not improve or have risk factors such as a history of cardiac disease or a family history of sudden cardiac death, chest pain, a QRS interval of more than 120 ms, a corrected QT interval of more than 450 ms or less than 300 ms, or syncope with no warning or while lying down, Dr. Tepper said. She declared no conflicts of interest.
SCOTTSDALE, ARIZ. – Exercise is a cornerstone of treating postural orthostatic tachycardia syndrome, according to Dr. Deborah Tepper.
“Exercise training is really the hot new way to treat this disorder. It’s been found to be very effective,” she said at a symposium sponsored by the American Headache Society.
Postural orthostatic tachycardia syndrome (POTS) is an autonomic disorder that causes symptoms resembling panic attacks, said Dr. Tepper, who is an internist at the Cleveland Clinic Neurological Center for Pain. Patients may report syncope, dizziness, palpitations, rapid or irregular breathing, fatigue, and chest pain, she said. But to be diagnosed with POTS, they must have an increase of at least 30 beats per minute within 10 minutes of standing or with the 60-degree tilt table test. Patients aged 12-19 years must have an increase in heart rate of at least 40 beats per minute. Blood pressure may drop or stay the same, but will not fall to the extent seen in orthostatic hypotension (that is, a systolic drop of 20 mm Hg or a diastolic fall of at least 10 mm Hg), Dr. Tepper noted.
Deconditioning, chronic fatigue, anxiety, dehydration, and various medications increase the frequency and severity of POTS-related symptoms, Dr. Tepper said. Patients sometimes severely restrict exercise in an effort to control symptoms, but lying on the couch or going to bed “is the worst thing you can do with POTS,” she said. Instead, patients should start a graded exercise program by swimming or exercising in recumbent or seated positions. Exercise training increases the aldosterone-to-renin ratio, can reduce migraine, improves overall health and stamina, supports independent functioning, and can help restore the sleep-wake cycle.
Increasing salt and fluid intake and wearing compression socks also can improve symptoms – often to the extent that patients will not need new medications. But this conservative approach is inadequate in patients with risky occupations and is less effective when patients have frequent episodes of syncope, Dr. Tepper said.
“Beta-blockers remain an option if salts, fluids, and patient education are not enough,” she added.
Beta-blockers inhibit epinephrine release and therefore can improve the migraine and anxiety symptoms that can occur in patients with POTS, although they should not be used in patients with asthma, Dr. Tepper emphasized.
Medications such as fludrocortisone and midodrine should be reserved for patients with recurrent or resistant symptoms, according to Dr. Tepper. “Midodrine can be helpful in some people, although the evidence overall rates it as low to moderate,” she said. Midodrine is a direct vasoconstrictor and alpha-adrenergic agonist that increases vasomotor tone, but also can exacerbate headaches and may cause supine hypertension, urinary retention, and insomnia, she noted. Fludrocortisone increases plasma volume, but worsens migraine, increases fluid retention, and cannot be used in diabetic patients, she said.
Clinicians should refer patients for a cardiology evaluation if they do not improve or have risk factors such as a history of cardiac disease or a family history of sudden cardiac death, chest pain, a QRS interval of more than 120 ms, a corrected QT interval of more than 450 ms or less than 300 ms, or syncope with no warning or while lying down, Dr. Tepper said. She declared no conflicts of interest.
SCOTTSDALE, ARIZ. – Exercise is a cornerstone of treating postural orthostatic tachycardia syndrome, according to Dr. Deborah Tepper.
“Exercise training is really the hot new way to treat this disorder. It’s been found to be very effective,” she said at a symposium sponsored by the American Headache Society.
Postural orthostatic tachycardia syndrome (POTS) is an autonomic disorder that causes symptoms resembling panic attacks, said Dr. Tepper, who is an internist at the Cleveland Clinic Neurological Center for Pain. Patients may report syncope, dizziness, palpitations, rapid or irregular breathing, fatigue, and chest pain, she said. But to be diagnosed with POTS, they must have an increase of at least 30 beats per minute within 10 minutes of standing or with the 60-degree tilt table test. Patients aged 12-19 years must have an increase in heart rate of at least 40 beats per minute. Blood pressure may drop or stay the same, but will not fall to the extent seen in orthostatic hypotension (that is, a systolic drop of 20 mm Hg or a diastolic fall of at least 10 mm Hg), Dr. Tepper noted.
Deconditioning, chronic fatigue, anxiety, dehydration, and various medications increase the frequency and severity of POTS-related symptoms, Dr. Tepper said. Patients sometimes severely restrict exercise in an effort to control symptoms, but lying on the couch or going to bed “is the worst thing you can do with POTS,” she said. Instead, patients should start a graded exercise program by swimming or exercising in recumbent or seated positions. Exercise training increases the aldosterone-to-renin ratio, can reduce migraine, improves overall health and stamina, supports independent functioning, and can help restore the sleep-wake cycle.
Increasing salt and fluid intake and wearing compression socks also can improve symptoms – often to the extent that patients will not need new medications. But this conservative approach is inadequate in patients with risky occupations and is less effective when patients have frequent episodes of syncope, Dr. Tepper said.
“Beta-blockers remain an option if salts, fluids, and patient education are not enough,” she added.
Beta-blockers inhibit epinephrine release and therefore can improve the migraine and anxiety symptoms that can occur in patients with POTS, although they should not be used in patients with asthma, Dr. Tepper emphasized.
Medications such as fludrocortisone and midodrine should be reserved for patients with recurrent or resistant symptoms, according to Dr. Tepper. “Midodrine can be helpful in some people, although the evidence overall rates it as low to moderate,” she said. Midodrine is a direct vasoconstrictor and alpha-adrenergic agonist that increases vasomotor tone, but also can exacerbate headaches and may cause supine hypertension, urinary retention, and insomnia, she noted. Fludrocortisone increases plasma volume, but worsens migraine, increases fluid retention, and cannot be used in diabetic patients, she said.
Clinicians should refer patients for a cardiology evaluation if they do not improve or have risk factors such as a history of cardiac disease or a family history of sudden cardiac death, chest pain, a QRS interval of more than 120 ms, a corrected QT interval of more than 450 ms or less than 300 ms, or syncope with no warning or while lying down, Dr. Tepper said. She declared no conflicts of interest.

ALS ice bucket challenge: It’s hard to argue with success
The ALS ice bucket challenge has taken the social media world by storm and surprised many by how fast and far it has spread. People simply make a video of themselves dumping a bucket of ice water on their heads and then post it on a social media site and challenge others to do the same within 24 hours or make a donation to ALS research (or both).
The stunt, which began early in the summer as a challenge unrelated to amyotrophic lateral sclerosis, has resonated with many people in the dog days of summer and has been helped by many celebrities taking up the challenge. It became linked to ALS when Peter Frates, a 29-year-old man with the disease, took the challenge – albeit by nodding his head to the song "Ice Ice Baby" instead of having ice water dumped on him – and asked others to do the same.

According to the ALS Association, as of Aug. 19, existing donors and more than 450,000 new donors have contributed $22.9 million since July 29, compared with $1.9 million during the same period last year. The ALS has a four-out-of-four stars rating on Charity Navigator, and an overall score of 90.73 out of 100. Overall, 72% of its expenses are spent on the programs and services it delivers, 11% on administration, and 17% on fundraising.
Some critics have suggested that the stunt promotes click and post activism, keeping people from doing real activism, or is "narcissism masked as altruism," but most people have embraced it as fun for a good cause.
I asked a few Clinical Neurology News editorial advisory board members to weigh in:
• Dr. Richard J. Caselli, professor of neurology at the Mayo Clinic, Scottsdale, Ariz.: "If it is raising money for ALS research, what’s not to like? Not everyone was destined to be a molecular biologist or clinical trialist, and this gives people a way to contribute that seems to be culturally in synch with the ‘social media’ community. ... The ice water is an interesting twist in that it implies that if you turn a blind eye to this cause you should punish yourself, and I suspect many people harbor feelings of at least slight guilt when they feel they are not contributing to worthwhile needs."

• Matthew Huentelman, Ph.D., associate professor of neurogenomics at the Translational Genomics Research Institute, Phoenix: "If a campaign works (and doesn’t harm) then it is hard to argue against. I think that any awareness campaign that actually gets a response from the White House has probably been a useful one. President Obama confirmed he would be donating but not doing the ice bucket thing. ... I suspect that a lot of research foundations are going to be having ‘tough’ conversations at this month’s board meetings as they compare their awareness and fundraising attempts to the simple ALS ice bucket challenge. It just demonstrates to all of us again that social media is relevant for both spreading the word and getting results. There are a few keys from this too: (1) visual "stuff" matters – short video clips is now how the world communicates; (2) challenging your friends/colleagues by name is important, too – it sort of forces a response from them; and (3) celebs are still key to pushing something viral in a truly short period of time."
(While you contemplate taking the ice bucket challenge, you might as well visit Dr. Huentelman’s social media project, MindCrowd, a site leveraging social media to recruit participants into a brain research study.)
The ALS ice bucket challenge has taken the social media world by storm and surprised many by how fast and far it has spread. People simply make a video of themselves dumping a bucket of ice water on their heads and then post it on a social media site and challenge others to do the same within 24 hours or make a donation to ALS research (or both).
The stunt, which began early in the summer as a challenge unrelated to amyotrophic lateral sclerosis, has resonated with many people in the dog days of summer and has been helped by many celebrities taking up the challenge. It became linked to ALS when Peter Frates, a 29-year-old man with the disease, took the challenge – albeit by nodding his head to the song "Ice Ice Baby" instead of having ice water dumped on him – and asked others to do the same.
According to the ALS Association, as of Aug. 19, existing donors and more than 450,000 new donors have contributed $22.9 million since July 29, compared with $1.9 million during the same period last year. The ALS has a four-out-of-four stars rating on Charity Navigator, and an overall score of 90.73 out of 100. Overall, 72% of its expenses are spent on the programs and services it delivers, 11% on administration, and 17% on fundraising.
Some critics have suggested that the stunt promotes click and post activism, keeping people from doing real activism, or is "narcissism masked as altruism," but most people have embraced it as fun for a good cause.
I asked a few Clinical Neurology News editorial advisory board members to weigh in:
• Dr. Richard J. Caselli, professor of neurology at the Mayo Clinic, Scottsdale, Ariz.: "If it is raising money for ALS research, what’s not to like? Not everyone was destined to be a molecular biologist or clinical trialist, and this gives people a way to contribute that seems to be culturally in synch with the ‘social media’ community. ... The ice water is an interesting twist in that it implies that if you turn a blind eye to this cause you should punish yourself, and I suspect many people harbor feelings of at least slight guilt when they feel they are not contributing to worthwhile needs."
• Matthew Huentelman, Ph.D., associate professor of neurogenomics at the Translational Genomics Research Institute, Phoenix: "If a campaign works (and doesn’t harm) then it is hard to argue against. I think that any awareness campaign that actually gets a response from the White House has probably been a useful one. President Obama confirmed he would be donating but not doing the ice bucket thing. ... I suspect that a lot of research foundations are going to be having ‘tough’ conversations at this month’s board meetings as they compare their awareness and fundraising attempts to the simple ALS ice bucket challenge. It just demonstrates to all of us again that social media is relevant for both spreading the word and getting results. There are a few keys from this too: (1) visual "stuff" matters – short video clips is now how the world communicates; (2) challenging your friends/colleagues by name is important, too – it sort of forces a response from them; and (3) celebs are still key to pushing something viral in a truly short period of time."
(While you contemplate taking the ice bucket challenge, you might as well visit Dr. Huentelman’s social media project, MindCrowd, a site leveraging social media to recruit participants into a brain research study.)
The ALS ice bucket challenge has taken the social media world by storm and surprised many by how fast and far it has spread. People simply make a video of themselves dumping a bucket of ice water on their heads and then post it on a social media site and challenge others to do the same within 24 hours or make a donation to ALS research (or both).
The stunt, which began early in the summer as a challenge unrelated to amyotrophic lateral sclerosis, has resonated with many people in the dog days of summer and has been helped by many celebrities taking up the challenge. It became linked to ALS when Peter Frates, a 29-year-old man with the disease, took the challenge – albeit by nodding his head to the song "Ice Ice Baby" instead of having ice water dumped on him – and asked others to do the same.
According to the ALS Association, as of Aug. 19, existing donors and more than 450,000 new donors have contributed $22.9 million since July 29, compared with $1.9 million during the same period last year. The ALS has a four-out-of-four stars rating on Charity Navigator, and an overall score of 90.73 out of 100. Overall, 72% of its expenses are spent on the programs and services it delivers, 11% on administration, and 17% on fundraising.
Some critics have suggested that the stunt promotes click and post activism, keeping people from doing real activism, or is "narcissism masked as altruism," but most people have embraced it as fun for a good cause.
I asked a few Clinical Neurology News editorial advisory board members to weigh in:
• Dr. Richard J. Caselli, professor of neurology at the Mayo Clinic, Scottsdale, Ariz.: "If it is raising money for ALS research, what’s not to like? Not everyone was destined to be a molecular biologist or clinical trialist, and this gives people a way to contribute that seems to be culturally in synch with the ‘social media’ community. ... The ice water is an interesting twist in that it implies that if you turn a blind eye to this cause you should punish yourself, and I suspect many people harbor feelings of at least slight guilt when they feel they are not contributing to worthwhile needs."
• Matthew Huentelman, Ph.D., associate professor of neurogenomics at the Translational Genomics Research Institute, Phoenix: "If a campaign works (and doesn’t harm) then it is hard to argue against. I think that any awareness campaign that actually gets a response from the White House has probably been a useful one. President Obama confirmed he would be donating but not doing the ice bucket thing. ... I suspect that a lot of research foundations are going to be having ‘tough’ conversations at this month’s board meetings as they compare their awareness and fundraising attempts to the simple ALS ice bucket challenge. It just demonstrates to all of us again that social media is relevant for both spreading the word and getting results. There are a few keys from this too: (1) visual "stuff" matters – short video clips is now how the world communicates; (2) challenging your friends/colleagues by name is important, too – it sort of forces a response from them; and (3) celebs are still key to pushing something viral in a truly short period of time."
(While you contemplate taking the ice bucket challenge, you might as well visit Dr. Huentelman’s social media project, MindCrowd, a site leveraging social media to recruit participants into a brain research study.)

Chikungunya vaccine safe, immunogenic in phase I trial
A novel chikungunya virus vaccine caused no serious adverse effects and was about as immunogenic as natural infection, authors of a phase I trial reported in the Lancet.
Eleven months after receiving the noninfectious viruslike particle (VLP) vaccine, trial participants had neutralizing antibody titers that resembled those seen in natural infection, said Dr. Lee-Jah Chang and associates at the National Institutes of Health in Bethesda, Md. (Lancet 2014 Aug. 15 [doi: 10.1016/S0140-6736(14)61185-5]).
Chikungunya virus infection is rarely fatal, but it causes fever and severe arthritis. The mosquito-born pathogen spread to the Americas in 2013, is now epidemic in the Caribbean, and has no approved vaccine or treatment.
To test a novel VLP vaccine candidate, researchers enrolled 25 healthy adults into a phase I, open-label, dose-escalation trial with doses of 10, 20, or 40 mcg administered at weeks 0, 4, and 20, the investigators reported. Participants were followed for 44 weeks after enrollment.
All groups had detectable neutralizing antibodies on ELISA (enzyme-linked immunosorbent assay) after the second dose, with geometric mean titers of the half-maximum inhibitory concentration of 2,688 in the 10-mcg group, 1,775 in the 20-mcg group, and 7,246 in the 40-mcg group. Antibody levels after the second dose substantially exceeded those measured after the first dose, with P values for differences ranging from .07 for the 10-mcg group to less than .0001 for the 40-mcg group, the investigators said. And titers did not significantly differ 4 weeks after the second and third doses, they added.
Participants reported no arthralgias or other serious adverse effects. Mild to moderate side effects occurred in four participants, and consisted of transient neutropenia (three cases) and transient increases in alanine aminotransferase (four cases), all of which resolved without clinical consequences, the investigators said.
"These clinical data represent an important step in vaccine development to combat this rapidly emerging pathogen," the researchers said. The next step is to test the vaccine in larger studies that include persons at risk of chikungunya virus infection, they added.
The trial was funded by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases; and the National Institutes of Health. The authors declared no competing interests.
"Although this VLP vaccine candidate exhibits a range of properties that suggest it would be a good vaccine option, there is always concern about whether a vaccine for a vector-borne virus will be licensed," said Dr. Ann M. Powers. "Development of vaccines for orphan agents is challenging, because the market might not be large enough to justify the investment."
Developing a vaccine in the United States costs an estimated $200-$500 million; nonetheless, vaccines are the most cost-effective way to prevent disease, she said. "In view of the burden of chikungunya outbreaks, which have affected up to 63% of local populations in a matter of months, the continued development of this VLP vaccine candidate, along with other vaccine options, should be encouraged."
A VLP contains the outer structural proteins of the virus that the immune system typically recognizes, Dr. Powers added. But it does not contain live genetic material, which confers a safety and manufacturing advantage because high-containment facilities are not needed for production.
Dr. Powers is with the division of vector-borne diseases at the U.S. Centers for Disease Control and Prevention. She declared that she had no competing interests. These remarks were excerpted from her editorial accompanying Dr. Chang’s report (Lancet 2014 Aug. 15 [doi: 10.1016/S0140-6736(14)61290-3]).
"Although this VLP vaccine candidate exhibits a range of properties that suggest it would be a good vaccine option, there is always concern about whether a vaccine for a vector-borne virus will be licensed," said Dr. Ann M. Powers. "Development of vaccines for orphan agents is challenging, because the market might not be large enough to justify the investment."
Developing a vaccine in the United States costs an estimated $200-$500 million; nonetheless, vaccines are the most cost-effective way to prevent disease, she said. "In view of the burden of chikungunya outbreaks, which have affected up to 63% of local populations in a matter of months, the continued development of this VLP vaccine candidate, along with other vaccine options, should be encouraged."
A VLP contains the outer structural proteins of the virus that the immune system typically recognizes, Dr. Powers added. But it does not contain live genetic material, which confers a safety and manufacturing advantage because high-containment facilities are not needed for production.
Dr. Powers is with the division of vector-borne diseases at the U.S. Centers for Disease Control and Prevention. She declared that she had no competing interests. These remarks were excerpted from her editorial accompanying Dr. Chang’s report (Lancet 2014 Aug. 15 [doi: 10.1016/S0140-6736(14)61290-3]).
"Although this VLP vaccine candidate exhibits a range of properties that suggest it would be a good vaccine option, there is always concern about whether a vaccine for a vector-borne virus will be licensed," said Dr. Ann M. Powers. "Development of vaccines for orphan agents is challenging, because the market might not be large enough to justify the investment."
Developing a vaccine in the United States costs an estimated $200-$500 million; nonetheless, vaccines are the most cost-effective way to prevent disease, she said. "In view of the burden of chikungunya outbreaks, which have affected up to 63% of local populations in a matter of months, the continued development of this VLP vaccine candidate, along with other vaccine options, should be encouraged."
A VLP contains the outer structural proteins of the virus that the immune system typically recognizes, Dr. Powers added. But it does not contain live genetic material, which confers a safety and manufacturing advantage because high-containment facilities are not needed for production.
Dr. Powers is with the division of vector-borne diseases at the U.S. Centers for Disease Control and Prevention. She declared that she had no competing interests. These remarks were excerpted from her editorial accompanying Dr. Chang’s report (Lancet 2014 Aug. 15 [doi: 10.1016/S0140-6736(14)61290-3]).
A novel chikungunya virus vaccine caused no serious adverse effects and was about as immunogenic as natural infection, authors of a phase I trial reported in the Lancet.
Eleven months after receiving the noninfectious viruslike particle (VLP) vaccine, trial participants had neutralizing antibody titers that resembled those seen in natural infection, said Dr. Lee-Jah Chang and associates at the National Institutes of Health in Bethesda, Md. (Lancet 2014 Aug. 15 [doi: 10.1016/S0140-6736(14)61185-5]).
Chikungunya virus infection is rarely fatal, but it causes fever and severe arthritis. The mosquito-born pathogen spread to the Americas in 2013, is now epidemic in the Caribbean, and has no approved vaccine or treatment.
To test a novel VLP vaccine candidate, researchers enrolled 25 healthy adults into a phase I, open-label, dose-escalation trial with doses of 10, 20, or 40 mcg administered at weeks 0, 4, and 20, the investigators reported. Participants were followed for 44 weeks after enrollment.
All groups had detectable neutralizing antibodies on ELISA (enzyme-linked immunosorbent assay) after the second dose, with geometric mean titers of the half-maximum inhibitory concentration of 2,688 in the 10-mcg group, 1,775 in the 20-mcg group, and 7,246 in the 40-mcg group. Antibody levels after the second dose substantially exceeded those measured after the first dose, with P values for differences ranging from .07 for the 10-mcg group to less than .0001 for the 40-mcg group, the investigators said. And titers did not significantly differ 4 weeks after the second and third doses, they added.
Participants reported no arthralgias or other serious adverse effects. Mild to moderate side effects occurred in four participants, and consisted of transient neutropenia (three cases) and transient increases in alanine aminotransferase (four cases), all of which resolved without clinical consequences, the investigators said.
"These clinical data represent an important step in vaccine development to combat this rapidly emerging pathogen," the researchers said. The next step is to test the vaccine in larger studies that include persons at risk of chikungunya virus infection, they added.
The trial was funded by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases; and the National Institutes of Health. The authors declared no competing interests.
A novel chikungunya virus vaccine caused no serious adverse effects and was about as immunogenic as natural infection, authors of a phase I trial reported in the Lancet.
Eleven months after receiving the noninfectious viruslike particle (VLP) vaccine, trial participants had neutralizing antibody titers that resembled those seen in natural infection, said Dr. Lee-Jah Chang and associates at the National Institutes of Health in Bethesda, Md. (Lancet 2014 Aug. 15 [doi: 10.1016/S0140-6736(14)61185-5]).
Chikungunya virus infection is rarely fatal, but it causes fever and severe arthritis. The mosquito-born pathogen spread to the Americas in 2013, is now epidemic in the Caribbean, and has no approved vaccine or treatment.
To test a novel VLP vaccine candidate, researchers enrolled 25 healthy adults into a phase I, open-label, dose-escalation trial with doses of 10, 20, or 40 mcg administered at weeks 0, 4, and 20, the investigators reported. Participants were followed for 44 weeks after enrollment.
All groups had detectable neutralizing antibodies on ELISA (enzyme-linked immunosorbent assay) after the second dose, with geometric mean titers of the half-maximum inhibitory concentration of 2,688 in the 10-mcg group, 1,775 in the 20-mcg group, and 7,246 in the 40-mcg group. Antibody levels after the second dose substantially exceeded those measured after the first dose, with P values for differences ranging from .07 for the 10-mcg group to less than .0001 for the 40-mcg group, the investigators said. And titers did not significantly differ 4 weeks after the second and third doses, they added.
Participants reported no arthralgias or other serious adverse effects. Mild to moderate side effects occurred in four participants, and consisted of transient neutropenia (three cases) and transient increases in alanine aminotransferase (four cases), all of which resolved without clinical consequences, the investigators said.
"These clinical data represent an important step in vaccine development to combat this rapidly emerging pathogen," the researchers said. The next step is to test the vaccine in larger studies that include persons at risk of chikungunya virus infection, they added.
The trial was funded by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases; and the National Institutes of Health. The authors declared no competing interests.
FROM THE LANCET
Key clinical point: A novel, noninfectious vaccine for chikungunya virus was highly immunogenic and caused no serious adverse effects in a phase I trial.
Major finding: All dosing groups developed detectable neutralizing antibodies after a second vaccination with a chikungunya virus vaccine (geometric mean titers of the half-maximum inhibitory concentration: 2,688 in the 10-mcg group, 1,775 in the 20-mcg group, and 7,246 in the 40-mcg group).
Data source: Phase I, dose-escalation, open-label clinical trial of noninfectious viruslike particle chikungunya vaccine in 25 healthy adults.
Disclosures: Funding sources included the Intramural Research Program of the Vaccine Research Center, the National Institute of Allergy and Infectious Diseases, and the National Institutes of Health. The authors declared no competing interests.
CDC: Prevalence of ALS is 4 per 100,000 in U.S.
The U.S. prevalence of amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease, is about 4 cases per 100,000 Americans, according to the Centers for Disease Control and Prevention.
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disorder that is usually fatal within 2-5 years of diagnosis. A hereditary form of the disease accounts for between 10% and 15% of cases. In the rest, the cause or causes are unknown, although chemical and infectious exposures are among the suspected triggers.
The new prevalence figures, which the CDC published in its Morbidity and Mortality Weekly Report issued July 25 (MMWR 2014;63[SS07]:1-14), represent the first national prevalence findings for ALS in the United States, and derive from surveillance begun in 2009 by the federal Agency for Toxic Substances and Disease Registry in Atlanta, where Dr. Paul Mehta led the investigation.
Dr. Mehta and his colleagues identified cases from Medicare, Medicaid, Veterans Heath Administration, and Veterans Benefits Administration databases, as well a secure public website through which ALS patients could self-report to the CDC by answering a series of screening questions.
Between October 2010 and the end of 2011, the registry identified 12,187 people 18 years and older with ALS, for a prevalence of 3.9 cases of ALS per 100,000 – findings that are consistent, the researchers said, with those from long-running European ALS registries.
As in other studies, men had a higher prevalence of ALS than did women (4.8 per 100,000 vs. 3.0 per 100,000). "Factors such as occupational history and environmental exposures might be associated with this finding," the researchers wrote in their analysis.
The prevalence of ALS among whites was more than double that of blacks (4.2 per vs. 2.0). "The reason for this difference in prevalence by race is unknown and needs to be investigated further," Dr. Mehta and his associates wrote. However, they noted, the race findings were also consistent with other studies.
The age group 70-79 was associated with the highest prevalence rate, at 17.0 per 100,000, followed by 60-69 at about 12.
Dr. Mehta and his colleagues acknowledged that their study had several limitations, including the fact that the ALS registry was relatively new; that ALS is not a notifiable disease in most states, making it difficult to capture all cases; that data errors or file duplication could have occurred; and that it was not possible to calculate ALS incidence, only prevalence, because most cases in the registry did not have a diagnosis date.
No conflicts of interest were mentioned in the report.
The U.S. prevalence of amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease, is about 4 cases per 100,000 Americans, according to the Centers for Disease Control and Prevention.
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disorder that is usually fatal within 2-5 years of diagnosis. A hereditary form of the disease accounts for between 10% and 15% of cases. In the rest, the cause or causes are unknown, although chemical and infectious exposures are among the suspected triggers.
The new prevalence figures, which the CDC published in its Morbidity and Mortality Weekly Report issued July 25 (MMWR 2014;63[SS07]:1-14), represent the first national prevalence findings for ALS in the United States, and derive from surveillance begun in 2009 by the federal Agency for Toxic Substances and Disease Registry in Atlanta, where Dr. Paul Mehta led the investigation.
Dr. Mehta and his colleagues identified cases from Medicare, Medicaid, Veterans Heath Administration, and Veterans Benefits Administration databases, as well a secure public website through which ALS patients could self-report to the CDC by answering a series of screening questions.
Between October 2010 and the end of 2011, the registry identified 12,187 people 18 years and older with ALS, for a prevalence of 3.9 cases of ALS per 100,000 – findings that are consistent, the researchers said, with those from long-running European ALS registries.
As in other studies, men had a higher prevalence of ALS than did women (4.8 per 100,000 vs. 3.0 per 100,000). "Factors such as occupational history and environmental exposures might be associated with this finding," the researchers wrote in their analysis.
The prevalence of ALS among whites was more than double that of blacks (4.2 per vs. 2.0). "The reason for this difference in prevalence by race is unknown and needs to be investigated further," Dr. Mehta and his associates wrote. However, they noted, the race findings were also consistent with other studies.
The age group 70-79 was associated with the highest prevalence rate, at 17.0 per 100,000, followed by 60-69 at about 12.
Dr. Mehta and his colleagues acknowledged that their study had several limitations, including the fact that the ALS registry was relatively new; that ALS is not a notifiable disease in most states, making it difficult to capture all cases; that data errors or file duplication could have occurred; and that it was not possible to calculate ALS incidence, only prevalence, because most cases in the registry did not have a diagnosis date.
No conflicts of interest were mentioned in the report.
The U.S. prevalence of amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease, is about 4 cases per 100,000 Americans, according to the Centers for Disease Control and Prevention.
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disorder that is usually fatal within 2-5 years of diagnosis. A hereditary form of the disease accounts for between 10% and 15% of cases. In the rest, the cause or causes are unknown, although chemical and infectious exposures are among the suspected triggers.
The new prevalence figures, which the CDC published in its Morbidity and Mortality Weekly Report issued July 25 (MMWR 2014;63[SS07]:1-14), represent the first national prevalence findings for ALS in the United States, and derive from surveillance begun in 2009 by the federal Agency for Toxic Substances and Disease Registry in Atlanta, where Dr. Paul Mehta led the investigation.
Dr. Mehta and his colleagues identified cases from Medicare, Medicaid, Veterans Heath Administration, and Veterans Benefits Administration databases, as well a secure public website through which ALS patients could self-report to the CDC by answering a series of screening questions.
Between October 2010 and the end of 2011, the registry identified 12,187 people 18 years and older with ALS, for a prevalence of 3.9 cases of ALS per 100,000 – findings that are consistent, the researchers said, with those from long-running European ALS registries.
As in other studies, men had a higher prevalence of ALS than did women (4.8 per 100,000 vs. 3.0 per 100,000). "Factors such as occupational history and environmental exposures might be associated with this finding," the researchers wrote in their analysis.
The prevalence of ALS among whites was more than double that of blacks (4.2 per vs. 2.0). "The reason for this difference in prevalence by race is unknown and needs to be investigated further," Dr. Mehta and his associates wrote. However, they noted, the race findings were also consistent with other studies.
The age group 70-79 was associated with the highest prevalence rate, at 17.0 per 100,000, followed by 60-69 at about 12.
Dr. Mehta and his colleagues acknowledged that their study had several limitations, including the fact that the ALS registry was relatively new; that ALS is not a notifiable disease in most states, making it difficult to capture all cases; that data errors or file duplication could have occurred; and that it was not possible to calculate ALS incidence, only prevalence, because most cases in the registry did not have a diagnosis date.
No conflicts of interest were mentioned in the report.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
Serum albumin, creatinine predicted survival in ALS
Serum levels of both albumin and creatinine proved to be independent biomarkers of disease severity in men and women with amyotrophic lateral sclerosis, with lower levels denoting more serious disease and a shorter survival time, according to a report published online July 21 in JAMA Neurology.
To identify any potential correlations between hematologic biomarkers and ALS severity, researchers analyzed data concerning 638 patients from a regional registry of people diagnosed during 2007-2011 in the Piemonte and Valle d’Aosta areas of Italy. The 352 men and 286 women underwent complete physical examinations at the time of diagnosis, which included tests for 17 serum biomarkers. The only two serum biomarkers found to correlate with ALS severity were albumin level, which reflected inflammation, and creatinine, which reflected muscle wasting, said Dr. Adriano Chio, professor of neuroscience, University of Torino, Turin, Italy, and his associates.
Both biomarkers showed an inverse dose-response relationship with clinical function at diagnosis in men and women. Both had sensitivity and specificity values at predicting 1-year mortality that were similar to those of "the best established prognostic factors" for ALS, such as forced vital capacity, age, and scores on the ALS Functional Rating Scale-Revised, the investigators said (JAMA Neurol. 2014 July 21 [doi:10.1001/jamaneurol.2014.1129]).
Dr. Chio and his colleagues performed a validation study in a cohort of 122 patients (54 men, 68 women) at all stages of ALS who were treated at an ALS tertiary care center in another area of Italy. This study confirmed the findings from the discovery cohort. "Both creatinine and albumin are reliable and easily detectable blood markers of the severity of motor dysfunction in ALS and could be used in defining patients’ prognosis at the time of diagnosis," they said.
This study was supported by the Italian Ministry of Health and the European Community’s Health Seventh Framework Programme. Dr. Chio reported serving on scientific advisory board for Biogen Idec and Cytokinetics
Serum levels of both albumin and creatinine proved to be independent biomarkers of disease severity in men and women with amyotrophic lateral sclerosis, with lower levels denoting more serious disease and a shorter survival time, according to a report published online July 21 in JAMA Neurology.
To identify any potential correlations between hematologic biomarkers and ALS severity, researchers analyzed data concerning 638 patients from a regional registry of people diagnosed during 2007-2011 in the Piemonte and Valle d’Aosta areas of Italy. The 352 men and 286 women underwent complete physical examinations at the time of diagnosis, which included tests for 17 serum biomarkers. The only two serum biomarkers found to correlate with ALS severity were albumin level, which reflected inflammation, and creatinine, which reflected muscle wasting, said Dr. Adriano Chio, professor of neuroscience, University of Torino, Turin, Italy, and his associates.
Both biomarkers showed an inverse dose-response relationship with clinical function at diagnosis in men and women. Both had sensitivity and specificity values at predicting 1-year mortality that were similar to those of "the best established prognostic factors" for ALS, such as forced vital capacity, age, and scores on the ALS Functional Rating Scale-Revised, the investigators said (JAMA Neurol. 2014 July 21 [doi:10.1001/jamaneurol.2014.1129]).
Dr. Chio and his colleagues performed a validation study in a cohort of 122 patients (54 men, 68 women) at all stages of ALS who were treated at an ALS tertiary care center in another area of Italy. This study confirmed the findings from the discovery cohort. "Both creatinine and albumin are reliable and easily detectable blood markers of the severity of motor dysfunction in ALS and could be used in defining patients’ prognosis at the time of diagnosis," they said.
This study was supported by the Italian Ministry of Health and the European Community’s Health Seventh Framework Programme. Dr. Chio reported serving on scientific advisory board for Biogen Idec and Cytokinetics
Serum levels of both albumin and creatinine proved to be independent biomarkers of disease severity in men and women with amyotrophic lateral sclerosis, with lower levels denoting more serious disease and a shorter survival time, according to a report published online July 21 in JAMA Neurology.
To identify any potential correlations between hematologic biomarkers and ALS severity, researchers analyzed data concerning 638 patients from a regional registry of people diagnosed during 2007-2011 in the Piemonte and Valle d’Aosta areas of Italy. The 352 men and 286 women underwent complete physical examinations at the time of diagnosis, which included tests for 17 serum biomarkers. The only two serum biomarkers found to correlate with ALS severity were albumin level, which reflected inflammation, and creatinine, which reflected muscle wasting, said Dr. Adriano Chio, professor of neuroscience, University of Torino, Turin, Italy, and his associates.
Both biomarkers showed an inverse dose-response relationship with clinical function at diagnosis in men and women. Both had sensitivity and specificity values at predicting 1-year mortality that were similar to those of "the best established prognostic factors" for ALS, such as forced vital capacity, age, and scores on the ALS Functional Rating Scale-Revised, the investigators said (JAMA Neurol. 2014 July 21 [doi:10.1001/jamaneurol.2014.1129]).
Dr. Chio and his colleagues performed a validation study in a cohort of 122 patients (54 men, 68 women) at all stages of ALS who were treated at an ALS tertiary care center in another area of Italy. This study confirmed the findings from the discovery cohort. "Both creatinine and albumin are reliable and easily detectable blood markers of the severity of motor dysfunction in ALS and could be used in defining patients’ prognosis at the time of diagnosis," they said.
This study was supported by the Italian Ministry of Health and the European Community’s Health Seventh Framework Programme. Dr. Chio reported serving on scientific advisory board for Biogen Idec and Cytokinetics
FROM JAMA NEUROLOGY
Key clinical point: Serum levels of both albumin and creatinine were independent biomarkers of ALS severity.
Major finding: At diagnosis, serum levels of both albumin and creatinine showed an inverse dose-response relationship with clinical function in men and women, with sensitivity and specificity values at predicting 1-year mortality that were similar to those of "the best established prognostic factors" for ALS.
Data source: A population-based cohort study of serum biomarkers in 638 ALS patients registered in an Italian regional database, and a validation cohort study of 122 patients with different stages of ALS seen consecutively at an ALS tertiary care center in another area of Italy.
Disclosures: This study was supported by the Italian Ministry of Health and the European Community’s Health Seventh Framework Programme. Dr. Chio reported serving on scientific advisory board for Biogen Idec and Cytokinetics.
High dietary omega-3 fatty acids are associated with lower ALS risk
Adults who consumed high levels of omega-3 polyunsaturated fatty acids showed a markedly reduced risk of developing amyotrophic lateral sclerosis in a pooled analysis of five large prospective cohort studies that assessed diet.
Diet-derived omega-3 polyunsaturated fatty acids (PUFAs) are known to have neuroprotective effects, and those present in neural plasma membranes can modulate oxidative stress, excitotoxicity, and inflammation. But no prospective studies have explored a possible relationship between omega-3 PUFA intake and amyotrophic lateral sclerosis (ALS) risk, according to Kathryn C. Fitzgerald of the department of nutrition, Harvard School of Public Health, Boston, and her associates.

In a study published July 14 in JAMA Neurology, Ms. Fitzgerald and her colleagues pooled data from the Health Professionals Follow-up Study, the Nurses’ Health Study, the Cancer Prevention Study II Nutrition Cohort, the Multiethnic Cohort Study, and the National Institutes of Health-AARP Diet and Health Study. A total of 995 ALS patients were identified among 1,002,082 participants in these studies. The five studies included detailed dietary information and tracked the occurrence of ALS in the study participants through the National Death Index.
Omega-3 PUFA intake in the highest quintile of consumption at a median of 2.11 g/day was associated with a 34% reduced risk of developing ALS, compared with the lowest quintile of consumption at a median of 0.94 g/day. This finding was consistent across all five studies. This means that adding 0.5% of energy from omega-3 PUFAs and maintaining a constant intake of omega-6 fatty acids while reducing the intake of other types of fat would reduce ALS risk by 34%. Consumption of alpha-linolenic acid, another PUFA, also was associated with significantly reduced risk of developing ALS. In contrast, consumption of omega-6 PUFAs, consumption of linolenic acid, total energy intake, and percentage of energy from other types of fat showed no association with ALS risk, the investigators said (JAMA Neurol. 2014 July 14 [doi:10.1001/jamaneurol.2014.1214]).
Foods that are rich in omega-3 PUFAs include fatty fish (salmon, sardines, tuna, herring) and fish oils; vegetable oils (corn, safflower, canola, soy, and flaxseed oils); and nuts and seeds (walnuts, chia seeds, butternuts, and sunflower seeds). Further studies are needed to confirm this protective effect in ALS and to determine whether patients who already have the disease would benefit from the addition of omega-3 PUFAs to their diets, Ms. Fitzgerald and her associates added.
The findings from Ms. Fitzgerald and her associates are persuasive and consistent with earlier suggestions that PUFAs may play a role in other neurodegenerative conditions, Dr. Michael Swash said in a related editorial (JAMA Neurol. 2014 July 14 [doi:10.1001/jamaneurol.2014.1894]).
"Ideas on long-term risk-susceptibility factors are very much welcomed in trying to unravel the mystery that is ALS. Now, in addition to genetic factors, there are the following five risk factors to work on: male sex, smoking status, BMI, physical exercise, and dietary intake of PUFAs," said Dr. Swash of the Royal London Hospital, Queen Mary University of London, and the Institute of Neuroscience at the University of Lisbon.
This study was supported by the National Institute of Neurological Diseases and Stroke, the National Cancer Institute, and the ALS Therapy Alliance Foundation. The study authors and Dr. Swash had no financial disclosures.
Adults who consumed high levels of omega-3 polyunsaturated fatty acids showed a markedly reduced risk of developing amyotrophic lateral sclerosis in a pooled analysis of five large prospective cohort studies that assessed diet.
Diet-derived omega-3 polyunsaturated fatty acids (PUFAs) are known to have neuroprotective effects, and those present in neural plasma membranes can modulate oxidative stress, excitotoxicity, and inflammation. But no prospective studies have explored a possible relationship between omega-3 PUFA intake and amyotrophic lateral sclerosis (ALS) risk, according to Kathryn C. Fitzgerald of the department of nutrition, Harvard School of Public Health, Boston, and her associates.
In a study published July 14 in JAMA Neurology, Ms. Fitzgerald and her colleagues pooled data from the Health Professionals Follow-up Study, the Nurses’ Health Study, the Cancer Prevention Study II Nutrition Cohort, the Multiethnic Cohort Study, and the National Institutes of Health-AARP Diet and Health Study. A total of 995 ALS patients were identified among 1,002,082 participants in these studies. The five studies included detailed dietary information and tracked the occurrence of ALS in the study participants through the National Death Index.
Omega-3 PUFA intake in the highest quintile of consumption at a median of 2.11 g/day was associated with a 34% reduced risk of developing ALS, compared with the lowest quintile of consumption at a median of 0.94 g/day. This finding was consistent across all five studies. This means that adding 0.5% of energy from omega-3 PUFAs and maintaining a constant intake of omega-6 fatty acids while reducing the intake of other types of fat would reduce ALS risk by 34%. Consumption of alpha-linolenic acid, another PUFA, also was associated with significantly reduced risk of developing ALS. In contrast, consumption of omega-6 PUFAs, consumption of linolenic acid, total energy intake, and percentage of energy from other types of fat showed no association with ALS risk, the investigators said (JAMA Neurol. 2014 July 14 [doi:10.1001/jamaneurol.2014.1214]).
Foods that are rich in omega-3 PUFAs include fatty fish (salmon, sardines, tuna, herring) and fish oils; vegetable oils (corn, safflower, canola, soy, and flaxseed oils); and nuts and seeds (walnuts, chia seeds, butternuts, and sunflower seeds). Further studies are needed to confirm this protective effect in ALS and to determine whether patients who already have the disease would benefit from the addition of omega-3 PUFAs to their diets, Ms. Fitzgerald and her associates added.
The findings from Ms. Fitzgerald and her associates are persuasive and consistent with earlier suggestions that PUFAs may play a role in other neurodegenerative conditions, Dr. Michael Swash said in a related editorial (JAMA Neurol. 2014 July 14 [doi:10.1001/jamaneurol.2014.1894]).
"Ideas on long-term risk-susceptibility factors are very much welcomed in trying to unravel the mystery that is ALS. Now, in addition to genetic factors, there are the following five risk factors to work on: male sex, smoking status, BMI, physical exercise, and dietary intake of PUFAs," said Dr. Swash of the Royal London Hospital, Queen Mary University of London, and the Institute of Neuroscience at the University of Lisbon.
This study was supported by the National Institute of Neurological Diseases and Stroke, the National Cancer Institute, and the ALS Therapy Alliance Foundation. The study authors and Dr. Swash had no financial disclosures.
Adults who consumed high levels of omega-3 polyunsaturated fatty acids showed a markedly reduced risk of developing amyotrophic lateral sclerosis in a pooled analysis of five large prospective cohort studies that assessed diet.
Diet-derived omega-3 polyunsaturated fatty acids (PUFAs) are known to have neuroprotective effects, and those present in neural plasma membranes can modulate oxidative stress, excitotoxicity, and inflammation. But no prospective studies have explored a possible relationship between omega-3 PUFA intake and amyotrophic lateral sclerosis (ALS) risk, according to Kathryn C. Fitzgerald of the department of nutrition, Harvard School of Public Health, Boston, and her associates.
In a study published July 14 in JAMA Neurology, Ms. Fitzgerald and her colleagues pooled data from the Health Professionals Follow-up Study, the Nurses’ Health Study, the Cancer Prevention Study II Nutrition Cohort, the Multiethnic Cohort Study, and the National Institutes of Health-AARP Diet and Health Study. A total of 995 ALS patients were identified among 1,002,082 participants in these studies. The five studies included detailed dietary information and tracked the occurrence of ALS in the study participants through the National Death Index.
Omega-3 PUFA intake in the highest quintile of consumption at a median of 2.11 g/day was associated with a 34% reduced risk of developing ALS, compared with the lowest quintile of consumption at a median of 0.94 g/day. This finding was consistent across all five studies. This means that adding 0.5% of energy from omega-3 PUFAs and maintaining a constant intake of omega-6 fatty acids while reducing the intake of other types of fat would reduce ALS risk by 34%. Consumption of alpha-linolenic acid, another PUFA, also was associated with significantly reduced risk of developing ALS. In contrast, consumption of omega-6 PUFAs, consumption of linolenic acid, total energy intake, and percentage of energy from other types of fat showed no association with ALS risk, the investigators said (JAMA Neurol. 2014 July 14 [doi:10.1001/jamaneurol.2014.1214]).
Foods that are rich in omega-3 PUFAs include fatty fish (salmon, sardines, tuna, herring) and fish oils; vegetable oils (corn, safflower, canola, soy, and flaxseed oils); and nuts and seeds (walnuts, chia seeds, butternuts, and sunflower seeds). Further studies are needed to confirm this protective effect in ALS and to determine whether patients who already have the disease would benefit from the addition of omega-3 PUFAs to their diets, Ms. Fitzgerald and her associates added.
The findings from Ms. Fitzgerald and her associates are persuasive and consistent with earlier suggestions that PUFAs may play a role in other neurodegenerative conditions, Dr. Michael Swash said in a related editorial (JAMA Neurol. 2014 July 14 [doi:10.1001/jamaneurol.2014.1894]).
"Ideas on long-term risk-susceptibility factors are very much welcomed in trying to unravel the mystery that is ALS. Now, in addition to genetic factors, there are the following five risk factors to work on: male sex, smoking status, BMI, physical exercise, and dietary intake of PUFAs," said Dr. Swash of the Royal London Hospital, Queen Mary University of London, and the Institute of Neuroscience at the University of Lisbon.
This study was supported by the National Institute of Neurological Diseases and Stroke, the National Cancer Institute, and the ALS Therapy Alliance Foundation. The study authors and Dr. Swash had no financial disclosures.
FROM JAMA NEUROLOGY
Key clinical point: Eating more omega-3 PUFAs, maintaining a constant intake of omega-6 fatty acids, and reducing the intake of other types of fat is associated with a reduced risk of ALS.
Major finding: High omega-3 PUFA intake was associated with a 34% reduction in the relative risk of developing ALS.
Data source: A pooled analysis of five large prospective cohorts totaling 1,002,082 participants to explore any association between dietary intake of various fatty acids and ALS risk.
Disclosures: This study was supported by the National Institute of Neurological Diseases and Stroke, the National Cancer Institute, and the ALS Therapy Alliance Foundation. The study authors and Dr. Swash had no financial disclosures.

Longer-term opioid use in workers’ comp cases highest in Louisiana
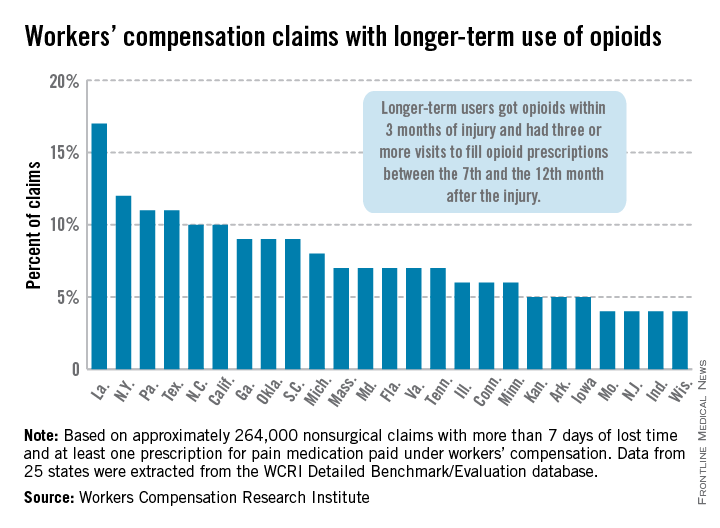
In Louisiana, opioid use lasted more than 6 months in 17% of nonsurgical workers’ compensation claims involving employees who received at least one prescription for pain medication, the Workers Compensation Research Institute reported.
In cases with more than 7 days of lost time, that was the highest rate seen among the 25 states in the study, with New York second at 12% and Pennsylvania and Texas tied for third at 11%. There were four states tied for the lowest rate, at 4%: Missouri, New Jersey, Indiana, and Wisconsin, according to the WCRI report.
Overall, use of narcotics for pain relief by injured workers in such cases ranged from 60% in New Jersey to 88% in Arkansas (median, 76%), while use of any pain medication ranged from 85% in Minnesota to 95% in Florida, Georgia, Tennessee, and Texas (median, 94%), the report showed.
The study involved claims with injuries that occurred from Oct. 1, 2009, through Sept. 30, 2010, with prescriptions filled through March 31, 2012. Longer-term users received a prescription for opioids within 3 months of their injury and had three or more visits to fill opioid prescriptions between the 7th and the 12th month after the injury.
The 25 states in the study "represent more than 70% of the workers’ compensation benefits paid in the United States," the WCRI noted.
The study was based on approximately 264,000 nonsurgical claims and more than 1.5 million prescriptions for pain medications. Data were extracted from the WCRI Detailed Benchmark/Evaluation database and consisted of detailed prescription transactions "collected from workers’ compensation payers and their medical bill review and pharmacy benefit management vendors," the report noted.
In Louisiana, opioid use lasted more than 6 months in 17% of nonsurgical workers’ compensation claims involving employees who received at least one prescription for pain medication, the Workers Compensation Research Institute reported.
In cases with more than 7 days of lost time, that was the highest rate seen among the 25 states in the study, with New York second at 12% and Pennsylvania and Texas tied for third at 11%. There were four states tied for the lowest rate, at 4%: Missouri, New Jersey, Indiana, and Wisconsin, according to the WCRI report.
Overall, use of narcotics for pain relief by injured workers in such cases ranged from 60% in New Jersey to 88% in Arkansas (median, 76%), while use of any pain medication ranged from 85% in Minnesota to 95% in Florida, Georgia, Tennessee, and Texas (median, 94%), the report showed.
The study involved claims with injuries that occurred from Oct. 1, 2009, through Sept. 30, 2010, with prescriptions filled through March 31, 2012. Longer-term users received a prescription for opioids within 3 months of their injury and had three or more visits to fill opioid prescriptions between the 7th and the 12th month after the injury.
The 25 states in the study "represent more than 70% of the workers’ compensation benefits paid in the United States," the WCRI noted.
The study was based on approximately 264,000 nonsurgical claims and more than 1.5 million prescriptions for pain medications. Data were extracted from the WCRI Detailed Benchmark/Evaluation database and consisted of detailed prescription transactions "collected from workers’ compensation payers and their medical bill review and pharmacy benefit management vendors," the report noted.
In Louisiana, opioid use lasted more than 6 months in 17% of nonsurgical workers’ compensation claims involving employees who received at least one prescription for pain medication, the Workers Compensation Research Institute reported.
In cases with more than 7 days of lost time, that was the highest rate seen among the 25 states in the study, with New York second at 12% and Pennsylvania and Texas tied for third at 11%. There were four states tied for the lowest rate, at 4%: Missouri, New Jersey, Indiana, and Wisconsin, according to the WCRI report.
Overall, use of narcotics for pain relief by injured workers in such cases ranged from 60% in New Jersey to 88% in Arkansas (median, 76%), while use of any pain medication ranged from 85% in Minnesota to 95% in Florida, Georgia, Tennessee, and Texas (median, 94%), the report showed.
The study involved claims with injuries that occurred from Oct. 1, 2009, through Sept. 30, 2010, with prescriptions filled through March 31, 2012. Longer-term users received a prescription for opioids within 3 months of their injury and had three or more visits to fill opioid prescriptions between the 7th and the 12th month after the injury.
The 25 states in the study "represent more than 70% of the workers’ compensation benefits paid in the United States," the WCRI noted.
The study was based on approximately 264,000 nonsurgical claims and more than 1.5 million prescriptions for pain medications. Data were extracted from the WCRI Detailed Benchmark/Evaluation database and consisted of detailed prescription transactions "collected from workers’ compensation payers and their medical bill review and pharmacy benefit management vendors," the report noted.

Studies hint at safety, efficacy of spinal muscular atrophy drug
PHILADELPHIA – The latest interim results from open-label studies of the investigational antisense oligonucleotide therapy ISIS-SMNRx for the treatment of patients with type 1, 2, or 3 spinal muscular atrophy support its safety and are starting to show its potential efficacy in treating the range of severity seen in the disease.
In two ongoing studies with up to 9 months of follow-up data, no safety or tolerability concerns arose with total doses of up to 18 mg in patients with type 2 or 3 spinal muscular atrophy (SMA) and in total doses of up to 48 mg in infants with type 1 SMA. Children aged 2-15 years with type 2 or 3 SMA had a dose- and time-dependent improvement in scores on the Hammersmith Functional Motor Scale-Expanded (HFMSE) that also correlated well with levels of SMN protein in cerebrospinal fluid. Infants with type 1 SMA achieved motor milestones on the Hammersmith Infant Neurological Exam that were consistent with increases in motor function test scores, according to investigators who presented the results at the annual meeting of the American Academy of Neurology.

"It’s very encouraging that we can do this safely and that the children tolerate the lumbar punctures, and there’s hope that the measures [used in the studies] are sensitive to change," said primary investigator Dr. Claudia Chiriboga, who presented the interim results of a study in patients with SMA types 2 or 3.
In that study, ISIS-SMNRx, an antisense oligonucleotide that promotes transcription of the full-length SMN protein from the SMN2 gene, was administered in an intrathecal bolus via lumbar puncture at points during a 3-month period; patients were then followed for 6 months. A total of eight patients received 3 mg at each dose (total dose, 9 mg); eight received 6 mg at each dose (total dose, 18 mg); and nine received 9 mg at each dose (18 mg total). Later, investigators added a 12-mg dose cohort that currently has eight patients enrolled, but results in that cohort are not yet available, said Dr. Chiriboga of the division of child neurology at Columbia University, New York.
The SMA type 2 and 3 patients included 10 patients with type 2 and 15 with type 3. They were medically stable and 2-15 years old, with a mean age of 7.5 years. Most (20) had three copies of the SMN2 gene; 4 had four copies and 1 had two copies. A majority of the patients (16) were nonambulatory.
None of the adverse events reported were considered related to the study drug, and most of the 143 adverse events were mild or moderate, the investigators found. Two severe adverse events were back pain and myalgia. Most of the adverse events were related to the lumbar punctures.
Scores on the HFMSE improved from baseline by a mean of 1.5 points in the 3-mg group, 2.3 points in the 6-mg group, and a statistically significant 3.7 points in the 9-mg group. SMN levels in cerebrospinal fluid at day 85 increased from baseline in all groups but were significantly increased in the 9-mg group only.
Additional secondary endpoints showed nonsignificant improvement of 22.7 m at 9 months on the 6-minute walk test in those who could walk, and an improvement of 2.3 points on an 18-point scale measuring upper limb function in weaker nonambulatory patients, but the open-label nature of the study and small numbers of patients make it difficult to interpret such findings, Dr. Chiriboga said.

"The feeling is that when there’s chronicity, like end-stage type of changes – severe scoliosis, for example – that those individuals don’t do as well. ... It’s not so much the age," Dr. Chiriboga said in an interview. Patients with type 3 disease also do better because they have more SMN2 to begin with, she said.
Similarly, in the ongoing open-label study of infants with type 1 SMA, ISIS-SMNRx was administered to 4 patients in 6-mg doses at days 1, 15, 85, and 253, and in 12-mg doses to 11 patients at the same time points. These infants were all aged 7 months or younger. Their mean age at symptom onset was 7 weeks, and they were enrolled in the study at a mean age of 18-21 weeks. All but one patient had two copies of the SMN2 gene, reported primary investigator Dr. Richard S. Finkel.
None of the adverse events in the infants were deemed to be related to ISIS-SMNRx. Of 14 severe adverse events, 11 were respiratory infections, and all were considered to be consistent with severe infant SMA, said Dr. Finkel, chief of the division of neurology at Nemours Children’s Hospital and professor of neurology at the University of Central Florida, both in Orlando.
One patient in the 6-mg group died accidentally, and another underwent permanent ventilation. Two of 11 patients in the 12-mg group died of pulmonary infection, and 1 required permanent ventilation (16 or more hours per day continuously for more than 2 weeks in the absence of an acute reversible illness), although 4 of the patients in this group have not yet received all their doses. At the last follow-up, or at the time of death or permanent ventilation, the median age was 14 months in the 6-mg group and 9.6 months in the 12-mg group (which has not been followed as long).
Scores on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) showed increases in 8 of 11 infants who had completed treatment and evaluation. The scores increased by a mean of 5.4 points overall and by 8.3 points in those in the 12-mg group. Incremental milestones on the Hammersmith Infant Neurological Exam were achieved by 9 of 11 infants, including 6 of 7 in the 12-mg group.
The median age at death or need for permanent ventilation is 10.5 months in infants with two SMN2 gene copies, and 85% reach this endpoint at 18 months. Scores on the CHOP-INTEND also declined by 1.27 points per year, according to a study of the natural history of type 1 SMA in 34 patients by Dr. Finkel and his colleagues that is under review for publication.
Compound muscle action potentials measured in the ulnar nerve–innervated abductor digiti minimi and peroneal nerve–innervated anterior tibialis were stable or increased in most infants, he said.
These encouraging results with ISIS-SMNRx have led Isis to begin plans for phase III trials in patients with SMA types 1-3, the investigators said.
The studies are funded by Isis Pharmaceuticals, the Department of Defense, and the National Institute of Neurological Disorders and Stroke. Neither Dr. Finkel nor Dr. Chiriboga had conflicts of interest. Some of the coauthors in each study were employees of Isis.
PHILADELPHIA – The latest interim results from open-label studies of the investigational antisense oligonucleotide therapy ISIS-SMNRx for the treatment of patients with type 1, 2, or 3 spinal muscular atrophy support its safety and are starting to show its potential efficacy in treating the range of severity seen in the disease.
In two ongoing studies with up to 9 months of follow-up data, no safety or tolerability concerns arose with total doses of up to 18 mg in patients with type 2 or 3 spinal muscular atrophy (SMA) and in total doses of up to 48 mg in infants with type 1 SMA. Children aged 2-15 years with type 2 or 3 SMA had a dose- and time-dependent improvement in scores on the Hammersmith Functional Motor Scale-Expanded (HFMSE) that also correlated well with levels of SMN protein in cerebrospinal fluid. Infants with type 1 SMA achieved motor milestones on the Hammersmith Infant Neurological Exam that were consistent with increases in motor function test scores, according to investigators who presented the results at the annual meeting of the American Academy of Neurology.
"It’s very encouraging that we can do this safely and that the children tolerate the lumbar punctures, and there’s hope that the measures [used in the studies] are sensitive to change," said primary investigator Dr. Claudia Chiriboga, who presented the interim results of a study in patients with SMA types 2 or 3.
In that study, ISIS-SMNRx, an antisense oligonucleotide that promotes transcription of the full-length SMN protein from the SMN2 gene, was administered in an intrathecal bolus via lumbar puncture at points during a 3-month period; patients were then followed for 6 months. A total of eight patients received 3 mg at each dose (total dose, 9 mg); eight received 6 mg at each dose (total dose, 18 mg); and nine received 9 mg at each dose (18 mg total). Later, investigators added a 12-mg dose cohort that currently has eight patients enrolled, but results in that cohort are not yet available, said Dr. Chiriboga of the division of child neurology at Columbia University, New York.
The SMA type 2 and 3 patients included 10 patients with type 2 and 15 with type 3. They were medically stable and 2-15 years old, with a mean age of 7.5 years. Most (20) had three copies of the SMN2 gene; 4 had four copies and 1 had two copies. A majority of the patients (16) were nonambulatory.
None of the adverse events reported were considered related to the study drug, and most of the 143 adverse events were mild or moderate, the investigators found. Two severe adverse events were back pain and myalgia. Most of the adverse events were related to the lumbar punctures.
Scores on the HFMSE improved from baseline by a mean of 1.5 points in the 3-mg group, 2.3 points in the 6-mg group, and a statistically significant 3.7 points in the 9-mg group. SMN levels in cerebrospinal fluid at day 85 increased from baseline in all groups but were significantly increased in the 9-mg group only.
Additional secondary endpoints showed nonsignificant improvement of 22.7 m at 9 months on the 6-minute walk test in those who could walk, and an improvement of 2.3 points on an 18-point scale measuring upper limb function in weaker nonambulatory patients, but the open-label nature of the study and small numbers of patients make it difficult to interpret such findings, Dr. Chiriboga said.
"The feeling is that when there’s chronicity, like end-stage type of changes – severe scoliosis, for example – that those individuals don’t do as well. ... It’s not so much the age," Dr. Chiriboga said in an interview. Patients with type 3 disease also do better because they have more SMN2 to begin with, she said.
Similarly, in the ongoing open-label study of infants with type 1 SMA, ISIS-SMNRx was administered to 4 patients in 6-mg doses at days 1, 15, 85, and 253, and in 12-mg doses to 11 patients at the same time points. These infants were all aged 7 months or younger. Their mean age at symptom onset was 7 weeks, and they were enrolled in the study at a mean age of 18-21 weeks. All but one patient had two copies of the SMN2 gene, reported primary investigator Dr. Richard S. Finkel.
None of the adverse events in the infants were deemed to be related to ISIS-SMNRx. Of 14 severe adverse events, 11 were respiratory infections, and all were considered to be consistent with severe infant SMA, said Dr. Finkel, chief of the division of neurology at Nemours Children’s Hospital and professor of neurology at the University of Central Florida, both in Orlando.
One patient in the 6-mg group died accidentally, and another underwent permanent ventilation. Two of 11 patients in the 12-mg group died of pulmonary infection, and 1 required permanent ventilation (16 or more hours per day continuously for more than 2 weeks in the absence of an acute reversible illness), although 4 of the patients in this group have not yet received all their doses. At the last follow-up, or at the time of death or permanent ventilation, the median age was 14 months in the 6-mg group and 9.6 months in the 12-mg group (which has not been followed as long).
Scores on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) showed increases in 8 of 11 infants who had completed treatment and evaluation. The scores increased by a mean of 5.4 points overall and by 8.3 points in those in the 12-mg group. Incremental milestones on the Hammersmith Infant Neurological Exam were achieved by 9 of 11 infants, including 6 of 7 in the 12-mg group.
The median age at death or need for permanent ventilation is 10.5 months in infants with two SMN2 gene copies, and 85% reach this endpoint at 18 months. Scores on the CHOP-INTEND also declined by 1.27 points per year, according to a study of the natural history of type 1 SMA in 34 patients by Dr. Finkel and his colleagues that is under review for publication.
Compound muscle action potentials measured in the ulnar nerve–innervated abductor digiti minimi and peroneal nerve–innervated anterior tibialis were stable or increased in most infants, he said.
These encouraging results with ISIS-SMNRx have led Isis to begin plans for phase III trials in patients with SMA types 1-3, the investigators said.
The studies are funded by Isis Pharmaceuticals, the Department of Defense, and the National Institute of Neurological Disorders and Stroke. Neither Dr. Finkel nor Dr. Chiriboga had conflicts of interest. Some of the coauthors in each study were employees of Isis.
PHILADELPHIA – The latest interim results from open-label studies of the investigational antisense oligonucleotide therapy ISIS-SMNRx for the treatment of patients with type 1, 2, or 3 spinal muscular atrophy support its safety and are starting to show its potential efficacy in treating the range of severity seen in the disease.
In two ongoing studies with up to 9 months of follow-up data, no safety or tolerability concerns arose with total doses of up to 18 mg in patients with type 2 or 3 spinal muscular atrophy (SMA) and in total doses of up to 48 mg in infants with type 1 SMA. Children aged 2-15 years with type 2 or 3 SMA had a dose- and time-dependent improvement in scores on the Hammersmith Functional Motor Scale-Expanded (HFMSE) that also correlated well with levels of SMN protein in cerebrospinal fluid. Infants with type 1 SMA achieved motor milestones on the Hammersmith Infant Neurological Exam that were consistent with increases in motor function test scores, according to investigators who presented the results at the annual meeting of the American Academy of Neurology.
"It’s very encouraging that we can do this safely and that the children tolerate the lumbar punctures, and there’s hope that the measures [used in the studies] are sensitive to change," said primary investigator Dr. Claudia Chiriboga, who presented the interim results of a study in patients with SMA types 2 or 3.
In that study, ISIS-SMNRx, an antisense oligonucleotide that promotes transcription of the full-length SMN protein from the SMN2 gene, was administered in an intrathecal bolus via lumbar puncture at points during a 3-month period; patients were then followed for 6 months. A total of eight patients received 3 mg at each dose (total dose, 9 mg); eight received 6 mg at each dose (total dose, 18 mg); and nine received 9 mg at each dose (18 mg total). Later, investigators added a 12-mg dose cohort that currently has eight patients enrolled, but results in that cohort are not yet available, said Dr. Chiriboga of the division of child neurology at Columbia University, New York.
The SMA type 2 and 3 patients included 10 patients with type 2 and 15 with type 3. They were medically stable and 2-15 years old, with a mean age of 7.5 years. Most (20) had three copies of the SMN2 gene; 4 had four copies and 1 had two copies. A majority of the patients (16) were nonambulatory.
None of the adverse events reported were considered related to the study drug, and most of the 143 adverse events were mild or moderate, the investigators found. Two severe adverse events were back pain and myalgia. Most of the adverse events were related to the lumbar punctures.
Scores on the HFMSE improved from baseline by a mean of 1.5 points in the 3-mg group, 2.3 points in the 6-mg group, and a statistically significant 3.7 points in the 9-mg group. SMN levels in cerebrospinal fluid at day 85 increased from baseline in all groups but were significantly increased in the 9-mg group only.
Additional secondary endpoints showed nonsignificant improvement of 22.7 m at 9 months on the 6-minute walk test in those who could walk, and an improvement of 2.3 points on an 18-point scale measuring upper limb function in weaker nonambulatory patients, but the open-label nature of the study and small numbers of patients make it difficult to interpret such findings, Dr. Chiriboga said.
"The feeling is that when there’s chronicity, like end-stage type of changes – severe scoliosis, for example – that those individuals don’t do as well. ... It’s not so much the age," Dr. Chiriboga said in an interview. Patients with type 3 disease also do better because they have more SMN2 to begin with, she said.
Similarly, in the ongoing open-label study of infants with type 1 SMA, ISIS-SMNRx was administered to 4 patients in 6-mg doses at days 1, 15, 85, and 253, and in 12-mg doses to 11 patients at the same time points. These infants were all aged 7 months or younger. Their mean age at symptom onset was 7 weeks, and they were enrolled in the study at a mean age of 18-21 weeks. All but one patient had two copies of the SMN2 gene, reported primary investigator Dr. Richard S. Finkel.
None of the adverse events in the infants were deemed to be related to ISIS-SMNRx. Of 14 severe adverse events, 11 were respiratory infections, and all were considered to be consistent with severe infant SMA, said Dr. Finkel, chief of the division of neurology at Nemours Children’s Hospital and professor of neurology at the University of Central Florida, both in Orlando.
One patient in the 6-mg group died accidentally, and another underwent permanent ventilation. Two of 11 patients in the 12-mg group died of pulmonary infection, and 1 required permanent ventilation (16 or more hours per day continuously for more than 2 weeks in the absence of an acute reversible illness), although 4 of the patients in this group have not yet received all their doses. At the last follow-up, or at the time of death or permanent ventilation, the median age was 14 months in the 6-mg group and 9.6 months in the 12-mg group (which has not been followed as long).
Scores on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) showed increases in 8 of 11 infants who had completed treatment and evaluation. The scores increased by a mean of 5.4 points overall and by 8.3 points in those in the 12-mg group. Incremental milestones on the Hammersmith Infant Neurological Exam were achieved by 9 of 11 infants, including 6 of 7 in the 12-mg group.
The median age at death or need for permanent ventilation is 10.5 months in infants with two SMN2 gene copies, and 85% reach this endpoint at 18 months. Scores on the CHOP-INTEND also declined by 1.27 points per year, according to a study of the natural history of type 1 SMA in 34 patients by Dr. Finkel and his colleagues that is under review for publication.
Compound muscle action potentials measured in the ulnar nerve–innervated abductor digiti minimi and peroneal nerve–innervated anterior tibialis were stable or increased in most infants, he said.
These encouraging results with ISIS-SMNRx have led Isis to begin plans for phase III trials in patients with SMA types 1-3, the investigators said.
The studies are funded by Isis Pharmaceuticals, the Department of Defense, and the National Institute of Neurological Disorders and Stroke. Neither Dr. Finkel nor Dr. Chiriboga had conflicts of interest. Some of the coauthors in each study were employees of Isis.
AT THE AAN 2014 ANNUAL MEETING

Eteplirsen showed safety, efficacy over 2 years in Duchenne muscular dystrophy
PHILADELPHIA – Eteplirsen safely maintained its beneficial effect on walking speeds for certain patients with Duchenne muscular dystrophy for more than 2 years and was the first drug to show stabilized diaphragm function over the same period.
The investigational gene therapy drug had a stabilizing effect on patients’ walking speed over the course of 120 weeks regardless of whether they started it earlier or later in the open-label extension of the initial 24-week, randomized trial, but those who started treatment earlier maintained a higher walking speed, Dr. Jerry R. Mendell said at the annual meeting of the American Academy of Neurology.

Coinvestigator Dr. Edward M. Kaye of Sarepta Therapeutics, the study sponsor, presented in-depth safety and pharmacokinetics data that showed no significant treatment-related adverse events with eteplirsen in the small study. No patients have discontinued or disrupted treatment, and no laboratory evidence of toxicity has been seen to date.
The initial 24-week, double-blind, randomized, placebo-controlled study involved four patients who were given the equivalent of 30 mg/kg eteplirsen weekly, four given 50 mg/kg eteplirsen weekly, and four given placebo. At the start of the open-label extension study, two of the placebo-treated patients were transitioned to 30 mg/kg weekly and two to 50 mg/kg weekly.
All patients underwent muscle biopsy at baseline, followed by biopsies at 12 weeks in two of the high-dose patients and in two of the placebo-treated patients, and at 24 weeks in the four patients taking 30 mg/kg weekly and in two placebo-treated patients.
"This design permitted a comparison between high dose over a shorter time interval and low dose over a longer time interval," said Dr. Mendell of the Center for Gene Therapy at Nationwide Children’s Hospital, Columbus, Ohio.
All patients underwent a biopsy at 1 year (48 weeks) in the open-label extension (Ann. Neurol. 2013;74:637-47).
By 48 weeks, the mean percentage of dystrophin-positive muscle fibers was 34% in the placebo/30 mg/kg delayed-treatment group, 43% in the placebo/50 mg/kg delayed-treatment group, 42% in the 50-mg/kg group, and 52% in the 30-mg/kg group.
Distance traveled on the 6-minute walk test began to diverge between active and placebo-treated patients at 12 weeks and stabilized at 24 weeks among those who received active treatment, but continued to decline until 36 weeks for placebo-treated patients who started treatment at 24 weeks.
At 120 weeks, the patients who had received continuous treatment with eteplirsen declined by a mean of 14 m on the 6-minute walk test since baseline, compared with a decline of 79 m in those who underwent delayed treatment with eteplirsen. In comparison, studies of the natural history of Duchenne muscular dystrophy (DMD) have shown steady declines on the 6-minute walk test, ranging from 30 to 115 m at 1 year and 97 to 125 m at 2 years.
There was "remarkable stability" in diaphragm function over the course of treatment with eteplirsen, "which is quite different from the natural history of untreated patients," Dr. Mendell said. In all 12 patients, maximal expiratory pressure remained stable, going from a mean of 90% of predicted at baseline to 95% at 120 weeks. The same was true for maximal inspiratory pressure, moving from 79% of expected at baseline to 80% at 120 weeks. In contrast, the natural history of untreated DMD shows that pulmonary function declines substantially over time: by 3.9%/year for maximal inspiratory pressure after starting at 90% of predicted at 9 years of age, and by 3.6%/year after starting at 45% of predicted at 9 years of age (Pediatr. Pulmonol. 2014;49:473-81).
Eteplirsen was created to help patients with a deletion of exon 51 in dystrophin, which causes a nonfunctional dystrophin protein. The drug is a charge-neutral phosphorodiamidate morpholino oligomer (PMO) that targets the 13% of DMD patients with this mutation by directing alternative splicing of the dystrophin gene through its ability to bind to exon 51 of dystrophin pre-mRNA. This restores transcription and translation of a truncated, yet functional, dystrophin protein, such as those found in Becker muscular dystrophy.
The neutral charge of eteplirsen helps it to avoid binding to serum proteins, which is one of the problems that have occurred with phosphorothioate antisense oligonucleotide-based drugs. Phosphorothioate antisense drugs have also been linked to immune activation, hepatotoxicity, thrombocytopenia, coagulopathy, renal toxicity, and proteinuria. But Dr. Kaye reported that eteplirsen is mainly excreted by the kidneys, has a half-life of about 3 hours, and showed no evidence of those problems. Only 13 of 453 urine protein assessments tested positive, but all were low, transient, and resolved spontaneously.
"The reason that this is important is there has actually been very little human data until recently, despite the fact that [PMOs have] been around for over 30 years. Most of the work has been done in animals and in research laboratories, and it’s only until recently that people have been exposed to the drug," Dr. Kaye said.
The study was funded by Sarepta Therapeutics. Dr. Kaye and two coauthors are employees of the company. Dr. Mendell had no relevant disclosures.
PHILADELPHIA – Eteplirsen safely maintained its beneficial effect on walking speeds for certain patients with Duchenne muscular dystrophy for more than 2 years and was the first drug to show stabilized diaphragm function over the same period.
The investigational gene therapy drug had a stabilizing effect on patients’ walking speed over the course of 120 weeks regardless of whether they started it earlier or later in the open-label extension of the initial 24-week, randomized trial, but those who started treatment earlier maintained a higher walking speed, Dr. Jerry R. Mendell said at the annual meeting of the American Academy of Neurology.
Coinvestigator Dr. Edward M. Kaye of Sarepta Therapeutics, the study sponsor, presented in-depth safety and pharmacokinetics data that showed no significant treatment-related adverse events with eteplirsen in the small study. No patients have discontinued or disrupted treatment, and no laboratory evidence of toxicity has been seen to date.
The initial 24-week, double-blind, randomized, placebo-controlled study involved four patients who were given the equivalent of 30 mg/kg eteplirsen weekly, four given 50 mg/kg eteplirsen weekly, and four given placebo. At the start of the open-label extension study, two of the placebo-treated patients were transitioned to 30 mg/kg weekly and two to 50 mg/kg weekly.
All patients underwent muscle biopsy at baseline, followed by biopsies at 12 weeks in two of the high-dose patients and in two of the placebo-treated patients, and at 24 weeks in the four patients taking 30 mg/kg weekly and in two placebo-treated patients.
"This design permitted a comparison between high dose over a shorter time interval and low dose over a longer time interval," said Dr. Mendell of the Center for Gene Therapy at Nationwide Children’s Hospital, Columbus, Ohio.
All patients underwent a biopsy at 1 year (48 weeks) in the open-label extension (Ann. Neurol. 2013;74:637-47).
By 48 weeks, the mean percentage of dystrophin-positive muscle fibers was 34% in the placebo/30 mg/kg delayed-treatment group, 43% in the placebo/50 mg/kg delayed-treatment group, 42% in the 50-mg/kg group, and 52% in the 30-mg/kg group.
Distance traveled on the 6-minute walk test began to diverge between active and placebo-treated patients at 12 weeks and stabilized at 24 weeks among those who received active treatment, but continued to decline until 36 weeks for placebo-treated patients who started treatment at 24 weeks.
At 120 weeks, the patients who had received continuous treatment with eteplirsen declined by a mean of 14 m on the 6-minute walk test since baseline, compared with a decline of 79 m in those who underwent delayed treatment with eteplirsen. In comparison, studies of the natural history of Duchenne muscular dystrophy (DMD) have shown steady declines on the 6-minute walk test, ranging from 30 to 115 m at 1 year and 97 to 125 m at 2 years.
There was "remarkable stability" in diaphragm function over the course of treatment with eteplirsen, "which is quite different from the natural history of untreated patients," Dr. Mendell said. In all 12 patients, maximal expiratory pressure remained stable, going from a mean of 90% of predicted at baseline to 95% at 120 weeks. The same was true for maximal inspiratory pressure, moving from 79% of expected at baseline to 80% at 120 weeks. In contrast, the natural history of untreated DMD shows that pulmonary function declines substantially over time: by 3.9%/year for maximal inspiratory pressure after starting at 90% of predicted at 9 years of age, and by 3.6%/year after starting at 45% of predicted at 9 years of age (Pediatr. Pulmonol. 2014;49:473-81).
Eteplirsen was created to help patients with a deletion of exon 51 in dystrophin, which causes a nonfunctional dystrophin protein. The drug is a charge-neutral phosphorodiamidate morpholino oligomer (PMO) that targets the 13% of DMD patients with this mutation by directing alternative splicing of the dystrophin gene through its ability to bind to exon 51 of dystrophin pre-mRNA. This restores transcription and translation of a truncated, yet functional, dystrophin protein, such as those found in Becker muscular dystrophy.
The neutral charge of eteplirsen helps it to avoid binding to serum proteins, which is one of the problems that have occurred with phosphorothioate antisense oligonucleotide-based drugs. Phosphorothioate antisense drugs have also been linked to immune activation, hepatotoxicity, thrombocytopenia, coagulopathy, renal toxicity, and proteinuria. But Dr. Kaye reported that eteplirsen is mainly excreted by the kidneys, has a half-life of about 3 hours, and showed no evidence of those problems. Only 13 of 453 urine protein assessments tested positive, but all were low, transient, and resolved spontaneously.
"The reason that this is important is there has actually been very little human data until recently, despite the fact that [PMOs have] been around for over 30 years. Most of the work has been done in animals and in research laboratories, and it’s only until recently that people have been exposed to the drug," Dr. Kaye said.
The study was funded by Sarepta Therapeutics. Dr. Kaye and two coauthors are employees of the company. Dr. Mendell had no relevant disclosures.
PHILADELPHIA – Eteplirsen safely maintained its beneficial effect on walking speeds for certain patients with Duchenne muscular dystrophy for more than 2 years and was the first drug to show stabilized diaphragm function over the same period.
The investigational gene therapy drug had a stabilizing effect on patients’ walking speed over the course of 120 weeks regardless of whether they started it earlier or later in the open-label extension of the initial 24-week, randomized trial, but those who started treatment earlier maintained a higher walking speed, Dr. Jerry R. Mendell said at the annual meeting of the American Academy of Neurology.
Coinvestigator Dr. Edward M. Kaye of Sarepta Therapeutics, the study sponsor, presented in-depth safety and pharmacokinetics data that showed no significant treatment-related adverse events with eteplirsen in the small study. No patients have discontinued or disrupted treatment, and no laboratory evidence of toxicity has been seen to date.
The initial 24-week, double-blind, randomized, placebo-controlled study involved four patients who were given the equivalent of 30 mg/kg eteplirsen weekly, four given 50 mg/kg eteplirsen weekly, and four given placebo. At the start of the open-label extension study, two of the placebo-treated patients were transitioned to 30 mg/kg weekly and two to 50 mg/kg weekly.
All patients underwent muscle biopsy at baseline, followed by biopsies at 12 weeks in two of the high-dose patients and in two of the placebo-treated patients, and at 24 weeks in the four patients taking 30 mg/kg weekly and in two placebo-treated patients.
"This design permitted a comparison between high dose over a shorter time interval and low dose over a longer time interval," said Dr. Mendell of the Center for Gene Therapy at Nationwide Children’s Hospital, Columbus, Ohio.
All patients underwent a biopsy at 1 year (48 weeks) in the open-label extension (Ann. Neurol. 2013;74:637-47).
By 48 weeks, the mean percentage of dystrophin-positive muscle fibers was 34% in the placebo/30 mg/kg delayed-treatment group, 43% in the placebo/50 mg/kg delayed-treatment group, 42% in the 50-mg/kg group, and 52% in the 30-mg/kg group.
Distance traveled on the 6-minute walk test began to diverge between active and placebo-treated patients at 12 weeks and stabilized at 24 weeks among those who received active treatment, but continued to decline until 36 weeks for placebo-treated patients who started treatment at 24 weeks.
At 120 weeks, the patients who had received continuous treatment with eteplirsen declined by a mean of 14 m on the 6-minute walk test since baseline, compared with a decline of 79 m in those who underwent delayed treatment with eteplirsen. In comparison, studies of the natural history of Duchenne muscular dystrophy (DMD) have shown steady declines on the 6-minute walk test, ranging from 30 to 115 m at 1 year and 97 to 125 m at 2 years.
There was "remarkable stability" in diaphragm function over the course of treatment with eteplirsen, "which is quite different from the natural history of untreated patients," Dr. Mendell said. In all 12 patients, maximal expiratory pressure remained stable, going from a mean of 90% of predicted at baseline to 95% at 120 weeks. The same was true for maximal inspiratory pressure, moving from 79% of expected at baseline to 80% at 120 weeks. In contrast, the natural history of untreated DMD shows that pulmonary function declines substantially over time: by 3.9%/year for maximal inspiratory pressure after starting at 90% of predicted at 9 years of age, and by 3.6%/year after starting at 45% of predicted at 9 years of age (Pediatr. Pulmonol. 2014;49:473-81).
Eteplirsen was created to help patients with a deletion of exon 51 in dystrophin, which causes a nonfunctional dystrophin protein. The drug is a charge-neutral phosphorodiamidate morpholino oligomer (PMO) that targets the 13% of DMD patients with this mutation by directing alternative splicing of the dystrophin gene through its ability to bind to exon 51 of dystrophin pre-mRNA. This restores transcription and translation of a truncated, yet functional, dystrophin protein, such as those found in Becker muscular dystrophy.
The neutral charge of eteplirsen helps it to avoid binding to serum proteins, which is one of the problems that have occurred with phosphorothioate antisense oligonucleotide-based drugs. Phosphorothioate antisense drugs have also been linked to immune activation, hepatotoxicity, thrombocytopenia, coagulopathy, renal toxicity, and proteinuria. But Dr. Kaye reported that eteplirsen is mainly excreted by the kidneys, has a half-life of about 3 hours, and showed no evidence of those problems. Only 13 of 453 urine protein assessments tested positive, but all were low, transient, and resolved spontaneously.
"The reason that this is important is there has actually been very little human data until recently, despite the fact that [PMOs have] been around for over 30 years. Most of the work has been done in animals and in research laboratories, and it’s only until recently that people have been exposed to the drug," Dr. Kaye said.
The study was funded by Sarepta Therapeutics. Dr. Kaye and two coauthors are employees of the company. Dr. Mendell had no relevant disclosures.
AT THE AAN 2014 ANNUAL MEETING
Key clinical point: Treatment with eteplirsen stabilized key clinical features of DMD and had no significant adverse events over 2 years.
Major finding: At 120 weeks, patients who received continuous treatment with eteplirsen had a mean decline of 14 m on the 6-minute walk test since baseline, compared with a decline of 79 m in those who underwent delayed treatment with eteplirsen.
Data source: An open-label extension of a 24-week, randomized, double-blind, placebo-controlled trial out to 120 weeks in 12 patients with DMD.
Disclosures: The study is funded by Sarepta Therapeutics. Dr. Kaye and two coauthors are employees of the company. Dr. Mendell had no relevant disclosures.

FDA panel considers human studies of modified oocytes for preventing disease
GAITHERSBURG, MD. – The first human studies evaluating the use of genetically modified oocytes to prevent the transmission of mitochondrial diseases could enroll women with diseases that are the most severe, tend to present in early childhood, and are relatively common for a mitochondrial disease, according to panelists at a meeting convened by the Food and Drug Administration to discuss the design of such trials and related issues.
At a meeting on Feb. 25 and 26, members of the FDA Cellular, Tissue, and Gene Therapies Advisory Committee mentioned two mitochondrial diseases in particular, Leigh’s disease and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes), that could be included in initial clinical trials evaluating the safety and efficacy of what the FDA refers to as "mitochondrial manipulation technologies."
This controversial approach, which is being developed to prevent maternal transmission of debilitating and often fatal mitochondrial diseases, entails removing the mitochondrial DNA from an affected woman’s oocyte or embryo and replacing it via assisted reproductive technologies with the mitochondrial DNA from the egg of a healthy donor.
The approach has been studied in animal and in vitro studies, but not yet in humans. However, researchers at Oregon Health and Science University (OHSU), Portland, say they are ready to start a clinical trial based on their results in macaque monkeys.
The FDA called the 2-day meeting to discuss potential clinical trials and to focus on the scientific, technologic, and clinical aspects of the technologies, but not to address public policy or ethical issues. In briefing documents posted before the meeting, the agency acknowledged that there are ethical and policy issues related to genetic modification of eggs and embryos, which can affect regulatory decisions, but added that these issues were "outside the scope" of this meeting.
Regarding clinical trial design and execution, panelists recommended that studies should closely monitor the fetus through gestation, and after birth and long-term follow-up, should include future generations, if female offspring are included. Several panelists supported including only male embryos to minimize the risk of a female passing on damaged DNA to future generations, while others said this would result in a lost opportunity to study the transgenerational risks of the technology.
Other recommendations included avoiding the enrollment of people at high risk of having a baby with a birth defect, or with comorbidities that could affect birth outcome, which would make it more difficult to evaluate the risks of the technologies. The use of controls, panelists said, was problematic, because of the variability in when and how mitochondrial diseases present and because of the relatively small populations of patients affected by these diseases. Historical controls could be used, but larger patient registries are needed, they said.
Panelists also recommended screening egg donors for mitochondrial diseases, and providing informed consent to children born to mothers in the trials when they turn age 18.
It is clear that there is a "deft group of creative, innovative investigators" who can perform these techniques, "which is a good start, but there are so many things we don’t know" that must be evaluated further in animal studies, said panelist Dr. David Keefe, who referred to thalidomide and diethylstilbestrol (DES) as historical examples of therapies that were thought to be promising but proved to have devastating effects.
Another concern Dr. Keefe raised was the possibility that a woman whose risk of having a baby without the inherited defect might be as high as 95% and that she might choose mitochondrial manipulation over preimplantation genetic diagnosis.
"A woman could be led down the primrose path towards a procedure that’s experimental and miss the opportunity to pursue a relatively well-established procedure," said Dr. Keefe, the Stanley H. Kaplan Professor, department of obstetrics and gynecology, New York University.
Another panelist, Dr. Katharine Wenstrom, professor of obstetrics and gynecology, Brown University, Providence, R.I., said that based on her experience with women with genetic diseases, these women "are very vulnerable, and my concern would be how to [provide] consent [for] somebody for whom a pregnancy would be very dangerous and [who] might not consider a pregnancy, but then given the opportunity to have this technique, might agree to a pregnancy that could actually be life threatening."
She also said that she was concerned about whether the technique could deplete mitochondria, which has been associated with several forms of cancer, and about the "inability to ensure that the technique has not inflicted some new abnormality" on the child.
Shoukhrat Mitalipov, Ph.D., whose research group at the Oregon Stem Cell Center at OHSU has tested the technology in macaque monkeys, said that their research cohort currently includes four subjects born through mitochondrial manipulation that are almost adults. To date, they have been healthy, with normal blood test results, and are no different from controls, showing that mitochondrial DNA in oocytes can be replaced.
The next step in their research is to recruit families who are carriers of early-onset mitochondrial DNA diseases who have had at least one affected child, recruit healthy egg donors, and then perform the procedure, followed by preimplantation genetic diagnosis of the embryo and/or prenatal diagnosis to "ensure complete mitochondrial DNA replacement and chromosomal normalcy," he said.
The panel was also asked to discuss the use of mitochondrial manipulation as a treatment for infertility. However, members considered this indication a far different type of application than preventing mitochondrial disease, which would have different inclusion criteria, controls, and risk-benefit evaluations, and several panelists raised particular concerns about the use of this technology for infertility.
"The idea we’re going to do anything to infertility patients involving mitochondria I think should be off the table," Dr. Keefe said, noting that there is "a very, very slippery slope when you’re dealing with human reproduction" in the United States, where licensure of infertility clinics is not required.
The controversies of this area of research, which some critics point out would result in a child with three genetically related parents, were not off limits to the open public hearing speakers, including Marcy Darnovsky, Ph.D., executive director of the Center for Genetics and Society.
"We want to avoid waking up in a world" where researchers, infertility clinics, governments, insurance companies, "or parents decide that they are going to try to engineer children with specific traits and even possibly [put] in motion a regime of high-tech consumer eugenics," she said.
GAITHERSBURG, MD. – The first human studies evaluating the use of genetically modified oocytes to prevent the transmission of mitochondrial diseases could enroll women with diseases that are the most severe, tend to present in early childhood, and are relatively common for a mitochondrial disease, according to panelists at a meeting convened by the Food and Drug Administration to discuss the design of such trials and related issues.
At a meeting on Feb. 25 and 26, members of the FDA Cellular, Tissue, and Gene Therapies Advisory Committee mentioned two mitochondrial diseases in particular, Leigh’s disease and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes), that could be included in initial clinical trials evaluating the safety and efficacy of what the FDA refers to as "mitochondrial manipulation technologies."
This controversial approach, which is being developed to prevent maternal transmission of debilitating and often fatal mitochondrial diseases, entails removing the mitochondrial DNA from an affected woman’s oocyte or embryo and replacing it via assisted reproductive technologies with the mitochondrial DNA from the egg of a healthy donor.
The approach has been studied in animal and in vitro studies, but not yet in humans. However, researchers at Oregon Health and Science University (OHSU), Portland, say they are ready to start a clinical trial based on their results in macaque monkeys.
The FDA called the 2-day meeting to discuss potential clinical trials and to focus on the scientific, technologic, and clinical aspects of the technologies, but not to address public policy or ethical issues. In briefing documents posted before the meeting, the agency acknowledged that there are ethical and policy issues related to genetic modification of eggs and embryos, which can affect regulatory decisions, but added that these issues were "outside the scope" of this meeting.
Regarding clinical trial design and execution, panelists recommended that studies should closely monitor the fetus through gestation, and after birth and long-term follow-up, should include future generations, if female offspring are included. Several panelists supported including only male embryos to minimize the risk of a female passing on damaged DNA to future generations, while others said this would result in a lost opportunity to study the transgenerational risks of the technology.
Other recommendations included avoiding the enrollment of people at high risk of having a baby with a birth defect, or with comorbidities that could affect birth outcome, which would make it more difficult to evaluate the risks of the technologies. The use of controls, panelists said, was problematic, because of the variability in when and how mitochondrial diseases present and because of the relatively small populations of patients affected by these diseases. Historical controls could be used, but larger patient registries are needed, they said.
Panelists also recommended screening egg donors for mitochondrial diseases, and providing informed consent to children born to mothers in the trials when they turn age 18.
It is clear that there is a "deft group of creative, innovative investigators" who can perform these techniques, "which is a good start, but there are so many things we don’t know" that must be evaluated further in animal studies, said panelist Dr. David Keefe, who referred to thalidomide and diethylstilbestrol (DES) as historical examples of therapies that were thought to be promising but proved to have devastating effects.
Another concern Dr. Keefe raised was the possibility that a woman whose risk of having a baby without the inherited defect might be as high as 95% and that she might choose mitochondrial manipulation over preimplantation genetic diagnosis.
"A woman could be led down the primrose path towards a procedure that’s experimental and miss the opportunity to pursue a relatively well-established procedure," said Dr. Keefe, the Stanley H. Kaplan Professor, department of obstetrics and gynecology, New York University.
Another panelist, Dr. Katharine Wenstrom, professor of obstetrics and gynecology, Brown University, Providence, R.I., said that based on her experience with women with genetic diseases, these women "are very vulnerable, and my concern would be how to [provide] consent [for] somebody for whom a pregnancy would be very dangerous and [who] might not consider a pregnancy, but then given the opportunity to have this technique, might agree to a pregnancy that could actually be life threatening."
She also said that she was concerned about whether the technique could deplete mitochondria, which has been associated with several forms of cancer, and about the "inability to ensure that the technique has not inflicted some new abnormality" on the child.
Shoukhrat Mitalipov, Ph.D., whose research group at the Oregon Stem Cell Center at OHSU has tested the technology in macaque monkeys, said that their research cohort currently includes four subjects born through mitochondrial manipulation that are almost adults. To date, they have been healthy, with normal blood test results, and are no different from controls, showing that mitochondrial DNA in oocytes can be replaced.
The next step in their research is to recruit families who are carriers of early-onset mitochondrial DNA diseases who have had at least one affected child, recruit healthy egg donors, and then perform the procedure, followed by preimplantation genetic diagnosis of the embryo and/or prenatal diagnosis to "ensure complete mitochondrial DNA replacement and chromosomal normalcy," he said.
The panel was also asked to discuss the use of mitochondrial manipulation as a treatment for infertility. However, members considered this indication a far different type of application than preventing mitochondrial disease, which would have different inclusion criteria, controls, and risk-benefit evaluations, and several panelists raised particular concerns about the use of this technology for infertility.
"The idea we’re going to do anything to infertility patients involving mitochondria I think should be off the table," Dr. Keefe said, noting that there is "a very, very slippery slope when you’re dealing with human reproduction" in the United States, where licensure of infertility clinics is not required.
The controversies of this area of research, which some critics point out would result in a child with three genetically related parents, were not off limits to the open public hearing speakers, including Marcy Darnovsky, Ph.D., executive director of the Center for Genetics and Society.
"We want to avoid waking up in a world" where researchers, infertility clinics, governments, insurance companies, "or parents decide that they are going to try to engineer children with specific traits and even possibly [put] in motion a regime of high-tech consumer eugenics," she said.
GAITHERSBURG, MD. – The first human studies evaluating the use of genetically modified oocytes to prevent the transmission of mitochondrial diseases could enroll women with diseases that are the most severe, tend to present in early childhood, and are relatively common for a mitochondrial disease, according to panelists at a meeting convened by the Food and Drug Administration to discuss the design of such trials and related issues.
At a meeting on Feb. 25 and 26, members of the FDA Cellular, Tissue, and Gene Therapies Advisory Committee mentioned two mitochondrial diseases in particular, Leigh’s disease and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes), that could be included in initial clinical trials evaluating the safety and efficacy of what the FDA refers to as "mitochondrial manipulation technologies."
This controversial approach, which is being developed to prevent maternal transmission of debilitating and often fatal mitochondrial diseases, entails removing the mitochondrial DNA from an affected woman’s oocyte or embryo and replacing it via assisted reproductive technologies with the mitochondrial DNA from the egg of a healthy donor.
The approach has been studied in animal and in vitro studies, but not yet in humans. However, researchers at Oregon Health and Science University (OHSU), Portland, say they are ready to start a clinical trial based on their results in macaque monkeys.
The FDA called the 2-day meeting to discuss potential clinical trials and to focus on the scientific, technologic, and clinical aspects of the technologies, but not to address public policy or ethical issues. In briefing documents posted before the meeting, the agency acknowledged that there are ethical and policy issues related to genetic modification of eggs and embryos, which can affect regulatory decisions, but added that these issues were "outside the scope" of this meeting.
Regarding clinical trial design and execution, panelists recommended that studies should closely monitor the fetus through gestation, and after birth and long-term follow-up, should include future generations, if female offspring are included. Several panelists supported including only male embryos to minimize the risk of a female passing on damaged DNA to future generations, while others said this would result in a lost opportunity to study the transgenerational risks of the technology.
Other recommendations included avoiding the enrollment of people at high risk of having a baby with a birth defect, or with comorbidities that could affect birth outcome, which would make it more difficult to evaluate the risks of the technologies. The use of controls, panelists said, was problematic, because of the variability in when and how mitochondrial diseases present and because of the relatively small populations of patients affected by these diseases. Historical controls could be used, but larger patient registries are needed, they said.
Panelists also recommended screening egg donors for mitochondrial diseases, and providing informed consent to children born to mothers in the trials when they turn age 18.
It is clear that there is a "deft group of creative, innovative investigators" who can perform these techniques, "which is a good start, but there are so many things we don’t know" that must be evaluated further in animal studies, said panelist Dr. David Keefe, who referred to thalidomide and diethylstilbestrol (DES) as historical examples of therapies that were thought to be promising but proved to have devastating effects.
Another concern Dr. Keefe raised was the possibility that a woman whose risk of having a baby without the inherited defect might be as high as 95% and that she might choose mitochondrial manipulation over preimplantation genetic diagnosis.
"A woman could be led down the primrose path towards a procedure that’s experimental and miss the opportunity to pursue a relatively well-established procedure," said Dr. Keefe, the Stanley H. Kaplan Professor, department of obstetrics and gynecology, New York University.
Another panelist, Dr. Katharine Wenstrom, professor of obstetrics and gynecology, Brown University, Providence, R.I., said that based on her experience with women with genetic diseases, these women "are very vulnerable, and my concern would be how to [provide] consent [for] somebody for whom a pregnancy would be very dangerous and [who] might not consider a pregnancy, but then given the opportunity to have this technique, might agree to a pregnancy that could actually be life threatening."
She also said that she was concerned about whether the technique could deplete mitochondria, which has been associated with several forms of cancer, and about the "inability to ensure that the technique has not inflicted some new abnormality" on the child.
Shoukhrat Mitalipov, Ph.D., whose research group at the Oregon Stem Cell Center at OHSU has tested the technology in macaque monkeys, said that their research cohort currently includes four subjects born through mitochondrial manipulation that are almost adults. To date, they have been healthy, with normal blood test results, and are no different from controls, showing that mitochondrial DNA in oocytes can be replaced.
The next step in their research is to recruit families who are carriers of early-onset mitochondrial DNA diseases who have had at least one affected child, recruit healthy egg donors, and then perform the procedure, followed by preimplantation genetic diagnosis of the embryo and/or prenatal diagnosis to "ensure complete mitochondrial DNA replacement and chromosomal normalcy," he said.
The panel was also asked to discuss the use of mitochondrial manipulation as a treatment for infertility. However, members considered this indication a far different type of application than preventing mitochondrial disease, which would have different inclusion criteria, controls, and risk-benefit evaluations, and several panelists raised particular concerns about the use of this technology for infertility.
"The idea we’re going to do anything to infertility patients involving mitochondria I think should be off the table," Dr. Keefe said, noting that there is "a very, very slippery slope when you’re dealing with human reproduction" in the United States, where licensure of infertility clinics is not required.
The controversies of this area of research, which some critics point out would result in a child with three genetically related parents, were not off limits to the open public hearing speakers, including Marcy Darnovsky, Ph.D., executive director of the Center for Genetics and Society.
"We want to avoid waking up in a world" where researchers, infertility clinics, governments, insurance companies, "or parents decide that they are going to try to engineer children with specific traits and even possibly [put] in motion a regime of high-tech consumer eugenics," she said.
AT AN FDA ADVISORY COMMITTEE MEETING