User login
Good news, bad news about HCV in kidney disease
SAN DIEGO – There’s good news and bad news about hepatitis C virus (HCV) in patients with chronic kidney disease (CKD): The new generation of drugs that cures HCV is effective in this population, but outbreaks of infection are still plaguing the nation’s dialysis clinics.

These perspectives came in a presentation about infections in CKD at Kidney Week 2018, sponsored by the American Society of Nephrology.
First, the good news about HCV. “Treatment is now feasible for all stages of chronic kidney disease,” said gastroenterologist Paul Martin, MD, of the University of Miami. “It was possible to achieve biological cure in 99% of patients, which is truly remarkable considering what a problem kidney patients were for hepatitis C until very recently.”
The key is to treat HCV with drug combinations that lower the risk of viral resistance. “These drugs are extremely well tolerated. They’re not like interferon or ribavirin,” he said, referring to a drug combo that was formerly used to treat HCV. “We can anticipate curing hepatitis C with a finite amount of therapy in virtually every patient we see, including those with kidney disease.”
In patients with CKD, all the new drugs are approved for glomerular filtration rates greater than 30 mL/min. Sofosbuvir (Sovaldi) is not approved for patients with a filtration rate under 30 mL/min, he said, but other options are available.
Ribavirin, he added, is no longer needed with current regimens.
Dr. Martin pointed to two studies that reveal the power of the new regimens against HCV in patients with CKD. One of the studies, a 2015 industry-funded report in the Lancet, found that “once-daily grazoprevir and elbasvir for 12 weeks had a low rate of adverse events and was effective in patients infected with HCV genotype 1 and stage 4-5 chronic kidney disease.” The other study, also funded by industry and published in 2017 in the New England Journal of Medicine, found that “treatment with glecaprevir and pibrentasvir for 12 weeks resulted in a high rate of sustained virologic response in patients with stage 4 or 5 chronic kidney disease and HCV infection.”
Meanwhile, there are signs that HCV treatment may boost survival in CKD patients on dialysis, Dr. Martin said.
In terms of bad news, Priti R. Patel, MD, MPH, a medical officer with the Centers for Disease Control and Prevention, warned that dialysis clinics are still seeing HCV outbreaks. “It’s a continuing problem,” she said. “What we hear about at the CDC is the tip of the iceberg.”
The CDC says it received word of 21 HCV outbreaks of two or more cases in dialysis clinics during 2008-2017. These affected 102 patients, and more than 3,000 patients were notified that they were at risk and should be screened.
One dialysis clinic in Philadelphia had 18 cases of HCV during 2008-2013; they were blamed on “multiple lapses in infection control ... including hand hygiene and glove use, vascular access care, medication preparation, cleaning, and disinfection.”
“There should be no more than one case that has to happen for a facility to detect that it has a problem and identify a solution,” Dr. Patel said.
Since acute HCV can appear without symptoms, every dialysis patients should be tested for HCV antibodies, she added. “If it’s positive, confirm it. If confirmed, they should be informed of their infection status and have an evaluation for treatment.”
Dr. Martin reported consulting for Bristol-Myers Squibb and AbbVie and receiving research funding from Gilead, Bristol-Myers Squibb, AbbVie, and Merck. Dr. Patel reported no disclosures.
SAN DIEGO – There’s good news and bad news about hepatitis C virus (HCV) in patients with chronic kidney disease (CKD): The new generation of drugs that cures HCV is effective in this population, but outbreaks of infection are still plaguing the nation’s dialysis clinics.
These perspectives came in a presentation about infections in CKD at Kidney Week 2018, sponsored by the American Society of Nephrology.
First, the good news about HCV. “Treatment is now feasible for all stages of chronic kidney disease,” said gastroenterologist Paul Martin, MD, of the University of Miami. “It was possible to achieve biological cure in 99% of patients, which is truly remarkable considering what a problem kidney patients were for hepatitis C until very recently.”
The key is to treat HCV with drug combinations that lower the risk of viral resistance. “These drugs are extremely well tolerated. They’re not like interferon or ribavirin,” he said, referring to a drug combo that was formerly used to treat HCV. “We can anticipate curing hepatitis C with a finite amount of therapy in virtually every patient we see, including those with kidney disease.”
In patients with CKD, all the new drugs are approved for glomerular filtration rates greater than 30 mL/min. Sofosbuvir (Sovaldi) is not approved for patients with a filtration rate under 30 mL/min, he said, but other options are available.
Ribavirin, he added, is no longer needed with current regimens.
Dr. Martin pointed to two studies that reveal the power of the new regimens against HCV in patients with CKD. One of the studies, a 2015 industry-funded report in the Lancet, found that “once-daily grazoprevir and elbasvir for 12 weeks had a low rate of adverse events and was effective in patients infected with HCV genotype 1 and stage 4-5 chronic kidney disease.” The other study, also funded by industry and published in 2017 in the New England Journal of Medicine, found that “treatment with glecaprevir and pibrentasvir for 12 weeks resulted in a high rate of sustained virologic response in patients with stage 4 or 5 chronic kidney disease and HCV infection.”
Meanwhile, there are signs that HCV treatment may boost survival in CKD patients on dialysis, Dr. Martin said.
In terms of bad news, Priti R. Patel, MD, MPH, a medical officer with the Centers for Disease Control and Prevention, warned that dialysis clinics are still seeing HCV outbreaks. “It’s a continuing problem,” she said. “What we hear about at the CDC is the tip of the iceberg.”
The CDC says it received word of 21 HCV outbreaks of two or more cases in dialysis clinics during 2008-2017. These affected 102 patients, and more than 3,000 patients were notified that they were at risk and should be screened.
One dialysis clinic in Philadelphia had 18 cases of HCV during 2008-2013; they were blamed on “multiple lapses in infection control ... including hand hygiene and glove use, vascular access care, medication preparation, cleaning, and disinfection.”
“There should be no more than one case that has to happen for a facility to detect that it has a problem and identify a solution,” Dr. Patel said.
Since acute HCV can appear without symptoms, every dialysis patients should be tested for HCV antibodies, she added. “If it’s positive, confirm it. If confirmed, they should be informed of their infection status and have an evaluation for treatment.”
Dr. Martin reported consulting for Bristol-Myers Squibb and AbbVie and receiving research funding from Gilead, Bristol-Myers Squibb, AbbVie, and Merck. Dr. Patel reported no disclosures.
SAN DIEGO – There’s good news and bad news about hepatitis C virus (HCV) in patients with chronic kidney disease (CKD): The new generation of drugs that cures HCV is effective in this population, but outbreaks of infection are still plaguing the nation’s dialysis clinics.
These perspectives came in a presentation about infections in CKD at Kidney Week 2018, sponsored by the American Society of Nephrology.
First, the good news about HCV. “Treatment is now feasible for all stages of chronic kidney disease,” said gastroenterologist Paul Martin, MD, of the University of Miami. “It was possible to achieve biological cure in 99% of patients, which is truly remarkable considering what a problem kidney patients were for hepatitis C until very recently.”
The key is to treat HCV with drug combinations that lower the risk of viral resistance. “These drugs are extremely well tolerated. They’re not like interferon or ribavirin,” he said, referring to a drug combo that was formerly used to treat HCV. “We can anticipate curing hepatitis C with a finite amount of therapy in virtually every patient we see, including those with kidney disease.”
In patients with CKD, all the new drugs are approved for glomerular filtration rates greater than 30 mL/min. Sofosbuvir (Sovaldi) is not approved for patients with a filtration rate under 30 mL/min, he said, but other options are available.
Ribavirin, he added, is no longer needed with current regimens.
Dr. Martin pointed to two studies that reveal the power of the new regimens against HCV in patients with CKD. One of the studies, a 2015 industry-funded report in the Lancet, found that “once-daily grazoprevir and elbasvir for 12 weeks had a low rate of adverse events and was effective in patients infected with HCV genotype 1 and stage 4-5 chronic kidney disease.” The other study, also funded by industry and published in 2017 in the New England Journal of Medicine, found that “treatment with glecaprevir and pibrentasvir for 12 weeks resulted in a high rate of sustained virologic response in patients with stage 4 or 5 chronic kidney disease and HCV infection.”
Meanwhile, there are signs that HCV treatment may boost survival in CKD patients on dialysis, Dr. Martin said.
In terms of bad news, Priti R. Patel, MD, MPH, a medical officer with the Centers for Disease Control and Prevention, warned that dialysis clinics are still seeing HCV outbreaks. “It’s a continuing problem,” she said. “What we hear about at the CDC is the tip of the iceberg.”
The CDC says it received word of 21 HCV outbreaks of two or more cases in dialysis clinics during 2008-2017. These affected 102 patients, and more than 3,000 patients were notified that they were at risk and should be screened.
One dialysis clinic in Philadelphia had 18 cases of HCV during 2008-2013; they were blamed on “multiple lapses in infection control ... including hand hygiene and glove use, vascular access care, medication preparation, cleaning, and disinfection.”
“There should be no more than one case that has to happen for a facility to detect that it has a problem and identify a solution,” Dr. Patel said.
Since acute HCV can appear without symptoms, every dialysis patients should be tested for HCV antibodies, she added. “If it’s positive, confirm it. If confirmed, they should be informed of their infection status and have an evaluation for treatment.”
Dr. Martin reported consulting for Bristol-Myers Squibb and AbbVie and receiving research funding from Gilead, Bristol-Myers Squibb, AbbVie, and Merck. Dr. Patel reported no disclosures.
EXPERT ANALYSIS FROM KIDNEY WEEK 2018
FDA authorizes emergency use of rapid fingerstick test for Ebola
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.

The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
Rate of STIs is rising, and many U.S. teens are sexually active
ORLANDO – Consider point-of-care testing and treat potentially infected partners when diagnosing and treating adolescents for STIs, Diane M. Straub, MD, MPH, said at the annual meeting of the American Academy of Pediatrics.

In addition, adolescents are sometimes reluctant to disclose their full sexual history to their health care provider, which can complicate diagnosis and treatment, noted Dr. Straub, professor of pediatrics at the University of South Florida, Tampa. “That sometimes takes a few questions,” but can be achieved by asking the same questions in different ways and emphasizing the clinical importance of testing.
According to the 2017 Youth Risk Behavior Surveillance survey, 40% of adolescents reported ever having sexual intercourse, with 20% of 9th-grade, 36% of 10th-grade, 47% of 11th-grade, and 57% of 12th-grade students reporting they had sexual intercourse. By gender, 41% of adolescent males and 38% of adolescent females reported ever having sexual intercourse; by race, 39% of white, 41% of Hispanic, and 46% of black participants reported any sexual activity. Overall, 10% of adolescents said they had four or more partners, 3% said they had intercourse before age 13 years, 54% used a condom the last time they had intercourse, and 7% said they were raped.
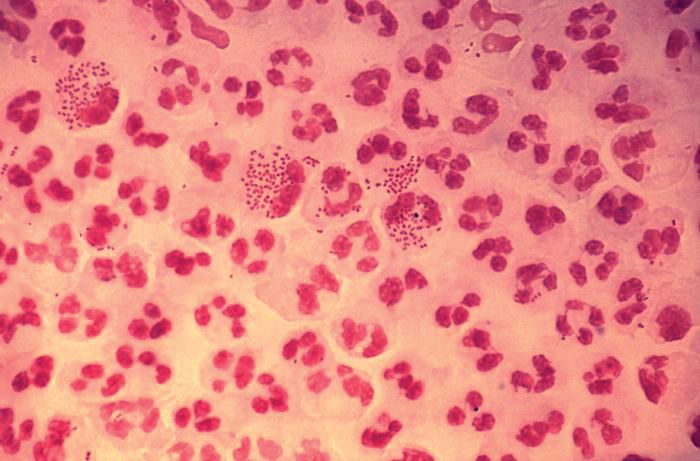
The rate of STIs in the United States is rising. There has been a sharp increase in the number of combined diagnoses of gonorrhea, syphilis, and chlamydia, with an increase from 1.8 million in 2013 to 2.3 million cases in 2017, according to the Centers for Disease Control and Prevention. During that same time period, gonorrhea increased 67% from 333,004 to 555,608 cases, syphilis (primary and secondary) rose 76% from 17,375 to 30,644 cases, and chlamydia increased 22% to 1.7 million cases.
According to a 2013 CDC infographic shown by Dr. Straub, young people in the United States aged 15-24 years old represent 27% of the total sexually active population but account for 50% of new STI cases each year. Persons in this population account for 70% of gonorrhea cases, 63% of chlamydia cases, 49% of human papillomavirus (HPV) cases, 45% of genital herpes cases, and 20% of syphilis cases.
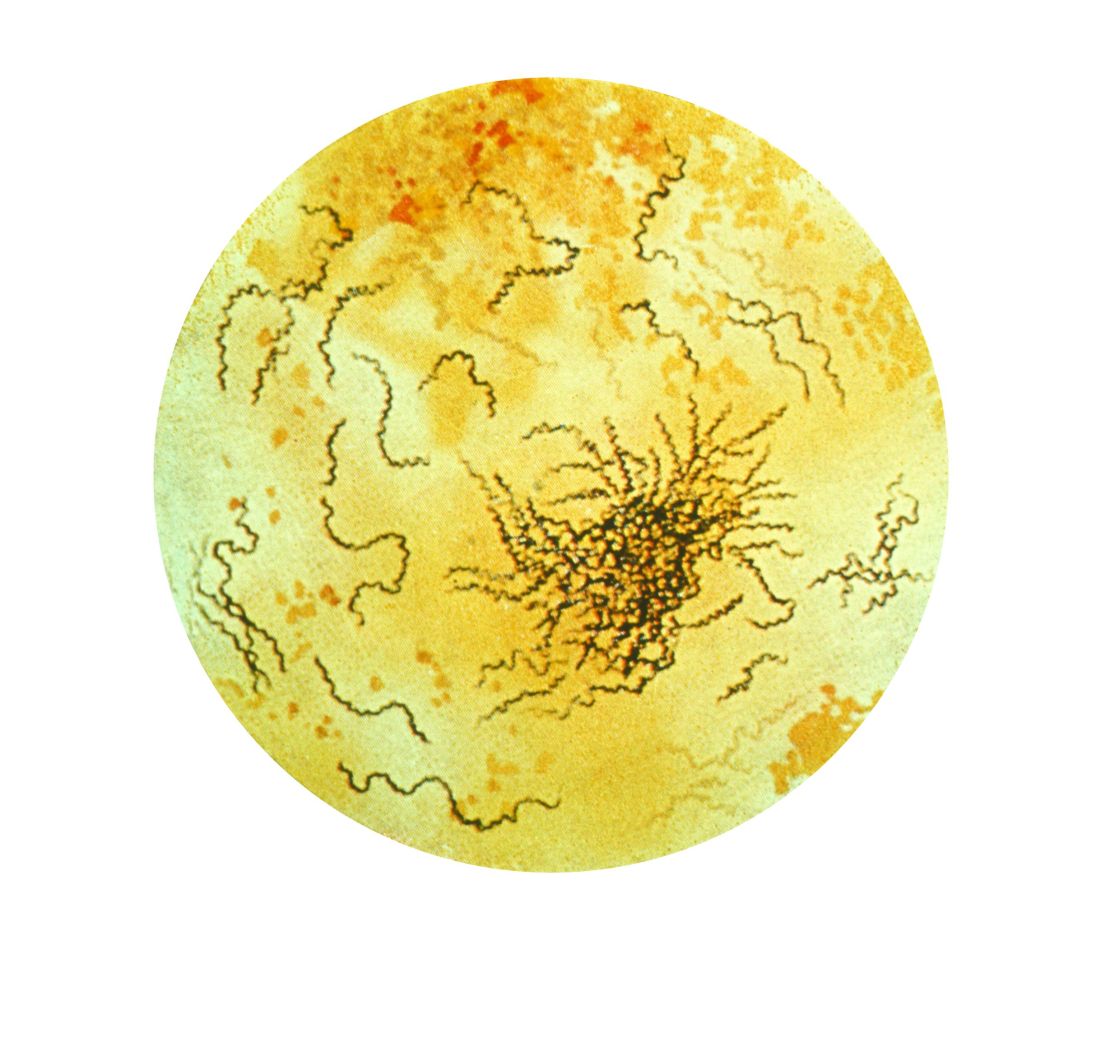
All sexually active females aged 25 years or younger should be screened for chlamydia and gonorrhea, as well as “at-risk” young men who have sex with men (YMSM), Dr. Straub said. All adolescent males and females aged over 13 years should be offered HIV screening, and HIV screening should be discussed “at least once.” And depending on how at risk each subpopulation is, health care providers should be have that conversation and offer screening multiple times.
Women who have sex with women (WSW) are a diverse population and should be treated based on their individual sexual identities, behaviors, and practices. “Most self-identified WSWs report having sex with men, so therefore adolescent WSWs and females with both male and female sex partners might be at increased risk for STIs, such as syphillis, chlamydia, and HPV as well as HIV, so you may want to adjust your screening accordingly,” she said.
Pregnant women, if at risk, should be screened for HIV, syphilis, hepatitis B, gonorrhea, and chlamydia.
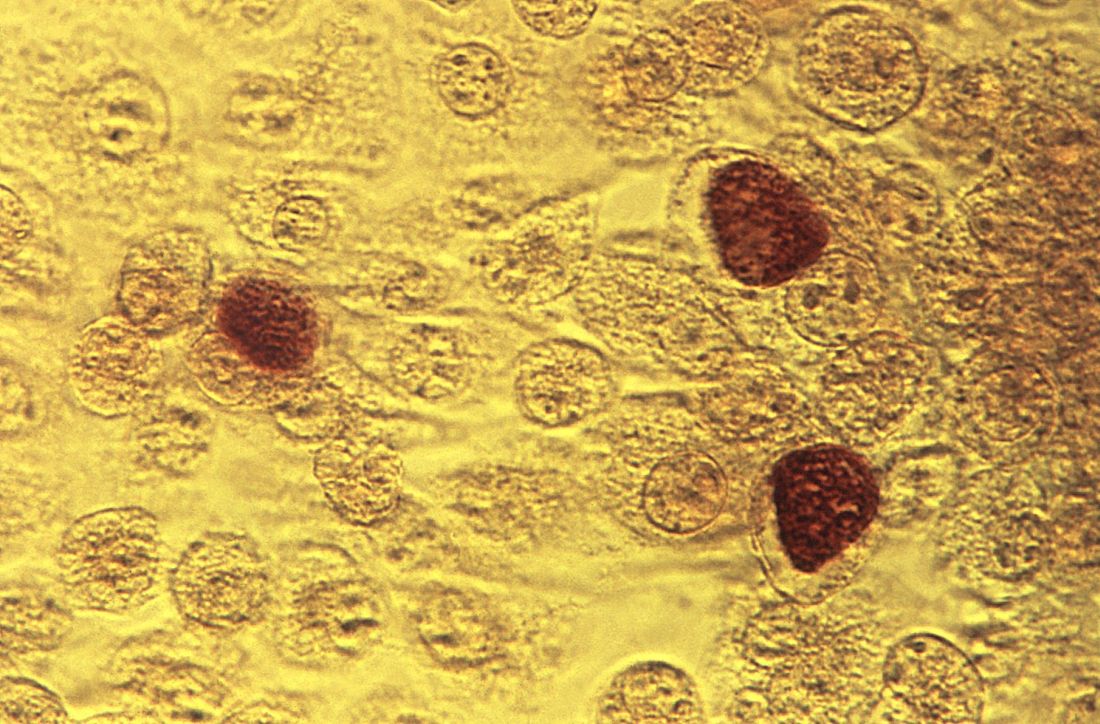
YMSM should have annual screenings for syphilis and HIV, screenings for chlamydia and gonorrhea by infection site; also consider herpes simplex virus serology and anal cytology in these patients, Dr. Straub said. They also should be screened for hepatitis B surface antigen, vaccinated for hepatitis A, hepatitis B and, if using drugs, screened* for hepatitis C virus.
Dr. Straub recommended licensed health care professionals who may treat minor patients review their state’s laws on minors and their legal ability to consent to treatment of STIs without the involvement of their parent or guardian, including disclosure of positive results and in the case of HIV care.
In places where index insured are allowed to find out about any services a beneficiary receives on their insurance, “this is a little problematic, because in some states, this is in direct conflict with the explanation of benefits requirement,” she said. “There are certain ways to get around that, but it’s really important for you to know what the statutes are where you’re practicing and where the breaches of confidentiality [are].”
Expedited partner therapy, or treating one or multiple partners of patients with an STI, is recommended for certain patients and infections, such as male partners of female patients with chlamydia and gonorrhea. While this is recommended less for YMSM because of a higher rate of concurrent infection, “if you have a young person who has partners who are unlikely to have access to care and get treated, it’s recommended you give that treatment to your index patient and to then treat their partners,” Dr. Straub said.
A recent and frequently updated resource on STI treatment can be found at the CDC website.
Dr. Straub reported no relevant conflicts of interest.
*This article was updated 1/11/19.
ORLANDO – Consider point-of-care testing and treat potentially infected partners when diagnosing and treating adolescents for STIs, Diane M. Straub, MD, MPH, said at the annual meeting of the American Academy of Pediatrics.
In addition, adolescents are sometimes reluctant to disclose their full sexual history to their health care provider, which can complicate diagnosis and treatment, noted Dr. Straub, professor of pediatrics at the University of South Florida, Tampa. “That sometimes takes a few questions,” but can be achieved by asking the same questions in different ways and emphasizing the clinical importance of testing.
According to the 2017 Youth Risk Behavior Surveillance survey, 40% of adolescents reported ever having sexual intercourse, with 20% of 9th-grade, 36% of 10th-grade, 47% of 11th-grade, and 57% of 12th-grade students reporting they had sexual intercourse. By gender, 41% of adolescent males and 38% of adolescent females reported ever having sexual intercourse; by race, 39% of white, 41% of Hispanic, and 46% of black participants reported any sexual activity. Overall, 10% of adolescents said they had four or more partners, 3% said they had intercourse before age 13 years, 54% used a condom the last time they had intercourse, and 7% said they were raped.
The rate of STIs in the United States is rising. There has been a sharp increase in the number of combined diagnoses of gonorrhea, syphilis, and chlamydia, with an increase from 1.8 million in 2013 to 2.3 million cases in 2017, according to the Centers for Disease Control and Prevention. During that same time period, gonorrhea increased 67% from 333,004 to 555,608 cases, syphilis (primary and secondary) rose 76% from 17,375 to 30,644 cases, and chlamydia increased 22% to 1.7 million cases.
According to a 2013 CDC infographic shown by Dr. Straub, young people in the United States aged 15-24 years old represent 27% of the total sexually active population but account for 50% of new STI cases each year. Persons in this population account for 70% of gonorrhea cases, 63% of chlamydia cases, 49% of human papillomavirus (HPV) cases, 45% of genital herpes cases, and 20% of syphilis cases.
All sexually active females aged 25 years or younger should be screened for chlamydia and gonorrhea, as well as “at-risk” young men who have sex with men (YMSM), Dr. Straub said. All adolescent males and females aged over 13 years should be offered HIV screening, and HIV screening should be discussed “at least once.” And depending on how at risk each subpopulation is, health care providers should be have that conversation and offer screening multiple times.
Women who have sex with women (WSW) are a diverse population and should be treated based on their individual sexual identities, behaviors, and practices. “Most self-identified WSWs report having sex with men, so therefore adolescent WSWs and females with both male and female sex partners might be at increased risk for STIs, such as syphillis, chlamydia, and HPV as well as HIV, so you may want to adjust your screening accordingly,” she said.
Pregnant women, if at risk, should be screened for HIV, syphilis, hepatitis B, gonorrhea, and chlamydia.
YMSM should have annual screenings for syphilis and HIV, screenings for chlamydia and gonorrhea by infection site; also consider herpes simplex virus serology and anal cytology in these patients, Dr. Straub said. They also should be screened for hepatitis B surface antigen, vaccinated for hepatitis A, hepatitis B and, if using drugs, screened* for hepatitis C virus.
Dr. Straub recommended licensed health care professionals who may treat minor patients review their state’s laws on minors and their legal ability to consent to treatment of STIs without the involvement of their parent or guardian, including disclosure of positive results and in the case of HIV care.
In places where index insured are allowed to find out about any services a beneficiary receives on their insurance, “this is a little problematic, because in some states, this is in direct conflict with the explanation of benefits requirement,” she said. “There are certain ways to get around that, but it’s really important for you to know what the statutes are where you’re practicing and where the breaches of confidentiality [are].”
Expedited partner therapy, or treating one or multiple partners of patients with an STI, is recommended for certain patients and infections, such as male partners of female patients with chlamydia and gonorrhea. While this is recommended less for YMSM because of a higher rate of concurrent infection, “if you have a young person who has partners who are unlikely to have access to care and get treated, it’s recommended you give that treatment to your index patient and to then treat their partners,” Dr. Straub said.
A recent and frequently updated resource on STI treatment can be found at the CDC website.
Dr. Straub reported no relevant conflicts of interest.
*This article was updated 1/11/19.
ORLANDO – Consider point-of-care testing and treat potentially infected partners when diagnosing and treating adolescents for STIs, Diane M. Straub, MD, MPH, said at the annual meeting of the American Academy of Pediatrics.
In addition, adolescents are sometimes reluctant to disclose their full sexual history to their health care provider, which can complicate diagnosis and treatment, noted Dr. Straub, professor of pediatrics at the University of South Florida, Tampa. “That sometimes takes a few questions,” but can be achieved by asking the same questions in different ways and emphasizing the clinical importance of testing.
According to the 2017 Youth Risk Behavior Surveillance survey, 40% of adolescents reported ever having sexual intercourse, with 20% of 9th-grade, 36% of 10th-grade, 47% of 11th-grade, and 57% of 12th-grade students reporting they had sexual intercourse. By gender, 41% of adolescent males and 38% of adolescent females reported ever having sexual intercourse; by race, 39% of white, 41% of Hispanic, and 46% of black participants reported any sexual activity. Overall, 10% of adolescents said they had four or more partners, 3% said they had intercourse before age 13 years, 54% used a condom the last time they had intercourse, and 7% said they were raped.
The rate of STIs in the United States is rising. There has been a sharp increase in the number of combined diagnoses of gonorrhea, syphilis, and chlamydia, with an increase from 1.8 million in 2013 to 2.3 million cases in 2017, according to the Centers for Disease Control and Prevention. During that same time period, gonorrhea increased 67% from 333,004 to 555,608 cases, syphilis (primary and secondary) rose 76% from 17,375 to 30,644 cases, and chlamydia increased 22% to 1.7 million cases.
According to a 2013 CDC infographic shown by Dr. Straub, young people in the United States aged 15-24 years old represent 27% of the total sexually active population but account for 50% of new STI cases each year. Persons in this population account for 70% of gonorrhea cases, 63% of chlamydia cases, 49% of human papillomavirus (HPV) cases, 45% of genital herpes cases, and 20% of syphilis cases.
All sexually active females aged 25 years or younger should be screened for chlamydia and gonorrhea, as well as “at-risk” young men who have sex with men (YMSM), Dr. Straub said. All adolescent males and females aged over 13 years should be offered HIV screening, and HIV screening should be discussed “at least once.” And depending on how at risk each subpopulation is, health care providers should be have that conversation and offer screening multiple times.
Women who have sex with women (WSW) are a diverse population and should be treated based on their individual sexual identities, behaviors, and practices. “Most self-identified WSWs report having sex with men, so therefore adolescent WSWs and females with both male and female sex partners might be at increased risk for STIs, such as syphillis, chlamydia, and HPV as well as HIV, so you may want to adjust your screening accordingly,” she said.
Pregnant women, if at risk, should be screened for HIV, syphilis, hepatitis B, gonorrhea, and chlamydia.
YMSM should have annual screenings for syphilis and HIV, screenings for chlamydia and gonorrhea by infection site; also consider herpes simplex virus serology and anal cytology in these patients, Dr. Straub said. They also should be screened for hepatitis B surface antigen, vaccinated for hepatitis A, hepatitis B and, if using drugs, screened* for hepatitis C virus.
Dr. Straub recommended licensed health care professionals who may treat minor patients review their state’s laws on minors and their legal ability to consent to treatment of STIs without the involvement of their parent or guardian, including disclosure of positive results and in the case of HIV care.
In places where index insured are allowed to find out about any services a beneficiary receives on their insurance, “this is a little problematic, because in some states, this is in direct conflict with the explanation of benefits requirement,” she said. “There are certain ways to get around that, but it’s really important for you to know what the statutes are where you’re practicing and where the breaches of confidentiality [are].”
Expedited partner therapy, or treating one or multiple partners of patients with an STI, is recommended for certain patients and infections, such as male partners of female patients with chlamydia and gonorrhea. While this is recommended less for YMSM because of a higher rate of concurrent infection, “if you have a young person who has partners who are unlikely to have access to care and get treated, it’s recommended you give that treatment to your index patient and to then treat their partners,” Dr. Straub said.
A recent and frequently updated resource on STI treatment can be found at the CDC website.
Dr. Straub reported no relevant conflicts of interest.
*This article was updated 1/11/19.
EXPERT ANALYSIS FROM AAP 18
Single-dose zoliflodacin successfully treats uncomplicated urogenital gonorrhea
A new antibiotic successfully treated uncomplicated urogenital and rectal gonococcal infections, but was not as effective as ceftriaxone in treating pharyngeal infections, according to the results of a randomized, phase 2 study.
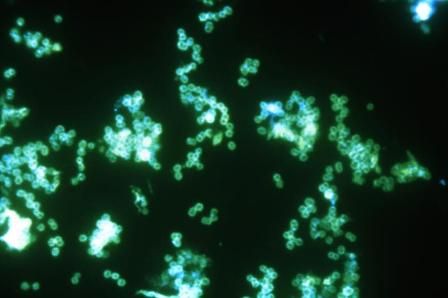
About 96% of patients with infection at urogenital sites had microbiologic cure with zoliflodacin, a novel antimicrobial agent that inhibits DNA biosynthesis. The cure rate was 100% for rectal infections, but was just 50%-82% for pharyngeal infections, though few participants in this study had infection at either of those sites.
The study investigators, led by Stephanie N. Taylor, MD, professor of medicine and microbiology at Louisiana State University, New Orleans, wrote that the need for new antimicrobial agents has been underscored by reports of multidrug-resistant Neisseria gonorrhoeae and the possibility of untreatable gonorrhea.
“This phase 2 trial creates equipoise for larger, more definitive studies of zoliflodacin,” Dr. Taylor and her coauthors wrote in the New England Journal of Medicine.
At this point, N. gonorrhoeae has developed resistance to every recommended antibiotic class, including cephalosporins and macrolides, they added.
Zoliflodacin (ETX0914) is an antimicrobial that has received fast-track designation from the Food and Drug Administration specifically for development as an oral treatment for gonococcal infections, the authors noted.
“The mechanism of action of zoliflodacin differs from currently available therapies in that it inhibits microbial biosynthesis by arresting the cleaved covalent gyrase complex and the formation of fused circular DNA required for biosynthesis,” they wrote.
Dr. Taylor and her colleagues studied single 2- and 3-gram doses of zoliflodacin in comparison with 500 mg of intramuscular ceftriaxone in 181 patients with uncomplicated urogenital gonorrhea enrolled in the open-label, randomized, phase 2 study between November 2014 and December 2015.
A total of 141 patients included in the microbiologic intention-to-treat analysis had confirmed positive urethral or cervical cultures. Cures were seen in 55 of 57 infections treated with 2 grams (96%) and 54 of 56 treated with 3 grams (96%) of zoliflodacin, and in 28 of 28 infections (100%) treated with ceftriaxone.
Of 15 confirmed rectal infections, 100% were cured, including 12 treated with zoliflodacin at 2 or 3 grams and 3 treated with ceftriaxone. Of 23 confirmed pharyngeal infections, cures were seen in 4 of 8 (50%) treated with 2 grams of zoliflodacin and 9 of 11 (82%) treated with 3 grams, compared with 4 of 4 cured (100%) with ceftriaxone.
That suggests zoliflodacin was not as effective as ceftriaxone in treating pharyngeal gonorrhea, which is generally considered more difficult to treat than infections at other sites, according to Dr. Taylor and her coauthors.
“Currently, this limitation has not curtailed recommendations for the use of drugs such as spectinomycin or fluoroquinolones for the treatment of gonorrhea,” they wrote.
The study was funded by the National Institutes of Health and Entasis Therapeutics. Dr. Taylor reported grants from the NIH during the study, and other disclosures related to a variety of pharma companies. Her coauthors reported disclosures related to AstraZeneca (parent company of Entasis, which is developing zoliflodacin) and other pharmaceutical companies.
SOURCE: Taylor SN et al. N Engl J Med. 2018 Nov 7; 379:1835-45.
This study represents a “step forward” in identifying new antimicrobial treatment options for patients with gonorrhea, according to Susan Blank, MD, and Demetre C. Daskalakis, MD.
“Given the challenges in clinical follow-up in this patient population, the single-dose regimen is promising,” Dr. Blank and Dr. Daskalakis wrote in a editorial.
While zoliflodacin has the potential to be an effective treatment for gonorrhea, its activity needs to be better defined, particularly in key anatomical sites of infection such as the pharynx, where limited activity was observed.
Progression of resistance of Neisseria gonorrhoeae is an “ever-present concern” given the history of the organism, the authors wrote.
“We are facing the real danger of multidrug-resistant, nearly untreatable gonorrhea,” they wrote. “To avoid untreatable cases of this high-incidence infection, we need to advance diagnostic technology and develop treatments with different mechanisms of action.”
Dr. Blank and Dr. Daskalakis are with the division of disease control in the New York City Department of Health and Mental Hygiene. Their editorial appears in the New England Journal of Medicine . Both reported having no conflicts of interest.
This study represents a “step forward” in identifying new antimicrobial treatment options for patients with gonorrhea, according to Susan Blank, MD, and Demetre C. Daskalakis, MD.
“Given the challenges in clinical follow-up in this patient population, the single-dose regimen is promising,” Dr. Blank and Dr. Daskalakis wrote in a editorial.
While zoliflodacin has the potential to be an effective treatment for gonorrhea, its activity needs to be better defined, particularly in key anatomical sites of infection such as the pharynx, where limited activity was observed.
Progression of resistance of Neisseria gonorrhoeae is an “ever-present concern” given the history of the organism, the authors wrote.
“We are facing the real danger of multidrug-resistant, nearly untreatable gonorrhea,” they wrote. “To avoid untreatable cases of this high-incidence infection, we need to advance diagnostic technology and develop treatments with different mechanisms of action.”
Dr. Blank and Dr. Daskalakis are with the division of disease control in the New York City Department of Health and Mental Hygiene. Their editorial appears in the New England Journal of Medicine . Both reported having no conflicts of interest.
This study represents a “step forward” in identifying new antimicrobial treatment options for patients with gonorrhea, according to Susan Blank, MD, and Demetre C. Daskalakis, MD.
“Given the challenges in clinical follow-up in this patient population, the single-dose regimen is promising,” Dr. Blank and Dr. Daskalakis wrote in a editorial.
While zoliflodacin has the potential to be an effective treatment for gonorrhea, its activity needs to be better defined, particularly in key anatomical sites of infection such as the pharynx, where limited activity was observed.
Progression of resistance of Neisseria gonorrhoeae is an “ever-present concern” given the history of the organism, the authors wrote.
“We are facing the real danger of multidrug-resistant, nearly untreatable gonorrhea,” they wrote. “To avoid untreatable cases of this high-incidence infection, we need to advance diagnostic technology and develop treatments with different mechanisms of action.”
Dr. Blank and Dr. Daskalakis are with the division of disease control in the New York City Department of Health and Mental Hygiene. Their editorial appears in the New England Journal of Medicine . Both reported having no conflicts of interest.
A new antibiotic successfully treated uncomplicated urogenital and rectal gonococcal infections, but was not as effective as ceftriaxone in treating pharyngeal infections, according to the results of a randomized, phase 2 study.
About 96% of patients with infection at urogenital sites had microbiologic cure with zoliflodacin, a novel antimicrobial agent that inhibits DNA biosynthesis. The cure rate was 100% for rectal infections, but was just 50%-82% for pharyngeal infections, though few participants in this study had infection at either of those sites.
The study investigators, led by Stephanie N. Taylor, MD, professor of medicine and microbiology at Louisiana State University, New Orleans, wrote that the need for new antimicrobial agents has been underscored by reports of multidrug-resistant Neisseria gonorrhoeae and the possibility of untreatable gonorrhea.
“This phase 2 trial creates equipoise for larger, more definitive studies of zoliflodacin,” Dr. Taylor and her coauthors wrote in the New England Journal of Medicine.
At this point, N. gonorrhoeae has developed resistance to every recommended antibiotic class, including cephalosporins and macrolides, they added.
Zoliflodacin (ETX0914) is an antimicrobial that has received fast-track designation from the Food and Drug Administration specifically for development as an oral treatment for gonococcal infections, the authors noted.
“The mechanism of action of zoliflodacin differs from currently available therapies in that it inhibits microbial biosynthesis by arresting the cleaved covalent gyrase complex and the formation of fused circular DNA required for biosynthesis,” they wrote.
Dr. Taylor and her colleagues studied single 2- and 3-gram doses of zoliflodacin in comparison with 500 mg of intramuscular ceftriaxone in 181 patients with uncomplicated urogenital gonorrhea enrolled in the open-label, randomized, phase 2 study between November 2014 and December 2015.
A total of 141 patients included in the microbiologic intention-to-treat analysis had confirmed positive urethral or cervical cultures. Cures were seen in 55 of 57 infections treated with 2 grams (96%) and 54 of 56 treated with 3 grams (96%) of zoliflodacin, and in 28 of 28 infections (100%) treated with ceftriaxone.
Of 15 confirmed rectal infections, 100% were cured, including 12 treated with zoliflodacin at 2 or 3 grams and 3 treated with ceftriaxone. Of 23 confirmed pharyngeal infections, cures were seen in 4 of 8 (50%) treated with 2 grams of zoliflodacin and 9 of 11 (82%) treated with 3 grams, compared with 4 of 4 cured (100%) with ceftriaxone.
That suggests zoliflodacin was not as effective as ceftriaxone in treating pharyngeal gonorrhea, which is generally considered more difficult to treat than infections at other sites, according to Dr. Taylor and her coauthors.
“Currently, this limitation has not curtailed recommendations for the use of drugs such as spectinomycin or fluoroquinolones for the treatment of gonorrhea,” they wrote.
The study was funded by the National Institutes of Health and Entasis Therapeutics. Dr. Taylor reported grants from the NIH during the study, and other disclosures related to a variety of pharma companies. Her coauthors reported disclosures related to AstraZeneca (parent company of Entasis, which is developing zoliflodacin) and other pharmaceutical companies.
SOURCE: Taylor SN et al. N Engl J Med. 2018 Nov 7; 379:1835-45.
A new antibiotic successfully treated uncomplicated urogenital and rectal gonococcal infections, but was not as effective as ceftriaxone in treating pharyngeal infections, according to the results of a randomized, phase 2 study.
About 96% of patients with infection at urogenital sites had microbiologic cure with zoliflodacin, a novel antimicrobial agent that inhibits DNA biosynthesis. The cure rate was 100% for rectal infections, but was just 50%-82% for pharyngeal infections, though few participants in this study had infection at either of those sites.
The study investigators, led by Stephanie N. Taylor, MD, professor of medicine and microbiology at Louisiana State University, New Orleans, wrote that the need for new antimicrobial agents has been underscored by reports of multidrug-resistant Neisseria gonorrhoeae and the possibility of untreatable gonorrhea.
“This phase 2 trial creates equipoise for larger, more definitive studies of zoliflodacin,” Dr. Taylor and her coauthors wrote in the New England Journal of Medicine.
At this point, N. gonorrhoeae has developed resistance to every recommended antibiotic class, including cephalosporins and macrolides, they added.
Zoliflodacin (ETX0914) is an antimicrobial that has received fast-track designation from the Food and Drug Administration specifically for development as an oral treatment for gonococcal infections, the authors noted.
“The mechanism of action of zoliflodacin differs from currently available therapies in that it inhibits microbial biosynthesis by arresting the cleaved covalent gyrase complex and the formation of fused circular DNA required for biosynthesis,” they wrote.
Dr. Taylor and her colleagues studied single 2- and 3-gram doses of zoliflodacin in comparison with 500 mg of intramuscular ceftriaxone in 181 patients with uncomplicated urogenital gonorrhea enrolled in the open-label, randomized, phase 2 study between November 2014 and December 2015.
A total of 141 patients included in the microbiologic intention-to-treat analysis had confirmed positive urethral or cervical cultures. Cures were seen in 55 of 57 infections treated with 2 grams (96%) and 54 of 56 treated with 3 grams (96%) of zoliflodacin, and in 28 of 28 infections (100%) treated with ceftriaxone.
Of 15 confirmed rectal infections, 100% were cured, including 12 treated with zoliflodacin at 2 or 3 grams and 3 treated with ceftriaxone. Of 23 confirmed pharyngeal infections, cures were seen in 4 of 8 (50%) treated with 2 grams of zoliflodacin and 9 of 11 (82%) treated with 3 grams, compared with 4 of 4 cured (100%) with ceftriaxone.
That suggests zoliflodacin was not as effective as ceftriaxone in treating pharyngeal gonorrhea, which is generally considered more difficult to treat than infections at other sites, according to Dr. Taylor and her coauthors.
“Currently, this limitation has not curtailed recommendations for the use of drugs such as spectinomycin or fluoroquinolones for the treatment of gonorrhea,” they wrote.
The study was funded by the National Institutes of Health and Entasis Therapeutics. Dr. Taylor reported grants from the NIH during the study, and other disclosures related to a variety of pharma companies. Her coauthors reported disclosures related to AstraZeneca (parent company of Entasis, which is developing zoliflodacin) and other pharmaceutical companies.
SOURCE: Taylor SN et al. N Engl J Med. 2018 Nov 7; 379:1835-45.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Cure rates of 96% and 100% were reported for urethral/cervical and rectal infections, respectively, and ranged from 50% to 82% in pharyngeal infections.
Study details: A randomized, open-label, phase 2 study including 181 patients with uncomplicated urogenital gonorrhea who received zoliflodacin or ceftriaxone.
Disclosures: The study authors reported disclosures related to AstraZeneca (parent company of Entasis Therapeutics, which is developing zoliflodacin) and other pharmaceutical companies.
Source: Taylor SN et al. N Engl J Med. 2018 Nov 7; 379:1835-45.
Discussing immunization with vaccine-hesitant parents requires caring, individualized approach
ORLANDO – Putting parents at ease, making vaccination the default option during discussions, appealing to their identity as a good parent, and focusing on positive word choice during discussions are the techniques two pediatricians have recommended using to get vaccine-hesitant parents to immunize their children.

“Your goal is to get parents to immunize their kids,” Katrina Saba, MD, of the Permanente Medical Group in Oakland, Calif., said during an interactive group panel at the annual meeting of the American Academy of Pediatrics. “Our goal is not to win a debate. You don’t have to correct every mistaken idea.”
“And really importantly, as we know, belief trumps science,” she added. “Their belief is so much stronger than our proof, and their belief will not be changed by evidence.”
Many parents who are vaccine- hesitant also belong to a social network that forms or reinforces their beliefs, and attacking those beliefs is the same as attacking their identity, Dr. Saba noted. “When you attack someone’s identity, you are immediately seen as not like them, and if you’re not like them, you’ve lost your strength in persuading them.”
Dr. Saba; Kenneth Hempstead, MD; and other pediatricians and educators in the Permanente Medical Group developed a framework for pediatricians and educators to talk with their patients about immunization at their center after California passed a law in 2013 that required health care professionals to discuss vaccines with patients and sign off that they had that discussion.
“We felt that, if we were going to be by law required to have that discussion, maybe we needed some new tools to have [the discussion] more effectively,” Dr. Saba said. “Because clearly, [what we were doing ] wasn’t working or there wouldn’t have been a need for that law.”

Dr. Hempstead explained the concerns of three major categories of vaccine-hesitant parents: those patients who are unsure of whether they should vaccinate, parents who wish to delay vaccination, and parents who refuse vaccination of their children.
Each parent requires a different approach for discussing the importance of vaccination based on their level of vaccine hesitancy, he said. For parents who are unsure, they may require general information about the safety and importance of vaccines.
Parents who delay immunization may have less trust in vaccines, have done research in their own social networks, and may present alternatives to a standard immunization schedule or want to omit certain vaccines from their child’s immunization schedule, he noted. Using the analogy of a car seat is one approach to identify the importance of vaccination to these parents: “Waiting to give the shots is like putting your baby in the car seat after you’ve already arrived at the store – the protection isn’t there for the most important part of the journey!”
In cases where parents refuse vaccination, you should not expect to change a parent’s mind in a single visit, but instead focus on building the patient-provider bond. However, presenting information the parent may have already seen, such as vaccination data from the Food and Drug Administration or Centers for Disease Control and Prevention, may alienate parents who identify with groups that share vaccine-hesitant viewpoints and erode your ability to persuade a parent’s intent to vaccinate.
A study by Nyhan et al. randomized parents to receive one of four pieces of interventions about the MMR vaccine: information from the CDC explaining the lack of evidence linking autism and the vaccine, information about the dangers of the diseases prevented by the vaccine, images of children who have had diseases prevented by the vaccine, and a “dramatic narrative” from a CDC fact sheet about a child who nearly died of measles. The researchers found no informational intervention helped in persuading to vaccinate in parents who had the “least favorable” attitudes toward the vaccine. And in the case of the dramatic narrative, there was an increased misperception about the MMR vaccine (Pediatrics. 2014;133[4]:e835-e842).
Dr. Hempstead and Dr. Saba outlined four rhetorical devices to include in conversations with patients about vaccination: cognitive ease, natural assumption, appeal to identity, and using advantageous terms.
Cognitive ease
Cognitive ease means creating an environment in which the patient is relaxed, comfortable, and more likely to be agreeable. Recognize when the tone shifts, and strive to maintain this calm and comfortable environment throughout the discussion. “If your blood pressure is coming up, that means theirs is, too,” Dr. Hempstead said.
Natural assumption
How you are offering the vaccination also matters, he added. Rather than asking whether a patient wants to vaccinate (“Have you thought about your flu vaccine this year?”), instead frame the discussion with vaccination as the default option (“Is your child due for a flu vaccination this year? Yes, he is. Let’s get that taken care of today”). Equating inaction with vaccination prevents the risk fallacy phenomenon from occurring in which, when given multiple options, people give equal weight to each option and may choose not to vaccinate, Dr. Hempstead noted.
Dr. Saba cited research that backed this approach. In a study by Opel et al., using a “presumptive” approach instead of a “participatory” approach when discussing a provider’s recommendation to vaccinate helped: The presumptive conversations had an odds ratio of 17.5, compared with the participatory approach. In cases in which parents resisted the provider’s recommendations, 50% of providers persisted with their original recommendations, and 47% of parents who initially resisted the recommendations agreed to vaccinate (Pediatrics. 2013;132[6]:1037-46).
Appeal to identity
Another strategy to use is appealing to the patient’s identity as a good parent and link the concept of vaccination with the good parent identity. Forging a new common identity with the parents through common beliefs – such as recognizing that networks to which parents belong are an important part of his or her identify – and reemphasizing the mutual desire to protect the child is another strategy.
Using advantageous terms
Positive terms, such as “protection,” “health,” “safety,” and “what’s best,” are much better words to use in conversation with parents and have more staying power than negative terms, like “autism” and “side effects,” Dr. Hempstead said.
“Stay with positive messaging,” he said. “Immediately coming back to the positive impact of this vaccine, why we care so much, why we’re doing this vaccine, is absolutely critical.”
Dr. Hempstead and Dr. Saba reported no relevant conflicts of interest.
ORLANDO – Putting parents at ease, making vaccination the default option during discussions, appealing to their identity as a good parent, and focusing on positive word choice during discussions are the techniques two pediatricians have recommended using to get vaccine-hesitant parents to immunize their children.
“Your goal is to get parents to immunize their kids,” Katrina Saba, MD, of the Permanente Medical Group in Oakland, Calif., said during an interactive group panel at the annual meeting of the American Academy of Pediatrics. “Our goal is not to win a debate. You don’t have to correct every mistaken idea.”
“And really importantly, as we know, belief trumps science,” she added. “Their belief is so much stronger than our proof, and their belief will not be changed by evidence.”
Many parents who are vaccine- hesitant also belong to a social network that forms or reinforces their beliefs, and attacking those beliefs is the same as attacking their identity, Dr. Saba noted. “When you attack someone’s identity, you are immediately seen as not like them, and if you’re not like them, you’ve lost your strength in persuading them.”
Dr. Saba; Kenneth Hempstead, MD; and other pediatricians and educators in the Permanente Medical Group developed a framework for pediatricians and educators to talk with their patients about immunization at their center after California passed a law in 2013 that required health care professionals to discuss vaccines with patients and sign off that they had that discussion.
“We felt that, if we were going to be by law required to have that discussion, maybe we needed some new tools to have [the discussion] more effectively,” Dr. Saba said. “Because clearly, [what we were doing ] wasn’t working or there wouldn’t have been a need for that law.”
Dr. Hempstead explained the concerns of three major categories of vaccine-hesitant parents: those patients who are unsure of whether they should vaccinate, parents who wish to delay vaccination, and parents who refuse vaccination of their children.
Each parent requires a different approach for discussing the importance of vaccination based on their level of vaccine hesitancy, he said. For parents who are unsure, they may require general information about the safety and importance of vaccines.
Parents who delay immunization may have less trust in vaccines, have done research in their own social networks, and may present alternatives to a standard immunization schedule or want to omit certain vaccines from their child’s immunization schedule, he noted. Using the analogy of a car seat is one approach to identify the importance of vaccination to these parents: “Waiting to give the shots is like putting your baby in the car seat after you’ve already arrived at the store – the protection isn’t there for the most important part of the journey!”
In cases where parents refuse vaccination, you should not expect to change a parent’s mind in a single visit, but instead focus on building the patient-provider bond. However, presenting information the parent may have already seen, such as vaccination data from the Food and Drug Administration or Centers for Disease Control and Prevention, may alienate parents who identify with groups that share vaccine-hesitant viewpoints and erode your ability to persuade a parent’s intent to vaccinate.
A study by Nyhan et al. randomized parents to receive one of four pieces of interventions about the MMR vaccine: information from the CDC explaining the lack of evidence linking autism and the vaccine, information about the dangers of the diseases prevented by the vaccine, images of children who have had diseases prevented by the vaccine, and a “dramatic narrative” from a CDC fact sheet about a child who nearly died of measles. The researchers found no informational intervention helped in persuading to vaccinate in parents who had the “least favorable” attitudes toward the vaccine. And in the case of the dramatic narrative, there was an increased misperception about the MMR vaccine (Pediatrics. 2014;133[4]:e835-e842).
Dr. Hempstead and Dr. Saba outlined four rhetorical devices to include in conversations with patients about vaccination: cognitive ease, natural assumption, appeal to identity, and using advantageous terms.
Cognitive ease
Cognitive ease means creating an environment in which the patient is relaxed, comfortable, and more likely to be agreeable. Recognize when the tone shifts, and strive to maintain this calm and comfortable environment throughout the discussion. “If your blood pressure is coming up, that means theirs is, too,” Dr. Hempstead said.
Natural assumption
How you are offering the vaccination also matters, he added. Rather than asking whether a patient wants to vaccinate (“Have you thought about your flu vaccine this year?”), instead frame the discussion with vaccination as the default option (“Is your child due for a flu vaccination this year? Yes, he is. Let’s get that taken care of today”). Equating inaction with vaccination prevents the risk fallacy phenomenon from occurring in which, when given multiple options, people give equal weight to each option and may choose not to vaccinate, Dr. Hempstead noted.
Dr. Saba cited research that backed this approach. In a study by Opel et al., using a “presumptive” approach instead of a “participatory” approach when discussing a provider’s recommendation to vaccinate helped: The presumptive conversations had an odds ratio of 17.5, compared with the participatory approach. In cases in which parents resisted the provider’s recommendations, 50% of providers persisted with their original recommendations, and 47% of parents who initially resisted the recommendations agreed to vaccinate (Pediatrics. 2013;132[6]:1037-46).
Appeal to identity
Another strategy to use is appealing to the patient’s identity as a good parent and link the concept of vaccination with the good parent identity. Forging a new common identity with the parents through common beliefs – such as recognizing that networks to which parents belong are an important part of his or her identify – and reemphasizing the mutual desire to protect the child is another strategy.
Using advantageous terms
Positive terms, such as “protection,” “health,” “safety,” and “what’s best,” are much better words to use in conversation with parents and have more staying power than negative terms, like “autism” and “side effects,” Dr. Hempstead said.
“Stay with positive messaging,” he said. “Immediately coming back to the positive impact of this vaccine, why we care so much, why we’re doing this vaccine, is absolutely critical.”
Dr. Hempstead and Dr. Saba reported no relevant conflicts of interest.
ORLANDO – Putting parents at ease, making vaccination the default option during discussions, appealing to their identity as a good parent, and focusing on positive word choice during discussions are the techniques two pediatricians have recommended using to get vaccine-hesitant parents to immunize their children.
“Your goal is to get parents to immunize their kids,” Katrina Saba, MD, of the Permanente Medical Group in Oakland, Calif., said during an interactive group panel at the annual meeting of the American Academy of Pediatrics. “Our goal is not to win a debate. You don’t have to correct every mistaken idea.”
“And really importantly, as we know, belief trumps science,” she added. “Their belief is so much stronger than our proof, and their belief will not be changed by evidence.”
Many parents who are vaccine- hesitant also belong to a social network that forms or reinforces their beliefs, and attacking those beliefs is the same as attacking their identity, Dr. Saba noted. “When you attack someone’s identity, you are immediately seen as not like them, and if you’re not like them, you’ve lost your strength in persuading them.”
Dr. Saba; Kenneth Hempstead, MD; and other pediatricians and educators in the Permanente Medical Group developed a framework for pediatricians and educators to talk with their patients about immunization at their center after California passed a law in 2013 that required health care professionals to discuss vaccines with patients and sign off that they had that discussion.
“We felt that, if we were going to be by law required to have that discussion, maybe we needed some new tools to have [the discussion] more effectively,” Dr. Saba said. “Because clearly, [what we were doing ] wasn’t working or there wouldn’t have been a need for that law.”
Dr. Hempstead explained the concerns of three major categories of vaccine-hesitant parents: those patients who are unsure of whether they should vaccinate, parents who wish to delay vaccination, and parents who refuse vaccination of their children.
Each parent requires a different approach for discussing the importance of vaccination based on their level of vaccine hesitancy, he said. For parents who are unsure, they may require general information about the safety and importance of vaccines.
Parents who delay immunization may have less trust in vaccines, have done research in their own social networks, and may present alternatives to a standard immunization schedule or want to omit certain vaccines from their child’s immunization schedule, he noted. Using the analogy of a car seat is one approach to identify the importance of vaccination to these parents: “Waiting to give the shots is like putting your baby in the car seat after you’ve already arrived at the store – the protection isn’t there for the most important part of the journey!”
In cases where parents refuse vaccination, you should not expect to change a parent’s mind in a single visit, but instead focus on building the patient-provider bond. However, presenting information the parent may have already seen, such as vaccination data from the Food and Drug Administration or Centers for Disease Control and Prevention, may alienate parents who identify with groups that share vaccine-hesitant viewpoints and erode your ability to persuade a parent’s intent to vaccinate.
A study by Nyhan et al. randomized parents to receive one of four pieces of interventions about the MMR vaccine: information from the CDC explaining the lack of evidence linking autism and the vaccine, information about the dangers of the diseases prevented by the vaccine, images of children who have had diseases prevented by the vaccine, and a “dramatic narrative” from a CDC fact sheet about a child who nearly died of measles. The researchers found no informational intervention helped in persuading to vaccinate in parents who had the “least favorable” attitudes toward the vaccine. And in the case of the dramatic narrative, there was an increased misperception about the MMR vaccine (Pediatrics. 2014;133[4]:e835-e842).
Dr. Hempstead and Dr. Saba outlined four rhetorical devices to include in conversations with patients about vaccination: cognitive ease, natural assumption, appeal to identity, and using advantageous terms.
Cognitive ease
Cognitive ease means creating an environment in which the patient is relaxed, comfortable, and more likely to be agreeable. Recognize when the tone shifts, and strive to maintain this calm and comfortable environment throughout the discussion. “If your blood pressure is coming up, that means theirs is, too,” Dr. Hempstead said.
Natural assumption
How you are offering the vaccination also matters, he added. Rather than asking whether a patient wants to vaccinate (“Have you thought about your flu vaccine this year?”), instead frame the discussion with vaccination as the default option (“Is your child due for a flu vaccination this year? Yes, he is. Let’s get that taken care of today”). Equating inaction with vaccination prevents the risk fallacy phenomenon from occurring in which, when given multiple options, people give equal weight to each option and may choose not to vaccinate, Dr. Hempstead noted.
Dr. Saba cited research that backed this approach. In a study by Opel et al., using a “presumptive” approach instead of a “participatory” approach when discussing a provider’s recommendation to vaccinate helped: The presumptive conversations had an odds ratio of 17.5, compared with the participatory approach. In cases in which parents resisted the provider’s recommendations, 50% of providers persisted with their original recommendations, and 47% of parents who initially resisted the recommendations agreed to vaccinate (Pediatrics. 2013;132[6]:1037-46).
Appeal to identity
Another strategy to use is appealing to the patient’s identity as a good parent and link the concept of vaccination with the good parent identity. Forging a new common identity with the parents through common beliefs – such as recognizing that networks to which parents belong are an important part of his or her identify – and reemphasizing the mutual desire to protect the child is another strategy.
Using advantageous terms
Positive terms, such as “protection,” “health,” “safety,” and “what’s best,” are much better words to use in conversation with parents and have more staying power than negative terms, like “autism” and “side effects,” Dr. Hempstead said.
“Stay with positive messaging,” he said. “Immediately coming back to the positive impact of this vaccine, why we care so much, why we’re doing this vaccine, is absolutely critical.”
Dr. Hempstead and Dr. Saba reported no relevant conflicts of interest.
EXPERT ANALYSIS FROM AAP 18
Study confirms advice to halt methotrexate when giving flu vaccine to RA patients
CHICAGO – Discontinuing methotrexate for 2 weeks in patients with RA starting the day they receive the seasonal influenza vaccine significantly improves the vaccine’s immunogenicity without aggravating disease activity, Kevin L. Winthrop, MD, reported at the annual meeting of the American College of Rheumatology.

“I think this is potentially clinically practice changing because now there are two studies showing the same thing,” said Dr. Winthrop, a professor of public health and preventive medicine at Oregon Health & Science University, Portland.
Based upon these prospective randomized studies, which he conducted together with investigators at Seoul National University in South Korea, initiating a 2-week halt of methotrexate on the day the influenza vaccine is given to patients with RA is now his routine practice, and he recommends other physicians do the same.
The first prospective, randomized trial included 199 RA patients on stable doses of methotrexate who were assigned to one of four groups in conjunction with seasonal influenza vaccination. One group continued their methotrexate as usual, the second stopped the drug for 1 month prior to vaccination and then restarted it at the time of vaccination, the third group halted methotrexate for 2 weeks before and 2 weeks after vaccination, and the fourth suspended methotrexate for 4 weeks starting on the day they got their flu shot. Everyone received trivalent influenza vaccine containing H1N1, H3N2, and B/Yamagata.
The lowest rate of satisfactory vaccine response as defined by at least a 300% titer increase 1 month after vaccination occurred in the group that continued their methotrexate as usual. The group that halted the drug for 2 weeks before and 2 weeks after influenza vaccination had a 51% satisfactory vaccine response against all three antigens, compared with a 31.5% rate in the methotrexate-as-usual group. RA flare rates ranged from 21% to 39% across the four study arms, differences that weren’t statistically significant (Ann Rheum Dis. 2017 Sep;76[9]:1559-65).
Next Dr. Winthrop and his colleagues conducted a confirmatory prospective, multicenter, randomized trial in which they sought to nail down the optimal duration and timing of methotrexate discontinuation. A total of 320 RA patients on stable doses of methotrexate were assigned to halt the drug for 2 weeks starting at the time they received a quadrivalent seasonal influenza vaccine containing H1N1, H3N2, B/Yamagata, and B/Victoria strains, or to continue their methotrexate throughout.
A satisfactory vaccine response was achieved in 75.5% of the group that discontinued the drug, significantly better than the 54.5% rate in the methotrexate continuers. The absolute difference in seroprotection was greater in patients who halted their methotrexate for 2 weeks after vaccination for all four antigens: an absolute 11% difference for H1N1, 16% for H3N2, 12% for B/Yamagata, and 15% for B/Victoria (Ann Rheum Dis. 2018 Jun;77[6]:898-904).
“It does seem to be a nice strategy. The percentage of people who flared their RA during their 2 weeks off methotrexate was very low, so there seems to be a good reason to do this,” according to Dr. Winthrop.
Some rheumatologists he has spoken with initially balked at the plausibility of the results.
“I had the same thought about these studies: It doesn’t make sense to me in terms of how methotrexate works, and why we would see this effect acutely by stopping methotrexate for just 2 weeks?” he said.
But then a coinvestigator drilled down deeper into the data and came up with the explanation: The benefit in terms of enhanced flu vaccine immunogenicity through temporary withholding of methotrexate was confined to the subgroup of RA patients with high baseline levels of B-cell activation factor (BAFF). In contrast, withholding methotrexate didn’t affect the vaccine response in patients with low or normal baseline BAFF (Ann Rheum Dis. 2018 Oct 8. doi: 10.1136/annrheumdis-2018-214025).
“I don’t know how to check anyone’s BAFF levels. I don’t think there’s a commercial test out there. But this does help explain why we saw this observation. So I think I would still hold everyone’s methotrexate for 2 weeks. That’s how I approach it. And they may get benefit from it, and they may not,” he said.
Dr. Winthrop reported having no financial conflicts regarding the study, which was funded by GC Pharma.
CHICAGO – Discontinuing methotrexate for 2 weeks in patients with RA starting the day they receive the seasonal influenza vaccine significantly improves the vaccine’s immunogenicity without aggravating disease activity, Kevin L. Winthrop, MD, reported at the annual meeting of the American College of Rheumatology.
“I think this is potentially clinically practice changing because now there are two studies showing the same thing,” said Dr. Winthrop, a professor of public health and preventive medicine at Oregon Health & Science University, Portland.
Based upon these prospective randomized studies, which he conducted together with investigators at Seoul National University in South Korea, initiating a 2-week halt of methotrexate on the day the influenza vaccine is given to patients with RA is now his routine practice, and he recommends other physicians do the same.
The first prospective, randomized trial included 199 RA patients on stable doses of methotrexate who were assigned to one of four groups in conjunction with seasonal influenza vaccination. One group continued their methotrexate as usual, the second stopped the drug for 1 month prior to vaccination and then restarted it at the time of vaccination, the third group halted methotrexate for 2 weeks before and 2 weeks after vaccination, and the fourth suspended methotrexate for 4 weeks starting on the day they got their flu shot. Everyone received trivalent influenza vaccine containing H1N1, H3N2, and B/Yamagata.
The lowest rate of satisfactory vaccine response as defined by at least a 300% titer increase 1 month after vaccination occurred in the group that continued their methotrexate as usual. The group that halted the drug for 2 weeks before and 2 weeks after influenza vaccination had a 51% satisfactory vaccine response against all three antigens, compared with a 31.5% rate in the methotrexate-as-usual group. RA flare rates ranged from 21% to 39% across the four study arms, differences that weren’t statistically significant (Ann Rheum Dis. 2017 Sep;76[9]:1559-65).
Next Dr. Winthrop and his colleagues conducted a confirmatory prospective, multicenter, randomized trial in which they sought to nail down the optimal duration and timing of methotrexate discontinuation. A total of 320 RA patients on stable doses of methotrexate were assigned to halt the drug for 2 weeks starting at the time they received a quadrivalent seasonal influenza vaccine containing H1N1, H3N2, B/Yamagata, and B/Victoria strains, or to continue their methotrexate throughout.
A satisfactory vaccine response was achieved in 75.5% of the group that discontinued the drug, significantly better than the 54.5% rate in the methotrexate continuers. The absolute difference in seroprotection was greater in patients who halted their methotrexate for 2 weeks after vaccination for all four antigens: an absolute 11% difference for H1N1, 16% for H3N2, 12% for B/Yamagata, and 15% for B/Victoria (Ann Rheum Dis. 2018 Jun;77[6]:898-904).
“It does seem to be a nice strategy. The percentage of people who flared their RA during their 2 weeks off methotrexate was very low, so there seems to be a good reason to do this,” according to Dr. Winthrop.
Some rheumatologists he has spoken with initially balked at the plausibility of the results.
“I had the same thought about these studies: It doesn’t make sense to me in terms of how methotrexate works, and why we would see this effect acutely by stopping methotrexate for just 2 weeks?” he said.
But then a coinvestigator drilled down deeper into the data and came up with the explanation: The benefit in terms of enhanced flu vaccine immunogenicity through temporary withholding of methotrexate was confined to the subgroup of RA patients with high baseline levels of B-cell activation factor (BAFF). In contrast, withholding methotrexate didn’t affect the vaccine response in patients with low or normal baseline BAFF (Ann Rheum Dis. 2018 Oct 8. doi: 10.1136/annrheumdis-2018-214025).
“I don’t know how to check anyone’s BAFF levels. I don’t think there’s a commercial test out there. But this does help explain why we saw this observation. So I think I would still hold everyone’s methotrexate for 2 weeks. That’s how I approach it. And they may get benefit from it, and they may not,” he said.
Dr. Winthrop reported having no financial conflicts regarding the study, which was funded by GC Pharma.
CHICAGO – Discontinuing methotrexate for 2 weeks in patients with RA starting the day they receive the seasonal influenza vaccine significantly improves the vaccine’s immunogenicity without aggravating disease activity, Kevin L. Winthrop, MD, reported at the annual meeting of the American College of Rheumatology.
“I think this is potentially clinically practice changing because now there are two studies showing the same thing,” said Dr. Winthrop, a professor of public health and preventive medicine at Oregon Health & Science University, Portland.
Based upon these prospective randomized studies, which he conducted together with investigators at Seoul National University in South Korea, initiating a 2-week halt of methotrexate on the day the influenza vaccine is given to patients with RA is now his routine practice, and he recommends other physicians do the same.
The first prospective, randomized trial included 199 RA patients on stable doses of methotrexate who were assigned to one of four groups in conjunction with seasonal influenza vaccination. One group continued their methotrexate as usual, the second stopped the drug for 1 month prior to vaccination and then restarted it at the time of vaccination, the third group halted methotrexate for 2 weeks before and 2 weeks after vaccination, and the fourth suspended methotrexate for 4 weeks starting on the day they got their flu shot. Everyone received trivalent influenza vaccine containing H1N1, H3N2, and B/Yamagata.
The lowest rate of satisfactory vaccine response as defined by at least a 300% titer increase 1 month after vaccination occurred in the group that continued their methotrexate as usual. The group that halted the drug for 2 weeks before and 2 weeks after influenza vaccination had a 51% satisfactory vaccine response against all three antigens, compared with a 31.5% rate in the methotrexate-as-usual group. RA flare rates ranged from 21% to 39% across the four study arms, differences that weren’t statistically significant (Ann Rheum Dis. 2017 Sep;76[9]:1559-65).
Next Dr. Winthrop and his colleagues conducted a confirmatory prospective, multicenter, randomized trial in which they sought to nail down the optimal duration and timing of methotrexate discontinuation. A total of 320 RA patients on stable doses of methotrexate were assigned to halt the drug for 2 weeks starting at the time they received a quadrivalent seasonal influenza vaccine containing H1N1, H3N2, B/Yamagata, and B/Victoria strains, or to continue their methotrexate throughout.
A satisfactory vaccine response was achieved in 75.5% of the group that discontinued the drug, significantly better than the 54.5% rate in the methotrexate continuers. The absolute difference in seroprotection was greater in patients who halted their methotrexate for 2 weeks after vaccination for all four antigens: an absolute 11% difference for H1N1, 16% for H3N2, 12% for B/Yamagata, and 15% for B/Victoria (Ann Rheum Dis. 2018 Jun;77[6]:898-904).
“It does seem to be a nice strategy. The percentage of people who flared their RA during their 2 weeks off methotrexate was very low, so there seems to be a good reason to do this,” according to Dr. Winthrop.
Some rheumatologists he has spoken with initially balked at the plausibility of the results.
“I had the same thought about these studies: It doesn’t make sense to me in terms of how methotrexate works, and why we would see this effect acutely by stopping methotrexate for just 2 weeks?” he said.
But then a coinvestigator drilled down deeper into the data and came up with the explanation: The benefit in terms of enhanced flu vaccine immunogenicity through temporary withholding of methotrexate was confined to the subgroup of RA patients with high baseline levels of B-cell activation factor (BAFF). In contrast, withholding methotrexate didn’t affect the vaccine response in patients with low or normal baseline BAFF (Ann Rheum Dis. 2018 Oct 8. doi: 10.1136/annrheumdis-2018-214025).
“I don’t know how to check anyone’s BAFF levels. I don’t think there’s a commercial test out there. But this does help explain why we saw this observation. So I think I would still hold everyone’s methotrexate for 2 weeks. That’s how I approach it. And they may get benefit from it, and they may not,” he said.
Dr. Winthrop reported having no financial conflicts regarding the study, which was funded by GC Pharma.
REPORTING FROM THE ACR ANNUAL MEETING
Struggling to reach an HCV vaccine
Currently, there is no effective hepatitis C virus (HCV) vaccine available despite numerous ongoing studies, according to the results of a review published in Gastroenterology.
In their article, Justin R. Bailey, MD, of Johns Hopkins University, Baltimore, and his colleagues reviewed the limited feasibility of applying traditional vaccine design to HCV and the problem of genetic diversity in the virus, as well as trials of vaccines designed to elicit T-cell responses.
One profound difficulty in the development and testing of an HCV vaccine is that the cohort most predictably at risk for high infection levels, people who inject drugs, are notoriously difficult to recruit, maintain consistent treatment, and follow up on – all necessary aspects of an appropriate vaccine trial.
Thus, at present, adjuvant envelope or core protein and virus-vectored nonstructural antigen vaccines have been tested only in healthy volunteers who are not at risk for HCV infection; viral vectors encoding nonstructural proteins remain the only vaccine strategy tested in truly at-risk individuals, according to Dr. Bailey and his colleagues.
“Although pharmaceutical companies invest in drug development, vaccine development requires investment from sources beyond government and charitable foundations. A prophylactic HCV vaccine is an important part of a successful strategy for global control. Although development is not easy, the quest is a worthy challenge,” Dr. Bailey and his colleagues concluded.
The authors reported having no conflicts.
SOURCE: Bailey JR et al. Gastroenterology. 2018 Sep 27. doi: 10.1053/j.gastro.2018.08.060.
Currently, there is no effective hepatitis C virus (HCV) vaccine available despite numerous ongoing studies, according to the results of a review published in Gastroenterology.
In their article, Justin R. Bailey, MD, of Johns Hopkins University, Baltimore, and his colleagues reviewed the limited feasibility of applying traditional vaccine design to HCV and the problem of genetic diversity in the virus, as well as trials of vaccines designed to elicit T-cell responses.
One profound difficulty in the development and testing of an HCV vaccine is that the cohort most predictably at risk for high infection levels, people who inject drugs, are notoriously difficult to recruit, maintain consistent treatment, and follow up on – all necessary aspects of an appropriate vaccine trial.
Thus, at present, adjuvant envelope or core protein and virus-vectored nonstructural antigen vaccines have been tested only in healthy volunteers who are not at risk for HCV infection; viral vectors encoding nonstructural proteins remain the only vaccine strategy tested in truly at-risk individuals, according to Dr. Bailey and his colleagues.
“Although pharmaceutical companies invest in drug development, vaccine development requires investment from sources beyond government and charitable foundations. A prophylactic HCV vaccine is an important part of a successful strategy for global control. Although development is not easy, the quest is a worthy challenge,” Dr. Bailey and his colleagues concluded.
The authors reported having no conflicts.
SOURCE: Bailey JR et al. Gastroenterology. 2018 Sep 27. doi: 10.1053/j.gastro.2018.08.060.
Currently, there is no effective hepatitis C virus (HCV) vaccine available despite numerous ongoing studies, according to the results of a review published in Gastroenterology.
In their article, Justin R. Bailey, MD, of Johns Hopkins University, Baltimore, and his colleagues reviewed the limited feasibility of applying traditional vaccine design to HCV and the problem of genetic diversity in the virus, as well as trials of vaccines designed to elicit T-cell responses.
One profound difficulty in the development and testing of an HCV vaccine is that the cohort most predictably at risk for high infection levels, people who inject drugs, are notoriously difficult to recruit, maintain consistent treatment, and follow up on – all necessary aspects of an appropriate vaccine trial.
Thus, at present, adjuvant envelope or core protein and virus-vectored nonstructural antigen vaccines have been tested only in healthy volunteers who are not at risk for HCV infection; viral vectors encoding nonstructural proteins remain the only vaccine strategy tested in truly at-risk individuals, according to Dr. Bailey and his colleagues.
“Although pharmaceutical companies invest in drug development, vaccine development requires investment from sources beyond government and charitable foundations. A prophylactic HCV vaccine is an important part of a successful strategy for global control. Although development is not easy, the quest is a worthy challenge,” Dr. Bailey and his colleagues concluded.
The authors reported having no conflicts.
SOURCE: Bailey JR et al. Gastroenterology. 2018 Sep 27. doi: 10.1053/j.gastro.2018.08.060.
FROM GASTROENTEROLOGY
Reviewing the state of HCV and HBV in children
The natural histories of hepatitis B virus (HBV) and hepatitis C virus (HCV) are very different in children, compared with their progress in adults, and depends on age at time of infection, mode of acquisition, ethnicity, and genotype, according to a review in a special pediatric issue of Clinics in Liver Disease.
Most children infected perinatally or vertically continue to be asymptomatic but are at uniquely higher risk of developing chronic viral hepatitis and progressing to liver cirrhosis and hepatocellular carcinoma (HCC), according to Krupa R. Mysore, MD, and Daniel H. Leung, MD, both of the Baylor College of Medicine, Houston. In addition, because the risk of progression to cancer along with such other liver damage is high in children, the reviewers stated that HCV and HBV can be classified as oncoviruses.
Their article assessed overall epidemiology, viral characteristics, and immune responses, as well as prevention, clinical manifestations, and current advances in the treatment of hepatitis B and C in children.
Because of the introduction of universal infant vaccination for HBV in the United States in 1991, the incidence of acute hepatitis B in U.S. children (those aged less than 19 years) has decreased from approximately 13.80/100,000 population (in children aged 10-19 years) in the 1980s to 0.34/100,000 population in 2002, Dr. Mysore and Dr. Leung wrote.
However, they added that those children who have chronic HBV remain at high risk for HCC, with a 100-fold greater incidence, compared with the HBV-negative population.
Similarly, HCV is a significant problem in children, with an estimated prevalence in the United States of 0.2% and 0.4% for children aged 6-11 years and 12-19 years, respectively. Vertical transmission from the mother is responsible for more than 60% of pediatric HCV infection and adds approximately 7,200 new cases in the United States yearly. Older children can acquire the virus through intravenous and intranasal drug use and high-risk sexual activity, they stated.
“Our understanding of the pathobiology and immunology of hepatitis B and C is unprecedented. As new antiviral therapies are being developed for the pediatric population, the differences in management and monitoring between children and adults with HBV and HCV are beginning to narrow but are still important,” the authors wrote.
They pointed out that soon-to-be-available treatments for HCV will be curative in children aged as young as 3 years. “[T]his will change the natural history of HCV and the prevalence of hepatocellular carcinoma over the next several decades for the better,” Dr. Mysore and Dr. Leung concluded.
They reported that they had no conflicts of interest to disclose.
SOURCE: Mysore KR et al. Clin Liver Dis. 2018; 22:703-22.
The natural histories of hepatitis B virus (HBV) and hepatitis C virus (HCV) are very different in children, compared with their progress in adults, and depends on age at time of infection, mode of acquisition, ethnicity, and genotype, according to a review in a special pediatric issue of Clinics in Liver Disease.
Most children infected perinatally or vertically continue to be asymptomatic but are at uniquely higher risk of developing chronic viral hepatitis and progressing to liver cirrhosis and hepatocellular carcinoma (HCC), according to Krupa R. Mysore, MD, and Daniel H. Leung, MD, both of the Baylor College of Medicine, Houston. In addition, because the risk of progression to cancer along with such other liver damage is high in children, the reviewers stated that HCV and HBV can be classified as oncoviruses.
Their article assessed overall epidemiology, viral characteristics, and immune responses, as well as prevention, clinical manifestations, and current advances in the treatment of hepatitis B and C in children.
Because of the introduction of universal infant vaccination for HBV in the United States in 1991, the incidence of acute hepatitis B in U.S. children (those aged less than 19 years) has decreased from approximately 13.80/100,000 population (in children aged 10-19 years) in the 1980s to 0.34/100,000 population in 2002, Dr. Mysore and Dr. Leung wrote.
However, they added that those children who have chronic HBV remain at high risk for HCC, with a 100-fold greater incidence, compared with the HBV-negative population.
Similarly, HCV is a significant problem in children, with an estimated prevalence in the United States of 0.2% and 0.4% for children aged 6-11 years and 12-19 years, respectively. Vertical transmission from the mother is responsible for more than 60% of pediatric HCV infection and adds approximately 7,200 new cases in the United States yearly. Older children can acquire the virus through intravenous and intranasal drug use and high-risk sexual activity, they stated.
“Our understanding of the pathobiology and immunology of hepatitis B and C is unprecedented. As new antiviral therapies are being developed for the pediatric population, the differences in management and monitoring between children and adults with HBV and HCV are beginning to narrow but are still important,” the authors wrote.
They pointed out that soon-to-be-available treatments for HCV will be curative in children aged as young as 3 years. “[T]his will change the natural history of HCV and the prevalence of hepatocellular carcinoma over the next several decades for the better,” Dr. Mysore and Dr. Leung concluded.
They reported that they had no conflicts of interest to disclose.
SOURCE: Mysore KR et al. Clin Liver Dis. 2018; 22:703-22.
The natural histories of hepatitis B virus (HBV) and hepatitis C virus (HCV) are very different in children, compared with their progress in adults, and depends on age at time of infection, mode of acquisition, ethnicity, and genotype, according to a review in a special pediatric issue of Clinics in Liver Disease.
Most children infected perinatally or vertically continue to be asymptomatic but are at uniquely higher risk of developing chronic viral hepatitis and progressing to liver cirrhosis and hepatocellular carcinoma (HCC), according to Krupa R. Mysore, MD, and Daniel H. Leung, MD, both of the Baylor College of Medicine, Houston. In addition, because the risk of progression to cancer along with such other liver damage is high in children, the reviewers stated that HCV and HBV can be classified as oncoviruses.
Their article assessed overall epidemiology, viral characteristics, and immune responses, as well as prevention, clinical manifestations, and current advances in the treatment of hepatitis B and C in children.
Because of the introduction of universal infant vaccination for HBV in the United States in 1991, the incidence of acute hepatitis B in U.S. children (those aged less than 19 years) has decreased from approximately 13.80/100,000 population (in children aged 10-19 years) in the 1980s to 0.34/100,000 population in 2002, Dr. Mysore and Dr. Leung wrote.
However, they added that those children who have chronic HBV remain at high risk for HCC, with a 100-fold greater incidence, compared with the HBV-negative population.
Similarly, HCV is a significant problem in children, with an estimated prevalence in the United States of 0.2% and 0.4% for children aged 6-11 years and 12-19 years, respectively. Vertical transmission from the mother is responsible for more than 60% of pediatric HCV infection and adds approximately 7,200 new cases in the United States yearly. Older children can acquire the virus through intravenous and intranasal drug use and high-risk sexual activity, they stated.
“Our understanding of the pathobiology and immunology of hepatitis B and C is unprecedented. As new antiviral therapies are being developed for the pediatric population, the differences in management and monitoring between children and adults with HBV and HCV are beginning to narrow but are still important,” the authors wrote.
They pointed out that soon-to-be-available treatments for HCV will be curative in children aged as young as 3 years. “[T]his will change the natural history of HCV and the prevalence of hepatocellular carcinoma over the next several decades for the better,” Dr. Mysore and Dr. Leung concluded.
They reported that they had no conflicts of interest to disclose.
SOURCE: Mysore KR et al. Clin Liver Dis. 2018; 22:703-22.
FROM CLINICS IN LIVER DISEASE
Pneumonia, COPD most common emergency care–sensitive conditions
SAN DIEGO – Emergency care–sensitive conditions – those for which timely access to high-quality emergency care impact morbidity and mortality—account for 14% of all ED visits, results from a large analysis of national data showed.

In previously published work, an eight-member expert panel identified 51 condition groups as emergency care–sensitive conditions (ECSCs), including asthma, cardiac arrest, cerebral infarction, and pneumonia. The purpose of the current study, published in Annals of Emergency Medicine and presented by Anita Vashi, MD, MPH, at the annual meeting of the American College of Emergency Physicians, was to provide the first national estimates of acute care utilization and the demographic characteristics of adults experiencing ECSCs, compare ECSC and non-ECSC ED visits, and assess patient- and hospital-level characteristics predictive of an ECSC-related ED visit.
Using the Nationwide Emergency Department Sample data set, Dr. Vashi, a physician investigator at the Center for Innovation to Implementation at the VA Palo Alto Health Care System, and her colleagues retrospectively evaluated all ED visits for patients aged 18 years and older from 2009 to 2014. The researchers used summary statistics to compare population characteristics across groups and multivariable logistic regression models to assess the odds of an ECSC-related ED visit with patient- and hospital-level characteristics.
Of the 622,725,542 estimated ED visits evaluated during the study period, 86,577,041 (14%) were ECSCs. Among these ECSC visits, 58% of patients were admitted for an average length of 3.2 days and an average charge of $2,240. The most frequent ECSC-related visits were for pneumonia (9%), chronic obstructive pulmonary disease (9%), asthma (7%), heart failure (7%), and sepsis (5%), but varied by age group.
Dr. Vashi and her colleagues found that ECSCs were more common among older adults, males, those who reside in low-income areas, those who reside in the South, and among metropolitan-based hospitals and nontrauma center hospitals. ECSCs also accounted for about 45% of all inpatient admissions.
Multivariate logistic regression analysis revealed that the odds of having an ECSC-related visit was highest among patients aged 65 years and older (odds ratio, 3.84), those on Medicare (OR, 1.37), those who resided in rural counties (OR, 1.21), and those who reside in the Western portion of the United States (OR, 1.11). Significant hospital-related factors related to ECSC visits included trauma centers (OR, 1.09), nonteaching hospitals (OR, 1.04), and EDs located in the wealthiest counties (OR, 1.02).
The researchers also found that 40% of patients who made ECSC-related ED visits were treated and discharged back to the community. “There is evidence of regional variability, suggesting the need for future research,” said Dr. Vashi, who also holds a faculty position in the department of emergency medicine at Stanford (Calif.) University. “We found no consistent relationship between insurance, income, and ED use for ECSC-related conditions. This suggests that ECSCs are not significantly influenced by socioeconomic factor and can serve as a reliable marker for acuity.”
The next steps in this research area, she added, are to create condition-specific measures related to morbidity, mortality, and posthospital events, as well as to analyze regional and hospital variations including correlation across conditions, and to compare performance across conditions and hospitals.
Dr. Vashi reported having no financial disclosures.
Source: Vashi A et al. Ann Emerg Med. 2018 Oct;72;4:S38. doi. 10.1016/j.annemergmed.2018.08.091.
SAN DIEGO – Emergency care–sensitive conditions – those for which timely access to high-quality emergency care impact morbidity and mortality—account for 14% of all ED visits, results from a large analysis of national data showed.
In previously published work, an eight-member expert panel identified 51 condition groups as emergency care–sensitive conditions (ECSCs), including asthma, cardiac arrest, cerebral infarction, and pneumonia. The purpose of the current study, published in Annals of Emergency Medicine and presented by Anita Vashi, MD, MPH, at the annual meeting of the American College of Emergency Physicians, was to provide the first national estimates of acute care utilization and the demographic characteristics of adults experiencing ECSCs, compare ECSC and non-ECSC ED visits, and assess patient- and hospital-level characteristics predictive of an ECSC-related ED visit.
Using the Nationwide Emergency Department Sample data set, Dr. Vashi, a physician investigator at the Center for Innovation to Implementation at the VA Palo Alto Health Care System, and her colleagues retrospectively evaluated all ED visits for patients aged 18 years and older from 2009 to 2014. The researchers used summary statistics to compare population characteristics across groups and multivariable logistic regression models to assess the odds of an ECSC-related ED visit with patient- and hospital-level characteristics.
Of the 622,725,542 estimated ED visits evaluated during the study period, 86,577,041 (14%) were ECSCs. Among these ECSC visits, 58% of patients were admitted for an average length of 3.2 days and an average charge of $2,240. The most frequent ECSC-related visits were for pneumonia (9%), chronic obstructive pulmonary disease (9%), asthma (7%), heart failure (7%), and sepsis (5%), but varied by age group.
Dr. Vashi and her colleagues found that ECSCs were more common among older adults, males, those who reside in low-income areas, those who reside in the South, and among metropolitan-based hospitals and nontrauma center hospitals. ECSCs also accounted for about 45% of all inpatient admissions.
Multivariate logistic regression analysis revealed that the odds of having an ECSC-related visit was highest among patients aged 65 years and older (odds ratio, 3.84), those on Medicare (OR, 1.37), those who resided in rural counties (OR, 1.21), and those who reside in the Western portion of the United States (OR, 1.11). Significant hospital-related factors related to ECSC visits included trauma centers (OR, 1.09), nonteaching hospitals (OR, 1.04), and EDs located in the wealthiest counties (OR, 1.02).
The researchers also found that 40% of patients who made ECSC-related ED visits were treated and discharged back to the community. “There is evidence of regional variability, suggesting the need for future research,” said Dr. Vashi, who also holds a faculty position in the department of emergency medicine at Stanford (Calif.) University. “We found no consistent relationship between insurance, income, and ED use for ECSC-related conditions. This suggests that ECSCs are not significantly influenced by socioeconomic factor and can serve as a reliable marker for acuity.”
The next steps in this research area, she added, are to create condition-specific measures related to morbidity, mortality, and posthospital events, as well as to analyze regional and hospital variations including correlation across conditions, and to compare performance across conditions and hospitals.
Dr. Vashi reported having no financial disclosures.
Source: Vashi A et al. Ann Emerg Med. 2018 Oct;72;4:S38. doi. 10.1016/j.annemergmed.2018.08.091.
SAN DIEGO – Emergency care–sensitive conditions – those for which timely access to high-quality emergency care impact morbidity and mortality—account for 14% of all ED visits, results from a large analysis of national data showed.
In previously published work, an eight-member expert panel identified 51 condition groups as emergency care–sensitive conditions (ECSCs), including asthma, cardiac arrest, cerebral infarction, and pneumonia. The purpose of the current study, published in Annals of Emergency Medicine and presented by Anita Vashi, MD, MPH, at the annual meeting of the American College of Emergency Physicians, was to provide the first national estimates of acute care utilization and the demographic characteristics of adults experiencing ECSCs, compare ECSC and non-ECSC ED visits, and assess patient- and hospital-level characteristics predictive of an ECSC-related ED visit.
Using the Nationwide Emergency Department Sample data set, Dr. Vashi, a physician investigator at the Center for Innovation to Implementation at the VA Palo Alto Health Care System, and her colleagues retrospectively evaluated all ED visits for patients aged 18 years and older from 2009 to 2014. The researchers used summary statistics to compare population characteristics across groups and multivariable logistic regression models to assess the odds of an ECSC-related ED visit with patient- and hospital-level characteristics.
Of the 622,725,542 estimated ED visits evaluated during the study period, 86,577,041 (14%) were ECSCs. Among these ECSC visits, 58% of patients were admitted for an average length of 3.2 days and an average charge of $2,240. The most frequent ECSC-related visits were for pneumonia (9%), chronic obstructive pulmonary disease (9%), asthma (7%), heart failure (7%), and sepsis (5%), but varied by age group.
Dr. Vashi and her colleagues found that ECSCs were more common among older adults, males, those who reside in low-income areas, those who reside in the South, and among metropolitan-based hospitals and nontrauma center hospitals. ECSCs also accounted for about 45% of all inpatient admissions.
Multivariate logistic regression analysis revealed that the odds of having an ECSC-related visit was highest among patients aged 65 years and older (odds ratio, 3.84), those on Medicare (OR, 1.37), those who resided in rural counties (OR, 1.21), and those who reside in the Western portion of the United States (OR, 1.11). Significant hospital-related factors related to ECSC visits included trauma centers (OR, 1.09), nonteaching hospitals (OR, 1.04), and EDs located in the wealthiest counties (OR, 1.02).
The researchers also found that 40% of patients who made ECSC-related ED visits were treated and discharged back to the community. “There is evidence of regional variability, suggesting the need for future research,” said Dr. Vashi, who also holds a faculty position in the department of emergency medicine at Stanford (Calif.) University. “We found no consistent relationship between insurance, income, and ED use for ECSC-related conditions. This suggests that ECSCs are not significantly influenced by socioeconomic factor and can serve as a reliable marker for acuity.”
The next steps in this research area, she added, are to create condition-specific measures related to morbidity, mortality, and posthospital events, as well as to analyze regional and hospital variations including correlation across conditions, and to compare performance across conditions and hospitals.
Dr. Vashi reported having no financial disclosures.
Source: Vashi A et al. Ann Emerg Med. 2018 Oct;72;4:S38. doi. 10.1016/j.annemergmed.2018.08.091.
REPORTING FROM ACEP18
Key clinical point: Emergency care–sensitive conditions (ECSCs) make up a significant proportion of ED visits.
Major finding: The most common ECSC-related visits were for pneumonia (9%), chronic obstructive pulmonary disease (9%), and asthma (7%).
Study details: A retrospective cohort study of more than 86.5 million ECSC-related ED visits.
Disclosures: Dr. Vashi reported having no financial disclosures.
Source: Vashi A et al. Ann Emerg Med. 2018 Oct;72;4:S38. doi. 10.1016/j.annemergmed.2018.08.091.
Antibiotics, antacids before age 2 linked to obesity
Acid-suppressing medications were also associated with childhood obesity, although to a lesser extent, according to results of the study, which included more than a quarter of a million children receiving care in the U.S. military health system.
Antibiotics and antacids are both microbiota-altering medications, researchers said, noting that obesity has been linked to variations in the native gut microbiota.
While the evidence from previous studies is conflicting on whether microbiota-altering medications may play a role in development of childhood obesity, this study does suggest such medications may lead to weight gain early in life, they said in the journal Gut.
“Providers should practice appropriate stewardship as the first-line response to these findings,” said Christopher M. Stark, MD, of the William Beaumont Army Medical Center, El Paso, Tex., and his co-authors.
This retrospective analysis included the largest cohort of pediatric patients ever studied for the link between antibiotics and obesity, according to Dr. Stark and colleagues, and was the first to look at the link between acid-suppressing medications and obesity in that age group.
The analysis included a total of 333,353 U.S. Department of Defense TRICARE beneficiaries born between October 2006 and September 2013 who were exposed to antibiotics, histamine-2-receptor antagonists (H2RAs), or proton pump inhibitors (PPIs) within the first 2 years of life.
Patients were followed past their initial exposure period, up to 8 years of age in some cases, investigators said.
Antibiotics were prescribed in 72.4% of those children, while H2RAs and PPIs were prescribed in 11.8% and 3.3%, respectively, with a substantial number of children receiving more than one of the medications of interest for this study.
A total of 46,993 (14.1%) of the children developed obesity. Of those obese children only 9,268, or 11%, had no antibiotic or acid suppressant prescriptions on record in the first 2 years of life, the reported data show.
Antibiotic prescriptions were associated with a 26% increase in obesity risk (unadjusted hazard ratio, 1.26; 95% CI, 1.23-1.28), investigators reported. That association strengthened steadily with the number of antibiotic prescriptions, with adjusted hazard ratios of 1.12 (95% CI, 1.09-1.15) for a single prescription, up to 1.42 (95% CI, 1.37-1.46) for 4 or more prescriptions
Likewise, H2RAs and PPIs were associated, albeit weakly, with an increased hazard of obesity, investigators said. The adjusted hazard ratios and 95% confidence intervals were 1.02 (1.01-1.03) for PPIs and 1.01 (1.004-1.02) for H2RA prescriptions.
The risk of obesity steadily climbed for those receiving multiple medications, researchers added, from a hazard ratio of 1.21 for one medication, 1.31 for two, and 1.42 for three, data show.
Dr. Stark and co-authors declared no competing interests related to the study.
SOURCE: Stark CM, et al. Gut. 2018 Oct 30. pii: gutjnl-2017-314971.
Acid-suppressing medications were also associated with childhood obesity, although to a lesser extent, according to results of the study, which included more than a quarter of a million children receiving care in the U.S. military health system.
Antibiotics and antacids are both microbiota-altering medications, researchers said, noting that obesity has been linked to variations in the native gut microbiota.
While the evidence from previous studies is conflicting on whether microbiota-altering medications may play a role in development of childhood obesity, this study does suggest such medications may lead to weight gain early in life, they said in the journal Gut.
“Providers should practice appropriate stewardship as the first-line response to these findings,” said Christopher M. Stark, MD, of the William Beaumont Army Medical Center, El Paso, Tex., and his co-authors.
This retrospective analysis included the largest cohort of pediatric patients ever studied for the link between antibiotics and obesity, according to Dr. Stark and colleagues, and was the first to look at the link between acid-suppressing medications and obesity in that age group.
The analysis included a total of 333,353 U.S. Department of Defense TRICARE beneficiaries born between October 2006 and September 2013 who were exposed to antibiotics, histamine-2-receptor antagonists (H2RAs), or proton pump inhibitors (PPIs) within the first 2 years of life.
Patients were followed past their initial exposure period, up to 8 years of age in some cases, investigators said.
Antibiotics were prescribed in 72.4% of those children, while H2RAs and PPIs were prescribed in 11.8% and 3.3%, respectively, with a substantial number of children receiving more than one of the medications of interest for this study.
A total of 46,993 (14.1%) of the children developed obesity. Of those obese children only 9,268, or 11%, had no antibiotic or acid suppressant prescriptions on record in the first 2 years of life, the reported data show.
Antibiotic prescriptions were associated with a 26% increase in obesity risk (unadjusted hazard ratio, 1.26; 95% CI, 1.23-1.28), investigators reported. That association strengthened steadily with the number of antibiotic prescriptions, with adjusted hazard ratios of 1.12 (95% CI, 1.09-1.15) for a single prescription, up to 1.42 (95% CI, 1.37-1.46) for 4 or more prescriptions
Likewise, H2RAs and PPIs were associated, albeit weakly, with an increased hazard of obesity, investigators said. The adjusted hazard ratios and 95% confidence intervals were 1.02 (1.01-1.03) for PPIs and 1.01 (1.004-1.02) for H2RA prescriptions.
The risk of obesity steadily climbed for those receiving multiple medications, researchers added, from a hazard ratio of 1.21 for one medication, 1.31 for two, and 1.42 for three, data show.
Dr. Stark and co-authors declared no competing interests related to the study.
SOURCE: Stark CM, et al. Gut. 2018 Oct 30. pii: gutjnl-2017-314971.
Acid-suppressing medications were also associated with childhood obesity, although to a lesser extent, according to results of the study, which included more than a quarter of a million children receiving care in the U.S. military health system.
Antibiotics and antacids are both microbiota-altering medications, researchers said, noting that obesity has been linked to variations in the native gut microbiota.
While the evidence from previous studies is conflicting on whether microbiota-altering medications may play a role in development of childhood obesity, this study does suggest such medications may lead to weight gain early in life, they said in the journal Gut.
“Providers should practice appropriate stewardship as the first-line response to these findings,” said Christopher M. Stark, MD, of the William Beaumont Army Medical Center, El Paso, Tex., and his co-authors.
This retrospective analysis included the largest cohort of pediatric patients ever studied for the link between antibiotics and obesity, according to Dr. Stark and colleagues, and was the first to look at the link between acid-suppressing medications and obesity in that age group.
The analysis included a total of 333,353 U.S. Department of Defense TRICARE beneficiaries born between October 2006 and September 2013 who were exposed to antibiotics, histamine-2-receptor antagonists (H2RAs), or proton pump inhibitors (PPIs) within the first 2 years of life.
Patients were followed past their initial exposure period, up to 8 years of age in some cases, investigators said.
Antibiotics were prescribed in 72.4% of those children, while H2RAs and PPIs were prescribed in 11.8% and 3.3%, respectively, with a substantial number of children receiving more than one of the medications of interest for this study.
A total of 46,993 (14.1%) of the children developed obesity. Of those obese children only 9,268, or 11%, had no antibiotic or acid suppressant prescriptions on record in the first 2 years of life, the reported data show.
Antibiotic prescriptions were associated with a 26% increase in obesity risk (unadjusted hazard ratio, 1.26; 95% CI, 1.23-1.28), investigators reported. That association strengthened steadily with the number of antibiotic prescriptions, with adjusted hazard ratios of 1.12 (95% CI, 1.09-1.15) for a single prescription, up to 1.42 (95% CI, 1.37-1.46) for 4 or more prescriptions
Likewise, H2RAs and PPIs were associated, albeit weakly, with an increased hazard of obesity, investigators said. The adjusted hazard ratios and 95% confidence intervals were 1.02 (1.01-1.03) for PPIs and 1.01 (1.004-1.02) for H2RA prescriptions.
The risk of obesity steadily climbed for those receiving multiple medications, researchers added, from a hazard ratio of 1.21 for one medication, 1.31 for two, and 1.42 for three, data show.
Dr. Stark and co-authors declared no competing interests related to the study.
SOURCE: Stark CM, et al. Gut. 2018 Oct 30. pii: gutjnl-2017-314971.
FROM GUT
Key clinical point: When prescribed early in life, antibiotics, and to a lesser extent antacids, were associated with childhood obesity.
Major finding: Antibiotic prescriptions were associated with a 26% increase in obesity risk (unadjusted hazard ratio, 1.26; 95% CI, 1.23-1.28).
Study details: Retrospective study of 333,353 children in the U.S. military health system exposed in the first two years of life to antibiotics, histamine-2-receptor antagonists, or proton pump inhibitors.
Disclosures: Study authors declared no competing interests related to the work.
Source: Stark CM, et al. Gut. 2018 Oct 30. pii: gutjnl-2017-314971.