User login
The evolution of office notes and the electronic medical record: The CAPS note
Until the advent of the electronic medical record (EMR), patient charts were filled with handwritten notes documenting visits to the office and read in linear fashion, starting with the patient’s perspective of the problem, then the objective findings of the physical examination, supporting objective data, and finally, the physician’s assessment and treatment plan.
The reliable subjective, objective, assessment, plan (SOAP) approach to notes first advocated by Lawrence Weed in the 1960s did a remarkable job of conveying the physician’s thought process, supporting data, and conclusions.1,2 The notes were brief by necessity, as the physician did not want to spend time writing extraneous information.
In the age of the EMR, large quantities of data are included in the patient notes that have no connection to or do not clearly convey the physician’s thought process. In 2013, 78% of office-based physicians were using EMRs, an increase from 18% in 2001 and an adoption rate accelerated by federal government policies.3,4 But many physicians still do not feel competent reading or writing notes in an EMR and still prefer to read succinct narrative notes.5
This problem is not unique to seasoned physicians. Medical students are also failing to learn how to appropriately document office visits in the EMR, as 52% of medical schools prohibit them from writing in patient charts.6
As a result, we believed that a reassessment of Dr. Weed’s problem-oriented approach to the medical record was required to streamline the EMR and facilitate the way information is conveyed between providers of the patient’s care. Too often, large quantities of laboratory, radiographic, and pathology results are dumped into the record, burying pertinent information about the physician’s thought process, assessment, and evaluation and treatment plan and making it difficult to quickly and efficiently determine the plan.
We recently adopted an approach to office notes that is a modification of the SOAP note. While physicians often gather subjective, objective, and laboratory information to deductively formulate a diagnosis, it is not necessary to document it in the traditional deductive format in the EMR when the information is readily accessible in other areas of the record. Furthermore, a deductive format in the modern EMR produces excessively lengthy notes that require pages of screen scrolling to find the key elements required for effective patient care. This is time-consuming and is a daily obstruction to patient care.
The format that we have been using for almost 10 years still allows the physician to adhere to the problem-oriented medical note philosophy. We call it the CAPS note, which stands for concern, assessment, plan, and supporting data. This approach allows others involved in the patient’s care to efficiently extract critical components (assessment and plan for a specifically stated problem) while still allowing the inclusion of supporting data for reference and for coding and billing.
The structure of the CAPS note is:
- Concern: The primary purpose of the patient’s visit, including the history of the present illness, as conveyed by the patient, and the current status of the concern.
- Assessment: A succinct definition of the patient’s concern along with an accompanying medical diagnosis.
- Plan: The clinician’s immediate and long-term intentions for addressing the patient’s concern or condition.
- Supporting objective and subjective information: All supporting objective data, starting with the physical examination, then the results of laboratory and radiographic tests, and any other information that contributed to the clinician’s medical reasoning. Then, subjective information is included, such as the patient’s past medical, surgical, family, and social histories; current medications; allergies; and a comprehensive review of systems.
This structure keeps the most important information at the top when the encounter is opened on the computer screen and eliminates the need for unnecessary scrolling and searching, not to mention frustration and delays in patient care. Other less pertinent information appears toward the bottom of the record.
THE APSO NOTE VS THE SOAP NOTE
Frustration over the difficulty of finding the most pertinent information in the EMR—the assessment and the plan—has led others to propose a rearrangement of the traditional SOAP note. The APSO (assessment, plan, subjective, objective) note7,8 was created for inpatient daily progress notes, a situation in which the patient’s concern is unlikely to change dramatically on a daily basis and was not intended for use in outpatient clinics.8 While the APSO format does allow colleagues rapid access to the physician’s assessment and plan, it abandons the patient-centered approach of Dr. Weed’s problem-oriented medical record in that it makes it more difficult to find why the patient initially sought care, how long the patient has had the problem, or if there were prior attempts to treat it. These critical details are buried in the bowels of the note.
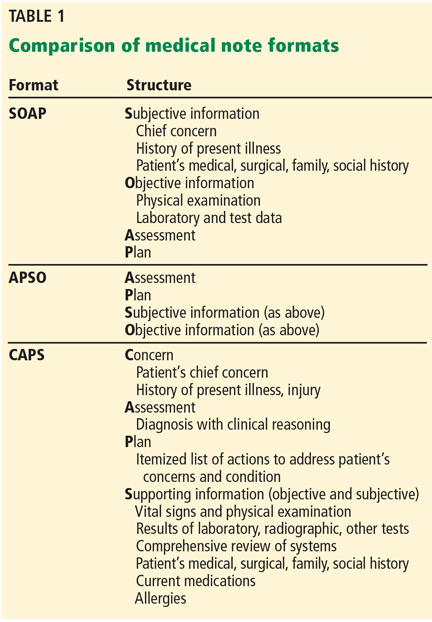
The advantage of the CAPS note (Table 1) is that it retains the patient-centered, problem-oriented spirit of the SOAP format, while moving potentially supportive yet distracting data fields to later in the note. Thus it is applicable to inpatient and outpatient settings.
In the inpatient setting, the fields remain in the same order, but the chief complaint is often the admitting diagnosis or surgical procedure, followed by a quick line on the interval history. The assessment and plan can then follow in much the same way as it would in the outpatient setting, and below that are the patient’s daily laboratory results, radiographic studies, physical examination findings, and any other relevant supporting data. This format allows rapid access to critical information needed by either consultants or cross-covering practitioners who primarily want to know why the patient was admitted, the status, and the primary team’s plan.
ANY TEMPLATE HAS LIMITATIONS
Any standardized template for progress notes in the EMR has limitations. The CAPS format would be easier for a hospital-based physician, who typically addresses one or a small number of concerns, than for an office-based general practitioner who may have to address a multitude of comorbidities in a single visit.
Also, different physicians use the EMR differently. For example, a survey of 1,088 physicians found that 60% of primary care physicians used templates (60%) vs only 34% of specialists, and that 38% of specialists relied mainly on dictation.9
The CAPS approach to the office visit note offers a blend of a template and free text, either typed or dictated, while keeping a structured format that permits others participating in the patient’s care to easily extract desired information. The template can easily be brought up in the patient’s chart, then by either typing or using voice-recognition software, the patient’s chief complaint, history of the present illness, assessment, and plan can be easily completed.
The CAPS format should continue to allow notes to fulfill medicolegal and billing obligations, but without cluttering true clinical reasoning. As more institutions adopt an open-notes policy, permitting patients to freely browse their own medical records, patients will benefit from a clearly structured clinical note that focuses on their problem and the practitioner’s solution. This provides patients a sense of validation and reassurance that the note starts with their concern and history, followed by the practitioner’s assessment and plan, so they can easily affirm that they were accurately heard and can identify the diagnosis given to them by the medical practitioner and the plan moving forward.
Since a return to succinct, albeit often illegible, handwritten clinic notes is impossible, our proposed method of documenting a clinic visit embraces the EMR with a concise yet comprehensive clinic note.
- Jacobs L. Interview with Lawrence Weed, MD—the father of the problem-oriented medical record looks ahead. Perm J 2009; 13:84–89.
- Cameron S, Turtle-Son I. Learning to write case notes using the SOAP format. JCD 2002; 80:286–292.
- Hsiao CJ, Hing E. Use and characteristics of electronic health record systems among office-based physician practices: United States, 2001-2013. NCHS Data Brief 2014; 143:1–8.
- Centers for Medicare & Medicaid Services (CMS). EHR incentive program. www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms/Basics.html. Accessed April 28, 2016.
- Han H, Lopp L. Writing and reading in the electronic health record: an entirely new world. Med Educ Online 2013; 18:1–7.
- Hammoud MM, Dalrymple JL, Christner JG, et al. Medical student documentation in electronic health records: a collaborative statement from the Alliance for Clinical Education. Teach Learn Med 2012; 24:257–266.
- Shoolin J, Ozeran L, Hamann C, Bria W 2nd. Association of Medical Directors of Information Systems consensus on inpatient electronic health record documentation. Appl Clin Inform 2013; 4:293–303.
- Hahn JS, Bernstein JA, McKenzie RB, King BJ, Longhurst CA. Rapid implementation of inpatient electronic physician documentation at an academic hospital. Appl Clin Inform 2012; 3:175–185.
- Pollard SE, Neri PM, Wilcox AR, et al. How physicians document outpatient visit notes in an electronic health record. Int J Med Inform 2013; 82:39–46.
Until the advent of the electronic medical record (EMR), patient charts were filled with handwritten notes documenting visits to the office and read in linear fashion, starting with the patient’s perspective of the problem, then the objective findings of the physical examination, supporting objective data, and finally, the physician’s assessment and treatment plan.
The reliable subjective, objective, assessment, plan (SOAP) approach to notes first advocated by Lawrence Weed in the 1960s did a remarkable job of conveying the physician’s thought process, supporting data, and conclusions.1,2 The notes were brief by necessity, as the physician did not want to spend time writing extraneous information.
In the age of the EMR, large quantities of data are included in the patient notes that have no connection to or do not clearly convey the physician’s thought process. In 2013, 78% of office-based physicians were using EMRs, an increase from 18% in 2001 and an adoption rate accelerated by federal government policies.3,4 But many physicians still do not feel competent reading or writing notes in an EMR and still prefer to read succinct narrative notes.5
This problem is not unique to seasoned physicians. Medical students are also failing to learn how to appropriately document office visits in the EMR, as 52% of medical schools prohibit them from writing in patient charts.6
As a result, we believed that a reassessment of Dr. Weed’s problem-oriented approach to the medical record was required to streamline the EMR and facilitate the way information is conveyed between providers of the patient’s care. Too often, large quantities of laboratory, radiographic, and pathology results are dumped into the record, burying pertinent information about the physician’s thought process, assessment, and evaluation and treatment plan and making it difficult to quickly and efficiently determine the plan.
We recently adopted an approach to office notes that is a modification of the SOAP note. While physicians often gather subjective, objective, and laboratory information to deductively formulate a diagnosis, it is not necessary to document it in the traditional deductive format in the EMR when the information is readily accessible in other areas of the record. Furthermore, a deductive format in the modern EMR produces excessively lengthy notes that require pages of screen scrolling to find the key elements required for effective patient care. This is time-consuming and is a daily obstruction to patient care.
The format that we have been using for almost 10 years still allows the physician to adhere to the problem-oriented medical note philosophy. We call it the CAPS note, which stands for concern, assessment, plan, and supporting data. This approach allows others involved in the patient’s care to efficiently extract critical components (assessment and plan for a specifically stated problem) while still allowing the inclusion of supporting data for reference and for coding and billing.
The structure of the CAPS note is:
- Concern: The primary purpose of the patient’s visit, including the history of the present illness, as conveyed by the patient, and the current status of the concern.
- Assessment: A succinct definition of the patient’s concern along with an accompanying medical diagnosis.
- Plan: The clinician’s immediate and long-term intentions for addressing the patient’s concern or condition.
- Supporting objective and subjective information: All supporting objective data, starting with the physical examination, then the results of laboratory and radiographic tests, and any other information that contributed to the clinician’s medical reasoning. Then, subjective information is included, such as the patient’s past medical, surgical, family, and social histories; current medications; allergies; and a comprehensive review of systems.
This structure keeps the most important information at the top when the encounter is opened on the computer screen and eliminates the need for unnecessary scrolling and searching, not to mention frustration and delays in patient care. Other less pertinent information appears toward the bottom of the record.
THE APSO NOTE VS THE SOAP NOTE
Frustration over the difficulty of finding the most pertinent information in the EMR—the assessment and the plan—has led others to propose a rearrangement of the traditional SOAP note. The APSO (assessment, plan, subjective, objective) note7,8 was created for inpatient daily progress notes, a situation in which the patient’s concern is unlikely to change dramatically on a daily basis and was not intended for use in outpatient clinics.8 While the APSO format does allow colleagues rapid access to the physician’s assessment and plan, it abandons the patient-centered approach of Dr. Weed’s problem-oriented medical record in that it makes it more difficult to find why the patient initially sought care, how long the patient has had the problem, or if there were prior attempts to treat it. These critical details are buried in the bowels of the note.
The advantage of the CAPS note (Table 1) is that it retains the patient-centered, problem-oriented spirit of the SOAP format, while moving potentially supportive yet distracting data fields to later in the note. Thus it is applicable to inpatient and outpatient settings.
In the inpatient setting, the fields remain in the same order, but the chief complaint is often the admitting diagnosis or surgical procedure, followed by a quick line on the interval history. The assessment and plan can then follow in much the same way as it would in the outpatient setting, and below that are the patient’s daily laboratory results, radiographic studies, physical examination findings, and any other relevant supporting data. This format allows rapid access to critical information needed by either consultants or cross-covering practitioners who primarily want to know why the patient was admitted, the status, and the primary team’s plan.
ANY TEMPLATE HAS LIMITATIONS
Any standardized template for progress notes in the EMR has limitations. The CAPS format would be easier for a hospital-based physician, who typically addresses one or a small number of concerns, than for an office-based general practitioner who may have to address a multitude of comorbidities in a single visit.
Also, different physicians use the EMR differently. For example, a survey of 1,088 physicians found that 60% of primary care physicians used templates (60%) vs only 34% of specialists, and that 38% of specialists relied mainly on dictation.9
The CAPS approach to the office visit note offers a blend of a template and free text, either typed or dictated, while keeping a structured format that permits others participating in the patient’s care to easily extract desired information. The template can easily be brought up in the patient’s chart, then by either typing or using voice-recognition software, the patient’s chief complaint, history of the present illness, assessment, and plan can be easily completed.
The CAPS format should continue to allow notes to fulfill medicolegal and billing obligations, but without cluttering true clinical reasoning. As more institutions adopt an open-notes policy, permitting patients to freely browse their own medical records, patients will benefit from a clearly structured clinical note that focuses on their problem and the practitioner’s solution. This provides patients a sense of validation and reassurance that the note starts with their concern and history, followed by the practitioner’s assessment and plan, so they can easily affirm that they were accurately heard and can identify the diagnosis given to them by the medical practitioner and the plan moving forward.
Since a return to succinct, albeit often illegible, handwritten clinic notes is impossible, our proposed method of documenting a clinic visit embraces the EMR with a concise yet comprehensive clinic note.
Until the advent of the electronic medical record (EMR), patient charts were filled with handwritten notes documenting visits to the office and read in linear fashion, starting with the patient’s perspective of the problem, then the objective findings of the physical examination, supporting objective data, and finally, the physician’s assessment and treatment plan.
The reliable subjective, objective, assessment, plan (SOAP) approach to notes first advocated by Lawrence Weed in the 1960s did a remarkable job of conveying the physician’s thought process, supporting data, and conclusions.1,2 The notes were brief by necessity, as the physician did not want to spend time writing extraneous information.
In the age of the EMR, large quantities of data are included in the patient notes that have no connection to or do not clearly convey the physician’s thought process. In 2013, 78% of office-based physicians were using EMRs, an increase from 18% in 2001 and an adoption rate accelerated by federal government policies.3,4 But many physicians still do not feel competent reading or writing notes in an EMR and still prefer to read succinct narrative notes.5
This problem is not unique to seasoned physicians. Medical students are also failing to learn how to appropriately document office visits in the EMR, as 52% of medical schools prohibit them from writing in patient charts.6
As a result, we believed that a reassessment of Dr. Weed’s problem-oriented approach to the medical record was required to streamline the EMR and facilitate the way information is conveyed between providers of the patient’s care. Too often, large quantities of laboratory, radiographic, and pathology results are dumped into the record, burying pertinent information about the physician’s thought process, assessment, and evaluation and treatment plan and making it difficult to quickly and efficiently determine the plan.
We recently adopted an approach to office notes that is a modification of the SOAP note. While physicians often gather subjective, objective, and laboratory information to deductively formulate a diagnosis, it is not necessary to document it in the traditional deductive format in the EMR when the information is readily accessible in other areas of the record. Furthermore, a deductive format in the modern EMR produces excessively lengthy notes that require pages of screen scrolling to find the key elements required for effective patient care. This is time-consuming and is a daily obstruction to patient care.
The format that we have been using for almost 10 years still allows the physician to adhere to the problem-oriented medical note philosophy. We call it the CAPS note, which stands for concern, assessment, plan, and supporting data. This approach allows others involved in the patient’s care to efficiently extract critical components (assessment and plan for a specifically stated problem) while still allowing the inclusion of supporting data for reference and for coding and billing.
The structure of the CAPS note is:
- Concern: The primary purpose of the patient’s visit, including the history of the present illness, as conveyed by the patient, and the current status of the concern.
- Assessment: A succinct definition of the patient’s concern along with an accompanying medical diagnosis.
- Plan: The clinician’s immediate and long-term intentions for addressing the patient’s concern or condition.
- Supporting objective and subjective information: All supporting objective data, starting with the physical examination, then the results of laboratory and radiographic tests, and any other information that contributed to the clinician’s medical reasoning. Then, subjective information is included, such as the patient’s past medical, surgical, family, and social histories; current medications; allergies; and a comprehensive review of systems.
This structure keeps the most important information at the top when the encounter is opened on the computer screen and eliminates the need for unnecessary scrolling and searching, not to mention frustration and delays in patient care. Other less pertinent information appears toward the bottom of the record.
THE APSO NOTE VS THE SOAP NOTE
Frustration over the difficulty of finding the most pertinent information in the EMR—the assessment and the plan—has led others to propose a rearrangement of the traditional SOAP note. The APSO (assessment, plan, subjective, objective) note7,8 was created for inpatient daily progress notes, a situation in which the patient’s concern is unlikely to change dramatically on a daily basis and was not intended for use in outpatient clinics.8 While the APSO format does allow colleagues rapid access to the physician’s assessment and plan, it abandons the patient-centered approach of Dr. Weed’s problem-oriented medical record in that it makes it more difficult to find why the patient initially sought care, how long the patient has had the problem, or if there were prior attempts to treat it. These critical details are buried in the bowels of the note.
The advantage of the CAPS note (Table 1) is that it retains the patient-centered, problem-oriented spirit of the SOAP format, while moving potentially supportive yet distracting data fields to later in the note. Thus it is applicable to inpatient and outpatient settings.
In the inpatient setting, the fields remain in the same order, but the chief complaint is often the admitting diagnosis or surgical procedure, followed by a quick line on the interval history. The assessment and plan can then follow in much the same way as it would in the outpatient setting, and below that are the patient’s daily laboratory results, radiographic studies, physical examination findings, and any other relevant supporting data. This format allows rapid access to critical information needed by either consultants or cross-covering practitioners who primarily want to know why the patient was admitted, the status, and the primary team’s plan.
ANY TEMPLATE HAS LIMITATIONS
Any standardized template for progress notes in the EMR has limitations. The CAPS format would be easier for a hospital-based physician, who typically addresses one or a small number of concerns, than for an office-based general practitioner who may have to address a multitude of comorbidities in a single visit.
Also, different physicians use the EMR differently. For example, a survey of 1,088 physicians found that 60% of primary care physicians used templates (60%) vs only 34% of specialists, and that 38% of specialists relied mainly on dictation.9
The CAPS approach to the office visit note offers a blend of a template and free text, either typed or dictated, while keeping a structured format that permits others participating in the patient’s care to easily extract desired information. The template can easily be brought up in the patient’s chart, then by either typing or using voice-recognition software, the patient’s chief complaint, history of the present illness, assessment, and plan can be easily completed.
The CAPS format should continue to allow notes to fulfill medicolegal and billing obligations, but without cluttering true clinical reasoning. As more institutions adopt an open-notes policy, permitting patients to freely browse their own medical records, patients will benefit from a clearly structured clinical note that focuses on their problem and the practitioner’s solution. This provides patients a sense of validation and reassurance that the note starts with their concern and history, followed by the practitioner’s assessment and plan, so they can easily affirm that they were accurately heard and can identify the diagnosis given to them by the medical practitioner and the plan moving forward.
Since a return to succinct, albeit often illegible, handwritten clinic notes is impossible, our proposed method of documenting a clinic visit embraces the EMR with a concise yet comprehensive clinic note.
- Jacobs L. Interview with Lawrence Weed, MD—the father of the problem-oriented medical record looks ahead. Perm J 2009; 13:84–89.
- Cameron S, Turtle-Son I. Learning to write case notes using the SOAP format. JCD 2002; 80:286–292.
- Hsiao CJ, Hing E. Use and characteristics of electronic health record systems among office-based physician practices: United States, 2001-2013. NCHS Data Brief 2014; 143:1–8.
- Centers for Medicare & Medicaid Services (CMS). EHR incentive program. www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms/Basics.html. Accessed April 28, 2016.
- Han H, Lopp L. Writing and reading in the electronic health record: an entirely new world. Med Educ Online 2013; 18:1–7.
- Hammoud MM, Dalrymple JL, Christner JG, et al. Medical student documentation in electronic health records: a collaborative statement from the Alliance for Clinical Education. Teach Learn Med 2012; 24:257–266.
- Shoolin J, Ozeran L, Hamann C, Bria W 2nd. Association of Medical Directors of Information Systems consensus on inpatient electronic health record documentation. Appl Clin Inform 2013; 4:293–303.
- Hahn JS, Bernstein JA, McKenzie RB, King BJ, Longhurst CA. Rapid implementation of inpatient electronic physician documentation at an academic hospital. Appl Clin Inform 2012; 3:175–185.
- Pollard SE, Neri PM, Wilcox AR, et al. How physicians document outpatient visit notes in an electronic health record. Int J Med Inform 2013; 82:39–46.
- Jacobs L. Interview with Lawrence Weed, MD—the father of the problem-oriented medical record looks ahead. Perm J 2009; 13:84–89.
- Cameron S, Turtle-Son I. Learning to write case notes using the SOAP format. JCD 2002; 80:286–292.
- Hsiao CJ, Hing E. Use and characteristics of electronic health record systems among office-based physician practices: United States, 2001-2013. NCHS Data Brief 2014; 143:1–8.
- Centers for Medicare & Medicaid Services (CMS). EHR incentive program. www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms/Basics.html. Accessed April 28, 2016.
- Han H, Lopp L. Writing and reading in the electronic health record: an entirely new world. Med Educ Online 2013; 18:1–7.
- Hammoud MM, Dalrymple JL, Christner JG, et al. Medical student documentation in electronic health records: a collaborative statement from the Alliance for Clinical Education. Teach Learn Med 2012; 24:257–266.
- Shoolin J, Ozeran L, Hamann C, Bria W 2nd. Association of Medical Directors of Information Systems consensus on inpatient electronic health record documentation. Appl Clin Inform 2013; 4:293–303.
- Hahn JS, Bernstein JA, McKenzie RB, King BJ, Longhurst CA. Rapid implementation of inpatient electronic physician documentation at an academic hospital. Appl Clin Inform 2012; 3:175–185.
- Pollard SE, Neri PM, Wilcox AR, et al. How physicians document outpatient visit notes in an electronic health record. Int J Med Inform 2013; 82:39–46.
KEY POINTS
- The CAPS format provides an advantage over the traditional approach by transferring potentially note-cluttering data that is available elsewhere in the EMR to the bottom of the note, allowing more efficient communication of the true purpose for the patient’s visit, the diagnosis, and the physician’s approach to resolving the patient’s concern.
- As healthcare systems allow patients to browse their electronic charts, the CAPS format shows them that their concern was heard accurately and clearly states the diagnosis and plan of care.

Are we causing anemia by ordering unnecessary blood tests?
A 68-year-old woman is admitted for community-acquired pneumonia. She receives antibiotics, and her condition begins to improve after 2 days. She has her blood drawn daily throughout her admission.
On hospital day 3, she complains of fatigue, and on day 4, laboratory results show that her hemoglobin and hematocrit values have fallen. To make sure this result is not spurious, her blood is drawn again to repeat the test. On day 5, her hemoglobin level has dropped to 7.0 g/dL, which is 2 g/dL lower than at admission, and she receives a transfusion.
On day 7, her hemoglobin level is stable at 8.5 g/dL, and her physicians decide to discharge her. The morning of her discharge, as a nurse is about to draw her blood, the patient asks, “Are all these blood tests really necessary?”
DO WE DRAW TOO MUCH BLOOD?
This case portrays a common occurrence. Significant amounts of blood are drawn from patients, especially in critical care. Clinical uncertainty drives most laboratory testing ordered by physicians. Too often, however, these tests lead to more testing and interventions, without a clear benefit to the patient.1
When blood testing leads to more testing, a patient’s hemoglobin and hematocrit can fall. Symptomatic iatrogenic anemia is associated with significant morbidity for patients with preexisting cardiopulmonary disease.
We draw much larger volumes of blood than most testing guidelines say are necessary. One author2 has noted that 50 to 60 mL of blood is removed for each set of tests, owing to the size of collection tubes, multiple reagents needed for each test, and the possibility that tests may need to be rerun. Yet about 3 mL of blood is sufficient to perform most laboratory tests even if the test needs to be rerun.2
CAN BLOOD DRAWS CAUSE ANEMIA?
A relationship between the volume of blood drawn and iatrogenic anemia was first described in 2005, when Thavendiranathan et al3 found that in adult patients on general medicine floors, the volume of blood drawn strongly predicted decreased hemoglobin and hematocrit levels. For every 100 mL of blood drawn, hemoglobin levels fell by an average of 0.7 g/dL, and 13.9% of the patients in the study had iron studies and fecal occult blood tests performed to investigate anemia.
Kurniali et al4 reported that during an average admission, 65% of patients experienced a drop in hemoglobin of 1.0 g/dL or more, and 49% developed anemia.
Salisbury et al,5 in 2011, studied 17,676 patients with acute myocardial infarction across 57 centers and found a correlation between the volume of blood taken and the development of anemia. On average, for every 50 mL of blood drawn, the risk of moderate to severe iatrogenic anemia increased by 18%. They also found significant variation in blood loss from testing in patients who developed moderate or severe anemia. The authors believed this indicated that moderate to severe anemia was more frequent at centers with higher than average diagnostic blood loss.5
This relationship has also been described in patients in intensive care, where it contributes to anemia of chronic disease. While anemia of critical illness is multifactorial, phlebotomy contributes to anemia in both short- and long-term stays in the intensive care unit.6
CHOOSING WISELY GUIDELINES
The Choosing Wisely initiative of the American Board of Internal Medicine Foundation collects recommendations by a number of medical specialty societies to reduce overuse of healthcare resources.7 The Critical Care Societies Collaborative recommends ordering diagnostic tests only when they answer specific clinical questions rather than routinely. The Society of Hospital Medicine also recommends against repeat complete blood cell count and blood chemistry testing because it may contribute to anemia, which is of particular concern in patients with cardiorespiratory disease.
POSSIBLE HARM
The Critical Care Societies Collaborative, in its Choosing Wisely Guidelines, specifically cites anemia as a potential harm of unnecessary phlebotomy, noting it may result in transfusion, with its associated risks and costs. In addition, aggressive investigation of incidental and nonpathologic results of routine studies is wasteful and exposes the patient to additional risks.
REDUCING PHLEBOTOMY DECREASES IATROGENIC ANEMIA
Since the relationship between excessive phlebotomy and iatrogenic anemia was described, hospitals have attempted to address the problem.
In 2011, Stuebing and Miner8 described an intervention in which the house staff and attending physicians on non-intensive care surgical services were given weekly reports of the cost of the laboratory services for the previous week. They found that simply making providers aware of the cost of their tests reduced the number of tests ordered and resulted in significant hospital savings.
Another strategy is to use pediatric collection tubes in adult patients. A 2008 study in which all blood samples were drawn using pediatric tubes reduced the blood volume removed per patient by almost 75% in inpatient and critical care patients, without the need for repeat blood draws.9 However, Kurniali et al found that the use of pediatric collection tubes did not significantly change hemoglobin fluctuations throughout patient hospital stays.4
Corson et al10 in 2015 described an intervention involving detailing, auditing, and giving feedback regarding the frequency of laboratory tests commonly ordered by a group of hospitalists. The intervention resulted in a modest reduction in the number of common laboratory tests ordered per patient day and in hospital costs, without any changes in the length of hospital stay, mortality rate, or readmission rate.10
THE CLINICAL BOTTOM LINE
As a general principle, diagnostic testing should be done to answer specific diagnostic questions and to guide management. Ordering of diagnostic tests should be decided on a day-to-day basis rather than scheduled automatically or done reflexively. In the case of blood draws, the volume of blood drawn is significantly increased by unnecessary testing, resulting in higher rates of hospital-acquired anemia.
- Ezzie ME, Aberegg SK, O’Brien JM Jr. Laboratory testing in the intensive care unit. Crit Care Clin 2007; 23:435–465.
- Stefanini M. Iatrogenic anemia (can it be prevented?). J Thromb Haemost 2014; 12:1591.
- Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med 2005; 20:520–524.
- Kurniali PC, Curry S, Brennan KW, et al. A retrospective study investigating the incidence and predisposing factors of hospital-acquired anemia. Anemia 2014; 2014:634582.
- Salisbury AC, Reid KJ, Alexander KP, et al. Diagnostic blood loss from phlebotomy and hospital-acquired anemia during acute myocardial infarction. Arch Intern Med 2011; 171:1646–1653.
- Walsh TS, Lee RJ, Maciver CR, et al. Anemia during and at discharge from intensive care: the impact of restrictive blood transfusion practice. Intensive Care Med 2006; 32:100–109.
- American Board of Internal Medicine Foundation. Choosing Wisely. www.abimfoundation.org/Initiatives/Choosing-Wisely.aspx. Accessed April 19, 2016.
- Stuebing EA, Miner TJ. Surgical vampires and rising health care expenditure: reducing the cost of daily phlebotomy. Arch Surg 2011; 146:524–527.
- Sanchez-Giron F, Alvarez-Mora F. Reduction of blood loss from laboratory testing in hospitalized adult patients using small-volume (pediatric) tubes. Arch Pathol Lab Med 2008; 132:1916–1919.
- Corson AH, Fan VS, White T, et al. A multifaceted hospitalist quality improvement intervention: decreased frequency of common labs. J Hosp Med 2015; 10:390–395.
A 68-year-old woman is admitted for community-acquired pneumonia. She receives antibiotics, and her condition begins to improve after 2 days. She has her blood drawn daily throughout her admission.
On hospital day 3, she complains of fatigue, and on day 4, laboratory results show that her hemoglobin and hematocrit values have fallen. To make sure this result is not spurious, her blood is drawn again to repeat the test. On day 5, her hemoglobin level has dropped to 7.0 g/dL, which is 2 g/dL lower than at admission, and she receives a transfusion.
On day 7, her hemoglobin level is stable at 8.5 g/dL, and her physicians decide to discharge her. The morning of her discharge, as a nurse is about to draw her blood, the patient asks, “Are all these blood tests really necessary?”
DO WE DRAW TOO MUCH BLOOD?
This case portrays a common occurrence. Significant amounts of blood are drawn from patients, especially in critical care. Clinical uncertainty drives most laboratory testing ordered by physicians. Too often, however, these tests lead to more testing and interventions, without a clear benefit to the patient.1
When blood testing leads to more testing, a patient’s hemoglobin and hematocrit can fall. Symptomatic iatrogenic anemia is associated with significant morbidity for patients with preexisting cardiopulmonary disease.
We draw much larger volumes of blood than most testing guidelines say are necessary. One author2 has noted that 50 to 60 mL of blood is removed for each set of tests, owing to the size of collection tubes, multiple reagents needed for each test, and the possibility that tests may need to be rerun. Yet about 3 mL of blood is sufficient to perform most laboratory tests even if the test needs to be rerun.2
CAN BLOOD DRAWS CAUSE ANEMIA?
A relationship between the volume of blood drawn and iatrogenic anemia was first described in 2005, when Thavendiranathan et al3 found that in adult patients on general medicine floors, the volume of blood drawn strongly predicted decreased hemoglobin and hematocrit levels. For every 100 mL of blood drawn, hemoglobin levels fell by an average of 0.7 g/dL, and 13.9% of the patients in the study had iron studies and fecal occult blood tests performed to investigate anemia.
Kurniali et al4 reported that during an average admission, 65% of patients experienced a drop in hemoglobin of 1.0 g/dL or more, and 49% developed anemia.
Salisbury et al,5 in 2011, studied 17,676 patients with acute myocardial infarction across 57 centers and found a correlation between the volume of blood taken and the development of anemia. On average, for every 50 mL of blood drawn, the risk of moderate to severe iatrogenic anemia increased by 18%. They also found significant variation in blood loss from testing in patients who developed moderate or severe anemia. The authors believed this indicated that moderate to severe anemia was more frequent at centers with higher than average diagnostic blood loss.5
This relationship has also been described in patients in intensive care, where it contributes to anemia of chronic disease. While anemia of critical illness is multifactorial, phlebotomy contributes to anemia in both short- and long-term stays in the intensive care unit.6
CHOOSING WISELY GUIDELINES
The Choosing Wisely initiative of the American Board of Internal Medicine Foundation collects recommendations by a number of medical specialty societies to reduce overuse of healthcare resources.7 The Critical Care Societies Collaborative recommends ordering diagnostic tests only when they answer specific clinical questions rather than routinely. The Society of Hospital Medicine also recommends against repeat complete blood cell count and blood chemistry testing because it may contribute to anemia, which is of particular concern in patients with cardiorespiratory disease.
POSSIBLE HARM
The Critical Care Societies Collaborative, in its Choosing Wisely Guidelines, specifically cites anemia as a potential harm of unnecessary phlebotomy, noting it may result in transfusion, with its associated risks and costs. In addition, aggressive investigation of incidental and nonpathologic results of routine studies is wasteful and exposes the patient to additional risks.
REDUCING PHLEBOTOMY DECREASES IATROGENIC ANEMIA
Since the relationship between excessive phlebotomy and iatrogenic anemia was described, hospitals have attempted to address the problem.
In 2011, Stuebing and Miner8 described an intervention in which the house staff and attending physicians on non-intensive care surgical services were given weekly reports of the cost of the laboratory services for the previous week. They found that simply making providers aware of the cost of their tests reduced the number of tests ordered and resulted in significant hospital savings.
Another strategy is to use pediatric collection tubes in adult patients. A 2008 study in which all blood samples were drawn using pediatric tubes reduced the blood volume removed per patient by almost 75% in inpatient and critical care patients, without the need for repeat blood draws.9 However, Kurniali et al found that the use of pediatric collection tubes did not significantly change hemoglobin fluctuations throughout patient hospital stays.4
Corson et al10 in 2015 described an intervention involving detailing, auditing, and giving feedback regarding the frequency of laboratory tests commonly ordered by a group of hospitalists. The intervention resulted in a modest reduction in the number of common laboratory tests ordered per patient day and in hospital costs, without any changes in the length of hospital stay, mortality rate, or readmission rate.10
THE CLINICAL BOTTOM LINE
As a general principle, diagnostic testing should be done to answer specific diagnostic questions and to guide management. Ordering of diagnostic tests should be decided on a day-to-day basis rather than scheduled automatically or done reflexively. In the case of blood draws, the volume of blood drawn is significantly increased by unnecessary testing, resulting in higher rates of hospital-acquired anemia.
A 68-year-old woman is admitted for community-acquired pneumonia. She receives antibiotics, and her condition begins to improve after 2 days. She has her blood drawn daily throughout her admission.
On hospital day 3, she complains of fatigue, and on day 4, laboratory results show that her hemoglobin and hematocrit values have fallen. To make sure this result is not spurious, her blood is drawn again to repeat the test. On day 5, her hemoglobin level has dropped to 7.0 g/dL, which is 2 g/dL lower than at admission, and she receives a transfusion.
On day 7, her hemoglobin level is stable at 8.5 g/dL, and her physicians decide to discharge her. The morning of her discharge, as a nurse is about to draw her blood, the patient asks, “Are all these blood tests really necessary?”
DO WE DRAW TOO MUCH BLOOD?
This case portrays a common occurrence. Significant amounts of blood are drawn from patients, especially in critical care. Clinical uncertainty drives most laboratory testing ordered by physicians. Too often, however, these tests lead to more testing and interventions, without a clear benefit to the patient.1
When blood testing leads to more testing, a patient’s hemoglobin and hematocrit can fall. Symptomatic iatrogenic anemia is associated with significant morbidity for patients with preexisting cardiopulmonary disease.
We draw much larger volumes of blood than most testing guidelines say are necessary. One author2 has noted that 50 to 60 mL of blood is removed for each set of tests, owing to the size of collection tubes, multiple reagents needed for each test, and the possibility that tests may need to be rerun. Yet about 3 mL of blood is sufficient to perform most laboratory tests even if the test needs to be rerun.2
CAN BLOOD DRAWS CAUSE ANEMIA?
A relationship between the volume of blood drawn and iatrogenic anemia was first described in 2005, when Thavendiranathan et al3 found that in adult patients on general medicine floors, the volume of blood drawn strongly predicted decreased hemoglobin and hematocrit levels. For every 100 mL of blood drawn, hemoglobin levels fell by an average of 0.7 g/dL, and 13.9% of the patients in the study had iron studies and fecal occult blood tests performed to investigate anemia.
Kurniali et al4 reported that during an average admission, 65% of patients experienced a drop in hemoglobin of 1.0 g/dL or more, and 49% developed anemia.
Salisbury et al,5 in 2011, studied 17,676 patients with acute myocardial infarction across 57 centers and found a correlation between the volume of blood taken and the development of anemia. On average, for every 50 mL of blood drawn, the risk of moderate to severe iatrogenic anemia increased by 18%. They also found significant variation in blood loss from testing in patients who developed moderate or severe anemia. The authors believed this indicated that moderate to severe anemia was more frequent at centers with higher than average diagnostic blood loss.5
This relationship has also been described in patients in intensive care, where it contributes to anemia of chronic disease. While anemia of critical illness is multifactorial, phlebotomy contributes to anemia in both short- and long-term stays in the intensive care unit.6
CHOOSING WISELY GUIDELINES
The Choosing Wisely initiative of the American Board of Internal Medicine Foundation collects recommendations by a number of medical specialty societies to reduce overuse of healthcare resources.7 The Critical Care Societies Collaborative recommends ordering diagnostic tests only when they answer specific clinical questions rather than routinely. The Society of Hospital Medicine also recommends against repeat complete blood cell count and blood chemistry testing because it may contribute to anemia, which is of particular concern in patients with cardiorespiratory disease.
POSSIBLE HARM
The Critical Care Societies Collaborative, in its Choosing Wisely Guidelines, specifically cites anemia as a potential harm of unnecessary phlebotomy, noting it may result in transfusion, with its associated risks and costs. In addition, aggressive investigation of incidental and nonpathologic results of routine studies is wasteful and exposes the patient to additional risks.
REDUCING PHLEBOTOMY DECREASES IATROGENIC ANEMIA
Since the relationship between excessive phlebotomy and iatrogenic anemia was described, hospitals have attempted to address the problem.
In 2011, Stuebing and Miner8 described an intervention in which the house staff and attending physicians on non-intensive care surgical services were given weekly reports of the cost of the laboratory services for the previous week. They found that simply making providers aware of the cost of their tests reduced the number of tests ordered and resulted in significant hospital savings.
Another strategy is to use pediatric collection tubes in adult patients. A 2008 study in which all blood samples were drawn using pediatric tubes reduced the blood volume removed per patient by almost 75% in inpatient and critical care patients, without the need for repeat blood draws.9 However, Kurniali et al found that the use of pediatric collection tubes did not significantly change hemoglobin fluctuations throughout patient hospital stays.4
Corson et al10 in 2015 described an intervention involving detailing, auditing, and giving feedback regarding the frequency of laboratory tests commonly ordered by a group of hospitalists. The intervention resulted in a modest reduction in the number of common laboratory tests ordered per patient day and in hospital costs, without any changes in the length of hospital stay, mortality rate, or readmission rate.10
THE CLINICAL BOTTOM LINE
As a general principle, diagnostic testing should be done to answer specific diagnostic questions and to guide management. Ordering of diagnostic tests should be decided on a day-to-day basis rather than scheduled automatically or done reflexively. In the case of blood draws, the volume of blood drawn is significantly increased by unnecessary testing, resulting in higher rates of hospital-acquired anemia.
- Ezzie ME, Aberegg SK, O’Brien JM Jr. Laboratory testing in the intensive care unit. Crit Care Clin 2007; 23:435–465.
- Stefanini M. Iatrogenic anemia (can it be prevented?). J Thromb Haemost 2014; 12:1591.
- Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med 2005; 20:520–524.
- Kurniali PC, Curry S, Brennan KW, et al. A retrospective study investigating the incidence and predisposing factors of hospital-acquired anemia. Anemia 2014; 2014:634582.
- Salisbury AC, Reid KJ, Alexander KP, et al. Diagnostic blood loss from phlebotomy and hospital-acquired anemia during acute myocardial infarction. Arch Intern Med 2011; 171:1646–1653.
- Walsh TS, Lee RJ, Maciver CR, et al. Anemia during and at discharge from intensive care: the impact of restrictive blood transfusion practice. Intensive Care Med 2006; 32:100–109.
- American Board of Internal Medicine Foundation. Choosing Wisely. www.abimfoundation.org/Initiatives/Choosing-Wisely.aspx. Accessed April 19, 2016.
- Stuebing EA, Miner TJ. Surgical vampires and rising health care expenditure: reducing the cost of daily phlebotomy. Arch Surg 2011; 146:524–527.
- Sanchez-Giron F, Alvarez-Mora F. Reduction of blood loss from laboratory testing in hospitalized adult patients using small-volume (pediatric) tubes. Arch Pathol Lab Med 2008; 132:1916–1919.
- Corson AH, Fan VS, White T, et al. A multifaceted hospitalist quality improvement intervention: decreased frequency of common labs. J Hosp Med 2015; 10:390–395.
- Ezzie ME, Aberegg SK, O’Brien JM Jr. Laboratory testing in the intensive care unit. Crit Care Clin 2007; 23:435–465.
- Stefanini M. Iatrogenic anemia (can it be prevented?). J Thromb Haemost 2014; 12:1591.
- Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med 2005; 20:520–524.
- Kurniali PC, Curry S, Brennan KW, et al. A retrospective study investigating the incidence and predisposing factors of hospital-acquired anemia. Anemia 2014; 2014:634582.
- Salisbury AC, Reid KJ, Alexander KP, et al. Diagnostic blood loss from phlebotomy and hospital-acquired anemia during acute myocardial infarction. Arch Intern Med 2011; 171:1646–1653.
- Walsh TS, Lee RJ, Maciver CR, et al. Anemia during and at discharge from intensive care: the impact of restrictive blood transfusion practice. Intensive Care Med 2006; 32:100–109.
- American Board of Internal Medicine Foundation. Choosing Wisely. www.abimfoundation.org/Initiatives/Choosing-Wisely.aspx. Accessed April 19, 2016.
- Stuebing EA, Miner TJ. Surgical vampires and rising health care expenditure: reducing the cost of daily phlebotomy. Arch Surg 2011; 146:524–527.
- Sanchez-Giron F, Alvarez-Mora F. Reduction of blood loss from laboratory testing in hospitalized adult patients using small-volume (pediatric) tubes. Arch Pathol Lab Med 2008; 132:1916–1919.
- Corson AH, Fan VS, White T, et al. A multifaceted hospitalist quality improvement intervention: decreased frequency of common labs. J Hosp Med 2015; 10:390–395.

Cognitive bias and diagnostic error
To the Editor: I appreciated the article on cognitive biases and diagnostic error by Mull et al in the November 2015 issue.1 They presented an excellent description of the pitfalls of diagnosis as reflected in a case of a patient misdiagnosed with heart failure who ultimately died of pulmonary tuberculosis complicated by pulmonary embolism (the latter possibly from using the wrong form of heparin). To the points they raised, I would like to add a few of my own about diagnosis in general and heart failure in particular.
First, any initial diagnosis not confirmed objectively within the first 24 hours should be questioned, and other possibilities should be investigated. I have found this to be essential for every day’s stay in the hospital and for every outpatient visit. The authors mention checklists as part of the solution to the problem of misdiagnosis, and I would suggest that confirmation of initial diagnoses be built into these checklists.
In the case of a presumptive diagnosis of an acute exacerbation of heart failure treated empirically with diuretics, the diagnosis should be confirmed by the next day’s response to the diuretics, ie, increased urine output, a lower respiratory rate, and a fall in the pro-B-type natriuretic peptide level. Moreover, a change in the radiographic appearance should be seen, and respiratory and pulmonary function should improve after the first 24 hours on oxygen supplementation plus diuretics. Daily patient weights are also critical in determining response to a diuretic, and are rarely done accurately. I order weights and review them daily for patients like this.
Second, it is good to look at things yourself, including the patient, medication lists, laboratory values, and radiographic films. The attending physician should look at the radiographs together with a senior radiologist. Seeing no improvement or change on the second hospital day, or seeing signs incompatible with heart failure, one could order computed tomography of the chest and begin to entertain pulmonary diagnoses.
Even vital signs can be questionable. For example, in the case presented here, with a temperature of 99°F, a heart rate of 105, and a pulse oxygenation saturation of 89%, a respiratory rate of 24 seems unbelievably low. In my experience, the respiratory rate is recorded erroneously most of the time unless it is recorded electronically or checked at the bedside by the physician using a timepiece with a sweep second-hand.
Additionally, I have found that ordering several days’ laboratory tests (eg, complete blood cell counts, chemistry panels) in advance, in many cases, risks missing important findings and wastes time, energy, and the patient’s blood. I have learned to evaluate each patient daily and to order the most pertinent laboratory tests. With electronic medical records, I can check laboratory results as soon as they are available.
- Mull N, Reilly JB, Myers JS. An elderly woman with ‘heart failure’: cognitive biases and diagnostic error. Cleve Clin J Med 2015; 82:745–753.
To the Editor: I appreciated the article on cognitive biases and diagnostic error by Mull et al in the November 2015 issue.1 They presented an excellent description of the pitfalls of diagnosis as reflected in a case of a patient misdiagnosed with heart failure who ultimately died of pulmonary tuberculosis complicated by pulmonary embolism (the latter possibly from using the wrong form of heparin). To the points they raised, I would like to add a few of my own about diagnosis in general and heart failure in particular.
First, any initial diagnosis not confirmed objectively within the first 24 hours should be questioned, and other possibilities should be investigated. I have found this to be essential for every day’s stay in the hospital and for every outpatient visit. The authors mention checklists as part of the solution to the problem of misdiagnosis, and I would suggest that confirmation of initial diagnoses be built into these checklists.
In the case of a presumptive diagnosis of an acute exacerbation of heart failure treated empirically with diuretics, the diagnosis should be confirmed by the next day’s response to the diuretics, ie, increased urine output, a lower respiratory rate, and a fall in the pro-B-type natriuretic peptide level. Moreover, a change in the radiographic appearance should be seen, and respiratory and pulmonary function should improve after the first 24 hours on oxygen supplementation plus diuretics. Daily patient weights are also critical in determining response to a diuretic, and are rarely done accurately. I order weights and review them daily for patients like this.
Second, it is good to look at things yourself, including the patient, medication lists, laboratory values, and radiographic films. The attending physician should look at the radiographs together with a senior radiologist. Seeing no improvement or change on the second hospital day, or seeing signs incompatible with heart failure, one could order computed tomography of the chest and begin to entertain pulmonary diagnoses.
Even vital signs can be questionable. For example, in the case presented here, with a temperature of 99°F, a heart rate of 105, and a pulse oxygenation saturation of 89%, a respiratory rate of 24 seems unbelievably low. In my experience, the respiratory rate is recorded erroneously most of the time unless it is recorded electronically or checked at the bedside by the physician using a timepiece with a sweep second-hand.
Additionally, I have found that ordering several days’ laboratory tests (eg, complete blood cell counts, chemistry panels) in advance, in many cases, risks missing important findings and wastes time, energy, and the patient’s blood. I have learned to evaluate each patient daily and to order the most pertinent laboratory tests. With electronic medical records, I can check laboratory results as soon as they are available.
To the Editor: I appreciated the article on cognitive biases and diagnostic error by Mull et al in the November 2015 issue.1 They presented an excellent description of the pitfalls of diagnosis as reflected in a case of a patient misdiagnosed with heart failure who ultimately died of pulmonary tuberculosis complicated by pulmonary embolism (the latter possibly from using the wrong form of heparin). To the points they raised, I would like to add a few of my own about diagnosis in general and heart failure in particular.
First, any initial diagnosis not confirmed objectively within the first 24 hours should be questioned, and other possibilities should be investigated. I have found this to be essential for every day’s stay in the hospital and for every outpatient visit. The authors mention checklists as part of the solution to the problem of misdiagnosis, and I would suggest that confirmation of initial diagnoses be built into these checklists.
In the case of a presumptive diagnosis of an acute exacerbation of heart failure treated empirically with diuretics, the diagnosis should be confirmed by the next day’s response to the diuretics, ie, increased urine output, a lower respiratory rate, and a fall in the pro-B-type natriuretic peptide level. Moreover, a change in the radiographic appearance should be seen, and respiratory and pulmonary function should improve after the first 24 hours on oxygen supplementation plus diuretics. Daily patient weights are also critical in determining response to a diuretic, and are rarely done accurately. I order weights and review them daily for patients like this.
Second, it is good to look at things yourself, including the patient, medication lists, laboratory values, and radiographic films. The attending physician should look at the radiographs together with a senior radiologist. Seeing no improvement or change on the second hospital day, or seeing signs incompatible with heart failure, one could order computed tomography of the chest and begin to entertain pulmonary diagnoses.
Even vital signs can be questionable. For example, in the case presented here, with a temperature of 99°F, a heart rate of 105, and a pulse oxygenation saturation of 89%, a respiratory rate of 24 seems unbelievably low. In my experience, the respiratory rate is recorded erroneously most of the time unless it is recorded electronically or checked at the bedside by the physician using a timepiece with a sweep second-hand.
Additionally, I have found that ordering several days’ laboratory tests (eg, complete blood cell counts, chemistry panels) in advance, in many cases, risks missing important findings and wastes time, energy, and the patient’s blood. I have learned to evaluate each patient daily and to order the most pertinent laboratory tests. With electronic medical records, I can check laboratory results as soon as they are available.
- Mull N, Reilly JB, Myers JS. An elderly woman with ‘heart failure’: cognitive biases and diagnostic error. Cleve Clin J Med 2015; 82:745–753.
- Mull N, Reilly JB, Myers JS. An elderly woman with ‘heart failure’: cognitive biases and diagnostic error. Cleve Clin J Med 2015; 82:745–753.
In reply: Cognitive bias and diagnostic error
In Reply: We thank Dr. Field for his insights and personal observations related to diagnosis and biases that contribute to diagnostic errors.
Dr. Field’s comment about the importance of revisiting one’s initial working diagnosis is consistent with our proposed diagnostic time out. A diagnostic time out can incorporate a short checklist and aid in debiasing clinicians when findings do not fit the case presentation, such as lack of response to diuretic therapy. Being mindful of slowing down and not necessarily rushing to judgment is another important component.1 Of note, the residents in our case did revisit their initial working diagnosis, as suggested by Dr. Field. Questions from learners have great potential to serve as debiasing instruments and should always be encouraged. Those who do not work with students can do the same by speaking with nurses or other members of the healthcare team, who offer observations that busy physicians might miss.
Our case highlights the problem that we lack objective criteria to diagnose symptomatic heart failure. While B-type natriuretic factor (BNP) has a strong negative predictive value, serial BNP measurements have not been established to be helpful in the management of heart failure.2 Although certain findings on chest radiography have strong positive and negative likelihood associations, the role of serial chest radiographs is less clear.3 Thus, heart failure remains a clinical diagnosis in current practice.
As Dr. Field points out, the accuracy and performance characteristics of diagnostic testing, such as the respiratory rate, need to be considered in conjunction with debiasing strategies to achieve higher diagnostic accuracy. Multiple factors can contribute to low-performing or misinterpreted diagnostic tests, and inaccurate vital signs have been shown to be similarly prone to potential error.4
Finally, we wholeheartedly agree with Dr. Field’s comment on unnecessary testing. High-value care is appropriate care. Using Bayesian reasoning to guide testing, monitoring the treatment course appropriately, and eliminating waste is highly likely to improve both value and diagnostic accuracy. Automated, ritual ordering of daily tests can indicate that thinking has been shut off, leaving clinicians susceptible to premature closure of the diagnostic process as well as the potential for “incidentalomas” to distract them from the right diagnosis, all the while leading to low-value care such as wasteful spending, patient dissatisfaction, and hospital-acquired anemia.5 We believe that deciding on a daily basis what the next day’s tests will be can be another powerful debiasing habit, one with benefits beyond diagnosis.
- Schiff GD. Minimizing diagnostic error: the importance of follow-up and feedback. Am J Med 2008; 121(suppl):S38–S42.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013; 128:e240–e327.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Philip KE, Pack E, Cambiano V, Rollmann H, Weil S, O’Beirne J. The accuracy of respiratory rate assessment by doctors in a London teaching hospital: a cross-sectional study. J Clin Monit Comput 2015; 29:455–460.
- Koch CG, Li L, Sun Z, et al. Hospital-acquired anemia: prevalence, outcomes, and healthcare implications. J Hosp Med 2013; 8:506–512.
In Reply: We thank Dr. Field for his insights and personal observations related to diagnosis and biases that contribute to diagnostic errors.
Dr. Field’s comment about the importance of revisiting one’s initial working diagnosis is consistent with our proposed diagnostic time out. A diagnostic time out can incorporate a short checklist and aid in debiasing clinicians when findings do not fit the case presentation, such as lack of response to diuretic therapy. Being mindful of slowing down and not necessarily rushing to judgment is another important component.1 Of note, the residents in our case did revisit their initial working diagnosis, as suggested by Dr. Field. Questions from learners have great potential to serve as debiasing instruments and should always be encouraged. Those who do not work with students can do the same by speaking with nurses or other members of the healthcare team, who offer observations that busy physicians might miss.
Our case highlights the problem that we lack objective criteria to diagnose symptomatic heart failure. While B-type natriuretic factor (BNP) has a strong negative predictive value, serial BNP measurements have not been established to be helpful in the management of heart failure.2 Although certain findings on chest radiography have strong positive and negative likelihood associations, the role of serial chest radiographs is less clear.3 Thus, heart failure remains a clinical diagnosis in current practice.
As Dr. Field points out, the accuracy and performance characteristics of diagnostic testing, such as the respiratory rate, need to be considered in conjunction with debiasing strategies to achieve higher diagnostic accuracy. Multiple factors can contribute to low-performing or misinterpreted diagnostic tests, and inaccurate vital signs have been shown to be similarly prone to potential error.4
Finally, we wholeheartedly agree with Dr. Field’s comment on unnecessary testing. High-value care is appropriate care. Using Bayesian reasoning to guide testing, monitoring the treatment course appropriately, and eliminating waste is highly likely to improve both value and diagnostic accuracy. Automated, ritual ordering of daily tests can indicate that thinking has been shut off, leaving clinicians susceptible to premature closure of the diagnostic process as well as the potential for “incidentalomas” to distract them from the right diagnosis, all the while leading to low-value care such as wasteful spending, patient dissatisfaction, and hospital-acquired anemia.5 We believe that deciding on a daily basis what the next day’s tests will be can be another powerful debiasing habit, one with benefits beyond diagnosis.
In Reply: We thank Dr. Field for his insights and personal observations related to diagnosis and biases that contribute to diagnostic errors.
Dr. Field’s comment about the importance of revisiting one’s initial working diagnosis is consistent with our proposed diagnostic time out. A diagnostic time out can incorporate a short checklist and aid in debiasing clinicians when findings do not fit the case presentation, such as lack of response to diuretic therapy. Being mindful of slowing down and not necessarily rushing to judgment is another important component.1 Of note, the residents in our case did revisit their initial working diagnosis, as suggested by Dr. Field. Questions from learners have great potential to serve as debiasing instruments and should always be encouraged. Those who do not work with students can do the same by speaking with nurses or other members of the healthcare team, who offer observations that busy physicians might miss.
Our case highlights the problem that we lack objective criteria to diagnose symptomatic heart failure. While B-type natriuretic factor (BNP) has a strong negative predictive value, serial BNP measurements have not been established to be helpful in the management of heart failure.2 Although certain findings on chest radiography have strong positive and negative likelihood associations, the role of serial chest radiographs is less clear.3 Thus, heart failure remains a clinical diagnosis in current practice.
As Dr. Field points out, the accuracy and performance characteristics of diagnostic testing, such as the respiratory rate, need to be considered in conjunction with debiasing strategies to achieve higher diagnostic accuracy. Multiple factors can contribute to low-performing or misinterpreted diagnostic tests, and inaccurate vital signs have been shown to be similarly prone to potential error.4
Finally, we wholeheartedly agree with Dr. Field’s comment on unnecessary testing. High-value care is appropriate care. Using Bayesian reasoning to guide testing, monitoring the treatment course appropriately, and eliminating waste is highly likely to improve both value and diagnostic accuracy. Automated, ritual ordering of daily tests can indicate that thinking has been shut off, leaving clinicians susceptible to premature closure of the diagnostic process as well as the potential for “incidentalomas” to distract them from the right diagnosis, all the while leading to low-value care such as wasteful spending, patient dissatisfaction, and hospital-acquired anemia.5 We believe that deciding on a daily basis what the next day’s tests will be can be another powerful debiasing habit, one with benefits beyond diagnosis.
- Schiff GD. Minimizing diagnostic error: the importance of follow-up and feedback. Am J Med 2008; 121(suppl):S38–S42.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013; 128:e240–e327.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Philip KE, Pack E, Cambiano V, Rollmann H, Weil S, O’Beirne J. The accuracy of respiratory rate assessment by doctors in a London teaching hospital: a cross-sectional study. J Clin Monit Comput 2015; 29:455–460.
- Koch CG, Li L, Sun Z, et al. Hospital-acquired anemia: prevalence, outcomes, and healthcare implications. J Hosp Med 2013; 8:506–512.
- Schiff GD. Minimizing diagnostic error: the importance of follow-up and feedback. Am J Med 2008; 121(suppl):S38–S42.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013; 128:e240–e327.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Philip KE, Pack E, Cambiano V, Rollmann H, Weil S, O’Beirne J. The accuracy of respiratory rate assessment by doctors in a London teaching hospital: a cross-sectional study. J Clin Monit Comput 2015; 29:455–460.
- Koch CG, Li L, Sun Z, et al. Hospital-acquired anemia: prevalence, outcomes, and healthcare implications. J Hosp Med 2013; 8:506–512.
Treating and preventing acute exacerbations of COPD
In contrast to stable chronic obstructive pulmonary disease (COPD),1 acute exacerbations of COPD pose special management challenges and can significantly increase the risk of morbidity and death and the cost of care.
This review addresses the definition and diagnosis of COPD exacerbations, disease burden and costs, etiology and pathogenesis, and management and prevention strategies.
DEFINITIONS ARE PROBLEMATIC
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines a COPD exacerbation as “an acute event characterized by a worsening of the patient’s respiratory symptoms that is beyond normal day-to-day variations and leads to a change in medication.”2 It further categorizes acute exacerbations by severity:
- Mild—treated with increased frequency of doses of existing medications
- Moderate—treated with corticosteroids or antibiotics, or both
- Severe—requires hospital utilization (either emergency room treatment or admission).
Although descriptive and useful for retrospective analyses, this current definition poses ambiguities for clinicians. Day-to-day variation in symptoms is not routinely assessed, so deviations from baseline may be difficult to detect. Although clinical tools are available for assessing symptoms in stable and exacerbated states (eg, the COPD assessment test3 and the Exacerbations of Chronic Pulmonary Disease Tool [EXACT]4), they have not been widely adopted in daily practice. Also, according to the current definition, the severity of an exacerbation can be classified only after the course of action is determined, so the severity is not helpful for forming a management strategy at bedside. In addition, physicians may have different thresholds for prescribing antibiotics and corticosteroids.
An earlier definition categorized a COPD exacerbation by the presence of its three cardinal symptoms (ie, increased shortness of breath, sputum volume, and purulence):
- Type I—all three symptoms present
- Type II—two symptoms present
- Type III—one symptom present, accompanied by at least one of the following: upper respiratory tract infection within the past 5 days, unexplained fever, increased wheezing or cough, or 20% increased respiratory rate or heart rate from baseline.
This older definition was successfully used in a prospective clinical trial to identify patients who benefited most from antibiotics for COPD exacerbations.5
Despite these caveats regarding a definition, most clinicians agree on the clinical presentation of a patient with COPD exacerbation: ie, having some combination of shortness of breath, increased sputum volume, and purulence. By the same token, patients with COPD who present with symptoms not typical of an exacerbation should be evaluated for another diagnosis. For instance, Tillie-Leblond et al6 reported that 49 (25%) of 197 patients hospitalized with an “unexplained” exacerbation of COPD were eventually diagnosed with pulmonary embolism.
EXACERBATIONS ARE COSTLY
The care of patients with COPD places a great burden on the healthcare system. Using multiple national databases, Ford et al7 estimated that medical costs in the United States in 2010 attributable to COPD and its complications were $32.1 billion.
The largest component of direct healthcare costs of COPD is exacerbations and subsequent hospitalizations.8 Data from a predominantly Medicare population indicate that the annualized mean COPD-related cost for a patient with no exacerbations was $1,425, compared with $12,765 for a patient with severe exacerbations.9 The investigators estimated that reducing exacerbations from two or more to none could save $5,125 per patient per year.
EXACERBATIONS AFFECT HEALTH BEYOND THE EVENT
COPD exacerbations are associated with a faster decline in lung function,10 reduced quality of life,11 and lost workdays.7 A single exacerbation may cause a decline in lung function and health status that may not return to baseline for several months, particularly if another exacerbation occurs within 6 months.12,13 COPD exacerbations have also been linked to poor clinical outcomes, including death.
In a prospective study in 304 men with COPD followed for 5 years, those who had three or more COPD exacerbations annually were four times as likely to die than patients who did not have an exacerbation.14 Nevertheless, the relationship with mortality may not be causal: Brusselle pointed out in an editorial15 that established mortality predictors for COPD do not include exacerbations, and symptomatic patients with COPD without any history of exacerbations are at greater risk of death than those who are asymptomatic but at high risk for exacerbations.
INFECTION + INFLAMMATION = EXACERBATION
An acute COPD exacerbation can be viewed as an acute inflammatory event superimposed on chronic inflammation associated with COPD. Inflammation in the airways increases resistance to air flow with consequent air trapping. Increased resistance and elastic load due to air trapping place respiratory muscles at a mechanical disadvantage and increase the work of breathing.
Infection starts the process
Infections, particularly bacterial and viral, are thought to be the major instigators of COPD exacerbation, although environmental factors such as air pollution may also play a role.16
Airway inflammation is markedly reduced when bacterial infection is eradicated. But if bacterial colonization continues, inflammatory markers remain elevated despite clinical resolution of the exacerbation.17 Desai et al18 found that patients with COPD and chronic bronchitis with bacterial colonization had a larger symptom burden than patients without colonization, even without an exacerbation.
Allergic profile increases risk
Although most studies indicate that infection is the main cause of exacerbations, clinicians should consider other mechanisms of inflammation on an individual basis. COPD exacerbations may be phenotyped by measuring inflammatory markers, perhaps as a starting point for tailored therapies.
Bafadhel et al19 studied 145 patients with COPD over the course of a year and recorded various biomarkers at baseline and during exacerbations. Exacerbations had an inflammatory profile that was predominantly bacterial in 37%, viral in 10%, and eosinophilic in 17%, and had limited changes in the inflammatory profile in 14%. The remaining episodes were combinations of categories. In another study,20 multivariate analysis conducted in two cohorts with COPD found that patients who had an allergic phenotype had more respiratory symptoms and a higher likelihood of COPD exacerbations.
Frequent COPD exacerbations are increasingly recognized as being associated with an asthma-COPD overlap syndrome, consisting of symptoms of increased airflow variability and incompletely reversible airflow obstruction.21
Inflammation as a marker of frequent exacerbations
Evidence is accumulating that supports systemic inflammation as a marker of frequent exacerbations. The Copenhagen Heart Study tested for baseline plasma C-reactive protein, fibrinogen, and white blood cell count in 6,574 stable patients with COPD.22 After multivariable adjustment, they found a significantly higher likelihood of having a subsequent exacerbation in patients who had all three biomarkers elevated (odds ratio [OR] 3.7, 95% confidence interval [CI] 1.9–7.4), even in patients with milder COPD and those without previous exacerbations.
Past exacerbations predict risk
The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints study23 found that a history of acute COPD exacerbation was the single best predictor of future exacerbations. This risk factor remained stable over 3 years and was present across the severity of COPD, ie, patients at lower GOLD stages who had a history of frequent exacerbations were likely to have exacerbations during follow-up.
EXACERBATION INCREASES CARDIOVASCULAR RISK
COPD exacerbations increase the risk of cardiovascular events, particularly myocardial infarction.24 During hospitalization for acute exacerbation of COPD, markers of myocardial injury and heart failure may be elevated and are a predictor of death.25
Patel et al26 measured arterial stiffness (aortic pulse wave velocity, a validated measure of cardiovascular risk) and cardiac biomarkers (troponin and N-terminal B-type natriuretic peptide) at baseline in 98 patients and longitudinally during and after a COPD exacerbation. In addition to increased levels of cardiac biomarkers, they found a significant rise in arterial stiffness during the exacerbation event without return to baseline levels over 35 days of follow-up. The arterial stiffness increase was related to airway inflammation as measured by sputum interleukin 6, particularly in patients with documented lower respiratory tract infection.
Retrospective analysis suggests a reduced all-cause mortality rate in COPD patients who are treated with beta-blockers.27
Recommendation. We recommend that patients already taking a selective beta-blocker continue to do so during a COPD exacerbation.
OUTPATIENT MANAGEMENT
Treatment with a combination of a corticosteroid, antibiotic, and bronchodilator addresses the underlying pathophysiologic processes of an acute exacerbation: inflammation, infection, and airway trapping.
Short course of a corticosteroid improves outcomes
A 10-day systemic course of a corticosteroid prescribed for COPD exacerbation before discharge from the emergency department was found to offer a small advantage over placebo for reducing treatment failure (unscheduled physician visits, return to emergency room for recurrent symptoms) and improving dyspnea scores and lung function.28 Even just a 3-day course improved measures of respiration (forced expiratory volume in the first second of expiration [FEV1] and arterial oxygenation) at days 3 and 10, and reduced treatment failures compared with placebo.29
Corticosteroid prescription should not be taken lightly, because adverse effects are common. In a systematic review, one adverse effect (hyperglycemia, weight gain, or insomnia) occurred for every five people treated.30
Identifying subgroups of patients most likely to benefit from corticosteroid treatment may be helpful. Corticosteroids may delay improvement in patients without eosinophilic inflammation and hasten recovery in those with more than 2% peripheral eosinophils.31 Siva et al32 found that limiting corticosteroids to patients with sputum eosinophilia reduced corticosteroid use and reduced severe exacerbations compared with standard care.32
Recommendation. For an acute exacerbation, we prescribe a short course of corticosteroids (eg, prednisone 40 mg daily for 5 to 7 days). Tapering dosing is probably unnecessary because adrenal insufficiency is uncommon before 2 weeks of corticosteroid exposure. Clinicians should weigh the merits of tapering (reduced corticosteroid exposure) against patient inconvenience and difficulty following complicated instructions.
Antibiotics help, but exact strategy uncertain
Although antibiotic therapy is one of the three pillars of COPD exacerbation management, the optimal antimicrobial agent, duration of therapy, and which patients will benefit remain areas of controversy and research. Thus far, large trials have been unable to definitely show the superiority of one antibiotic over another.33,34
A 1987 randomized controlled trial5 of antibiotic therapy in acute exacerbation of COPD found the greatest benefit to patients who had all three cardinal symptoms (ie, increased shortness of breath, sputum volume, and purulence), with less marked but still significant improvement in patients with two symptoms. In a 2012 multicenter trial35 patients with mild to moderate COPD experiencing an exacerbation were treated with either combined amoxicillin and clavulanate or placebo if they had one of the three cardinal symptoms. The antibiotic group had a significantly higher clinical cure rate at days 9 to 11 (74.1% vs 59.9%) as well as a longer time until the next exacerbation (233 vs 160 days).

Recommendation. Optimal antibiotic management of COPD exacerbations may also depend on risk factors. For patients with at least two cardinal symptoms, we favor a scheme akin to one proposed for treating community-acquired pneumonia (Table 1).16,36
INPATIENT MANAGEMENT
Corticosteroids improve outcomes
A Department of Veterans Affairs cooperative trial37 randomized 271 patients hospitalized with COPD exacerbation to receive either corticosteroids (intravenous followed by oral) or placebo for either 2 weeks or 8 weeks. Corticosteroid recipients had lower rates of treatment failure at 30 and 90 days, defined as death from any cause, need for mechanical ventilation, readmission, or intensification of pharmacologic therapy. Corticosteroid therapy also reduced hospital length of stay and improved the rate of recovery. The longer corticosteroid course was associated with a higher rate of adverse effects.
Oral corticosteroids not inferior to intravenous
Using the same end point of treatment failure as the Veterans Affairs cooperative trial, deJong et al38 demonstrated that prednisone 60 mg by mouth was not inferior to intravenous prednisone. Neither trial demonstrated a difference in mortality between corticosteroid use and placebo.
Short course of a corticosteroid not inferior to a long course
In 2013, the Reduction in the Use of Corticosteroids in Exacerbated COPD (REDUCE) trial39 randomized 314 patients presenting with an acute COPD exacerbation (92% requiring hospital admission) to oral prednisone 40 mg daily for either 5 days or 14 days. They found that the short course was noninferior in preventing exacerbations over the ensuing 6 months in terms of death and the need for mechanical ventilation.
Recommendation. Our threshold for initiating systemic corticosteroid therapy is lower in hospitalized patients than in outpatients. We recommend the regimen of the REDUCE trial: prednisone 40 mg daily for 5 days.
Corticosteroids for patients on ventilatory support
Severe COPD exacerbations requiring admission to intensive care are a significant source of morbidity and mortality, and the strategy of corticosteroid treatment is still under investigation.
Intravenous corticosteroids are effective. A multicenter trial40 in 354 patients requiring either invasive or noninvasive mechanical ventilation randomized them to treatment with either intravenous methylprednisolone (tapered) or placebo. Treatment was associated with fewer mechanical ventilation days and a lower rate of noninvasive ventilation failure.
Low-dose oral corticosteroids ineffective. In contrast, an open-label trial41 of patients requiring ventilatory support and randomized to either oral prednisone (1 mg/kg for up to 10 days) or usual care found no difference in intensive care length of stay or noninvasive ventilation failure. This study used the oral route and smaller doses, and its open-label design might have introduced bias.
Lower-dose steroids better than high-dose. A 2014 cohort study of 17,239 patients admitted to the ICU with acute exacerbations of COPD evaluated outcomes of treatment with high methylprednisolone dosages (> 240 mg per day) vs lower dosages, using propensity score matching.42 No mortality difference was found between the groups. The lower dosage group (median methylprednisolone dose 100 mg per day) had shorter hospital and intensive care unit stays, shorter duration of noninvasive positive pressure ventilation, less need for insulin therapy, and fewer fungal infections.
Antibiotics for hospitalized patients
Only scarce data are available on the use of antibiotics for patients hospitalized with COPD exacerbation. In a study of patients hospitalized with COPD exacerbations, adding doxycycline to corticosteroids led to better clinical success and cure rates at 10 days compared with placebo, but the primary end point of clinical success at 30 days was not different between the two groups.43
BRONCHODILATORS: A MAINSTAY OF COPD TREATMENT
Bronchodilators are an important part of treatment of COPD exacerbations in inpatient and outpatient settings.
Nebulized beta-2 agonists are given every 1 to 4 hours. Albuterol at a 2.5-mg dose in each nebulization was found to be as effective as 5 mg for length of hospital stay and recovery of lung function in patients with an acute exacerbation of COPD.44
Adding an anticholinergic may help. Nebulized anticholinergics can be given alone or combined with beta-2 agonists. Whether long-acting bronchodilators should be used to manage COPD patients hospitalized with an exacerbation requires further inquiry. In an observational study with historical controls, Drescher and colleagues45 found cost savings and shorter hospital stays if tiotropium (a long-acting anticholinergic) was added to the respiratory care protocol, which also included formoterol (a long-acting beta-2 agonist).
OXYGEN: TITRATED APPROACH SAFER
Oxygen should be supplied during a COPD exacerbation to ensure adequate oxyhemoglobin saturation. Caution is needed to avoid hyperoxemic hypercapnia, particularly in patients with severe COPD and propensity to ventilatory failure. The routine administration of oxygen at high concentrations during a COPD exacerbation has been associated with a higher mortality rate than with a titrated oxygen approach.46 Long-term oxygen treatment started at discharge or as outpatient therapy is associated with reduced hospital admissions and shorter hospital stays for acute exacerbations of COPD.47
VENTILATION SUPPORT
Noninvasive positive-pressure ventilation is a useful adjunct to treatment of COPD exacerbations with evidence of ventilatory failure (ie, acute respiratory acidosis), helping to offset the work of breathing until respiratory system mechanics improve. Keenan et al48 reviewed 15 randomized controlled trials, involving 636 patients, of noninvasive positive-pressure ventilation in the setting of COPD exacerbation. They concluded that noninvasive positive-pressure ventilation reduced the in-hospital mortality rate and length of stay compared with standard therapy. Noninvasive positive-pressure ventilation is most useful in patients with severe COPD exacerbations and acute respiratory acidosis (pH < 7.35).49
Intubation and mechanical ventilation. Although no standards exist for determining which COPD exacerbations may be too severe for noninvasive positive-pressure ventilation, intubation is clearly indicated for impending respiratory failure or hemodynamic instability. Other factors to consider include the greater likelihood of noninvasive positive-pressure ventilation failure in patients with severe respiratory acidosis (pH < 7.25 is associated with a > 50% failure rate) and in those with no improvement in acidosis or respiratory rate during the first hour after initiation of noninvasive positive-pressure ventilation.50
PREVENTING EXACERBATIONS

Recent data indicate that COPD exacerbations can often be prevented (Table 2).
Inhaled pharmacotherapy
Inhaled pharmacotherapeutic agents, singly or in combination, reduce the frequency of COPD exacerbations.
Combined long-acting beta-2 agonist and corticosteroid is better than single-agent therapy. In 2007, the Towards a Revolution in COPD Health (TORCH) trial51 evaluated outpatient therapy in more than 6,000 patients worldwide with either an inhaled long-acting beta-2 agonist (salmeterol), an inhaled corticosteroid (fluticasone), both drugs in combination, or placebo. Patients had baseline prebronchodilator FEV1 of less than 60% and were followed for 3 years. No difference was found between the groups in the primary end point of deaths, but the annualized rate of moderate to severe exacerbations was reduced by 25% in the group that received combination therapy vs placebo. Combination therapy showed superior efficacy over individual drug therapy in preventing exacerbations. Treatment with the inhaled corticosteroid, whether alone or in combination with salmeterol, increased the risk of pneumonia.
A long-acting antimuscarinic agent is better than placebo. In 2008, the Understanding Potential Long-Term Impacts on Function With Tiotropium (UPLIFT) trial52 randomized nearly 6,000 patients with COPD and a postbronchodilator FEV1 of less than 70% to placebo or tiotropium, a long-acting antimuscarinic agent. Tiotropium reduced the exacerbation rate by 14% compared with placebo and improved quality of life.
Antimuscarinics may be better than beta-2 agonists. Head-to-head comparisons suggest that long-acting antimuscarinic agents are preferable to long-acting beta-2 agonists for preventing COPD exacerbations.53,54
Triple therapy: evidence is mixed. For patients with severe symptomatic COPD and frequent exacerbations, triple therapy with a combination of an inhaled long-acting antimuscarinic agent, an inhaled long-acting beta-2 agonist, and an inhaled corticosteroid has been suggested.
Data to support this practice are limited. In the Canadian Optimal Trial,55 the rate of exacerbations was not different between tiotropium alone, tiotropium plus salmeterol, and triple therapy. However, the rate of hospitalization for severe exacerbation was lower with triple therapy than tiotropium alone. A large, retrospective cohort study also supported triple therapy by finding reduced mortality, hospitalizations, and need for oral corticosteroid bursts compared to combination therapy with an inhaled long-acting beta-2 agonist and an inhaled corticosteroid.56
The drawback of triple therapy is an increased incidence of pneumonia associated with combined beta-2 agonist and corticosteroids, most likely due to the corticosteroid component.51 The risk appears to be higher for higher potency corticosteroids, eg, fluticasone.57
In 2014, the Withdrawal of Inhaled Steroids During Optimised Bronchodilator Management (WISDOM) trial58 randomized nearly 2,500 patients with a history of COPD exacerbation receiving triple therapy consisting of tiotropium, salmeterol, and inhaled fluticasone to either continue treatment or withdraw the corticosteroid for 3 months. The investigators defined an annualized exacerbation rate of 1.2 (ie, a 20% increase) as the upper limit of the confidence interval for an acceptable therapeutic margin of noninferiority. The study showed that the risk of moderate to severe exacerbations with combined tiotropium and salmeterol was noninferior to triple therapy.
Nevertheless, caution is advised when removing the corticosteroid component from triple therapy. The trial demonstrated a worsening in overall health status, some reduction in lung function, and a transient increase in severe exacerbations in the withdrawal group. Patients with increased symptom burden at baseline and a history of severe exacerbations may not be optimal candidates for this strategy.
Roflumilast is effective but has side effects
Roflumilast, an oral phosphodiesterase 4 inhibitor, is an anti-inflammatory drug without bronchodilator properties. In randomized controlled trials, the drug was associated with a 17% reduction in acute exacerbations compared with placebo.59
Adding roflumilast to either a long-acting beta-2 agonist or a long-acting antimuscarinic agent resulted in a 6% to 8% further reduction in the proportion of patients with exacerbation.60,61 Martinez et al61 found that roflumilast added to a regimen of a long-acting beta-2 agonist plus an inhaled corticosteroid reduced moderate to severe exacerbations by 14.2%, even in the presence of tiotropium. Compared with placebo, roflumilast treatment reduced exacerbations necessitating hospitalizations by 23.9%.
The FDA has approved oral roflumilast 500 µg once daily to prevent COPD exacerbations.
Roflumilast is frequently associated with side effects, including gastrointestinal symptoms (chiefly diarrhea), weight loss, and psychiatric effects. A benefit-to-harm study in 2014 concluded that using the drug is only favorable for patients who have a high risk of severe exacerbations, ie, those who have a greater than 22% baseline risk of having at least one exacerbation annually.62
Recommendation. Roflumilast should be reserved for patients who have severe COPD with a chronic bronchitis phenotype (ie, with cough and sputum production) and repeated exacerbations despite an optimal regimen of an inhaled corticosteroid, long-acting beta-2 agonist, and long-acting antimuscarinic agent.
Macrolide antibiotics: Role unclear
Macrolide antibiotics have anti-inflammatory and immunomodulatory activities.
Azithromycin: fewer exacerbations but some side effects. A multicenter trial63 in 1,142 COPD patients randomized to either oral azithromycin 250 mg daily or placebo found a 27% reduction in the risk of COPD exacerbation in the intervention arm. No differences were found between the groups in mortality, hospitalizations, emergency department visits, or respiratory failure. Hearing loss and increased macrolide resistance were noted in the intervention arm. In a secondary subgroup analysis,64 no difference in efficacy was found by sex, history of chronic bronchitis, oxygen use, or concomitant COPD treatment.
The COPD: Influence of Macrolides on Exacerbation Frequency in Patients trial65 helped refine patient selection for macrolide therapy. In this single-center study, 92 patients with COPD and at least three exacerbations during the year prior to enrollment were randomized to receive either azithromycin 500 mg three times weekly or placebo. Exacerbations in the intervention group were markedly reduced (42%) with no difference in hospitalization rate.
The place of macrolide antibiotics in the treatment strategy of COPD is unclear, and they are not currently part of the GOLD guidelines. Still unknown is the incremental benefit of adding them to existing preventive regimens, cardiovascular safety, side effects, and potential effects on the resident microbial flora.
Other antibiotics have also been investigated for efficacy in preventing exacerbations.
Moxifloxacin: fewer exacerbations. The Pulsed Moxifloxacin Usage and Its Long-term Impact on the Reduction of Subsequent Exacerbations study66 randomized more than 1,000 patients with stable COPD to receive either moxifloxacin 400 mg or placebo daily for 5 days repeated every 8 weeks for six courses. Frequent assessment during the treatment period and for 6 months afterward revealed a reduced exacerbation rate in the intervention group but without benefit in hospitalization rate, mortality, lung function, or health status.
Recommendation. Azithromycin (either 250 mg daily or 500 mg three times weekly) can be considered for patients who have repeated COPD exacerbations despite an optimal regimen of an inhaled corticosteroid, inhaled long-acting beta-2 agonist, and inhaled long-acting antimuscarinic agent. The need to continue azithromycin should be reassessed yearly.
Mucolytics
Greatest benefit to patients not taking inhaled corticosteroids. Mucolytic agents help clear airway secretions by reducing viscosity. N-acetylcysteine and carbocysteine (not available in the United States) also have antioxidant properties that may counteract oxidant stress associated with acute COPD exacerbations.
The Bronchitis Randomized on NAC Cost-Utility Study (BRONCUS)67 randomized 523 COPD patients to N-acetylcysteine 600 mg daily or placebo. After 3 years of follow-up, no differences were found in the rate of exacerbations, lung function decline, and quality of life. Subgroup analysis suggested a reduction in exacerbations for patients who were not taking inhaled corticosteroids.
The Effect of Carbocisteine on Acute Exacerbation of Chronic Obstructive Pulmonary Disease (PEACE) study randomized more than 700 patients from multiple centers in China who had COPD and a recent history of exacerbations; they found a 25% lower exacerbation rate over 1 year with carbocysteine vs placebo.68 Most of the patients (83%) were not on inhaled corticosteroids, which complemented findings of the BRONCUS trial.
The Effect of High Dose N-acetylcysteine on Air Trapping and Airway Resistance of COPD (HIACE) study randomized 120 patients with stable COPD in a hospital in Hong Kong to either oral N-acetylcysteine (600 mg twice daily) or placebo and found a reduced exacerbation rate of exacerbations. Patients were matched at baseline for inhaled corticosteroid use.69
In 2014, the Twice Daily N-acetylcysteine 600 mg for Exacerbations of Chronic Obstructive Pulmonary Disease (PANTHEON) study70 randomized 1,006 patients from multiple hospitals in China with a history of moderate to severe COPD and exacerbations to receive either N-acetylcysteine 600 mg twice daily or placebo for 1 year. They found a 22% reduction in exacerbations in the treatment group vs placebo.
GOLD guidelines2 recommend mucolytics for patients with severe COPD and exacerbations when inhaled corticosteroids are not available or affordable.
Recommendation. Mucolytics may be useful for patients with difficulty expectorating and with a history of exacerbations despite appropriate inhaled therapy.
OTHER INTERVENTIONS CAN HELP
Pulmonary rehabilitation provides multiple benefits
Pulmonary rehabilitation increases exercise tolerance and reduces symptom burden in patients with stable COPD. It is also a multidisciplinary effort that may help reinforce adherence to medications, enhance COPD education, and provide closer medical surveillance to patients at high risk for recurrent exacerbations.
A small randomized controlled trial71 prescribed pulmonary rehabilitation on discharge for a COPD exacerbation and found sustainable improvements in exercise capacity and health status after 3 months.
In a later study,72 the same group started pulmonary rehabilitation within a week of hospital discharge and found reduced hospital readmissions over a 3-month period.
Smoking cessation is always worth advocating
A large observational cohort study concluded that current smokers were at a higher risk for COPD exacerbations compared with former smokers.73 Although there are no randomized controlled trials that assess the effects of smoking cessation at the time of COPD exacerbation, we recommend seizing the opportunity to implement this important intervention.
Vaccinations: Influenza and pneumococcal
Influenza vaccination is associated with reduced incidence of hospitalization among patients with cardiopulmonary disease.74 A meta-analysis of randomized clinical trials of influenza vaccination for patients with COPD75 reported significantly fewer exacerbations from vaccination, mostly owing to fewer episodes occurring after 3 to 4 weeks, coinciding with anticipated vaccine-induced immune protection. Furumoto and colleagues76 reported an added benefit of combined vaccination with 23-valent pneumococcal polysaccharide vaccine and influenza vaccine in reducing hospital admissions over influenza vaccination alone. We also recommend providing the 13-valent pneumococcal conjugate vaccine to patients with COPD, particularly for those older than 65, consistent with CDC recommendations.77
- Hatipoglu U, Aboussouan LS. Chronic obstructive pulmonary disease: an update for the primary physician. Cleve Clin J Med 2014; 81:373–383.
- Global initiative for chronic obstructive lung disease. Pocket Guide to COPD Diagnosis, Management, and Prevention. A Guide for Health Care Professionals. Updated 2016. http://www.goldcopd.org/uploads/users/files/WatermarkedPocket%20Guide%202016(2).pdf. Accessed March 7, 2016.
- Gupta N, Pinto LM, Morogan A, Bourbeau J. The COPD assessment test: a systematic review. Eur Respir J 2014; 44:873–884.
- Leidy NK, Wilcox TK, Jones PW, et al; EXACT-PRO Study Group. Development of the EXAcerbations of chronic obstructive pulmonary disease tool (EXACT): a patient-reported outcome (PRO) measure. Value Health 2010; 13:965–975.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Tillie-Leblond I, Marquette CH, Perez T, et al. Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: prevalence and risk factors. Ann Intern Med 2006; 144:390–396.
- Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged ≥ 18 years in the United States for 2010 and projections through 2020. Chest 2015; 147:31–45.
- Toy EL, Gallagher KF, Stanley EL, Swensen AR, Duh MS. The economic impact of exacerbations of chronic obstructive pulmonary disease and exacerbation definition: a review. COPD 2010; 7:214–228.
- Pasquale MK, Sun SX, Song F, Hartnett HJ, Stemkowski SA. Impact of exacerbations on health care cost and resource utilization in chronic obstructive pulmonary disease patients with chronic bronchitis from a predominantly Medicare population. Int J Chron Obstruct Pulmon Dis 2012; 7:757–764.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157:1418–1422.
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 161:1608–1613.
- Spencer S, Calverley PM, Sherwood Burge P, Jones PW; ISOLDE Study Group, Inhaled Steroids in Obstructive Lung Disease. Health status deterioration in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:122–128.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Brusselle G. Why doesn’t reducing exacerbations decrease COPD mortality? Lancet Respir Med 2014; 2:681–683.
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359:2355–2365.
- White AJ, Gompertz S, Bayley DL, et al. Resolution of bronchial inflammation is related to bacterial eradication following treatment of exacerbations of chronic bronchitis. Thorax 2003; 58:680–685.
- Desai H, Eschberger K, Wrona C, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc 2014; 11:303–309.
- Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184:662–671.
- Jamieson DB, Matsui EC, Belli A, et al. Effects of allergic phenotype on respiratory symptoms and exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:187–192.
- Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax 2009; 64:728–735.
- Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA 2013; 309:2353–2361.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Donaldson GC, Hurst JR, Smith CJ, Hubbard RB, Wedzicha JA. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest 2010; 137:1091–1097.
- Chang CL, Robinson SC, Mills GD, et al. Biochemical markers of cardiac dysfunction predict mortality in acute exacerbations of COPD. Thorax 2011; 66:764–768.
- Patel AR, Kowlessar BS, Donaldson GC, et al. Cardiovascular risk, myocardial injury, and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:1091–1099.
- Dransfield MT, Rowe SM, Johnson JE, Bailey WC, Gerald LB. Use of beta blockers and the risk of death in hospitalised patients with acute exacerbations of COPD. Thorax 2008; 63:301–305.
- Thompson WH, Nielson CP, Carvalho P, Charan NB, Crowley JJ. Controlled trial of oral prednisone in outpatients with acute COPD exacerbation. Am J Respir Crit Care Med 1996; 154:407–412.
- Aaron SD, Vandemheen KL, Hebert P, et al. Outpatient oral prednisone after emergency treatment of chronic obstructive pulmonary disease. N Engl J Med 2003; 348:2618–2625.
- Walters JA, Gibson PG, Wood-Baker R, Hannay M, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2009; 1:CD001288.
- Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med 2012; 186:48–55.
- Siva R, Green RH, Brightling CE, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J 2007; 29:906–913.
- Wilson R, Allegra L, Huchon G, et al; MOSAIC Study Group. Short-term and long-term outcomes of moxifloxacin compared to standard antibiotic treatment in acute exacerbations of chronic bronchitis. Chest 2004; 125:953–964.
- Wilson R, Anzueto A, Miravitlles M, et al. Moxifloxacin versus amoxicillin/clavulanic acid in outpatient acute exacerbations of COPD: MAESTRAL results. Eur Respir J 2012; 40:17–27.
- Llor C, Moragas A, Hernandez S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Anzueto A. Primary care management of chronic obstructive pulmonary disease to reduce exacerbations and their consequences. Am J Med Sci 2010; 340:309–318.
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med 1999; 340:1941–1947.
- de Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: a randomized, controlled, double-blind study. Chest 2007; 132:1741–1747.
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 2013; 309:2223–2231.
- Alia I, de la Cal MA, Esteban A, et al. Efficacy of corticosteroid therapy in patients with an acute exacerbation of chronic obstructive pulmonary disease receiving ventilatory support. Arch Intern Med 2011; 171:1939–1946.
- Abroug F, Ouanes-Besbes L, Fkih-Hassen M, et al. Prednisone in COPD exacerbation requiring ventilatory support: an open-label randomised evaluation. Eur Respir J 2014; 43:717–724.
- Kiser TH, Allen RR, Valuck RJ, Moss M, Vandivier RW. Outcomes associated with corticosteroid dosage in critically ill patients with acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 189:1052–1064.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Nair S, Thomas E, Pearson SB, Henry MT. A randomized controlled trial to assess the optimal dose and effect of nebulized albuterol in acute exacerbations of COPD. Chest 2005; 128:48–54.
- Drescher GS, Carnathan BJ, Imus S, Colice GL. Incorporating tiotropium into a respiratory therapist-directed bronchodilator protocol for managing in-patients with COPD exacerbations decreases bronchodilator costs. Respir Care 2008; 53:1678–1684.
- Austin MA, Wills KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ 2010; 341:c5462.
- Ringbaek TJ, Viskum K, Lange P. Does long-term oxygen therapy reduce hospitalisation in hypoxaemic chronic obstructive pulmonary disease? Eur Respir J 2002; 20:38–42.
- Keenan SP, Sinuff T, Cook DJ, Hill NS. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive-pressure ventilation? A systematic review of the literature. Ann Intern Med 2003; 138:861–870.
- Quon BS, Gan WQ, Sin DD. Contemporary management of acute exacerbations of COPD: a systematic review and metaanalysis. Chest 2008; 133:756–766.
- Sinuff T, Keenan SP; Department of Medicine, McMaster University. Clinical practice guideline for the use of noninvasive positive pressure ventilation in COPD patients with acute respiratory failure. J Crit Care 2004; 19:82–91.
- Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Vogelmeier C, Hederer B, Glaab T, et al; POET-COPD Investigators. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011; 364:1093–1103.
- Decramer ML, Chapman KR, Dahl R, et al; INVIGORATE investigators. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013; 1:524–533.
- Aaron SD, Vandemheen KL, Fergusson D, et al; Canadian Thoracic Society/Canadian Respiratory Clinical Research Consortium. Tiotropium in combination with placebo, salmeterol, or fluticasone-salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2007; 146:545–555.
- Short PM, Williamson PA, Elder DH, Lipworth SI, Schembri S, Lipworth BJ. The impact of tiotropium on mortality and exacerbations when added to inhaled corticosteroids and long-acting beta-agonist therapy in COPD. Chest 2012; 141:81–86.
- Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax 2013; 68:1029–1036.
- Magnussen H, Disse B, Rodriguez-Roisin R, et al; WISDOM Investigators. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med 2014; 371:1285–1294.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385:857–866.
- Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax 2014; 69:616–622.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014; 189:1503–1508.
- Uzun S, Djamin RS, Kluytmans JA, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2014; 2:361–368.
- Sethi S, Jones PW, Theron MS, et al; PULSE Study group. Pulsed moxifloxacin for the prevention of exacerbations of chronic obstructive pulmonary disease: a randomized controlled trial. Respir Res 2010; 11:10.
- Decramer M, Rutten-van Molken M, Dekhuijzen PN, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized On NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet 2005; 365:1552–1560.
- Zheng JP, Kang J, Huang SG, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE study): a randomised placebo-controlled study. Lancet 2008; 371:2013–2018.
- Tse HN, Raiteri L, Wong KY, et al. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 2013; 144:106–118.
- Zheng JP, Wen FQ, Bai CX, et al; PANTHEON study group. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2014; 2:187–194.
- Man WD, Polkey MI, Donaldson N, Gray BJ, Moxham J. Community pulmonary rehabilitation after hospitalisation for acute exacerbations of chronic obstructive pulmonary disease: randomised controlled study. BMJ 2004; 329:1209.
- Seymour JM, Moore L, Jolley CJ, et al. Outpatient pulmonary rehabilitation following acute exacerbations of COPD. Thorax 2010; 65:423–428.
- Au DH, Bryson CL, Chien JW, et al. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med 2009; 24:457–463.
- Seo YB, Hong KW, Kim IS, et al. Effectiveness of the influenza vaccine at preventing hospitalization due to acute lower respiratory infection and exacerbation of chronic cardiopulmonary disease in Korea during 2010-2011. Vaccine 2013; 31:1426–1430.
- Poole PJ, Chacko E, Wood-Baker RW, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2006; 1:CD002733.
- Furumoto A, Ohkusa Y, Chen M, et al. Additive effect of pneumococcal vaccine and influenza vaccine on acute exacerbation in patients with chronic lung disease. Vaccine 2008; 26:4284–4289.
- Tomczyk S, Bennett NM, Stoecker C, et al; Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥65 years: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2014; 63:822–825.
In contrast to stable chronic obstructive pulmonary disease (COPD),1 acute exacerbations of COPD pose special management challenges and can significantly increase the risk of morbidity and death and the cost of care.
This review addresses the definition and diagnosis of COPD exacerbations, disease burden and costs, etiology and pathogenesis, and management and prevention strategies.
DEFINITIONS ARE PROBLEMATIC
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines a COPD exacerbation as “an acute event characterized by a worsening of the patient’s respiratory symptoms that is beyond normal day-to-day variations and leads to a change in medication.”2 It further categorizes acute exacerbations by severity:
- Mild—treated with increased frequency of doses of existing medications
- Moderate—treated with corticosteroids or antibiotics, or both
- Severe—requires hospital utilization (either emergency room treatment or admission).
Although descriptive and useful for retrospective analyses, this current definition poses ambiguities for clinicians. Day-to-day variation in symptoms is not routinely assessed, so deviations from baseline may be difficult to detect. Although clinical tools are available for assessing symptoms in stable and exacerbated states (eg, the COPD assessment test3 and the Exacerbations of Chronic Pulmonary Disease Tool [EXACT]4), they have not been widely adopted in daily practice. Also, according to the current definition, the severity of an exacerbation can be classified only after the course of action is determined, so the severity is not helpful for forming a management strategy at bedside. In addition, physicians may have different thresholds for prescribing antibiotics and corticosteroids.
An earlier definition categorized a COPD exacerbation by the presence of its three cardinal symptoms (ie, increased shortness of breath, sputum volume, and purulence):
- Type I—all three symptoms present
- Type II—two symptoms present
- Type III—one symptom present, accompanied by at least one of the following: upper respiratory tract infection within the past 5 days, unexplained fever, increased wheezing or cough, or 20% increased respiratory rate or heart rate from baseline.
This older definition was successfully used in a prospective clinical trial to identify patients who benefited most from antibiotics for COPD exacerbations.5
Despite these caveats regarding a definition, most clinicians agree on the clinical presentation of a patient with COPD exacerbation: ie, having some combination of shortness of breath, increased sputum volume, and purulence. By the same token, patients with COPD who present with symptoms not typical of an exacerbation should be evaluated for another diagnosis. For instance, Tillie-Leblond et al6 reported that 49 (25%) of 197 patients hospitalized with an “unexplained” exacerbation of COPD were eventually diagnosed with pulmonary embolism.
EXACERBATIONS ARE COSTLY
The care of patients with COPD places a great burden on the healthcare system. Using multiple national databases, Ford et al7 estimated that medical costs in the United States in 2010 attributable to COPD and its complications were $32.1 billion.
The largest component of direct healthcare costs of COPD is exacerbations and subsequent hospitalizations.8 Data from a predominantly Medicare population indicate that the annualized mean COPD-related cost for a patient with no exacerbations was $1,425, compared with $12,765 for a patient with severe exacerbations.9 The investigators estimated that reducing exacerbations from two or more to none could save $5,125 per patient per year.
EXACERBATIONS AFFECT HEALTH BEYOND THE EVENT
COPD exacerbations are associated with a faster decline in lung function,10 reduced quality of life,11 and lost workdays.7 A single exacerbation may cause a decline in lung function and health status that may not return to baseline for several months, particularly if another exacerbation occurs within 6 months.12,13 COPD exacerbations have also been linked to poor clinical outcomes, including death.
In a prospective study in 304 men with COPD followed for 5 years, those who had three or more COPD exacerbations annually were four times as likely to die than patients who did not have an exacerbation.14 Nevertheless, the relationship with mortality may not be causal: Brusselle pointed out in an editorial15 that established mortality predictors for COPD do not include exacerbations, and symptomatic patients with COPD without any history of exacerbations are at greater risk of death than those who are asymptomatic but at high risk for exacerbations.
INFECTION + INFLAMMATION = EXACERBATION
An acute COPD exacerbation can be viewed as an acute inflammatory event superimposed on chronic inflammation associated with COPD. Inflammation in the airways increases resistance to air flow with consequent air trapping. Increased resistance and elastic load due to air trapping place respiratory muscles at a mechanical disadvantage and increase the work of breathing.
Infection starts the process
Infections, particularly bacterial and viral, are thought to be the major instigators of COPD exacerbation, although environmental factors such as air pollution may also play a role.16
Airway inflammation is markedly reduced when bacterial infection is eradicated. But if bacterial colonization continues, inflammatory markers remain elevated despite clinical resolution of the exacerbation.17 Desai et al18 found that patients with COPD and chronic bronchitis with bacterial colonization had a larger symptom burden than patients without colonization, even without an exacerbation.
Allergic profile increases risk
Although most studies indicate that infection is the main cause of exacerbations, clinicians should consider other mechanisms of inflammation on an individual basis. COPD exacerbations may be phenotyped by measuring inflammatory markers, perhaps as a starting point for tailored therapies.
Bafadhel et al19 studied 145 patients with COPD over the course of a year and recorded various biomarkers at baseline and during exacerbations. Exacerbations had an inflammatory profile that was predominantly bacterial in 37%, viral in 10%, and eosinophilic in 17%, and had limited changes in the inflammatory profile in 14%. The remaining episodes were combinations of categories. In another study,20 multivariate analysis conducted in two cohorts with COPD found that patients who had an allergic phenotype had more respiratory symptoms and a higher likelihood of COPD exacerbations.
Frequent COPD exacerbations are increasingly recognized as being associated with an asthma-COPD overlap syndrome, consisting of symptoms of increased airflow variability and incompletely reversible airflow obstruction.21
Inflammation as a marker of frequent exacerbations
Evidence is accumulating that supports systemic inflammation as a marker of frequent exacerbations. The Copenhagen Heart Study tested for baseline plasma C-reactive protein, fibrinogen, and white blood cell count in 6,574 stable patients with COPD.22 After multivariable adjustment, they found a significantly higher likelihood of having a subsequent exacerbation in patients who had all three biomarkers elevated (odds ratio [OR] 3.7, 95% confidence interval [CI] 1.9–7.4), even in patients with milder COPD and those without previous exacerbations.
Past exacerbations predict risk
The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints study23 found that a history of acute COPD exacerbation was the single best predictor of future exacerbations. This risk factor remained stable over 3 years and was present across the severity of COPD, ie, patients at lower GOLD stages who had a history of frequent exacerbations were likely to have exacerbations during follow-up.
EXACERBATION INCREASES CARDIOVASCULAR RISK
COPD exacerbations increase the risk of cardiovascular events, particularly myocardial infarction.24 During hospitalization for acute exacerbation of COPD, markers of myocardial injury and heart failure may be elevated and are a predictor of death.25
Patel et al26 measured arterial stiffness (aortic pulse wave velocity, a validated measure of cardiovascular risk) and cardiac biomarkers (troponin and N-terminal B-type natriuretic peptide) at baseline in 98 patients and longitudinally during and after a COPD exacerbation. In addition to increased levels of cardiac biomarkers, they found a significant rise in arterial stiffness during the exacerbation event without return to baseline levels over 35 days of follow-up. The arterial stiffness increase was related to airway inflammation as measured by sputum interleukin 6, particularly in patients with documented lower respiratory tract infection.
Retrospective analysis suggests a reduced all-cause mortality rate in COPD patients who are treated with beta-blockers.27
Recommendation. We recommend that patients already taking a selective beta-blocker continue to do so during a COPD exacerbation.
OUTPATIENT MANAGEMENT
Treatment with a combination of a corticosteroid, antibiotic, and bronchodilator addresses the underlying pathophysiologic processes of an acute exacerbation: inflammation, infection, and airway trapping.
Short course of a corticosteroid improves outcomes
A 10-day systemic course of a corticosteroid prescribed for COPD exacerbation before discharge from the emergency department was found to offer a small advantage over placebo for reducing treatment failure (unscheduled physician visits, return to emergency room for recurrent symptoms) and improving dyspnea scores and lung function.28 Even just a 3-day course improved measures of respiration (forced expiratory volume in the first second of expiration [FEV1] and arterial oxygenation) at days 3 and 10, and reduced treatment failures compared with placebo.29
Corticosteroid prescription should not be taken lightly, because adverse effects are common. In a systematic review, one adverse effect (hyperglycemia, weight gain, or insomnia) occurred for every five people treated.30
Identifying subgroups of patients most likely to benefit from corticosteroid treatment may be helpful. Corticosteroids may delay improvement in patients without eosinophilic inflammation and hasten recovery in those with more than 2% peripheral eosinophils.31 Siva et al32 found that limiting corticosteroids to patients with sputum eosinophilia reduced corticosteroid use and reduced severe exacerbations compared with standard care.32
Recommendation. For an acute exacerbation, we prescribe a short course of corticosteroids (eg, prednisone 40 mg daily for 5 to 7 days). Tapering dosing is probably unnecessary because adrenal insufficiency is uncommon before 2 weeks of corticosteroid exposure. Clinicians should weigh the merits of tapering (reduced corticosteroid exposure) against patient inconvenience and difficulty following complicated instructions.
Antibiotics help, but exact strategy uncertain
Although antibiotic therapy is one of the three pillars of COPD exacerbation management, the optimal antimicrobial agent, duration of therapy, and which patients will benefit remain areas of controversy and research. Thus far, large trials have been unable to definitely show the superiority of one antibiotic over another.33,34
A 1987 randomized controlled trial5 of antibiotic therapy in acute exacerbation of COPD found the greatest benefit to patients who had all three cardinal symptoms (ie, increased shortness of breath, sputum volume, and purulence), with less marked but still significant improvement in patients with two symptoms. In a 2012 multicenter trial35 patients with mild to moderate COPD experiencing an exacerbation were treated with either combined amoxicillin and clavulanate or placebo if they had one of the three cardinal symptoms. The antibiotic group had a significantly higher clinical cure rate at days 9 to 11 (74.1% vs 59.9%) as well as a longer time until the next exacerbation (233 vs 160 days).
Recommendation. Optimal antibiotic management of COPD exacerbations may also depend on risk factors. For patients with at least two cardinal symptoms, we favor a scheme akin to one proposed for treating community-acquired pneumonia (Table 1).16,36
INPATIENT MANAGEMENT
Corticosteroids improve outcomes
A Department of Veterans Affairs cooperative trial37 randomized 271 patients hospitalized with COPD exacerbation to receive either corticosteroids (intravenous followed by oral) or placebo for either 2 weeks or 8 weeks. Corticosteroid recipients had lower rates of treatment failure at 30 and 90 days, defined as death from any cause, need for mechanical ventilation, readmission, or intensification of pharmacologic therapy. Corticosteroid therapy also reduced hospital length of stay and improved the rate of recovery. The longer corticosteroid course was associated with a higher rate of adverse effects.
Oral corticosteroids not inferior to intravenous
Using the same end point of treatment failure as the Veterans Affairs cooperative trial, deJong et al38 demonstrated that prednisone 60 mg by mouth was not inferior to intravenous prednisone. Neither trial demonstrated a difference in mortality between corticosteroid use and placebo.
Short course of a corticosteroid not inferior to a long course
In 2013, the Reduction in the Use of Corticosteroids in Exacerbated COPD (REDUCE) trial39 randomized 314 patients presenting with an acute COPD exacerbation (92% requiring hospital admission) to oral prednisone 40 mg daily for either 5 days or 14 days. They found that the short course was noninferior in preventing exacerbations over the ensuing 6 months in terms of death and the need for mechanical ventilation.
Recommendation. Our threshold for initiating systemic corticosteroid therapy is lower in hospitalized patients than in outpatients. We recommend the regimen of the REDUCE trial: prednisone 40 mg daily for 5 days.
Corticosteroids for patients on ventilatory support
Severe COPD exacerbations requiring admission to intensive care are a significant source of morbidity and mortality, and the strategy of corticosteroid treatment is still under investigation.
Intravenous corticosteroids are effective. A multicenter trial40 in 354 patients requiring either invasive or noninvasive mechanical ventilation randomized them to treatment with either intravenous methylprednisolone (tapered) or placebo. Treatment was associated with fewer mechanical ventilation days and a lower rate of noninvasive ventilation failure.
Low-dose oral corticosteroids ineffective. In contrast, an open-label trial41 of patients requiring ventilatory support and randomized to either oral prednisone (1 mg/kg for up to 10 days) or usual care found no difference in intensive care length of stay or noninvasive ventilation failure. This study used the oral route and smaller doses, and its open-label design might have introduced bias.
Lower-dose steroids better than high-dose. A 2014 cohort study of 17,239 patients admitted to the ICU with acute exacerbations of COPD evaluated outcomes of treatment with high methylprednisolone dosages (> 240 mg per day) vs lower dosages, using propensity score matching.42 No mortality difference was found between the groups. The lower dosage group (median methylprednisolone dose 100 mg per day) had shorter hospital and intensive care unit stays, shorter duration of noninvasive positive pressure ventilation, less need for insulin therapy, and fewer fungal infections.
Antibiotics for hospitalized patients
Only scarce data are available on the use of antibiotics for patients hospitalized with COPD exacerbation. In a study of patients hospitalized with COPD exacerbations, adding doxycycline to corticosteroids led to better clinical success and cure rates at 10 days compared with placebo, but the primary end point of clinical success at 30 days was not different between the two groups.43
BRONCHODILATORS: A MAINSTAY OF COPD TREATMENT
Bronchodilators are an important part of treatment of COPD exacerbations in inpatient and outpatient settings.
Nebulized beta-2 agonists are given every 1 to 4 hours. Albuterol at a 2.5-mg dose in each nebulization was found to be as effective as 5 mg for length of hospital stay and recovery of lung function in patients with an acute exacerbation of COPD.44
Adding an anticholinergic may help. Nebulized anticholinergics can be given alone or combined with beta-2 agonists. Whether long-acting bronchodilators should be used to manage COPD patients hospitalized with an exacerbation requires further inquiry. In an observational study with historical controls, Drescher and colleagues45 found cost savings and shorter hospital stays if tiotropium (a long-acting anticholinergic) was added to the respiratory care protocol, which also included formoterol (a long-acting beta-2 agonist).
OXYGEN: TITRATED APPROACH SAFER
Oxygen should be supplied during a COPD exacerbation to ensure adequate oxyhemoglobin saturation. Caution is needed to avoid hyperoxemic hypercapnia, particularly in patients with severe COPD and propensity to ventilatory failure. The routine administration of oxygen at high concentrations during a COPD exacerbation has been associated with a higher mortality rate than with a titrated oxygen approach.46 Long-term oxygen treatment started at discharge or as outpatient therapy is associated with reduced hospital admissions and shorter hospital stays for acute exacerbations of COPD.47
VENTILATION SUPPORT
Noninvasive positive-pressure ventilation is a useful adjunct to treatment of COPD exacerbations with evidence of ventilatory failure (ie, acute respiratory acidosis), helping to offset the work of breathing until respiratory system mechanics improve. Keenan et al48 reviewed 15 randomized controlled trials, involving 636 patients, of noninvasive positive-pressure ventilation in the setting of COPD exacerbation. They concluded that noninvasive positive-pressure ventilation reduced the in-hospital mortality rate and length of stay compared with standard therapy. Noninvasive positive-pressure ventilation is most useful in patients with severe COPD exacerbations and acute respiratory acidosis (pH < 7.35).49
Intubation and mechanical ventilation. Although no standards exist for determining which COPD exacerbations may be too severe for noninvasive positive-pressure ventilation, intubation is clearly indicated for impending respiratory failure or hemodynamic instability. Other factors to consider include the greater likelihood of noninvasive positive-pressure ventilation failure in patients with severe respiratory acidosis (pH < 7.25 is associated with a > 50% failure rate) and in those with no improvement in acidosis or respiratory rate during the first hour after initiation of noninvasive positive-pressure ventilation.50
PREVENTING EXACERBATIONS
Recent data indicate that COPD exacerbations can often be prevented (Table 2).
Inhaled pharmacotherapy
Inhaled pharmacotherapeutic agents, singly or in combination, reduce the frequency of COPD exacerbations.
Combined long-acting beta-2 agonist and corticosteroid is better than single-agent therapy. In 2007, the Towards a Revolution in COPD Health (TORCH) trial51 evaluated outpatient therapy in more than 6,000 patients worldwide with either an inhaled long-acting beta-2 agonist (salmeterol), an inhaled corticosteroid (fluticasone), both drugs in combination, or placebo. Patients had baseline prebronchodilator FEV1 of less than 60% and were followed for 3 years. No difference was found between the groups in the primary end point of deaths, but the annualized rate of moderate to severe exacerbations was reduced by 25% in the group that received combination therapy vs placebo. Combination therapy showed superior efficacy over individual drug therapy in preventing exacerbations. Treatment with the inhaled corticosteroid, whether alone or in combination with salmeterol, increased the risk of pneumonia.
A long-acting antimuscarinic agent is better than placebo. In 2008, the Understanding Potential Long-Term Impacts on Function With Tiotropium (UPLIFT) trial52 randomized nearly 6,000 patients with COPD and a postbronchodilator FEV1 of less than 70% to placebo or tiotropium, a long-acting antimuscarinic agent. Tiotropium reduced the exacerbation rate by 14% compared with placebo and improved quality of life.
Antimuscarinics may be better than beta-2 agonists. Head-to-head comparisons suggest that long-acting antimuscarinic agents are preferable to long-acting beta-2 agonists for preventing COPD exacerbations.53,54
Triple therapy: evidence is mixed. For patients with severe symptomatic COPD and frequent exacerbations, triple therapy with a combination of an inhaled long-acting antimuscarinic agent, an inhaled long-acting beta-2 agonist, and an inhaled corticosteroid has been suggested.
Data to support this practice are limited. In the Canadian Optimal Trial,55 the rate of exacerbations was not different between tiotropium alone, tiotropium plus salmeterol, and triple therapy. However, the rate of hospitalization for severe exacerbation was lower with triple therapy than tiotropium alone. A large, retrospective cohort study also supported triple therapy by finding reduced mortality, hospitalizations, and need for oral corticosteroid bursts compared to combination therapy with an inhaled long-acting beta-2 agonist and an inhaled corticosteroid.56
The drawback of triple therapy is an increased incidence of pneumonia associated with combined beta-2 agonist and corticosteroids, most likely due to the corticosteroid component.51 The risk appears to be higher for higher potency corticosteroids, eg, fluticasone.57
In 2014, the Withdrawal of Inhaled Steroids During Optimised Bronchodilator Management (WISDOM) trial58 randomized nearly 2,500 patients with a history of COPD exacerbation receiving triple therapy consisting of tiotropium, salmeterol, and inhaled fluticasone to either continue treatment or withdraw the corticosteroid for 3 months. The investigators defined an annualized exacerbation rate of 1.2 (ie, a 20% increase) as the upper limit of the confidence interval for an acceptable therapeutic margin of noninferiority. The study showed that the risk of moderate to severe exacerbations with combined tiotropium and salmeterol was noninferior to triple therapy.
Nevertheless, caution is advised when removing the corticosteroid component from triple therapy. The trial demonstrated a worsening in overall health status, some reduction in lung function, and a transient increase in severe exacerbations in the withdrawal group. Patients with increased symptom burden at baseline and a history of severe exacerbations may not be optimal candidates for this strategy.
Roflumilast is effective but has side effects
Roflumilast, an oral phosphodiesterase 4 inhibitor, is an anti-inflammatory drug without bronchodilator properties. In randomized controlled trials, the drug was associated with a 17% reduction in acute exacerbations compared with placebo.59
Adding roflumilast to either a long-acting beta-2 agonist or a long-acting antimuscarinic agent resulted in a 6% to 8% further reduction in the proportion of patients with exacerbation.60,61 Martinez et al61 found that roflumilast added to a regimen of a long-acting beta-2 agonist plus an inhaled corticosteroid reduced moderate to severe exacerbations by 14.2%, even in the presence of tiotropium. Compared with placebo, roflumilast treatment reduced exacerbations necessitating hospitalizations by 23.9%.
The FDA has approved oral roflumilast 500 µg once daily to prevent COPD exacerbations.
Roflumilast is frequently associated with side effects, including gastrointestinal symptoms (chiefly diarrhea), weight loss, and psychiatric effects. A benefit-to-harm study in 2014 concluded that using the drug is only favorable for patients who have a high risk of severe exacerbations, ie, those who have a greater than 22% baseline risk of having at least one exacerbation annually.62
Recommendation. Roflumilast should be reserved for patients who have severe COPD with a chronic bronchitis phenotype (ie, with cough and sputum production) and repeated exacerbations despite an optimal regimen of an inhaled corticosteroid, long-acting beta-2 agonist, and long-acting antimuscarinic agent.
Macrolide antibiotics: Role unclear
Macrolide antibiotics have anti-inflammatory and immunomodulatory activities.
Azithromycin: fewer exacerbations but some side effects. A multicenter trial63 in 1,142 COPD patients randomized to either oral azithromycin 250 mg daily or placebo found a 27% reduction in the risk of COPD exacerbation in the intervention arm. No differences were found between the groups in mortality, hospitalizations, emergency department visits, or respiratory failure. Hearing loss and increased macrolide resistance were noted in the intervention arm. In a secondary subgroup analysis,64 no difference in efficacy was found by sex, history of chronic bronchitis, oxygen use, or concomitant COPD treatment.
The COPD: Influence of Macrolides on Exacerbation Frequency in Patients trial65 helped refine patient selection for macrolide therapy. In this single-center study, 92 patients with COPD and at least three exacerbations during the year prior to enrollment were randomized to receive either azithromycin 500 mg three times weekly or placebo. Exacerbations in the intervention group were markedly reduced (42%) with no difference in hospitalization rate.
The place of macrolide antibiotics in the treatment strategy of COPD is unclear, and they are not currently part of the GOLD guidelines. Still unknown is the incremental benefit of adding them to existing preventive regimens, cardiovascular safety, side effects, and potential effects on the resident microbial flora.
Other antibiotics have also been investigated for efficacy in preventing exacerbations.
Moxifloxacin: fewer exacerbations. The Pulsed Moxifloxacin Usage and Its Long-term Impact on the Reduction of Subsequent Exacerbations study66 randomized more than 1,000 patients with stable COPD to receive either moxifloxacin 400 mg or placebo daily for 5 days repeated every 8 weeks for six courses. Frequent assessment during the treatment period and for 6 months afterward revealed a reduced exacerbation rate in the intervention group but without benefit in hospitalization rate, mortality, lung function, or health status.
Recommendation. Azithromycin (either 250 mg daily or 500 mg three times weekly) can be considered for patients who have repeated COPD exacerbations despite an optimal regimen of an inhaled corticosteroid, inhaled long-acting beta-2 agonist, and inhaled long-acting antimuscarinic agent. The need to continue azithromycin should be reassessed yearly.
Mucolytics
Greatest benefit to patients not taking inhaled corticosteroids. Mucolytic agents help clear airway secretions by reducing viscosity. N-acetylcysteine and carbocysteine (not available in the United States) also have antioxidant properties that may counteract oxidant stress associated with acute COPD exacerbations.
The Bronchitis Randomized on NAC Cost-Utility Study (BRONCUS)67 randomized 523 COPD patients to N-acetylcysteine 600 mg daily or placebo. After 3 years of follow-up, no differences were found in the rate of exacerbations, lung function decline, and quality of life. Subgroup analysis suggested a reduction in exacerbations for patients who were not taking inhaled corticosteroids.
The Effect of Carbocisteine on Acute Exacerbation of Chronic Obstructive Pulmonary Disease (PEACE) study randomized more than 700 patients from multiple centers in China who had COPD and a recent history of exacerbations; they found a 25% lower exacerbation rate over 1 year with carbocysteine vs placebo.68 Most of the patients (83%) were not on inhaled corticosteroids, which complemented findings of the BRONCUS trial.
The Effect of High Dose N-acetylcysteine on Air Trapping and Airway Resistance of COPD (HIACE) study randomized 120 patients with stable COPD in a hospital in Hong Kong to either oral N-acetylcysteine (600 mg twice daily) or placebo and found a reduced exacerbation rate of exacerbations. Patients were matched at baseline for inhaled corticosteroid use.69
In 2014, the Twice Daily N-acetylcysteine 600 mg for Exacerbations of Chronic Obstructive Pulmonary Disease (PANTHEON) study70 randomized 1,006 patients from multiple hospitals in China with a history of moderate to severe COPD and exacerbations to receive either N-acetylcysteine 600 mg twice daily or placebo for 1 year. They found a 22% reduction in exacerbations in the treatment group vs placebo.
GOLD guidelines2 recommend mucolytics for patients with severe COPD and exacerbations when inhaled corticosteroids are not available or affordable.
Recommendation. Mucolytics may be useful for patients with difficulty expectorating and with a history of exacerbations despite appropriate inhaled therapy.
OTHER INTERVENTIONS CAN HELP
Pulmonary rehabilitation provides multiple benefits
Pulmonary rehabilitation increases exercise tolerance and reduces symptom burden in patients with stable COPD. It is also a multidisciplinary effort that may help reinforce adherence to medications, enhance COPD education, and provide closer medical surveillance to patients at high risk for recurrent exacerbations.
A small randomized controlled trial71 prescribed pulmonary rehabilitation on discharge for a COPD exacerbation and found sustainable improvements in exercise capacity and health status after 3 months.
In a later study,72 the same group started pulmonary rehabilitation within a week of hospital discharge and found reduced hospital readmissions over a 3-month period.
Smoking cessation is always worth advocating
A large observational cohort study concluded that current smokers were at a higher risk for COPD exacerbations compared with former smokers.73 Although there are no randomized controlled trials that assess the effects of smoking cessation at the time of COPD exacerbation, we recommend seizing the opportunity to implement this important intervention.
Vaccinations: Influenza and pneumococcal
Influenza vaccination is associated with reduced incidence of hospitalization among patients with cardiopulmonary disease.74 A meta-analysis of randomized clinical trials of influenza vaccination for patients with COPD75 reported significantly fewer exacerbations from vaccination, mostly owing to fewer episodes occurring after 3 to 4 weeks, coinciding with anticipated vaccine-induced immune protection. Furumoto and colleagues76 reported an added benefit of combined vaccination with 23-valent pneumococcal polysaccharide vaccine and influenza vaccine in reducing hospital admissions over influenza vaccination alone. We also recommend providing the 13-valent pneumococcal conjugate vaccine to patients with COPD, particularly for those older than 65, consistent with CDC recommendations.77
In contrast to stable chronic obstructive pulmonary disease (COPD),1 acute exacerbations of COPD pose special management challenges and can significantly increase the risk of morbidity and death and the cost of care.
This review addresses the definition and diagnosis of COPD exacerbations, disease burden and costs, etiology and pathogenesis, and management and prevention strategies.
DEFINITIONS ARE PROBLEMATIC
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines a COPD exacerbation as “an acute event characterized by a worsening of the patient’s respiratory symptoms that is beyond normal day-to-day variations and leads to a change in medication.”2 It further categorizes acute exacerbations by severity:
- Mild—treated with increased frequency of doses of existing medications
- Moderate—treated with corticosteroids or antibiotics, or both
- Severe—requires hospital utilization (either emergency room treatment or admission).
Although descriptive and useful for retrospective analyses, this current definition poses ambiguities for clinicians. Day-to-day variation in symptoms is not routinely assessed, so deviations from baseline may be difficult to detect. Although clinical tools are available for assessing symptoms in stable and exacerbated states (eg, the COPD assessment test3 and the Exacerbations of Chronic Pulmonary Disease Tool [EXACT]4), they have not been widely adopted in daily practice. Also, according to the current definition, the severity of an exacerbation can be classified only after the course of action is determined, so the severity is not helpful for forming a management strategy at bedside. In addition, physicians may have different thresholds for prescribing antibiotics and corticosteroids.
An earlier definition categorized a COPD exacerbation by the presence of its three cardinal symptoms (ie, increased shortness of breath, sputum volume, and purulence):
- Type I—all three symptoms present
- Type II—two symptoms present
- Type III—one symptom present, accompanied by at least one of the following: upper respiratory tract infection within the past 5 days, unexplained fever, increased wheezing or cough, or 20% increased respiratory rate or heart rate from baseline.
This older definition was successfully used in a prospective clinical trial to identify patients who benefited most from antibiotics for COPD exacerbations.5
Despite these caveats regarding a definition, most clinicians agree on the clinical presentation of a patient with COPD exacerbation: ie, having some combination of shortness of breath, increased sputum volume, and purulence. By the same token, patients with COPD who present with symptoms not typical of an exacerbation should be evaluated for another diagnosis. For instance, Tillie-Leblond et al6 reported that 49 (25%) of 197 patients hospitalized with an “unexplained” exacerbation of COPD were eventually diagnosed with pulmonary embolism.
EXACERBATIONS ARE COSTLY
The care of patients with COPD places a great burden on the healthcare system. Using multiple national databases, Ford et al7 estimated that medical costs in the United States in 2010 attributable to COPD and its complications were $32.1 billion.
The largest component of direct healthcare costs of COPD is exacerbations and subsequent hospitalizations.8 Data from a predominantly Medicare population indicate that the annualized mean COPD-related cost for a patient with no exacerbations was $1,425, compared with $12,765 for a patient with severe exacerbations.9 The investigators estimated that reducing exacerbations from two or more to none could save $5,125 per patient per year.
EXACERBATIONS AFFECT HEALTH BEYOND THE EVENT
COPD exacerbations are associated with a faster decline in lung function,10 reduced quality of life,11 and lost workdays.7 A single exacerbation may cause a decline in lung function and health status that may not return to baseline for several months, particularly if another exacerbation occurs within 6 months.12,13 COPD exacerbations have also been linked to poor clinical outcomes, including death.
In a prospective study in 304 men with COPD followed for 5 years, those who had three or more COPD exacerbations annually were four times as likely to die than patients who did not have an exacerbation.14 Nevertheless, the relationship with mortality may not be causal: Brusselle pointed out in an editorial15 that established mortality predictors for COPD do not include exacerbations, and symptomatic patients with COPD without any history of exacerbations are at greater risk of death than those who are asymptomatic but at high risk for exacerbations.
INFECTION + INFLAMMATION = EXACERBATION
An acute COPD exacerbation can be viewed as an acute inflammatory event superimposed on chronic inflammation associated with COPD. Inflammation in the airways increases resistance to air flow with consequent air trapping. Increased resistance and elastic load due to air trapping place respiratory muscles at a mechanical disadvantage and increase the work of breathing.
Infection starts the process
Infections, particularly bacterial and viral, are thought to be the major instigators of COPD exacerbation, although environmental factors such as air pollution may also play a role.16
Airway inflammation is markedly reduced when bacterial infection is eradicated. But if bacterial colonization continues, inflammatory markers remain elevated despite clinical resolution of the exacerbation.17 Desai et al18 found that patients with COPD and chronic bronchitis with bacterial colonization had a larger symptom burden than patients without colonization, even without an exacerbation.
Allergic profile increases risk
Although most studies indicate that infection is the main cause of exacerbations, clinicians should consider other mechanisms of inflammation on an individual basis. COPD exacerbations may be phenotyped by measuring inflammatory markers, perhaps as a starting point for tailored therapies.
Bafadhel et al19 studied 145 patients with COPD over the course of a year and recorded various biomarkers at baseline and during exacerbations. Exacerbations had an inflammatory profile that was predominantly bacterial in 37%, viral in 10%, and eosinophilic in 17%, and had limited changes in the inflammatory profile in 14%. The remaining episodes were combinations of categories. In another study,20 multivariate analysis conducted in two cohorts with COPD found that patients who had an allergic phenotype had more respiratory symptoms and a higher likelihood of COPD exacerbations.
Frequent COPD exacerbations are increasingly recognized as being associated with an asthma-COPD overlap syndrome, consisting of symptoms of increased airflow variability and incompletely reversible airflow obstruction.21
Inflammation as a marker of frequent exacerbations
Evidence is accumulating that supports systemic inflammation as a marker of frequent exacerbations. The Copenhagen Heart Study tested for baseline plasma C-reactive protein, fibrinogen, and white blood cell count in 6,574 stable patients with COPD.22 After multivariable adjustment, they found a significantly higher likelihood of having a subsequent exacerbation in patients who had all three biomarkers elevated (odds ratio [OR] 3.7, 95% confidence interval [CI] 1.9–7.4), even in patients with milder COPD and those without previous exacerbations.
Past exacerbations predict risk
The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints study23 found that a history of acute COPD exacerbation was the single best predictor of future exacerbations. This risk factor remained stable over 3 years and was present across the severity of COPD, ie, patients at lower GOLD stages who had a history of frequent exacerbations were likely to have exacerbations during follow-up.
EXACERBATION INCREASES CARDIOVASCULAR RISK
COPD exacerbations increase the risk of cardiovascular events, particularly myocardial infarction.24 During hospitalization for acute exacerbation of COPD, markers of myocardial injury and heart failure may be elevated and are a predictor of death.25
Patel et al26 measured arterial stiffness (aortic pulse wave velocity, a validated measure of cardiovascular risk) and cardiac biomarkers (troponin and N-terminal B-type natriuretic peptide) at baseline in 98 patients and longitudinally during and after a COPD exacerbation. In addition to increased levels of cardiac biomarkers, they found a significant rise in arterial stiffness during the exacerbation event without return to baseline levels over 35 days of follow-up. The arterial stiffness increase was related to airway inflammation as measured by sputum interleukin 6, particularly in patients with documented lower respiratory tract infection.
Retrospective analysis suggests a reduced all-cause mortality rate in COPD patients who are treated with beta-blockers.27
Recommendation. We recommend that patients already taking a selective beta-blocker continue to do so during a COPD exacerbation.
OUTPATIENT MANAGEMENT
Treatment with a combination of a corticosteroid, antibiotic, and bronchodilator addresses the underlying pathophysiologic processes of an acute exacerbation: inflammation, infection, and airway trapping.
Short course of a corticosteroid improves outcomes
A 10-day systemic course of a corticosteroid prescribed for COPD exacerbation before discharge from the emergency department was found to offer a small advantage over placebo for reducing treatment failure (unscheduled physician visits, return to emergency room for recurrent symptoms) and improving dyspnea scores and lung function.28 Even just a 3-day course improved measures of respiration (forced expiratory volume in the first second of expiration [FEV1] and arterial oxygenation) at days 3 and 10, and reduced treatment failures compared with placebo.29
Corticosteroid prescription should not be taken lightly, because adverse effects are common. In a systematic review, one adverse effect (hyperglycemia, weight gain, or insomnia) occurred for every five people treated.30
Identifying subgroups of patients most likely to benefit from corticosteroid treatment may be helpful. Corticosteroids may delay improvement in patients without eosinophilic inflammation and hasten recovery in those with more than 2% peripheral eosinophils.31 Siva et al32 found that limiting corticosteroids to patients with sputum eosinophilia reduced corticosteroid use and reduced severe exacerbations compared with standard care.32
Recommendation. For an acute exacerbation, we prescribe a short course of corticosteroids (eg, prednisone 40 mg daily for 5 to 7 days). Tapering dosing is probably unnecessary because adrenal insufficiency is uncommon before 2 weeks of corticosteroid exposure. Clinicians should weigh the merits of tapering (reduced corticosteroid exposure) against patient inconvenience and difficulty following complicated instructions.
Antibiotics help, but exact strategy uncertain
Although antibiotic therapy is one of the three pillars of COPD exacerbation management, the optimal antimicrobial agent, duration of therapy, and which patients will benefit remain areas of controversy and research. Thus far, large trials have been unable to definitely show the superiority of one antibiotic over another.33,34
A 1987 randomized controlled trial5 of antibiotic therapy in acute exacerbation of COPD found the greatest benefit to patients who had all three cardinal symptoms (ie, increased shortness of breath, sputum volume, and purulence), with less marked but still significant improvement in patients with two symptoms. In a 2012 multicenter trial35 patients with mild to moderate COPD experiencing an exacerbation were treated with either combined amoxicillin and clavulanate or placebo if they had one of the three cardinal symptoms. The antibiotic group had a significantly higher clinical cure rate at days 9 to 11 (74.1% vs 59.9%) as well as a longer time until the next exacerbation (233 vs 160 days).
Recommendation. Optimal antibiotic management of COPD exacerbations may also depend on risk factors. For patients with at least two cardinal symptoms, we favor a scheme akin to one proposed for treating community-acquired pneumonia (Table 1).16,36
INPATIENT MANAGEMENT
Corticosteroids improve outcomes
A Department of Veterans Affairs cooperative trial37 randomized 271 patients hospitalized with COPD exacerbation to receive either corticosteroids (intravenous followed by oral) or placebo for either 2 weeks or 8 weeks. Corticosteroid recipients had lower rates of treatment failure at 30 and 90 days, defined as death from any cause, need for mechanical ventilation, readmission, or intensification of pharmacologic therapy. Corticosteroid therapy also reduced hospital length of stay and improved the rate of recovery. The longer corticosteroid course was associated with a higher rate of adverse effects.
Oral corticosteroids not inferior to intravenous
Using the same end point of treatment failure as the Veterans Affairs cooperative trial, deJong et al38 demonstrated that prednisone 60 mg by mouth was not inferior to intravenous prednisone. Neither trial demonstrated a difference in mortality between corticosteroid use and placebo.
Short course of a corticosteroid not inferior to a long course
In 2013, the Reduction in the Use of Corticosteroids in Exacerbated COPD (REDUCE) trial39 randomized 314 patients presenting with an acute COPD exacerbation (92% requiring hospital admission) to oral prednisone 40 mg daily for either 5 days or 14 days. They found that the short course was noninferior in preventing exacerbations over the ensuing 6 months in terms of death and the need for mechanical ventilation.
Recommendation. Our threshold for initiating systemic corticosteroid therapy is lower in hospitalized patients than in outpatients. We recommend the regimen of the REDUCE trial: prednisone 40 mg daily for 5 days.
Corticosteroids for patients on ventilatory support
Severe COPD exacerbations requiring admission to intensive care are a significant source of morbidity and mortality, and the strategy of corticosteroid treatment is still under investigation.
Intravenous corticosteroids are effective. A multicenter trial40 in 354 patients requiring either invasive or noninvasive mechanical ventilation randomized them to treatment with either intravenous methylprednisolone (tapered) or placebo. Treatment was associated with fewer mechanical ventilation days and a lower rate of noninvasive ventilation failure.
Low-dose oral corticosteroids ineffective. In contrast, an open-label trial41 of patients requiring ventilatory support and randomized to either oral prednisone (1 mg/kg for up to 10 days) or usual care found no difference in intensive care length of stay or noninvasive ventilation failure. This study used the oral route and smaller doses, and its open-label design might have introduced bias.
Lower-dose steroids better than high-dose. A 2014 cohort study of 17,239 patients admitted to the ICU with acute exacerbations of COPD evaluated outcomes of treatment with high methylprednisolone dosages (> 240 mg per day) vs lower dosages, using propensity score matching.42 No mortality difference was found between the groups. The lower dosage group (median methylprednisolone dose 100 mg per day) had shorter hospital and intensive care unit stays, shorter duration of noninvasive positive pressure ventilation, less need for insulin therapy, and fewer fungal infections.
Antibiotics for hospitalized patients
Only scarce data are available on the use of antibiotics for patients hospitalized with COPD exacerbation. In a study of patients hospitalized with COPD exacerbations, adding doxycycline to corticosteroids led to better clinical success and cure rates at 10 days compared with placebo, but the primary end point of clinical success at 30 days was not different between the two groups.43
BRONCHODILATORS: A MAINSTAY OF COPD TREATMENT
Bronchodilators are an important part of treatment of COPD exacerbations in inpatient and outpatient settings.
Nebulized beta-2 agonists are given every 1 to 4 hours. Albuterol at a 2.5-mg dose in each nebulization was found to be as effective as 5 mg for length of hospital stay and recovery of lung function in patients with an acute exacerbation of COPD.44
Adding an anticholinergic may help. Nebulized anticholinergics can be given alone or combined with beta-2 agonists. Whether long-acting bronchodilators should be used to manage COPD patients hospitalized with an exacerbation requires further inquiry. In an observational study with historical controls, Drescher and colleagues45 found cost savings and shorter hospital stays if tiotropium (a long-acting anticholinergic) was added to the respiratory care protocol, which also included formoterol (a long-acting beta-2 agonist).
OXYGEN: TITRATED APPROACH SAFER
Oxygen should be supplied during a COPD exacerbation to ensure adequate oxyhemoglobin saturation. Caution is needed to avoid hyperoxemic hypercapnia, particularly in patients with severe COPD and propensity to ventilatory failure. The routine administration of oxygen at high concentrations during a COPD exacerbation has been associated with a higher mortality rate than with a titrated oxygen approach.46 Long-term oxygen treatment started at discharge or as outpatient therapy is associated with reduced hospital admissions and shorter hospital stays for acute exacerbations of COPD.47
VENTILATION SUPPORT
Noninvasive positive-pressure ventilation is a useful adjunct to treatment of COPD exacerbations with evidence of ventilatory failure (ie, acute respiratory acidosis), helping to offset the work of breathing until respiratory system mechanics improve. Keenan et al48 reviewed 15 randomized controlled trials, involving 636 patients, of noninvasive positive-pressure ventilation in the setting of COPD exacerbation. They concluded that noninvasive positive-pressure ventilation reduced the in-hospital mortality rate and length of stay compared with standard therapy. Noninvasive positive-pressure ventilation is most useful in patients with severe COPD exacerbations and acute respiratory acidosis (pH < 7.35).49
Intubation and mechanical ventilation. Although no standards exist for determining which COPD exacerbations may be too severe for noninvasive positive-pressure ventilation, intubation is clearly indicated for impending respiratory failure or hemodynamic instability. Other factors to consider include the greater likelihood of noninvasive positive-pressure ventilation failure in patients with severe respiratory acidosis (pH < 7.25 is associated with a > 50% failure rate) and in those with no improvement in acidosis or respiratory rate during the first hour after initiation of noninvasive positive-pressure ventilation.50
PREVENTING EXACERBATIONS
Recent data indicate that COPD exacerbations can often be prevented (Table 2).
Inhaled pharmacotherapy
Inhaled pharmacotherapeutic agents, singly or in combination, reduce the frequency of COPD exacerbations.
Combined long-acting beta-2 agonist and corticosteroid is better than single-agent therapy. In 2007, the Towards a Revolution in COPD Health (TORCH) trial51 evaluated outpatient therapy in more than 6,000 patients worldwide with either an inhaled long-acting beta-2 agonist (salmeterol), an inhaled corticosteroid (fluticasone), both drugs in combination, or placebo. Patients had baseline prebronchodilator FEV1 of less than 60% and were followed for 3 years. No difference was found between the groups in the primary end point of deaths, but the annualized rate of moderate to severe exacerbations was reduced by 25% in the group that received combination therapy vs placebo. Combination therapy showed superior efficacy over individual drug therapy in preventing exacerbations. Treatment with the inhaled corticosteroid, whether alone or in combination with salmeterol, increased the risk of pneumonia.
A long-acting antimuscarinic agent is better than placebo. In 2008, the Understanding Potential Long-Term Impacts on Function With Tiotropium (UPLIFT) trial52 randomized nearly 6,000 patients with COPD and a postbronchodilator FEV1 of less than 70% to placebo or tiotropium, a long-acting antimuscarinic agent. Tiotropium reduced the exacerbation rate by 14% compared with placebo and improved quality of life.
Antimuscarinics may be better than beta-2 agonists. Head-to-head comparisons suggest that long-acting antimuscarinic agents are preferable to long-acting beta-2 agonists for preventing COPD exacerbations.53,54
Triple therapy: evidence is mixed. For patients with severe symptomatic COPD and frequent exacerbations, triple therapy with a combination of an inhaled long-acting antimuscarinic agent, an inhaled long-acting beta-2 agonist, and an inhaled corticosteroid has been suggested.
Data to support this practice are limited. In the Canadian Optimal Trial,55 the rate of exacerbations was not different between tiotropium alone, tiotropium plus salmeterol, and triple therapy. However, the rate of hospitalization for severe exacerbation was lower with triple therapy than tiotropium alone. A large, retrospective cohort study also supported triple therapy by finding reduced mortality, hospitalizations, and need for oral corticosteroid bursts compared to combination therapy with an inhaled long-acting beta-2 agonist and an inhaled corticosteroid.56
The drawback of triple therapy is an increased incidence of pneumonia associated with combined beta-2 agonist and corticosteroids, most likely due to the corticosteroid component.51 The risk appears to be higher for higher potency corticosteroids, eg, fluticasone.57
In 2014, the Withdrawal of Inhaled Steroids During Optimised Bronchodilator Management (WISDOM) trial58 randomized nearly 2,500 patients with a history of COPD exacerbation receiving triple therapy consisting of tiotropium, salmeterol, and inhaled fluticasone to either continue treatment or withdraw the corticosteroid for 3 months. The investigators defined an annualized exacerbation rate of 1.2 (ie, a 20% increase) as the upper limit of the confidence interval for an acceptable therapeutic margin of noninferiority. The study showed that the risk of moderate to severe exacerbations with combined tiotropium and salmeterol was noninferior to triple therapy.
Nevertheless, caution is advised when removing the corticosteroid component from triple therapy. The trial demonstrated a worsening in overall health status, some reduction in lung function, and a transient increase in severe exacerbations in the withdrawal group. Patients with increased symptom burden at baseline and a history of severe exacerbations may not be optimal candidates for this strategy.
Roflumilast is effective but has side effects
Roflumilast, an oral phosphodiesterase 4 inhibitor, is an anti-inflammatory drug without bronchodilator properties. In randomized controlled trials, the drug was associated with a 17% reduction in acute exacerbations compared with placebo.59
Adding roflumilast to either a long-acting beta-2 agonist or a long-acting antimuscarinic agent resulted in a 6% to 8% further reduction in the proportion of patients with exacerbation.60,61 Martinez et al61 found that roflumilast added to a regimen of a long-acting beta-2 agonist plus an inhaled corticosteroid reduced moderate to severe exacerbations by 14.2%, even in the presence of tiotropium. Compared with placebo, roflumilast treatment reduced exacerbations necessitating hospitalizations by 23.9%.
The FDA has approved oral roflumilast 500 µg once daily to prevent COPD exacerbations.
Roflumilast is frequently associated with side effects, including gastrointestinal symptoms (chiefly diarrhea), weight loss, and psychiatric effects. A benefit-to-harm study in 2014 concluded that using the drug is only favorable for patients who have a high risk of severe exacerbations, ie, those who have a greater than 22% baseline risk of having at least one exacerbation annually.62
Recommendation. Roflumilast should be reserved for patients who have severe COPD with a chronic bronchitis phenotype (ie, with cough and sputum production) and repeated exacerbations despite an optimal regimen of an inhaled corticosteroid, long-acting beta-2 agonist, and long-acting antimuscarinic agent.
Macrolide antibiotics: Role unclear
Macrolide antibiotics have anti-inflammatory and immunomodulatory activities.
Azithromycin: fewer exacerbations but some side effects. A multicenter trial63 in 1,142 COPD patients randomized to either oral azithromycin 250 mg daily or placebo found a 27% reduction in the risk of COPD exacerbation in the intervention arm. No differences were found between the groups in mortality, hospitalizations, emergency department visits, or respiratory failure. Hearing loss and increased macrolide resistance were noted in the intervention arm. In a secondary subgroup analysis,64 no difference in efficacy was found by sex, history of chronic bronchitis, oxygen use, or concomitant COPD treatment.
The COPD: Influence of Macrolides on Exacerbation Frequency in Patients trial65 helped refine patient selection for macrolide therapy. In this single-center study, 92 patients with COPD and at least three exacerbations during the year prior to enrollment were randomized to receive either azithromycin 500 mg three times weekly or placebo. Exacerbations in the intervention group were markedly reduced (42%) with no difference in hospitalization rate.
The place of macrolide antibiotics in the treatment strategy of COPD is unclear, and they are not currently part of the GOLD guidelines. Still unknown is the incremental benefit of adding them to existing preventive regimens, cardiovascular safety, side effects, and potential effects on the resident microbial flora.
Other antibiotics have also been investigated for efficacy in preventing exacerbations.
Moxifloxacin: fewer exacerbations. The Pulsed Moxifloxacin Usage and Its Long-term Impact on the Reduction of Subsequent Exacerbations study66 randomized more than 1,000 patients with stable COPD to receive either moxifloxacin 400 mg or placebo daily for 5 days repeated every 8 weeks for six courses. Frequent assessment during the treatment period and for 6 months afterward revealed a reduced exacerbation rate in the intervention group but without benefit in hospitalization rate, mortality, lung function, or health status.
Recommendation. Azithromycin (either 250 mg daily or 500 mg three times weekly) can be considered for patients who have repeated COPD exacerbations despite an optimal regimen of an inhaled corticosteroid, inhaled long-acting beta-2 agonist, and inhaled long-acting antimuscarinic agent. The need to continue azithromycin should be reassessed yearly.
Mucolytics
Greatest benefit to patients not taking inhaled corticosteroids. Mucolytic agents help clear airway secretions by reducing viscosity. N-acetylcysteine and carbocysteine (not available in the United States) also have antioxidant properties that may counteract oxidant stress associated with acute COPD exacerbations.
The Bronchitis Randomized on NAC Cost-Utility Study (BRONCUS)67 randomized 523 COPD patients to N-acetylcysteine 600 mg daily or placebo. After 3 years of follow-up, no differences were found in the rate of exacerbations, lung function decline, and quality of life. Subgroup analysis suggested a reduction in exacerbations for patients who were not taking inhaled corticosteroids.
The Effect of Carbocisteine on Acute Exacerbation of Chronic Obstructive Pulmonary Disease (PEACE) study randomized more than 700 patients from multiple centers in China who had COPD and a recent history of exacerbations; they found a 25% lower exacerbation rate over 1 year with carbocysteine vs placebo.68 Most of the patients (83%) were not on inhaled corticosteroids, which complemented findings of the BRONCUS trial.
The Effect of High Dose N-acetylcysteine on Air Trapping and Airway Resistance of COPD (HIACE) study randomized 120 patients with stable COPD in a hospital in Hong Kong to either oral N-acetylcysteine (600 mg twice daily) or placebo and found a reduced exacerbation rate of exacerbations. Patients were matched at baseline for inhaled corticosteroid use.69
In 2014, the Twice Daily N-acetylcysteine 600 mg for Exacerbations of Chronic Obstructive Pulmonary Disease (PANTHEON) study70 randomized 1,006 patients from multiple hospitals in China with a history of moderate to severe COPD and exacerbations to receive either N-acetylcysteine 600 mg twice daily or placebo for 1 year. They found a 22% reduction in exacerbations in the treatment group vs placebo.
GOLD guidelines2 recommend mucolytics for patients with severe COPD and exacerbations when inhaled corticosteroids are not available or affordable.
Recommendation. Mucolytics may be useful for patients with difficulty expectorating and with a history of exacerbations despite appropriate inhaled therapy.
OTHER INTERVENTIONS CAN HELP
Pulmonary rehabilitation provides multiple benefits
Pulmonary rehabilitation increases exercise tolerance and reduces symptom burden in patients with stable COPD. It is also a multidisciplinary effort that may help reinforce adherence to medications, enhance COPD education, and provide closer medical surveillance to patients at high risk for recurrent exacerbations.
A small randomized controlled trial71 prescribed pulmonary rehabilitation on discharge for a COPD exacerbation and found sustainable improvements in exercise capacity and health status after 3 months.
In a later study,72 the same group started pulmonary rehabilitation within a week of hospital discharge and found reduced hospital readmissions over a 3-month period.
Smoking cessation is always worth advocating
A large observational cohort study concluded that current smokers were at a higher risk for COPD exacerbations compared with former smokers.73 Although there are no randomized controlled trials that assess the effects of smoking cessation at the time of COPD exacerbation, we recommend seizing the opportunity to implement this important intervention.
Vaccinations: Influenza and pneumococcal
Influenza vaccination is associated with reduced incidence of hospitalization among patients with cardiopulmonary disease.74 A meta-analysis of randomized clinical trials of influenza vaccination for patients with COPD75 reported significantly fewer exacerbations from vaccination, mostly owing to fewer episodes occurring after 3 to 4 weeks, coinciding with anticipated vaccine-induced immune protection. Furumoto and colleagues76 reported an added benefit of combined vaccination with 23-valent pneumococcal polysaccharide vaccine and influenza vaccine in reducing hospital admissions over influenza vaccination alone. We also recommend providing the 13-valent pneumococcal conjugate vaccine to patients with COPD, particularly for those older than 65, consistent with CDC recommendations.77
- Hatipoglu U, Aboussouan LS. Chronic obstructive pulmonary disease: an update for the primary physician. Cleve Clin J Med 2014; 81:373–383.
- Global initiative for chronic obstructive lung disease. Pocket Guide to COPD Diagnosis, Management, and Prevention. A Guide for Health Care Professionals. Updated 2016. http://www.goldcopd.org/uploads/users/files/WatermarkedPocket%20Guide%202016(2).pdf. Accessed March 7, 2016.
- Gupta N, Pinto LM, Morogan A, Bourbeau J. The COPD assessment test: a systematic review. Eur Respir J 2014; 44:873–884.
- Leidy NK, Wilcox TK, Jones PW, et al; EXACT-PRO Study Group. Development of the EXAcerbations of chronic obstructive pulmonary disease tool (EXACT): a patient-reported outcome (PRO) measure. Value Health 2010; 13:965–975.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Tillie-Leblond I, Marquette CH, Perez T, et al. Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: prevalence and risk factors. Ann Intern Med 2006; 144:390–396.
- Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged ≥ 18 years in the United States for 2010 and projections through 2020. Chest 2015; 147:31–45.
- Toy EL, Gallagher KF, Stanley EL, Swensen AR, Duh MS. The economic impact of exacerbations of chronic obstructive pulmonary disease and exacerbation definition: a review. COPD 2010; 7:214–228.
- Pasquale MK, Sun SX, Song F, Hartnett HJ, Stemkowski SA. Impact of exacerbations on health care cost and resource utilization in chronic obstructive pulmonary disease patients with chronic bronchitis from a predominantly Medicare population. Int J Chron Obstruct Pulmon Dis 2012; 7:757–764.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157:1418–1422.
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 161:1608–1613.
- Spencer S, Calverley PM, Sherwood Burge P, Jones PW; ISOLDE Study Group, Inhaled Steroids in Obstructive Lung Disease. Health status deterioration in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:122–128.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Brusselle G. Why doesn’t reducing exacerbations decrease COPD mortality? Lancet Respir Med 2014; 2:681–683.
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359:2355–2365.
- White AJ, Gompertz S, Bayley DL, et al. Resolution of bronchial inflammation is related to bacterial eradication following treatment of exacerbations of chronic bronchitis. Thorax 2003; 58:680–685.
- Desai H, Eschberger K, Wrona C, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc 2014; 11:303–309.
- Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184:662–671.
- Jamieson DB, Matsui EC, Belli A, et al. Effects of allergic phenotype on respiratory symptoms and exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:187–192.
- Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax 2009; 64:728–735.
- Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA 2013; 309:2353–2361.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Donaldson GC, Hurst JR, Smith CJ, Hubbard RB, Wedzicha JA. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest 2010; 137:1091–1097.
- Chang CL, Robinson SC, Mills GD, et al. Biochemical markers of cardiac dysfunction predict mortality in acute exacerbations of COPD. Thorax 2011; 66:764–768.
- Patel AR, Kowlessar BS, Donaldson GC, et al. Cardiovascular risk, myocardial injury, and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:1091–1099.
- Dransfield MT, Rowe SM, Johnson JE, Bailey WC, Gerald LB. Use of beta blockers and the risk of death in hospitalised patients with acute exacerbations of COPD. Thorax 2008; 63:301–305.
- Thompson WH, Nielson CP, Carvalho P, Charan NB, Crowley JJ. Controlled trial of oral prednisone in outpatients with acute COPD exacerbation. Am J Respir Crit Care Med 1996; 154:407–412.
- Aaron SD, Vandemheen KL, Hebert P, et al. Outpatient oral prednisone after emergency treatment of chronic obstructive pulmonary disease. N Engl J Med 2003; 348:2618–2625.
- Walters JA, Gibson PG, Wood-Baker R, Hannay M, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2009; 1:CD001288.
- Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med 2012; 186:48–55.
- Siva R, Green RH, Brightling CE, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J 2007; 29:906–913.
- Wilson R, Allegra L, Huchon G, et al; MOSAIC Study Group. Short-term and long-term outcomes of moxifloxacin compared to standard antibiotic treatment in acute exacerbations of chronic bronchitis. Chest 2004; 125:953–964.
- Wilson R, Anzueto A, Miravitlles M, et al. Moxifloxacin versus amoxicillin/clavulanic acid in outpatient acute exacerbations of COPD: MAESTRAL results. Eur Respir J 2012; 40:17–27.
- Llor C, Moragas A, Hernandez S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Anzueto A. Primary care management of chronic obstructive pulmonary disease to reduce exacerbations and their consequences. Am J Med Sci 2010; 340:309–318.
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med 1999; 340:1941–1947.
- de Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: a randomized, controlled, double-blind study. Chest 2007; 132:1741–1747.
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 2013; 309:2223–2231.
- Alia I, de la Cal MA, Esteban A, et al. Efficacy of corticosteroid therapy in patients with an acute exacerbation of chronic obstructive pulmonary disease receiving ventilatory support. Arch Intern Med 2011; 171:1939–1946.
- Abroug F, Ouanes-Besbes L, Fkih-Hassen M, et al. Prednisone in COPD exacerbation requiring ventilatory support: an open-label randomised evaluation. Eur Respir J 2014; 43:717–724.
- Kiser TH, Allen RR, Valuck RJ, Moss M, Vandivier RW. Outcomes associated with corticosteroid dosage in critically ill patients with acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 189:1052–1064.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Nair S, Thomas E, Pearson SB, Henry MT. A randomized controlled trial to assess the optimal dose and effect of nebulized albuterol in acute exacerbations of COPD. Chest 2005; 128:48–54.
- Drescher GS, Carnathan BJ, Imus S, Colice GL. Incorporating tiotropium into a respiratory therapist-directed bronchodilator protocol for managing in-patients with COPD exacerbations decreases bronchodilator costs. Respir Care 2008; 53:1678–1684.
- Austin MA, Wills KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ 2010; 341:c5462.
- Ringbaek TJ, Viskum K, Lange P. Does long-term oxygen therapy reduce hospitalisation in hypoxaemic chronic obstructive pulmonary disease? Eur Respir J 2002; 20:38–42.
- Keenan SP, Sinuff T, Cook DJ, Hill NS. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive-pressure ventilation? A systematic review of the literature. Ann Intern Med 2003; 138:861–870.
- Quon BS, Gan WQ, Sin DD. Contemporary management of acute exacerbations of COPD: a systematic review and metaanalysis. Chest 2008; 133:756–766.
- Sinuff T, Keenan SP; Department of Medicine, McMaster University. Clinical practice guideline for the use of noninvasive positive pressure ventilation in COPD patients with acute respiratory failure. J Crit Care 2004; 19:82–91.
- Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Vogelmeier C, Hederer B, Glaab T, et al; POET-COPD Investigators. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011; 364:1093–1103.
- Decramer ML, Chapman KR, Dahl R, et al; INVIGORATE investigators. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013; 1:524–533.
- Aaron SD, Vandemheen KL, Fergusson D, et al; Canadian Thoracic Society/Canadian Respiratory Clinical Research Consortium. Tiotropium in combination with placebo, salmeterol, or fluticasone-salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2007; 146:545–555.
- Short PM, Williamson PA, Elder DH, Lipworth SI, Schembri S, Lipworth BJ. The impact of tiotropium on mortality and exacerbations when added to inhaled corticosteroids and long-acting beta-agonist therapy in COPD. Chest 2012; 141:81–86.
- Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax 2013; 68:1029–1036.
- Magnussen H, Disse B, Rodriguez-Roisin R, et al; WISDOM Investigators. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med 2014; 371:1285–1294.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385:857–866.
- Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax 2014; 69:616–622.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014; 189:1503–1508.
- Uzun S, Djamin RS, Kluytmans JA, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2014; 2:361–368.
- Sethi S, Jones PW, Theron MS, et al; PULSE Study group. Pulsed moxifloxacin for the prevention of exacerbations of chronic obstructive pulmonary disease: a randomized controlled trial. Respir Res 2010; 11:10.
- Decramer M, Rutten-van Molken M, Dekhuijzen PN, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized On NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet 2005; 365:1552–1560.
- Zheng JP, Kang J, Huang SG, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE study): a randomised placebo-controlled study. Lancet 2008; 371:2013–2018.
- Tse HN, Raiteri L, Wong KY, et al. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 2013; 144:106–118.
- Zheng JP, Wen FQ, Bai CX, et al; PANTHEON study group. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2014; 2:187–194.
- Man WD, Polkey MI, Donaldson N, Gray BJ, Moxham J. Community pulmonary rehabilitation after hospitalisation for acute exacerbations of chronic obstructive pulmonary disease: randomised controlled study. BMJ 2004; 329:1209.
- Seymour JM, Moore L, Jolley CJ, et al. Outpatient pulmonary rehabilitation following acute exacerbations of COPD. Thorax 2010; 65:423–428.
- Au DH, Bryson CL, Chien JW, et al. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med 2009; 24:457–463.
- Seo YB, Hong KW, Kim IS, et al. Effectiveness of the influenza vaccine at preventing hospitalization due to acute lower respiratory infection and exacerbation of chronic cardiopulmonary disease in Korea during 2010-2011. Vaccine 2013; 31:1426–1430.
- Poole PJ, Chacko E, Wood-Baker RW, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2006; 1:CD002733.
- Furumoto A, Ohkusa Y, Chen M, et al. Additive effect of pneumococcal vaccine and influenza vaccine on acute exacerbation in patients with chronic lung disease. Vaccine 2008; 26:4284–4289.
- Tomczyk S, Bennett NM, Stoecker C, et al; Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥65 years: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2014; 63:822–825.
- Hatipoglu U, Aboussouan LS. Chronic obstructive pulmonary disease: an update for the primary physician. Cleve Clin J Med 2014; 81:373–383.
- Global initiative for chronic obstructive lung disease. Pocket Guide to COPD Diagnosis, Management, and Prevention. A Guide for Health Care Professionals. Updated 2016. http://www.goldcopd.org/uploads/users/files/WatermarkedPocket%20Guide%202016(2).pdf. Accessed March 7, 2016.
- Gupta N, Pinto LM, Morogan A, Bourbeau J. The COPD assessment test: a systematic review. Eur Respir J 2014; 44:873–884.
- Leidy NK, Wilcox TK, Jones PW, et al; EXACT-PRO Study Group. Development of the EXAcerbations of chronic obstructive pulmonary disease tool (EXACT): a patient-reported outcome (PRO) measure. Value Health 2010; 13:965–975.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Tillie-Leblond I, Marquette CH, Perez T, et al. Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: prevalence and risk factors. Ann Intern Med 2006; 144:390–396.
- Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged ≥ 18 years in the United States for 2010 and projections through 2020. Chest 2015; 147:31–45.
- Toy EL, Gallagher KF, Stanley EL, Swensen AR, Duh MS. The economic impact of exacerbations of chronic obstructive pulmonary disease and exacerbation definition: a review. COPD 2010; 7:214–228.
- Pasquale MK, Sun SX, Song F, Hartnett HJ, Stemkowski SA. Impact of exacerbations on health care cost and resource utilization in chronic obstructive pulmonary disease patients with chronic bronchitis from a predominantly Medicare population. Int J Chron Obstruct Pulmon Dis 2012; 7:757–764.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157:1418–1422.
- Seemungal TA, Donaldson GC, Bhowmik A, Jeffries DJ, Wedzicha JA. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 161:1608–1613.
- Spencer S, Calverley PM, Sherwood Burge P, Jones PW; ISOLDE Study Group, Inhaled Steroids in Obstructive Lung Disease. Health status deterioration in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:122–128.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Brusselle G. Why doesn’t reducing exacerbations decrease COPD mortality? Lancet Respir Med 2014; 2:681–683.
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359:2355–2365.
- White AJ, Gompertz S, Bayley DL, et al. Resolution of bronchial inflammation is related to bacterial eradication following treatment of exacerbations of chronic bronchitis. Thorax 2003; 58:680–685.
- Desai H, Eschberger K, Wrona C, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc 2014; 11:303–309.
- Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med 2011; 184:662–671.
- Jamieson DB, Matsui EC, Belli A, et al. Effects of allergic phenotype on respiratory symptoms and exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:187–192.
- Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax 2009; 64:728–735.
- Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA 2013; 309:2353–2361.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Donaldson GC, Hurst JR, Smith CJ, Hubbard RB, Wedzicha JA. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest 2010; 137:1091–1097.
- Chang CL, Robinson SC, Mills GD, et al. Biochemical markers of cardiac dysfunction predict mortality in acute exacerbations of COPD. Thorax 2011; 66:764–768.
- Patel AR, Kowlessar BS, Donaldson GC, et al. Cardiovascular risk, myocardial injury, and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:1091–1099.
- Dransfield MT, Rowe SM, Johnson JE, Bailey WC, Gerald LB. Use of beta blockers and the risk of death in hospitalised patients with acute exacerbations of COPD. Thorax 2008; 63:301–305.
- Thompson WH, Nielson CP, Carvalho P, Charan NB, Crowley JJ. Controlled trial of oral prednisone in outpatients with acute COPD exacerbation. Am J Respir Crit Care Med 1996; 154:407–412.
- Aaron SD, Vandemheen KL, Hebert P, et al. Outpatient oral prednisone after emergency treatment of chronic obstructive pulmonary disease. N Engl J Med 2003; 348:2618–2625.
- Walters JA, Gibson PG, Wood-Baker R, Hannay M, Walters EH. Systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2009; 1:CD001288.
- Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med 2012; 186:48–55.
- Siva R, Green RH, Brightling CE, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. Eur Respir J 2007; 29:906–913.
- Wilson R, Allegra L, Huchon G, et al; MOSAIC Study Group. Short-term and long-term outcomes of moxifloxacin compared to standard antibiotic treatment in acute exacerbations of chronic bronchitis. Chest 2004; 125:953–964.
- Wilson R, Anzueto A, Miravitlles M, et al. Moxifloxacin versus amoxicillin/clavulanic acid in outpatient acute exacerbations of COPD: MAESTRAL results. Eur Respir J 2012; 40:17–27.
- Llor C, Moragas A, Hernandez S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Anzueto A. Primary care management of chronic obstructive pulmonary disease to reduce exacerbations and their consequences. Am J Med Sci 2010; 340:309–318.
- Niewoehner DE, Erbland ML, Deupree RH, et al. Effect of systemic glucocorticoids on exacerbations of chronic obstructive pulmonary disease. Department of Veterans Affairs Cooperative Study Group. N Engl J Med 1999; 340:1941–1947.
- de Jong YP, Uil SM, Grotjohan HP, Postma DS, Kerstjens HA, van den Berg JW. Oral or IV prednisolone in the treatment of COPD exacerbations: a randomized, controlled, double-blind study. Chest 2007; 132:1741–1747.
- Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs conventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE randomized clinical trial. JAMA 2013; 309:2223–2231.
- Alia I, de la Cal MA, Esteban A, et al. Efficacy of corticosteroid therapy in patients with an acute exacerbation of chronic obstructive pulmonary disease receiving ventilatory support. Arch Intern Med 2011; 171:1939–1946.
- Abroug F, Ouanes-Besbes L, Fkih-Hassen M, et al. Prednisone in COPD exacerbation requiring ventilatory support: an open-label randomised evaluation. Eur Respir J 2014; 43:717–724.
- Kiser TH, Allen RR, Valuck RJ, Moss M, Vandivier RW. Outcomes associated with corticosteroid dosage in critically ill patients with acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 189:1052–1064.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Nair S, Thomas E, Pearson SB, Henry MT. A randomized controlled trial to assess the optimal dose and effect of nebulized albuterol in acute exacerbations of COPD. Chest 2005; 128:48–54.
- Drescher GS, Carnathan BJ, Imus S, Colice GL. Incorporating tiotropium into a respiratory therapist-directed bronchodilator protocol for managing in-patients with COPD exacerbations decreases bronchodilator costs. Respir Care 2008; 53:1678–1684.
- Austin MA, Wills KE, Blizzard L, Walters EH, Wood-Baker R. Effect of high flow oxygen on mortality in chronic obstructive pulmonary disease patients in prehospital setting: randomised controlled trial. BMJ 2010; 341:c5462.
- Ringbaek TJ, Viskum K, Lange P. Does long-term oxygen therapy reduce hospitalisation in hypoxaemic chronic obstructive pulmonary disease? Eur Respir J 2002; 20:38–42.
- Keenan SP, Sinuff T, Cook DJ, Hill NS. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive-pressure ventilation? A systematic review of the literature. Ann Intern Med 2003; 138:861–870.
- Quon BS, Gan WQ, Sin DD. Contemporary management of acute exacerbations of COPD: a systematic review and metaanalysis. Chest 2008; 133:756–766.
- Sinuff T, Keenan SP; Department of Medicine, McMaster University. Clinical practice guideline for the use of noninvasive positive pressure ventilation in COPD patients with acute respiratory failure. J Crit Care 2004; 19:82–91.
- Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Vogelmeier C, Hederer B, Glaab T, et al; POET-COPD Investigators. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011; 364:1093–1103.
- Decramer ML, Chapman KR, Dahl R, et al; INVIGORATE investigators. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013; 1:524–533.
- Aaron SD, Vandemheen KL, Fergusson D, et al; Canadian Thoracic Society/Canadian Respiratory Clinical Research Consortium. Tiotropium in combination with placebo, salmeterol, or fluticasone-salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2007; 146:545–555.
- Short PM, Williamson PA, Elder DH, Lipworth SI, Schembri S, Lipworth BJ. The impact of tiotropium on mortality and exacerbations when added to inhaled corticosteroids and long-acting beta-agonist therapy in COPD. Chest 2012; 141:81–86.
- Suissa S, Patenaude V, Lapi F, Ernst P. Inhaled corticosteroids in COPD and the risk of serious pneumonia. Thorax 2013; 68:1029–1036.
- Magnussen H, Disse B, Rodriguez-Roisin R, et al; WISDOM Investigators. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med 2014; 371:1285–1294.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Martinez FJ, Calverley PM, Goehring UM, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet 2015; 385:857–866.
- Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflumilast in moderate to severe COPD. Thorax 2014; 69:616–622.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014; 189:1503–1508.
- Uzun S, Djamin RS, Kluytmans JA, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2014; 2:361–368.
- Sethi S, Jones PW, Theron MS, et al; PULSE Study group. Pulsed moxifloxacin for the prevention of exacerbations of chronic obstructive pulmonary disease: a randomized controlled trial. Respir Res 2010; 11:10.
- Decramer M, Rutten-van Molken M, Dekhuijzen PN, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized On NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet 2005; 365:1552–1560.
- Zheng JP, Kang J, Huang SG, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE study): a randomised placebo-controlled study. Lancet 2008; 371:2013–2018.
- Tse HN, Raiteri L, Wong KY, et al. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest 2013; 144:106–118.
- Zheng JP, Wen FQ, Bai CX, et al; PANTHEON study group. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2014; 2:187–194.
- Man WD, Polkey MI, Donaldson N, Gray BJ, Moxham J. Community pulmonary rehabilitation after hospitalisation for acute exacerbations of chronic obstructive pulmonary disease: randomised controlled study. BMJ 2004; 329:1209.
- Seymour JM, Moore L, Jolley CJ, et al. Outpatient pulmonary rehabilitation following acute exacerbations of COPD. Thorax 2010; 65:423–428.
- Au DH, Bryson CL, Chien JW, et al. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med 2009; 24:457–463.
- Seo YB, Hong KW, Kim IS, et al. Effectiveness of the influenza vaccine at preventing hospitalization due to acute lower respiratory infection and exacerbation of chronic cardiopulmonary disease in Korea during 2010-2011. Vaccine 2013; 31:1426–1430.
- Poole PJ, Chacko E, Wood-Baker RW, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2006; 1:CD002733.
- Furumoto A, Ohkusa Y, Chen M, et al. Additive effect of pneumococcal vaccine and influenza vaccine on acute exacerbation in patients with chronic lung disease. Vaccine 2008; 26:4284–4289.
- Tomczyk S, Bennett NM, Stoecker C, et al; Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine among adults aged ≥65 years: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2014; 63:822–825.
KEY POINTS
- COPD exacerbations usually start with an infection.
- A short course of corticosteroids (eg, prednisone 40 mg daily for 5 to 7 days) improves outcomes with low risk.
- The choice of antibiotic depends on severity and frequency of exacerbations and the patient’s age and condition.
- Inhaled albuterol 2.5 mg, every 1 to 4 hours, should be prescribed with or without a nebulized anticholinergic.
- Ventilation support is important for patients with acute respiratory acidosis (pH < 7.35).
- Exacerbations can be prevented with some combination of inhaled agents (long-acting beta-2 agonist, corticosteroid, long-acting antimuscarinic), roflumilast (an oral phosphodiesterase inhibitor), and a mucolytic, depending on the patient’s needs.
Managing diabetes in hospitalized patients with chronic kidney disease
Managing glycemic control in hospitalized patients with chronic kidney disease (CKD) and diabetes mellitus is a challenge, with no published guidelines. In this setting, avoiding hypoglycemia takes precedence over meeting strict blood glucose targets. Optimal management is essential to reduce hypoglycemia and the risk of death from cardiovascular disease.1
This article reviews the evidence to guide diabetes management in hospitalized patients with CKD, focusing on blood glucose monitoring, insulin dosing, and concerns about other diabetic agents.
FOCUS ON AVOIDING HYPOGLYCEMIA
CKD is common, estimated to affect more than 50 million people worldwide.2 Diabetes mellitus is the primary cause of kidney failure in 45% of dialysis patients with CKD.
Tight control comes with a cost
Hyperglycemia in hospitalized patients is associated with a higher risk of death, a higher risk of infections, and a longer hospital stay.3,4 In 2001, Van den Berghe et al5 found that intensive insulin therapy reduced the mortality rate in critically ill patients in the surgical intensive care unit. But subsequent studies6,7 found that intensive insulin therapy to achieve tight glycemic control increased rates of morbidity and mortality without adding clinical benefit.
Randomized clinical trials in outpatients have shown that tight control of blood glucose levels reduces microvascular and macrovascular complications in patients with type 1 diabetes.8–10 In the Diabetes Control and Complications Trial,9 compared with conventional therapy, intensive insulin therapy reduced the incidence of retinopathy progression (4.7 vs 1.2 cases per 100 patient-years, number needed to treat [NNT] = 3 for 10 years) and clinical neuropathy (9.8 vs 3.1 per 100 patient-years, NNT = 1.5 for 10 years). The long-term likelihood of a cardiovascular event was also significantly lower in the intensive treatment group (0.38 vs 0.80 events per 100 patient-years).9
Similarly, in the Epidemiology of Diabetes Interventions and Complications follow-up study, the intensive therapy group had fewer cardiovascular deaths.11 On the other hand, the risk of severe hypoglycemia and subsequent coma or seizure was significantly higher in the intensive therapy group than in the conventional therapy group (16.3 vs 5.4 per 100 patient-years).8
CKD increases hypoglycemia risk

Moen et al12 found that the incidence of hypoglycemia was significantly higher in patients with CKD (estimated glomerular filtration rate [GFR] < 60 mL/min) with or without diabetes, and that patients with both conditions were at greatest risk (Figure 1). Multiple factors contribute to the increased risk of hypoglycemia: patients with advanced CKD tend to have poor nutrition, resulting in reduced glycogen stores, and a smaller renal mass reduces renal gluconeogenesis and decreases the elimination of insulin and oral antidiabetic agents.
After the onset of diabetic nephropathy, progression of renal complications and overall life expectancy are influenced by earlier glycemic control.8 Development of diabetic nephropathy is commonly accompanied by changes in metabolic control, particularly an increased risk of hypoglycemia.13 In addition, episodes of severe hypoglycemia constitute an independent cardiovascular risk factor.14
Aggressive glycemic control in hospitalized patients, particularly those with advanced CKD, is associated with a risk of hypoglycemia without overall improvement in outcomes.15 Elderly patients with type 2 diabetes are similar to patients with CKD in that they have a reduced GFR and are thus more sensitive to insulin. In both groups, intensifying glycemic control, especially in the hospital, is associated with more frequent episodes of severe hypoglycemia.16 The focus should be not only on maintaining optimal blood glucose concentration, but also on preventing hypoglycemia.
‘Burnt-out’ diabetes

Paradoxically, patients with end-stage renal disease and type 2 diabetes often experience altered glucose homeostasis with markedly improved glycemic control. They may attain normoglycemia and normalization of hemoglobin A1c, a condition known as “burnt-out” diabetes. Its precise mechanism is not understood and its significance remains unclear (Table 1).17
HEMOGLOBIN A1c CAN BE FALSELY HIGH OR FALSELY LOW
Hemoglobin A1c measurement is used to diagnose diabetes and to assess long-term glycemic control. It is a measure of the fraction of hemoglobin that has been glycated by exposure to glucose. Because the average lifespan of a red cell is 120 days, the hemoglobin A1c value reflects the mean blood glucose concentration over the preceding 3 months.
But hemoglobin A1c measurement has limitations: any condition that alters the lifespan of erythrocytes leads to higher or lower hemoglobin A1c levels. Hemoglobin A1c levels are also affected by kidney dysfunction, hemolysis, and acidosis.18
Falsely high hemoglobin A1c levels are associated with conditions that prolong the lifespan of erythrocytes, such as asplenia. Iron deficiency also increases the average age of circulating red cells because of reduced red cell production. For patients in whom blood glucose measurements do not correlate with hemoglobin A1c measurements, iron deficiency anemia should be considered before altering a treatment regimen.
Falsely low hemoglobin A1c levels are associated with conditions of more rapid erythrocyte turnover, such as autoimmune hemolytic anemia, hereditary spherocytosis, and acute blood loss anemia. In patients with CKD, recombinant erythropoietin treatment lowers hemoglobin A1c levels by increasing the number of immature red cells, which are less likely to glycosylate.19
Morgan et al20 compared the association between hemoglobin A1c and blood glucose levels in diabetic patients with moderate to severe CKD not requiring dialysis and in diabetic patients with normal renal function and found no difference between these two groups, suggesting that hemoglobin A1c is reliable in this setting. But study results conflict for patients on dialysis, making the usefulness of hemoglobin A1c testing for those patients less clear. In one study, hemoglobin A1c testing underestimated glycemic control,20 but other studies found that glycemic control was overestimated.21,22
Alternatives to hemoglobin A1c
Other measures of long-term glycemic control such as fructosamine and glycated albumin levels are sometimes used in conditions in which hemoglobin A1c may not be reliable.
Albumin also undergoes glycation when exposed to glucose. Glycated albumin appears to be a better measure of glycemic control in patients with CKD and diabetes than serum fructosamine,23 which has failed to show a significant correlation with blood glucose levels in patients with CKD.24 However, because serum albumin has a short half-life, glycated albumin reflects glycemic control in only the approximately 1 to 2 weeks before sampling,25 so monthly monitoring is required.
Glycated albumin levels may be reduced due to increased albumin turnover in patients with nephrotic-range proteinuria and in diabetic patients on peritoneal dialysis. Several issues remain unclear, such as the appropriate target level of glycated albumin and at what stage of CKD it should replace hemoglobin A1c testing. If an improved assay that is unaffected by changes in serum albumin becomes available, it may be appropriate to use glycated albumin measurements to assess long-term glycemic control for patients with CKD.
In general, therapeutic decisions to achieve optimum glycemic control in patients with diabetes and CKD should be based on hemoglobin A1c testing, multiple glucose measurements, and patient symptoms of hypoglycemia or hyperglycemia. The best measure for assessing glycemic control in hospitalized patients with CKD remains multiple blood glucose testing daily.
INSULIN THERAPY PREFERRED
Although several studies have evaluated inpatient glycemic control,26–29 no guidelines have been published for hospitalized patients with diabetes and CKD. Insulin therapy is preferred for achieving glycemic control in acutely ill or hospitalized patients with diabetes. Oral hypoglycemic agents should be discontinued.
Regardless of the form of insulin chosen to treat diabetes, caution is needed for patients with kidney disease. During hospitalization, clinical changes are expected owing to illness and differences in caloric intake and physical activity, resulting in altered insulin sensitivity. Insulin-treated hospitalized patients require individualized care, including multiple daily blood glucose tests and insulin therapy modifications for ideal glycemic control.
For surgical or medical intensive care patients on insulin therapy, the target blood glucose level before meals should be 140 mg/dL, and the target random level should be less than 180 mg/dL.15,26–29
Basal-bolus insulin
Sliding-scale therapy should be avoided as the only method for glycemic control. Instead, scheduled subcutaneous basal insulin once or twice daily combined with rapid- or short-acting insulin with meals is recommended.
Basal-bolus insulin therapy, one of the most advanced and flexible insulin replacement therapies, mimics endogenous insulin release and offers great advantages in diabetes care. Using mealtime bolus insulin permits variation in the amount of food eaten; more insulin can be taken with a larger meal and less with smaller meals. A bolus approach offers the flexibility of administering rapid-acting insulin immediately after meals when oral intake is variable.
Individualize insulin therapy
Optimizing glycemic control requires an understanding of the altered pharmacokinetics and pharmacodynamics of insulin in patients with diabetic nephropathy. Table 2 shows the pharmacokinetic profiles of insulin preparations in healthy people. Analogue insulins, which are manufactured by recombinant DNA technology, have conformational changes in the insulin molecule that alter their pharmacokinetics and pharmacodynamics. The rapid-acting analogue insulins are absorbed quickly, making them suitable for postprandial glucose control.
Changes in GFR are associated with altered pharmacokinetics and pharmacodynamics of insulin,30,31 but unlike for oral antidiabetic agents, these properties are not well characterized for insulin preparations in patients with renal insufficiency.13,32–36
CKD may reduce insulin clearance. Rave et al32 reported that the clearance of regular human insulin was reduced by 30% to 40% in patients with type 1 diabetes and a mean estimated GFR of 54 mL/min. They found that the metabolic activity of insulin lispro was more robust than that of short-acting regular human insulin in patients with diabetic nephropathy. In another study, patients with diabetes treated with insulin aspart did not show any significant change in the required insulin dosage in relation to the renal filtration rate.34 Biesenbach et al33 found a 38% reduction in insulin requirements in patients with type 1 diabetes as estimated GFR decreased from 80 mL/min to 10 mL/min. Further studies are required to better understand the safety of insulin in treating hospitalized patients with diabetes and renal insufficiency.

Few studies have compared the pharmacodynamics of long-acting insulins in relation to declining renal function. The long-acting analogue insulins have less of a peak than human insulin and thus better mimic endogenous insulin secretion. For insulin detemir, Lindholm and Jacobsen found no significant differences in the pharmacokinetics related to the stages of CKD.35 When using the long-acting insulins glargine or detemir, one should consider giving much lower doses (half the initial starting dosage) and titrating the dosage until target fasting glucose concentrations are reached to prevent hypoglycemia.
Table 3 summarizes recommended insulin dosage adjustments in CKD based on the literature and our clinical experience.
Considerations for dialysis patients
Subcutaneously administered insulin is eliminated renally, unlike endogenous insulin, which undergoes first-pass metabolism in the liver.13,37 As renal function declines, insulin clearance decreases and the insulin dosage must be reduced to prevent hypoglycemia.
Patients on hemodialysis or peritoneal dialysis pose a challenge for insulin dosing. Hemodialysis improves insulin sensitivity but also increases insulin clearance, making it difficult to determine insulin requirements. Sobngwi et al38 conducted a study in diabetic patients with end-stage renal disease on hemodialysis, using a 24-hour euglycemic clamp. They found that exogenous basal insulin requirements were 25% lower on the day after hemodialysis compared with the day before, but premeal insulin requirements stayed the same.
Peritoneal dialysis exposes patients to a high glucose load via the peritoneum, which can worsen insulin resistance. Intraperitoneal administration of insulin during peritoneal dialysis provides a more physiologic effect than subcutaneous administration: it prevents fluctuations of blood glucose and the formation of insulin antibodies. But insulin requirements are higher owing to a dilutional effect and to insulin binding to the plastic surface of the dialysis fluid reservoir.39
GLYCEMIC CONTROL FOR PROCEDURES
No guidelines have been established regarding the optimal blood glucose range for diabetic patients with CKD undergoing diagnostic or surgical procedures. Given the risk of hypoglycemia in such settings, less-stringent targets are reasonable, ie, premeal blood glucose levels of 140 mg/dL and random blood glucose levels of less than 180 mg/dL.
Before surgery, consideration should be given to the type of diabetes, surgical procedure, and metabolic control. Patients on insulin detemir or glargine as part of a basal-bolus regimen with rapid-acting insulin may safely be given the full dose of their basal insulin the night before or the morning of their procedure. However, patients on neutral protamine Hagedorn (NPH) insulin as a part of their basal-bolus regimen should receive half of their usual dose due to a difference in pharmacokinetic profile compared with insulin glargine or detemir.
In insulin-treated patients undergoing prolonged procedures (eg, coronary artery bypass grafting, transplant):
- Discontinue subcutaneous insulin and start an intravenous insulin infusion, titrated to maintain a blood glucose range of 140 to 180 mg/dL
- Subcutaneous insulin management may be acceptable for patients undergoing shorter outpatient procedures
- Supplemental subcutaneous doses of short- or rapid-acting insulin preparations can be given for blood glucose elevation greater than 180 mg/dL.
AVOID ORAL AGENTS AND NONINSULIN INJECTABLES
Oral antidiabetic agents and noninsulin injectables (Table 4) should generally be avoided in hospitalized patients, especially for those with decompensated heart failure, renal insufficiency, hypoperfusion, or chronic pulmonary disease, or for those given intravenous contrast. Most oral medications used to treat diabetes are affected by reduced kidney function, resulting in prolonged drug exposure and increased risk of hypoglycemia in patients with moderate to severe CKD (stages 3–5).
Metformin, a biguanide, is contraindicated in patients with high serum creatinine levels (> 1.5 mg/dL in men, > 1.4 mg/dL in women) because of the theoretical risk of lactic acidosis.40
Sulfonylurea clearance depends on kidney function.41 Severe prolonged episodes of hypoglycemia have been reported in dialysis patients taking these drugs, except with glipizide, which carries a lower risk.41,42
Repaglinide, a nonsulfonylurea insulin secretagogue, can be used in CKD stages 3 to 4 without any dosage adjustment.43
Thiazolidinediones have been reported to slow the progression of diabetic kidney disease independent of glycemic control.44 Adverse effects include fluid retention, edema, and congestive heart failure. Thiazolidinediones should not be used in patients with New York Heart Association class 3 or 4 heart failure,45 and so should not be prescribed in the hospital except for patients who are clinically stable or ready for discharge.
Quick-release bromocriptine, a dopamine receptor agonist, has been shown to be effective in lowering fasting plasma glucose levels and hemoglobin A1c, and improving glucose tolerance in obese patients with type 2 diabetes, although its usefulness in hospitalized patients with diabetes is not known.46,47
Dipeptidyl peptidase inhibitors. Sitagliptin and saxagliptin have been shown to be safe and effective in hospitalized patients with type 2 diabetes.48 However, except for linagliptin, dose reduction is recommended in patients with CKD stage 3 and higher.49–52
GLP-1 receptor agonists. Drugs of this class are potent agents for the reduction of glucose in the outpatient setting but are relatively contraindicated if the GFR is less than 30 mL/min, and they are currently not used in the hospital.
BLOOD GLUCOSE MONITORING IN HOSPITALIZED PATIENTS
Bedside blood glucose monitoring is recommended for all hospitalized patients with known diabetes with or without CKD, those with newly recognized hyperglycemia, and those who receive therapy associated with high risk for hyperglycemia, such as glucocorticoid therapy and enteral and parenteral nutrition. For patients on scheduled diets, fingerstick blood glucose monitoring is recommended before meals and at bedtime. In patients with no oral intake or on continuous enteral or parenteral nutrition, blood glucose monitoring every 4 to 6 hours is recommended. More frequent monitoring (eg, adding a 3:00 am check) may be prudent in patients with CKD.
Continuous glucose monitoring systems use a sensor inserted under the skin and transmit information via radio to a wireless monitor. Such systems are more expensive than conventional glucose monitoring but may enable better glucose control by providing real-time glucose measurements, with levels displayed at 5-minute or 1-minute intervals. Marshall et al53 confirmed this technology’s accuracy and precision in uremic patients on dialysis.
Considerations for peritoneal dialysis
For patients on peritoneal dialysis, glucose in the dialysate exacerbates hyperglycemia. Dialysis solutions with the glucose polymer icodextrin as the osmotic agent instead of glucose have been suggested to reduce glucose exposure.
Glucose monitoring systems measure interstitial fluid glucose by the glucose oxidase reaction and therefore are not affected by icodextrin. However, icodextrin is converted to maltose, a disaccharide composed of two glucose molecules, which can cause spuriously high readings in devices that use test strips containing the enzymes glucose dehydrogenase pyrroloquinoline quinone or glucose dye oxidoreductase. Spurious hyperglycemia may lead to giving too much insulin, in turn leading to symptomatic hypoglycemia.
Clinicians caring for patients receiving icodextrin should ensure that the glucose monitoring system uses only test strips that contain glucose oxidase, glucose dehydrogenase-nicotinamide adenine dinucleotide, or glucose dehydrogenase-flavin adenine dinucleotide, which are not affected by icodextrin.54
IMPROVING QUALITY
Hospitalized patients face many barriers to optimal glycemic control. Less experienced practitioners tend to have insufficient knowledge of insulin preparations and appropriate insulin dosing. Also, diabetes is often listed as a secondary diagnosis and so may be overlooked by the inpatient care team.
Educational programs should be instituted to overcome these barriers and improve knowledge related to inpatient diabetes care. When necessary, the appropriate use of consultants is important in hospitalized settings to improve quality and make hospital care more efficient and cost-effective.
No national benchmarks currently exist for inpatient diabetes care, and they need to be developed to ensure best practices. Physicians should take the initiative to remedy this by collaborating with other healthcare providers, such as dedicated diabetes educators, nursing staff, pharmacists, registered dietitians, and physicians with expertise in diabetes management, with the aim of achieving optimum glycemic control and minimizing hypoglycemia.
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
- Newman DJ, Mattock MB, Dawnay AB, et al. Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol Assess 2005; 9:iii–vi, xiii–163.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Golden SH, Peart-Vigilance C, Kao WH, Brancati FL. Perioperative glycemic control and the risk of infectious complications in a cohort of adults with diabetes. Diabetes Care 1999; 22:1408–1414.
- Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Brunkhorst FM, Engel C, Bloos F, et al; German Competence Network Sepsis (SepNet). Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Effect of intensive diabetes management on macrovascular events and risk factors in the Diabetes Control and Complications Trial. Am J Cardiol 1995; 75:894–903.
- Nathan DM, Lachin J, Cleary P, et al; Diabetes Control and Complications Trial; Epidemiology of Diabetes Interventions and Complications Research Group. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med 2003; 348:2294–2303.
- Writing Group for the DCCT/EDIC Research Group; Orchard TJ, Nathan DM, Zinman B, et al. Association between 7 years of intensive treatment of type 1 diabetes and long-term mortality. JAMA 2015; 313:45–53.
- Moen MF, Zhan M, Hsu VD, et al. Frequency of hypoglycemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol 2009; 4:1121–1127.
- Iglesias P, Díez J. Insulin therapy in renal disease. Diabetes Obes Metab 2008; 10:811–823.
- Zoungas S, Patel A, Chalmers J, et al; ADVANCE Collaborative Group. Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010; 363:1410–1418.
- Moghissi ES, Korytkowski MT, DiNardo M, et al; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Kovesdy CP, Park JC, Kalantar-Zadeh K. Glycemic control and burnt-out diabetes in ESRD. Semin Dial 2010; 23:148–156.
- De Marchi S, Cecchin E, Camurri C, et al. Origin of glycosylated hemoglobin A1 in chronic renal failure. Int J Artif Organs 1983; 6:77–82.
- Brown JN, Kemp DW, Brice KR. Class effect of erythropoietin therapy on hemoglobin A(1c) in a patient with diabetes mellitus and chronic kidney disease not undergoing hemodialysis. Pharmacotherapy 2009; 29:468–472.
- Morgan L, Marenah CB, Jeffcoate WJ, Morgan AG. Glycated proteins as indices of glycemic control in diabetic patients with chronic renal failure. Diabet Med 1996; 13:514–519.
- Peacock TP, Shihabi ZK, Bleyer AJ, et al. Comparison of glycated albumin and hemoglobin A(1c) levels in diabetic subjects on hemodialysis. Kidney Int 2008; 73:1062–1068.
- Joy MS, Cefalu WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Mittman N, Desiraju B, Fazil I, et al. Serum fructosamine versus glycosylated hemoglobin as an index of glycemic control, hospitalization, and infection in diabetic hemodialysis patients. Kidney Int 2010; 78(suppl 117):S41–S45.
- Alskar O, Korelli J, Duffull SB. A pharmacokinetic model for the glycation of albumin. J Pharmacokinet Pharmacodyn 2012; 39:273–282.
- Qaseem A, Humphrey LL, Chou R, Snow V, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Use of intensive insulin therapy for the management of glycemic control in hospitalized patients: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2011; 154:260–267.
- Murad MH, Coburn JA, Coto-Yglesias F, et al. Glycemic control in non-critically ill hospitalized patients: a systematic review and meta-analysis. J Clin Endocrinol Metab 2012; 97:49–58.
- Bogun M, Inzucchi SE. Inpatient management of diabetes and hyperglycemia. Clin Ther 2013; 35:724–733.
- Miller DB. Glycemic targets in hospital and barriers to attaining them. Can J Diabetes 2014; 38:74–78.
- Eidemak I, Feldt-Rasmussen B, Kanstrup IL, Nielsen SL, Schmitz O, Strandgaard S. Insulin resistance and hyperinsulinaemia in mild to moderate progressive chronic renal failure and its association with aerobic work capacity. Diabetologia 1995; 38:565–572.
- Svensson M, Yu Z, Eriksson J. A small reduction in glomerular filtration is accompanied by insulin resistance in type I diabetes patients with diabetic nephropathy. Eur J Clin Invest 2002; 32:100–109.
- Rave K, Heise T, Pfutzner A, Heinemann L, Sawicki P. Impact of diabetic nephropathy on pharmacodynamics and pharmacokinetic properties of insulin in type I diabetic patients. Diabetes Care 2001; 24:886–890.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Holmes G, Galitz L, Hu P, Lyness W. Pharmacokinetics of insulin aspart in obesity, renal impairment, or hepatic impairment. Br J Clin Pharmacol 2005; 60:469–476.
- Lindholm A, Jacobsen LV. Clinical pharmacokinetics and pharmacodynamics of insulin aspart. Clin Pharmacokinet 2001; 40:641–659.
- Bolli GB, Hahn AD, Schmidt R, et al. Plasma exposure to insulin glargine and its metabolites M1 and M2 after subcutaneous injection of therapeutic and supratherapeutic doses of glargine in subjects with type 1 diabetes. Diabetes Care 2012; 35:2626–2630.
- Nielsen S. Time course and kinetics of proximal tubular processing of insulin. Am J Physiol 1992; 262:F813–F822.
- Sobngwi E, Enoru S, Ashuntantang G, et al. Day-to-day variation of insulin requirements of patients with type 2 diabetes and end-stage renal disease undergoing maintenance hemodialysis. Diabetes Care 2010; 33:1409–1412.
- Quellhorst E. Insulin therapy during peritoneal dialysis: pros and cons of various forms of administration. J Am Soc Nephrol 2002; 13(suppl 1):S92–S96.
- Davidson MB, Peters AL. An overview of metformin in the treatment of type 2 diabetes mellitus. Am J Med 1997; 102:99–110.
- Ahmed Z, Simon B, Choudhury D. Management of diabetes in patients with chronic kidney disease. Postgrad Med 2009; 121:52–60.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26(suppl 4):73–85.
- Hasslacher C; Multinational Repaglinide Renal Study Group. Safety and efficacy of repaglinide in type 2 diabetic patients with and without impaired renal function. Diabetes Care 2003; 26:886–891.
- Iglesias P, Dies JJ. Peroxisome proliferator-activated receptor gamma agonists in renal disease. Eur J Endocrinol 2006; 154:613–621.
- Hollenberg NK. Considerations for management of fluid dynamic issues associated with thiazolidinediones. Am J Med 2003; 115(suppl. 8A) 111S–115S.
- Kamath V, Jones CN, Yip JC, et al. Effects of a quick-release form of bromocriptine (Ergoset) on fasting and postprandial plasma glucose, insulin, lipid, and lipoprotein concentrations in obese nondiabetic hyperinsulinemic women. Diabetes Care 1997; 20:1697–1701.
- Pijl H, Ohashi S, Matsuda M, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care 2000; 23:1154–1161.
- Umpierrez GE, Gianchandani R, Smiley D, et al. Safety and efficacy of sitagliptin therapy for the inpatient management of general medicine and surgery patients with type 2 diabetes: a pilot, randomized, controlled study. Diabetes Care 2013; 36:3430–3435.
- Chan JC, Scott R, Arjona Ferreira JC, et al. Safety and efficacy of sitagliptin in patients with type 2 diabetes and chronic renal insufficiency. Diabetes Obes Metab 2008; 10:545–555.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Onglyza package insert. www.azpicentral.com/onglyza/pi_onglyza.pdf. Accessed March 8, 2016.
- Gallwitz B. Safety and efficacy of linagliptin in type 2 diabetes patients with common renal and cardiovascular risk factors. Ther Adv Endocrinol Metab 2013; 4:95–105.
- Marshall J, Jennings P, Scott A, Fluck RJ, McIntyre CW. Glycemic control in diabetic CAPD patients assessed by continuous glucose monitoring system (CGMS). Kidney Int 2003; 64:1480–1486.
- Schleis TG. Interference of maltose, icodextrin, galactose, or xylose with some blood glucose monitoring systems. Pharmacotherapy 2007; 27:1313–1321.
Managing glycemic control in hospitalized patients with chronic kidney disease (CKD) and diabetes mellitus is a challenge, with no published guidelines. In this setting, avoiding hypoglycemia takes precedence over meeting strict blood glucose targets. Optimal management is essential to reduce hypoglycemia and the risk of death from cardiovascular disease.1
This article reviews the evidence to guide diabetes management in hospitalized patients with CKD, focusing on blood glucose monitoring, insulin dosing, and concerns about other diabetic agents.
FOCUS ON AVOIDING HYPOGLYCEMIA
CKD is common, estimated to affect more than 50 million people worldwide.2 Diabetes mellitus is the primary cause of kidney failure in 45% of dialysis patients with CKD.
Tight control comes with a cost
Hyperglycemia in hospitalized patients is associated with a higher risk of death, a higher risk of infections, and a longer hospital stay.3,4 In 2001, Van den Berghe et al5 found that intensive insulin therapy reduced the mortality rate in critically ill patients in the surgical intensive care unit. But subsequent studies6,7 found that intensive insulin therapy to achieve tight glycemic control increased rates of morbidity and mortality without adding clinical benefit.
Randomized clinical trials in outpatients have shown that tight control of blood glucose levels reduces microvascular and macrovascular complications in patients with type 1 diabetes.8–10 In the Diabetes Control and Complications Trial,9 compared with conventional therapy, intensive insulin therapy reduced the incidence of retinopathy progression (4.7 vs 1.2 cases per 100 patient-years, number needed to treat [NNT] = 3 for 10 years) and clinical neuropathy (9.8 vs 3.1 per 100 patient-years, NNT = 1.5 for 10 years). The long-term likelihood of a cardiovascular event was also significantly lower in the intensive treatment group (0.38 vs 0.80 events per 100 patient-years).9
Similarly, in the Epidemiology of Diabetes Interventions and Complications follow-up study, the intensive therapy group had fewer cardiovascular deaths.11 On the other hand, the risk of severe hypoglycemia and subsequent coma or seizure was significantly higher in the intensive therapy group than in the conventional therapy group (16.3 vs 5.4 per 100 patient-years).8
CKD increases hypoglycemia risk
Moen et al12 found that the incidence of hypoglycemia was significantly higher in patients with CKD (estimated glomerular filtration rate [GFR] < 60 mL/min) with or without diabetes, and that patients with both conditions were at greatest risk (Figure 1). Multiple factors contribute to the increased risk of hypoglycemia: patients with advanced CKD tend to have poor nutrition, resulting in reduced glycogen stores, and a smaller renal mass reduces renal gluconeogenesis and decreases the elimination of insulin and oral antidiabetic agents.
After the onset of diabetic nephropathy, progression of renal complications and overall life expectancy are influenced by earlier glycemic control.8 Development of diabetic nephropathy is commonly accompanied by changes in metabolic control, particularly an increased risk of hypoglycemia.13 In addition, episodes of severe hypoglycemia constitute an independent cardiovascular risk factor.14
Aggressive glycemic control in hospitalized patients, particularly those with advanced CKD, is associated with a risk of hypoglycemia without overall improvement in outcomes.15 Elderly patients with type 2 diabetes are similar to patients with CKD in that they have a reduced GFR and are thus more sensitive to insulin. In both groups, intensifying glycemic control, especially in the hospital, is associated with more frequent episodes of severe hypoglycemia.16 The focus should be not only on maintaining optimal blood glucose concentration, but also on preventing hypoglycemia.
‘Burnt-out’ diabetes
Paradoxically, patients with end-stage renal disease and type 2 diabetes often experience altered glucose homeostasis with markedly improved glycemic control. They may attain normoglycemia and normalization of hemoglobin A1c, a condition known as “burnt-out” diabetes. Its precise mechanism is not understood and its significance remains unclear (Table 1).17
HEMOGLOBIN A1c CAN BE FALSELY HIGH OR FALSELY LOW
Hemoglobin A1c measurement is used to diagnose diabetes and to assess long-term glycemic control. It is a measure of the fraction of hemoglobin that has been glycated by exposure to glucose. Because the average lifespan of a red cell is 120 days, the hemoglobin A1c value reflects the mean blood glucose concentration over the preceding 3 months.
But hemoglobin A1c measurement has limitations: any condition that alters the lifespan of erythrocytes leads to higher or lower hemoglobin A1c levels. Hemoglobin A1c levels are also affected by kidney dysfunction, hemolysis, and acidosis.18
Falsely high hemoglobin A1c levels are associated with conditions that prolong the lifespan of erythrocytes, such as asplenia. Iron deficiency also increases the average age of circulating red cells because of reduced red cell production. For patients in whom blood glucose measurements do not correlate with hemoglobin A1c measurements, iron deficiency anemia should be considered before altering a treatment regimen.
Falsely low hemoglobin A1c levels are associated with conditions of more rapid erythrocyte turnover, such as autoimmune hemolytic anemia, hereditary spherocytosis, and acute blood loss anemia. In patients with CKD, recombinant erythropoietin treatment lowers hemoglobin A1c levels by increasing the number of immature red cells, which are less likely to glycosylate.19
Morgan et al20 compared the association between hemoglobin A1c and blood glucose levels in diabetic patients with moderate to severe CKD not requiring dialysis and in diabetic patients with normal renal function and found no difference between these two groups, suggesting that hemoglobin A1c is reliable in this setting. But study results conflict for patients on dialysis, making the usefulness of hemoglobin A1c testing for those patients less clear. In one study, hemoglobin A1c testing underestimated glycemic control,20 but other studies found that glycemic control was overestimated.21,22
Alternatives to hemoglobin A1c
Other measures of long-term glycemic control such as fructosamine and glycated albumin levels are sometimes used in conditions in which hemoglobin A1c may not be reliable.
Albumin also undergoes glycation when exposed to glucose. Glycated albumin appears to be a better measure of glycemic control in patients with CKD and diabetes than serum fructosamine,23 which has failed to show a significant correlation with blood glucose levels in patients with CKD.24 However, because serum albumin has a short half-life, glycated albumin reflects glycemic control in only the approximately 1 to 2 weeks before sampling,25 so monthly monitoring is required.
Glycated albumin levels may be reduced due to increased albumin turnover in patients with nephrotic-range proteinuria and in diabetic patients on peritoneal dialysis. Several issues remain unclear, such as the appropriate target level of glycated albumin and at what stage of CKD it should replace hemoglobin A1c testing. If an improved assay that is unaffected by changes in serum albumin becomes available, it may be appropriate to use glycated albumin measurements to assess long-term glycemic control for patients with CKD.
In general, therapeutic decisions to achieve optimum glycemic control in patients with diabetes and CKD should be based on hemoglobin A1c testing, multiple glucose measurements, and patient symptoms of hypoglycemia or hyperglycemia. The best measure for assessing glycemic control in hospitalized patients with CKD remains multiple blood glucose testing daily.
INSULIN THERAPY PREFERRED
Although several studies have evaluated inpatient glycemic control,26–29 no guidelines have been published for hospitalized patients with diabetes and CKD. Insulin therapy is preferred for achieving glycemic control in acutely ill or hospitalized patients with diabetes. Oral hypoglycemic agents should be discontinued.
Regardless of the form of insulin chosen to treat diabetes, caution is needed for patients with kidney disease. During hospitalization, clinical changes are expected owing to illness and differences in caloric intake and physical activity, resulting in altered insulin sensitivity. Insulin-treated hospitalized patients require individualized care, including multiple daily blood glucose tests and insulin therapy modifications for ideal glycemic control.
For surgical or medical intensive care patients on insulin therapy, the target blood glucose level before meals should be 140 mg/dL, and the target random level should be less than 180 mg/dL.15,26–29
Basal-bolus insulin
Sliding-scale therapy should be avoided as the only method for glycemic control. Instead, scheduled subcutaneous basal insulin once or twice daily combined with rapid- or short-acting insulin with meals is recommended.
Basal-bolus insulin therapy, one of the most advanced and flexible insulin replacement therapies, mimics endogenous insulin release and offers great advantages in diabetes care. Using mealtime bolus insulin permits variation in the amount of food eaten; more insulin can be taken with a larger meal and less with smaller meals. A bolus approach offers the flexibility of administering rapid-acting insulin immediately after meals when oral intake is variable.
Individualize insulin therapy
Optimizing glycemic control requires an understanding of the altered pharmacokinetics and pharmacodynamics of insulin in patients with diabetic nephropathy. Table 2 shows the pharmacokinetic profiles of insulin preparations in healthy people. Analogue insulins, which are manufactured by recombinant DNA technology, have conformational changes in the insulin molecule that alter their pharmacokinetics and pharmacodynamics. The rapid-acting analogue insulins are absorbed quickly, making them suitable for postprandial glucose control.
Changes in GFR are associated with altered pharmacokinetics and pharmacodynamics of insulin,30,31 but unlike for oral antidiabetic agents, these properties are not well characterized for insulin preparations in patients with renal insufficiency.13,32–36
CKD may reduce insulin clearance. Rave et al32 reported that the clearance of regular human insulin was reduced by 30% to 40% in patients with type 1 diabetes and a mean estimated GFR of 54 mL/min. They found that the metabolic activity of insulin lispro was more robust than that of short-acting regular human insulin in patients with diabetic nephropathy. In another study, patients with diabetes treated with insulin aspart did not show any significant change in the required insulin dosage in relation to the renal filtration rate.34 Biesenbach et al33 found a 38% reduction in insulin requirements in patients with type 1 diabetes as estimated GFR decreased from 80 mL/min to 10 mL/min. Further studies are required to better understand the safety of insulin in treating hospitalized patients with diabetes and renal insufficiency.
Few studies have compared the pharmacodynamics of long-acting insulins in relation to declining renal function. The long-acting analogue insulins have less of a peak than human insulin and thus better mimic endogenous insulin secretion. For insulin detemir, Lindholm and Jacobsen found no significant differences in the pharmacokinetics related to the stages of CKD.35 When using the long-acting insulins glargine or detemir, one should consider giving much lower doses (half the initial starting dosage) and titrating the dosage until target fasting glucose concentrations are reached to prevent hypoglycemia.
Table 3 summarizes recommended insulin dosage adjustments in CKD based on the literature and our clinical experience.
Considerations for dialysis patients
Subcutaneously administered insulin is eliminated renally, unlike endogenous insulin, which undergoes first-pass metabolism in the liver.13,37 As renal function declines, insulin clearance decreases and the insulin dosage must be reduced to prevent hypoglycemia.
Patients on hemodialysis or peritoneal dialysis pose a challenge for insulin dosing. Hemodialysis improves insulin sensitivity but also increases insulin clearance, making it difficult to determine insulin requirements. Sobngwi et al38 conducted a study in diabetic patients with end-stage renal disease on hemodialysis, using a 24-hour euglycemic clamp. They found that exogenous basal insulin requirements were 25% lower on the day after hemodialysis compared with the day before, but premeal insulin requirements stayed the same.
Peritoneal dialysis exposes patients to a high glucose load via the peritoneum, which can worsen insulin resistance. Intraperitoneal administration of insulin during peritoneal dialysis provides a more physiologic effect than subcutaneous administration: it prevents fluctuations of blood glucose and the formation of insulin antibodies. But insulin requirements are higher owing to a dilutional effect and to insulin binding to the plastic surface of the dialysis fluid reservoir.39
GLYCEMIC CONTROL FOR PROCEDURES
No guidelines have been established regarding the optimal blood glucose range for diabetic patients with CKD undergoing diagnostic or surgical procedures. Given the risk of hypoglycemia in such settings, less-stringent targets are reasonable, ie, premeal blood glucose levels of 140 mg/dL and random blood glucose levels of less than 180 mg/dL.
Before surgery, consideration should be given to the type of diabetes, surgical procedure, and metabolic control. Patients on insulin detemir or glargine as part of a basal-bolus regimen with rapid-acting insulin may safely be given the full dose of their basal insulin the night before or the morning of their procedure. However, patients on neutral protamine Hagedorn (NPH) insulin as a part of their basal-bolus regimen should receive half of their usual dose due to a difference in pharmacokinetic profile compared with insulin glargine or detemir.
In insulin-treated patients undergoing prolonged procedures (eg, coronary artery bypass grafting, transplant):
- Discontinue subcutaneous insulin and start an intravenous insulin infusion, titrated to maintain a blood glucose range of 140 to 180 mg/dL
- Subcutaneous insulin management may be acceptable for patients undergoing shorter outpatient procedures
- Supplemental subcutaneous doses of short- or rapid-acting insulin preparations can be given for blood glucose elevation greater than 180 mg/dL.
AVOID ORAL AGENTS AND NONINSULIN INJECTABLES
Oral antidiabetic agents and noninsulin injectables (Table 4) should generally be avoided in hospitalized patients, especially for those with decompensated heart failure, renal insufficiency, hypoperfusion, or chronic pulmonary disease, or for those given intravenous contrast. Most oral medications used to treat diabetes are affected by reduced kidney function, resulting in prolonged drug exposure and increased risk of hypoglycemia in patients with moderate to severe CKD (stages 3–5).
Metformin, a biguanide, is contraindicated in patients with high serum creatinine levels (> 1.5 mg/dL in men, > 1.4 mg/dL in women) because of the theoretical risk of lactic acidosis.40
Sulfonylurea clearance depends on kidney function.41 Severe prolonged episodes of hypoglycemia have been reported in dialysis patients taking these drugs, except with glipizide, which carries a lower risk.41,42
Repaglinide, a nonsulfonylurea insulin secretagogue, can be used in CKD stages 3 to 4 without any dosage adjustment.43
Thiazolidinediones have been reported to slow the progression of diabetic kidney disease independent of glycemic control.44 Adverse effects include fluid retention, edema, and congestive heart failure. Thiazolidinediones should not be used in patients with New York Heart Association class 3 or 4 heart failure,45 and so should not be prescribed in the hospital except for patients who are clinically stable or ready for discharge.
Quick-release bromocriptine, a dopamine receptor agonist, has been shown to be effective in lowering fasting plasma glucose levels and hemoglobin A1c, and improving glucose tolerance in obese patients with type 2 diabetes, although its usefulness in hospitalized patients with diabetes is not known.46,47
Dipeptidyl peptidase inhibitors. Sitagliptin and saxagliptin have been shown to be safe and effective in hospitalized patients with type 2 diabetes.48 However, except for linagliptin, dose reduction is recommended in patients with CKD stage 3 and higher.49–52
GLP-1 receptor agonists. Drugs of this class are potent agents for the reduction of glucose in the outpatient setting but are relatively contraindicated if the GFR is less than 30 mL/min, and they are currently not used in the hospital.
BLOOD GLUCOSE MONITORING IN HOSPITALIZED PATIENTS
Bedside blood glucose monitoring is recommended for all hospitalized patients with known diabetes with or without CKD, those with newly recognized hyperglycemia, and those who receive therapy associated with high risk for hyperglycemia, such as glucocorticoid therapy and enteral and parenteral nutrition. For patients on scheduled diets, fingerstick blood glucose monitoring is recommended before meals and at bedtime. In patients with no oral intake or on continuous enteral or parenteral nutrition, blood glucose monitoring every 4 to 6 hours is recommended. More frequent monitoring (eg, adding a 3:00 am check) may be prudent in patients with CKD.
Continuous glucose monitoring systems use a sensor inserted under the skin and transmit information via radio to a wireless monitor. Such systems are more expensive than conventional glucose monitoring but may enable better glucose control by providing real-time glucose measurements, with levels displayed at 5-minute or 1-minute intervals. Marshall et al53 confirmed this technology’s accuracy and precision in uremic patients on dialysis.
Considerations for peritoneal dialysis
For patients on peritoneal dialysis, glucose in the dialysate exacerbates hyperglycemia. Dialysis solutions with the glucose polymer icodextrin as the osmotic agent instead of glucose have been suggested to reduce glucose exposure.
Glucose monitoring systems measure interstitial fluid glucose by the glucose oxidase reaction and therefore are not affected by icodextrin. However, icodextrin is converted to maltose, a disaccharide composed of two glucose molecules, which can cause spuriously high readings in devices that use test strips containing the enzymes glucose dehydrogenase pyrroloquinoline quinone or glucose dye oxidoreductase. Spurious hyperglycemia may lead to giving too much insulin, in turn leading to symptomatic hypoglycemia.
Clinicians caring for patients receiving icodextrin should ensure that the glucose monitoring system uses only test strips that contain glucose oxidase, glucose dehydrogenase-nicotinamide adenine dinucleotide, or glucose dehydrogenase-flavin adenine dinucleotide, which are not affected by icodextrin.54
IMPROVING QUALITY
Hospitalized patients face many barriers to optimal glycemic control. Less experienced practitioners tend to have insufficient knowledge of insulin preparations and appropriate insulin dosing. Also, diabetes is often listed as a secondary diagnosis and so may be overlooked by the inpatient care team.
Educational programs should be instituted to overcome these barriers and improve knowledge related to inpatient diabetes care. When necessary, the appropriate use of consultants is important in hospitalized settings to improve quality and make hospital care more efficient and cost-effective.
No national benchmarks currently exist for inpatient diabetes care, and they need to be developed to ensure best practices. Physicians should take the initiative to remedy this by collaborating with other healthcare providers, such as dedicated diabetes educators, nursing staff, pharmacists, registered dietitians, and physicians with expertise in diabetes management, with the aim of achieving optimum glycemic control and minimizing hypoglycemia.
Managing glycemic control in hospitalized patients with chronic kidney disease (CKD) and diabetes mellitus is a challenge, with no published guidelines. In this setting, avoiding hypoglycemia takes precedence over meeting strict blood glucose targets. Optimal management is essential to reduce hypoglycemia and the risk of death from cardiovascular disease.1
This article reviews the evidence to guide diabetes management in hospitalized patients with CKD, focusing on blood glucose monitoring, insulin dosing, and concerns about other diabetic agents.
FOCUS ON AVOIDING HYPOGLYCEMIA
CKD is common, estimated to affect more than 50 million people worldwide.2 Diabetes mellitus is the primary cause of kidney failure in 45% of dialysis patients with CKD.
Tight control comes with a cost
Hyperglycemia in hospitalized patients is associated with a higher risk of death, a higher risk of infections, and a longer hospital stay.3,4 In 2001, Van den Berghe et al5 found that intensive insulin therapy reduced the mortality rate in critically ill patients in the surgical intensive care unit. But subsequent studies6,7 found that intensive insulin therapy to achieve tight glycemic control increased rates of morbidity and mortality without adding clinical benefit.
Randomized clinical trials in outpatients have shown that tight control of blood glucose levels reduces microvascular and macrovascular complications in patients with type 1 diabetes.8–10 In the Diabetes Control and Complications Trial,9 compared with conventional therapy, intensive insulin therapy reduced the incidence of retinopathy progression (4.7 vs 1.2 cases per 100 patient-years, number needed to treat [NNT] = 3 for 10 years) and clinical neuropathy (9.8 vs 3.1 per 100 patient-years, NNT = 1.5 for 10 years). The long-term likelihood of a cardiovascular event was also significantly lower in the intensive treatment group (0.38 vs 0.80 events per 100 patient-years).9
Similarly, in the Epidemiology of Diabetes Interventions and Complications follow-up study, the intensive therapy group had fewer cardiovascular deaths.11 On the other hand, the risk of severe hypoglycemia and subsequent coma or seizure was significantly higher in the intensive therapy group than in the conventional therapy group (16.3 vs 5.4 per 100 patient-years).8
CKD increases hypoglycemia risk
Moen et al12 found that the incidence of hypoglycemia was significantly higher in patients with CKD (estimated glomerular filtration rate [GFR] < 60 mL/min) with or without diabetes, and that patients with both conditions were at greatest risk (Figure 1). Multiple factors contribute to the increased risk of hypoglycemia: patients with advanced CKD tend to have poor nutrition, resulting in reduced glycogen stores, and a smaller renal mass reduces renal gluconeogenesis and decreases the elimination of insulin and oral antidiabetic agents.
After the onset of diabetic nephropathy, progression of renal complications and overall life expectancy are influenced by earlier glycemic control.8 Development of diabetic nephropathy is commonly accompanied by changes in metabolic control, particularly an increased risk of hypoglycemia.13 In addition, episodes of severe hypoglycemia constitute an independent cardiovascular risk factor.14
Aggressive glycemic control in hospitalized patients, particularly those with advanced CKD, is associated with a risk of hypoglycemia without overall improvement in outcomes.15 Elderly patients with type 2 diabetes are similar to patients with CKD in that they have a reduced GFR and are thus more sensitive to insulin. In both groups, intensifying glycemic control, especially in the hospital, is associated with more frequent episodes of severe hypoglycemia.16 The focus should be not only on maintaining optimal blood glucose concentration, but also on preventing hypoglycemia.
‘Burnt-out’ diabetes
Paradoxically, patients with end-stage renal disease and type 2 diabetes often experience altered glucose homeostasis with markedly improved glycemic control. They may attain normoglycemia and normalization of hemoglobin A1c, a condition known as “burnt-out” diabetes. Its precise mechanism is not understood and its significance remains unclear (Table 1).17
HEMOGLOBIN A1c CAN BE FALSELY HIGH OR FALSELY LOW
Hemoglobin A1c measurement is used to diagnose diabetes and to assess long-term glycemic control. It is a measure of the fraction of hemoglobin that has been glycated by exposure to glucose. Because the average lifespan of a red cell is 120 days, the hemoglobin A1c value reflects the mean blood glucose concentration over the preceding 3 months.
But hemoglobin A1c measurement has limitations: any condition that alters the lifespan of erythrocytes leads to higher or lower hemoglobin A1c levels. Hemoglobin A1c levels are also affected by kidney dysfunction, hemolysis, and acidosis.18
Falsely high hemoglobin A1c levels are associated with conditions that prolong the lifespan of erythrocytes, such as asplenia. Iron deficiency also increases the average age of circulating red cells because of reduced red cell production. For patients in whom blood glucose measurements do not correlate with hemoglobin A1c measurements, iron deficiency anemia should be considered before altering a treatment regimen.
Falsely low hemoglobin A1c levels are associated with conditions of more rapid erythrocyte turnover, such as autoimmune hemolytic anemia, hereditary spherocytosis, and acute blood loss anemia. In patients with CKD, recombinant erythropoietin treatment lowers hemoglobin A1c levels by increasing the number of immature red cells, which are less likely to glycosylate.19
Morgan et al20 compared the association between hemoglobin A1c and blood glucose levels in diabetic patients with moderate to severe CKD not requiring dialysis and in diabetic patients with normal renal function and found no difference between these two groups, suggesting that hemoglobin A1c is reliable in this setting. But study results conflict for patients on dialysis, making the usefulness of hemoglobin A1c testing for those patients less clear. In one study, hemoglobin A1c testing underestimated glycemic control,20 but other studies found that glycemic control was overestimated.21,22
Alternatives to hemoglobin A1c
Other measures of long-term glycemic control such as fructosamine and glycated albumin levels are sometimes used in conditions in which hemoglobin A1c may not be reliable.
Albumin also undergoes glycation when exposed to glucose. Glycated albumin appears to be a better measure of glycemic control in patients with CKD and diabetes than serum fructosamine,23 which has failed to show a significant correlation with blood glucose levels in patients with CKD.24 However, because serum albumin has a short half-life, glycated albumin reflects glycemic control in only the approximately 1 to 2 weeks before sampling,25 so monthly monitoring is required.
Glycated albumin levels may be reduced due to increased albumin turnover in patients with nephrotic-range proteinuria and in diabetic patients on peritoneal dialysis. Several issues remain unclear, such as the appropriate target level of glycated albumin and at what stage of CKD it should replace hemoglobin A1c testing. If an improved assay that is unaffected by changes in serum albumin becomes available, it may be appropriate to use glycated albumin measurements to assess long-term glycemic control for patients with CKD.
In general, therapeutic decisions to achieve optimum glycemic control in patients with diabetes and CKD should be based on hemoglobin A1c testing, multiple glucose measurements, and patient symptoms of hypoglycemia or hyperglycemia. The best measure for assessing glycemic control in hospitalized patients with CKD remains multiple blood glucose testing daily.
INSULIN THERAPY PREFERRED
Although several studies have evaluated inpatient glycemic control,26–29 no guidelines have been published for hospitalized patients with diabetes and CKD. Insulin therapy is preferred for achieving glycemic control in acutely ill or hospitalized patients with diabetes. Oral hypoglycemic agents should be discontinued.
Regardless of the form of insulin chosen to treat diabetes, caution is needed for patients with kidney disease. During hospitalization, clinical changes are expected owing to illness and differences in caloric intake and physical activity, resulting in altered insulin sensitivity. Insulin-treated hospitalized patients require individualized care, including multiple daily blood glucose tests and insulin therapy modifications for ideal glycemic control.
For surgical or medical intensive care patients on insulin therapy, the target blood glucose level before meals should be 140 mg/dL, and the target random level should be less than 180 mg/dL.15,26–29
Basal-bolus insulin
Sliding-scale therapy should be avoided as the only method for glycemic control. Instead, scheduled subcutaneous basal insulin once or twice daily combined with rapid- or short-acting insulin with meals is recommended.
Basal-bolus insulin therapy, one of the most advanced and flexible insulin replacement therapies, mimics endogenous insulin release and offers great advantages in diabetes care. Using mealtime bolus insulin permits variation in the amount of food eaten; more insulin can be taken with a larger meal and less with smaller meals. A bolus approach offers the flexibility of administering rapid-acting insulin immediately after meals when oral intake is variable.
Individualize insulin therapy
Optimizing glycemic control requires an understanding of the altered pharmacokinetics and pharmacodynamics of insulin in patients with diabetic nephropathy. Table 2 shows the pharmacokinetic profiles of insulin preparations in healthy people. Analogue insulins, which are manufactured by recombinant DNA technology, have conformational changes in the insulin molecule that alter their pharmacokinetics and pharmacodynamics. The rapid-acting analogue insulins are absorbed quickly, making them suitable for postprandial glucose control.
Changes in GFR are associated with altered pharmacokinetics and pharmacodynamics of insulin,30,31 but unlike for oral antidiabetic agents, these properties are not well characterized for insulin preparations in patients with renal insufficiency.13,32–36
CKD may reduce insulin clearance. Rave et al32 reported that the clearance of regular human insulin was reduced by 30% to 40% in patients with type 1 diabetes and a mean estimated GFR of 54 mL/min. They found that the metabolic activity of insulin lispro was more robust than that of short-acting regular human insulin in patients with diabetic nephropathy. In another study, patients with diabetes treated with insulin aspart did not show any significant change in the required insulin dosage in relation to the renal filtration rate.34 Biesenbach et al33 found a 38% reduction in insulin requirements in patients with type 1 diabetes as estimated GFR decreased from 80 mL/min to 10 mL/min. Further studies are required to better understand the safety of insulin in treating hospitalized patients with diabetes and renal insufficiency.
Few studies have compared the pharmacodynamics of long-acting insulins in relation to declining renal function. The long-acting analogue insulins have less of a peak than human insulin and thus better mimic endogenous insulin secretion. For insulin detemir, Lindholm and Jacobsen found no significant differences in the pharmacokinetics related to the stages of CKD.35 When using the long-acting insulins glargine or detemir, one should consider giving much lower doses (half the initial starting dosage) and titrating the dosage until target fasting glucose concentrations are reached to prevent hypoglycemia.
Table 3 summarizes recommended insulin dosage adjustments in CKD based on the literature and our clinical experience.
Considerations for dialysis patients
Subcutaneously administered insulin is eliminated renally, unlike endogenous insulin, which undergoes first-pass metabolism in the liver.13,37 As renal function declines, insulin clearance decreases and the insulin dosage must be reduced to prevent hypoglycemia.
Patients on hemodialysis or peritoneal dialysis pose a challenge for insulin dosing. Hemodialysis improves insulin sensitivity but also increases insulin clearance, making it difficult to determine insulin requirements. Sobngwi et al38 conducted a study in diabetic patients with end-stage renal disease on hemodialysis, using a 24-hour euglycemic clamp. They found that exogenous basal insulin requirements were 25% lower on the day after hemodialysis compared with the day before, but premeal insulin requirements stayed the same.
Peritoneal dialysis exposes patients to a high glucose load via the peritoneum, which can worsen insulin resistance. Intraperitoneal administration of insulin during peritoneal dialysis provides a more physiologic effect than subcutaneous administration: it prevents fluctuations of blood glucose and the formation of insulin antibodies. But insulin requirements are higher owing to a dilutional effect and to insulin binding to the plastic surface of the dialysis fluid reservoir.39
GLYCEMIC CONTROL FOR PROCEDURES
No guidelines have been established regarding the optimal blood glucose range for diabetic patients with CKD undergoing diagnostic or surgical procedures. Given the risk of hypoglycemia in such settings, less-stringent targets are reasonable, ie, premeal blood glucose levels of 140 mg/dL and random blood glucose levels of less than 180 mg/dL.
Before surgery, consideration should be given to the type of diabetes, surgical procedure, and metabolic control. Patients on insulin detemir or glargine as part of a basal-bolus regimen with rapid-acting insulin may safely be given the full dose of their basal insulin the night before or the morning of their procedure. However, patients on neutral protamine Hagedorn (NPH) insulin as a part of their basal-bolus regimen should receive half of their usual dose due to a difference in pharmacokinetic profile compared with insulin glargine or detemir.
In insulin-treated patients undergoing prolonged procedures (eg, coronary artery bypass grafting, transplant):
- Discontinue subcutaneous insulin and start an intravenous insulin infusion, titrated to maintain a blood glucose range of 140 to 180 mg/dL
- Subcutaneous insulin management may be acceptable for patients undergoing shorter outpatient procedures
- Supplemental subcutaneous doses of short- or rapid-acting insulin preparations can be given for blood glucose elevation greater than 180 mg/dL.
AVOID ORAL AGENTS AND NONINSULIN INJECTABLES
Oral antidiabetic agents and noninsulin injectables (Table 4) should generally be avoided in hospitalized patients, especially for those with decompensated heart failure, renal insufficiency, hypoperfusion, or chronic pulmonary disease, or for those given intravenous contrast. Most oral medications used to treat diabetes are affected by reduced kidney function, resulting in prolonged drug exposure and increased risk of hypoglycemia in patients with moderate to severe CKD (stages 3–5).
Metformin, a biguanide, is contraindicated in patients with high serum creatinine levels (> 1.5 mg/dL in men, > 1.4 mg/dL in women) because of the theoretical risk of lactic acidosis.40
Sulfonylurea clearance depends on kidney function.41 Severe prolonged episodes of hypoglycemia have been reported in dialysis patients taking these drugs, except with glipizide, which carries a lower risk.41,42
Repaglinide, a nonsulfonylurea insulin secretagogue, can be used in CKD stages 3 to 4 without any dosage adjustment.43
Thiazolidinediones have been reported to slow the progression of diabetic kidney disease independent of glycemic control.44 Adverse effects include fluid retention, edema, and congestive heart failure. Thiazolidinediones should not be used in patients with New York Heart Association class 3 or 4 heart failure,45 and so should not be prescribed in the hospital except for patients who are clinically stable or ready for discharge.
Quick-release bromocriptine, a dopamine receptor agonist, has been shown to be effective in lowering fasting plasma glucose levels and hemoglobin A1c, and improving glucose tolerance in obese patients with type 2 diabetes, although its usefulness in hospitalized patients with diabetes is not known.46,47
Dipeptidyl peptidase inhibitors. Sitagliptin and saxagliptin have been shown to be safe and effective in hospitalized patients with type 2 diabetes.48 However, except for linagliptin, dose reduction is recommended in patients with CKD stage 3 and higher.49–52
GLP-1 receptor agonists. Drugs of this class are potent agents for the reduction of glucose in the outpatient setting but are relatively contraindicated if the GFR is less than 30 mL/min, and they are currently not used in the hospital.
BLOOD GLUCOSE MONITORING IN HOSPITALIZED PATIENTS
Bedside blood glucose monitoring is recommended for all hospitalized patients with known diabetes with or without CKD, those with newly recognized hyperglycemia, and those who receive therapy associated with high risk for hyperglycemia, such as glucocorticoid therapy and enteral and parenteral nutrition. For patients on scheduled diets, fingerstick blood glucose monitoring is recommended before meals and at bedtime. In patients with no oral intake or on continuous enteral or parenteral nutrition, blood glucose monitoring every 4 to 6 hours is recommended. More frequent monitoring (eg, adding a 3:00 am check) may be prudent in patients with CKD.
Continuous glucose monitoring systems use a sensor inserted under the skin and transmit information via radio to a wireless monitor. Such systems are more expensive than conventional glucose monitoring but may enable better glucose control by providing real-time glucose measurements, with levels displayed at 5-minute or 1-minute intervals. Marshall et al53 confirmed this technology’s accuracy and precision in uremic patients on dialysis.
Considerations for peritoneal dialysis
For patients on peritoneal dialysis, glucose in the dialysate exacerbates hyperglycemia. Dialysis solutions with the glucose polymer icodextrin as the osmotic agent instead of glucose have been suggested to reduce glucose exposure.
Glucose monitoring systems measure interstitial fluid glucose by the glucose oxidase reaction and therefore are not affected by icodextrin. However, icodextrin is converted to maltose, a disaccharide composed of two glucose molecules, which can cause spuriously high readings in devices that use test strips containing the enzymes glucose dehydrogenase pyrroloquinoline quinone or glucose dye oxidoreductase. Spurious hyperglycemia may lead to giving too much insulin, in turn leading to symptomatic hypoglycemia.
Clinicians caring for patients receiving icodextrin should ensure that the glucose monitoring system uses only test strips that contain glucose oxidase, glucose dehydrogenase-nicotinamide adenine dinucleotide, or glucose dehydrogenase-flavin adenine dinucleotide, which are not affected by icodextrin.54
IMPROVING QUALITY
Hospitalized patients face many barriers to optimal glycemic control. Less experienced practitioners tend to have insufficient knowledge of insulin preparations and appropriate insulin dosing. Also, diabetes is often listed as a secondary diagnosis and so may be overlooked by the inpatient care team.
Educational programs should be instituted to overcome these barriers and improve knowledge related to inpatient diabetes care. When necessary, the appropriate use of consultants is important in hospitalized settings to improve quality and make hospital care more efficient and cost-effective.
No national benchmarks currently exist for inpatient diabetes care, and they need to be developed to ensure best practices. Physicians should take the initiative to remedy this by collaborating with other healthcare providers, such as dedicated diabetes educators, nursing staff, pharmacists, registered dietitians, and physicians with expertise in diabetes management, with the aim of achieving optimum glycemic control and minimizing hypoglycemia.
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
- Newman DJ, Mattock MB, Dawnay AB, et al. Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol Assess 2005; 9:iii–vi, xiii–163.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Golden SH, Peart-Vigilance C, Kao WH, Brancati FL. Perioperative glycemic control and the risk of infectious complications in a cohort of adults with diabetes. Diabetes Care 1999; 22:1408–1414.
- Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Brunkhorst FM, Engel C, Bloos F, et al; German Competence Network Sepsis (SepNet). Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Effect of intensive diabetes management on macrovascular events and risk factors in the Diabetes Control and Complications Trial. Am J Cardiol 1995; 75:894–903.
- Nathan DM, Lachin J, Cleary P, et al; Diabetes Control and Complications Trial; Epidemiology of Diabetes Interventions and Complications Research Group. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med 2003; 348:2294–2303.
- Writing Group for the DCCT/EDIC Research Group; Orchard TJ, Nathan DM, Zinman B, et al. Association between 7 years of intensive treatment of type 1 diabetes and long-term mortality. JAMA 2015; 313:45–53.
- Moen MF, Zhan M, Hsu VD, et al. Frequency of hypoglycemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol 2009; 4:1121–1127.
- Iglesias P, Díez J. Insulin therapy in renal disease. Diabetes Obes Metab 2008; 10:811–823.
- Zoungas S, Patel A, Chalmers J, et al; ADVANCE Collaborative Group. Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010; 363:1410–1418.
- Moghissi ES, Korytkowski MT, DiNardo M, et al; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Kovesdy CP, Park JC, Kalantar-Zadeh K. Glycemic control and burnt-out diabetes in ESRD. Semin Dial 2010; 23:148–156.
- De Marchi S, Cecchin E, Camurri C, et al. Origin of glycosylated hemoglobin A1 in chronic renal failure. Int J Artif Organs 1983; 6:77–82.
- Brown JN, Kemp DW, Brice KR. Class effect of erythropoietin therapy on hemoglobin A(1c) in a patient with diabetes mellitus and chronic kidney disease not undergoing hemodialysis. Pharmacotherapy 2009; 29:468–472.
- Morgan L, Marenah CB, Jeffcoate WJ, Morgan AG. Glycated proteins as indices of glycemic control in diabetic patients with chronic renal failure. Diabet Med 1996; 13:514–519.
- Peacock TP, Shihabi ZK, Bleyer AJ, et al. Comparison of glycated albumin and hemoglobin A(1c) levels in diabetic subjects on hemodialysis. Kidney Int 2008; 73:1062–1068.
- Joy MS, Cefalu WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Mittman N, Desiraju B, Fazil I, et al. Serum fructosamine versus glycosylated hemoglobin as an index of glycemic control, hospitalization, and infection in diabetic hemodialysis patients. Kidney Int 2010; 78(suppl 117):S41–S45.
- Alskar O, Korelli J, Duffull SB. A pharmacokinetic model for the glycation of albumin. J Pharmacokinet Pharmacodyn 2012; 39:273–282.
- Qaseem A, Humphrey LL, Chou R, Snow V, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Use of intensive insulin therapy for the management of glycemic control in hospitalized patients: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2011; 154:260–267.
- Murad MH, Coburn JA, Coto-Yglesias F, et al. Glycemic control in non-critically ill hospitalized patients: a systematic review and meta-analysis. J Clin Endocrinol Metab 2012; 97:49–58.
- Bogun M, Inzucchi SE. Inpatient management of diabetes and hyperglycemia. Clin Ther 2013; 35:724–733.
- Miller DB. Glycemic targets in hospital and barriers to attaining them. Can J Diabetes 2014; 38:74–78.
- Eidemak I, Feldt-Rasmussen B, Kanstrup IL, Nielsen SL, Schmitz O, Strandgaard S. Insulin resistance and hyperinsulinaemia in mild to moderate progressive chronic renal failure and its association with aerobic work capacity. Diabetologia 1995; 38:565–572.
- Svensson M, Yu Z, Eriksson J. A small reduction in glomerular filtration is accompanied by insulin resistance in type I diabetes patients with diabetic nephropathy. Eur J Clin Invest 2002; 32:100–109.
- Rave K, Heise T, Pfutzner A, Heinemann L, Sawicki P. Impact of diabetic nephropathy on pharmacodynamics and pharmacokinetic properties of insulin in type I diabetic patients. Diabetes Care 2001; 24:886–890.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Holmes G, Galitz L, Hu P, Lyness W. Pharmacokinetics of insulin aspart in obesity, renal impairment, or hepatic impairment. Br J Clin Pharmacol 2005; 60:469–476.
- Lindholm A, Jacobsen LV. Clinical pharmacokinetics and pharmacodynamics of insulin aspart. Clin Pharmacokinet 2001; 40:641–659.
- Bolli GB, Hahn AD, Schmidt R, et al. Plasma exposure to insulin glargine and its metabolites M1 and M2 after subcutaneous injection of therapeutic and supratherapeutic doses of glargine in subjects with type 1 diabetes. Diabetes Care 2012; 35:2626–2630.
- Nielsen S. Time course and kinetics of proximal tubular processing of insulin. Am J Physiol 1992; 262:F813–F822.
- Sobngwi E, Enoru S, Ashuntantang G, et al. Day-to-day variation of insulin requirements of patients with type 2 diabetes and end-stage renal disease undergoing maintenance hemodialysis. Diabetes Care 2010; 33:1409–1412.
- Quellhorst E. Insulin therapy during peritoneal dialysis: pros and cons of various forms of administration. J Am Soc Nephrol 2002; 13(suppl 1):S92–S96.
- Davidson MB, Peters AL. An overview of metformin in the treatment of type 2 diabetes mellitus. Am J Med 1997; 102:99–110.
- Ahmed Z, Simon B, Choudhury D. Management of diabetes in patients with chronic kidney disease. Postgrad Med 2009; 121:52–60.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26(suppl 4):73–85.
- Hasslacher C; Multinational Repaglinide Renal Study Group. Safety and efficacy of repaglinide in type 2 diabetic patients with and without impaired renal function. Diabetes Care 2003; 26:886–891.
- Iglesias P, Dies JJ. Peroxisome proliferator-activated receptor gamma agonists in renal disease. Eur J Endocrinol 2006; 154:613–621.
- Hollenberg NK. Considerations for management of fluid dynamic issues associated with thiazolidinediones. Am J Med 2003; 115(suppl. 8A) 111S–115S.
- Kamath V, Jones CN, Yip JC, et al. Effects of a quick-release form of bromocriptine (Ergoset) on fasting and postprandial plasma glucose, insulin, lipid, and lipoprotein concentrations in obese nondiabetic hyperinsulinemic women. Diabetes Care 1997; 20:1697–1701.
- Pijl H, Ohashi S, Matsuda M, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care 2000; 23:1154–1161.
- Umpierrez GE, Gianchandani R, Smiley D, et al. Safety and efficacy of sitagliptin therapy for the inpatient management of general medicine and surgery patients with type 2 diabetes: a pilot, randomized, controlled study. Diabetes Care 2013; 36:3430–3435.
- Chan JC, Scott R, Arjona Ferreira JC, et al. Safety and efficacy of sitagliptin in patients with type 2 diabetes and chronic renal insufficiency. Diabetes Obes Metab 2008; 10:545–555.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Onglyza package insert. www.azpicentral.com/onglyza/pi_onglyza.pdf. Accessed March 8, 2016.
- Gallwitz B. Safety and efficacy of linagliptin in type 2 diabetes patients with common renal and cardiovascular risk factors. Ther Adv Endocrinol Metab 2013; 4:95–105.
- Marshall J, Jennings P, Scott A, Fluck RJ, McIntyre CW. Glycemic control in diabetic CAPD patients assessed by continuous glucose monitoring system (CGMS). Kidney Int 2003; 64:1480–1486.
- Schleis TG. Interference of maltose, icodextrin, galactose, or xylose with some blood glucose monitoring systems. Pharmacotherapy 2007; 27:1313–1321.
- Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351:1296–1305.
- Newman DJ, Mattock MB, Dawnay AB, et al. Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol Assess 2005; 9:iii–vi, xiii–163.
- Umpierrez GE, Isaacs SD, Bazargan N, You X, Thaler LM, Kitabchi AE. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002; 87:978–982.
- Golden SH, Peart-Vigilance C, Kao WH, Brancati FL. Perioperative glycemic control and the risk of infectious complications in a cohort of adults with diabetes. Diabetes Care 1999; 22:1408–1414.
- Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367.
- NICE-SUGAR Study Investigators; Finfer S, Chittock DR, Su SY, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Brunkhorst FM, Engel C, Bloos F, et al; German Competence Network Sepsis (SepNet). Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Effect of intensive diabetes management on macrovascular events and risk factors in the Diabetes Control and Complications Trial. Am J Cardiol 1995; 75:894–903.
- Nathan DM, Lachin J, Cleary P, et al; Diabetes Control and Complications Trial; Epidemiology of Diabetes Interventions and Complications Research Group. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med 2003; 348:2294–2303.
- Writing Group for the DCCT/EDIC Research Group; Orchard TJ, Nathan DM, Zinman B, et al. Association between 7 years of intensive treatment of type 1 diabetes and long-term mortality. JAMA 2015; 313:45–53.
- Moen MF, Zhan M, Hsu VD, et al. Frequency of hypoglycemia and its significance in chronic kidney disease. Clin J Am Soc Nephrol 2009; 4:1121–1127.
- Iglesias P, Díez J. Insulin therapy in renal disease. Diabetes Obes Metab 2008; 10:811–823.
- Zoungas S, Patel A, Chalmers J, et al; ADVANCE Collaborative Group. Severe hypoglycemia and risks of vascular events and death. N Engl J Med 2010; 363:1410–1418.
- Moghissi ES, Korytkowski MT, DiNardo M, et al; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care 2009; 32:1119–1131.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- Kovesdy CP, Park JC, Kalantar-Zadeh K. Glycemic control and burnt-out diabetes in ESRD. Semin Dial 2010; 23:148–156.
- De Marchi S, Cecchin E, Camurri C, et al. Origin of glycosylated hemoglobin A1 in chronic renal failure. Int J Artif Organs 1983; 6:77–82.
- Brown JN, Kemp DW, Brice KR. Class effect of erythropoietin therapy on hemoglobin A(1c) in a patient with diabetes mellitus and chronic kidney disease not undergoing hemodialysis. Pharmacotherapy 2009; 29:468–472.
- Morgan L, Marenah CB, Jeffcoate WJ, Morgan AG. Glycated proteins as indices of glycemic control in diabetic patients with chronic renal failure. Diabet Med 1996; 13:514–519.
- Peacock TP, Shihabi ZK, Bleyer AJ, et al. Comparison of glycated albumin and hemoglobin A(1c) levels in diabetic subjects on hemodialysis. Kidney Int 2008; 73:1062–1068.
- Joy MS, Cefalu WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Mittman N, Desiraju B, Fazil I, et al. Serum fructosamine versus glycosylated hemoglobin as an index of glycemic control, hospitalization, and infection in diabetic hemodialysis patients. Kidney Int 2010; 78(suppl 117):S41–S45.
- Alskar O, Korelli J, Duffull SB. A pharmacokinetic model for the glycation of albumin. J Pharmacokinet Pharmacodyn 2012; 39:273–282.
- Qaseem A, Humphrey LL, Chou R, Snow V, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Use of intensive insulin therapy for the management of glycemic control in hospitalized patients: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2011; 154:260–267.
- Murad MH, Coburn JA, Coto-Yglesias F, et al. Glycemic control in non-critically ill hospitalized patients: a systematic review and meta-analysis. J Clin Endocrinol Metab 2012; 97:49–58.
- Bogun M, Inzucchi SE. Inpatient management of diabetes and hyperglycemia. Clin Ther 2013; 35:724–733.
- Miller DB. Glycemic targets in hospital and barriers to attaining them. Can J Diabetes 2014; 38:74–78.
- Eidemak I, Feldt-Rasmussen B, Kanstrup IL, Nielsen SL, Schmitz O, Strandgaard S. Insulin resistance and hyperinsulinaemia in mild to moderate progressive chronic renal failure and its association with aerobic work capacity. Diabetologia 1995; 38:565–572.
- Svensson M, Yu Z, Eriksson J. A small reduction in glomerular filtration is accompanied by insulin resistance in type I diabetes patients with diabetic nephropathy. Eur J Clin Invest 2002; 32:100–109.
- Rave K, Heise T, Pfutzner A, Heinemann L, Sawicki P. Impact of diabetic nephropathy on pharmacodynamics and pharmacokinetic properties of insulin in type I diabetic patients. Diabetes Care 2001; 24:886–890.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Holmes G, Galitz L, Hu P, Lyness W. Pharmacokinetics of insulin aspart in obesity, renal impairment, or hepatic impairment. Br J Clin Pharmacol 2005; 60:469–476.
- Lindholm A, Jacobsen LV. Clinical pharmacokinetics and pharmacodynamics of insulin aspart. Clin Pharmacokinet 2001; 40:641–659.
- Bolli GB, Hahn AD, Schmidt R, et al. Plasma exposure to insulin glargine and its metabolites M1 and M2 after subcutaneous injection of therapeutic and supratherapeutic doses of glargine in subjects with type 1 diabetes. Diabetes Care 2012; 35:2626–2630.
- Nielsen S. Time course and kinetics of proximal tubular processing of insulin. Am J Physiol 1992; 262:F813–F822.
- Sobngwi E, Enoru S, Ashuntantang G, et al. Day-to-day variation of insulin requirements of patients with type 2 diabetes and end-stage renal disease undergoing maintenance hemodialysis. Diabetes Care 2010; 33:1409–1412.
- Quellhorst E. Insulin therapy during peritoneal dialysis: pros and cons of various forms of administration. J Am Soc Nephrol 2002; 13(suppl 1):S92–S96.
- Davidson MB, Peters AL. An overview of metformin in the treatment of type 2 diabetes mellitus. Am J Med 1997; 102:99–110.
- Ahmed Z, Simon B, Choudhury D. Management of diabetes in patients with chronic kidney disease. Postgrad Med 2009; 121:52–60.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26(suppl 4):73–85.
- Hasslacher C; Multinational Repaglinide Renal Study Group. Safety and efficacy of repaglinide in type 2 diabetic patients with and without impaired renal function. Diabetes Care 2003; 26:886–891.
- Iglesias P, Dies JJ. Peroxisome proliferator-activated receptor gamma agonists in renal disease. Eur J Endocrinol 2006; 154:613–621.
- Hollenberg NK. Considerations for management of fluid dynamic issues associated with thiazolidinediones. Am J Med 2003; 115(suppl. 8A) 111S–115S.
- Kamath V, Jones CN, Yip JC, et al. Effects of a quick-release form of bromocriptine (Ergoset) on fasting and postprandial plasma glucose, insulin, lipid, and lipoprotein concentrations in obese nondiabetic hyperinsulinemic women. Diabetes Care 1997; 20:1697–1701.
- Pijl H, Ohashi S, Matsuda M, et al. Bromocriptine: a novel approach to the treatment of type 2 diabetes. Diabetes Care 2000; 23:1154–1161.
- Umpierrez GE, Gianchandani R, Smiley D, et al. Safety and efficacy of sitagliptin therapy for the inpatient management of general medicine and surgery patients with type 2 diabetes: a pilot, randomized, controlled study. Diabetes Care 2013; 36:3430–3435.
- Chan JC, Scott R, Arjona Ferreira JC, et al. Safety and efficacy of sitagliptin in patients with type 2 diabetes and chronic renal insufficiency. Diabetes Obes Metab 2008; 10:545–555.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Onglyza package insert. www.azpicentral.com/onglyza/pi_onglyza.pdf. Accessed March 8, 2016.
- Gallwitz B. Safety and efficacy of linagliptin in type 2 diabetes patients with common renal and cardiovascular risk factors. Ther Adv Endocrinol Metab 2013; 4:95–105.
- Marshall J, Jennings P, Scott A, Fluck RJ, McIntyre CW. Glycemic control in diabetic CAPD patients assessed by continuous glucose monitoring system (CGMS). Kidney Int 2003; 64:1480–1486.
- Schleis TG. Interference of maltose, icodextrin, galactose, or xylose with some blood glucose monitoring systems. Pharmacotherapy 2007; 27:1313–1321.
KEY POINTS
- Hemoglobin A1c values are often unreliable in patients with end-stage renal disease; close monitoring by fingerstick testing or a continuous monitoring system is recommended during hospitalization.
- Insulin is the preferred treatment for hospitalized patients with diabetes; oral antidiabetic agents should be avoided.
- Blood glucose targets for hospitalized patients with diabetes or stress hyperglycemia should be less than 140 mg/dL before meals, and random values should be less than 180 mg/dL.
- A basal-bolus insulin approach is flexible and mimics endogenous insulin release.
- Many insulin-treated patients with type 2 diabetes and CKD stop needing insulin as kidney disease progresses.
Can patients with infectious endocarditis be safely anticoagulated?
Managing anticoagulation in patients with infectious endocarditis requires an individualized approach, using a careful risk-benefit assessment on a case-by-case basis. There is a dearth of high-quality evidence; consequently, the recommendations also vary according to the clinical situation.
Newly diagnosed native valve infectious endocarditis in itself is not an indication for anticoagulation.1–3 The question of whether to anticoagulate arises in patients who have a preexisting or coexisting indication for anticoagulation such as atrial fibrillation, deep vein thrombosis, pulmonary embolism, or a mechanical prosthetic heart valve. The question becomes yet more complex in patients with cerebrovascular complications and a coexistent strong indication for anticoagulation, creating what is often a very thorny dilemma.
Based on a review of available evidence, recommendations for anticoagulation in patients with infectious endocarditis are summarized below.
AVAILABLE EVIDENCE IS SCARCE AND MIXED
Earlier observational studies suggested a significant risk of cerebral hemorrhage with anticoagulation in patients with native valve endocarditis, although none of these studies were recent (some of them took place in the 1940s), and none are methodologically compelling.4–8 Consequently, some experts have expressed skepticism regarding their findings, particularly in recent years.
In part, this skepticism arises from studies that showed a lower incidence of cerebrovascular complications and smaller vegetation size in patients with prosthetic valve infectious endocarditis, studies in which many of the patients received anticoagulation therapy.9,10 The mechanism responsible for this effect is theorized to be that the vegetation is an amalgam of destroyed cells, platelets, and fibrin, with anticoagulation preventing this aggregation from further growth and propagation.
How great is the benefit or the potential harm?
Some experts argue that the incidence of ischemic stroke with hemorrhagic transformation in patients with infectious endocarditis receiving anticoagulation is overestimated. According to this view, the beneficial effects of anticoagulation at least counterbalance the potential harmful effects.
In addition to the studies cited above, recent studies have shown that patients on anticoagulation tend to have smaller vegetations and fewer cerebrovascular complications.11–13 Snygg-Martin et al11 and Rasmussen et al12 found not only that cerebrovascular complications were less common in patients already on anticoagulation at the time infectious endocarditis was diagnosed, but also that no increase in the rate of hemorrhagic lesions was reported.
These were all nonrandomized studies, and most of the patients in them had native valve infectious endocarditis diagnosed at an early stage. Importantly, these studies found that the beneficial effects of anticoagulation were only present if the patient was receiving warfarin before infectious endocarditis was diagnosed and antibiotic therapy was initiated. No benefits from anticoagulation were demonstrated once antimicrobial therapy was begun.
Similarly, Anavekar et al14 showed that embolic events occurred significantly less often in those who were currently on continuous daily antiplatelet therapy, suggesting that receiving antiplatelet agents at baseline protects against cardioembolic events in patients who develop infective endocarditis. However, the only randomized trial examining the initiation of antiplatelet therapy in patients diagnosed with infectious endocarditis receiving antibiotic treatment showed that adding aspirin did not reduce the risk of embolic events and was associated with a trend toward increased risk of bleeding.15
A recent large cohort study suggested that infectious endocarditis patients who receive anticoagulation therapy may have a higher incidence of cerebrovascular complications (hazard ratio 1.31, 95% confidence interval 1.00–1.72, P = .048), with a particular association of anticoagulation therapy with intracranial bleeding (hazard ratio 2.71, 95% confidence interval 1.54–4.76, P = .001).16
Another provocative link supported by the same study was a higher incidence of hemorrhagic complications with anticoagulation in patients with infectious endocarditis caused by Staphylococcus aureus, an association also suggested by older data from Tornos et al,8 but not seen in a study by Rasmussen et al.12
Continuing anticoagulation is an individualized decision
The benefit or harm of anticoagulation in patients with infectious endocarditis may be determined at least in part by a complex mix of factors including the valve involved (embolic events are more common with mitral valve vegetations than with aortic valve vegetations), vegetation size (higher risk if > 1 cm), mobility of vegetations, and perhaps the virulence of the causative organism.16,17 The fact that antimicrobial therapy obviates any beneficial effect of anticoagulation speaks strongly against starting anticoagulation therapy in infectious endocarditis patients with the sole purpose of reducing stroke risk.

Without large randomized trials to better delineate the risks and benefits of continuing preexisting anticoagulation in all patients with infectious endocarditis, patients already receiving anticoagulants need a careful, individualized risk-benefit assessment. Current guidelines agree that newly diagnosed infectious endocarditis per se is not an indication for anticoagulation or aspirin therapy (Table 1).1–3
TAKE-HOME POINTS
- Starting antiplatelet and anticoagulation therapy for the sole purpose of stroke prevention is not recommended in patients with newly diagnosed infectious endocarditis.
- In most cases, anticoagulation and antiplatelet therapy should be temporarily discontinued in patients with infectious endocarditis and stroke or suspected stroke.
- Patients need careful assessment on a case-by-case basis, and the presence of risk factors predisposing patients to cerebrovascular complications (eg, large or very mobile vegetations, causative pathogens such as S aureus or Candida spp) may prompt temporary suspension of anticoagulation and antiplatelet therapy.
- If there is a clear preexisting or coexisting indication for these agents and surgery is not anticipated, consider continuing antiplatelet and anticoagulant therapy in patients with infectious endocarditis, provided they lack the risk factors described above and stroke has been excluded.
- If there is a clear preexisting or coexisting indication for these agents and surgery is being considered, consider using a short-acting anticoagulant such as intravenous or low-molecular weight heparin as a bridge to surgery.
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132:1435–1486.
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis. Rev Esp Cardiol (Engl Ed) 2016; 69:69.
- Whitlock RP, Sun JC, Fremes SE, Rubens FD, Teoh KH. Antithrombotic and thrombolytic therapy for valvular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141:e576S–e600S.
- Delahaye JP, Poncet P, Malquarti V, Beaune J, Garé JP, Mann JM. Cerebrovascular accidents in infective endocarditis: role of anticoagulation. Eur Heart J 1990; 11:1074–1078.
- Loewe L. The combined use of anti-infectives and anticoagulants in the treatment of subacute bacterial endocarditis. Bull N Y Acad Med 1945; 21:59–86.
- Priest WS, Smith JM, McGee GC. The effect of anticoagulants on the penicillin therapy and the pathologic lesions of subacute bacterial endocarditis. N Engl J Med 1946; 235:699–706.
- Pruitt AA, Rubin RH, Karchmer AW, Duncan GW. Neurologic complications of bacterial endocarditis. Medicine 1978; 57:329–343.
- Tornos P, Almirante B, Mirabet S, Permanyer G, Pahissa A, Soler-Soler J. Infective endocarditis due to Staphylococcus aureus: deleterious effect of anticoagulant therapy. Arch Intern Med 1999; 159:473–475.
- Wilson WR, Geraci JE, Danielson GK, et al. Anticoagulant therapy and central nervous system complications in patients with prosthetic valve endocarditis. Circulation 1978; 57:1004–1007.
- Schulz R, Werner GS, Fuchs JB, et al. Clinical outcome and echocardiographic findings of native and prosthetic valve endocarditis in the 1990’s. Eur Heart J 1996; 17:281–288.
- Snygg-Martin U, Rasmussen RV, Hassager C, Bruun NE, Andersson R, Olaison L. Warfarin therapy and incidence of cerebrovascular complications in left-sided native valve endocarditis. Eur J Clin Microbiol Infect Dis 2011; 30:151–157.
- Rasmussen RV, Snygg-Martin U, Olaison L, et al. Major cerebral events in Staphylococcus aureus infective endocarditis: is anticoagulant therapy safe? Cardiology 2009; 114:284–291.
- Yau JW, Lee P, Wilson A, Jenkins AJ. Prosthetic valve endocarditis: what is the evidence for anticoagulant therapy? Intern Med J 2011; 41:795–797.
- Anavekar NS, Tleyjeh IM, Mirzoyev Z, et al. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis 2007; 44:1180–1186.
- Chan KL, Dumesnil JG, Cujec B, et al. A randomized trial of aspirin on the risk of embolic events in patients with infective endocarditis. J Am Coll Cardiol 2003; 42:775–780.
- Garcia-Cabrera E, Fernández-Hidalgo N, Almirante B, et al. Neurological complications of infective endocarditis: risk factors, outcome, and impact of cardiac surgery: a multicenter observational study. Circulation 2013; 127:2272–2284.
- Thuny F, Di Salvo G, Belliard O, et al. Risk of embolism and death in infective endocarditis: prognostic value of echocardiography: a prospective multicenter study. Circulation 2005; 112:69–75.
Managing anticoagulation in patients with infectious endocarditis requires an individualized approach, using a careful risk-benefit assessment on a case-by-case basis. There is a dearth of high-quality evidence; consequently, the recommendations also vary according to the clinical situation.
Newly diagnosed native valve infectious endocarditis in itself is not an indication for anticoagulation.1–3 The question of whether to anticoagulate arises in patients who have a preexisting or coexisting indication for anticoagulation such as atrial fibrillation, deep vein thrombosis, pulmonary embolism, or a mechanical prosthetic heart valve. The question becomes yet more complex in patients with cerebrovascular complications and a coexistent strong indication for anticoagulation, creating what is often a very thorny dilemma.
Based on a review of available evidence, recommendations for anticoagulation in patients with infectious endocarditis are summarized below.
AVAILABLE EVIDENCE IS SCARCE AND MIXED
Earlier observational studies suggested a significant risk of cerebral hemorrhage with anticoagulation in patients with native valve endocarditis, although none of these studies were recent (some of them took place in the 1940s), and none are methodologically compelling.4–8 Consequently, some experts have expressed skepticism regarding their findings, particularly in recent years.
In part, this skepticism arises from studies that showed a lower incidence of cerebrovascular complications and smaller vegetation size in patients with prosthetic valve infectious endocarditis, studies in which many of the patients received anticoagulation therapy.9,10 The mechanism responsible for this effect is theorized to be that the vegetation is an amalgam of destroyed cells, platelets, and fibrin, with anticoagulation preventing this aggregation from further growth and propagation.
How great is the benefit or the potential harm?
Some experts argue that the incidence of ischemic stroke with hemorrhagic transformation in patients with infectious endocarditis receiving anticoagulation is overestimated. According to this view, the beneficial effects of anticoagulation at least counterbalance the potential harmful effects.
In addition to the studies cited above, recent studies have shown that patients on anticoagulation tend to have smaller vegetations and fewer cerebrovascular complications.11–13 Snygg-Martin et al11 and Rasmussen et al12 found not only that cerebrovascular complications were less common in patients already on anticoagulation at the time infectious endocarditis was diagnosed, but also that no increase in the rate of hemorrhagic lesions was reported.
These were all nonrandomized studies, and most of the patients in them had native valve infectious endocarditis diagnosed at an early stage. Importantly, these studies found that the beneficial effects of anticoagulation were only present if the patient was receiving warfarin before infectious endocarditis was diagnosed and antibiotic therapy was initiated. No benefits from anticoagulation were demonstrated once antimicrobial therapy was begun.
Similarly, Anavekar et al14 showed that embolic events occurred significantly less often in those who were currently on continuous daily antiplatelet therapy, suggesting that receiving antiplatelet agents at baseline protects against cardioembolic events in patients who develop infective endocarditis. However, the only randomized trial examining the initiation of antiplatelet therapy in patients diagnosed with infectious endocarditis receiving antibiotic treatment showed that adding aspirin did not reduce the risk of embolic events and was associated with a trend toward increased risk of bleeding.15
A recent large cohort study suggested that infectious endocarditis patients who receive anticoagulation therapy may have a higher incidence of cerebrovascular complications (hazard ratio 1.31, 95% confidence interval 1.00–1.72, P = .048), with a particular association of anticoagulation therapy with intracranial bleeding (hazard ratio 2.71, 95% confidence interval 1.54–4.76, P = .001).16
Another provocative link supported by the same study was a higher incidence of hemorrhagic complications with anticoagulation in patients with infectious endocarditis caused by Staphylococcus aureus, an association also suggested by older data from Tornos et al,8 but not seen in a study by Rasmussen et al.12
Continuing anticoagulation is an individualized decision
The benefit or harm of anticoagulation in patients with infectious endocarditis may be determined at least in part by a complex mix of factors including the valve involved (embolic events are more common with mitral valve vegetations than with aortic valve vegetations), vegetation size (higher risk if > 1 cm), mobility of vegetations, and perhaps the virulence of the causative organism.16,17 The fact that antimicrobial therapy obviates any beneficial effect of anticoagulation speaks strongly against starting anticoagulation therapy in infectious endocarditis patients with the sole purpose of reducing stroke risk.
Without large randomized trials to better delineate the risks and benefits of continuing preexisting anticoagulation in all patients with infectious endocarditis, patients already receiving anticoagulants need a careful, individualized risk-benefit assessment. Current guidelines agree that newly diagnosed infectious endocarditis per se is not an indication for anticoagulation or aspirin therapy (Table 1).1–3
TAKE-HOME POINTS
- Starting antiplatelet and anticoagulation therapy for the sole purpose of stroke prevention is not recommended in patients with newly diagnosed infectious endocarditis.
- In most cases, anticoagulation and antiplatelet therapy should be temporarily discontinued in patients with infectious endocarditis and stroke or suspected stroke.
- Patients need careful assessment on a case-by-case basis, and the presence of risk factors predisposing patients to cerebrovascular complications (eg, large or very mobile vegetations, causative pathogens such as S aureus or Candida spp) may prompt temporary suspension of anticoagulation and antiplatelet therapy.
- If there is a clear preexisting or coexisting indication for these agents and surgery is not anticipated, consider continuing antiplatelet and anticoagulant therapy in patients with infectious endocarditis, provided they lack the risk factors described above and stroke has been excluded.
- If there is a clear preexisting or coexisting indication for these agents and surgery is being considered, consider using a short-acting anticoagulant such as intravenous or low-molecular weight heparin as a bridge to surgery.
Managing anticoagulation in patients with infectious endocarditis requires an individualized approach, using a careful risk-benefit assessment on a case-by-case basis. There is a dearth of high-quality evidence; consequently, the recommendations also vary according to the clinical situation.
Newly diagnosed native valve infectious endocarditis in itself is not an indication for anticoagulation.1–3 The question of whether to anticoagulate arises in patients who have a preexisting or coexisting indication for anticoagulation such as atrial fibrillation, deep vein thrombosis, pulmonary embolism, or a mechanical prosthetic heart valve. The question becomes yet more complex in patients with cerebrovascular complications and a coexistent strong indication for anticoagulation, creating what is often a very thorny dilemma.
Based on a review of available evidence, recommendations for anticoagulation in patients with infectious endocarditis are summarized below.
AVAILABLE EVIDENCE IS SCARCE AND MIXED
Earlier observational studies suggested a significant risk of cerebral hemorrhage with anticoagulation in patients with native valve endocarditis, although none of these studies were recent (some of them took place in the 1940s), and none are methodologically compelling.4–8 Consequently, some experts have expressed skepticism regarding their findings, particularly in recent years.
In part, this skepticism arises from studies that showed a lower incidence of cerebrovascular complications and smaller vegetation size in patients with prosthetic valve infectious endocarditis, studies in which many of the patients received anticoagulation therapy.9,10 The mechanism responsible for this effect is theorized to be that the vegetation is an amalgam of destroyed cells, platelets, and fibrin, with anticoagulation preventing this aggregation from further growth and propagation.
How great is the benefit or the potential harm?
Some experts argue that the incidence of ischemic stroke with hemorrhagic transformation in patients with infectious endocarditis receiving anticoagulation is overestimated. According to this view, the beneficial effects of anticoagulation at least counterbalance the potential harmful effects.
In addition to the studies cited above, recent studies have shown that patients on anticoagulation tend to have smaller vegetations and fewer cerebrovascular complications.11–13 Snygg-Martin et al11 and Rasmussen et al12 found not only that cerebrovascular complications were less common in patients already on anticoagulation at the time infectious endocarditis was diagnosed, but also that no increase in the rate of hemorrhagic lesions was reported.
These were all nonrandomized studies, and most of the patients in them had native valve infectious endocarditis diagnosed at an early stage. Importantly, these studies found that the beneficial effects of anticoagulation were only present if the patient was receiving warfarin before infectious endocarditis was diagnosed and antibiotic therapy was initiated. No benefits from anticoagulation were demonstrated once antimicrobial therapy was begun.
Similarly, Anavekar et al14 showed that embolic events occurred significantly less often in those who were currently on continuous daily antiplatelet therapy, suggesting that receiving antiplatelet agents at baseline protects against cardioembolic events in patients who develop infective endocarditis. However, the only randomized trial examining the initiation of antiplatelet therapy in patients diagnosed with infectious endocarditis receiving antibiotic treatment showed that adding aspirin did not reduce the risk of embolic events and was associated with a trend toward increased risk of bleeding.15
A recent large cohort study suggested that infectious endocarditis patients who receive anticoagulation therapy may have a higher incidence of cerebrovascular complications (hazard ratio 1.31, 95% confidence interval 1.00–1.72, P = .048), with a particular association of anticoagulation therapy with intracranial bleeding (hazard ratio 2.71, 95% confidence interval 1.54–4.76, P = .001).16
Another provocative link supported by the same study was a higher incidence of hemorrhagic complications with anticoagulation in patients with infectious endocarditis caused by Staphylococcus aureus, an association also suggested by older data from Tornos et al,8 but not seen in a study by Rasmussen et al.12
Continuing anticoagulation is an individualized decision
The benefit or harm of anticoagulation in patients with infectious endocarditis may be determined at least in part by a complex mix of factors including the valve involved (embolic events are more common with mitral valve vegetations than with aortic valve vegetations), vegetation size (higher risk if > 1 cm), mobility of vegetations, and perhaps the virulence of the causative organism.16,17 The fact that antimicrobial therapy obviates any beneficial effect of anticoagulation speaks strongly against starting anticoagulation therapy in infectious endocarditis patients with the sole purpose of reducing stroke risk.
Without large randomized trials to better delineate the risks and benefits of continuing preexisting anticoagulation in all patients with infectious endocarditis, patients already receiving anticoagulants need a careful, individualized risk-benefit assessment. Current guidelines agree that newly diagnosed infectious endocarditis per se is not an indication for anticoagulation or aspirin therapy (Table 1).1–3
TAKE-HOME POINTS
- Starting antiplatelet and anticoagulation therapy for the sole purpose of stroke prevention is not recommended in patients with newly diagnosed infectious endocarditis.
- In most cases, anticoagulation and antiplatelet therapy should be temporarily discontinued in patients with infectious endocarditis and stroke or suspected stroke.
- Patients need careful assessment on a case-by-case basis, and the presence of risk factors predisposing patients to cerebrovascular complications (eg, large or very mobile vegetations, causative pathogens such as S aureus or Candida spp) may prompt temporary suspension of anticoagulation and antiplatelet therapy.
- If there is a clear preexisting or coexisting indication for these agents and surgery is not anticipated, consider continuing antiplatelet and anticoagulant therapy in patients with infectious endocarditis, provided they lack the risk factors described above and stroke has been excluded.
- If there is a clear preexisting or coexisting indication for these agents and surgery is being considered, consider using a short-acting anticoagulant such as intravenous or low-molecular weight heparin as a bridge to surgery.
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132:1435–1486.
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis. Rev Esp Cardiol (Engl Ed) 2016; 69:69.
- Whitlock RP, Sun JC, Fremes SE, Rubens FD, Teoh KH. Antithrombotic and thrombolytic therapy for valvular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141:e576S–e600S.
- Delahaye JP, Poncet P, Malquarti V, Beaune J, Garé JP, Mann JM. Cerebrovascular accidents in infective endocarditis: role of anticoagulation. Eur Heart J 1990; 11:1074–1078.
- Loewe L. The combined use of anti-infectives and anticoagulants in the treatment of subacute bacterial endocarditis. Bull N Y Acad Med 1945; 21:59–86.
- Priest WS, Smith JM, McGee GC. The effect of anticoagulants on the penicillin therapy and the pathologic lesions of subacute bacterial endocarditis. N Engl J Med 1946; 235:699–706.
- Pruitt AA, Rubin RH, Karchmer AW, Duncan GW. Neurologic complications of bacterial endocarditis. Medicine 1978; 57:329–343.
- Tornos P, Almirante B, Mirabet S, Permanyer G, Pahissa A, Soler-Soler J. Infective endocarditis due to Staphylococcus aureus: deleterious effect of anticoagulant therapy. Arch Intern Med 1999; 159:473–475.
- Wilson WR, Geraci JE, Danielson GK, et al. Anticoagulant therapy and central nervous system complications in patients with prosthetic valve endocarditis. Circulation 1978; 57:1004–1007.
- Schulz R, Werner GS, Fuchs JB, et al. Clinical outcome and echocardiographic findings of native and prosthetic valve endocarditis in the 1990’s. Eur Heart J 1996; 17:281–288.
- Snygg-Martin U, Rasmussen RV, Hassager C, Bruun NE, Andersson R, Olaison L. Warfarin therapy and incidence of cerebrovascular complications in left-sided native valve endocarditis. Eur J Clin Microbiol Infect Dis 2011; 30:151–157.
- Rasmussen RV, Snygg-Martin U, Olaison L, et al. Major cerebral events in Staphylococcus aureus infective endocarditis: is anticoagulant therapy safe? Cardiology 2009; 114:284–291.
- Yau JW, Lee P, Wilson A, Jenkins AJ. Prosthetic valve endocarditis: what is the evidence for anticoagulant therapy? Intern Med J 2011; 41:795–797.
- Anavekar NS, Tleyjeh IM, Mirzoyev Z, et al. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis 2007; 44:1180–1186.
- Chan KL, Dumesnil JG, Cujec B, et al. A randomized trial of aspirin on the risk of embolic events in patients with infective endocarditis. J Am Coll Cardiol 2003; 42:775–780.
- Garcia-Cabrera E, Fernández-Hidalgo N, Almirante B, et al. Neurological complications of infective endocarditis: risk factors, outcome, and impact of cardiac surgery: a multicenter observational study. Circulation 2013; 127:2272–2284.
- Thuny F, Di Salvo G, Belliard O, et al. Risk of embolism and death in infective endocarditis: prognostic value of echocardiography: a prospective multicenter study. Circulation 2005; 112:69–75.
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation 2015; 132:1435–1486.
- Habib G, Lancellotti P, Antunes MJ, et al. 2015 ESC guidelines for the management of infective endocarditis. Rev Esp Cardiol (Engl Ed) 2016; 69:69.
- Whitlock RP, Sun JC, Fremes SE, Rubens FD, Teoh KH. Antithrombotic and thrombolytic therapy for valvular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141:e576S–e600S.
- Delahaye JP, Poncet P, Malquarti V, Beaune J, Garé JP, Mann JM. Cerebrovascular accidents in infective endocarditis: role of anticoagulation. Eur Heart J 1990; 11:1074–1078.
- Loewe L. The combined use of anti-infectives and anticoagulants in the treatment of subacute bacterial endocarditis. Bull N Y Acad Med 1945; 21:59–86.
- Priest WS, Smith JM, McGee GC. The effect of anticoagulants on the penicillin therapy and the pathologic lesions of subacute bacterial endocarditis. N Engl J Med 1946; 235:699–706.
- Pruitt AA, Rubin RH, Karchmer AW, Duncan GW. Neurologic complications of bacterial endocarditis. Medicine 1978; 57:329–343.
- Tornos P, Almirante B, Mirabet S, Permanyer G, Pahissa A, Soler-Soler J. Infective endocarditis due to Staphylococcus aureus: deleterious effect of anticoagulant therapy. Arch Intern Med 1999; 159:473–475.
- Wilson WR, Geraci JE, Danielson GK, et al. Anticoagulant therapy and central nervous system complications in patients with prosthetic valve endocarditis. Circulation 1978; 57:1004–1007.
- Schulz R, Werner GS, Fuchs JB, et al. Clinical outcome and echocardiographic findings of native and prosthetic valve endocarditis in the 1990’s. Eur Heart J 1996; 17:281–288.
- Snygg-Martin U, Rasmussen RV, Hassager C, Bruun NE, Andersson R, Olaison L. Warfarin therapy and incidence of cerebrovascular complications in left-sided native valve endocarditis. Eur J Clin Microbiol Infect Dis 2011; 30:151–157.
- Rasmussen RV, Snygg-Martin U, Olaison L, et al. Major cerebral events in Staphylococcus aureus infective endocarditis: is anticoagulant therapy safe? Cardiology 2009; 114:284–291.
- Yau JW, Lee P, Wilson A, Jenkins AJ. Prosthetic valve endocarditis: what is the evidence for anticoagulant therapy? Intern Med J 2011; 41:795–797.
- Anavekar NS, Tleyjeh IM, Mirzoyev Z, et al. Impact of prior antiplatelet therapy on risk of embolism in infective endocarditis. Clin Infect Dis 2007; 44:1180–1186.
- Chan KL, Dumesnil JG, Cujec B, et al. A randomized trial of aspirin on the risk of embolic events in patients with infective endocarditis. J Am Coll Cardiol 2003; 42:775–780.
- Garcia-Cabrera E, Fernández-Hidalgo N, Almirante B, et al. Neurological complications of infective endocarditis: risk factors, outcome, and impact of cardiac surgery: a multicenter observational study. Circulation 2013; 127:2272–2284.
- Thuny F, Di Salvo G, Belliard O, et al. Risk of embolism and death in infective endocarditis: prognostic value of echocardiography: a prospective multicenter study. Circulation 2005; 112:69–75.
When we need to remember that it is more than a job

“I am forever humbled.” So said a heart failure specialist on rounds when I was a resident in the intensive care unit several decades ago. He was talking about the perpetual mismatch between a physician’s level of knowledge and the unpredictability inherent in the management and outcome of critically ill patients. His words ring true for me nearly every day. We should never think we are so smart that we are truly in control of our patients’ outcomes or that we don’t make mistakes—but we also cannot become so paralyzed by the awareness of our limitations that we don’t make decisions.
I have spoken those same powerful words many times on teaching rounds. I also frequently push them to the back of my mind. As a consultant at a major medical center, I am supposed to know. It is a fine line we walk.
I know I am not alone in harboring these self-doubts. Ready access to online information does little to assuage the concern that we can never know enough. Have I ordered enough diagnostic tests to be sure? Have I ordered too many tests and thus will be penalized for providing cost-ineffective care? Should we follow generic guidelines, or deviate from the guidelines based on our clinical instincts, our own interpretation of the literature, and the patient’s unique circumstances and desires?
And then what happens when we make wrong decisions, or even the right decisions that result in a poor patient outcome, which of course is at some point inevitable? We are told to be open about errors, to be honest and transparent about our limitations, to throw down our elaborate emotional and intellectual defensive shields and expose our vulnerability.
But what do we experience emotionally when we are named in a malpractice suit? We may have done all that we thought we could do: we responsibly explored the diagnostic and therapeutic options, provided empathetic care, and listened to the voice of the patient. Yet an adverse outcome still occurred. The practice of medicine is indeed humbling. We feel crushed. We revisit the patient’s care in a vivid perpetual replay loop in our head. Maybe we didn’t evaluate all the options as we should have. If we had been a bit smarter, a bit more efficient, maybe the outcome would have been different.
Then during a deposition, the plaintiff’s counsel points out the temporal and documentary inconsistencies in the electronic medical record: “Doctor, you say you saw the patient at 2:00 pm, but there was no note finalized until 10:00 pm…and why was your documented physical exam exactly the same as the one from the day before and exactly the same as that of the resident who saw the patient that afternoon?” We now feel crushed, totally vulnerable, emotionally trampled, and often isolated and disconnected from our patients and peers. The intellectualized humility becomes transformed into a sense of inadequacy. Why should I keep doing this?
In this issue of the Journal, experienced malpractice attorney Kevin Giordano explores aspects of the malpractice process as they relate to the physicians he defends. He notes how the electronic medical record, a tool ostensibly in place to improve communication and the sharing of medical information between caregivers and patients, can be our worst enemy in a courtroom. He discusses the pressures of our complicated healthcare system that promote documentation errors that he must try to explain away to the jury in our defense, demonstrating that these documentation errors do not necessarily mirror the care and caring of the named physicians. This is critically important information for us to understand and to act on for our personal protection, but it is not his most important message to us.
Mr. Giordano is a sincere, empathetic, and proficient professional. He has spoken for and to many physicians. He has listened to us and observed our behaviors. And as he has defended many of us in a court of law, he has learned to diagnose in his clients the damage that can persist following involvement in a malpractice case and the emotional scars the malpractice experience leaves on physicians. He emphasizes that we must not let the event of a malpractice suit force us to withdraw and strip us of our connection and engagement to patients. If anything, he and Drs. Susan Rehm and Bradford Borden, in an accompanying editorial, urge us to keep in mind that our personal engagement with patients and the mindful practice of medicine is our raison d’être as physicians.
I am continuously humbled by the breadth of the pathology, clinical medicine, and social challenges that I encounter on a daily basis, armed with limited knowledge and experience. It is intellectually rewarding to make an arcane diagnosis or to see an individualized therapy work as I had hoped. But I agree with Mr. Giordano that it is the genuine engagement with patients that provides us with the real joy in the practice of medicine and pushes us to deliver care at a consistently proficient level. We must not forget that, even in the face of significant and emotionally challenging events such as being named in a malpractice suit. It is the nature of our engagement with our patients and our colleagues that make what we do more than a job.
As more physicians in the United States become employed by health systems, I hope that the administrative leaders within these systems truly recognize these issues. As they struggle to balance the provision of safe high-quality care to patients with their increasingly complex financial spreadsheets, I hope that the emotional health of their physician employees is not forgotten. And not just after a malpractice suit.
“I am forever humbled.” So said a heart failure specialist on rounds when I was a resident in the intensive care unit several decades ago. He was talking about the perpetual mismatch between a physician’s level of knowledge and the unpredictability inherent in the management and outcome of critically ill patients. His words ring true for me nearly every day. We should never think we are so smart that we are truly in control of our patients’ outcomes or that we don’t make mistakes—but we also cannot become so paralyzed by the awareness of our limitations that we don’t make decisions.
I have spoken those same powerful words many times on teaching rounds. I also frequently push them to the back of my mind. As a consultant at a major medical center, I am supposed to know. It is a fine line we walk.
I know I am not alone in harboring these self-doubts. Ready access to online information does little to assuage the concern that we can never know enough. Have I ordered enough diagnostic tests to be sure? Have I ordered too many tests and thus will be penalized for providing cost-ineffective care? Should we follow generic guidelines, or deviate from the guidelines based on our clinical instincts, our own interpretation of the literature, and the patient’s unique circumstances and desires?
And then what happens when we make wrong decisions, or even the right decisions that result in a poor patient outcome, which of course is at some point inevitable? We are told to be open about errors, to be honest and transparent about our limitations, to throw down our elaborate emotional and intellectual defensive shields and expose our vulnerability.
But what do we experience emotionally when we are named in a malpractice suit? We may have done all that we thought we could do: we responsibly explored the diagnostic and therapeutic options, provided empathetic care, and listened to the voice of the patient. Yet an adverse outcome still occurred. The practice of medicine is indeed humbling. We feel crushed. We revisit the patient’s care in a vivid perpetual replay loop in our head. Maybe we didn’t evaluate all the options as we should have. If we had been a bit smarter, a bit more efficient, maybe the outcome would have been different.
Then during a deposition, the plaintiff’s counsel points out the temporal and documentary inconsistencies in the electronic medical record: “Doctor, you say you saw the patient at 2:00 pm, but there was no note finalized until 10:00 pm…and why was your documented physical exam exactly the same as the one from the day before and exactly the same as that of the resident who saw the patient that afternoon?” We now feel crushed, totally vulnerable, emotionally trampled, and often isolated and disconnected from our patients and peers. The intellectualized humility becomes transformed into a sense of inadequacy. Why should I keep doing this?
In this issue of the Journal, experienced malpractice attorney Kevin Giordano explores aspects of the malpractice process as they relate to the physicians he defends. He notes how the electronic medical record, a tool ostensibly in place to improve communication and the sharing of medical information between caregivers and patients, can be our worst enemy in a courtroom. He discusses the pressures of our complicated healthcare system that promote documentation errors that he must try to explain away to the jury in our defense, demonstrating that these documentation errors do not necessarily mirror the care and caring of the named physicians. This is critically important information for us to understand and to act on for our personal protection, but it is not his most important message to us.
Mr. Giordano is a sincere, empathetic, and proficient professional. He has spoken for and to many physicians. He has listened to us and observed our behaviors. And as he has defended many of us in a court of law, he has learned to diagnose in his clients the damage that can persist following involvement in a malpractice case and the emotional scars the malpractice experience leaves on physicians. He emphasizes that we must not let the event of a malpractice suit force us to withdraw and strip us of our connection and engagement to patients. If anything, he and Drs. Susan Rehm and Bradford Borden, in an accompanying editorial, urge us to keep in mind that our personal engagement with patients and the mindful practice of medicine is our raison d’être as physicians.
I am continuously humbled by the breadth of the pathology, clinical medicine, and social challenges that I encounter on a daily basis, armed with limited knowledge and experience. It is intellectually rewarding to make an arcane diagnosis or to see an individualized therapy work as I had hoped. But I agree with Mr. Giordano that it is the genuine engagement with patients that provides us with the real joy in the practice of medicine and pushes us to deliver care at a consistently proficient level. We must not forget that, even in the face of significant and emotionally challenging events such as being named in a malpractice suit. It is the nature of our engagement with our patients and our colleagues that make what we do more than a job.
As more physicians in the United States become employed by health systems, I hope that the administrative leaders within these systems truly recognize these issues. As they struggle to balance the provision of safe high-quality care to patients with their increasingly complex financial spreadsheets, I hope that the emotional health of their physician employees is not forgotten. And not just after a malpractice suit.
“I am forever humbled.” So said a heart failure specialist on rounds when I was a resident in the intensive care unit several decades ago. He was talking about the perpetual mismatch between a physician’s level of knowledge and the unpredictability inherent in the management and outcome of critically ill patients. His words ring true for me nearly every day. We should never think we are so smart that we are truly in control of our patients’ outcomes or that we don’t make mistakes—but we also cannot become so paralyzed by the awareness of our limitations that we don’t make decisions.
I have spoken those same powerful words many times on teaching rounds. I also frequently push them to the back of my mind. As a consultant at a major medical center, I am supposed to know. It is a fine line we walk.
I know I am not alone in harboring these self-doubts. Ready access to online information does little to assuage the concern that we can never know enough. Have I ordered enough diagnostic tests to be sure? Have I ordered too many tests and thus will be penalized for providing cost-ineffective care? Should we follow generic guidelines, or deviate from the guidelines based on our clinical instincts, our own interpretation of the literature, and the patient’s unique circumstances and desires?
And then what happens when we make wrong decisions, or even the right decisions that result in a poor patient outcome, which of course is at some point inevitable? We are told to be open about errors, to be honest and transparent about our limitations, to throw down our elaborate emotional and intellectual defensive shields and expose our vulnerability.
But what do we experience emotionally when we are named in a malpractice suit? We may have done all that we thought we could do: we responsibly explored the diagnostic and therapeutic options, provided empathetic care, and listened to the voice of the patient. Yet an adverse outcome still occurred. The practice of medicine is indeed humbling. We feel crushed. We revisit the patient’s care in a vivid perpetual replay loop in our head. Maybe we didn’t evaluate all the options as we should have. If we had been a bit smarter, a bit more efficient, maybe the outcome would have been different.
Then during a deposition, the plaintiff’s counsel points out the temporal and documentary inconsistencies in the electronic medical record: “Doctor, you say you saw the patient at 2:00 pm, but there was no note finalized until 10:00 pm…and why was your documented physical exam exactly the same as the one from the day before and exactly the same as that of the resident who saw the patient that afternoon?” We now feel crushed, totally vulnerable, emotionally trampled, and often isolated and disconnected from our patients and peers. The intellectualized humility becomes transformed into a sense of inadequacy. Why should I keep doing this?
In this issue of the Journal, experienced malpractice attorney Kevin Giordano explores aspects of the malpractice process as they relate to the physicians he defends. He notes how the electronic medical record, a tool ostensibly in place to improve communication and the sharing of medical information between caregivers and patients, can be our worst enemy in a courtroom. He discusses the pressures of our complicated healthcare system that promote documentation errors that he must try to explain away to the jury in our defense, demonstrating that these documentation errors do not necessarily mirror the care and caring of the named physicians. This is critically important information for us to understand and to act on for our personal protection, but it is not his most important message to us.
Mr. Giordano is a sincere, empathetic, and proficient professional. He has spoken for and to many physicians. He has listened to us and observed our behaviors. And as he has defended many of us in a court of law, he has learned to diagnose in his clients the damage that can persist following involvement in a malpractice case and the emotional scars the malpractice experience leaves on physicians. He emphasizes that we must not let the event of a malpractice suit force us to withdraw and strip us of our connection and engagement to patients. If anything, he and Drs. Susan Rehm and Bradford Borden, in an accompanying editorial, urge us to keep in mind that our personal engagement with patients and the mindful practice of medicine is our raison d’être as physicians.
I am continuously humbled by the breadth of the pathology, clinical medicine, and social challenges that I encounter on a daily basis, armed with limited knowledge and experience. It is intellectually rewarding to make an arcane diagnosis or to see an individualized therapy work as I had hoped. But I agree with Mr. Giordano that it is the genuine engagement with patients that provides us with the real joy in the practice of medicine and pushes us to deliver care at a consistently proficient level. We must not forget that, even in the face of significant and emotionally challenging events such as being named in a malpractice suit. It is the nature of our engagement with our patients and our colleagues that make what we do more than a job.
As more physicians in the United States become employed by health systems, I hope that the administrative leaders within these systems truly recognize these issues. As they struggle to balance the provision of safe high-quality care to patients with their increasingly complex financial spreadsheets, I hope that the emotional health of their physician employees is not forgotten. And not just after a malpractice suit.
The emotional impact of a malpractice suit on physicians: Maintaining resilience
Physicians who have been involved in malpractice actions are all too familiar with the range of emotions they experience during the process. Anxiety, fear, frustration, remorse, self-doubt, shame, betrayal, anger…no pleasant feelings here. Add malpractice stress to the high level of pressure experienced at home and at work, and crisis looms.
In his commentary in this issue, Kevin Giordano states, “it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team toward disconnection.”1
Because of the nature of our work as physicians, we are isolated, and malpractice isolates us further. Because of embarrassment, we avoid talking with our colleagues and managers. Legal counsel reminds us to correspond with no one about the details of the case. Spouses and friends may offer support, but it is difficult— perhaps impossible—to be reassured.
Isolation fuels our self-doubt and erodes our confidence, leading us to focus on what may go wrong, rather than on healing. Every decision is fraught with anxiety, and efficiency evaporates. Paralysis may set in, leading to disengagement from patient care and increasing the chance of further problems.
IT TAKES RESILIENCE TO THRIVE
It takes resilience to thrive in today’s pressure-cooker healthcare environment, let alone in the setting of malpractice stress. Resilient people are able to face reality and see a better future, put things into perspective, and bounce back from adversity.2 Resilience, a trait that protects against stress and burnout, is relevant at the personal, managerial, and system levels. Though this definition is not specific to caregiver or malpractice stress, it is applicable. It is an essential component of wellness and requires perpetual attention to self-care for successful maintenance.
Studies of physicians who have avoided burnout reveal remarkably consistent qualities, including finding gratification related to work, maintaining useful habits and practices, and developing attitudes that keep them resilient.3 Rather than adding activities to their full schedules, these physicians stayed resilient through mindfulness of various aspects of their daily lives. Interactions with colleagues—discussing cases, treatments, and outcomes (including errors)—proved vital. Professional development, encompassing activities such as continuing education, coaching, mentoring, and counseling, was recognized as an important self-directed resilience measure. Maintenance of relationships with family and friends, cultivation of leisure-time activities, and appreciation of the need for personal reflection time were traits often found in resilient physicians.
FOSTERING RESILIENCE
As part of the Mayo Clinic’s biannual survey of its physicians, Shanafelt et al4 studied relationships between qualities of physician leaders and burnout and satisfaction among the physicians they supervised. Many of the desirable leadership traits were related to building relationships through respectful communication, along with provision of opportunities for personal and professional development. The acknowledgment that resilient, healthy physicians are satisfied, productive, and able to provide safer and higher-quality patient care should lead to the establishment of physician wellness as a “dashboard metric.” This makes priorities clear by rewarding managers who foster self-care and resilience among their staff.
Likewise, at the healthcare system level, Beckman5 recognized that organizations can provide opportunities to promote resilience among caregivers. Organizational initiatives that set the stage for resilience include:
- Curricula to enhance communication with patients, coworkers, and family
- “Best practices” for efficient and effective patient care
- Self-care through health insurance incentives and educational sessions
- Accessible, affordable, and confidential behavioral health support
- Time for self-care activities during the workday
- Coaching and mentoring programs
- Narrative-and-reflection groups and mindfulness training.5
Through an atmosphere of support for resilience, organizations provide a place for physicians to maintain a sense of meaning and purpose in their work. For individuals facing malpractice action, this infrastructure can be used to weather the storm. As Mr. Giordano writes, staying engaged “may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.”1 We must pay attention to developing individual physicians, educating managers, and building systems so that caregivers can remain engaged and resilient. It may help those affected by malpractice stress, and perhaps as importantly, it may reduce the chance of future “disconnection” leading to recourse in the legal system.
- Giordano KC. It is not the critic’s voice that should count. Cleve Clin J Med 2016; 83:174–176.
- Coutu DL. How resilience works. Harv Bus Rev 2002; 80(5):46–55.
- Zwack J, Schweitzer J. If every fifth physician is affected by burnout, what about the other four? Acad Med 2013; 88:382–389.
- Shanafelt TD, Gorringe G, Menaker R, et al. Impact of organizational leadership on physician burnout and satisfaction. Mayo Clin Proc 2015; 90:432–440.
- Beckman H. The role of medical culture in the journey to resilience. Acad Med 2015; 90:710–712.
Physicians who have been involved in malpractice actions are all too familiar with the range of emotions they experience during the process. Anxiety, fear, frustration, remorse, self-doubt, shame, betrayal, anger…no pleasant feelings here. Add malpractice stress to the high level of pressure experienced at home and at work, and crisis looms.
In his commentary in this issue, Kevin Giordano states, “it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team toward disconnection.”1
Because of the nature of our work as physicians, we are isolated, and malpractice isolates us further. Because of embarrassment, we avoid talking with our colleagues and managers. Legal counsel reminds us to correspond with no one about the details of the case. Spouses and friends may offer support, but it is difficult— perhaps impossible—to be reassured.
Isolation fuels our self-doubt and erodes our confidence, leading us to focus on what may go wrong, rather than on healing. Every decision is fraught with anxiety, and efficiency evaporates. Paralysis may set in, leading to disengagement from patient care and increasing the chance of further problems.
IT TAKES RESILIENCE TO THRIVE
It takes resilience to thrive in today’s pressure-cooker healthcare environment, let alone in the setting of malpractice stress. Resilient people are able to face reality and see a better future, put things into perspective, and bounce back from adversity.2 Resilience, a trait that protects against stress and burnout, is relevant at the personal, managerial, and system levels. Though this definition is not specific to caregiver or malpractice stress, it is applicable. It is an essential component of wellness and requires perpetual attention to self-care for successful maintenance.
Studies of physicians who have avoided burnout reveal remarkably consistent qualities, including finding gratification related to work, maintaining useful habits and practices, and developing attitudes that keep them resilient.3 Rather than adding activities to their full schedules, these physicians stayed resilient through mindfulness of various aspects of their daily lives. Interactions with colleagues—discussing cases, treatments, and outcomes (including errors)—proved vital. Professional development, encompassing activities such as continuing education, coaching, mentoring, and counseling, was recognized as an important self-directed resilience measure. Maintenance of relationships with family and friends, cultivation of leisure-time activities, and appreciation of the need for personal reflection time were traits often found in resilient physicians.
FOSTERING RESILIENCE
As part of the Mayo Clinic’s biannual survey of its physicians, Shanafelt et al4 studied relationships between qualities of physician leaders and burnout and satisfaction among the physicians they supervised. Many of the desirable leadership traits were related to building relationships through respectful communication, along with provision of opportunities for personal and professional development. The acknowledgment that resilient, healthy physicians are satisfied, productive, and able to provide safer and higher-quality patient care should lead to the establishment of physician wellness as a “dashboard metric.” This makes priorities clear by rewarding managers who foster self-care and resilience among their staff.
Likewise, at the healthcare system level, Beckman5 recognized that organizations can provide opportunities to promote resilience among caregivers. Organizational initiatives that set the stage for resilience include:
- Curricula to enhance communication with patients, coworkers, and family
- “Best practices” for efficient and effective patient care
- Self-care through health insurance incentives and educational sessions
- Accessible, affordable, and confidential behavioral health support
- Time for self-care activities during the workday
- Coaching and mentoring programs
- Narrative-and-reflection groups and mindfulness training.5
Through an atmosphere of support for resilience, organizations provide a place for physicians to maintain a sense of meaning and purpose in their work. For individuals facing malpractice action, this infrastructure can be used to weather the storm. As Mr. Giordano writes, staying engaged “may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.”1 We must pay attention to developing individual physicians, educating managers, and building systems so that caregivers can remain engaged and resilient. It may help those affected by malpractice stress, and perhaps as importantly, it may reduce the chance of future “disconnection” leading to recourse in the legal system.
Physicians who have been involved in malpractice actions are all too familiar with the range of emotions they experience during the process. Anxiety, fear, frustration, remorse, self-doubt, shame, betrayal, anger…no pleasant feelings here. Add malpractice stress to the high level of pressure experienced at home and at work, and crisis looms.
In his commentary in this issue, Kevin Giordano states, “it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team toward disconnection.”1
Because of the nature of our work as physicians, we are isolated, and malpractice isolates us further. Because of embarrassment, we avoid talking with our colleagues and managers. Legal counsel reminds us to correspond with no one about the details of the case. Spouses and friends may offer support, but it is difficult— perhaps impossible—to be reassured.
Isolation fuels our self-doubt and erodes our confidence, leading us to focus on what may go wrong, rather than on healing. Every decision is fraught with anxiety, and efficiency evaporates. Paralysis may set in, leading to disengagement from patient care and increasing the chance of further problems.
IT TAKES RESILIENCE TO THRIVE
It takes resilience to thrive in today’s pressure-cooker healthcare environment, let alone in the setting of malpractice stress. Resilient people are able to face reality and see a better future, put things into perspective, and bounce back from adversity.2 Resilience, a trait that protects against stress and burnout, is relevant at the personal, managerial, and system levels. Though this definition is not specific to caregiver or malpractice stress, it is applicable. It is an essential component of wellness and requires perpetual attention to self-care for successful maintenance.
Studies of physicians who have avoided burnout reveal remarkably consistent qualities, including finding gratification related to work, maintaining useful habits and practices, and developing attitudes that keep them resilient.3 Rather than adding activities to their full schedules, these physicians stayed resilient through mindfulness of various aspects of their daily lives. Interactions with colleagues—discussing cases, treatments, and outcomes (including errors)—proved vital. Professional development, encompassing activities such as continuing education, coaching, mentoring, and counseling, was recognized as an important self-directed resilience measure. Maintenance of relationships with family and friends, cultivation of leisure-time activities, and appreciation of the need for personal reflection time were traits often found in resilient physicians.
FOSTERING RESILIENCE
As part of the Mayo Clinic’s biannual survey of its physicians, Shanafelt et al4 studied relationships between qualities of physician leaders and burnout and satisfaction among the physicians they supervised. Many of the desirable leadership traits were related to building relationships through respectful communication, along with provision of opportunities for personal and professional development. The acknowledgment that resilient, healthy physicians are satisfied, productive, and able to provide safer and higher-quality patient care should lead to the establishment of physician wellness as a “dashboard metric.” This makes priorities clear by rewarding managers who foster self-care and resilience among their staff.
Likewise, at the healthcare system level, Beckman5 recognized that organizations can provide opportunities to promote resilience among caregivers. Organizational initiatives that set the stage for resilience include:
- Curricula to enhance communication with patients, coworkers, and family
- “Best practices” for efficient and effective patient care
- Self-care through health insurance incentives and educational sessions
- Accessible, affordable, and confidential behavioral health support
- Time for self-care activities during the workday
- Coaching and mentoring programs
- Narrative-and-reflection groups and mindfulness training.5
Through an atmosphere of support for resilience, organizations provide a place for physicians to maintain a sense of meaning and purpose in their work. For individuals facing malpractice action, this infrastructure can be used to weather the storm. As Mr. Giordano writes, staying engaged “may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.”1 We must pay attention to developing individual physicians, educating managers, and building systems so that caregivers can remain engaged and resilient. It may help those affected by malpractice stress, and perhaps as importantly, it may reduce the chance of future “disconnection” leading to recourse in the legal system.
- Giordano KC. It is not the critic’s voice that should count. Cleve Clin J Med 2016; 83:174–176.
- Coutu DL. How resilience works. Harv Bus Rev 2002; 80(5):46–55.
- Zwack J, Schweitzer J. If every fifth physician is affected by burnout, what about the other four? Acad Med 2013; 88:382–389.
- Shanafelt TD, Gorringe G, Menaker R, et al. Impact of organizational leadership on physician burnout and satisfaction. Mayo Clin Proc 2015; 90:432–440.
- Beckman H. The role of medical culture in the journey to resilience. Acad Med 2015; 90:710–712.
- Giordano KC. It is not the critic’s voice that should count. Cleve Clin J Med 2016; 83:174–176.
- Coutu DL. How resilience works. Harv Bus Rev 2002; 80(5):46–55.
- Zwack J, Schweitzer J. If every fifth physician is affected by burnout, what about the other four? Acad Med 2013; 88:382–389.
- Shanafelt TD, Gorringe G, Menaker R, et al. Impact of organizational leadership on physician burnout and satisfaction. Mayo Clin Proc 2015; 90:432–440.
- Beckman H. The role of medical culture in the journey to resilience. Acad Med 2015; 90:710–712.

It is not the critic’s voice that should count
During my 25 years as a defense attorney, I have seen the traumatic impact that the allegation of medical malpractice can have on healthcare providers. And I have seen many times that in the aftermath of a case it remains difficult, if not impossible, for the practitioner to return to the clinical setting unscarred by the process. Although vindication by the jury provides some solace, by itself it does not create healing. Instead, the critic’s voice continues to resonate long after the trial.
During a lawsuit, physicians and other providers are commonly confronted with incidental imperfections in the care they provided, errors in their documentation, or both. Consequently, a provider’s perception of events and ultimately the meaning derived from the experience is shaped less by the valid defenses and opinions of the supportive defense experts than by the inconsequential flaws and errors that can often be found in any medical record.
A RECENT CASE
Recently, I defended a hospital team consisting of a hospitalist, trauma surgeon, three residents, and a nurse. The case involved a 74-year-old man who was admitted to the hospital with pancreatitis of unknown cause. Six days after admission, he died of complications of acute respiratory distress syndrome. The team was accused of causing the patient’s death. Specifically, the plaintiff alleged that although the patient’s liver enzyme levels were improving, his condition was deteriorating, and he ultimately developed hemorrhagic pancreatitis. It was the plaintiff’s contention that proper ongoing evaluation, including computed tomographic imaging, would have led to treatment that would have avoided the worsening of pancreatitis, development of an ileus, and ultimately the insult to his bowel and lungs that they claim caused acute respiratory distress syndrome and death. The patient was survived by his wife and their three children. After his death, hospital representatives and the hospitalist met with her in an effort to explain the events that led to her husband’s death. Unfortunately, these discussions did not ameliorate her feelings of loss and anger. She filed a lawsuit, and 4 years later, the case went to trial.
During the trial, the plaintiff’s attorney highlighted errors in the electronic medical record. Entries had been cut and pasted, saving time, but without updating information that had changed in the interim. The inaccuracies included “assessment: worsening pancreatitis” on a day it was considered to have improved. Another entry contained “persistent fever” on a day when no fever was present. Other mistakes involved notes that contained care plans made after morning rounds that were not revised later in the day after changes in the patient’s condition necessitated a change in the plan. In fact, most references to medication dosing in the progress notes on the last 2 days did not match the medication dosing documented in the medication administration record.
In the end, the plaintiff’s counsel did not convince the jury that the healthcare team had been negligent, but unfortunately, she planted doubt in the minds of the caregivers themselves. Perhaps in part, these doubts were the result of having to defend a bad outcome in the face of criticism that was based solely in retrospect. But the providers’ doubts seemed mostly to emanate from the inadequacies in their documentation as they observed how every entry in a far-from-perfect medical record was scrutinized and then manipulated to challenge its textual integrity—and to portray the healthcare team as unengaged and substandard clinicians.
Despite the team’s high level of engagement and the quality of care they provided, any imperfection—whether a documentation error or a minor omission in some aspect of the care provided to this complex patient—became a source of self-doubt and self-criticism.
THE ELECTRONIC MEDICAL RECORD: A MIXED BLESSING
Documentation failures have long been used to “prove” that physicians are disconnected from the clinical situation. The electronic medical record has not proved to be a strong shield against malpractice allegations. In fact, because the electronic medical record absorbs more of the physician’s time and that of the care team’s members, efforts to save time through work-arounds and shortcuts have increased the risk of errors in entering information.
For instance, drop-down menus have led to wrong selections. Cutting and pasting has led to entries that contain data superseded by clinical events, thus creating contradictions within the record itself, and worse, with the physician’s own testimony pertaining to the basis of the clinical decision-making. And boilerplate language has created difficulties when the language does not completely fit the context or when inapplicable verbiage that fills itself in automatically goes unedited. An emergency department physician I represented at trial had to awkwardly explain that some of the data reported in his physical exam findings were inaccurate because of programmed language and should have been deleted; he had no explanation for his oversight.
But my experience has been that juries can forgive imperfections in documentation and even incidental aspects of care. They want to trust that the clinician was there for, and there with, the patient. This emphasis is what allowed us to defend the case involving the patient with pancreatitis. Clinical judgment means being engaged enough to choose what you pay attention to and to process the data you receive.
Unfortunately, the electronic medical record seems designed more for billing and for guarding against claims of fraud than for communication among clinicians or documenting clinically significant events. Many clinicians believe that redundancy and standardized phraseology have weakened the meaningful use of the medical record, as the clinical information is now of questionable reliability or value or is simply hard to find. Consequently, the electronic medical record has become less effective as a communication tool for providing continuity of care.
More importantly, the electronic medical record too often places the physician in front of a computer, so that the computer becomes the focus, not the patient. Studies suggest that the way the electronic medical record is currently used in the examination room affects the quality of physician-patient communication as well as the physician’s cognitive processing of information. Unless the physician is alert and attuned, the electronic medical record can be a barrier to connection. This not only creates the potential for mistakes, but it can also cause patients to question the quality of care they are getting and to distrust the level of the provider’s engagement. In this context, the likelihood that the patient retains an attorney increases when a bad outcome occurs, avoidable or not.
WHAT PATIENTS WANT FROM PHYSICIANS
When I first began seeing my own primary care physician, her office was 5 minutes from my home. Then she relocated to a practice 15 minutes away. And then, because of office consolidation and acquisition, her office was relocated 40 minutes away.
So why do I still go to her? Her training is not better than that of most internists, and my medical history is not so complex that I require more care than most 55-year-old men. I am only speculating, but I would guess that she is not the most financially productive physician in her group. I know that her transition to the electronic medical record has been difficult. Recently, I asked her about it. Except in some situations, she does not type while taking a history, and she stays totally away from the computer while in the examination room with me. She sits a couple of feet from me, and it feels like the days before the electronic medical record. She is clearly more comfortable listening and taking notes first and worrying about the electronic record later. I imagine she stays later to do her notes than most of the other physicians, or she finishes them at home.
The reason I continue to see her as my primary care physician is that she remains totally engaged during my office visit. What tells me that is not just her avoidance of the computer or her body language, but the depth of questions she asks. My responses often prompt her to look back at an earlier office note, and she will then ask follow-up questions to confirm what she had previously recorded. Her examination is thorough, with testing to confirm and retesting to be sure. Doing this may mean that she has difficulty meeting financial or administrative benchmarks established by her practice. I don’t know. But I have no doubt that the likelihood of her missing something in her clinical care is small, and what I suspect is even smaller is the risk that one of her patients would bring a lawsuit against her, given the time she takes to listen and remain connected throughout the office visit.
STAYING CONNECTED, IN SPITE OF EVERYTHING
My point is not to suggest that everyone must conform to the same practice philosophy, particularly with the economic pressures in the medical field. What I am suggesting is that it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team towards disconnection. Quality healthcare means making every effort to remain engaged at all times with your patient’s care, which will reduce the likelihood of a bad outcome and may preserve the physician-patient relationship even when a bad outcome occurs.
In the end, perhaps it is not possible to avoid being named as a defendant in a malpractice case, just as it is not possible to avoid all bad medical outcomes despite exceptional care. In law, as in medicine, there are always factors beyond your control. My aspiration is to find a pathway to get providers through the system unbroken—also not an easy task. But one thing I know is true: the more you can stay engaged in the care you provide and in your documentation, the more you will preclude a plaintiff’s attorney from exploiting the effects of the forces within the system that drive providers toward disconnection. As long as you stay engaged and supported by the knowledge that the care provided was appropriate, it is my hope that the voice of the critic will not count as much in the aftermath of a malpractice case. But more importantly, it may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.
During my 25 years as a defense attorney, I have seen the traumatic impact that the allegation of medical malpractice can have on healthcare providers. And I have seen many times that in the aftermath of a case it remains difficult, if not impossible, for the practitioner to return to the clinical setting unscarred by the process. Although vindication by the jury provides some solace, by itself it does not create healing. Instead, the critic’s voice continues to resonate long after the trial.
During a lawsuit, physicians and other providers are commonly confronted with incidental imperfections in the care they provided, errors in their documentation, or both. Consequently, a provider’s perception of events and ultimately the meaning derived from the experience is shaped less by the valid defenses and opinions of the supportive defense experts than by the inconsequential flaws and errors that can often be found in any medical record.
A RECENT CASE
Recently, I defended a hospital team consisting of a hospitalist, trauma surgeon, three residents, and a nurse. The case involved a 74-year-old man who was admitted to the hospital with pancreatitis of unknown cause. Six days after admission, he died of complications of acute respiratory distress syndrome. The team was accused of causing the patient’s death. Specifically, the plaintiff alleged that although the patient’s liver enzyme levels were improving, his condition was deteriorating, and he ultimately developed hemorrhagic pancreatitis. It was the plaintiff’s contention that proper ongoing evaluation, including computed tomographic imaging, would have led to treatment that would have avoided the worsening of pancreatitis, development of an ileus, and ultimately the insult to his bowel and lungs that they claim caused acute respiratory distress syndrome and death. The patient was survived by his wife and their three children. After his death, hospital representatives and the hospitalist met with her in an effort to explain the events that led to her husband’s death. Unfortunately, these discussions did not ameliorate her feelings of loss and anger. She filed a lawsuit, and 4 years later, the case went to trial.
During the trial, the plaintiff’s attorney highlighted errors in the electronic medical record. Entries had been cut and pasted, saving time, but without updating information that had changed in the interim. The inaccuracies included “assessment: worsening pancreatitis” on a day it was considered to have improved. Another entry contained “persistent fever” on a day when no fever was present. Other mistakes involved notes that contained care plans made after morning rounds that were not revised later in the day after changes in the patient’s condition necessitated a change in the plan. In fact, most references to medication dosing in the progress notes on the last 2 days did not match the medication dosing documented in the medication administration record.
In the end, the plaintiff’s counsel did not convince the jury that the healthcare team had been negligent, but unfortunately, she planted doubt in the minds of the caregivers themselves. Perhaps in part, these doubts were the result of having to defend a bad outcome in the face of criticism that was based solely in retrospect. But the providers’ doubts seemed mostly to emanate from the inadequacies in their documentation as they observed how every entry in a far-from-perfect medical record was scrutinized and then manipulated to challenge its textual integrity—and to portray the healthcare team as unengaged and substandard clinicians.
Despite the team’s high level of engagement and the quality of care they provided, any imperfection—whether a documentation error or a minor omission in some aspect of the care provided to this complex patient—became a source of self-doubt and self-criticism.
THE ELECTRONIC MEDICAL RECORD: A MIXED BLESSING
Documentation failures have long been used to “prove” that physicians are disconnected from the clinical situation. The electronic medical record has not proved to be a strong shield against malpractice allegations. In fact, because the electronic medical record absorbs more of the physician’s time and that of the care team’s members, efforts to save time through work-arounds and shortcuts have increased the risk of errors in entering information.
For instance, drop-down menus have led to wrong selections. Cutting and pasting has led to entries that contain data superseded by clinical events, thus creating contradictions within the record itself, and worse, with the physician’s own testimony pertaining to the basis of the clinical decision-making. And boilerplate language has created difficulties when the language does not completely fit the context or when inapplicable verbiage that fills itself in automatically goes unedited. An emergency department physician I represented at trial had to awkwardly explain that some of the data reported in his physical exam findings were inaccurate because of programmed language and should have been deleted; he had no explanation for his oversight.
But my experience has been that juries can forgive imperfections in documentation and even incidental aspects of care. They want to trust that the clinician was there for, and there with, the patient. This emphasis is what allowed us to defend the case involving the patient with pancreatitis. Clinical judgment means being engaged enough to choose what you pay attention to and to process the data you receive.
Unfortunately, the electronic medical record seems designed more for billing and for guarding against claims of fraud than for communication among clinicians or documenting clinically significant events. Many clinicians believe that redundancy and standardized phraseology have weakened the meaningful use of the medical record, as the clinical information is now of questionable reliability or value or is simply hard to find. Consequently, the electronic medical record has become less effective as a communication tool for providing continuity of care.
More importantly, the electronic medical record too often places the physician in front of a computer, so that the computer becomes the focus, not the patient. Studies suggest that the way the electronic medical record is currently used in the examination room affects the quality of physician-patient communication as well as the physician’s cognitive processing of information. Unless the physician is alert and attuned, the electronic medical record can be a barrier to connection. This not only creates the potential for mistakes, but it can also cause patients to question the quality of care they are getting and to distrust the level of the provider’s engagement. In this context, the likelihood that the patient retains an attorney increases when a bad outcome occurs, avoidable or not.
WHAT PATIENTS WANT FROM PHYSICIANS
When I first began seeing my own primary care physician, her office was 5 minutes from my home. Then she relocated to a practice 15 minutes away. And then, because of office consolidation and acquisition, her office was relocated 40 minutes away.
So why do I still go to her? Her training is not better than that of most internists, and my medical history is not so complex that I require more care than most 55-year-old men. I am only speculating, but I would guess that she is not the most financially productive physician in her group. I know that her transition to the electronic medical record has been difficult. Recently, I asked her about it. Except in some situations, she does not type while taking a history, and she stays totally away from the computer while in the examination room with me. She sits a couple of feet from me, and it feels like the days before the electronic medical record. She is clearly more comfortable listening and taking notes first and worrying about the electronic record later. I imagine she stays later to do her notes than most of the other physicians, or she finishes them at home.
The reason I continue to see her as my primary care physician is that she remains totally engaged during my office visit. What tells me that is not just her avoidance of the computer or her body language, but the depth of questions she asks. My responses often prompt her to look back at an earlier office note, and she will then ask follow-up questions to confirm what she had previously recorded. Her examination is thorough, with testing to confirm and retesting to be sure. Doing this may mean that she has difficulty meeting financial or administrative benchmarks established by her practice. I don’t know. But I have no doubt that the likelihood of her missing something in her clinical care is small, and what I suspect is even smaller is the risk that one of her patients would bring a lawsuit against her, given the time she takes to listen and remain connected throughout the office visit.
STAYING CONNECTED, IN SPITE OF EVERYTHING
My point is not to suggest that everyone must conform to the same practice philosophy, particularly with the economic pressures in the medical field. What I am suggesting is that it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team towards disconnection. Quality healthcare means making every effort to remain engaged at all times with your patient’s care, which will reduce the likelihood of a bad outcome and may preserve the physician-patient relationship even when a bad outcome occurs.
In the end, perhaps it is not possible to avoid being named as a defendant in a malpractice case, just as it is not possible to avoid all bad medical outcomes despite exceptional care. In law, as in medicine, there are always factors beyond your control. My aspiration is to find a pathway to get providers through the system unbroken—also not an easy task. But one thing I know is true: the more you can stay engaged in the care you provide and in your documentation, the more you will preclude a plaintiff’s attorney from exploiting the effects of the forces within the system that drive providers toward disconnection. As long as you stay engaged and supported by the knowledge that the care provided was appropriate, it is my hope that the voice of the critic will not count as much in the aftermath of a malpractice case. But more importantly, it may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.
During my 25 years as a defense attorney, I have seen the traumatic impact that the allegation of medical malpractice can have on healthcare providers. And I have seen many times that in the aftermath of a case it remains difficult, if not impossible, for the practitioner to return to the clinical setting unscarred by the process. Although vindication by the jury provides some solace, by itself it does not create healing. Instead, the critic’s voice continues to resonate long after the trial.
During a lawsuit, physicians and other providers are commonly confronted with incidental imperfections in the care they provided, errors in their documentation, or both. Consequently, a provider’s perception of events and ultimately the meaning derived from the experience is shaped less by the valid defenses and opinions of the supportive defense experts than by the inconsequential flaws and errors that can often be found in any medical record.
A RECENT CASE
Recently, I defended a hospital team consisting of a hospitalist, trauma surgeon, three residents, and a nurse. The case involved a 74-year-old man who was admitted to the hospital with pancreatitis of unknown cause. Six days after admission, he died of complications of acute respiratory distress syndrome. The team was accused of causing the patient’s death. Specifically, the plaintiff alleged that although the patient’s liver enzyme levels were improving, his condition was deteriorating, and he ultimately developed hemorrhagic pancreatitis. It was the plaintiff’s contention that proper ongoing evaluation, including computed tomographic imaging, would have led to treatment that would have avoided the worsening of pancreatitis, development of an ileus, and ultimately the insult to his bowel and lungs that they claim caused acute respiratory distress syndrome and death. The patient was survived by his wife and their three children. After his death, hospital representatives and the hospitalist met with her in an effort to explain the events that led to her husband’s death. Unfortunately, these discussions did not ameliorate her feelings of loss and anger. She filed a lawsuit, and 4 years later, the case went to trial.
During the trial, the plaintiff’s attorney highlighted errors in the electronic medical record. Entries had been cut and pasted, saving time, but without updating information that had changed in the interim. The inaccuracies included “assessment: worsening pancreatitis” on a day it was considered to have improved. Another entry contained “persistent fever” on a day when no fever was present. Other mistakes involved notes that contained care plans made after morning rounds that were not revised later in the day after changes in the patient’s condition necessitated a change in the plan. In fact, most references to medication dosing in the progress notes on the last 2 days did not match the medication dosing documented in the medication administration record.
In the end, the plaintiff’s counsel did not convince the jury that the healthcare team had been negligent, but unfortunately, she planted doubt in the minds of the caregivers themselves. Perhaps in part, these doubts were the result of having to defend a bad outcome in the face of criticism that was based solely in retrospect. But the providers’ doubts seemed mostly to emanate from the inadequacies in their documentation as they observed how every entry in a far-from-perfect medical record was scrutinized and then manipulated to challenge its textual integrity—and to portray the healthcare team as unengaged and substandard clinicians.
Despite the team’s high level of engagement and the quality of care they provided, any imperfection—whether a documentation error or a minor omission in some aspect of the care provided to this complex patient—became a source of self-doubt and self-criticism.
THE ELECTRONIC MEDICAL RECORD: A MIXED BLESSING
Documentation failures have long been used to “prove” that physicians are disconnected from the clinical situation. The electronic medical record has not proved to be a strong shield against malpractice allegations. In fact, because the electronic medical record absorbs more of the physician’s time and that of the care team’s members, efforts to save time through work-arounds and shortcuts have increased the risk of errors in entering information.
For instance, drop-down menus have led to wrong selections. Cutting and pasting has led to entries that contain data superseded by clinical events, thus creating contradictions within the record itself, and worse, with the physician’s own testimony pertaining to the basis of the clinical decision-making. And boilerplate language has created difficulties when the language does not completely fit the context or when inapplicable verbiage that fills itself in automatically goes unedited. An emergency department physician I represented at trial had to awkwardly explain that some of the data reported in his physical exam findings were inaccurate because of programmed language and should have been deleted; he had no explanation for his oversight.
But my experience has been that juries can forgive imperfections in documentation and even incidental aspects of care. They want to trust that the clinician was there for, and there with, the patient. This emphasis is what allowed us to defend the case involving the patient with pancreatitis. Clinical judgment means being engaged enough to choose what you pay attention to and to process the data you receive.
Unfortunately, the electronic medical record seems designed more for billing and for guarding against claims of fraud than for communication among clinicians or documenting clinically significant events. Many clinicians believe that redundancy and standardized phraseology have weakened the meaningful use of the medical record, as the clinical information is now of questionable reliability or value or is simply hard to find. Consequently, the electronic medical record has become less effective as a communication tool for providing continuity of care.
More importantly, the electronic medical record too often places the physician in front of a computer, so that the computer becomes the focus, not the patient. Studies suggest that the way the electronic medical record is currently used in the examination room affects the quality of physician-patient communication as well as the physician’s cognitive processing of information. Unless the physician is alert and attuned, the electronic medical record can be a barrier to connection. This not only creates the potential for mistakes, but it can also cause patients to question the quality of care they are getting and to distrust the level of the provider’s engagement. In this context, the likelihood that the patient retains an attorney increases when a bad outcome occurs, avoidable or not.
WHAT PATIENTS WANT FROM PHYSICIANS
When I first began seeing my own primary care physician, her office was 5 minutes from my home. Then she relocated to a practice 15 minutes away. And then, because of office consolidation and acquisition, her office was relocated 40 minutes away.
So why do I still go to her? Her training is not better than that of most internists, and my medical history is not so complex that I require more care than most 55-year-old men. I am only speculating, but I would guess that she is not the most financially productive physician in her group. I know that her transition to the electronic medical record has been difficult. Recently, I asked her about it. Except in some situations, she does not type while taking a history, and she stays totally away from the computer while in the examination room with me. She sits a couple of feet from me, and it feels like the days before the electronic medical record. She is clearly more comfortable listening and taking notes first and worrying about the electronic record later. I imagine she stays later to do her notes than most of the other physicians, or she finishes them at home.
The reason I continue to see her as my primary care physician is that she remains totally engaged during my office visit. What tells me that is not just her avoidance of the computer or her body language, but the depth of questions she asks. My responses often prompt her to look back at an earlier office note, and she will then ask follow-up questions to confirm what she had previously recorded. Her examination is thorough, with testing to confirm and retesting to be sure. Doing this may mean that she has difficulty meeting financial or administrative benchmarks established by her practice. I don’t know. But I have no doubt that the likelihood of her missing something in her clinical care is small, and what I suspect is even smaller is the risk that one of her patients would bring a lawsuit against her, given the time she takes to listen and remain connected throughout the office visit.
STAYING CONNECTED, IN SPITE OF EVERYTHING
My point is not to suggest that everyone must conform to the same practice philosophy, particularly with the economic pressures in the medical field. What I am suggesting is that it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team towards disconnection. Quality healthcare means making every effort to remain engaged at all times with your patient’s care, which will reduce the likelihood of a bad outcome and may preserve the physician-patient relationship even when a bad outcome occurs.
In the end, perhaps it is not possible to avoid being named as a defendant in a malpractice case, just as it is not possible to avoid all bad medical outcomes despite exceptional care. In law, as in medicine, there are always factors beyond your control. My aspiration is to find a pathway to get providers through the system unbroken—also not an easy task. But one thing I know is true: the more you can stay engaged in the care you provide and in your documentation, the more you will preclude a plaintiff’s attorney from exploiting the effects of the forces within the system that drive providers toward disconnection. As long as you stay engaged and supported by the knowledge that the care provided was appropriate, it is my hope that the voice of the critic will not count as much in the aftermath of a malpractice case. But more importantly, it may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.
