User login
Can’t Shake This Feeling
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
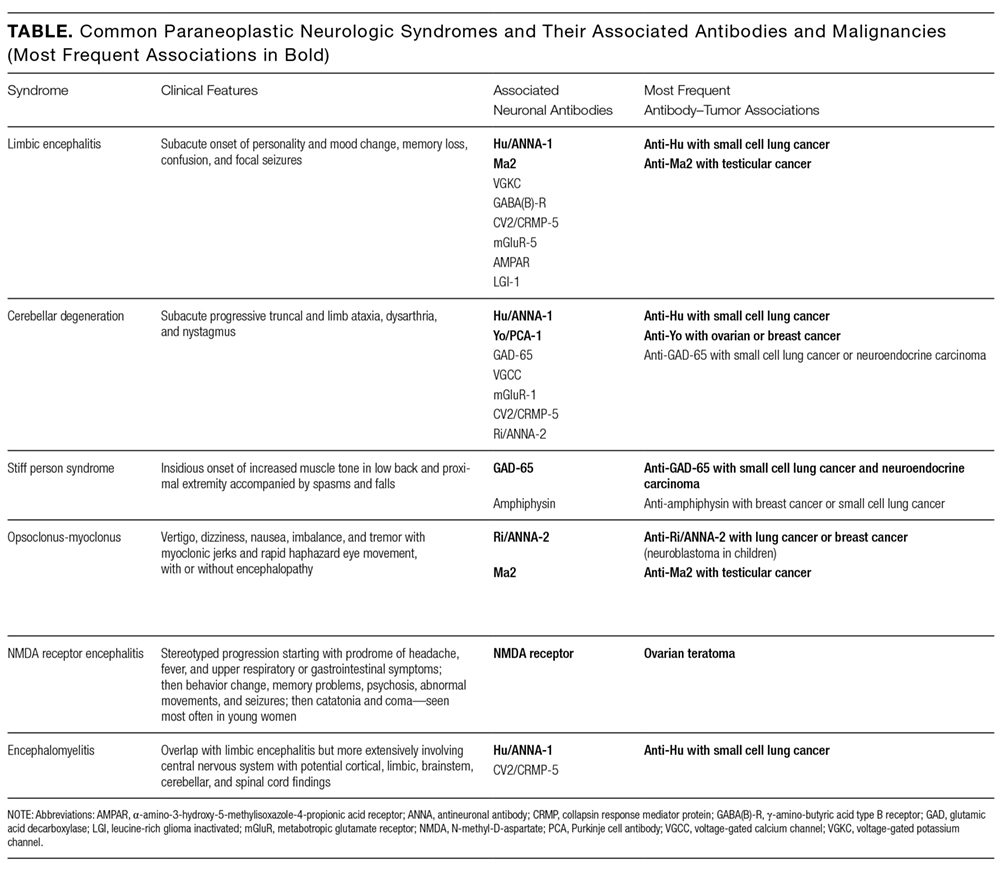
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
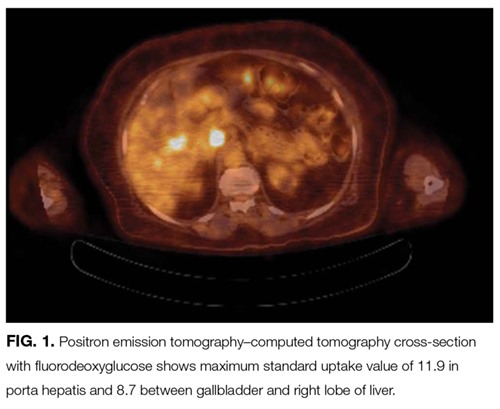
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
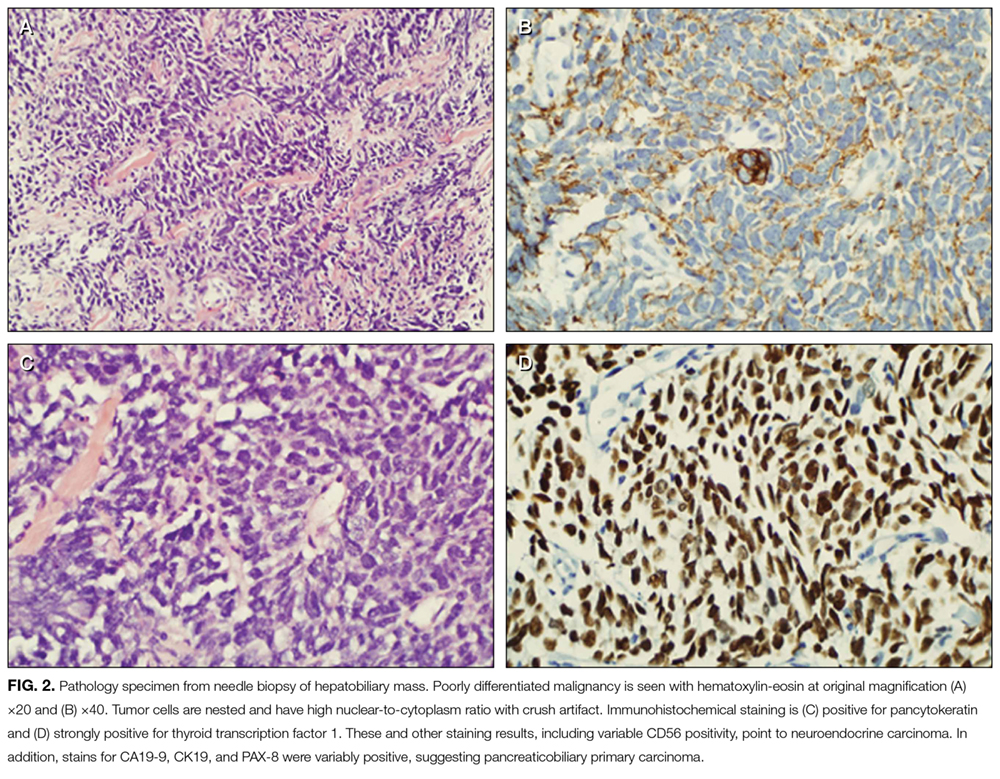
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
© 2017 Society of Hospital Medicine
Rendered speechless
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.


The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.
The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.
The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
© 2017 Society of Hospital Medicine
Unnerving
A 73‐year‐old African American man presented to his primary care physician's office concerned about several years of muscle cramps throughout his body as if his nerves were jumping and 1 month of bilateral arm weakness.
For the past 10 years, he had experienced intermittent cramping in his calves and thighs, described as a slow tightening of the muscles associated with mild pain. Initially, the cramps lasted less than 5 minutes, occurred every few days at various times of the day, and might awaken him from sleep. They happened more often following periods of inactivity and on occasion would resolve after playing golf. In recent weeks, the sensations became more frequent, more diffuse, and lasted up to several hours. He described them as a shivering. They began to affect his biceps, pectorals, deltoids, forearms, back, and calves, and would occur unrelated to activity or inactivity. He denied sensory disturbances, facial twitching or facial weakness, diplopia, dysarthria, dysphagia, dyspnea, changes in bowel or bladder function, unexplained lapses of consciousness, fevers, or weight loss.
Long‐standing cramping is nonspecific and may reflect transient electrolyte derangements or muscle overuse. However, the more recent change in frequency, duration, and quality of these sensations, along with the reported weakness, raises concern for a process involving the peripheral nervous system. It will be important to differentiate cramping from other abnormal movements such as fasciculations, tremor, or myoclonus, and to determine whether there is objective weakness on the neurological examination.
His past medical history was significant for coronary artery disease with an ST‐segment elevation myocardial infarction several years prior, which was treated with a drug‐eluting stent. He was also diagnosed with essential thrombocythemia at the time of his myocardial infarction and tested positive for the JAK2 mutation. He was treated for several years with hydroxyurea following his diagnosis of essential thrombocythemia. Hydroxyurea had been discontinued 6 months prior due to cytopenias. The remainder of his history was significant for hypertension, chronic kidney disease stage 3, and prediabetes.
Medications were clopidogrel, atorvastatin, metoprolol, lisinopril, and hydrochlorothiazide. He did not use tobacco nor consume alcohol or illicit drugs, and he drank caffeine only occasionally. He had no family history of neurologic disorders.
Apart from his use of statins, which often affect muscles (and less commonly the nerves), the past medical history provides minimal additional insights into the cause of his symptoms. If weakness is detected on physical exam, the next step would be to distinguish upper (central) from a lower motor neuron (peripheral) localization. A diffuse problem involving all 4 limbs is generally more likely to arise from a disorder of a lower motor neuron (LMN) structure (anterior horn cell, nerve, neuromuscular junction, or muscle). To explain bilateral symptoms of the upper and lower limbs, an upper motor neuron (UMN) disease would have to affect the bilateral brain or cervical cord, a somewhat less likely possibility given the cramps described. It would also be quite unusual to have weakness of central nervous system origin without sensory deficits.
On physical examination, the patient was well‐appearing and in no apparent distress. Temperature was 98.1, blood pressure 134/84, pulse 110 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 100% while breathing ambient air. There was no lymphadenopathy. Lung, heart, abdominal, and skin exams were unremarkable. He was alert and oriented. His speech was without dysarthria. Examinations of the cranial nerves were intact. No tongue atrophy or fasciculations were noted. No pooling of secretions was appreciated in the oropharynx. Examination of the musculature revealed normal tone, strength, and bulk. However, there were diffuse fasciculations present, most prominent in the bilateral biceps, pectorals, deltoids, forearms, upper back, and calves. Sensation to light touch, temperature, and vibration were intact. Babinski's sign was absent, and deep tendon reflexes were normal, except at the ankles where they were reduced. Coordination and gait were normal.
The exam is notable for diffuse fasciculations, defined as spontaneous local involuntary muscle contraction and relaxation, which is often visible. Benign fasciculations are extremely common, with up to 70% of otherwise healthy adults experiencing them, and may be brought on by physical exertion. Men experience these benign fasciculations more frequently than women, and they can occur at any age and persist throughout life. Fasciculations may point to LMN disease, usually localizing to the anterior horn cell (for instance in amyotrophic lateral sclerosis [ALS]), muscle, or nerve disorders (including diffuse polyneuropathy). The presence of fasciculations in patients without other complaints and an otherwise normal physical examination supports benign fasciculations. The presence of neurologic deficits, however, such as weakness or reflex loss, is worrisome for another etiology. The absence of sensory changes makes anterior horn cell disease or myopathy most likely, as pure motor neuropathies are uncommon.
The fasciculations in this patient are most prominent in the proximal muscles, which may indicate a primary muscle disorder. Myopathies are typically characterized by diffuse symmetric weakness that is more proximal than distal, with no changes in sensation or deep tendon reflexes. One muscle disease characterized by fasciculations and cramping is periodic paralysis, which is often associated with potassium abnormalities or thyroid dysfunction caused by specific channelopathies. However, patients with this disorder typically present with episodic crises in contrast to the constant symptoms in this case.
Given the accelerated tempo of this patient's symptoms, further diagnostic evaluation should include basic laboratory testing including electrolytes, creatinine kinase, and thyrotropin. If these initial tests fail to reveal an etiology, the next study of choice would be an electromyography (EMG) and nerve conduction study (NCS), which can definitively localize the disorder to and within the peripheral nervous system. Unlike in UMN disease, in which neuroimaging is crucial, imaging is unlikely to be informative in patients with an LMN disorder.
Results of a complete blood count demonstrated a platelet count of 590,000/L (normal 140400 K/L), but was otherwise normal. Sodium, potassium, magnesium, and calcium levels were normal as were alanine aminotransferase, thyrotropin, and urinalysis. The hemoglobin A1c was 6.4%. Serum creatinine kinase was 157 U/L (normal170 U/L), and the serum creatinine was 1.7mg/dL, similar to previous results. The erythrocyte sedimentation rate was 58mm/h (normal 020mm/h). Magnetic resonance imaging (MRI) (Figure 1) of the cervical spine demonstrated diffuse disc desiccation, and multilevel spondylosis most prominent at C4C5 and C5C6, with severe central canal stenosis and neural foraminal narrowing.
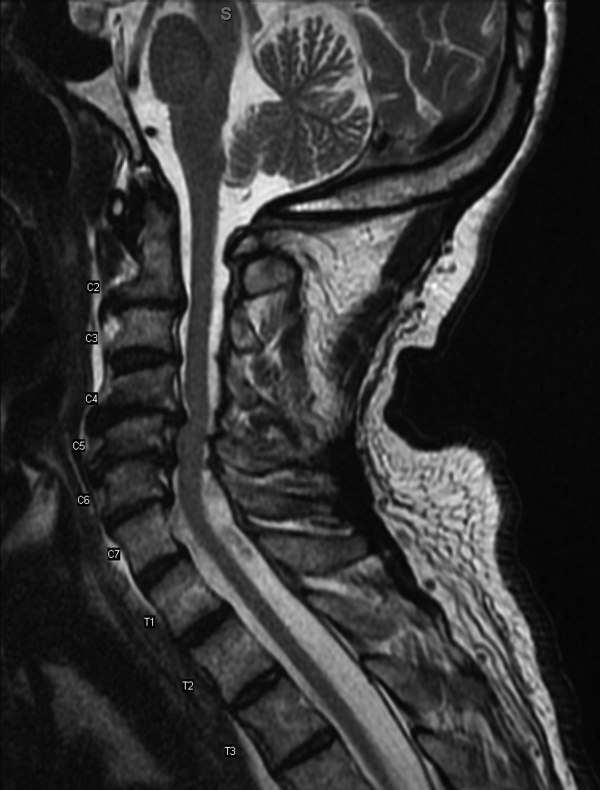
The routine laboratory tests do not point to an obvious cause of this man's symptoms. As expected, the MRI findings do not explain diffuse fascinations in all limbs with no sensory disturbance. Further evaluation should include EMG and NCS.
NCS of the median and ulnar nerves demonstrated minimally reduced conduction velocities and markedly prolonged latencies. Sensory responses were absent. No conduction block was detected. EMG demonstrated fasciculations of the right extensor digitorum and first dorsal interossei, as well as decreased amplitude and decreased recruitment of motor units.
Abnormalities on the EMG testing can reflect either a primary muscle disorder or muscle derangement resulting from disease of the nerves. NCSs are important to differentiate these 2 possibilities. This patient's NCS indicates that the primary process is a diffuse motor and sensory neuropathy, not myopathy. The lack of sensory findings on physical examination emphasizes the ability of electrodiagnostic testing to extend the clinical neurologic examination in some cases. Markedly prolonged latencies and more modest reduction in amplitude support a motor and sensory neuropathy mostly due to demyelination, rather than axonal loss, and that it is more severe in the lower limbs.
Among the demyelinating neuropathies, acute inflammatory demyelinating neuropathy (Guillain‐Barr syndrome), is the most commonly recognized. The prolonged time course of this patient's illness excludes this possibility. Chronic inflammatory demyelinating polyneuropathy is also very unlikely in the absence of conduction block on NCS. Demyelinating neuropathies may also result from antibody‐mediated nerve injury. The serum paraprotein most commonly involved is immunoglobulin M (IgM), as is detected in neuropathy due to antibodies to myelin‐associated glycoprotein (anti‐MAG) neuropathy. Another variant is the ganglioside monosialic acid antibody (anti‐GM1) associated with a rare disease called multifocal motor neuropathy (MMN), an important condition to recognize because symptoms of this illness may mimic the presentation of ALS, often with fasciculations and weakness. Unlike ALS, MMN is very responsive to treatment. Other antibody‐mediated neuropathies are much rarer. In this patient, MMN is unlikely because sensory nerves are affected in addition to motor nerves.
Because NCSs also indicate some axonal loss, it would be reasonable to screen for vitamin deficiencies, human immunodeficiency virus, and viral hepatitis. The pattern here is more symmetric and confluent than would be expected if he had mononeuritic multiplex from vasculitis.
Vitamin E level was normal. Vitamin B12 level was 323 pg/mL (normal>200 pg/mL), methylmalonate 0.3mol/L (normal 00.3mol/L). Antibodies to human immunodeficiency virus and surface antibody and antigen to hepatitis B were not detected. Cryoglobulins, anti‐nuclear antibody, and antibodies to myeloperoxidase and proteinase 3 were not detected. Serum antibodies to tissue transglutaminase and Borrelia burgdorferi were not detectable. Serum protein electrophoresis demonstrated 2 small spikes in the gamma region. Quantitative serum immunoglobulin levels were normal except for IgM, which was elevated at 1.7g/dL (normal0.19g/dL). Serum free light chains showed a kappa component of 43.7mg/L (normal 319mg/L), a lambda component of 13.8mg/L (normal 526mg/L), and a kappa/lambda ratio of 3.17mg/L (normal 0.261.65mg/L).
The differential for a symmetric demyelinating neuropathy is quite narrow, and tests for vasculitis, celiac disease, and Lyme disease are not necessary. To pursue the cause of the elevated IgM, specific serum testing should be obtained for anti‐MAG antibodies. Many cases of anti‐MAG neuropathy are associated with an underlying lymphoproliferative disorder. As such, additional imaging to identify occult lymphoma is warranted.
Anti‐GM1 and asialoganglioside were not detectable. The anti‐MAG IgM titer was >102,400 (normal1:1600). Abdominal ultrasound showed normal sized kidneys with normal cortical echogenicity and no splenomegaly. Computed tomography with contrast was not performed due to chronic kidney disease.
Treatment for anti‐MAG neuropathy is evolving rapidly as our understanding of the entity improves. Cyclophosphamide, intravenous immune globulin, and plasmapheresis have been the traditional treatments, but in the past decade, favorable experiences with rituximab have led some to try this medication earlier in the course. Prognosis can be favorable in many patients.
Over the next 2 months he continued to have fasciculations. He developed progressive generalized weakness, an unsteady gait, required a walker for mobility, and began to have trouble with his activities of daily living. His cognition remained intact. There was no pooling of secretions. Serial neurologic examinations demonstrated persistent fasciculations, progressive atrophy, most notably in the intrinsic hand muscles and legs, and progressive weakness of all limbs, worse in the distal muscle groups. Deep tendon reflexes remained preserved, except at the ankles where they were absent. Sensory exam showed stocking diminution to temperature up to his knees and elbows. Romberg sign was present, and he could not walk without support. He was started on rituximab, and after 4 weeks his condition continued to deteriorate.
The response to rituximab may be delayed. Alternatively, his disease may have an underlying cause such as occult lymphoma not yet identified, which would require treatment to control the neuropathy. Because of the potential association between lymphoma in some patients with anti‐MAG neuropathy, and because he is not responding to immunotherapy, whole body imaging with positron emission tomography and bone marrow biopsy should be performed.
CD19 levels indicated an appropriate B cell response to rituximab, but the anti‐MAG titer remained elevated at >102,400. He received additional doses of rituximab, but continued to decline. A bone marrow biopsy was considered, but the patient opted to forgo the procedure. After several months of rituximab, he developed mild dysarthria and dysphagia and was hospitalized for plasma exchange. After 5 sessions of plasma exchange, he showed no improvement and was discharged to a rehabilitation facility. Over the ensuing months, he became restricted to wheel chair or bed and eventually opted for comfort measures. He died after an aspiration pneumonia 15 months after his initial visit to his physician. Permission for an autopsy was not granted.
COMMENTARY
When encountering patients with involuntary muscle movements, hospitalists must recognize potential serious underlying disorders and implement a cost‐effective evaluation strategy. Fasciculations are a common finding that represent involuntary discharges of a motor unit, with a wide array of causes including radiculopathies, neuropathies, metabolic disturbances, and motor neuron diseases.[1, 2] Useful clues might point to a probable cause, such as a statin‐induced myopathy in patients with concomitant myalgias, or hypokalemia in patients on loop diuretics. Confinement of fasciculations to specific anatomic structures may be useful, as in carpal tunnel syndrome, where fasciculations would only be expected distal to median nerve compression. Features such as sensory loss, muscle atrophy, or abnormal reflexes should alert the clinician to a possible neurologic lesion.
Although fasciculations rarely reflect serious underlying pathology, the presence of neurologic deficits, such as muscle weakness, abnormal reflexes, or sensory loss, should prompt further investigation.[3] Because fasciculations typically point to an abnormality of LMN structures, a reasonable approach is to measure serum electrolytes, creatinine kinase, and thyrotropin to evaluate for myopathy. If these tests are unrevealing, the next step would be to perform EMG and NCS to help localize the lesion among the LMN structures. Muscle localization could then be pursued with muscle biopsy. Alternatively, when electrodiagnostic testing indicates peripheral nerve pathology, further evaluation is guided by the type of neuropathy: demyelinating, axonal, or mixed. If electrodiagnostic and clinical findings are unrevealing, the patient is diagnosed with benign fasciculations.
Demyelinating neuropathy, as seen in our patient, is relevant to hospitalists for several reasons. First, the list of diagnostic possibilities is narrow, allowing hospitalists to forgo many unnecessary laboratory tests and brain MRI. Second, unlike many axonal neuropathies, demyelinating neuropathies are potentially reversible if recognized early and promptly treated. Third, demyelinating neuropathy may involve the diaphragm, necessitating vigilance for neuromuscular respiratory failure. Finally, hospitalists need to be aware that some demyelinating neuropathies are associated with underlying malignancy, and identifying and treating the primary cancer may be critical to ameliorating the neuropathy.[4, 5, 6, 7, 8]
IgM paraproteinemia, with or without an underlying malignancy, is 1 type of demyelinating neuropathy that is potentially reversible with early treatment. The typical patient is exemplified by the case presented in this report: an older man who experiences symmetric, gradually worsening sensory disturbances and ataxia over months to years.[9] Motor deficits may progress more rapidly, prompting patients to seek hospital care.[7, 10] The hallmark of NCS in anti‐MAG disease is a demyelinating pattern with a predominance of distal abnormalities including marked prolongation of distal motor latencies and reductions in conduction velocities and sensory action potentials.[9] Findings of areflexia or conduction block should prompt consideration of other etiologies, such as acute or chronic inflammatory demyelinating polyneuropathy.
For unclear reasons, IgM is more likely than other immunoglobulins to cause neuropathy. Although IgM accounts for only 17% of monoclonal gammopathies, IgM is detected in 50% to 70% of patients who have both monoclonal gammopathy and peripheral neuropathy.[11] Approximately half of the patients with IgM‐associated neuropathy produce antibodies to MAG.[11, 12] Several lines of evidence have firmly established the causative role of anti‐MAG antibodies.[13]
Because the majority of patients with anti‐MAG neuropathy will have no malignant source of IgM paraprotein identified, it is unclear how extensively to search for occult malignancy. A reasonable approach is to perform a bone marrow biopsy to distinguish underlying IgM monoclonal gammopathy of undetermined significance from Waldenstrom's macroglobulinemia.[14] Bone marrow analysis may also detect B‐cell lymphoma, primary amyloidosis, chronic lymphocytic leukemia, and hairy cell leukemia, which have been described in cases of anti‐MAG syndrome.[4, 5, 6, 7, 8] There are no reports of anti‐MAG neuropathy linked to either essential thrombocythemia nor hydroxyurea use.
The goals of treatment in anti‐MAG neuropathy are to deplete monoclonal B cells and to reduce antibody levels. Although it is reported that approximately half of patients will improve with some form of immunotherapy, a Cochrane review of randomized controlled trials of treatments for anti‐MAG neuropathy (including plasma exchange, intravenous immunoglobulin [IVIG], rituximab, corticosteroids, and chemotherapy) concluded that evidence is lacking to recommend 1 treatment over another.[15] European guidelines suggest deferring therapy unless progressive or severe neuropathy is present, in which case IVIG, plasma exchange, or rituximab may be tried.[14] In patients with underlying malignancy, treatment of the hematologic disorder may improve the neuropathy.[4, 8]
Although fasciculations and peripheral neuropathy typically present in outpatient settings, they can be harbingers of more dire diagnoses that prompt patients to seek hospitalization. A sequential and cost‐effective approach can allow the astute hospitalist to pinpoint the diagnosis in what might otherwise be an unnerving case.
KEY TEACHING POINTS
- Fasciculations are extremely common and usually benign, but may indicate a more serious neurologic process, especially when accompanied by weakness or other neurologic symptoms.
- Localizing neurologic deficits to upper motor neuron or lower motor neuron structures guides further evaluation.
- Central nervous system imaging is not indicated in demyelinating neuropathies.
- Bone marrow biopsy and cross‐sectional imaging to evaluate for malignancy should be considered in patients with anti‐MAG neuropathy who fail to improve despite therapy.
- , . Fasciculations: what do we know of their significance? J Neurol Sci. 1997;152(suppl 1):S43–S48.
- , . Muscle fasciculations in a healthy population. Arch Neurol. 1963;9:363–367.
- . Muscle pain, fatigue, and fasciculations. Neurol Clin. 1997;15(3):697–709.
- , , , et al. Monocytoid B cell lymphoma associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Acta Haematol. 2001;106(3):130–132.
- . Anti‐myelin‐associated glycoprotein peripheral neuropathy as the only presentation of low grade lymphoma: a case report. Cases J. 2009;2(1):6370–6373.
- , , , et al. Antibodies to myelin‐associated glycoprotein (anti‐Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve. 2008;37(4):490–495.
- , , , et al. Heterogeneity of polyneuropathy associated with anti‐MAG antibodies. J Immunol Res. 2015;2015(3):450391–450399.
- , , , et al. Hairy cell leukaemia complicated by anti‐MAG paraproteinemic demyelinating neuropathy: resolution of neurological syndrome after cladribrine treatment. Leuk Res. 2007;31(6):873–876.
- , , , , , . Neuropathy associated with “benign” anti‐myelin‐associated glycoprotein IgM gammopathy: clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J Neurol. 1996;243(1):34–43.
- , , , et al. IgM MGUS anti‐MAG neuropathy with predominant muscle weakness and extensive muscle atrophy. Muscle Nerve. 2010;42(3):433–435.
- , , , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- , . Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26(2):55–65.
- , . Effector mechanisms in anti‐MAG antibody‐mediated and other demyelinating neuropathies. J Neurol Sci. 2004;220(1–2):127–129.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15(3):185–195.
- , . Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.
A 73‐year‐old African American man presented to his primary care physician's office concerned about several years of muscle cramps throughout his body as if his nerves were jumping and 1 month of bilateral arm weakness.
For the past 10 years, he had experienced intermittent cramping in his calves and thighs, described as a slow tightening of the muscles associated with mild pain. Initially, the cramps lasted less than 5 minutes, occurred every few days at various times of the day, and might awaken him from sleep. They happened more often following periods of inactivity and on occasion would resolve after playing golf. In recent weeks, the sensations became more frequent, more diffuse, and lasted up to several hours. He described them as a shivering. They began to affect his biceps, pectorals, deltoids, forearms, back, and calves, and would occur unrelated to activity or inactivity. He denied sensory disturbances, facial twitching or facial weakness, diplopia, dysarthria, dysphagia, dyspnea, changes in bowel or bladder function, unexplained lapses of consciousness, fevers, or weight loss.
Long‐standing cramping is nonspecific and may reflect transient electrolyte derangements or muscle overuse. However, the more recent change in frequency, duration, and quality of these sensations, along with the reported weakness, raises concern for a process involving the peripheral nervous system. It will be important to differentiate cramping from other abnormal movements such as fasciculations, tremor, or myoclonus, and to determine whether there is objective weakness on the neurological examination.
His past medical history was significant for coronary artery disease with an ST‐segment elevation myocardial infarction several years prior, which was treated with a drug‐eluting stent. He was also diagnosed with essential thrombocythemia at the time of his myocardial infarction and tested positive for the JAK2 mutation. He was treated for several years with hydroxyurea following his diagnosis of essential thrombocythemia. Hydroxyurea had been discontinued 6 months prior due to cytopenias. The remainder of his history was significant for hypertension, chronic kidney disease stage 3, and prediabetes.
Medications were clopidogrel, atorvastatin, metoprolol, lisinopril, and hydrochlorothiazide. He did not use tobacco nor consume alcohol or illicit drugs, and he drank caffeine only occasionally. He had no family history of neurologic disorders.
Apart from his use of statins, which often affect muscles (and less commonly the nerves), the past medical history provides minimal additional insights into the cause of his symptoms. If weakness is detected on physical exam, the next step would be to distinguish upper (central) from a lower motor neuron (peripheral) localization. A diffuse problem involving all 4 limbs is generally more likely to arise from a disorder of a lower motor neuron (LMN) structure (anterior horn cell, nerve, neuromuscular junction, or muscle). To explain bilateral symptoms of the upper and lower limbs, an upper motor neuron (UMN) disease would have to affect the bilateral brain or cervical cord, a somewhat less likely possibility given the cramps described. It would also be quite unusual to have weakness of central nervous system origin without sensory deficits.
On physical examination, the patient was well‐appearing and in no apparent distress. Temperature was 98.1, blood pressure 134/84, pulse 110 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 100% while breathing ambient air. There was no lymphadenopathy. Lung, heart, abdominal, and skin exams were unremarkable. He was alert and oriented. His speech was without dysarthria. Examinations of the cranial nerves were intact. No tongue atrophy or fasciculations were noted. No pooling of secretions was appreciated in the oropharynx. Examination of the musculature revealed normal tone, strength, and bulk. However, there were diffuse fasciculations present, most prominent in the bilateral biceps, pectorals, deltoids, forearms, upper back, and calves. Sensation to light touch, temperature, and vibration were intact. Babinski's sign was absent, and deep tendon reflexes were normal, except at the ankles where they were reduced. Coordination and gait were normal.
The exam is notable for diffuse fasciculations, defined as spontaneous local involuntary muscle contraction and relaxation, which is often visible. Benign fasciculations are extremely common, with up to 70% of otherwise healthy adults experiencing them, and may be brought on by physical exertion. Men experience these benign fasciculations more frequently than women, and they can occur at any age and persist throughout life. Fasciculations may point to LMN disease, usually localizing to the anterior horn cell (for instance in amyotrophic lateral sclerosis [ALS]), muscle, or nerve disorders (including diffuse polyneuropathy). The presence of fasciculations in patients without other complaints and an otherwise normal physical examination supports benign fasciculations. The presence of neurologic deficits, however, such as weakness or reflex loss, is worrisome for another etiology. The absence of sensory changes makes anterior horn cell disease or myopathy most likely, as pure motor neuropathies are uncommon.
The fasciculations in this patient are most prominent in the proximal muscles, which may indicate a primary muscle disorder. Myopathies are typically characterized by diffuse symmetric weakness that is more proximal than distal, with no changes in sensation or deep tendon reflexes. One muscle disease characterized by fasciculations and cramping is periodic paralysis, which is often associated with potassium abnormalities or thyroid dysfunction caused by specific channelopathies. However, patients with this disorder typically present with episodic crises in contrast to the constant symptoms in this case.
Given the accelerated tempo of this patient's symptoms, further diagnostic evaluation should include basic laboratory testing including electrolytes, creatinine kinase, and thyrotropin. If these initial tests fail to reveal an etiology, the next study of choice would be an electromyography (EMG) and nerve conduction study (NCS), which can definitively localize the disorder to and within the peripheral nervous system. Unlike in UMN disease, in which neuroimaging is crucial, imaging is unlikely to be informative in patients with an LMN disorder.
Results of a complete blood count demonstrated a platelet count of 590,000/L (normal 140400 K/L), but was otherwise normal. Sodium, potassium, magnesium, and calcium levels were normal as were alanine aminotransferase, thyrotropin, and urinalysis. The hemoglobin A1c was 6.4%. Serum creatinine kinase was 157 U/L (normal170 U/L), and the serum creatinine was 1.7mg/dL, similar to previous results. The erythrocyte sedimentation rate was 58mm/h (normal 020mm/h). Magnetic resonance imaging (MRI) (Figure 1) of the cervical spine demonstrated diffuse disc desiccation, and multilevel spondylosis most prominent at C4C5 and C5C6, with severe central canal stenosis and neural foraminal narrowing.
The routine laboratory tests do not point to an obvious cause of this man's symptoms. As expected, the MRI findings do not explain diffuse fascinations in all limbs with no sensory disturbance. Further evaluation should include EMG and NCS.
NCS of the median and ulnar nerves demonstrated minimally reduced conduction velocities and markedly prolonged latencies. Sensory responses were absent. No conduction block was detected. EMG demonstrated fasciculations of the right extensor digitorum and first dorsal interossei, as well as decreased amplitude and decreased recruitment of motor units.
Abnormalities on the EMG testing can reflect either a primary muscle disorder or muscle derangement resulting from disease of the nerves. NCSs are important to differentiate these 2 possibilities. This patient's NCS indicates that the primary process is a diffuse motor and sensory neuropathy, not myopathy. The lack of sensory findings on physical examination emphasizes the ability of electrodiagnostic testing to extend the clinical neurologic examination in some cases. Markedly prolonged latencies and more modest reduction in amplitude support a motor and sensory neuropathy mostly due to demyelination, rather than axonal loss, and that it is more severe in the lower limbs.
Among the demyelinating neuropathies, acute inflammatory demyelinating neuropathy (Guillain‐Barr syndrome), is the most commonly recognized. The prolonged time course of this patient's illness excludes this possibility. Chronic inflammatory demyelinating polyneuropathy is also very unlikely in the absence of conduction block on NCS. Demyelinating neuropathies may also result from antibody‐mediated nerve injury. The serum paraprotein most commonly involved is immunoglobulin M (IgM), as is detected in neuropathy due to antibodies to myelin‐associated glycoprotein (anti‐MAG) neuropathy. Another variant is the ganglioside monosialic acid antibody (anti‐GM1) associated with a rare disease called multifocal motor neuropathy (MMN), an important condition to recognize because symptoms of this illness may mimic the presentation of ALS, often with fasciculations and weakness. Unlike ALS, MMN is very responsive to treatment. Other antibody‐mediated neuropathies are much rarer. In this patient, MMN is unlikely because sensory nerves are affected in addition to motor nerves.
Because NCSs also indicate some axonal loss, it would be reasonable to screen for vitamin deficiencies, human immunodeficiency virus, and viral hepatitis. The pattern here is more symmetric and confluent than would be expected if he had mononeuritic multiplex from vasculitis.
Vitamin E level was normal. Vitamin B12 level was 323 pg/mL (normal>200 pg/mL), methylmalonate 0.3mol/L (normal 00.3mol/L). Antibodies to human immunodeficiency virus and surface antibody and antigen to hepatitis B were not detected. Cryoglobulins, anti‐nuclear antibody, and antibodies to myeloperoxidase and proteinase 3 were not detected. Serum antibodies to tissue transglutaminase and Borrelia burgdorferi were not detectable. Serum protein electrophoresis demonstrated 2 small spikes in the gamma region. Quantitative serum immunoglobulin levels were normal except for IgM, which was elevated at 1.7g/dL (normal0.19g/dL). Serum free light chains showed a kappa component of 43.7mg/L (normal 319mg/L), a lambda component of 13.8mg/L (normal 526mg/L), and a kappa/lambda ratio of 3.17mg/L (normal 0.261.65mg/L).
The differential for a symmetric demyelinating neuropathy is quite narrow, and tests for vasculitis, celiac disease, and Lyme disease are not necessary. To pursue the cause of the elevated IgM, specific serum testing should be obtained for anti‐MAG antibodies. Many cases of anti‐MAG neuropathy are associated with an underlying lymphoproliferative disorder. As such, additional imaging to identify occult lymphoma is warranted.
Anti‐GM1 and asialoganglioside were not detectable. The anti‐MAG IgM titer was >102,400 (normal1:1600). Abdominal ultrasound showed normal sized kidneys with normal cortical echogenicity and no splenomegaly. Computed tomography with contrast was not performed due to chronic kidney disease.
Treatment for anti‐MAG neuropathy is evolving rapidly as our understanding of the entity improves. Cyclophosphamide, intravenous immune globulin, and plasmapheresis have been the traditional treatments, but in the past decade, favorable experiences with rituximab have led some to try this medication earlier in the course. Prognosis can be favorable in many patients.
Over the next 2 months he continued to have fasciculations. He developed progressive generalized weakness, an unsteady gait, required a walker for mobility, and began to have trouble with his activities of daily living. His cognition remained intact. There was no pooling of secretions. Serial neurologic examinations demonstrated persistent fasciculations, progressive atrophy, most notably in the intrinsic hand muscles and legs, and progressive weakness of all limbs, worse in the distal muscle groups. Deep tendon reflexes remained preserved, except at the ankles where they were absent. Sensory exam showed stocking diminution to temperature up to his knees and elbows. Romberg sign was present, and he could not walk without support. He was started on rituximab, and after 4 weeks his condition continued to deteriorate.
The response to rituximab may be delayed. Alternatively, his disease may have an underlying cause such as occult lymphoma not yet identified, which would require treatment to control the neuropathy. Because of the potential association between lymphoma in some patients with anti‐MAG neuropathy, and because he is not responding to immunotherapy, whole body imaging with positron emission tomography and bone marrow biopsy should be performed.
CD19 levels indicated an appropriate B cell response to rituximab, but the anti‐MAG titer remained elevated at >102,400. He received additional doses of rituximab, but continued to decline. A bone marrow biopsy was considered, but the patient opted to forgo the procedure. After several months of rituximab, he developed mild dysarthria and dysphagia and was hospitalized for plasma exchange. After 5 sessions of plasma exchange, he showed no improvement and was discharged to a rehabilitation facility. Over the ensuing months, he became restricted to wheel chair or bed and eventually opted for comfort measures. He died after an aspiration pneumonia 15 months after his initial visit to his physician. Permission for an autopsy was not granted.
COMMENTARY
When encountering patients with involuntary muscle movements, hospitalists must recognize potential serious underlying disorders and implement a cost‐effective evaluation strategy. Fasciculations are a common finding that represent involuntary discharges of a motor unit, with a wide array of causes including radiculopathies, neuropathies, metabolic disturbances, and motor neuron diseases.[1, 2] Useful clues might point to a probable cause, such as a statin‐induced myopathy in patients with concomitant myalgias, or hypokalemia in patients on loop diuretics. Confinement of fasciculations to specific anatomic structures may be useful, as in carpal tunnel syndrome, where fasciculations would only be expected distal to median nerve compression. Features such as sensory loss, muscle atrophy, or abnormal reflexes should alert the clinician to a possible neurologic lesion.
Although fasciculations rarely reflect serious underlying pathology, the presence of neurologic deficits, such as muscle weakness, abnormal reflexes, or sensory loss, should prompt further investigation.[3] Because fasciculations typically point to an abnormality of LMN structures, a reasonable approach is to measure serum electrolytes, creatinine kinase, and thyrotropin to evaluate for myopathy. If these tests are unrevealing, the next step would be to perform EMG and NCS to help localize the lesion among the LMN structures. Muscle localization could then be pursued with muscle biopsy. Alternatively, when electrodiagnostic testing indicates peripheral nerve pathology, further evaluation is guided by the type of neuropathy: demyelinating, axonal, or mixed. If electrodiagnostic and clinical findings are unrevealing, the patient is diagnosed with benign fasciculations.
Demyelinating neuropathy, as seen in our patient, is relevant to hospitalists for several reasons. First, the list of diagnostic possibilities is narrow, allowing hospitalists to forgo many unnecessary laboratory tests and brain MRI. Second, unlike many axonal neuropathies, demyelinating neuropathies are potentially reversible if recognized early and promptly treated. Third, demyelinating neuropathy may involve the diaphragm, necessitating vigilance for neuromuscular respiratory failure. Finally, hospitalists need to be aware that some demyelinating neuropathies are associated with underlying malignancy, and identifying and treating the primary cancer may be critical to ameliorating the neuropathy.[4, 5, 6, 7, 8]
IgM paraproteinemia, with or without an underlying malignancy, is 1 type of demyelinating neuropathy that is potentially reversible with early treatment. The typical patient is exemplified by the case presented in this report: an older man who experiences symmetric, gradually worsening sensory disturbances and ataxia over months to years.[9] Motor deficits may progress more rapidly, prompting patients to seek hospital care.[7, 10] The hallmark of NCS in anti‐MAG disease is a demyelinating pattern with a predominance of distal abnormalities including marked prolongation of distal motor latencies and reductions in conduction velocities and sensory action potentials.[9] Findings of areflexia or conduction block should prompt consideration of other etiologies, such as acute or chronic inflammatory demyelinating polyneuropathy.
For unclear reasons, IgM is more likely than other immunoglobulins to cause neuropathy. Although IgM accounts for only 17% of monoclonal gammopathies, IgM is detected in 50% to 70% of patients who have both monoclonal gammopathy and peripheral neuropathy.[11] Approximately half of the patients with IgM‐associated neuropathy produce antibodies to MAG.[11, 12] Several lines of evidence have firmly established the causative role of anti‐MAG antibodies.[13]
Because the majority of patients with anti‐MAG neuropathy will have no malignant source of IgM paraprotein identified, it is unclear how extensively to search for occult malignancy. A reasonable approach is to perform a bone marrow biopsy to distinguish underlying IgM monoclonal gammopathy of undetermined significance from Waldenstrom's macroglobulinemia.[14] Bone marrow analysis may also detect B‐cell lymphoma, primary amyloidosis, chronic lymphocytic leukemia, and hairy cell leukemia, which have been described in cases of anti‐MAG syndrome.[4, 5, 6, 7, 8] There are no reports of anti‐MAG neuropathy linked to either essential thrombocythemia nor hydroxyurea use.
The goals of treatment in anti‐MAG neuropathy are to deplete monoclonal B cells and to reduce antibody levels. Although it is reported that approximately half of patients will improve with some form of immunotherapy, a Cochrane review of randomized controlled trials of treatments for anti‐MAG neuropathy (including plasma exchange, intravenous immunoglobulin [IVIG], rituximab, corticosteroids, and chemotherapy) concluded that evidence is lacking to recommend 1 treatment over another.[15] European guidelines suggest deferring therapy unless progressive or severe neuropathy is present, in which case IVIG, plasma exchange, or rituximab may be tried.[14] In patients with underlying malignancy, treatment of the hematologic disorder may improve the neuropathy.[4, 8]
Although fasciculations and peripheral neuropathy typically present in outpatient settings, they can be harbingers of more dire diagnoses that prompt patients to seek hospitalization. A sequential and cost‐effective approach can allow the astute hospitalist to pinpoint the diagnosis in what might otherwise be an unnerving case.
KEY TEACHING POINTS
- Fasciculations are extremely common and usually benign, but may indicate a more serious neurologic process, especially when accompanied by weakness or other neurologic symptoms.
- Localizing neurologic deficits to upper motor neuron or lower motor neuron structures guides further evaluation.
- Central nervous system imaging is not indicated in demyelinating neuropathies.
- Bone marrow biopsy and cross‐sectional imaging to evaluate for malignancy should be considered in patients with anti‐MAG neuropathy who fail to improve despite therapy.
A 73‐year‐old African American man presented to his primary care physician's office concerned about several years of muscle cramps throughout his body as if his nerves were jumping and 1 month of bilateral arm weakness.
For the past 10 years, he had experienced intermittent cramping in his calves and thighs, described as a slow tightening of the muscles associated with mild pain. Initially, the cramps lasted less than 5 minutes, occurred every few days at various times of the day, and might awaken him from sleep. They happened more often following periods of inactivity and on occasion would resolve after playing golf. In recent weeks, the sensations became more frequent, more diffuse, and lasted up to several hours. He described them as a shivering. They began to affect his biceps, pectorals, deltoids, forearms, back, and calves, and would occur unrelated to activity or inactivity. He denied sensory disturbances, facial twitching or facial weakness, diplopia, dysarthria, dysphagia, dyspnea, changes in bowel or bladder function, unexplained lapses of consciousness, fevers, or weight loss.
Long‐standing cramping is nonspecific and may reflect transient electrolyte derangements or muscle overuse. However, the more recent change in frequency, duration, and quality of these sensations, along with the reported weakness, raises concern for a process involving the peripheral nervous system. It will be important to differentiate cramping from other abnormal movements such as fasciculations, tremor, or myoclonus, and to determine whether there is objective weakness on the neurological examination.
His past medical history was significant for coronary artery disease with an ST‐segment elevation myocardial infarction several years prior, which was treated with a drug‐eluting stent. He was also diagnosed with essential thrombocythemia at the time of his myocardial infarction and tested positive for the JAK2 mutation. He was treated for several years with hydroxyurea following his diagnosis of essential thrombocythemia. Hydroxyurea had been discontinued 6 months prior due to cytopenias. The remainder of his history was significant for hypertension, chronic kidney disease stage 3, and prediabetes.
Medications were clopidogrel, atorvastatin, metoprolol, lisinopril, and hydrochlorothiazide. He did not use tobacco nor consume alcohol or illicit drugs, and he drank caffeine only occasionally. He had no family history of neurologic disorders.
Apart from his use of statins, which often affect muscles (and less commonly the nerves), the past medical history provides minimal additional insights into the cause of his symptoms. If weakness is detected on physical exam, the next step would be to distinguish upper (central) from a lower motor neuron (peripheral) localization. A diffuse problem involving all 4 limbs is generally more likely to arise from a disorder of a lower motor neuron (LMN) structure (anterior horn cell, nerve, neuromuscular junction, or muscle). To explain bilateral symptoms of the upper and lower limbs, an upper motor neuron (UMN) disease would have to affect the bilateral brain or cervical cord, a somewhat less likely possibility given the cramps described. It would also be quite unusual to have weakness of central nervous system origin without sensory deficits.
On physical examination, the patient was well‐appearing and in no apparent distress. Temperature was 98.1, blood pressure 134/84, pulse 110 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation was 100% while breathing ambient air. There was no lymphadenopathy. Lung, heart, abdominal, and skin exams were unremarkable. He was alert and oriented. His speech was without dysarthria. Examinations of the cranial nerves were intact. No tongue atrophy or fasciculations were noted. No pooling of secretions was appreciated in the oropharynx. Examination of the musculature revealed normal tone, strength, and bulk. However, there were diffuse fasciculations present, most prominent in the bilateral biceps, pectorals, deltoids, forearms, upper back, and calves. Sensation to light touch, temperature, and vibration were intact. Babinski's sign was absent, and deep tendon reflexes were normal, except at the ankles where they were reduced. Coordination and gait were normal.
The exam is notable for diffuse fasciculations, defined as spontaneous local involuntary muscle contraction and relaxation, which is often visible. Benign fasciculations are extremely common, with up to 70% of otherwise healthy adults experiencing them, and may be brought on by physical exertion. Men experience these benign fasciculations more frequently than women, and they can occur at any age and persist throughout life. Fasciculations may point to LMN disease, usually localizing to the anterior horn cell (for instance in amyotrophic lateral sclerosis [ALS]), muscle, or nerve disorders (including diffuse polyneuropathy). The presence of fasciculations in patients without other complaints and an otherwise normal physical examination supports benign fasciculations. The presence of neurologic deficits, however, such as weakness or reflex loss, is worrisome for another etiology. The absence of sensory changes makes anterior horn cell disease or myopathy most likely, as pure motor neuropathies are uncommon.
The fasciculations in this patient are most prominent in the proximal muscles, which may indicate a primary muscle disorder. Myopathies are typically characterized by diffuse symmetric weakness that is more proximal than distal, with no changes in sensation or deep tendon reflexes. One muscle disease characterized by fasciculations and cramping is periodic paralysis, which is often associated with potassium abnormalities or thyroid dysfunction caused by specific channelopathies. However, patients with this disorder typically present with episodic crises in contrast to the constant symptoms in this case.
Given the accelerated tempo of this patient's symptoms, further diagnostic evaluation should include basic laboratory testing including electrolytes, creatinine kinase, and thyrotropin. If these initial tests fail to reveal an etiology, the next study of choice would be an electromyography (EMG) and nerve conduction study (NCS), which can definitively localize the disorder to and within the peripheral nervous system. Unlike in UMN disease, in which neuroimaging is crucial, imaging is unlikely to be informative in patients with an LMN disorder.
Results of a complete blood count demonstrated a platelet count of 590,000/L (normal 140400 K/L), but was otherwise normal. Sodium, potassium, magnesium, and calcium levels were normal as were alanine aminotransferase, thyrotropin, and urinalysis. The hemoglobin A1c was 6.4%. Serum creatinine kinase was 157 U/L (normal170 U/L), and the serum creatinine was 1.7mg/dL, similar to previous results. The erythrocyte sedimentation rate was 58mm/h (normal 020mm/h). Magnetic resonance imaging (MRI) (Figure 1) of the cervical spine demonstrated diffuse disc desiccation, and multilevel spondylosis most prominent at C4C5 and C5C6, with severe central canal stenosis and neural foraminal narrowing.
The routine laboratory tests do not point to an obvious cause of this man's symptoms. As expected, the MRI findings do not explain diffuse fascinations in all limbs with no sensory disturbance. Further evaluation should include EMG and NCS.
NCS of the median and ulnar nerves demonstrated minimally reduced conduction velocities and markedly prolonged latencies. Sensory responses were absent. No conduction block was detected. EMG demonstrated fasciculations of the right extensor digitorum and first dorsal interossei, as well as decreased amplitude and decreased recruitment of motor units.
Abnormalities on the EMG testing can reflect either a primary muscle disorder or muscle derangement resulting from disease of the nerves. NCSs are important to differentiate these 2 possibilities. This patient's NCS indicates that the primary process is a diffuse motor and sensory neuropathy, not myopathy. The lack of sensory findings on physical examination emphasizes the ability of electrodiagnostic testing to extend the clinical neurologic examination in some cases. Markedly prolonged latencies and more modest reduction in amplitude support a motor and sensory neuropathy mostly due to demyelination, rather than axonal loss, and that it is more severe in the lower limbs.
Among the demyelinating neuropathies, acute inflammatory demyelinating neuropathy (Guillain‐Barr syndrome), is the most commonly recognized. The prolonged time course of this patient's illness excludes this possibility. Chronic inflammatory demyelinating polyneuropathy is also very unlikely in the absence of conduction block on NCS. Demyelinating neuropathies may also result from antibody‐mediated nerve injury. The serum paraprotein most commonly involved is immunoglobulin M (IgM), as is detected in neuropathy due to antibodies to myelin‐associated glycoprotein (anti‐MAG) neuropathy. Another variant is the ganglioside monosialic acid antibody (anti‐GM1) associated with a rare disease called multifocal motor neuropathy (MMN), an important condition to recognize because symptoms of this illness may mimic the presentation of ALS, often with fasciculations and weakness. Unlike ALS, MMN is very responsive to treatment. Other antibody‐mediated neuropathies are much rarer. In this patient, MMN is unlikely because sensory nerves are affected in addition to motor nerves.
Because NCSs also indicate some axonal loss, it would be reasonable to screen for vitamin deficiencies, human immunodeficiency virus, and viral hepatitis. The pattern here is more symmetric and confluent than would be expected if he had mononeuritic multiplex from vasculitis.
Vitamin E level was normal. Vitamin B12 level was 323 pg/mL (normal>200 pg/mL), methylmalonate 0.3mol/L (normal 00.3mol/L). Antibodies to human immunodeficiency virus and surface antibody and antigen to hepatitis B were not detected. Cryoglobulins, anti‐nuclear antibody, and antibodies to myeloperoxidase and proteinase 3 were not detected. Serum antibodies to tissue transglutaminase and Borrelia burgdorferi were not detectable. Serum protein electrophoresis demonstrated 2 small spikes in the gamma region. Quantitative serum immunoglobulin levels were normal except for IgM, which was elevated at 1.7g/dL (normal0.19g/dL). Serum free light chains showed a kappa component of 43.7mg/L (normal 319mg/L), a lambda component of 13.8mg/L (normal 526mg/L), and a kappa/lambda ratio of 3.17mg/L (normal 0.261.65mg/L).
The differential for a symmetric demyelinating neuropathy is quite narrow, and tests for vasculitis, celiac disease, and Lyme disease are not necessary. To pursue the cause of the elevated IgM, specific serum testing should be obtained for anti‐MAG antibodies. Many cases of anti‐MAG neuropathy are associated with an underlying lymphoproliferative disorder. As such, additional imaging to identify occult lymphoma is warranted.
Anti‐GM1 and asialoganglioside were not detectable. The anti‐MAG IgM titer was >102,400 (normal1:1600). Abdominal ultrasound showed normal sized kidneys with normal cortical echogenicity and no splenomegaly. Computed tomography with contrast was not performed due to chronic kidney disease.
Treatment for anti‐MAG neuropathy is evolving rapidly as our understanding of the entity improves. Cyclophosphamide, intravenous immune globulin, and plasmapheresis have been the traditional treatments, but in the past decade, favorable experiences with rituximab have led some to try this medication earlier in the course. Prognosis can be favorable in many patients.
Over the next 2 months he continued to have fasciculations. He developed progressive generalized weakness, an unsteady gait, required a walker for mobility, and began to have trouble with his activities of daily living. His cognition remained intact. There was no pooling of secretions. Serial neurologic examinations demonstrated persistent fasciculations, progressive atrophy, most notably in the intrinsic hand muscles and legs, and progressive weakness of all limbs, worse in the distal muscle groups. Deep tendon reflexes remained preserved, except at the ankles where they were absent. Sensory exam showed stocking diminution to temperature up to his knees and elbows. Romberg sign was present, and he could not walk without support. He was started on rituximab, and after 4 weeks his condition continued to deteriorate.
The response to rituximab may be delayed. Alternatively, his disease may have an underlying cause such as occult lymphoma not yet identified, which would require treatment to control the neuropathy. Because of the potential association between lymphoma in some patients with anti‐MAG neuropathy, and because he is not responding to immunotherapy, whole body imaging with positron emission tomography and bone marrow biopsy should be performed.
CD19 levels indicated an appropriate B cell response to rituximab, but the anti‐MAG titer remained elevated at >102,400. He received additional doses of rituximab, but continued to decline. A bone marrow biopsy was considered, but the patient opted to forgo the procedure. After several months of rituximab, he developed mild dysarthria and dysphagia and was hospitalized for plasma exchange. After 5 sessions of plasma exchange, he showed no improvement and was discharged to a rehabilitation facility. Over the ensuing months, he became restricted to wheel chair or bed and eventually opted for comfort measures. He died after an aspiration pneumonia 15 months after his initial visit to his physician. Permission for an autopsy was not granted.
COMMENTARY
When encountering patients with involuntary muscle movements, hospitalists must recognize potential serious underlying disorders and implement a cost‐effective evaluation strategy. Fasciculations are a common finding that represent involuntary discharges of a motor unit, with a wide array of causes including radiculopathies, neuropathies, metabolic disturbances, and motor neuron diseases.[1, 2] Useful clues might point to a probable cause, such as a statin‐induced myopathy in patients with concomitant myalgias, or hypokalemia in patients on loop diuretics. Confinement of fasciculations to specific anatomic structures may be useful, as in carpal tunnel syndrome, where fasciculations would only be expected distal to median nerve compression. Features such as sensory loss, muscle atrophy, or abnormal reflexes should alert the clinician to a possible neurologic lesion.
Although fasciculations rarely reflect serious underlying pathology, the presence of neurologic deficits, such as muscle weakness, abnormal reflexes, or sensory loss, should prompt further investigation.[3] Because fasciculations typically point to an abnormality of LMN structures, a reasonable approach is to measure serum electrolytes, creatinine kinase, and thyrotropin to evaluate for myopathy. If these tests are unrevealing, the next step would be to perform EMG and NCS to help localize the lesion among the LMN structures. Muscle localization could then be pursued with muscle biopsy. Alternatively, when electrodiagnostic testing indicates peripheral nerve pathology, further evaluation is guided by the type of neuropathy: demyelinating, axonal, or mixed. If electrodiagnostic and clinical findings are unrevealing, the patient is diagnosed with benign fasciculations.
Demyelinating neuropathy, as seen in our patient, is relevant to hospitalists for several reasons. First, the list of diagnostic possibilities is narrow, allowing hospitalists to forgo many unnecessary laboratory tests and brain MRI. Second, unlike many axonal neuropathies, demyelinating neuropathies are potentially reversible if recognized early and promptly treated. Third, demyelinating neuropathy may involve the diaphragm, necessitating vigilance for neuromuscular respiratory failure. Finally, hospitalists need to be aware that some demyelinating neuropathies are associated with underlying malignancy, and identifying and treating the primary cancer may be critical to ameliorating the neuropathy.[4, 5, 6, 7, 8]
IgM paraproteinemia, with or without an underlying malignancy, is 1 type of demyelinating neuropathy that is potentially reversible with early treatment. The typical patient is exemplified by the case presented in this report: an older man who experiences symmetric, gradually worsening sensory disturbances and ataxia over months to years.[9] Motor deficits may progress more rapidly, prompting patients to seek hospital care.[7, 10] The hallmark of NCS in anti‐MAG disease is a demyelinating pattern with a predominance of distal abnormalities including marked prolongation of distal motor latencies and reductions in conduction velocities and sensory action potentials.[9] Findings of areflexia or conduction block should prompt consideration of other etiologies, such as acute or chronic inflammatory demyelinating polyneuropathy.
For unclear reasons, IgM is more likely than other immunoglobulins to cause neuropathy. Although IgM accounts for only 17% of monoclonal gammopathies, IgM is detected in 50% to 70% of patients who have both monoclonal gammopathy and peripheral neuropathy.[11] Approximately half of the patients with IgM‐associated neuropathy produce antibodies to MAG.[11, 12] Several lines of evidence have firmly established the causative role of anti‐MAG antibodies.[13]
Because the majority of patients with anti‐MAG neuropathy will have no malignant source of IgM paraprotein identified, it is unclear how extensively to search for occult malignancy. A reasonable approach is to perform a bone marrow biopsy to distinguish underlying IgM monoclonal gammopathy of undetermined significance from Waldenstrom's macroglobulinemia.[14] Bone marrow analysis may also detect B‐cell lymphoma, primary amyloidosis, chronic lymphocytic leukemia, and hairy cell leukemia, which have been described in cases of anti‐MAG syndrome.[4, 5, 6, 7, 8] There are no reports of anti‐MAG neuropathy linked to either essential thrombocythemia nor hydroxyurea use.
The goals of treatment in anti‐MAG neuropathy are to deplete monoclonal B cells and to reduce antibody levels. Although it is reported that approximately half of patients will improve with some form of immunotherapy, a Cochrane review of randomized controlled trials of treatments for anti‐MAG neuropathy (including plasma exchange, intravenous immunoglobulin [IVIG], rituximab, corticosteroids, and chemotherapy) concluded that evidence is lacking to recommend 1 treatment over another.[15] European guidelines suggest deferring therapy unless progressive or severe neuropathy is present, in which case IVIG, plasma exchange, or rituximab may be tried.[14] In patients with underlying malignancy, treatment of the hematologic disorder may improve the neuropathy.[4, 8]
Although fasciculations and peripheral neuropathy typically present in outpatient settings, they can be harbingers of more dire diagnoses that prompt patients to seek hospitalization. A sequential and cost‐effective approach can allow the astute hospitalist to pinpoint the diagnosis in what might otherwise be an unnerving case.
KEY TEACHING POINTS
- Fasciculations are extremely common and usually benign, but may indicate a more serious neurologic process, especially when accompanied by weakness or other neurologic symptoms.
- Localizing neurologic deficits to upper motor neuron or lower motor neuron structures guides further evaluation.
- Central nervous system imaging is not indicated in demyelinating neuropathies.
- Bone marrow biopsy and cross‐sectional imaging to evaluate for malignancy should be considered in patients with anti‐MAG neuropathy who fail to improve despite therapy.
- , . Fasciculations: what do we know of their significance? J Neurol Sci. 1997;152(suppl 1):S43–S48.
- , . Muscle fasciculations in a healthy population. Arch Neurol. 1963;9:363–367.
- . Muscle pain, fatigue, and fasciculations. Neurol Clin. 1997;15(3):697–709.
- , , , et al. Monocytoid B cell lymphoma associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Acta Haematol. 2001;106(3):130–132.
- . Anti‐myelin‐associated glycoprotein peripheral neuropathy as the only presentation of low grade lymphoma: a case report. Cases J. 2009;2(1):6370–6373.
- , , , et al. Antibodies to myelin‐associated glycoprotein (anti‐Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve. 2008;37(4):490–495.
- , , , et al. Heterogeneity of polyneuropathy associated with anti‐MAG antibodies. J Immunol Res. 2015;2015(3):450391–450399.
- , , , et al. Hairy cell leukaemia complicated by anti‐MAG paraproteinemic demyelinating neuropathy: resolution of neurological syndrome after cladribrine treatment. Leuk Res. 2007;31(6):873–876.
- , , , , , . Neuropathy associated with “benign” anti‐myelin‐associated glycoprotein IgM gammopathy: clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J Neurol. 1996;243(1):34–43.
- , , , et al. IgM MGUS anti‐MAG neuropathy with predominant muscle weakness and extensive muscle atrophy. Muscle Nerve. 2010;42(3):433–435.
- , , , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- , . Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26(2):55–65.
- , . Effector mechanisms in anti‐MAG antibody‐mediated and other demyelinating neuropathies. J Neurol Sci. 2004;220(1–2):127–129.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15(3):185–195.
- , . Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.
- , . Fasciculations: what do we know of their significance? J Neurol Sci. 1997;152(suppl 1):S43–S48.
- , . Muscle fasciculations in a healthy population. Arch Neurol. 1963;9:363–367.
- . Muscle pain, fatigue, and fasciculations. Neurol Clin. 1997;15(3):697–709.
- , , , et al. Monocytoid B cell lymphoma associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Acta Haematol. 2001;106(3):130–132.
- . Anti‐myelin‐associated glycoprotein peripheral neuropathy as the only presentation of low grade lymphoma: a case report. Cases J. 2009;2(1):6370–6373.
- , , , et al. Antibodies to myelin‐associated glycoprotein (anti‐Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve. 2008;37(4):490–495.
- , , , et al. Heterogeneity of polyneuropathy associated with anti‐MAG antibodies. J Immunol Res. 2015;2015(3):450391–450399.
- , , , et al. Hairy cell leukaemia complicated by anti‐MAG paraproteinemic demyelinating neuropathy: resolution of neurological syndrome after cladribrine treatment. Leuk Res. 2007;31(6):873–876.
- , , , , , . Neuropathy associated with “benign” anti‐myelin‐associated glycoprotein IgM gammopathy: clinical, immunological, neurophysiological pathological findings and response to treatment in 33 cases. J Neurol. 1996;243(1):34–43.
- , , , et al. IgM MGUS anti‐MAG neuropathy with predominant muscle weakness and extensive muscle atrophy. Muscle Nerve. 2010;42(3):433–435.
- , , , et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- , . Review of peripheral neuropathy in plasma cell disorders. Hematol Oncol. 2008;26(2):55–65.
- , . Effector mechanisms in anti‐MAG antibody‐mediated and other demyelinating neuropathies. J Neurol Sci. 2004;220(1–2):127–129.
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15(3):185–195.
- , . Immunotherapy for IgM anti‐myelin‐associated glycoprotein paraprotein‐associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;5:CD002827.
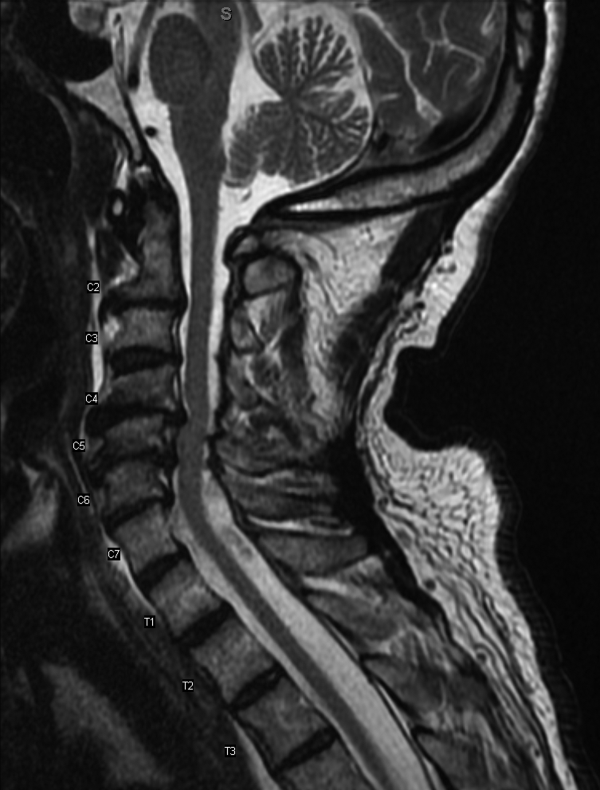
Common neurologic emergencies for nonneurologists: When minutes count
Neurologic emergencies such as acute stroke, status epilepticus, subarachnoid hemorrhage, neuromuscular weakness, and spinal cord injury affect millions of Americans yearly.1,2 These conditions can be difficult to diagnose, and delays in recognition and treatment can have devastating results. Consequently, it is important for nonneurologists to be able to quickly recognize these conditions and initiate timely management, often while awaiting neurologic consultation.
Here, we review how to recognize and treat these common, serious conditions.
ACUTE ISCHEMIC STROKE: TIME IS OF THE ESSENCE
Stroke is the fourth leading cause of death in the United States and is one of the most common causes of disability worldwide.3–5 About 85% of strokes are ischemic, resulting from diminished vascular supply to the brain. Symptoms such as facial droop, unilateral weakness or numbness, aphasia, gaze deviation, and unsteadiness of gait may be seen. Time is of the essence, as all currently available interventions are safe and effective only within defined time windows.
Diagnosis and assessment
When acute ischemic stroke is suspected, the clinical history, time of onset, and basic neurologic examination should be obtained quickly.
The National Institutes of Health (NIH) stroke scale is an objective marker for assessing stroke severity as well as evolution of disease and should be obtained in all stroke patients. Scores range from 0 (best) to 42 (worst) (www.ninds.nih.gov/doctors/NIH_Stroke_Scale.pdf).
Time of onset of symptoms is essential to determine, since it guides eligibility for acute therapies. Clinicians should ascertain the last time the patient was seen to be neurologically well in order to estimate this time window as closely as possible.
Laboratory tests should include a fingerstick blood glucose measurement, coagulation studies, complete blood cell count, and basic metabolic profile.
Computed tomography (CT) of the head without contrast should be obtained immediately to exclude acute hemorrhage and any alternative diagnoses that could explain the patient’s symptoms. Acute brain ischemia is often not apparent on CT during the first few hours of injury. Therefore, a patient presenting with new focal neurologic deficits and an unremarkable result on CT of the head should be treated as having had an acute ischemic stroke, and interventional therapies should be considered.

Stroke mimics should be considered and treated, as appropriate (Table 1).
Acute management of ischemic stroke
Acute treatment should not be delayed by obtaining chest radiography, inserting a Foley catheter, or obtaining an electrocardiogram. The longer the time that elapses before treatment, the worse the functional outcome, underscoring the need for rapid decision-making.6–8
Lowering the head of the bed may provide benefit by promoting blood flow to ischemic brain tissue.9 However, this should not be done in patients with significantly elevated intracerebral pressure and concern for herniation.
Permissive hypertension (antihypertensive treatment only for blood pressure greater than 220/110 mm Hg) should be allowed per national guidelines to provide adequate perfusion to brain areas at risk of injury.10
Tissue plasminogen activator. Patients with ischemic stroke who present within 3 hours of symptom onset should be considered for intravenous administration of tissue plasminogen activator (tPA), a safe and effective therapy with nearly 2 decades of evidence to support its use.10 The treating physician should carefully review the risks and benefits of this therapy.
To receive tPA, the patient must have all of the following:
- Clinical diagnosis of ischemic stroke with measurable neurologic deficit
- Onset of symptoms within the past 3 hours
- Age 18 or older.
The patient must not have any of the following:
- Significant stroke within the past 3 months
- Severe traumatic head injury within the past 3 months
- History of significant intracerebral hemorrhage
- Previously ruptured arteriovenous malformation or intracranial aneurysm
- Central nervous system neoplasm
- Arterial puncture at a noncompressible site within the past 7 days
- Evidence of hemorrhage on CT of the head
- Evidence of ischemia in greater than 33% of the cerebral hemisphere on head CT
- History and symptoms strongly suggesting subarachnoid hemorrhage
- Persistent hypertension (systolic pressure ≥ 185 mm Hg or diastolic pressure ≥ 110 mm Hg)
- Evidence of acute significant bleeding (external or internal)
- Hypoglycemia—ie, serum glucose less than 50 mg/dL (< 2.8 mmol/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Significant coagulopathy (international normalized ratio > 1.7, prothrombin time > 15 seconds, or abnormally elevated activated partial thromboplastin time)
- Current use of a factor Xa inhibitor or direct thrombin inhibitor.
Relative contraindications:
- Minor or rapidly resolving symptoms
- Major surgery or trauma within the past 14 days
- Gastrointestinal or urinary tract bleeding within the past 21 days
- Myocardial infarction in the past 3 months
- Unruptured intracranial aneurysm
- Seizure occurring at stroke onset
- Pregnancy.
If these criteria are satisfied, tPA should be given at a dose of 0.9 mg/kg intravenously over 60 minutes. Ten percent of the dose should be given as an initial bolus, followed by a constant infusion of the remaining 90% over 1 hour.
If tPA is given, the blood pressure must be kept lower than 185/110 mm Hg to minimize the risk of symptomatic intracerebral hemorrhage.
A subset of patients may benefit from receiving intravenous tPA between 3 and 4.5 hours after the onset of stroke symptoms. These include patients who are no more than 80 years old, who have not recently used oral anticoagulants, who do not have severe neurologic injury (ie, do not have NIH Stroke Scale scores > 25), and who do not have diabetes mellitus or a history of ischemic stroke.11 Although many hospitals have such a protocol for tPA up to 4.5 hours after the onset of stroke symptoms, this time window is not currently approved by the US Food and Drug Administration.
Intra-arterial therapy. Based on recent trials, some patients may benefit further from intra-arterial thrombolysis or mechanical thrombectomy, both delivered during catheter-based cerebral angiography, independent of intravenous tPA administration.12,13 These patients should be evaluated on a case-by-case basis by a neurologist and neurointerventional team. Time windows for these treatments generally extend to 6 hours from stroke onset and perhaps even longer in some situations (eg, basilar artery occlusion).
An antiplatelet agent should be started quickly in all stroke patients who do not receive tPA. Patients who receive tPA can begin receiving an antiplatelet agent 24 hours afterward.
Unfractionated heparin. There is no evidence to support the use of unfractionated heparin in most cases of acute ischemic stroke.10
Glucose control (in the range of 140–180 mg/dL) and fever control remain essential elements of post-acute stroke care to provide additional protection to the damaged brain.
For ischemic stroke due to atrial fibrillation
In ischemic stroke due to atrial fibrillation, early anticoagulation should be considered, based on the CHA2DS2-VASC risk of ischemic stroke vs the HAS-BLED risk of hemorrhage (calculators available at www.mdcalc.com).
In general, anticoagulation may be withheld during the first 72 hours while further stroke workup and evaluation of extent of injury are carried out, as there is an increased risk of hemorrhagic transformation of the ischemic stroke. Often, anticoagulation is resumed at a full dose between 72 hours and 2 weeks of the ischemic stroke.
ACUTE HEMORRHAGIC STROKE: BLOOD PRESSURE, COAGULATION

Approximately 15% of strokes are caused by intracerebral hemorrhage, which can be detected with noncontrast head CT with a sensitivity of 98.6% within 6 hours of the onset of bleeding.14 A common underlying cause of intracerebral hemorrhage is chronic poorly controlled hypertension, causing rupture of damaged (or “lipohyalinized”) vessels with resultant blood extravasation into the brain parenchyma. Other causes are less common (Table 2).
Treatment of acute hemorrhagic stroke
Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy or anticoagulation, and sometimes intracranial pressure control. There is little role for surgery in most cases, based on findings of randomized trials.15
Blood pressure control. Many studies have investigated optimal blood pressure goals in acute intracerebral hemorrhage. Recent data suggest that early aggressive therapy, targeting a systolic blood pressure goal less than 140 mm Hg within the first hour, is safe and can lead to better functional outcomes than a more conservative blood-pressure-lowering target.16 Rapid-onset, short-acting antihypertensive agents in intravenous form, such as nicardipine and labetalol, are frequently used. Of note, this treatment strategy for hemorrhagic stroke is in direct contrast to the treatment of ischemic stroke, in which permissive hypertension (blood pressure goal < 220/110 mm Hg) is often pursued.
Reversal of any coagulation abnormalities should be done quickly in intracranial hemorrhage. Warfarin use has been shown to be a strong independent predictor of intracranial hemorrhage expansion, which increases the risk of death.17,18
Increasingly, agents other than vitamin K or fresh-frozen plasma are being used to rapidly reverse anticoagulation, including prothrombin complex concentrate (available in three- and four-factor preparations) and recombinant factor VIIa. While four-factor prothrombin complex concentrate and recombinant factor VIIa have been shown to be more efficacious than fresh-frozen plasma, there are limited data directly comparing these newer reversal agents against each other.19 The use of these medications is limited by availability and practitioner familiarity.20–22
Reversing anticoagulation due to target-specific oral anticoagulants. The acute management of intracranial hemorrhage in patients taking the new target-specific oral anticoagulants (eg, dabigatran, apixaban, rivaroxaban, edoxaban) remains challenging. Laboratory tests such as factor Xa levels are not readily available in many institutions and do not provide results in a timely fashion, and in the interim, acute hemorrhage and clinical deterioration may occur. Management strategies involve giving fresh-frozen plasma, prothrombin complex concentrate, and consideration of hemodialysis.23 Dabigatran reversal with idarucizumab has recently been shown to have efficacy.24
Vigilance for elevated intracranial pressure. Intracranial hemorrhage can occasionally cause elevated intracranial pressure, which should be treated rapidly. Any acute decline in mental status in a patient with intracranial hemorrhage requires emergency imaging to evaluate for expansion of hemorrhage.
SUBARACHNOID HEMORRHAGE
The sudden onset of a “thunderclap” headache (often described by patients as “the worst headache of my life”) suggests subarachnoid hemorrhage.
In contrast to intracranial hemorrhage, in subarachnoid hemorrhage blood collects mainly in the cerebral spinal fluid-containing spaces surrounding the brain, leading to a higher incidence of hydrocephalus from impaired drainage of cerebrospinal fluid. Nontraumatic subarachnoid hemorrhage is most often caused by rupture of an intracranial aneurysm, which can be a devastating event, with death rates approaching 50%.25
Diagnosis of subarachnoid hemorrhage
Noncontrast CT of the head is the main modality for diagnosing subarachnoid hemorrhage. Blood within the subarachnoid space is demonstrable in 92% of cases if CT is performed within the first 24 hours of hemorrhage, with an initial sensitivity of about 95% within the first 6 hours of onset.14,26,27 The longer CT is delayed, the lower the sensitivity.
Some studies suggest that a protocol of CT followed by CT angiography can safely exclude aneurysmal subarachnoid hemorrhage and obviate the need for lumbar puncture. However, further research is required to validate this approach.28
Lumbar puncture. If clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT is negative, lumbar puncture must be performed for cerebrospinal fluid analysis.29 Xanthochromia (a yellowish pigmentation of the cerebrospinal fluid due to the degeneration of blood products that occurs within 8 to 12 hours of bleeding) should raise the alarm for subarachnoid hemorrhage; this sign may be present up to 4 weeks after the bleeding event.30
If lumbar puncture is contraindicated, then aneurysmal subarachnoid hemorrhage has not been ruled out, and further neurologic consultation should be pursued.
Management of subarachnoid hemorrhage
Early management of blood pressure for a ruptured intracranial aneurysm follows strategies similar to those for intracranial hemorrhage. Further investigation is rapidly directed toward an underlying vascular malformation, with intracranial vessel imaging such as CT angiography, magnetic resonance angiography, or the gold standard test—catheter-based cerebral angiography.
Aneurysms are treated (or “secured”) either by surgical clipping or by endovascular coiling. Endovascular coiling is preferable in cases in which both can be safely attempted.31 If the facility lacks the resources to do these procedures, the patient should be referred to a nearby tertiary care center.
INTRACRANIAL HYPERTENSION: DANGER OF BRAIN HERNIATION
A number of conditions can cause an acute intracranial pressure elevation. The danger of brain herniation requires that therapies be implemented rapidly to prevent catastrophic neurologic injury. In many situations, nonneurologists are the first responders and therefore should be familiar with basic intracranial pressure management.
Initial symptoms of acute rise in intracranial pressure
As intracranial pressure rises, pressure is typically equally distributed throughout the cranial vault, leading to dysfunction of the ascending reticular activating system, which clinically manifests as the inability to stay alert despite varying degrees of noxious stimulation. Progressive cranial neuropathies (often starting with pupillary abnormalities) and coma are often seen in this setting as the upper brainstem begins to be compressed.
Initial assessment and treatment of elevated intracranial pressure
Noncontrast CT of the head is often obtained immediately when acutely elevated intracranial pressure is suspected. If clinical examination and radiographic findings are consistent with intracranial hypertension, prompt measures can be started at the bedside.
Elevate the head of the bed to 30 degrees to promote venous drainage and reduce intracranial pressure. (In contrast, most other hemodynamically unstable patients are placed flat or in the Trendelenburg position.)
Intubation should be done quickly in cases of airway compromise, and hyperventilation should be started with a goal Paco2 of 30 to 35 mm Hg. This hypocarbic strategy promotes cerebral vasoconstriction and a transient decrease in intracranial pressure.
Hyperosmolar therapy allows for transient intracranial volume decompression and is the mainstay of emergency medical treatment of intracranial hypertension. Mannitol is a hyperosmolar polysaccharide that promotes osmotic diuresis and removes excessive cerebral water. In the acute setting, it can be given as an intravenous bolus of 1 to 2 g/kg through a peripheral intravenous line, followed by a bolus every 4 to 6 hours. Hypotension can occur after diuresis, and renal function should be closely monitored since frequent mannitol use can promote acute tubular necrosis. In patients who are anuric, the medication is typically not used.
Hypertonic saline (typically 3% sodium chloride, though different concentrations are available) is an alternative that helps draw interstitial fluid into the intravascular space, decreasing cerebral edema and maintaining hemodynamic stability. Relative contraindications include congestive heart failure or renal failure leading to pulmonary edema from volume overload. Hypertonic saline can be given as a bolus or a constant infusion. Some institutions have rapid access to 23.4% saline, which can be given as a 30-mL bolus but typically requires a central venous catheter for rapid infusion.
Comatose patients with radiographic findings of hydrocephalus, epidural or subdural hematoma, or mass effect with midline shift warrant prompt neurosurgical consultation for further surgical measures of intracranial pressure control and monitoring.
The ‘blown’ pupil

The physician should be concerned about elevated intracranial pressure if a patient has mydriasis, ie, an abnormally dilated (“blown”) pupil, which is a worrisome sign in the setting of true intracranial hypertension. However, many different processes can cause mydriasis and should be kept in mind when evaluating this finding (Table 3).32 If radiographic findings do not suggest elevated intracranial pressure, further workup into these other processes should be pursued.
STATUS EPILEPTICUS: SEIZURE CONTROL IS IMPORTANT
A continuous unremitting seizure lasting longer than 5 minutes or recurrent seizure activity in a patient who does not regain consciousness between seizures should be treated as status epilepticus. All seizure types carry the risk of progressing to status epilepticus, and responsiveness to antiepileptic drug therapy is inversely related to the duration of seizures. It is imperative that seizure activity be treated early and aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.33

Once the ABCs of emergency stabilization have been performed (ie, airway, breathing, circulation), antiepileptic drug therapy should start immediately using established algorithms (Figure 1).34–36 During the course of treatment, the reliability of the neurologic examination may be limited due to medication effects or continued status epilepticus, making continuous video electroencephalographic monitoring often necessary to guide further therapy in patients who are not rapidly recovering.34–38
Once status epilepticus has resolved, further investigation into the underlying cause should be pursued quickly, especially in patients without a previous diagnosis of epilepsy. Head CT with contrast or magnetic resonance imaging can be used to look for any structural abnormality that may explain seizures. Basic laboratory tests including toxicology screening can identify a common trigger such as hypoglycemia or stimulant use. Fever or other possible signs of meningitis should be investigated further with cerebrospinal fluid analysis.
SPINAL CORD INJURY
Acute spinal cord injury can lead to substantial long-term neurologic impairment and should be suspected in any patient presenting with focal motor loss, sensory loss, or both with sparing of the cranial nerves and mental status. Causes of injury include compression (traumatic or nontraumatic) and inflammatory and noninflammatory myelopathies.
The location of the injury can be inferred by analyzing the symptoms, which can point to the cord level and indicate whether the anterior or posterior of the cord is involved. Anterior cord injury tends to affect the descending corticospinal and pyramidal tracts, resulting in motor deficits and weakness. Posterior cord injury involves the dorsal columns, leading to deficits of vibration sensation and proprioception. High cervical cord injuries tend to involve varying degrees of quadriparesis, sensory loss, and sometimes respiratory compromise. A clinical history of bilateral lower-extremity weakness, a “band-like” sensory complaint around the lower chest or abdomen, or both, can suggest thoracic cord involvement. Symptoms isolated to one or both lower extremities along with lower back pain and bowel or bladder involvement may point to injury of the lumbosacral cord.
Basic management of spinal cord injury includes decompression of the bladder and initial protection against further injury with a stabilizing collar or brace.
Magnetic resonance imaging with and without contrast is the ideal study to evaluate injuries to the spinal cord itself. While CT is helpful in identifying bony disease of the spinal column (eg, evaluating traumatic fractures), it is not helpful in viewing intrinsic cord pathology.
Traumatic myelopathy
Traumatic spinal cord injury is usually suggested by the clinical history and confirmed with CT. In this setting, early consultation with a neurosurgeon is required to prevent permanent cord injury.
Guidelines suggest maintaining a mean arterial pressure greater than 85 to 90 mm Hg for the first 7 days after traumatic spinal cord injury, a particular problem in the setting of hemodynamic instability, which can accompany lesions above the midthoracic level.39,40
Patients with vertebral body misalignment should be placed in an appropriate stabilizing collar or brace until a medically trained professional deems it appropriate to discontinue the device, or until surgical stabilization is performed.
Methylprednisone is a controversial intervention for acute spinal cord trauma, lacking clear benefit in meta-analyses.41
Nontraumatic compressive myelopathy
Patients with nontraumatic compressive myelopathy tend to present with varying degrees of back pain and worsening sensorimotor function. The differential diagnosis includes epidural abscesses, hematoma, metastatic neoplasm, and osteophyte compression (Table 4). The clinical history helps to guide therapy and should involve assessment for previous spinal column injury, immunocompromised state, travel history (which provides information on risks of exposure to a variety of diseases, including infections), and constitutional symptoms such as fever and weight loss.
Epidural abscess can have devastating results if missed. Red flags such as recent illness, intravenous drug use, focal back pain, fever, worsening numbness or weakness, and bowel or bladder incontinence should raise suspicion of this disorder. Emergency magnetic resonance imaging is required to diagnose this condition, and treatment involves urgent administration of antibiotics and consideration of surgical drainage.
Noncompressive myelopathies
There are numerous causes of noncompressive spinal cord injury (Table 4), and the etiology may be inflammatory (eg, “myelitis”) or noninflammatory. The diagnostic workup may require both magnetic resonance imaging and cerebrospinal fluid analysis. Acute disease-targeted therapy is rarely indicated and can be deferred until a full diagnostic workup has been completed.
NEUROMUSCULAR DISEASE: IS VENTILATION NEEDED?

Diseases involving the motor components of the peripheral nervous system (Table 5) share the common risk of causing ventilatory failure due to weakness of the diaphragm, intercostal muscles, and upper-airway muscles. Clinicians need to be aware of this risk and view these disorders as neurologic emergencies.
Determining when these patients require mechanical intubation is a challenge. Serial measurements of maximum inspiratory force and vital capacity are important and can be accomplished quickly at the bedside by a respiratory therapist. A maximum inspiratory force less than –30 cm H2O or a vital capacity less than 20 mL/kg, or both, are worrisome markers that raise concern for impending ventilatory failure. Serial measurements can detect changes in these values that might indicate the need for elective intubation. In any patient presenting with weakness of the limbs, these measurements are an important step in the initial evaluation.
Myasthenic crisis
Myasthenia gravis is caused by autoantibodies directed against postsynaptic acetylcholine receptors. Patients demonstrate muscle weakness, usually in a proximal pattern, with fatigue, respiratory distress, nasal speech, ophthalmoparesis, and dysphagia. Exacerbations can occur as a response to recent infection, surgery, or medications such as neuromuscular blocking agents or aminoglycosides.
Myasthenic crisis, while uncommon, is a life-threatening emergency characterized by bulbar or respiratory failure secondary to muscle weakness. It can occur in patients already diagnosed with myasthenia gravis or may be the initial manifestation of the disease.42–49 Intubation and mechanical ventilation are frequently required. Postoperative myasthenic patients in whom extubation has been delayed more than 24 hours should be considered in crisis.45
The diagnosis of myasthenia gravis can be made by serum autoantibody testing, electromyography, and nerve conduction studies (with repetitive stimulation) or administration of edrophonium in patients with obvious ptosis.
The mainstay of therapy for myasthenic crisis is either intravenous immunoglobulin at a dose of 2 g/kg over 2 to 5 days or plasmapheresis (5–7 exchanges over 7–14 days). Corticosteroids are not recommended in myasthenic crisis in patients who are not intubated, as they can potentiate an initial worsening of crisis. Once the patient begins to show clinical improvement, outpatient pyridostigmine and immunosuppressive medications can be resumed at a low dose and titrated as tolerated.
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome)
Acute inflammatory demyelinating polyneuropathy is an autoimmune disorder involving autoantibodies against axons or myelin in the peripheral nervous system.
This disease should be suspected in a patient who is developing worsening muscle weakness (usually with areflexia) over the course of days to weeks. Occasionally, a recent diarrheal or other systemic infectious trigger can be identified. Blood pressure instability and cardiac arrhythmia can also be seen due to autonomic nerve involvement. Although classically described as an “ascending paralysis,” other variants of this disease have distinct clinical presentations (eg, the descending paralysis, ataxia, areflexia, ophthalmoparesis of the Miller Fisher syndrome).
Acute inflammatory demyelinating polyneuropathy is diagnosed by electromyography and nerve conduction studies. A cerebrospinal fluid profile demonstrating elevated protein and few white blood cells is typical.
Treatment, as in myasthenic crisis, involves intravenous immunoglobulin or plasmapheresis. Corticosteroids are ineffective. Anticipation of ventilatory failure and expectant intubation is essential, given the progressive nature of the disorder.50
- Pitts SR, Niska RW, Xu J, Burt CW. National hospital ambulatory medical care survey: 2006 emergency department summary. Natl Health Stat Report 2008; 7:1–38.
- McMullan JT, Knight WA, Clark JF, Beyette FR, Pancioli A. Time-critical neurological emergencies: the unfulfilled role for point-of-care testing. Int J Emerg Med 2010; 3:127–131.
- Centers for Disease Control and Prevention (CDC). Prevalence of stroke: United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2012; 61:379–382.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2095–2128.
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1,160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2163–2196.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 1995; 333:1581–1587.
- Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363:768–774.
- Saver JL, Fonarrow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013; 309:2480–2488.
- Wojner-Alexander AW, Garami Z, Chernyshev OY, Alexandrov AV. Heads down: flat positioning improves blood flow velocity in acute ischemic stroke. Neurology 2005; 64:1354–1357.
- Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Hacke W, Kaste M, Bluhmki E, et al; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1317–1329.
- Berkhemer OA, Fransen PSS, Beumer D, et al; MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Eng J Med 2015; 372:11–20.
- Campbell BC, Mitchell PJ, Kleinig TJ, et al; EXTEND-IA Investigators. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med 2015; 372:1009–1018.
- Backes D, Rinkel GJ, Kemperman H, Linn FH, Vergouwen MD. Time-dependent test characteristics of head computed tomography in patients suspected of nontraumatic subarachnoid hemorrhage. Stroke 2012; 43:2115–2119.
- Mendelow AD, Gregson BA, Fernandes HM, et al; STICH investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 2005; 365: 387–397.
- Anderson CS, Helley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Flibotte JJ, Hagan N, O'Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 2004; 63:1059–1064.
- Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006; 66:1175–1181.
- Woo CH, Patel N, Conell C, et al. Rapid warfarin reversal in the setting of intracranial hemorrhage: a comparison of plasma, recombinant activated factor VII, and prothrombin complex concentrate. World Neurosurg 2014; 81:110–115.
- Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007; 38:2001–2023.
- Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke 2006, 37:151–155.
- Chapman SA, Irwin ED, Beal AL, Kulinski NM, Hutson KE, Thorson MA. Prothrombin complex concentrate versus standard therapies for INR reversal in trauma patients receiving warfarin. Ann Pharmacother 2011; 45:869–875.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373:511-520.
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25:1342–1347.
- Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 1990; 73:18–36.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Vermuelen M, Hasan D, Blijenberg BG, Hijdra A, van Gijn J. Xanthochromia after subarachnoid haemorrhage needs no revisitation. J Neurol Neurosurg Psychiatry 1989; 52:826–828.
- Molyneaux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid hemorrhage trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Caglayan HZ, Colpak IA, Kansu T. A diagnostic challenge: dilated pupil. Curr Opin Ophthalmol 2013; 24:550–557.
- Brophy GM, Bell R, Claassen J, et al; Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
- Chang CW, Bleck TP. Status epilepticus. Neurol Clin 1995; 13:529–548.
- Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia 1993; 34(suppl 1):S2–S11.
- Leppick IE. Status epilepticus: the next decade. Neurology 1990; 40(suppl 2):4–9.
- Aranda A, Foucart G, Ducassé JL, Grolleau S, McGonigal A, Valton L. Generalized convulsive status epilepticus management in adults: a cohort study with evaluation of professional practice. Epilepsia 2010; 51:2159–2167.
- DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39:833–840.
- Casha S, Christie S. A systematic review of intensive cardiopulmonary management after spinal cord injury. J Neurotrauma 2011; 28:1479–1495.
- Walters BC, Hadley MN, Hurlbert RJ, et al; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 2013; 60(suppl 1):82–91.
- Hurlbert RJ, Hadley MN, Walters BC, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery 2013; 72(suppl 2):93–105.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci 1981; 377:670–677.
- Belack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis 2002; 4:40–42.
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM 2009; 102:97–107.
- Mayer SA. Intensive care of the myasthenic patient. Neurology 1997; 48(suppl 5):70S–75S.
- Jani-Acsadi A, Lisak RP. Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 2007; 261:127–133.
- Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J 2008; 101:63–69.
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol 2005; 7:129–141.
- Godoy DA, Vaz de Mello LJ, Masotti L, Napoli MD. The myasthenic patient in crisis: an update of the management in neurointensive care unit. Arq Neuropsiquiatr 2013; 71:627–639.
- Hughes RA, Wijdicks EF, Benson E, et al; Multidisciplinary Consensus Group. Supportive care for patients with Guillain-Barré syndrome: Arch Neurol 2005; 62:1194–1198.
Neurologic emergencies such as acute stroke, status epilepticus, subarachnoid hemorrhage, neuromuscular weakness, and spinal cord injury affect millions of Americans yearly.1,2 These conditions can be difficult to diagnose, and delays in recognition and treatment can have devastating results. Consequently, it is important for nonneurologists to be able to quickly recognize these conditions and initiate timely management, often while awaiting neurologic consultation.
Here, we review how to recognize and treat these common, serious conditions.
ACUTE ISCHEMIC STROKE: TIME IS OF THE ESSENCE
Stroke is the fourth leading cause of death in the United States and is one of the most common causes of disability worldwide.3–5 About 85% of strokes are ischemic, resulting from diminished vascular supply to the brain. Symptoms such as facial droop, unilateral weakness or numbness, aphasia, gaze deviation, and unsteadiness of gait may be seen. Time is of the essence, as all currently available interventions are safe and effective only within defined time windows.
Diagnosis and assessment
When acute ischemic stroke is suspected, the clinical history, time of onset, and basic neurologic examination should be obtained quickly.
The National Institutes of Health (NIH) stroke scale is an objective marker for assessing stroke severity as well as evolution of disease and should be obtained in all stroke patients. Scores range from 0 (best) to 42 (worst) (www.ninds.nih.gov/doctors/NIH_Stroke_Scale.pdf).
Time of onset of symptoms is essential to determine, since it guides eligibility for acute therapies. Clinicians should ascertain the last time the patient was seen to be neurologically well in order to estimate this time window as closely as possible.
Laboratory tests should include a fingerstick blood glucose measurement, coagulation studies, complete blood cell count, and basic metabolic profile.
Computed tomography (CT) of the head without contrast should be obtained immediately to exclude acute hemorrhage and any alternative diagnoses that could explain the patient’s symptoms. Acute brain ischemia is often not apparent on CT during the first few hours of injury. Therefore, a patient presenting with new focal neurologic deficits and an unremarkable result on CT of the head should be treated as having had an acute ischemic stroke, and interventional therapies should be considered.
Stroke mimics should be considered and treated, as appropriate (Table 1).
Acute management of ischemic stroke
Acute treatment should not be delayed by obtaining chest radiography, inserting a Foley catheter, or obtaining an electrocardiogram. The longer the time that elapses before treatment, the worse the functional outcome, underscoring the need for rapid decision-making.6–8
Lowering the head of the bed may provide benefit by promoting blood flow to ischemic brain tissue.9 However, this should not be done in patients with significantly elevated intracerebral pressure and concern for herniation.
Permissive hypertension (antihypertensive treatment only for blood pressure greater than 220/110 mm Hg) should be allowed per national guidelines to provide adequate perfusion to brain areas at risk of injury.10
Tissue plasminogen activator. Patients with ischemic stroke who present within 3 hours of symptom onset should be considered for intravenous administration of tissue plasminogen activator (tPA), a safe and effective therapy with nearly 2 decades of evidence to support its use.10 The treating physician should carefully review the risks and benefits of this therapy.
To receive tPA, the patient must have all of the following:
- Clinical diagnosis of ischemic stroke with measurable neurologic deficit
- Onset of symptoms within the past 3 hours
- Age 18 or older.
The patient must not have any of the following:
- Significant stroke within the past 3 months
- Severe traumatic head injury within the past 3 months
- History of significant intracerebral hemorrhage
- Previously ruptured arteriovenous malformation or intracranial aneurysm
- Central nervous system neoplasm
- Arterial puncture at a noncompressible site within the past 7 days
- Evidence of hemorrhage on CT of the head
- Evidence of ischemia in greater than 33% of the cerebral hemisphere on head CT
- History and symptoms strongly suggesting subarachnoid hemorrhage
- Persistent hypertension (systolic pressure ≥ 185 mm Hg or diastolic pressure ≥ 110 mm Hg)
- Evidence of acute significant bleeding (external or internal)
- Hypoglycemia—ie, serum glucose less than 50 mg/dL (< 2.8 mmol/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Significant coagulopathy (international normalized ratio > 1.7, prothrombin time > 15 seconds, or abnormally elevated activated partial thromboplastin time)
- Current use of a factor Xa inhibitor or direct thrombin inhibitor.
Relative contraindications:
- Minor or rapidly resolving symptoms
- Major surgery or trauma within the past 14 days
- Gastrointestinal or urinary tract bleeding within the past 21 days
- Myocardial infarction in the past 3 months
- Unruptured intracranial aneurysm
- Seizure occurring at stroke onset
- Pregnancy.
If these criteria are satisfied, tPA should be given at a dose of 0.9 mg/kg intravenously over 60 minutes. Ten percent of the dose should be given as an initial bolus, followed by a constant infusion of the remaining 90% over 1 hour.
If tPA is given, the blood pressure must be kept lower than 185/110 mm Hg to minimize the risk of symptomatic intracerebral hemorrhage.
A subset of patients may benefit from receiving intravenous tPA between 3 and 4.5 hours after the onset of stroke symptoms. These include patients who are no more than 80 years old, who have not recently used oral anticoagulants, who do not have severe neurologic injury (ie, do not have NIH Stroke Scale scores > 25), and who do not have diabetes mellitus or a history of ischemic stroke.11 Although many hospitals have such a protocol for tPA up to 4.5 hours after the onset of stroke symptoms, this time window is not currently approved by the US Food and Drug Administration.
Intra-arterial therapy. Based on recent trials, some patients may benefit further from intra-arterial thrombolysis or mechanical thrombectomy, both delivered during catheter-based cerebral angiography, independent of intravenous tPA administration.12,13 These patients should be evaluated on a case-by-case basis by a neurologist and neurointerventional team. Time windows for these treatments generally extend to 6 hours from stroke onset and perhaps even longer in some situations (eg, basilar artery occlusion).
An antiplatelet agent should be started quickly in all stroke patients who do not receive tPA. Patients who receive tPA can begin receiving an antiplatelet agent 24 hours afterward.
Unfractionated heparin. There is no evidence to support the use of unfractionated heparin in most cases of acute ischemic stroke.10
Glucose control (in the range of 140–180 mg/dL) and fever control remain essential elements of post-acute stroke care to provide additional protection to the damaged brain.
For ischemic stroke due to atrial fibrillation
In ischemic stroke due to atrial fibrillation, early anticoagulation should be considered, based on the CHA2DS2-VASC risk of ischemic stroke vs the HAS-BLED risk of hemorrhage (calculators available at www.mdcalc.com).
In general, anticoagulation may be withheld during the first 72 hours while further stroke workup and evaluation of extent of injury are carried out, as there is an increased risk of hemorrhagic transformation of the ischemic stroke. Often, anticoagulation is resumed at a full dose between 72 hours and 2 weeks of the ischemic stroke.
ACUTE HEMORRHAGIC STROKE: BLOOD PRESSURE, COAGULATION
Approximately 15% of strokes are caused by intracerebral hemorrhage, which can be detected with noncontrast head CT with a sensitivity of 98.6% within 6 hours of the onset of bleeding.14 A common underlying cause of intracerebral hemorrhage is chronic poorly controlled hypertension, causing rupture of damaged (or “lipohyalinized”) vessels with resultant blood extravasation into the brain parenchyma. Other causes are less common (Table 2).
Treatment of acute hemorrhagic stroke
Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy or anticoagulation, and sometimes intracranial pressure control. There is little role for surgery in most cases, based on findings of randomized trials.15
Blood pressure control. Many studies have investigated optimal blood pressure goals in acute intracerebral hemorrhage. Recent data suggest that early aggressive therapy, targeting a systolic blood pressure goal less than 140 mm Hg within the first hour, is safe and can lead to better functional outcomes than a more conservative blood-pressure-lowering target.16 Rapid-onset, short-acting antihypertensive agents in intravenous form, such as nicardipine and labetalol, are frequently used. Of note, this treatment strategy for hemorrhagic stroke is in direct contrast to the treatment of ischemic stroke, in which permissive hypertension (blood pressure goal < 220/110 mm Hg) is often pursued.
Reversal of any coagulation abnormalities should be done quickly in intracranial hemorrhage. Warfarin use has been shown to be a strong independent predictor of intracranial hemorrhage expansion, which increases the risk of death.17,18
Increasingly, agents other than vitamin K or fresh-frozen plasma are being used to rapidly reverse anticoagulation, including prothrombin complex concentrate (available in three- and four-factor preparations) and recombinant factor VIIa. While four-factor prothrombin complex concentrate and recombinant factor VIIa have been shown to be more efficacious than fresh-frozen plasma, there are limited data directly comparing these newer reversal agents against each other.19 The use of these medications is limited by availability and practitioner familiarity.20–22
Reversing anticoagulation due to target-specific oral anticoagulants. The acute management of intracranial hemorrhage in patients taking the new target-specific oral anticoagulants (eg, dabigatran, apixaban, rivaroxaban, edoxaban) remains challenging. Laboratory tests such as factor Xa levels are not readily available in many institutions and do not provide results in a timely fashion, and in the interim, acute hemorrhage and clinical deterioration may occur. Management strategies involve giving fresh-frozen plasma, prothrombin complex concentrate, and consideration of hemodialysis.23 Dabigatran reversal with idarucizumab has recently been shown to have efficacy.24
Vigilance for elevated intracranial pressure. Intracranial hemorrhage can occasionally cause elevated intracranial pressure, which should be treated rapidly. Any acute decline in mental status in a patient with intracranial hemorrhage requires emergency imaging to evaluate for expansion of hemorrhage.
SUBARACHNOID HEMORRHAGE
The sudden onset of a “thunderclap” headache (often described by patients as “the worst headache of my life”) suggests subarachnoid hemorrhage.
In contrast to intracranial hemorrhage, in subarachnoid hemorrhage blood collects mainly in the cerebral spinal fluid-containing spaces surrounding the brain, leading to a higher incidence of hydrocephalus from impaired drainage of cerebrospinal fluid. Nontraumatic subarachnoid hemorrhage is most often caused by rupture of an intracranial aneurysm, which can be a devastating event, with death rates approaching 50%.25
Diagnosis of subarachnoid hemorrhage
Noncontrast CT of the head is the main modality for diagnosing subarachnoid hemorrhage. Blood within the subarachnoid space is demonstrable in 92% of cases if CT is performed within the first 24 hours of hemorrhage, with an initial sensitivity of about 95% within the first 6 hours of onset.14,26,27 The longer CT is delayed, the lower the sensitivity.
Some studies suggest that a protocol of CT followed by CT angiography can safely exclude aneurysmal subarachnoid hemorrhage and obviate the need for lumbar puncture. However, further research is required to validate this approach.28
Lumbar puncture. If clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT is negative, lumbar puncture must be performed for cerebrospinal fluid analysis.29 Xanthochromia (a yellowish pigmentation of the cerebrospinal fluid due to the degeneration of blood products that occurs within 8 to 12 hours of bleeding) should raise the alarm for subarachnoid hemorrhage; this sign may be present up to 4 weeks after the bleeding event.30
If lumbar puncture is contraindicated, then aneurysmal subarachnoid hemorrhage has not been ruled out, and further neurologic consultation should be pursued.
Management of subarachnoid hemorrhage
Early management of blood pressure for a ruptured intracranial aneurysm follows strategies similar to those for intracranial hemorrhage. Further investigation is rapidly directed toward an underlying vascular malformation, with intracranial vessel imaging such as CT angiography, magnetic resonance angiography, or the gold standard test—catheter-based cerebral angiography.
Aneurysms are treated (or “secured”) either by surgical clipping or by endovascular coiling. Endovascular coiling is preferable in cases in which both can be safely attempted.31 If the facility lacks the resources to do these procedures, the patient should be referred to a nearby tertiary care center.
INTRACRANIAL HYPERTENSION: DANGER OF BRAIN HERNIATION
A number of conditions can cause an acute intracranial pressure elevation. The danger of brain herniation requires that therapies be implemented rapidly to prevent catastrophic neurologic injury. In many situations, nonneurologists are the first responders and therefore should be familiar with basic intracranial pressure management.
Initial symptoms of acute rise in intracranial pressure
As intracranial pressure rises, pressure is typically equally distributed throughout the cranial vault, leading to dysfunction of the ascending reticular activating system, which clinically manifests as the inability to stay alert despite varying degrees of noxious stimulation. Progressive cranial neuropathies (often starting with pupillary abnormalities) and coma are often seen in this setting as the upper brainstem begins to be compressed.
Initial assessment and treatment of elevated intracranial pressure
Noncontrast CT of the head is often obtained immediately when acutely elevated intracranial pressure is suspected. If clinical examination and radiographic findings are consistent with intracranial hypertension, prompt measures can be started at the bedside.
Elevate the head of the bed to 30 degrees to promote venous drainage and reduce intracranial pressure. (In contrast, most other hemodynamically unstable patients are placed flat or in the Trendelenburg position.)
Intubation should be done quickly in cases of airway compromise, and hyperventilation should be started with a goal Paco2 of 30 to 35 mm Hg. This hypocarbic strategy promotes cerebral vasoconstriction and a transient decrease in intracranial pressure.
Hyperosmolar therapy allows for transient intracranial volume decompression and is the mainstay of emergency medical treatment of intracranial hypertension. Mannitol is a hyperosmolar polysaccharide that promotes osmotic diuresis and removes excessive cerebral water. In the acute setting, it can be given as an intravenous bolus of 1 to 2 g/kg through a peripheral intravenous line, followed by a bolus every 4 to 6 hours. Hypotension can occur after diuresis, and renal function should be closely monitored since frequent mannitol use can promote acute tubular necrosis. In patients who are anuric, the medication is typically not used.
Hypertonic saline (typically 3% sodium chloride, though different concentrations are available) is an alternative that helps draw interstitial fluid into the intravascular space, decreasing cerebral edema and maintaining hemodynamic stability. Relative contraindications include congestive heart failure or renal failure leading to pulmonary edema from volume overload. Hypertonic saline can be given as a bolus or a constant infusion. Some institutions have rapid access to 23.4% saline, which can be given as a 30-mL bolus but typically requires a central venous catheter for rapid infusion.
Comatose patients with radiographic findings of hydrocephalus, epidural or subdural hematoma, or mass effect with midline shift warrant prompt neurosurgical consultation for further surgical measures of intracranial pressure control and monitoring.
The ‘blown’ pupil
The physician should be concerned about elevated intracranial pressure if a patient has mydriasis, ie, an abnormally dilated (“blown”) pupil, which is a worrisome sign in the setting of true intracranial hypertension. However, many different processes can cause mydriasis and should be kept in mind when evaluating this finding (Table 3).32 If radiographic findings do not suggest elevated intracranial pressure, further workup into these other processes should be pursued.
STATUS EPILEPTICUS: SEIZURE CONTROL IS IMPORTANT
A continuous unremitting seizure lasting longer than 5 minutes or recurrent seizure activity in a patient who does not regain consciousness between seizures should be treated as status epilepticus. All seizure types carry the risk of progressing to status epilepticus, and responsiveness to antiepileptic drug therapy is inversely related to the duration of seizures. It is imperative that seizure activity be treated early and aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.33
Once the ABCs of emergency stabilization have been performed (ie, airway, breathing, circulation), antiepileptic drug therapy should start immediately using established algorithms (Figure 1).34–36 During the course of treatment, the reliability of the neurologic examination may be limited due to medication effects or continued status epilepticus, making continuous video electroencephalographic monitoring often necessary to guide further therapy in patients who are not rapidly recovering.34–38
Once status epilepticus has resolved, further investigation into the underlying cause should be pursued quickly, especially in patients without a previous diagnosis of epilepsy. Head CT with contrast or magnetic resonance imaging can be used to look for any structural abnormality that may explain seizures. Basic laboratory tests including toxicology screening can identify a common trigger such as hypoglycemia or stimulant use. Fever or other possible signs of meningitis should be investigated further with cerebrospinal fluid analysis.
SPINAL CORD INJURY
Acute spinal cord injury can lead to substantial long-term neurologic impairment and should be suspected in any patient presenting with focal motor loss, sensory loss, or both with sparing of the cranial nerves and mental status. Causes of injury include compression (traumatic or nontraumatic) and inflammatory and noninflammatory myelopathies.
The location of the injury can be inferred by analyzing the symptoms, which can point to the cord level and indicate whether the anterior or posterior of the cord is involved. Anterior cord injury tends to affect the descending corticospinal and pyramidal tracts, resulting in motor deficits and weakness. Posterior cord injury involves the dorsal columns, leading to deficits of vibration sensation and proprioception. High cervical cord injuries tend to involve varying degrees of quadriparesis, sensory loss, and sometimes respiratory compromise. A clinical history of bilateral lower-extremity weakness, a “band-like” sensory complaint around the lower chest or abdomen, or both, can suggest thoracic cord involvement. Symptoms isolated to one or both lower extremities along with lower back pain and bowel or bladder involvement may point to injury of the lumbosacral cord.
Basic management of spinal cord injury includes decompression of the bladder and initial protection against further injury with a stabilizing collar or brace.
Magnetic resonance imaging with and without contrast is the ideal study to evaluate injuries to the spinal cord itself. While CT is helpful in identifying bony disease of the spinal column (eg, evaluating traumatic fractures), it is not helpful in viewing intrinsic cord pathology.
Traumatic myelopathy
Traumatic spinal cord injury is usually suggested by the clinical history and confirmed with CT. In this setting, early consultation with a neurosurgeon is required to prevent permanent cord injury.
Guidelines suggest maintaining a mean arterial pressure greater than 85 to 90 mm Hg for the first 7 days after traumatic spinal cord injury, a particular problem in the setting of hemodynamic instability, which can accompany lesions above the midthoracic level.39,40
Patients with vertebral body misalignment should be placed in an appropriate stabilizing collar or brace until a medically trained professional deems it appropriate to discontinue the device, or until surgical stabilization is performed.
Methylprednisone is a controversial intervention for acute spinal cord trauma, lacking clear benefit in meta-analyses.41
Nontraumatic compressive myelopathy
Patients with nontraumatic compressive myelopathy tend to present with varying degrees of back pain and worsening sensorimotor function. The differential diagnosis includes epidural abscesses, hematoma, metastatic neoplasm, and osteophyte compression (Table 4). The clinical history helps to guide therapy and should involve assessment for previous spinal column injury, immunocompromised state, travel history (which provides information on risks of exposure to a variety of diseases, including infections), and constitutional symptoms such as fever and weight loss.
Epidural abscess can have devastating results if missed. Red flags such as recent illness, intravenous drug use, focal back pain, fever, worsening numbness or weakness, and bowel or bladder incontinence should raise suspicion of this disorder. Emergency magnetic resonance imaging is required to diagnose this condition, and treatment involves urgent administration of antibiotics and consideration of surgical drainage.
Noncompressive myelopathies
There are numerous causes of noncompressive spinal cord injury (Table 4), and the etiology may be inflammatory (eg, “myelitis”) or noninflammatory. The diagnostic workup may require both magnetic resonance imaging and cerebrospinal fluid analysis. Acute disease-targeted therapy is rarely indicated and can be deferred until a full diagnostic workup has been completed.
NEUROMUSCULAR DISEASE: IS VENTILATION NEEDED?
Diseases involving the motor components of the peripheral nervous system (Table 5) share the common risk of causing ventilatory failure due to weakness of the diaphragm, intercostal muscles, and upper-airway muscles. Clinicians need to be aware of this risk and view these disorders as neurologic emergencies.
Determining when these patients require mechanical intubation is a challenge. Serial measurements of maximum inspiratory force and vital capacity are important and can be accomplished quickly at the bedside by a respiratory therapist. A maximum inspiratory force less than –30 cm H2O or a vital capacity less than 20 mL/kg, or both, are worrisome markers that raise concern for impending ventilatory failure. Serial measurements can detect changes in these values that might indicate the need for elective intubation. In any patient presenting with weakness of the limbs, these measurements are an important step in the initial evaluation.
Myasthenic crisis
Myasthenia gravis is caused by autoantibodies directed against postsynaptic acetylcholine receptors. Patients demonstrate muscle weakness, usually in a proximal pattern, with fatigue, respiratory distress, nasal speech, ophthalmoparesis, and dysphagia. Exacerbations can occur as a response to recent infection, surgery, or medications such as neuromuscular blocking agents or aminoglycosides.
Myasthenic crisis, while uncommon, is a life-threatening emergency characterized by bulbar or respiratory failure secondary to muscle weakness. It can occur in patients already diagnosed with myasthenia gravis or may be the initial manifestation of the disease.42–49 Intubation and mechanical ventilation are frequently required. Postoperative myasthenic patients in whom extubation has been delayed more than 24 hours should be considered in crisis.45
The diagnosis of myasthenia gravis can be made by serum autoantibody testing, electromyography, and nerve conduction studies (with repetitive stimulation) or administration of edrophonium in patients with obvious ptosis.
The mainstay of therapy for myasthenic crisis is either intravenous immunoglobulin at a dose of 2 g/kg over 2 to 5 days or plasmapheresis (5–7 exchanges over 7–14 days). Corticosteroids are not recommended in myasthenic crisis in patients who are not intubated, as they can potentiate an initial worsening of crisis. Once the patient begins to show clinical improvement, outpatient pyridostigmine and immunosuppressive medications can be resumed at a low dose and titrated as tolerated.
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome)
Acute inflammatory demyelinating polyneuropathy is an autoimmune disorder involving autoantibodies against axons or myelin in the peripheral nervous system.
This disease should be suspected in a patient who is developing worsening muscle weakness (usually with areflexia) over the course of days to weeks. Occasionally, a recent diarrheal or other systemic infectious trigger can be identified. Blood pressure instability and cardiac arrhythmia can also be seen due to autonomic nerve involvement. Although classically described as an “ascending paralysis,” other variants of this disease have distinct clinical presentations (eg, the descending paralysis, ataxia, areflexia, ophthalmoparesis of the Miller Fisher syndrome).
Acute inflammatory demyelinating polyneuropathy is diagnosed by electromyography and nerve conduction studies. A cerebrospinal fluid profile demonstrating elevated protein and few white blood cells is typical.
Treatment, as in myasthenic crisis, involves intravenous immunoglobulin or plasmapheresis. Corticosteroids are ineffective. Anticipation of ventilatory failure and expectant intubation is essential, given the progressive nature of the disorder.50
Neurologic emergencies such as acute stroke, status epilepticus, subarachnoid hemorrhage, neuromuscular weakness, and spinal cord injury affect millions of Americans yearly.1,2 These conditions can be difficult to diagnose, and delays in recognition and treatment can have devastating results. Consequently, it is important for nonneurologists to be able to quickly recognize these conditions and initiate timely management, often while awaiting neurologic consultation.
Here, we review how to recognize and treat these common, serious conditions.
ACUTE ISCHEMIC STROKE: TIME IS OF THE ESSENCE
Stroke is the fourth leading cause of death in the United States and is one of the most common causes of disability worldwide.3–5 About 85% of strokes are ischemic, resulting from diminished vascular supply to the brain. Symptoms such as facial droop, unilateral weakness or numbness, aphasia, gaze deviation, and unsteadiness of gait may be seen. Time is of the essence, as all currently available interventions are safe and effective only within defined time windows.
Diagnosis and assessment
When acute ischemic stroke is suspected, the clinical history, time of onset, and basic neurologic examination should be obtained quickly.
The National Institutes of Health (NIH) stroke scale is an objective marker for assessing stroke severity as well as evolution of disease and should be obtained in all stroke patients. Scores range from 0 (best) to 42 (worst) (www.ninds.nih.gov/doctors/NIH_Stroke_Scale.pdf).
Time of onset of symptoms is essential to determine, since it guides eligibility for acute therapies. Clinicians should ascertain the last time the patient was seen to be neurologically well in order to estimate this time window as closely as possible.
Laboratory tests should include a fingerstick blood glucose measurement, coagulation studies, complete blood cell count, and basic metabolic profile.
Computed tomography (CT) of the head without contrast should be obtained immediately to exclude acute hemorrhage and any alternative diagnoses that could explain the patient’s symptoms. Acute brain ischemia is often not apparent on CT during the first few hours of injury. Therefore, a patient presenting with new focal neurologic deficits and an unremarkable result on CT of the head should be treated as having had an acute ischemic stroke, and interventional therapies should be considered.
Stroke mimics should be considered and treated, as appropriate (Table 1).
Acute management of ischemic stroke
Acute treatment should not be delayed by obtaining chest radiography, inserting a Foley catheter, or obtaining an electrocardiogram. The longer the time that elapses before treatment, the worse the functional outcome, underscoring the need for rapid decision-making.6–8
Lowering the head of the bed may provide benefit by promoting blood flow to ischemic brain tissue.9 However, this should not be done in patients with significantly elevated intracerebral pressure and concern for herniation.
Permissive hypertension (antihypertensive treatment only for blood pressure greater than 220/110 mm Hg) should be allowed per national guidelines to provide adequate perfusion to brain areas at risk of injury.10
Tissue plasminogen activator. Patients with ischemic stroke who present within 3 hours of symptom onset should be considered for intravenous administration of tissue plasminogen activator (tPA), a safe and effective therapy with nearly 2 decades of evidence to support its use.10 The treating physician should carefully review the risks and benefits of this therapy.
To receive tPA, the patient must have all of the following:
- Clinical diagnosis of ischemic stroke with measurable neurologic deficit
- Onset of symptoms within the past 3 hours
- Age 18 or older.
The patient must not have any of the following:
- Significant stroke within the past 3 months
- Severe traumatic head injury within the past 3 months
- History of significant intracerebral hemorrhage
- Previously ruptured arteriovenous malformation or intracranial aneurysm
- Central nervous system neoplasm
- Arterial puncture at a noncompressible site within the past 7 days
- Evidence of hemorrhage on CT of the head
- Evidence of ischemia in greater than 33% of the cerebral hemisphere on head CT
- History and symptoms strongly suggesting subarachnoid hemorrhage
- Persistent hypertension (systolic pressure ≥ 185 mm Hg or diastolic pressure ≥ 110 mm Hg)
- Evidence of acute significant bleeding (external or internal)
- Hypoglycemia—ie, serum glucose less than 50 mg/dL (< 2.8 mmol/L)
- Thrombocytopenia (platelet count < 100 × 109/L)
- Significant coagulopathy (international normalized ratio > 1.7, prothrombin time > 15 seconds, or abnormally elevated activated partial thromboplastin time)
- Current use of a factor Xa inhibitor or direct thrombin inhibitor.
Relative contraindications:
- Minor or rapidly resolving symptoms
- Major surgery or trauma within the past 14 days
- Gastrointestinal or urinary tract bleeding within the past 21 days
- Myocardial infarction in the past 3 months
- Unruptured intracranial aneurysm
- Seizure occurring at stroke onset
- Pregnancy.
If these criteria are satisfied, tPA should be given at a dose of 0.9 mg/kg intravenously over 60 minutes. Ten percent of the dose should be given as an initial bolus, followed by a constant infusion of the remaining 90% over 1 hour.
If tPA is given, the blood pressure must be kept lower than 185/110 mm Hg to minimize the risk of symptomatic intracerebral hemorrhage.
A subset of patients may benefit from receiving intravenous tPA between 3 and 4.5 hours after the onset of stroke symptoms. These include patients who are no more than 80 years old, who have not recently used oral anticoagulants, who do not have severe neurologic injury (ie, do not have NIH Stroke Scale scores > 25), and who do not have diabetes mellitus or a history of ischemic stroke.11 Although many hospitals have such a protocol for tPA up to 4.5 hours after the onset of stroke symptoms, this time window is not currently approved by the US Food and Drug Administration.
Intra-arterial therapy. Based on recent trials, some patients may benefit further from intra-arterial thrombolysis or mechanical thrombectomy, both delivered during catheter-based cerebral angiography, independent of intravenous tPA administration.12,13 These patients should be evaluated on a case-by-case basis by a neurologist and neurointerventional team. Time windows for these treatments generally extend to 6 hours from stroke onset and perhaps even longer in some situations (eg, basilar artery occlusion).
An antiplatelet agent should be started quickly in all stroke patients who do not receive tPA. Patients who receive tPA can begin receiving an antiplatelet agent 24 hours afterward.
Unfractionated heparin. There is no evidence to support the use of unfractionated heparin in most cases of acute ischemic stroke.10
Glucose control (in the range of 140–180 mg/dL) and fever control remain essential elements of post-acute stroke care to provide additional protection to the damaged brain.
For ischemic stroke due to atrial fibrillation
In ischemic stroke due to atrial fibrillation, early anticoagulation should be considered, based on the CHA2DS2-VASC risk of ischemic stroke vs the HAS-BLED risk of hemorrhage (calculators available at www.mdcalc.com).
In general, anticoagulation may be withheld during the first 72 hours while further stroke workup and evaluation of extent of injury are carried out, as there is an increased risk of hemorrhagic transformation of the ischemic stroke. Often, anticoagulation is resumed at a full dose between 72 hours and 2 weeks of the ischemic stroke.
ACUTE HEMORRHAGIC STROKE: BLOOD PRESSURE, COAGULATION
Approximately 15% of strokes are caused by intracerebral hemorrhage, which can be detected with noncontrast head CT with a sensitivity of 98.6% within 6 hours of the onset of bleeding.14 A common underlying cause of intracerebral hemorrhage is chronic poorly controlled hypertension, causing rupture of damaged (or “lipohyalinized”) vessels with resultant blood extravasation into the brain parenchyma. Other causes are less common (Table 2).
Treatment of acute hemorrhagic stroke
Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy or anticoagulation, and sometimes intracranial pressure control. There is little role for surgery in most cases, based on findings of randomized trials.15
Blood pressure control. Many studies have investigated optimal blood pressure goals in acute intracerebral hemorrhage. Recent data suggest that early aggressive therapy, targeting a systolic blood pressure goal less than 140 mm Hg within the first hour, is safe and can lead to better functional outcomes than a more conservative blood-pressure-lowering target.16 Rapid-onset, short-acting antihypertensive agents in intravenous form, such as nicardipine and labetalol, are frequently used. Of note, this treatment strategy for hemorrhagic stroke is in direct contrast to the treatment of ischemic stroke, in which permissive hypertension (blood pressure goal < 220/110 mm Hg) is often pursued.
Reversal of any coagulation abnormalities should be done quickly in intracranial hemorrhage. Warfarin use has been shown to be a strong independent predictor of intracranial hemorrhage expansion, which increases the risk of death.17,18
Increasingly, agents other than vitamin K or fresh-frozen plasma are being used to rapidly reverse anticoagulation, including prothrombin complex concentrate (available in three- and four-factor preparations) and recombinant factor VIIa. While four-factor prothrombin complex concentrate and recombinant factor VIIa have been shown to be more efficacious than fresh-frozen plasma, there are limited data directly comparing these newer reversal agents against each other.19 The use of these medications is limited by availability and practitioner familiarity.20–22
Reversing anticoagulation due to target-specific oral anticoagulants. The acute management of intracranial hemorrhage in patients taking the new target-specific oral anticoagulants (eg, dabigatran, apixaban, rivaroxaban, edoxaban) remains challenging. Laboratory tests such as factor Xa levels are not readily available in many institutions and do not provide results in a timely fashion, and in the interim, acute hemorrhage and clinical deterioration may occur. Management strategies involve giving fresh-frozen plasma, prothrombin complex concentrate, and consideration of hemodialysis.23 Dabigatran reversal with idarucizumab has recently been shown to have efficacy.24
Vigilance for elevated intracranial pressure. Intracranial hemorrhage can occasionally cause elevated intracranial pressure, which should be treated rapidly. Any acute decline in mental status in a patient with intracranial hemorrhage requires emergency imaging to evaluate for expansion of hemorrhage.
SUBARACHNOID HEMORRHAGE
The sudden onset of a “thunderclap” headache (often described by patients as “the worst headache of my life”) suggests subarachnoid hemorrhage.
In contrast to intracranial hemorrhage, in subarachnoid hemorrhage blood collects mainly in the cerebral spinal fluid-containing spaces surrounding the brain, leading to a higher incidence of hydrocephalus from impaired drainage of cerebrospinal fluid. Nontraumatic subarachnoid hemorrhage is most often caused by rupture of an intracranial aneurysm, which can be a devastating event, with death rates approaching 50%.25
Diagnosis of subarachnoid hemorrhage
Noncontrast CT of the head is the main modality for diagnosing subarachnoid hemorrhage. Blood within the subarachnoid space is demonstrable in 92% of cases if CT is performed within the first 24 hours of hemorrhage, with an initial sensitivity of about 95% within the first 6 hours of onset.14,26,27 The longer CT is delayed, the lower the sensitivity.
Some studies suggest that a protocol of CT followed by CT angiography can safely exclude aneurysmal subarachnoid hemorrhage and obviate the need for lumbar puncture. However, further research is required to validate this approach.28
Lumbar puncture. If clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT is negative, lumbar puncture must be performed for cerebrospinal fluid analysis.29 Xanthochromia (a yellowish pigmentation of the cerebrospinal fluid due to the degeneration of blood products that occurs within 8 to 12 hours of bleeding) should raise the alarm for subarachnoid hemorrhage; this sign may be present up to 4 weeks after the bleeding event.30
If lumbar puncture is contraindicated, then aneurysmal subarachnoid hemorrhage has not been ruled out, and further neurologic consultation should be pursued.
Management of subarachnoid hemorrhage
Early management of blood pressure for a ruptured intracranial aneurysm follows strategies similar to those for intracranial hemorrhage. Further investigation is rapidly directed toward an underlying vascular malformation, with intracranial vessel imaging such as CT angiography, magnetic resonance angiography, or the gold standard test—catheter-based cerebral angiography.
Aneurysms are treated (or “secured”) either by surgical clipping or by endovascular coiling. Endovascular coiling is preferable in cases in which both can be safely attempted.31 If the facility lacks the resources to do these procedures, the patient should be referred to a nearby tertiary care center.
INTRACRANIAL HYPERTENSION: DANGER OF BRAIN HERNIATION
A number of conditions can cause an acute intracranial pressure elevation. The danger of brain herniation requires that therapies be implemented rapidly to prevent catastrophic neurologic injury. In many situations, nonneurologists are the first responders and therefore should be familiar with basic intracranial pressure management.
Initial symptoms of acute rise in intracranial pressure
As intracranial pressure rises, pressure is typically equally distributed throughout the cranial vault, leading to dysfunction of the ascending reticular activating system, which clinically manifests as the inability to stay alert despite varying degrees of noxious stimulation. Progressive cranial neuropathies (often starting with pupillary abnormalities) and coma are often seen in this setting as the upper brainstem begins to be compressed.
Initial assessment and treatment of elevated intracranial pressure
Noncontrast CT of the head is often obtained immediately when acutely elevated intracranial pressure is suspected. If clinical examination and radiographic findings are consistent with intracranial hypertension, prompt measures can be started at the bedside.
Elevate the head of the bed to 30 degrees to promote venous drainage and reduce intracranial pressure. (In contrast, most other hemodynamically unstable patients are placed flat or in the Trendelenburg position.)
Intubation should be done quickly in cases of airway compromise, and hyperventilation should be started with a goal Paco2 of 30 to 35 mm Hg. This hypocarbic strategy promotes cerebral vasoconstriction and a transient decrease in intracranial pressure.
Hyperosmolar therapy allows for transient intracranial volume decompression and is the mainstay of emergency medical treatment of intracranial hypertension. Mannitol is a hyperosmolar polysaccharide that promotes osmotic diuresis and removes excessive cerebral water. In the acute setting, it can be given as an intravenous bolus of 1 to 2 g/kg through a peripheral intravenous line, followed by a bolus every 4 to 6 hours. Hypotension can occur after diuresis, and renal function should be closely monitored since frequent mannitol use can promote acute tubular necrosis. In patients who are anuric, the medication is typically not used.
Hypertonic saline (typically 3% sodium chloride, though different concentrations are available) is an alternative that helps draw interstitial fluid into the intravascular space, decreasing cerebral edema and maintaining hemodynamic stability. Relative contraindications include congestive heart failure or renal failure leading to pulmonary edema from volume overload. Hypertonic saline can be given as a bolus or a constant infusion. Some institutions have rapid access to 23.4% saline, which can be given as a 30-mL bolus but typically requires a central venous catheter for rapid infusion.
Comatose patients with radiographic findings of hydrocephalus, epidural or subdural hematoma, or mass effect with midline shift warrant prompt neurosurgical consultation for further surgical measures of intracranial pressure control and monitoring.
The ‘blown’ pupil
The physician should be concerned about elevated intracranial pressure if a patient has mydriasis, ie, an abnormally dilated (“blown”) pupil, which is a worrisome sign in the setting of true intracranial hypertension. However, many different processes can cause mydriasis and should be kept in mind when evaluating this finding (Table 3).32 If radiographic findings do not suggest elevated intracranial pressure, further workup into these other processes should be pursued.
STATUS EPILEPTICUS: SEIZURE CONTROL IS IMPORTANT
A continuous unremitting seizure lasting longer than 5 minutes or recurrent seizure activity in a patient who does not regain consciousness between seizures should be treated as status epilepticus. All seizure types carry the risk of progressing to status epilepticus, and responsiveness to antiepileptic drug therapy is inversely related to the duration of seizures. It is imperative that seizure activity be treated early and aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.33
Once the ABCs of emergency stabilization have been performed (ie, airway, breathing, circulation), antiepileptic drug therapy should start immediately using established algorithms (Figure 1).34–36 During the course of treatment, the reliability of the neurologic examination may be limited due to medication effects or continued status epilepticus, making continuous video electroencephalographic monitoring often necessary to guide further therapy in patients who are not rapidly recovering.34–38
Once status epilepticus has resolved, further investigation into the underlying cause should be pursued quickly, especially in patients without a previous diagnosis of epilepsy. Head CT with contrast or magnetic resonance imaging can be used to look for any structural abnormality that may explain seizures. Basic laboratory tests including toxicology screening can identify a common trigger such as hypoglycemia or stimulant use. Fever or other possible signs of meningitis should be investigated further with cerebrospinal fluid analysis.
SPINAL CORD INJURY
Acute spinal cord injury can lead to substantial long-term neurologic impairment and should be suspected in any patient presenting with focal motor loss, sensory loss, or both with sparing of the cranial nerves and mental status. Causes of injury include compression (traumatic or nontraumatic) and inflammatory and noninflammatory myelopathies.
The location of the injury can be inferred by analyzing the symptoms, which can point to the cord level and indicate whether the anterior or posterior of the cord is involved. Anterior cord injury tends to affect the descending corticospinal and pyramidal tracts, resulting in motor deficits and weakness. Posterior cord injury involves the dorsal columns, leading to deficits of vibration sensation and proprioception. High cervical cord injuries tend to involve varying degrees of quadriparesis, sensory loss, and sometimes respiratory compromise. A clinical history of bilateral lower-extremity weakness, a “band-like” sensory complaint around the lower chest or abdomen, or both, can suggest thoracic cord involvement. Symptoms isolated to one or both lower extremities along with lower back pain and bowel or bladder involvement may point to injury of the lumbosacral cord.
Basic management of spinal cord injury includes decompression of the bladder and initial protection against further injury with a stabilizing collar or brace.
Magnetic resonance imaging with and without contrast is the ideal study to evaluate injuries to the spinal cord itself. While CT is helpful in identifying bony disease of the spinal column (eg, evaluating traumatic fractures), it is not helpful in viewing intrinsic cord pathology.
Traumatic myelopathy
Traumatic spinal cord injury is usually suggested by the clinical history and confirmed with CT. In this setting, early consultation with a neurosurgeon is required to prevent permanent cord injury.
Guidelines suggest maintaining a mean arterial pressure greater than 85 to 90 mm Hg for the first 7 days after traumatic spinal cord injury, a particular problem in the setting of hemodynamic instability, which can accompany lesions above the midthoracic level.39,40
Patients with vertebral body misalignment should be placed in an appropriate stabilizing collar or brace until a medically trained professional deems it appropriate to discontinue the device, or until surgical stabilization is performed.
Methylprednisone is a controversial intervention for acute spinal cord trauma, lacking clear benefit in meta-analyses.41
Nontraumatic compressive myelopathy
Patients with nontraumatic compressive myelopathy tend to present with varying degrees of back pain and worsening sensorimotor function. The differential diagnosis includes epidural abscesses, hematoma, metastatic neoplasm, and osteophyte compression (Table 4). The clinical history helps to guide therapy and should involve assessment for previous spinal column injury, immunocompromised state, travel history (which provides information on risks of exposure to a variety of diseases, including infections), and constitutional symptoms such as fever and weight loss.
Epidural abscess can have devastating results if missed. Red flags such as recent illness, intravenous drug use, focal back pain, fever, worsening numbness or weakness, and bowel or bladder incontinence should raise suspicion of this disorder. Emergency magnetic resonance imaging is required to diagnose this condition, and treatment involves urgent administration of antibiotics and consideration of surgical drainage.
Noncompressive myelopathies
There are numerous causes of noncompressive spinal cord injury (Table 4), and the etiology may be inflammatory (eg, “myelitis”) or noninflammatory. The diagnostic workup may require both magnetic resonance imaging and cerebrospinal fluid analysis. Acute disease-targeted therapy is rarely indicated and can be deferred until a full diagnostic workup has been completed.
NEUROMUSCULAR DISEASE: IS VENTILATION NEEDED?
Diseases involving the motor components of the peripheral nervous system (Table 5) share the common risk of causing ventilatory failure due to weakness of the diaphragm, intercostal muscles, and upper-airway muscles. Clinicians need to be aware of this risk and view these disorders as neurologic emergencies.
Determining when these patients require mechanical intubation is a challenge. Serial measurements of maximum inspiratory force and vital capacity are important and can be accomplished quickly at the bedside by a respiratory therapist. A maximum inspiratory force less than –30 cm H2O or a vital capacity less than 20 mL/kg, or both, are worrisome markers that raise concern for impending ventilatory failure. Serial measurements can detect changes in these values that might indicate the need for elective intubation. In any patient presenting with weakness of the limbs, these measurements are an important step in the initial evaluation.
Myasthenic crisis
Myasthenia gravis is caused by autoantibodies directed against postsynaptic acetylcholine receptors. Patients demonstrate muscle weakness, usually in a proximal pattern, with fatigue, respiratory distress, nasal speech, ophthalmoparesis, and dysphagia. Exacerbations can occur as a response to recent infection, surgery, or medications such as neuromuscular blocking agents or aminoglycosides.
Myasthenic crisis, while uncommon, is a life-threatening emergency characterized by bulbar or respiratory failure secondary to muscle weakness. It can occur in patients already diagnosed with myasthenia gravis or may be the initial manifestation of the disease.42–49 Intubation and mechanical ventilation are frequently required. Postoperative myasthenic patients in whom extubation has been delayed more than 24 hours should be considered in crisis.45
The diagnosis of myasthenia gravis can be made by serum autoantibody testing, electromyography, and nerve conduction studies (with repetitive stimulation) or administration of edrophonium in patients with obvious ptosis.
The mainstay of therapy for myasthenic crisis is either intravenous immunoglobulin at a dose of 2 g/kg over 2 to 5 days or plasmapheresis (5–7 exchanges over 7–14 days). Corticosteroids are not recommended in myasthenic crisis in patients who are not intubated, as they can potentiate an initial worsening of crisis. Once the patient begins to show clinical improvement, outpatient pyridostigmine and immunosuppressive medications can be resumed at a low dose and titrated as tolerated.
Acute inflammatory demyelinating polyneuropathy (Guillain-Barré syndrome)
Acute inflammatory demyelinating polyneuropathy is an autoimmune disorder involving autoantibodies against axons or myelin in the peripheral nervous system.
This disease should be suspected in a patient who is developing worsening muscle weakness (usually with areflexia) over the course of days to weeks. Occasionally, a recent diarrheal or other systemic infectious trigger can be identified. Blood pressure instability and cardiac arrhythmia can also be seen due to autonomic nerve involvement. Although classically described as an “ascending paralysis,” other variants of this disease have distinct clinical presentations (eg, the descending paralysis, ataxia, areflexia, ophthalmoparesis of the Miller Fisher syndrome).
Acute inflammatory demyelinating polyneuropathy is diagnosed by electromyography and nerve conduction studies. A cerebrospinal fluid profile demonstrating elevated protein and few white blood cells is typical.
Treatment, as in myasthenic crisis, involves intravenous immunoglobulin or plasmapheresis. Corticosteroids are ineffective. Anticipation of ventilatory failure and expectant intubation is essential, given the progressive nature of the disorder.50
- Pitts SR, Niska RW, Xu J, Burt CW. National hospital ambulatory medical care survey: 2006 emergency department summary. Natl Health Stat Report 2008; 7:1–38.
- McMullan JT, Knight WA, Clark JF, Beyette FR, Pancioli A. Time-critical neurological emergencies: the unfulfilled role for point-of-care testing. Int J Emerg Med 2010; 3:127–131.
- Centers for Disease Control and Prevention (CDC). Prevalence of stroke: United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2012; 61:379–382.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2095–2128.
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1,160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2163–2196.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 1995; 333:1581–1587.
- Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363:768–774.
- Saver JL, Fonarrow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013; 309:2480–2488.
- Wojner-Alexander AW, Garami Z, Chernyshev OY, Alexandrov AV. Heads down: flat positioning improves blood flow velocity in acute ischemic stroke. Neurology 2005; 64:1354–1357.
- Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Hacke W, Kaste M, Bluhmki E, et al; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1317–1329.
- Berkhemer OA, Fransen PSS, Beumer D, et al; MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Eng J Med 2015; 372:11–20.
- Campbell BC, Mitchell PJ, Kleinig TJ, et al; EXTEND-IA Investigators. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med 2015; 372:1009–1018.
- Backes D, Rinkel GJ, Kemperman H, Linn FH, Vergouwen MD. Time-dependent test characteristics of head computed tomography in patients suspected of nontraumatic subarachnoid hemorrhage. Stroke 2012; 43:2115–2119.
- Mendelow AD, Gregson BA, Fernandes HM, et al; STICH investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 2005; 365: 387–397.
- Anderson CS, Helley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Flibotte JJ, Hagan N, O'Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 2004; 63:1059–1064.
- Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006; 66:1175–1181.
- Woo CH, Patel N, Conell C, et al. Rapid warfarin reversal in the setting of intracranial hemorrhage: a comparison of plasma, recombinant activated factor VII, and prothrombin complex concentrate. World Neurosurg 2014; 81:110–115.
- Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007; 38:2001–2023.
- Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke 2006, 37:151–155.
- Chapman SA, Irwin ED, Beal AL, Kulinski NM, Hutson KE, Thorson MA. Prothrombin complex concentrate versus standard therapies for INR reversal in trauma patients receiving warfarin. Ann Pharmacother 2011; 45:869–875.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373:511-520.
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25:1342–1347.
- Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 1990; 73:18–36.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Vermuelen M, Hasan D, Blijenberg BG, Hijdra A, van Gijn J. Xanthochromia after subarachnoid haemorrhage needs no revisitation. J Neurol Neurosurg Psychiatry 1989; 52:826–828.
- Molyneaux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid hemorrhage trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Caglayan HZ, Colpak IA, Kansu T. A diagnostic challenge: dilated pupil. Curr Opin Ophthalmol 2013; 24:550–557.
- Brophy GM, Bell R, Claassen J, et al; Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
- Chang CW, Bleck TP. Status epilepticus. Neurol Clin 1995; 13:529–548.
- Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia 1993; 34(suppl 1):S2–S11.
- Leppick IE. Status epilepticus: the next decade. Neurology 1990; 40(suppl 2):4–9.
- Aranda A, Foucart G, Ducassé JL, Grolleau S, McGonigal A, Valton L. Generalized convulsive status epilepticus management in adults: a cohort study with evaluation of professional practice. Epilepsia 2010; 51:2159–2167.
- DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39:833–840.
- Casha S, Christie S. A systematic review of intensive cardiopulmonary management after spinal cord injury. J Neurotrauma 2011; 28:1479–1495.
- Walters BC, Hadley MN, Hurlbert RJ, et al; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 2013; 60(suppl 1):82–91.
- Hurlbert RJ, Hadley MN, Walters BC, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery 2013; 72(suppl 2):93–105.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci 1981; 377:670–677.
- Belack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis 2002; 4:40–42.
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM 2009; 102:97–107.
- Mayer SA. Intensive care of the myasthenic patient. Neurology 1997; 48(suppl 5):70S–75S.
- Jani-Acsadi A, Lisak RP. Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 2007; 261:127–133.
- Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J 2008; 101:63–69.
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol 2005; 7:129–141.
- Godoy DA, Vaz de Mello LJ, Masotti L, Napoli MD. The myasthenic patient in crisis: an update of the management in neurointensive care unit. Arq Neuropsiquiatr 2013; 71:627–639.
- Hughes RA, Wijdicks EF, Benson E, et al; Multidisciplinary Consensus Group. Supportive care for patients with Guillain-Barré syndrome: Arch Neurol 2005; 62:1194–1198.
- Pitts SR, Niska RW, Xu J, Burt CW. National hospital ambulatory medical care survey: 2006 emergency department summary. Natl Health Stat Report 2008; 7:1–38.
- McMullan JT, Knight WA, Clark JF, Beyette FR, Pancioli A. Time-critical neurological emergencies: the unfulfilled role for point-of-care testing. Int J Emerg Med 2010; 3:127–131.
- Centers for Disease Control and Prevention (CDC). Prevalence of stroke: United States, 2006–2010. MMWR Morb Mortal Wkly Rep 2012; 61:379–382.
- Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2095–2128.
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1,160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet 2012; 380:2163–2196.
- Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 1995; 333:1581–1587.
- Hacke W, Donnan G, Fieschi C, et al; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363:768–774.
- Saver JL, Fonarrow GC, Smith EE, et al. Time to treatment with intravenous tissue plasminogen activator and outcome from acute ischemic stroke. JAMA 2013; 309:2480–2488.
- Wojner-Alexander AW, Garami Z, Chernyshev OY, Alexandrov AV. Heads down: flat positioning improves blood flow velocity in acute ischemic stroke. Neurology 2005; 64:1354–1357.
- Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013; 44:870–947.
- Hacke W, Kaste M, Bluhmki E, et al; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008; 359:1317–1329.
- Berkhemer OA, Fransen PSS, Beumer D, et al; MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N Eng J Med 2015; 372:11–20.
- Campbell BC, Mitchell PJ, Kleinig TJ, et al; EXTEND-IA Investigators. Endovascular therapy for ischemic stroke with perfusion-imaging selection. N Engl J Med 2015; 372:1009–1018.
- Backes D, Rinkel GJ, Kemperman H, Linn FH, Vergouwen MD. Time-dependent test characteristics of head computed tomography in patients suspected of nontraumatic subarachnoid hemorrhage. Stroke 2012; 43:2115–2119.
- Mendelow AD, Gregson BA, Fernandes HM, et al; STICH investigators. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet 2005; 365: 387–397.
- Anderson CS, Helley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Flibotte JJ, Hagan N, O'Donnell J, Greenberg SM, Rosand J. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology 2004; 63:1059–1064.
- Davis SM, Broderick J, Hennerici M, et al; Recombinant Activated Factor VII Intracerebral Hemorrhage Trial Investigators. Hematoma growth is a determinant of mortality and poor outcome after intracerebral hemorrhage. Neurology 2006; 66:1175–1181.
- Woo CH, Patel N, Conell C, et al. Rapid warfarin reversal in the setting of intracranial hemorrhage: a comparison of plasma, recombinant activated factor VII, and prothrombin complex concentrate. World Neurosurg 2014; 81:110–115.
- Broderick J, Connolly S, Feldmann E, et al; American Heart Association; American Stroke Association Stroke Council; High Blood Pressure Research Council; Quality of Care and Outcomes in Research Interdisciplinary Working Group. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke 2007; 38:2001–2023.
- Goldstein JN, Thomas SH, Frontiero V, et al. Timing of fresh frozen plasma administration and rapid correction of coagulopathy in warfarin-related intracerebral hemorrhage. Stroke 2006, 37:151–155.
- Chapman SA, Irwin ED, Beal AL, Kulinski NM, Hutson KE, Thorson MA. Prothrombin complex concentrate versus standard therapies for INR reversal in trauma patients receiving warfarin. Ann Pharmacother 2011; 45:869–875.
- Fawole A, Daw HA, Crowther MA. Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban. Cleve Clin J Med 2013; 80:443–451.
- Pollack CV Jr, Reilly PA, Eikelboom J, et al. Idarucizumab for dabigatran reversal. N Engl J Med 2015; 373:511-520.
- Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent bleeding are the major causes of death following subarachnoid hemorrhage. Stroke 1994; 25:1342–1347.
- Kassell NF, Torner JC, Haley EC Jr, Jane JA, Adams HP, Kongable GL. The international cooperative study on the timing of aneurysm surgery. Part 1: overall management results. J Neurosurg 1990; 73:18–36.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Vermuelen M, Hasan D, Blijenberg BG, Hijdra A, van Gijn J. Xanthochromia after subarachnoid haemorrhage needs no revisitation. J Neurol Neurosurg Psychiatry 1989; 52:826–828.
- Molyneaux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid hemorrhage trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Caglayan HZ, Colpak IA, Kansu T. A diagnostic challenge: dilated pupil. Curr Opin Ophthalmol 2013; 24:550–557.
- Brophy GM, Bell R, Claassen J, et al; Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012; 17:3–23.
- Chang CW, Bleck TP. Status epilepticus. Neurol Clin 1995; 13:529–548.
- Treiman DM. Generalized convulsive status epilepticus in the adult. Epilepsia 1993; 34(suppl 1):S2–S11.
- Leppick IE. Status epilepticus: the next decade. Neurology 1990; 40(suppl 2):4–9.
- Aranda A, Foucart G, Ducassé JL, Grolleau S, McGonigal A, Valton L. Generalized convulsive status epilepticus management in adults: a cohort study with evaluation of professional practice. Epilepsia 2010; 51:2159–2167.
- DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39:833–840.
- Casha S, Christie S. A systematic review of intensive cardiopulmonary management after spinal cord injury. J Neurotrauma 2011; 28:1479–1495.
- Walters BC, Hadley MN, Hurlbert RJ, et al; American Association of Neurological Surgeons; Congress of Neurological Surgeons. Guidelines for the management of acute cervical spine and spinal cord injuries: 2013 update. Neurosurgery 2013; 60(suppl 1):82–91.
- Hurlbert RJ, Hadley MN, Walters BC, et al. Pharmacological therapy for acute spinal cord injury. Neurosurgery 2013; 72(suppl 2):93–105.
- Cohen MS, Younger D. Aspects of the natural history of myasthenia gravis: crisis and death. Ann NY Acad Sci 1981; 377:670–677.
- Belack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromuscul Dis 2002; 4:40–42.
- Chaudhuri A, Behan PO. Myasthenic crisis. QJM 2009; 102:97–107.
- Mayer SA. Intensive care of the myasthenic patient. Neurology 1997; 48(suppl 5):70S–75S.
- Jani-Acsadi A, Lisak RP. Myasthenic crisis: guidelines for prevention and treatment. J Neurol Sci 2007; 261:127–133.
- Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J 2008; 101:63–69.
- Ahmed S, Kirmani JF, Janjua N, et al. An update on myasthenic crisis. Curr Treat Options Neurol 2005; 7:129–141.
- Godoy DA, Vaz de Mello LJ, Masotti L, Napoli MD. The myasthenic patient in crisis: an update of the management in neurointensive care unit. Arq Neuropsiquiatr 2013; 71:627–639.
- Hughes RA, Wijdicks EF, Benson E, et al; Multidisciplinary Consensus Group. Supportive care for patients with Guillain-Barré syndrome: Arch Neurol 2005; 62:1194–1198.
KEY POINTS
- Patients with possible acute ischemic stroke should be assessed quickly to see if they should receive tissue plasminogen activator, which should be started within 3 hours of stroke onset. Computed tomography (CT) of the head without contrast should be done immediately to rule out acute hemorrhagic stroke.
- Acute treatment of intracerebral hemorrhage includes blood pressure control, reversal of underlying coagulopathy, and sometimes intracranial pressure control.
- If the clinical suspicion of subarachnoid hemorrhage remains strong even though initial CT was negative, lumbar puncture is mandatory.
- Hyperosmolar therapy is the mainstay of emergency medical treatment of intracranial hypertension.
- Seizure activity must be treated aggressively to prevent recalcitrant seizure activity, neuronal damage, and progression to status epilepticus.
Ischemic Stroke Workup
After entertaining the possibility of acute intervention, the majority of hospitalists efforts in the management of patients with ischemic stroke involve identifying an etiology and initiating secondary prevention strategies. Other than evaluating stroke risk factors, workup has traditionally involved extracranial and intracranial vessel imaging, cardiac telemetry, and echocardiography. Even after exhaustive searches, no cause for stroke is found in nearly 25% of cases, leading to a recent focus on determining why these so‐called cryptogenic strokes happen and how to prevent their recurrence.[1, 2]
Echocardiography is commonly obtained in most patients with ischemic stroke, but its yield is probably modest at best. Although transesophageal echocardiography (TEE) may be superior to transthoracic echocardiography (TTE) for determining an etiology of stroke, whether these findings substantially change management remains debatable.[3, 4] In this issue of the Journal of Hospital Medicine, Marino and colleagues examined the yield of TEE in patients without a known cause of ischemic stroke following a normal TTE.[5] A possible cause of stroke was identified in 42%, including aortic plaques and patent foramen ovale (PFO), but in only 1 patient did this discovery change management.
Secondary prevention strategies in ischemic stroke outside of atrial fibrillation now almost exclusively involve antiplatelet medications.[6] Studies of secondary prevention in aortic arch atheromas, patients with depressed systolic function, and those with PFO have failed to demonstrate any strategy that is superior to antiplatelets, and therefore the bar is high to show that any TEE findings impact treatment other than obvious and rare smoking guns such as a rare valvular lesion, cardiac tumor, or atrial thrombus.[7, 8, 9]
What is more of a recent headline in stroke workup is the increasing emphasis on long‐term cardiac monitoring following discharge to detect those with atrial fibrillation, which likely comprise between 15% and 20% of cryptogenic stroke patients.[10] Finding atrial fibrillation clearly changes management and therefore has a higher yield than the vast majority of possible findings on echocardiography. Perhaps in patients in whom a TEE is being considered, extended monitoring should happen first as an outpatient, followed by TEE if the stroke etiology remains obscure. On the other hand, severe left atrial enlargement, thrombus in the atrium, or atrial spontaneous echo contrast (smoke) are features on echocardiography that might raise the suspicion of atrial fibrillation so high that anticoagulation could be considered while long‐term monitoring is being used to definitively prove an atrial arrhythmia.
The current study does have some limitations other than those inherent to its retrospective design. Patients were only included if they were older than 50 years. Some have advocated using TEE as the echocardiogram of choice in young stroke patients due to its perhaps higher yield in these individuals; this study does not address this strategy. At institutions such as ours, an abnormal TTE in a cryptogenic stroke patient is followed by a TEE, and this study again does not alter this approach, because only those with a normal TTE were included. The definition of a normal TTE used in the study was so narrow, including normal left ventricular systolic function, that a majority of stroke patients with vascular risk factors such as hypertension would have likely been excluded. Determining what features and quality of a TTE are so definitive that a TEE is not necessary will be an important thrust of additional research. However, because TEE shows a better view of the left atrial appendage, the aortic arch, and is probably a better shunt study compared with TTE, it is not clear if a normal TTE will ever be adequate to prevent this second more invasive study in selected patients.
At the heart of the matter for health systems is the cost‐effectiveness of any screening approach used to determine the etiology of acute ischemic stroke. Studies are underway that will likely demonstrate that long‐term monitoring for atrial fibrillation will be worth it. Although it is dubious that TEE would ever fall into the same category due to its low yield, one might imagine a scenario, as our workup for cryptogenic stroke becomes more and more complicated, where obtaining a TEE is cost‐effective simply because it gives an answer and therefore can halt further testing. Perhaps at the end of the day, a TEE will just allow us to say to our stroke patients that enough is enough.
Disclosures: Dr. Josephson receives personal compensation as Editor‐in‐Chief of the New England Journal of Medicine Journal Watch Neurology and in an editorial capacity for Continuum Audio.
- , , , et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13:429–438.
- , , , et al. Incidence, outcome, risk factors, and long‐term prognosis of cryptogenic transient ischaemic attack and ischaemic stroke: a population‐based study. Lancet Neurol. 2015;14:903–913.
- , , , et al. Transesophageal echocardiography is superior to transthoracic echocardiography in management of patients of any age with transient ischemic attack or stroke. Stroke. 2006;37:2531–2534.
- , , , , , . Transesophageal echocardiography in patients with cryptogenic ischemic stroke: a systematic review. Am Heart J. 2014;168:706–712.
- , , , , . Impact of transesophageal echocardiography on clinical management of patients over age 50 with cryptogenic stroke and normal transthoracic echocardiogram. J Hosp Med. 2016;11(2):95–98.
- , , , et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–2236.
- , , , et al. Clopidogrel plus aspirin versus warfarin in patients with stroke and aortic arch plaques. Stroke. 2014;45:1248–1257.
- , , , et al. Warfarin and aspirin in patients with heart failure and sinus rhythm. N Engl J Med. 2012;366:1859–1869.
- , , , et al. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med. 2012;366:991–999.
- , , , et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370:2467–2477.
After entertaining the possibility of acute intervention, the majority of hospitalists efforts in the management of patients with ischemic stroke involve identifying an etiology and initiating secondary prevention strategies. Other than evaluating stroke risk factors, workup has traditionally involved extracranial and intracranial vessel imaging, cardiac telemetry, and echocardiography. Even after exhaustive searches, no cause for stroke is found in nearly 25% of cases, leading to a recent focus on determining why these so‐called cryptogenic strokes happen and how to prevent their recurrence.[1, 2]
Echocardiography is commonly obtained in most patients with ischemic stroke, but its yield is probably modest at best. Although transesophageal echocardiography (TEE) may be superior to transthoracic echocardiography (TTE) for determining an etiology of stroke, whether these findings substantially change management remains debatable.[3, 4] In this issue of the Journal of Hospital Medicine, Marino and colleagues examined the yield of TEE in patients without a known cause of ischemic stroke following a normal TTE.[5] A possible cause of stroke was identified in 42%, including aortic plaques and patent foramen ovale (PFO), but in only 1 patient did this discovery change management.
Secondary prevention strategies in ischemic stroke outside of atrial fibrillation now almost exclusively involve antiplatelet medications.[6] Studies of secondary prevention in aortic arch atheromas, patients with depressed systolic function, and those with PFO have failed to demonstrate any strategy that is superior to antiplatelets, and therefore the bar is high to show that any TEE findings impact treatment other than obvious and rare smoking guns such as a rare valvular lesion, cardiac tumor, or atrial thrombus.[7, 8, 9]
What is more of a recent headline in stroke workup is the increasing emphasis on long‐term cardiac monitoring following discharge to detect those with atrial fibrillation, which likely comprise between 15% and 20% of cryptogenic stroke patients.[10] Finding atrial fibrillation clearly changes management and therefore has a higher yield than the vast majority of possible findings on echocardiography. Perhaps in patients in whom a TEE is being considered, extended monitoring should happen first as an outpatient, followed by TEE if the stroke etiology remains obscure. On the other hand, severe left atrial enlargement, thrombus in the atrium, or atrial spontaneous echo contrast (smoke) are features on echocardiography that might raise the suspicion of atrial fibrillation so high that anticoagulation could be considered while long‐term monitoring is being used to definitively prove an atrial arrhythmia.
The current study does have some limitations other than those inherent to its retrospective design. Patients were only included if they were older than 50 years. Some have advocated using TEE as the echocardiogram of choice in young stroke patients due to its perhaps higher yield in these individuals; this study does not address this strategy. At institutions such as ours, an abnormal TTE in a cryptogenic stroke patient is followed by a TEE, and this study again does not alter this approach, because only those with a normal TTE were included. The definition of a normal TTE used in the study was so narrow, including normal left ventricular systolic function, that a majority of stroke patients with vascular risk factors such as hypertension would have likely been excluded. Determining what features and quality of a TTE are so definitive that a TEE is not necessary will be an important thrust of additional research. However, because TEE shows a better view of the left atrial appendage, the aortic arch, and is probably a better shunt study compared with TTE, it is not clear if a normal TTE will ever be adequate to prevent this second more invasive study in selected patients.
At the heart of the matter for health systems is the cost‐effectiveness of any screening approach used to determine the etiology of acute ischemic stroke. Studies are underway that will likely demonstrate that long‐term monitoring for atrial fibrillation will be worth it. Although it is dubious that TEE would ever fall into the same category due to its low yield, one might imagine a scenario, as our workup for cryptogenic stroke becomes more and more complicated, where obtaining a TEE is cost‐effective simply because it gives an answer and therefore can halt further testing. Perhaps at the end of the day, a TEE will just allow us to say to our stroke patients that enough is enough.
Disclosures: Dr. Josephson receives personal compensation as Editor‐in‐Chief of the New England Journal of Medicine Journal Watch Neurology and in an editorial capacity for Continuum Audio.
After entertaining the possibility of acute intervention, the majority of hospitalists efforts in the management of patients with ischemic stroke involve identifying an etiology and initiating secondary prevention strategies. Other than evaluating stroke risk factors, workup has traditionally involved extracranial and intracranial vessel imaging, cardiac telemetry, and echocardiography. Even after exhaustive searches, no cause for stroke is found in nearly 25% of cases, leading to a recent focus on determining why these so‐called cryptogenic strokes happen and how to prevent their recurrence.[1, 2]
Echocardiography is commonly obtained in most patients with ischemic stroke, but its yield is probably modest at best. Although transesophageal echocardiography (TEE) may be superior to transthoracic echocardiography (TTE) for determining an etiology of stroke, whether these findings substantially change management remains debatable.[3, 4] In this issue of the Journal of Hospital Medicine, Marino and colleagues examined the yield of TEE in patients without a known cause of ischemic stroke following a normal TTE.[5] A possible cause of stroke was identified in 42%, including aortic plaques and patent foramen ovale (PFO), but in only 1 patient did this discovery change management.
Secondary prevention strategies in ischemic stroke outside of atrial fibrillation now almost exclusively involve antiplatelet medications.[6] Studies of secondary prevention in aortic arch atheromas, patients with depressed systolic function, and those with PFO have failed to demonstrate any strategy that is superior to antiplatelets, and therefore the bar is high to show that any TEE findings impact treatment other than obvious and rare smoking guns such as a rare valvular lesion, cardiac tumor, or atrial thrombus.[7, 8, 9]
What is more of a recent headline in stroke workup is the increasing emphasis on long‐term cardiac monitoring following discharge to detect those with atrial fibrillation, which likely comprise between 15% and 20% of cryptogenic stroke patients.[10] Finding atrial fibrillation clearly changes management and therefore has a higher yield than the vast majority of possible findings on echocardiography. Perhaps in patients in whom a TEE is being considered, extended monitoring should happen first as an outpatient, followed by TEE if the stroke etiology remains obscure. On the other hand, severe left atrial enlargement, thrombus in the atrium, or atrial spontaneous echo contrast (smoke) are features on echocardiography that might raise the suspicion of atrial fibrillation so high that anticoagulation could be considered while long‐term monitoring is being used to definitively prove an atrial arrhythmia.
The current study does have some limitations other than those inherent to its retrospective design. Patients were only included if they were older than 50 years. Some have advocated using TEE as the echocardiogram of choice in young stroke patients due to its perhaps higher yield in these individuals; this study does not address this strategy. At institutions such as ours, an abnormal TTE in a cryptogenic stroke patient is followed by a TEE, and this study again does not alter this approach, because only those with a normal TTE were included. The definition of a normal TTE used in the study was so narrow, including normal left ventricular systolic function, that a majority of stroke patients with vascular risk factors such as hypertension would have likely been excluded. Determining what features and quality of a TTE are so definitive that a TEE is not necessary will be an important thrust of additional research. However, because TEE shows a better view of the left atrial appendage, the aortic arch, and is probably a better shunt study compared with TTE, it is not clear if a normal TTE will ever be adequate to prevent this second more invasive study in selected patients.
At the heart of the matter for health systems is the cost‐effectiveness of any screening approach used to determine the etiology of acute ischemic stroke. Studies are underway that will likely demonstrate that long‐term monitoring for atrial fibrillation will be worth it. Although it is dubious that TEE would ever fall into the same category due to its low yield, one might imagine a scenario, as our workup for cryptogenic stroke becomes more and more complicated, where obtaining a TEE is cost‐effective simply because it gives an answer and therefore can halt further testing. Perhaps at the end of the day, a TEE will just allow us to say to our stroke patients that enough is enough.
Disclosures: Dr. Josephson receives personal compensation as Editor‐in‐Chief of the New England Journal of Medicine Journal Watch Neurology and in an editorial capacity for Continuum Audio.
- , , , et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13:429–438.
- , , , et al. Incidence, outcome, risk factors, and long‐term prognosis of cryptogenic transient ischaemic attack and ischaemic stroke: a population‐based study. Lancet Neurol. 2015;14:903–913.
- , , , et al. Transesophageal echocardiography is superior to transthoracic echocardiography in management of patients of any age with transient ischemic attack or stroke. Stroke. 2006;37:2531–2534.
- , , , , , . Transesophageal echocardiography in patients with cryptogenic ischemic stroke: a systematic review. Am Heart J. 2014;168:706–712.
- , , , , . Impact of transesophageal echocardiography on clinical management of patients over age 50 with cryptogenic stroke and normal transthoracic echocardiogram. J Hosp Med. 2016;11(2):95–98.
- , , , et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–2236.
- , , , et al. Clopidogrel plus aspirin versus warfarin in patients with stroke and aortic arch plaques. Stroke. 2014;45:1248–1257.
- , , , et al. Warfarin and aspirin in patients with heart failure and sinus rhythm. N Engl J Med. 2012;366:1859–1869.
- , , , et al. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med. 2012;366:991–999.
- , , , et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370:2467–2477.
- , , , et al. Embolic strokes of undetermined source: the case for a new clinical construct. Lancet Neurol. 2014;13:429–438.
- , , , et al. Incidence, outcome, risk factors, and long‐term prognosis of cryptogenic transient ischaemic attack and ischaemic stroke: a population‐based study. Lancet Neurol. 2015;14:903–913.
- , , , et al. Transesophageal echocardiography is superior to transthoracic echocardiography in management of patients of any age with transient ischemic attack or stroke. Stroke. 2006;37:2531–2534.
- , , , , , . Transesophageal echocardiography in patients with cryptogenic ischemic stroke: a systematic review. Am Heart J. 2014;168:706–712.
- , , , , . Impact of transesophageal echocardiography on clinical management of patients over age 50 with cryptogenic stroke and normal transthoracic echocardiogram. J Hosp Med. 2016;11(2):95–98.
- , , , et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–2236.
- , , , et al. Clopidogrel plus aspirin versus warfarin in patients with stroke and aortic arch plaques. Stroke. 2014;45:1248–1257.
- , , , et al. Warfarin and aspirin in patients with heart failure and sinus rhythm. N Engl J Med. 2012;366:1859–1869.
- , , , et al. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med. 2012;366:991–999.
- , , , et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370:2467–2477.
A Validated Delirium Prediction Rule
Delirium is characterized by fluctuating disturbances in cognition and consciousness and is a common complication of hospitalization in medical and surgical patients. Studies estimate the prevalence of delirium in hospitalized patients[1] to be 14% to 56%, and up to 70% in critically ill elderly patients.[2] Estimates of total healthcare costs associated with delirium range from $38 to $152 billion per year in the United States.[3] Delirious patients are more likely to be discharged to a nursing home and have increased hospital mortality and longer lengths of stay.[4, 5, 6] Recent data suggest long‐term effects of delirium including cognitive impairments up to 1 year following the illness[7] and an increased likelihood of developing[8] or worsening dementia.[9]
It is estimated that one‐third of hospital‐acquired delirium cases could be prevented with appropriate interventions.[10] A prediction rule that easily and accurately identifies high‐risk patients upon admission could therefore have a substantial clinical impact. In addition, a prediction rule could be used to identify patients in whom new targeted interventions for delirium prevention could be investigated. A number of risk factors for delirium have been identified, including older age, preexisting cognitive dysfunction, vision and hearing impairment, severe illness, dehydration, electrolyte abnormalities, overmedication, and alcohol abuse.[11, 12, 13, 14, 15, 16] Existing prediction rules using various combinations of these measures have been limited by their complexity,[17] do not predict incident delirium,[18, 19] or are restricted to surgical[20, 21, 22] or intensive care[23] patients and therefore are not broadly applicable to the general medical population, which is particularly susceptible to developing delirium.
We conducted this study to develop a simple, efficient, and accurate prediction rule for hospital‐acquired delirium in adult medical inpatients assessed at the time of admission. Our a priori hypothesis was that a delirium prediction rule would consist of a combination of known risk factors and most likely incorporate old age, illness severity, and preexisting cognitive dysfunction.
METHODS
Design and Setting
This was a prospective cohort study with a derivation phase from May 2010 to November 2010 at 2 hospitals at the University of California, San Francisco (UCSF) (Moffitt‐Long and Mount Zion Hospitals) and a validation phase from October 2011 to March 2012 at the San Francisco Veterans Affairs Medical Center (SFVAMC).
Participants and Measurements
Subject identification, recruitment, and inclusion and exclusion criteria were identical for the derivation and validation cohorts. Subjects were identified by reviewing daily admission logs. All non‐intensive care unit patients aged 50 years or older admitted through the emergency department to the medicine, cardiology, or neurology services were screened for eligibility through chart review or in person within 24 hours of admission by a trained research assistant. One research assistant, a college graduate, conducted all screening for the derivation cohort, and 2 research assistants, 1 a fourth‐year medical student and the other a third‐year psychology graduate student, conducted screening for the validation cohort. In‐person screening included an assessment for delirium using the long version of the Confusion Assessment Method (CAM).[24] To minimize the possibility of enrolling delirious subjects, research assistants were instructed to notify the study supervisor (V.C.D.), a board‐certified neurologist, to discuss every case in which any yes checkbox was marked on the CAM score sheet. Subjects delirious upon initial evaluation, admitted for alcohol withdrawal, admitted for comfort care, who were aphasic or who could not speak English were excluded. For all patients, or if they were unable to provide consent, their surrogates provided written informed consent, and the study was approved by the institutional review boards at UCSF and SFVAMC.
In the derivation cohort, 1241 patients were screened, and 439 were eligible for enrollment. Of these, 180 declined, 50 were discharged prior to the first follow‐up visit, and 209 were included. In the validation cohort, 420 patients were screened, and 368 were eligible for enrollment. Of these, 144 declined, 59 were discharged prior to the first follow‐up visit, and 165 were included.
Baseline data regarding known delirium risk factors[11, 12, 13, 14, 15, 16] were collected from subjects in the derivation cohort. Cognitive performance was assessed with the Mini Mental Status Examination (MMSE),[25] forward digit span,[26] and clock draw.[27] Permission for administration of the MMSE was granted by Psychological Assessment Resources, Inc., and each administration was paid for. A structured interview was conducted with validated questions regarding visual and hearing impairment, pain, mobility, place of residence, and alcohol, tobacco, and drug use.[28, 29, 30, 31] A whisper test for hearing loss was performed.[32] Subjects' charts were reviewed for demographic, clinical, and laboratory data. Illness severity was assessed by asking each subject's nurse to rate their patient on a scale from not ill to mildly ill, moderately ill, severely ill, or moribund.[33] Each nurse was shown these 5 choices, but more specific definitions of what each level of illness severity meant were not provided. We chose this method to assess illness severity because this rating scale was incorporated into a previous validated and widely cited delirium prediction rule.[17] This illness severity scale has been validated as a predictor of outcomes and correlates with other measures of illness severity and comorbidity when graded by physicians.[33, 34] Nurse and physician ratings of illness severity have been shown to be comparable,[35] and therefore if the scale were incorporated into the prediction rule it would allow nurses to perform it independently. In the validation cohort, only data required to complete the baseline CAM and apply the prediction rule were collected.
Assessment of Outcomes
All subjects were assessed for delirium daily for 6 days after enrollment or until discharge, whichever came first. Follow‐up was limited to 6 days, based on the assumption that delirium occurring beyond 1 week is more likely due to events during the hospitalization as opposed to factors measurable at admission. Delirium was assessed using the short CAM, an internationally recognized and validated tool.[24] To complete the CAM during follow‐up visits, subjects and their nurses were interviewed using a written script, and an MMSE and forward digit span were performed.
Daily follow‐up assessments were performed by research assistants who were not blinded to the initial assessment but who, in the validation phase, were blinded to the prediction rule score. Some weekend follow‐ups were performed by postgraduate year 2, 3, or 4 neurology residents, or internal medicine faculty experienced in the assessment of delirium and blinded to both the initial assessment and prediction rule score. Neurology residents and internists read the CAM training manual and were educated in the administration and scoring of the CAM by 1 of the senior investigators (V.C.D.) prior to their first shift; these nonstudy personnel covered 17 of 189 days of follow‐up in the derivation cohort and 21 of 169 days of follow‐up in the validation cohort. To maximize sensitivity of delirium detection, for any change in cognition, MMSE score, or forward digit span compared to baseline, a board‐certified neurologist blinded to the initial assessment was notified to discuss the case and validate the diagnosis of delirium in person (derivation cohort) or over the phone (validation cohort). All research assistants were trained by a board‐certified neurologist (V.C.D.) in the administration and interpretation of the CAM using published methods prior to enrollment of any subjects.[36] Training included the performance of independent long‐version CAMs by the trainer and the trainee on a series of delirious and nondelirious patients until there was consistent agreement for each item on the CAM in 5 consecutive patients. In addition, a board‐certified neurologist supervised the first 5 administrations of the CAM performed by each research assistant.
Statistical Analysis
Sample size for the derivation cohort was based on the predicted ability to detect a difference in rates of delirium among those with and without cognitive impairment, the strongest risk factor for delirium. Using a [2] test with an of 0.05 and of 0.80, we estimated we would need to enroll 260 subjects, assuming a prevalence of cognitive dysfunction in our cohort of 10% and an estimated rate of delirium of 24% and 6% among those with and without cognitive dysfunction respectively.[14, 16, 17, 20] We were unable to reach enrollment targets because of a short funding period and slower than expected recruitment.
To construct the prediction rule in the derivation cohort, all variables were dichotomized. Age was dichotomized at 80 years because old age is a known risk factor for delirium, and only 1 of 46 subjects between the ages of 70 and 80 years became delirious in the derivation cohort. Components of the MMSE were dichotomized as correct/emncorrect, with a correct response requiring perfect performance based on expert consensus. For 3 subjects who would not attempt to spell world backward (2 in the derivation and 1 in the validation cohort), their score on serial 7s was used instead. The total MMSE score was not used because our objective was to develop a prediction rule using elements that could be assessed quickly in the fast‐paced environment of the hospital. Illness severity was dichotomized at moderate or worse/mild or better because there were only 15 subjects in the severe illness category, and the majority of delirium (22 outcomes) occurred in the moderate illness category. High blood urea nitrogen:creatinine ratio was defined as >18.[37]
The association between predictor variables and occurrence of delirium was analyzed using univariate logistic regression. A forward stepwise logistic regression was then performed using the variables associated with the outcome at a significance level of P<0.05 in univariate analysis. Variables were eligible for addition to the multivariable model if they were associated with the outcome at a significance level of <0.05. The 4 independent predictors thus identified were combined into a prediction rule by assigning each predictor 1 point if present. The performance of the prediction rule was assessed by using Cuzick's nonparametric test for a trend across groups ordered by score.[38]
The prediction rule was tested in the validation cohort using the nonparametric test for trend. Receiver operating characteristic (ROC) curves were compared between the derivation and validation cohorts. All statistical analysis was performed using Stata software (StataCorp, College Station, TX).
RESULTS
The derivation cohort consisted of elderly patients (mean age, 68.0811.96 years; interquartile range, 5096 years), and included more males than females (54.1% vs 45.9%). Subjects were predominantly white (73.7%) and lived at home (90%) (Table 1). The mean admission MMSE score was 27.0 (standard deviation [SD], 3.4; range, 730). Median follow‐up was 2 days (interquartile range, 13). Delirium developed in 12% (n=25) of the cohort.
| Derivation Cohort, N=209 | Validation Cohort, N=165 | |
|---|---|---|
| ||
| Gender, No. (%) | ||
| Male | 113 (54) | 157 (95) |
| Female | 96 (46) | 8 (4.8) |
| Race, No. (%) | ||
| White | 154 (74) | 125 (76) |
| African American | 34 (16) | 25 (15) |
| Asian | 21 (10.0) | 13 (7.9) |
| Native American | 0 | 2 (1.2) |
| Illness severity, No. (%) | ||
| Not ill | 1 (0.5) | 0 |
| Mildly ill | 49 (23) | 62 (38) |
| Moderately ill | 129 (62) | 86 (52) |
| Severely ill | 15 (7.2) | 17 (10) |
| Moribund | 0 | 0 |
| Living situation, No. (%) | ||
| Home | 188 (90) | 147 (89) |
| Assisted living | 11 (5.3) | 6 (3.6) |
| Hotel | 4 (1.9) | 5 (3.0) |
| SNF | 1 (0.5) | 3 (1.8) |
| Homeless | 4 (1.9) | 4 (2.4) |
| Developed delirium | 25 (12) | 14 (8.5) |
Univariate analysis of the derivation study identified 10 variables significantly associated (P<0.05) with delirium (Table 2). Predictors of delirium included abnormal scores on 4 subtests of the MMSE, low score on the Mini‐Cog, living in an assisted living or skilled nursing facility, moderate to severe illness, old age, a past history of dementia, and hearing loss as assessed by the whisper test. These predictors were then entered into a stepwise logistic regression analysis that identified 4 independent predictors of delirium (Table 3).
| Variable | No. (%) Without Delirium | No. (%) With Delirium | Odds Ratio | P Value | 95% Confidence Interval |
|---|---|---|---|---|---|
| |||||
| Age 80 years | 30 (16) | 13 (52) | 5.6 | <0.001 | 2.313.4 |
| Male sex | 99 (54) | 14 (56) | 1.1 | 0.84 | 0.52.5 |
| White race | 135 (73) | 19 (76) | 1.2 | 0.78 | 0.433.1 |
| Score <5 on date questions of MMSE | 37 (20) | 12 (48) | 3.7 | 0.003 | 1.68.7 |
| Score <5 on place questions of MMSE | 50 (27) | 14 (56) | 3.4 | 0.005 | 1.58.0 |
| Score <3 on MMSE recall | 89 (48) | 18 (72) | 2.7 | 0.03 | 1.16.9 |
| Score <5 on MMSE W‐O‐R‐L‐D backward | 37 (20) | 13 (52) | 4.3 | 0.001 | 1.810.2 |
| Score 0 on MMSE pentagon copy, n=203 | 53 (30) | 12 (48) | 2.2 | 0.07 | 0.935.1 |
| Score 0 on clock draw, n=203 | 70 (39) | 15 (60) | 2.3 | 0.05 | 0.985.4 |
| MiniCog score 02, n=203[27] | 46 (26) | 12 (48) | 2.7 | 0.03 | 1.16.2 |
| Self‐rated vision fair, poor, or very poor | 55 (30) | 8 (32) | 1.1 | 0.83 | 0.452.7 |
| Endorses hearing loss | 89 (48) | 12 (48) | 0.99 | 0.97 | 0.432.3 |
| Uses hearing aid | 19 (10) | 2 (8) | 0.76 | 0.72 | 0.173.5 |
| Fails whisper test in either ear | 39 (21) | 10 (40) | 2.5 | 0.04 | 1.05.9 |
| Prior episode of delirium per patient or informant | 70 (38) | 13 (52) | 1.8 | 0.19 | 0.764.1 |
| Dementia in past medical history | 3 (2) | 3 (12) | 8.2 | 0.01 | 1.643.3 |
| Depression in past medical history | 16 (9) | 1 (4) | 0.44 | 0.43 | 0.063.5 |
| Lives in assisted living or SNF | 8 (4) | 4 (16) | 4.2 | 0.03 | 1.215.1 |
| Endorses pain | 82 (45) | 7 (28) | 0.48 | 0.12 | 0.191.2 |
| Less than independent for transfers | 11 (6) | 3 (12) | 2.1 | 0.27 | 0.568.3 |
| Less than independent for mobility on a level surface | 36 (20) | 7 (28) | 1.6 | 0.33 | 0.624.1 |
| Score of 24 on CAGE questionnaire[29] | 5 (3) | 0 (0) | No outcomes | ||
| Drinks any alcohol | 84 (46) | 10 (40) | 0.79 | 0.60 | 0.341.9 |
| Current smoker | 20 (11) | 2 (8) | 0.71 | 0.66 | 0.164.1 |
| Uses illicit drugs | 13 (7) | 2 (8) | 1.2 | 0.83 | 0.255.6 |
| Moderately or severely ill on nursing assessment, n=194 | 121 (71) | 23 (96) | 9.3 | 0.031 | 1.270.9 |
| Fever | 8 (4) | 0 (0) | No outcomes | ||
| Serum sodium <134mmol/L | 38 (21) | 3 (12) | 0.52 | 0.31 | 0.151.8 |
| WBC count>10109/L, n=208 | 57 (31) | 6 (24) | 0.70 | 0.47 | 0.261.8 |
| AST>41 U/L, n=131 | 27 (23) | 2 (15) | 0.61 | 0.54 | 0.132.9 |
| BUN:Cr>18, n=208 | 66 (36) | 13 (52) | 1.9 | 0.13 | 0.834.5 |
| Infection as admission diagnosis | 28 (15) | 4 (16) | 1.1 | 0.92 | 0.343.3 |
| Variable | Odds Ratio | 95% Confidence Interval | P Value | Points Toward AWOL Score |
|---|---|---|---|---|
| Age 80 years | 5.7 | 2.115.6 | 0.001 | 1 |
| Unable to correctly spell world backward | 3.5 | 1.39.6 | 0.01 | 1 |
| Not oriented to city, state, county, hospital name, and floor | 2.9 | 1.17.9 | 0.03 | 1 |
| Nursing illness severity assessment of moderately ill, severely ill, or moribund (as opposed to not ill or mildly ill) | 10.5 | 1.386.9 | 0.03 | 1 |
These 4 independent predictors were assigned 1 point each if present to create a prediction rule with a range of possible scores from 0 to 4. There was a significant trend predicting higher rates of delirium with higher scores, with no subjects who scored 0 becoming delirious, compared to 40% of those subjects scoring 3 or 4 (P for trend<0.001) (Table 4).
| Derivation Cohorta | Validation Cohort | Combined Cohorts | ||||
|---|---|---|---|---|---|---|
| AWOL Score | Not Delirious | Delirious | Not Delirious | Delirious | Not Delirious | Delirious |
| ||||||
| 0 | 26 (100%) | 0 (0%) | 24 (96%) | 1 (4%) | 49 (98%) | 1 (2%) |
| 1 | 86 (95%) | 5 (5%) | 57 (97%) | 2 (3%) | 136 (96%) | 5 (4%) |
| 2 | 41 (85%) | 7 (15%) | 44 (90%) | 5 (10%) | 92 (86%) | 15 (14%) |
| 3 | 17 (74%) | 6 (26%) | 22 (79%) | 6 (21%) | 40 (80%) | 10 (20%) |
| 4 | 0 (0%) | 6 (100%) | 4 (100%) | 0 (0%) | 4 (36%) | 7 (64%) |
| Total | 170 | 24 | 151 | 14 | 321 | 38 |
| P<0.001 | P=0.025 | P<0.001 | ||||
The validation cohort consisted of adults with a mean age of 70.7210.6 years, (interquartile range, 5194 years), who were predominantly white (75.8%) and overwhelmingly male (95.2%) (Table 1). The mean admission MMSE score was 26.75 (SD, 2.8; range, 1730). Median follow‐up was 2 days (interquartile range, 15). Delirium developed in 8.5% (n=14) of the cohort. In the validation cohort, 4% of subjects with a score of 0 became delirious, whereas 19% of those scoring 3 or 4 became delirious (P for trend 0.025) (Table 4).
ROC curves were compared for the derivation and validation cohorts. The area under the ROC curve for the derivation cohort (0.81, 95% confidence interval [CI]: 0.720.90) was slightly better than that in the validation cohort (0.69, 95% CI: 0.540.83), but the difference did not reach statistical significance (P=0.14) (Figure 1).

DISCUSSION
We derived and validated a prediction rule to assess the risk of developing delirium in hospitalized adult medical patients. Four variables easily assessed on admission in a screen lasting less than 2 minutes were independently associated with the development of delirium. The prediction rule can be remembered with the following mnemonic: AWOL (Age80 years; unable to spell World backward; not fully Oriented to place; and moderate or severe iLlness severity).
It is estimated up to a third of hospital acquired delirium cases can be prevented.[10] Recent guidelines recommend the use of a multicomponent intervention to prevent delirium and provide evidence that such a strategy would be cost‐effective.[39] Nevertheless, such interventions are resource intense, requiring specialized nurse training and staffing[40] and have not been widely implemented. Acute care for the elderly units, where interventions to prevent delirium might logically be implemented, also require physical remodeling to provide carpeted hallways, handrails, and elevated toilet seats and door levers.[41] A method of risk stratification to identify the patients who would benefit most from resource‐intensive prevention strategies would be valuable.
The AWOL tool may provide a practical alternative to existing delirium prediction rules for adult medical inpatients. Because it can be completed by a nurse in <2 minutes, the AWOL tool may be easier to apply and disseminate than a previously described score relying on the MMSE, Acute Physiology and Chronic Health Evaluation scores, and measured visual acuity.[17] Two other tools, 1 based on chart abstraction[18] and the other based on clinical variables measured at admission,[19] are similarly easy to apply but only predict prevalent and not incident delirium, making them less clinically useful.
This study's strengths include its prospective cohort design and the derivation and validation being performed in different hospitals. The derivation cohort consisted of patients admitted to a tertiary care academic medical center or an affiliated hospital where routine mixed gender general medical patients are treated, whereas validation was performed at the SFVAMC, where patients are predominantly older men with a high incidence of vascular risk factors. The outcome was assessed on a daily basis, and the likelihood any cases were missed was low. Although there is some potential for bias because the outcome was assessed by a research assistant not blinded to baseline characteristics, this was mitigated by having each outcome validated by a blinded neurologist and in the validation cohort having the research assistant blinded to the AWOL score. Other strengths are the broad inclusion criteria, with both middle‐aged and elderly patients having a wide range of medical and neurological conditions, allowing for wide application of the results. Although many studies of delirium focus on patients over age 70 years, we chose to include patients aged 50 years or older because hospital‐acquired delirium still occurs in this age group (17 of 195 [8%] patients aged 5069 years became delirious in this study), and risk factors such as severe illness and cognitive dysfunction are likely to be predictors of delirium even at younger ages. Additionally, the inclusion of nurses' clinical judgment to assess illness severity using a straightforward rating scale allows bedside nurses to readily administer the prediction rule in practice.[34]
This study has several potential limitations. The number of outcomes in the derivation cohort was small compared to the number of predictors chosen for the prediction rule. This could potentially have led to overfitting the model in the derivation cohort and thus an overly optimistic estimation of the model's performance. In the validation cohort, the area under the ROC curve was lower than in the derivation cohort, and although the difference did not reach statistical significance, this may have been due to the small sample size. In addition, none of the 4 subjects with an AWOL score of 4 became delirious, potentially reflecting poor calibration of the prediction rule. However, the trend of higher rates of delirium among subjects with higher AWOL scores still reached statistical significance, and the prediction rule demonstrated good discrimination between patients at high and low risk for developing delirium.
To test whether a better prediction tool could be derived from our data, we combined the derivation and validation cohorts and repeated a stepwise multivariable logistic regression with the same variables used for derivation of the AWOL tool (with the exception of the whisper test of hearing and a past medical history of dementia, because these data were not collected in the validation cohort). This model produced the same 4 independent predictors of delirium used in the AWOL tool. We then used bootstrapping to internally validate the prediction rule, suggesting that the predictors in the AWOL tool were the best fit for the available data. However, given the small number of outcomes in our study, the AWOL tool may benefit from further validation in a larger independent cohort to more precisely calibrate the number of expected outcomes with each score.
Although the majority of medical inpatients were eligible for enrollment in our study, some populations were excluded, and our results may not generalize to these populations. Non‐English speaking patients were excluded to preserve the validity of our study instruments. In addition, patients with profound aphasia or an admission diagnosis of alcohol withdrawal were excluded. Patients discharged on the first day of their hospitalization were excluded either because they were discharged prior to screening or prior to their first follow‐up visit. Therefore, our results may only be valid in patients who remained in the hospital for over 24 hours. In addition, because we only included medical patients, our results cannot necessarily be generalized to the surgical population.
Finally, parts of the prediction rule (orientation and spelling world backward) are also components of the CAM and were used in the assessment of the outcome, and this may introduce a potential tautology: if patients are disoriented or have poor attention because they cannot spell world backward at admission, they already have fulfilled part of the criteria for delirium. However, a diagnosis of delirium using the CAM involves a comprehensive patient and caregiver interview, and in addition to poor attention, requires the presence of an acute change in mental status and disorganized thinking or altered level of consciousness. Therefore, it is possible, and common, for patients to be disoriented to place and/or unable to spell world backward, yet not be delirious, and predicting a subsequent change in cognition during the hospitalization is still clinically important. It is possible the AWOL tool works by identifying patients with impaired attention and subclinical delirium, but one could argue this makes a strong case for its validity because these patients especially should be triaged to an inpatient unit that specializes in delirium prevention. It is also possible the cognitive tasks that are part of the AWOL tool detect preexisting cognitive impairment, which is in turn a major risk factor for delirium.
Recognizing and classifying the risk of delirium during hospitalization is imperative, considering the illness' significant contribution to healthcare costs, morbidity, and mortality. The cost‐effectiveness of proven interventions to detect and prevent delirium could be magnified with focused implementation in those patients at highest risk.[39, 40, 41] Further research is required to determine whether the combination of delirium prediction rules such as those developed here and prevention strategies will result in decreased rates of delirium and economic savings for the healthcare system.
Acknowledgments
The following University of California, San Francisco neurology residents provided follow‐up of study subjects on weekends and were financially compensated: Amar Dhand, MD, DPhil; Tim West, MD; Sarah Shalev, MD; Karen DaSilva, MD; Mark Burish, MD, PhD; Maggie Waung, MD, PhD; Raquel Gardner, MD; Molly Burnett, MD; Adam Ziemann, MD, PhD; Kathryn Kvam, MD; Neel Singhal, MD, PhD; James Orengo, MD, PhD; Kelly Mills, MD; and Joanna Hellmuth, MD, MHS. The authors are grateful to Dr. Douglas Bauer for assisting with the study design.
Disclosures
Drs. Douglas, Hessler, Dhaliwal, Betjemann, Lucatorto, Johnston, Josephson, and Ms. Fukuda and Ms. Alameddine have no conflicts of interest or financial disclosures. This research was made possible by the Ruth E. Raskin Fund and a UCSF Dean's Research Scholarship. These funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
- , , . Occurrence and outcome of delirium in medical in‐patients: a systematic literature review. Age Ageing. 2006;35(4):350–364.
- , , , , , . Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591–598.
- , , , , . One‐year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27–32.
- , , , , . Does delirium contribute to poor hospital outcomes? A three‐site epidemiologic study. J Gen Intern Med. 1998;13(4):234–242.
- , , , , , . Delirium duration and mortality in lightly sedated, mechanically ventilated intensive care patients. Crit Care Med. 2010;38(12):2311–2318.
- , , , et al. Delirium epidemiology in critical care (DECCA): an international study. Crit Care. 2010;14(6):R210.
- , , , et al. Delirium as a predictor of long‐term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–1520.
- , , , , , . Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta‐analysis. JAMA. 2010;304(4):443–451.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156(12):848–856.
- , , , et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669–676.
- , , , et al. Simple cognitive testing (Mini‐Cog) predicts in‐hospital delirium in the elderly. J Am Geriatr Soc. 2007;55(2):314–316.
- , , . A prospective study of delirium in hospitalized elderly. JAMA. 1990;263(8):1097–1101.
- , . Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275(11):852–857.
- , , , , , . Risk factors for delirium at discharge: development and validation of a predictive model. Arch Intern Med. 2007;167(13):1406–1413.
- , . Delirium in vascular surgery. Eur J Vasc Endovasc Surg. 2007;34(2):131–134.
- , , , , , . Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42(8):809–815.
- , , , , . A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119(6):474–481.
- , , , , , . Validation of a medical record‐based delirium risk assessment. J Am Geriatr Soc. 2011;59(suppl 2):S289–S294.
- , , , et al. Derivation and validation of a clinical prediction rule for delirium in patients admitted to a medical ward: an observational study. BMJ Open. 2012;2(5) pii: e001599.
- , , , et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134–139.
- , , , , , . Prediction of postoperative delirium after abdominal surgery in the elderly. J Anesth. 2009;23(1):51–56.
- , , , et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229–236.
- , , , et al. Development and validation of PRE‐DELIRIC (PREdiction of DELIRium in ICu patients) delirium prediction model for intensive care patients: observational multicentre study. BMJ. 2012;344:e420.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , . “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198.
- . Wechsler Memory Scale‐III. New York, NY: Psychological Corp.; 1997.
- , , , , . The mini‐cog: a cognitive 'vital signs' measure for dementia screening in multi‐lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021–1027.
- , . Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65.
- , , . The CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry. 1974;131(10):1121–1123.
- , , , , , . Is the NEI‐VFQ‐25 a useful tool in identifying visual impairment in an elderly population? BMC Ophthalmol. 2006;6:24.
- , , , et al. Validation of self‐reported hearing loss. The Blue Mountains Hearing Study. Int J Epidemiol. 2001;30(6):1371–1378.
- , , . Does this patient have hearing impairment? JAMA. 2006;295(4):416–428.
- , , , , , . Realizing the potential of clinical judgment: a real‐time strategy for predicting outcomes and cost for medical inpatients. Am J Med. 2000;109(3):189–195.
- , , , , , . Assessing illness severity: does clinical judgment work? J Chronic Dis. 1986;39(6):439–452.
- , , , , , . Prognostication in acutely admitted older patients by nurses and physicians. J Gen Intern Med. 2008;23(11):1883–1889.
- . The Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, CT: Yale University School of Medicine; 2003.
- , , , . Acute confusional states and dementia in the elderly: the role of dehydration/volume depletion, physical illness and age. Age Ageing. 1980;9(3):137–146.
- . A Wilcoxon‐type test for trend. Stat Med. 1985;4(1):87–90.
- , , , . Synopsis of the National Institute for Health and Clinical Excellence guideline for prevention of delirium. Ann Intern Med. 2011;154(11):746–751.
- , , , , . The Hospital Elder Life Program: a model of care to prevent cognitive and functional decline in older hospitalized patients. Hospital Elder Life Program. J Am Geriatr Soc. 2000;48(12):1697–1706.
- , , , , . A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338–1344.
Delirium is characterized by fluctuating disturbances in cognition and consciousness and is a common complication of hospitalization in medical and surgical patients. Studies estimate the prevalence of delirium in hospitalized patients[1] to be 14% to 56%, and up to 70% in critically ill elderly patients.[2] Estimates of total healthcare costs associated with delirium range from $38 to $152 billion per year in the United States.[3] Delirious patients are more likely to be discharged to a nursing home and have increased hospital mortality and longer lengths of stay.[4, 5, 6] Recent data suggest long‐term effects of delirium including cognitive impairments up to 1 year following the illness[7] and an increased likelihood of developing[8] or worsening dementia.[9]
It is estimated that one‐third of hospital‐acquired delirium cases could be prevented with appropriate interventions.[10] A prediction rule that easily and accurately identifies high‐risk patients upon admission could therefore have a substantial clinical impact. In addition, a prediction rule could be used to identify patients in whom new targeted interventions for delirium prevention could be investigated. A number of risk factors for delirium have been identified, including older age, preexisting cognitive dysfunction, vision and hearing impairment, severe illness, dehydration, electrolyte abnormalities, overmedication, and alcohol abuse.[11, 12, 13, 14, 15, 16] Existing prediction rules using various combinations of these measures have been limited by their complexity,[17] do not predict incident delirium,[18, 19] or are restricted to surgical[20, 21, 22] or intensive care[23] patients and therefore are not broadly applicable to the general medical population, which is particularly susceptible to developing delirium.
We conducted this study to develop a simple, efficient, and accurate prediction rule for hospital‐acquired delirium in adult medical inpatients assessed at the time of admission. Our a priori hypothesis was that a delirium prediction rule would consist of a combination of known risk factors and most likely incorporate old age, illness severity, and preexisting cognitive dysfunction.
METHODS
Design and Setting
This was a prospective cohort study with a derivation phase from May 2010 to November 2010 at 2 hospitals at the University of California, San Francisco (UCSF) (Moffitt‐Long and Mount Zion Hospitals) and a validation phase from October 2011 to March 2012 at the San Francisco Veterans Affairs Medical Center (SFVAMC).
Participants and Measurements
Subject identification, recruitment, and inclusion and exclusion criteria were identical for the derivation and validation cohorts. Subjects were identified by reviewing daily admission logs. All non‐intensive care unit patients aged 50 years or older admitted through the emergency department to the medicine, cardiology, or neurology services were screened for eligibility through chart review or in person within 24 hours of admission by a trained research assistant. One research assistant, a college graduate, conducted all screening for the derivation cohort, and 2 research assistants, 1 a fourth‐year medical student and the other a third‐year psychology graduate student, conducted screening for the validation cohort. In‐person screening included an assessment for delirium using the long version of the Confusion Assessment Method (CAM).[24] To minimize the possibility of enrolling delirious subjects, research assistants were instructed to notify the study supervisor (V.C.D.), a board‐certified neurologist, to discuss every case in which any yes checkbox was marked on the CAM score sheet. Subjects delirious upon initial evaluation, admitted for alcohol withdrawal, admitted for comfort care, who were aphasic or who could not speak English were excluded. For all patients, or if they were unable to provide consent, their surrogates provided written informed consent, and the study was approved by the institutional review boards at UCSF and SFVAMC.
In the derivation cohort, 1241 patients were screened, and 439 were eligible for enrollment. Of these, 180 declined, 50 were discharged prior to the first follow‐up visit, and 209 were included. In the validation cohort, 420 patients were screened, and 368 were eligible for enrollment. Of these, 144 declined, 59 were discharged prior to the first follow‐up visit, and 165 were included.
Baseline data regarding known delirium risk factors[11, 12, 13, 14, 15, 16] were collected from subjects in the derivation cohort. Cognitive performance was assessed with the Mini Mental Status Examination (MMSE),[25] forward digit span,[26] and clock draw.[27] Permission for administration of the MMSE was granted by Psychological Assessment Resources, Inc., and each administration was paid for. A structured interview was conducted with validated questions regarding visual and hearing impairment, pain, mobility, place of residence, and alcohol, tobacco, and drug use.[28, 29, 30, 31] A whisper test for hearing loss was performed.[32] Subjects' charts were reviewed for demographic, clinical, and laboratory data. Illness severity was assessed by asking each subject's nurse to rate their patient on a scale from not ill to mildly ill, moderately ill, severely ill, or moribund.[33] Each nurse was shown these 5 choices, but more specific definitions of what each level of illness severity meant were not provided. We chose this method to assess illness severity because this rating scale was incorporated into a previous validated and widely cited delirium prediction rule.[17] This illness severity scale has been validated as a predictor of outcomes and correlates with other measures of illness severity and comorbidity when graded by physicians.[33, 34] Nurse and physician ratings of illness severity have been shown to be comparable,[35] and therefore if the scale were incorporated into the prediction rule it would allow nurses to perform it independently. In the validation cohort, only data required to complete the baseline CAM and apply the prediction rule were collected.
Assessment of Outcomes
All subjects were assessed for delirium daily for 6 days after enrollment or until discharge, whichever came first. Follow‐up was limited to 6 days, based on the assumption that delirium occurring beyond 1 week is more likely due to events during the hospitalization as opposed to factors measurable at admission. Delirium was assessed using the short CAM, an internationally recognized and validated tool.[24] To complete the CAM during follow‐up visits, subjects and their nurses were interviewed using a written script, and an MMSE and forward digit span were performed.
Daily follow‐up assessments were performed by research assistants who were not blinded to the initial assessment but who, in the validation phase, were blinded to the prediction rule score. Some weekend follow‐ups were performed by postgraduate year 2, 3, or 4 neurology residents, or internal medicine faculty experienced in the assessment of delirium and blinded to both the initial assessment and prediction rule score. Neurology residents and internists read the CAM training manual and were educated in the administration and scoring of the CAM by 1 of the senior investigators (V.C.D.) prior to their first shift; these nonstudy personnel covered 17 of 189 days of follow‐up in the derivation cohort and 21 of 169 days of follow‐up in the validation cohort. To maximize sensitivity of delirium detection, for any change in cognition, MMSE score, or forward digit span compared to baseline, a board‐certified neurologist blinded to the initial assessment was notified to discuss the case and validate the diagnosis of delirium in person (derivation cohort) or over the phone (validation cohort). All research assistants were trained by a board‐certified neurologist (V.C.D.) in the administration and interpretation of the CAM using published methods prior to enrollment of any subjects.[36] Training included the performance of independent long‐version CAMs by the trainer and the trainee on a series of delirious and nondelirious patients until there was consistent agreement for each item on the CAM in 5 consecutive patients. In addition, a board‐certified neurologist supervised the first 5 administrations of the CAM performed by each research assistant.
Statistical Analysis
Sample size for the derivation cohort was based on the predicted ability to detect a difference in rates of delirium among those with and without cognitive impairment, the strongest risk factor for delirium. Using a [2] test with an of 0.05 and of 0.80, we estimated we would need to enroll 260 subjects, assuming a prevalence of cognitive dysfunction in our cohort of 10% and an estimated rate of delirium of 24% and 6% among those with and without cognitive dysfunction respectively.[14, 16, 17, 20] We were unable to reach enrollment targets because of a short funding period and slower than expected recruitment.
To construct the prediction rule in the derivation cohort, all variables were dichotomized. Age was dichotomized at 80 years because old age is a known risk factor for delirium, and only 1 of 46 subjects between the ages of 70 and 80 years became delirious in the derivation cohort. Components of the MMSE were dichotomized as correct/emncorrect, with a correct response requiring perfect performance based on expert consensus. For 3 subjects who would not attempt to spell world backward (2 in the derivation and 1 in the validation cohort), their score on serial 7s was used instead. The total MMSE score was not used because our objective was to develop a prediction rule using elements that could be assessed quickly in the fast‐paced environment of the hospital. Illness severity was dichotomized at moderate or worse/mild or better because there were only 15 subjects in the severe illness category, and the majority of delirium (22 outcomes) occurred in the moderate illness category. High blood urea nitrogen:creatinine ratio was defined as >18.[37]
The association between predictor variables and occurrence of delirium was analyzed using univariate logistic regression. A forward stepwise logistic regression was then performed using the variables associated with the outcome at a significance level of P<0.05 in univariate analysis. Variables were eligible for addition to the multivariable model if they were associated with the outcome at a significance level of <0.05. The 4 independent predictors thus identified were combined into a prediction rule by assigning each predictor 1 point if present. The performance of the prediction rule was assessed by using Cuzick's nonparametric test for a trend across groups ordered by score.[38]
The prediction rule was tested in the validation cohort using the nonparametric test for trend. Receiver operating characteristic (ROC) curves were compared between the derivation and validation cohorts. All statistical analysis was performed using Stata software (StataCorp, College Station, TX).
RESULTS
The derivation cohort consisted of elderly patients (mean age, 68.0811.96 years; interquartile range, 5096 years), and included more males than females (54.1% vs 45.9%). Subjects were predominantly white (73.7%) and lived at home (90%) (Table 1). The mean admission MMSE score was 27.0 (standard deviation [SD], 3.4; range, 730). Median follow‐up was 2 days (interquartile range, 13). Delirium developed in 12% (n=25) of the cohort.
| Derivation Cohort, N=209 | Validation Cohort, N=165 | |
|---|---|---|
| ||
| Gender, No. (%) | ||
| Male | 113 (54) | 157 (95) |
| Female | 96 (46) | 8 (4.8) |
| Race, No. (%) | ||
| White | 154 (74) | 125 (76) |
| African American | 34 (16) | 25 (15) |
| Asian | 21 (10.0) | 13 (7.9) |
| Native American | 0 | 2 (1.2) |
| Illness severity, No. (%) | ||
| Not ill | 1 (0.5) | 0 |
| Mildly ill | 49 (23) | 62 (38) |
| Moderately ill | 129 (62) | 86 (52) |
| Severely ill | 15 (7.2) | 17 (10) |
| Moribund | 0 | 0 |
| Living situation, No. (%) | ||
| Home | 188 (90) | 147 (89) |
| Assisted living | 11 (5.3) | 6 (3.6) |
| Hotel | 4 (1.9) | 5 (3.0) |
| SNF | 1 (0.5) | 3 (1.8) |
| Homeless | 4 (1.9) | 4 (2.4) |
| Developed delirium | 25 (12) | 14 (8.5) |
Univariate analysis of the derivation study identified 10 variables significantly associated (P<0.05) with delirium (Table 2). Predictors of delirium included abnormal scores on 4 subtests of the MMSE, low score on the Mini‐Cog, living in an assisted living or skilled nursing facility, moderate to severe illness, old age, a past history of dementia, and hearing loss as assessed by the whisper test. These predictors were then entered into a stepwise logistic regression analysis that identified 4 independent predictors of delirium (Table 3).
| Variable | No. (%) Without Delirium | No. (%) With Delirium | Odds Ratio | P Value | 95% Confidence Interval |
|---|---|---|---|---|---|
| |||||
| Age 80 years | 30 (16) | 13 (52) | 5.6 | <0.001 | 2.313.4 |
| Male sex | 99 (54) | 14 (56) | 1.1 | 0.84 | 0.52.5 |
| White race | 135 (73) | 19 (76) | 1.2 | 0.78 | 0.433.1 |
| Score <5 on date questions of MMSE | 37 (20) | 12 (48) | 3.7 | 0.003 | 1.68.7 |
| Score <5 on place questions of MMSE | 50 (27) | 14 (56) | 3.4 | 0.005 | 1.58.0 |
| Score <3 on MMSE recall | 89 (48) | 18 (72) | 2.7 | 0.03 | 1.16.9 |
| Score <5 on MMSE W‐O‐R‐L‐D backward | 37 (20) | 13 (52) | 4.3 | 0.001 | 1.810.2 |
| Score 0 on MMSE pentagon copy, n=203 | 53 (30) | 12 (48) | 2.2 | 0.07 | 0.935.1 |
| Score 0 on clock draw, n=203 | 70 (39) | 15 (60) | 2.3 | 0.05 | 0.985.4 |
| MiniCog score 02, n=203[27] | 46 (26) | 12 (48) | 2.7 | 0.03 | 1.16.2 |
| Self‐rated vision fair, poor, or very poor | 55 (30) | 8 (32) | 1.1 | 0.83 | 0.452.7 |
| Endorses hearing loss | 89 (48) | 12 (48) | 0.99 | 0.97 | 0.432.3 |
| Uses hearing aid | 19 (10) | 2 (8) | 0.76 | 0.72 | 0.173.5 |
| Fails whisper test in either ear | 39 (21) | 10 (40) | 2.5 | 0.04 | 1.05.9 |
| Prior episode of delirium per patient or informant | 70 (38) | 13 (52) | 1.8 | 0.19 | 0.764.1 |
| Dementia in past medical history | 3 (2) | 3 (12) | 8.2 | 0.01 | 1.643.3 |
| Depression in past medical history | 16 (9) | 1 (4) | 0.44 | 0.43 | 0.063.5 |
| Lives in assisted living or SNF | 8 (4) | 4 (16) | 4.2 | 0.03 | 1.215.1 |
| Endorses pain | 82 (45) | 7 (28) | 0.48 | 0.12 | 0.191.2 |
| Less than independent for transfers | 11 (6) | 3 (12) | 2.1 | 0.27 | 0.568.3 |
| Less than independent for mobility on a level surface | 36 (20) | 7 (28) | 1.6 | 0.33 | 0.624.1 |
| Score of 24 on CAGE questionnaire[29] | 5 (3) | 0 (0) | No outcomes | ||
| Drinks any alcohol | 84 (46) | 10 (40) | 0.79 | 0.60 | 0.341.9 |
| Current smoker | 20 (11) | 2 (8) | 0.71 | 0.66 | 0.164.1 |
| Uses illicit drugs | 13 (7) | 2 (8) | 1.2 | 0.83 | 0.255.6 |
| Moderately or severely ill on nursing assessment, n=194 | 121 (71) | 23 (96) | 9.3 | 0.031 | 1.270.9 |
| Fever | 8 (4) | 0 (0) | No outcomes | ||
| Serum sodium <134mmol/L | 38 (21) | 3 (12) | 0.52 | 0.31 | 0.151.8 |
| WBC count>10109/L, n=208 | 57 (31) | 6 (24) | 0.70 | 0.47 | 0.261.8 |
| AST>41 U/L, n=131 | 27 (23) | 2 (15) | 0.61 | 0.54 | 0.132.9 |
| BUN:Cr>18, n=208 | 66 (36) | 13 (52) | 1.9 | 0.13 | 0.834.5 |
| Infection as admission diagnosis | 28 (15) | 4 (16) | 1.1 | 0.92 | 0.343.3 |
| Variable | Odds Ratio | 95% Confidence Interval | P Value | Points Toward AWOL Score |
|---|---|---|---|---|
| Age 80 years | 5.7 | 2.115.6 | 0.001 | 1 |
| Unable to correctly spell world backward | 3.5 | 1.39.6 | 0.01 | 1 |
| Not oriented to city, state, county, hospital name, and floor | 2.9 | 1.17.9 | 0.03 | 1 |
| Nursing illness severity assessment of moderately ill, severely ill, or moribund (as opposed to not ill or mildly ill) | 10.5 | 1.386.9 | 0.03 | 1 |
These 4 independent predictors were assigned 1 point each if present to create a prediction rule with a range of possible scores from 0 to 4. There was a significant trend predicting higher rates of delirium with higher scores, with no subjects who scored 0 becoming delirious, compared to 40% of those subjects scoring 3 or 4 (P for trend<0.001) (Table 4).
| Derivation Cohorta | Validation Cohort | Combined Cohorts | ||||
|---|---|---|---|---|---|---|
| AWOL Score | Not Delirious | Delirious | Not Delirious | Delirious | Not Delirious | Delirious |
| ||||||
| 0 | 26 (100%) | 0 (0%) | 24 (96%) | 1 (4%) | 49 (98%) | 1 (2%) |
| 1 | 86 (95%) | 5 (5%) | 57 (97%) | 2 (3%) | 136 (96%) | 5 (4%) |
| 2 | 41 (85%) | 7 (15%) | 44 (90%) | 5 (10%) | 92 (86%) | 15 (14%) |
| 3 | 17 (74%) | 6 (26%) | 22 (79%) | 6 (21%) | 40 (80%) | 10 (20%) |
| 4 | 0 (0%) | 6 (100%) | 4 (100%) | 0 (0%) | 4 (36%) | 7 (64%) |
| Total | 170 | 24 | 151 | 14 | 321 | 38 |
| P<0.001 | P=0.025 | P<0.001 | ||||
The validation cohort consisted of adults with a mean age of 70.7210.6 years, (interquartile range, 5194 years), who were predominantly white (75.8%) and overwhelmingly male (95.2%) (Table 1). The mean admission MMSE score was 26.75 (SD, 2.8; range, 1730). Median follow‐up was 2 days (interquartile range, 15). Delirium developed in 8.5% (n=14) of the cohort. In the validation cohort, 4% of subjects with a score of 0 became delirious, whereas 19% of those scoring 3 or 4 became delirious (P for trend 0.025) (Table 4).
ROC curves were compared for the derivation and validation cohorts. The area under the ROC curve for the derivation cohort (0.81, 95% confidence interval [CI]: 0.720.90) was slightly better than that in the validation cohort (0.69, 95% CI: 0.540.83), but the difference did not reach statistical significance (P=0.14) (Figure 1).
DISCUSSION
We derived and validated a prediction rule to assess the risk of developing delirium in hospitalized adult medical patients. Four variables easily assessed on admission in a screen lasting less than 2 minutes were independently associated with the development of delirium. The prediction rule can be remembered with the following mnemonic: AWOL (Age80 years; unable to spell World backward; not fully Oriented to place; and moderate or severe iLlness severity).
It is estimated up to a third of hospital acquired delirium cases can be prevented.[10] Recent guidelines recommend the use of a multicomponent intervention to prevent delirium and provide evidence that such a strategy would be cost‐effective.[39] Nevertheless, such interventions are resource intense, requiring specialized nurse training and staffing[40] and have not been widely implemented. Acute care for the elderly units, where interventions to prevent delirium might logically be implemented, also require physical remodeling to provide carpeted hallways, handrails, and elevated toilet seats and door levers.[41] A method of risk stratification to identify the patients who would benefit most from resource‐intensive prevention strategies would be valuable.
The AWOL tool may provide a practical alternative to existing delirium prediction rules for adult medical inpatients. Because it can be completed by a nurse in <2 minutes, the AWOL tool may be easier to apply and disseminate than a previously described score relying on the MMSE, Acute Physiology and Chronic Health Evaluation scores, and measured visual acuity.[17] Two other tools, 1 based on chart abstraction[18] and the other based on clinical variables measured at admission,[19] are similarly easy to apply but only predict prevalent and not incident delirium, making them less clinically useful.
This study's strengths include its prospective cohort design and the derivation and validation being performed in different hospitals. The derivation cohort consisted of patients admitted to a tertiary care academic medical center or an affiliated hospital where routine mixed gender general medical patients are treated, whereas validation was performed at the SFVAMC, where patients are predominantly older men with a high incidence of vascular risk factors. The outcome was assessed on a daily basis, and the likelihood any cases were missed was low. Although there is some potential for bias because the outcome was assessed by a research assistant not blinded to baseline characteristics, this was mitigated by having each outcome validated by a blinded neurologist and in the validation cohort having the research assistant blinded to the AWOL score. Other strengths are the broad inclusion criteria, with both middle‐aged and elderly patients having a wide range of medical and neurological conditions, allowing for wide application of the results. Although many studies of delirium focus on patients over age 70 years, we chose to include patients aged 50 years or older because hospital‐acquired delirium still occurs in this age group (17 of 195 [8%] patients aged 5069 years became delirious in this study), and risk factors such as severe illness and cognitive dysfunction are likely to be predictors of delirium even at younger ages. Additionally, the inclusion of nurses' clinical judgment to assess illness severity using a straightforward rating scale allows bedside nurses to readily administer the prediction rule in practice.[34]
This study has several potential limitations. The number of outcomes in the derivation cohort was small compared to the number of predictors chosen for the prediction rule. This could potentially have led to overfitting the model in the derivation cohort and thus an overly optimistic estimation of the model's performance. In the validation cohort, the area under the ROC curve was lower than in the derivation cohort, and although the difference did not reach statistical significance, this may have been due to the small sample size. In addition, none of the 4 subjects with an AWOL score of 4 became delirious, potentially reflecting poor calibration of the prediction rule. However, the trend of higher rates of delirium among subjects with higher AWOL scores still reached statistical significance, and the prediction rule demonstrated good discrimination between patients at high and low risk for developing delirium.
To test whether a better prediction tool could be derived from our data, we combined the derivation and validation cohorts and repeated a stepwise multivariable logistic regression with the same variables used for derivation of the AWOL tool (with the exception of the whisper test of hearing and a past medical history of dementia, because these data were not collected in the validation cohort). This model produced the same 4 independent predictors of delirium used in the AWOL tool. We then used bootstrapping to internally validate the prediction rule, suggesting that the predictors in the AWOL tool were the best fit for the available data. However, given the small number of outcomes in our study, the AWOL tool may benefit from further validation in a larger independent cohort to more precisely calibrate the number of expected outcomes with each score.
Although the majority of medical inpatients were eligible for enrollment in our study, some populations were excluded, and our results may not generalize to these populations. Non‐English speaking patients were excluded to preserve the validity of our study instruments. In addition, patients with profound aphasia or an admission diagnosis of alcohol withdrawal were excluded. Patients discharged on the first day of their hospitalization were excluded either because they were discharged prior to screening or prior to their first follow‐up visit. Therefore, our results may only be valid in patients who remained in the hospital for over 24 hours. In addition, because we only included medical patients, our results cannot necessarily be generalized to the surgical population.
Finally, parts of the prediction rule (orientation and spelling world backward) are also components of the CAM and were used in the assessment of the outcome, and this may introduce a potential tautology: if patients are disoriented or have poor attention because they cannot spell world backward at admission, they already have fulfilled part of the criteria for delirium. However, a diagnosis of delirium using the CAM involves a comprehensive patient and caregiver interview, and in addition to poor attention, requires the presence of an acute change in mental status and disorganized thinking or altered level of consciousness. Therefore, it is possible, and common, for patients to be disoriented to place and/or unable to spell world backward, yet not be delirious, and predicting a subsequent change in cognition during the hospitalization is still clinically important. It is possible the AWOL tool works by identifying patients with impaired attention and subclinical delirium, but one could argue this makes a strong case for its validity because these patients especially should be triaged to an inpatient unit that specializes in delirium prevention. It is also possible the cognitive tasks that are part of the AWOL tool detect preexisting cognitive impairment, which is in turn a major risk factor for delirium.
Recognizing and classifying the risk of delirium during hospitalization is imperative, considering the illness' significant contribution to healthcare costs, morbidity, and mortality. The cost‐effectiveness of proven interventions to detect and prevent delirium could be magnified with focused implementation in those patients at highest risk.[39, 40, 41] Further research is required to determine whether the combination of delirium prediction rules such as those developed here and prevention strategies will result in decreased rates of delirium and economic savings for the healthcare system.
Acknowledgments
The following University of California, San Francisco neurology residents provided follow‐up of study subjects on weekends and were financially compensated: Amar Dhand, MD, DPhil; Tim West, MD; Sarah Shalev, MD; Karen DaSilva, MD; Mark Burish, MD, PhD; Maggie Waung, MD, PhD; Raquel Gardner, MD; Molly Burnett, MD; Adam Ziemann, MD, PhD; Kathryn Kvam, MD; Neel Singhal, MD, PhD; James Orengo, MD, PhD; Kelly Mills, MD; and Joanna Hellmuth, MD, MHS. The authors are grateful to Dr. Douglas Bauer for assisting with the study design.
Disclosures
Drs. Douglas, Hessler, Dhaliwal, Betjemann, Lucatorto, Johnston, Josephson, and Ms. Fukuda and Ms. Alameddine have no conflicts of interest or financial disclosures. This research was made possible by the Ruth E. Raskin Fund and a UCSF Dean's Research Scholarship. These funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Delirium is characterized by fluctuating disturbances in cognition and consciousness and is a common complication of hospitalization in medical and surgical patients. Studies estimate the prevalence of delirium in hospitalized patients[1] to be 14% to 56%, and up to 70% in critically ill elderly patients.[2] Estimates of total healthcare costs associated with delirium range from $38 to $152 billion per year in the United States.[3] Delirious patients are more likely to be discharged to a nursing home and have increased hospital mortality and longer lengths of stay.[4, 5, 6] Recent data suggest long‐term effects of delirium including cognitive impairments up to 1 year following the illness[7] and an increased likelihood of developing[8] or worsening dementia.[9]
It is estimated that one‐third of hospital‐acquired delirium cases could be prevented with appropriate interventions.[10] A prediction rule that easily and accurately identifies high‐risk patients upon admission could therefore have a substantial clinical impact. In addition, a prediction rule could be used to identify patients in whom new targeted interventions for delirium prevention could be investigated. A number of risk factors for delirium have been identified, including older age, preexisting cognitive dysfunction, vision and hearing impairment, severe illness, dehydration, electrolyte abnormalities, overmedication, and alcohol abuse.[11, 12, 13, 14, 15, 16] Existing prediction rules using various combinations of these measures have been limited by their complexity,[17] do not predict incident delirium,[18, 19] or are restricted to surgical[20, 21, 22] or intensive care[23] patients and therefore are not broadly applicable to the general medical population, which is particularly susceptible to developing delirium.
We conducted this study to develop a simple, efficient, and accurate prediction rule for hospital‐acquired delirium in adult medical inpatients assessed at the time of admission. Our a priori hypothesis was that a delirium prediction rule would consist of a combination of known risk factors and most likely incorporate old age, illness severity, and preexisting cognitive dysfunction.
METHODS
Design and Setting
This was a prospective cohort study with a derivation phase from May 2010 to November 2010 at 2 hospitals at the University of California, San Francisco (UCSF) (Moffitt‐Long and Mount Zion Hospitals) and a validation phase from October 2011 to March 2012 at the San Francisco Veterans Affairs Medical Center (SFVAMC).
Participants and Measurements
Subject identification, recruitment, and inclusion and exclusion criteria were identical for the derivation and validation cohorts. Subjects were identified by reviewing daily admission logs. All non‐intensive care unit patients aged 50 years or older admitted through the emergency department to the medicine, cardiology, or neurology services were screened for eligibility through chart review or in person within 24 hours of admission by a trained research assistant. One research assistant, a college graduate, conducted all screening for the derivation cohort, and 2 research assistants, 1 a fourth‐year medical student and the other a third‐year psychology graduate student, conducted screening for the validation cohort. In‐person screening included an assessment for delirium using the long version of the Confusion Assessment Method (CAM).[24] To minimize the possibility of enrolling delirious subjects, research assistants were instructed to notify the study supervisor (V.C.D.), a board‐certified neurologist, to discuss every case in which any yes checkbox was marked on the CAM score sheet. Subjects delirious upon initial evaluation, admitted for alcohol withdrawal, admitted for comfort care, who were aphasic or who could not speak English were excluded. For all patients, or if they were unable to provide consent, their surrogates provided written informed consent, and the study was approved by the institutional review boards at UCSF and SFVAMC.
In the derivation cohort, 1241 patients were screened, and 439 were eligible for enrollment. Of these, 180 declined, 50 were discharged prior to the first follow‐up visit, and 209 were included. In the validation cohort, 420 patients were screened, and 368 were eligible for enrollment. Of these, 144 declined, 59 were discharged prior to the first follow‐up visit, and 165 were included.
Baseline data regarding known delirium risk factors[11, 12, 13, 14, 15, 16] were collected from subjects in the derivation cohort. Cognitive performance was assessed with the Mini Mental Status Examination (MMSE),[25] forward digit span,[26] and clock draw.[27] Permission for administration of the MMSE was granted by Psychological Assessment Resources, Inc., and each administration was paid for. A structured interview was conducted with validated questions regarding visual and hearing impairment, pain, mobility, place of residence, and alcohol, tobacco, and drug use.[28, 29, 30, 31] A whisper test for hearing loss was performed.[32] Subjects' charts were reviewed for demographic, clinical, and laboratory data. Illness severity was assessed by asking each subject's nurse to rate their patient on a scale from not ill to mildly ill, moderately ill, severely ill, or moribund.[33] Each nurse was shown these 5 choices, but more specific definitions of what each level of illness severity meant were not provided. We chose this method to assess illness severity because this rating scale was incorporated into a previous validated and widely cited delirium prediction rule.[17] This illness severity scale has been validated as a predictor of outcomes and correlates with other measures of illness severity and comorbidity when graded by physicians.[33, 34] Nurse and physician ratings of illness severity have been shown to be comparable,[35] and therefore if the scale were incorporated into the prediction rule it would allow nurses to perform it independently. In the validation cohort, only data required to complete the baseline CAM and apply the prediction rule were collected.
Assessment of Outcomes
All subjects were assessed for delirium daily for 6 days after enrollment or until discharge, whichever came first. Follow‐up was limited to 6 days, based on the assumption that delirium occurring beyond 1 week is more likely due to events during the hospitalization as opposed to factors measurable at admission. Delirium was assessed using the short CAM, an internationally recognized and validated tool.[24] To complete the CAM during follow‐up visits, subjects and their nurses were interviewed using a written script, and an MMSE and forward digit span were performed.
Daily follow‐up assessments were performed by research assistants who were not blinded to the initial assessment but who, in the validation phase, were blinded to the prediction rule score. Some weekend follow‐ups were performed by postgraduate year 2, 3, or 4 neurology residents, or internal medicine faculty experienced in the assessment of delirium and blinded to both the initial assessment and prediction rule score. Neurology residents and internists read the CAM training manual and were educated in the administration and scoring of the CAM by 1 of the senior investigators (V.C.D.) prior to their first shift; these nonstudy personnel covered 17 of 189 days of follow‐up in the derivation cohort and 21 of 169 days of follow‐up in the validation cohort. To maximize sensitivity of delirium detection, for any change in cognition, MMSE score, or forward digit span compared to baseline, a board‐certified neurologist blinded to the initial assessment was notified to discuss the case and validate the diagnosis of delirium in person (derivation cohort) or over the phone (validation cohort). All research assistants were trained by a board‐certified neurologist (V.C.D.) in the administration and interpretation of the CAM using published methods prior to enrollment of any subjects.[36] Training included the performance of independent long‐version CAMs by the trainer and the trainee on a series of delirious and nondelirious patients until there was consistent agreement for each item on the CAM in 5 consecutive patients. In addition, a board‐certified neurologist supervised the first 5 administrations of the CAM performed by each research assistant.
Statistical Analysis
Sample size for the derivation cohort was based on the predicted ability to detect a difference in rates of delirium among those with and without cognitive impairment, the strongest risk factor for delirium. Using a [2] test with an of 0.05 and of 0.80, we estimated we would need to enroll 260 subjects, assuming a prevalence of cognitive dysfunction in our cohort of 10% and an estimated rate of delirium of 24% and 6% among those with and without cognitive dysfunction respectively.[14, 16, 17, 20] We were unable to reach enrollment targets because of a short funding period and slower than expected recruitment.
To construct the prediction rule in the derivation cohort, all variables were dichotomized. Age was dichotomized at 80 years because old age is a known risk factor for delirium, and only 1 of 46 subjects between the ages of 70 and 80 years became delirious in the derivation cohort. Components of the MMSE were dichotomized as correct/emncorrect, with a correct response requiring perfect performance based on expert consensus. For 3 subjects who would not attempt to spell world backward (2 in the derivation and 1 in the validation cohort), their score on serial 7s was used instead. The total MMSE score was not used because our objective was to develop a prediction rule using elements that could be assessed quickly in the fast‐paced environment of the hospital. Illness severity was dichotomized at moderate or worse/mild or better because there were only 15 subjects in the severe illness category, and the majority of delirium (22 outcomes) occurred in the moderate illness category. High blood urea nitrogen:creatinine ratio was defined as >18.[37]
The association between predictor variables and occurrence of delirium was analyzed using univariate logistic regression. A forward stepwise logistic regression was then performed using the variables associated with the outcome at a significance level of P<0.05 in univariate analysis. Variables were eligible for addition to the multivariable model if they were associated with the outcome at a significance level of <0.05. The 4 independent predictors thus identified were combined into a prediction rule by assigning each predictor 1 point if present. The performance of the prediction rule was assessed by using Cuzick's nonparametric test for a trend across groups ordered by score.[38]
The prediction rule was tested in the validation cohort using the nonparametric test for trend. Receiver operating characteristic (ROC) curves were compared between the derivation and validation cohorts. All statistical analysis was performed using Stata software (StataCorp, College Station, TX).
RESULTS
The derivation cohort consisted of elderly patients (mean age, 68.0811.96 years; interquartile range, 5096 years), and included more males than females (54.1% vs 45.9%). Subjects were predominantly white (73.7%) and lived at home (90%) (Table 1). The mean admission MMSE score was 27.0 (standard deviation [SD], 3.4; range, 730). Median follow‐up was 2 days (interquartile range, 13). Delirium developed in 12% (n=25) of the cohort.
| Derivation Cohort, N=209 | Validation Cohort, N=165 | |
|---|---|---|
| ||
| Gender, No. (%) | ||
| Male | 113 (54) | 157 (95) |
| Female | 96 (46) | 8 (4.8) |
| Race, No. (%) | ||
| White | 154 (74) | 125 (76) |
| African American | 34 (16) | 25 (15) |
| Asian | 21 (10.0) | 13 (7.9) |
| Native American | 0 | 2 (1.2) |
| Illness severity, No. (%) | ||
| Not ill | 1 (0.5) | 0 |
| Mildly ill | 49 (23) | 62 (38) |
| Moderately ill | 129 (62) | 86 (52) |
| Severely ill | 15 (7.2) | 17 (10) |
| Moribund | 0 | 0 |
| Living situation, No. (%) | ||
| Home | 188 (90) | 147 (89) |
| Assisted living | 11 (5.3) | 6 (3.6) |
| Hotel | 4 (1.9) | 5 (3.0) |
| SNF | 1 (0.5) | 3 (1.8) |
| Homeless | 4 (1.9) | 4 (2.4) |
| Developed delirium | 25 (12) | 14 (8.5) |
Univariate analysis of the derivation study identified 10 variables significantly associated (P<0.05) with delirium (Table 2). Predictors of delirium included abnormal scores on 4 subtests of the MMSE, low score on the Mini‐Cog, living in an assisted living or skilled nursing facility, moderate to severe illness, old age, a past history of dementia, and hearing loss as assessed by the whisper test. These predictors were then entered into a stepwise logistic regression analysis that identified 4 independent predictors of delirium (Table 3).
| Variable | No. (%) Without Delirium | No. (%) With Delirium | Odds Ratio | P Value | 95% Confidence Interval |
|---|---|---|---|---|---|
| |||||
| Age 80 years | 30 (16) | 13 (52) | 5.6 | <0.001 | 2.313.4 |
| Male sex | 99 (54) | 14 (56) | 1.1 | 0.84 | 0.52.5 |
| White race | 135 (73) | 19 (76) | 1.2 | 0.78 | 0.433.1 |
| Score <5 on date questions of MMSE | 37 (20) | 12 (48) | 3.7 | 0.003 | 1.68.7 |
| Score <5 on place questions of MMSE | 50 (27) | 14 (56) | 3.4 | 0.005 | 1.58.0 |
| Score <3 on MMSE recall | 89 (48) | 18 (72) | 2.7 | 0.03 | 1.16.9 |
| Score <5 on MMSE W‐O‐R‐L‐D backward | 37 (20) | 13 (52) | 4.3 | 0.001 | 1.810.2 |
| Score 0 on MMSE pentagon copy, n=203 | 53 (30) | 12 (48) | 2.2 | 0.07 | 0.935.1 |
| Score 0 on clock draw, n=203 | 70 (39) | 15 (60) | 2.3 | 0.05 | 0.985.4 |
| MiniCog score 02, n=203[27] | 46 (26) | 12 (48) | 2.7 | 0.03 | 1.16.2 |
| Self‐rated vision fair, poor, or very poor | 55 (30) | 8 (32) | 1.1 | 0.83 | 0.452.7 |
| Endorses hearing loss | 89 (48) | 12 (48) | 0.99 | 0.97 | 0.432.3 |
| Uses hearing aid | 19 (10) | 2 (8) | 0.76 | 0.72 | 0.173.5 |
| Fails whisper test in either ear | 39 (21) | 10 (40) | 2.5 | 0.04 | 1.05.9 |
| Prior episode of delirium per patient or informant | 70 (38) | 13 (52) | 1.8 | 0.19 | 0.764.1 |
| Dementia in past medical history | 3 (2) | 3 (12) | 8.2 | 0.01 | 1.643.3 |
| Depression in past medical history | 16 (9) | 1 (4) | 0.44 | 0.43 | 0.063.5 |
| Lives in assisted living or SNF | 8 (4) | 4 (16) | 4.2 | 0.03 | 1.215.1 |
| Endorses pain | 82 (45) | 7 (28) | 0.48 | 0.12 | 0.191.2 |
| Less than independent for transfers | 11 (6) | 3 (12) | 2.1 | 0.27 | 0.568.3 |
| Less than independent for mobility on a level surface | 36 (20) | 7 (28) | 1.6 | 0.33 | 0.624.1 |
| Score of 24 on CAGE questionnaire[29] | 5 (3) | 0 (0) | No outcomes | ||
| Drinks any alcohol | 84 (46) | 10 (40) | 0.79 | 0.60 | 0.341.9 |
| Current smoker | 20 (11) | 2 (8) | 0.71 | 0.66 | 0.164.1 |
| Uses illicit drugs | 13 (7) | 2 (8) | 1.2 | 0.83 | 0.255.6 |
| Moderately or severely ill on nursing assessment, n=194 | 121 (71) | 23 (96) | 9.3 | 0.031 | 1.270.9 |
| Fever | 8 (4) | 0 (0) | No outcomes | ||
| Serum sodium <134mmol/L | 38 (21) | 3 (12) | 0.52 | 0.31 | 0.151.8 |
| WBC count>10109/L, n=208 | 57 (31) | 6 (24) | 0.70 | 0.47 | 0.261.8 |
| AST>41 U/L, n=131 | 27 (23) | 2 (15) | 0.61 | 0.54 | 0.132.9 |
| BUN:Cr>18, n=208 | 66 (36) | 13 (52) | 1.9 | 0.13 | 0.834.5 |
| Infection as admission diagnosis | 28 (15) | 4 (16) | 1.1 | 0.92 | 0.343.3 |
| Variable | Odds Ratio | 95% Confidence Interval | P Value | Points Toward AWOL Score |
|---|---|---|---|---|
| Age 80 years | 5.7 | 2.115.6 | 0.001 | 1 |
| Unable to correctly spell world backward | 3.5 | 1.39.6 | 0.01 | 1 |
| Not oriented to city, state, county, hospital name, and floor | 2.9 | 1.17.9 | 0.03 | 1 |
| Nursing illness severity assessment of moderately ill, severely ill, or moribund (as opposed to not ill or mildly ill) | 10.5 | 1.386.9 | 0.03 | 1 |
These 4 independent predictors were assigned 1 point each if present to create a prediction rule with a range of possible scores from 0 to 4. There was a significant trend predicting higher rates of delirium with higher scores, with no subjects who scored 0 becoming delirious, compared to 40% of those subjects scoring 3 or 4 (P for trend<0.001) (Table 4).
| Derivation Cohorta | Validation Cohort | Combined Cohorts | ||||
|---|---|---|---|---|---|---|
| AWOL Score | Not Delirious | Delirious | Not Delirious | Delirious | Not Delirious | Delirious |
| ||||||
| 0 | 26 (100%) | 0 (0%) | 24 (96%) | 1 (4%) | 49 (98%) | 1 (2%) |
| 1 | 86 (95%) | 5 (5%) | 57 (97%) | 2 (3%) | 136 (96%) | 5 (4%) |
| 2 | 41 (85%) | 7 (15%) | 44 (90%) | 5 (10%) | 92 (86%) | 15 (14%) |
| 3 | 17 (74%) | 6 (26%) | 22 (79%) | 6 (21%) | 40 (80%) | 10 (20%) |
| 4 | 0 (0%) | 6 (100%) | 4 (100%) | 0 (0%) | 4 (36%) | 7 (64%) |
| Total | 170 | 24 | 151 | 14 | 321 | 38 |
| P<0.001 | P=0.025 | P<0.001 | ||||
The validation cohort consisted of adults with a mean age of 70.7210.6 years, (interquartile range, 5194 years), who were predominantly white (75.8%) and overwhelmingly male (95.2%) (Table 1). The mean admission MMSE score was 26.75 (SD, 2.8; range, 1730). Median follow‐up was 2 days (interquartile range, 15). Delirium developed in 8.5% (n=14) of the cohort. In the validation cohort, 4% of subjects with a score of 0 became delirious, whereas 19% of those scoring 3 or 4 became delirious (P for trend 0.025) (Table 4).
ROC curves were compared for the derivation and validation cohorts. The area under the ROC curve for the derivation cohort (0.81, 95% confidence interval [CI]: 0.720.90) was slightly better than that in the validation cohort (0.69, 95% CI: 0.540.83), but the difference did not reach statistical significance (P=0.14) (Figure 1).
DISCUSSION
We derived and validated a prediction rule to assess the risk of developing delirium in hospitalized adult medical patients. Four variables easily assessed on admission in a screen lasting less than 2 minutes were independently associated with the development of delirium. The prediction rule can be remembered with the following mnemonic: AWOL (Age80 years; unable to spell World backward; not fully Oriented to place; and moderate or severe iLlness severity).
It is estimated up to a third of hospital acquired delirium cases can be prevented.[10] Recent guidelines recommend the use of a multicomponent intervention to prevent delirium and provide evidence that such a strategy would be cost‐effective.[39] Nevertheless, such interventions are resource intense, requiring specialized nurse training and staffing[40] and have not been widely implemented. Acute care for the elderly units, where interventions to prevent delirium might logically be implemented, also require physical remodeling to provide carpeted hallways, handrails, and elevated toilet seats and door levers.[41] A method of risk stratification to identify the patients who would benefit most from resource‐intensive prevention strategies would be valuable.
The AWOL tool may provide a practical alternative to existing delirium prediction rules for adult medical inpatients. Because it can be completed by a nurse in <2 minutes, the AWOL tool may be easier to apply and disseminate than a previously described score relying on the MMSE, Acute Physiology and Chronic Health Evaluation scores, and measured visual acuity.[17] Two other tools, 1 based on chart abstraction[18] and the other based on clinical variables measured at admission,[19] are similarly easy to apply but only predict prevalent and not incident delirium, making them less clinically useful.
This study's strengths include its prospective cohort design and the derivation and validation being performed in different hospitals. The derivation cohort consisted of patients admitted to a tertiary care academic medical center or an affiliated hospital where routine mixed gender general medical patients are treated, whereas validation was performed at the SFVAMC, where patients are predominantly older men with a high incidence of vascular risk factors. The outcome was assessed on a daily basis, and the likelihood any cases were missed was low. Although there is some potential for bias because the outcome was assessed by a research assistant not blinded to baseline characteristics, this was mitigated by having each outcome validated by a blinded neurologist and in the validation cohort having the research assistant blinded to the AWOL score. Other strengths are the broad inclusion criteria, with both middle‐aged and elderly patients having a wide range of medical and neurological conditions, allowing for wide application of the results. Although many studies of delirium focus on patients over age 70 years, we chose to include patients aged 50 years or older because hospital‐acquired delirium still occurs in this age group (17 of 195 [8%] patients aged 5069 years became delirious in this study), and risk factors such as severe illness and cognitive dysfunction are likely to be predictors of delirium even at younger ages. Additionally, the inclusion of nurses' clinical judgment to assess illness severity using a straightforward rating scale allows bedside nurses to readily administer the prediction rule in practice.[34]
This study has several potential limitations. The number of outcomes in the derivation cohort was small compared to the number of predictors chosen for the prediction rule. This could potentially have led to overfitting the model in the derivation cohort and thus an overly optimistic estimation of the model's performance. In the validation cohort, the area under the ROC curve was lower than in the derivation cohort, and although the difference did not reach statistical significance, this may have been due to the small sample size. In addition, none of the 4 subjects with an AWOL score of 4 became delirious, potentially reflecting poor calibration of the prediction rule. However, the trend of higher rates of delirium among subjects with higher AWOL scores still reached statistical significance, and the prediction rule demonstrated good discrimination between patients at high and low risk for developing delirium.
To test whether a better prediction tool could be derived from our data, we combined the derivation and validation cohorts and repeated a stepwise multivariable logistic regression with the same variables used for derivation of the AWOL tool (with the exception of the whisper test of hearing and a past medical history of dementia, because these data were not collected in the validation cohort). This model produced the same 4 independent predictors of delirium used in the AWOL tool. We then used bootstrapping to internally validate the prediction rule, suggesting that the predictors in the AWOL tool were the best fit for the available data. However, given the small number of outcomes in our study, the AWOL tool may benefit from further validation in a larger independent cohort to more precisely calibrate the number of expected outcomes with each score.
Although the majority of medical inpatients were eligible for enrollment in our study, some populations were excluded, and our results may not generalize to these populations. Non‐English speaking patients were excluded to preserve the validity of our study instruments. In addition, patients with profound aphasia or an admission diagnosis of alcohol withdrawal were excluded. Patients discharged on the first day of their hospitalization were excluded either because they were discharged prior to screening or prior to their first follow‐up visit. Therefore, our results may only be valid in patients who remained in the hospital for over 24 hours. In addition, because we only included medical patients, our results cannot necessarily be generalized to the surgical population.
Finally, parts of the prediction rule (orientation and spelling world backward) are also components of the CAM and were used in the assessment of the outcome, and this may introduce a potential tautology: if patients are disoriented or have poor attention because they cannot spell world backward at admission, they already have fulfilled part of the criteria for delirium. However, a diagnosis of delirium using the CAM involves a comprehensive patient and caregiver interview, and in addition to poor attention, requires the presence of an acute change in mental status and disorganized thinking or altered level of consciousness. Therefore, it is possible, and common, for patients to be disoriented to place and/or unable to spell world backward, yet not be delirious, and predicting a subsequent change in cognition during the hospitalization is still clinically important. It is possible the AWOL tool works by identifying patients with impaired attention and subclinical delirium, but one could argue this makes a strong case for its validity because these patients especially should be triaged to an inpatient unit that specializes in delirium prevention. It is also possible the cognitive tasks that are part of the AWOL tool detect preexisting cognitive impairment, which is in turn a major risk factor for delirium.
Recognizing and classifying the risk of delirium during hospitalization is imperative, considering the illness' significant contribution to healthcare costs, morbidity, and mortality. The cost‐effectiveness of proven interventions to detect and prevent delirium could be magnified with focused implementation in those patients at highest risk.[39, 40, 41] Further research is required to determine whether the combination of delirium prediction rules such as those developed here and prevention strategies will result in decreased rates of delirium and economic savings for the healthcare system.
Acknowledgments
The following University of California, San Francisco neurology residents provided follow‐up of study subjects on weekends and were financially compensated: Amar Dhand, MD, DPhil; Tim West, MD; Sarah Shalev, MD; Karen DaSilva, MD; Mark Burish, MD, PhD; Maggie Waung, MD, PhD; Raquel Gardner, MD; Molly Burnett, MD; Adam Ziemann, MD, PhD; Kathryn Kvam, MD; Neel Singhal, MD, PhD; James Orengo, MD, PhD; Kelly Mills, MD; and Joanna Hellmuth, MD, MHS. The authors are grateful to Dr. Douglas Bauer for assisting with the study design.
Disclosures
Drs. Douglas, Hessler, Dhaliwal, Betjemann, Lucatorto, Johnston, Josephson, and Ms. Fukuda and Ms. Alameddine have no conflicts of interest or financial disclosures. This research was made possible by the Ruth E. Raskin Fund and a UCSF Dean's Research Scholarship. These funding agencies had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
- , , . Occurrence and outcome of delirium in medical in‐patients: a systematic literature review. Age Ageing. 2006;35(4):350–364.
- , , , , , . Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591–598.
- , , , , . One‐year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27–32.
- , , , , . Does delirium contribute to poor hospital outcomes? A three‐site epidemiologic study. J Gen Intern Med. 1998;13(4):234–242.
- , , , , , . Delirium duration and mortality in lightly sedated, mechanically ventilated intensive care patients. Crit Care Med. 2010;38(12):2311–2318.
- , , , et al. Delirium epidemiology in critical care (DECCA): an international study. Crit Care. 2010;14(6):R210.
- , , , et al. Delirium as a predictor of long‐term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–1520.
- , , , , , . Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta‐analysis. JAMA. 2010;304(4):443–451.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156(12):848–856.
- , , , et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669–676.
- , , , et al. Simple cognitive testing (Mini‐Cog) predicts in‐hospital delirium in the elderly. J Am Geriatr Soc. 2007;55(2):314–316.
- , , . A prospective study of delirium in hospitalized elderly. JAMA. 1990;263(8):1097–1101.
- , . Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275(11):852–857.
- , , , , , . Risk factors for delirium at discharge: development and validation of a predictive model. Arch Intern Med. 2007;167(13):1406–1413.
- , . Delirium in vascular surgery. Eur J Vasc Endovasc Surg. 2007;34(2):131–134.
- , , , , , . Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42(8):809–815.
- , , , , . A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119(6):474–481.
- , , , , , . Validation of a medical record‐based delirium risk assessment. J Am Geriatr Soc. 2011;59(suppl 2):S289–S294.
- , , , et al. Derivation and validation of a clinical prediction rule for delirium in patients admitted to a medical ward: an observational study. BMJ Open. 2012;2(5) pii: e001599.
- , , , et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134–139.
- , , , , , . Prediction of postoperative delirium after abdominal surgery in the elderly. J Anesth. 2009;23(1):51–56.
- , , , et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229–236.
- , , , et al. Development and validation of PRE‐DELIRIC (PREdiction of DELIRium in ICu patients) delirium prediction model for intensive care patients: observational multicentre study. BMJ. 2012;344:e420.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , . “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198.
- . Wechsler Memory Scale‐III. New York, NY: Psychological Corp.; 1997.
- , , , , . The mini‐cog: a cognitive 'vital signs' measure for dementia screening in multi‐lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021–1027.
- , . Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65.
- , , . The CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry. 1974;131(10):1121–1123.
- , , , , , . Is the NEI‐VFQ‐25 a useful tool in identifying visual impairment in an elderly population? BMC Ophthalmol. 2006;6:24.
- , , , et al. Validation of self‐reported hearing loss. The Blue Mountains Hearing Study. Int J Epidemiol. 2001;30(6):1371–1378.
- , , . Does this patient have hearing impairment? JAMA. 2006;295(4):416–428.
- , , , , , . Realizing the potential of clinical judgment: a real‐time strategy for predicting outcomes and cost for medical inpatients. Am J Med. 2000;109(3):189–195.
- , , , , , . Assessing illness severity: does clinical judgment work? J Chronic Dis. 1986;39(6):439–452.
- , , , , , . Prognostication in acutely admitted older patients by nurses and physicians. J Gen Intern Med. 2008;23(11):1883–1889.
- . The Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, CT: Yale University School of Medicine; 2003.
- , , , . Acute confusional states and dementia in the elderly: the role of dehydration/volume depletion, physical illness and age. Age Ageing. 1980;9(3):137–146.
- . A Wilcoxon‐type test for trend. Stat Med. 1985;4(1):87–90.
- , , , . Synopsis of the National Institute for Health and Clinical Excellence guideline for prevention of delirium. Ann Intern Med. 2011;154(11):746–751.
- , , , , . The Hospital Elder Life Program: a model of care to prevent cognitive and functional decline in older hospitalized patients. Hospital Elder Life Program. J Am Geriatr Soc. 2000;48(12):1697–1706.
- , , , , . A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338–1344.
- , , . Occurrence and outcome of delirium in medical in‐patients: a systematic literature review. Age Ageing. 2006;35(4):350–364.
- , , , , , . Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc. 2003;51(5):591–598.
- , , , , . One‐year health care costs associated with delirium in the elderly population. Arch Intern Med. 2008;168(1):27–32.
- , , , , . Does delirium contribute to poor hospital outcomes? A three‐site epidemiologic study. J Gen Intern Med. 1998;13(4):234–242.
- , , , , , . Delirium duration and mortality in lightly sedated, mechanically ventilated intensive care patients. Crit Care Med. 2010;38(12):2311–2318.
- , , , et al. Delirium epidemiology in critical care (DECCA): an international study. Crit Care. 2010;14(6):R210.
- , , , et al. Delirium as a predictor of long‐term cognitive impairment in survivors of critical illness. Crit Care Med. 2010;38(7):1513–1520.
- , , , , , . Delirium in elderly patients and the risk of postdischarge mortality, institutionalization, and dementia: a meta‐analysis. JAMA. 2010;304(4):443–451.
- , , , et al. Adverse outcomes after hospitalization and delirium in persons with Alzheimer disease. Ann Intern Med. 2012;156(12):848–856.
- , , , et al. A multicomponent intervention to prevent delirium in hospitalized older patients. N Engl J Med. 1999;340(9):669–676.
- , , , et al. Simple cognitive testing (Mini‐Cog) predicts in‐hospital delirium in the elderly. J Am Geriatr Soc. 2007;55(2):314–316.
- , , . A prospective study of delirium in hospitalized elderly. JAMA. 1990;263(8):1097–1101.
- , . Precipitating factors for delirium in hospitalized elderly persons. Predictive model and interrelationship with baseline vulnerability. JAMA. 1996;275(11):852–857.
- , , , , , . Risk factors for delirium at discharge: development and validation of a predictive model. Arch Intern Med. 2007;167(13):1406–1413.
- , . Delirium in vascular surgery. Eur J Vasc Endovasc Surg. 2007;34(2):131–134.
- , , , , , . Delirium in hospitalized older persons: outcomes and predictors. J Am Geriatr Soc. 1994;42(8):809–815.
- , , , , . A predictive model for delirium in hospitalized elderly medical patients based on admission characteristics. Ann Intern Med. 1993;119(6):474–481.
- , , , , , . Validation of a medical record‐based delirium risk assessment. J Am Geriatr Soc. 2011;59(suppl 2):S289–S294.
- , , , et al. Derivation and validation of a clinical prediction rule for delirium in patients admitted to a medical ward: an observational study. BMJ Open. 2012;2(5) pii: e001599.
- , , , et al. A clinical prediction rule for delirium after elective noncardiac surgery. JAMA. 1994;271(2):134–139.
- , , , , , . Prediction of postoperative delirium after abdominal surgery in the elderly. J Anesth. 2009;23(1):51–56.
- , , , et al. Derivation and validation of a preoperative prediction rule for delirium after cardiac surgery. Circulation. 2009;119(2):229–236.
- , , , et al. Development and validation of PRE‐DELIRIC (PREdiction of DELIRium in ICu patients) delirium prediction model for intensive care patients: observational multicentre study. BMJ. 2012;344:e420.
- , , , , , . Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med. 1990;113(12):941–948.
- , , . “Mini‐mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198.
- . Wechsler Memory Scale‐III. New York, NY: Psychological Corp.; 1997.
- , , , , . The mini‐cog: a cognitive 'vital signs' measure for dementia screening in multi‐lingual elderly. Int J Geriatr Psychiatry. 2000;15(11):1021–1027.
- , . Functional evaluation: the Barthel index. Md State Med J. 1965;14:61–65.
- , , . The CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry. 1974;131(10):1121–1123.
- , , , , , . Is the NEI‐VFQ‐25 a useful tool in identifying visual impairment in an elderly population? BMC Ophthalmol. 2006;6:24.
- , , , et al. Validation of self‐reported hearing loss. The Blue Mountains Hearing Study. Int J Epidemiol. 2001;30(6):1371–1378.
- , , . Does this patient have hearing impairment? JAMA. 2006;295(4):416–428.
- , , , , , . Realizing the potential of clinical judgment: a real‐time strategy for predicting outcomes and cost for medical inpatients. Am J Med. 2000;109(3):189–195.
- , , , , , . Assessing illness severity: does clinical judgment work? J Chronic Dis. 1986;39(6):439–452.
- , , , , , . Prognostication in acutely admitted older patients by nurses and physicians. J Gen Intern Med. 2008;23(11):1883–1889.
- . The Confusion Assessment Method (CAM): Training Manual and Coding Guide. New Haven, CT: Yale University School of Medicine; 2003.
- , , , . Acute confusional states and dementia in the elderly: the role of dehydration/volume depletion, physical illness and age. Age Ageing. 1980;9(3):137–146.
- . A Wilcoxon‐type test for trend. Stat Med. 1985;4(1):87–90.
- , , , . Synopsis of the National Institute for Health and Clinical Excellence guideline for prevention of delirium. Ann Intern Med. 2011;154(11):746–751.
- , , , , . The Hospital Elder Life Program: a model of care to prevent cognitive and functional decline in older hospitalized patients. Hospital Elder Life Program. J Am Geriatr Soc. 2000;48(12):1697–1706.
- , , , , . A randomized trial of care in a hospital medical unit especially designed to improve the functional outcomes of acutely ill older patients. N Engl J Med. 1995;332(20):1338–1344.
Copyright © 2013 Society of Hospital Medicine
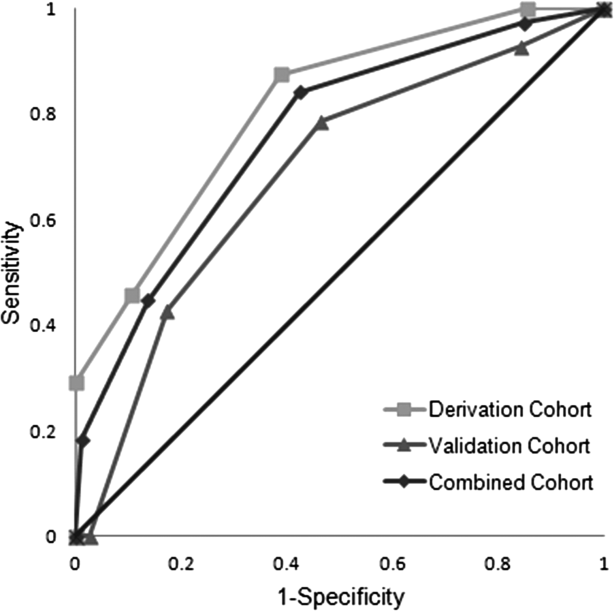
The Third Time's the Charm
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
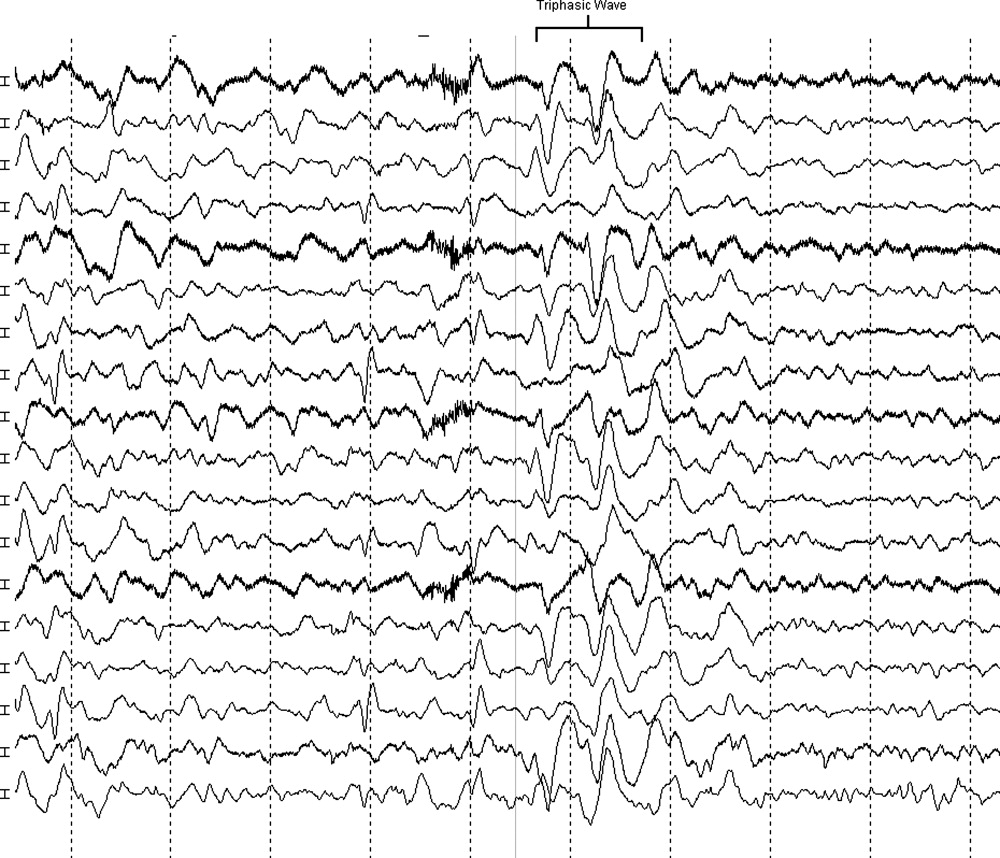
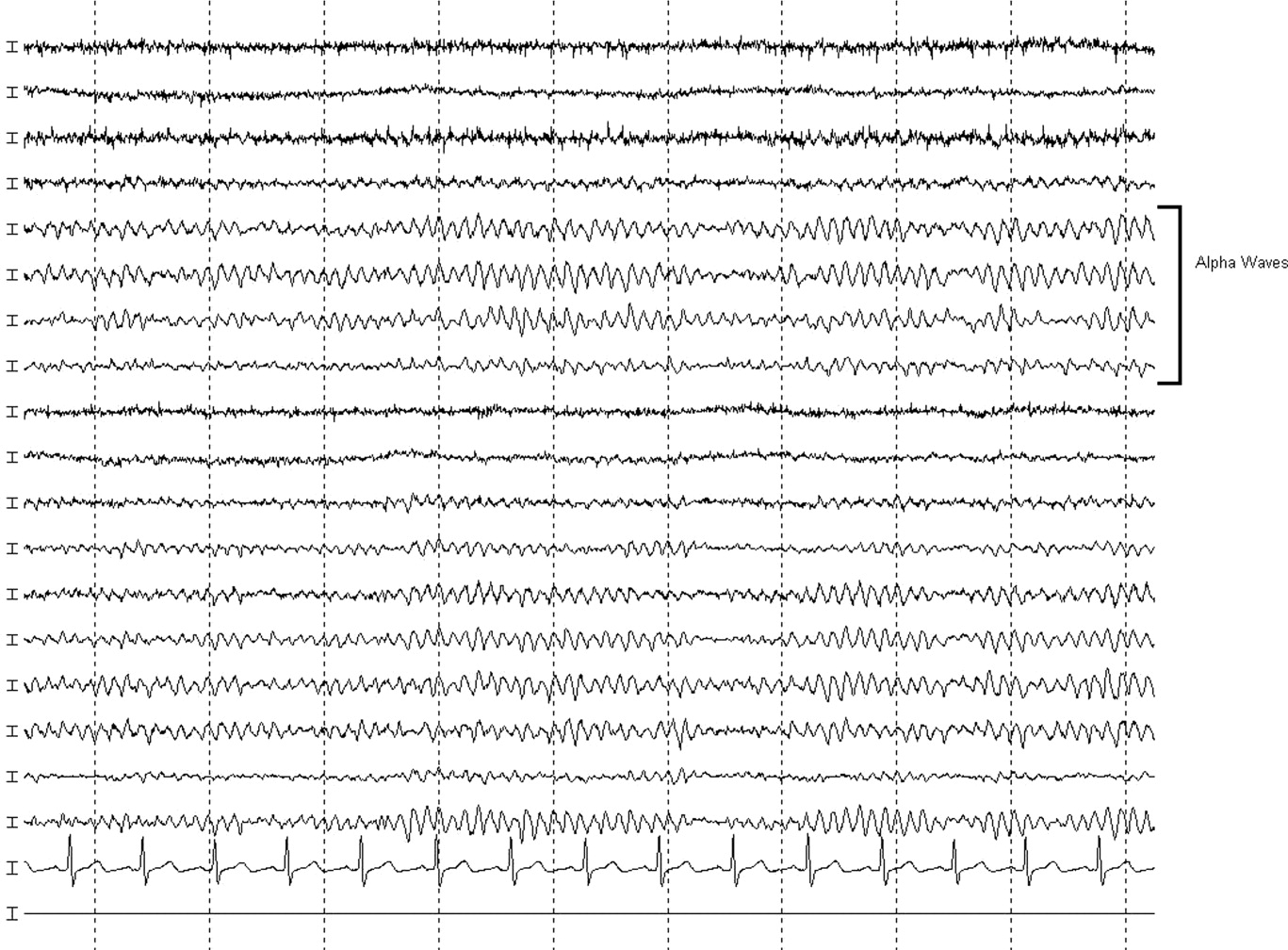
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, 5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, 10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, 5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, 10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, 5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, 10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.