User login
Tissue Isn’t the Issue
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
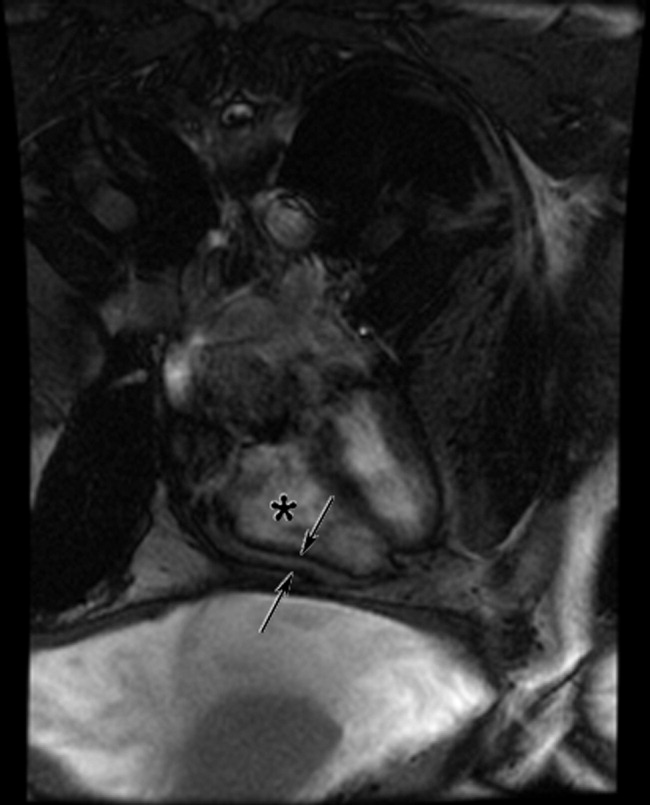
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.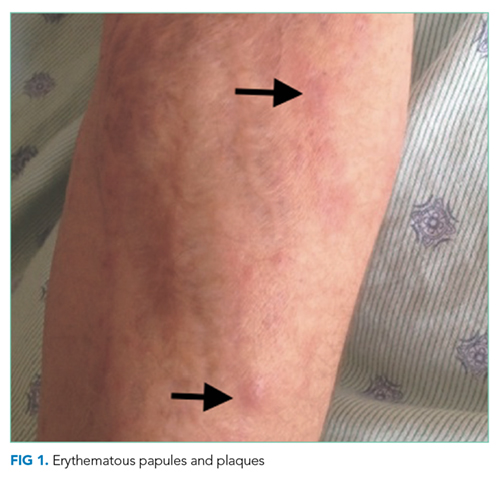
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower

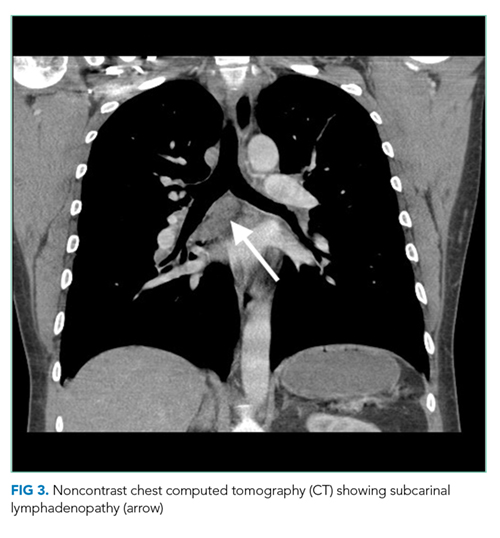
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
A 43-year-old man with a history of asplenia, hepatitis C, and nephrolithiasis reported right-flank pain. He described severe, sharp pain that came in waves and radiated to the right groin, associated with nausea and nonbloody emesis. He noted “pink urine” but no dysuria. He had 4prior similar episodes during which he had passed kidney stones, although stone analysis had never been performed. He denied having fevers or chills.
The patient had been involved in a remote motor vehicle accident complicated by splenic laceration, for which he underwent splenectomy. He was appropriately immunized. The patient also suffered from bipolar affective disorder and untreated chronic hepatitis C infection with no evidence of cirrhosis. He smoked one pack of tobacco per day for the last 10 years and reported distant alcohol and methamphetamine use.
Right-flank pain can arise from conditions affecting the lower thorax (effusion, pneumonia, pulmonary embolism), abdomen (hepatobiliary or intestinal disease), retroperitoneum (hemorrhage or infection), musculoskeletal system, peripheral nerves (herpes zoster), or the genitourinary system (pyelonephritis). Pain radiating to the groin, discolored urine (suggesting hematuria), and history of kidney stones increase the likelihood of renal colic from nephrolithiasis.
Less commonly, flank pain and hematuria may present as initial symptoms of renal cell carcinoma, renal infarction, or aortic dissection. The patient’s immunosuppression from asplenia and active injection drug use could predispose him to septic emboli to his kidneys. Prior trauma causing aortic injury could predispose himto subsequent dissection.
The patient appeared well with a heart rate of 100 beats per minute, blood pressure 122/76 mmHg, temperature 36.8°C, respiratory rate 16 breaths per minute, and oxygen saturation 96% on room air. His cardiopulmonary and abdominal examinations were normal, and he had no costovertebral angle tenderness. His skin was warm and dry without rashes. His white blood cell (WBC) count was 26,000/μL; absolute neutrophil count was 22,000/μL. Serum chemistries were normal, including creatinine 0.63 mg/dL, calcium 8.8 mg/dL, and phosphorus 3.1 mg/dL. Lactate was 0.8 mmol/L (reference range: 0-2.0 mmol/L). Urinalysis revealed large ketones, >50 red blood cells (RBC) per high power field (HPF), <5 WBC per HPF, 1+ calcium oxalate crystals and pH 6.0. A bedside ultrasound showed mild right hydronephrosis. Computed tomography (CT) with intravenous contrast of his abdomen and pelvis demonstrated diffuse, mildly prominent subcentimeter mesenteric lymphadenopathy and no kidney stones. He was treated with intravenous fluids and pain control, and was discharged with a presumptive diagnosis of a passed kidney stone.
A passed stone would not explain this degree of leukocytosis. The CT results reduce the likelihood of a renal neoplasm, renal infarction, or pyelonephritis. Mesenteric lymphadenopathy is nonspecific, but it may signal underlying infection or malignancy with spread to lymph nodes, or it may be part of a systemic disorder causing generalized lymphadenopathy. Malignant causes of mesenteric lymphadenopathy (with no apparent primary tumor) include testicular cancer, lymphoma, and primary urogenital neoplasms.
The lower extremity nodules are consistent with erythema nodosum, which may be observed in numerous infectious and noninfectious illnesses. The rapid tempo of this febrile illness mandates early consideration of infection. Splenectomized patients are at risk for overwhelming post-splenectomy infection from encapsulated organisms, although this risk is significantly mitigated with appropriate immunization. The patient is at risk of bacterial endocarditis, which could explain his fevers and polyarthritis, although plaques, pustules, and oral ulcers would be unusual. Disseminated gonococcal infection causes fevers, oral lesions, polyarthritis and pustular skin lesions, but plaques are uncommon. Disseminated mycobacterial and fungal infections may cause oral ulcers, but affected patients tend to be severely ill and have profound immunosuppression. Secondary syphilis may account for many of the findings; however, oral ulcers would be unusual, and the rash tends to be more widespread, with a predilection for the palms and soles. Human immunodeficiency virus (HIV) can cause oral ulcers and is the chief viral etiology to consider.
Noninfectious illnesses to consider include neoplasms and connective tissue diseases. Malignancy would be unlikely to manifest this abruptly or produce a paraneoplastic disorder with these features.
The patient described severe fatigue and drenching night sweats for two months prior to admission. He denied dyspnea or cough. He was born in the southwestern United States and had lived in California for almost a decade. He had been incarcerated for a few years and released three years prior. He had intermittently lived in homeless shelters, but currently lived alone in downtown San Francisco. He had traveled remotely to the Caribbean, and more recently traveled frequently to the Central Valley in California. The patient formerly worked as a pipe-fitter and welder. He denied animal exposure or recent sick contacts. He was sexually active with women, and intermittently used barrier protection.
His years in the southwestern United States may have exposed the patient to blastomycosis or histoplasmosis; both can mimic mycobacterial disease. Blastomycosis demonstrates a slightly stronger predilection for spreading to the bones, genitourinary tract, and central nervous system, whereas histoplasmosis is a more frequent cause of polyarthrtitis and mesenteric adenopathy. The patient’s travel to the Central Valley, California raises the possibility of coccidioidomycosis, which typically starts with pulmonary disease prior to dissemination to bones, skin, and other less common sites. Pipe-fitters are predisposed to asbestos-related illnesses, including lung cancer and mesothelioma, which would not explain this patient’s presentation. Incarceration and high-risk sexual practices increase his risk for tuberculosis, HIV, and syphilis. Widespread skin involvement is more characteristic of syphilis or primary HIV infection than of disseminated fungal or mycobacterial infection.
WBC measured 29,000/uL with a neutrophilic predominance. His peripheral blood smear was unremarkable. A comprehensive metabolic panel was normal. Lactate dehydrogenase (LDH) was 317 U/L (reference range 140-280 U/L). Erythrocyte sedimentation rate (ESR) was 39 mm/hr (reference range < 20 mm/hr) and C-reactive protein (CRP) was 66 mg/L (reference range <6.3 mg/L). Blood, urine, and throat cultures were sent. Chest radiograph showed clear lungs without adenopathy. Ankle and knee radiographs identified small effusions bilaterally without bony abnormalities. CT of his brain showed a small, hypodense lesion in the right lacrimal gland. A lumbar puncture with cerebrospinal fluid (CSF) analysis showed absence of RBCs; WBC, 2/µL; protein, 35 mg/dL; glucose, 62 mg/dL; negative gram stain. CSF bacterial and fungal cultures, venereal disease research laboratory (VDRL), herpes simplex virus polymerase chain reaction (HSV PCR), and cryptococcal antigen were sent for laboratory analysis. The patient was started on vancomycin and aztreonam.
Lesions of the lacrimal gland feature multiple causes, including autoimmune diseases (Sjögren’s, Behçet’s disease), granulomatous diseases (sarcoidosis, granulomatosis with polyangiitis), neoplasms (salivary gland tumors, lymphoma), and infections. Initiating broad-spectrum antibiotics is reasonable while awaiting additional information from blood and urine cultures, serologies for HIV and syphilis, and purified protein derivative or interferon-gamma release assay (IGRA).
If these tests fail to reveal a diagnosis, the search for atypical infections and noninfectious possibilities should expand.
The patient continued to have intermittent fevers, sweats, and malaise over the next 3 days. All bacterial and fungal cultures remained negative, and antibiotics were discontinued. Rheumatoid factor, anticyclic citrullinated peptide, antinuclear antibody, and cryoglobulins were negative. Serum C3, C4, and angiotensin-converting enzyme (ACE) levels were normal. A rapid plasma reagin (RPR), HIV antibody, IGRA, and serum antibodies for Coccidioides, histoplasmosis, and West Nile virus were negative. Urine nucleic acid amplification testing for gonorrhea and chlamydia was negative. CSF VDRL, HSV PCR and cryptococcal antigen were negative. HSV culture from an oral ulcer showed no growth. The patient had a reactive hepatitis C antibody with a viral load of 3 million virus equivalents/mL.
The additional test results lower
The most likely diagnosis is Löfgren’s syndrome, a variant of sarcoidosis characterized by erythema nodosum, bilateral hilar lymphadenopathy, and polyarthralgias or polyarthritis. Löfgren’s syndrome may include fevers, uveitis, widespread skin lesions and other systemic manifestations. Sarcoidosis could explain the lacrimal gland lesion, and could manifest with recurrent kidney stones. Oral lesions may occur in sarcoidosis. A normal serum ACE level may be observed in up to half of patients. The lack of visualized granulomas on the submental node FNA may reflect sampling error, lower likelihood of visualizing granulomas on FNA (compared with excisional biopsy), or biopsy location (hilar nodes are more likely to demonstrate sarcoid granulomas).
Although Löfgren’s syndrome is often self-limited, treatment can ameliorate symptoms. Nonsteroidal anti-inflammatory medication can be tried first, with prednisone reserved for refractory cases.
The constellation of bilateral hilar adenopathy, arthritis, and erythema nodosum was consistent with Löfgren’s syndrome, further supported by granulomatous infiltrates on biopsy. The patient’s symptoms resolved with naproxen. He was scheduled for follow-up in dermatology and rheumatology clinics and was referred to hepatology for management of hepatitis C.
COMMENTARY
Sarcoidosis is a multisystem granulomatous disease of unclear etiology. The disease derives its name from Boeck’s 1899 report describing benign cutaneous lesions that resembled sarcomas.1 Sarcoidosis most commonly manifests as bilateral hilar adenopathy and pulmonary infiltrates, but may impact any tissue or organ, including the eyes, nonhilar lymph nodes, liver, spleen, joints, mucous membranes, and skin. Nephrolithiasis may result from hypercalcemia and/or hypercalciuria (related to granulomatous production of 1,25 vitamin D) and can be the presenting feature of sarcoidosis.2 Less common presentations include neurologic sarcoidosis (which can present with seizures, aseptic meningitis, encephalopathy, neuroendocrine dysfunction, myelopathy and peripheral neuropathies), cardiac sarcoidosis (which may present with arrhythmias, valvular dysfunction, heart failure, ischemia, or pericardial disease), and Heerfordt syndrome (the constellation of parotid gland enlargement, facial palsy, anterior uveitis, and fever). Sarcoidosis may mimic other diseases, including malignancy, idiopathic pulmonary fibrosis, and infiltrative tuberculosis.3 Sarcoidosis-like reactions have occurred in response to malignancy and medications.4
The patient’s rash demonstrated a predilection for areas of prior scarring, which has a limited differential diagnosis. Keloids and hypertrophic scars occur at sites of former surgical wounds, lacerations, or areas of inflammation. Pruritic urticarial papules and plaques of pregnancy (PUPPP) is a benign inflammatory condition where papules cluster in areas of prior striae. Cutaneous lesions of Behçet’s syndrome display pathergy, where pustular response is observed at sites of injury. Granulomatous infiltration in sarcoidosis may demonstrate a predilection for scars and tattoos (ie, scar or tattoo sarcoidosis).5 Sarcoidosis can have other cutaneous manifestations, including psoriaform, ulcerative, or erythrodermic lesions; subcutaneous nodules; scarring or nonscarring alopecia; and lupus pernio – violaceous, nodular and plaque-like lesions on the nose, earlobes, cheeks, and digits.5
Löfgren’s syndrome is a distinct variant of sarcoidosis.In 1952, Dr. Löfgren described a case series of patients with bilateral hilar lymphadenopathy and coexisting erythema nodosum and polyarthralgia.6 The epidemiology favors young women.7 Patients with Löfgren’s syndrome present acutely (as in this case), which differs from the typical subacute course observed with sarcoidosis. In addition to the classic presentation described above, patients with Löfgren’s syndrome may demonstrate additional manifestations of sarcoidosis, including fevers, peripheral adenopathy, arthritis, and granulomatous skin lesions. Painful symptoms may require short-term anti-inflammatory treatments. Most patients do not require systemic immunosuppression. Symptoms usually decrease over several months, and the majority of patients experience complete remission within years. Rare recurrences have been described up to several years.8
In confirming the diagnosis of sarcoidosis, current guidelines recommend exclusion of other diseases that present similarly, a work-up that generally includes compatible laboratory tests and imaging, and histologic demonstration of noncaseating granulomas.9 However, Löfgren’s syndrome is a notable exception. The constellation of fever, bilateral hilar adenopathy, polyarthralgia, and erythema nodosum suffices to diagnose Löfgren’s syndrome as long as the disease remits rapidly and spontaneously.9 Thus, in this case, although granulomatous infiltrates were confirmed on biopsy, the diagnosis of Löfgren’s syndrome could have been based on clinical and radiologic features alone.
KEY LEARNING POINTS
- Sarcoidosis is a multisystem granulomatous disease that most commonly presents with bilateral hilar adenopathy and pulmonary infiltrates but can also present atypically, including with nephrolithiasis from hypercalcemia, neurologic syndromes, and cardiac involvement.
- Löfgren’s syndrome, a variant of sarcoidosis, is characterized by relatively acute onset of fevers, erythema nodosum, bilateral hilar adenopathy, and polyarthralgia or polyarthritis. Most patients recover and manifest complete remission.
- A limited differential exists for rashes with a predilection for areas of tattoos and prior scarring, including keloids, PUPPP, Behçet’s disease, and granulomatous infiltration.
Disclosure
There are no conflicts of interest or financial disclosures to report.
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
1. Multiple Benign Sarcoids of the Skin. JAMA. 1899;XXXIII(26):1620-1621.
2. Rizzato G, Fraioli P, Montemurro L. Nephrolithiasis as a presenting feature of chronic sarcoidosis. Thorax. 1995;50(5):555-559. PubMed
3. Romanov V. Atypical variants of clinical course of sarcoidosis. Eur Respir J. 2014;44(58):3782. PubMed
4. Arish N, Kuint R, Sapir E, et al. Characteristics of Sarcoidosis in Patients with Previous Malignancy: Causality or Coincidence? Respiration. 2017;93(4):247-252. PubMed
5. Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25(3):295-302. PubMed
6. Löfgren S. The Bilateral Hilar Lymphoma Syndrome. Acta Med Scand. 1952;142(4):265-273. PubMed
7. Mañá J, Gómez-Vaquero C, Montero A et al. Löfgren’s syndrome revisited: a study of 186 patients. Am J Med. 1999;107(3):240-245. PubMed
8. Gran J, Bohmer E. Acute Sarcoid Arthritis: A Favourable Outcome? Scand J Rheumatol. 1996;25(2):70-73. PubMed
9. American Thoracic Society. Statement on Sarcoidosis. Am J Respir Crit Care Med. 1999;160:736-755.Otate voluptiatia qui aut iur, utendi quiae incipis m PubMed
© 2018 Society of Hospital Medicine
Morbo Serpentino
A 58-year-old Danish man presented to an urgent care center due to several months of gradually worsening fatigue, weight loss, abdominal pain, and changes in vision . His abdominal pain was diffuse, constant, and moderate in severity. There was no association with meals, and he reported no nausea, vomiting, or change in bowel movements. He also said his vision in both eyes was blurry, but denied diplopia and said the blurring did not improve when either eye w as closed. He denied dysphagia, headache, focal weakness, or sensitivity to bright lights.
Fatigue and weight loss in a middle-aged man are nonspecific complaints that mainly help to alert the clinician that there may be a serious, systemic process lurking. Constant abdominal pain without nausea, vomiting, or change in bowel movements makes intestinal obstruction or a motility disorder less likely. Given that the pain is diffuse, it raises the possibility of an intraperitoneal process or a process within an organ that is irritating the peritoneum.
Worsening of vision can result from disorders anywhere along the visual pathway, including the cornea (keratitis or corneal edema from glaucoma), anterior chamber (uveitis or hyphema), lens (cataracts, dislocations, hyperglycemia), vitreous humor (uveitis), retina (infections, ischemia, detachment, diabetic retinopathy), macula (degenerative disease), optic nerve (optic neuritis), optic chiasm, and the visual projections through the hemispheres to the occipital lobes. To narrow the differential diagnosis, it would be important to inquire about prior eye problems, to measure visual acuity and intraocular pressure, to perform fundoscopic and slit-lamp exams to detect retinal and anterior chamber disorders, respectively, and to assess visual fields. An afferent pupillary defect would suggest optic nerve pathology.
Disorders that could unify the constitutional, abdominal, and visual symptoms include systemic inflammatory diseases, such as sarcoidosis (which has an increased incidence among Northern Europeans), tuberculosis, or cancer. While diabetes mellitus could explain his visual problems, weight loss, and fatigue, the absence of polyuria, polydipsia, or polyphagia argues against this possibility.
The patient had hypercholesterolemia and type 2 diabetes mellitus. Medications were metformin, atorvastatin, and glimepiride. He was a former smoker with 23 pack-years and had quit over 5 years prior. He had not traveled outside of Denmark in 2 years and had no pets at home. He reported being monogamous with his same-sex partner for the past 25 years. He had no significant family history, and he worked at a local hospital as a nurse. He denied any previous ocular history.
On examination, the pulse was 67 beats per minute, temperature was 36.7 degrees Celsius, respiratory rate was 16 breaths per minute, oxygen saturation was 99% while breathing ambient air, and blood pressure was 132/78. Oropharynx demonstrated no thrush or other lesions. The heart rhythm was regular and there were no murmurs. Lungs were clear to auscultation bilaterally. Abdominal exam was normal except for mild tenderness upon palpation in all quadrants, but no masses, organomegaly, rigidity, or rebound tenderness were present. Skin examination revealed several subcutaneous nodules measuring up to 0.5 cm in diameter overlying the right and left posterolateral chest walls. T he nodules were rubbery, pink, nontender, and not warm nor fluctuant. Visual acuity was reduced in both eyes. Extraocular movements were intact, and the pupils reacted to light and accommodated appropriately. The sclerae were injected bilaterally. The remainder of the cranial nerves and neurologic exam were normal. Due to the vision loss , the patient was referred to an ophthalmologist who diagnosed bilateral anterior uveitis.
Though monogamous with his male partner for many years, it is mandatory to consider complications of human immunodeficiency virus infection (HIV ). The absence of oral lesions indicative of a low CD4 count, such as oral hairy leukoplakia or thrush, does not rule out HIV disease. Additional history about his work as a nurse might shed light on his risk of infection, such as airborne exposure to tuberculosis or acquisition of blood-borne pathogens through a needle stick injury. His unremarkable vital signs support the chronicity of his medical condition.
Uveitis can result from numerous causes. When confined to the eye, uncommon hereditary and acquired causes are less likely . In many patients, uveitis arises in the setting of systemic infection or inflammation. The numerous infectious causes of uveitis include syphilis, tuberculosis, toxoplasmosis, cat scratch disease, and viruses such as HIV, West Nile, and Ebola. Among the inflammatory diseases that can cause uveitis are sarcoidosis, inflammatory bowel disease, systemic lupus erythematosus, Behçet disease, and Sjogren syndrome.
Several of these conditions, including tuberculosis and syphilis, may also cause subcutaneous nodules. Both tuberculosis and syphilis can cause skin and gastrointestinal disease. Sarcoidosis could involve the skin, peritoneum, and uvea, and is a possibility in this patient. The dermatologic conditions associated with sarcoidosis are protean and include granulomatous inflammation and nongranulomatous processes such as erythema nodosum. Usually the nodules of erythema nodosum are tender, red or purple, and located on the lower extremities. The lack of tenderness points away from erythema nodosum in this patient. Metastatic cancer can disseminate to the subcutaneous tissue, and the patient’s smoking history and age mandate we consider malignancy. However, skin metastases tend to be hard, not rubbery.
A cost-effective evaluation at this point would include syphilis serologies, HIV testing, testing for tuberculosis with either a purified protein derivative test or interferon gamma release assay, chest radiography, and biopsy of 1 of the lesions on his back.
Laboratory data showed 12,400 white blood cells per cubic milliliter (64% neutrophils, 24% lymphocytes, 9% monocytes, 2% eosinophils, 1% basophils), hemoglobin 7.9 g/dL, mean corpuscular volume 85 fL, platelets 476,000 per cubic milliliter , C-reactive protein 43 mg/ d L (normal < 8 mg/L), gamma-glutamyl-transferase 554 IU/L (normal range 0-45), alkaline phosphatase 865 U/L (normal range 60-200), and erythrocyte sedimentation rate (ESR) 71 mm per hour. International normalized ratio was 1.0, albumin was 3.0 mg/dL, activated partial thromboplastin time was 32 seconds (normal 22 to 35 seconds), and bilirubin was 0.3 mg/dL. Antibodies to HIV , hepatitis C, and hepatitis B surface antigen were not detectable. Electrocardiography ( ECG ) was normal. Plain radiograph of the chest demonstrated multiple nodular lesions bilaterally measuring up to 1 cm with no cavitation. There was a left pleural effusion.
The history and exam findings indicate a serious inflammatory condition affecting his lungs, pleura, eyes, skin, liver, and possibly his peritoneum. In this context, the elevated C-reactive protein and ESR are not helpful in differentiating inflammatory from infectious causes. The constellation of uveitis, pulmonary and cutaneous nodules, and marked abnormalities of liver tests in a middle-aged man of Northern European origin points us toward sarcoidosis. Pleural effusions are not common with sarcoidosis but may occur. However, to avoid premature closure, it is important to consider other possibilities.
Metastatic cancer, including lymphoma, could cause pulmonary and cutaneous nodules and liver involvement, but the chronic time course and uveitis are not consistent with malignancy. Tuberculosis is still a consideration, though one would have expected him to report fevers, night sweats, and, perhaps, exposure to patients with pulmonary tuberculosis in his job as a nurse. Multiple solid pulmonary nodules are also uncommon with pulmonary tuberculosis. Fungal infections such as histoplasmosis can cause skin lesions and pulmonary nodules but do not fit well with uveitis.
At this point, “ tissue is the issue.” A skin nodule would be the easiest site to biopsy. If skin biopsy was not diagnostic, computed tomography (CT) of his chest and abdomen should be performed to identify the next most accessible site for biopsy.
Esophagogastroduodenoscopy (EGD) and colonoscopy showed normal findings, and random biopsies from the stomach and colon were normal. CT of the chest, abdomen, and pelvis performed with the administration of intravenous contrast showed multiple solid opacities in both lung fields up to 1 cm, with enlarged mediastinal and retroperitoneal lymph nodes measuring 1 to 3 cm in diameter, a left pleural effusion, wall thickening in the right colon, and several nonspecific hypodensities in the liver. A punch biopsy taken from the right chest wall lesion demonstrated chronic inflammation without granulomas. The patient underwent CT-guided biopsy of 1 of the right-sided lung nodules, which revealed noncaseating granulomatous inflammation, fibrosis, and necrosis. Neither biopsy contained malignant cells, and additional stains revealed no bacteria, fungi, or acid fast bacilli.
The retroperitoneal and mediastinal adenopathy are indicative of a widely disseminated inflammatory process. Lymphoma continues to be a concern, though uveitis as an initial presenting problem would very unusual. Although biopsy of the chest wall lesion failed to demonstrate granulomatous inflammation, the most parsimonious explanation is that the skin and lung nodules are both related to a single systemic process.
Granulomas form in an attempt to wall off offending agents, whether foreign antigens (talc, certain medications), infectious agents, or self-antigens. Review of histopathology and microbiologic studies are useful first steps. Stains for bacteria, fungi, or acid-fast organisms may diagnose an infectious cause, such as tuberculosis, leprosy, syphilis, fungi, or cat scratch disease. Granulomas in association with vascular inflammation would indicate vasculitis. Other autoimmune considerations include sarcoidosis and Crohn disease. Noncaseating granulomas are typically found in sarcoidosis, cat scratch disease, syphilis, leprosy, or Crohn disease, but do not entirely exclude tuberculosis.
The negative infectious studies and lack of classic features of Crohn disease or other autoimmune diseases further point to sarcoidosis as the etiology of this patient’s illness. A Norwegian dermatologist first described the pathology of sarcoidosis based upon specimens taken from skin nodules. He thought the lesions were sarcoma and described them as, “ multiple benign sarcoid of the skin,” which is where the name “ sarcoidosis” originated.
Diagnosing sarcoidosis requires excluding other mimickers. Additional testing should include syphilis serologies, rheumatoid factor, and antineutrophilic cytoplasmic antibodies. The latter is associated with granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, either of which may produce granulomatous inflammation of the lungs, skin, and uvea.
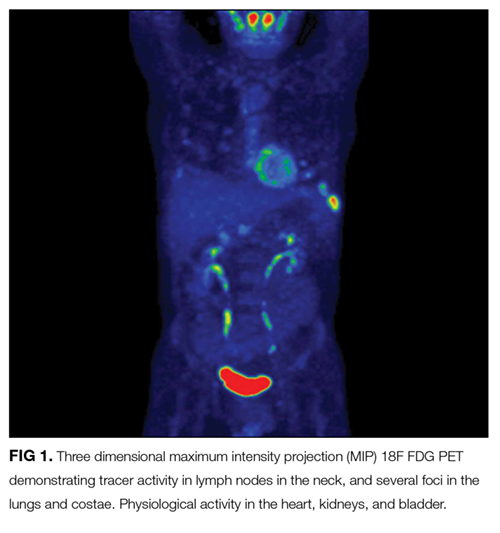
At this juncture, PET-CT represents a costly and unnecessary test that does not narrow our diagnostic possibilities sufficiently to justify its use. Osteolytic lesions would be unusual in sarcoidosis and more likely in lymphoma or infectious processes such as tuberculosis. Tests for syphilis and tuberculosis are required, and are a fraction of the cost of a PET-CT.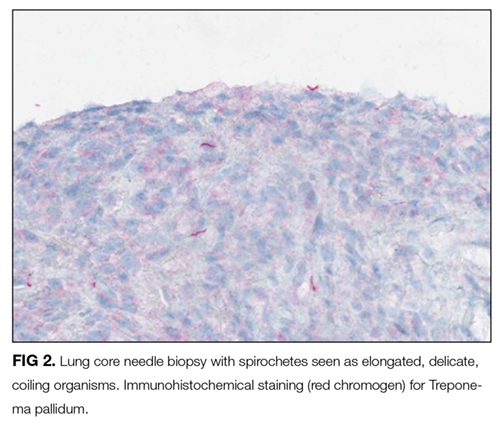
With the biopsy revealing spirochetes, and the positive results of a nontreponemal test (RPR) and confirmatory treponemal results, the diagnosis of syphilis is firmly established. Uveitis indicates neurosyphilis and warrants a longer course of intravenous penicillin. Lumbar puncture should be performed.
A lumbar puncture was performed. Cerebrospinal fluid (CSF) contained 9 white blood cells and 73 red blood cells per cubic milliliter; protein concentration was 73 mg/dL, and glucose was 116 mg/dL. Polymerase chain reaction for T. pallidum was negative. Transthoracic ECG and magnetic resonance imaging of the brain were normal. The patient was treated with intravenous penicillin G at 5 million units 4 times daily for 15 days. A PET-CT scan 3 months later revealed complete resolution of the subcutaneous, pulmonary, liver lesions, lymphadenopathy, and uveitis. Repeat treponemal serologies demonstrated a greater than 4-fold decline in titers.
DISCUSSION
Syphilis is a sexually transmitted disease with increasing incidence worldwide. Untreated infection progresses through 3 stages. The primary stage is characterized by the appearance of a painless chancre after an incubation period of 2 to 3 weeks. Four to 8 weeks later, the secondary stage emerges as a systemic infection, often heralded by a maculopapular rash with desquamation, frequently involving the soles and palms. Hepatitis, iridocyclitis, and early neurosyphilis may also be seen at this stage. Subsequently, syphilis becomes latent. One-third of patients with untreated latent syphilis will develop tertiary syphilis, typified by late neurosyphilis (tabes dorsalis and general paresis), cardiovascular disease (aortitis), or gummatous disease. 1
Gummas are destructive granulomatous lesions that typically present indolently, may occur singly or multiply, and may involve almost any organ. It has been suggested that gummas are the immune system’s defense to slow the bacteria after attempts to kill it have failed. Histologically, gummas are hyalinized nodules with surrounding granulomatous infiltrate of lymphocytes, plasma cells, and multinucleated giant cells with or without necrosis . In the preantibiotic era, gummas were seen in approximately 15% of infected patients, with a latency of 1 to 46 years after primary infection. 2 Penicillin led to a drastic reduction in gummas until the HIV epidemic, which led to the resurgence of gummas at a drastically shortened interval following primary syphilis. 3
Most commonly, gummas affect the skin and bones. In the skin , lesions may be superficial or deep and may progress into ulcerative nodules. In the bones, destructive gummas have a characteristic “moth-eaten” appearance. Less common sequelae of gummas incude gummatous hepatitis, perforated nasal septum (saddle nose deformity), or hard palate erosions. 2,4 R arely, syphilis involves the lungs, appearing as nodules, infiltrates, or pleural effusion. 5
Ocular manifestations occur in approximately 5% of patients with syphilis, more often in secondary and tertiary stages, and are strongly associated with a spread to the central nervous system. Syphilis may affect any structure of the eye, with anterior uveitis as the most frequent manifestation. Partial or complete vision loss is identified in approximately half of the patients with ocular syphilis and may be completely reversed by appropriate treatment. Ophthalmologic findings such as optic neuritis and papilledema imply advanced illness , as do Argyll-Robertson pupils (small pupils that are poorly reactive to light , but with preserved accommodation and convergence). 6,7 The treatment of ocular syphilis is identical to that of neurosyphilis. The Centers for Disease Control and Prevention recommends CSF analysis in any patient with ocular syphilis. Abnormal results should prompt repeat lumbar puncture every 3 to 6 months following treatment until the CSF results normalize. 8
The diagnosis of syphilis relies on indirect serologic tests. T. pallidum cannot be cultured in vitro, and techniques to identify spirochetes directly by using darkfield microscopy or DNA amplification via polymerase chain reaction are limited by availability or by poor sensitivity in advanced syphilis. 1 Imaging modalities including PET cannot reliably differentiate syphilis from other infectious and noninfectious mimickers. 9 F ortunately, syphilis infection can be diagnosed accurately based on reactive treponemal and nontreponemal serum tests. Nontreponemal tests, such as the RPR and Venereal Disease Research Laboratory, have traditionally been utilized as first-line evaluation, followed by a confirmatory treponemal test. However, nontreponemal tests may be nonreactive in a few settings: very early or very late in infection, and in individuals previously treated for syphilis. Thus, newer “reverse testing” algorithms utilize more sensitive and less expensive treponemal tests as the first test, followed by nontreponemal tests if the initial treponemal test is reactive. 8 Regardless of the testing sequence, in patients with no prior history of syphilis, reactive results on both treponemal and nontreponemal assays firmly establish a diagnosis of syphilis, obviating the need for more invasive and costly testing.
In patients with unexplained systemic illness, clinicians should have a low threshold to test for syphilis. Testing should be extended to certain asymptomatic individuals at higher risk of infection, including men who have sex with men, sexual partners of patients infected with syphilis, individuals with HIV or sexually-transmitted diseases, and others with high-risk sexual behavior or a history of sexually-transmitted diseases. 8 As the discussant points out, earlier consideration of and testing for syphilis would have spared the patient from unnecessary and costly EGD, colonoscopy, PET-CT scanning, and 3 biopsies.
Syphilis has been known to be a horribly destructive disease for centuries, earning the moniker “morbo serpentino” (serpentine disease) from the Spanish physician Ruiz Diaz de Isla in the 1500s. 10 In the modern era, physicians must remember to consider the diagnosis of syphilis in order to effectively mitigate the harm from this resurgent disease when it attacks our patients.
TEACHING POINTS
- Syphilis, the great imposter, is rising in incidence and should be on the differential diagnosis in all patients with unexplained multisystem inflammatory disease.
- A cost-effective diagnostic approach to syphilis entails serologic testing with treponemal and nontreponemal assays.
- Unexplained granulomas, especially in the skin, bone, or liver, should prompt consideration of gummatous syphilis.
- Ocular syphilis may involve any part of the visual tract and is treated the same as neurosyphilis.
Disclosure
Dr. Weinreich has received payment for lectures from Boehringer er Ingelheim, Astra Zeneca, TEVA and Novartis in 2016. All other contributors have nothing to report.
1. French P. Syphilis. BMJ. 2007;334:143-147. PubMed
2. Singh AE, Romanowski B. Syphilis: Review with emphasis on clinical, epidemiologic, and some biologic features. Clin Micriobio Rev. 1999;12(2):187-209. PubMed
3. Karp G, Schlaeffer F, Jotkowitz A, Riesenberg K. Syphilis and HIV co-infection. Eur J Int Med. 2009; 20:9-13. PubMed
4. Pilozzi-Edmonds L, Kong LY, Szabo J, Birnbaum LM. Rapid progression to gummatous syphilitic hepatitis and neurosyphilis in a patient with newly diagnosed HIV. Int J STD AIDS. 2014;26(13)985-987. PubMed
5. David G, Perpoint T, Boibieux A, et al. Secondary pulmonary syphilis: report of a likely case and literature review. Clin Infect Dis. 2006;42(3):e11-e15. PubMed
6. Moradi A, Salek S, Daniel E, et al. Clinical features and incidence rates of ocular complications in patients with ocular syphilis. Am J Ophthalmol. 2015;159:334-343. PubMed
7. Aldave AJ, King JA, Cunningham ET Jr. Ocular syphilis. Curr Opin Ophthalmol. 2001;12:433-441. PubMed
8. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137. PubMed
9. Lin M, Darwish B, Chu J. Neurosyphilitic gumma on F18-2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography: An old disease investigated with new technology. J Clin Neurosc. 2009;16:410-412. PubMed
10. de Ricon‐Ferraz A. Early work on syphilis: Diaz de Ysla’s treatise on the serpentine disease of Hispaniola Island. Int J Dermatol. 1999;38(3):222-227. PubMed
A 58-year-old Danish man presented to an urgent care center due to several months of gradually worsening fatigue, weight loss, abdominal pain, and changes in vision . His abdominal pain was diffuse, constant, and moderate in severity. There was no association with meals, and he reported no nausea, vomiting, or change in bowel movements. He also said his vision in both eyes was blurry, but denied diplopia and said the blurring did not improve when either eye w as closed. He denied dysphagia, headache, focal weakness, or sensitivity to bright lights.
Fatigue and weight loss in a middle-aged man are nonspecific complaints that mainly help to alert the clinician that there may be a serious, systemic process lurking. Constant abdominal pain without nausea, vomiting, or change in bowel movements makes intestinal obstruction or a motility disorder less likely. Given that the pain is diffuse, it raises the possibility of an intraperitoneal process or a process within an organ that is irritating the peritoneum.
Worsening of vision can result from disorders anywhere along the visual pathway, including the cornea (keratitis or corneal edema from glaucoma), anterior chamber (uveitis or hyphema), lens (cataracts, dislocations, hyperglycemia), vitreous humor (uveitis), retina (infections, ischemia, detachment, diabetic retinopathy), macula (degenerative disease), optic nerve (optic neuritis), optic chiasm, and the visual projections through the hemispheres to the occipital lobes. To narrow the differential diagnosis, it would be important to inquire about prior eye problems, to measure visual acuity and intraocular pressure, to perform fundoscopic and slit-lamp exams to detect retinal and anterior chamber disorders, respectively, and to assess visual fields. An afferent pupillary defect would suggest optic nerve pathology.
Disorders that could unify the constitutional, abdominal, and visual symptoms include systemic inflammatory diseases, such as sarcoidosis (which has an increased incidence among Northern Europeans), tuberculosis, or cancer. While diabetes mellitus could explain his visual problems, weight loss, and fatigue, the absence of polyuria, polydipsia, or polyphagia argues against this possibility.
The patient had hypercholesterolemia and type 2 diabetes mellitus. Medications were metformin, atorvastatin, and glimepiride. He was a former smoker with 23 pack-years and had quit over 5 years prior. He had not traveled outside of Denmark in 2 years and had no pets at home. He reported being monogamous with his same-sex partner for the past 25 years. He had no significant family history, and he worked at a local hospital as a nurse. He denied any previous ocular history.
On examination, the pulse was 67 beats per minute, temperature was 36.7 degrees Celsius, respiratory rate was 16 breaths per minute, oxygen saturation was 99% while breathing ambient air, and blood pressure was 132/78. Oropharynx demonstrated no thrush or other lesions. The heart rhythm was regular and there were no murmurs. Lungs were clear to auscultation bilaterally. Abdominal exam was normal except for mild tenderness upon palpation in all quadrants, but no masses, organomegaly, rigidity, or rebound tenderness were present. Skin examination revealed several subcutaneous nodules measuring up to 0.5 cm in diameter overlying the right and left posterolateral chest walls. T he nodules were rubbery, pink, nontender, and not warm nor fluctuant. Visual acuity was reduced in both eyes. Extraocular movements were intact, and the pupils reacted to light and accommodated appropriately. The sclerae were injected bilaterally. The remainder of the cranial nerves and neurologic exam were normal. Due to the vision loss , the patient was referred to an ophthalmologist who diagnosed bilateral anterior uveitis.
Though monogamous with his male partner for many years, it is mandatory to consider complications of human immunodeficiency virus infection (HIV ). The absence of oral lesions indicative of a low CD4 count, such as oral hairy leukoplakia or thrush, does not rule out HIV disease. Additional history about his work as a nurse might shed light on his risk of infection, such as airborne exposure to tuberculosis or acquisition of blood-borne pathogens through a needle stick injury. His unremarkable vital signs support the chronicity of his medical condition.
Uveitis can result from numerous causes. When confined to the eye, uncommon hereditary and acquired causes are less likely . In many patients, uveitis arises in the setting of systemic infection or inflammation. The numerous infectious causes of uveitis include syphilis, tuberculosis, toxoplasmosis, cat scratch disease, and viruses such as HIV, West Nile, and Ebola. Among the inflammatory diseases that can cause uveitis are sarcoidosis, inflammatory bowel disease, systemic lupus erythematosus, Behçet disease, and Sjogren syndrome.
Several of these conditions, including tuberculosis and syphilis, may also cause subcutaneous nodules. Both tuberculosis and syphilis can cause skin and gastrointestinal disease. Sarcoidosis could involve the skin, peritoneum, and uvea, and is a possibility in this patient. The dermatologic conditions associated with sarcoidosis are protean and include granulomatous inflammation and nongranulomatous processes such as erythema nodosum. Usually the nodules of erythema nodosum are tender, red or purple, and located on the lower extremities. The lack of tenderness points away from erythema nodosum in this patient. Metastatic cancer can disseminate to the subcutaneous tissue, and the patient’s smoking history and age mandate we consider malignancy. However, skin metastases tend to be hard, not rubbery.
A cost-effective evaluation at this point would include syphilis serologies, HIV testing, testing for tuberculosis with either a purified protein derivative test or interferon gamma release assay, chest radiography, and biopsy of 1 of the lesions on his back.
Laboratory data showed 12,400 white blood cells per cubic milliliter (64% neutrophils, 24% lymphocytes, 9% monocytes, 2% eosinophils, 1% basophils), hemoglobin 7.9 g/dL, mean corpuscular volume 85 fL, platelets 476,000 per cubic milliliter , C-reactive protein 43 mg/ d L (normal < 8 mg/L), gamma-glutamyl-transferase 554 IU/L (normal range 0-45), alkaline phosphatase 865 U/L (normal range 60-200), and erythrocyte sedimentation rate (ESR) 71 mm per hour. International normalized ratio was 1.0, albumin was 3.0 mg/dL, activated partial thromboplastin time was 32 seconds (normal 22 to 35 seconds), and bilirubin was 0.3 mg/dL. Antibodies to HIV , hepatitis C, and hepatitis B surface antigen were not detectable. Electrocardiography ( ECG ) was normal. Plain radiograph of the chest demonstrated multiple nodular lesions bilaterally measuring up to 1 cm with no cavitation. There was a left pleural effusion.
The history and exam findings indicate a serious inflammatory condition affecting his lungs, pleura, eyes, skin, liver, and possibly his peritoneum. In this context, the elevated C-reactive protein and ESR are not helpful in differentiating inflammatory from infectious causes. The constellation of uveitis, pulmonary and cutaneous nodules, and marked abnormalities of liver tests in a middle-aged man of Northern European origin points us toward sarcoidosis. Pleural effusions are not common with sarcoidosis but may occur. However, to avoid premature closure, it is important to consider other possibilities.
Metastatic cancer, including lymphoma, could cause pulmonary and cutaneous nodules and liver involvement, but the chronic time course and uveitis are not consistent with malignancy. Tuberculosis is still a consideration, though one would have expected him to report fevers, night sweats, and, perhaps, exposure to patients with pulmonary tuberculosis in his job as a nurse. Multiple solid pulmonary nodules are also uncommon with pulmonary tuberculosis. Fungal infections such as histoplasmosis can cause skin lesions and pulmonary nodules but do not fit well with uveitis.
At this point, “ tissue is the issue.” A skin nodule would be the easiest site to biopsy. If skin biopsy was not diagnostic, computed tomography (CT) of his chest and abdomen should be performed to identify the next most accessible site for biopsy.
Esophagogastroduodenoscopy (EGD) and colonoscopy showed normal findings, and random biopsies from the stomach and colon were normal. CT of the chest, abdomen, and pelvis performed with the administration of intravenous contrast showed multiple solid opacities in both lung fields up to 1 cm, with enlarged mediastinal and retroperitoneal lymph nodes measuring 1 to 3 cm in diameter, a left pleural effusion, wall thickening in the right colon, and several nonspecific hypodensities in the liver. A punch biopsy taken from the right chest wall lesion demonstrated chronic inflammation without granulomas. The patient underwent CT-guided biopsy of 1 of the right-sided lung nodules, which revealed noncaseating granulomatous inflammation, fibrosis, and necrosis. Neither biopsy contained malignant cells, and additional stains revealed no bacteria, fungi, or acid fast bacilli.
The retroperitoneal and mediastinal adenopathy are indicative of a widely disseminated inflammatory process. Lymphoma continues to be a concern, though uveitis as an initial presenting problem would very unusual. Although biopsy of the chest wall lesion failed to demonstrate granulomatous inflammation, the most parsimonious explanation is that the skin and lung nodules are both related to a single systemic process.
Granulomas form in an attempt to wall off offending agents, whether foreign antigens (talc, certain medications), infectious agents, or self-antigens. Review of histopathology and microbiologic studies are useful first steps. Stains for bacteria, fungi, or acid-fast organisms may diagnose an infectious cause, such as tuberculosis, leprosy, syphilis, fungi, or cat scratch disease. Granulomas in association with vascular inflammation would indicate vasculitis. Other autoimmune considerations include sarcoidosis and Crohn disease. Noncaseating granulomas are typically found in sarcoidosis, cat scratch disease, syphilis, leprosy, or Crohn disease, but do not entirely exclude tuberculosis.
The negative infectious studies and lack of classic features of Crohn disease or other autoimmune diseases further point to sarcoidosis as the etiology of this patient’s illness. A Norwegian dermatologist first described the pathology of sarcoidosis based upon specimens taken from skin nodules. He thought the lesions were sarcoma and described them as, “ multiple benign sarcoid of the skin,” which is where the name “ sarcoidosis” originated.
Diagnosing sarcoidosis requires excluding other mimickers. Additional testing should include syphilis serologies, rheumatoid factor, and antineutrophilic cytoplasmic antibodies. The latter is associated with granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, either of which may produce granulomatous inflammation of the lungs, skin, and uvea.
At this juncture, PET-CT represents a costly and unnecessary test that does not narrow our diagnostic possibilities sufficiently to justify its use. Osteolytic lesions would be unusual in sarcoidosis and more likely in lymphoma or infectious processes such as tuberculosis. Tests for syphilis and tuberculosis are required, and are a fraction of the cost of a PET-CT.
With the biopsy revealing spirochetes, and the positive results of a nontreponemal test (RPR) and confirmatory treponemal results, the diagnosis of syphilis is firmly established. Uveitis indicates neurosyphilis and warrants a longer course of intravenous penicillin. Lumbar puncture should be performed.
A lumbar puncture was performed. Cerebrospinal fluid (CSF) contained 9 white blood cells and 73 red blood cells per cubic milliliter; protein concentration was 73 mg/dL, and glucose was 116 mg/dL. Polymerase chain reaction for T. pallidum was negative. Transthoracic ECG and magnetic resonance imaging of the brain were normal. The patient was treated with intravenous penicillin G at 5 million units 4 times daily for 15 days. A PET-CT scan 3 months later revealed complete resolution of the subcutaneous, pulmonary, liver lesions, lymphadenopathy, and uveitis. Repeat treponemal serologies demonstrated a greater than 4-fold decline in titers.
DISCUSSION
Syphilis is a sexually transmitted disease with increasing incidence worldwide. Untreated infection progresses through 3 stages. The primary stage is characterized by the appearance of a painless chancre after an incubation period of 2 to 3 weeks. Four to 8 weeks later, the secondary stage emerges as a systemic infection, often heralded by a maculopapular rash with desquamation, frequently involving the soles and palms. Hepatitis, iridocyclitis, and early neurosyphilis may also be seen at this stage. Subsequently, syphilis becomes latent. One-third of patients with untreated latent syphilis will develop tertiary syphilis, typified by late neurosyphilis (tabes dorsalis and general paresis), cardiovascular disease (aortitis), or gummatous disease. 1
Gummas are destructive granulomatous lesions that typically present indolently, may occur singly or multiply, and may involve almost any organ. It has been suggested that gummas are the immune system’s defense to slow the bacteria after attempts to kill it have failed. Histologically, gummas are hyalinized nodules with surrounding granulomatous infiltrate of lymphocytes, plasma cells, and multinucleated giant cells with or without necrosis . In the preantibiotic era, gummas were seen in approximately 15% of infected patients, with a latency of 1 to 46 years after primary infection. 2 Penicillin led to a drastic reduction in gummas until the HIV epidemic, which led to the resurgence of gummas at a drastically shortened interval following primary syphilis. 3
Most commonly, gummas affect the skin and bones. In the skin , lesions may be superficial or deep and may progress into ulcerative nodules. In the bones, destructive gummas have a characteristic “moth-eaten” appearance. Less common sequelae of gummas incude gummatous hepatitis, perforated nasal septum (saddle nose deformity), or hard palate erosions. 2,4 R arely, syphilis involves the lungs, appearing as nodules, infiltrates, or pleural effusion. 5
Ocular manifestations occur in approximately 5% of patients with syphilis, more often in secondary and tertiary stages, and are strongly associated with a spread to the central nervous system. Syphilis may affect any structure of the eye, with anterior uveitis as the most frequent manifestation. Partial or complete vision loss is identified in approximately half of the patients with ocular syphilis and may be completely reversed by appropriate treatment. Ophthalmologic findings such as optic neuritis and papilledema imply advanced illness , as do Argyll-Robertson pupils (small pupils that are poorly reactive to light , but with preserved accommodation and convergence). 6,7 The treatment of ocular syphilis is identical to that of neurosyphilis. The Centers for Disease Control and Prevention recommends CSF analysis in any patient with ocular syphilis. Abnormal results should prompt repeat lumbar puncture every 3 to 6 months following treatment until the CSF results normalize. 8
The diagnosis of syphilis relies on indirect serologic tests. T. pallidum cannot be cultured in vitro, and techniques to identify spirochetes directly by using darkfield microscopy or DNA amplification via polymerase chain reaction are limited by availability or by poor sensitivity in advanced syphilis. 1 Imaging modalities including PET cannot reliably differentiate syphilis from other infectious and noninfectious mimickers. 9 F ortunately, syphilis infection can be diagnosed accurately based on reactive treponemal and nontreponemal serum tests. Nontreponemal tests, such as the RPR and Venereal Disease Research Laboratory, have traditionally been utilized as first-line evaluation, followed by a confirmatory treponemal test. However, nontreponemal tests may be nonreactive in a few settings: very early or very late in infection, and in individuals previously treated for syphilis. Thus, newer “reverse testing” algorithms utilize more sensitive and less expensive treponemal tests as the first test, followed by nontreponemal tests if the initial treponemal test is reactive. 8 Regardless of the testing sequence, in patients with no prior history of syphilis, reactive results on both treponemal and nontreponemal assays firmly establish a diagnosis of syphilis, obviating the need for more invasive and costly testing.
In patients with unexplained systemic illness, clinicians should have a low threshold to test for syphilis. Testing should be extended to certain asymptomatic individuals at higher risk of infection, including men who have sex with men, sexual partners of patients infected with syphilis, individuals with HIV or sexually-transmitted diseases, and others with high-risk sexual behavior or a history of sexually-transmitted diseases. 8 As the discussant points out, earlier consideration of and testing for syphilis would have spared the patient from unnecessary and costly EGD, colonoscopy, PET-CT scanning, and 3 biopsies.
Syphilis has been known to be a horribly destructive disease for centuries, earning the moniker “morbo serpentino” (serpentine disease) from the Spanish physician Ruiz Diaz de Isla in the 1500s. 10 In the modern era, physicians must remember to consider the diagnosis of syphilis in order to effectively mitigate the harm from this resurgent disease when it attacks our patients.
TEACHING POINTS
- Syphilis, the great imposter, is rising in incidence and should be on the differential diagnosis in all patients with unexplained multisystem inflammatory disease.
- A cost-effective diagnostic approach to syphilis entails serologic testing with treponemal and nontreponemal assays.
- Unexplained granulomas, especially in the skin, bone, or liver, should prompt consideration of gummatous syphilis.
- Ocular syphilis may involve any part of the visual tract and is treated the same as neurosyphilis.
Disclosure
Dr. Weinreich has received payment for lectures from Boehringer er Ingelheim, Astra Zeneca, TEVA and Novartis in 2016. All other contributors have nothing to report.
A 58-year-old Danish man presented to an urgent care center due to several months of gradually worsening fatigue, weight loss, abdominal pain, and changes in vision . His abdominal pain was diffuse, constant, and moderate in severity. There was no association with meals, and he reported no nausea, vomiting, or change in bowel movements. He also said his vision in both eyes was blurry, but denied diplopia and said the blurring did not improve when either eye w as closed. He denied dysphagia, headache, focal weakness, or sensitivity to bright lights.
Fatigue and weight loss in a middle-aged man are nonspecific complaints that mainly help to alert the clinician that there may be a serious, systemic process lurking. Constant abdominal pain without nausea, vomiting, or change in bowel movements makes intestinal obstruction or a motility disorder less likely. Given that the pain is diffuse, it raises the possibility of an intraperitoneal process or a process within an organ that is irritating the peritoneum.
Worsening of vision can result from disorders anywhere along the visual pathway, including the cornea (keratitis or corneal edema from glaucoma), anterior chamber (uveitis or hyphema), lens (cataracts, dislocations, hyperglycemia), vitreous humor (uveitis), retina (infections, ischemia, detachment, diabetic retinopathy), macula (degenerative disease), optic nerve (optic neuritis), optic chiasm, and the visual projections through the hemispheres to the occipital lobes. To narrow the differential diagnosis, it would be important to inquire about prior eye problems, to measure visual acuity and intraocular pressure, to perform fundoscopic and slit-lamp exams to detect retinal and anterior chamber disorders, respectively, and to assess visual fields. An afferent pupillary defect would suggest optic nerve pathology.
Disorders that could unify the constitutional, abdominal, and visual symptoms include systemic inflammatory diseases, such as sarcoidosis (which has an increased incidence among Northern Europeans), tuberculosis, or cancer. While diabetes mellitus could explain his visual problems, weight loss, and fatigue, the absence of polyuria, polydipsia, or polyphagia argues against this possibility.
The patient had hypercholesterolemia and type 2 diabetes mellitus. Medications were metformin, atorvastatin, and glimepiride. He was a former smoker with 23 pack-years and had quit over 5 years prior. He had not traveled outside of Denmark in 2 years and had no pets at home. He reported being monogamous with his same-sex partner for the past 25 years. He had no significant family history, and he worked at a local hospital as a nurse. He denied any previous ocular history.
On examination, the pulse was 67 beats per minute, temperature was 36.7 degrees Celsius, respiratory rate was 16 breaths per minute, oxygen saturation was 99% while breathing ambient air, and blood pressure was 132/78. Oropharynx demonstrated no thrush or other lesions. The heart rhythm was regular and there were no murmurs. Lungs were clear to auscultation bilaterally. Abdominal exam was normal except for mild tenderness upon palpation in all quadrants, but no masses, organomegaly, rigidity, or rebound tenderness were present. Skin examination revealed several subcutaneous nodules measuring up to 0.5 cm in diameter overlying the right and left posterolateral chest walls. T he nodules were rubbery, pink, nontender, and not warm nor fluctuant. Visual acuity was reduced in both eyes. Extraocular movements were intact, and the pupils reacted to light and accommodated appropriately. The sclerae were injected bilaterally. The remainder of the cranial nerves and neurologic exam were normal. Due to the vision loss , the patient was referred to an ophthalmologist who diagnosed bilateral anterior uveitis.
Though monogamous with his male partner for many years, it is mandatory to consider complications of human immunodeficiency virus infection (HIV ). The absence of oral lesions indicative of a low CD4 count, such as oral hairy leukoplakia or thrush, does not rule out HIV disease. Additional history about his work as a nurse might shed light on his risk of infection, such as airborne exposure to tuberculosis or acquisition of blood-borne pathogens through a needle stick injury. His unremarkable vital signs support the chronicity of his medical condition.
Uveitis can result from numerous causes. When confined to the eye, uncommon hereditary and acquired causes are less likely . In many patients, uveitis arises in the setting of systemic infection or inflammation. The numerous infectious causes of uveitis include syphilis, tuberculosis, toxoplasmosis, cat scratch disease, and viruses such as HIV, West Nile, and Ebola. Among the inflammatory diseases that can cause uveitis are sarcoidosis, inflammatory bowel disease, systemic lupus erythematosus, Behçet disease, and Sjogren syndrome.
Several of these conditions, including tuberculosis and syphilis, may also cause subcutaneous nodules. Both tuberculosis and syphilis can cause skin and gastrointestinal disease. Sarcoidosis could involve the skin, peritoneum, and uvea, and is a possibility in this patient. The dermatologic conditions associated with sarcoidosis are protean and include granulomatous inflammation and nongranulomatous processes such as erythema nodosum. Usually the nodules of erythema nodosum are tender, red or purple, and located on the lower extremities. The lack of tenderness points away from erythema nodosum in this patient. Metastatic cancer can disseminate to the subcutaneous tissue, and the patient’s smoking history and age mandate we consider malignancy. However, skin metastases tend to be hard, not rubbery.
A cost-effective evaluation at this point would include syphilis serologies, HIV testing, testing for tuberculosis with either a purified protein derivative test or interferon gamma release assay, chest radiography, and biopsy of 1 of the lesions on his back.
Laboratory data showed 12,400 white blood cells per cubic milliliter (64% neutrophils, 24% lymphocytes, 9% monocytes, 2% eosinophils, 1% basophils), hemoglobin 7.9 g/dL, mean corpuscular volume 85 fL, platelets 476,000 per cubic milliliter , C-reactive protein 43 mg/ d L (normal < 8 mg/L), gamma-glutamyl-transferase 554 IU/L (normal range 0-45), alkaline phosphatase 865 U/L (normal range 60-200), and erythrocyte sedimentation rate (ESR) 71 mm per hour. International normalized ratio was 1.0, albumin was 3.0 mg/dL, activated partial thromboplastin time was 32 seconds (normal 22 to 35 seconds), and bilirubin was 0.3 mg/dL. Antibodies to HIV , hepatitis C, and hepatitis B surface antigen were not detectable. Electrocardiography ( ECG ) was normal. Plain radiograph of the chest demonstrated multiple nodular lesions bilaterally measuring up to 1 cm with no cavitation. There was a left pleural effusion.
The history and exam findings indicate a serious inflammatory condition affecting his lungs, pleura, eyes, skin, liver, and possibly his peritoneum. In this context, the elevated C-reactive protein and ESR are not helpful in differentiating inflammatory from infectious causes. The constellation of uveitis, pulmonary and cutaneous nodules, and marked abnormalities of liver tests in a middle-aged man of Northern European origin points us toward sarcoidosis. Pleural effusions are not common with sarcoidosis but may occur. However, to avoid premature closure, it is important to consider other possibilities.
Metastatic cancer, including lymphoma, could cause pulmonary and cutaneous nodules and liver involvement, but the chronic time course and uveitis are not consistent with malignancy. Tuberculosis is still a consideration, though one would have expected him to report fevers, night sweats, and, perhaps, exposure to patients with pulmonary tuberculosis in his job as a nurse. Multiple solid pulmonary nodules are also uncommon with pulmonary tuberculosis. Fungal infections such as histoplasmosis can cause skin lesions and pulmonary nodules but do not fit well with uveitis.
At this point, “ tissue is the issue.” A skin nodule would be the easiest site to biopsy. If skin biopsy was not diagnostic, computed tomography (CT) of his chest and abdomen should be performed to identify the next most accessible site for biopsy.
Esophagogastroduodenoscopy (EGD) and colonoscopy showed normal findings, and random biopsies from the stomach and colon were normal. CT of the chest, abdomen, and pelvis performed with the administration of intravenous contrast showed multiple solid opacities in both lung fields up to 1 cm, with enlarged mediastinal and retroperitoneal lymph nodes measuring 1 to 3 cm in diameter, a left pleural effusion, wall thickening in the right colon, and several nonspecific hypodensities in the liver. A punch biopsy taken from the right chest wall lesion demonstrated chronic inflammation without granulomas. The patient underwent CT-guided biopsy of 1 of the right-sided lung nodules, which revealed noncaseating granulomatous inflammation, fibrosis, and necrosis. Neither biopsy contained malignant cells, and additional stains revealed no bacteria, fungi, or acid fast bacilli.
The retroperitoneal and mediastinal adenopathy are indicative of a widely disseminated inflammatory process. Lymphoma continues to be a concern, though uveitis as an initial presenting problem would very unusual. Although biopsy of the chest wall lesion failed to demonstrate granulomatous inflammation, the most parsimonious explanation is that the skin and lung nodules are both related to a single systemic process.
Granulomas form in an attempt to wall off offending agents, whether foreign antigens (talc, certain medications), infectious agents, or self-antigens. Review of histopathology and microbiologic studies are useful first steps. Stains for bacteria, fungi, or acid-fast organisms may diagnose an infectious cause, such as tuberculosis, leprosy, syphilis, fungi, or cat scratch disease. Granulomas in association with vascular inflammation would indicate vasculitis. Other autoimmune considerations include sarcoidosis and Crohn disease. Noncaseating granulomas are typically found in sarcoidosis, cat scratch disease, syphilis, leprosy, or Crohn disease, but do not entirely exclude tuberculosis.
The negative infectious studies and lack of classic features of Crohn disease or other autoimmune diseases further point to sarcoidosis as the etiology of this patient’s illness. A Norwegian dermatologist first described the pathology of sarcoidosis based upon specimens taken from skin nodules. He thought the lesions were sarcoma and described them as, “ multiple benign sarcoid of the skin,” which is where the name “ sarcoidosis” originated.
Diagnosing sarcoidosis requires excluding other mimickers. Additional testing should include syphilis serologies, rheumatoid factor, and antineutrophilic cytoplasmic antibodies. The latter is associated with granulomatosis with polyangiitis and eosinophilic granulomatosis with polyangiitis, either of which may produce granulomatous inflammation of the lungs, skin, and uvea.
At this juncture, PET-CT represents a costly and unnecessary test that does not narrow our diagnostic possibilities sufficiently to justify its use. Osteolytic lesions would be unusual in sarcoidosis and more likely in lymphoma or infectious processes such as tuberculosis. Tests for syphilis and tuberculosis are required, and are a fraction of the cost of a PET-CT.
With the biopsy revealing spirochetes, and the positive results of a nontreponemal test (RPR) and confirmatory treponemal results, the diagnosis of syphilis is firmly established. Uveitis indicates neurosyphilis and warrants a longer course of intravenous penicillin. Lumbar puncture should be performed.
A lumbar puncture was performed. Cerebrospinal fluid (CSF) contained 9 white blood cells and 73 red blood cells per cubic milliliter; protein concentration was 73 mg/dL, and glucose was 116 mg/dL. Polymerase chain reaction for T. pallidum was negative. Transthoracic ECG and magnetic resonance imaging of the brain were normal. The patient was treated with intravenous penicillin G at 5 million units 4 times daily for 15 days. A PET-CT scan 3 months later revealed complete resolution of the subcutaneous, pulmonary, liver lesions, lymphadenopathy, and uveitis. Repeat treponemal serologies demonstrated a greater than 4-fold decline in titers.
DISCUSSION
Syphilis is a sexually transmitted disease with increasing incidence worldwide. Untreated infection progresses through 3 stages. The primary stage is characterized by the appearance of a painless chancre after an incubation period of 2 to 3 weeks. Four to 8 weeks later, the secondary stage emerges as a systemic infection, often heralded by a maculopapular rash with desquamation, frequently involving the soles and palms. Hepatitis, iridocyclitis, and early neurosyphilis may also be seen at this stage. Subsequently, syphilis becomes latent. One-third of patients with untreated latent syphilis will develop tertiary syphilis, typified by late neurosyphilis (tabes dorsalis and general paresis), cardiovascular disease (aortitis), or gummatous disease. 1
Gummas are destructive granulomatous lesions that typically present indolently, may occur singly or multiply, and may involve almost any organ. It has been suggested that gummas are the immune system’s defense to slow the bacteria after attempts to kill it have failed. Histologically, gummas are hyalinized nodules with surrounding granulomatous infiltrate of lymphocytes, plasma cells, and multinucleated giant cells with or without necrosis . In the preantibiotic era, gummas were seen in approximately 15% of infected patients, with a latency of 1 to 46 years after primary infection. 2 Penicillin led to a drastic reduction in gummas until the HIV epidemic, which led to the resurgence of gummas at a drastically shortened interval following primary syphilis. 3
Most commonly, gummas affect the skin and bones. In the skin , lesions may be superficial or deep and may progress into ulcerative nodules. In the bones, destructive gummas have a characteristic “moth-eaten” appearance. Less common sequelae of gummas incude gummatous hepatitis, perforated nasal septum (saddle nose deformity), or hard palate erosions. 2,4 R arely, syphilis involves the lungs, appearing as nodules, infiltrates, or pleural effusion. 5
Ocular manifestations occur in approximately 5% of patients with syphilis, more often in secondary and tertiary stages, and are strongly associated with a spread to the central nervous system. Syphilis may affect any structure of the eye, with anterior uveitis as the most frequent manifestation. Partial or complete vision loss is identified in approximately half of the patients with ocular syphilis and may be completely reversed by appropriate treatment. Ophthalmologic findings such as optic neuritis and papilledema imply advanced illness , as do Argyll-Robertson pupils (small pupils that are poorly reactive to light , but with preserved accommodation and convergence). 6,7 The treatment of ocular syphilis is identical to that of neurosyphilis. The Centers for Disease Control and Prevention recommends CSF analysis in any patient with ocular syphilis. Abnormal results should prompt repeat lumbar puncture every 3 to 6 months following treatment until the CSF results normalize. 8
The diagnosis of syphilis relies on indirect serologic tests. T. pallidum cannot be cultured in vitro, and techniques to identify spirochetes directly by using darkfield microscopy or DNA amplification via polymerase chain reaction are limited by availability or by poor sensitivity in advanced syphilis. 1 Imaging modalities including PET cannot reliably differentiate syphilis from other infectious and noninfectious mimickers. 9 F ortunately, syphilis infection can be diagnosed accurately based on reactive treponemal and nontreponemal serum tests. Nontreponemal tests, such as the RPR and Venereal Disease Research Laboratory, have traditionally been utilized as first-line evaluation, followed by a confirmatory treponemal test. However, nontreponemal tests may be nonreactive in a few settings: very early or very late in infection, and in individuals previously treated for syphilis. Thus, newer “reverse testing” algorithms utilize more sensitive and less expensive treponemal tests as the first test, followed by nontreponemal tests if the initial treponemal test is reactive. 8 Regardless of the testing sequence, in patients with no prior history of syphilis, reactive results on both treponemal and nontreponemal assays firmly establish a diagnosis of syphilis, obviating the need for more invasive and costly testing.
In patients with unexplained systemic illness, clinicians should have a low threshold to test for syphilis. Testing should be extended to certain asymptomatic individuals at higher risk of infection, including men who have sex with men, sexual partners of patients infected with syphilis, individuals with HIV or sexually-transmitted diseases, and others with high-risk sexual behavior or a history of sexually-transmitted diseases. 8 As the discussant points out, earlier consideration of and testing for syphilis would have spared the patient from unnecessary and costly EGD, colonoscopy, PET-CT scanning, and 3 biopsies.
Syphilis has been known to be a horribly destructive disease for centuries, earning the moniker “morbo serpentino” (serpentine disease) from the Spanish physician Ruiz Diaz de Isla in the 1500s. 10 In the modern era, physicians must remember to consider the diagnosis of syphilis in order to effectively mitigate the harm from this resurgent disease when it attacks our patients.
TEACHING POINTS
- Syphilis, the great imposter, is rising in incidence and should be on the differential diagnosis in all patients with unexplained multisystem inflammatory disease.
- A cost-effective diagnostic approach to syphilis entails serologic testing with treponemal and nontreponemal assays.
- Unexplained granulomas, especially in the skin, bone, or liver, should prompt consideration of gummatous syphilis.
- Ocular syphilis may involve any part of the visual tract and is treated the same as neurosyphilis.
Disclosure
Dr. Weinreich has received payment for lectures from Boehringer er Ingelheim, Astra Zeneca, TEVA and Novartis in 2016. All other contributors have nothing to report.
1. French P. Syphilis. BMJ. 2007;334:143-147. PubMed
2. Singh AE, Romanowski B. Syphilis: Review with emphasis on clinical, epidemiologic, and some biologic features. Clin Micriobio Rev. 1999;12(2):187-209. PubMed
3. Karp G, Schlaeffer F, Jotkowitz A, Riesenberg K. Syphilis and HIV co-infection. Eur J Int Med. 2009; 20:9-13. PubMed
4. Pilozzi-Edmonds L, Kong LY, Szabo J, Birnbaum LM. Rapid progression to gummatous syphilitic hepatitis and neurosyphilis in a patient with newly diagnosed HIV. Int J STD AIDS. 2014;26(13)985-987. PubMed
5. David G, Perpoint T, Boibieux A, et al. Secondary pulmonary syphilis: report of a likely case and literature review. Clin Infect Dis. 2006;42(3):e11-e15. PubMed
6. Moradi A, Salek S, Daniel E, et al. Clinical features and incidence rates of ocular complications in patients with ocular syphilis. Am J Ophthalmol. 2015;159:334-343. PubMed
7. Aldave AJ, King JA, Cunningham ET Jr. Ocular syphilis. Curr Opin Ophthalmol. 2001;12:433-441. PubMed
8. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137. PubMed
9. Lin M, Darwish B, Chu J. Neurosyphilitic gumma on F18-2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography: An old disease investigated with new technology. J Clin Neurosc. 2009;16:410-412. PubMed
10. de Ricon‐Ferraz A. Early work on syphilis: Diaz de Ysla’s treatise on the serpentine disease of Hispaniola Island. Int J Dermatol. 1999;38(3):222-227. PubMed
1. French P. Syphilis. BMJ. 2007;334:143-147. PubMed
2. Singh AE, Romanowski B. Syphilis: Review with emphasis on clinical, epidemiologic, and some biologic features. Clin Micriobio Rev. 1999;12(2):187-209. PubMed
3. Karp G, Schlaeffer F, Jotkowitz A, Riesenberg K. Syphilis and HIV co-infection. Eur J Int Med. 2009; 20:9-13. PubMed
4. Pilozzi-Edmonds L, Kong LY, Szabo J, Birnbaum LM. Rapid progression to gummatous syphilitic hepatitis and neurosyphilis in a patient with newly diagnosed HIV. Int J STD AIDS. 2014;26(13)985-987. PubMed
5. David G, Perpoint T, Boibieux A, et al. Secondary pulmonary syphilis: report of a likely case and literature review. Clin Infect Dis. 2006;42(3):e11-e15. PubMed
6. Moradi A, Salek S, Daniel E, et al. Clinical features and incidence rates of ocular complications in patients with ocular syphilis. Am J Ophthalmol. 2015;159:334-343. PubMed
7. Aldave AJ, King JA, Cunningham ET Jr. Ocular syphilis. Curr Opin Ophthalmol. 2001;12:433-441. PubMed
8. Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137. PubMed
9. Lin M, Darwish B, Chu J. Neurosyphilitic gumma on F18-2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography: An old disease investigated with new technology. J Clin Neurosc. 2009;16:410-412. PubMed
10. de Ricon‐Ferraz A. Early work on syphilis: Diaz de Ysla’s treatise on the serpentine disease of Hispaniola Island. Int J Dermatol. 1999;38(3):222-227. PubMed
© 2017 Society of Hospital Medicine
Can’t Shake This Feeling
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
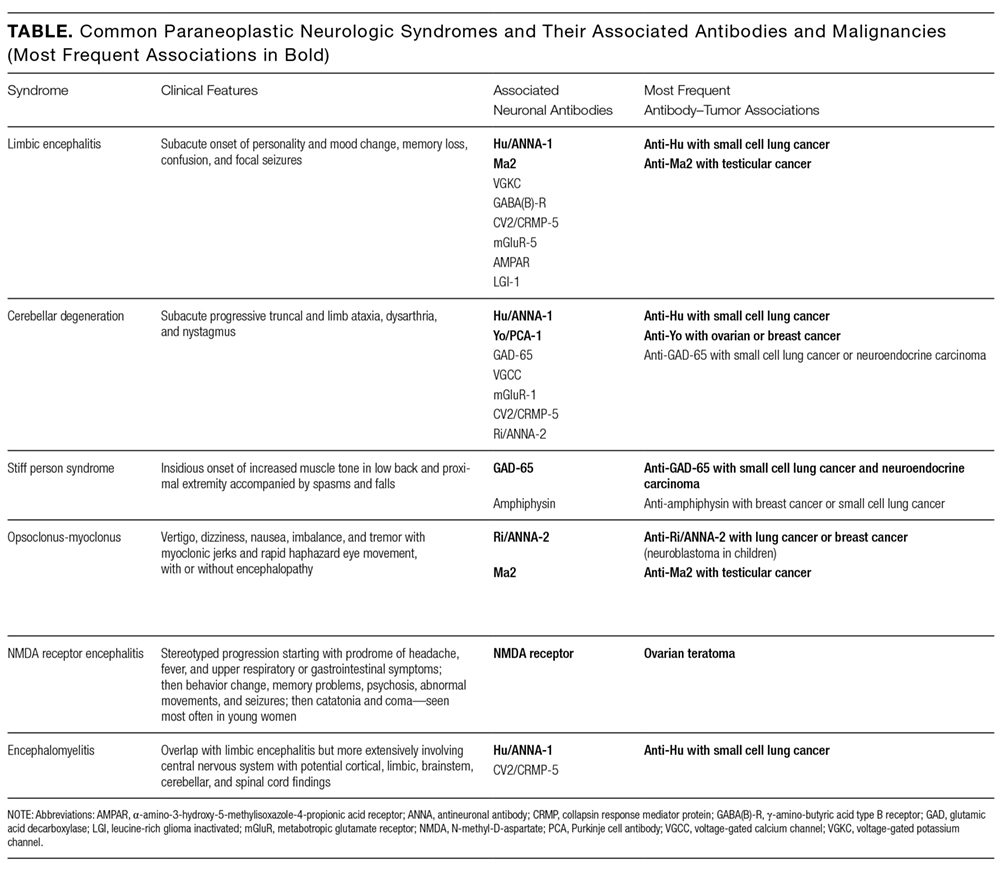
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 78-year-old woman presented to her primary care physician with a 2-month history of progressive leg weakness. She reported walking difficulty caused by occasional “buckling” of the knees.
The knee buckling may be a clue to the quadriceps muscle weakness. The quadriceps straightens and locks the knee when the foot is being planted. Weakness of this muscle can result in the knee giving way. Isolated quadriceps weakness, which is uncommon, typically is caused by lower motor neuron issues, such as femoral neuropathy, L4–L5 radiculopathy, lumbosacral plexopathy, and primary muscle diseases, including inclusion body myositis.
The patient had diabetes mellitus and hypertension. Her medications were insulin glargine, metformin, glipizide, lisinopril, atorvastatin, and aspirin, and she was taking vitamin D and calcium. None of these was recently changed or added. Aside from having the weakness, the patient was in her usual state of health and had no other complaints. She denied weight changes, fevers, night sweats, and fatigue. She was widowed, lived with her daughter, had no pets, never used tobacco, and did not drink alcohol or use illicit drugs. There was no family history of neuromuscular disorders, and both of her parents died of natural causes at advanced ages.
The physical examination revealed no knee deformities, and the patient had good active range of motion of both knees and normal strength throughout her limbs. Plain radiographs of the knees showed mild medial compartment osteoarthritis. The patient was advised to stop atorvastatin.
Patients who take atorvastatin and other statins (3-hydroxy-3-methyl-glutaryl-co-enzyme A reductase inhibitors) can experience a spectrum of muscle disease, from myalgias and weakness to (rare) overt myositis with rhabdomyolysis. Statin-induced myopathy tends to affect larger proximal muscles, can occur at any time during the period the medication is being used, and usually resolves within weeks after discontinuation. Given this patient’s preserved strength, it was reasonable to manage her conservatively.
One month later, she presented to another hospital’s emergency department with increasing weakness in the lower extremities and new loss of balance requiring use of a walker for ambulation. She thought the weakness was confined to her legs and was affecting her thigh muscles more than her calves or feet. She reported fatigue, decreased appetite, and an unintentional 15-pound weight loss. She denied diarrhea, back pain, bowel and bladder function problems, sensation changes, myalgias, and arthralgias. She reported no swallowing or vision problems, rashes, Raynaud disease symptoms, photosensitivity, dry eyes or mouth, recent falls or trauma, fevers, night sweats, recent illness, or travel.
On physical examination, the patient’s temperature was 98.2°F, blood pressure 120/84 mm Hg, pulse 73 beats per minute, respiratory rate 16 breaths per minute, and oxygen saturation 99% with ambient air. The patient was obese and not in distress. She was alert, oriented, and able to follow multistep instructions. Cranial nerve examination was normal. The patient had mild weakness in her bilateral deltoids and bilateral hip flexors but full strength in all other muscle groups. Deep tendon reflexes were normal in the biceps and patella and reduced in the ankles. The Babinski sign was absent. Throughout the lower extremities, sensation was intact to light touch; there was no saddle anesthesia. Finger–nose–finger testing showed slight dysmetria in the left upper extremity. Because of her imbalance, the patient needed help to stand up; once upright, though, she was able to take 3 steps forward and backward with use of a walker. Her stride length was diminished, and her gait unsteady and wide based.
Serum chemistry panel was normal, creatinine level 0.47 mg/dL, and albumin level 4.0 g/dL. White blood cell (WBC) count was 8100/mm3, hemoglobin level 12 g/dL, and platelet count 287,000/mm3. Alanine aminotransferase (ALT) level was 74 U/L (reference range, 0-36 U/L), alkaline phosphatase level 41 U/L (reference range, 37-117 U/L), and total bilirubin level 0.4 mg/dL (reference range, 0.2-1.2 mg/dL). Prothrombin time and thyrotropin were normal. Creatine kinase (CK) level was 2328 U/L (reference range, <200 U/L). Erythrocyte sedimentation rate was 17 mm/h, and C-reactive protein level 0.1 mg/L. Urinalysis (dipstick testing) detected no myoglobin, and there were no casts. Plain radiograph of the chest was normal.
The elevated CK indicates muscle disease, and, in the absence of other findings of liver disease, the ALT elevation likely has a muscle origin as well. The differential diagnosis for elevated CK includes myopathy caused either by infection, trauma, ischemia, or a toxin (medication included) or by a hereditary, metabolic, endocrinologic, or inflammatory disorder. There is no history of trauma, strenuous exertion, or muscle toxin other than the statin, and the progression of symptoms after medication discontinuation argues against statin myopathy. The laboratory test results rule out derangement of potassium, calcium, phosphorus, magnesium, vitamin D, or thyroid function as the cause of the myopathy. The absence of fever, absence of diffuse organ involvement, and normal inflammatory markers point away from a systemic infection or vasculitis. The inflammatory myopathies dermatomyositis and polymyositis classically produce proximal muscle weakness and are possibilities in this case, but one would expect the inflammatory markers to be elevated in these conditions. Malignancy related to dermatomyositis or to paraneoplastic syndrome may account for the myopathy.
The data provided do not identify a unifying diagnosis. To look for an inflammatory myopathy, such as dermatomyositis or polymyositis, it is reasonable to perform electromyography (EMG) to delineate the location of muscle involvement and identify a site for tissue biopsy. As no obvious toxins or metabolic conditions explain the dysmetria, magnetic resonance imaging (MRI) of the brain should be performed to evaluate for lesions in the cerebellum.
The patient was admitted to the hospital. On T2-weighted and FLAIR (fluid attenuation inversion recovery) sequences, MRI of the brain showed a few scattered subcortical and periventricular white matter hyperintense foci bilaterally. Antibodies to human immunodeficiency virus 1 and 2, and Treponema pallidum immunoglobulins G and M, were not detected. Serum levels of lactate dehydrogenase, vitamin B 12 , angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, and anti–cyclic citrullinated peptide IgG were normal.
The brain imaging excludes a space-occupying lesion in the cerebellum but does not identify the cause of dysmetria. Toxic-metabolic conditions, such as alcohol toxicity, vitamin B12 deficiency, anoxia, and toxicity of certain medications, may impair cerebellar function (MRI findings may be normal), but none of these is present. Other disorders that attack the central nervous system (CNS) (again, brain imaging may show minimal abnormalities) include infections, early-stage neurodegenerative illnesses, and antibody-associated disorders (eg, autoimmune diseases, postinfectious and paraneoplastic conditions).
Four days after intravenous fluids were started, the patient’s CK level improved, but her weakness persisted. There was no evidence of peripheral neuropathy on lower extremity nerve conduction studies. EMG revealed few fibrillations and positive sharp waves in proximal limb muscles and thoracic paraspinal muscles. Deltoid, biceps, and tensor fasciae latae showed shorter duration complex motor units with early recruitment. The patient declined muscle biopsy. A rheumatologist was consulted, and prednisone 60 mg/d was started for possible inflammatory myopathy. The patient was discharged to a skilled nursing facility for physical therapy.
The fibrillations and positive sharp waves on EMG can be seen in both neuropathy (from denervation) and myopathy. The normal nerve conduction studies make localization to the nerve unlikely. In addition, the shorter duration motor units with early recruitment on EMG are characteristic of a myopathy. Despite the ongoing myopathy, the improved CK level suggests the muscle disease is playing a minimal role in the patient’s current illness. Prescribing corticosteroids for a presumed inflammatory myopathy without a clear diagnosis is risky, as steroids may render subsequent biopsy results unreliable, may themselves cause myopathy, and expose the patient to the side effects of immunosuppression.
One month later, the patient saw her rheumatologist. Although she had tapered the prednisone down to 10 mg/d, she had not returned to baseline strength, was still using a walker, and reported increased difficulty keeping her head raised. She also noted 2 new symptoms: speech slurring and, in both hands, a tremor that made it difficult to hold objects.
Examination revealed a fine tremor in both arms. There were no skin lesions, and the joints were normal. The patient was oriented to name, place, and date. Memory testing was 3 for 3 on register but 0 for 3 on recall. She was unable to perform serial 7s and able to spell backward only 3 of the 5 letters in the word world . Speech was dysarthric and scanning in quality. On extraocular movements, she demonstrated poor smooth pursuit. Examination of the head and neck was significant for nearly constant head titubation and weak neck flexors. Upper extremity strength was normal. Mild weakness was noted in both hip flexors. Deep tendon reflexes were preserved except at the ankle, where they were reduced. Finger–nose–finger testing revealed marked dysmetria, more pronounced on the left, and there was mild bilateral heel-to-shin dysmetria.
Diffuse myopathy cannot account for the patient’s impaired cognition or progressive cerebellar findings, which now include head titubation and scanning speech. As more than a month has elapsed since the brain imaging was performed, MRI could be repeated for evidence of infection, malignancy, inflammation, or demyelination. More important, lumbar puncture is indicated to exclude infection and, with flow cytometry, cytology, and measurement of oligoclonal bands and IgG index, to assess for autoimmune or paraneoplastic antibody-mediated disorders.
The patient was readmitted. On repeat brain MRI, there were no new significant findings. Complete blood cell count and chemistry panel results were unremarkable. Erythrocyte sedimentation rate and C-reactive protein level remained normal. CK level was 451 U/L, and ALT level 29 U/L.
Lumbar puncture was performed. Opening pressure was 14.5 cm of water, and cerebrospinal fluid (CSF) was clear and colorless. There were 3 red blood cells/mm 3 and no WBCs. Glucose level was 94 mg/dL, and protein level 74 mg/dL. CSF IgG synthesis rate was normal, flow cytometry revealed no abnormal clonal populations, and cytology was negative for malignancy. Two unique oligoclonal bands were found in the CSF.
The absence of WBCs in the CSF excludes CNS infection. The patient’s main problem is an inflammatory CNS process as defined by presence of oligoclonal bands in the CSF, compared with their absence in the serum. Autoimmune, neoplastic, and paraneoplastic disorders could explain these bands. There was no evidence of systemic autoimmune illness. The patient has not had a recent infection that could result in postinfectious demyelination, and her clinical and imaging features are not suggestive of a demyelinating disorder, such as multiple sclerosis. Of the neoplastic possibilities, lymphoma with CNS involvement may be difficult to detect initially; this diagnosis, however, is not supported by the unremarkable MRI, flow cytometry, and cytology findings. In paraneoplastic syndromes, the CSF may include antibodies that react to antigens in the brain or cerebellum.
At this point, evaluation for malignancy should involve mammography, imaging of the chest, abdomen, and pelvis, and colorectal screening. Testing should also include measurement of serum and CSF autoantibodies associated with paraneoplastic cerebellar degeneration. The expanding list of paraneoplastic antibodies that may attack the cerebellum includes anti-Hu (often associated with small cell lung cancer), anti-Yo (associated with ovarian or breast cancer), anti-aquaporin 4, antibodies to the voltage-gated potassium channel, and anti–glutamic acid decarboxylase (anti-GAD).
Mammography and breast examination findings were normal. Computed tomography (CT) of the chest showed no adenopathy, nodules, or masses. Abdomen CT showed nonspecific prominence of the gallbladder wall. Flexible sigmoidoscopy revealed no masses, only thickened folds in the sigmoid colon; results of multiple colon biopsy tests were normal. Carcinoembryonic antigen level was 2.0 μg/L, and CA-125 level 5 U/mL. Serum GAD-65 antibodies were elevated (>30 nmol/L).
Anti-GAD is mostly known as the antibody associated with type 1 diabetes mellitus (T1DM). In rare instances, even in patients without a history of diabetes, anti-GAD antibodies may lead to an autoimmune attack on the brain, particularly the cerebellum, as part of an idiopathic autoimmune disorder or as a paraneoplastic syndrome. In either case, treatment involves corticosteroids, intravenous Ig, or plasma exchange. When the autoimmune attack is associated with malignancy, treatment response is poorer, unless the malignancy is successfully managed. The next steps are intravenous Ig or plasma exchange and positron emission tomography–CT (PET-CT) assessing for an underlying neoplasm that may have been too small to be detected with routine CT.
DISCUSSION
When clinical, MRI, and CSF findings suggest PNS, the next step in establishing the diagnosis is testing for neuronal antibodies. Testing should be performed for a comprehensive panel of antibodies in both serum and CSF.3,4 Testing for a single antibody can miss potential cases because various syndromes may be associated with multiple antibodies. In addition, presence of multiple antibodies (vs a single antibody) is a better predictor of cancer type.5,6 Sensitivity can be optimized by examining both serum and CSF, as in some cases, the antibody is identified in only one of these fluids.7,8 An identified antibody predicts the underlying malignancies most likely involved. For example, presence of anti-Hu antibodies is associated most often with small cell lung cancer, whereas presence of anti-Yo antibodies correlates with cancers of the breast, ovary, and lung. When the evaluation does not identify an underlying malignancy and PNS is suspected, PET-CT can be successfully used to detect an occult malignancy in 20% to 56% of patients.8-10
According to reports, at least 17 autoantibodies, including classic Purkinje cell cytoplasmic antibody type 1 (anti-Yo), antineuronal nuclear antibody type 1 (anti-Hu), and GAD-65 antibody, attack antigens in the cerebellum.11 GAD-65, an enzyme expressed in the brain and pancreatic β cells, is a soluble synaptic protein that produces the inhibitory neurotransmitter γ-amino-butyric acid (GABA).12 Inhibition of GAD-65 in cerebellar tissue leads to decreased expression of GABA, resulting in extensive cerebellar deficits, such as those in the present case. Anti-GAD-65 antibodies have been associated with various disease processes. For example, anti-GAD-65 is found in the serum of 80% of patients with insulin-dependent T1DM.13 GAD-65 antibodies may also be detected in patients with stiff person syndrome (Table) and in patients with cerebellar ataxia caused by a paraneoplastic or autoimmune syndrome.14,15
Paraneoplastic anti-GAD cerebellar ataxia is very rare. It occurs at a median age of 60 years, affects men more often than women, and has an extremely poor prognosis.11,16 Underlying cancers identified in patients with this ataxia include solid organ tumors, lymphoma, and neuroendocrine carcinoma.17 The present case of anti-GAD-65 cerebellar ataxia is the first reported in a patient with biliary tract neuroendocrine carcinoma. Given the rarity of the disease and the advanced stage of illness when the condition is detected, optimal treatment is unknown. As extrapolated from management of other PNSs, recommended treatments are intravenous Ig, plasma exchange, steroids, and other immunosuppressants, as well as control of the underlying neoplasm.11
The discussant in this case couldn’t shake the feeling that there was more to the patient’s illness than statin or inflammatory myopathy. It was the patient’s shaking itself—the dysmetric limb and truncal titubation—that provided a clue to the cerebellar localization and ultimately led to the discovery of a paraneoplastic disorder linked to anatomically remote neuroendocrine cancer.
KEY TEACHING POINTS
- The differential diagnosis for cerebellar deficits associated with normal brain MRI includes infection, toxic-metabolic insults (alcohol toxicity, vitamin B12 deficiency, medication toxicity), anoxia, early neurodegenerative illness, and antibody-mediated disorders, such as autoimmune, postinfectious, and paraneoplastic syndromes.
- Hospitalists should suspect a PNS when a patient with known cancer develops unexplained neurologic deficits or when evaluation for neurologic symptoms identifies an inflammatory CSF profile that cannot be explained by a demyelinating disorder or an infection.
- Hospitalists should familiarize themselves with the classic PNS presentations, including limbic encephalitis, cerebellar degeneration, stiff person syndrome, opsoclonus-myoclonus, NMDA receptor encephalitis, and encephalomyelitis.
- Suspicion for PNS may be confirmed by the presence of paraneoplastic antibodies in CSF or serum. When routine evaluation fails to identify cancer, PET-CT should be performed.
Disclosure
Nothing to report.
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
1. Darnell RB, Posner JB. Paraneoplastic syndromes and the nervous system. N Engl J Med. 2003;3(4):287-288. PubMed
2. Psimaras D, Carpentier AF, Rossi C; PNS Euronetwork. Cerebrospinal fluid study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatry. 2010;81(1):42-45. PubMed
3. Lancaster E, Damlau J. Neuronal autoantigens—pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8(7):380-390. PubMed
4. McKeon A. Paraneoplastic and other autoimmune disorders of the central nervous system. Neurohospitalist. 2012;3(2):53-64. PubMed
5. Kannoth S. Paraneoplastic neurologic syndrome: a practical approach. Ann Indian Acad Neurol. 2012;15(1):6-12. PubMed
6. Hoftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol. 2015;27(6):489-495. PubMed
7. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology. 2011;76(12):1108-1110. PubMed
8. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography–computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol. 2010;67(3):322-329. PubMed
9. Titulaer MJ, Soffietti R, Dalmau J, et al; European Federation of Neurological Societies. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18(1):19-e3. PubMed
10. Basu S, Alavi A. Role of FDG-PET in the clinical management of paraneoplastic neurological syndrome: detection of the underlying malignancy and the brain PET-MRI correlates. Mol Imaging Biol. 2008;10(3):131-137. PubMed
11. Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol. 2015;72(11):1304-1312. PubMed
12. Tohid H. Anti-glutamic acid decarboxylase antibody positive neurological syndromes. Neurosciences. 2016;21(3):215-222. PubMed
13. Asakura T, Yoshida S, Maeshima A, et al. Small cell lung cancer expressing glutamate decarboxylase with latent autoimmune diabetes in adults. Intern Med. 2015;54(23):3035-3037. PubMed
14. Agarwal P, Ichaporia N. Glutamic acid decarboxylase antibody-positive paraneoplastic stiff limb syndrome associated with carcinoma of the breast. Neurol India. 2010;58(3):449-451. PubMed
15. Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147-165. PubMed
16. Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. PubMed
17. Hernandez-Echebarria L, Saiz A, Ares A, et al. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology. 2006;66(3):450-451. PubMed
© 2017 Society of Hospital Medicine
More Than a Mnemonic
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2 20 mm Hg, PaO2 127 mm Hg, and HCO3 5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of 5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal 120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2 20 mm Hg, PaO2 127 mm Hg, and HCO3 5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of 5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal 120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2 20 mm Hg, PaO2 127 mm Hg, and HCO3 5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of 5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal 120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
The Heart of the Matter
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
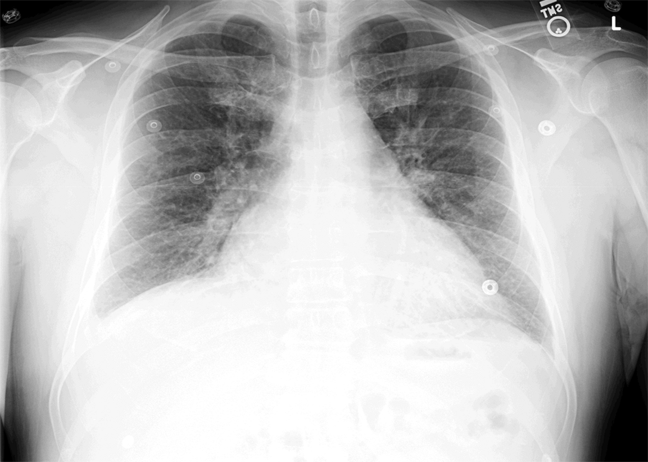
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
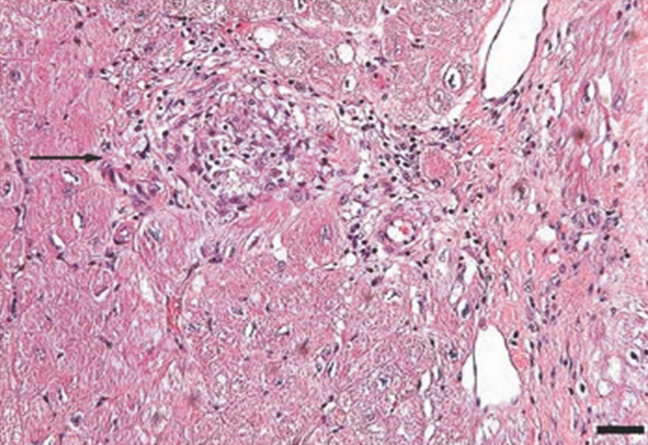
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
Overcome by Weakness
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, 5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
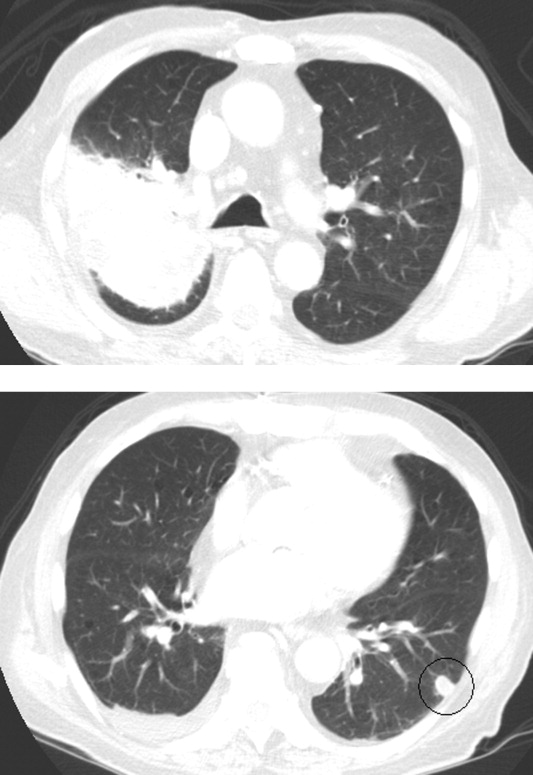
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.

Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, 5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.
Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, 5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.
Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
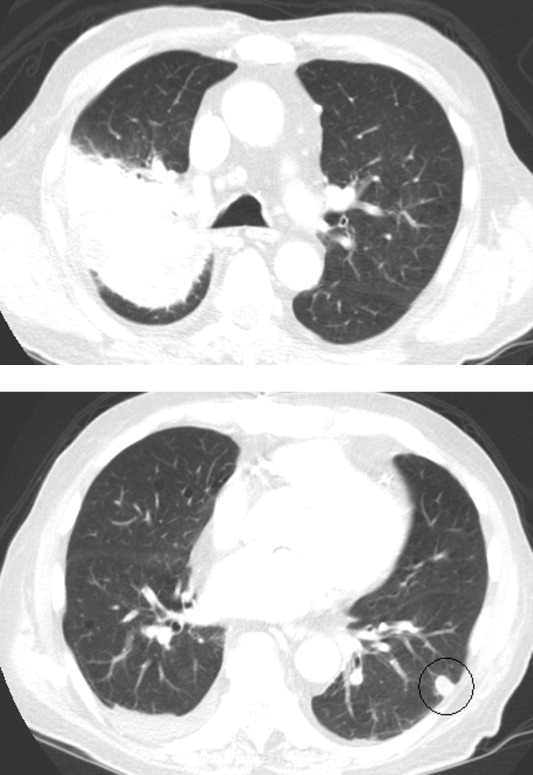
In the Face of It All
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, 57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, 2), and lambda was 4.25 mg/dL (normal range, 2).
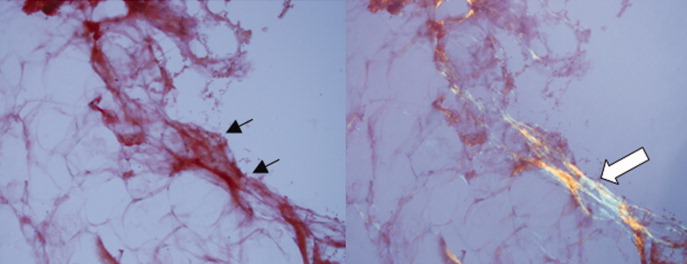
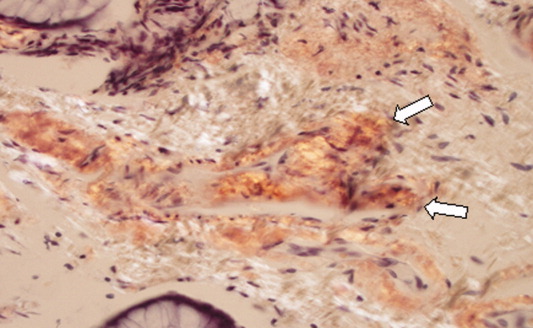
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, 57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, 2), and lambda was 4.25 mg/dL (normal range, 2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, 57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, 2), and lambda was 4.25 mg/dL (normal range, 2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
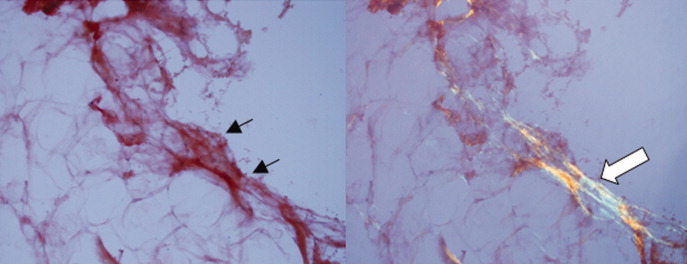
One Hundred Years Later
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 36 year‐old male physician was admitted to a Baltimore hospital in April 1907 with weight loss, weakness, arthralgias, and abdominal distension that had progressed over 5 years.
In 1907, major causes of unexplained weight loss included tuberculosis, hyperthyroidism, cancer, and diabetes. Arthralgias and weakness are not specific. The insidious progression over 5 years narrows the infectious possibilities; tuberculosis and syphilis are important considerations. Since surgical removal was the main treatment for malignancy in 1907, a history of prior surgery might point to a previously diagnosed malignancy that is now progressing.
Five years earlier, while visiting Turkey as a medical missionary, he first noted the onset of arthralgias that lasted 6 to 8 hours and occurred 3 to 4 times per week. Over time, these attacks lasted up to 24 hours and became associated with warmth, swelling, and tenderness of both small and large joints. He gradually lost weight and strength. One year prior to arrival in the hospital, he developed a cough productive of yellow sputum. Seven months prior, he returned from Turkey to Atlanta and noticed an increase in his cough, along with fevers of 100 Fahrenheit and night sweats.
The primacy of the arthralgias in this illness lead me to consider primary rheumatic diseases, and multisystem diseases (including infections) with a predominant skeletal component. In 1907, tests for lupus and the rheumatoid factor were not available. Neither skeletal remains nor works of art provide evidence that rheumatoid arthritis existed until the 19th century, whereas ankylosing spondylitis, gout, and rickets were present by then.
As a medical missionary, he might have acquired a disease endemic to the areas he visited, or the travel history may be a red herring. Familial Mediterranean fever, though prevalent in Turkey and a cause of arthralgias accompanied by recurrent attacks of abdominal pain and fever, is not an acquired disease. Behcet's disease, also known as Silk Trader's Route disease, is found in descendents of the countries that comprised the ancient Silk Route from Japan to the Middle East and may cause arthritis along with oral ulcers, genital lesions, pathergy or uveitis. I would inquire about his ancestry and fevers before dismissing these possibilities.
Although 5 years would be unusually long for tuberculosis to go unrecognized, a physician in the first half of the 20th century would place tuberculosis near the top of possible diagnoses. In 1930, a time when the population of the United States was considerably less, there were over 300,000 cases of tuberculosis. Physicians, and in particular pathologists since autopsies were more commonly performed, often died from tuberculosis since streptomycin, the first antituberculous medication, did not arrive until 1944. At the turn of the 20th century, the ability to detect tubercle bacilli was quite good. Thus, I would include tuberculous peritonitis as a cause of the progressive abdominal symptoms in this physician. In approximately one‐third of patients with tuberculous peritonitis there is evidence of pulmonary disease, and I would try to culture tuberculosis in samples of sputum, a test then so common it probably rivaled our frequent complete blood counts in popularity.
Six months prior, examinations of sputum were negative for tubercle bacilli. Four months prior to arrival, the patient moved to New Mexico. His cough improved but he continued to lose weight and had diarrhea consisting of 3 to 4 loose or semiformed bowel movements per day. Three months prior to admission, he noted an increase in abdominal girth along with right lower quadrant fullness. One month prior, he noted painful swelling and warmth in both ankles as well as dyspnea with exertion.
The increased abdominal girth in the context of chronic illness might be due to ascites, adenopathy, visceromegaly, or mass lesions such as a neoplasm or abscess. If ascites is the cause, one would need to consider primary hepatic disorders, as well as extrahepatic diseases that could progress over years. Infection with hepatitis A virus does not cause chronic liver disease. Hepatitis B, in those days, was referred to as serum hepatitis, and a serum marker for the B virusthe Australia antigenwas not identified until 1967. Cardiac causes of ascites include congestive heart failure and constrictive pericarditis, the latter an important consideration because it is potentially curable. Also, constrictive pericarditis can present as an indolent weight‐losing disease because of chronic visceral congestion. Other considerations include nephrotic syndrome, infection, and neoplasm, including mesothelioma.
Abdominal distention might also be seen with a smoldering abscess. In addition to an appendiceal process, the travel and right lower quadrant localization reminds us to consider ameboma. This patient surely was in an area where amebiasis was endemic, and amebomaa chronic inflammatory form of infection with E. histolytica not associated with diarrhea or liver cystsmay mimic cecal carcinoma. Exertional dyspnea suggests at least the possibility of cardiac disease. Despite the negative sputum cultures, tuberculosis remains high on the list as a cause of constrictive pericarditis or peritonitis, either of which may occur in the absence of active pulmonary disease.
Past medical history included measles and whooping cough as a child, mild pleurisy at age 14, mild influenza 7 years previously. The patient had a tonsillectomy as a child and had a portion of his inferior turbinate bone removed in an attempt to relieve a nasal condition.
On physical exam, the patient was thin and the skin over his face and hands was deep brown. His temperature was 101.5 Fahrenheit, the heart rate was 100, and the respiratory rate was 24. Small lymph nodes were palpable in the axillary and epitrochlear areas. His thorax moved asymmetrically, with less movement on the left apex and slight dullness to percussion in that area. The pulmonic component of the second heart sound was mildly accentuated. The abdomen displayed fullness and tympany, most pronounced in the right lower quadrant without hepatosplenomegaly. The left ankle was swollen, and the overlying skin was tense, shiny, and hot. On both lower legs, areas of discoloration and slight induration were observed, felt to be consistent with faded erythema nodosum.
Though pleurisy has numerous causes, its presence raises the specter of tuberculosis again. The nasal condition triggers thoughts of Wegener's granulomatosis or lethal midline granuloma, both unlikely diagnoses here. The pulmonary exam suggests an apical process, such as tuberculosis, and the accentuated pulmonic heart sound implies pulmonary hypertension, which could be due to a number of chronic pulmonary diseases. The epitrochlear nodes are of interest since lymphoma and Hodgkin's disease rarely involve this area; syphilis and human immunodeficiency virus (HIV) are a few of the chronic diseases that may involve this lymph node region. More helpful is the absence of hepatosplenomegaly, since many indolent malignancies and infections would be expected to enlarge these organs by this point.
Monoarticular arthritis is often due to infection, and less likely due to rheumatoid disease. When rheumatoid arthritis flares, the entire skeleton flares, not single joints. Given the indolence and this single joint involvement, tuberculosis again comes to mind.
I would next want to obtain a plain chest radiograph, looking for evidence of tuberculosis. As with any test, one should ask how this will change management. In 1907, antituberculous medications were not available, so therapy was directed at lowering oxygen tension in the primary site of infection; for example, pulmonary disease was addressed via pneumothorax. If the chest radiograph provides little hint of tuberculosis, then consideration must be given to exploratory surgery of the abdomen given the focal abnormality in the right lower quadrant.
A peripheral blood smear revealed a hypochromic microcytic anemia. The total red blood cell count was 4.468 million/mm3 (normal range for men is 4.52‐5.90 million/mm3), white blood cell count was 8180/mm3, including 80% granulocytes and 9% eosinophils. On gross inspection, the stool was clay‐colored, and stool microscopy demonstrated large numbers of neutral fat droplets, but no ova, parasites, or tubercle bacilli. Urinalysis revealed no albumin or casts, and the bones were normal on ankle radiographs. Another sample of sputum revealed no tubercle bacilli, and intradermal placement of tuberculin provoked no reaction.
His negative tuberculin skin reaction is unusual for that era, because of the prevalence of tuberculosis. Most likely, he is anergic because of his severe underlying illness, and the absent reaction is thus not all that helpful a clue. Multiple negative sputum examinations lower the possibility of pulmonary, but not extrapulmonary, tuberculosis. The absence of bony destruction on ankle radiographs lowers my suspicion for tuberculous arthritis.
The excess stool fat implies steatogenic diarrhea from malabsorption, and 2 categories here are pancreatic and luminal diseases. Of these 2, pancreatic etiologies produce more severe malabsorption. We do not hear mention of jaundice, however, and I cannot see how to link the pancreas to the arthritis. A chronic infection which may produce malabsorption and eosinophilia is strongyloidiasis, endemic in the southeastern United States. However, this patient did not manifest the most common finding of chronic strongyloidiasis, namely asthma. Adrenal insufficiency, as might result from disseminated tuberculosis, is associated with increased skin pigmentation, diarrhea, and eosinophilia. However, the diarrhea of adrenal insufficiency is not malabsorptive, and serum electrolytes and cortisol tests were not available then to confirm this diagnosis antemortem.
In an attempt to identify a unifying cause of chronic arthritis, malabsorption, and increased skin pigmentation, I must consider Whipple's disease first and foremost. Physicians then were strapped and observation was often the default mode of the day. Given the abdominal findings, an exploratory laparotomy would be warranted if his condition deteriorated.
Despite forced oral feedings, the patient continued to lose weight, from his normal of 175 pounds to a nadir of 145 pounds. Because of worsening abdominal distention, the patient underwent exploratory abdominal surgery on the twenty‐first hospital day. Intraoperatively, no ascites was seen, but his mesenteric lymph nodes were hard and markedly enlarged. The abdomen was closed without further intervention. Two days after the surgery, the patient abruptly developed dyspnea. His respirations were 40 per minute, heart rate was 120, and he had minimal rales at the lung bases without findings of consolidation. He died 2 hours later, on the twenty‐third hospital day, and an autopsy was performed.
The final event may have been a pulmonary embolism. As for the adenopathy, lymphoma and tuberculosis are possible. Heavy chain disease, an unusual lymphoproliferative disorder found in persons from the old Silk Trader's Route from the Middle East to the Orient, is a remote prospect. However, 5 years is just too indolent for most cancers and would be very unusual for tuberculosis. I think the findings support Whipple's disease, and I wonder if this was the first reported case.
On postmortem examination, the abdominal adenopathy was striking. The small intestine contained enlarged villi with thickened submucosa, and the mesenteric nodes were enlarged with fat deposits and abnormal foamy cells. Within these foamy cells, microscopy revealed numerous rod‐shaped organisms. All studies were negative for tuberculosis, and although the pathologist, Dr. George Hoyt Whipple, suspected an infectious etiology, he offered the name intestinal lipodystrophy to emphasize the striking small intestinal changes he witnessed at autopsy, and which are the hallmarks of the disease that now bears his name. Whipple also shared the 1934 Nobel Prize in Physiology or Medicine with Minot and Murphy for their discovery that a nutritional substance in liver, now known as vitamin B12, was beneficial in treating pernicious anemia.
COMMENTARY
This is the index case of Whipple's disease, summarized from the original 1907 description.1 George Hoyt Whipple, then a pathologist at Johns Hopkins, highlights the value of keen observation and a well‐done case report in describing a new disease entity. One of the roles of case reports is to detail the features of an unknown disease. In this capacity, Whipple's summary is exemplary. His achievement was having the openness of mind to realize he was witnessing something novel, and to take the first step on the road to discovery. Although Whipple suspected he was staring at a unique disease, he could not pinpoint the culprit bacteria and he had trouble squaring the extraintestinal findings with the marked intestinal anomalies. It was left to decades of input from others to confirm the association of arthralgias, eosinophilia, skin hyperpigmentation, and cardiac valve abnormalities with intestinal malabsorption, and to culture the infectious agent.
In his discussion, Whipple recognized he was confronted with a novel clinical entity. Prior to surgery, pulmonary and mesenteric tuberculosis were suspected, based on the fevers, weight loss, cough, fat malabsorption, and lymphadenopathy. However, he felt the left apical exam was more representative of retraction from prior disease than active infection. He was also bothered by the negative skin reaction and sputum tests. At surgery, the pronounced adenopathy suggested sarcoma or Hodgkin's disease but postmortem examination eliminated these possibilities. At autopsy, the abdominal findings were most striking. The small intestine demonstrated enlarged villi with thickened submucosa and markedly enlarged mesenteric lymph glands containing large fat deposits and distinctly abnormal foamy cells. These foamy macrophages contained great numbers of rod‐shaped organisms resembling the tubercle bacillus. However, all tests were negative for tuberculosis, and the lungs contained no active disease. Though he suspected an infectious etiology, Whipple offered the name intestinal lipodystrophy to emphasize the striking small intestinal pathology.
Although Whipple had surmised a novel infectious agent in 1907, it took almost a century to isolate the causative microbe. Granules within foamy macrophages of the small intestine were detected on periodic acid‐Schiff (PAS) staining in 1949.2 Similar PAS‐positive granules were soon discovered in other tissues and fluid, providing a plausible explanation for the systemic features of the disease.3 Electron microscopy confirmed the presence of infectious bacilli in 1961,4 ushering in the era of antimicrobial treatment for this disease. More recently, using polymerase chain reaction (PCR), a unique bacterial 16S ribosomal RNA gene was isolated in patients with Whipple's disease.5, 6 Phylogenetically classified with the actinobacteria, Tropheryma whipplei (fom the Greek trophe, nourishment, and eryma, barrier) was ultimately subcultured in 2000,7 and immunodetection testing became possible. Using this technique, the archived pathology specimens from the 1907 index case demonstrated numerous intracellular bacteria in the lamina propria, closing the loop started by Whipple nearly a century earlier.8
Whipple's index case report described most of the manifestations of the disease we are familiar with today. As in the original description, arthralgias are the most common initial symptom and may precede diagnosis by a mean of 8 years. Other cardinal features include weight loss, abdominal pain and steatorrhea due to small intestinal involvement. Table 1 summarizes the important signs and symptoms of Whipple's disease.9, 10 One notable manifestation missing in Whipple's report is central nervous system involvement. Central nervous system (CNS) disease ranges from cognitive deficits to encephalitis and focal defects, and may occur years after treatment and without concomitant intestinal symptoms.
| Clinical Feature | Comment |
|---|---|
| |
| Cardinal features (present in 60% to 90%) | |
| Arthropathy | Most common initial symptom, preceding diagnosis by a mean of 8 years. Migratory, nonerosive, mainly in the peripheral joints. |
| Weight loss | |
| Diarrhea | Usually steatorrhea, may be associated with pain or occult blood in the stool |
| Other common features (present in 20% to 45%) | |
| Fever | |
| Lymphadenopathy | May present as a palpable mass |
| Increased skin pigmentation | Mechanism unknown (evidence of adrenal insufficiency has not been found in Whipple's) |
| Cardiac disease | Culture‐negative endocarditis |
| Hypotension | |
| Peripheral edema | |
| Uncommon clinical features | |
| Central nervous system involvement | May be global (dementia, personality change, sleep disturbance) or focal (cranial neuropathy, nystagmus) |
| Eye disease | Uveitis, retinitis |
| Hepatosplenomegaly | |
| Polyserositis | |
| Ascites | |
A remaining mystery is why this pathogen results only rarely in clinical disease. Caucasians comprise the majority of infected patients, and men are affected 8 times more often than women. An overrepresentation of HLA‐B27 suggests a genetic predisposition, though its role in pathogenesis is unclear. T. whipplei has been identified by PCR methods in asymptomatic individuals, implying additional abnormalities must be present in susceptible hosts for symptoms to occur following colonization.11 The exact immune defects are speculative, and immunodeficiency states (such as HIV) have not been consistently identified in patients with Whipple's disease.
The cornerstone of diagnosing Whipple's disease is upper endoscopy with duodenal biopsy. Flattening of the villi and markedly increased PAS‐positive staining of lamina propria macrophages are strongly suggestive of the diagnosis. PAS‐positive staining is not unique to T. whipplei, however. In patients with profound immunodeficiency, Mycobacterium avium intracellulare may stain positive with PAS. Since Whipple's disease is only rarely associated with HIV, a negative HIV test would favor a diagnosis of Whipple's disease. Electron microscopy may distinguish T. whipplei from its mimickers by morphology. For extraintestinal disease, PCR testing on samples from infected tissue has been found to be a reliable diagnostic aid.9
Given the rarity of the disease, controlled clinical trials addressing optimal treatment are lacking. Current recommendations include initial therapy for 14 days with an agent that crosses the blood‐brain barrier (eg, ceftriaxone) to reduce the incidence of CNS disease. This is then followed by a year or more of oral antimicrobial therapy with trimethoprim‐sulfamethoxazole or a tetracycline.9 While most patients respond within 2 to 3 weeks, relapse may occur in as many as one‐third of patients.
Historical case reports reinforce the case‐based learning paradigm. As the discussant remarks, observation was all too often the only recourse for physicians a century ago. In recounting the 7‐year progression of disease in 1 individual, Whipple provides a unique window into the natural evolution of the key features of this systemic disease. Viewed through the prism of Whipple's eyes, we can recall the striking lymphoid hyperplasia and unusual organisms in the small intestine, cementing our understanding of the pathogenesis of this disorder. Revisiting past cases allows us to learn of and learn from the past.
Teaching Points
-
Whipple's disease should be considered in patients with unexplained arthralgias accompanied by weight loss, malabsorption, and abdominal pain.
-
For suspected intestinal Whipple's disease, diagnosis is best made by duodenal biopsy demonstrating PAS‐positive staining in lamina propria macrophages.
-
Systemic manifestations of Whipple's disease include culture‐negative endocarditis and CNS disease. PCR testing of involved sites for T. whipplei is recommended to confirm extraintestinal disease.
- .A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues.Bull Johns Hopkins Hosp.1907;18:382–391.
- .The tinctoral demonstration of a glycoprotein in Whipple's disease.Proc Soc Exp Biol Med.1949;72:225–227.
- ,,.Whipple's disease: clinical, biochemical, and histopathologic features and assessment of treatment in 29 patients.Mayo Clin Proc.1988;63:539–551.
- ,.Combined electron and light microscopy in Whipple's disease: demonstration of “bacillary bodies” in the intestine.Bull Johns Hopkins Hosp.1961;109:80–98.
- ,,,.Phylogeny of the Whipple's disease‐associated bacterium.Lancet.1991;338:474–475.
- ,,,.Identification of the uncultured bacillus of Whipple's disease.N Engl J Med.1992;327:293–301.
- ,,, et al.Cultivation of the bacillus of Whipple's disease.N Engl J Med.2000;342:620–625.
- ,,,.Immunodetection of Tropheryma whipplei in intestinal tissue from Dr. Whipple's 1907 patient.N Engl J Med.2003;348:1411– 1412.
- ,.Whipple's disease.Lancet.2003;361:239–246.
- ,,, et al.Diagnostic guidelines in central nervous system Whipple's disease.Ann Neurol.1996;40:561–568.
- ,,, et al.PCR‐positive tests for Tropheryma whippleii in patients without Whipple's disease.Lancet.1999;353:2214.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 36 year‐old male physician was admitted to a Baltimore hospital in April 1907 with weight loss, weakness, arthralgias, and abdominal distension that had progressed over 5 years.
In 1907, major causes of unexplained weight loss included tuberculosis, hyperthyroidism, cancer, and diabetes. Arthralgias and weakness are not specific. The insidious progression over 5 years narrows the infectious possibilities; tuberculosis and syphilis are important considerations. Since surgical removal was the main treatment for malignancy in 1907, a history of prior surgery might point to a previously diagnosed malignancy that is now progressing.
Five years earlier, while visiting Turkey as a medical missionary, he first noted the onset of arthralgias that lasted 6 to 8 hours and occurred 3 to 4 times per week. Over time, these attacks lasted up to 24 hours and became associated with warmth, swelling, and tenderness of both small and large joints. He gradually lost weight and strength. One year prior to arrival in the hospital, he developed a cough productive of yellow sputum. Seven months prior, he returned from Turkey to Atlanta and noticed an increase in his cough, along with fevers of 100 Fahrenheit and night sweats.
The primacy of the arthralgias in this illness lead me to consider primary rheumatic diseases, and multisystem diseases (including infections) with a predominant skeletal component. In 1907, tests for lupus and the rheumatoid factor were not available. Neither skeletal remains nor works of art provide evidence that rheumatoid arthritis existed until the 19th century, whereas ankylosing spondylitis, gout, and rickets were present by then.
As a medical missionary, he might have acquired a disease endemic to the areas he visited, or the travel history may be a red herring. Familial Mediterranean fever, though prevalent in Turkey and a cause of arthralgias accompanied by recurrent attacks of abdominal pain and fever, is not an acquired disease. Behcet's disease, also known as Silk Trader's Route disease, is found in descendents of the countries that comprised the ancient Silk Route from Japan to the Middle East and may cause arthritis along with oral ulcers, genital lesions, pathergy or uveitis. I would inquire about his ancestry and fevers before dismissing these possibilities.
Although 5 years would be unusually long for tuberculosis to go unrecognized, a physician in the first half of the 20th century would place tuberculosis near the top of possible diagnoses. In 1930, a time when the population of the United States was considerably less, there were over 300,000 cases of tuberculosis. Physicians, and in particular pathologists since autopsies were more commonly performed, often died from tuberculosis since streptomycin, the first antituberculous medication, did not arrive until 1944. At the turn of the 20th century, the ability to detect tubercle bacilli was quite good. Thus, I would include tuberculous peritonitis as a cause of the progressive abdominal symptoms in this physician. In approximately one‐third of patients with tuberculous peritonitis there is evidence of pulmonary disease, and I would try to culture tuberculosis in samples of sputum, a test then so common it probably rivaled our frequent complete blood counts in popularity.
Six months prior, examinations of sputum were negative for tubercle bacilli. Four months prior to arrival, the patient moved to New Mexico. His cough improved but he continued to lose weight and had diarrhea consisting of 3 to 4 loose or semiformed bowel movements per day. Three months prior to admission, he noted an increase in abdominal girth along with right lower quadrant fullness. One month prior, he noted painful swelling and warmth in both ankles as well as dyspnea with exertion.
The increased abdominal girth in the context of chronic illness might be due to ascites, adenopathy, visceromegaly, or mass lesions such as a neoplasm or abscess. If ascites is the cause, one would need to consider primary hepatic disorders, as well as extrahepatic diseases that could progress over years. Infection with hepatitis A virus does not cause chronic liver disease. Hepatitis B, in those days, was referred to as serum hepatitis, and a serum marker for the B virusthe Australia antigenwas not identified until 1967. Cardiac causes of ascites include congestive heart failure and constrictive pericarditis, the latter an important consideration because it is potentially curable. Also, constrictive pericarditis can present as an indolent weight‐losing disease because of chronic visceral congestion. Other considerations include nephrotic syndrome, infection, and neoplasm, including mesothelioma.
Abdominal distention might also be seen with a smoldering abscess. In addition to an appendiceal process, the travel and right lower quadrant localization reminds us to consider ameboma. This patient surely was in an area where amebiasis was endemic, and amebomaa chronic inflammatory form of infection with E. histolytica not associated with diarrhea or liver cystsmay mimic cecal carcinoma. Exertional dyspnea suggests at least the possibility of cardiac disease. Despite the negative sputum cultures, tuberculosis remains high on the list as a cause of constrictive pericarditis or peritonitis, either of which may occur in the absence of active pulmonary disease.
Past medical history included measles and whooping cough as a child, mild pleurisy at age 14, mild influenza 7 years previously. The patient had a tonsillectomy as a child and had a portion of his inferior turbinate bone removed in an attempt to relieve a nasal condition.
On physical exam, the patient was thin and the skin over his face and hands was deep brown. His temperature was 101.5 Fahrenheit, the heart rate was 100, and the respiratory rate was 24. Small lymph nodes were palpable in the axillary and epitrochlear areas. His thorax moved asymmetrically, with less movement on the left apex and slight dullness to percussion in that area. The pulmonic component of the second heart sound was mildly accentuated. The abdomen displayed fullness and tympany, most pronounced in the right lower quadrant without hepatosplenomegaly. The left ankle was swollen, and the overlying skin was tense, shiny, and hot. On both lower legs, areas of discoloration and slight induration were observed, felt to be consistent with faded erythema nodosum.
Though pleurisy has numerous causes, its presence raises the specter of tuberculosis again. The nasal condition triggers thoughts of Wegener's granulomatosis or lethal midline granuloma, both unlikely diagnoses here. The pulmonary exam suggests an apical process, such as tuberculosis, and the accentuated pulmonic heart sound implies pulmonary hypertension, which could be due to a number of chronic pulmonary diseases. The epitrochlear nodes are of interest since lymphoma and Hodgkin's disease rarely involve this area; syphilis and human immunodeficiency virus (HIV) are a few of the chronic diseases that may involve this lymph node region. More helpful is the absence of hepatosplenomegaly, since many indolent malignancies and infections would be expected to enlarge these organs by this point.
Monoarticular arthritis is often due to infection, and less likely due to rheumatoid disease. When rheumatoid arthritis flares, the entire skeleton flares, not single joints. Given the indolence and this single joint involvement, tuberculosis again comes to mind.
I would next want to obtain a plain chest radiograph, looking for evidence of tuberculosis. As with any test, one should ask how this will change management. In 1907, antituberculous medications were not available, so therapy was directed at lowering oxygen tension in the primary site of infection; for example, pulmonary disease was addressed via pneumothorax. If the chest radiograph provides little hint of tuberculosis, then consideration must be given to exploratory surgery of the abdomen given the focal abnormality in the right lower quadrant.
A peripheral blood smear revealed a hypochromic microcytic anemia. The total red blood cell count was 4.468 million/mm3 (normal range for men is 4.52‐5.90 million/mm3), white blood cell count was 8180/mm3, including 80% granulocytes and 9% eosinophils. On gross inspection, the stool was clay‐colored, and stool microscopy demonstrated large numbers of neutral fat droplets, but no ova, parasites, or tubercle bacilli. Urinalysis revealed no albumin or casts, and the bones were normal on ankle radiographs. Another sample of sputum revealed no tubercle bacilli, and intradermal placement of tuberculin provoked no reaction.
His negative tuberculin skin reaction is unusual for that era, because of the prevalence of tuberculosis. Most likely, he is anergic because of his severe underlying illness, and the absent reaction is thus not all that helpful a clue. Multiple negative sputum examinations lower the possibility of pulmonary, but not extrapulmonary, tuberculosis. The absence of bony destruction on ankle radiographs lowers my suspicion for tuberculous arthritis.
The excess stool fat implies steatogenic diarrhea from malabsorption, and 2 categories here are pancreatic and luminal diseases. Of these 2, pancreatic etiologies produce more severe malabsorption. We do not hear mention of jaundice, however, and I cannot see how to link the pancreas to the arthritis. A chronic infection which may produce malabsorption and eosinophilia is strongyloidiasis, endemic in the southeastern United States. However, this patient did not manifest the most common finding of chronic strongyloidiasis, namely asthma. Adrenal insufficiency, as might result from disseminated tuberculosis, is associated with increased skin pigmentation, diarrhea, and eosinophilia. However, the diarrhea of adrenal insufficiency is not malabsorptive, and serum electrolytes and cortisol tests were not available then to confirm this diagnosis antemortem.
In an attempt to identify a unifying cause of chronic arthritis, malabsorption, and increased skin pigmentation, I must consider Whipple's disease first and foremost. Physicians then were strapped and observation was often the default mode of the day. Given the abdominal findings, an exploratory laparotomy would be warranted if his condition deteriorated.
Despite forced oral feedings, the patient continued to lose weight, from his normal of 175 pounds to a nadir of 145 pounds. Because of worsening abdominal distention, the patient underwent exploratory abdominal surgery on the twenty‐first hospital day. Intraoperatively, no ascites was seen, but his mesenteric lymph nodes were hard and markedly enlarged. The abdomen was closed without further intervention. Two days after the surgery, the patient abruptly developed dyspnea. His respirations were 40 per minute, heart rate was 120, and he had minimal rales at the lung bases without findings of consolidation. He died 2 hours later, on the twenty‐third hospital day, and an autopsy was performed.
The final event may have been a pulmonary embolism. As for the adenopathy, lymphoma and tuberculosis are possible. Heavy chain disease, an unusual lymphoproliferative disorder found in persons from the old Silk Trader's Route from the Middle East to the Orient, is a remote prospect. However, 5 years is just too indolent for most cancers and would be very unusual for tuberculosis. I think the findings support Whipple's disease, and I wonder if this was the first reported case.
On postmortem examination, the abdominal adenopathy was striking. The small intestine contained enlarged villi with thickened submucosa, and the mesenteric nodes were enlarged with fat deposits and abnormal foamy cells. Within these foamy cells, microscopy revealed numerous rod‐shaped organisms. All studies were negative for tuberculosis, and although the pathologist, Dr. George Hoyt Whipple, suspected an infectious etiology, he offered the name intestinal lipodystrophy to emphasize the striking small intestinal changes he witnessed at autopsy, and which are the hallmarks of the disease that now bears his name. Whipple also shared the 1934 Nobel Prize in Physiology or Medicine with Minot and Murphy for their discovery that a nutritional substance in liver, now known as vitamin B12, was beneficial in treating pernicious anemia.
COMMENTARY
This is the index case of Whipple's disease, summarized from the original 1907 description.1 George Hoyt Whipple, then a pathologist at Johns Hopkins, highlights the value of keen observation and a well‐done case report in describing a new disease entity. One of the roles of case reports is to detail the features of an unknown disease. In this capacity, Whipple's summary is exemplary. His achievement was having the openness of mind to realize he was witnessing something novel, and to take the first step on the road to discovery. Although Whipple suspected he was staring at a unique disease, he could not pinpoint the culprit bacteria and he had trouble squaring the extraintestinal findings with the marked intestinal anomalies. It was left to decades of input from others to confirm the association of arthralgias, eosinophilia, skin hyperpigmentation, and cardiac valve abnormalities with intestinal malabsorption, and to culture the infectious agent.
In his discussion, Whipple recognized he was confronted with a novel clinical entity. Prior to surgery, pulmonary and mesenteric tuberculosis were suspected, based on the fevers, weight loss, cough, fat malabsorption, and lymphadenopathy. However, he felt the left apical exam was more representative of retraction from prior disease than active infection. He was also bothered by the negative skin reaction and sputum tests. At surgery, the pronounced adenopathy suggested sarcoma or Hodgkin's disease but postmortem examination eliminated these possibilities. At autopsy, the abdominal findings were most striking. The small intestine demonstrated enlarged villi with thickened submucosa and markedly enlarged mesenteric lymph glands containing large fat deposits and distinctly abnormal foamy cells. These foamy macrophages contained great numbers of rod‐shaped organisms resembling the tubercle bacillus. However, all tests were negative for tuberculosis, and the lungs contained no active disease. Though he suspected an infectious etiology, Whipple offered the name intestinal lipodystrophy to emphasize the striking small intestinal pathology.
Although Whipple had surmised a novel infectious agent in 1907, it took almost a century to isolate the causative microbe. Granules within foamy macrophages of the small intestine were detected on periodic acid‐Schiff (PAS) staining in 1949.2 Similar PAS‐positive granules were soon discovered in other tissues and fluid, providing a plausible explanation for the systemic features of the disease.3 Electron microscopy confirmed the presence of infectious bacilli in 1961,4 ushering in the era of antimicrobial treatment for this disease. More recently, using polymerase chain reaction (PCR), a unique bacterial 16S ribosomal RNA gene was isolated in patients with Whipple's disease.5, 6 Phylogenetically classified with the actinobacteria, Tropheryma whipplei (fom the Greek trophe, nourishment, and eryma, barrier) was ultimately subcultured in 2000,7 and immunodetection testing became possible. Using this technique, the archived pathology specimens from the 1907 index case demonstrated numerous intracellular bacteria in the lamina propria, closing the loop started by Whipple nearly a century earlier.8
Whipple's index case report described most of the manifestations of the disease we are familiar with today. As in the original description, arthralgias are the most common initial symptom and may precede diagnosis by a mean of 8 years. Other cardinal features include weight loss, abdominal pain and steatorrhea due to small intestinal involvement. Table 1 summarizes the important signs and symptoms of Whipple's disease.9, 10 One notable manifestation missing in Whipple's report is central nervous system involvement. Central nervous system (CNS) disease ranges from cognitive deficits to encephalitis and focal defects, and may occur years after treatment and without concomitant intestinal symptoms.
| Clinical Feature | Comment |
|---|---|
| |
| Cardinal features (present in 60% to 90%) | |
| Arthropathy | Most common initial symptom, preceding diagnosis by a mean of 8 years. Migratory, nonerosive, mainly in the peripheral joints. |
| Weight loss | |
| Diarrhea | Usually steatorrhea, may be associated with pain or occult blood in the stool |
| Other common features (present in 20% to 45%) | |
| Fever | |
| Lymphadenopathy | May present as a palpable mass |
| Increased skin pigmentation | Mechanism unknown (evidence of adrenal insufficiency has not been found in Whipple's) |
| Cardiac disease | Culture‐negative endocarditis |
| Hypotension | |
| Peripheral edema | |
| Uncommon clinical features | |
| Central nervous system involvement | May be global (dementia, personality change, sleep disturbance) or focal (cranial neuropathy, nystagmus) |
| Eye disease | Uveitis, retinitis |
| Hepatosplenomegaly | |
| Polyserositis | |
| Ascites | |
A remaining mystery is why this pathogen results only rarely in clinical disease. Caucasians comprise the majority of infected patients, and men are affected 8 times more often than women. An overrepresentation of HLA‐B27 suggests a genetic predisposition, though its role in pathogenesis is unclear. T. whipplei has been identified by PCR methods in asymptomatic individuals, implying additional abnormalities must be present in susceptible hosts for symptoms to occur following colonization.11 The exact immune defects are speculative, and immunodeficiency states (such as HIV) have not been consistently identified in patients with Whipple's disease.
The cornerstone of diagnosing Whipple's disease is upper endoscopy with duodenal biopsy. Flattening of the villi and markedly increased PAS‐positive staining of lamina propria macrophages are strongly suggestive of the diagnosis. PAS‐positive staining is not unique to T. whipplei, however. In patients with profound immunodeficiency, Mycobacterium avium intracellulare may stain positive with PAS. Since Whipple's disease is only rarely associated with HIV, a negative HIV test would favor a diagnosis of Whipple's disease. Electron microscopy may distinguish T. whipplei from its mimickers by morphology. For extraintestinal disease, PCR testing on samples from infected tissue has been found to be a reliable diagnostic aid.9
Given the rarity of the disease, controlled clinical trials addressing optimal treatment are lacking. Current recommendations include initial therapy for 14 days with an agent that crosses the blood‐brain barrier (eg, ceftriaxone) to reduce the incidence of CNS disease. This is then followed by a year or more of oral antimicrobial therapy with trimethoprim‐sulfamethoxazole or a tetracycline.9 While most patients respond within 2 to 3 weeks, relapse may occur in as many as one‐third of patients.
Historical case reports reinforce the case‐based learning paradigm. As the discussant remarks, observation was all too often the only recourse for physicians a century ago. In recounting the 7‐year progression of disease in 1 individual, Whipple provides a unique window into the natural evolution of the key features of this systemic disease. Viewed through the prism of Whipple's eyes, we can recall the striking lymphoid hyperplasia and unusual organisms in the small intestine, cementing our understanding of the pathogenesis of this disorder. Revisiting past cases allows us to learn of and learn from the past.
Teaching Points
-
Whipple's disease should be considered in patients with unexplained arthralgias accompanied by weight loss, malabsorption, and abdominal pain.
-
For suspected intestinal Whipple's disease, diagnosis is best made by duodenal biopsy demonstrating PAS‐positive staining in lamina propria macrophages.
-
Systemic manifestations of Whipple's disease include culture‐negative endocarditis and CNS disease. PCR testing of involved sites for T. whipplei is recommended to confirm extraintestinal disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 36 year‐old male physician was admitted to a Baltimore hospital in April 1907 with weight loss, weakness, arthralgias, and abdominal distension that had progressed over 5 years.
In 1907, major causes of unexplained weight loss included tuberculosis, hyperthyroidism, cancer, and diabetes. Arthralgias and weakness are not specific. The insidious progression over 5 years narrows the infectious possibilities; tuberculosis and syphilis are important considerations. Since surgical removal was the main treatment for malignancy in 1907, a history of prior surgery might point to a previously diagnosed malignancy that is now progressing.
Five years earlier, while visiting Turkey as a medical missionary, he first noted the onset of arthralgias that lasted 6 to 8 hours and occurred 3 to 4 times per week. Over time, these attacks lasted up to 24 hours and became associated with warmth, swelling, and tenderness of both small and large joints. He gradually lost weight and strength. One year prior to arrival in the hospital, he developed a cough productive of yellow sputum. Seven months prior, he returned from Turkey to Atlanta and noticed an increase in his cough, along with fevers of 100 Fahrenheit and night sweats.
The primacy of the arthralgias in this illness lead me to consider primary rheumatic diseases, and multisystem diseases (including infections) with a predominant skeletal component. In 1907, tests for lupus and the rheumatoid factor were not available. Neither skeletal remains nor works of art provide evidence that rheumatoid arthritis existed until the 19th century, whereas ankylosing spondylitis, gout, and rickets were present by then.
As a medical missionary, he might have acquired a disease endemic to the areas he visited, or the travel history may be a red herring. Familial Mediterranean fever, though prevalent in Turkey and a cause of arthralgias accompanied by recurrent attacks of abdominal pain and fever, is not an acquired disease. Behcet's disease, also known as Silk Trader's Route disease, is found in descendents of the countries that comprised the ancient Silk Route from Japan to the Middle East and may cause arthritis along with oral ulcers, genital lesions, pathergy or uveitis. I would inquire about his ancestry and fevers before dismissing these possibilities.
Although 5 years would be unusually long for tuberculosis to go unrecognized, a physician in the first half of the 20th century would place tuberculosis near the top of possible diagnoses. In 1930, a time when the population of the United States was considerably less, there were over 300,000 cases of tuberculosis. Physicians, and in particular pathologists since autopsies were more commonly performed, often died from tuberculosis since streptomycin, the first antituberculous medication, did not arrive until 1944. At the turn of the 20th century, the ability to detect tubercle bacilli was quite good. Thus, I would include tuberculous peritonitis as a cause of the progressive abdominal symptoms in this physician. In approximately one‐third of patients with tuberculous peritonitis there is evidence of pulmonary disease, and I would try to culture tuberculosis in samples of sputum, a test then so common it probably rivaled our frequent complete blood counts in popularity.
Six months prior, examinations of sputum were negative for tubercle bacilli. Four months prior to arrival, the patient moved to New Mexico. His cough improved but he continued to lose weight and had diarrhea consisting of 3 to 4 loose or semiformed bowel movements per day. Three months prior to admission, he noted an increase in abdominal girth along with right lower quadrant fullness. One month prior, he noted painful swelling and warmth in both ankles as well as dyspnea with exertion.
The increased abdominal girth in the context of chronic illness might be due to ascites, adenopathy, visceromegaly, or mass lesions such as a neoplasm or abscess. If ascites is the cause, one would need to consider primary hepatic disorders, as well as extrahepatic diseases that could progress over years. Infection with hepatitis A virus does not cause chronic liver disease. Hepatitis B, in those days, was referred to as serum hepatitis, and a serum marker for the B virusthe Australia antigenwas not identified until 1967. Cardiac causes of ascites include congestive heart failure and constrictive pericarditis, the latter an important consideration because it is potentially curable. Also, constrictive pericarditis can present as an indolent weight‐losing disease because of chronic visceral congestion. Other considerations include nephrotic syndrome, infection, and neoplasm, including mesothelioma.
Abdominal distention might also be seen with a smoldering abscess. In addition to an appendiceal process, the travel and right lower quadrant localization reminds us to consider ameboma. This patient surely was in an area where amebiasis was endemic, and amebomaa chronic inflammatory form of infection with E. histolytica not associated with diarrhea or liver cystsmay mimic cecal carcinoma. Exertional dyspnea suggests at least the possibility of cardiac disease. Despite the negative sputum cultures, tuberculosis remains high on the list as a cause of constrictive pericarditis or peritonitis, either of which may occur in the absence of active pulmonary disease.
Past medical history included measles and whooping cough as a child, mild pleurisy at age 14, mild influenza 7 years previously. The patient had a tonsillectomy as a child and had a portion of his inferior turbinate bone removed in an attempt to relieve a nasal condition.
On physical exam, the patient was thin and the skin over his face and hands was deep brown. His temperature was 101.5 Fahrenheit, the heart rate was 100, and the respiratory rate was 24. Small lymph nodes were palpable in the axillary and epitrochlear areas. His thorax moved asymmetrically, with less movement on the left apex and slight dullness to percussion in that area. The pulmonic component of the second heart sound was mildly accentuated. The abdomen displayed fullness and tympany, most pronounced in the right lower quadrant without hepatosplenomegaly. The left ankle was swollen, and the overlying skin was tense, shiny, and hot. On both lower legs, areas of discoloration and slight induration were observed, felt to be consistent with faded erythema nodosum.
Though pleurisy has numerous causes, its presence raises the specter of tuberculosis again. The nasal condition triggers thoughts of Wegener's granulomatosis or lethal midline granuloma, both unlikely diagnoses here. The pulmonary exam suggests an apical process, such as tuberculosis, and the accentuated pulmonic heart sound implies pulmonary hypertension, which could be due to a number of chronic pulmonary diseases. The epitrochlear nodes are of interest since lymphoma and Hodgkin's disease rarely involve this area; syphilis and human immunodeficiency virus (HIV) are a few of the chronic diseases that may involve this lymph node region. More helpful is the absence of hepatosplenomegaly, since many indolent malignancies and infections would be expected to enlarge these organs by this point.
Monoarticular arthritis is often due to infection, and less likely due to rheumatoid disease. When rheumatoid arthritis flares, the entire skeleton flares, not single joints. Given the indolence and this single joint involvement, tuberculosis again comes to mind.
I would next want to obtain a plain chest radiograph, looking for evidence of tuberculosis. As with any test, one should ask how this will change management. In 1907, antituberculous medications were not available, so therapy was directed at lowering oxygen tension in the primary site of infection; for example, pulmonary disease was addressed via pneumothorax. If the chest radiograph provides little hint of tuberculosis, then consideration must be given to exploratory surgery of the abdomen given the focal abnormality in the right lower quadrant.
A peripheral blood smear revealed a hypochromic microcytic anemia. The total red blood cell count was 4.468 million/mm3 (normal range for men is 4.52‐5.90 million/mm3), white blood cell count was 8180/mm3, including 80% granulocytes and 9% eosinophils. On gross inspection, the stool was clay‐colored, and stool microscopy demonstrated large numbers of neutral fat droplets, but no ova, parasites, or tubercle bacilli. Urinalysis revealed no albumin or casts, and the bones were normal on ankle radiographs. Another sample of sputum revealed no tubercle bacilli, and intradermal placement of tuberculin provoked no reaction.
His negative tuberculin skin reaction is unusual for that era, because of the prevalence of tuberculosis. Most likely, he is anergic because of his severe underlying illness, and the absent reaction is thus not all that helpful a clue. Multiple negative sputum examinations lower the possibility of pulmonary, but not extrapulmonary, tuberculosis. The absence of bony destruction on ankle radiographs lowers my suspicion for tuberculous arthritis.
The excess stool fat implies steatogenic diarrhea from malabsorption, and 2 categories here are pancreatic and luminal diseases. Of these 2, pancreatic etiologies produce more severe malabsorption. We do not hear mention of jaundice, however, and I cannot see how to link the pancreas to the arthritis. A chronic infection which may produce malabsorption and eosinophilia is strongyloidiasis, endemic in the southeastern United States. However, this patient did not manifest the most common finding of chronic strongyloidiasis, namely asthma. Adrenal insufficiency, as might result from disseminated tuberculosis, is associated with increased skin pigmentation, diarrhea, and eosinophilia. However, the diarrhea of adrenal insufficiency is not malabsorptive, and serum electrolytes and cortisol tests were not available then to confirm this diagnosis antemortem.
In an attempt to identify a unifying cause of chronic arthritis, malabsorption, and increased skin pigmentation, I must consider Whipple's disease first and foremost. Physicians then were strapped and observation was often the default mode of the day. Given the abdominal findings, an exploratory laparotomy would be warranted if his condition deteriorated.
Despite forced oral feedings, the patient continued to lose weight, from his normal of 175 pounds to a nadir of 145 pounds. Because of worsening abdominal distention, the patient underwent exploratory abdominal surgery on the twenty‐first hospital day. Intraoperatively, no ascites was seen, but his mesenteric lymph nodes were hard and markedly enlarged. The abdomen was closed without further intervention. Two days after the surgery, the patient abruptly developed dyspnea. His respirations were 40 per minute, heart rate was 120, and he had minimal rales at the lung bases without findings of consolidation. He died 2 hours later, on the twenty‐third hospital day, and an autopsy was performed.
The final event may have been a pulmonary embolism. As for the adenopathy, lymphoma and tuberculosis are possible. Heavy chain disease, an unusual lymphoproliferative disorder found in persons from the old Silk Trader's Route from the Middle East to the Orient, is a remote prospect. However, 5 years is just too indolent for most cancers and would be very unusual for tuberculosis. I think the findings support Whipple's disease, and I wonder if this was the first reported case.
On postmortem examination, the abdominal adenopathy was striking. The small intestine contained enlarged villi with thickened submucosa, and the mesenteric nodes were enlarged with fat deposits and abnormal foamy cells. Within these foamy cells, microscopy revealed numerous rod‐shaped organisms. All studies were negative for tuberculosis, and although the pathologist, Dr. George Hoyt Whipple, suspected an infectious etiology, he offered the name intestinal lipodystrophy to emphasize the striking small intestinal changes he witnessed at autopsy, and which are the hallmarks of the disease that now bears his name. Whipple also shared the 1934 Nobel Prize in Physiology or Medicine with Minot and Murphy for their discovery that a nutritional substance in liver, now known as vitamin B12, was beneficial in treating pernicious anemia.
COMMENTARY
This is the index case of Whipple's disease, summarized from the original 1907 description.1 George Hoyt Whipple, then a pathologist at Johns Hopkins, highlights the value of keen observation and a well‐done case report in describing a new disease entity. One of the roles of case reports is to detail the features of an unknown disease. In this capacity, Whipple's summary is exemplary. His achievement was having the openness of mind to realize he was witnessing something novel, and to take the first step on the road to discovery. Although Whipple suspected he was staring at a unique disease, he could not pinpoint the culprit bacteria and he had trouble squaring the extraintestinal findings with the marked intestinal anomalies. It was left to decades of input from others to confirm the association of arthralgias, eosinophilia, skin hyperpigmentation, and cardiac valve abnormalities with intestinal malabsorption, and to culture the infectious agent.
In his discussion, Whipple recognized he was confronted with a novel clinical entity. Prior to surgery, pulmonary and mesenteric tuberculosis were suspected, based on the fevers, weight loss, cough, fat malabsorption, and lymphadenopathy. However, he felt the left apical exam was more representative of retraction from prior disease than active infection. He was also bothered by the negative skin reaction and sputum tests. At surgery, the pronounced adenopathy suggested sarcoma or Hodgkin's disease but postmortem examination eliminated these possibilities. At autopsy, the abdominal findings were most striking. The small intestine demonstrated enlarged villi with thickened submucosa and markedly enlarged mesenteric lymph glands containing large fat deposits and distinctly abnormal foamy cells. These foamy macrophages contained great numbers of rod‐shaped organisms resembling the tubercle bacillus. However, all tests were negative for tuberculosis, and the lungs contained no active disease. Though he suspected an infectious etiology, Whipple offered the name intestinal lipodystrophy to emphasize the striking small intestinal pathology.
Although Whipple had surmised a novel infectious agent in 1907, it took almost a century to isolate the causative microbe. Granules within foamy macrophages of the small intestine were detected on periodic acid‐Schiff (PAS) staining in 1949.2 Similar PAS‐positive granules were soon discovered in other tissues and fluid, providing a plausible explanation for the systemic features of the disease.3 Electron microscopy confirmed the presence of infectious bacilli in 1961,4 ushering in the era of antimicrobial treatment for this disease. More recently, using polymerase chain reaction (PCR), a unique bacterial 16S ribosomal RNA gene was isolated in patients with Whipple's disease.5, 6 Phylogenetically classified with the actinobacteria, Tropheryma whipplei (fom the Greek trophe, nourishment, and eryma, barrier) was ultimately subcultured in 2000,7 and immunodetection testing became possible. Using this technique, the archived pathology specimens from the 1907 index case demonstrated numerous intracellular bacteria in the lamina propria, closing the loop started by Whipple nearly a century earlier.8
Whipple's index case report described most of the manifestations of the disease we are familiar with today. As in the original description, arthralgias are the most common initial symptom and may precede diagnosis by a mean of 8 years. Other cardinal features include weight loss, abdominal pain and steatorrhea due to small intestinal involvement. Table 1 summarizes the important signs and symptoms of Whipple's disease.9, 10 One notable manifestation missing in Whipple's report is central nervous system involvement. Central nervous system (CNS) disease ranges from cognitive deficits to encephalitis and focal defects, and may occur years after treatment and without concomitant intestinal symptoms.
| Clinical Feature | Comment |
|---|---|
| |
| Cardinal features (present in 60% to 90%) | |
| Arthropathy | Most common initial symptom, preceding diagnosis by a mean of 8 years. Migratory, nonerosive, mainly in the peripheral joints. |
| Weight loss | |
| Diarrhea | Usually steatorrhea, may be associated with pain or occult blood in the stool |
| Other common features (present in 20% to 45%) | |
| Fever | |
| Lymphadenopathy | May present as a palpable mass |
| Increased skin pigmentation | Mechanism unknown (evidence of adrenal insufficiency has not been found in Whipple's) |
| Cardiac disease | Culture‐negative endocarditis |
| Hypotension | |
| Peripheral edema | |
| Uncommon clinical features | |
| Central nervous system involvement | May be global (dementia, personality change, sleep disturbance) or focal (cranial neuropathy, nystagmus) |
| Eye disease | Uveitis, retinitis |
| Hepatosplenomegaly | |
| Polyserositis | |
| Ascites | |
A remaining mystery is why this pathogen results only rarely in clinical disease. Caucasians comprise the majority of infected patients, and men are affected 8 times more often than women. An overrepresentation of HLA‐B27 suggests a genetic predisposition, though its role in pathogenesis is unclear. T. whipplei has been identified by PCR methods in asymptomatic individuals, implying additional abnormalities must be present in susceptible hosts for symptoms to occur following colonization.11 The exact immune defects are speculative, and immunodeficiency states (such as HIV) have not been consistently identified in patients with Whipple's disease.
The cornerstone of diagnosing Whipple's disease is upper endoscopy with duodenal biopsy. Flattening of the villi and markedly increased PAS‐positive staining of lamina propria macrophages are strongly suggestive of the diagnosis. PAS‐positive staining is not unique to T. whipplei, however. In patients with profound immunodeficiency, Mycobacterium avium intracellulare may stain positive with PAS. Since Whipple's disease is only rarely associated with HIV, a negative HIV test would favor a diagnosis of Whipple's disease. Electron microscopy may distinguish T. whipplei from its mimickers by morphology. For extraintestinal disease, PCR testing on samples from infected tissue has been found to be a reliable diagnostic aid.9
Given the rarity of the disease, controlled clinical trials addressing optimal treatment are lacking. Current recommendations include initial therapy for 14 days with an agent that crosses the blood‐brain barrier (eg, ceftriaxone) to reduce the incidence of CNS disease. This is then followed by a year or more of oral antimicrobial therapy with trimethoprim‐sulfamethoxazole or a tetracycline.9 While most patients respond within 2 to 3 weeks, relapse may occur in as many as one‐third of patients.
Historical case reports reinforce the case‐based learning paradigm. As the discussant remarks, observation was all too often the only recourse for physicians a century ago. In recounting the 7‐year progression of disease in 1 individual, Whipple provides a unique window into the natural evolution of the key features of this systemic disease. Viewed through the prism of Whipple's eyes, we can recall the striking lymphoid hyperplasia and unusual organisms in the small intestine, cementing our understanding of the pathogenesis of this disorder. Revisiting past cases allows us to learn of and learn from the past.
Teaching Points
-
Whipple's disease should be considered in patients with unexplained arthralgias accompanied by weight loss, malabsorption, and abdominal pain.
-
For suspected intestinal Whipple's disease, diagnosis is best made by duodenal biopsy demonstrating PAS‐positive staining in lamina propria macrophages.
-
Systemic manifestations of Whipple's disease include culture‐negative endocarditis and CNS disease. PCR testing of involved sites for T. whipplei is recommended to confirm extraintestinal disease.
- .A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues.Bull Johns Hopkins Hosp.1907;18:382–391.
- .The tinctoral demonstration of a glycoprotein in Whipple's disease.Proc Soc Exp Biol Med.1949;72:225–227.
- ,,.Whipple's disease: clinical, biochemical, and histopathologic features and assessment of treatment in 29 patients.Mayo Clin Proc.1988;63:539–551.
- ,.Combined electron and light microscopy in Whipple's disease: demonstration of “bacillary bodies” in the intestine.Bull Johns Hopkins Hosp.1961;109:80–98.
- ,,,.Phylogeny of the Whipple's disease‐associated bacterium.Lancet.1991;338:474–475.
- ,,,.Identification of the uncultured bacillus of Whipple's disease.N Engl J Med.1992;327:293–301.
- ,,, et al.Cultivation of the bacillus of Whipple's disease.N Engl J Med.2000;342:620–625.
- ,,,.Immunodetection of Tropheryma whipplei in intestinal tissue from Dr. Whipple's 1907 patient.N Engl J Med.2003;348:1411– 1412.
- ,.Whipple's disease.Lancet.2003;361:239–246.
- ,,, et al.Diagnostic guidelines in central nervous system Whipple's disease.Ann Neurol.1996;40:561–568.
- ,,, et al.PCR‐positive tests for Tropheryma whippleii in patients without Whipple's disease.Lancet.1999;353:2214.
- .A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues.Bull Johns Hopkins Hosp.1907;18:382–391.
- .The tinctoral demonstration of a glycoprotein in Whipple's disease.Proc Soc Exp Biol Med.1949;72:225–227.
- ,,.Whipple's disease: clinical, biochemical, and histopathologic features and assessment of treatment in 29 patients.Mayo Clin Proc.1988;63:539–551.
- ,.Combined electron and light microscopy in Whipple's disease: demonstration of “bacillary bodies” in the intestine.Bull Johns Hopkins Hosp.1961;109:80–98.
- ,,,.Phylogeny of the Whipple's disease‐associated bacterium.Lancet.1991;338:474–475.
- ,,,.Identification of the uncultured bacillus of Whipple's disease.N Engl J Med.1992;327:293–301.
- ,,, et al.Cultivation of the bacillus of Whipple's disease.N Engl J Med.2000;342:620–625.
- ,,,.Immunodetection of Tropheryma whipplei in intestinal tissue from Dr. Whipple's 1907 patient.N Engl J Med.2003;348:1411– 1412.
- ,.Whipple's disease.Lancet.2003;361:239–246.
- ,,, et al.Diagnostic guidelines in central nervous system Whipple's disease.Ann Neurol.1996;40:561–568.
- ,,, et al.PCR‐positive tests for Tropheryma whippleii in patients without Whipple's disease.Lancet.1999;353:2214.
Diagnosis of Exclusion
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
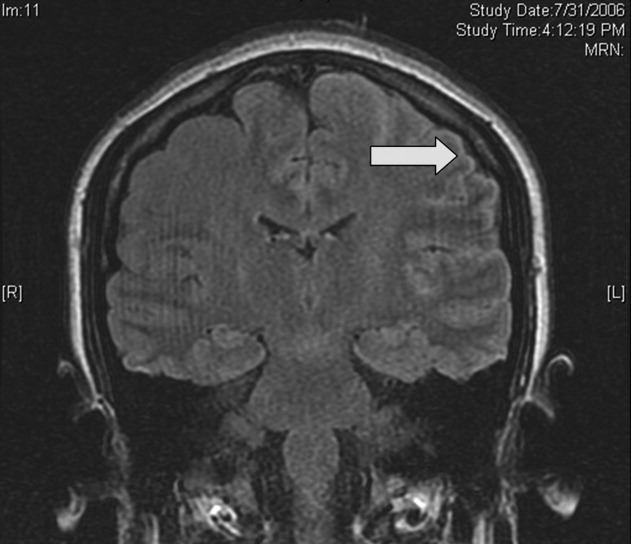
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
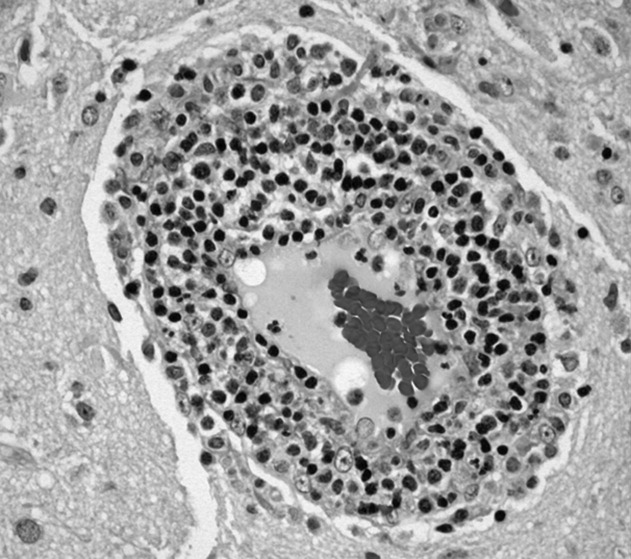
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
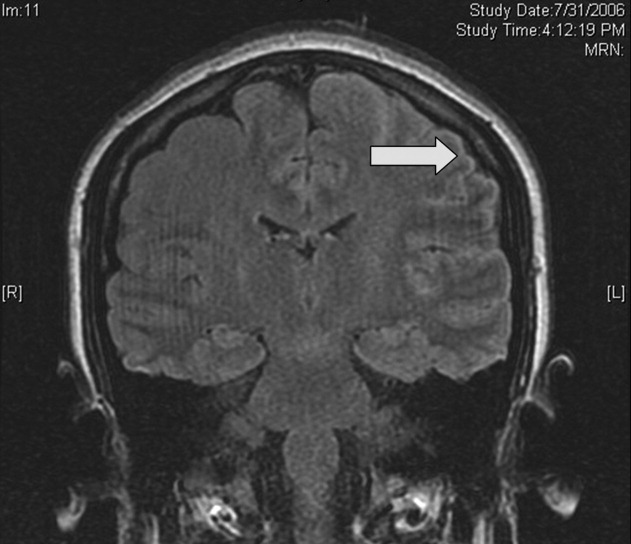
Thinking Inside the Box
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.