User login
Diagnosis of Exclusion
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
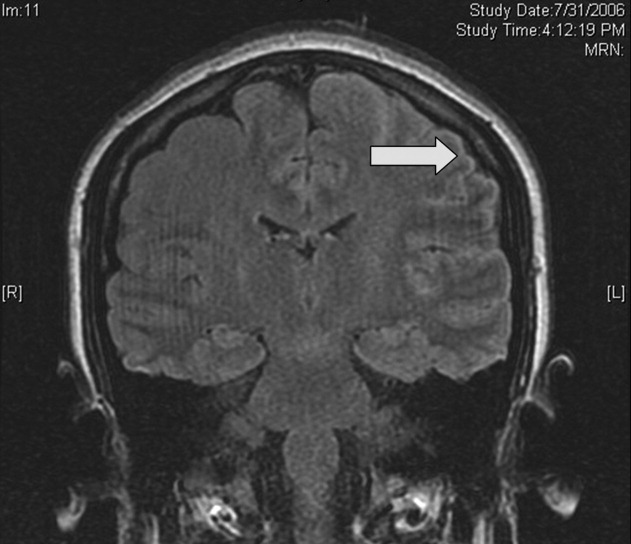
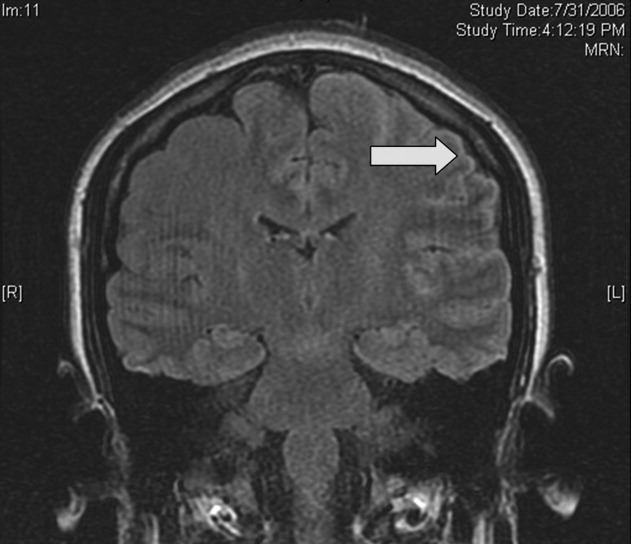
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
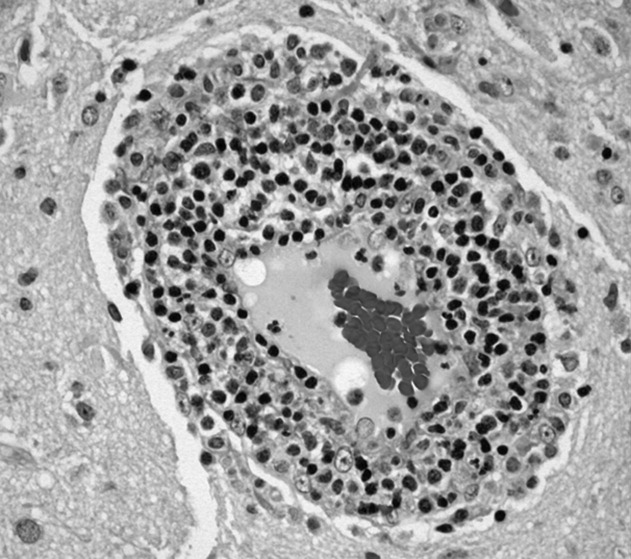
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.