User login
The Heart of the Matter
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
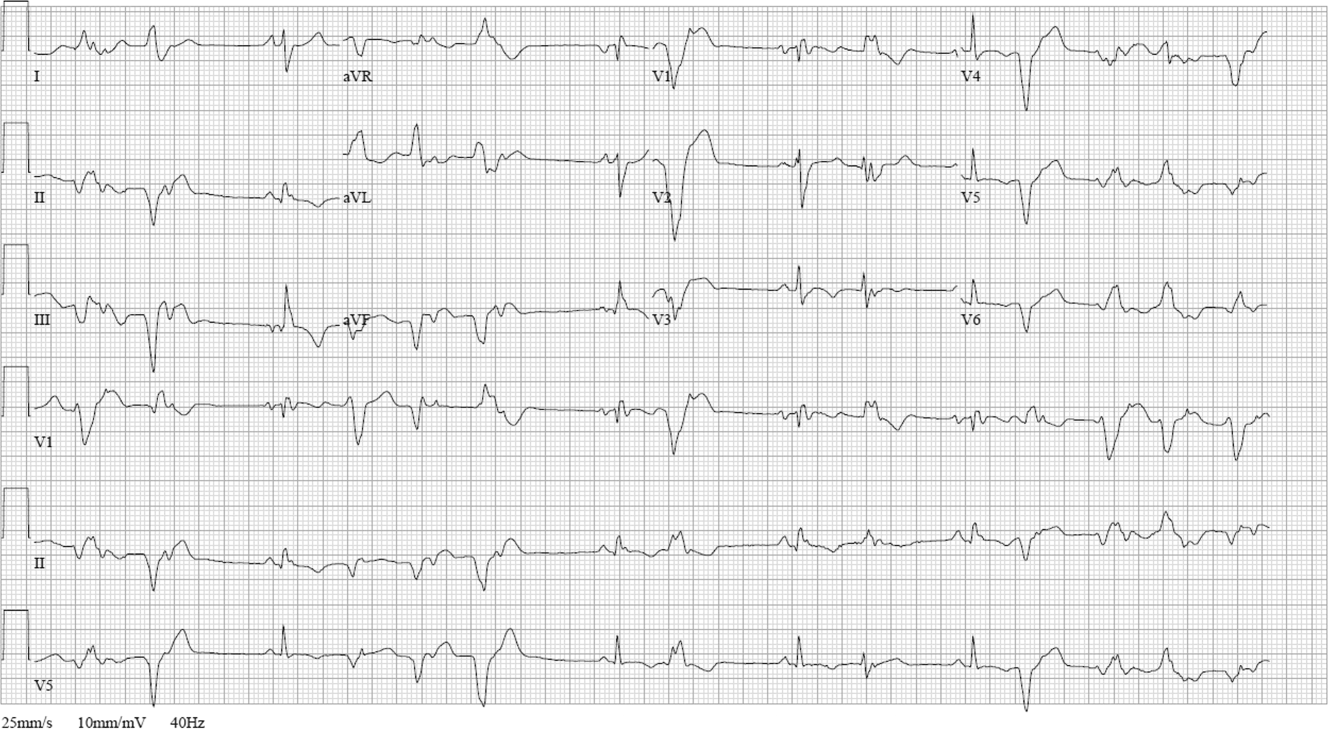
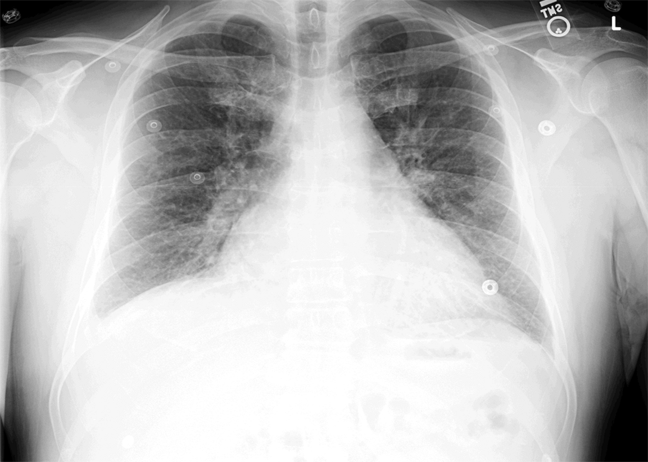
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
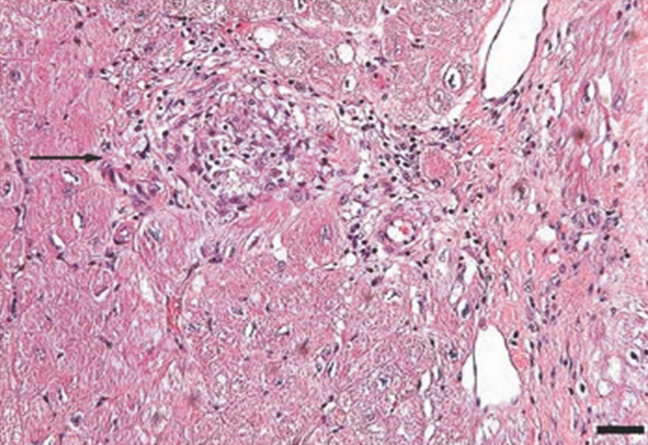
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.