User login
Dr. Fauci: Feds may ease indoor mask mandates soon
Federal guidance on indoor mask use may change soon, Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, said on May 9.
He was asked whether it’s time to start relaxing indoor mask requirements.
“I think so, and I think you’re going to probably be seeing that as we go along and as more people get vaccinated,” Dr. Fauci said on ABC News’s This Week.Nearly 150 million adults in the United States – or about 58% of the adult population – have received at least one COVID-19 vaccine dose, according to the latest CDC tally. About 113 million adults, or 44%, are considered fully vaccinated.
“The CDC will be, you know, almost in real time … updating their recommendations and their guidelines,” Dr. Fauci said.
In April, the CDC relaxed its guidance for those who have been vaccinated against COVID-19. Those who have gotten a shot don’t need to wear a mask outdoors or in small indoor gatherings with other vaccinated people, but both vaccinated and unvaccinated people are still advised to wear masks in indoor public spaces.
“We do need to start being more liberal as we get more people vaccinated,” Dr. Fauci said. “As you get more people vaccinated, the number of cases per day will absolutely go down.”
The United States is averaging about 43,000 cases per day, he said, adding that the cases need to be “much, much lower.” When the case numbers drop and vaccination numbers increase, the risk of infection will fall dramatically indoors and outdoors, he said.
Even after the pandemic, though, wearing masks could become a seasonal habit, Dr. Fauci said May 9 on NBC News’s Meet the Press.“I think people have gotten used to the fact that wearing masks, clearly if you look at the data, it diminishes respiratory diseases. We’ve had practically a nonexistent flu season this year,” he said.
“So it is conceivable that as we go on, a year or 2 or more from now, that during certain seasonal periods when you have respiratory-borne viruses like the flu, people might actually elect to wear masks to diminish the likelihood that you’ll spread these respiratory-borne diseases,” he said.
Dr. Fauci was asked about indoor mask guidelines on May 9 after former FDA Commissioner Scott Gottlieb, MD, said face mask requirements should be relaxed.
“Certainly outdoors, we shouldn’t be putting limits on gatherings anymore,” Dr. Gottlieb said on CBS News’s Face the Nation.“The states where prevalence is low, vaccination rates are high, we have good testing in place, and we’re identifying infections, I think we could start lifting these restrictions indoors as well, on a broad basis,” he said.
Lifting pandemic-related restrictions in areas where they’re no longer necessary could also encourage people to implement them again if cases increase during future surges, such as this fall or winter, Dr. Gottlieb said.
At the same time, Americans should continue to follow CDC guidance and wait for new guidelines before changing their indoor mask use, Jeffrey Zients, the White House COVID-19 response coordinator, said on CNN’s State of the Union on May 9.
“We all want to get back to a normal lifestyle,” he said. “I think we’re on the path to do that, but stay disciplined, and let’s take advantage of the new privilege of being vaccinated and not wearing masks outdoors, for example, unless you’re in a crowded place.”
Mr. Zients pointed to President Joe Biden’s goal for 70% of adults to receive at least one vaccine dose by July 4.
“As we all move toward that 70% goal, there will be more and more advantages to being vaccinated,” he said. “And if you’re not vaccinated, you’re not protected.”
A version of this article first appeared on WebMD.com.
Federal guidance on indoor mask use may change soon, Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, said on May 9.
He was asked whether it’s time to start relaxing indoor mask requirements.
“I think so, and I think you’re going to probably be seeing that as we go along and as more people get vaccinated,” Dr. Fauci said on ABC News’s This Week.Nearly 150 million adults in the United States – or about 58% of the adult population – have received at least one COVID-19 vaccine dose, according to the latest CDC tally. About 113 million adults, or 44%, are considered fully vaccinated.
“The CDC will be, you know, almost in real time … updating their recommendations and their guidelines,” Dr. Fauci said.
In April, the CDC relaxed its guidance for those who have been vaccinated against COVID-19. Those who have gotten a shot don’t need to wear a mask outdoors or in small indoor gatherings with other vaccinated people, but both vaccinated and unvaccinated people are still advised to wear masks in indoor public spaces.
“We do need to start being more liberal as we get more people vaccinated,” Dr. Fauci said. “As you get more people vaccinated, the number of cases per day will absolutely go down.”
The United States is averaging about 43,000 cases per day, he said, adding that the cases need to be “much, much lower.” When the case numbers drop and vaccination numbers increase, the risk of infection will fall dramatically indoors and outdoors, he said.
Even after the pandemic, though, wearing masks could become a seasonal habit, Dr. Fauci said May 9 on NBC News’s Meet the Press.“I think people have gotten used to the fact that wearing masks, clearly if you look at the data, it diminishes respiratory diseases. We’ve had practically a nonexistent flu season this year,” he said.
“So it is conceivable that as we go on, a year or 2 or more from now, that during certain seasonal periods when you have respiratory-borne viruses like the flu, people might actually elect to wear masks to diminish the likelihood that you’ll spread these respiratory-borne diseases,” he said.
Dr. Fauci was asked about indoor mask guidelines on May 9 after former FDA Commissioner Scott Gottlieb, MD, said face mask requirements should be relaxed.
“Certainly outdoors, we shouldn’t be putting limits on gatherings anymore,” Dr. Gottlieb said on CBS News’s Face the Nation.“The states where prevalence is low, vaccination rates are high, we have good testing in place, and we’re identifying infections, I think we could start lifting these restrictions indoors as well, on a broad basis,” he said.
Lifting pandemic-related restrictions in areas where they’re no longer necessary could also encourage people to implement them again if cases increase during future surges, such as this fall or winter, Dr. Gottlieb said.
At the same time, Americans should continue to follow CDC guidance and wait for new guidelines before changing their indoor mask use, Jeffrey Zients, the White House COVID-19 response coordinator, said on CNN’s State of the Union on May 9.
“We all want to get back to a normal lifestyle,” he said. “I think we’re on the path to do that, but stay disciplined, and let’s take advantage of the new privilege of being vaccinated and not wearing masks outdoors, for example, unless you’re in a crowded place.”
Mr. Zients pointed to President Joe Biden’s goal for 70% of adults to receive at least one vaccine dose by July 4.
“As we all move toward that 70% goal, there will be more and more advantages to being vaccinated,” he said. “And if you’re not vaccinated, you’re not protected.”
A version of this article first appeared on WebMD.com.
Federal guidance on indoor mask use may change soon, Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, said on May 9.
He was asked whether it’s time to start relaxing indoor mask requirements.
“I think so, and I think you’re going to probably be seeing that as we go along and as more people get vaccinated,” Dr. Fauci said on ABC News’s This Week.Nearly 150 million adults in the United States – or about 58% of the adult population – have received at least one COVID-19 vaccine dose, according to the latest CDC tally. About 113 million adults, or 44%, are considered fully vaccinated.
“The CDC will be, you know, almost in real time … updating their recommendations and their guidelines,” Dr. Fauci said.
In April, the CDC relaxed its guidance for those who have been vaccinated against COVID-19. Those who have gotten a shot don’t need to wear a mask outdoors or in small indoor gatherings with other vaccinated people, but both vaccinated and unvaccinated people are still advised to wear masks in indoor public spaces.
“We do need to start being more liberal as we get more people vaccinated,” Dr. Fauci said. “As you get more people vaccinated, the number of cases per day will absolutely go down.”
The United States is averaging about 43,000 cases per day, he said, adding that the cases need to be “much, much lower.” When the case numbers drop and vaccination numbers increase, the risk of infection will fall dramatically indoors and outdoors, he said.
Even after the pandemic, though, wearing masks could become a seasonal habit, Dr. Fauci said May 9 on NBC News’s Meet the Press.“I think people have gotten used to the fact that wearing masks, clearly if you look at the data, it diminishes respiratory diseases. We’ve had practically a nonexistent flu season this year,” he said.
“So it is conceivable that as we go on, a year or 2 or more from now, that during certain seasonal periods when you have respiratory-borne viruses like the flu, people might actually elect to wear masks to diminish the likelihood that you’ll spread these respiratory-borne diseases,” he said.
Dr. Fauci was asked about indoor mask guidelines on May 9 after former FDA Commissioner Scott Gottlieb, MD, said face mask requirements should be relaxed.
“Certainly outdoors, we shouldn’t be putting limits on gatherings anymore,” Dr. Gottlieb said on CBS News’s Face the Nation.“The states where prevalence is low, vaccination rates are high, we have good testing in place, and we’re identifying infections, I think we could start lifting these restrictions indoors as well, on a broad basis,” he said.
Lifting pandemic-related restrictions in areas where they’re no longer necessary could also encourage people to implement them again if cases increase during future surges, such as this fall or winter, Dr. Gottlieb said.
At the same time, Americans should continue to follow CDC guidance and wait for new guidelines before changing their indoor mask use, Jeffrey Zients, the White House COVID-19 response coordinator, said on CNN’s State of the Union on May 9.
“We all want to get back to a normal lifestyle,” he said. “I think we’re on the path to do that, but stay disciplined, and let’s take advantage of the new privilege of being vaccinated and not wearing masks outdoors, for example, unless you’re in a crowded place.”
Mr. Zients pointed to President Joe Biden’s goal for 70% of adults to receive at least one vaccine dose by July 4.
“As we all move toward that 70% goal, there will be more and more advantages to being vaccinated,” he said. “And if you’re not vaccinated, you’re not protected.”
A version of this article first appeared on WebMD.com.
FDA moves to ban menthol in cigarettes
The Food and Drug Administration said that within a year it will ban menthol in cigarettes and ban all flavors including menthol in cigars.

Menthol makes it easier to start smoking, and also enhances the effects of nicotine, making it more addictive and harder to quit, the FDA said in announcing its actions on Thursday.
Nineteen organizations – including the American Academy of Pediatrics, American Cancer Society, American College of Chest Physicians, American Medical Association, American Heart Association, and the National Medical Association – have pushed the FDA to ban menthol for years. The agency banned all flavors in cigarettes in 2009 but did not take any action against menthol. In 2013, the groups filed a petition demanding that the FDA ban menthol, too. The agency responded months later with a notice that it would start the process.
But it never took any action. Action on Smoking and Health and the African American Tobacco Control Leadership Council, later joined by the AMA and the NMA, sued in 2020 to compel the agency to do something. Now it has finally agreed to act.
The African American Tobacco Control Leadership Council welcomed the move but said the fight is not over and encouraged tobacco control activists to fight to ban menthol tobacco products at the local, state and federal level. “We know that this rule-making process could take years and we know that the tobacco industry will continue to do everything in their power to derail any attempt to remove their deadly products from the market,” Phillip Gardiner, MD, council cochair, said in a statement.
The AMA is urging the FDA to quickly implement the ban and remove the products “without further delay,” AMA President Susan R. Bailey, MD, said in a statement.
“FDA’s long-awaited decision to take action to eliminate menthol flavoring in cigarettes and all flavors in cigars ends a decades-long deference to the tobacco industry, which has repeatedly demonstrated its willingness to profit from products that result in death,” Lisa Lacasse, president of the American Cancer Society Cancer Action Network, said in her own statement.
Ms. Lacasse said banning menthol will help eliminate health disparities. She said 86% of Black people who smoke use menthol cigarettes, compared with 46% of Hispanic people who smoke, 39% of Asian people who smoke, and 29% of White people who smoke. “FDA’s actions today send a clear message that Big Tobacco’s strategy to profit off addicting Black communities will no longer be tolerated,” she said.
Not all groups are on board, however. The American Civil Liberties Union and several other organizations wrote to the country’s top health officials urging them to reconsider.
“Such a ban will trigger criminal penalties which will disproportionately impact people of color, as well as prioritize criminalization over public health and harm reduction,” the letter says. “A ban will also lead to unconstitutional policing and other negative interactions with local law enforcement.”
The letter calls the proposed ban “well intentioned,” but said any effort to reduce death and disease from tobacco “must avoid solutions that will create yet another reason for armed police to engage citizens on the street based on pretext or conduct that does not pose a threat to public safety.”
Instead of a ban, the organizations said, policy makers should consider increased education for adults and minors, stop-smoking programs, and increased funding for health centers in communities of color.
The Biden administration, however, pressed the point that banning menthol will bring many positives. Acting FDA Commissioner Janet Woodcock, MD said in a statement that banning menthol “will help significantly reduce youth initiation, increase the chances of smoking cessation among current smokers, and address health disparities experienced by communities of color, low-income populations, and LGBTQ-plus individuals, all of whom are far more likely to use these tobacco products.”
The FDA cited data showing that, in the first year or so after a ban goes into effect, an additional 923,000 smokers would quit, including 230,000 African Americans. Another study suggests that 633,000 deaths would be averted, including 237,000 Black Americans.
Dr. Woodcock added that, “armed with strong scientific evidence, and with full support from the [Biden] administration, we believe these actions will launch us on a trajectory toward ending tobacco-related disease and death in the U.S.”
The FDA estimates that 18.6 million Americans who are current smokers use menthol cigarettes, with a disproportionately high number being Black people. Menthol cigarette use among Black and Hispanic youth increased from 2011 to 2018, but declined for non-Hispanic White youth.
Flavored mass-produced cigars and cigarillos are disproportionately popular among youth, especially non-Hispanic Black high school students, who in 2020 reported past 30-day cigar smoking at levels twice as high as their White counterparts, said the FDA. Three-quarters of 12- to 17-year-olds reported they smoke cigars because they like the flavors. In 2020, more young people tried a cigar every day than tried a cigarette, reports the agency.
“This long-overdue decision will protect future generations of young people from nicotine addiction, especially Black children and communities, which have disproportionately suffered from menthol tobacco use due to targeted efforts from the tobacco industry,” Lee Savio Beers, MD, president of the American Academy of Pediatrics, said in a statement.
The FDA’s announcement “is only a first step that must be followed with urgent, comprehensive action to remove these flavored products from the market,” he said.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration said that within a year it will ban menthol in cigarettes and ban all flavors including menthol in cigars.
Menthol makes it easier to start smoking, and also enhances the effects of nicotine, making it more addictive and harder to quit, the FDA said in announcing its actions on Thursday.
Nineteen organizations – including the American Academy of Pediatrics, American Cancer Society, American College of Chest Physicians, American Medical Association, American Heart Association, and the National Medical Association – have pushed the FDA to ban menthol for years. The agency banned all flavors in cigarettes in 2009 but did not take any action against menthol. In 2013, the groups filed a petition demanding that the FDA ban menthol, too. The agency responded months later with a notice that it would start the process.
But it never took any action. Action on Smoking and Health and the African American Tobacco Control Leadership Council, later joined by the AMA and the NMA, sued in 2020 to compel the agency to do something. Now it has finally agreed to act.
The African American Tobacco Control Leadership Council welcomed the move but said the fight is not over and encouraged tobacco control activists to fight to ban menthol tobacco products at the local, state and federal level. “We know that this rule-making process could take years and we know that the tobacco industry will continue to do everything in their power to derail any attempt to remove their deadly products from the market,” Phillip Gardiner, MD, council cochair, said in a statement.
The AMA is urging the FDA to quickly implement the ban and remove the products “without further delay,” AMA President Susan R. Bailey, MD, said in a statement.
“FDA’s long-awaited decision to take action to eliminate menthol flavoring in cigarettes and all flavors in cigars ends a decades-long deference to the tobacco industry, which has repeatedly demonstrated its willingness to profit from products that result in death,” Lisa Lacasse, president of the American Cancer Society Cancer Action Network, said in her own statement.
Ms. Lacasse said banning menthol will help eliminate health disparities. She said 86% of Black people who smoke use menthol cigarettes, compared with 46% of Hispanic people who smoke, 39% of Asian people who smoke, and 29% of White people who smoke. “FDA’s actions today send a clear message that Big Tobacco’s strategy to profit off addicting Black communities will no longer be tolerated,” she said.
Not all groups are on board, however. The American Civil Liberties Union and several other organizations wrote to the country’s top health officials urging them to reconsider.
“Such a ban will trigger criminal penalties which will disproportionately impact people of color, as well as prioritize criminalization over public health and harm reduction,” the letter says. “A ban will also lead to unconstitutional policing and other negative interactions with local law enforcement.”
The letter calls the proposed ban “well intentioned,” but said any effort to reduce death and disease from tobacco “must avoid solutions that will create yet another reason for armed police to engage citizens on the street based on pretext or conduct that does not pose a threat to public safety.”
Instead of a ban, the organizations said, policy makers should consider increased education for adults and minors, stop-smoking programs, and increased funding for health centers in communities of color.
The Biden administration, however, pressed the point that banning menthol will bring many positives. Acting FDA Commissioner Janet Woodcock, MD said in a statement that banning menthol “will help significantly reduce youth initiation, increase the chances of smoking cessation among current smokers, and address health disparities experienced by communities of color, low-income populations, and LGBTQ-plus individuals, all of whom are far more likely to use these tobacco products.”
The FDA cited data showing that, in the first year or so after a ban goes into effect, an additional 923,000 smokers would quit, including 230,000 African Americans. Another study suggests that 633,000 deaths would be averted, including 237,000 Black Americans.
Dr. Woodcock added that, “armed with strong scientific evidence, and with full support from the [Biden] administration, we believe these actions will launch us on a trajectory toward ending tobacco-related disease and death in the U.S.”
The FDA estimates that 18.6 million Americans who are current smokers use menthol cigarettes, with a disproportionately high number being Black people. Menthol cigarette use among Black and Hispanic youth increased from 2011 to 2018, but declined for non-Hispanic White youth.
Flavored mass-produced cigars and cigarillos are disproportionately popular among youth, especially non-Hispanic Black high school students, who in 2020 reported past 30-day cigar smoking at levels twice as high as their White counterparts, said the FDA. Three-quarters of 12- to 17-year-olds reported they smoke cigars because they like the flavors. In 2020, more young people tried a cigar every day than tried a cigarette, reports the agency.
“This long-overdue decision will protect future generations of young people from nicotine addiction, especially Black children and communities, which have disproportionately suffered from menthol tobacco use due to targeted efforts from the tobacco industry,” Lee Savio Beers, MD, president of the American Academy of Pediatrics, said in a statement.
The FDA’s announcement “is only a first step that must be followed with urgent, comprehensive action to remove these flavored products from the market,” he said.
A version of this article first appeared on WebMD.com.
The Food and Drug Administration said that within a year it will ban menthol in cigarettes and ban all flavors including menthol in cigars.
Menthol makes it easier to start smoking, and also enhances the effects of nicotine, making it more addictive and harder to quit, the FDA said in announcing its actions on Thursday.
Nineteen organizations – including the American Academy of Pediatrics, American Cancer Society, American College of Chest Physicians, American Medical Association, American Heart Association, and the National Medical Association – have pushed the FDA to ban menthol for years. The agency banned all flavors in cigarettes in 2009 but did not take any action against menthol. In 2013, the groups filed a petition demanding that the FDA ban menthol, too. The agency responded months later with a notice that it would start the process.
But it never took any action. Action on Smoking and Health and the African American Tobacco Control Leadership Council, later joined by the AMA and the NMA, sued in 2020 to compel the agency to do something. Now it has finally agreed to act.
The African American Tobacco Control Leadership Council welcomed the move but said the fight is not over and encouraged tobacco control activists to fight to ban menthol tobacco products at the local, state and federal level. “We know that this rule-making process could take years and we know that the tobacco industry will continue to do everything in their power to derail any attempt to remove their deadly products from the market,” Phillip Gardiner, MD, council cochair, said in a statement.
The AMA is urging the FDA to quickly implement the ban and remove the products “without further delay,” AMA President Susan R. Bailey, MD, said in a statement.
“FDA’s long-awaited decision to take action to eliminate menthol flavoring in cigarettes and all flavors in cigars ends a decades-long deference to the tobacco industry, which has repeatedly demonstrated its willingness to profit from products that result in death,” Lisa Lacasse, president of the American Cancer Society Cancer Action Network, said in her own statement.
Ms. Lacasse said banning menthol will help eliminate health disparities. She said 86% of Black people who smoke use menthol cigarettes, compared with 46% of Hispanic people who smoke, 39% of Asian people who smoke, and 29% of White people who smoke. “FDA’s actions today send a clear message that Big Tobacco’s strategy to profit off addicting Black communities will no longer be tolerated,” she said.
Not all groups are on board, however. The American Civil Liberties Union and several other organizations wrote to the country’s top health officials urging them to reconsider.
“Such a ban will trigger criminal penalties which will disproportionately impact people of color, as well as prioritize criminalization over public health and harm reduction,” the letter says. “A ban will also lead to unconstitutional policing and other negative interactions with local law enforcement.”
The letter calls the proposed ban “well intentioned,” but said any effort to reduce death and disease from tobacco “must avoid solutions that will create yet another reason for armed police to engage citizens on the street based on pretext or conduct that does not pose a threat to public safety.”
Instead of a ban, the organizations said, policy makers should consider increased education for adults and minors, stop-smoking programs, and increased funding for health centers in communities of color.
The Biden administration, however, pressed the point that banning menthol will bring many positives. Acting FDA Commissioner Janet Woodcock, MD said in a statement that banning menthol “will help significantly reduce youth initiation, increase the chances of smoking cessation among current smokers, and address health disparities experienced by communities of color, low-income populations, and LGBTQ-plus individuals, all of whom are far more likely to use these tobacco products.”
The FDA cited data showing that, in the first year or so after a ban goes into effect, an additional 923,000 smokers would quit, including 230,000 African Americans. Another study suggests that 633,000 deaths would be averted, including 237,000 Black Americans.
Dr. Woodcock added that, “armed with strong scientific evidence, and with full support from the [Biden] administration, we believe these actions will launch us on a trajectory toward ending tobacco-related disease and death in the U.S.”
The FDA estimates that 18.6 million Americans who are current smokers use menthol cigarettes, with a disproportionately high number being Black people. Menthol cigarette use among Black and Hispanic youth increased from 2011 to 2018, but declined for non-Hispanic White youth.
Flavored mass-produced cigars and cigarillos are disproportionately popular among youth, especially non-Hispanic Black high school students, who in 2020 reported past 30-day cigar smoking at levels twice as high as their White counterparts, said the FDA. Three-quarters of 12- to 17-year-olds reported they smoke cigars because they like the flavors. In 2020, more young people tried a cigar every day than tried a cigarette, reports the agency.
“This long-overdue decision will protect future generations of young people from nicotine addiction, especially Black children and communities, which have disproportionately suffered from menthol tobacco use due to targeted efforts from the tobacco industry,” Lee Savio Beers, MD, president of the American Academy of Pediatrics, said in a statement.
The FDA’s announcement “is only a first step that must be followed with urgent, comprehensive action to remove these flavored products from the market,” he said.
A version of this article first appeared on WebMD.com.
CDC guidelines coming on long COVID
The Centers for Disease Control and Prevention is finalizing new guidelines to help clinicians diagnose and manage long COVID, or postacute sequelae of SARS-CoV-2 infection.
In a day-long congressional hearing on April 28, John Brooks, MD, a medical epidemiologist at the CDC’s division of HIV/AIDS prevention, testified that the guidelines were going through the clearance process at the agency, but would be forthcoming.
“They should be coming out very shortly,” Dr. Brooks said.
The guidelines, which were developed in collaboration with newly established long-COVID clinics and patient advocacy groups, will “illustrate how to diagnose and begin to pull together what we know about management,” of the complex condition, he said.
For many doctors and patients who are struggling to understand symptoms that persist for months after the initial viral infection, the guidelines can’t come soon enough.
National Institutes of Health Director Francis Collins, MD, PhD, who also testified at the hearing, estimated that as many as 3 million people could be left with chronic health problems after even mild COVID infections.
“I can’t overstate how serious this issue is for the health of our nation,” he said.
Dr. Collins said his estimate was based on studies showing that roughly 10% of people who get COVID could be affected by this and whose “long-term course is uncertain,” he said. So far, more than 32 million Americans are known to have been infected with the new coronavirus.
“We need to make sure we put our arms around them and bring answers and care to them,” said Rep. Anna Eshoo (D-Calif.), chairwoman of the Subcommittee on Health.
Jennifer Possick, MD, who directs the post-COVID recovery program at Yale New Haven (Conn.) Hospital, testified that the tidal wave of patients she and her colleagues were seeing was overwhelming.
“We are a well-resourced program at an academic medical center, but we are swamped by the need in our community. This year, we have seen more patients with post COVID-19 conditions in our clinic alone than we have new cases of asthma and COPD combined,” she said. “The magnitude of the challenge is daunting.”
Dr. Possick estimated that there are “over 60” clinics in the United States that have started to treat long-COVID patients, but said they are grassroots efforts and all very different from each other.
“Whoever had the resources, had the time, [and] was able to take the initiative and forge to the relationships because most of them are multidisciplinary, did so,” she said.
Patients testify
Several representatives shared moving personal stories of loved ones or staffers who remained ill months after a COVID diagnosis.
Rep. Ann Kuster, from New Hampshire, talked about her 34-year-old niece, a member of the U.S. Ski Team, who had COVID just over a year ago and “continues to struggle with everything, even the simplest activities of daily living” she said. “She has to choose between taking a shower or making dinner. I’m so proud of her for hanging in there.”
Long-COVID patients invited to testify by the subcommittee described months of disability that left them with soaring medical bills and no ability to work to pay them.
“I am now a poor, Black, disabled woman, living with long COVID,” said Chimere Smith, who said she had been a school teacher in Baltimore. “Saying it aloud makes it no more easy to accept.”
She said COVID had affected her ability to think clearly and caused debilitating fatigue, which prevented her from working. She said she lost her vision for almost 5 months because doctors misdiagnosed a cataract caused by long COVID as dry eye.
“If I did not have a loving family, I [would] be speaking to you today [from] my car, the only property I now own.”
Ms. Smith said that long-COVID clinics, which are mostly housed within academic medical centers, were not going to be accessible for all long-haulers, who are disproportionately women of color. She has started a clinic, based out of her church, to help other patients from her community.
“No one wants to hear that long COVID has decimated my life or the lives of other black women in less than a year,” Ms. Smith said. “We’ve just been waiting and hoping for compassionate doctors and politicians who would acknowledge us.”
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention is finalizing new guidelines to help clinicians diagnose and manage long COVID, or postacute sequelae of SARS-CoV-2 infection.
In a day-long congressional hearing on April 28, John Brooks, MD, a medical epidemiologist at the CDC’s division of HIV/AIDS prevention, testified that the guidelines were going through the clearance process at the agency, but would be forthcoming.
“They should be coming out very shortly,” Dr. Brooks said.
The guidelines, which were developed in collaboration with newly established long-COVID clinics and patient advocacy groups, will “illustrate how to diagnose and begin to pull together what we know about management,” of the complex condition, he said.
For many doctors and patients who are struggling to understand symptoms that persist for months after the initial viral infection, the guidelines can’t come soon enough.
National Institutes of Health Director Francis Collins, MD, PhD, who also testified at the hearing, estimated that as many as 3 million people could be left with chronic health problems after even mild COVID infections.
“I can’t overstate how serious this issue is for the health of our nation,” he said.
Dr. Collins said his estimate was based on studies showing that roughly 10% of people who get COVID could be affected by this and whose “long-term course is uncertain,” he said. So far, more than 32 million Americans are known to have been infected with the new coronavirus.
“We need to make sure we put our arms around them and bring answers and care to them,” said Rep. Anna Eshoo (D-Calif.), chairwoman of the Subcommittee on Health.
Jennifer Possick, MD, who directs the post-COVID recovery program at Yale New Haven (Conn.) Hospital, testified that the tidal wave of patients she and her colleagues were seeing was overwhelming.
“We are a well-resourced program at an academic medical center, but we are swamped by the need in our community. This year, we have seen more patients with post COVID-19 conditions in our clinic alone than we have new cases of asthma and COPD combined,” she said. “The magnitude of the challenge is daunting.”
Dr. Possick estimated that there are “over 60” clinics in the United States that have started to treat long-COVID patients, but said they are grassroots efforts and all very different from each other.
“Whoever had the resources, had the time, [and] was able to take the initiative and forge to the relationships because most of them are multidisciplinary, did so,” she said.
Patients testify
Several representatives shared moving personal stories of loved ones or staffers who remained ill months after a COVID diagnosis.
Rep. Ann Kuster, from New Hampshire, talked about her 34-year-old niece, a member of the U.S. Ski Team, who had COVID just over a year ago and “continues to struggle with everything, even the simplest activities of daily living” she said. “She has to choose between taking a shower or making dinner. I’m so proud of her for hanging in there.”
Long-COVID patients invited to testify by the subcommittee described months of disability that left them with soaring medical bills and no ability to work to pay them.
“I am now a poor, Black, disabled woman, living with long COVID,” said Chimere Smith, who said she had been a school teacher in Baltimore. “Saying it aloud makes it no more easy to accept.”
She said COVID had affected her ability to think clearly and caused debilitating fatigue, which prevented her from working. She said she lost her vision for almost 5 months because doctors misdiagnosed a cataract caused by long COVID as dry eye.
“If I did not have a loving family, I [would] be speaking to you today [from] my car, the only property I now own.”
Ms. Smith said that long-COVID clinics, which are mostly housed within academic medical centers, were not going to be accessible for all long-haulers, who are disproportionately women of color. She has started a clinic, based out of her church, to help other patients from her community.
“No one wants to hear that long COVID has decimated my life or the lives of other black women in less than a year,” Ms. Smith said. “We’ve just been waiting and hoping for compassionate doctors and politicians who would acknowledge us.”
A version of this article first appeared on Medscape.com.
The Centers for Disease Control and Prevention is finalizing new guidelines to help clinicians diagnose and manage long COVID, or postacute sequelae of SARS-CoV-2 infection.
In a day-long congressional hearing on April 28, John Brooks, MD, a medical epidemiologist at the CDC’s division of HIV/AIDS prevention, testified that the guidelines were going through the clearance process at the agency, but would be forthcoming.
“They should be coming out very shortly,” Dr. Brooks said.
The guidelines, which were developed in collaboration with newly established long-COVID clinics and patient advocacy groups, will “illustrate how to diagnose and begin to pull together what we know about management,” of the complex condition, he said.
For many doctors and patients who are struggling to understand symptoms that persist for months after the initial viral infection, the guidelines can’t come soon enough.
National Institutes of Health Director Francis Collins, MD, PhD, who also testified at the hearing, estimated that as many as 3 million people could be left with chronic health problems after even mild COVID infections.
“I can’t overstate how serious this issue is for the health of our nation,” he said.
Dr. Collins said his estimate was based on studies showing that roughly 10% of people who get COVID could be affected by this and whose “long-term course is uncertain,” he said. So far, more than 32 million Americans are known to have been infected with the new coronavirus.
“We need to make sure we put our arms around them and bring answers and care to them,” said Rep. Anna Eshoo (D-Calif.), chairwoman of the Subcommittee on Health.
Jennifer Possick, MD, who directs the post-COVID recovery program at Yale New Haven (Conn.) Hospital, testified that the tidal wave of patients she and her colleagues were seeing was overwhelming.
“We are a well-resourced program at an academic medical center, but we are swamped by the need in our community. This year, we have seen more patients with post COVID-19 conditions in our clinic alone than we have new cases of asthma and COPD combined,” she said. “The magnitude of the challenge is daunting.”
Dr. Possick estimated that there are “over 60” clinics in the United States that have started to treat long-COVID patients, but said they are grassroots efforts and all very different from each other.
“Whoever had the resources, had the time, [and] was able to take the initiative and forge to the relationships because most of them are multidisciplinary, did so,” she said.
Patients testify
Several representatives shared moving personal stories of loved ones or staffers who remained ill months after a COVID diagnosis.
Rep. Ann Kuster, from New Hampshire, talked about her 34-year-old niece, a member of the U.S. Ski Team, who had COVID just over a year ago and “continues to struggle with everything, even the simplest activities of daily living” she said. “She has to choose between taking a shower or making dinner. I’m so proud of her for hanging in there.”
Long-COVID patients invited to testify by the subcommittee described months of disability that left them with soaring medical bills and no ability to work to pay them.
“I am now a poor, Black, disabled woman, living with long COVID,” said Chimere Smith, who said she had been a school teacher in Baltimore. “Saying it aloud makes it no more easy to accept.”
She said COVID had affected her ability to think clearly and caused debilitating fatigue, which prevented her from working. She said she lost her vision for almost 5 months because doctors misdiagnosed a cataract caused by long COVID as dry eye.
“If I did not have a loving family, I [would] be speaking to you today [from] my car, the only property I now own.”
Ms. Smith said that long-COVID clinics, which are mostly housed within academic medical centers, were not going to be accessible for all long-haulers, who are disproportionately women of color. She has started a clinic, based out of her church, to help other patients from her community.
“No one wants to hear that long COVID has decimated my life or the lives of other black women in less than a year,” Ms. Smith said. “We’ve just been waiting and hoping for compassionate doctors and politicians who would acknowledge us.”
A version of this article first appeared on Medscape.com.
Feds lift pause of J&J COVID vaccine, add new warning
Use of the Johnson & Johnson COVID-19 vaccine should resume in the United States for all adults, the Food and Drug Administration and Centers for Disease Contol and Prevention said April 23, although health care providers should warn patients of the risk of developing the rare and serious blood clots that caused the agencies to pause the vaccine’s distribution earlier this month.
“What we are seeing is the overall rate of events was 1.9 cases per million people. In women 18 to 29 years there was an approximate 7 cases per million. The risk is even lower in women over the age of 50 at .9 cases per million,” CDC Director Rochelle Walensky, MD, said in a news briefing the same day.
In the end, the potential benefits of the vaccine far outweighed its risks.
“In terms of benefits, we found that for every 1 million doses of this vaccine, the J&J vaccine could prevent over 650 hospitalizations and 12 deaths among women ages 18-49,” Dr. Walensky said. The potential benefits to women over 50 were even greater: It could prevent 4,700 hospitalizations and 650 deaths.
“In the end, this vaccine was shown to be safe and effective for the vast majority of people,” Dr. Walensky said.
The recommendation to continue the vaccine’s rollout came barely 2 hours after a CDC Advisory Committee on Immunization Practices voted to recommend the pause be lifted. The vote was 10-4 with one abstention.
The decision also includes instructions for the warning directed at women under 50 who have an increased risk of a rare but serious blood clot disorder called thrombosis with thrombocytopenia syndrome (TTS).
As of April 21, 15 cases of TTS, all in women and 13 of them in women under 50, have been confirmed among 7.98 million doses of the J&J vaccine administered in the United States. Three women have died.
The FDA and CDC recommended the pause on April 13 after reports that 6 women developed a blood clotting disorder 6 to 13 days after they received the J&J vaccine.
William Schaffner, MD, an infectious disease expert at Vanderbilt University in Nashville, and a non-voting ACIP member, said in an interview the panel made the right recommendation.
He applauded both the decision to restart the vaccine and the updated warning information that “will explain [TTS] more fully to people, particularly women, who are coming to be vaccinated.”
As to women in the risk group needing to have a choice of vaccines, Dr. Schaffner said that will be addressed differently across the country.
“Every provider will not have alternative vaccines in their location so there will be many different ways to do this. You may have to get this information and select which site you’re going to depending on which vaccine is available if this matter is important to you,” he noted.
ACIP made the decision after a 6-hour emergency meeting to hear evidence on the Johnson & Johnson vaccine's protective benefits against COVID-19 vs. risk of TTS.
In the CDC-FDA press briefing, Dr. Walensky pointed out that over the past few days, as regulators have reviewed the rare events, newly identified patients had been treated appropriately, without the use of heparin, which is not advised for treating TTS.
As a result, regulators felt as if their messages had gotten out to doctors who now knew how to take special precautions when treating patients with the disorder.
She said the Johnson & Johnson shot remained an important option because it was convenient to give and easier to store than the other vaccines currently authorized in the United States.
Peter Marks, MD, the director of FDA’s Center for Biologics Evaluation and Research, said the agency had already added information describing the risk of the rare clotting disorder to its fact sheets for patients and doctors.
Janet Woodcock, MD, acting commissioner of the FDA, said vaccination centers could resume giving the “one and done” shots as early as April 24.
This article was updated April 24, 2021, and first appeared on WebMD.com.
Use of the Johnson & Johnson COVID-19 vaccine should resume in the United States for all adults, the Food and Drug Administration and Centers for Disease Contol and Prevention said April 23, although health care providers should warn patients of the risk of developing the rare and serious blood clots that caused the agencies to pause the vaccine’s distribution earlier this month.
“What we are seeing is the overall rate of events was 1.9 cases per million people. In women 18 to 29 years there was an approximate 7 cases per million. The risk is even lower in women over the age of 50 at .9 cases per million,” CDC Director Rochelle Walensky, MD, said in a news briefing the same day.
In the end, the potential benefits of the vaccine far outweighed its risks.
“In terms of benefits, we found that for every 1 million doses of this vaccine, the J&J vaccine could prevent over 650 hospitalizations and 12 deaths among women ages 18-49,” Dr. Walensky said. The potential benefits to women over 50 were even greater: It could prevent 4,700 hospitalizations and 650 deaths.
“In the end, this vaccine was shown to be safe and effective for the vast majority of people,” Dr. Walensky said.
The recommendation to continue the vaccine’s rollout came barely 2 hours after a CDC Advisory Committee on Immunization Practices voted to recommend the pause be lifted. The vote was 10-4 with one abstention.
The decision also includes instructions for the warning directed at women under 50 who have an increased risk of a rare but serious blood clot disorder called thrombosis with thrombocytopenia syndrome (TTS).
As of April 21, 15 cases of TTS, all in women and 13 of them in women under 50, have been confirmed among 7.98 million doses of the J&J vaccine administered in the United States. Three women have died.
The FDA and CDC recommended the pause on April 13 after reports that 6 women developed a blood clotting disorder 6 to 13 days after they received the J&J vaccine.
William Schaffner, MD, an infectious disease expert at Vanderbilt University in Nashville, and a non-voting ACIP member, said in an interview the panel made the right recommendation.
He applauded both the decision to restart the vaccine and the updated warning information that “will explain [TTS] more fully to people, particularly women, who are coming to be vaccinated.”
As to women in the risk group needing to have a choice of vaccines, Dr. Schaffner said that will be addressed differently across the country.
“Every provider will not have alternative vaccines in their location so there will be many different ways to do this. You may have to get this information and select which site you’re going to depending on which vaccine is available if this matter is important to you,” he noted.
ACIP made the decision after a 6-hour emergency meeting to hear evidence on the Johnson & Johnson vaccine's protective benefits against COVID-19 vs. risk of TTS.
In the CDC-FDA press briefing, Dr. Walensky pointed out that over the past few days, as regulators have reviewed the rare events, newly identified patients had been treated appropriately, without the use of heparin, which is not advised for treating TTS.
As a result, regulators felt as if their messages had gotten out to doctors who now knew how to take special precautions when treating patients with the disorder.
She said the Johnson & Johnson shot remained an important option because it was convenient to give and easier to store than the other vaccines currently authorized in the United States.
Peter Marks, MD, the director of FDA’s Center for Biologics Evaluation and Research, said the agency had already added information describing the risk of the rare clotting disorder to its fact sheets for patients and doctors.
Janet Woodcock, MD, acting commissioner of the FDA, said vaccination centers could resume giving the “one and done” shots as early as April 24.
This article was updated April 24, 2021, and first appeared on WebMD.com.
Use of the Johnson & Johnson COVID-19 vaccine should resume in the United States for all adults, the Food and Drug Administration and Centers for Disease Contol and Prevention said April 23, although health care providers should warn patients of the risk of developing the rare and serious blood clots that caused the agencies to pause the vaccine’s distribution earlier this month.
“What we are seeing is the overall rate of events was 1.9 cases per million people. In women 18 to 29 years there was an approximate 7 cases per million. The risk is even lower in women over the age of 50 at .9 cases per million,” CDC Director Rochelle Walensky, MD, said in a news briefing the same day.
In the end, the potential benefits of the vaccine far outweighed its risks.
“In terms of benefits, we found that for every 1 million doses of this vaccine, the J&J vaccine could prevent over 650 hospitalizations and 12 deaths among women ages 18-49,” Dr. Walensky said. The potential benefits to women over 50 were even greater: It could prevent 4,700 hospitalizations and 650 deaths.
“In the end, this vaccine was shown to be safe and effective for the vast majority of people,” Dr. Walensky said.
The recommendation to continue the vaccine’s rollout came barely 2 hours after a CDC Advisory Committee on Immunization Practices voted to recommend the pause be lifted. The vote was 10-4 with one abstention.
The decision also includes instructions for the warning directed at women under 50 who have an increased risk of a rare but serious blood clot disorder called thrombosis with thrombocytopenia syndrome (TTS).
As of April 21, 15 cases of TTS, all in women and 13 of them in women under 50, have been confirmed among 7.98 million doses of the J&J vaccine administered in the United States. Three women have died.
The FDA and CDC recommended the pause on April 13 after reports that 6 women developed a blood clotting disorder 6 to 13 days after they received the J&J vaccine.
William Schaffner, MD, an infectious disease expert at Vanderbilt University in Nashville, and a non-voting ACIP member, said in an interview the panel made the right recommendation.
He applauded both the decision to restart the vaccine and the updated warning information that “will explain [TTS] more fully to people, particularly women, who are coming to be vaccinated.”
As to women in the risk group needing to have a choice of vaccines, Dr. Schaffner said that will be addressed differently across the country.
“Every provider will not have alternative vaccines in their location so there will be many different ways to do this. You may have to get this information and select which site you’re going to depending on which vaccine is available if this matter is important to you,” he noted.
ACIP made the decision after a 6-hour emergency meeting to hear evidence on the Johnson & Johnson vaccine's protective benefits against COVID-19 vs. risk of TTS.
In the CDC-FDA press briefing, Dr. Walensky pointed out that over the past few days, as regulators have reviewed the rare events, newly identified patients had been treated appropriately, without the use of heparin, which is not advised for treating TTS.
As a result, regulators felt as if their messages had gotten out to doctors who now knew how to take special precautions when treating patients with the disorder.
She said the Johnson & Johnson shot remained an important option because it was convenient to give and easier to store than the other vaccines currently authorized in the United States.
Peter Marks, MD, the director of FDA’s Center for Biologics Evaluation and Research, said the agency had already added information describing the risk of the rare clotting disorder to its fact sheets for patients and doctors.
Janet Woodcock, MD, acting commissioner of the FDA, said vaccination centers could resume giving the “one and done” shots as early as April 24.
This article was updated April 24, 2021, and first appeared on WebMD.com.
HHS proposes overturning Title X ‘gag’ rule
The Department of Health & Human Services has proposed overturning rules issued during the Trump administration that effectively prohibit clinicians at Title X–funded health clinics from discussing abortion or referring patients for abortions.
HHS proposed the overhaul of the Title X regulations on April 14. The previous administration’s 2019 rules “have undermined the public health of the population the program is meant to serve,” HHS said in the introduction to its proposal.
Medical organizations and reproductive health specialists lauded the move.
“Clinicians providing care to patients must be empowered to share the full spectrum of accurate medical information necessary to ensure that their patients are able to make timely, fully informed medical decisions,” Maureen G. Phipps, MD, MPH, CEO of the American College of Obstetricians and Gynecologists, said in a statement. “This means transparent, respectful, evidence-based conversations about contraception and abortion care. The proposed rule will ensure that those conversations can once again happen without restrictions, interference, or threat of financial loss.”
“Providers of comprehensive reproductive health care, including abortion care, base their relationships with their patients on trust,” Physicians for Reproductive Health President and CEO Jamila Perritt, MD, said in a statement. “The Title X gag rule went against everything we knew as providers of ethical, evidence-based health care by forcing providers at Title X funded clinics to withhold information that their patients needed and requested.”
HHS said that, since 2019, more than 1,000 Title X–funded service sites (25% of the total) have withdrawn from the program. Currently, Title X services – which include family planning, STI testing, cancer screening, and HIV testing and treatment – are not available in six states and are only available on a limited basis in six additional states. Planned Parenthood fully withdrew from Title X.
HHS said that tens of thousands fewer birth control implant procedures have been performed and that hundreds of thousands fewer Pap tests and a half-million or more fewer tests for chlamydia and gonorrhea have been conducted. In addition, the reduction in services may have led to up to 181,477 unintended pregnancies, HHS said.
The closure of sites and decreased availability of services have also exacerbated health inequities, according to the department.
The loss of services “has been especially felt by those already facing disproportionate barriers to accessing care, including the Black, Latinx and Indigenous communities that have also suffered the most harm during the COVID-19 pandemic,” agreed Dr. Phipps.
The new regulation proposes to “ensure access to equitable, affordable, client-centered, quality family-planning services for all clients, especially for low-income clients,” HHS said.
The proposed change in the rules “brings us one step closer to restoring access to necessary care for millions of low-income and uninsured patients who depend on Title X for family planning services,” American Medical Association President Susan R. Bailey, MD, said in a statement. “We are pleased that the Biden administration shares our commitment to undoing this dangerous and discriminatory ‘gag rule,’ and look forward to its elimination through any means necessary to achieve the best outcome for patients and physicians – improving the health of our nation.”
Planned Parenthood also applauded the move, and the HIV Medicine Association thanked the Biden administration for its proposal, which it called “a major step to improve #HealthEquity for all people in this country,” in a tweet.
March for Life, an antiabortion group, however, said it strongly opposed the HHS proposal. The rules “appear specifically designed to bring America’s largest abortion provider, Planned Parenthood, back into the taxpayer-funded program and keep prolife organizations out,” said the group in a tweet.
“Abortion is neither health care nor family planning, and the Title X program should not be funding it,” said the group.
The Title X program does not pay for abortions, however.
The Trump administration rules prohibit abortion referrals and impose counseling standards for pregnant patients and what the Guttmacher Institute called “unnecessary and stringent requirements for the physical and financial separation of Title X–funded activities from a range of abortion-related activities.”
The new rules would reestablish regulations from 2000, with some new additions. For instance, the program will “formally integrate elements of quality family-planning services guidelines developed by [Centers for Disease Control and Prevention] and Office of Population Affairs,” tweeted Alina Salganicoff, director of women’s health policy at the Kaiser Family Foundation. “That means that higher standards for providing family planning will be required,” she tweeted. In addition, sites that offer natural family planning and abstinence “will only be able to participate if they offer referrals to other providers that offer clients access to the contraceptive of their choice.”
The proposed rules are open for public comment for 30 days. They could be made final by the fall. The Kaiser Family Foundation reports that many sites could be ready to return to the program by then, especially since the recently passed coronavirus relief package, the American Rescue Plan, included a $50 million supplemental appropriation for Title X.
The 2019 rules remain in effect in the meantime, although the U.S. Supreme Court agreed in February to hear a challenge mounted by 21 states, the city of Baltimore, and organizations that included the AMA and Planned Parenthood. Those plaintiffs have requested that the case be dismissed, but it currently remains on the docket.
Not all medical providers are likely to support the new rules if they go into effect. The American Association of Pro-Life Obstetricians and Gynecologists, the Christian Medical and Dental Associations, and the Catholic Medical Association filed motions in the Supreme Court case to defend the Trump regulations.
A version of this article first appeared on Medscape.com.
The Department of Health & Human Services has proposed overturning rules issued during the Trump administration that effectively prohibit clinicians at Title X–funded health clinics from discussing abortion or referring patients for abortions.
HHS proposed the overhaul of the Title X regulations on April 14. The previous administration’s 2019 rules “have undermined the public health of the population the program is meant to serve,” HHS said in the introduction to its proposal.
Medical organizations and reproductive health specialists lauded the move.
“Clinicians providing care to patients must be empowered to share the full spectrum of accurate medical information necessary to ensure that their patients are able to make timely, fully informed medical decisions,” Maureen G. Phipps, MD, MPH, CEO of the American College of Obstetricians and Gynecologists, said in a statement. “This means transparent, respectful, evidence-based conversations about contraception and abortion care. The proposed rule will ensure that those conversations can once again happen without restrictions, interference, or threat of financial loss.”
“Providers of comprehensive reproductive health care, including abortion care, base their relationships with their patients on trust,” Physicians for Reproductive Health President and CEO Jamila Perritt, MD, said in a statement. “The Title X gag rule went against everything we knew as providers of ethical, evidence-based health care by forcing providers at Title X funded clinics to withhold information that their patients needed and requested.”
HHS said that, since 2019, more than 1,000 Title X–funded service sites (25% of the total) have withdrawn from the program. Currently, Title X services – which include family planning, STI testing, cancer screening, and HIV testing and treatment – are not available in six states and are only available on a limited basis in six additional states. Planned Parenthood fully withdrew from Title X.
HHS said that tens of thousands fewer birth control implant procedures have been performed and that hundreds of thousands fewer Pap tests and a half-million or more fewer tests for chlamydia and gonorrhea have been conducted. In addition, the reduction in services may have led to up to 181,477 unintended pregnancies, HHS said.
The closure of sites and decreased availability of services have also exacerbated health inequities, according to the department.
The loss of services “has been especially felt by those already facing disproportionate barriers to accessing care, including the Black, Latinx and Indigenous communities that have also suffered the most harm during the COVID-19 pandemic,” agreed Dr. Phipps.
The new regulation proposes to “ensure access to equitable, affordable, client-centered, quality family-planning services for all clients, especially for low-income clients,” HHS said.
The proposed change in the rules “brings us one step closer to restoring access to necessary care for millions of low-income and uninsured patients who depend on Title X for family planning services,” American Medical Association President Susan R. Bailey, MD, said in a statement. “We are pleased that the Biden administration shares our commitment to undoing this dangerous and discriminatory ‘gag rule,’ and look forward to its elimination through any means necessary to achieve the best outcome for patients and physicians – improving the health of our nation.”
Planned Parenthood also applauded the move, and the HIV Medicine Association thanked the Biden administration for its proposal, which it called “a major step to improve #HealthEquity for all people in this country,” in a tweet.
March for Life, an antiabortion group, however, said it strongly opposed the HHS proposal. The rules “appear specifically designed to bring America’s largest abortion provider, Planned Parenthood, back into the taxpayer-funded program and keep prolife organizations out,” said the group in a tweet.
“Abortion is neither health care nor family planning, and the Title X program should not be funding it,” said the group.
The Title X program does not pay for abortions, however.
The Trump administration rules prohibit abortion referrals and impose counseling standards for pregnant patients and what the Guttmacher Institute called “unnecessary and stringent requirements for the physical and financial separation of Title X–funded activities from a range of abortion-related activities.”
The new rules would reestablish regulations from 2000, with some new additions. For instance, the program will “formally integrate elements of quality family-planning services guidelines developed by [Centers for Disease Control and Prevention] and Office of Population Affairs,” tweeted Alina Salganicoff, director of women’s health policy at the Kaiser Family Foundation. “That means that higher standards for providing family planning will be required,” she tweeted. In addition, sites that offer natural family planning and abstinence “will only be able to participate if they offer referrals to other providers that offer clients access to the contraceptive of their choice.”
The proposed rules are open for public comment for 30 days. They could be made final by the fall. The Kaiser Family Foundation reports that many sites could be ready to return to the program by then, especially since the recently passed coronavirus relief package, the American Rescue Plan, included a $50 million supplemental appropriation for Title X.
The 2019 rules remain in effect in the meantime, although the U.S. Supreme Court agreed in February to hear a challenge mounted by 21 states, the city of Baltimore, and organizations that included the AMA and Planned Parenthood. Those plaintiffs have requested that the case be dismissed, but it currently remains on the docket.
Not all medical providers are likely to support the new rules if they go into effect. The American Association of Pro-Life Obstetricians and Gynecologists, the Christian Medical and Dental Associations, and the Catholic Medical Association filed motions in the Supreme Court case to defend the Trump regulations.
A version of this article first appeared on Medscape.com.
The Department of Health & Human Services has proposed overturning rules issued during the Trump administration that effectively prohibit clinicians at Title X–funded health clinics from discussing abortion or referring patients for abortions.
HHS proposed the overhaul of the Title X regulations on April 14. The previous administration’s 2019 rules “have undermined the public health of the population the program is meant to serve,” HHS said in the introduction to its proposal.
Medical organizations and reproductive health specialists lauded the move.
“Clinicians providing care to patients must be empowered to share the full spectrum of accurate medical information necessary to ensure that their patients are able to make timely, fully informed medical decisions,” Maureen G. Phipps, MD, MPH, CEO of the American College of Obstetricians and Gynecologists, said in a statement. “This means transparent, respectful, evidence-based conversations about contraception and abortion care. The proposed rule will ensure that those conversations can once again happen without restrictions, interference, or threat of financial loss.”
“Providers of comprehensive reproductive health care, including abortion care, base their relationships with their patients on trust,” Physicians for Reproductive Health President and CEO Jamila Perritt, MD, said in a statement. “The Title X gag rule went against everything we knew as providers of ethical, evidence-based health care by forcing providers at Title X funded clinics to withhold information that their patients needed and requested.”
HHS said that, since 2019, more than 1,000 Title X–funded service sites (25% of the total) have withdrawn from the program. Currently, Title X services – which include family planning, STI testing, cancer screening, and HIV testing and treatment – are not available in six states and are only available on a limited basis in six additional states. Planned Parenthood fully withdrew from Title X.
HHS said that tens of thousands fewer birth control implant procedures have been performed and that hundreds of thousands fewer Pap tests and a half-million or more fewer tests for chlamydia and gonorrhea have been conducted. In addition, the reduction in services may have led to up to 181,477 unintended pregnancies, HHS said.
The closure of sites and decreased availability of services have also exacerbated health inequities, according to the department.
The loss of services “has been especially felt by those already facing disproportionate barriers to accessing care, including the Black, Latinx and Indigenous communities that have also suffered the most harm during the COVID-19 pandemic,” agreed Dr. Phipps.
The new regulation proposes to “ensure access to equitable, affordable, client-centered, quality family-planning services for all clients, especially for low-income clients,” HHS said.
The proposed change in the rules “brings us one step closer to restoring access to necessary care for millions of low-income and uninsured patients who depend on Title X for family planning services,” American Medical Association President Susan R. Bailey, MD, said in a statement. “We are pleased that the Biden administration shares our commitment to undoing this dangerous and discriminatory ‘gag rule,’ and look forward to its elimination through any means necessary to achieve the best outcome for patients and physicians – improving the health of our nation.”
Planned Parenthood also applauded the move, and the HIV Medicine Association thanked the Biden administration for its proposal, which it called “a major step to improve #HealthEquity for all people in this country,” in a tweet.
March for Life, an antiabortion group, however, said it strongly opposed the HHS proposal. The rules “appear specifically designed to bring America’s largest abortion provider, Planned Parenthood, back into the taxpayer-funded program and keep prolife organizations out,” said the group in a tweet.
“Abortion is neither health care nor family planning, and the Title X program should not be funding it,” said the group.
The Title X program does not pay for abortions, however.
The Trump administration rules prohibit abortion referrals and impose counseling standards for pregnant patients and what the Guttmacher Institute called “unnecessary and stringent requirements for the physical and financial separation of Title X–funded activities from a range of abortion-related activities.”
The new rules would reestablish regulations from 2000, with some new additions. For instance, the program will “formally integrate elements of quality family-planning services guidelines developed by [Centers for Disease Control and Prevention] and Office of Population Affairs,” tweeted Alina Salganicoff, director of women’s health policy at the Kaiser Family Foundation. “That means that higher standards for providing family planning will be required,” she tweeted. In addition, sites that offer natural family planning and abstinence “will only be able to participate if they offer referrals to other providers that offer clients access to the contraceptive of their choice.”
The proposed rules are open for public comment for 30 days. They could be made final by the fall. The Kaiser Family Foundation reports that many sites could be ready to return to the program by then, especially since the recently passed coronavirus relief package, the American Rescue Plan, included a $50 million supplemental appropriation for Title X.
The 2019 rules remain in effect in the meantime, although the U.S. Supreme Court agreed in February to hear a challenge mounted by 21 states, the city of Baltimore, and organizations that included the AMA and Planned Parenthood. Those plaintiffs have requested that the case be dismissed, but it currently remains on the docket.
Not all medical providers are likely to support the new rules if they go into effect. The American Association of Pro-Life Obstetricians and Gynecologists, the Christian Medical and Dental Associations, and the Catholic Medical Association filed motions in the Supreme Court case to defend the Trump regulations.
A version of this article first appeared on Medscape.com.
Comparison of Dermatologist Ratings on Health Care–Specific and General Consumer Websites
Health care–specific (eg, Healthgrades, Zocdoc, Vitals, WebMD) and general consumer websites (eg, Google, Yelp) are popular platforms for patients to find physicians, schedule appointments, and review physician experiences. Patients find ratings on these websites more trustworthy than standardized surveys distributed by hospitals, but many physicians do not trust the reviews on these sites. For example, in a survey of both physicians (n=828) and patients (n=494), 36% of physicians trusted online reviews compared to 57% of patients.1 The objective of this study was to determine if health care–specific or general consumer websites more accurately reflect overall patient sentiment. This knowledge can help physicians who are seeking to improve the patient experience understand which websites have more accurate and trustworthy reviews.
Methods
A list of dermatologists from the top 10 most and least dermatologist–dense areas in the United States was compiled to examine different physician populations.2 Equal numbers of male and female dermatologists were randomly selected from the most dense areas. All physicians were included from the least dense areas because of limited sample size. Ratings were collected from websites most likely to appear on the first page of a Google search for a physician name, as these are most likely to be seen by patients. Descriptive statistics were generated to describe the study population; mean and median physician rating (using a scale of 1–5); SD; and minimum, maximum, and interquartile ranges. Spearman correlation coefficients were generated to examine the strength of association between ratings from website pairs. P<.05 was considered statistically significant, with analyses performed in R (3.6.2) for Windows (the R Foundation).
Results
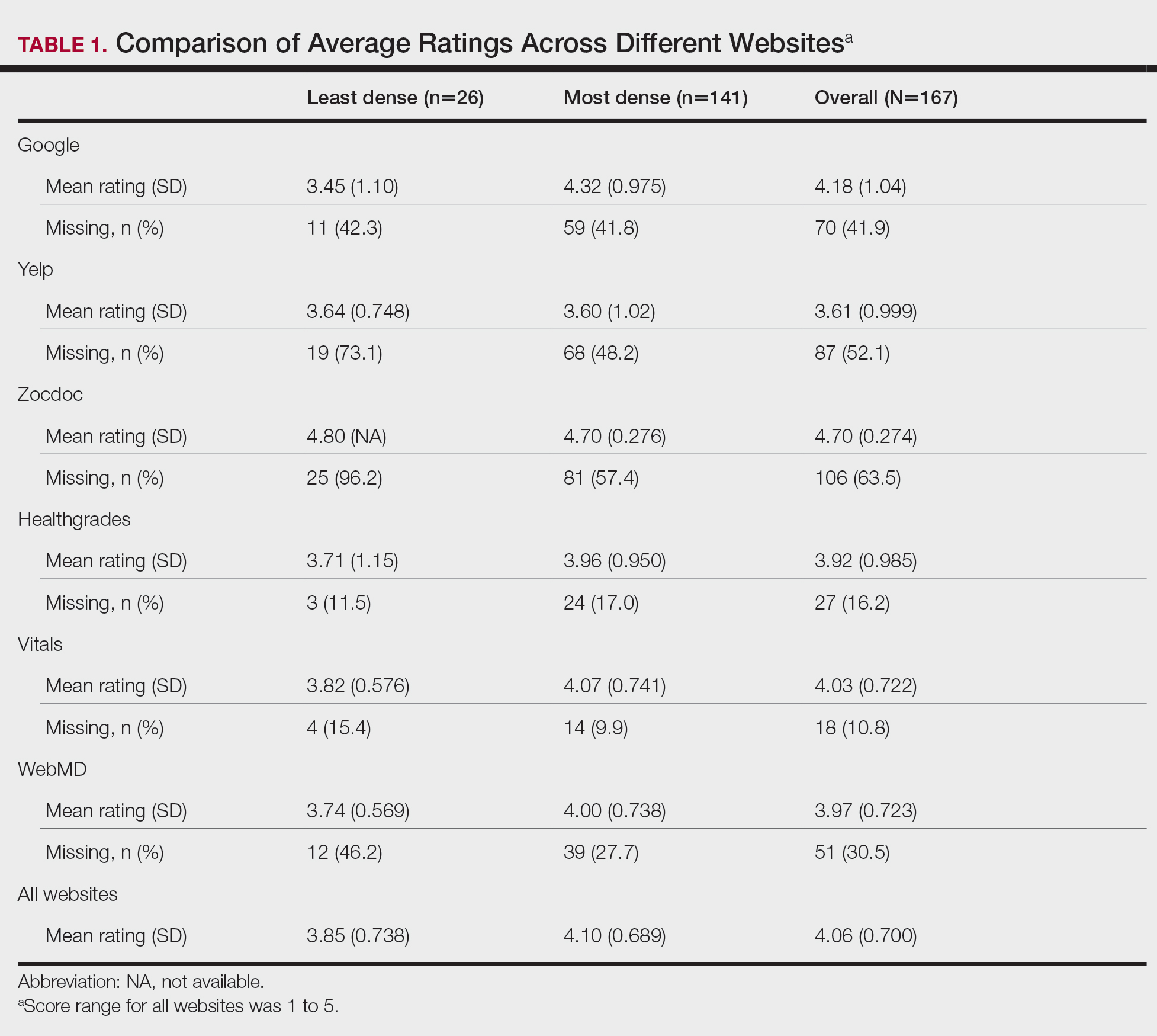
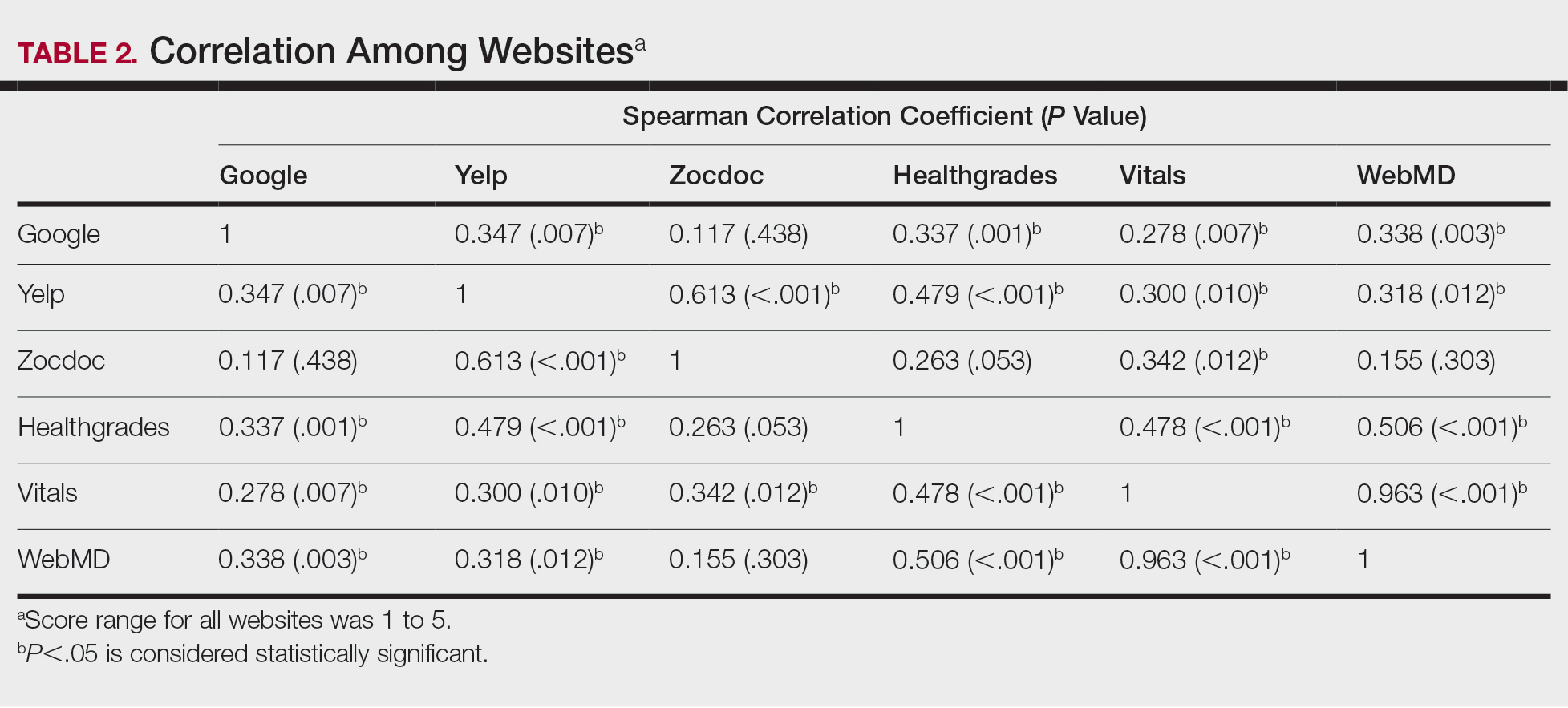
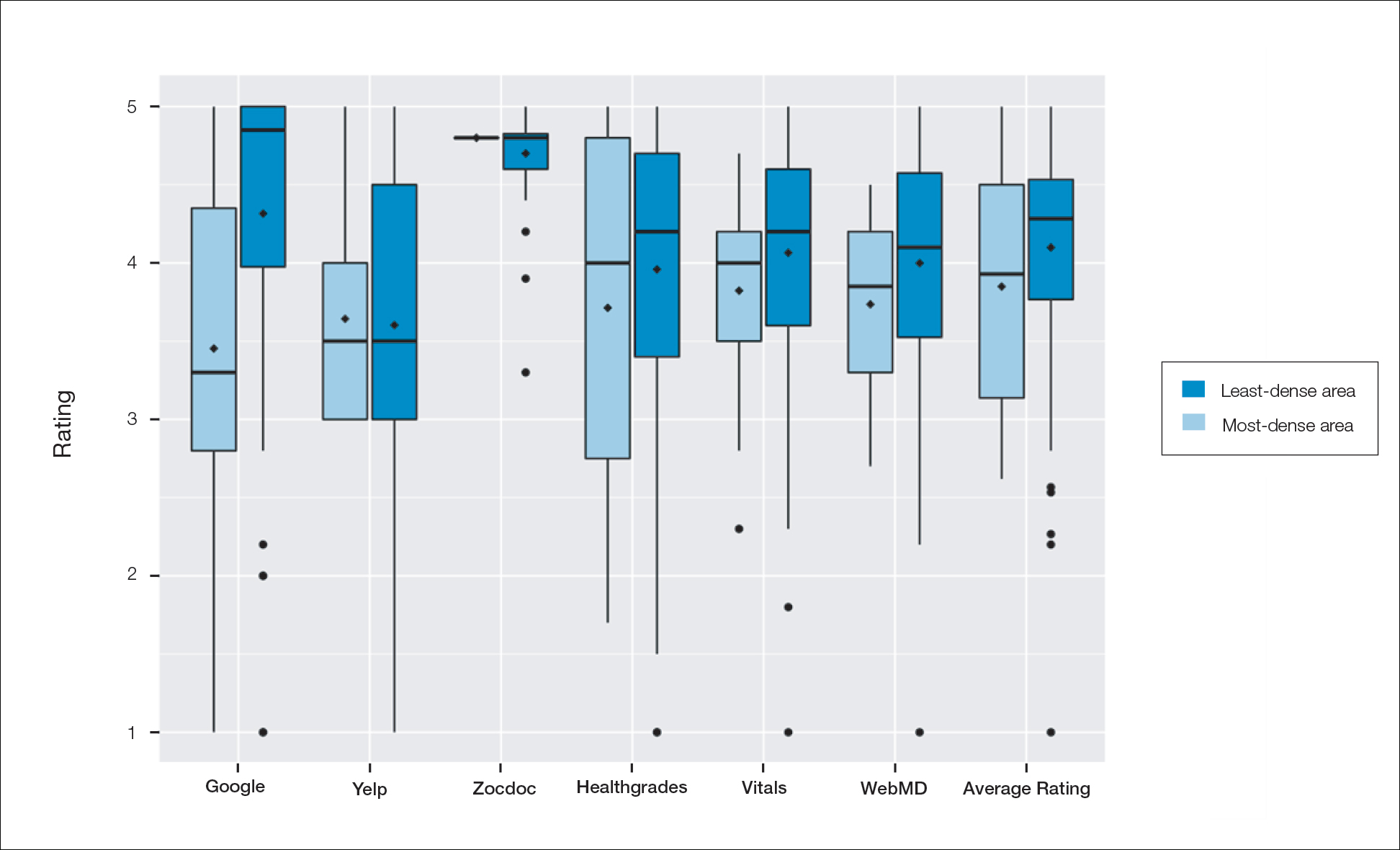
A total of 167 representative physicians were included in this analysis; 141 from the most dense areas, and 26 from the least dense areas. The lowest average ratings for the entire sample and most dermatologist–dense areas were found on Yelp (3.61 and 3.60, respectively), and the lowest ratings in the least dermatologist–dense areas were found on Google (3.45)(Table 1). Correlation coefficient values were lowest for Zocdoc and Healthgrades (0.263) and highest for Vitals and WebMD (0.963)(Table 2). The health care–specific sites were closer to the overall average (4.06) than the general consumer sites (eFigure).
Comment
Although dermatologist ratings on each site had a broad range, we found that patients typically expressed negative interactions on general consumer websites rather than health care–specific websites. When comparing the ratings of the same group of dermatologists across different sites, ratings on health care–specific sites had a higher degree of correlation, with physician ratings more similar between 2 health care–specific sites and less similar between a health care–specific and a general consumer website. This pattern was consistent in both dermatologist-dense and dermatologist-poor areas, despite patients having varying levels of access to dermatologic care and medical resources and potentially different regional preferences of consumer websites. Taken together, these findings imply that health care–specific websites more consistently reflect overall patient sentiment.
Although one 2016 study comparing reviews of dermatology practices on Zocdoc and Yelp also demonstrated lower average ratings on Yelp,3 our study suggests that this trend is not isolated to these 2 sites but can be seen when comparing many health care–specific sites vs general consumer sites.
Our study compared ratings of dermatologists among popular websites to understand those that are most representative of patient attitudes toward physicians. These findings are important because online reviews reflect the entire patient experience, not just the patient-physician interaction, which may explain why physician scores on standardized questionnaires, such as Press Ganey surveys, do not correlate well with their online reviews.4 In a study comparing 98 physicians with negative online ratings to 82 physicians in similar departments with positive ratings, there was no significant difference in scores on patient-physician interaction questions on the Press Ganey survey.5 However, physicians who received negative online reviews scored lower on Press Ganey questions related to nonphysician interactions (eg, office cleanliness, interactions with staff).
The current study was subject to several limitations. Our analysis included all physicians in our random selection without accounting for those physicians with a greater online presence who might be more cognizant of these ratings and try to manipulate them through a reputation-management company or public relations consultant.
Conclusion
Our study suggests that consumer websites are not primarily used by disgruntled patients wishing to express grievances; instead, on average, most physicians received positive reviews. Furthermore, health care–specific websites show a higher degree of concordance than and may more accurately reflect overall patient attitudes toward their physicians than general consumer sites. Reviews from these health care–specific sites may be more helpful than general consumer websites in allowing physicians to understand patient sentiment and improve patient experiences.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38:e257-e262. doi:10.3928/01477447-20150402-52
- Waqas B, Cooley V, Lipner SR. Association of sex, location, and experience with online patient ratings of dermatologists. J Am Acad Dermatol. 2020;83:954-955.
- Smith RJ, Lipoff JB. Evaluation of dermatology practice online reviews: lessons from qualitative analysis. JAMA Dermatol. 2016;152:153-157. doi:10.1001/jamadermatol.2015.3950
- Chen J, Presson A, Zhang C, et al. Online physician review websites poorly correlate to a validated metric of patient satisfaction. J Surg Res. 2018;227:1-6.
- Widmer RJ, Maurer MJ, Nayar VR, et al. Online physician reviews do not reflect patient satisfaction survey responses. Mayo Clinic Proc. 2018;93:453-457.
Health care–specific (eg, Healthgrades, Zocdoc, Vitals, WebMD) and general consumer websites (eg, Google, Yelp) are popular platforms for patients to find physicians, schedule appointments, and review physician experiences. Patients find ratings on these websites more trustworthy than standardized surveys distributed by hospitals, but many physicians do not trust the reviews on these sites. For example, in a survey of both physicians (n=828) and patients (n=494), 36% of physicians trusted online reviews compared to 57% of patients.1 The objective of this study was to determine if health care–specific or general consumer websites more accurately reflect overall patient sentiment. This knowledge can help physicians who are seeking to improve the patient experience understand which websites have more accurate and trustworthy reviews.
Methods
A list of dermatologists from the top 10 most and least dermatologist–dense areas in the United States was compiled to examine different physician populations.2 Equal numbers of male and female dermatologists were randomly selected from the most dense areas. All physicians were included from the least dense areas because of limited sample size. Ratings were collected from websites most likely to appear on the first page of a Google search for a physician name, as these are most likely to be seen by patients. Descriptive statistics were generated to describe the study population; mean and median physician rating (using a scale of 1–5); SD; and minimum, maximum, and interquartile ranges. Spearman correlation coefficients were generated to examine the strength of association between ratings from website pairs. P<.05 was considered statistically significant, with analyses performed in R (3.6.2) for Windows (the R Foundation).
Results
A total of 167 representative physicians were included in this analysis; 141 from the most dense areas, and 26 from the least dense areas. The lowest average ratings for the entire sample and most dermatologist–dense areas were found on Yelp (3.61 and 3.60, respectively), and the lowest ratings in the least dermatologist–dense areas were found on Google (3.45)(Table 1). Correlation coefficient values were lowest for Zocdoc and Healthgrades (0.263) and highest for Vitals and WebMD (0.963)(Table 2). The health care–specific sites were closer to the overall average (4.06) than the general consumer sites (eFigure).
Comment
Although dermatologist ratings on each site had a broad range, we found that patients typically expressed negative interactions on general consumer websites rather than health care–specific websites. When comparing the ratings of the same group of dermatologists across different sites, ratings on health care–specific sites had a higher degree of correlation, with physician ratings more similar between 2 health care–specific sites and less similar between a health care–specific and a general consumer website. This pattern was consistent in both dermatologist-dense and dermatologist-poor areas, despite patients having varying levels of access to dermatologic care and medical resources and potentially different regional preferences of consumer websites. Taken together, these findings imply that health care–specific websites more consistently reflect overall patient sentiment.
Although one 2016 study comparing reviews of dermatology practices on Zocdoc and Yelp also demonstrated lower average ratings on Yelp,3 our study suggests that this trend is not isolated to these 2 sites but can be seen when comparing many health care–specific sites vs general consumer sites.
Our study compared ratings of dermatologists among popular websites to understand those that are most representative of patient attitudes toward physicians. These findings are important because online reviews reflect the entire patient experience, not just the patient-physician interaction, which may explain why physician scores on standardized questionnaires, such as Press Ganey surveys, do not correlate well with their online reviews.4 In a study comparing 98 physicians with negative online ratings to 82 physicians in similar departments with positive ratings, there was no significant difference in scores on patient-physician interaction questions on the Press Ganey survey.5 However, physicians who received negative online reviews scored lower on Press Ganey questions related to nonphysician interactions (eg, office cleanliness, interactions with staff).
The current study was subject to several limitations. Our analysis included all physicians in our random selection without accounting for those physicians with a greater online presence who might be more cognizant of these ratings and try to manipulate them through a reputation-management company or public relations consultant.
Conclusion
Our study suggests that consumer websites are not primarily used by disgruntled patients wishing to express grievances; instead, on average, most physicians received positive reviews. Furthermore, health care–specific websites show a higher degree of concordance than and may more accurately reflect overall patient attitudes toward their physicians than general consumer sites. Reviews from these health care–specific sites may be more helpful than general consumer websites in allowing physicians to understand patient sentiment and improve patient experiences.
Health care–specific (eg, Healthgrades, Zocdoc, Vitals, WebMD) and general consumer websites (eg, Google, Yelp) are popular platforms for patients to find physicians, schedule appointments, and review physician experiences. Patients find ratings on these websites more trustworthy than standardized surveys distributed by hospitals, but many physicians do not trust the reviews on these sites. For example, in a survey of both physicians (n=828) and patients (n=494), 36% of physicians trusted online reviews compared to 57% of patients.1 The objective of this study was to determine if health care–specific or general consumer websites more accurately reflect overall patient sentiment. This knowledge can help physicians who are seeking to improve the patient experience understand which websites have more accurate and trustworthy reviews.
Methods
A list of dermatologists from the top 10 most and least dermatologist–dense areas in the United States was compiled to examine different physician populations.2 Equal numbers of male and female dermatologists were randomly selected from the most dense areas. All physicians were included from the least dense areas because of limited sample size. Ratings were collected from websites most likely to appear on the first page of a Google search for a physician name, as these are most likely to be seen by patients. Descriptive statistics were generated to describe the study population; mean and median physician rating (using a scale of 1–5); SD; and minimum, maximum, and interquartile ranges. Spearman correlation coefficients were generated to examine the strength of association between ratings from website pairs. P<.05 was considered statistically significant, with analyses performed in R (3.6.2) for Windows (the R Foundation).
Results
A total of 167 representative physicians were included in this analysis; 141 from the most dense areas, and 26 from the least dense areas. The lowest average ratings for the entire sample and most dermatologist–dense areas were found on Yelp (3.61 and 3.60, respectively), and the lowest ratings in the least dermatologist–dense areas were found on Google (3.45)(Table 1). Correlation coefficient values were lowest for Zocdoc and Healthgrades (0.263) and highest for Vitals and WebMD (0.963)(Table 2). The health care–specific sites were closer to the overall average (4.06) than the general consumer sites (eFigure).
Comment
Although dermatologist ratings on each site had a broad range, we found that patients typically expressed negative interactions on general consumer websites rather than health care–specific websites. When comparing the ratings of the same group of dermatologists across different sites, ratings on health care–specific sites had a higher degree of correlation, with physician ratings more similar between 2 health care–specific sites and less similar between a health care–specific and a general consumer website. This pattern was consistent in both dermatologist-dense and dermatologist-poor areas, despite patients having varying levels of access to dermatologic care and medical resources and potentially different regional preferences of consumer websites. Taken together, these findings imply that health care–specific websites more consistently reflect overall patient sentiment.
Although one 2016 study comparing reviews of dermatology practices on Zocdoc and Yelp also demonstrated lower average ratings on Yelp,3 our study suggests that this trend is not isolated to these 2 sites but can be seen when comparing many health care–specific sites vs general consumer sites.
Our study compared ratings of dermatologists among popular websites to understand those that are most representative of patient attitudes toward physicians. These findings are important because online reviews reflect the entire patient experience, not just the patient-physician interaction, which may explain why physician scores on standardized questionnaires, such as Press Ganey surveys, do not correlate well with their online reviews.4 In a study comparing 98 physicians with negative online ratings to 82 physicians in similar departments with positive ratings, there was no significant difference in scores on patient-physician interaction questions on the Press Ganey survey.5 However, physicians who received negative online reviews scored lower on Press Ganey questions related to nonphysician interactions (eg, office cleanliness, interactions with staff).
The current study was subject to several limitations. Our analysis included all physicians in our random selection without accounting for those physicians with a greater online presence who might be more cognizant of these ratings and try to manipulate them through a reputation-management company or public relations consultant.
Conclusion
Our study suggests that consumer websites are not primarily used by disgruntled patients wishing to express grievances; instead, on average, most physicians received positive reviews. Furthermore, health care–specific websites show a higher degree of concordance than and may more accurately reflect overall patient attitudes toward their physicians than general consumer sites. Reviews from these health care–specific sites may be more helpful than general consumer websites in allowing physicians to understand patient sentiment and improve patient experiences.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38:e257-e262. doi:10.3928/01477447-20150402-52
- Waqas B, Cooley V, Lipner SR. Association of sex, location, and experience with online patient ratings of dermatologists. J Am Acad Dermatol. 2020;83:954-955.
- Smith RJ, Lipoff JB. Evaluation of dermatology practice online reviews: lessons from qualitative analysis. JAMA Dermatol. 2016;152:153-157. doi:10.1001/jamadermatol.2015.3950
- Chen J, Presson A, Zhang C, et al. Online physician review websites poorly correlate to a validated metric of patient satisfaction. J Surg Res. 2018;227:1-6.
- Widmer RJ, Maurer MJ, Nayar VR, et al. Online physician reviews do not reflect patient satisfaction survey responses. Mayo Clinic Proc. 2018;93:453-457.
- Frost C, Mesfin A. Online reviews of orthopedic surgeons: an emerging trend. Orthopedics. 2015;38:e257-e262. doi:10.3928/01477447-20150402-52
- Waqas B, Cooley V, Lipner SR. Association of sex, location, and experience with online patient ratings of dermatologists. J Am Acad Dermatol. 2020;83:954-955.
- Smith RJ, Lipoff JB. Evaluation of dermatology practice online reviews: lessons from qualitative analysis. JAMA Dermatol. 2016;152:153-157. doi:10.1001/jamadermatol.2015.3950
- Chen J, Presson A, Zhang C, et al. Online physician review websites poorly correlate to a validated metric of patient satisfaction. J Surg Res. 2018;227:1-6.
- Widmer RJ, Maurer MJ, Nayar VR, et al. Online physician reviews do not reflect patient satisfaction survey responses. Mayo Clinic Proc. 2018;93:453-457.
Practice Points
- Online physician-rating websites are commonly used by patients to find physicians and review experiences.
- Health care–specific sites may more accurately reflect patient sentiment than general consumer sites.
- Dermatologists can use health care–specific sites to understand patient sentiment and learn how to improve patient experiences.
List of COVID-19 high-risk comorbidities expanded
The list of medical according to the Centers for Disease Control and Prevention.

The CDC’s latest list consists of 17 conditions or groups of related conditions that may increase patients’ risk of developing severe outcomes of COVID-19, the CDC said on a web page intended for the general public.
On a separate page, the CDC defines severe outcomes “as hospitalization, admission to the intensive care unit, intubation or mechanical ventilation, or death.”
Asthma is included in the newly expanded list with other chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis; the list’s heart disease entry covers coronary artery disease, heart failure, cardiomyopathies, and hypertension, the CDC said.
The list of medical according to the Centers for Disease Control and Prevention.
The CDC’s latest list consists of 17 conditions or groups of related conditions that may increase patients’ risk of developing severe outcomes of COVID-19, the CDC said on a web page intended for the general public.
On a separate page, the CDC defines severe outcomes “as hospitalization, admission to the intensive care unit, intubation or mechanical ventilation, or death.”
Asthma is included in the newly expanded list with other chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis; the list’s heart disease entry covers coronary artery disease, heart failure, cardiomyopathies, and hypertension, the CDC said.
The list of medical according to the Centers for Disease Control and Prevention.
The CDC’s latest list consists of 17 conditions or groups of related conditions that may increase patients’ risk of developing severe outcomes of COVID-19, the CDC said on a web page intended for the general public.
On a separate page, the CDC defines severe outcomes “as hospitalization, admission to the intensive care unit, intubation or mechanical ventilation, or death.”
Asthma is included in the newly expanded list with other chronic lung diseases such as chronic obstructive pulmonary disease and cystic fibrosis; the list’s heart disease entry covers coronary artery disease, heart failure, cardiomyopathies, and hypertension, the CDC said.
COVID-19 Monoclonal Antibody Infusions: A Multidisciplinary Initiative to Operationalize EUA Novel Treatment Options
From Mount Sinai Medical Center, Miami Beach, FL.
Abstract
Objective: To develop and implement a process for administering COVID-19 monoclonal antibody infusions for outpatients with mild or moderate COVID-19 at high risk for hospitalization, using multidisciplinary collaboration, US Food and Drug Administration (FDA) guidance, and infection prevention standards.
Methods: When monoclonal antibody therapy became available for mild or moderate COVID-19 outpatients via Emergency Use Authorization (EUA), our institution sought to provide this therapy option to our patients. We describe the process for planning, implementing, and maintaining a successful program for administering novel therapies based on FDA guidance and infection prevention standards. Key components of our implementation process were multidisciplinary planning involving decision makers and stakeholders; setting realistic goals in the process; team communication; and measuring and reporting quality improvement on a regular basis.
Results: A total of 790 COVID-19 monoclonal antibody infusions were administered from November 20, 2020 to March 5, 2021. Steps to minimize the likelihood of adverse drug reactions were implemented and a low incidence (< 1%) has occurred. There has been no concern from staff regarding infection during the process. Rarely, patients have raised cost-related concerns, typically due to incomplete communication regarding billing prior to the infusion. Patients, families, nursing staff, physicians, pharmacy, and hospital administration have expressed satisfaction with the program.
Conclusion: This process can provide a template for other hospitals or health care delivery facilities to provide novel therapies to patients with mild or moderate COVID-19 in a safe and effective manner.
Keywords: COVID-19; monoclonal antibody; infusion; emergency use authorization.
SARS-CoV-2 and the disease it causes, COVID-19, have transformed from scientific vernacular to common household terms. It began with a cluster of pneumonia cases of unknown etiology in December 2019 in Wuhan, China, with physicians there reporting a novel coronavirus strain (2019-nCoV), now referred to as SARS-CoV-2. Rapid spread of this virus resulted in the World Health Organization (WHO) declaring an international public health emergency. Since this time, the virus has evolved into a worldwide pandemic. COVID-19 has dramatically impacted our society, resulting in more than 2.63 million global deaths as of this writing, of which more than 527,000 deaths have occurred in the United States.1 This novel virus has resulted in a flurry of literature, research, therapies, and collaboration across multiple disciplines in an effort to prevent, treat, and mitigate cases and complications of this disease.
On November 9, 2020, and November 21, 2020, the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for 2 novel COVID-19 monoclonal therapies, bamlanivimab2-3 and casirivimab/imdevimab,3-4 respectively. The EUAs granted permission for these therapies to be administered for the treatment of mild to moderate COVID-19 in adult and pediatric patients (≥ 12 years and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The therapies work by targeting the SARS-CoV-2 spike protein and subsequent attachment to human angiotensin-converting enzyme 2 receptors. Clinical trial data leading to the EUA demonstrated a reduction in viral load, safe outcome, and most importantly, fewer hospitalization and emergency room visits, as compared to the placebo group.5-7 The use of monoclonal antibodies is not new and gained recognition during the Ebola crisis, when the monoclonal antibody to the Ebola virus showed a significant survival benefit.8 Providing monoclonal antibody therapy soon after symptom onset aligns with a shift from the onset of the pandemic to the current focus on the administration of pharmaceutical therapy early in the disease course. This shift prevents progression to severe COVID-19, with the goal of reducing patient mortality, hospitalizations, and strain on health care systems.
The availability of novel neutralizing monoclonal antibodies for COVID-19 led to discussions of how to incorporate these therapies as new options for patients. Our institution networked with colleagues from multiple disciplines to discuss processes and policies for the safe administration of the monoclonal antibody infusion therapies. Federal health leaders urge more use of monoclonal antibodies, but many hospitals have been unable to successfully implement infusions due to staff and logistical challenges.9 This article presents a viable process that hospitals can use to provide these novel therapies to outpatients with mild to moderate COVID-19.
The Mount Sinai Medical Center, Florida Experience
Mount Sinai Medical Center in Miami Beach, Florida, is the largest private, independent, not-for-profit teaching hospital in South Florida, comprising 672 licensed beds and supporting 150,000 emergency department (ED) visits annually. Per the EUA criteria for use, COVID-19 monoclonal antibody therapies are not authorized for patients who are hospitalized or who require oxygen therapy due to COVID-19. Therefore, options for outpatient administration needed to be evaluated. Directly following the first EUA press release, a task force of key stakeholders was assembled to brainstorm and develop a process to offer this therapy to the community. A multidisciplinary task force with representation from the ED, nursing, primary care, hospital medicine, pharmacy, risk management, billing, information technology, infection prevention, and senior level leadership participated (Table).

The task force reviewed institutional outpatient locations to determine whether offering this service would be feasible (eg, ED, ambulatory care facilities, cancer center). The ED was selected because it would offer the largest array of appointment times to meet the community needs with around-the-clock availability. While Mount Sinai Medical Center offers care in 3 emergency center locations in Aventura, Hialeah, and Miami Beach, it was determined to initiate the infusions at the main campus center in Miami Beach only. The main campus affords an onsite pharmacy with suitable staffing to prepare the anticipated volume of infusions in a timely manner, as both therapies have short stabilities following preparation. Thus, it was decided that patients from freestanding emergency centers in Aventura and Hialeah would be moved to the Miami Beach ED location to receive therapy. Operating at a single site also allowed for more rapid implementation, monitoring, and ability to make modifications more easily. Discussions for the possible expansion of COVID-19 monoclonal antibody infusions at satellite locations are underway.

On November 20, 2020, 11 days after the formation of the multidisciplinary task force, the first COVID-19 monoclonal infusion was successfully administered. Figure 1 depicts the timeline from assessment to program implementation. Critical to implementation was the involvement of decision makers from all necessary departments early in the planning process to ensure that standard operating procedures were followed and that the patients, community, and organization had a positive experience. This allowed for simultaneous planning of electronic health record (Epic; EHR) builds, departmental workflows, and staff education, as described in the following section. Figure 2 shows the patient safety activities included in the implementation process.

Key Stakeholder Involvement and Workflow
On the day of bamlanivimab EUA release, email communication was shared among hospital leadership with details of the press release. Departments were quickly involved to initiate a task force to assess if and how this therapy could be offered at Mount Sinai Medical Center. The following sections explain the role of each stakeholder and their essential role to operationalize these novel EUA treatment options. The task force was organized and led by our chief medical officer and chief nursing officer.
Information Technology
Medication Ordering and Documentation EHR and Smart Pumps. Early in the pandemic, the antimicrobial stewardship (ASP) clinical coordinator became the designated point person for pharmacy assessment of novel COVID-19 therapies. As such, this pharmacist began reviewing the bamlanivimab and, later, the casirivimab/imdevimab EUA Fact Sheet for Health Care Providers. All necessary elements for the complete and safe ordering and dispensing of the medication were developed and reviewed by pharmacy administration and ED nursing leadership for input, prior to submitting to the information technology team for implementation. Building the COVID-19 monoclonal medication records into the EHR allowed for detailed direction (ie, administration and preparation instructions) to be consistently applied. The medication records were also built into hospital smart pumps so that nurses could access prepopulated, accurate volumes and infusion rates to minimize errors.
Order Set Development. The pharmacy medication build was added to a comprehensive order set (Figure 3), which was then developed to guide prescribers and standardize the process around ordering of COVID-19 monoclonal therapies. While these therapies are new, oncology monoclonal therapies are regularly administered to outpatients at Mount Sinai Cancer Center. The cancer center was therefore consulted on their process surrounding best practices in administration of monoclonal antibody therapies. This included protocols for medications used in pretreatment and management of hypersensitivity reactions and potential adverse drug reactions of both COVID-19 monoclonal therapies. These medication orders were selected by default in the order set to ensure that all patients received premedications aimed at minimizing the risk of hypersensitivity reaction, and had as-needed medication orders, in the event a hypersensitivity reaction occurred. Reducing hypersensitivity reaction risk is important as well to increase the likelihood that the patient would receive full therapy, as management of this adverse drug reactions involves possible cessation of therapy depending on the level of severity. The pharmacy department also ensured these medications were stocked in ED automated dispensing cabinets to promote quick access. In addition to the aforementioned nursing orders, we added EUA criteria for use and hyperlinks to the Fact Sheets for Patients and Caregivers and Health Care Providers for each monoclonal therapy, and restricted ordering to ED physicians, nurse practitioners, and physician assistants.

The order set underwent multidisciplinary review by pharmacy administration, the chair of emergency medicine, physicians, and ED nursing leadership prior to presentation and approval by the Pharmacy and Therapeutics Committee. Lastly, at time of implementation, the order set was added to the ED preference list, preventing inpatient access. Additionally, as a patient safety action, free- standing orders of COVID-19 monoclonal therapies were disabled, so providers could only order therapies via the approved, comprehensive order set.
Preliminary Assessment Tool. A provider assessment tool was developed to document patient-specific EUA criteria for use during initial assessment (Figure 4). This tool serves as a checklist and is visible to the full multidisciplinary team in the patient’s EHR. It is used as a resource at the time of pharmacist verification and ED physician assessment to ensure criteria for use are met.

Outpatient Offices
Patient Referral. Patients with symptoms or concerns of COVID-19 exposure can make physician appointments via telemedicine or in person at Mount Sinai Medical Center’s primary care and specialty offices. At the time of patient encounter, physicians suspecting a COVID-19 diagnosis will refer patients for outpatient COVID-19 polymerase chain reaction (PCR) laboratory testing, which has an approximate 24-hour turnaround to results. Physicians also assess whether the patient meets EUA criteria for use, pending results of testing. In the event a patient meets EUA criteria for use, the physician provides patient counseling and requests verbal consent. Following this, the physician enters a note in the EHR describing the patient’s condition, criteria for use evaluation, and the patient’s verbal agreement to therapy. This preliminary screening is beneficial to begin planning with both the patient and ED to minimize delays. Patients are notified of the results of their test once available. If the COVID-19 PCR test returns positive, the physician will call the ED at the main campus and schedule the patient for COVID-19 monoclonal therapy. As the desired timeframe for administering COVID-19 monoclonal therapies is within less than 10 days of symptom onset, timely scheduling of appointments is crucial. Infusion appointments are typically provided the same or next day. The patients are informed that they must bring documentation of their positive COVID-19 PCR test to their ED visit. Lastly, because patients are pretreated with medication that may potentially impair driving, they are instructed that they cannot drive themselves home; ride shares also are not allowed in order to limit the spread of infection.
Emergency Department
Patient Arrival and Screening. A COVID-19 patient can be evaluated in the ED 1 of 2 ways. The first option is via outpatient office referral, as described previously. Upon arrival to the ED, a second screening is performed to ensure the patient still meets EUA criteria for use and the positive COVID-19 PCR test result is confirmed. If the patient no longer meets criteria, the patient is triaged accordingly, including evaluation for higher-level care (eg, supplemental oxygen, hospital admission). The second optoion is via new patient walk-ins without outpatient physician referral (Figure 4). In these cases, an initial screening is performed, documenting EUA criteria for use in the preliminary assessment (Figure 5). Physicians will consider an outside COVID-19 test as valid, so long as documentation is readily available confirming a positive PCR result. Otherwise, an in-house COVID-19 PCR test will be performed, which has a 2-hour turnaround time.

Infusion Schedule. The ED offers a total of 16 COVID-19 monoclonal infusions slots daily. These are broken up into 4 infusion time blocks (eg, 8
Patient Education. Prior to administration of the monoclonal therapy, physician and nursing staff obtain a formal, written patient consent for therapy and provide patients with the option of participating in the institutional review board (IRB) approved study. Details of this are discussed in the risk management and IRB sections of the article. Nursing staff also provides the medication-specific Fact Sheet for Patients and Caregivers in either Spanish or English, which is also included as a hyperlink on the COVID-19 Monoclonal Antibody Order Set for ease of access. Interpreter services are available for patients who speak other languages. An ED decentralized pharmacist is also available onsite Monday through Friday from 12
Infusion Ordering. Once the patient is ready to begin therapy, the he/she is brought to a dedicated overflow area of the ED. There are few, if any, patients in this location, and it is adjacent to the main emergency center for easy access by the patients, nurses, pharmacists, and physicians. The physician then enters orders in the EHR using the COVID-19 Monoclonal Antibody Order Set (Figure 3). Three discrete questions were built into the medication order: (1) Was patient consent obtained? (2) Was the Fact Sheet for Patient/Caregiver provided to the patient? (3) Is the patient COVID-19 PCR-positive? These questions were built as hard stops so that the medication orders cannot be placed without a response. This serves as another double-check to ensure processes are followed and helps facilitate timely verification by the pharmacist.
Medication Administration. One nurse is dedicated to administering the monoclonal therapies scheduled at 8
Pharmacy Department
Medication Receipt Process. Inventory is currently allocated biweekly from the state department of health and will soon be transitioning to a direct order system. The pharmacy technician in charge of deliveries notifies the pharmacy Antimicrobial Stewardship Program (ASP) clinical coordinator upon receipt of the monoclonal therapies. Bamlanivimab is supplied as 1 vial per dose, whereas casirivimab/imdevimab is supplied as 4 vials or 8 vials per dose, depending how it is shipped. To reduce the likelihood of medication errors, the ASP clinical coordinator assembles each of the casirivimab/imdevimab vials into kits, where 1 kit equals 1 dose. Labels are then affixed to each kit indicating the medication name, number of vials which equal a full dose, and pharmacist signature. The kits are stored in a dedicated refrigerator, and inventory logs are affixed to the outside of the refrigerator and updated daily. This inventory is also communicated daily to ED physician, nursing, and pharmacy leadership, as well as the director of patient safety, who reports weekly usage to the state Department of Health and Human Services. These weekly reports are used to determine allocation amounts.
Medication Verification and Delivery. The Mount Sinai Medical Center pharmacist staffing model consists of centralized order entry and specialized, decentralized positions. All orders are verified by the ED pharmacist when scheduled (not a 24/7 service) and by the designated pharmacist for all other times. At the time of medication verification, the pharmacist documents patient-specific EUA criteria for use and confirms that consent was obtained and the Fact Sheet for Patients/Caregivers was provided. A pharmacist intervention was developed to assist with this documentation. Pharmacists input smart text “.COVIDmonoclonal” and a drop-down menu of EUA criteria for use appears. The pharmacist reviews the patient care notes and medication order question responses to ascertain this information, contacting the ED prescriber if further clarification is required. This verification serves as another check to ensure processes put in place are followed. Lastly, intravenous preparation and delivery are electronically recorded in the EHR, and the medications require nursing signature at the time of delivery to ensure a formal chain of custody.
Risk Management
At Mount Sinai Medical Center, all EUA and investigational therapies require patient consent. Consistent with this requirement, a COVID-19 monoclonal specific consent was developed by risk management. This is provided to every patient receiving a COVID-19 monoclonal infusion, in addition to the FDA EUA Fact Sheet for Patients and Caregivers, and documented as part of their EHR. The questions providers must answer are built into the order set to ensure this process is followed and these patient safety checks are incorporated into the workflow.
Billing and Finance Department
In alignment with Mount Sinai Medical Center’s mission to provide high-quality health care to its diverse community through teaching, research, charity care, and financial responsibility, it was determined that this therapy would be provided to all patients regardless of insurance type, including those who are uninsured. The billing and finance department was consulted prior to this service being offered, to provide patients with accurate and pertinent information. The billing and finance department provided guidance on how to document patient encounters at time of registration to facilitate appropriate billing. At this time, the medication is free of charge, but nonmedication-related ED fees apply. This is explained to patients so there is a clear understanding prior to booking their appointment.
Infection Prevention
As patients receiving COVID-19 monoclonal therapies can transmit the virus to others, measures to ensure protection for other patients and staff are vital. To minimize exposure, specific nursing and physician staff from the ED are assigned to the treatment of these patients, and patients receive infusions and postobservation monitoring in a designated wing of the ED. Additionally, all staff who interact with these patients are required to don full personal protective equipment. This includes not only physicians and nurses but all specialties such as physician assistants, nurse practitioners, pharmacists, and laboratory technicians. Moreover, patients are not permitted to go home in a ride share and are counseled on Centers for Disease Control and Prevention quarantining following infusion.
Measurement of Process and Outcomes and Reporting
IRB approval was sought and obtained early during initiation of this service, allowing study consent to be offered to patients at the time general consent was obtained, which maximized patient recruitment and streamlined workflow. The study is a prospective observational research study to determine the impact of administration of COVID-19 monoclonal antibody therapy on length of symptoms, chronic illness, and rate of hospitalization. Most patients were eager to participate and offer their assistance to the scientific community during this pandemic.
Staff Education
In order to successfully implement this multidisciplinary EUA treatment option, comprehensive staff education was paramount after the workflow was developed. Prior to the first day of infusions, nurses and pharmacists were provided education during multiple huddle announcements. The pharmacy team also provided screen captures via email to the pharmacists so they could become familiar with the order set, intervention documentation, and location of the preliminary assessment of EUA criteria for use at the time of order verification. The emergency medicine department chair and chief medical officer also provided education via several virtual meetings and email to referring physicians (specialists and primary care) and residents in the emergency centers involved in COVID-19 monoclonal therapy-related patient care.
Factors Contributing to Success
We believe the reasons for continued success of this process are multifactorial and include the following key elements. Multidisciplinary planning, which included decision makers and all stakeholders, began at the time the idea was conceived. This allowed quick implementation of this service by efficiently navigating barriers to engaging impacted staff early on. Throughout this process, the authors set realistic step-wise goals. While navigating through the many details to implementation described, we also kept in mind the big picture, which was to provide this potentially lifesaving therapy to as many qualifying members of our community as possible. This included being flexible with the process and adapting when needed to achieve this ultimate goal. A focus on safety remained a priority to minimize possible errors and enhance patient and staff satisfaction. The optimization of the EHR streamlined workflow, provided point-of-care resources, and enhanced patient safety. Additionally, the target date set for implementation allowed staff and department leads adequate time to plan for and anticipate the changes. Serving only 1 patient on the first day allowed time for staff to experience this new process hands-on and provided opportunity for focused education. This team communication was essential to implementing this project, including staff training of processes and procedures prior to go-live. Early incorporation of IRB approval allowed the experience to be assessed and considered for contribution to the scientific literature to tackle this novel virus that has impacted our communities locally, nationally, and abroad. Moreover, continued measurement and reporting on a regular basis leads to performance improvement. The process outlined here can be adapted to incorporate other new therapies in the future, such as the recent February 9, 2021, EUA of the COVID-19 monoclonal antibody combination bamlanivimab and etesevimab.10
Conclusion
We administered 790 COVID-19 monoclonal antibody infusions between November 20, 2020 and March 5, 2021. Steps to minimize the likelihood of hypersensitivity reactions were implemented, and a low incidence (< 1%) has been observed. There has been no incidence of infection, concern from staff about infection prevention, or risk of infection during the processes. There have been very infrequent cost-related concerns raised by patients, typically due to incomplete communication regarding billing prior to the infusion. To address these issues, staff education has been provided to enhance patient instruction on this topic. The program has provided patient and family satisfaction, as well nursing, physician, pharmacist, clinical staff, and hospital administration pride and gratification. Setting up a new program to provide a 4-hour patient encounter to infuse therapy to high-risk patients with COVID-19 requires commitment and effort. This article describes the experience, ideas, and formula others may consider using to set up such a program. Through networking and formal phone calls and meetings about monoclonal antibody therapy, we have heard about other institutions who have not been able to institute this program due to various barriers to implementation. We hope our experience serves as a resource for others to provide this therapy to their patients and expand access in an effort to mitigate COVID-19 consequences and cases affecting our communities.
Corresponding author: Kathleen Jodoin, PharmD, BCPS, Mount Sinai Medical Center, 4300 Alton Rd, Miami Beach, FL 33140; [email protected].
Financial disclosures: None.
1. COVID Data Tracker. Center for Disease Control and Prevention. https://covid.cdc.gov/covid-data-tracker/#global-counts-rates. Accessed March 12, 2021.
2. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/143603/download
3. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19 | FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. Accessed February 14, 2021.
4. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. US Food and Drug Administration. Updated December 2020. Accessed March 9, 2021. https://www.fda.gov/media/143892/download
5. Chen P, Nirula A, Heller B, et al. SARS-CoV-2 Neutralizing antibody LY-CoV555 in outpatients with COVID-19. N Engl J Med. 2021;384(3):229-237. doi:10.1056/NEJMoa2029849
6. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. 10.1JAMA. 2021;325(7):632-644. doi:10.1001/jama.2021.0202
7. Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. 10.1N Engl J Med. 2021;384:238-251. doi:10.1056/nejmoa2035002
8. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. 10.1N Engl J Med. 2019;381:2293-2303. doi:10.1056/NEJMoa1910993
9. Boyle, P. Can an experimental treatment keep COVID-19 patients out of hospitals? Association of American Medical Colleges. January 29, 2021. Accessed March 9, 2021. https://www.aamc.org/news-insights/can-experimental-treatment-keep-covid-19-patients-out-hospitals
10. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/145802/download
From Mount Sinai Medical Center, Miami Beach, FL.
Abstract
Objective: To develop and implement a process for administering COVID-19 monoclonal antibody infusions for outpatients with mild or moderate COVID-19 at high risk for hospitalization, using multidisciplinary collaboration, US Food and Drug Administration (FDA) guidance, and infection prevention standards.
Methods: When monoclonal antibody therapy became available for mild or moderate COVID-19 outpatients via Emergency Use Authorization (EUA), our institution sought to provide this therapy option to our patients. We describe the process for planning, implementing, and maintaining a successful program for administering novel therapies based on FDA guidance and infection prevention standards. Key components of our implementation process were multidisciplinary planning involving decision makers and stakeholders; setting realistic goals in the process; team communication; and measuring and reporting quality improvement on a regular basis.
Results: A total of 790 COVID-19 monoclonal antibody infusions were administered from November 20, 2020 to March 5, 2021. Steps to minimize the likelihood of adverse drug reactions were implemented and a low incidence (< 1%) has occurred. There has been no concern from staff regarding infection during the process. Rarely, patients have raised cost-related concerns, typically due to incomplete communication regarding billing prior to the infusion. Patients, families, nursing staff, physicians, pharmacy, and hospital administration have expressed satisfaction with the program.
Conclusion: This process can provide a template for other hospitals or health care delivery facilities to provide novel therapies to patients with mild or moderate COVID-19 in a safe and effective manner.
Keywords: COVID-19; monoclonal antibody; infusion; emergency use authorization.
SARS-CoV-2 and the disease it causes, COVID-19, have transformed from scientific vernacular to common household terms. It began with a cluster of pneumonia cases of unknown etiology in December 2019 in Wuhan, China, with physicians there reporting a novel coronavirus strain (2019-nCoV), now referred to as SARS-CoV-2. Rapid spread of this virus resulted in the World Health Organization (WHO) declaring an international public health emergency. Since this time, the virus has evolved into a worldwide pandemic. COVID-19 has dramatically impacted our society, resulting in more than 2.63 million global deaths as of this writing, of which more than 527,000 deaths have occurred in the United States.1 This novel virus has resulted in a flurry of literature, research, therapies, and collaboration across multiple disciplines in an effort to prevent, treat, and mitigate cases and complications of this disease.
On November 9, 2020, and November 21, 2020, the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for 2 novel COVID-19 monoclonal therapies, bamlanivimab2-3 and casirivimab/imdevimab,3-4 respectively. The EUAs granted permission for these therapies to be administered for the treatment of mild to moderate COVID-19 in adult and pediatric patients (≥ 12 years and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The therapies work by targeting the SARS-CoV-2 spike protein and subsequent attachment to human angiotensin-converting enzyme 2 receptors. Clinical trial data leading to the EUA demonstrated a reduction in viral load, safe outcome, and most importantly, fewer hospitalization and emergency room visits, as compared to the placebo group.5-7 The use of monoclonal antibodies is not new and gained recognition during the Ebola crisis, when the monoclonal antibody to the Ebola virus showed a significant survival benefit.8 Providing monoclonal antibody therapy soon after symptom onset aligns with a shift from the onset of the pandemic to the current focus on the administration of pharmaceutical therapy early in the disease course. This shift prevents progression to severe COVID-19, with the goal of reducing patient mortality, hospitalizations, and strain on health care systems.
The availability of novel neutralizing monoclonal antibodies for COVID-19 led to discussions of how to incorporate these therapies as new options for patients. Our institution networked with colleagues from multiple disciplines to discuss processes and policies for the safe administration of the monoclonal antibody infusion therapies. Federal health leaders urge more use of monoclonal antibodies, but many hospitals have been unable to successfully implement infusions due to staff and logistical challenges.9 This article presents a viable process that hospitals can use to provide these novel therapies to outpatients with mild to moderate COVID-19.
The Mount Sinai Medical Center, Florida Experience
Mount Sinai Medical Center in Miami Beach, Florida, is the largest private, independent, not-for-profit teaching hospital in South Florida, comprising 672 licensed beds and supporting 150,000 emergency department (ED) visits annually. Per the EUA criteria for use, COVID-19 monoclonal antibody therapies are not authorized for patients who are hospitalized or who require oxygen therapy due to COVID-19. Therefore, options for outpatient administration needed to be evaluated. Directly following the first EUA press release, a task force of key stakeholders was assembled to brainstorm and develop a process to offer this therapy to the community. A multidisciplinary task force with representation from the ED, nursing, primary care, hospital medicine, pharmacy, risk management, billing, information technology, infection prevention, and senior level leadership participated (Table).
The task force reviewed institutional outpatient locations to determine whether offering this service would be feasible (eg, ED, ambulatory care facilities, cancer center). The ED was selected because it would offer the largest array of appointment times to meet the community needs with around-the-clock availability. While Mount Sinai Medical Center offers care in 3 emergency center locations in Aventura, Hialeah, and Miami Beach, it was determined to initiate the infusions at the main campus center in Miami Beach only. The main campus affords an onsite pharmacy with suitable staffing to prepare the anticipated volume of infusions in a timely manner, as both therapies have short stabilities following preparation. Thus, it was decided that patients from freestanding emergency centers in Aventura and Hialeah would be moved to the Miami Beach ED location to receive therapy. Operating at a single site also allowed for more rapid implementation, monitoring, and ability to make modifications more easily. Discussions for the possible expansion of COVID-19 monoclonal antibody infusions at satellite locations are underway.
On November 20, 2020, 11 days after the formation of the multidisciplinary task force, the first COVID-19 monoclonal infusion was successfully administered. Figure 1 depicts the timeline from assessment to program implementation. Critical to implementation was the involvement of decision makers from all necessary departments early in the planning process to ensure that standard operating procedures were followed and that the patients, community, and organization had a positive experience. This allowed for simultaneous planning of electronic health record (Epic; EHR) builds, departmental workflows, and staff education, as described in the following section. Figure 2 shows the patient safety activities included in the implementation process.
Key Stakeholder Involvement and Workflow
On the day of bamlanivimab EUA release, email communication was shared among hospital leadership with details of the press release. Departments were quickly involved to initiate a task force to assess if and how this therapy could be offered at Mount Sinai Medical Center. The following sections explain the role of each stakeholder and their essential role to operationalize these novel EUA treatment options. The task force was organized and led by our chief medical officer and chief nursing officer.
Information Technology
Medication Ordering and Documentation EHR and Smart Pumps. Early in the pandemic, the antimicrobial stewardship (ASP) clinical coordinator became the designated point person for pharmacy assessment of novel COVID-19 therapies. As such, this pharmacist began reviewing the bamlanivimab and, later, the casirivimab/imdevimab EUA Fact Sheet for Health Care Providers. All necessary elements for the complete and safe ordering and dispensing of the medication were developed and reviewed by pharmacy administration and ED nursing leadership for input, prior to submitting to the information technology team for implementation. Building the COVID-19 monoclonal medication records into the EHR allowed for detailed direction (ie, administration and preparation instructions) to be consistently applied. The medication records were also built into hospital smart pumps so that nurses could access prepopulated, accurate volumes and infusion rates to minimize errors.
Order Set Development. The pharmacy medication build was added to a comprehensive order set (Figure 3), which was then developed to guide prescribers and standardize the process around ordering of COVID-19 monoclonal therapies. While these therapies are new, oncology monoclonal therapies are regularly administered to outpatients at Mount Sinai Cancer Center. The cancer center was therefore consulted on their process surrounding best practices in administration of monoclonal antibody therapies. This included protocols for medications used in pretreatment and management of hypersensitivity reactions and potential adverse drug reactions of both COVID-19 monoclonal therapies. These medication orders were selected by default in the order set to ensure that all patients received premedications aimed at minimizing the risk of hypersensitivity reaction, and had as-needed medication orders, in the event a hypersensitivity reaction occurred. Reducing hypersensitivity reaction risk is important as well to increase the likelihood that the patient would receive full therapy, as management of this adverse drug reactions involves possible cessation of therapy depending on the level of severity. The pharmacy department also ensured these medications were stocked in ED automated dispensing cabinets to promote quick access. In addition to the aforementioned nursing orders, we added EUA criteria for use and hyperlinks to the Fact Sheets for Patients and Caregivers and Health Care Providers for each monoclonal therapy, and restricted ordering to ED physicians, nurse practitioners, and physician assistants.
The order set underwent multidisciplinary review by pharmacy administration, the chair of emergency medicine, physicians, and ED nursing leadership prior to presentation and approval by the Pharmacy and Therapeutics Committee. Lastly, at time of implementation, the order set was added to the ED preference list, preventing inpatient access. Additionally, as a patient safety action, free- standing orders of COVID-19 monoclonal therapies were disabled, so providers could only order therapies via the approved, comprehensive order set.
Preliminary Assessment Tool. A provider assessment tool was developed to document patient-specific EUA criteria for use during initial assessment (Figure 4). This tool serves as a checklist and is visible to the full multidisciplinary team in the patient’s EHR. It is used as a resource at the time of pharmacist verification and ED physician assessment to ensure criteria for use are met.
Outpatient Offices
Patient Referral. Patients with symptoms or concerns of COVID-19 exposure can make physician appointments via telemedicine or in person at Mount Sinai Medical Center’s primary care and specialty offices. At the time of patient encounter, physicians suspecting a COVID-19 diagnosis will refer patients for outpatient COVID-19 polymerase chain reaction (PCR) laboratory testing, which has an approximate 24-hour turnaround to results. Physicians also assess whether the patient meets EUA criteria for use, pending results of testing. In the event a patient meets EUA criteria for use, the physician provides patient counseling and requests verbal consent. Following this, the physician enters a note in the EHR describing the patient’s condition, criteria for use evaluation, and the patient’s verbal agreement to therapy. This preliminary screening is beneficial to begin planning with both the patient and ED to minimize delays. Patients are notified of the results of their test once available. If the COVID-19 PCR test returns positive, the physician will call the ED at the main campus and schedule the patient for COVID-19 monoclonal therapy. As the desired timeframe for administering COVID-19 monoclonal therapies is within less than 10 days of symptom onset, timely scheduling of appointments is crucial. Infusion appointments are typically provided the same or next day. The patients are informed that they must bring documentation of their positive COVID-19 PCR test to their ED visit. Lastly, because patients are pretreated with medication that may potentially impair driving, they are instructed that they cannot drive themselves home; ride shares also are not allowed in order to limit the spread of infection.
Emergency Department
Patient Arrival and Screening. A COVID-19 patient can be evaluated in the ED 1 of 2 ways. The first option is via outpatient office referral, as described previously. Upon arrival to the ED, a second screening is performed to ensure the patient still meets EUA criteria for use and the positive COVID-19 PCR test result is confirmed. If the patient no longer meets criteria, the patient is triaged accordingly, including evaluation for higher-level care (eg, supplemental oxygen, hospital admission). The second optoion is via new patient walk-ins without outpatient physician referral (Figure 4). In these cases, an initial screening is performed, documenting EUA criteria for use in the preliminary assessment (Figure 5). Physicians will consider an outside COVID-19 test as valid, so long as documentation is readily available confirming a positive PCR result. Otherwise, an in-house COVID-19 PCR test will be performed, which has a 2-hour turnaround time.
Infusion Schedule. The ED offers a total of 16 COVID-19 monoclonal infusions slots daily. These are broken up into 4 infusion time blocks (eg, 8
Patient Education. Prior to administration of the monoclonal therapy, physician and nursing staff obtain a formal, written patient consent for therapy and provide patients with the option of participating in the institutional review board (IRB) approved study. Details of this are discussed in the risk management and IRB sections of the article. Nursing staff also provides the medication-specific Fact Sheet for Patients and Caregivers in either Spanish or English, which is also included as a hyperlink on the COVID-19 Monoclonal Antibody Order Set for ease of access. Interpreter services are available for patients who speak other languages. An ED decentralized pharmacist is also available onsite Monday through Friday from 12
Infusion Ordering. Once the patient is ready to begin therapy, the he/she is brought to a dedicated overflow area of the ED. There are few, if any, patients in this location, and it is adjacent to the main emergency center for easy access by the patients, nurses, pharmacists, and physicians. The physician then enters orders in the EHR using the COVID-19 Monoclonal Antibody Order Set (Figure 3). Three discrete questions were built into the medication order: (1) Was patient consent obtained? (2) Was the Fact Sheet for Patient/Caregiver provided to the patient? (3) Is the patient COVID-19 PCR-positive? These questions were built as hard stops so that the medication orders cannot be placed without a response. This serves as another double-check to ensure processes are followed and helps facilitate timely verification by the pharmacist.
Medication Administration. One nurse is dedicated to administering the monoclonal therapies scheduled at 8
Pharmacy Department
Medication Receipt Process. Inventory is currently allocated biweekly from the state department of health and will soon be transitioning to a direct order system. The pharmacy technician in charge of deliveries notifies the pharmacy Antimicrobial Stewardship Program (ASP) clinical coordinator upon receipt of the monoclonal therapies. Bamlanivimab is supplied as 1 vial per dose, whereas casirivimab/imdevimab is supplied as 4 vials or 8 vials per dose, depending how it is shipped. To reduce the likelihood of medication errors, the ASP clinical coordinator assembles each of the casirivimab/imdevimab vials into kits, where 1 kit equals 1 dose. Labels are then affixed to each kit indicating the medication name, number of vials which equal a full dose, and pharmacist signature. The kits are stored in a dedicated refrigerator, and inventory logs are affixed to the outside of the refrigerator and updated daily. This inventory is also communicated daily to ED physician, nursing, and pharmacy leadership, as well as the director of patient safety, who reports weekly usage to the state Department of Health and Human Services. These weekly reports are used to determine allocation amounts.
Medication Verification and Delivery. The Mount Sinai Medical Center pharmacist staffing model consists of centralized order entry and specialized, decentralized positions. All orders are verified by the ED pharmacist when scheduled (not a 24/7 service) and by the designated pharmacist for all other times. At the time of medication verification, the pharmacist documents patient-specific EUA criteria for use and confirms that consent was obtained and the Fact Sheet for Patients/Caregivers was provided. A pharmacist intervention was developed to assist with this documentation. Pharmacists input smart text “.COVIDmonoclonal” and a drop-down menu of EUA criteria for use appears. The pharmacist reviews the patient care notes and medication order question responses to ascertain this information, contacting the ED prescriber if further clarification is required. This verification serves as another check to ensure processes put in place are followed. Lastly, intravenous preparation and delivery are electronically recorded in the EHR, and the medications require nursing signature at the time of delivery to ensure a formal chain of custody.
Risk Management
At Mount Sinai Medical Center, all EUA and investigational therapies require patient consent. Consistent with this requirement, a COVID-19 monoclonal specific consent was developed by risk management. This is provided to every patient receiving a COVID-19 monoclonal infusion, in addition to the FDA EUA Fact Sheet for Patients and Caregivers, and documented as part of their EHR. The questions providers must answer are built into the order set to ensure this process is followed and these patient safety checks are incorporated into the workflow.
Billing and Finance Department
In alignment with Mount Sinai Medical Center’s mission to provide high-quality health care to its diverse community through teaching, research, charity care, and financial responsibility, it was determined that this therapy would be provided to all patients regardless of insurance type, including those who are uninsured. The billing and finance department was consulted prior to this service being offered, to provide patients with accurate and pertinent information. The billing and finance department provided guidance on how to document patient encounters at time of registration to facilitate appropriate billing. At this time, the medication is free of charge, but nonmedication-related ED fees apply. This is explained to patients so there is a clear understanding prior to booking their appointment.
Infection Prevention
As patients receiving COVID-19 monoclonal therapies can transmit the virus to others, measures to ensure protection for other patients and staff are vital. To minimize exposure, specific nursing and physician staff from the ED are assigned to the treatment of these patients, and patients receive infusions and postobservation monitoring in a designated wing of the ED. Additionally, all staff who interact with these patients are required to don full personal protective equipment. This includes not only physicians and nurses but all specialties such as physician assistants, nurse practitioners, pharmacists, and laboratory technicians. Moreover, patients are not permitted to go home in a ride share and are counseled on Centers for Disease Control and Prevention quarantining following infusion.
Measurement of Process and Outcomes and Reporting
IRB approval was sought and obtained early during initiation of this service, allowing study consent to be offered to patients at the time general consent was obtained, which maximized patient recruitment and streamlined workflow. The study is a prospective observational research study to determine the impact of administration of COVID-19 monoclonal antibody therapy on length of symptoms, chronic illness, and rate of hospitalization. Most patients were eager to participate and offer their assistance to the scientific community during this pandemic.
Staff Education
In order to successfully implement this multidisciplinary EUA treatment option, comprehensive staff education was paramount after the workflow was developed. Prior to the first day of infusions, nurses and pharmacists were provided education during multiple huddle announcements. The pharmacy team also provided screen captures via email to the pharmacists so they could become familiar with the order set, intervention documentation, and location of the preliminary assessment of EUA criteria for use at the time of order verification. The emergency medicine department chair and chief medical officer also provided education via several virtual meetings and email to referring physicians (specialists and primary care) and residents in the emergency centers involved in COVID-19 monoclonal therapy-related patient care.
Factors Contributing to Success
We believe the reasons for continued success of this process are multifactorial and include the following key elements. Multidisciplinary planning, which included decision makers and all stakeholders, began at the time the idea was conceived. This allowed quick implementation of this service by efficiently navigating barriers to engaging impacted staff early on. Throughout this process, the authors set realistic step-wise goals. While navigating through the many details to implementation described, we also kept in mind the big picture, which was to provide this potentially lifesaving therapy to as many qualifying members of our community as possible. This included being flexible with the process and adapting when needed to achieve this ultimate goal. A focus on safety remained a priority to minimize possible errors and enhance patient and staff satisfaction. The optimization of the EHR streamlined workflow, provided point-of-care resources, and enhanced patient safety. Additionally, the target date set for implementation allowed staff and department leads adequate time to plan for and anticipate the changes. Serving only 1 patient on the first day allowed time for staff to experience this new process hands-on and provided opportunity for focused education. This team communication was essential to implementing this project, including staff training of processes and procedures prior to go-live. Early incorporation of IRB approval allowed the experience to be assessed and considered for contribution to the scientific literature to tackle this novel virus that has impacted our communities locally, nationally, and abroad. Moreover, continued measurement and reporting on a regular basis leads to performance improvement. The process outlined here can be adapted to incorporate other new therapies in the future, such as the recent February 9, 2021, EUA of the COVID-19 monoclonal antibody combination bamlanivimab and etesevimab.10
Conclusion
We administered 790 COVID-19 monoclonal antibody infusions between November 20, 2020 and March 5, 2021. Steps to minimize the likelihood of hypersensitivity reactions were implemented, and a low incidence (< 1%) has been observed. There has been no incidence of infection, concern from staff about infection prevention, or risk of infection during the processes. There have been very infrequent cost-related concerns raised by patients, typically due to incomplete communication regarding billing prior to the infusion. To address these issues, staff education has been provided to enhance patient instruction on this topic. The program has provided patient and family satisfaction, as well nursing, physician, pharmacist, clinical staff, and hospital administration pride and gratification. Setting up a new program to provide a 4-hour patient encounter to infuse therapy to high-risk patients with COVID-19 requires commitment and effort. This article describes the experience, ideas, and formula others may consider using to set up such a program. Through networking and formal phone calls and meetings about monoclonal antibody therapy, we have heard about other institutions who have not been able to institute this program due to various barriers to implementation. We hope our experience serves as a resource for others to provide this therapy to their patients and expand access in an effort to mitigate COVID-19 consequences and cases affecting our communities.
Corresponding author: Kathleen Jodoin, PharmD, BCPS, Mount Sinai Medical Center, 4300 Alton Rd, Miami Beach, FL 33140; [email protected].
Financial disclosures: None.
From Mount Sinai Medical Center, Miami Beach, FL.
Abstract
Objective: To develop and implement a process for administering COVID-19 monoclonal antibody infusions for outpatients with mild or moderate COVID-19 at high risk for hospitalization, using multidisciplinary collaboration, US Food and Drug Administration (FDA) guidance, and infection prevention standards.
Methods: When monoclonal antibody therapy became available for mild or moderate COVID-19 outpatients via Emergency Use Authorization (EUA), our institution sought to provide this therapy option to our patients. We describe the process for planning, implementing, and maintaining a successful program for administering novel therapies based on FDA guidance and infection prevention standards. Key components of our implementation process were multidisciplinary planning involving decision makers and stakeholders; setting realistic goals in the process; team communication; and measuring and reporting quality improvement on a regular basis.
Results: A total of 790 COVID-19 monoclonal antibody infusions were administered from November 20, 2020 to March 5, 2021. Steps to minimize the likelihood of adverse drug reactions were implemented and a low incidence (< 1%) has occurred. There has been no concern from staff regarding infection during the process. Rarely, patients have raised cost-related concerns, typically due to incomplete communication regarding billing prior to the infusion. Patients, families, nursing staff, physicians, pharmacy, and hospital administration have expressed satisfaction with the program.
Conclusion: This process can provide a template for other hospitals or health care delivery facilities to provide novel therapies to patients with mild or moderate COVID-19 in a safe and effective manner.
Keywords: COVID-19; monoclonal antibody; infusion; emergency use authorization.
SARS-CoV-2 and the disease it causes, COVID-19, have transformed from scientific vernacular to common household terms. It began with a cluster of pneumonia cases of unknown etiology in December 2019 in Wuhan, China, with physicians there reporting a novel coronavirus strain (2019-nCoV), now referred to as SARS-CoV-2. Rapid spread of this virus resulted in the World Health Organization (WHO) declaring an international public health emergency. Since this time, the virus has evolved into a worldwide pandemic. COVID-19 has dramatically impacted our society, resulting in more than 2.63 million global deaths as of this writing, of which more than 527,000 deaths have occurred in the United States.1 This novel virus has resulted in a flurry of literature, research, therapies, and collaboration across multiple disciplines in an effort to prevent, treat, and mitigate cases and complications of this disease.
On November 9, 2020, and November 21, 2020, the US Food and Drug Administration (FDA) issued Emergency Use Authorizations (EUA) for 2 novel COVID-19 monoclonal therapies, bamlanivimab2-3 and casirivimab/imdevimab,3-4 respectively. The EUAs granted permission for these therapies to be administered for the treatment of mild to moderate COVID-19 in adult and pediatric patients (≥ 12 years and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19 and/or hospitalization. The therapies work by targeting the SARS-CoV-2 spike protein and subsequent attachment to human angiotensin-converting enzyme 2 receptors. Clinical trial data leading to the EUA demonstrated a reduction in viral load, safe outcome, and most importantly, fewer hospitalization and emergency room visits, as compared to the placebo group.5-7 The use of monoclonal antibodies is not new and gained recognition during the Ebola crisis, when the monoclonal antibody to the Ebola virus showed a significant survival benefit.8 Providing monoclonal antibody therapy soon after symptom onset aligns with a shift from the onset of the pandemic to the current focus on the administration of pharmaceutical therapy early in the disease course. This shift prevents progression to severe COVID-19, with the goal of reducing patient mortality, hospitalizations, and strain on health care systems.
The availability of novel neutralizing monoclonal antibodies for COVID-19 led to discussions of how to incorporate these therapies as new options for patients. Our institution networked with colleagues from multiple disciplines to discuss processes and policies for the safe administration of the monoclonal antibody infusion therapies. Federal health leaders urge more use of monoclonal antibodies, but many hospitals have been unable to successfully implement infusions due to staff and logistical challenges.9 This article presents a viable process that hospitals can use to provide these novel therapies to outpatients with mild to moderate COVID-19.
The Mount Sinai Medical Center, Florida Experience
Mount Sinai Medical Center in Miami Beach, Florida, is the largest private, independent, not-for-profit teaching hospital in South Florida, comprising 672 licensed beds and supporting 150,000 emergency department (ED) visits annually. Per the EUA criteria for use, COVID-19 monoclonal antibody therapies are not authorized for patients who are hospitalized or who require oxygen therapy due to COVID-19. Therefore, options for outpatient administration needed to be evaluated. Directly following the first EUA press release, a task force of key stakeholders was assembled to brainstorm and develop a process to offer this therapy to the community. A multidisciplinary task force with representation from the ED, nursing, primary care, hospital medicine, pharmacy, risk management, billing, information technology, infection prevention, and senior level leadership participated (Table).
The task force reviewed institutional outpatient locations to determine whether offering this service would be feasible (eg, ED, ambulatory care facilities, cancer center). The ED was selected because it would offer the largest array of appointment times to meet the community needs with around-the-clock availability. While Mount Sinai Medical Center offers care in 3 emergency center locations in Aventura, Hialeah, and Miami Beach, it was determined to initiate the infusions at the main campus center in Miami Beach only. The main campus affords an onsite pharmacy with suitable staffing to prepare the anticipated volume of infusions in a timely manner, as both therapies have short stabilities following preparation. Thus, it was decided that patients from freestanding emergency centers in Aventura and Hialeah would be moved to the Miami Beach ED location to receive therapy. Operating at a single site also allowed for more rapid implementation, monitoring, and ability to make modifications more easily. Discussions for the possible expansion of COVID-19 monoclonal antibody infusions at satellite locations are underway.
On November 20, 2020, 11 days after the formation of the multidisciplinary task force, the first COVID-19 monoclonal infusion was successfully administered. Figure 1 depicts the timeline from assessment to program implementation. Critical to implementation was the involvement of decision makers from all necessary departments early in the planning process to ensure that standard operating procedures were followed and that the patients, community, and organization had a positive experience. This allowed for simultaneous planning of electronic health record (Epic; EHR) builds, departmental workflows, and staff education, as described in the following section. Figure 2 shows the patient safety activities included in the implementation process.
Key Stakeholder Involvement and Workflow
On the day of bamlanivimab EUA release, email communication was shared among hospital leadership with details of the press release. Departments were quickly involved to initiate a task force to assess if and how this therapy could be offered at Mount Sinai Medical Center. The following sections explain the role of each stakeholder and their essential role to operationalize these novel EUA treatment options. The task force was organized and led by our chief medical officer and chief nursing officer.
Information Technology
Medication Ordering and Documentation EHR and Smart Pumps. Early in the pandemic, the antimicrobial stewardship (ASP) clinical coordinator became the designated point person for pharmacy assessment of novel COVID-19 therapies. As such, this pharmacist began reviewing the bamlanivimab and, later, the casirivimab/imdevimab EUA Fact Sheet for Health Care Providers. All necessary elements for the complete and safe ordering and dispensing of the medication were developed and reviewed by pharmacy administration and ED nursing leadership for input, prior to submitting to the information technology team for implementation. Building the COVID-19 monoclonal medication records into the EHR allowed for detailed direction (ie, administration and preparation instructions) to be consistently applied. The medication records were also built into hospital smart pumps so that nurses could access prepopulated, accurate volumes and infusion rates to minimize errors.
Order Set Development. The pharmacy medication build was added to a comprehensive order set (Figure 3), which was then developed to guide prescribers and standardize the process around ordering of COVID-19 monoclonal therapies. While these therapies are new, oncology monoclonal therapies are regularly administered to outpatients at Mount Sinai Cancer Center. The cancer center was therefore consulted on their process surrounding best practices in administration of monoclonal antibody therapies. This included protocols for medications used in pretreatment and management of hypersensitivity reactions and potential adverse drug reactions of both COVID-19 monoclonal therapies. These medication orders were selected by default in the order set to ensure that all patients received premedications aimed at minimizing the risk of hypersensitivity reaction, and had as-needed medication orders, in the event a hypersensitivity reaction occurred. Reducing hypersensitivity reaction risk is important as well to increase the likelihood that the patient would receive full therapy, as management of this adverse drug reactions involves possible cessation of therapy depending on the level of severity. The pharmacy department also ensured these medications were stocked in ED automated dispensing cabinets to promote quick access. In addition to the aforementioned nursing orders, we added EUA criteria for use and hyperlinks to the Fact Sheets for Patients and Caregivers and Health Care Providers for each monoclonal therapy, and restricted ordering to ED physicians, nurse practitioners, and physician assistants.
The order set underwent multidisciplinary review by pharmacy administration, the chair of emergency medicine, physicians, and ED nursing leadership prior to presentation and approval by the Pharmacy and Therapeutics Committee. Lastly, at time of implementation, the order set was added to the ED preference list, preventing inpatient access. Additionally, as a patient safety action, free- standing orders of COVID-19 monoclonal therapies were disabled, so providers could only order therapies via the approved, comprehensive order set.
Preliminary Assessment Tool. A provider assessment tool was developed to document patient-specific EUA criteria for use during initial assessment (Figure 4). This tool serves as a checklist and is visible to the full multidisciplinary team in the patient’s EHR. It is used as a resource at the time of pharmacist verification and ED physician assessment to ensure criteria for use are met.
Outpatient Offices
Patient Referral. Patients with symptoms or concerns of COVID-19 exposure can make physician appointments via telemedicine or in person at Mount Sinai Medical Center’s primary care and specialty offices. At the time of patient encounter, physicians suspecting a COVID-19 diagnosis will refer patients for outpatient COVID-19 polymerase chain reaction (PCR) laboratory testing, which has an approximate 24-hour turnaround to results. Physicians also assess whether the patient meets EUA criteria for use, pending results of testing. In the event a patient meets EUA criteria for use, the physician provides patient counseling and requests verbal consent. Following this, the physician enters a note in the EHR describing the patient’s condition, criteria for use evaluation, and the patient’s verbal agreement to therapy. This preliminary screening is beneficial to begin planning with both the patient and ED to minimize delays. Patients are notified of the results of their test once available. If the COVID-19 PCR test returns positive, the physician will call the ED at the main campus and schedule the patient for COVID-19 monoclonal therapy. As the desired timeframe for administering COVID-19 monoclonal therapies is within less than 10 days of symptom onset, timely scheduling of appointments is crucial. Infusion appointments are typically provided the same or next day. The patients are informed that they must bring documentation of their positive COVID-19 PCR test to their ED visit. Lastly, because patients are pretreated with medication that may potentially impair driving, they are instructed that they cannot drive themselves home; ride shares also are not allowed in order to limit the spread of infection.
Emergency Department
Patient Arrival and Screening. A COVID-19 patient can be evaluated in the ED 1 of 2 ways. The first option is via outpatient office referral, as described previously. Upon arrival to the ED, a second screening is performed to ensure the patient still meets EUA criteria for use and the positive COVID-19 PCR test result is confirmed. If the patient no longer meets criteria, the patient is triaged accordingly, including evaluation for higher-level care (eg, supplemental oxygen, hospital admission). The second optoion is via new patient walk-ins without outpatient physician referral (Figure 4). In these cases, an initial screening is performed, documenting EUA criteria for use in the preliminary assessment (Figure 5). Physicians will consider an outside COVID-19 test as valid, so long as documentation is readily available confirming a positive PCR result. Otherwise, an in-house COVID-19 PCR test will be performed, which has a 2-hour turnaround time.
Infusion Schedule. The ED offers a total of 16 COVID-19 monoclonal infusions slots daily. These are broken up into 4 infusion time blocks (eg, 8
Patient Education. Prior to administration of the monoclonal therapy, physician and nursing staff obtain a formal, written patient consent for therapy and provide patients with the option of participating in the institutional review board (IRB) approved study. Details of this are discussed in the risk management and IRB sections of the article. Nursing staff also provides the medication-specific Fact Sheet for Patients and Caregivers in either Spanish or English, which is also included as a hyperlink on the COVID-19 Monoclonal Antibody Order Set for ease of access. Interpreter services are available for patients who speak other languages. An ED decentralized pharmacist is also available onsite Monday through Friday from 12
Infusion Ordering. Once the patient is ready to begin therapy, the he/she is brought to a dedicated overflow area of the ED. There are few, if any, patients in this location, and it is adjacent to the main emergency center for easy access by the patients, nurses, pharmacists, and physicians. The physician then enters orders in the EHR using the COVID-19 Monoclonal Antibody Order Set (Figure 3). Three discrete questions were built into the medication order: (1) Was patient consent obtained? (2) Was the Fact Sheet for Patient/Caregiver provided to the patient? (3) Is the patient COVID-19 PCR-positive? These questions were built as hard stops so that the medication orders cannot be placed without a response. This serves as another double-check to ensure processes are followed and helps facilitate timely verification by the pharmacist.
Medication Administration. One nurse is dedicated to administering the monoclonal therapies scheduled at 8
Pharmacy Department
Medication Receipt Process. Inventory is currently allocated biweekly from the state department of health and will soon be transitioning to a direct order system. The pharmacy technician in charge of deliveries notifies the pharmacy Antimicrobial Stewardship Program (ASP) clinical coordinator upon receipt of the monoclonal therapies. Bamlanivimab is supplied as 1 vial per dose, whereas casirivimab/imdevimab is supplied as 4 vials or 8 vials per dose, depending how it is shipped. To reduce the likelihood of medication errors, the ASP clinical coordinator assembles each of the casirivimab/imdevimab vials into kits, where 1 kit equals 1 dose. Labels are then affixed to each kit indicating the medication name, number of vials which equal a full dose, and pharmacist signature. The kits are stored in a dedicated refrigerator, and inventory logs are affixed to the outside of the refrigerator and updated daily. This inventory is also communicated daily to ED physician, nursing, and pharmacy leadership, as well as the director of patient safety, who reports weekly usage to the state Department of Health and Human Services. These weekly reports are used to determine allocation amounts.
Medication Verification and Delivery. The Mount Sinai Medical Center pharmacist staffing model consists of centralized order entry and specialized, decentralized positions. All orders are verified by the ED pharmacist when scheduled (not a 24/7 service) and by the designated pharmacist for all other times. At the time of medication verification, the pharmacist documents patient-specific EUA criteria for use and confirms that consent was obtained and the Fact Sheet for Patients/Caregivers was provided. A pharmacist intervention was developed to assist with this documentation. Pharmacists input smart text “.COVIDmonoclonal” and a drop-down menu of EUA criteria for use appears. The pharmacist reviews the patient care notes and medication order question responses to ascertain this information, contacting the ED prescriber if further clarification is required. This verification serves as another check to ensure processes put in place are followed. Lastly, intravenous preparation and delivery are electronically recorded in the EHR, and the medications require nursing signature at the time of delivery to ensure a formal chain of custody.
Risk Management
At Mount Sinai Medical Center, all EUA and investigational therapies require patient consent. Consistent with this requirement, a COVID-19 monoclonal specific consent was developed by risk management. This is provided to every patient receiving a COVID-19 monoclonal infusion, in addition to the FDA EUA Fact Sheet for Patients and Caregivers, and documented as part of their EHR. The questions providers must answer are built into the order set to ensure this process is followed and these patient safety checks are incorporated into the workflow.
Billing and Finance Department
In alignment with Mount Sinai Medical Center’s mission to provide high-quality health care to its diverse community through teaching, research, charity care, and financial responsibility, it was determined that this therapy would be provided to all patients regardless of insurance type, including those who are uninsured. The billing and finance department was consulted prior to this service being offered, to provide patients with accurate and pertinent information. The billing and finance department provided guidance on how to document patient encounters at time of registration to facilitate appropriate billing. At this time, the medication is free of charge, but nonmedication-related ED fees apply. This is explained to patients so there is a clear understanding prior to booking their appointment.
Infection Prevention
As patients receiving COVID-19 monoclonal therapies can transmit the virus to others, measures to ensure protection for other patients and staff are vital. To minimize exposure, specific nursing and physician staff from the ED are assigned to the treatment of these patients, and patients receive infusions and postobservation monitoring in a designated wing of the ED. Additionally, all staff who interact with these patients are required to don full personal protective equipment. This includes not only physicians and nurses but all specialties such as physician assistants, nurse practitioners, pharmacists, and laboratory technicians. Moreover, patients are not permitted to go home in a ride share and are counseled on Centers for Disease Control and Prevention quarantining following infusion.
Measurement of Process and Outcomes and Reporting
IRB approval was sought and obtained early during initiation of this service, allowing study consent to be offered to patients at the time general consent was obtained, which maximized patient recruitment and streamlined workflow. The study is a prospective observational research study to determine the impact of administration of COVID-19 monoclonal antibody therapy on length of symptoms, chronic illness, and rate of hospitalization. Most patients were eager to participate and offer their assistance to the scientific community during this pandemic.
Staff Education
In order to successfully implement this multidisciplinary EUA treatment option, comprehensive staff education was paramount after the workflow was developed. Prior to the first day of infusions, nurses and pharmacists were provided education during multiple huddle announcements. The pharmacy team also provided screen captures via email to the pharmacists so they could become familiar with the order set, intervention documentation, and location of the preliminary assessment of EUA criteria for use at the time of order verification. The emergency medicine department chair and chief medical officer also provided education via several virtual meetings and email to referring physicians (specialists and primary care) and residents in the emergency centers involved in COVID-19 monoclonal therapy-related patient care.
Factors Contributing to Success
We believe the reasons for continued success of this process are multifactorial and include the following key elements. Multidisciplinary planning, which included decision makers and all stakeholders, began at the time the idea was conceived. This allowed quick implementation of this service by efficiently navigating barriers to engaging impacted staff early on. Throughout this process, the authors set realistic step-wise goals. While navigating through the many details to implementation described, we also kept in mind the big picture, which was to provide this potentially lifesaving therapy to as many qualifying members of our community as possible. This included being flexible with the process and adapting when needed to achieve this ultimate goal. A focus on safety remained a priority to minimize possible errors and enhance patient and staff satisfaction. The optimization of the EHR streamlined workflow, provided point-of-care resources, and enhanced patient safety. Additionally, the target date set for implementation allowed staff and department leads adequate time to plan for and anticipate the changes. Serving only 1 patient on the first day allowed time for staff to experience this new process hands-on and provided opportunity for focused education. This team communication was essential to implementing this project, including staff training of processes and procedures prior to go-live. Early incorporation of IRB approval allowed the experience to be assessed and considered for contribution to the scientific literature to tackle this novel virus that has impacted our communities locally, nationally, and abroad. Moreover, continued measurement and reporting on a regular basis leads to performance improvement. The process outlined here can be adapted to incorporate other new therapies in the future, such as the recent February 9, 2021, EUA of the COVID-19 monoclonal antibody combination bamlanivimab and etesevimab.10
Conclusion
We administered 790 COVID-19 monoclonal antibody infusions between November 20, 2020 and March 5, 2021. Steps to minimize the likelihood of hypersensitivity reactions were implemented, and a low incidence (< 1%) has been observed. There has been no incidence of infection, concern from staff about infection prevention, or risk of infection during the processes. There have been very infrequent cost-related concerns raised by patients, typically due to incomplete communication regarding billing prior to the infusion. To address these issues, staff education has been provided to enhance patient instruction on this topic. The program has provided patient and family satisfaction, as well nursing, physician, pharmacist, clinical staff, and hospital administration pride and gratification. Setting up a new program to provide a 4-hour patient encounter to infuse therapy to high-risk patients with COVID-19 requires commitment and effort. This article describes the experience, ideas, and formula others may consider using to set up such a program. Through networking and formal phone calls and meetings about monoclonal antibody therapy, we have heard about other institutions who have not been able to institute this program due to various barriers to implementation. We hope our experience serves as a resource for others to provide this therapy to their patients and expand access in an effort to mitigate COVID-19 consequences and cases affecting our communities.
Corresponding author: Kathleen Jodoin, PharmD, BCPS, Mount Sinai Medical Center, 4300 Alton Rd, Miami Beach, FL 33140; [email protected].
Financial disclosures: None.
1. COVID Data Tracker. Center for Disease Control and Prevention. https://covid.cdc.gov/covid-data-tracker/#global-counts-rates. Accessed March 12, 2021.
2. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/143603/download
3. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19 | FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. Accessed February 14, 2021.
4. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. US Food and Drug Administration. Updated December 2020. Accessed March 9, 2021. https://www.fda.gov/media/143892/download
5. Chen P, Nirula A, Heller B, et al. SARS-CoV-2 Neutralizing antibody LY-CoV555 in outpatients with COVID-19. N Engl J Med. 2021;384(3):229-237. doi:10.1056/NEJMoa2029849
6. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. 10.1JAMA. 2021;325(7):632-644. doi:10.1001/jama.2021.0202
7. Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. 10.1N Engl J Med. 2021;384:238-251. doi:10.1056/nejmoa2035002
8. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. 10.1N Engl J Med. 2019;381:2293-2303. doi:10.1056/NEJMoa1910993
9. Boyle, P. Can an experimental treatment keep COVID-19 patients out of hospitals? Association of American Medical Colleges. January 29, 2021. Accessed March 9, 2021. https://www.aamc.org/news-insights/can-experimental-treatment-keep-covid-19-patients-out-hospitals
10. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/145802/download
1. COVID Data Tracker. Center for Disease Control and Prevention. https://covid.cdc.gov/covid-data-tracker/#global-counts-rates. Accessed March 12, 2021.
2. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/143603/download
3. Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19 | FDA. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19. Accessed February 14, 2021.
4. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Casirivimab and Imdevimab. US Food and Drug Administration. Updated December 2020. Accessed March 9, 2021. https://www.fda.gov/media/143892/download
5. Chen P, Nirula A, Heller B, et al. SARS-CoV-2 Neutralizing antibody LY-CoV555 in outpatients with COVID-19. N Engl J Med. 2021;384(3):229-237. doi:10.1056/NEJMoa2029849
6. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. 10.1JAMA. 2021;325(7):632-644. doi:10.1001/jama.2021.0202
7. Weinreich DM, Sivapalasingam S, Norton T, et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. 10.1N Engl J Med. 2021;384:238-251. doi:10.1056/nejmoa2035002
8. Mulangu S, Dodd LE, Davey RT Jr, et al. A randomized, controlled trial of Ebola virus disease therapeutics. 10.1N Engl J Med. 2019;381:2293-2303. doi:10.1056/NEJMoa1910993
9. Boyle, P. Can an experimental treatment keep COVID-19 patients out of hospitals? Association of American Medical Colleges. January 29, 2021. Accessed March 9, 2021. https://www.aamc.org/news-insights/can-experimental-treatment-keep-covid-19-patients-out-hospitals
10. Fact Sheet for Health Care Providers Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab. US Food and Drug Administration. Updated February 2021. Accessed March 9, 2021. https://www.fda.gov/media/145802/download

Implementing the AMI READMITS Risk Assessment Score to Increase Referrals Among Patients With Type I Myocardial Infarction
From The Johns Hopkins Hospital, Baltimore, MD (Dr. Muganlinskaya and Dr. Skojec, retired); The George Washington University, Washington, DC (Dr. Posey); and Johns Hopkins University, Baltimore, MD (Dr. Resar).
Abstract
Objective: Assessing the risk characteristics of patients with acute myocardial infarction (MI) can help providers make appropriate referral decisions. This quality improvement project sought to improve timely, appropriate referrals among patients with type I MI by adding a risk assessment, the AMI READMITS score, to the existing referral protocol.
Methods: Patients’ chart data were analyzed to assess changes in referrals and timely follow-up appointments from pre-intervention to intervention. A survey assessed providers’ satisfaction with the new referral protocol.
Results: Among 57 patients (n = 29 preintervention; n = 28 intervention), documented referrals increased significantly from 66% to 89% (χ2 = 4.571, df = 1, P = 0.033); and timely appointments increased by 10%, which was not significant (χ2 = 3.550, df = 2, P = 0.169). Most providers agreed that the new protocol was easy to use, useful in making referral decisions, and improved the referral process. All agreed the risk score should be incorporated into electronic clinical notes. Provider opinions related to implementing the risk score in clinical practice were mixed. Qualitative feedback suggests this was due to limited validation of the AMI READMITS score in reducing readmissions.
Conclusions: Our risk-based referral protocol helped to increase appropriate referrals among patients with type I MI. Provider adoption may be enhanced by incorporating the protocol into electronic clinical notes. Research to further validate the accuracy of the AMI READMITS score in predicting readmissions may support adoption of the protocol in clinical practice.
Keywords: quality improvement; type I myocardial infarction; referral process; readmission risk; risk assessment; chart review.
Early follow-up after discharge is an important strategy to reduce the risk of unplanned hospital readmissions among patients with various conditions.1-3 While patient confounding factors, such as chronic health problems, environment, socioeconomic status, and literacy, make it difficult to avoid all unplanned readmissions, early follow-up may help providers identify and appropriately manage some health-related issues, and as such is a pivotal element of a readmission prevention strategy.4 There is evidence that patients with non-ST elevation myocardial infarction (NSTEMI) who have an outpatient appointment with a physician within 7 days after discharge have a lower risk of 30-day readmission.5
Our hospital’s postmyocardial infarction clinic was created to prevent unplanned readmissions within 30 days after discharge among patients with type I myocardial infarction (MI). Since inception, the number of referrals has been much lower than expected. In 2018, the total number of patients discharged from the hospital with type I MI and any troponin I level above 0.40 ng/mL was 313. Most of these patients were discharged from the hospital’s cardiac units; however, only 91 referrals were made. To increase referrals, the cardiology nurse practitioners (NPs) developed a post-MI referral protocol (Figure 1). However, this protocol was not consistently used and referrals to the clinic remained low.

Evidence-based risk assessment tools have the potential to increase effective patient management. For example, cardiology providers at the hospital utilize various scores, such as CHA2DS2-VASc6 and the Society of Thoracic Surgery risk score,7 to plan patient management. Among the scores used to predict unplanned readmissions for MI patients, the most promising is the AMI READMITS score.8 Unlike other nonspecific prediction models, the AMI READMITS score was developed based on variables extracted from the electronic health records (EHRs) of patients who were hospitalized for MI and readmitted within 30 days after discharge. Recognizing the potential to increase referrals by integrating an MI-specific risk assessment, this quality improvement study modified the existing referral protocol to include the patients’ AMI READMITS score and recommendations for follow-up.
Currently, there are no clear recommendations on how soon after discharge patients with MI should undergo follow-up. As research data vary, we selected 7 days follow-up for patients from high risk groups based on the “See you in 7” initiative for patients with heart failure (HF) and MI,9,10 as well as evidence that patients with NSTEMI have a lower risk of 30-day readmission if they have follow-up within 7 days after discharge5; and we selected 14 days follow-up for patients from low-risk groups based on evidence that postdischarge follow-up within 14 days reduces risk of 30-day readmission in patients with acute myocardial infarction (AMI) and/or acutely decompensated HF.11
Methods
This project was designed to answer the following question: For adult patients with type I MI, does implementation of a readmission risk assessment referral protocol increase the percentage of referrals and appointments scheduled within a recommended time? Anticipated outcomes included: (1) increased referrals to a cardiologist or the post-MI clinic; (2) increased scheduled follow-up appointments within 7 to 14 days; (3) provider satisfaction with the usability and usefulness of the new protocol; and (4) consistent provider adoption of the new risk assessment referral protocol.
To evaluate the degree to which these outcomes were achieved, we reviewed patient charts for 2 months prior and 2 months during implementation of the new referral protocol. As shown in Figure 2, the new protocol added the following process steps to the existing protocol: calculation of the AMI READMITS score, recommendations for follow-up based on patients’ risk score, and guidance to refer patients to the post-MI clinic if patients did not have an appointment with a cardiologist within 7 to 14 days after discharge. Patients’ risk assessment scores were obtained from forms completed by clinicians during the intervention. Clinician’s perceptions related to the usability and usefulness of the new protocol and feedback related to its long-term adoption were assessed using a descriptive survey.

The institutional review board classified this project as a quality improvement project. To avoid potential loss of patient privacy, no identifiable data were collected, a unique identifier unrelated to patients’ records was generated for each patient, and data were saved on a password-protected cardiology office computer.
Population
The project population included all adult patients (≥ 18 years old) with type I MI who were admitted or transferred to the hospital, had a percutaneous coronary intervention (PCI), or were managed without PCI and discharged from the hospital’s cardiac care unit (CCU) and progressive cardiac care unit (PCCU). The criteria for type I MI included the “detection of a rise and/or fall of cardiac troponin with at least 1 value above the 99th percentile and with at least 1 of the following: symptoms of acute myocardial ischemia; new ischemic electrocardiographic (ECG) changes; development of new pathological Q waves; imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology; identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy.”12 The study excluded patients with type I MI who were referred for coronary bypass surgery.
Intervention
The revised risk assessment protocol was implemented within the CCU and PCCU. The lead investigator met with each provider to discuss the role of the post-MI clinic, current referral rates, the purpose of the project, and the new referral process to be completed during the project for each patient discharged with type I MI. Cardiology NPs, fellows, and residents were asked to use the risk-assessment form to calculate patients’ risk for readmission, and refer patients to the post-MI clinic if an appointment with a cardiologist was not available within 7 to 14 days after discharge. Every week during the intervention phase, the investigator sent reminder emails to ensure form completion. Providers were asked to calculate and write the score, the discharge and referral dates, where referrals were made (a cardiologist or the post-MI clinic), date of appointment, and reason for not scheduling an appointment or not referring on the risk assessment form, and to drop the completed forms in specific labeled boxes located at the CCU and PCCU work stations. The investigator collected the completed forms weekly. When the number of discharged patients did not match the number of completed forms, the investigator followed up with discharging providers to understand why.
Data and Data Collection
Data to determine whether the use of the new protocol increased discharge referrals among patients with type I MI within the recommended timeframes were collected by electronic chart review. Data included discharging unit, patients’ age, gender, admission and discharge date, diagnosis, referral to a cardiologist and the post-MI clinic, and appointment date. Clinical data needed to calculate the AMI READMITS score was also collected: PCI within 24 hours, serum creatinine, systolic blood pressure (SBP), brain natriuretic peptide (BNP), and diabetes status.
Data to assess provider satisfaction with the usability and usefulness of the new protocol were gathered through an online survey. The survey included 1 question related to the providers’ role, 1 question asking whether they used the risk assessment for each patient, and 5 Likert-items assessing the ease of usage. An additional open-ended question asked providers to share feedback related to integrating the AMI READMITS risk assessment score to the post-MI referral protocol long term.
To evaluate how consistently providers utilized the new referral protocol when discharging patients with type I MI, the number of completed forms was compared with the number of those patients who were discharged.
Statistical Analysis
Descriptive statistics were used to summarize patient demographics and to calculate the frequency of referrals before and during the intervention. Chi-square statistics were calculated to determine whether the change in percentage of referrals and timely referrals was significant. Descriptive statistics were used to determine the level of provider satisfaction related to each survey item. A content analysis method was used to synthesize themes from the open-ended question asking clinicians to share their feedback related to the new protocol.
Results
Fifty-seven patients met the study inclusion criteria: 29 patients during the preintervention phase and 28 patients during the intervention phase. There were 35 male (61.4%) and 22 female (38.6%) patients. Twenty-five patients (43.9%) were from age groups 41 through 60 years and 61 through 80 years, respectively, representing the majority of included patients. Seven patients (12.3%) were from the 81 years and older age group. There were no patients in the age group 18 through 40 years. Based on the AMI READMITS score calculation, 57.9% (n = 33) patients were from a low-risk group (includes extremely low and low risk for readmission) and 42.1% (n = 24) were from a high-risk group (includes moderate, high, and extremely high risk for readmission).
Provider adoption of the new protocol during the intervention was high. Referral forms were completed for 82% (n = 23) of the 28 patients during the intervention. Analysis findings showed a statistically significant increase in documented referrals after implementing the new referral protocol. During the preintervention phase, 66% (n = 19) of patients with type I MI were referred to see a cardiologist or an NP at a post-MI clinic and there was no documented referral for 34% (n = 10) of patients. During the intervention phase, 89% (n = 25) of patients were referred and there was no documented referral for 11% (n = 3) of patients. Chi-square results indicated that the increase in referrals was significant (χ2 = 4.571, df = 1, P = 0.033).
Data analysis examined whether patient referrals fell within the recommended timeframe of 7 days for the high-risk group (included moderate-to-extremely high risk) and 14 days for the low-risk group (included low-to-extremely low risk). During the preintervention phase, 31% (n = 9) of patient referrals were scheduled as recommended; 28% (n = 8) of patient referrals were scheduled but delayed; and there was no referral date documented for 41% (n = 12) of patients. During the intervention phase, referrals scheduled as recommended increased to 53% (n = 15); 25% (n = 7) of referrals were scheduled but delayed; and there was no referral date documented for 21.4% (n = 6) of patients. The change in appointments scheduled as recommended was not significant (χ2 = 3.550, df = 2, P = 0.169).
Surveys were emailed to 25 cardiology fellows and 3 cardiology NPs who participated in this study. Eighteen of the 28 clinicians (15 cardiology fellows and 3 cardiology NPs) responded for a response rate of 64%. One of several residents who rotated through the CCU and PCCU during the intervention also completed the survey, for a total of 19 participants. When asked if the protocol was easy to use, 79% agreed or strongly agreed. Eighteen of the 19 participants (95%) agreed or strongly agreed that the protocol was useful in making referral decisions. Sixty-eight percent agreed or strongly agreed that the AMI READMITS risk assessment score improves referral process. All participants agreed or strongly agreed that there should be an option to incorporate the AMI READMITS risk assessment score into electronic clinical notes. When asked whether the AMI READMITS risk score should be implemented in clinical practice, responses were mixed (Figure 3). A common theme among the 4 participants who responded with comments was the need for additional data to validate the usefulness of the AMI READMITS to reduce readmissions. In addition, 1 participant commented that “manual calculation [of the risk score] is not ideal.”

Discussion
This project demonstrated that implementing an evidence-based referral protocol integrating the AMI-READMITS score can increase timely postdischarge referrals among patients with type I MI. The percentage of appropriately scheduled appointments increased during the intervention phase; however, a relatively high number of appointments were scheduled outside of the recommended timeframe, similar to preintervention. Thus, while the new protocol increased referrals and provider documentation of these referrals, it appears that challenges in scheduling timely referral appointments remained. This project did not examine the reasons for delayed appointments.
The survey findings indicated that providers were generally satisfied with the usability and usefulness of the new risk assessment protocol. A large majority agreed or strongly agreed that it was easy to use and useful in making referral decisions, and most agreed or strongly agreed that it improves the referral process. Mixed opinions regarding implementing the AMI READMITS score in clinical practice, combined with qualitative findings, suggest that a lack of external validation of the AMI READMITS presents a barrier to its long-term adoption. All providers who participated in the survey agreed or strongly agreed that the risk assessment should be incorporated into electronic clinical notes. We have begun the process of working with the EHR vendor to automate the AMI risk-assessment within the referral work-flow, which will provide an opportunity for a follow-up quality improvement study.
This quality improvement project has several limitations. First, it implemented a small change in 2 inpatient units at 1 hospital using a simple pre- posttest design. Therefore, the findings are not generalizable to other settings. Prior to the intervention, some referrals may have been made without documentation. While the authors were able to trace undocumented referrals for patients who were referred to the post-MI clinic or to a cardiologist affiliated with the hospital, some patients may have been referred to cardiologists who were not affiliated with the hospital. Another limitation was that the self-created provider survey used was not tested in other clinical settings; thus, it cannot be determined whether the sensitivity and specificity of the survey questions are high. In addition, the clinical providers who participated in the study knew the study team, which may have influenced their behavior during the study period. Furthermore, the identified improvement in clinicians’ referral practices may not be sustainable due to the complexity and effort required to manually calculate the risk score. This limitation could be eliminated by integrating the risk score calculation into the EHR.
Conclusion
Early follow-up after discharge plays an important role in supporting patients’ self-management of some risk factors (ie, diet, weight, and smoking) and identifying gaps in postdischarge care which may lead to readmission. This project provides evidence that integrating the AMI READMITS risk assessment score into the referral process can help to guide discharge decision-making and increase timely, appropriate referrals for patients with MI. Integration of a specific risk assessment, such as the AMI READMITS, within the post-MI referral protocol may help clinicians make more efficient, educated referral decisions. Future studies should explore more specifically how and why the new protocol impacts clinicians’ decision-making and behavior related to post-MI referrals. In addition, future studies should investigate challenges associated with scheduling postdischarge appointments. It will be important to investigate how integration of the new protocol within the EHR may increase efficiency, consistency, and provider satisfaction with the new referral process. Additional research investigating the effects of the AMI READMITS score on readmissions reduction will be important to promote long-term adoption of the improved referral protocol in clinical practice.
Acknowledgments: The authors thank Shelly Conaway, ANP-BC, MSN, Angela Street, ANP-BC, MSN, Andrew Geis, ACNP-BC, MSN, Richard P. Jones II, MD, Eunice Young, MD, Joy Rothwell, MSN, RN-BC, Allison Olazo, MBA, MSN, RN-BC, Elizabeth Heck, RN-BC, and Matthew Trojanowski, MHA, MS, RRT, CSSBB for their support of this study.
Corresponding author: Nailya Muganlinskaya, DNP, MPH, ACNP-BC, MSN, The Johns Hopkins Hospital, 1800 Orleans St, Baltimore, MD 21287; [email protected].
Financial disclosures: None.
1. Why it is important to improve care transitions? Society of Hospital Medicine. Accessed June 15, 2020. https://www.hospitalmedicine.org/clinical-topics/care-transitions/
2. Tong L, Arnold T, Yang J, et al. The association between outpatient follow-up visits and all-cause non-elective 30-day readmissions: a retrospective observational cohort study. PloS One. 2018;13(7):e0200691.
3. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-22.
4. Health Research & Educational Trust. Preventable Readmissions Change Package. American Hospital Association. Updated December 2015. Accessed June 10, 2020. https://www.aha.org/sites/default/files/hiin/HRETHEN_ChangePackage_Readmissions.pd
5. Tung Y-C, Chang G-M, Chang H-Y, Yu T-H. Relationship between early physician follow-up and 30-day readmission after acute myocardial infarction and heart failure. Plos One. 2017;12(1):e0170061.
6. Kaplan RM, Koehler J, Zieger PD, et al. Stroke risk as a function of atrial fibrillation duration and CHA2DS2-VASc score. Circulation. 2019;140(20):1639-46.
7. Balan P, Zhao Y, Johnson S, et al. The Society of Thoracic Surgery Risk Score as a predictor of 30-day mortality in transcatheter vs surgical aortic valve replacement: a single-center experience and its implications for the development of a TAVR risk-prediction model. J Invasive Cardiol. 2017;29(3):109-14.
8. Smith LN, Makam AN, Darden D, et al. Acute myocardial infarction readmission risk prediction models: A systematic review of model performance. Circ Cardiovasc Qual Outcomes9.9. 2018;11(1):e003885.
9. Baker H, Oliver-McNeil S, Deng L, Hummel SL. See you in 7: regional hospital collaboration and outcomes in Medicare heart failure patients. JACC Heart Fail. 2015;3(10):765-73.
10. Batten A, Jaeger C, Griffen D, et al. See you in 7: improving acute myocardial infarction follow-up care. BMJ Open Qual. 2018;7(2):e000296.
11. Lee DW, Armistead L, Coleman H, et al. Abstract 15387: Post-discharge follow-up within 14 days reduces 30-day hospital readmission rates in patients with acute myocardial infarction and/or acutely decompensated heart failure. Circulation. 2018;134 (1):A 15387.
12. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Circulation. 2018;138 (20):e:618-51.
From The Johns Hopkins Hospital, Baltimore, MD (Dr. Muganlinskaya and Dr. Skojec, retired); The George Washington University, Washington, DC (Dr. Posey); and Johns Hopkins University, Baltimore, MD (Dr. Resar).
Abstract
Objective: Assessing the risk characteristics of patients with acute myocardial infarction (MI) can help providers make appropriate referral decisions. This quality improvement project sought to improve timely, appropriate referrals among patients with type I MI by adding a risk assessment, the AMI READMITS score, to the existing referral protocol.
Methods: Patients’ chart data were analyzed to assess changes in referrals and timely follow-up appointments from pre-intervention to intervention. A survey assessed providers’ satisfaction with the new referral protocol.
Results: Among 57 patients (n = 29 preintervention; n = 28 intervention), documented referrals increased significantly from 66% to 89% (χ2 = 4.571, df = 1, P = 0.033); and timely appointments increased by 10%, which was not significant (χ2 = 3.550, df = 2, P = 0.169). Most providers agreed that the new protocol was easy to use, useful in making referral decisions, and improved the referral process. All agreed the risk score should be incorporated into electronic clinical notes. Provider opinions related to implementing the risk score in clinical practice were mixed. Qualitative feedback suggests this was due to limited validation of the AMI READMITS score in reducing readmissions.
Conclusions: Our risk-based referral protocol helped to increase appropriate referrals among patients with type I MI. Provider adoption may be enhanced by incorporating the protocol into electronic clinical notes. Research to further validate the accuracy of the AMI READMITS score in predicting readmissions may support adoption of the protocol in clinical practice.
Keywords: quality improvement; type I myocardial infarction; referral process; readmission risk; risk assessment; chart review.
Early follow-up after discharge is an important strategy to reduce the risk of unplanned hospital readmissions among patients with various conditions.1-3 While patient confounding factors, such as chronic health problems, environment, socioeconomic status, and literacy, make it difficult to avoid all unplanned readmissions, early follow-up may help providers identify and appropriately manage some health-related issues, and as such is a pivotal element of a readmission prevention strategy.4 There is evidence that patients with non-ST elevation myocardial infarction (NSTEMI) who have an outpatient appointment with a physician within 7 days after discharge have a lower risk of 30-day readmission.5
Our hospital’s postmyocardial infarction clinic was created to prevent unplanned readmissions within 30 days after discharge among patients with type I myocardial infarction (MI). Since inception, the number of referrals has been much lower than expected. In 2018, the total number of patients discharged from the hospital with type I MI and any troponin I level above 0.40 ng/mL was 313. Most of these patients were discharged from the hospital’s cardiac units; however, only 91 referrals were made. To increase referrals, the cardiology nurse practitioners (NPs) developed a post-MI referral protocol (Figure 1). However, this protocol was not consistently used and referrals to the clinic remained low.
Evidence-based risk assessment tools have the potential to increase effective patient management. For example, cardiology providers at the hospital utilize various scores, such as CHA2DS2-VASc6 and the Society of Thoracic Surgery risk score,7 to plan patient management. Among the scores used to predict unplanned readmissions for MI patients, the most promising is the AMI READMITS score.8 Unlike other nonspecific prediction models, the AMI READMITS score was developed based on variables extracted from the electronic health records (EHRs) of patients who were hospitalized for MI and readmitted within 30 days after discharge. Recognizing the potential to increase referrals by integrating an MI-specific risk assessment, this quality improvement study modified the existing referral protocol to include the patients’ AMI READMITS score and recommendations for follow-up.
Currently, there are no clear recommendations on how soon after discharge patients with MI should undergo follow-up. As research data vary, we selected 7 days follow-up for patients from high risk groups based on the “See you in 7” initiative for patients with heart failure (HF) and MI,9,10 as well as evidence that patients with NSTEMI have a lower risk of 30-day readmission if they have follow-up within 7 days after discharge5; and we selected 14 days follow-up for patients from low-risk groups based on evidence that postdischarge follow-up within 14 days reduces risk of 30-day readmission in patients with acute myocardial infarction (AMI) and/or acutely decompensated HF.11
Methods
This project was designed to answer the following question: For adult patients with type I MI, does implementation of a readmission risk assessment referral protocol increase the percentage of referrals and appointments scheduled within a recommended time? Anticipated outcomes included: (1) increased referrals to a cardiologist or the post-MI clinic; (2) increased scheduled follow-up appointments within 7 to 14 days; (3) provider satisfaction with the usability and usefulness of the new protocol; and (4) consistent provider adoption of the new risk assessment referral protocol.
To evaluate the degree to which these outcomes were achieved, we reviewed patient charts for 2 months prior and 2 months during implementation of the new referral protocol. As shown in Figure 2, the new protocol added the following process steps to the existing protocol: calculation of the AMI READMITS score, recommendations for follow-up based on patients’ risk score, and guidance to refer patients to the post-MI clinic if patients did not have an appointment with a cardiologist within 7 to 14 days after discharge. Patients’ risk assessment scores were obtained from forms completed by clinicians during the intervention. Clinician’s perceptions related to the usability and usefulness of the new protocol and feedback related to its long-term adoption were assessed using a descriptive survey.
The institutional review board classified this project as a quality improvement project. To avoid potential loss of patient privacy, no identifiable data were collected, a unique identifier unrelated to patients’ records was generated for each patient, and data were saved on a password-protected cardiology office computer.
Population
The project population included all adult patients (≥ 18 years old) with type I MI who were admitted or transferred to the hospital, had a percutaneous coronary intervention (PCI), or were managed without PCI and discharged from the hospital’s cardiac care unit (CCU) and progressive cardiac care unit (PCCU). The criteria for type I MI included the “detection of a rise and/or fall of cardiac troponin with at least 1 value above the 99th percentile and with at least 1 of the following: symptoms of acute myocardial ischemia; new ischemic electrocardiographic (ECG) changes; development of new pathological Q waves; imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology; identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy.”12 The study excluded patients with type I MI who were referred for coronary bypass surgery.
Intervention
The revised risk assessment protocol was implemented within the CCU and PCCU. The lead investigator met with each provider to discuss the role of the post-MI clinic, current referral rates, the purpose of the project, and the new referral process to be completed during the project for each patient discharged with type I MI. Cardiology NPs, fellows, and residents were asked to use the risk-assessment form to calculate patients’ risk for readmission, and refer patients to the post-MI clinic if an appointment with a cardiologist was not available within 7 to 14 days after discharge. Every week during the intervention phase, the investigator sent reminder emails to ensure form completion. Providers were asked to calculate and write the score, the discharge and referral dates, where referrals were made (a cardiologist or the post-MI clinic), date of appointment, and reason for not scheduling an appointment or not referring on the risk assessment form, and to drop the completed forms in specific labeled boxes located at the CCU and PCCU work stations. The investigator collected the completed forms weekly. When the number of discharged patients did not match the number of completed forms, the investigator followed up with discharging providers to understand why.
Data and Data Collection
Data to determine whether the use of the new protocol increased discharge referrals among patients with type I MI within the recommended timeframes were collected by electronic chart review. Data included discharging unit, patients’ age, gender, admission and discharge date, diagnosis, referral to a cardiologist and the post-MI clinic, and appointment date. Clinical data needed to calculate the AMI READMITS score was also collected: PCI within 24 hours, serum creatinine, systolic blood pressure (SBP), brain natriuretic peptide (BNP), and diabetes status.
Data to assess provider satisfaction with the usability and usefulness of the new protocol were gathered through an online survey. The survey included 1 question related to the providers’ role, 1 question asking whether they used the risk assessment for each patient, and 5 Likert-items assessing the ease of usage. An additional open-ended question asked providers to share feedback related to integrating the AMI READMITS risk assessment score to the post-MI referral protocol long term.
To evaluate how consistently providers utilized the new referral protocol when discharging patients with type I MI, the number of completed forms was compared with the number of those patients who were discharged.
Statistical Analysis
Descriptive statistics were used to summarize patient demographics and to calculate the frequency of referrals before and during the intervention. Chi-square statistics were calculated to determine whether the change in percentage of referrals and timely referrals was significant. Descriptive statistics were used to determine the level of provider satisfaction related to each survey item. A content analysis method was used to synthesize themes from the open-ended question asking clinicians to share their feedback related to the new protocol.
Results
Fifty-seven patients met the study inclusion criteria: 29 patients during the preintervention phase and 28 patients during the intervention phase. There were 35 male (61.4%) and 22 female (38.6%) patients. Twenty-five patients (43.9%) were from age groups 41 through 60 years and 61 through 80 years, respectively, representing the majority of included patients. Seven patients (12.3%) were from the 81 years and older age group. There were no patients in the age group 18 through 40 years. Based on the AMI READMITS score calculation, 57.9% (n = 33) patients were from a low-risk group (includes extremely low and low risk for readmission) and 42.1% (n = 24) were from a high-risk group (includes moderate, high, and extremely high risk for readmission).
Provider adoption of the new protocol during the intervention was high. Referral forms were completed for 82% (n = 23) of the 28 patients during the intervention. Analysis findings showed a statistically significant increase in documented referrals after implementing the new referral protocol. During the preintervention phase, 66% (n = 19) of patients with type I MI were referred to see a cardiologist or an NP at a post-MI clinic and there was no documented referral for 34% (n = 10) of patients. During the intervention phase, 89% (n = 25) of patients were referred and there was no documented referral for 11% (n = 3) of patients. Chi-square results indicated that the increase in referrals was significant (χ2 = 4.571, df = 1, P = 0.033).
Data analysis examined whether patient referrals fell within the recommended timeframe of 7 days for the high-risk group (included moderate-to-extremely high risk) and 14 days for the low-risk group (included low-to-extremely low risk). During the preintervention phase, 31% (n = 9) of patient referrals were scheduled as recommended; 28% (n = 8) of patient referrals were scheduled but delayed; and there was no referral date documented for 41% (n = 12) of patients. During the intervention phase, referrals scheduled as recommended increased to 53% (n = 15); 25% (n = 7) of referrals were scheduled but delayed; and there was no referral date documented for 21.4% (n = 6) of patients. The change in appointments scheduled as recommended was not significant (χ2 = 3.550, df = 2, P = 0.169).
Surveys were emailed to 25 cardiology fellows and 3 cardiology NPs who participated in this study. Eighteen of the 28 clinicians (15 cardiology fellows and 3 cardiology NPs) responded for a response rate of 64%. One of several residents who rotated through the CCU and PCCU during the intervention also completed the survey, for a total of 19 participants. When asked if the protocol was easy to use, 79% agreed or strongly agreed. Eighteen of the 19 participants (95%) agreed or strongly agreed that the protocol was useful in making referral decisions. Sixty-eight percent agreed or strongly agreed that the AMI READMITS risk assessment score improves referral process. All participants agreed or strongly agreed that there should be an option to incorporate the AMI READMITS risk assessment score into electronic clinical notes. When asked whether the AMI READMITS risk score should be implemented in clinical practice, responses were mixed (Figure 3). A common theme among the 4 participants who responded with comments was the need for additional data to validate the usefulness of the AMI READMITS to reduce readmissions. In addition, 1 participant commented that “manual calculation [of the risk score] is not ideal.”
Discussion
This project demonstrated that implementing an evidence-based referral protocol integrating the AMI-READMITS score can increase timely postdischarge referrals among patients with type I MI. The percentage of appropriately scheduled appointments increased during the intervention phase; however, a relatively high number of appointments were scheduled outside of the recommended timeframe, similar to preintervention. Thus, while the new protocol increased referrals and provider documentation of these referrals, it appears that challenges in scheduling timely referral appointments remained. This project did not examine the reasons for delayed appointments.
The survey findings indicated that providers were generally satisfied with the usability and usefulness of the new risk assessment protocol. A large majority agreed or strongly agreed that it was easy to use and useful in making referral decisions, and most agreed or strongly agreed that it improves the referral process. Mixed opinions regarding implementing the AMI READMITS score in clinical practice, combined with qualitative findings, suggest that a lack of external validation of the AMI READMITS presents a barrier to its long-term adoption. All providers who participated in the survey agreed or strongly agreed that the risk assessment should be incorporated into electronic clinical notes. We have begun the process of working with the EHR vendor to automate the AMI risk-assessment within the referral work-flow, which will provide an opportunity for a follow-up quality improvement study.
This quality improvement project has several limitations. First, it implemented a small change in 2 inpatient units at 1 hospital using a simple pre- posttest design. Therefore, the findings are not generalizable to other settings. Prior to the intervention, some referrals may have been made without documentation. While the authors were able to trace undocumented referrals for patients who were referred to the post-MI clinic or to a cardiologist affiliated with the hospital, some patients may have been referred to cardiologists who were not affiliated with the hospital. Another limitation was that the self-created provider survey used was not tested in other clinical settings; thus, it cannot be determined whether the sensitivity and specificity of the survey questions are high. In addition, the clinical providers who participated in the study knew the study team, which may have influenced their behavior during the study period. Furthermore, the identified improvement in clinicians’ referral practices may not be sustainable due to the complexity and effort required to manually calculate the risk score. This limitation could be eliminated by integrating the risk score calculation into the EHR.
Conclusion
Early follow-up after discharge plays an important role in supporting patients’ self-management of some risk factors (ie, diet, weight, and smoking) and identifying gaps in postdischarge care which may lead to readmission. This project provides evidence that integrating the AMI READMITS risk assessment score into the referral process can help to guide discharge decision-making and increase timely, appropriate referrals for patients with MI. Integration of a specific risk assessment, such as the AMI READMITS, within the post-MI referral protocol may help clinicians make more efficient, educated referral decisions. Future studies should explore more specifically how and why the new protocol impacts clinicians’ decision-making and behavior related to post-MI referrals. In addition, future studies should investigate challenges associated with scheduling postdischarge appointments. It will be important to investigate how integration of the new protocol within the EHR may increase efficiency, consistency, and provider satisfaction with the new referral process. Additional research investigating the effects of the AMI READMITS score on readmissions reduction will be important to promote long-term adoption of the improved referral protocol in clinical practice.
Acknowledgments: The authors thank Shelly Conaway, ANP-BC, MSN, Angela Street, ANP-BC, MSN, Andrew Geis, ACNP-BC, MSN, Richard P. Jones II, MD, Eunice Young, MD, Joy Rothwell, MSN, RN-BC, Allison Olazo, MBA, MSN, RN-BC, Elizabeth Heck, RN-BC, and Matthew Trojanowski, MHA, MS, RRT, CSSBB for their support of this study.
Corresponding author: Nailya Muganlinskaya, DNP, MPH, ACNP-BC, MSN, The Johns Hopkins Hospital, 1800 Orleans St, Baltimore, MD 21287; [email protected].
Financial disclosures: None.
From The Johns Hopkins Hospital, Baltimore, MD (Dr. Muganlinskaya and Dr. Skojec, retired); The George Washington University, Washington, DC (Dr. Posey); and Johns Hopkins University, Baltimore, MD (Dr. Resar).
Abstract
Objective: Assessing the risk characteristics of patients with acute myocardial infarction (MI) can help providers make appropriate referral decisions. This quality improvement project sought to improve timely, appropriate referrals among patients with type I MI by adding a risk assessment, the AMI READMITS score, to the existing referral protocol.
Methods: Patients’ chart data were analyzed to assess changes in referrals and timely follow-up appointments from pre-intervention to intervention. A survey assessed providers’ satisfaction with the new referral protocol.
Results: Among 57 patients (n = 29 preintervention; n = 28 intervention), documented referrals increased significantly from 66% to 89% (χ2 = 4.571, df = 1, P = 0.033); and timely appointments increased by 10%, which was not significant (χ2 = 3.550, df = 2, P = 0.169). Most providers agreed that the new protocol was easy to use, useful in making referral decisions, and improved the referral process. All agreed the risk score should be incorporated into electronic clinical notes. Provider opinions related to implementing the risk score in clinical practice were mixed. Qualitative feedback suggests this was due to limited validation of the AMI READMITS score in reducing readmissions.
Conclusions: Our risk-based referral protocol helped to increase appropriate referrals among patients with type I MI. Provider adoption may be enhanced by incorporating the protocol into electronic clinical notes. Research to further validate the accuracy of the AMI READMITS score in predicting readmissions may support adoption of the protocol in clinical practice.
Keywords: quality improvement; type I myocardial infarction; referral process; readmission risk; risk assessment; chart review.
Early follow-up after discharge is an important strategy to reduce the risk of unplanned hospital readmissions among patients with various conditions.1-3 While patient confounding factors, such as chronic health problems, environment, socioeconomic status, and literacy, make it difficult to avoid all unplanned readmissions, early follow-up may help providers identify and appropriately manage some health-related issues, and as such is a pivotal element of a readmission prevention strategy.4 There is evidence that patients with non-ST elevation myocardial infarction (NSTEMI) who have an outpatient appointment with a physician within 7 days after discharge have a lower risk of 30-day readmission.5
Our hospital’s postmyocardial infarction clinic was created to prevent unplanned readmissions within 30 days after discharge among patients with type I myocardial infarction (MI). Since inception, the number of referrals has been much lower than expected. In 2018, the total number of patients discharged from the hospital with type I MI and any troponin I level above 0.40 ng/mL was 313. Most of these patients were discharged from the hospital’s cardiac units; however, only 91 referrals were made. To increase referrals, the cardiology nurse practitioners (NPs) developed a post-MI referral protocol (Figure 1). However, this protocol was not consistently used and referrals to the clinic remained low.
Evidence-based risk assessment tools have the potential to increase effective patient management. For example, cardiology providers at the hospital utilize various scores, such as CHA2DS2-VASc6 and the Society of Thoracic Surgery risk score,7 to plan patient management. Among the scores used to predict unplanned readmissions for MI patients, the most promising is the AMI READMITS score.8 Unlike other nonspecific prediction models, the AMI READMITS score was developed based on variables extracted from the electronic health records (EHRs) of patients who were hospitalized for MI and readmitted within 30 days after discharge. Recognizing the potential to increase referrals by integrating an MI-specific risk assessment, this quality improvement study modified the existing referral protocol to include the patients’ AMI READMITS score and recommendations for follow-up.
Currently, there are no clear recommendations on how soon after discharge patients with MI should undergo follow-up. As research data vary, we selected 7 days follow-up for patients from high risk groups based on the “See you in 7” initiative for patients with heart failure (HF) and MI,9,10 as well as evidence that patients with NSTEMI have a lower risk of 30-day readmission if they have follow-up within 7 days after discharge5; and we selected 14 days follow-up for patients from low-risk groups based on evidence that postdischarge follow-up within 14 days reduces risk of 30-day readmission in patients with acute myocardial infarction (AMI) and/or acutely decompensated HF.11
Methods
This project was designed to answer the following question: For adult patients with type I MI, does implementation of a readmission risk assessment referral protocol increase the percentage of referrals and appointments scheduled within a recommended time? Anticipated outcomes included: (1) increased referrals to a cardiologist or the post-MI clinic; (2) increased scheduled follow-up appointments within 7 to 14 days; (3) provider satisfaction with the usability and usefulness of the new protocol; and (4) consistent provider adoption of the new risk assessment referral protocol.
To evaluate the degree to which these outcomes were achieved, we reviewed patient charts for 2 months prior and 2 months during implementation of the new referral protocol. As shown in Figure 2, the new protocol added the following process steps to the existing protocol: calculation of the AMI READMITS score, recommendations for follow-up based on patients’ risk score, and guidance to refer patients to the post-MI clinic if patients did not have an appointment with a cardiologist within 7 to 14 days after discharge. Patients’ risk assessment scores were obtained from forms completed by clinicians during the intervention. Clinician’s perceptions related to the usability and usefulness of the new protocol and feedback related to its long-term adoption were assessed using a descriptive survey.
The institutional review board classified this project as a quality improvement project. To avoid potential loss of patient privacy, no identifiable data were collected, a unique identifier unrelated to patients’ records was generated for each patient, and data were saved on a password-protected cardiology office computer.
Population
The project population included all adult patients (≥ 18 years old) with type I MI who were admitted or transferred to the hospital, had a percutaneous coronary intervention (PCI), or were managed without PCI and discharged from the hospital’s cardiac care unit (CCU) and progressive cardiac care unit (PCCU). The criteria for type I MI included the “detection of a rise and/or fall of cardiac troponin with at least 1 value above the 99th percentile and with at least 1 of the following: symptoms of acute myocardial ischemia; new ischemic electrocardiographic (ECG) changes; development of new pathological Q waves; imaging evidence of new loss of viable myocardium or new regional wall motion abnormality in a pattern consistent with an ischemic etiology; identification of a coronary thrombus by angiography including intracoronary imaging or by autopsy.”12 The study excluded patients with type I MI who were referred for coronary bypass surgery.
Intervention
The revised risk assessment protocol was implemented within the CCU and PCCU. The lead investigator met with each provider to discuss the role of the post-MI clinic, current referral rates, the purpose of the project, and the new referral process to be completed during the project for each patient discharged with type I MI. Cardiology NPs, fellows, and residents were asked to use the risk-assessment form to calculate patients’ risk for readmission, and refer patients to the post-MI clinic if an appointment with a cardiologist was not available within 7 to 14 days after discharge. Every week during the intervention phase, the investigator sent reminder emails to ensure form completion. Providers were asked to calculate and write the score, the discharge and referral dates, where referrals were made (a cardiologist or the post-MI clinic), date of appointment, and reason for not scheduling an appointment or not referring on the risk assessment form, and to drop the completed forms in specific labeled boxes located at the CCU and PCCU work stations. The investigator collected the completed forms weekly. When the number of discharged patients did not match the number of completed forms, the investigator followed up with discharging providers to understand why.
Data and Data Collection
Data to determine whether the use of the new protocol increased discharge referrals among patients with type I MI within the recommended timeframes were collected by electronic chart review. Data included discharging unit, patients’ age, gender, admission and discharge date, diagnosis, referral to a cardiologist and the post-MI clinic, and appointment date. Clinical data needed to calculate the AMI READMITS score was also collected: PCI within 24 hours, serum creatinine, systolic blood pressure (SBP), brain natriuretic peptide (BNP), and diabetes status.
Data to assess provider satisfaction with the usability and usefulness of the new protocol were gathered through an online survey. The survey included 1 question related to the providers’ role, 1 question asking whether they used the risk assessment for each patient, and 5 Likert-items assessing the ease of usage. An additional open-ended question asked providers to share feedback related to integrating the AMI READMITS risk assessment score to the post-MI referral protocol long term.
To evaluate how consistently providers utilized the new referral protocol when discharging patients with type I MI, the number of completed forms was compared with the number of those patients who were discharged.
Statistical Analysis
Descriptive statistics were used to summarize patient demographics and to calculate the frequency of referrals before and during the intervention. Chi-square statistics were calculated to determine whether the change in percentage of referrals and timely referrals was significant. Descriptive statistics were used to determine the level of provider satisfaction related to each survey item. A content analysis method was used to synthesize themes from the open-ended question asking clinicians to share their feedback related to the new protocol.
Results
Fifty-seven patients met the study inclusion criteria: 29 patients during the preintervention phase and 28 patients during the intervention phase. There were 35 male (61.4%) and 22 female (38.6%) patients. Twenty-five patients (43.9%) were from age groups 41 through 60 years and 61 through 80 years, respectively, representing the majority of included patients. Seven patients (12.3%) were from the 81 years and older age group. There were no patients in the age group 18 through 40 years. Based on the AMI READMITS score calculation, 57.9% (n = 33) patients were from a low-risk group (includes extremely low and low risk for readmission) and 42.1% (n = 24) were from a high-risk group (includes moderate, high, and extremely high risk for readmission).
Provider adoption of the new protocol during the intervention was high. Referral forms were completed for 82% (n = 23) of the 28 patients during the intervention. Analysis findings showed a statistically significant increase in documented referrals after implementing the new referral protocol. During the preintervention phase, 66% (n = 19) of patients with type I MI were referred to see a cardiologist or an NP at a post-MI clinic and there was no documented referral for 34% (n = 10) of patients. During the intervention phase, 89% (n = 25) of patients were referred and there was no documented referral for 11% (n = 3) of patients. Chi-square results indicated that the increase in referrals was significant (χ2 = 4.571, df = 1, P = 0.033).
Data analysis examined whether patient referrals fell within the recommended timeframe of 7 days for the high-risk group (included moderate-to-extremely high risk) and 14 days for the low-risk group (included low-to-extremely low risk). During the preintervention phase, 31% (n = 9) of patient referrals were scheduled as recommended; 28% (n = 8) of patient referrals were scheduled but delayed; and there was no referral date documented for 41% (n = 12) of patients. During the intervention phase, referrals scheduled as recommended increased to 53% (n = 15); 25% (n = 7) of referrals were scheduled but delayed; and there was no referral date documented for 21.4% (n = 6) of patients. The change in appointments scheduled as recommended was not significant (χ2 = 3.550, df = 2, P = 0.169).
Surveys were emailed to 25 cardiology fellows and 3 cardiology NPs who participated in this study. Eighteen of the 28 clinicians (15 cardiology fellows and 3 cardiology NPs) responded for a response rate of 64%. One of several residents who rotated through the CCU and PCCU during the intervention also completed the survey, for a total of 19 participants. When asked if the protocol was easy to use, 79% agreed or strongly agreed. Eighteen of the 19 participants (95%) agreed or strongly agreed that the protocol was useful in making referral decisions. Sixty-eight percent agreed or strongly agreed that the AMI READMITS risk assessment score improves referral process. All participants agreed or strongly agreed that there should be an option to incorporate the AMI READMITS risk assessment score into electronic clinical notes. When asked whether the AMI READMITS risk score should be implemented in clinical practice, responses were mixed (Figure 3). A common theme among the 4 participants who responded with comments was the need for additional data to validate the usefulness of the AMI READMITS to reduce readmissions. In addition, 1 participant commented that “manual calculation [of the risk score] is not ideal.”
Discussion
This project demonstrated that implementing an evidence-based referral protocol integrating the AMI-READMITS score can increase timely postdischarge referrals among patients with type I MI. The percentage of appropriately scheduled appointments increased during the intervention phase; however, a relatively high number of appointments were scheduled outside of the recommended timeframe, similar to preintervention. Thus, while the new protocol increased referrals and provider documentation of these referrals, it appears that challenges in scheduling timely referral appointments remained. This project did not examine the reasons for delayed appointments.
The survey findings indicated that providers were generally satisfied with the usability and usefulness of the new risk assessment protocol. A large majority agreed or strongly agreed that it was easy to use and useful in making referral decisions, and most agreed or strongly agreed that it improves the referral process. Mixed opinions regarding implementing the AMI READMITS score in clinical practice, combined with qualitative findings, suggest that a lack of external validation of the AMI READMITS presents a barrier to its long-term adoption. All providers who participated in the survey agreed or strongly agreed that the risk assessment should be incorporated into electronic clinical notes. We have begun the process of working with the EHR vendor to automate the AMI risk-assessment within the referral work-flow, which will provide an opportunity for a follow-up quality improvement study.
This quality improvement project has several limitations. First, it implemented a small change in 2 inpatient units at 1 hospital using a simple pre- posttest design. Therefore, the findings are not generalizable to other settings. Prior to the intervention, some referrals may have been made without documentation. While the authors were able to trace undocumented referrals for patients who were referred to the post-MI clinic or to a cardiologist affiliated with the hospital, some patients may have been referred to cardiologists who were not affiliated with the hospital. Another limitation was that the self-created provider survey used was not tested in other clinical settings; thus, it cannot be determined whether the sensitivity and specificity of the survey questions are high. In addition, the clinical providers who participated in the study knew the study team, which may have influenced their behavior during the study period. Furthermore, the identified improvement in clinicians’ referral practices may not be sustainable due to the complexity and effort required to manually calculate the risk score. This limitation could be eliminated by integrating the risk score calculation into the EHR.
Conclusion
Early follow-up after discharge plays an important role in supporting patients’ self-management of some risk factors (ie, diet, weight, and smoking) and identifying gaps in postdischarge care which may lead to readmission. This project provides evidence that integrating the AMI READMITS risk assessment score into the referral process can help to guide discharge decision-making and increase timely, appropriate referrals for patients with MI. Integration of a specific risk assessment, such as the AMI READMITS, within the post-MI referral protocol may help clinicians make more efficient, educated referral decisions. Future studies should explore more specifically how and why the new protocol impacts clinicians’ decision-making and behavior related to post-MI referrals. In addition, future studies should investigate challenges associated with scheduling postdischarge appointments. It will be important to investigate how integration of the new protocol within the EHR may increase efficiency, consistency, and provider satisfaction with the new referral process. Additional research investigating the effects of the AMI READMITS score on readmissions reduction will be important to promote long-term adoption of the improved referral protocol in clinical practice.
Acknowledgments: The authors thank Shelly Conaway, ANP-BC, MSN, Angela Street, ANP-BC, MSN, Andrew Geis, ACNP-BC, MSN, Richard P. Jones II, MD, Eunice Young, MD, Joy Rothwell, MSN, RN-BC, Allison Olazo, MBA, MSN, RN-BC, Elizabeth Heck, RN-BC, and Matthew Trojanowski, MHA, MS, RRT, CSSBB for their support of this study.
Corresponding author: Nailya Muganlinskaya, DNP, MPH, ACNP-BC, MSN, The Johns Hopkins Hospital, 1800 Orleans St, Baltimore, MD 21287; [email protected].
Financial disclosures: None.
1. Why it is important to improve care transitions? Society of Hospital Medicine. Accessed June 15, 2020. https://www.hospitalmedicine.org/clinical-topics/care-transitions/
2. Tong L, Arnold T, Yang J, et al. The association between outpatient follow-up visits and all-cause non-elective 30-day readmissions: a retrospective observational cohort study. PloS One. 2018;13(7):e0200691.
3. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-22.
4. Health Research & Educational Trust. Preventable Readmissions Change Package. American Hospital Association. Updated December 2015. Accessed June 10, 2020. https://www.aha.org/sites/default/files/hiin/HRETHEN_ChangePackage_Readmissions.pd
5. Tung Y-C, Chang G-M, Chang H-Y, Yu T-H. Relationship between early physician follow-up and 30-day readmission after acute myocardial infarction and heart failure. Plos One. 2017;12(1):e0170061.
6. Kaplan RM, Koehler J, Zieger PD, et al. Stroke risk as a function of atrial fibrillation duration and CHA2DS2-VASc score. Circulation. 2019;140(20):1639-46.
7. Balan P, Zhao Y, Johnson S, et al. The Society of Thoracic Surgery Risk Score as a predictor of 30-day mortality in transcatheter vs surgical aortic valve replacement: a single-center experience and its implications for the development of a TAVR risk-prediction model. J Invasive Cardiol. 2017;29(3):109-14.
8. Smith LN, Makam AN, Darden D, et al. Acute myocardial infarction readmission risk prediction models: A systematic review of model performance. Circ Cardiovasc Qual Outcomes9.9. 2018;11(1):e003885.
9. Baker H, Oliver-McNeil S, Deng L, Hummel SL. See you in 7: regional hospital collaboration and outcomes in Medicare heart failure patients. JACC Heart Fail. 2015;3(10):765-73.
10. Batten A, Jaeger C, Griffen D, et al. See you in 7: improving acute myocardial infarction follow-up care. BMJ Open Qual. 2018;7(2):e000296.
11. Lee DW, Armistead L, Coleman H, et al. Abstract 15387: Post-discharge follow-up within 14 days reduces 30-day hospital readmission rates in patients with acute myocardial infarction and/or acutely decompensated heart failure. Circulation. 2018;134 (1):A 15387.
12. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Circulation. 2018;138 (20):e:618-51.
1. Why it is important to improve care transitions? Society of Hospital Medicine. Accessed June 15, 2020. https://www.hospitalmedicine.org/clinical-topics/care-transitions/
2. Tong L, Arnold T, Yang J, et al. The association between outpatient follow-up visits and all-cause non-elective 30-day readmissions: a retrospective observational cohort study. PloS One. 2018;13(7):e0200691.
3. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-22.
4. Health Research & Educational Trust. Preventable Readmissions Change Package. American Hospital Association. Updated December 2015. Accessed June 10, 2020. https://www.aha.org/sites/default/files/hiin/HRETHEN_ChangePackage_Readmissions.pd
5. Tung Y-C, Chang G-M, Chang H-Y, Yu T-H. Relationship between early physician follow-up and 30-day readmission after acute myocardial infarction and heart failure. Plos One. 2017;12(1):e0170061.
6. Kaplan RM, Koehler J, Zieger PD, et al. Stroke risk as a function of atrial fibrillation duration and CHA2DS2-VASc score. Circulation. 2019;140(20):1639-46.
7. Balan P, Zhao Y, Johnson S, et al. The Society of Thoracic Surgery Risk Score as a predictor of 30-day mortality in transcatheter vs surgical aortic valve replacement: a single-center experience and its implications for the development of a TAVR risk-prediction model. J Invasive Cardiol. 2017;29(3):109-14.
8. Smith LN, Makam AN, Darden D, et al. Acute myocardial infarction readmission risk prediction models: A systematic review of model performance. Circ Cardiovasc Qual Outcomes9.9. 2018;11(1):e003885.
9. Baker H, Oliver-McNeil S, Deng L, Hummel SL. See you in 7: regional hospital collaboration and outcomes in Medicare heart failure patients. JACC Heart Fail. 2015;3(10):765-73.
10. Batten A, Jaeger C, Griffen D, et al. See you in 7: improving acute myocardial infarction follow-up care. BMJ Open Qual. 2018;7(2):e000296.
11. Lee DW, Armistead L, Coleman H, et al. Abstract 15387: Post-discharge follow-up within 14 days reduces 30-day hospital readmission rates in patients with acute myocardial infarction and/or acutely decompensated heart failure. Circulation. 2018;134 (1):A 15387.
12. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Circulation. 2018;138 (20):e:618-51.
FDA warning letters target OTC cannabidiol product claims for pain relief
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.

In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”
The Food and Drug Administration has warned two manufacturers about illegal marketing of drugs containing cannabidiol (CBD) for over-the-counter use without an approved new drug application, for using substandard manufacturing processes, and for failure to comply with current good manufacturing practices. These warnings add to 51 previous warning letters issued by the FDA since 2015 to other manufacturers of products containing CBD who were violating the Federal Food, Drug, and Cosmetic Act.
In a news release, the agency explained that its two most recent letters, sent to Honest Globe Inc. on March 15 and BioLyte Laboratories LLC on March 18, were issued because CBD has “known pharmacologic effects on humans, with demonstrated risks, it cannot be legally marketed as an inactive ingredient in OTC drug products that are not reviewed and approved by the FDA.” They also describe the companies’ failures to comply with current good manufacturing practices.
“The FDA continues to alert the public to potential safety and efficacy concerns with unapproved CBD products sold online and in stores across the country,” FDA Principal Deputy Commissioner Amy P. Abernethy, MD, PhD, said in the release. “It’s important that consumers understand that the FDA has only approved one drug containing CBD as an ingredient [Epidiolex]. These other, unapproved, CBD products may have dangerous health impacts and side effects. We remain focused on exploring potential pathways for CBD products to be lawfully marketed while also educating the public about these outstanding questions of CBD’s safety. Meanwhile, we will continue to monitor and take action, as needed, against companies that unlawfully market their products – prioritizing those that pose a risk to public health.”
The specific products from Santa Ana, Calif.–based Honest Globe that the FDA called unapproved new drugs and misbranded under the Federal Food, Drug, and Cosmetic Act included Elixicure Original Pain Relief and Elixicure Lavender Pain Relief, both of which were described as containing CBD. Products from Grand Rapids, Mich.–based BioLyte Laboratories LLC that the FDA similarly cited for violations included Silver Gel, Silver Gel with Aloe, Silver Liquid Supplement, Therapeutic Pain Gel, Pain Relief Cream, and Magnesium Oil Spray.
The agency has asked the two companies to respond to its letters within 15 working days, “stating how they will address these violations or providing their reasoning and supporting information as to why they believe these products are not in violation of the law. Failure to adequately address the violations promptly may result in legal action, including product seizure and/or injunction.”