User login
Studies highlight diagnostic and treatment challenges in hidradenitis suppurativa
SCOTTSDALE, ARIZ. – Patients with hidradenitis suppurativa (HS) may be misdiagnosed when they see providers who are not dermatologists – as is usually the case during the initial years of their disease, according to a large analysis of medical claims data.
The findings highlight the need for visual diagnostic aids and specific guidelines for treating HS that target nondermatologists, Melissa Butt, MPH, of Penn State Hershey (Pa.) Medical Center, said during an interview at the annual meeting of the Society for Investigative Dermatology. She presented the findings during a poster session at the meeting.

HS is a chronic inflammatory disease of the hair follicles that affects 0.5%-4% of people in the United States. In past studies, up to 12 years elapsed between disease onset and diagnosis, in part because patients often cannot readily access dermatologists, Ms. Butt said. To better understand patterns of health care use during the years leading up to HS diagnosis, she and her colleagues used MarketScan data to identify 1,733 patients with HS-specific medical care claims filed in 2012 and 2013. Then they looked back at medical claims for these patients during 2008 through 2011, before the patients were diagnosed with HS. The cohort averaged 37 years of age (standard deviation, 15 years), and 73% were female.
Among 239,892 claims filed before patients were diagnosed with HS, 11,381 (4.7%) included codes for other diseases of the skin and subcutaneous tissues, Ms. Butt said. Dermatologists filed only 31% of these skin-specific claims, while 69% were filed by other providers, such as family practitioners, internists, emergency department physicians, and acute care hospitalists.
Notably, about two-thirds of the skin-specific diagnostic codes could have represented a misdiagnosis of HS. These codes included conditions such as abscesses, carbuncles, local infections, ulcers, and diseases of the sebaceous glands.
The fact that 78% of visits occurred in offices and other outpatient settings further underscores the need to improve the detection and care of HS in these environments, Ms. Butt said. Given current national shortages of dermatologists, visual HS diagnostic aids and “detailed, multistep clinical practice guidelines” for nondermatologists could help improve care of HS while patients wait to see the specialists, she added.
A second poster presented at the meeting provided results of a study on the use and impact of antibiotics in the treatment of HS. Alexander Fischer of Johns Hopkins University, Baltimore, and his associates studied antibiotic prescriptions and bacterial cultures from the lesions of 239 patients with HS who were treated at Johns Hopkins medical facilities between 2010 and 2015. Not only were 51% of HS patients on antibiotics at the time of culture, but these patients’ lesions were significantly more likely to contain antibiotic-resistant bacteria than were those of patients not on antibiotics.
Strikingly, Proteus species were isolated from nearly half of patients on trimethoprim-sulfamethoxazole (TMP/SMX), and 88% of colonies were resistant to TMP/SMX, while only 13% of cultures from untreated patients grew Proteus (P less than .001) and all were TMP/SMX-susceptible (P less than .001). Likewise, 100% of methicillin-resistant Staphylococcus aureus (MRSA) strains from patients prescribed ciprofloxacin were resistant to it, compared with a 10% background rate of ciprofloxacin resistance among MRSA from patients not taking antibiotics (P = .04). In addition, the proportion of other S. aureus strains that were clindamycin-resistant was higher when patients were taking this antibiotic than when they were not (63% versus 17%; P = .03).
The results “raise questions” about whether antibiotics should be used in HS patients who are not clearly benefiting from them, according to the researchers.
The authors of both studies reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Patients with hidradenitis suppurativa (HS) may be misdiagnosed when they see providers who are not dermatologists – as is usually the case during the initial years of their disease, according to a large analysis of medical claims data.
The findings highlight the need for visual diagnostic aids and specific guidelines for treating HS that target nondermatologists, Melissa Butt, MPH, of Penn State Hershey (Pa.) Medical Center, said during an interview at the annual meeting of the Society for Investigative Dermatology. She presented the findings during a poster session at the meeting.
HS is a chronic inflammatory disease of the hair follicles that affects 0.5%-4% of people in the United States. In past studies, up to 12 years elapsed between disease onset and diagnosis, in part because patients often cannot readily access dermatologists, Ms. Butt said. To better understand patterns of health care use during the years leading up to HS diagnosis, she and her colleagues used MarketScan data to identify 1,733 patients with HS-specific medical care claims filed in 2012 and 2013. Then they looked back at medical claims for these patients during 2008 through 2011, before the patients were diagnosed with HS. The cohort averaged 37 years of age (standard deviation, 15 years), and 73% were female.
Among 239,892 claims filed before patients were diagnosed with HS, 11,381 (4.7%) included codes for other diseases of the skin and subcutaneous tissues, Ms. Butt said. Dermatologists filed only 31% of these skin-specific claims, while 69% were filed by other providers, such as family practitioners, internists, emergency department physicians, and acute care hospitalists.
Notably, about two-thirds of the skin-specific diagnostic codes could have represented a misdiagnosis of HS. These codes included conditions such as abscesses, carbuncles, local infections, ulcers, and diseases of the sebaceous glands.
The fact that 78% of visits occurred in offices and other outpatient settings further underscores the need to improve the detection and care of HS in these environments, Ms. Butt said. Given current national shortages of dermatologists, visual HS diagnostic aids and “detailed, multistep clinical practice guidelines” for nondermatologists could help improve care of HS while patients wait to see the specialists, she added.
A second poster presented at the meeting provided results of a study on the use and impact of antibiotics in the treatment of HS. Alexander Fischer of Johns Hopkins University, Baltimore, and his associates studied antibiotic prescriptions and bacterial cultures from the lesions of 239 patients with HS who were treated at Johns Hopkins medical facilities between 2010 and 2015. Not only were 51% of HS patients on antibiotics at the time of culture, but these patients’ lesions were significantly more likely to contain antibiotic-resistant bacteria than were those of patients not on antibiotics.
Strikingly, Proteus species were isolated from nearly half of patients on trimethoprim-sulfamethoxazole (TMP/SMX), and 88% of colonies were resistant to TMP/SMX, while only 13% of cultures from untreated patients grew Proteus (P less than .001) and all were TMP/SMX-susceptible (P less than .001). Likewise, 100% of methicillin-resistant Staphylococcus aureus (MRSA) strains from patients prescribed ciprofloxacin were resistant to it, compared with a 10% background rate of ciprofloxacin resistance among MRSA from patients not taking antibiotics (P = .04). In addition, the proportion of other S. aureus strains that were clindamycin-resistant was higher when patients were taking this antibiotic than when they were not (63% versus 17%; P = .03).
The results “raise questions” about whether antibiotics should be used in HS patients who are not clearly benefiting from them, according to the researchers.
The authors of both studies reported no funding sources and had no disclosures.
SCOTTSDALE, ARIZ. – Patients with hidradenitis suppurativa (HS) may be misdiagnosed when they see providers who are not dermatologists – as is usually the case during the initial years of their disease, according to a large analysis of medical claims data.
The findings highlight the need for visual diagnostic aids and specific guidelines for treating HS that target nondermatologists, Melissa Butt, MPH, of Penn State Hershey (Pa.) Medical Center, said during an interview at the annual meeting of the Society for Investigative Dermatology. She presented the findings during a poster session at the meeting.
HS is a chronic inflammatory disease of the hair follicles that affects 0.5%-4% of people in the United States. In past studies, up to 12 years elapsed between disease onset and diagnosis, in part because patients often cannot readily access dermatologists, Ms. Butt said. To better understand patterns of health care use during the years leading up to HS diagnosis, she and her colleagues used MarketScan data to identify 1,733 patients with HS-specific medical care claims filed in 2012 and 2013. Then they looked back at medical claims for these patients during 2008 through 2011, before the patients were diagnosed with HS. The cohort averaged 37 years of age (standard deviation, 15 years), and 73% were female.
Among 239,892 claims filed before patients were diagnosed with HS, 11,381 (4.7%) included codes for other diseases of the skin and subcutaneous tissues, Ms. Butt said. Dermatologists filed only 31% of these skin-specific claims, while 69% were filed by other providers, such as family practitioners, internists, emergency department physicians, and acute care hospitalists.
Notably, about two-thirds of the skin-specific diagnostic codes could have represented a misdiagnosis of HS. These codes included conditions such as abscesses, carbuncles, local infections, ulcers, and diseases of the sebaceous glands.
The fact that 78% of visits occurred in offices and other outpatient settings further underscores the need to improve the detection and care of HS in these environments, Ms. Butt said. Given current national shortages of dermatologists, visual HS diagnostic aids and “detailed, multistep clinical practice guidelines” for nondermatologists could help improve care of HS while patients wait to see the specialists, she added.
A second poster presented at the meeting provided results of a study on the use and impact of antibiotics in the treatment of HS. Alexander Fischer of Johns Hopkins University, Baltimore, and his associates studied antibiotic prescriptions and bacterial cultures from the lesions of 239 patients with HS who were treated at Johns Hopkins medical facilities between 2010 and 2015. Not only were 51% of HS patients on antibiotics at the time of culture, but these patients’ lesions were significantly more likely to contain antibiotic-resistant bacteria than were those of patients not on antibiotics.
Strikingly, Proteus species were isolated from nearly half of patients on trimethoprim-sulfamethoxazole (TMP/SMX), and 88% of colonies were resistant to TMP/SMX, while only 13% of cultures from untreated patients grew Proteus (P less than .001) and all were TMP/SMX-susceptible (P less than .001). Likewise, 100% of methicillin-resistant Staphylococcus aureus (MRSA) strains from patients prescribed ciprofloxacin were resistant to it, compared with a 10% background rate of ciprofloxacin resistance among MRSA from patients not taking antibiotics (P = .04). In addition, the proportion of other S. aureus strains that were clindamycin-resistant was higher when patients were taking this antibiotic than when they were not (63% versus 17%; P = .03).
The results “raise questions” about whether antibiotics should be used in HS patients who are not clearly benefiting from them, according to the researchers.
The authors of both studies reported no funding sources and had no disclosures.
AT THE 2016 SID ANNUAL MEETING
Key clinical point: Two studies underscored current challenges in diagnosing and treating hidradenitis suppurativa (HS).
Major finding: HS was usually diagnosed in outpatient settings by nondermatologists who often initially filed claims for carbuncles, ulcers, and other conditions that are confused with HS. In a separate study, antibiotic-resistant bacteria were significantly more prevalent in the lesions of HS patients who were receiving antibiotics than in patients who were not taking antibiotics.
Data source: A medical claims analysis of 1,733 patients with HS, and a study of antibiotic prescriptions and bacterial cultures from 239 patients with HS.
Disclosures: The authors of both studies reported no funding sources and had no disclosures.

Patch of Hair Loss on the Scalp
The Diagnosis: Temporal Triangular Alopecia
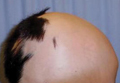
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
The Diagnosis: Temporal Triangular Alopecia
Temporal triangular alopecia (TTA), also known as congenital triangular alopecia, was first described in the early 1900s.1 It presents clinically as a triangular-shaped area of nonscarring alopecia either unilaterally or bilaterally. Limited clinical data suggest that most unilateral cases are on the left frontotemporal region of the scalp. In bilateral cases, there may be asymmetry in size of the area involved.2 Dermatoscopically, TTA is characterized by decreased terminal hair follicle density as well as the presence of vellus hairs with an absence of inflammation.3 The majority of TTA is noted between birth and 6 years of life with the areas staying stable thereafter. Large areas of TTA may suggest cerebello-trigeminal-dermal dysplasia (Gomez-Lopez-Hernandez syndrome), a rare neurocutaneous syndrome characterized by rhombencephalosynapsis, trigeminal anesthesia, and parietooccipital alopecia (Online Mendelian Inheritance in Man 601853).4 Although TTA is largely idiopathic, it has been suggested that the trait may be paradominant, whereby a postzygotic loss of the wild-type allele in a heterozygotic state causes triangular alopecia and reflects hamartomatous mosaicism.5 It also is an important mimicker of alopecia areata. Correct identification prevents unnecessary treatment to the areas of the scalp. Hair restoration surgery has been reported as a tool to treat this disorder.6
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
- Tosti A. Congenital triangular alopecia. report of fourteen cases. J Am Acad Dermatol. 1987;16:991-993.
- Armstrong DK, Burrows D. Congenital triangular alopecia. Pediatr Dermatol. 1996;13:394-396.
- Iorizzo M, Pazzaglia M, Starace M, et al. Videodermoscopy: a useful tool for diagnosing congenital triangular alopecia. Pediatr Dermatol. 2008;25:652-654.
- Assoly P, Happle R. A hairy paradox: congenital triangular alopecia with a central hair tuft. Dermatology. 2010;221:107-109.
- Happle R. Congenital triangular alopecia may be categorized as a paradominant trait. Eur J Dermatol. 2003;13:346-347.
- Wu WY, Otberg N, Kang H, et al. Successful treatment of temporal triangular alopecia by hair restoration surgery using follicular unit transplantation. Dermatol Surg. 2009;35:1307-1310.
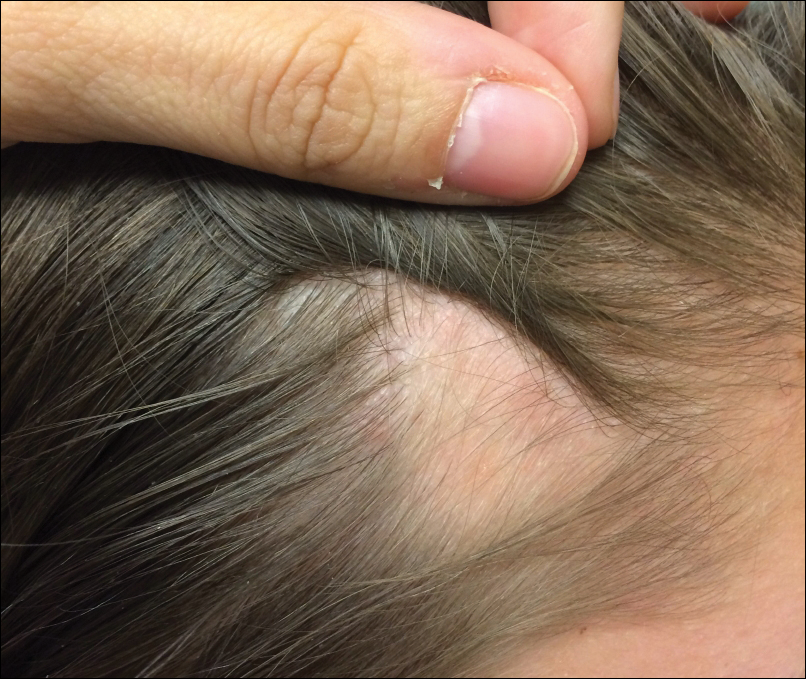
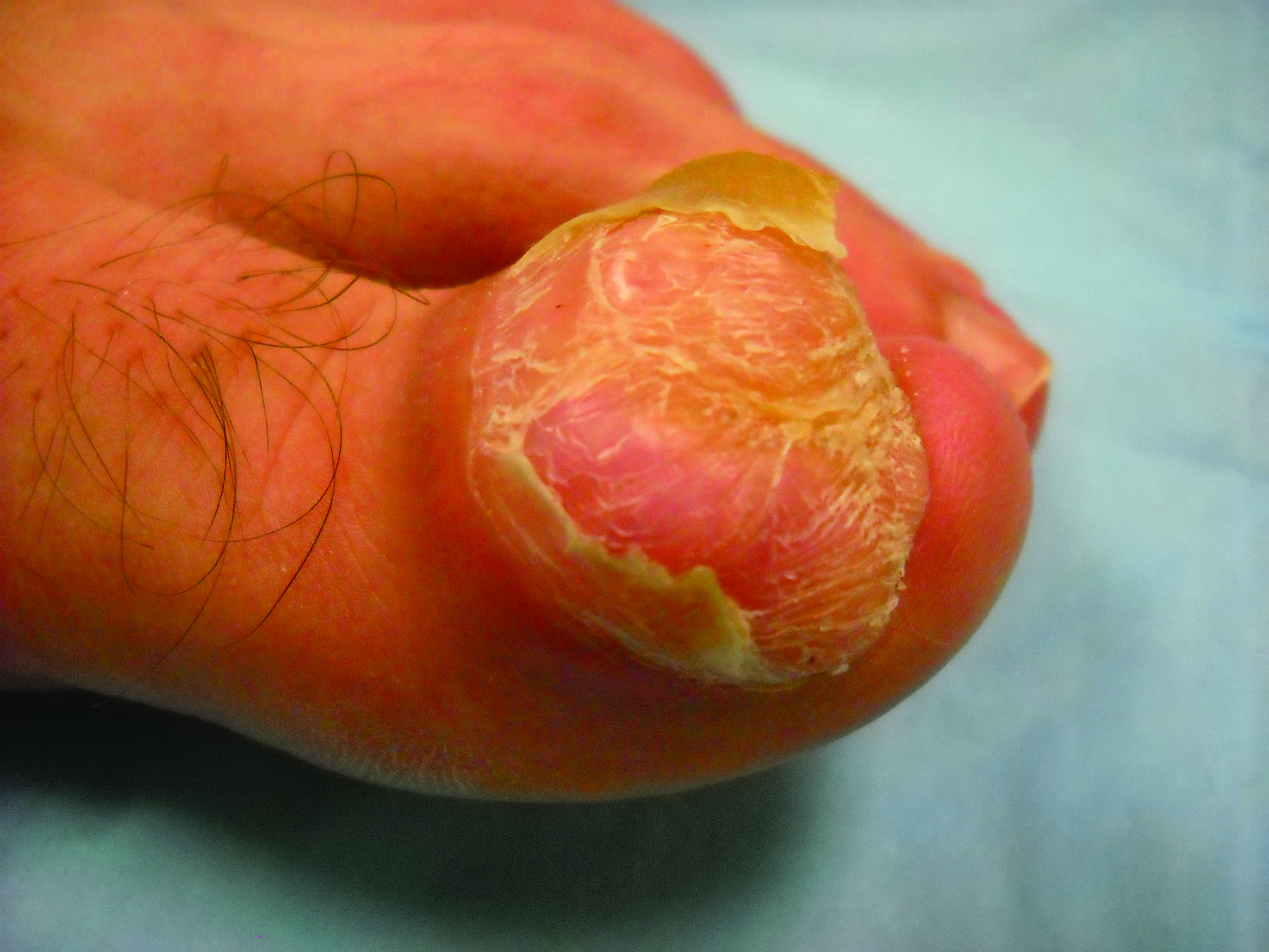
An 11-year-old girl presented for evaluation of a patch of hair loss on the right parietal scalp that had been present and stable for 2.5 years. Physical examination revealed a unilateral area of hair loss that was triangular in shape on the right parietal/temporal region, measuring 2.1×2.2 cm. Dermatoscope examination showed vellus hairs throughout. A hair-pull test was negative and the patient confirmed that the area had never been completely smooth. There were no associated symptoms and no family history of autoimmune disease or hair loss. Prior to presentation, the patient underwent a trial of intralesional steroids and topical steroids to the area without effect.
Clinical Characteristics and HLA Alleles of a Family With Simultaneously Occurring Alopecia Areata
Alopecia areata (AA) presents as sudden, nonscarring, recurrent hair loss characterized by well-circumscribed hairless patches. Although AA may be observed on any hair-bearing areas of the body, the most commonly affected sites are the scalp, beard area, eyebrows, and eyelashes.1 The incidence of AA is 1% to 2% in the general population and it is more common in males than females younger than 40 years.2 Although the majority of patients present with self-limited and well-circumscribed hairless patches that resolve within 2 years, 7% to 10% display a chronic and severe prognosis.3
The etiopathogenesis of AA is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.2 Alopecia areata is observed to occur sporadically in most patients. Family history has been found in 3% to 42% of cases, but simultaneous occurrence of AA in family members is rare.4 In this case series, we present 4 cases of active AA lesions occurring simultaneously in a family who also had associated psychologic disorders.
Case Series
Patient 1 (Proband)
An 11-year-old boy presented with a 6-year history of ongoing AA with recurrent improvement and relapses on the scalp, eyebrows, and eyelashes. Various topical and oral medications had been prescribed by several outside dermatologists; however, these treatments provided minimal benefit and resulted in the recurrence of AA. Dermatologic examination revealed hair loss on the entire frontal, parietal, and temporal regions of the scalp, as well as half of the occipital region and one-third of the lateral side of the eyebrows (Figure 1). Psychological evaluation revealed introvert personality characteristics, lack of self-confidence, and signs of depression and anxiety.
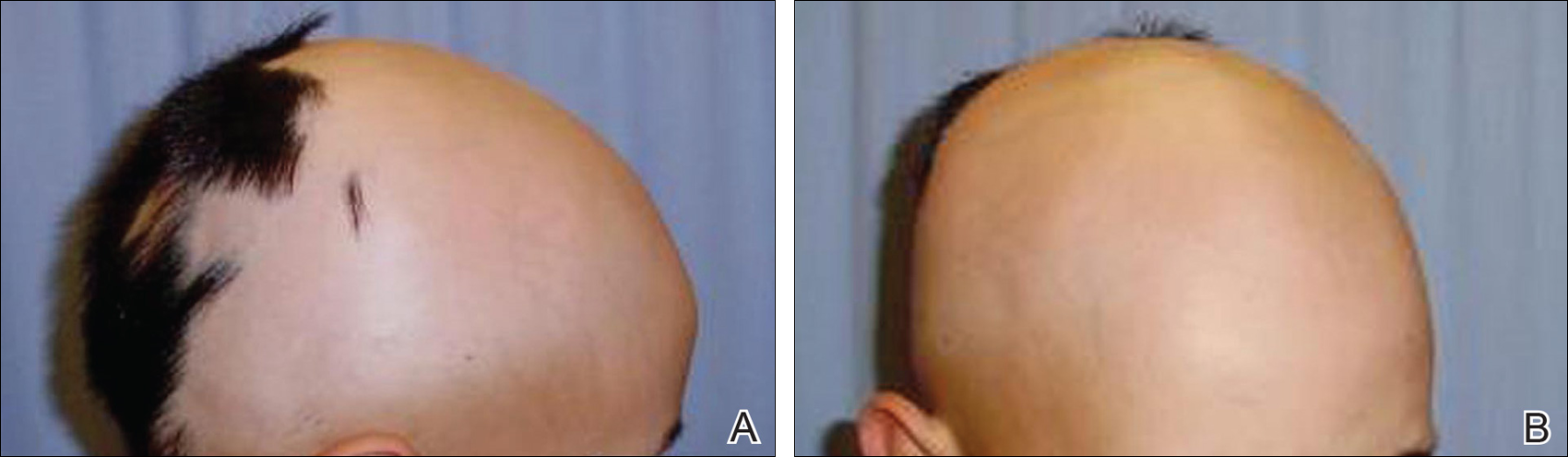
Patient 2 (Proband’s Father)
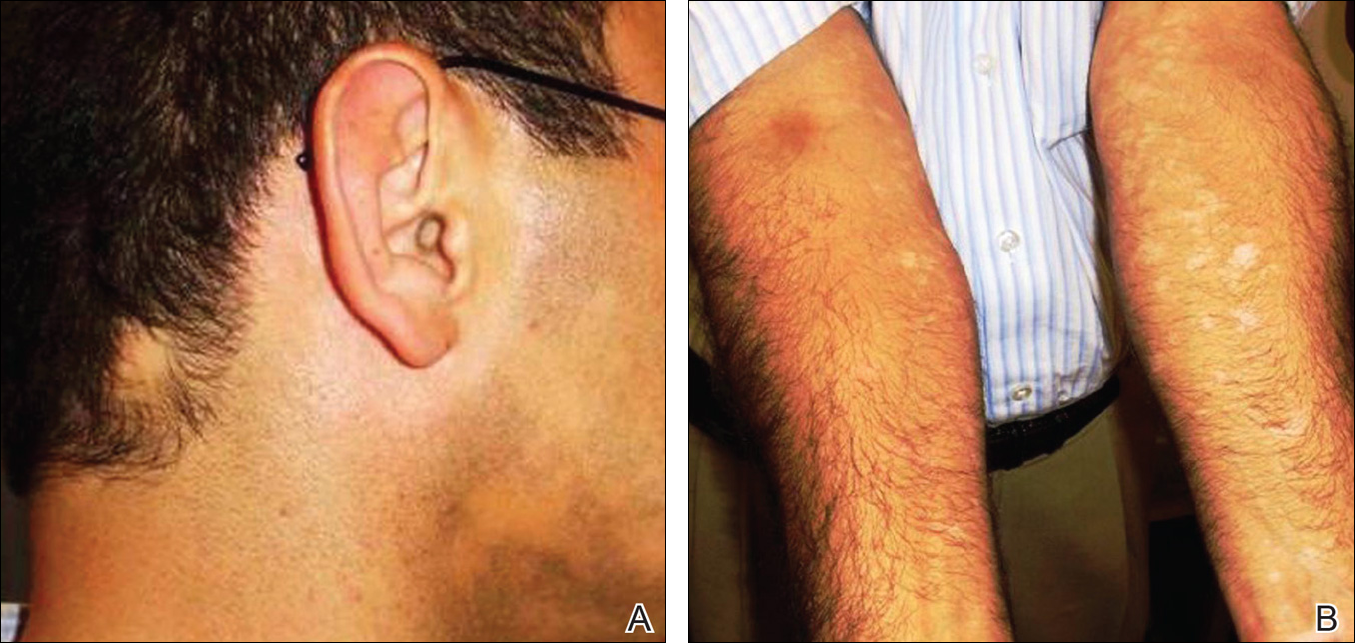
A 38-year-old man presented with a 16-year history of recurrent loss and regrowth of hair on the scalp and beard area and white spots on the penis and arms. He previously had not undergone any treatments. Dermatologic examination revealed well-circumscribed, 1- to 4-cm, hairless patches on the occipital region of the scalp and in the beard area (Figure 2A) and multiple, 2- to 10-mm, vitiliginous lesions on both forearms (Figure 2B) and the penis. The patient had been unemployed for 6 months. Psychological evaluation revealed obsessive-compulsive disorder and obsessive-compulsive personality disorder.
Patient 3 (Proband’s Mother)
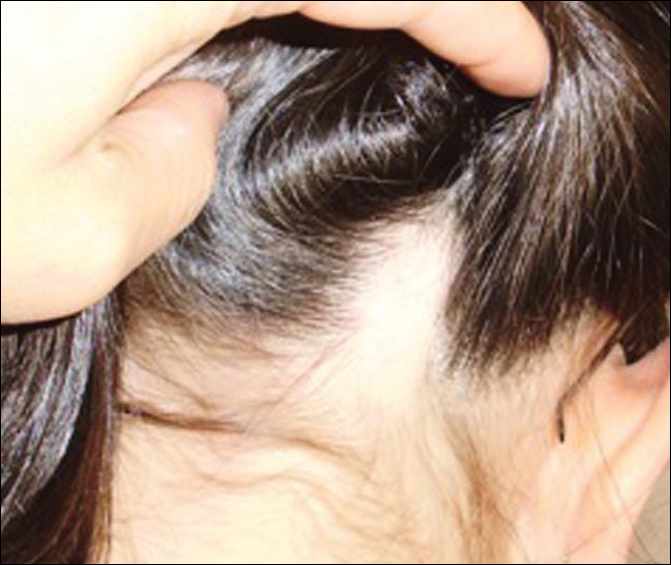
A 32-year-old woman presented with a 3-year history of chronic AA. She previously had not undergone any treatments. Dermatologic examination revealed 2 well-circumscribed, 3- to 4-cm patches of hair loss on the occipital and left temporal regions of the scalp (Figure 3). Psychological evaluation revealed obsessive-compulsive personality disorder and depression. The patient did not have any autoimmune diseases.

Patient 4 (Proband’s Sister)
A 10-year-old girl presented with a 6-year history of recurrent, self-limited AA on various areas of scalp. She previously had not undergone any treatments. Dermatologic examination revealed a 3-cm hairless patch on the occipital region of the scalp (Figure 4). Psychiatric evaluation revealed narcissistic personality disorder, anxiety, and lack of self-confidence.
Laboratory Evaluation and HLA Antigen DNA Typing
Laboratory testing including complete blood cell count; liver, kidney, and thyroid function; and vitamin B12, zinc, folic acid, and fasting blood sugar levels were performed in all patients.
HLA antigen DNA typing was performed by polymerase chain reaction with sequence-specific primers in all patients after informed consent was obtained.
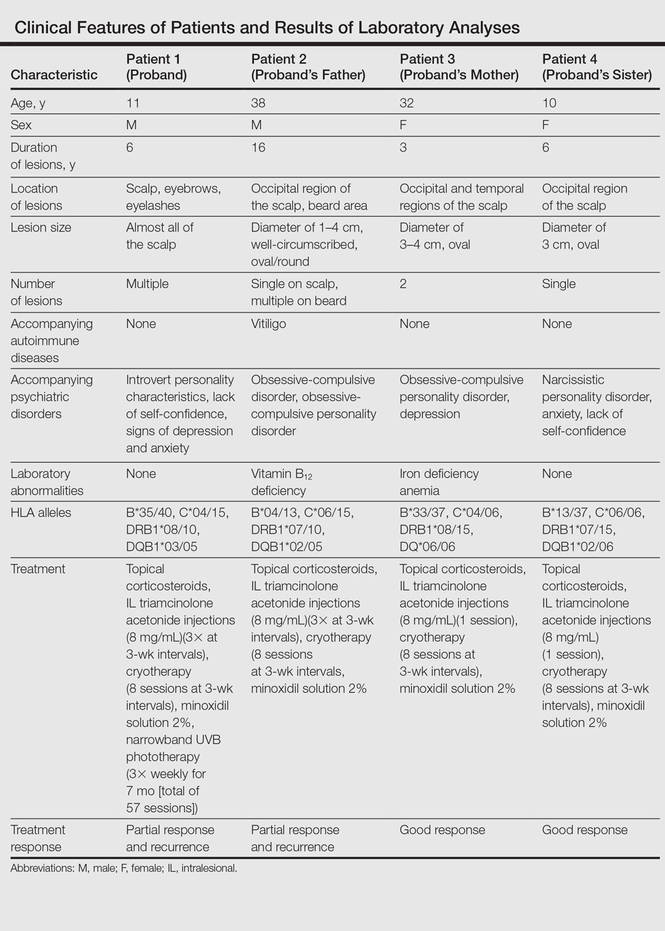
Clinical and laboratory examinations revealed no symptoms or findings of Epstein-Barr virus and cytomegalovirus infections, cicatricial alopecia, or connective tissue diseases in any of the patients. HLA antigen DNA typing revealed the following HLA alleles: B*35/40, C*04/15, DRB1*08/10, and DQB1*03/05 in patient 1; B*04/13, C*06/15, DRB1*07/10, and DQB1*02/05 in patient 2; B*33/37, C*04/06, DRB1*08/15, and DQ*06/06 in patient 3; B*13/37, C*06/06, DRB1*07/15, and DQB1*02/06 in patient 4.
Laboratory testing revealed vitamin B12 deficiency in patient 2 and iron deficiency anemia in patient 3; all other laboratory tests were within reference range. Antithyroglobulin and antithyroid peroxidase autoantibodies were all negative. Clinical features and laboratory analyses for all patients are summarized in the Table.
Treatment
All patients were recommended psychiatric therapy and started on dermatologic treatments. Topical corticosteroids, intralesional triamcinolone acetonide (8 mg/mL) injections into areas of hair loss, 8 total sessions of cryotherapy administered at 3-week intervals, and minoxidil solution 2% were administered respectively to all 4 patients. Alopecia areata in patients 3 and 4 completely regressed; however, no benefit was observed in patients 1 and 2 after 1 year of treatment. Because there was no response to the prior interventions, patient 1 was started on treatment with cyclosporine 2.5 mg/kg twice daily. However, therapy was discontinued after 1 month and treatment with narrowband UVB (3 times per week for 7 months [total of 57 sessions]) and topical corticosteroids were initiated (Table). The patient partially benefited from these regimens and recurrence was observed during the course of the treatment.
Although it was recommended that all 4 patients undergo psychiatric treatment and follow-up regularly with a psychiatrist, the patients declined. After approximately 1 year of dermatologic treatment, all 4 patients were lost to follow-up.
Comment
The etiopathogenesis of AA is unclear, but there is strong evidence suggesting that it is a T-cell–mediated autoimmune disease targeting the hair follicles. Common association of AA with autoimmune diseases such as vitiligo and thyroiditis support the immunological origin of the disease.3 In our case, patient 2 had AA along with vitiligo, but no associated autoimmune diseases (eg, vitiligo, diabetes mellitus, pernicious anemia, thyroid diseases) were noted in the other patients. Genetic and environmental factors are known to be influential as much as immune dysfunction in the etiology of AA.2
The presence of family history in 20% of patients supports the genetic predisposition of AA.4 In a genetic study by Martinez-Mir et al,5 susceptibility loci for AA were demonstrated on chromosomes 6, 10, 16, and 18. HLA antigen alleles, which provide predisposition to AA, have been investigated and associations with many different HLA antigens have been described for AA. In these studies, a relationship between AA and HLA class I antigens was not determined. Notable results mainly focused on HLA class II antigens.6-8 Colombe et al7 and Marques Da Costa et al8 demonstrated that long-lasting alopecia totalis or alopecia universalis (AT/AU) patients had a strong relationship with HLA-DRB1*1104; DRB1*04/05 was reported to be the most frequent HLA group among all patients with AA.6-10 In contrast, we did not detect these alleles in our patients. Colombe et al7,11 noted that HLA-DQB1*03 is a marker for both patch-type AA and AT/AU. Colombe et al10 showed that HLA-DQB1*03 was present in more than 80% of patients (N=286) with long-lasting AA. Barahmani et al9 confirmed a strong association between HLA-DQB1*0301, DRB1*1104, and AT/AU. In our patients, we detected HLA-DQB1*03/05 in patient 1 who had the earliest onset and most severe presentation of AA. In some studies, HLA-DRB1*03 was found to be less frequent in patients with AA, and this allele was suggested to be a protective factor.6,12 However, this allele was not detected in any of our patients.
The association of HLA alleles and AA has been investigated in Turkish patients with AA.13-15 Akar et al13 and Kavak et al14 detected that the frequency of HLA-DQB1*03 allele was remarkably higher in patients with AA than in healthy controls. These results were consistent with Colombe et al.10 On the other hand, Kavak et al14 reported that the frequency of HLA-DR16 was decreased in the patient group with AA. In another study, the frequency of HLA-B62 was increased in patients with AA compared to healthy controls.15 The HLA-DQB1*03 allele was found to be associated with AA in only patient 1 in our case series, and HLA alleles were not commonly shared among the 4 patients. Additionally, lack of consanguinity between patients 2 and 3 (the parents) also suggested that genetic factors were not involved in our familial cases.
Blaumeiser et al16 reported a lifetime risk of 7.4% in parents and 7.1% in siblings of 206 AA patients; however, because these studies investigated the presence of AA in any given life period of the family members, their results do not reflect frequency of simultaneous AA presence within one family. In a literature search using PubMed, Google Scholar, and other national databases for the terms alopecia areata as well as family, sibling, concurrently, concomitant, co-existent, and simultaneously, only 2 cases involving a husband and wife and 1 case of 2 siblings who concurrently had AA have been previously reported.17,18 Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.19 Among our patients, despite different age of onset and duration, AA was simultaneously present in the entire family.
Moreover, Rodriguez et al20 reported that the concordance rate of AA in identical twins was 42% and dizygotic twins was 10%. Environmental factors and infections also have been implicated in the etiology of AA. Infections caused by viruses such as cytomegalovirus and Epstein-Barr virus have been thought to be potential triggering factors; however, no evidence has been found.21,22 The clinical and laboratory examinations in our study did not reveal any presence and/or history of any known infectious disease, and there was no history of contact with water infected by acrylamide or a similar chemical.
Various life events and intense psychological stress may play an important role in triggering AA. Depression, hysteria, psychopathic deviance, psychasthenia, schizophrenia, anxiety, health concerns, bizarre thoughts, and family problems were found to be more frequent in patients with AA than healthy controls.23 The most common psychological disorders associated with AA are generalized anxiety disorder, major depressive disorder, adjustment disorders, and phobias.1,24 Ruiz-Doblado et al25 determined the presence of psychiatric comorbidities in 66% (21/32) of AA cases. Chu et al26 reported that the differences in ages of onset of AA revealed differences in psychiatric comorbidities. The risk for depression was higher in patients with AA younger than 20 years. An increased rate of anxiety was detected with patients with an onset of AA between the ages of 20 and 39 years. Obsessive-compulsive disorder and anxiety were more common in patients aged 40 to 59 years. Interestingly, the investigators also observed that approximately 50% of psychiatric disorders occurred prior to onset of AA.26 One study showed higher rates of stressful life events in children than in controls.27 Ghanizadeh24 reported at least 1 psychiatric disorder in 78% (11/14) of children and adolescents with AA. In the same study, obsessive-compulsive disorder was found to be the second common condition following major depression in AA.24
In our patients, psychiatric evaluations revealed obsessive-compulsive personality disorder in patients 2 and 3, depression in patient 3, and symptoms of anxiety with a lack of self-confidence in patients 1 and 4. Psychiatric disorders affecting the entire family may stem from unemployment of the father. Similar to the results noted in prior studies, depression, the most commonly associated psychiatric disorder of AA, was present in 2 of 4 patients. Obsessive-compulsive disorder, the second most common psychiatric disorder among AA patients, was present in patients 2 and 3. These results indicate that AA may be associated with shared stressful events and psychiatric disorders. Therefore, in addition to dermatologic treatment, it was recommended that all patients undergo psychiatric treatment and follow-up regularly with a psychiatrist; however, the patients declined. At the end of a 1-year treatment period and follow-up, resistance to therapy with minimal recovery followed by a rapid recurrence was determined in patients 1 and 2.
Conclusion
This report demonstrated that familial AA was strongly associated with psychological disorders that were detected in all patients. In our patients, HLA alleles did not seem to have a role in the development of familial AA. These results suggest that HLA was not associated with AA triggered by psychological stress. We believe that psychological disorders and stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
- García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, et al. Alopecia areata, stress and psychiatric disorders: a review. J Dermatol. 1999;26:625-632.
- Bhat YJ, Manzoor S, Khan AR, et al. Trace element levels in alopecia areata. Indian J Dermatol Venereol Leprol. 2009;75:29-31.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol. 2004;14:364-370.
- Green J, Sinclair RD. Genetics of alopecia areata. Australas J Dermatol. 2000;41:213-218.
- Martinez-Mir A, Zlotogorski A, Gordon D, et al.Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316-328.
- Entz P, Blaumeiser B, Betz RC, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006;16:363-367.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II alleles in long-standing alopecia totalis/alopecia universalis and long-standing patchy alopecia areata differentiate these two clinical groups. J Invest Dermatol. 1995;104(suppl 5):4-5.
- Marques Da Costa C, Dupont E, Van der Cruys M, et al. Earlier occurrence of severe alopecia areata in HLA-DRB1*11-positive patients. Dermatology. 2006;213:12-14.
- Barahmani N, de Andrade M, Slusser JP, et al. Human leukocyte antigen class II alleles are associated with risk of alopecia areata. J Invest Dermatol. 2008;128:240-243.
- Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999;4:216-219.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5, pt 1):757-764.
- Broniarczyk-Dyła G, Prusińska-Bratoś M, Dubla-Berner M, et al. The protective role of the HLA-DR locus in patients with various clinical types of alopecia areata. Arch Immunol Ther Exp (Warsz). 2002;50:333-336.
- Akar A, Orkunuglu E, Sengul A, et al. HLA class II alleles in patients with alopecia areata. Eur J Dermatol. 2002;12:236-239.
- Kavak A, Baykal C, Ozarmagan G, et al. HLA in alopecia areata. Int J Dermatol. 2000;30:589-592.
- Aliagaoglu C, Pirim I, Atasoy M, et al. Association between alopecia areata and HLA class I and II in Turkey. J Dermatol. 2005;32:711-714.
- Blaumeiser B, Goot I, Fimmers R, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006;54:627-632.
- Zalka AD, Byarlay JA, Goldsmith LA. Alopecia a deux: simultaneous occurrence of alopecia in a husband and wife. Arch Dermatol. 1994;130:390-392.
- Menon R, Kiran C. Concomitant presentation of alopecia areata in siblings: a rare occurrence. Int J Trichology. 2012;4:86-88.
- Valsecchi R, Vicari O, Frigeni A, et al. Familial alopecia areata-genetic susceptibility or coincidence? Acta Derm Venereol (Stockh). 1985;65:175-177.
- Rodriguez TA, Fernandes KE, Dresser KL, et al. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. 2010;62:525-527.
- Rodriguez TA, Duvic M. Onset of alopecia areata after Epstein Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137-139.
- Offidani A, Amerio P, Bernardini ML, et al. Role of cytomegalovirus replication in alopecia areata pathogenesis. J Cutan Med Surg. 2000;4:63-65.
- Alfani S, Antinone V, Mozzetta A, et al. Psychological status of patients with alopecia areata. Acta Derm Venereol. 2012;92:304-306.
- Ghanizadeh A. Comorbidity of psychiatric disorders in children and adolescents with alopecia areata in a child and adolescent psychiatry clinical sample. Int J Dermatol. 2008;47:1118-1120.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol. 2003;42:434-437.
- Chu SY, Chen YJ, Tseng WC, et al. Psychiatric comorbidities in patients with alopecia areata in Taiwan: a case-control study. Br J Dermatol. 2012;166:525-531.
- Manolache L, Petrescu-Seceleanu D, Benea V. Alopecia areata and stressful events in children. J Eur Acad Dermatol Venereol. 2009;23:107-109.
Alopecia areata (AA) presents as sudden, nonscarring, recurrent hair loss characterized by well-circumscribed hairless patches. Although AA may be observed on any hair-bearing areas of the body, the most commonly affected sites are the scalp, beard area, eyebrows, and eyelashes.1 The incidence of AA is 1% to 2% in the general population and it is more common in males than females younger than 40 years.2 Although the majority of patients present with self-limited and well-circumscribed hairless patches that resolve within 2 years, 7% to 10% display a chronic and severe prognosis.3
The etiopathogenesis of AA is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.2 Alopecia areata is observed to occur sporadically in most patients. Family history has been found in 3% to 42% of cases, but simultaneous occurrence of AA in family members is rare.4 In this case series, we present 4 cases of active AA lesions occurring simultaneously in a family who also had associated psychologic disorders.
Case Series
Patient 1 (Proband)
An 11-year-old boy presented with a 6-year history of ongoing AA with recurrent improvement and relapses on the scalp, eyebrows, and eyelashes. Various topical and oral medications had been prescribed by several outside dermatologists; however, these treatments provided minimal benefit and resulted in the recurrence of AA. Dermatologic examination revealed hair loss on the entire frontal, parietal, and temporal regions of the scalp, as well as half of the occipital region and one-third of the lateral side of the eyebrows (Figure 1). Psychological evaluation revealed introvert personality characteristics, lack of self-confidence, and signs of depression and anxiety.
Patient 2 (Proband’s Father)
A 38-year-old man presented with a 16-year history of recurrent loss and regrowth of hair on the scalp and beard area and white spots on the penis and arms. He previously had not undergone any treatments. Dermatologic examination revealed well-circumscribed, 1- to 4-cm, hairless patches on the occipital region of the scalp and in the beard area (Figure 2A) and multiple, 2- to 10-mm, vitiliginous lesions on both forearms (Figure 2B) and the penis. The patient had been unemployed for 6 months. Psychological evaluation revealed obsessive-compulsive disorder and obsessive-compulsive personality disorder.
Patient 3 (Proband’s Mother)
A 32-year-old woman presented with a 3-year history of chronic AA. She previously had not undergone any treatments. Dermatologic examination revealed 2 well-circumscribed, 3- to 4-cm patches of hair loss on the occipital and left temporal regions of the scalp (Figure 3). Psychological evaluation revealed obsessive-compulsive personality disorder and depression. The patient did not have any autoimmune diseases.
Patient 4 (Proband’s Sister)
A 10-year-old girl presented with a 6-year history of recurrent, self-limited AA on various areas of scalp. She previously had not undergone any treatments. Dermatologic examination revealed a 3-cm hairless patch on the occipital region of the scalp (Figure 4). Psychiatric evaluation revealed narcissistic personality disorder, anxiety, and lack of self-confidence.
Laboratory Evaluation and HLA Antigen DNA Typing
Laboratory testing including complete blood cell count; liver, kidney, and thyroid function; and vitamin B12, zinc, folic acid, and fasting blood sugar levels were performed in all patients.
HLA antigen DNA typing was performed by polymerase chain reaction with sequence-specific primers in all patients after informed consent was obtained.
Clinical and laboratory examinations revealed no symptoms or findings of Epstein-Barr virus and cytomegalovirus infections, cicatricial alopecia, or connective tissue diseases in any of the patients. HLA antigen DNA typing revealed the following HLA alleles: B*35/40, C*04/15, DRB1*08/10, and DQB1*03/05 in patient 1; B*04/13, C*06/15, DRB1*07/10, and DQB1*02/05 in patient 2; B*33/37, C*04/06, DRB1*08/15, and DQ*06/06 in patient 3; B*13/37, C*06/06, DRB1*07/15, and DQB1*02/06 in patient 4.
Laboratory testing revealed vitamin B12 deficiency in patient 2 and iron deficiency anemia in patient 3; all other laboratory tests were within reference range. Antithyroglobulin and antithyroid peroxidase autoantibodies were all negative. Clinical features and laboratory analyses for all patients are summarized in the Table.
Treatment
All patients were recommended psychiatric therapy and started on dermatologic treatments. Topical corticosteroids, intralesional triamcinolone acetonide (8 mg/mL) injections into areas of hair loss, 8 total sessions of cryotherapy administered at 3-week intervals, and minoxidil solution 2% were administered respectively to all 4 patients. Alopecia areata in patients 3 and 4 completely regressed; however, no benefit was observed in patients 1 and 2 after 1 year of treatment. Because there was no response to the prior interventions, patient 1 was started on treatment with cyclosporine 2.5 mg/kg twice daily. However, therapy was discontinued after 1 month and treatment with narrowband UVB (3 times per week for 7 months [total of 57 sessions]) and topical corticosteroids were initiated (Table). The patient partially benefited from these regimens and recurrence was observed during the course of the treatment.
Although it was recommended that all 4 patients undergo psychiatric treatment and follow-up regularly with a psychiatrist, the patients declined. After approximately 1 year of dermatologic treatment, all 4 patients were lost to follow-up.
Comment
The etiopathogenesis of AA is unclear, but there is strong evidence suggesting that it is a T-cell–mediated autoimmune disease targeting the hair follicles. Common association of AA with autoimmune diseases such as vitiligo and thyroiditis support the immunological origin of the disease.3 In our case, patient 2 had AA along with vitiligo, but no associated autoimmune diseases (eg, vitiligo, diabetes mellitus, pernicious anemia, thyroid diseases) were noted in the other patients. Genetic and environmental factors are known to be influential as much as immune dysfunction in the etiology of AA.2
The presence of family history in 20% of patients supports the genetic predisposition of AA.4 In a genetic study by Martinez-Mir et al,5 susceptibility loci for AA were demonstrated on chromosomes 6, 10, 16, and 18. HLA antigen alleles, which provide predisposition to AA, have been investigated and associations with many different HLA antigens have been described for AA. In these studies, a relationship between AA and HLA class I antigens was not determined. Notable results mainly focused on HLA class II antigens.6-8 Colombe et al7 and Marques Da Costa et al8 demonstrated that long-lasting alopecia totalis or alopecia universalis (AT/AU) patients had a strong relationship with HLA-DRB1*1104; DRB1*04/05 was reported to be the most frequent HLA group among all patients with AA.6-10 In contrast, we did not detect these alleles in our patients. Colombe et al7,11 noted that HLA-DQB1*03 is a marker for both patch-type AA and AT/AU. Colombe et al10 showed that HLA-DQB1*03 was present in more than 80% of patients (N=286) with long-lasting AA. Barahmani et al9 confirmed a strong association between HLA-DQB1*0301, DRB1*1104, and AT/AU. In our patients, we detected HLA-DQB1*03/05 in patient 1 who had the earliest onset and most severe presentation of AA. In some studies, HLA-DRB1*03 was found to be less frequent in patients with AA, and this allele was suggested to be a protective factor.6,12 However, this allele was not detected in any of our patients.
The association of HLA alleles and AA has been investigated in Turkish patients with AA.13-15 Akar et al13 and Kavak et al14 detected that the frequency of HLA-DQB1*03 allele was remarkably higher in patients with AA than in healthy controls. These results were consistent with Colombe et al.10 On the other hand, Kavak et al14 reported that the frequency of HLA-DR16 was decreased in the patient group with AA. In another study, the frequency of HLA-B62 was increased in patients with AA compared to healthy controls.15 The HLA-DQB1*03 allele was found to be associated with AA in only patient 1 in our case series, and HLA alleles were not commonly shared among the 4 patients. Additionally, lack of consanguinity between patients 2 and 3 (the parents) also suggested that genetic factors were not involved in our familial cases.
Blaumeiser et al16 reported a lifetime risk of 7.4% in parents and 7.1% in siblings of 206 AA patients; however, because these studies investigated the presence of AA in any given life period of the family members, their results do not reflect frequency of simultaneous AA presence within one family. In a literature search using PubMed, Google Scholar, and other national databases for the terms alopecia areata as well as family, sibling, concurrently, concomitant, co-existent, and simultaneously, only 2 cases involving a husband and wife and 1 case of 2 siblings who concurrently had AA have been previously reported.17,18 Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.19 Among our patients, despite different age of onset and duration, AA was simultaneously present in the entire family.
Moreover, Rodriguez et al20 reported that the concordance rate of AA in identical twins was 42% and dizygotic twins was 10%. Environmental factors and infections also have been implicated in the etiology of AA. Infections caused by viruses such as cytomegalovirus and Epstein-Barr virus have been thought to be potential triggering factors; however, no evidence has been found.21,22 The clinical and laboratory examinations in our study did not reveal any presence and/or history of any known infectious disease, and there was no history of contact with water infected by acrylamide or a similar chemical.
Various life events and intense psychological stress may play an important role in triggering AA. Depression, hysteria, psychopathic deviance, psychasthenia, schizophrenia, anxiety, health concerns, bizarre thoughts, and family problems were found to be more frequent in patients with AA than healthy controls.23 The most common psychological disorders associated with AA are generalized anxiety disorder, major depressive disorder, adjustment disorders, and phobias.1,24 Ruiz-Doblado et al25 determined the presence of psychiatric comorbidities in 66% (21/32) of AA cases. Chu et al26 reported that the differences in ages of onset of AA revealed differences in psychiatric comorbidities. The risk for depression was higher in patients with AA younger than 20 years. An increased rate of anxiety was detected with patients with an onset of AA between the ages of 20 and 39 years. Obsessive-compulsive disorder and anxiety were more common in patients aged 40 to 59 years. Interestingly, the investigators also observed that approximately 50% of psychiatric disorders occurred prior to onset of AA.26 One study showed higher rates of stressful life events in children than in controls.27 Ghanizadeh24 reported at least 1 psychiatric disorder in 78% (11/14) of children and adolescents with AA. In the same study, obsessive-compulsive disorder was found to be the second common condition following major depression in AA.24
In our patients, psychiatric evaluations revealed obsessive-compulsive personality disorder in patients 2 and 3, depression in patient 3, and symptoms of anxiety with a lack of self-confidence in patients 1 and 4. Psychiatric disorders affecting the entire family may stem from unemployment of the father. Similar to the results noted in prior studies, depression, the most commonly associated psychiatric disorder of AA, was present in 2 of 4 patients. Obsessive-compulsive disorder, the second most common psychiatric disorder among AA patients, was present in patients 2 and 3. These results indicate that AA may be associated with shared stressful events and psychiatric disorders. Therefore, in addition to dermatologic treatment, it was recommended that all patients undergo psychiatric treatment and follow-up regularly with a psychiatrist; however, the patients declined. At the end of a 1-year treatment period and follow-up, resistance to therapy with minimal recovery followed by a rapid recurrence was determined in patients 1 and 2.
Conclusion
This report demonstrated that familial AA was strongly associated with psychological disorders that were detected in all patients. In our patients, HLA alleles did not seem to have a role in the development of familial AA. These results suggest that HLA was not associated with AA triggered by psychological stress. We believe that psychological disorders and stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
Alopecia areata (AA) presents as sudden, nonscarring, recurrent hair loss characterized by well-circumscribed hairless patches. Although AA may be observed on any hair-bearing areas of the body, the most commonly affected sites are the scalp, beard area, eyebrows, and eyelashes.1 The incidence of AA is 1% to 2% in the general population and it is more common in males than females younger than 40 years.2 Although the majority of patients present with self-limited and well-circumscribed hairless patches that resolve within 2 years, 7% to 10% display a chronic and severe prognosis.3
The etiopathogenesis of AA is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.2 Alopecia areata is observed to occur sporadically in most patients. Family history has been found in 3% to 42% of cases, but simultaneous occurrence of AA in family members is rare.4 In this case series, we present 4 cases of active AA lesions occurring simultaneously in a family who also had associated psychologic disorders.
Case Series
Patient 1 (Proband)
An 11-year-old boy presented with a 6-year history of ongoing AA with recurrent improvement and relapses on the scalp, eyebrows, and eyelashes. Various topical and oral medications had been prescribed by several outside dermatologists; however, these treatments provided minimal benefit and resulted in the recurrence of AA. Dermatologic examination revealed hair loss on the entire frontal, parietal, and temporal regions of the scalp, as well as half of the occipital region and one-third of the lateral side of the eyebrows (Figure 1). Psychological evaluation revealed introvert personality characteristics, lack of self-confidence, and signs of depression and anxiety.
Patient 2 (Proband’s Father)
A 38-year-old man presented with a 16-year history of recurrent loss and regrowth of hair on the scalp and beard area and white spots on the penis and arms. He previously had not undergone any treatments. Dermatologic examination revealed well-circumscribed, 1- to 4-cm, hairless patches on the occipital region of the scalp and in the beard area (Figure 2A) and multiple, 2- to 10-mm, vitiliginous lesions on both forearms (Figure 2B) and the penis. The patient had been unemployed for 6 months. Psychological evaluation revealed obsessive-compulsive disorder and obsessive-compulsive personality disorder.
Patient 3 (Proband’s Mother)
A 32-year-old woman presented with a 3-year history of chronic AA. She previously had not undergone any treatments. Dermatologic examination revealed 2 well-circumscribed, 3- to 4-cm patches of hair loss on the occipital and left temporal regions of the scalp (Figure 3). Psychological evaluation revealed obsessive-compulsive personality disorder and depression. The patient did not have any autoimmune diseases.
Patient 4 (Proband’s Sister)
A 10-year-old girl presented with a 6-year history of recurrent, self-limited AA on various areas of scalp. She previously had not undergone any treatments. Dermatologic examination revealed a 3-cm hairless patch on the occipital region of the scalp (Figure 4). Psychiatric evaluation revealed narcissistic personality disorder, anxiety, and lack of self-confidence.
Laboratory Evaluation and HLA Antigen DNA Typing
Laboratory testing including complete blood cell count; liver, kidney, and thyroid function; and vitamin B12, zinc, folic acid, and fasting blood sugar levels were performed in all patients.
HLA antigen DNA typing was performed by polymerase chain reaction with sequence-specific primers in all patients after informed consent was obtained.
Clinical and laboratory examinations revealed no symptoms or findings of Epstein-Barr virus and cytomegalovirus infections, cicatricial alopecia, or connective tissue diseases in any of the patients. HLA antigen DNA typing revealed the following HLA alleles: B*35/40, C*04/15, DRB1*08/10, and DQB1*03/05 in patient 1; B*04/13, C*06/15, DRB1*07/10, and DQB1*02/05 in patient 2; B*33/37, C*04/06, DRB1*08/15, and DQ*06/06 in patient 3; B*13/37, C*06/06, DRB1*07/15, and DQB1*02/06 in patient 4.
Laboratory testing revealed vitamin B12 deficiency in patient 2 and iron deficiency anemia in patient 3; all other laboratory tests were within reference range. Antithyroglobulin and antithyroid peroxidase autoantibodies were all negative. Clinical features and laboratory analyses for all patients are summarized in the Table.
Treatment
All patients were recommended psychiatric therapy and started on dermatologic treatments. Topical corticosteroids, intralesional triamcinolone acetonide (8 mg/mL) injections into areas of hair loss, 8 total sessions of cryotherapy administered at 3-week intervals, and minoxidil solution 2% were administered respectively to all 4 patients. Alopecia areata in patients 3 and 4 completely regressed; however, no benefit was observed in patients 1 and 2 after 1 year of treatment. Because there was no response to the prior interventions, patient 1 was started on treatment with cyclosporine 2.5 mg/kg twice daily. However, therapy was discontinued after 1 month and treatment with narrowband UVB (3 times per week for 7 months [total of 57 sessions]) and topical corticosteroids were initiated (Table). The patient partially benefited from these regimens and recurrence was observed during the course of the treatment.
Although it was recommended that all 4 patients undergo psychiatric treatment and follow-up regularly with a psychiatrist, the patients declined. After approximately 1 year of dermatologic treatment, all 4 patients were lost to follow-up.
Comment
The etiopathogenesis of AA is unclear, but there is strong evidence suggesting that it is a T-cell–mediated autoimmune disease targeting the hair follicles. Common association of AA with autoimmune diseases such as vitiligo and thyroiditis support the immunological origin of the disease.3 In our case, patient 2 had AA along with vitiligo, but no associated autoimmune diseases (eg, vitiligo, diabetes mellitus, pernicious anemia, thyroid diseases) were noted in the other patients. Genetic and environmental factors are known to be influential as much as immune dysfunction in the etiology of AA.2
The presence of family history in 20% of patients supports the genetic predisposition of AA.4 In a genetic study by Martinez-Mir et al,5 susceptibility loci for AA were demonstrated on chromosomes 6, 10, 16, and 18. HLA antigen alleles, which provide predisposition to AA, have been investigated and associations with many different HLA antigens have been described for AA. In these studies, a relationship between AA and HLA class I antigens was not determined. Notable results mainly focused on HLA class II antigens.6-8 Colombe et al7 and Marques Da Costa et al8 demonstrated that long-lasting alopecia totalis or alopecia universalis (AT/AU) patients had a strong relationship with HLA-DRB1*1104; DRB1*04/05 was reported to be the most frequent HLA group among all patients with AA.6-10 In contrast, we did not detect these alleles in our patients. Colombe et al7,11 noted that HLA-DQB1*03 is a marker for both patch-type AA and AT/AU. Colombe et al10 showed that HLA-DQB1*03 was present in more than 80% of patients (N=286) with long-lasting AA. Barahmani et al9 confirmed a strong association between HLA-DQB1*0301, DRB1*1104, and AT/AU. In our patients, we detected HLA-DQB1*03/05 in patient 1 who had the earliest onset and most severe presentation of AA. In some studies, HLA-DRB1*03 was found to be less frequent in patients with AA, and this allele was suggested to be a protective factor.6,12 However, this allele was not detected in any of our patients.
The association of HLA alleles and AA has been investigated in Turkish patients with AA.13-15 Akar et al13 and Kavak et al14 detected that the frequency of HLA-DQB1*03 allele was remarkably higher in patients with AA than in healthy controls. These results were consistent with Colombe et al.10 On the other hand, Kavak et al14 reported that the frequency of HLA-DR16 was decreased in the patient group with AA. In another study, the frequency of HLA-B62 was increased in patients with AA compared to healthy controls.15 The HLA-DQB1*03 allele was found to be associated with AA in only patient 1 in our case series, and HLA alleles were not commonly shared among the 4 patients. Additionally, lack of consanguinity between patients 2 and 3 (the parents) also suggested that genetic factors were not involved in our familial cases.
Blaumeiser et al16 reported a lifetime risk of 7.4% in parents and 7.1% in siblings of 206 AA patients; however, because these studies investigated the presence of AA in any given life period of the family members, their results do not reflect frequency of simultaneous AA presence within one family. In a literature search using PubMed, Google Scholar, and other national databases for the terms alopecia areata as well as family, sibling, concurrently, concomitant, co-existent, and simultaneously, only 2 cases involving a husband and wife and 1 case of 2 siblings who concurrently had AA have been previously reported.17,18 Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.19 Among our patients, despite different age of onset and duration, AA was simultaneously present in the entire family.
Moreover, Rodriguez et al20 reported that the concordance rate of AA in identical twins was 42% and dizygotic twins was 10%. Environmental factors and infections also have been implicated in the etiology of AA. Infections caused by viruses such as cytomegalovirus and Epstein-Barr virus have been thought to be potential triggering factors; however, no evidence has been found.21,22 The clinical and laboratory examinations in our study did not reveal any presence and/or history of any known infectious disease, and there was no history of contact with water infected by acrylamide or a similar chemical.
Various life events and intense psychological stress may play an important role in triggering AA. Depression, hysteria, psychopathic deviance, psychasthenia, schizophrenia, anxiety, health concerns, bizarre thoughts, and family problems were found to be more frequent in patients with AA than healthy controls.23 The most common psychological disorders associated with AA are generalized anxiety disorder, major depressive disorder, adjustment disorders, and phobias.1,24 Ruiz-Doblado et al25 determined the presence of psychiatric comorbidities in 66% (21/32) of AA cases. Chu et al26 reported that the differences in ages of onset of AA revealed differences in psychiatric comorbidities. The risk for depression was higher in patients with AA younger than 20 years. An increased rate of anxiety was detected with patients with an onset of AA between the ages of 20 and 39 years. Obsessive-compulsive disorder and anxiety were more common in patients aged 40 to 59 years. Interestingly, the investigators also observed that approximately 50% of psychiatric disorders occurred prior to onset of AA.26 One study showed higher rates of stressful life events in children than in controls.27 Ghanizadeh24 reported at least 1 psychiatric disorder in 78% (11/14) of children and adolescents with AA. In the same study, obsessive-compulsive disorder was found to be the second common condition following major depression in AA.24
In our patients, psychiatric evaluations revealed obsessive-compulsive personality disorder in patients 2 and 3, depression in patient 3, and symptoms of anxiety with a lack of self-confidence in patients 1 and 4. Psychiatric disorders affecting the entire family may stem from unemployment of the father. Similar to the results noted in prior studies, depression, the most commonly associated psychiatric disorder of AA, was present in 2 of 4 patients. Obsessive-compulsive disorder, the second most common psychiatric disorder among AA patients, was present in patients 2 and 3. These results indicate that AA may be associated with shared stressful events and psychiatric disorders. Therefore, in addition to dermatologic treatment, it was recommended that all patients undergo psychiatric treatment and follow-up regularly with a psychiatrist; however, the patients declined. At the end of a 1-year treatment period and follow-up, resistance to therapy with minimal recovery followed by a rapid recurrence was determined in patients 1 and 2.
Conclusion
This report demonstrated that familial AA was strongly associated with psychological disorders that were detected in all patients. In our patients, HLA alleles did not seem to have a role in the development of familial AA. These results suggest that HLA was not associated with AA triggered by psychological stress. We believe that psychological disorders and stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
- García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, et al. Alopecia areata, stress and psychiatric disorders: a review. J Dermatol. 1999;26:625-632.
- Bhat YJ, Manzoor S, Khan AR, et al. Trace element levels in alopecia areata. Indian J Dermatol Venereol Leprol. 2009;75:29-31.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol. 2004;14:364-370.
- Green J, Sinclair RD. Genetics of alopecia areata. Australas J Dermatol. 2000;41:213-218.
- Martinez-Mir A, Zlotogorski A, Gordon D, et al.Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316-328.
- Entz P, Blaumeiser B, Betz RC, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006;16:363-367.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II alleles in long-standing alopecia totalis/alopecia universalis and long-standing patchy alopecia areata differentiate these two clinical groups. J Invest Dermatol. 1995;104(suppl 5):4-5.
- Marques Da Costa C, Dupont E, Van der Cruys M, et al. Earlier occurrence of severe alopecia areata in HLA-DRB1*11-positive patients. Dermatology. 2006;213:12-14.
- Barahmani N, de Andrade M, Slusser JP, et al. Human leukocyte antigen class II alleles are associated with risk of alopecia areata. J Invest Dermatol. 2008;128:240-243.
- Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999;4:216-219.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5, pt 1):757-764.
- Broniarczyk-Dyła G, Prusińska-Bratoś M, Dubla-Berner M, et al. The protective role of the HLA-DR locus in patients with various clinical types of alopecia areata. Arch Immunol Ther Exp (Warsz). 2002;50:333-336.
- Akar A, Orkunuglu E, Sengul A, et al. HLA class II alleles in patients with alopecia areata. Eur J Dermatol. 2002;12:236-239.
- Kavak A, Baykal C, Ozarmagan G, et al. HLA in alopecia areata. Int J Dermatol. 2000;30:589-592.
- Aliagaoglu C, Pirim I, Atasoy M, et al. Association between alopecia areata and HLA class I and II in Turkey. J Dermatol. 2005;32:711-714.
- Blaumeiser B, Goot I, Fimmers R, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006;54:627-632.
- Zalka AD, Byarlay JA, Goldsmith LA. Alopecia a deux: simultaneous occurrence of alopecia in a husband and wife. Arch Dermatol. 1994;130:390-392.
- Menon R, Kiran C. Concomitant presentation of alopecia areata in siblings: a rare occurrence. Int J Trichology. 2012;4:86-88.
- Valsecchi R, Vicari O, Frigeni A, et al. Familial alopecia areata-genetic susceptibility or coincidence? Acta Derm Venereol (Stockh). 1985;65:175-177.
- Rodriguez TA, Fernandes KE, Dresser KL, et al. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. 2010;62:525-527.
- Rodriguez TA, Duvic M. Onset of alopecia areata after Epstein Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137-139.
- Offidani A, Amerio P, Bernardini ML, et al. Role of cytomegalovirus replication in alopecia areata pathogenesis. J Cutan Med Surg. 2000;4:63-65.
- Alfani S, Antinone V, Mozzetta A, et al. Psychological status of patients with alopecia areata. Acta Derm Venereol. 2012;92:304-306.
- Ghanizadeh A. Comorbidity of psychiatric disorders in children and adolescents with alopecia areata in a child and adolescent psychiatry clinical sample. Int J Dermatol. 2008;47:1118-1120.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol. 2003;42:434-437.
- Chu SY, Chen YJ, Tseng WC, et al. Psychiatric comorbidities in patients with alopecia areata in Taiwan: a case-control study. Br J Dermatol. 2012;166:525-531.
- Manolache L, Petrescu-Seceleanu D, Benea V. Alopecia areata and stressful events in children. J Eur Acad Dermatol Venereol. 2009;23:107-109.
- García-Hernández MJ, Ruiz-Doblado S, Rodriguez-Pichardo A, et al. Alopecia areata, stress and psychiatric disorders: a review. J Dermatol. 1999;26:625-632.
- Bhat YJ, Manzoor S, Khan AR, et al. Trace element levels in alopecia areata. Indian J Dermatol Venereol Leprol. 2009;75:29-31.
- Alexis AF, Dudda-Subramanya R, Sinha AA. Alopecia areata: autoimmune basis of hair loss. Eur J Dermatol. 2004;14:364-370.
- Green J, Sinclair RD. Genetics of alopecia areata. Australas J Dermatol. 2000;41:213-218.
- Martinez-Mir A, Zlotogorski A, Gordon D, et al.Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316-328.
- Entz P, Blaumeiser B, Betz RC, et al. Investigation of the HLA-DRB1 locus in alopecia areata. Eur J Dermatol. 2006;16:363-367.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II alleles in long-standing alopecia totalis/alopecia universalis and long-standing patchy alopecia areata differentiate these two clinical groups. J Invest Dermatol. 1995;104(suppl 5):4-5.
- Marques Da Costa C, Dupont E, Van der Cruys M, et al. Earlier occurrence of severe alopecia areata in HLA-DRB1*11-positive patients. Dermatology. 2006;213:12-14.
- Barahmani N, de Andrade M, Slusser JP, et al. Human leukocyte antigen class II alleles are associated with risk of alopecia areata. J Invest Dermatol. 2008;128:240-243.
- Colombe BW, Lou CD, Price VH. The genetic basis of alopecia areata: HLA associations with patchy alopecia areata versus alopecia totalis and alopecia universalis. J Investig Dermatol Symp Proc. 1999;4:216-219.
- Colombe BW, Price VH, Khoury EL, et al. HLA class II antigen associations help to define two types of alopecia areata. J Am Acad Dermatol. 1995;33(5, pt 1):757-764.
- Broniarczyk-Dyła G, Prusińska-Bratoś M, Dubla-Berner M, et al. The protective role of the HLA-DR locus in patients with various clinical types of alopecia areata. Arch Immunol Ther Exp (Warsz). 2002;50:333-336.
- Akar A, Orkunuglu E, Sengul A, et al. HLA class II alleles in patients with alopecia areata. Eur J Dermatol. 2002;12:236-239.
- Kavak A, Baykal C, Ozarmagan G, et al. HLA in alopecia areata. Int J Dermatol. 2000;30:589-592.
- Aliagaoglu C, Pirim I, Atasoy M, et al. Association between alopecia areata and HLA class I and II in Turkey. J Dermatol. 2005;32:711-714.
- Blaumeiser B, Goot I, Fimmers R, et al. Familial aggregation of alopecia areata. J Am Acad Dermatol. 2006;54:627-632.
- Zalka AD, Byarlay JA, Goldsmith LA. Alopecia a deux: simultaneous occurrence of alopecia in a husband and wife. Arch Dermatol. 1994;130:390-392.
- Menon R, Kiran C. Concomitant presentation of alopecia areata in siblings: a rare occurrence. Int J Trichology. 2012;4:86-88.
- Valsecchi R, Vicari O, Frigeni A, et al. Familial alopecia areata-genetic susceptibility or coincidence? Acta Derm Venereol (Stockh). 1985;65:175-177.
- Rodriguez TA, Fernandes KE, Dresser KL, et al. Concordance rate of alopecia areata in identical twins supports both genetic and environmental factors. J Am Acad Dermatol. 2010;62:525-527.
- Rodriguez TA, Duvic M. Onset of alopecia areata after Epstein Barr virus infectious mononucleosis. J Am Acad Dermatol. 2008;59:137-139.
- Offidani A, Amerio P, Bernardini ML, et al. Role of cytomegalovirus replication in alopecia areata pathogenesis. J Cutan Med Surg. 2000;4:63-65.
- Alfani S, Antinone V, Mozzetta A, et al. Psychological status of patients with alopecia areata. Acta Derm Venereol. 2012;92:304-306.
- Ghanizadeh A. Comorbidity of psychiatric disorders in children and adolescents with alopecia areata in a child and adolescent psychiatry clinical sample. Int J Dermatol. 2008;47:1118-1120.
- Ruiz-Doblado S, Carrizosa A, Garcia-Hernandez MJ. Alopecia areata: psychiatric comorbidity and adjustment to illness. Int J Dermatol. 2003;42:434-437.
- Chu SY, Chen YJ, Tseng WC, et al. Psychiatric comorbidities in patients with alopecia areata in Taiwan: a case-control study. Br J Dermatol. 2012;166:525-531.
- Manolache L, Petrescu-Seceleanu D, Benea V. Alopecia areata and stressful events in children. J Eur Acad Dermatol Venereol. 2009;23:107-109.
Practice Points
- The etiopathogenesis of alopecia areata (AA) is not clearly understood, but its occurrence and progression can involve immune dysfunction, genetic predisposition, infections, and physical and psychological trauma.
- Alopecia areata is observed to occur sporadically in most patients. Simultaneous presence of AA in more than 3 members of the same family is rare, and these cases have been observed in different generations and time periods.
- HLA antigen alleles, which provide predisposition to AA, have been investigated, and associations with many different HLA antigens have been described for AA. In previous studies, HLA-DQB1*03 allele was reported as the most common HLA allele in patients with AA.
- Psychological disorders and shared stressful life events may play an important role in the occurrence of AA and lead to the development of resistance against treatment in familial and resistant AA cases.
VIDEO: The ins and outs of JAK ihibitors for alopecia
NEWPORT BEACH, CALIF. – The promise of Janus kinase (JAK) inhibitors for alopecia seems to be holding up in the practice of Dr. Natasha Mesinkovska, a dermatologist at the University of California, Irvine.
There’s been much excitement about JAK inhibitors since Yale researchers reported in 2014 that tofacitinib (Xeljanz), a JAK inhibitor approved in the United States for rheumatoid arthritis, appeared to grow a full head of hair, plus body hair, in an essentially hairless 25-year-old man with plaque psoriasis. JAK inhibitors have been under investigation for alopecia ever since. Meanwhile, they are being used off label for hair loss around the country.
In her own practice, Dr. Mesinkovska estimates that about two-thirds of patients have some degree of hair regrowth, with particularly satisfying results in men. About 40 of her alopecia patients have opted for JAK inhibitors so far.
In an interview at the Summit in Aesthetic Medicine, Dr. Mesinkovska shared her insights and tips, as well as promising alopecia results for the psoriasis biologic ustekinumab (Stelara), an interleukin-12 and -23 antagonist. “This is a very exciting time for alopecia areata,” she said.
The Summit in Aesthetic Medicine is held by the Global Academy for Medical Education. Global Academy and this news organization are owned by the same company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NEWPORT BEACH, CALIF. – The promise of Janus kinase (JAK) inhibitors for alopecia seems to be holding up in the practice of Dr. Natasha Mesinkovska, a dermatologist at the University of California, Irvine.
There’s been much excitement about JAK inhibitors since Yale researchers reported in 2014 that tofacitinib (Xeljanz), a JAK inhibitor approved in the United States for rheumatoid arthritis, appeared to grow a full head of hair, plus body hair, in an essentially hairless 25-year-old man with plaque psoriasis. JAK inhibitors have been under investigation for alopecia ever since. Meanwhile, they are being used off label for hair loss around the country.
In her own practice, Dr. Mesinkovska estimates that about two-thirds of patients have some degree of hair regrowth, with particularly satisfying results in men. About 40 of her alopecia patients have opted for JAK inhibitors so far.
In an interview at the Summit in Aesthetic Medicine, Dr. Mesinkovska shared her insights and tips, as well as promising alopecia results for the psoriasis biologic ustekinumab (Stelara), an interleukin-12 and -23 antagonist. “This is a very exciting time for alopecia areata,” she said.
The Summit in Aesthetic Medicine is held by the Global Academy for Medical Education. Global Academy and this news organization are owned by the same company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
NEWPORT BEACH, CALIF. – The promise of Janus kinase (JAK) inhibitors for alopecia seems to be holding up in the practice of Dr. Natasha Mesinkovska, a dermatologist at the University of California, Irvine.
There’s been much excitement about JAK inhibitors since Yale researchers reported in 2014 that tofacitinib (Xeljanz), a JAK inhibitor approved in the United States for rheumatoid arthritis, appeared to grow a full head of hair, plus body hair, in an essentially hairless 25-year-old man with plaque psoriasis. JAK inhibitors have been under investigation for alopecia ever since. Meanwhile, they are being used off label for hair loss around the country.
In her own practice, Dr. Mesinkovska estimates that about two-thirds of patients have some degree of hair regrowth, with particularly satisfying results in men. About 40 of her alopecia patients have opted for JAK inhibitors so far.
In an interview at the Summit in Aesthetic Medicine, Dr. Mesinkovska shared her insights and tips, as well as promising alopecia results for the psoriasis biologic ustekinumab (Stelara), an interleukin-12 and -23 antagonist. “This is a very exciting time for alopecia areata,” she said.
The Summit in Aesthetic Medicine is held by the Global Academy for Medical Education. Global Academy and this news organization are owned by the same company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM THE SUMMIT IN AESTHETIC MEDICINE

Keys to alopecia areata might lie in gut microbiome
SCOTTSDALE, ARIZ. – Wiping out the gut microbiome with antibiotics prevented alopecia areata in a study of mice, providing evidence that the gut microbiome may play a role in alopecia, Dr. James Chen reported at the annual meeting of the Society for Investigative Dermatology.
The finding shows that the bacterial culprits in alopecia “reside in the gut microbiome, and not in the skin,” said Dr. Chen, a postdoctoral research fellow in medical genetics at Columbia University, New York.
Alopecia areata is mediated by autoreactive NKG2D+ CD8+ T cells. Aberrations in the human microbiome underlie several other autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, and type I diabetes, Dr. Chen noted. “The gut microbiome also has been linked to skin conditions, such as acne, psoriasis, and atopic dermatitis,” he added. “So we asked, if we deplete this microbiome with an antibiotic cocktail, do we see an effect on alopecia?”
To find out, he and his coinvestigators grafted skin from C3H/Hej mice, which spontaneously develop alopecia, onto healthy younger mice, causing them to develop alopecia 6-10 weeks later. “Strikingly, we found that treating unaffected mice with an oral antibiotic cocktail prior to grafting completely prevented the development of alopecia areata, and this remained true through 15 weeks,” he said. “This is the first evidence that the gut microbiome could be implicated in alopecia, based on the absence of the phenotype that we see in treated mice.”
The researchers also evaluated whether the skin microbiomes of antibiotic-treated and control mice differed, and determined that the skin samples resembled each other in terms of overall bacterial load and bacterial taxonomic clustering patterns. That suggests that the skin microbiome is not involved in alopecia areata, Dr. Chen said.
Finally, the investigators transferred NKG2D+ CD8+ T cells from the cutaneous lymph nodes of alopecic mice to normal mice that had been pretreated with antibiotics. The treated mice had little infiltration of these T cells into the skin, and lower overall T-cell levels than control mice, Dr. Chen reported.
The investigators are now testing combinations of antibiotics and fecal transplants to pinpoint which gut bacteria make mice susceptible to hair loss. Doing so “will have significant implications on both our understanding of alopecia areata susceptibility, as well as actionable therapeutic targets for treatment” in humans, Dr. Chen said.
The study was funded by the National Institutes of Health, the Medical Research Council, the Dermatology Foundation, Locks of Love Foundation, and NYSTEM (New York State Stem Cell Science). Dr. Chen had no financial disclosures.
SCOTTSDALE, ARIZ. – Wiping out the gut microbiome with antibiotics prevented alopecia areata in a study of mice, providing evidence that the gut microbiome may play a role in alopecia, Dr. James Chen reported at the annual meeting of the Society for Investigative Dermatology.
The finding shows that the bacterial culprits in alopecia “reside in the gut microbiome, and not in the skin,” said Dr. Chen, a postdoctoral research fellow in medical genetics at Columbia University, New York.
Alopecia areata is mediated by autoreactive NKG2D+ CD8+ T cells. Aberrations in the human microbiome underlie several other autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, and type I diabetes, Dr. Chen noted. “The gut microbiome also has been linked to skin conditions, such as acne, psoriasis, and atopic dermatitis,” he added. “So we asked, if we deplete this microbiome with an antibiotic cocktail, do we see an effect on alopecia?”
To find out, he and his coinvestigators grafted skin from C3H/Hej mice, which spontaneously develop alopecia, onto healthy younger mice, causing them to develop alopecia 6-10 weeks later. “Strikingly, we found that treating unaffected mice with an oral antibiotic cocktail prior to grafting completely prevented the development of alopecia areata, and this remained true through 15 weeks,” he said. “This is the first evidence that the gut microbiome could be implicated in alopecia, based on the absence of the phenotype that we see in treated mice.”
The researchers also evaluated whether the skin microbiomes of antibiotic-treated and control mice differed, and determined that the skin samples resembled each other in terms of overall bacterial load and bacterial taxonomic clustering patterns. That suggests that the skin microbiome is not involved in alopecia areata, Dr. Chen said.
Finally, the investigators transferred NKG2D+ CD8+ T cells from the cutaneous lymph nodes of alopecic mice to normal mice that had been pretreated with antibiotics. The treated mice had little infiltration of these T cells into the skin, and lower overall T-cell levels than control mice, Dr. Chen reported.
The investigators are now testing combinations of antibiotics and fecal transplants to pinpoint which gut bacteria make mice susceptible to hair loss. Doing so “will have significant implications on both our understanding of alopecia areata susceptibility, as well as actionable therapeutic targets for treatment” in humans, Dr. Chen said.
The study was funded by the National Institutes of Health, the Medical Research Council, the Dermatology Foundation, Locks of Love Foundation, and NYSTEM (New York State Stem Cell Science). Dr. Chen had no financial disclosures.
SCOTTSDALE, ARIZ. – Wiping out the gut microbiome with antibiotics prevented alopecia areata in a study of mice, providing evidence that the gut microbiome may play a role in alopecia, Dr. James Chen reported at the annual meeting of the Society for Investigative Dermatology.
The finding shows that the bacterial culprits in alopecia “reside in the gut microbiome, and not in the skin,” said Dr. Chen, a postdoctoral research fellow in medical genetics at Columbia University, New York.
Alopecia areata is mediated by autoreactive NKG2D+ CD8+ T cells. Aberrations in the human microbiome underlie several other autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, and type I diabetes, Dr. Chen noted. “The gut microbiome also has been linked to skin conditions, such as acne, psoriasis, and atopic dermatitis,” he added. “So we asked, if we deplete this microbiome with an antibiotic cocktail, do we see an effect on alopecia?”
To find out, he and his coinvestigators grafted skin from C3H/Hej mice, which spontaneously develop alopecia, onto healthy younger mice, causing them to develop alopecia 6-10 weeks later. “Strikingly, we found that treating unaffected mice with an oral antibiotic cocktail prior to grafting completely prevented the development of alopecia areata, and this remained true through 15 weeks,” he said. “This is the first evidence that the gut microbiome could be implicated in alopecia, based on the absence of the phenotype that we see in treated mice.”
The researchers also evaluated whether the skin microbiomes of antibiotic-treated and control mice differed, and determined that the skin samples resembled each other in terms of overall bacterial load and bacterial taxonomic clustering patterns. That suggests that the skin microbiome is not involved in alopecia areata, Dr. Chen said.
Finally, the investigators transferred NKG2D+ CD8+ T cells from the cutaneous lymph nodes of alopecic mice to normal mice that had been pretreated with antibiotics. The treated mice had little infiltration of these T cells into the skin, and lower overall T-cell levels than control mice, Dr. Chen reported.
The investigators are now testing combinations of antibiotics and fecal transplants to pinpoint which gut bacteria make mice susceptible to hair loss. Doing so “will have significant implications on both our understanding of alopecia areata susceptibility, as well as actionable therapeutic targets for treatment” in humans, Dr. Chen said.
The study was funded by the National Institutes of Health, the Medical Research Council, the Dermatology Foundation, Locks of Love Foundation, and NYSTEM (New York State Stem Cell Science). Dr. Chen had no financial disclosures.
AT THE 2016 SID ANNUAL MEETING
Key clinical point: Using antibiotics to eliminate the gut microbiome in mice prevented them from developing alopecia.
Major finding: The mice also had lower levels of cytotoxic T-cell infiltration into the skin, compared with alopecic controls.
Data source: A study of C3H/Hej (alopecic) mice and healthy young mice that received skin grafts from the alopecic phenotype.
Disclosures: The study was funded by the National Institutes of Health, the Medical Research Council, the Dermatology Foundation, Locks of Love Foundation, and NYSTEM (New York State Stem Cell Science). Dr. Chen had no financial disclosures.
JAK inhibitor improves alopecia, with caveats
SCOTTSDALE, ARIZ. – The Janus kinase (JAK) inhibitor tofacitinib dramatically improved several cases of alopecia areata (AA), although some patients relapsed to worse than baseline after completing treatment in a small open label pilot trial.
“Regrowth was demonstrated in 11 out of 12 patients on tofacitinib. Seven out of 12 patients achieved more than 50% regrowth,” Dr. Shawn Sidharthan reported at the annual meeting of the Society for Investigative Dermatology.
Worldwide, alopecia areata, which is caused by immune-mediated destruction of hair follicles, has a lifetime incidence of about 2% (Clin Cosmet Investig Dermatol. 2015;8:397-403). But there are no Food and Drug Administration–approved treatments for AA. Tofacitinib (Xeljanz), which is approved by the FDA for moderate to severe rheumatoid arthritis in adults, is a JAK1 and JAK3 inhibitor that curbs the interferon-gamma response inflammatory pathway, said Dr. Sidharthan of the department of dermatology and genetics at Columbia University, New York.
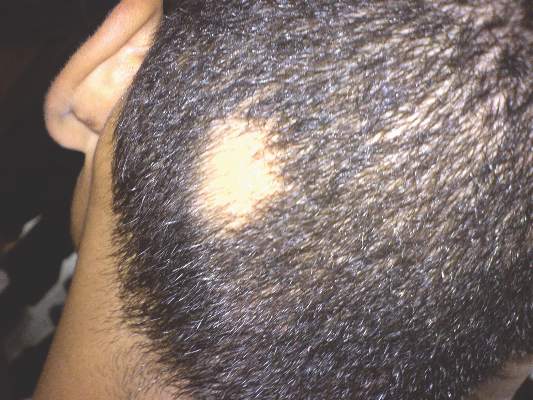
AA shares the same interferon response pathway, and tofacitinib prevented alopecia in mice and led to hair regrowth in a patient with alopecia universalis, he noted.
The single-arm trial included seven patients with moderate to severe patchy AA and five patients with alopecia totalis or alopecia universalis. Patients were treated for 6 months. They initially received 5 mg tofacitinib orally twice daily, which was increased to 10 mg twice daily to improve response. The investigators evaluated patients based on SALT (Severity of Alopecia Tool) scores and the Alopecia Areata Disease Activity Index (ALADIN), which uses three-dimensional bioinformatics to identify groups of genes linked to alopecia.
Seven of 12 patients experienced at least 50% regrowth, including six patients who only improved on 10 mg tofacitinib twice daily, Dr. Sidharthan said. Three additional patients “had good regrowth, but not 50%,” he reported. Among the two remaining patients, one had full regrowth, but dropped out of the study because of uncontrolled hypertension, and one patient with alopecia universalis had little or no regrowth.
Notably, two patients began shedding hair after stopping tofacitinib during the observation period of the study, and their final SALT scores were worse than baseline, Dr. Sidharthan said.
Laboratory monitoring of the cohort revealed no severe adverse events, but one patient paused treatment because of thrombocytopenia. The patient’s platelet count normalized after 2 weeks off tofacitinib, and remained normal when the dose was gradually increased to 10 mg twice daily. Another patient developed leukocytosis that resolved during the off-treatment observation period. One patient who did not comply with instructions to avoid alcohol had elevated liver function tests and was taken off the study. Two patients experienced self-limiting diarrhea, and one patient developed trace hematuria, Dr. Sidharthan noted.
In the study, ALADIN scores correlated with clinical response, he said.
He and his coinvestigators concluded that the overall results “provide a strong rationale for larger clinical trials using JAK inhibitors in alopecia areata,” he said.
Dr. Sidharthan noted that another oral JAK inhibitor, ruxolitinib (Jakafi), led to nearly full hair regrowth in three patients with alopecia in a Columbia University study (Nat Med. 2014 Sep; 20[9]:1043-9).
The Locks of Love Foundation funded the research. Dr. Sidharthan, a clinical research fellow in dermatology at Columbia, had no disclosures.
SCOTTSDALE, ARIZ. – The Janus kinase (JAK) inhibitor tofacitinib dramatically improved several cases of alopecia areata (AA), although some patients relapsed to worse than baseline after completing treatment in a small open label pilot trial.
“Regrowth was demonstrated in 11 out of 12 patients on tofacitinib. Seven out of 12 patients achieved more than 50% regrowth,” Dr. Shawn Sidharthan reported at the annual meeting of the Society for Investigative Dermatology.
Worldwide, alopecia areata, which is caused by immune-mediated destruction of hair follicles, has a lifetime incidence of about 2% (Clin Cosmet Investig Dermatol. 2015;8:397-403). But there are no Food and Drug Administration–approved treatments for AA. Tofacitinib (Xeljanz), which is approved by the FDA for moderate to severe rheumatoid arthritis in adults, is a JAK1 and JAK3 inhibitor that curbs the interferon-gamma response inflammatory pathway, said Dr. Sidharthan of the department of dermatology and genetics at Columbia University, New York.
AA shares the same interferon response pathway, and tofacitinib prevented alopecia in mice and led to hair regrowth in a patient with alopecia universalis, he noted.
The single-arm trial included seven patients with moderate to severe patchy AA and five patients with alopecia totalis or alopecia universalis. Patients were treated for 6 months. They initially received 5 mg tofacitinib orally twice daily, which was increased to 10 mg twice daily to improve response. The investigators evaluated patients based on SALT (Severity of Alopecia Tool) scores and the Alopecia Areata Disease Activity Index (ALADIN), which uses three-dimensional bioinformatics to identify groups of genes linked to alopecia.
Seven of 12 patients experienced at least 50% regrowth, including six patients who only improved on 10 mg tofacitinib twice daily, Dr. Sidharthan said. Three additional patients “had good regrowth, but not 50%,” he reported. Among the two remaining patients, one had full regrowth, but dropped out of the study because of uncontrolled hypertension, and one patient with alopecia universalis had little or no regrowth.
Notably, two patients began shedding hair after stopping tofacitinib during the observation period of the study, and their final SALT scores were worse than baseline, Dr. Sidharthan said.
Laboratory monitoring of the cohort revealed no severe adverse events, but one patient paused treatment because of thrombocytopenia. The patient’s platelet count normalized after 2 weeks off tofacitinib, and remained normal when the dose was gradually increased to 10 mg twice daily. Another patient developed leukocytosis that resolved during the off-treatment observation period. One patient who did not comply with instructions to avoid alcohol had elevated liver function tests and was taken off the study. Two patients experienced self-limiting diarrhea, and one patient developed trace hematuria, Dr. Sidharthan noted.
In the study, ALADIN scores correlated with clinical response, he said.
He and his coinvestigators concluded that the overall results “provide a strong rationale for larger clinical trials using JAK inhibitors in alopecia areata,” he said.
Dr. Sidharthan noted that another oral JAK inhibitor, ruxolitinib (Jakafi), led to nearly full hair regrowth in three patients with alopecia in a Columbia University study (Nat Med. 2014 Sep; 20[9]:1043-9).
The Locks of Love Foundation funded the research. Dr. Sidharthan, a clinical research fellow in dermatology at Columbia, had no disclosures.
SCOTTSDALE, ARIZ. – The Janus kinase (JAK) inhibitor tofacitinib dramatically improved several cases of alopecia areata (AA), although some patients relapsed to worse than baseline after completing treatment in a small open label pilot trial.
“Regrowth was demonstrated in 11 out of 12 patients on tofacitinib. Seven out of 12 patients achieved more than 50% regrowth,” Dr. Shawn Sidharthan reported at the annual meeting of the Society for Investigative Dermatology.
Worldwide, alopecia areata, which is caused by immune-mediated destruction of hair follicles, has a lifetime incidence of about 2% (Clin Cosmet Investig Dermatol. 2015;8:397-403). But there are no Food and Drug Administration–approved treatments for AA. Tofacitinib (Xeljanz), which is approved by the FDA for moderate to severe rheumatoid arthritis in adults, is a JAK1 and JAK3 inhibitor that curbs the interferon-gamma response inflammatory pathway, said Dr. Sidharthan of the department of dermatology and genetics at Columbia University, New York.
AA shares the same interferon response pathway, and tofacitinib prevented alopecia in mice and led to hair regrowth in a patient with alopecia universalis, he noted.
The single-arm trial included seven patients with moderate to severe patchy AA and five patients with alopecia totalis or alopecia universalis. Patients were treated for 6 months. They initially received 5 mg tofacitinib orally twice daily, which was increased to 10 mg twice daily to improve response. The investigators evaluated patients based on SALT (Severity of Alopecia Tool) scores and the Alopecia Areata Disease Activity Index (ALADIN), which uses three-dimensional bioinformatics to identify groups of genes linked to alopecia.
Seven of 12 patients experienced at least 50% regrowth, including six patients who only improved on 10 mg tofacitinib twice daily, Dr. Sidharthan said. Three additional patients “had good regrowth, but not 50%,” he reported. Among the two remaining patients, one had full regrowth, but dropped out of the study because of uncontrolled hypertension, and one patient with alopecia universalis had little or no regrowth.
Notably, two patients began shedding hair after stopping tofacitinib during the observation period of the study, and their final SALT scores were worse than baseline, Dr. Sidharthan said.
Laboratory monitoring of the cohort revealed no severe adverse events, but one patient paused treatment because of thrombocytopenia. The patient’s platelet count normalized after 2 weeks off tofacitinib, and remained normal when the dose was gradually increased to 10 mg twice daily. Another patient developed leukocytosis that resolved during the off-treatment observation period. One patient who did not comply with instructions to avoid alcohol had elevated liver function tests and was taken off the study. Two patients experienced self-limiting diarrhea, and one patient developed trace hematuria, Dr. Sidharthan noted.
In the study, ALADIN scores correlated with clinical response, he said.
He and his coinvestigators concluded that the overall results “provide a strong rationale for larger clinical trials using JAK inhibitors in alopecia areata,” he said.
Dr. Sidharthan noted that another oral JAK inhibitor, ruxolitinib (Jakafi), led to nearly full hair regrowth in three patients with alopecia in a Columbia University study (Nat Med. 2014 Sep; 20[9]:1043-9).
The Locks of Love Foundation funded the research. Dr. Sidharthan, a clinical research fellow in dermatology at Columbia, had no disclosures.
AT THE 2016 SID ANNUAL MEETING
Key clinical point: Tofacitinib dramatically improved several cases of alopecia areata, but some patients relapsed after stopping treatment.
Major finding: Eleven of 12 patients experienced regrowth, including seven with at least 50% regrowth, but two patients relapsed to worse than baseline after stopping treatment.
Data source: The single-center open-label pilot trial evaluated tofacitinib in 12 patients with alopecia areata, totalis, or universalis.
Disclosures: The Locks of Love Foundation funded the research. Dr. Shawn Sidharthan had no disclosures.
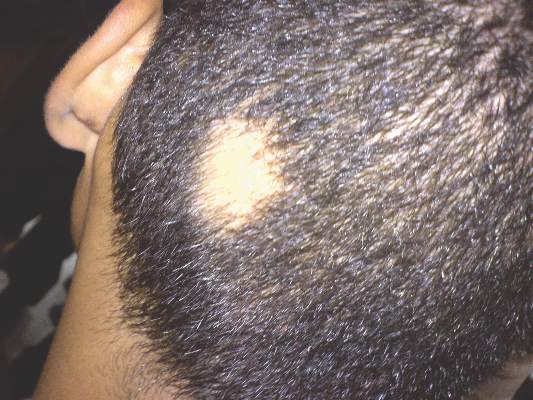
Skin Lesions in Patients Treated With Imatinib Mesylate: A 5-Year Prospective Study
Imatinib mesylate (IM) represents the first-line treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GISTs). Its pharmacological activity is related to a specific action on several tyrosine kinases in different tumors, including Bcr-Abl in CML, c-Kit (CD117) in GIST, and platelet-derived growth factor receptor in dermatofibrosarcoma protuberans.1,2
Imatinib mesylate has been shown to improve progression-free survival and overall survival2; however, it also has several side effects. Among the adverse effects (AEs), less than 10% are nonhematologic, such as nausea, vomiting, diarrhea, muscle cramps, and cutaneous reactions.3,4
We followed patients who were treated with IM for 5 years to identify AEs of therapy.
Methods
The aim of this prospective study was to identify and collect data regarding IM cutaneous side effects so that clinicians can detect AEs early and differentiate them from AEs caused by other medications. All patients were subjected to a median of 5 years’ follow-up. We included all the patients treated with IM and excluded patients who had a history of eczematous dermatitis, psoriasis, renal impairment, or dyshidrosis palmoplantar. Before starting IM, all patients presented for a dermatologic visit. They were subsequently evaluated every 3 months.
The incidence rate was defined as the ratio of patients with cutaneous side effects and the total patients treated with IM. Furthermore, we calculated the ratio between each class of patient with a specific cutaneous manifestation and the entire cohort of patients with cutaneous side effects related to IM.
When necessary, microbiological, serological, and histopathological analyses were performed.
Results
In 60 months, we followed 220 patients treated with IM. Among them, 55 (25%) developed cutaneous side effects (35 males; 20 females). The incidence rate of the patients with cutaneous side effects was 1:4. The median age of the entire cohort was 52.5 years. Fifty patients were being treated for CML and 5 for GISTs. All patients received IM at a dosage of 400 mg daily.
The following skin diseases were observed in patients treated with IM (Table): 19 patients with maculopapular rash with pruritus (no maculopapular rash without pruritus was detected), 7 patients with eczematous dermatitis such as stasis dermatitis and seborrheic dermatitis, 6 patients with onychodystrophy melanonychia (Figure 1), 5 patients with psoriasis, 5 patients with skin cancers including basal cell carcinoma (BCC)(Figure 2), 3 patients with periorbital edema (Figure 3), 3 patients with mycosis, 3 patients with dermatofibromas, 2 patients with dyshidrosis palmoplantar, 1 patient with pityriasis rosea–like eruption (Figure 4), and 1 patient with actinic keratoses on the face. No hypopigmentation or hyperpigmentation, excluding the individual case of melanonychia, was observed.
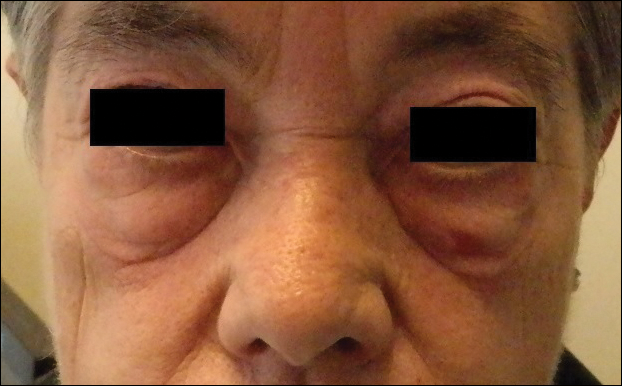
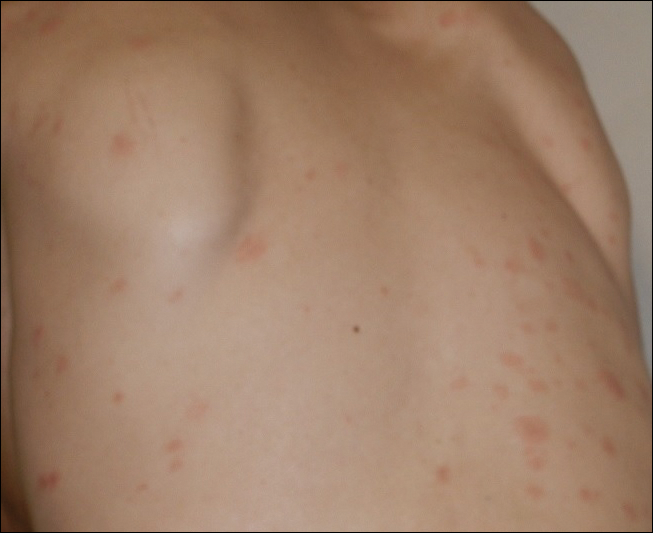
All cutaneous diseases reported in this study appeared after IM therapy (median, 3.8 months). The median time to onset for each cutaneous disorder is reported in the Table. During the first dermatologic visit before starting IM therapy, none of the patients showed any of these cutaneous diseases.
The adverse cutaneous reactions were treated with appropriate drugs. Generally, eczematous dermatitis was treated using topical steroids, emollients, and oral antihistamines. In patients with maculopapular rash with pruritus, oral corticosteroids (eg, betamethasone 3 mg daily or prednisolone 1 mg/kg) in association with antihistamine was necessary. Psoriasis was completely improved with topical betamethasone 0.5 mg and calcipotriol 50 µg. Skin cancers were treated with surgical excision with histologic examination. All treatments are outlined in the Table.
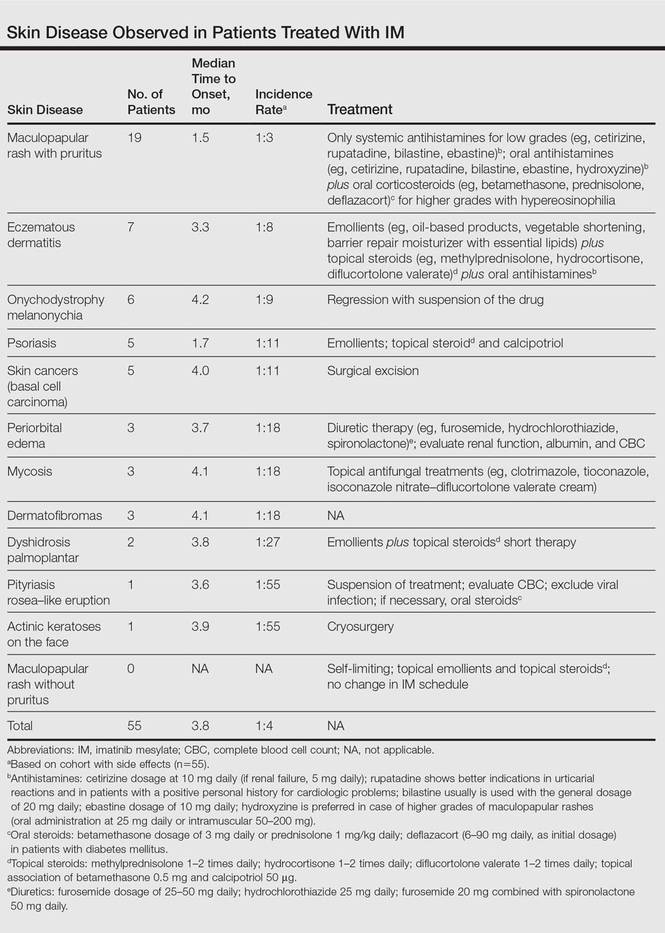
Imatinib mesylate therapy was suspended in 2 patients with maculopapular rash with moderate to severe pruritus; however, despite the temporary suspension of the drug and the appropriate therapies (corticosteroids and antihistamines), cutaneous side effects reappeared 7 to 10 days after therapy resumed. Therefore, the treatment was permanently suspended in these 2 cases and IM was replaced with erlotinib, a second-generation Bcr-Abl tyrosine kinase inhibitor.
Comment
The introduction of IM for the treatment of GIST and CML has changed the history of these diseases. The drug typically is well tolerated and few patients have reported severe AEs. Mild skin reactions are relatively frequent, ranging from 7% to 21% of patients treated.3 In our case, the percentage was relatively higher (25%), likely because of close monitoring of patients, with an increase in the incidence rate.
Imatinib mesylate cutaneous reactions are dose dependent.4 Indeed, in all our cases, the cutaneous reactions arose with an IM dosage of 400 mg daily, which is compatible with the definition of dose-independent cutaneous AEs.
The most common cutaneous AEs reported in the literature were swelling/edema and maculopapular rash. Swelling is the most common AE described during therapy with IM with an incidence of 63% to 84%.5 Swelling often involves the periorbital area and occurs approximately 6 weeks after starting IM. Although its pathogenesis is uncertain, it has been shown that IM blocks the platelet-derived growth factor receptor expressed on blood vessels that regulates the transportation transcapillary. The inhibition of this receptor can lead to increased pore pressure, resulting in edema and erythema. Maculopapular eruptions (50% of cases) often affect the trunk and the limbs and are accompanied by pruritus. Commonly, these rashes arise after 9 weeks of IM therapy. These eruptions are self-limiting and only topical emollients and steroids are required, without any change in IM schedule. To treat maculopapular eruptions with pruritus, oral steroids and antihistamines may be helpful, without suspending IM treatment. When grade 2 or 3 pruriginous maculopapular eruptions arise, the suspension of IM combined with steroids and antihistamines may be necessary. When the readministration of IM is required, it is mandatory to start IM at a lower dose (50–100 mg/d), administering prednisolone 0.5 to 1.0 mg/kg daily. Then, the steroid gradually can be tapered.6 Critical cutaneous AEs that are resistant to supportive measures warrant suspension of IM therapy. However, the incidence of this event is small (<1% of all patients).7
Regarding severe cutaneous AEs from IM therapy, Hsiao et al8 reported the case of Stevens-Johnson syndrome. In this case, IM was immediately stopped and systemic steroids were started. Rarely, erythroderma (grade 4 toxicity) can develop for which a prompt and perpetual suspension of IM is necessary and supportive care therapy with oral and topical steroids is recommended.9
Hyperpigmentation induced by IM, mostly in patients with Fitzpatrick skin types V to VI and with a general prevalence of 16% to 40% in treated patients, often is related to a mutation of c-Kit or other kinases that are activated rather than inhibited by the drug, resulting in overstimulation of melanogenesis.10 The prevalence of Fitzpatrick skin types I to III determined the absence of pigmentation changes in our cohort, excluding melanonychia. Hyperpigmentation was observed in the skin as well as the appendages such as nails, resulting in melanonychia (Figure 1). However, Brazzelli et al11 reported hypopigmentation in 5 white patients treated with IM; furthermore, they found a direct correlation between hypopigmentation and development of skin cancers in these patients. The susceptibility to develop skin cancers may persist, even without a clear manifestation of hypopigmentation, as reported in the current analysis. We documented BCC in 5 patients, 1 patient developed actinic keratoses, and 3 patients developed dermatofibromas. However, these neoplasms probably were not provoked by IM. On the contrary, we did not note squamous cell carcinoma, which was reported by Baskaynak et al12 in 2 CML patients treated with IM.
The administration of IM can be associated with exacerbation of psoriasis. Paradoxically, in genetically predisposed individuals, tumor necrosis factor α (TNF-α) antagonists, such as IM, seem to induce psoriasis, producing IFN-α rather than TNF-α and increasing inflammation.13 In fact, some research shows induction of psoriasis by anti–TNF-α drugs.14-16 Two cases of IM associated with psoriasis have been reported, and both cases represented an exacerbation of previously diagnosed psoriasis.13,17 On the contrary, in our analysis we reported 5 cases of psoriasis vulgaris induced by IM administration. Our patients developed cutaneous psoriatic lesions approximately 1.7 months after the start of IM therapy.
The pityriasis rosea–like eruption (Figure 4) presented as nonpruritic, erythematous, scaly patches on the trunk and extremities, and arose 3.6 months after the start of treatment. This particular cutaneous AE is rare. In 3 case reports, the IM dosage also was 400 mg daily.18-20 The pathophysiology of this rare skin reaction stems from the pharmacological effect of IM rather than a hypersensitivity reaction.18
Deininger et al7 reported that patients with a high basophil count (>20%) rarely show urticarial eruptions after IM due to histamine release from basophils. Premedication with an antihistamine was helpful and the urticarial eruption resolved after normalization in basophil count.7
Given the importance of IM for patients who have limited therapeutic alternatives for their disease and the ability to safely treat the cutaneous AEs, as demonstrated in our analysis, the suspension of IM for dermatological complications is necessary only in rare cases, as shown by the low number of patients (n=2) who had to discontinue therapy. The cutaneous AEs should be diagnosed and treated early with less impact on chemotherapy treatments. The administration of IM should involve a coordinated effort among oncologists and dermatologists to prevent important complications.
- Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-1037.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Breccia M, Carmosimo I, Russo E, et al. Early and tardive skin adverse events in chronic myeloid leukaemia patients treated with imatinib. Eur J Haematol. 2005;74:121-123.
- Ugurel S, Hildebrand R, Dippel E, et al. Dose dependent severe cutaneous reactions to imatinib. Br J Cancer. 2003;88:1157-1159.
- Valeyrie L, Bastuji-Garin S, Revuz J, et al. Adverse cutaneous reactions to imatinib (STI571) in Philadelphia chromosome-positive leukaemias: a prospective study of 54 patients. J Am Acad Dermatol. 2003;48:201-206.
- Scott LC, White JD, Reid R, et al. Management of skin toxicity related to the use of imatinibnmesylate (STI571, GlivecTM) for advanced stage gastrointestinal stromal tumors. Sarcoma. 2005;9:157-160.
- Deininger MW, O’Brien SG, Ford JM, et al. Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol. 2003;21:1637-1647.
- Hsiao LT, Chung HM, Lin JT, et al. Stevens-Johnson syndrome after treatment with STI571: a case report. Br J Haematol. 2002;117:620-622.
- Sehgal VN, Srivastava G, Sardana K. Erythroderma/exfoliative dermatitis: a synopsis. Int J Dermatol. 2004;43:39-47.
- Pietras K, Pahler J, Bergers G, et al. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19.
- Brazzelli V, Prestinari F, Barbagallo T, et al. A long-term time course of colorimetric assessment of the effects of imatinib mesylate on skin pigmentation: a study of five patients. J Eur Acad Dermatol Venerol. 2007;21:384-387.
- Baskaynak G, Kreuzer KA, Schwarz M, et al. Squamous cutaneous epithelial cell carcinoma in two CML patients with progressive disease under imatinib treatment. Eur J Haematol. 2003;70:231-234.
- Cheng H, Geist DE, Piperdi M, et al. Management of imatinib-related exacerbation of psoriasis in a patient with a gastrointestinal stromal tumor. Australas J Dermatol. 2009;50:41-43.
- Faillace C, Duarte GV, Cunha RS, et al. Severe infliximab-induced psoriasis treated with adalimumab switching. Int J Dermatol. 2013;52:234-238.
- Iborra M, Beltrán B, Bastida G, et al. Infliximab and adalimumab-induced psoriasis in Crohn’s disease: a aradoxical side effect. J Crohns Colitis. 2011;5:157-161.
- Fernandes IC, Torres T, Sanches M, et al. Psoriasis induced by infliximab. Acta Med Port. 2011;24:709-712.
- Woo SM, Huh CH, Park KC, et al. Exacerbation of psoriasis in a chronic myelogenous leukemia patient treated with imatinib. J Dermatol. 2007;34:724-726.
- Brazzelli V, Prestinari F, Roveda E, et al. Pytiriasis rosea-like eruption during treatment with imatinib mesylate. description of 3 cases. J Am Acad Dermatol. 2005;53:240-243.
- Konstantapoulos K, Papadogianni A, Dimopoulou M, et al. Pytriasis rosea associated with imatinib (STI571, Gleevec). Dermatology. 2002;205:172-173.
- Cho AY, Kim DH, Im M, et al. Pityriasis rosealike drug eruption induced by imatinib mesylate (Gleevec). Ann Dermatol. 2011;23(suppl 3):360-363.
Imatinib mesylate (IM) represents the first-line treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GISTs). Its pharmacological activity is related to a specific action on several tyrosine kinases in different tumors, including Bcr-Abl in CML, c-Kit (CD117) in GIST, and platelet-derived growth factor receptor in dermatofibrosarcoma protuberans.1,2
Imatinib mesylate has been shown to improve progression-free survival and overall survival2; however, it also has several side effects. Among the adverse effects (AEs), less than 10% are nonhematologic, such as nausea, vomiting, diarrhea, muscle cramps, and cutaneous reactions.3,4
We followed patients who were treated with IM for 5 years to identify AEs of therapy.
Methods
The aim of this prospective study was to identify and collect data regarding IM cutaneous side effects so that clinicians can detect AEs early and differentiate them from AEs caused by other medications. All patients were subjected to a median of 5 years’ follow-up. We included all the patients treated with IM and excluded patients who had a history of eczematous dermatitis, psoriasis, renal impairment, or dyshidrosis palmoplantar. Before starting IM, all patients presented for a dermatologic visit. They were subsequently evaluated every 3 months.
The incidence rate was defined as the ratio of patients with cutaneous side effects and the total patients treated with IM. Furthermore, we calculated the ratio between each class of patient with a specific cutaneous manifestation and the entire cohort of patients with cutaneous side effects related to IM.
When necessary, microbiological, serological, and histopathological analyses were performed.
Results
In 60 months, we followed 220 patients treated with IM. Among them, 55 (25%) developed cutaneous side effects (35 males; 20 females). The incidence rate of the patients with cutaneous side effects was 1:4. The median age of the entire cohort was 52.5 years. Fifty patients were being treated for CML and 5 for GISTs. All patients received IM at a dosage of 400 mg daily.
The following skin diseases were observed in patients treated with IM (Table): 19 patients with maculopapular rash with pruritus (no maculopapular rash without pruritus was detected), 7 patients with eczematous dermatitis such as stasis dermatitis and seborrheic dermatitis, 6 patients with onychodystrophy melanonychia (Figure 1), 5 patients with psoriasis, 5 patients with skin cancers including basal cell carcinoma (BCC)(Figure 2), 3 patients with periorbital edema (Figure 3), 3 patients with mycosis, 3 patients with dermatofibromas, 2 patients with dyshidrosis palmoplantar, 1 patient with pityriasis rosea–like eruption (Figure 4), and 1 patient with actinic keratoses on the face. No hypopigmentation or hyperpigmentation, excluding the individual case of melanonychia, was observed.
All cutaneous diseases reported in this study appeared after IM therapy (median, 3.8 months). The median time to onset for each cutaneous disorder is reported in the Table. During the first dermatologic visit before starting IM therapy, none of the patients showed any of these cutaneous diseases.
The adverse cutaneous reactions were treated with appropriate drugs. Generally, eczematous dermatitis was treated using topical steroids, emollients, and oral antihistamines. In patients with maculopapular rash with pruritus, oral corticosteroids (eg, betamethasone 3 mg daily or prednisolone 1 mg/kg) in association with antihistamine was necessary. Psoriasis was completely improved with topical betamethasone 0.5 mg and calcipotriol 50 µg. Skin cancers were treated with surgical excision with histologic examination. All treatments are outlined in the Table.
Imatinib mesylate therapy was suspended in 2 patients with maculopapular rash with moderate to severe pruritus; however, despite the temporary suspension of the drug and the appropriate therapies (corticosteroids and antihistamines), cutaneous side effects reappeared 7 to 10 days after therapy resumed. Therefore, the treatment was permanently suspended in these 2 cases and IM was replaced with erlotinib, a second-generation Bcr-Abl tyrosine kinase inhibitor.
Comment
The introduction of IM for the treatment of GIST and CML has changed the history of these diseases. The drug typically is well tolerated and few patients have reported severe AEs. Mild skin reactions are relatively frequent, ranging from 7% to 21% of patients treated.3 In our case, the percentage was relatively higher (25%), likely because of close monitoring of patients, with an increase in the incidence rate.
Imatinib mesylate cutaneous reactions are dose dependent.4 Indeed, in all our cases, the cutaneous reactions arose with an IM dosage of 400 mg daily, which is compatible with the definition of dose-independent cutaneous AEs.
The most common cutaneous AEs reported in the literature were swelling/edema and maculopapular rash. Swelling is the most common AE described during therapy with IM with an incidence of 63% to 84%.5 Swelling often involves the periorbital area and occurs approximately 6 weeks after starting IM. Although its pathogenesis is uncertain, it has been shown that IM blocks the platelet-derived growth factor receptor expressed on blood vessels that regulates the transportation transcapillary. The inhibition of this receptor can lead to increased pore pressure, resulting in edema and erythema. Maculopapular eruptions (50% of cases) often affect the trunk and the limbs and are accompanied by pruritus. Commonly, these rashes arise after 9 weeks of IM therapy. These eruptions are self-limiting and only topical emollients and steroids are required, without any change in IM schedule. To treat maculopapular eruptions with pruritus, oral steroids and antihistamines may be helpful, without suspending IM treatment. When grade 2 or 3 pruriginous maculopapular eruptions arise, the suspension of IM combined with steroids and antihistamines may be necessary. When the readministration of IM is required, it is mandatory to start IM at a lower dose (50–100 mg/d), administering prednisolone 0.5 to 1.0 mg/kg daily. Then, the steroid gradually can be tapered.6 Critical cutaneous AEs that are resistant to supportive measures warrant suspension of IM therapy. However, the incidence of this event is small (<1% of all patients).7
Regarding severe cutaneous AEs from IM therapy, Hsiao et al8 reported the case of Stevens-Johnson syndrome. In this case, IM was immediately stopped and systemic steroids were started. Rarely, erythroderma (grade 4 toxicity) can develop for which a prompt and perpetual suspension of IM is necessary and supportive care therapy with oral and topical steroids is recommended.9
Hyperpigmentation induced by IM, mostly in patients with Fitzpatrick skin types V to VI and with a general prevalence of 16% to 40% in treated patients, often is related to a mutation of c-Kit or other kinases that are activated rather than inhibited by the drug, resulting in overstimulation of melanogenesis.10 The prevalence of Fitzpatrick skin types I to III determined the absence of pigmentation changes in our cohort, excluding melanonychia. Hyperpigmentation was observed in the skin as well as the appendages such as nails, resulting in melanonychia (Figure 1). However, Brazzelli et al11 reported hypopigmentation in 5 white patients treated with IM; furthermore, they found a direct correlation between hypopigmentation and development of skin cancers in these patients. The susceptibility to develop skin cancers may persist, even without a clear manifestation of hypopigmentation, as reported in the current analysis. We documented BCC in 5 patients, 1 patient developed actinic keratoses, and 3 patients developed dermatofibromas. However, these neoplasms probably were not provoked by IM. On the contrary, we did not note squamous cell carcinoma, which was reported by Baskaynak et al12 in 2 CML patients treated with IM.
The administration of IM can be associated with exacerbation of psoriasis. Paradoxically, in genetically predisposed individuals, tumor necrosis factor α (TNF-α) antagonists, such as IM, seem to induce psoriasis, producing IFN-α rather than TNF-α and increasing inflammation.13 In fact, some research shows induction of psoriasis by anti–TNF-α drugs.14-16 Two cases of IM associated with psoriasis have been reported, and both cases represented an exacerbation of previously diagnosed psoriasis.13,17 On the contrary, in our analysis we reported 5 cases of psoriasis vulgaris induced by IM administration. Our patients developed cutaneous psoriatic lesions approximately 1.7 months after the start of IM therapy.
The pityriasis rosea–like eruption (Figure 4) presented as nonpruritic, erythematous, scaly patches on the trunk and extremities, and arose 3.6 months after the start of treatment. This particular cutaneous AE is rare. In 3 case reports, the IM dosage also was 400 mg daily.18-20 The pathophysiology of this rare skin reaction stems from the pharmacological effect of IM rather than a hypersensitivity reaction.18
Deininger et al7 reported that patients with a high basophil count (>20%) rarely show urticarial eruptions after IM due to histamine release from basophils. Premedication with an antihistamine was helpful and the urticarial eruption resolved after normalization in basophil count.7
Given the importance of IM for patients who have limited therapeutic alternatives for their disease and the ability to safely treat the cutaneous AEs, as demonstrated in our analysis, the suspension of IM for dermatological complications is necessary only in rare cases, as shown by the low number of patients (n=2) who had to discontinue therapy. The cutaneous AEs should be diagnosed and treated early with less impact on chemotherapy treatments. The administration of IM should involve a coordinated effort among oncologists and dermatologists to prevent important complications.
Imatinib mesylate (IM) represents the first-line treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumors (GISTs). Its pharmacological activity is related to a specific action on several tyrosine kinases in different tumors, including Bcr-Abl in CML, c-Kit (CD117) in GIST, and platelet-derived growth factor receptor in dermatofibrosarcoma protuberans.1,2
Imatinib mesylate has been shown to improve progression-free survival and overall survival2; however, it also has several side effects. Among the adverse effects (AEs), less than 10% are nonhematologic, such as nausea, vomiting, diarrhea, muscle cramps, and cutaneous reactions.3,4
We followed patients who were treated with IM for 5 years to identify AEs of therapy.
Methods
The aim of this prospective study was to identify and collect data regarding IM cutaneous side effects so that clinicians can detect AEs early and differentiate them from AEs caused by other medications. All patients were subjected to a median of 5 years’ follow-up. We included all the patients treated with IM and excluded patients who had a history of eczematous dermatitis, psoriasis, renal impairment, or dyshidrosis palmoplantar. Before starting IM, all patients presented for a dermatologic visit. They were subsequently evaluated every 3 months.
The incidence rate was defined as the ratio of patients with cutaneous side effects and the total patients treated with IM. Furthermore, we calculated the ratio between each class of patient with a specific cutaneous manifestation and the entire cohort of patients with cutaneous side effects related to IM.
When necessary, microbiological, serological, and histopathological analyses were performed.
Results
In 60 months, we followed 220 patients treated with IM. Among them, 55 (25%) developed cutaneous side effects (35 males; 20 females). The incidence rate of the patients with cutaneous side effects was 1:4. The median age of the entire cohort was 52.5 years. Fifty patients were being treated for CML and 5 for GISTs. All patients received IM at a dosage of 400 mg daily.
The following skin diseases were observed in patients treated with IM (Table): 19 patients with maculopapular rash with pruritus (no maculopapular rash without pruritus was detected), 7 patients with eczematous dermatitis such as stasis dermatitis and seborrheic dermatitis, 6 patients with onychodystrophy melanonychia (Figure 1), 5 patients with psoriasis, 5 patients with skin cancers including basal cell carcinoma (BCC)(Figure 2), 3 patients with periorbital edema (Figure 3), 3 patients with mycosis, 3 patients with dermatofibromas, 2 patients with dyshidrosis palmoplantar, 1 patient with pityriasis rosea–like eruption (Figure 4), and 1 patient with actinic keratoses on the face. No hypopigmentation or hyperpigmentation, excluding the individual case of melanonychia, was observed.
All cutaneous diseases reported in this study appeared after IM therapy (median, 3.8 months). The median time to onset for each cutaneous disorder is reported in the Table. During the first dermatologic visit before starting IM therapy, none of the patients showed any of these cutaneous diseases.
The adverse cutaneous reactions were treated with appropriate drugs. Generally, eczematous dermatitis was treated using topical steroids, emollients, and oral antihistamines. In patients with maculopapular rash with pruritus, oral corticosteroids (eg, betamethasone 3 mg daily or prednisolone 1 mg/kg) in association with antihistamine was necessary. Psoriasis was completely improved with topical betamethasone 0.5 mg and calcipotriol 50 µg. Skin cancers were treated with surgical excision with histologic examination. All treatments are outlined in the Table.
Imatinib mesylate therapy was suspended in 2 patients with maculopapular rash with moderate to severe pruritus; however, despite the temporary suspension of the drug and the appropriate therapies (corticosteroids and antihistamines), cutaneous side effects reappeared 7 to 10 days after therapy resumed. Therefore, the treatment was permanently suspended in these 2 cases and IM was replaced with erlotinib, a second-generation Bcr-Abl tyrosine kinase inhibitor.
Comment
The introduction of IM for the treatment of GIST and CML has changed the history of these diseases. The drug typically is well tolerated and few patients have reported severe AEs. Mild skin reactions are relatively frequent, ranging from 7% to 21% of patients treated.3 In our case, the percentage was relatively higher (25%), likely because of close monitoring of patients, with an increase in the incidence rate.
Imatinib mesylate cutaneous reactions are dose dependent.4 Indeed, in all our cases, the cutaneous reactions arose with an IM dosage of 400 mg daily, which is compatible with the definition of dose-independent cutaneous AEs.
The most common cutaneous AEs reported in the literature were swelling/edema and maculopapular rash. Swelling is the most common AE described during therapy with IM with an incidence of 63% to 84%.5 Swelling often involves the periorbital area and occurs approximately 6 weeks after starting IM. Although its pathogenesis is uncertain, it has been shown that IM blocks the platelet-derived growth factor receptor expressed on blood vessels that regulates the transportation transcapillary. The inhibition of this receptor can lead to increased pore pressure, resulting in edema and erythema. Maculopapular eruptions (50% of cases) often affect the trunk and the limbs and are accompanied by pruritus. Commonly, these rashes arise after 9 weeks of IM therapy. These eruptions are self-limiting and only topical emollients and steroids are required, without any change in IM schedule. To treat maculopapular eruptions with pruritus, oral steroids and antihistamines may be helpful, without suspending IM treatment. When grade 2 or 3 pruriginous maculopapular eruptions arise, the suspension of IM combined with steroids and antihistamines may be necessary. When the readministration of IM is required, it is mandatory to start IM at a lower dose (50–100 mg/d), administering prednisolone 0.5 to 1.0 mg/kg daily. Then, the steroid gradually can be tapered.6 Critical cutaneous AEs that are resistant to supportive measures warrant suspension of IM therapy. However, the incidence of this event is small (<1% of all patients).7
Regarding severe cutaneous AEs from IM therapy, Hsiao et al8 reported the case of Stevens-Johnson syndrome. In this case, IM was immediately stopped and systemic steroids were started. Rarely, erythroderma (grade 4 toxicity) can develop for which a prompt and perpetual suspension of IM is necessary and supportive care therapy with oral and topical steroids is recommended.9
Hyperpigmentation induced by IM, mostly in patients with Fitzpatrick skin types V to VI and with a general prevalence of 16% to 40% in treated patients, often is related to a mutation of c-Kit or other kinases that are activated rather than inhibited by the drug, resulting in overstimulation of melanogenesis.10 The prevalence of Fitzpatrick skin types I to III determined the absence of pigmentation changes in our cohort, excluding melanonychia. Hyperpigmentation was observed in the skin as well as the appendages such as nails, resulting in melanonychia (Figure 1). However, Brazzelli et al11 reported hypopigmentation in 5 white patients treated with IM; furthermore, they found a direct correlation between hypopigmentation and development of skin cancers in these patients. The susceptibility to develop skin cancers may persist, even without a clear manifestation of hypopigmentation, as reported in the current analysis. We documented BCC in 5 patients, 1 patient developed actinic keratoses, and 3 patients developed dermatofibromas. However, these neoplasms probably were not provoked by IM. On the contrary, we did not note squamous cell carcinoma, which was reported by Baskaynak et al12 in 2 CML patients treated with IM.
The administration of IM can be associated with exacerbation of psoriasis. Paradoxically, in genetically predisposed individuals, tumor necrosis factor α (TNF-α) antagonists, such as IM, seem to induce psoriasis, producing IFN-α rather than TNF-α and increasing inflammation.13 In fact, some research shows induction of psoriasis by anti–TNF-α drugs.14-16 Two cases of IM associated with psoriasis have been reported, and both cases represented an exacerbation of previously diagnosed psoriasis.13,17 On the contrary, in our analysis we reported 5 cases of psoriasis vulgaris induced by IM administration. Our patients developed cutaneous psoriatic lesions approximately 1.7 months after the start of IM therapy.
The pityriasis rosea–like eruption (Figure 4) presented as nonpruritic, erythematous, scaly patches on the trunk and extremities, and arose 3.6 months after the start of treatment. This particular cutaneous AE is rare. In 3 case reports, the IM dosage also was 400 mg daily.18-20 The pathophysiology of this rare skin reaction stems from the pharmacological effect of IM rather than a hypersensitivity reaction.18
Deininger et al7 reported that patients with a high basophil count (>20%) rarely show urticarial eruptions after IM due to histamine release from basophils. Premedication with an antihistamine was helpful and the urticarial eruption resolved after normalization in basophil count.7
Given the importance of IM for patients who have limited therapeutic alternatives for their disease and the ability to safely treat the cutaneous AEs, as demonstrated in our analysis, the suspension of IM for dermatological complications is necessary only in rare cases, as shown by the low number of patients (n=2) who had to discontinue therapy. The cutaneous AEs should be diagnosed and treated early with less impact on chemotherapy treatments. The administration of IM should involve a coordinated effort among oncologists and dermatologists to prevent important complications.
- Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-1037.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Breccia M, Carmosimo I, Russo E, et al. Early and tardive skin adverse events in chronic myeloid leukaemia patients treated with imatinib. Eur J Haematol. 2005;74:121-123.
- Ugurel S, Hildebrand R, Dippel E, et al. Dose dependent severe cutaneous reactions to imatinib. Br J Cancer. 2003;88:1157-1159.
- Valeyrie L, Bastuji-Garin S, Revuz J, et al. Adverse cutaneous reactions to imatinib (STI571) in Philadelphia chromosome-positive leukaemias: a prospective study of 54 patients. J Am Acad Dermatol. 2003;48:201-206.
- Scott LC, White JD, Reid R, et al. Management of skin toxicity related to the use of imatinibnmesylate (STI571, GlivecTM) for advanced stage gastrointestinal stromal tumors. Sarcoma. 2005;9:157-160.
- Deininger MW, O’Brien SG, Ford JM, et al. Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol. 2003;21:1637-1647.
- Hsiao LT, Chung HM, Lin JT, et al. Stevens-Johnson syndrome after treatment with STI571: a case report. Br J Haematol. 2002;117:620-622.
- Sehgal VN, Srivastava G, Sardana K. Erythroderma/exfoliative dermatitis: a synopsis. Int J Dermatol. 2004;43:39-47.
- Pietras K, Pahler J, Bergers G, et al. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19.
- Brazzelli V, Prestinari F, Barbagallo T, et al. A long-term time course of colorimetric assessment of the effects of imatinib mesylate on skin pigmentation: a study of five patients. J Eur Acad Dermatol Venerol. 2007;21:384-387.
- Baskaynak G, Kreuzer KA, Schwarz M, et al. Squamous cutaneous epithelial cell carcinoma in two CML patients with progressive disease under imatinib treatment. Eur J Haematol. 2003;70:231-234.
- Cheng H, Geist DE, Piperdi M, et al. Management of imatinib-related exacerbation of psoriasis in a patient with a gastrointestinal stromal tumor. Australas J Dermatol. 2009;50:41-43.
- Faillace C, Duarte GV, Cunha RS, et al. Severe infliximab-induced psoriasis treated with adalimumab switching. Int J Dermatol. 2013;52:234-238.
- Iborra M, Beltrán B, Bastida G, et al. Infliximab and adalimumab-induced psoriasis in Crohn’s disease: a aradoxical side effect. J Crohns Colitis. 2011;5:157-161.
- Fernandes IC, Torres T, Sanches M, et al. Psoriasis induced by infliximab. Acta Med Port. 2011;24:709-712.
- Woo SM, Huh CH, Park KC, et al. Exacerbation of psoriasis in a chronic myelogenous leukemia patient treated with imatinib. J Dermatol. 2007;34:724-726.
- Brazzelli V, Prestinari F, Roveda E, et al. Pytiriasis rosea-like eruption during treatment with imatinib mesylate. description of 3 cases. J Am Acad Dermatol. 2005;53:240-243.
- Konstantapoulos K, Papadogianni A, Dimopoulou M, et al. Pytriasis rosea associated with imatinib (STI571, Gleevec). Dermatology. 2002;205:172-173.
- Cho AY, Kim DH, Im M, et al. Pityriasis rosealike drug eruption induced by imatinib mesylate (Gleevec). Ann Dermatol. 2011;23(suppl 3):360-363.
- Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-1037.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Breccia M, Carmosimo I, Russo E, et al. Early and tardive skin adverse events in chronic myeloid leukaemia patients treated with imatinib. Eur J Haematol. 2005;74:121-123.
- Ugurel S, Hildebrand R, Dippel E, et al. Dose dependent severe cutaneous reactions to imatinib. Br J Cancer. 2003;88:1157-1159.
- Valeyrie L, Bastuji-Garin S, Revuz J, et al. Adverse cutaneous reactions to imatinib (STI571) in Philadelphia chromosome-positive leukaemias: a prospective study of 54 patients. J Am Acad Dermatol. 2003;48:201-206.
- Scott LC, White JD, Reid R, et al. Management of skin toxicity related to the use of imatinibnmesylate (STI571, GlivecTM) for advanced stage gastrointestinal stromal tumors. Sarcoma. 2005;9:157-160.
- Deininger MW, O’Brien SG, Ford JM, et al. Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol. 2003;21:1637-1647.
- Hsiao LT, Chung HM, Lin JT, et al. Stevens-Johnson syndrome after treatment with STI571: a case report. Br J Haematol. 2002;117:620-622.
- Sehgal VN, Srivastava G, Sardana K. Erythroderma/exfoliative dermatitis: a synopsis. Int J Dermatol. 2004;43:39-47.
- Pietras K, Pahler J, Bergers G, et al. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19.
- Brazzelli V, Prestinari F, Barbagallo T, et al. A long-term time course of colorimetric assessment of the effects of imatinib mesylate on skin pigmentation: a study of five patients. J Eur Acad Dermatol Venerol. 2007;21:384-387.
- Baskaynak G, Kreuzer KA, Schwarz M, et al. Squamous cutaneous epithelial cell carcinoma in two CML patients with progressive disease under imatinib treatment. Eur J Haematol. 2003;70:231-234.
- Cheng H, Geist DE, Piperdi M, et al. Management of imatinib-related exacerbation of psoriasis in a patient with a gastrointestinal stromal tumor. Australas J Dermatol. 2009;50:41-43.
- Faillace C, Duarte GV, Cunha RS, et al. Severe infliximab-induced psoriasis treated with adalimumab switching. Int J Dermatol. 2013;52:234-238.
- Iborra M, Beltrán B, Bastida G, et al. Infliximab and adalimumab-induced psoriasis in Crohn’s disease: a aradoxical side effect. J Crohns Colitis. 2011;5:157-161.
- Fernandes IC, Torres T, Sanches M, et al. Psoriasis induced by infliximab. Acta Med Port. 2011;24:709-712.
- Woo SM, Huh CH, Park KC, et al. Exacerbation of psoriasis in a chronic myelogenous leukemia patient treated with imatinib. J Dermatol. 2007;34:724-726.
- Brazzelli V, Prestinari F, Roveda E, et al. Pytiriasis rosea-like eruption during treatment with imatinib mesylate. description of 3 cases. J Am Acad Dermatol. 2005;53:240-243.
- Konstantapoulos K, Papadogianni A, Dimopoulou M, et al. Pytriasis rosea associated with imatinib (STI571, Gleevec). Dermatology. 2002;205:172-173.
- Cho AY, Kim DH, Im M, et al. Pityriasis rosealike drug eruption induced by imatinib mesylate (Gleevec). Ann Dermatol. 2011;23(suppl 3):360-363.
Practice Points
- The most common cutaneous adverse reactions from imatinib mesylate (IM) are swelling and edema.
- Maculopapular rash with pruritus is one of the most common side effects from IM and can be effectively treated with oral or systemic antihistamines.
- The onset of periorbital edema requires a complete evaluation of renal function.
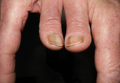
Spontaneous Repigmentation of Silvery Hair in an Infant With Congenital Hydrops Fetalis and Hypoproteinemia
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
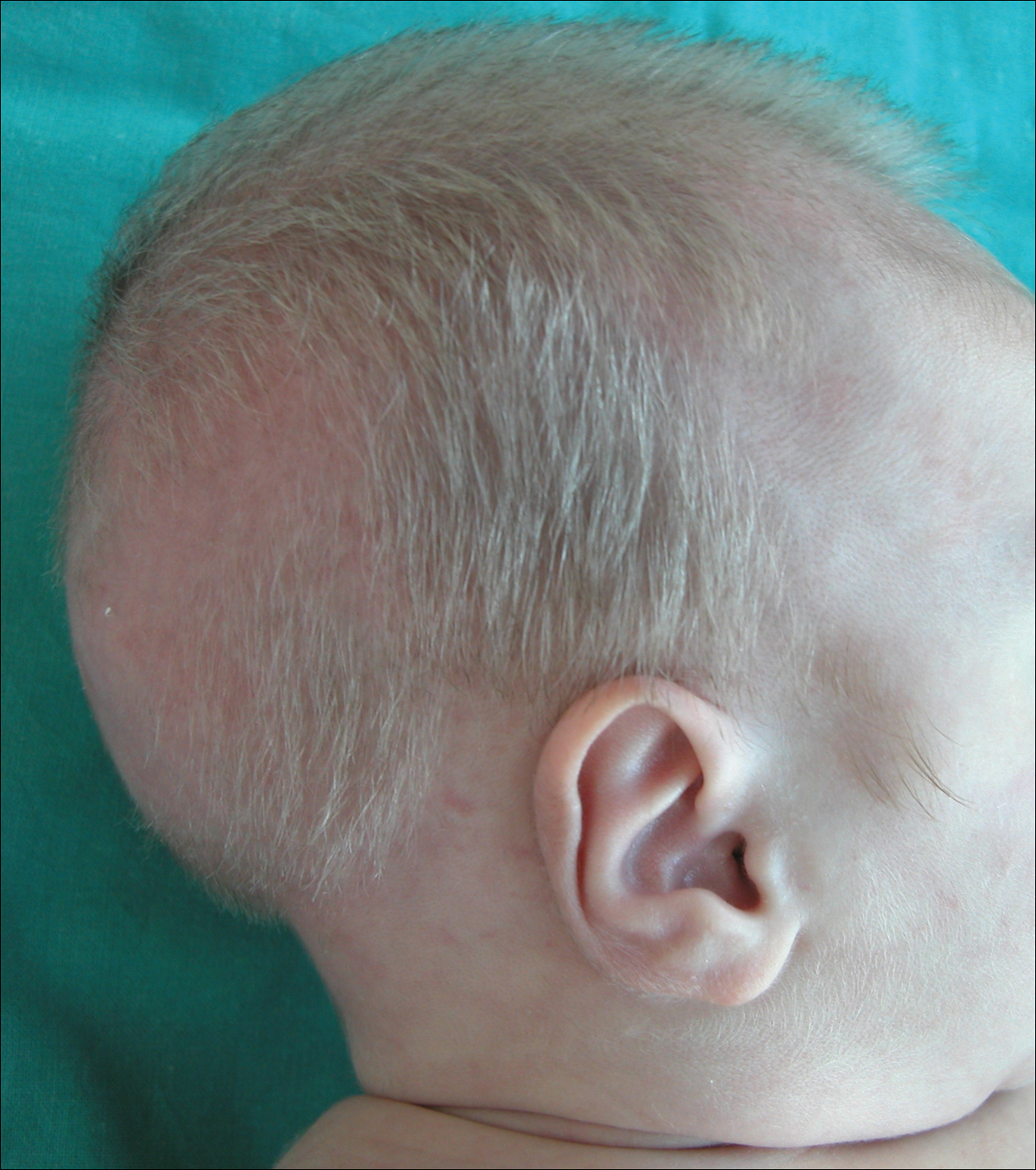
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
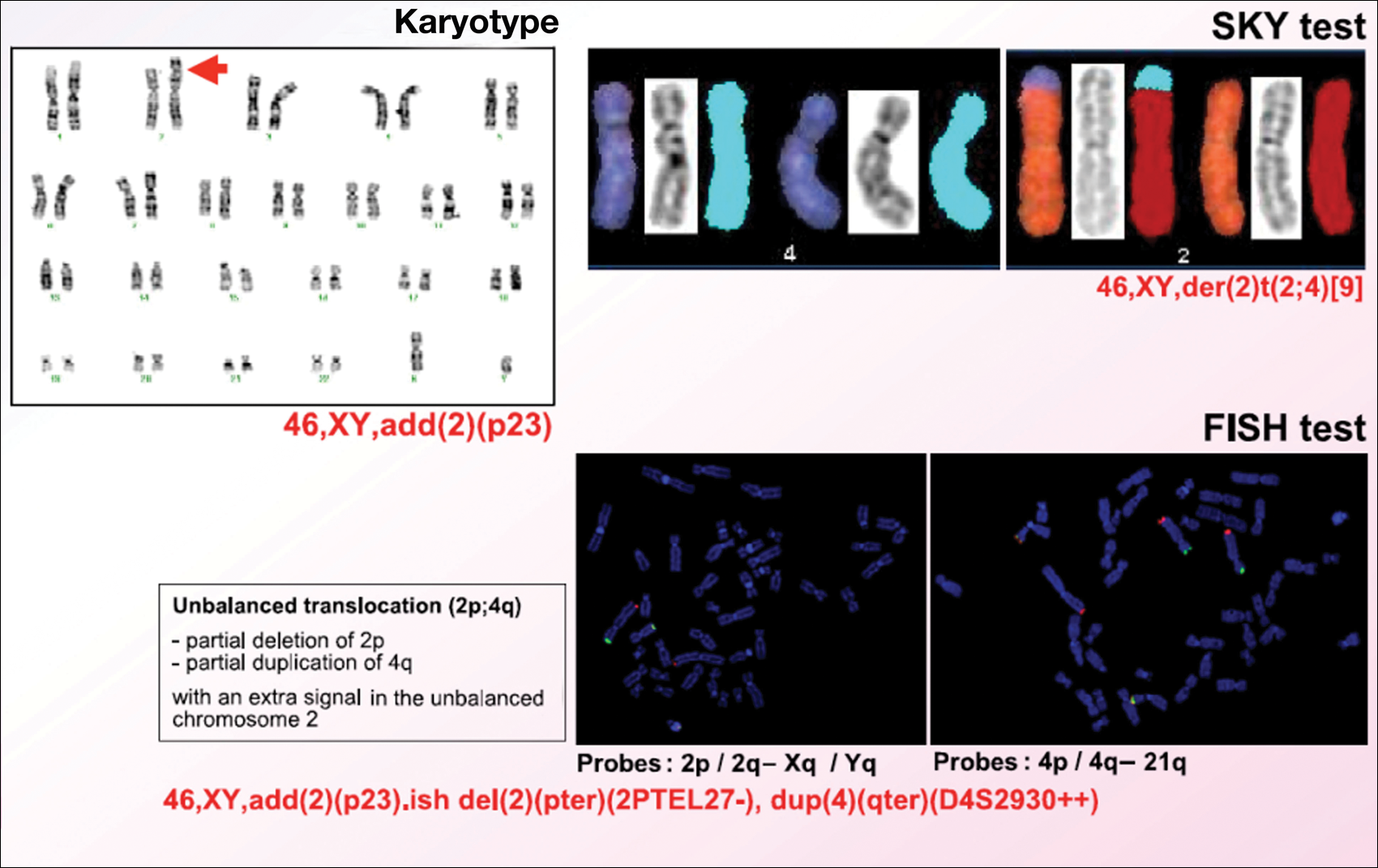
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.


Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
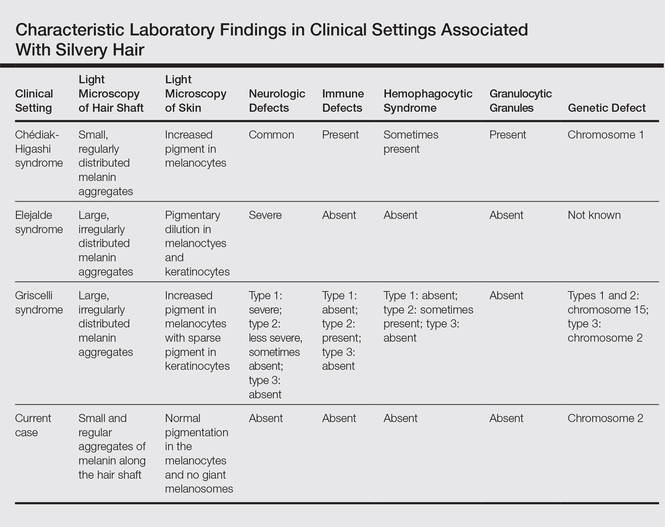
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
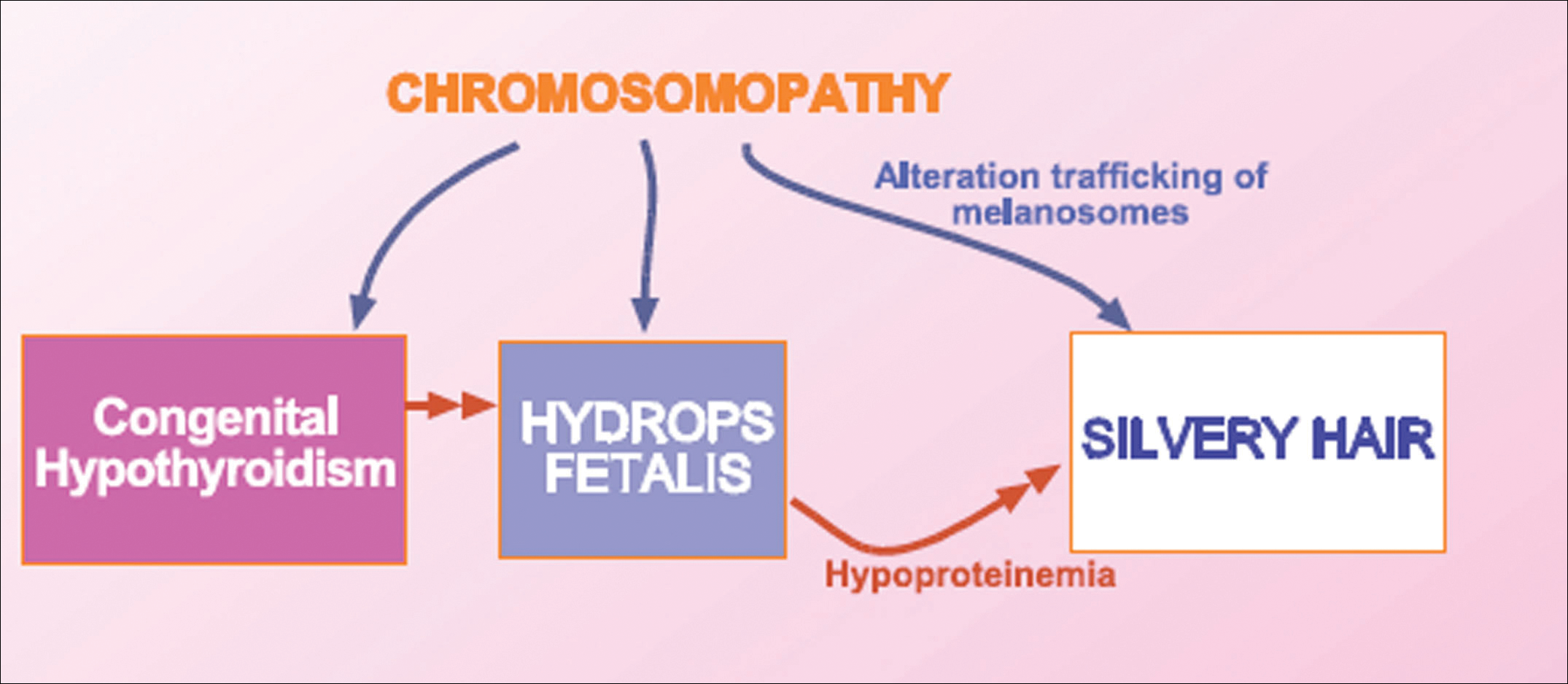
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
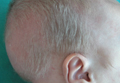
Onychomadesis Following Hand-foot-and-mouth Disease
To the Editor:
Onychomadesis is characterized by separation of the nail plate from the matrix due to a temporary arrest in nail matrix activity. Hand-foot-and-mouth disease (HFMD) is a relatively common viral infection, especially in children. Although the relationship between onychomadesis and HFMD has been noted, there are few reports in the literature.1-9 We present 2 cases of onychomadesis following HFMD in Taiwanese siblings.
A 3-year-old girl presented with proximal nail plate detachment from the proximal nail fold on the bilateral great toenails (Figure 1) and a transverse whole-thickness sulcus on the bilateral thumbnails (Figure 2) of several weeks’ duration. Her 6-year-old sister had similar nail changes. Hand-foot-and-mouth disease was diagnosed about 4 weeks prior to nail changes. The mother reported that only the younger sister experienced fever. There was no history of notable medication intake, nail trauma, periungual erythema, vesicular lesion, or dermatitis. In both patients, the nail changes were temporary with spontaneous normal nail plate regrowth several months later. A diagnosis of onychomadesis was made.
The etiology of onychomadesis includes drug ingestion, especially chemotherapy; severe systemic diseases; high fever; infection, including viral illnesses such as influenza, measles, and HFMD; and idiopathic onychomadesis.1,2,5,10 In 2000, Clementz and Mancini1 reported 5 children with nail matrix arrest following HFMD and suggested an epidemic caused by the same virus strain. Bernier et al2 reported another 4 cases and suggested more than one viral strain may have been implicated in the nail matrix arrest. Although these authors list HFMD as one of the causes of onychomadesis,1,2 the number of cases reported was small; however, studies with a larger number of cases and even outbreak have been reported more recently.3-8 Salazar et al3 reported an onychomadesis outbreak associated with HFMD in Valencia, Spain, in 2008 (N=298). This outbreak primarily was caused by coxsackievirus (CV) A10 (49% of cases).5 Another onychomadesis outbreak occurred in Saragossa, Spain, in 2008, and CV B1, B2, and unidentified nonpoliovirus enterovirus were isolated.6 Outbreaks also occurred in Finland in 2008, and the causative agents were identified as CV A6 and A10.7,8 The latency period for onychomadesis following HFMD was 1 to 2 months (mean, 40 days), and the majority of cases occurred in patients younger than 6 years.1-5 Not all of the nails were involved; in one report, each patient shed only 4 nails on average.6
Although there is a definite relationship between HFMD and onychomadesis, the mechanism is still unclear. Some authors claim that nail matrix arrest is caused by high fever10; however, we found that 40% (2/5)1 to 63% (10/16)4 of reported cases did not have a fever. Additionally, only 1 of our patients had fever. Therefore high fever–induced nail matrix arrest is not a reasonable explanation. Davia et al5 observed no relationship between onychomadesis and the severity of HFMD. In addition, no serious complications of HFMD were mentioned in prior reports.
We propose that HFMD-related onychomadesis is caused by the viral infection itself, rather than by severe systemic disease.1-5,7 Certain viral strains associated with HFMD can induce arrest of nail matrix activity. Osterback et al7 detected CV A6 in shed nail fragments and suggested that virus replication damaged the nail matrix and resulted in temporary nail dystrophy. This hypothesis can explain that only some nails, not all, were involved. In our cases, we noted an incomplete and slanted cleft on the thumbnail (Figure 2). We also found that incomplete onychomadesis appeared in the clinical photograph from a prior report.5 The slanted cleft in our case may be caused by secondary external force after original incomplete onychomadesis or a different rate of nail regrowth because of different intensity of nail matrix damage. The phenomenon of incomplete onychomadesis in the same nail further suggests the mechanism of onychomadesis following HFMD is localized nail matrix damage.
In conclusion, we report 2 cases of onychomadesis associated with HFMD. Our report highlights that there is no racial difference in post-HFMD onychomadesis. These cases highlight that HFMD is an important cause of onychomadesis, especially in children. We suggest that certain viral strains associated with HFMD may specifically arrest nail matrix growth activity, regardless of fever or disease severity.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Bernier V, Labreze C, Bury F, et al. Nail matrix arrest in the course of hand, foot and mouth disease. Eur J Pediatr. 2001;160:649-651.
- Salazar A, Febrer I, Guiral S, et al. Onychomadesis outbreak in Valencia, Spain, June 2008. Euro Surveill. 2008;13:18917.
- Redondo Granado MJ, Torres Hinojal MC, Izquierdo López B. Post viral onychomadesis outbreak in Valladolid [in Spanish]. An Pediatr (Barc). 2009;71:436-439.
- Davia JL, Bel PH, Ninet VZ, et al. Onychomadesis outbreak in Valencia, Spain associated with hand, foot, and mouth disease caused by enteroviruses. Pediatr Dermatol. 2011;28:1-5.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Habif TP. Nail diseases. In: Habif TP, ed. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:947-973.
To the Editor:
Onychomadesis is characterized by separation of the nail plate from the matrix due to a temporary arrest in nail matrix activity. Hand-foot-and-mouth disease (HFMD) is a relatively common viral infection, especially in children. Although the relationship between onychomadesis and HFMD has been noted, there are few reports in the literature.1-9 We present 2 cases of onychomadesis following HFMD in Taiwanese siblings.
A 3-year-old girl presented with proximal nail plate detachment from the proximal nail fold on the bilateral great toenails (Figure 1) and a transverse whole-thickness sulcus on the bilateral thumbnails (Figure 2) of several weeks’ duration. Her 6-year-old sister had similar nail changes. Hand-foot-and-mouth disease was diagnosed about 4 weeks prior to nail changes. The mother reported that only the younger sister experienced fever. There was no history of notable medication intake, nail trauma, periungual erythema, vesicular lesion, or dermatitis. In both patients, the nail changes were temporary with spontaneous normal nail plate regrowth several months later. A diagnosis of onychomadesis was made.
The etiology of onychomadesis includes drug ingestion, especially chemotherapy; severe systemic diseases; high fever; infection, including viral illnesses such as influenza, measles, and HFMD; and idiopathic onychomadesis.1,2,5,10 In 2000, Clementz and Mancini1 reported 5 children with nail matrix arrest following HFMD and suggested an epidemic caused by the same virus strain. Bernier et al2 reported another 4 cases and suggested more than one viral strain may have been implicated in the nail matrix arrest. Although these authors list HFMD as one of the causes of onychomadesis,1,2 the number of cases reported was small; however, studies with a larger number of cases and even outbreak have been reported more recently.3-8 Salazar et al3 reported an onychomadesis outbreak associated with HFMD in Valencia, Spain, in 2008 (N=298). This outbreak primarily was caused by coxsackievirus (CV) A10 (49% of cases).5 Another onychomadesis outbreak occurred in Saragossa, Spain, in 2008, and CV B1, B2, and unidentified nonpoliovirus enterovirus were isolated.6 Outbreaks also occurred in Finland in 2008, and the causative agents were identified as CV A6 and A10.7,8 The latency period for onychomadesis following HFMD was 1 to 2 months (mean, 40 days), and the majority of cases occurred in patients younger than 6 years.1-5 Not all of the nails were involved; in one report, each patient shed only 4 nails on average.6
Although there is a definite relationship between HFMD and onychomadesis, the mechanism is still unclear. Some authors claim that nail matrix arrest is caused by high fever10; however, we found that 40% (2/5)1 to 63% (10/16)4 of reported cases did not have a fever. Additionally, only 1 of our patients had fever. Therefore high fever–induced nail matrix arrest is not a reasonable explanation. Davia et al5 observed no relationship between onychomadesis and the severity of HFMD. In addition, no serious complications of HFMD were mentioned in prior reports.
We propose that HFMD-related onychomadesis is caused by the viral infection itself, rather than by severe systemic disease.1-5,7 Certain viral strains associated with HFMD can induce arrest of nail matrix activity. Osterback et al7 detected CV A6 in shed nail fragments and suggested that virus replication damaged the nail matrix and resulted in temporary nail dystrophy. This hypothesis can explain that only some nails, not all, were involved. In our cases, we noted an incomplete and slanted cleft on the thumbnail (Figure 2). We also found that incomplete onychomadesis appeared in the clinical photograph from a prior report.5 The slanted cleft in our case may be caused by secondary external force after original incomplete onychomadesis or a different rate of nail regrowth because of different intensity of nail matrix damage. The phenomenon of incomplete onychomadesis in the same nail further suggests the mechanism of onychomadesis following HFMD is localized nail matrix damage.
In conclusion, we report 2 cases of onychomadesis associated with HFMD. Our report highlights that there is no racial difference in post-HFMD onychomadesis. These cases highlight that HFMD is an important cause of onychomadesis, especially in children. We suggest that certain viral strains associated with HFMD may specifically arrest nail matrix growth activity, regardless of fever or disease severity.
To the Editor:
Onychomadesis is characterized by separation of the nail plate from the matrix due to a temporary arrest in nail matrix activity. Hand-foot-and-mouth disease (HFMD) is a relatively common viral infection, especially in children. Although the relationship between onychomadesis and HFMD has been noted, there are few reports in the literature.1-9 We present 2 cases of onychomadesis following HFMD in Taiwanese siblings.
A 3-year-old girl presented with proximal nail plate detachment from the proximal nail fold on the bilateral great toenails (Figure 1) and a transverse whole-thickness sulcus on the bilateral thumbnails (Figure 2) of several weeks’ duration. Her 6-year-old sister had similar nail changes. Hand-foot-and-mouth disease was diagnosed about 4 weeks prior to nail changes. The mother reported that only the younger sister experienced fever. There was no history of notable medication intake, nail trauma, periungual erythema, vesicular lesion, or dermatitis. In both patients, the nail changes were temporary with spontaneous normal nail plate regrowth several months later. A diagnosis of onychomadesis was made.
The etiology of onychomadesis includes drug ingestion, especially chemotherapy; severe systemic diseases; high fever; infection, including viral illnesses such as influenza, measles, and HFMD; and idiopathic onychomadesis.1,2,5,10 In 2000, Clementz and Mancini1 reported 5 children with nail matrix arrest following HFMD and suggested an epidemic caused by the same virus strain. Bernier et al2 reported another 4 cases and suggested more than one viral strain may have been implicated in the nail matrix arrest. Although these authors list HFMD as one of the causes of onychomadesis,1,2 the number of cases reported was small; however, studies with a larger number of cases and even outbreak have been reported more recently.3-8 Salazar et al3 reported an onychomadesis outbreak associated with HFMD in Valencia, Spain, in 2008 (N=298). This outbreak primarily was caused by coxsackievirus (CV) A10 (49% of cases).5 Another onychomadesis outbreak occurred in Saragossa, Spain, in 2008, and CV B1, B2, and unidentified nonpoliovirus enterovirus were isolated.6 Outbreaks also occurred in Finland in 2008, and the causative agents were identified as CV A6 and A10.7,8 The latency period for onychomadesis following HFMD was 1 to 2 months (mean, 40 days), and the majority of cases occurred in patients younger than 6 years.1-5 Not all of the nails were involved; in one report, each patient shed only 4 nails on average.6
Although there is a definite relationship between HFMD and onychomadesis, the mechanism is still unclear. Some authors claim that nail matrix arrest is caused by high fever10; however, we found that 40% (2/5)1 to 63% (10/16)4 of reported cases did not have a fever. Additionally, only 1 of our patients had fever. Therefore high fever–induced nail matrix arrest is not a reasonable explanation. Davia et al5 observed no relationship between onychomadesis and the severity of HFMD. In addition, no serious complications of HFMD were mentioned in prior reports.
We propose that HFMD-related onychomadesis is caused by the viral infection itself, rather than by severe systemic disease.1-5,7 Certain viral strains associated with HFMD can induce arrest of nail matrix activity. Osterback et al7 detected CV A6 in shed nail fragments and suggested that virus replication damaged the nail matrix and resulted in temporary nail dystrophy. This hypothesis can explain that only some nails, not all, were involved. In our cases, we noted an incomplete and slanted cleft on the thumbnail (Figure 2). We also found that incomplete onychomadesis appeared in the clinical photograph from a prior report.5 The slanted cleft in our case may be caused by secondary external force after original incomplete onychomadesis or a different rate of nail regrowth because of different intensity of nail matrix damage. The phenomenon of incomplete onychomadesis in the same nail further suggests the mechanism of onychomadesis following HFMD is localized nail matrix damage.
In conclusion, we report 2 cases of onychomadesis associated with HFMD. Our report highlights that there is no racial difference in post-HFMD onychomadesis. These cases highlight that HFMD is an important cause of onychomadesis, especially in children. We suggest that certain viral strains associated with HFMD may specifically arrest nail matrix growth activity, regardless of fever or disease severity.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Bernier V, Labreze C, Bury F, et al. Nail matrix arrest in the course of hand, foot and mouth disease. Eur J Pediatr. 2001;160:649-651.
- Salazar A, Febrer I, Guiral S, et al. Onychomadesis outbreak in Valencia, Spain, June 2008. Euro Surveill. 2008;13:18917.
- Redondo Granado MJ, Torres Hinojal MC, Izquierdo López B. Post viral onychomadesis outbreak in Valladolid [in Spanish]. An Pediatr (Barc). 2009;71:436-439.
- Davia JL, Bel PH, Ninet VZ, et al. Onychomadesis outbreak in Valencia, Spain associated with hand, foot, and mouth disease caused by enteroviruses. Pediatr Dermatol. 2011;28:1-5.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Habif TP. Nail diseases. In: Habif TP, ed. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:947-973.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Bernier V, Labreze C, Bury F, et al. Nail matrix arrest in the course of hand, foot and mouth disease. Eur J Pediatr. 2001;160:649-651.
- Salazar A, Febrer I, Guiral S, et al. Onychomadesis outbreak in Valencia, Spain, June 2008. Euro Surveill. 2008;13:18917.
- Redondo Granado MJ, Torres Hinojal MC, Izquierdo López B. Post viral onychomadesis outbreak in Valladolid [in Spanish]. An Pediatr (Barc). 2009;71:436-439.
- Davia JL, Bel PH, Ninet VZ, et al. Onychomadesis outbreak in Valencia, Spain associated with hand, foot, and mouth disease caused by enteroviruses. Pediatr Dermatol. 2011;28:1-5.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Habif TP. Nail diseases. In: Habif TP, ed. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Philadelphia, PA: Mosby/Elsevier; 2010:947-973.
Practice Points
- Onychomadesis is a late complication of hand-foot-and-mouth disease (HFMD) with a latency period of 1 to 2 months.
- Although the mechanism between onychomadesis and HFMD is still unclear, we propose that it is caused by the viral infection itself rather than severe systemic disease.
Toe Nodule Obliterating the Nail Bed
The Diagnosis: Superficial Acral Fibromyxoma
Superficial acral fibromyxoma (SAF) was first described in 2001 by Fetsch et al.1 Subsequently, the term digital fibromyxoma was proposed in 2012 by Hollmann et al2 to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. The benign growth typically presents as a painless or tender nodule in middle-aged adults with a slight male predominance (1.3:1 ratio).1,2 In a case series (N=124) described by Hollmann et al,2 9 of 25 patients (36%) who had imaging studies showed bone involvement by an erosive or lytic lesion. Reports of SAF with bone involvement also have been described in the radiologic and orthopedic surgery literature.3,4 Radiographically, the soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs (Figure 1).3
Histologically, SAFs are moderately cellular with spindled or stellate fibroblastlike cells within a myxoid or collagenous matrix (Figure 2).1 The vasculature is mildly accentuated and an increase in mast cells usually is observed. The nuclei have a low degree of atypia with few mitotic figures, and the stellate cells exhibit positive immunohistochemical staining for CD34 (Figure 3), epithelial membrane antigen, and CD99.1 Hollmann et al2 found that 66 of 95 tumors (69.5%) infiltrated the dermal collagen, 26 (27.4%) infiltrated fat, and 3 (3.2%) invaded bone. Of the 47 cases that were evaluated on follow-up, 10 tumors (21.3%) recurred locally (all near the nail unit of the fingers or toes) after a mean interval of 27 months. Although invasion of underlying tissues and recurrence of the tumor has been demonstrated, this growth is considered benign. The histologic differential diagnosis includes neurofibroma, myxoma, fibroma, low-grade fibromyxoid sarcoma, dermatofibroma, superficial angiomyxoma, and dermatofibrosarcoma protuberans.2
The primary treatment of SAF is local excision. The incidence of local recurrence found in the case series by Hollmann et al2 was directly linked to positive margins after the first excision (10/47 [21.3%] recurrent lesions had positive margins). To date, there are no known reports of metastatic disease in SAF.2 Our case manifested with a late recurrence of the tumor and bone involvement requiring surgical excision, which illustrates the role of adjuvant imaging and close follow-up following excision of any soft-tissue tumors of the fingers and toes that have been histologically confirmed as SAF, particularly those of the periungual region.
|
|
|
|
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma (a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes.) Hum Pathol. 2001;32:704-714.
2. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
3. Varikatt W, Soper J, Simmon G, et al. Superficial acral fibromyxoma: a report of two cases with radiological findings. Skeletal Radiol. 2008;37:499-503.
4. Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx. a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
The Diagnosis: Superficial Acral Fibromyxoma
Superficial acral fibromyxoma (SAF) was first described in 2001 by Fetsch et al.1 Subsequently, the term digital fibromyxoma was proposed in 2012 by Hollmann et al2 to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. The benign growth typically presents as a painless or tender nodule in middle-aged adults with a slight male predominance (1.3:1 ratio).1,2 In a case series (N=124) described by Hollmann et al,2 9 of 25 patients (36%) who had imaging studies showed bone involvement by an erosive or lytic lesion. Reports of SAF with bone involvement also have been described in the radiologic and orthopedic surgery literature.3,4 Radiographically, the soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs (Figure 1).3
Histologically, SAFs are moderately cellular with spindled or stellate fibroblastlike cells within a myxoid or collagenous matrix (Figure 2).1 The vasculature is mildly accentuated and an increase in mast cells usually is observed. The nuclei have a low degree of atypia with few mitotic figures, and the stellate cells exhibit positive immunohistochemical staining for CD34 (Figure 3), epithelial membrane antigen, and CD99.1 Hollmann et al2 found that 66 of 95 tumors (69.5%) infiltrated the dermal collagen, 26 (27.4%) infiltrated fat, and 3 (3.2%) invaded bone. Of the 47 cases that were evaluated on follow-up, 10 tumors (21.3%) recurred locally (all near the nail unit of the fingers or toes) after a mean interval of 27 months. Although invasion of underlying tissues and recurrence of the tumor has been demonstrated, this growth is considered benign. The histologic differential diagnosis includes neurofibroma, myxoma, fibroma, low-grade fibromyxoid sarcoma, dermatofibroma, superficial angiomyxoma, and dermatofibrosarcoma protuberans.2
The primary treatment of SAF is local excision. The incidence of local recurrence found in the case series by Hollmann et al2 was directly linked to positive margins after the first excision (10/47 [21.3%] recurrent lesions had positive margins). To date, there are no known reports of metastatic disease in SAF.2 Our case manifested with a late recurrence of the tumor and bone involvement requiring surgical excision, which illustrates the role of adjuvant imaging and close follow-up following excision of any soft-tissue tumors of the fingers and toes that have been histologically confirmed as SAF, particularly those of the periungual region.
|
|
|
|
|
|
The Diagnosis: Superficial Acral Fibromyxoma
Superficial acral fibromyxoma (SAF) was first described in 2001 by Fetsch et al.1 Subsequently, the term digital fibromyxoma was proposed in 2012 by Hollmann et al2 to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. The benign growth typically presents as a painless or tender nodule in middle-aged adults with a slight male predominance (1.3:1 ratio).1,2 In a case series (N=124) described by Hollmann et al,2 9 of 25 patients (36%) who had imaging studies showed bone involvement by an erosive or lytic lesion. Reports of SAF with bone involvement also have been described in the radiologic and orthopedic surgery literature.3,4 Radiographically, the soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs (Figure 1).3
Histologically, SAFs are moderately cellular with spindled or stellate fibroblastlike cells within a myxoid or collagenous matrix (Figure 2).1 The vasculature is mildly accentuated and an increase in mast cells usually is observed. The nuclei have a low degree of atypia with few mitotic figures, and the stellate cells exhibit positive immunohistochemical staining for CD34 (Figure 3), epithelial membrane antigen, and CD99.1 Hollmann et al2 found that 66 of 95 tumors (69.5%) infiltrated the dermal collagen, 26 (27.4%) infiltrated fat, and 3 (3.2%) invaded bone. Of the 47 cases that were evaluated on follow-up, 10 tumors (21.3%) recurred locally (all near the nail unit of the fingers or toes) after a mean interval of 27 months. Although invasion of underlying tissues and recurrence of the tumor has been demonstrated, this growth is considered benign. The histologic differential diagnosis includes neurofibroma, myxoma, fibroma, low-grade fibromyxoid sarcoma, dermatofibroma, superficial angiomyxoma, and dermatofibrosarcoma protuberans.2
The primary treatment of SAF is local excision. The incidence of local recurrence found in the case series by Hollmann et al2 was directly linked to positive margins after the first excision (10/47 [21.3%] recurrent lesions had positive margins). To date, there are no known reports of metastatic disease in SAF.2 Our case manifested with a late recurrence of the tumor and bone involvement requiring surgical excision, which illustrates the role of adjuvant imaging and close follow-up following excision of any soft-tissue tumors of the fingers and toes that have been histologically confirmed as SAF, particularly those of the periungual region.
|
|
|
|
|
|
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma (a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes.) Hum Pathol. 2001;32:704-714.
2. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
3. Varikatt W, Soper J, Simmon G, et al. Superficial acral fibromyxoma: a report of two cases with radiological findings. Skeletal Radiol. 2008;37:499-503.
4. Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx. a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma (a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes.) Hum Pathol. 2001;32:704-714.
2. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
3. Varikatt W, Soper J, Simmon G, et al. Superficial acral fibromyxoma: a report of two cases with radiological findings. Skeletal Radiol. 2008;37:499-503.
4. Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx. a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
A generally healthy 30-year-old man presented with a 3-cm exophytic, yellowish red, subungual nodule of the left great toe of 1 year’s duration that was obliterating the nail plate. Ten years prior, a similar nodule in the same location was removed via laser by a podiatrist. Medical records were not retrievable, but the patient reported that he was told the excised lesion was a benign tumor. Plain radiographs were performed at the current presentation and demonstrated an inferior cortical lucency of the distal phalanx as well as a lucency over the nail bed region with extension of calcification to the soft tissues. Magnetic resonance imaging showed a mass with a proximal to distal maximum dimension of 2.1 cm that involved the dorsal surface of the proximal phalanx. Magnetic resonance imaging also demonstrated bone erosion from the overlying mass. A 4-mm incisional punch biopsy was performed prior to surgical excision.