User login
Menstrual manipulation: Options for suppressing the cycle
If they wish, women can have more control over when and if they menstruate. By using hormonal contraceptives in extended or continuous regimens, they can have their period less often, a practice called menstrual manipulation or menstrual suppression.
Actually, with the help of their clinicians, women have been doing this for years. But now that several products have been approved by the US Food and Drug Administration (FDA) specifically for use in extended or continuous regimens, the practice has become more widely accepted.
Reasons for suppressing menstrual flow range from avoiding bleeding during a particular event (eg, a wedding, graduation, or sports competition) to finding relief from dysmenorrhea or reducing or eliminating menstruation in the treatment of endometriosis, migraine, and other medical conditions exacerbated by hormonal changes around the time of menses.1 Alternatively, some women may practice menstrual manipulation for no other reason than to simply avoid menstruation.
MENSTRUAL DISORDERS ARE TROUBLESOME, COMMON
Each year in the United States, menstrual disorders such as dysmenorrhea (painful menstruation), menorrhagia (excessive or frequent menstruation), metrorrhagia (irregular menstruation), menometrorrhagia (excessive and irregular menstruation), and premenstrual syndrome affect nearly 2.5 million women age 18 to 50 years.2 Menstrual disorders are the leading cause of gynecologic morbidity in the United States, outnumbering adnexal masses (the second most common cause) by a factor of three.2 In addition, these disorders extend into the workplace, costing US industry about 8% of its total wage bill.3
A BRIEF HISTORY OF CONTRACEPTIVE DEVELOPMENT
The idea of using progestins for birth control was first advanced in the 1950s by Dr. Gregory Pincus, who proposed a regimen of 21 days of active drug followed by 7 drug-free days to allow withdrawal bleeding, mimicking the natural cycle.4 This “21/7” regimen was designed to follow the lunar cycle in the hope it would be, in the words of Dr. John Rock, “a morally permissible variant of the rhythm method,”5 thereby making it acceptable to women, clinicians, and the Catholic Church.
In 1977, Loudon et al6 reported the results of a study in which women took active pills for 84 days instead of 21 days, which reduced the frequency of menstruation to every 3 months. Since then, extending the active pills beyond 21 days to avoid menses and other hormone-withdrawal symptoms has become popular in clinical practice, and many studies have investigated the extended or continuous use of oral and other forms of contraception to delay menses.7–18
CURRENT METHODS OF MENSTRUAL MANIPULATION
A variety of available products prevent conception by altering the menstrual cycle:
- Oral estrogen-progestin contraceptive pills
- A drug-releasing intrauterine device
- Depot medroxyprogesterone acetate injections
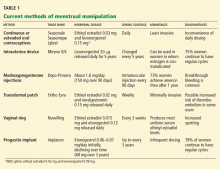
- A contraceptive vaginal ring
- An implantable etonogestrel contraceptive.
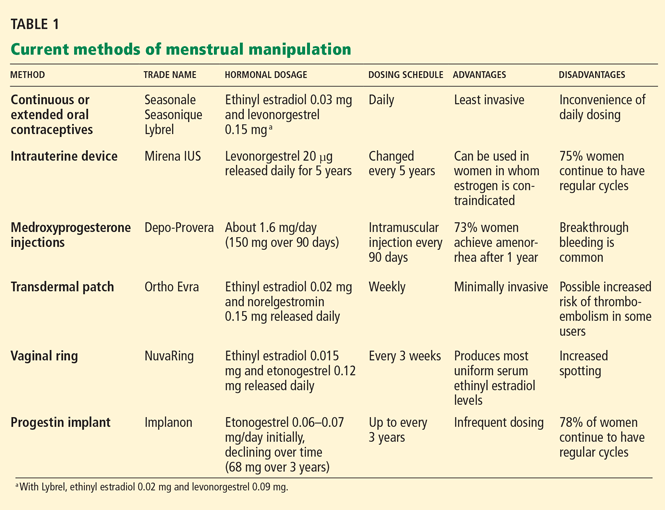
Their use in menstrual manipulation is summarized in Table 1.
Oral contraceptive pills
The most common way to manipulate the menstrual cycle is to extend the time between hormone-free weeks in an oral contraceptive regimen.
If the patient is young, you can prescribe a monophasic 21/7 oral contraceptive and tell her to take one active pill every day for 21 days and then start a new pack and keep taking active pills for up to 84 consecutive days, skipping the placebo pills until she wants to have her menstrual period. She can choose which week to have it: if the scheduled 12th week of an extended-cycle oral contraceptive regimen is inconvenient, she can plan it for week 10, or week 9, or whichever week is convenient.
The rationale for using an 84-day (12-week) cycle is that it still provides four periods per year, alleviating fears of hypertrophic endometrium.19
In this scenario, unscheduled or breakthrough bleeding can be managed by taking a “double-up pill” from a spare pack on any day breakthrough bleeding occurs and until it resolves. Menstrual periods should not be planned for intervals shorter than 21 days, owing to the risk of ovulation. Missed days of pills or use of placebo pills should also not exceed 7 days to prevent escape ovulation. 20
In some women with endometriosis and other medical reasons, continuous oral contraception with no placebo week can be prescribed.
Unfortunately, the downside to suppressing withdrawal bleeding is unscheduled or “breakthrough” bleeding. The best way to treat this unscheduled bleeding is not known. Patients who are not sexually active can be reassured that the goal of an atrophic endometrium can still be achieved, with resultant pill amenorrhea (particularly useful for those with severe dysmenorrhea or other reasons to want to avoid flow). Patients could also try to manage flow by periodically taking a 3- to 5-day break from hormone-containing pills to allow flow. They can also try switching to another oral contraceptive that has a different progestin that would spiral the arterioles of the endometrium more tightly and thus more aggressively induce atrophy.13,17,21 For instance, levonorgestrel is 10 to 20 times more potent than norethindrone. Choosing a pill with a higher monophasic dosing of levonorgestrel or a similar progestin may minimize unscheduled bleeding.
Currently, several oral contraceptives are approved for use in an extended regimen.
Seasonale was the first oral contraceptive marketed in the United States with an extended active regimen.22 It comes in a pack of 84 pills containing ethinyl estradiol 0.03 mg and levonorgestrel 0.15 mg, plus 7 placebo pills.
Seasonique is similar to Seasonale, but instead of placebo pills it has seven pills that contain ethinyl estradiol 0.010 mg.
Lybrel is a low-dose combination containing ethinyl estradiol 0.02 mg and levonorgestrel 0.09 mg. Packaged as an entire year’s worth of active pills to be taken continuously for 365 days without a placebo phase or pillfree interval,23 it is the only FDA-approved continuous oral contraceptive available in the United States.
An intrauterine device
Intrauterine devices were originally developed as contraceptives. The addition of a progestin to these devices has been shown to reduce heavy menstrual bleeding by up to 90%.24,25
Mirena IUS, a levonorgestrel-releasing device, is the only medicated intrauterine device that is currently available in the United States. (“IUS” stands for “intrauterine system.”) It was recently approved by the FDA to treat heavy menstrual bleeding in women who use intrauterine contraception as their method of pregnancy prevention.26 About 50% of women who use this device develop amenorrhea within 6 months of insertion, while 25% report oligomenorrhea.27
The Mirena device can be left in the uterus for up to 5 years. It may be a good choice for inducing amenorrhea in women with hemostatic disorders or in whom estrogen either is contraindicated or causes health concerns.18 The copper intrauterine device (Paragard; Duramed Pharmaceuticals Inc., Pomona, NY) remains a viable option for those who cannot or do not tolerate hormonal therapy. However, Mirena may provide less unscheduled bleeding than the copper intrauterine device.
Depot medroxyprogesterone acetate injections
Depo-Provera (depot medroxyprogesterone acetate) injections are given at 90-day intervals. 28 This contraceptive method inhibits ovulation and decidualizes the endometrium, thereby reducing or eliminating uterine bleeding. 29
While new users may initially experience excessive prolonged bleeding (10 or more days) while shedding their existing lining, the rate of amenorrhea has been shown to increase over time as the lining atrophies.30 Thus, prolonged use of this agent reduces the frequency of menstruation as well as menstruation-related symptoms.
Depot medroxyprogesterone acetate is ideal for patients whose menstrual periods pose a significant hygiene problem (eg, developmentally challenged girls). In our experience, the injections can be given at shorter intervals to induce atrophy of the endometrium quickly. In this scenario, the clinician might give an injection every 4 to 6 weeks for two or three doses to induce amenorrhea and then return to every-12-week dosing.
The main risk when using medroxyprogesterone injections to induce amenorrhea is the potential for bone loss. Users of this method have been shown to have lower mean bone mineral density31–33 and significantly higher levels of biomarkers of bone formation and resorption32,34 than nonusers. However, these changes are similar to those seen in breastfeeding women,35 are reversible with cessation, 36 and are not associated with increased fracture risk.37 In adolescent girls, pregnancy poses similar risks to the bones, with longerterm consequences.
Medroxyprogesterone can also stimulate appetite, causing 10 to 20 kg of weight gain in adolescents and women who are already obese and have trouble with appetite regulation.38 Slender users tend not to gain weight, however.
Given this information, depot medroxyprogesterone acetate appears to be a cost-effective contraceptive option that should be considered in the context of the clinical situation and preference of each patient.
Transdermal contraceptive patch
Ortho Evra, a transdermal patch, is designed to deliver ethinyl estradiol 0.02 mg and norelgestromin 0.150 mg daily.39 It is usually applied weekly for 3 weeks, followed by a patch-free week to induce regular monthly withdrawal bleeding.
Extended use of the patch to manipulate menstruation is an off-label use. In the only trial evaluating extended use of the patch, amenorrhea occurred in 12% of users, but unscheduled bleeding and spotting were common. 16
Although there is some evidence that the long-term use of the patch may increase the risk of venous thromboembolism,40,41 the risk in women who use the patch has been found to be similar to that in women using an oral contraceptive.42 However, serum ethinyl estradiol levels have been found to be higher with the use of the weekly patch than with oral contraceptives or the contraceptive vaginal ring39; as a result, many physicians are hesitant to recommend its continuous use.
Pending further data about the safety profile of this contraceptive, the World Health Organization Medical Eligibility Criteria for Contraceptive Use suggest that the same guidelines for the prescription of combination oral contraceptives should also apply to the patch.43
Contraceptive vaginal ring
NuvaRing, a contraceptive vaginal ring, releases a daily dose of ethinyl estradiol 0.015 mg and etonogestrel 0.12 mg.10 It is inserted, left in for 21 days, and then removed and left out for 7 days, during which withdrawal bleeding occurs.10
Vaginal administration has been shown to allow low, continuous dosing, which results in more stable serum concentrations than with the patch or oral contraceptives.39 In the only trial comparing an extended vaginal ring regimen and the traditional 28-day regimen, extended use resulted in fewer overall days of bleeding than monthly use, but with more unscheduled spotting.15
The most common side effects include headache, vaginitis, and leukorrhea,44 but there is no evidence of bacteriologic or cytologic changes in the cervicovaginal epithelium, even with extended use.45,46
Etonogestrel implantable contraceptive
Implanon, a single-rod progestin implant, is available in the United States and elsewhere. It is placed subdermally in the inner upper arm and provides contraception for as long as 3 years.
Implanon contains 68 mg of the progestin etonogestrel, which it slowly releases over time, initially at 0.06 to 0.07 mg/day, decreasing to 0.035 to 0.045 mg/day at the end of the first year, to 0.03 to 0.04 mg/day at the end of the second year, and then to 0.025 to 0.03 mg/day at the end of the third year.47
The amount of vaginal bleeding associated with the use of the implant is generally modest, but the pattern tends to be unpredictable. 48 In addition, because amenorrhea is reported as a side effect in only 22% of women during the first 2 years of its use,48 the progestin implant is a less satisfactory means of menstrual suppression than the other methods discussed above.
BENEFITS OF MENSTRUAL MANIPULATION
Menstrual manipulation has a number of benefits in terms of both overall health and lifestyle.
For most women, using a long-acting hormonal contraceptive carries low risks and substantial health benefits. Women who take oral contraceptives are less likely to develop osteoporosis, ovarian or endometrial cancer, benign breast changes, or pelvic inflammatory disease. 49 Long-term use of an oral contraceptive can also preserve fertility by reducing and delaying the incidence of endometriosis,50 and is effective at treating acne vulgaris, which tends to be common among patients with polycystic ovary syndrome.51,52 In addition, this practice can be used to reduce overall blood loss, an application that is particularly important in women with a bleeding diathesis such as von Willebrand disease, who frequently suffer from menorrhagia.53
Reduced menstruation may also prove more convenient during particular occasions, such as vacations and athletic activities. Specifically, it may be useful to women serving in the military. In a study by Schneider et al,54 a cohort of 83 female cadets reported a significant perceived impact of premenstrual and menstrual-related symptoms on academic, physical, and military activities, as well as difficulties in obtaining, changing, and disposing of menstrual materials in a military setting. Likewise, reduced menstrual frequency or amenorrhea may play an important role in female athletes, who reportedly use oral contraceptives to control premenstrual symptoms, to protect bone health, and to manipulate the menstrual cycle in order to maximize performance.55
Adolescent girls are another group who may benefit from reduced or absent menses, once they have reached near-final height. By practicing menstrual suppression, girls can avoid dysmenorrhea and the inconvenience of menstruation during the school day, when their access to painkillers, sanitary pads or tampons, and a change of clothes may be limited. 56 Clinicians who discuss with teenage patients the benefits of innovative hormonal contraceptive schedules that reduce menstrual frequency may be able to improve the quality of life for these young women.
In a very short girl just after menarche, care must be taken not to start a hormonal method too early so as not to prematurely close epiphyses and stunt final height; after menarche, most girls still have 1 to 4 inches of potential growth. For a young lady 4 feet 11 inches tall, that extra inch may be important.
Finally, menstrual manipulation may also find a niche among the developmentally challenged. Women with cognitive impairment and physical disabilities may have difficulty with hygienic practice around menses. For a number of years, contraceptives have been used to manage menstrual hygiene in patients with catamenial (ie, menstrual) epilepsy, and to address caregiver concerns in women with severe mental retardation, with improved behavior noted in some patients.57–59 In this setting, an agent that suppresses menses and also provides contraception, especially for those girls and women at risk of abuse, may offer substantial benefits.
DISADVANTAGES OF MENSTRUAL MANIPULATION
Rates of adverse events and of discontinuation of extended and continuous oral contraceptive regimens are comparable with those reported for cyclic regimens, except for higher rates of breakthrough bleeding.
In a trial of continuous oral contraceptive use in more than 2,000 patients, 396 (18.5%) withdrew from the study as a result of bothersome uterine bleeding.60 However, while breakthrough bleeding often occurs during the first few months of extended oral contraceptive use, it usually decreases with each successive cycle of therapy and is comparable to that reported by patients on the conventional oral contraceptive regimen by the fourth extended cycle.12
CONTRACEPTIVE EFFICACY
The efficacy of extended and continuous oral contraceptive regimens is comparable with that of cyclic regimens.12,60,61 One reason for this may be better adherence to continuous regimens: women using this regimen have been shown to miss fewer pills than those on a cyclic regimen, especially during the critical first week of the pill pack.21
Several studies have shown that some women ovulate during the standard 21/7 oral contraceptive regimen even if they do not miss any pills or take pills off-schedule, putting them at greater risk of pregnancy.62 Large studies evaluating the efficacy of an extendedcycle regimen have shown a pregnancy rate during the 1-year study period that was either comparable with61 or lower than12,60 rates with standard regimens.
Heterosexual couples need to be advised to use condoms to further reduce the already low failure rate and to prevent sexually transmitted diseases.
ACCEPTABILITY OF MENSTRUAL MANIPULATION
Ever since the earliest trial of an extended oral contraceptive regimen, participants have expressed a favorable response to the resulting decrease in menstrual frequency; in the 1977 study by Loudon et al,6 patients on the extended regimen cited infrequent periods (82%), fewer menstrual problems (20%), and easier pill-taking (19%) as favorable features.
In 1999, den Tonkelaar and Oddens63 surveyed 1,300 Dutch women about their preferred frequency of menstruation and found that about 70% between the ages of 15 and 49 preferred a frequency of between every 3 months and never. A similar survey in the United States indicated that 58% preferred a bleeding frequency of either every 3 months or never to more frequent periods.64
While patients find menstrual manipulation generally acceptable, clinician approval has been more varied. Loudon et al reported that “the doctors and nurses on the clinic staff were less enthusiastic about this regimen than the volunteers themselves.”6 In a survey of 222 clinicians,65 90% of responders reported ever having prescribed extended or continuous dosing regimens to adolescents, and 33% reported that extended cycles made up more than 10% of their total oral contraceptive prescriptions.
Myths and misperceptions about menstrual manipulation abound. Many clinicians believe that routine use of an extended or continuous oral contraceptive regimen is inadvisable, despite the lack of evidence to support this notion.66 Therefore, many care providers need more education about the practice and benefits of menstrual manipulation.
THE RIGHT METHOD FOR THE RIGHT PATIENT
Manipulation and suppression of menstruation through continuous or extended use of oral contraceptives or by other means may have a number of advantages to women, including fewer menstrual-related syndromes, reduced absenteeism from work or school, and greater overall satisfaction.
For women whose goal is to reduce but not necessarily to eliminate monthly bleeding, the cyclic use of estrogen-progestin contraception (rather than progestins alone or continuous use of combined hormonal preparations) is suggested.

Clinicians should not overestimate the risks of oral contraceptives and other hormonal methods, but rather educate themselves so that they can utilize menstrual manipulation safely to match the individual patient’s needs.
- Association of Reproductive Health Professionals. Extended and continuous use of contraceptives to reduce menstruation. September 2004. http://www.arhp.org/publications-and-resources/clinical-proceedings/reduce-menses. Accessed May 17, 2010.
- Kjerulff KH, Erickson BA, Langenberg PW. Chronic gynecological conditions reported by US women: findings from the National Health Interview Survey, 1984 to 1992. Am J Public Health 1996; 86:195–199.
- Thomas SL, Ellertson C. Nuisance or natural and healthy: should monthly menstruation be optional for women? Lancet 2000; 355:922–924.
- Connell EB. Contraception in the prepill era. Contraception 1999; 59(suppl 1):7S–10S.
- Marks LV. Sexual chemistry: a history of the contraceptive pill. New Haven, CT: Yale University Press, 2001.
- Loudon NB, Foxwell M, Potts DM, Guild AL, Short RV. Acceptability of an oral contraceptive that reduces the frequency of menstruation: the tri-cycle pill regimen. Br Med J 1977; 2:487–490.
- Sulak PJ, Cressman BE, Waldrop E, Holleman S, Kuehl TJ. Extending the duration of active oral contraceptive pills to manage hormone withdrawal symptoms. Obstet Gynecol 1997; 89:179–183.
- Long-term reversible contraception. Twelve years of experience with the TCu380A and TCu220C. Contraception 1997; 56:341–352.
- Miller L, Notter KM. Menstrual reduction with extended use of combination oral contraceptive pills: randomized controlled trial. Obstet Gynecol 2001; 98:771–778.
- Mulders TM, Dieben TO. Use of the novel combined contraceptive vaginal ring NuvaRing for ovulation inhibition. Fertil Steril 2001; 75:865–870.
- Stanford JB, Mikolajczyk RT. Mechanisms of action of intrauterine devices: update and estimation of postfertilization effects. Am J Obstet Gynecol 2002; 187:1699–1708.
- Anderson FD, Hait H. A multicenter, randomized study of an extended cycle oral contraceptive. Contraception 2003; 68:89–96.
- Miller L, Hughes JP. Continuous combination oral contraceptive pills to eliminate withdrawal bleeding: a randomized trial. Obstet Gynecol 2003; 101:653–661.
- Sillem M, Schneidereit R, Heithecker R, Mueck AO. Use of an oral contraceptive containing drospirenone in an extended regimen. Eur J Contracept Reprod Health Care 2003; 8:162–169.
- Miller L, Verhoeven CH, Hout J. Extended regimens of the contraceptive vaginal ring: a randomized trial. Obstet Gynecol 2005; 106:473–482.
- Stewart FH, Kaunitz AM, Laguardia KD, Karvois DL, Fisher AC, Friedman AJ. Extended use of transdermal norelgestromin/ethinyl estradiol: a randomized trial. Obstet Gynecol 2005; 105:1389–1396.
- Sulak PJ, Kuehl TJ, Coffee A, Willis S. Prospective analysis of occurrence and management of breakthrough bleeding during an extended oral contraceptive regimen. Am J Obstet Gynecol 2006; 195:935–941.
- Lukes AS, Reardon B, Arepally G. Use of the levonorgestrel– releasing intrauterine system in women with hemostatic disorders. Fertil Steril 2008; 90:673–677.
- Anderson FD, Feldman R, Reape KZ. Endometrial effects of a 91-day extended-regimen oral contraceptive with low-dose estrogen in place of placebo. Contraception 2008; 77:91–96.
- Wright KP, Johnson JV. Evaluation of extended and continuous use oral contraceptives. Ther Clin Risk Manag 2008; 4:905–911.
- Edelman AB, Gallo MF, Jensen JT, Nichols MD, Schulz KF, Grimes DA. Continuous or extended cycle vs. cyclic use of combined oral contraceptives for contraception. Cochrane Database Syst Rev 2005; 3:CD004695.
- Sulak PJ, Kuehl TJ, Ortiz M, Shull BL. Acceptance of altering the standard 21-day/7-day oral contraceptive regimen to delay menses and reduce hormone withdrawal symptoms. Am J Obstet Gynecol 2002; 186:1142–1149.
- Turok D. The quest for better contraception: future methods. Obstet Gynecol Clin North Am 2007; 34:137–166.
- Bergqvist A, Rybo G. Treatment of menorrhagia with intrauterine release of progesterone. Br J Obstet Gynaecol 1983; 90:255–258.
- Andersson K, Odlind V, Rybo G. Levonorgestrel-releasing and copper-releasing (Nova T) IUDs during five years of use: a randomized comparative trial. Contraception 1994; 49:56–72.
- US Food and Drug Administration. FDA Approves Additional Use for IUD Mirena to Treat Heavy Menstrual Bleeding in IUD Users. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm184747.htm. Accessed May 17, 2010.
- Hidalgo M, Bahamondes L, Perrotti M, Diaz J, Dantas-Monteiro C, Petta C. Bleeding patterns and clinical performance of the levonorgestrel-releasing intrauterine system (Mirena) up to two years. Contraception 2002; 65:129–132.
- Schwallie PC, Assenzo JR. Contraceptive use—efficacy study utilizing medroxyprogesterone acetate administered as an intramuscular injection once every 90 days. Fertil Steril 1973; 24:331–339.
- Kaunitz AM. Injectable contraception. New and existing options. Obstet Gynecol Clin North Am 2000; 27:741–780.
- Mainwaring R, Hales HA, Stevenson K, et al. Metabolic parameter, bleeding, and weight changes in US women using progestin only contraceptives. Contraception 1995; 51:149–153.
- Curtis KM, Martins SL. Progestogen-only contraception and bone mineral density: a systematic review. Contraception 2006; 73:470–487.
- Shaarawy M, El-Mallah SY, Seoudi S, Hassan M, Mohsen IA. Effects of the long-term use of depot medroxyprogesterone acetate as hormonal contraceptive on bone mineral density and biochemical markers of bone remodeling. Contraception 2006; 74:297–302.
- Cromer BA, Bonny AE, Stager M, et al. Bone mineral density in adolescent females using injectable or oral contraceptives: a 24-month prospective study. Fertil Steril 2008; 90:2060–2067.
- Rome E, Ziegler J, Secic M, et al. Bone biochemical markers in adolescent girls using either depot medroxyprogesterone acetate or an oral contraceptive. J Pediatr Adolesc Gynecol 2004; 17:373–377.
- More C, Bettembuk P, Bhattoa HP, Balogh A. The effects of pregnancy and lactation on bone mineral density. Osteoporos Int 2001; 12:732–737.
- Kaunitz AM, Miller PD, Rice VM, Ross D, McClung MR. Bone mineral density in women aged 25-35 years receiving depot medroxyprogesterone acetate: recovery following discontinuation. Contraception 2006; 74:90–99.
- Guilbert ER, Brown JP, Kaunitz AM, et al. The use of depot-medroxyprogesterone acetate in contraception and its potential impact on skeletal health. Contraception 2009; 79:167–177.
- Bonny AE, Ziegler J, Harvey R, Debanne SM, Secic M, Cromer BA. Weight gain in obese and nonobese adolescent girls initiating depot medroxyprogesterone, oral contraceptive pills, or no hormonal contraceptive method. Arch Pediatr Adolesc Med 2006; 160:40–45.
- van den Heuvel MW, van Bragt AJ, Alnabawy AK, Kaptein MC. Comparison of ethinylestradiol pharmacokinetics in three hormonal contraceptive formulations: the vaginal ring, the transdermal patch and an oral contraceptive. Contraception 2005; 72:168–174.
- Douketis JD, Ginsberg JS, Holbrook A, Crowther M, Duku EK, Burrows RF. A reevaluation of the risk for venous thromboembolism with the use of oral contraceptives and hormone replacement therapy. Arch Intern Med 1997; 157:1522–1530.
- Cole JA, Norman H, Doherty M, Walker AM. Venous thromboembolism, myocardial infarction, and stroke among transdermal contraceptive system users. Obstet Gynecol 2007; 109:339–346.
- Jick S, Kaye JA, Li L, Jick H. Further results on the risk of nonfatal venous thromboembolism in users of the contraceptive transdermal patch compared to users of oral contraceptives containing norgestimate and 35 microg of ethinyl estradiol. Contraception 2007; 76:4–7.
- World Health Organization. Medical Eligibility Criteria for Contraceptive Use. 3rd ed. Geneva: Reproductive Health and Research, World Health Organization; 2004.
- Dieben TO, Roumen FJ, Apter D. Efficacy, cycle control, and user acceptability of a novel combined contraceptive vaginal ring. Obstet Gynecol 2002; 100:585–593.
- Davies GC, Feng LX, Newton JR, Dieben TO, Coelingh-Bennink HJ. The effects of a combined contraceptive vaginal ring releasing ethinyloestradiol and 3-ketodesogestrel on vaginal flora. Contraception 1992; 45:511–518.
- Roumen FJ, Boon ME, van Velzen D, Dieben TO, Coelingh Bennink HJ. The cervico-vaginal epithelium during 20 cycles’ use of a combined contraceptive vaginal ring. Hum Reprod 1996; 11:2443–2448.
- Wenzl R, van Beek A, Schnabel P, Huber J. Pharmacokinetics of etonogestrel released from the contraceptive implant Implanon. Contraception 1998; 58:283–288.
- Darney P, Patel A, Rosen K, Shapiro LS, Kaunitz AM. Safety and efficacy of a single-rod etonogestrel implant (Implanon): results from 11 international clinical trials. Fertil Steril 2009; 91:1646–1653.
- Jensen JT, Speroff L. Health benefits of oral contraceptives. Obstet Gynecol Clin North Am 2000; 27:705–721.
- Seracchioli R, Mabrouk M, Frascà C, et al. Long-term cyclic and continuous oral contraceptive therapy and endometrioma recurrence: a randomized controlled trial. Fertil Steril 2010; 93:52–56.
- Falsetti L, Dordoni D, Gastaldi C, Gastaldi A. A new association of ethinylestradiol (0.035 mg) cyproterone acetate (2 mg) in the therapy of polycystic ovary syndrome. Acta Eur Fertil 1986; 17:19–25.
- Koltun W, Lucky AW, Thiboutot D, et al. Efficacy and safety of 3 mg drospirenone/20 mcg ethinylestradiol oral contraceptive administered in 24/4 regimen in the treatment of acne vulgaris: a randomized, double-blind, placebo-controlled trial. Contraception 2008; 77:249–256.
- Kadir RA, Sabin CA, Pollard D, Lee CA, Economides DL. Quality of life during menstruation in patients with inherited bleeding disorders. Haemophilia 1998; 4:836–841.
- Schneider MB, Fisher M, Friedman SB, Bijur PE, Toffler AP. Menstrual and premenstrual issues in female military cadets: a unique population with significant concerns. J Pediatr Adolesc Gynecol 1999; 12:195–201.
- Bennell K, White S, Crossley K. The oral contraceptive pill: a revolution for sportswomen? Br J Sports Med 1999; 33:231–238.
- Kaplowitz PB, Oberfield SE. Reexamination of the age limit for defining when puberty is precocious in girls in the United States: implications for evaluation and treatment. Drug and Therapeutics and Executive Committees of the Lawson Wilkins Pediatric Endocrine Society. Pediatrics 1999; 104:936–941.
- Roxburgh DR, West MJ. The use of norethisterone to suppress menstruation in the intellectually severely retarded woman. Med J Aust 1973; 2:310–313.
- Egan TM, Siegert RJ, Fairley NA. Use of hormonal contraceptives in an institutional setting: reasons for use, consent and safety in women with psychiatric and intellectual disabilities. N Z Med J 1993; 106:338–341.
- Pillai M, O’Brien K, Hill E. The levonorgestrel intrauterine system (Mirena) for the treatment of menstrual problems in adolescents with medical disorders, or physical or learning disabilities. BJOG 2010; 117:216–221.
- Archer DF, Jensen JT, Johnson JV, Borisute H, Grubb GS, Constantine GD. Evaluation of a continuous regimen of levonorgestrel/ethinyl estradiol: phase 3 study results. Contraception 2006; 74:439–445.
- Kroll R, Reape KZ, Margolis M. The efficacy and safety of a low-dose, 91-day, extended-regimen oral contraceptive with continuous ethinyl estradiol. Contraception 2010; 81:41–48.
- Archer DF. Menstrual-cycle-related symptoms: a review of the rationale for continuous use of oral contraceptives. Contraception 2006; 74:359–366.
- den Tonkelaar I, Oddens BJ. Preferred frequency and characteristics of menstrual bleeding in relation to reproductive status, oral contraceptive use, and hormone replacement therapy use. Contraception 1999; 59:357–362.
- Edelman A, Lew R, Cwiak C, Nichols M, Jensen J. Acceptability of contraceptive-induced amenorrhea in a racially diverse group of US women. Contraception 2007; 75:450–453.
- Gerschultz KL, Sucato GS, Hennon TR, Murray PJ, Gold MA. Extended cycling of combined hormonal contraceptives in adolescents: physician views and prescribing practices. J Adolesc Health 2007; 40:151–157.
- Frankovich RJ, Lebrun CM. Menstrual cycle, contraception, and performance. Clin Sports Med 2000; 19:251–271.
- Speroff L, Darney PD. A Clinical Guide for Contraception. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Kaunitz AM. Long-acting contraceptive options. Int J Fertil Menopausal Stud 1996; 41:69–76.
- US Food and Drug Administration. Guidance for Industry Labeling for Combined Oral Contraceptives, 2004. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075075.pdfAccessed May 17, 2010.
If they wish, women can have more control over when and if they menstruate. By using hormonal contraceptives in extended or continuous regimens, they can have their period less often, a practice called menstrual manipulation or menstrual suppression.
Actually, with the help of their clinicians, women have been doing this for years. But now that several products have been approved by the US Food and Drug Administration (FDA) specifically for use in extended or continuous regimens, the practice has become more widely accepted.
Reasons for suppressing menstrual flow range from avoiding bleeding during a particular event (eg, a wedding, graduation, or sports competition) to finding relief from dysmenorrhea or reducing or eliminating menstruation in the treatment of endometriosis, migraine, and other medical conditions exacerbated by hormonal changes around the time of menses.1 Alternatively, some women may practice menstrual manipulation for no other reason than to simply avoid menstruation.
MENSTRUAL DISORDERS ARE TROUBLESOME, COMMON
Each year in the United States, menstrual disorders such as dysmenorrhea (painful menstruation), menorrhagia (excessive or frequent menstruation), metrorrhagia (irregular menstruation), menometrorrhagia (excessive and irregular menstruation), and premenstrual syndrome affect nearly 2.5 million women age 18 to 50 years.2 Menstrual disorders are the leading cause of gynecologic morbidity in the United States, outnumbering adnexal masses (the second most common cause) by a factor of three.2 In addition, these disorders extend into the workplace, costing US industry about 8% of its total wage bill.3
A BRIEF HISTORY OF CONTRACEPTIVE DEVELOPMENT
The idea of using progestins for birth control was first advanced in the 1950s by Dr. Gregory Pincus, who proposed a regimen of 21 days of active drug followed by 7 drug-free days to allow withdrawal bleeding, mimicking the natural cycle.4 This “21/7” regimen was designed to follow the lunar cycle in the hope it would be, in the words of Dr. John Rock, “a morally permissible variant of the rhythm method,”5 thereby making it acceptable to women, clinicians, and the Catholic Church.
In 1977, Loudon et al6 reported the results of a study in which women took active pills for 84 days instead of 21 days, which reduced the frequency of menstruation to every 3 months. Since then, extending the active pills beyond 21 days to avoid menses and other hormone-withdrawal symptoms has become popular in clinical practice, and many studies have investigated the extended or continuous use of oral and other forms of contraception to delay menses.7–18
CURRENT METHODS OF MENSTRUAL MANIPULATION
A variety of available products prevent conception by altering the menstrual cycle:
- Oral estrogen-progestin contraceptive pills
- A drug-releasing intrauterine device
- Depot medroxyprogesterone acetate injections
- A contraceptive vaginal ring
- An implantable etonogestrel contraceptive.
Their use in menstrual manipulation is summarized in Table 1.
Oral contraceptive pills
The most common way to manipulate the menstrual cycle is to extend the time between hormone-free weeks in an oral contraceptive regimen.
If the patient is young, you can prescribe a monophasic 21/7 oral contraceptive and tell her to take one active pill every day for 21 days and then start a new pack and keep taking active pills for up to 84 consecutive days, skipping the placebo pills until she wants to have her menstrual period. She can choose which week to have it: if the scheduled 12th week of an extended-cycle oral contraceptive regimen is inconvenient, she can plan it for week 10, or week 9, or whichever week is convenient.
The rationale for using an 84-day (12-week) cycle is that it still provides four periods per year, alleviating fears of hypertrophic endometrium.19
In this scenario, unscheduled or breakthrough bleeding can be managed by taking a “double-up pill” from a spare pack on any day breakthrough bleeding occurs and until it resolves. Menstrual periods should not be planned for intervals shorter than 21 days, owing to the risk of ovulation. Missed days of pills or use of placebo pills should also not exceed 7 days to prevent escape ovulation. 20
In some women with endometriosis and other medical reasons, continuous oral contraception with no placebo week can be prescribed.
Unfortunately, the downside to suppressing withdrawal bleeding is unscheduled or “breakthrough” bleeding. The best way to treat this unscheduled bleeding is not known. Patients who are not sexually active can be reassured that the goal of an atrophic endometrium can still be achieved, with resultant pill amenorrhea (particularly useful for those with severe dysmenorrhea or other reasons to want to avoid flow). Patients could also try to manage flow by periodically taking a 3- to 5-day break from hormone-containing pills to allow flow. They can also try switching to another oral contraceptive that has a different progestin that would spiral the arterioles of the endometrium more tightly and thus more aggressively induce atrophy.13,17,21 For instance, levonorgestrel is 10 to 20 times more potent than norethindrone. Choosing a pill with a higher monophasic dosing of levonorgestrel or a similar progestin may minimize unscheduled bleeding.
Currently, several oral contraceptives are approved for use in an extended regimen.
Seasonale was the first oral contraceptive marketed in the United States with an extended active regimen.22 It comes in a pack of 84 pills containing ethinyl estradiol 0.03 mg and levonorgestrel 0.15 mg, plus 7 placebo pills.
Seasonique is similar to Seasonale, but instead of placebo pills it has seven pills that contain ethinyl estradiol 0.010 mg.
Lybrel is a low-dose combination containing ethinyl estradiol 0.02 mg and levonorgestrel 0.09 mg. Packaged as an entire year’s worth of active pills to be taken continuously for 365 days without a placebo phase or pillfree interval,23 it is the only FDA-approved continuous oral contraceptive available in the United States.
An intrauterine device
Intrauterine devices were originally developed as contraceptives. The addition of a progestin to these devices has been shown to reduce heavy menstrual bleeding by up to 90%.24,25
Mirena IUS, a levonorgestrel-releasing device, is the only medicated intrauterine device that is currently available in the United States. (“IUS” stands for “intrauterine system.”) It was recently approved by the FDA to treat heavy menstrual bleeding in women who use intrauterine contraception as their method of pregnancy prevention.26 About 50% of women who use this device develop amenorrhea within 6 months of insertion, while 25% report oligomenorrhea.27
The Mirena device can be left in the uterus for up to 5 years. It may be a good choice for inducing amenorrhea in women with hemostatic disorders or in whom estrogen either is contraindicated or causes health concerns.18 The copper intrauterine device (Paragard; Duramed Pharmaceuticals Inc., Pomona, NY) remains a viable option for those who cannot or do not tolerate hormonal therapy. However, Mirena may provide less unscheduled bleeding than the copper intrauterine device.
Depot medroxyprogesterone acetate injections
Depo-Provera (depot medroxyprogesterone acetate) injections are given at 90-day intervals. 28 This contraceptive method inhibits ovulation and decidualizes the endometrium, thereby reducing or eliminating uterine bleeding. 29
While new users may initially experience excessive prolonged bleeding (10 or more days) while shedding their existing lining, the rate of amenorrhea has been shown to increase over time as the lining atrophies.30 Thus, prolonged use of this agent reduces the frequency of menstruation as well as menstruation-related symptoms.
Depot medroxyprogesterone acetate is ideal for patients whose menstrual periods pose a significant hygiene problem (eg, developmentally challenged girls). In our experience, the injections can be given at shorter intervals to induce atrophy of the endometrium quickly. In this scenario, the clinician might give an injection every 4 to 6 weeks for two or three doses to induce amenorrhea and then return to every-12-week dosing.
The main risk when using medroxyprogesterone injections to induce amenorrhea is the potential for bone loss. Users of this method have been shown to have lower mean bone mineral density31–33 and significantly higher levels of biomarkers of bone formation and resorption32,34 than nonusers. However, these changes are similar to those seen in breastfeeding women,35 are reversible with cessation, 36 and are not associated with increased fracture risk.37 In adolescent girls, pregnancy poses similar risks to the bones, with longerterm consequences.
Medroxyprogesterone can also stimulate appetite, causing 10 to 20 kg of weight gain in adolescents and women who are already obese and have trouble with appetite regulation.38 Slender users tend not to gain weight, however.
Given this information, depot medroxyprogesterone acetate appears to be a cost-effective contraceptive option that should be considered in the context of the clinical situation and preference of each patient.
Transdermal contraceptive patch
Ortho Evra, a transdermal patch, is designed to deliver ethinyl estradiol 0.02 mg and norelgestromin 0.150 mg daily.39 It is usually applied weekly for 3 weeks, followed by a patch-free week to induce regular monthly withdrawal bleeding.
Extended use of the patch to manipulate menstruation is an off-label use. In the only trial evaluating extended use of the patch, amenorrhea occurred in 12% of users, but unscheduled bleeding and spotting were common. 16
Although there is some evidence that the long-term use of the patch may increase the risk of venous thromboembolism,40,41 the risk in women who use the patch has been found to be similar to that in women using an oral contraceptive.42 However, serum ethinyl estradiol levels have been found to be higher with the use of the weekly patch than with oral contraceptives or the contraceptive vaginal ring39; as a result, many physicians are hesitant to recommend its continuous use.
Pending further data about the safety profile of this contraceptive, the World Health Organization Medical Eligibility Criteria for Contraceptive Use suggest that the same guidelines for the prescription of combination oral contraceptives should also apply to the patch.43
Contraceptive vaginal ring
NuvaRing, a contraceptive vaginal ring, releases a daily dose of ethinyl estradiol 0.015 mg and etonogestrel 0.12 mg.10 It is inserted, left in for 21 days, and then removed and left out for 7 days, during which withdrawal bleeding occurs.10
Vaginal administration has been shown to allow low, continuous dosing, which results in more stable serum concentrations than with the patch or oral contraceptives.39 In the only trial comparing an extended vaginal ring regimen and the traditional 28-day regimen, extended use resulted in fewer overall days of bleeding than monthly use, but with more unscheduled spotting.15
The most common side effects include headache, vaginitis, and leukorrhea,44 but there is no evidence of bacteriologic or cytologic changes in the cervicovaginal epithelium, even with extended use.45,46
Etonogestrel implantable contraceptive
Implanon, a single-rod progestin implant, is available in the United States and elsewhere. It is placed subdermally in the inner upper arm and provides contraception for as long as 3 years.
Implanon contains 68 mg of the progestin etonogestrel, which it slowly releases over time, initially at 0.06 to 0.07 mg/day, decreasing to 0.035 to 0.045 mg/day at the end of the first year, to 0.03 to 0.04 mg/day at the end of the second year, and then to 0.025 to 0.03 mg/day at the end of the third year.47
The amount of vaginal bleeding associated with the use of the implant is generally modest, but the pattern tends to be unpredictable. 48 In addition, because amenorrhea is reported as a side effect in only 22% of women during the first 2 years of its use,48 the progestin implant is a less satisfactory means of menstrual suppression than the other methods discussed above.
BENEFITS OF MENSTRUAL MANIPULATION
Menstrual manipulation has a number of benefits in terms of both overall health and lifestyle.
For most women, using a long-acting hormonal contraceptive carries low risks and substantial health benefits. Women who take oral contraceptives are less likely to develop osteoporosis, ovarian or endometrial cancer, benign breast changes, or pelvic inflammatory disease. 49 Long-term use of an oral contraceptive can also preserve fertility by reducing and delaying the incidence of endometriosis,50 and is effective at treating acne vulgaris, which tends to be common among patients with polycystic ovary syndrome.51,52 In addition, this practice can be used to reduce overall blood loss, an application that is particularly important in women with a bleeding diathesis such as von Willebrand disease, who frequently suffer from menorrhagia.53
Reduced menstruation may also prove more convenient during particular occasions, such as vacations and athletic activities. Specifically, it may be useful to women serving in the military. In a study by Schneider et al,54 a cohort of 83 female cadets reported a significant perceived impact of premenstrual and menstrual-related symptoms on academic, physical, and military activities, as well as difficulties in obtaining, changing, and disposing of menstrual materials in a military setting. Likewise, reduced menstrual frequency or amenorrhea may play an important role in female athletes, who reportedly use oral contraceptives to control premenstrual symptoms, to protect bone health, and to manipulate the menstrual cycle in order to maximize performance.55
Adolescent girls are another group who may benefit from reduced or absent menses, once they have reached near-final height. By practicing menstrual suppression, girls can avoid dysmenorrhea and the inconvenience of menstruation during the school day, when their access to painkillers, sanitary pads or tampons, and a change of clothes may be limited. 56 Clinicians who discuss with teenage patients the benefits of innovative hormonal contraceptive schedules that reduce menstrual frequency may be able to improve the quality of life for these young women.
In a very short girl just after menarche, care must be taken not to start a hormonal method too early so as not to prematurely close epiphyses and stunt final height; after menarche, most girls still have 1 to 4 inches of potential growth. For a young lady 4 feet 11 inches tall, that extra inch may be important.
Finally, menstrual manipulation may also find a niche among the developmentally challenged. Women with cognitive impairment and physical disabilities may have difficulty with hygienic practice around menses. For a number of years, contraceptives have been used to manage menstrual hygiene in patients with catamenial (ie, menstrual) epilepsy, and to address caregiver concerns in women with severe mental retardation, with improved behavior noted in some patients.57–59 In this setting, an agent that suppresses menses and also provides contraception, especially for those girls and women at risk of abuse, may offer substantial benefits.
DISADVANTAGES OF MENSTRUAL MANIPULATION
Rates of adverse events and of discontinuation of extended and continuous oral contraceptive regimens are comparable with those reported for cyclic regimens, except for higher rates of breakthrough bleeding.
In a trial of continuous oral contraceptive use in more than 2,000 patients, 396 (18.5%) withdrew from the study as a result of bothersome uterine bleeding.60 However, while breakthrough bleeding often occurs during the first few months of extended oral contraceptive use, it usually decreases with each successive cycle of therapy and is comparable to that reported by patients on the conventional oral contraceptive regimen by the fourth extended cycle.12
CONTRACEPTIVE EFFICACY
The efficacy of extended and continuous oral contraceptive regimens is comparable with that of cyclic regimens.12,60,61 One reason for this may be better adherence to continuous regimens: women using this regimen have been shown to miss fewer pills than those on a cyclic regimen, especially during the critical first week of the pill pack.21
Several studies have shown that some women ovulate during the standard 21/7 oral contraceptive regimen even if they do not miss any pills or take pills off-schedule, putting them at greater risk of pregnancy.62 Large studies evaluating the efficacy of an extendedcycle regimen have shown a pregnancy rate during the 1-year study period that was either comparable with61 or lower than12,60 rates with standard regimens.
Heterosexual couples need to be advised to use condoms to further reduce the already low failure rate and to prevent sexually transmitted diseases.
ACCEPTABILITY OF MENSTRUAL MANIPULATION
Ever since the earliest trial of an extended oral contraceptive regimen, participants have expressed a favorable response to the resulting decrease in menstrual frequency; in the 1977 study by Loudon et al,6 patients on the extended regimen cited infrequent periods (82%), fewer menstrual problems (20%), and easier pill-taking (19%) as favorable features.
In 1999, den Tonkelaar and Oddens63 surveyed 1,300 Dutch women about their preferred frequency of menstruation and found that about 70% between the ages of 15 and 49 preferred a frequency of between every 3 months and never. A similar survey in the United States indicated that 58% preferred a bleeding frequency of either every 3 months or never to more frequent periods.64
While patients find menstrual manipulation generally acceptable, clinician approval has been more varied. Loudon et al reported that “the doctors and nurses on the clinic staff were less enthusiastic about this regimen than the volunteers themselves.”6 In a survey of 222 clinicians,65 90% of responders reported ever having prescribed extended or continuous dosing regimens to adolescents, and 33% reported that extended cycles made up more than 10% of their total oral contraceptive prescriptions.
Myths and misperceptions about menstrual manipulation abound. Many clinicians believe that routine use of an extended or continuous oral contraceptive regimen is inadvisable, despite the lack of evidence to support this notion.66 Therefore, many care providers need more education about the practice and benefits of menstrual manipulation.
THE RIGHT METHOD FOR THE RIGHT PATIENT
Manipulation and suppression of menstruation through continuous or extended use of oral contraceptives or by other means may have a number of advantages to women, including fewer menstrual-related syndromes, reduced absenteeism from work or school, and greater overall satisfaction.
For women whose goal is to reduce but not necessarily to eliminate monthly bleeding, the cyclic use of estrogen-progestin contraception (rather than progestins alone or continuous use of combined hormonal preparations) is suggested.
Clinicians should not overestimate the risks of oral contraceptives and other hormonal methods, but rather educate themselves so that they can utilize menstrual manipulation safely to match the individual patient’s needs.
If they wish, women can have more control over when and if they menstruate. By using hormonal contraceptives in extended or continuous regimens, they can have their period less often, a practice called menstrual manipulation or menstrual suppression.
Actually, with the help of their clinicians, women have been doing this for years. But now that several products have been approved by the US Food and Drug Administration (FDA) specifically for use in extended or continuous regimens, the practice has become more widely accepted.
Reasons for suppressing menstrual flow range from avoiding bleeding during a particular event (eg, a wedding, graduation, or sports competition) to finding relief from dysmenorrhea or reducing or eliminating menstruation in the treatment of endometriosis, migraine, and other medical conditions exacerbated by hormonal changes around the time of menses.1 Alternatively, some women may practice menstrual manipulation for no other reason than to simply avoid menstruation.
MENSTRUAL DISORDERS ARE TROUBLESOME, COMMON
Each year in the United States, menstrual disorders such as dysmenorrhea (painful menstruation), menorrhagia (excessive or frequent menstruation), metrorrhagia (irregular menstruation), menometrorrhagia (excessive and irregular menstruation), and premenstrual syndrome affect nearly 2.5 million women age 18 to 50 years.2 Menstrual disorders are the leading cause of gynecologic morbidity in the United States, outnumbering adnexal masses (the second most common cause) by a factor of three.2 In addition, these disorders extend into the workplace, costing US industry about 8% of its total wage bill.3
A BRIEF HISTORY OF CONTRACEPTIVE DEVELOPMENT
The idea of using progestins for birth control was first advanced in the 1950s by Dr. Gregory Pincus, who proposed a regimen of 21 days of active drug followed by 7 drug-free days to allow withdrawal bleeding, mimicking the natural cycle.4 This “21/7” regimen was designed to follow the lunar cycle in the hope it would be, in the words of Dr. John Rock, “a morally permissible variant of the rhythm method,”5 thereby making it acceptable to women, clinicians, and the Catholic Church.
In 1977, Loudon et al6 reported the results of a study in which women took active pills for 84 days instead of 21 days, which reduced the frequency of menstruation to every 3 months. Since then, extending the active pills beyond 21 days to avoid menses and other hormone-withdrawal symptoms has become popular in clinical practice, and many studies have investigated the extended or continuous use of oral and other forms of contraception to delay menses.7–18
CURRENT METHODS OF MENSTRUAL MANIPULATION
A variety of available products prevent conception by altering the menstrual cycle:
- Oral estrogen-progestin contraceptive pills
- A drug-releasing intrauterine device
- Depot medroxyprogesterone acetate injections
- A contraceptive vaginal ring
- An implantable etonogestrel contraceptive.
Their use in menstrual manipulation is summarized in Table 1.
Oral contraceptive pills
The most common way to manipulate the menstrual cycle is to extend the time between hormone-free weeks in an oral contraceptive regimen.
If the patient is young, you can prescribe a monophasic 21/7 oral contraceptive and tell her to take one active pill every day for 21 days and then start a new pack and keep taking active pills for up to 84 consecutive days, skipping the placebo pills until she wants to have her menstrual period. She can choose which week to have it: if the scheduled 12th week of an extended-cycle oral contraceptive regimen is inconvenient, she can plan it for week 10, or week 9, or whichever week is convenient.
The rationale for using an 84-day (12-week) cycle is that it still provides four periods per year, alleviating fears of hypertrophic endometrium.19
In this scenario, unscheduled or breakthrough bleeding can be managed by taking a “double-up pill” from a spare pack on any day breakthrough bleeding occurs and until it resolves. Menstrual periods should not be planned for intervals shorter than 21 days, owing to the risk of ovulation. Missed days of pills or use of placebo pills should also not exceed 7 days to prevent escape ovulation. 20
In some women with endometriosis and other medical reasons, continuous oral contraception with no placebo week can be prescribed.
Unfortunately, the downside to suppressing withdrawal bleeding is unscheduled or “breakthrough” bleeding. The best way to treat this unscheduled bleeding is not known. Patients who are not sexually active can be reassured that the goal of an atrophic endometrium can still be achieved, with resultant pill amenorrhea (particularly useful for those with severe dysmenorrhea or other reasons to want to avoid flow). Patients could also try to manage flow by periodically taking a 3- to 5-day break from hormone-containing pills to allow flow. They can also try switching to another oral contraceptive that has a different progestin that would spiral the arterioles of the endometrium more tightly and thus more aggressively induce atrophy.13,17,21 For instance, levonorgestrel is 10 to 20 times more potent than norethindrone. Choosing a pill with a higher monophasic dosing of levonorgestrel or a similar progestin may minimize unscheduled bleeding.
Currently, several oral contraceptives are approved for use in an extended regimen.
Seasonale was the first oral contraceptive marketed in the United States with an extended active regimen.22 It comes in a pack of 84 pills containing ethinyl estradiol 0.03 mg and levonorgestrel 0.15 mg, plus 7 placebo pills.
Seasonique is similar to Seasonale, but instead of placebo pills it has seven pills that contain ethinyl estradiol 0.010 mg.
Lybrel is a low-dose combination containing ethinyl estradiol 0.02 mg and levonorgestrel 0.09 mg. Packaged as an entire year’s worth of active pills to be taken continuously for 365 days without a placebo phase or pillfree interval,23 it is the only FDA-approved continuous oral contraceptive available in the United States.
An intrauterine device
Intrauterine devices were originally developed as contraceptives. The addition of a progestin to these devices has been shown to reduce heavy menstrual bleeding by up to 90%.24,25
Mirena IUS, a levonorgestrel-releasing device, is the only medicated intrauterine device that is currently available in the United States. (“IUS” stands for “intrauterine system.”) It was recently approved by the FDA to treat heavy menstrual bleeding in women who use intrauterine contraception as their method of pregnancy prevention.26 About 50% of women who use this device develop amenorrhea within 6 months of insertion, while 25% report oligomenorrhea.27
The Mirena device can be left in the uterus for up to 5 years. It may be a good choice for inducing amenorrhea in women with hemostatic disorders or in whom estrogen either is contraindicated or causes health concerns.18 The copper intrauterine device (Paragard; Duramed Pharmaceuticals Inc., Pomona, NY) remains a viable option for those who cannot or do not tolerate hormonal therapy. However, Mirena may provide less unscheduled bleeding than the copper intrauterine device.
Depot medroxyprogesterone acetate injections
Depo-Provera (depot medroxyprogesterone acetate) injections are given at 90-day intervals. 28 This contraceptive method inhibits ovulation and decidualizes the endometrium, thereby reducing or eliminating uterine bleeding. 29
While new users may initially experience excessive prolonged bleeding (10 or more days) while shedding their existing lining, the rate of amenorrhea has been shown to increase over time as the lining atrophies.30 Thus, prolonged use of this agent reduces the frequency of menstruation as well as menstruation-related symptoms.
Depot medroxyprogesterone acetate is ideal for patients whose menstrual periods pose a significant hygiene problem (eg, developmentally challenged girls). In our experience, the injections can be given at shorter intervals to induce atrophy of the endometrium quickly. In this scenario, the clinician might give an injection every 4 to 6 weeks for two or three doses to induce amenorrhea and then return to every-12-week dosing.
The main risk when using medroxyprogesterone injections to induce amenorrhea is the potential for bone loss. Users of this method have been shown to have lower mean bone mineral density31–33 and significantly higher levels of biomarkers of bone formation and resorption32,34 than nonusers. However, these changes are similar to those seen in breastfeeding women,35 are reversible with cessation, 36 and are not associated with increased fracture risk.37 In adolescent girls, pregnancy poses similar risks to the bones, with longerterm consequences.
Medroxyprogesterone can also stimulate appetite, causing 10 to 20 kg of weight gain in adolescents and women who are already obese and have trouble with appetite regulation.38 Slender users tend not to gain weight, however.
Given this information, depot medroxyprogesterone acetate appears to be a cost-effective contraceptive option that should be considered in the context of the clinical situation and preference of each patient.
Transdermal contraceptive patch
Ortho Evra, a transdermal patch, is designed to deliver ethinyl estradiol 0.02 mg and norelgestromin 0.150 mg daily.39 It is usually applied weekly for 3 weeks, followed by a patch-free week to induce regular monthly withdrawal bleeding.
Extended use of the patch to manipulate menstruation is an off-label use. In the only trial evaluating extended use of the patch, amenorrhea occurred in 12% of users, but unscheduled bleeding and spotting were common. 16
Although there is some evidence that the long-term use of the patch may increase the risk of venous thromboembolism,40,41 the risk in women who use the patch has been found to be similar to that in women using an oral contraceptive.42 However, serum ethinyl estradiol levels have been found to be higher with the use of the weekly patch than with oral contraceptives or the contraceptive vaginal ring39; as a result, many physicians are hesitant to recommend its continuous use.
Pending further data about the safety profile of this contraceptive, the World Health Organization Medical Eligibility Criteria for Contraceptive Use suggest that the same guidelines for the prescription of combination oral contraceptives should also apply to the patch.43
Contraceptive vaginal ring
NuvaRing, a contraceptive vaginal ring, releases a daily dose of ethinyl estradiol 0.015 mg and etonogestrel 0.12 mg.10 It is inserted, left in for 21 days, and then removed and left out for 7 days, during which withdrawal bleeding occurs.10
Vaginal administration has been shown to allow low, continuous dosing, which results in more stable serum concentrations than with the patch or oral contraceptives.39 In the only trial comparing an extended vaginal ring regimen and the traditional 28-day regimen, extended use resulted in fewer overall days of bleeding than monthly use, but with more unscheduled spotting.15
The most common side effects include headache, vaginitis, and leukorrhea,44 but there is no evidence of bacteriologic or cytologic changes in the cervicovaginal epithelium, even with extended use.45,46
Etonogestrel implantable contraceptive
Implanon, a single-rod progestin implant, is available in the United States and elsewhere. It is placed subdermally in the inner upper arm and provides contraception for as long as 3 years.
Implanon contains 68 mg of the progestin etonogestrel, which it slowly releases over time, initially at 0.06 to 0.07 mg/day, decreasing to 0.035 to 0.045 mg/day at the end of the first year, to 0.03 to 0.04 mg/day at the end of the second year, and then to 0.025 to 0.03 mg/day at the end of the third year.47
The amount of vaginal bleeding associated with the use of the implant is generally modest, but the pattern tends to be unpredictable. 48 In addition, because amenorrhea is reported as a side effect in only 22% of women during the first 2 years of its use,48 the progestin implant is a less satisfactory means of menstrual suppression than the other methods discussed above.
BENEFITS OF MENSTRUAL MANIPULATION
Menstrual manipulation has a number of benefits in terms of both overall health and lifestyle.
For most women, using a long-acting hormonal contraceptive carries low risks and substantial health benefits. Women who take oral contraceptives are less likely to develop osteoporosis, ovarian or endometrial cancer, benign breast changes, or pelvic inflammatory disease. 49 Long-term use of an oral contraceptive can also preserve fertility by reducing and delaying the incidence of endometriosis,50 and is effective at treating acne vulgaris, which tends to be common among patients with polycystic ovary syndrome.51,52 In addition, this practice can be used to reduce overall blood loss, an application that is particularly important in women with a bleeding diathesis such as von Willebrand disease, who frequently suffer from menorrhagia.53
Reduced menstruation may also prove more convenient during particular occasions, such as vacations and athletic activities. Specifically, it may be useful to women serving in the military. In a study by Schneider et al,54 a cohort of 83 female cadets reported a significant perceived impact of premenstrual and menstrual-related symptoms on academic, physical, and military activities, as well as difficulties in obtaining, changing, and disposing of menstrual materials in a military setting. Likewise, reduced menstrual frequency or amenorrhea may play an important role in female athletes, who reportedly use oral contraceptives to control premenstrual symptoms, to protect bone health, and to manipulate the menstrual cycle in order to maximize performance.55
Adolescent girls are another group who may benefit from reduced or absent menses, once they have reached near-final height. By practicing menstrual suppression, girls can avoid dysmenorrhea and the inconvenience of menstruation during the school day, when their access to painkillers, sanitary pads or tampons, and a change of clothes may be limited. 56 Clinicians who discuss with teenage patients the benefits of innovative hormonal contraceptive schedules that reduce menstrual frequency may be able to improve the quality of life for these young women.
In a very short girl just after menarche, care must be taken not to start a hormonal method too early so as not to prematurely close epiphyses and stunt final height; after menarche, most girls still have 1 to 4 inches of potential growth. For a young lady 4 feet 11 inches tall, that extra inch may be important.
Finally, menstrual manipulation may also find a niche among the developmentally challenged. Women with cognitive impairment and physical disabilities may have difficulty with hygienic practice around menses. For a number of years, contraceptives have been used to manage menstrual hygiene in patients with catamenial (ie, menstrual) epilepsy, and to address caregiver concerns in women with severe mental retardation, with improved behavior noted in some patients.57–59 In this setting, an agent that suppresses menses and also provides contraception, especially for those girls and women at risk of abuse, may offer substantial benefits.
DISADVANTAGES OF MENSTRUAL MANIPULATION
Rates of adverse events and of discontinuation of extended and continuous oral contraceptive regimens are comparable with those reported for cyclic regimens, except for higher rates of breakthrough bleeding.
In a trial of continuous oral contraceptive use in more than 2,000 patients, 396 (18.5%) withdrew from the study as a result of bothersome uterine bleeding.60 However, while breakthrough bleeding often occurs during the first few months of extended oral contraceptive use, it usually decreases with each successive cycle of therapy and is comparable to that reported by patients on the conventional oral contraceptive regimen by the fourth extended cycle.12
CONTRACEPTIVE EFFICACY
The efficacy of extended and continuous oral contraceptive regimens is comparable with that of cyclic regimens.12,60,61 One reason for this may be better adherence to continuous regimens: women using this regimen have been shown to miss fewer pills than those on a cyclic regimen, especially during the critical first week of the pill pack.21
Several studies have shown that some women ovulate during the standard 21/7 oral contraceptive regimen even if they do not miss any pills or take pills off-schedule, putting them at greater risk of pregnancy.62 Large studies evaluating the efficacy of an extendedcycle regimen have shown a pregnancy rate during the 1-year study period that was either comparable with61 or lower than12,60 rates with standard regimens.
Heterosexual couples need to be advised to use condoms to further reduce the already low failure rate and to prevent sexually transmitted diseases.
ACCEPTABILITY OF MENSTRUAL MANIPULATION
Ever since the earliest trial of an extended oral contraceptive regimen, participants have expressed a favorable response to the resulting decrease in menstrual frequency; in the 1977 study by Loudon et al,6 patients on the extended regimen cited infrequent periods (82%), fewer menstrual problems (20%), and easier pill-taking (19%) as favorable features.
In 1999, den Tonkelaar and Oddens63 surveyed 1,300 Dutch women about their preferred frequency of menstruation and found that about 70% between the ages of 15 and 49 preferred a frequency of between every 3 months and never. A similar survey in the United States indicated that 58% preferred a bleeding frequency of either every 3 months or never to more frequent periods.64
While patients find menstrual manipulation generally acceptable, clinician approval has been more varied. Loudon et al reported that “the doctors and nurses on the clinic staff were less enthusiastic about this regimen than the volunteers themselves.”6 In a survey of 222 clinicians,65 90% of responders reported ever having prescribed extended or continuous dosing regimens to adolescents, and 33% reported that extended cycles made up more than 10% of their total oral contraceptive prescriptions.
Myths and misperceptions about menstrual manipulation abound. Many clinicians believe that routine use of an extended or continuous oral contraceptive regimen is inadvisable, despite the lack of evidence to support this notion.66 Therefore, many care providers need more education about the practice and benefits of menstrual manipulation.
THE RIGHT METHOD FOR THE RIGHT PATIENT
Manipulation and suppression of menstruation through continuous or extended use of oral contraceptives or by other means may have a number of advantages to women, including fewer menstrual-related syndromes, reduced absenteeism from work or school, and greater overall satisfaction.
For women whose goal is to reduce but not necessarily to eliminate monthly bleeding, the cyclic use of estrogen-progestin contraception (rather than progestins alone or continuous use of combined hormonal preparations) is suggested.
Clinicians should not overestimate the risks of oral contraceptives and other hormonal methods, but rather educate themselves so that they can utilize menstrual manipulation safely to match the individual patient’s needs.
- Association of Reproductive Health Professionals. Extended and continuous use of contraceptives to reduce menstruation. September 2004. http://www.arhp.org/publications-and-resources/clinical-proceedings/reduce-menses. Accessed May 17, 2010.
- Kjerulff KH, Erickson BA, Langenberg PW. Chronic gynecological conditions reported by US women: findings from the National Health Interview Survey, 1984 to 1992. Am J Public Health 1996; 86:195–199.
- Thomas SL, Ellertson C. Nuisance or natural and healthy: should monthly menstruation be optional for women? Lancet 2000; 355:922–924.
- Connell EB. Contraception in the prepill era. Contraception 1999; 59(suppl 1):7S–10S.
- Marks LV. Sexual chemistry: a history of the contraceptive pill. New Haven, CT: Yale University Press, 2001.
- Loudon NB, Foxwell M, Potts DM, Guild AL, Short RV. Acceptability of an oral contraceptive that reduces the frequency of menstruation: the tri-cycle pill regimen. Br Med J 1977; 2:487–490.
- Sulak PJ, Cressman BE, Waldrop E, Holleman S, Kuehl TJ. Extending the duration of active oral contraceptive pills to manage hormone withdrawal symptoms. Obstet Gynecol 1997; 89:179–183.
- Long-term reversible contraception. Twelve years of experience with the TCu380A and TCu220C. Contraception 1997; 56:341–352.
- Miller L, Notter KM. Menstrual reduction with extended use of combination oral contraceptive pills: randomized controlled trial. Obstet Gynecol 2001; 98:771–778.
- Mulders TM, Dieben TO. Use of the novel combined contraceptive vaginal ring NuvaRing for ovulation inhibition. Fertil Steril 2001; 75:865–870.
- Stanford JB, Mikolajczyk RT. Mechanisms of action of intrauterine devices: update and estimation of postfertilization effects. Am J Obstet Gynecol 2002; 187:1699–1708.
- Anderson FD, Hait H. A multicenter, randomized study of an extended cycle oral contraceptive. Contraception 2003; 68:89–96.
- Miller L, Hughes JP. Continuous combination oral contraceptive pills to eliminate withdrawal bleeding: a randomized trial. Obstet Gynecol 2003; 101:653–661.
- Sillem M, Schneidereit R, Heithecker R, Mueck AO. Use of an oral contraceptive containing drospirenone in an extended regimen. Eur J Contracept Reprod Health Care 2003; 8:162–169.
- Miller L, Verhoeven CH, Hout J. Extended regimens of the contraceptive vaginal ring: a randomized trial. Obstet Gynecol 2005; 106:473–482.
- Stewart FH, Kaunitz AM, Laguardia KD, Karvois DL, Fisher AC, Friedman AJ. Extended use of transdermal norelgestromin/ethinyl estradiol: a randomized trial. Obstet Gynecol 2005; 105:1389–1396.
- Sulak PJ, Kuehl TJ, Coffee A, Willis S. Prospective analysis of occurrence and management of breakthrough bleeding during an extended oral contraceptive regimen. Am J Obstet Gynecol 2006; 195:935–941.
- Lukes AS, Reardon B, Arepally G. Use of the levonorgestrel– releasing intrauterine system in women with hemostatic disorders. Fertil Steril 2008; 90:673–677.
- Anderson FD, Feldman R, Reape KZ. Endometrial effects of a 91-day extended-regimen oral contraceptive with low-dose estrogen in place of placebo. Contraception 2008; 77:91–96.
- Wright KP, Johnson JV. Evaluation of extended and continuous use oral contraceptives. Ther Clin Risk Manag 2008; 4:905–911.
- Edelman AB, Gallo MF, Jensen JT, Nichols MD, Schulz KF, Grimes DA. Continuous or extended cycle vs. cyclic use of combined oral contraceptives for contraception. Cochrane Database Syst Rev 2005; 3:CD004695.
- Sulak PJ, Kuehl TJ, Ortiz M, Shull BL. Acceptance of altering the standard 21-day/7-day oral contraceptive regimen to delay menses and reduce hormone withdrawal symptoms. Am J Obstet Gynecol 2002; 186:1142–1149.
- Turok D. The quest for better contraception: future methods. Obstet Gynecol Clin North Am 2007; 34:137–166.
- Bergqvist A, Rybo G. Treatment of menorrhagia with intrauterine release of progesterone. Br J Obstet Gynaecol 1983; 90:255–258.
- Andersson K, Odlind V, Rybo G. Levonorgestrel-releasing and copper-releasing (Nova T) IUDs during five years of use: a randomized comparative trial. Contraception 1994; 49:56–72.
- US Food and Drug Administration. FDA Approves Additional Use for IUD Mirena to Treat Heavy Menstrual Bleeding in IUD Users. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm184747.htm. Accessed May 17, 2010.
- Hidalgo M, Bahamondes L, Perrotti M, Diaz J, Dantas-Monteiro C, Petta C. Bleeding patterns and clinical performance of the levonorgestrel-releasing intrauterine system (Mirena) up to two years. Contraception 2002; 65:129–132.
- Schwallie PC, Assenzo JR. Contraceptive use—efficacy study utilizing medroxyprogesterone acetate administered as an intramuscular injection once every 90 days. Fertil Steril 1973; 24:331–339.
- Kaunitz AM. Injectable contraception. New and existing options. Obstet Gynecol Clin North Am 2000; 27:741–780.
- Mainwaring R, Hales HA, Stevenson K, et al. Metabolic parameter, bleeding, and weight changes in US women using progestin only contraceptives. Contraception 1995; 51:149–153.
- Curtis KM, Martins SL. Progestogen-only contraception and bone mineral density: a systematic review. Contraception 2006; 73:470–487.
- Shaarawy M, El-Mallah SY, Seoudi S, Hassan M, Mohsen IA. Effects of the long-term use of depot medroxyprogesterone acetate as hormonal contraceptive on bone mineral density and biochemical markers of bone remodeling. Contraception 2006; 74:297–302.
- Cromer BA, Bonny AE, Stager M, et al. Bone mineral density in adolescent females using injectable or oral contraceptives: a 24-month prospective study. Fertil Steril 2008; 90:2060–2067.
- Rome E, Ziegler J, Secic M, et al. Bone biochemical markers in adolescent girls using either depot medroxyprogesterone acetate or an oral contraceptive. J Pediatr Adolesc Gynecol 2004; 17:373–377.
- More C, Bettembuk P, Bhattoa HP, Balogh A. The effects of pregnancy and lactation on bone mineral density. Osteoporos Int 2001; 12:732–737.
- Kaunitz AM, Miller PD, Rice VM, Ross D, McClung MR. Bone mineral density in women aged 25-35 years receiving depot medroxyprogesterone acetate: recovery following discontinuation. Contraception 2006; 74:90–99.
- Guilbert ER, Brown JP, Kaunitz AM, et al. The use of depot-medroxyprogesterone acetate in contraception and its potential impact on skeletal health. Contraception 2009; 79:167–177.
- Bonny AE, Ziegler J, Harvey R, Debanne SM, Secic M, Cromer BA. Weight gain in obese and nonobese adolescent girls initiating depot medroxyprogesterone, oral contraceptive pills, or no hormonal contraceptive method. Arch Pediatr Adolesc Med 2006; 160:40–45.
- van den Heuvel MW, van Bragt AJ, Alnabawy AK, Kaptein MC. Comparison of ethinylestradiol pharmacokinetics in three hormonal contraceptive formulations: the vaginal ring, the transdermal patch and an oral contraceptive. Contraception 2005; 72:168–174.
- Douketis JD, Ginsberg JS, Holbrook A, Crowther M, Duku EK, Burrows RF. A reevaluation of the risk for venous thromboembolism with the use of oral contraceptives and hormone replacement therapy. Arch Intern Med 1997; 157:1522–1530.
- Cole JA, Norman H, Doherty M, Walker AM. Venous thromboembolism, myocardial infarction, and stroke among transdermal contraceptive system users. Obstet Gynecol 2007; 109:339–346.
- Jick S, Kaye JA, Li L, Jick H. Further results on the risk of nonfatal venous thromboembolism in users of the contraceptive transdermal patch compared to users of oral contraceptives containing norgestimate and 35 microg of ethinyl estradiol. Contraception 2007; 76:4–7.
- World Health Organization. Medical Eligibility Criteria for Contraceptive Use. 3rd ed. Geneva: Reproductive Health and Research, World Health Organization; 2004.
- Dieben TO, Roumen FJ, Apter D. Efficacy, cycle control, and user acceptability of a novel combined contraceptive vaginal ring. Obstet Gynecol 2002; 100:585–593.
- Davies GC, Feng LX, Newton JR, Dieben TO, Coelingh-Bennink HJ. The effects of a combined contraceptive vaginal ring releasing ethinyloestradiol and 3-ketodesogestrel on vaginal flora. Contraception 1992; 45:511–518.
- Roumen FJ, Boon ME, van Velzen D, Dieben TO, Coelingh Bennink HJ. The cervico-vaginal epithelium during 20 cycles’ use of a combined contraceptive vaginal ring. Hum Reprod 1996; 11:2443–2448.
- Wenzl R, van Beek A, Schnabel P, Huber J. Pharmacokinetics of etonogestrel released from the contraceptive implant Implanon. Contraception 1998; 58:283–288.
- Darney P, Patel A, Rosen K, Shapiro LS, Kaunitz AM. Safety and efficacy of a single-rod etonogestrel implant (Implanon): results from 11 international clinical trials. Fertil Steril 2009; 91:1646–1653.
- Jensen JT, Speroff L. Health benefits of oral contraceptives. Obstet Gynecol Clin North Am 2000; 27:705–721.
- Seracchioli R, Mabrouk M, Frascà C, et al. Long-term cyclic and continuous oral contraceptive therapy and endometrioma recurrence: a randomized controlled trial. Fertil Steril 2010; 93:52–56.
- Falsetti L, Dordoni D, Gastaldi C, Gastaldi A. A new association of ethinylestradiol (0.035 mg) cyproterone acetate (2 mg) in the therapy of polycystic ovary syndrome. Acta Eur Fertil 1986; 17:19–25.
- Koltun W, Lucky AW, Thiboutot D, et al. Efficacy and safety of 3 mg drospirenone/20 mcg ethinylestradiol oral contraceptive administered in 24/4 regimen in the treatment of acne vulgaris: a randomized, double-blind, placebo-controlled trial. Contraception 2008; 77:249–256.
- Kadir RA, Sabin CA, Pollard D, Lee CA, Economides DL. Quality of life during menstruation in patients with inherited bleeding disorders. Haemophilia 1998; 4:836–841.
- Schneider MB, Fisher M, Friedman SB, Bijur PE, Toffler AP. Menstrual and premenstrual issues in female military cadets: a unique population with significant concerns. J Pediatr Adolesc Gynecol 1999; 12:195–201.
- Bennell K, White S, Crossley K. The oral contraceptive pill: a revolution for sportswomen? Br J Sports Med 1999; 33:231–238.
- Kaplowitz PB, Oberfield SE. Reexamination of the age limit for defining when puberty is precocious in girls in the United States: implications for evaluation and treatment. Drug and Therapeutics and Executive Committees of the Lawson Wilkins Pediatric Endocrine Society. Pediatrics 1999; 104:936–941.
- Roxburgh DR, West MJ. The use of norethisterone to suppress menstruation in the intellectually severely retarded woman. Med J Aust 1973; 2:310–313.
- Egan TM, Siegert RJ, Fairley NA. Use of hormonal contraceptives in an institutional setting: reasons for use, consent and safety in women with psychiatric and intellectual disabilities. N Z Med J 1993; 106:338–341.
- Pillai M, O’Brien K, Hill E. The levonorgestrel intrauterine system (Mirena) for the treatment of menstrual problems in adolescents with medical disorders, or physical or learning disabilities. BJOG 2010; 117:216–221.
- Archer DF, Jensen JT, Johnson JV, Borisute H, Grubb GS, Constantine GD. Evaluation of a continuous regimen of levonorgestrel/ethinyl estradiol: phase 3 study results. Contraception 2006; 74:439–445.
- Kroll R, Reape KZ, Margolis M. The efficacy and safety of a low-dose, 91-day, extended-regimen oral contraceptive with continuous ethinyl estradiol. Contraception 2010; 81:41–48.
- Archer DF. Menstrual-cycle-related symptoms: a review of the rationale for continuous use of oral contraceptives. Contraception 2006; 74:359–366.
- den Tonkelaar I, Oddens BJ. Preferred frequency and characteristics of menstrual bleeding in relation to reproductive status, oral contraceptive use, and hormone replacement therapy use. Contraception 1999; 59:357–362.
- Edelman A, Lew R, Cwiak C, Nichols M, Jensen J. Acceptability of contraceptive-induced amenorrhea in a racially diverse group of US women. Contraception 2007; 75:450–453.
- Gerschultz KL, Sucato GS, Hennon TR, Murray PJ, Gold MA. Extended cycling of combined hormonal contraceptives in adolescents: physician views and prescribing practices. J Adolesc Health 2007; 40:151–157.
- Frankovich RJ, Lebrun CM. Menstrual cycle, contraception, and performance. Clin Sports Med 2000; 19:251–271.
- Speroff L, Darney PD. A Clinical Guide for Contraception. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Kaunitz AM. Long-acting contraceptive options. Int J Fertil Menopausal Stud 1996; 41:69–76.
- US Food and Drug Administration. Guidance for Industry Labeling for Combined Oral Contraceptives, 2004. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075075.pdfAccessed May 17, 2010.
- Association of Reproductive Health Professionals. Extended and continuous use of contraceptives to reduce menstruation. September 2004. http://www.arhp.org/publications-and-resources/clinical-proceedings/reduce-menses. Accessed May 17, 2010.
- Kjerulff KH, Erickson BA, Langenberg PW. Chronic gynecological conditions reported by US women: findings from the National Health Interview Survey, 1984 to 1992. Am J Public Health 1996; 86:195–199.
- Thomas SL, Ellertson C. Nuisance or natural and healthy: should monthly menstruation be optional for women? Lancet 2000; 355:922–924.
- Connell EB. Contraception in the prepill era. Contraception 1999; 59(suppl 1):7S–10S.
- Marks LV. Sexual chemistry: a history of the contraceptive pill. New Haven, CT: Yale University Press, 2001.
- Loudon NB, Foxwell M, Potts DM, Guild AL, Short RV. Acceptability of an oral contraceptive that reduces the frequency of menstruation: the tri-cycle pill regimen. Br Med J 1977; 2:487–490.
- Sulak PJ, Cressman BE, Waldrop E, Holleman S, Kuehl TJ. Extending the duration of active oral contraceptive pills to manage hormone withdrawal symptoms. Obstet Gynecol 1997; 89:179–183.
- Long-term reversible contraception. Twelve years of experience with the TCu380A and TCu220C. Contraception 1997; 56:341–352.
- Miller L, Notter KM. Menstrual reduction with extended use of combination oral contraceptive pills: randomized controlled trial. Obstet Gynecol 2001; 98:771–778.
- Mulders TM, Dieben TO. Use of the novel combined contraceptive vaginal ring NuvaRing for ovulation inhibition. Fertil Steril 2001; 75:865–870.
- Stanford JB, Mikolajczyk RT. Mechanisms of action of intrauterine devices: update and estimation of postfertilization effects. Am J Obstet Gynecol 2002; 187:1699–1708.
- Anderson FD, Hait H. A multicenter, randomized study of an extended cycle oral contraceptive. Contraception 2003; 68:89–96.
- Miller L, Hughes JP. Continuous combination oral contraceptive pills to eliminate withdrawal bleeding: a randomized trial. Obstet Gynecol 2003; 101:653–661.
- Sillem M, Schneidereit R, Heithecker R, Mueck AO. Use of an oral contraceptive containing drospirenone in an extended regimen. Eur J Contracept Reprod Health Care 2003; 8:162–169.
- Miller L, Verhoeven CH, Hout J. Extended regimens of the contraceptive vaginal ring: a randomized trial. Obstet Gynecol 2005; 106:473–482.
- Stewart FH, Kaunitz AM, Laguardia KD, Karvois DL, Fisher AC, Friedman AJ. Extended use of transdermal norelgestromin/ethinyl estradiol: a randomized trial. Obstet Gynecol 2005; 105:1389–1396.
- Sulak PJ, Kuehl TJ, Coffee A, Willis S. Prospective analysis of occurrence and management of breakthrough bleeding during an extended oral contraceptive regimen. Am J Obstet Gynecol 2006; 195:935–941.
- Lukes AS, Reardon B, Arepally G. Use of the levonorgestrel– releasing intrauterine system in women with hemostatic disorders. Fertil Steril 2008; 90:673–677.
- Anderson FD, Feldman R, Reape KZ. Endometrial effects of a 91-day extended-regimen oral contraceptive with low-dose estrogen in place of placebo. Contraception 2008; 77:91–96.
- Wright KP, Johnson JV. Evaluation of extended and continuous use oral contraceptives. Ther Clin Risk Manag 2008; 4:905–911.
- Edelman AB, Gallo MF, Jensen JT, Nichols MD, Schulz KF, Grimes DA. Continuous or extended cycle vs. cyclic use of combined oral contraceptives for contraception. Cochrane Database Syst Rev 2005; 3:CD004695.
- Sulak PJ, Kuehl TJ, Ortiz M, Shull BL. Acceptance of altering the standard 21-day/7-day oral contraceptive regimen to delay menses and reduce hormone withdrawal symptoms. Am J Obstet Gynecol 2002; 186:1142–1149.
- Turok D. The quest for better contraception: future methods. Obstet Gynecol Clin North Am 2007; 34:137–166.
- Bergqvist A, Rybo G. Treatment of menorrhagia with intrauterine release of progesterone. Br J Obstet Gynaecol 1983; 90:255–258.
- Andersson K, Odlind V, Rybo G. Levonorgestrel-releasing and copper-releasing (Nova T) IUDs during five years of use: a randomized comparative trial. Contraception 1994; 49:56–72.
- US Food and Drug Administration. FDA Approves Additional Use for IUD Mirena to Treat Heavy Menstrual Bleeding in IUD Users. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm184747.htm. Accessed May 17, 2010.
- Hidalgo M, Bahamondes L, Perrotti M, Diaz J, Dantas-Monteiro C, Petta C. Bleeding patterns and clinical performance of the levonorgestrel-releasing intrauterine system (Mirena) up to two years. Contraception 2002; 65:129–132.
- Schwallie PC, Assenzo JR. Contraceptive use—efficacy study utilizing medroxyprogesterone acetate administered as an intramuscular injection once every 90 days. Fertil Steril 1973; 24:331–339.
- Kaunitz AM. Injectable contraception. New and existing options. Obstet Gynecol Clin North Am 2000; 27:741–780.
- Mainwaring R, Hales HA, Stevenson K, et al. Metabolic parameter, bleeding, and weight changes in US women using progestin only contraceptives. Contraception 1995; 51:149–153.
- Curtis KM, Martins SL. Progestogen-only contraception and bone mineral density: a systematic review. Contraception 2006; 73:470–487.
- Shaarawy M, El-Mallah SY, Seoudi S, Hassan M, Mohsen IA. Effects of the long-term use of depot medroxyprogesterone acetate as hormonal contraceptive on bone mineral density and biochemical markers of bone remodeling. Contraception 2006; 74:297–302.
- Cromer BA, Bonny AE, Stager M, et al. Bone mineral density in adolescent females using injectable or oral contraceptives: a 24-month prospective study. Fertil Steril 2008; 90:2060–2067.
- Rome E, Ziegler J, Secic M, et al. Bone biochemical markers in adolescent girls using either depot medroxyprogesterone acetate or an oral contraceptive. J Pediatr Adolesc Gynecol 2004; 17:373–377.
- More C, Bettembuk P, Bhattoa HP, Balogh A. The effects of pregnancy and lactation on bone mineral density. Osteoporos Int 2001; 12:732–737.
- Kaunitz AM, Miller PD, Rice VM, Ross D, McClung MR. Bone mineral density in women aged 25-35 years receiving depot medroxyprogesterone acetate: recovery following discontinuation. Contraception 2006; 74:90–99.
- Guilbert ER, Brown JP, Kaunitz AM, et al. The use of depot-medroxyprogesterone acetate in contraception and its potential impact on skeletal health. Contraception 2009; 79:167–177.
- Bonny AE, Ziegler J, Harvey R, Debanne SM, Secic M, Cromer BA. Weight gain in obese and nonobese adolescent girls initiating depot medroxyprogesterone, oral contraceptive pills, or no hormonal contraceptive method. Arch Pediatr Adolesc Med 2006; 160:40–45.
- van den Heuvel MW, van Bragt AJ, Alnabawy AK, Kaptein MC. Comparison of ethinylestradiol pharmacokinetics in three hormonal contraceptive formulations: the vaginal ring, the transdermal patch and an oral contraceptive. Contraception 2005; 72:168–174.
- Douketis JD, Ginsberg JS, Holbrook A, Crowther M, Duku EK, Burrows RF. A reevaluation of the risk for venous thromboembolism with the use of oral contraceptives and hormone replacement therapy. Arch Intern Med 1997; 157:1522–1530.
- Cole JA, Norman H, Doherty M, Walker AM. Venous thromboembolism, myocardial infarction, and stroke among transdermal contraceptive system users. Obstet Gynecol 2007; 109:339–346.
- Jick S, Kaye JA, Li L, Jick H. Further results on the risk of nonfatal venous thromboembolism in users of the contraceptive transdermal patch compared to users of oral contraceptives containing norgestimate and 35 microg of ethinyl estradiol. Contraception 2007; 76:4–7.
- World Health Organization. Medical Eligibility Criteria for Contraceptive Use. 3rd ed. Geneva: Reproductive Health and Research, World Health Organization; 2004.
- Dieben TO, Roumen FJ, Apter D. Efficacy, cycle control, and user acceptability of a novel combined contraceptive vaginal ring. Obstet Gynecol 2002; 100:585–593.
- Davies GC, Feng LX, Newton JR, Dieben TO, Coelingh-Bennink HJ. The effects of a combined contraceptive vaginal ring releasing ethinyloestradiol and 3-ketodesogestrel on vaginal flora. Contraception 1992; 45:511–518.
- Roumen FJ, Boon ME, van Velzen D, Dieben TO, Coelingh Bennink HJ. The cervico-vaginal epithelium during 20 cycles’ use of a combined contraceptive vaginal ring. Hum Reprod 1996; 11:2443–2448.
- Wenzl R, van Beek A, Schnabel P, Huber J. Pharmacokinetics of etonogestrel released from the contraceptive implant Implanon. Contraception 1998; 58:283–288.
- Darney P, Patel A, Rosen K, Shapiro LS, Kaunitz AM. Safety and efficacy of a single-rod etonogestrel implant (Implanon): results from 11 international clinical trials. Fertil Steril 2009; 91:1646–1653.
- Jensen JT, Speroff L. Health benefits of oral contraceptives. Obstet Gynecol Clin North Am 2000; 27:705–721.
- Seracchioli R, Mabrouk M, Frascà C, et al. Long-term cyclic and continuous oral contraceptive therapy and endometrioma recurrence: a randomized controlled trial. Fertil Steril 2010; 93:52–56.
- Falsetti L, Dordoni D, Gastaldi C, Gastaldi A. A new association of ethinylestradiol (0.035 mg) cyproterone acetate (2 mg) in the therapy of polycystic ovary syndrome. Acta Eur Fertil 1986; 17:19–25.
- Koltun W, Lucky AW, Thiboutot D, et al. Efficacy and safety of 3 mg drospirenone/20 mcg ethinylestradiol oral contraceptive administered in 24/4 regimen in the treatment of acne vulgaris: a randomized, double-blind, placebo-controlled trial. Contraception 2008; 77:249–256.
- Kadir RA, Sabin CA, Pollard D, Lee CA, Economides DL. Quality of life during menstruation in patients with inherited bleeding disorders. Haemophilia 1998; 4:836–841.
- Schneider MB, Fisher M, Friedman SB, Bijur PE, Toffler AP. Menstrual and premenstrual issues in female military cadets: a unique population with significant concerns. J Pediatr Adolesc Gynecol 1999; 12:195–201.
- Bennell K, White S, Crossley K. The oral contraceptive pill: a revolution for sportswomen? Br J Sports Med 1999; 33:231–238.
- Kaplowitz PB, Oberfield SE. Reexamination of the age limit for defining when puberty is precocious in girls in the United States: implications for evaluation and treatment. Drug and Therapeutics and Executive Committees of the Lawson Wilkins Pediatric Endocrine Society. Pediatrics 1999; 104:936–941.
- Roxburgh DR, West MJ. The use of norethisterone to suppress menstruation in the intellectually severely retarded woman. Med J Aust 1973; 2:310–313.
- Egan TM, Siegert RJ, Fairley NA. Use of hormonal contraceptives in an institutional setting: reasons for use, consent and safety in women with psychiatric and intellectual disabilities. N Z Med J 1993; 106:338–341.
- Pillai M, O’Brien K, Hill E. The levonorgestrel intrauterine system (Mirena) for the treatment of menstrual problems in adolescents with medical disorders, or physical or learning disabilities. BJOG 2010; 117:216–221.
- Archer DF, Jensen JT, Johnson JV, Borisute H, Grubb GS, Constantine GD. Evaluation of a continuous regimen of levonorgestrel/ethinyl estradiol: phase 3 study results. Contraception 2006; 74:439–445.
- Kroll R, Reape KZ, Margolis M. The efficacy and safety of a low-dose, 91-day, extended-regimen oral contraceptive with continuous ethinyl estradiol. Contraception 2010; 81:41–48.
- Archer DF. Menstrual-cycle-related symptoms: a review of the rationale for continuous use of oral contraceptives. Contraception 2006; 74:359–366.
- den Tonkelaar I, Oddens BJ. Preferred frequency and characteristics of menstrual bleeding in relation to reproductive status, oral contraceptive use, and hormone replacement therapy use. Contraception 1999; 59:357–362.
- Edelman A, Lew R, Cwiak C, Nichols M, Jensen J. Acceptability of contraceptive-induced amenorrhea in a racially diverse group of US women. Contraception 2007; 75:450–453.
- Gerschultz KL, Sucato GS, Hennon TR, Murray PJ, Gold MA. Extended cycling of combined hormonal contraceptives in adolescents: physician views and prescribing practices. J Adolesc Health 2007; 40:151–157.
- Frankovich RJ, Lebrun CM. Menstrual cycle, contraception, and performance. Clin Sports Med 2000; 19:251–271.
- Speroff L, Darney PD. A Clinical Guide for Contraception. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Kaunitz AM. Long-acting contraceptive options. Int J Fertil Menopausal Stud 1996; 41:69–76.
- US Food and Drug Administration. Guidance for Industry Labeling for Combined Oral Contraceptives, 2004. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm075075.pdfAccessed May 17, 2010.
KEY POINTS
- The options for menstrual manipulation are extended or continuous regimens of oral, transdermal, or vaginal hormonal contraceptives; a levonorgestrel-releasing intrauterine device; a progestin implant; and depot medroxyprogesterone injections.
- Benefits include fewer menstrual-related syndromes, less absenteeism from work or school, and greater overall satisfaction. Medical indications for it are conditions exacerbated by hormonal changes around the time of menses.
- The main disadvantage is a higher rate of breakthrough bleeding.
- Myths and misperceptions about menstrual manipulation persist; some physicians believe it is somehow inadvisable.
Coenzyme Q10: A therapy for hypertension and statin-induced myalgia?
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
Coenzyme Q10 supplements have been purported to be effective for treating a variety of disorders,1,2 in particular hypertension and statin-induced myalgia.
Several studies3–7 found that coenzyme Q10 supplementation significantly lowered blood pressure in hypertensive patients. Moreover, some trials have demonstrated that statin therapy reduces serum or muscle levels of coenzyme Q10,8–14 prompting investigations to determine whether coenzyme Q10 deficiency is related to statin-induced muscle pain.15–17
In this review, we discuss the efficacy and safety of coenzyme Q10 supplementation in patients with hypertension and those taking statins, and some of the caveats about using supplements that are not approved by the US Food and Drug Administration (FDA), as well as the bioavailability and quality of available formulations.
WHAT IS COENZYME Q10?
Coenzyme Q10, also known as coenzyme Q, ubidecarenone, and ubiquinone, is found in all human cells, with the highest concentrations in the heart, liver, kidney, and pancreas.1,2 It is a potent antioxidant, a membrane stabilizer, and an integral cofactor in the mitochondrial respiratory chain, helping to generate adenosine triphosphate, the major cellular energy source.1,2,18 It may also regulate genes associated with cell metabolism.19
RATIONALE FOR SUPPLEMENTATION
Coenzyme Q10 supplementation has been used, recommended, or studied in heart failure, hypertension, parkinsonism, mitochondrial encephalomyopathies, and other ailments.
In hypertension
Depending on the class, various antihypertensive drugs can have adverse effects such as depression, cough, and cardiac and renal dysfunction. 20,21 Furthermore, many patients need to take more than one drug to control their blood pressure, increasing their risk of side effects. Some researchers believe coenzyme Q10 supplementation may reduce the need to take multiple antihypertensive drugs.5
Coenzyme Q10 appears to lower blood pressure. The exact mechanism is not known, but one theory is that it reduces peripheral resistance by preserving nitric oxide.21 Nitric oxide relaxes peripheral arteries, lowering blood pressure. In some forms of hypertension, superoxide radicals that inactivate nitric oxide are overproduced; coenzyme Q10, with its antioxidant effects, may prevent the inactivation of nitric oxide by these free radicals. Alternatively, coenzyme Q10 may boost the production of the prostaglandin prostacyclin (PGI2) a potent vasodilator and inhibitor of platelet aggregation, or it may enhance the sensitivity of arterial smooth muscle to PGI2, or both.1,22
In patients taking statins
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), first-line agents for lowering cholesterol levels to prevent cardiovascular disease, are some of the most commonly prescribed medications.23,24 However, statin therapy carries a risk of myopathy, which can range from muscle aches to rhabdomyolysis. 23,24
In a clinical advisory,25 the American College of Cardiology, the American Heart Association, and the National Heart, Lung, and Blood Institute recommend that patients on statin therapy who experience muscle soreness, tenderness, or pain with serum creatine kinase levels 3 to 10 times the upper limit of normal should have their creatine kinase level checked weekly. If the level is 3 to 10 times the upper limit of normal, statin therapy may be continued, but if it exceeds 10 times the upper limit, then statins and other potential offending agents (eg, niacin, fibrate) need to be discontinued.
Statins inhibit the synthesis of cholesterol by reducing the production of mevalonate, a precursor of both cholesterol and coenzyme Q10. Since both cholesterol and coenzyme Q10 are produced by the same pathway, it is not surprising that statins have been reported to reduce serum and muscle coenzyme Q10 levels.9–14 However, one study did not report a significant reduction of coenzyme Q10 levels in muscle tissue in patients treated with simvastatin 20 mg for 6 months.26
Nonetheless, researchers have hypothesized that a reduction in coenzyme Q10 levels in muscle tissue causes mitochondrial dysfunction, which could increase the risk of statininduced myopathy,13–17 and some believe that treatment with coenzyme Q10 may reduce myalgic symptoms and allow patients to remain on statin therapy.13,24
Researchers have investigated the potential of coenzyme Q10 supplementation to reduce or prevent statin-induced myopathy.15–17 (More on this below.)
Interestingly, a randomized, placebo-controlled trial27 found that 6 months of daily therapy with simvastatin (Zocor) 20 mg or pravastatin (Pravachol) 40 mg lowered systolic and diastolic blood pressure significantly in patients with no documented history of cardiovascular disease or diabetes. A possible mechanism of statin-induced blood pressure reduction is the up-regulation of endothelial nitric oxide synthetase, a potent vasodilator. Coenzyme Q10 levels were not assessed during this study. Whether coenzyme Q10 supplementation used to treat statin-induced myalgia enhances or inhibits the antihypertensive effects of statins is not yet known.
EVIDENCE OF EFFECTIVENESS IN HYPERTENSION
Rosenfeldt et al28 performed a meta-analysis and found that some trials documented statistically significant reductions in diastolic or systolic blood pressure or both, while others reported negligible effects.3,29 In one small trial,30 blood pressures actually went up in patients taking coenzyme Q10. Coenzyme Q10 dosages and length of therapy varied from study to study in the meta-analysis. Only minor adverse effects such as gastrointestinal upset and headache were reported.
Yamagami et al3 randomly assigned 20 patients with hypertension and a low coenzyme Q10 level to receive 100 mg of coenzyme Q10 or placebo daily for 12 weeks. Patients continued their usual antihypertensive regimen during the study period. Blood pressures, coenzyme Q10 levels, and antihypertensive drugs used were comparable between the study groups.
After 12 weeks of therapy, the mean coenzyme Q10 level in the active-treatment group had more than doubled, from 0.704 to 1.597 μg/mL. This group also experienced a statistically significant drop in systolic blood pressure, from 167 mm Hg at baseline to 148 mm Hg at 12 weeks. In the placebo group, the systolic blood pressure was 168 mm Hg at baseline and 164 mm Hg at 12 weeks; the change was not statistically significant. Diastolic pressure was not significantly lower at 12 weeks than at baseline in either group.
The authors concluded that coenzyme Q10 supplementation brought a mild reduction in high blood pressure in patients who had low coenzyme Q10 serum levels.
Digiesi et al31 randomized 18 patients with essential hypertension to receive either coenzyme Q10 100 mg or placebo daily for 10 weeks. All antihypertensive therapy was discontinued at baseline. After the first 10 weeks, patients went through a 2-week washout period and then were switched to the opposite therapy for an additional 10 weeks. Mean baseline blood pressure values were 167 mm Hg systolic and 103 mm Hg diastolic.
Those taking the supplement had a statistically significant decrease in systolic and diastolic pressures (P < .001). The antihypertensive effect was noted in the 3rd or 4th week of active treatment and persisted for the duration of therapy. The effects dissipated 7 to 10 days after coenzyme Q10 was stopped.
Langsjoen et al5 evaluated the effects of adding coenzyme Q10 to the antihypertensive drug regimen of 109 patients who had a primary diagnosis of essential hypertension in a prospective observational study. Patients with hypertension as a secondary diagnosis and other cardiovascular diseases were excluded. Variable doses of coenzyme Q10 were given, adjusted according to clinical response and to achieve serum levels greater than 2.0 μg/mL. The average dose was 225 mg/day; the mean serum level attained was 3.02 μg/mL.
Over several months, patients taking the supplement had a reduction in mean systolic pressure from 159 mm Hg at baseline to 147 mm Hg (P < .001), and a reduction in mean diastolic pressure from 94 to 85 mm Hg (P < .001). Thirty-seven percent of patients were able to discontinue one antihypertensive drug, 11% discontinued two drugs, and 4% were able to stop taking three drugs. However, 46% remained on the same antihypertensive regimen, and 3% needed an additional drug.
Singh et al6 randomized 64 patients who had coronary artery disease and who had been on antihypertensive drugs for more than 1 year to receive either B-complex vitamins or coenzyme Q10 (hydrosoluble Q-Gel) 60 mg orally once daily for 8 weeks. Five patients were not available for follow-up; therefore, only 59 patients were evaluated. Fifty-five (93%) of the 59 patients were taking only one antihypertensive drug. Initial antihypertensive drug use was similar between study groups and was continued throughout the trial.
After 8 weeks of therapy, the coenzyme Q10 group had significantly lower systolic and diastolic blood pressure than the placebo group (P < .05 for both). There was also a statistically significant decrease in the dosage of antihypertensive drugs in the coenzyme Q10 group but not in the placebo group (P < .05), reflecting coenzyme Q10’s additive antihypertensive effect.
Burke et al7 randomized 41 men and 35 women with isolated systolic hypertension (systolic pressure 150–170 mm Hg, diastolic pressure < 90 mm Hg) to receive a twice-daily dose of 60 mg of emulsified coenzyme Q10 (hydrosoluble Q-Gel) with 150 IU of vitamin E or placebo containing vitamin E alone for 12 weeks. The study also included 5 men and 4 women with normal blood pressure, all of whom received coenzyme Q10. A total of 80 patients completed treatment. The primary goal of the study was to determine the efficacy of coenzyme Q10 in the treatment of isolated systolic hypertension in patients without comorbid conditions. Blood pressures were monitored twice a week during the trial, by the same nurse.
After 12 weeks of treatment, the mean reduction in systolic pressure in hypertensive patients on coenzyme Q10 was 17.8 ± 7.3 mm Hg. There were no significant changes in diastolic pressure in any study group with treatment. Patients with isolated systolic hypertension who were taking coenzyme Q10 had a statistically significant reduction in systolic pressure compared with baseline and placebo (P < .01 for both). Approximately 55% of patients on coenzyme Q10 achieved a reduction in systolic pressure of 4 mm Hg or greater, while 45% did not respond to therapy. The mean plasma coenzyme Q10 level of the treatment group increased from 0.47 ± 0.19 μg/mL to 2.69 ± 0.54 μg/mL after 12 weeks; however, the study did not have the statistical power to demonstrate a relationship between coenzyme Q10 levels and changes in blood pressure. Twenty-seven (34%) of the 80 patients were taking a statin while on coenzyme Q10 therapy.
STUDIES IN STATIN-INDUCED MYOPATHY
Thibault et al32 and Kim et al33 reported that patients taking lovastatin (Mevacor) at dosages as high as 35 mg/kg/day to inhibit tumor growth achieved symptomatic relief of statin-induced musculoskeletal toxicity after coenzyme Q10 supplementation.
Caso et al15 performed a small pilot study in 32 patients to determine if coenzyme Q10 supplementation would improve myalgic symptoms in patients treated with statins. In this double-blind, randomized trial, patients received either coenzyme Q10 100 mg/day or vitamin E 400 IU/day for 30 days. The extent of muscle pain and its interference with daily activities were determined before and after therapy using the Brief Pain Inventory Questionnaire. The statins were atorvastatin (Lipitor) 10 mg or 20 mg, lovastatin 40 mg, pravastatin 40 mg, and simvastatin 10, 20, 40, and 80 mg. Five patients in the coenzyme Q10 group and four patients in the vitamin E group were taking nonsteroidal anti-inflammatory drugs before and during the trial. The intensity of muscle pain and its interference with daily activities were similar between study groups before the start of therapy.
After 30 days of treatment with coenzyme Q10, the pain intensity had decreased significantly from baseline (P < .001). In contrast, no change in pain intensity from baseline was noted in patients receiving vitamin E. The Pain Severity Score was significantly different between study groups, favoring the coenzyme Q10 group (P < .001). Sixteen of 18 patients on coenzyme Q10 reported a reduction in pain, while only 3 of 14 patients on vitamin E reported a similar response. Also, the interference of pain with daily activities significantly improved with coenzyme Q10 (P < .02), whereas vitamin E did not have a significant impact on this.
Young et al17 randomized 44 patients with prior statin-induced myalgia to receive increasing doses of simvastatin (10–40 mg/day) in combination with either coenzyme Q10 (Q-Gel) 200 mg/day or placebo. The primary goal was to determine if coenzyme Q10 supplementation would help improve statin tolerance in patients with a history of statininduced myalgia. Plasma coenzyme Q10 and lipid levels were measured at baseline and at the end of the study. The intensity of myalgia was assessed with a visual analogue scale.
At 12 weeks, the coenzyme Q10 plasma level was significantly higher in the treatment group than in the placebo group (P < .001). However, no differences were noted between groups in the number of patients who tolerated the 40-mg/day simvastatin dose (P = .34) or in the number of patients who remained on any simvastatin dose (P = .47). Additionally, myalgia scores did not differ between groups (P = .63). The authors acknowledged that there were only small increases in the myalgia pain scores reported in either group. Therefore, patients in the treatment group may not have experienced sufficiently severe muscle pain to have benefited from coenzyme Q10 supplementation.
IS COENZYME Q10 SAFE?
Studies have indicated that these supplements are well tolerated, with relatively few adverse effects or potential drug interactions.1,2,34
The FDA does not routinely assess the purity or quality of over-the-counter coenzyme Q10 products.35 However, the United States Pharmacopeia (USP) does test dietary supplements to make sure that they are not mislabeled and that they do not contain contaminants. 36
A USP-verified dietary supplement should:
- Contain the exact ingredients listed on the label in the listed potency and amounts
- Not include harmful levels of certain contaminants such as lead, mercury, pesticides, or bacteria
- Appropriately disintegrate and release its contents into the body within a specified period of time
- Be produced using the FDA’s current Good Manufacturing Practices.36
Side effects, contraindications, warnings
Coenzyme Q10 is a relatively safe dietary supplement. It is contraindicated in patients who are allergic to it or to any of its components.2 Most clinical trials have not reported significant adverse effects that necessitated stopping therapy.34 However, gastrointestinal effects such as abdominal discomfort, nausea, vomiting, diarrhea, and anorexia have occurred.1,2,34 Allergic rash and headache have also been reported.1,2,34 In addition, coenzyme Q10’s antiplatelet effect may increase the risk of bleeding. 37,38 It undergoes biotransformation in the liver and is eliminated primarily via the biliary tract,39 so it can accumulate in patients with hepatic impairment or biliary obstruction.
Interactions with drugs
Coenzyme Q10’s effects on platelet function may increase the risk of bleeding in patients taking antiplatelet drugs such as aspirin or clopidogrel (Plavix).37,38 On the other hand, since it acts like vitamin K, it may counteract the anticoagulant effects of warfarin (Coumadin). 1,2,40
Coenzyme Q10 may have an additive antihypertensive effect when given with antihypertensive drugs.41
Coenzyme Q10 may improve beta-cell function and enhance insulin sensitivity, which may reduce insulin requirements for diabetic patients.42,43
SLOWLY ABSORBED
Coenzyme Q10 is absorbed slowly from the gastrointestinal tract, possibly because it has a high molecular weight and is not very watersoluble. 39
One pharmacokinetic study found that after a single 100-mg oral dose of coenzyme Q10, the mean peak plasma levels of about 1 μg/mL occurred between 5 and 10 hours (mean 6.5 hours).44 Coenzyme Q10 100 mg given orally three times daily produced a mean steadystate plasma level of 5.4 μg/mL; about 90% of this steady-state concentration was achieved after 4 days.39
Some formulations have significantly better oral bioavailability and therefore produce higher plasma levels. Soft-gel capsules, especially those with vegetable oil or vitamin E, may have better absorption.43
A pharmacokinetic study showed that the area under the curve of the plasma coenzyme Q10 concentration was more than twice as high with Q-Gel soft-gel capsules, a completely solubilized formulation, than with softgel capsules with an oil suspension, powderfilled hard-shell capsules, or regular tablets.45 Another study reported that colloidal-Q10, a formulation contained in VESIsorb (a novel drug delivery system sold as CoQsource) had greater bioavailability than solubilized and oil-based preparations.46 Commercially available solubilized preparations containing ubiquinol, a metabolized form of coenzyme Q10, have been shown to produce higher serum levels than solubilized products.47
Of note: unless the manufacturer claims that its product is water-soluble, the USP does not evaluate its dissolution rate.48 Therefore, USP-verified coenzyme Q10 products that are not water-soluble may have lower bioavailability than their solubilized counterparts.
Dry dosage forms of coenzyme Q10 (eg, tablets, capsules) may be more readily absorbed if taken with a fatty meal.43
SLOWLY ELIMINATED
Taken orally, coenzyme Q10 has a low clearance rate, with an elimination half-life of about 34 hours.39
After absorption, exogenous coenzyme Q10 is taken up by chylomicrons that transport it to the liver, where it is incorporated into verylow-density lipoproteins. It is then distributed to various organs, including the adrenal glands, spleen, kidneys, lungs, and heart. Coenzyme Q10 is eliminated primarily via the biliary tract. About 60% of an oral dose is eliminated in the feces during chronic oral administration.39
TWICE-DAILY DOSING
A typical daily dose of coenzyme Q10 for treating hypertension is 120 to 200 mg, usually given orally in two divided doses.1 For statininduced myopathy, 100 to 200 mg orally daily has been used.1
Coenzyme Q10 is given in divided doses to enhance its absorption and to minimize gastrointestinal effects.1,43 Taking it with a fatty meal may also increase its absorption.43
Since solubilized forms of coenzyme Q10 and ubiquinol have significantly greater bioavailability than nonsolubilized forms, the therapeutic dose of these formulations may be lower.47
MONITORING DURING TREATMENT
Without supplementation, the mean serum level of endogenous coenzyme Q10 has been reported to be 0.99 ± 0.30 mg/L (range 0.55– 1.87).18 Serum levels above 2 μg/mL have been associated with significant reductions in blood pressure.5,7,28
The possible effects of coenzyme Q10 on blood pressure, blood glucose levels, serum creatine kinase levels, and myopathic symptoms should be kept in mind when monitoring patients who have hypertension,41 diabetes,41,42 or statin-induced myalgia.15,17 Coenzyme Q10’s possible potentiating effects on antiplatelet drugs and its inhibitory effect on warfarin should be kept in mind as well.
COST VARIES
Coenzyme Q10 is available in different dosage forms (eg, regular and rapid-release softgel capsules, regular and chewable tablets, chewable wafers, and liquid) from a variety of manufacturers. Products come in different strengths, typically ranging from 30 to 400 mg. USP-verified formulations are listed at www.usp.org/USPVerified/dietarySupplements/under “Verified Supplements.” Only USP-verified products that claim to be water-soluble meet USP dissolution requirements.
The cost varies, depending on the vendor. In general, dosage forms with greater bioavailability, such as Q-Gel and ubiquinol supplements, are more expensive. For example, a regimen of 60 mg twice daily of regular-release coenzyme Q capsules may cost approximately $20 per month, compared with $60 per month for the same supply of Q-Gel Ultra capsules. However, in some cases, supplements that produce higher serum levels may be more cost-effective.
CURRENT ROLE IN THERAPY
As an antihypertensive adjunct
Several small clinical trials have shown that coenzyme Q10 supplementation can lower blood pressure. The supplements were reported to be safe and well tolerated. Moreover, some patients with essential hypertension who were taking coenzyme Q10 were able to discontinue one or more antihypertensive drugs. A significant reduction in blood pressure with use of coenzyme Q10 would be expected to reduce the adverse consequences of hypertension in the same manner as conventional antihypertensive agents.
However, no large, double-blind, randomized study has evaluated the impact of coenzyme Q10 when taken with other antihypertensive drugs (eg, angiotensin-converting enzyme inhibitors, beta-blockers, diuretics) on specific clinical end points such as the incidence of stroke or death from a major cardiac event. Furthermore, its effects on cardiac function, exercise tolerance, and quality of life have not been determined.
The bottom line. In some cases, it seems reasonable to recommend this product as an adjunct to conventional antihypertensive therapy. Larger, well-designed clinical trials of coenzyme Q10’s antihypertensive effects on specific clinical end points such as the risk of stroke or myocardial infarction are needed to define its true therapeutic value.
As a treatment for statin-induced myalgia
Clinical evidence supporting coenzyme Q10’s use in the treatment of statin-induced myopathy is limited. Whether coenzyme Q10 is depleted from muscle tissue during statin therapy has not been confirmed. Supplementation helped reduce the severity of musculoskeletal effects of megadoses of lovastatin. However, clinical trials of coenzyme Q10 in the treatment of myalgia associated with antilipidemic statin doses did not consistently report significant improvement. Nevertheless, coenzyme Q10 has been shown to be relatively safe, with few adverse effects.
The bottom line. In some cases, coenzyme Q could be considered as a possible treatment for statin-induced myalgia, pending large-scale studies to determine if it is truly effective for this purpose.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
- Jelin JM, Gregory PJ, et al. Natural medicines comprehensive database/compiled by the editors of Pharmacist’s Letter, Prescriber’s Letter. 11th ed. Stockton, CA: Therapeutic Research Faculty; 2009:452–457.
- Fetrow CW, Avila JR. Professional’s Handbook of Complementary & Alternative Medicines. 2nd ed. Springhouse, PA: Springhouse; 2001:211–215.
- Yamagami T, Takagi M, Akagami H, et al. Effect of coenzyme Q10 on essential hypertension, a double-blind controlled study. In:Folkers K, Yamamura Y, editors. Biomedical and Clinical Aspects of Coenzyme Q10: Proceedings of the Fifth International Symposium on the Biomedical and Clinical Aspects of Coenzyme Q10, vol 5. Amsterdam: Elsevier Science Publishers; 1986:337–343.
- Digiesi V, Cantini F, Oradei A, et al. Coenzyme Q10 in essential hypertension. Mol Aspects Med 1994; 15(suppl):S257–S263.
- Langsjoen P, Langsjoen P, Willis R, Folkers K. Treatment of essential hypertension with coenzyme Q10. Mol Aspects Med 1994; 15(suppl):S265–S272.
- Singh RB, Niaz MA, Rastogi SS, Shukla PK, Thakur AS. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens 1999; 13:203–208.
- Burke BE, Neuenschwander R, Olson RD. Randomized, double-blind, placebo-controlled trial of coenzyme Q10 in isolated systolic hypertension. South Med J 2001; 94:1112–1117.
- De Pinieux G, Chariot P, Ammi-Saïd M, et al. Lipidlowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol 1996; 42:333–337.
- Mortensen SA, Leth A, Agner E, Rohde M. Dose-related decrease of serum coenzyme Q10 during treatment with HMG-CoA reductase inhibitors. Mol Aspects Med 1997; 18(suppl):S137–S144.
- Ghirlanda G, Oradei A, Manto A, et al. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J Clin Pharmacol 1993; 33:226–229.
- Folkers K, Langsjoen P, Willis R, et al. Lovastatin decreases coenzyme Q10 levels in humans. Proc Natl Acad Sci U S A 1990; 87:8931–8934.
- Watts GF, Castelluccio C, Rice-Evans C, Taub NA, Baum H, Quinn PJ. Plasma coenzyme Q10 (ubiquinone) concentrations in patients treated with simvastatin. J Clin Pathol 1993; 46:1055–1057.
- Lamperti C, Naini AB, Lucchini V, et al. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol 2005; 62:1709–1712.
- Päivä H, Thelen KM, Van Coster R, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther 2005; 78:60–68.
- Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am J Cardiol 2007; 99:1409–1412.
- Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol 2007; 49:2231–2237.
- Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol 2007; 100:1400–1403.
- Berthold HK, Naini A, Di Mauro S, et al. Effect of ezetimibe and/or simvastatin on coenzyme Q10 levels in plasma: a randomised trial. Drug Saf 2006; 29:703–712.
- Groneberg DA, Kindermann B, Althammer M, et al. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int J Biochem Cell Biol 2005; 37:1208–1218.
- Hadj A, Pepe S, Rosenfeldt F. The clinical application of metabolic therapy for cardiovascular disease. Heart Lung Circ 2007; 16(suppl 3):S56–S64.
- Pepe S, Marasco SF, Haas SJ, Sheeran FL, Krum H, Rosenfeldt FL. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007; 7(suppl 1):S154–S167.
- Lönnrot K, Pörsti I, Alho H, Wu X, Hervonen A, Tolvanen JP. Control of arterial tone after long-term coenzyme Q10 supplementation in senescent rats. Br J Pharmacol 1998; 124:1500–1506.
- Sewright KA, Clarkson PM, Thompson PD. Statin myopathy: incidence, risk factors, and pathophysiology. Curr Atheroscler Rep 2007; 9:389–396.
- Radcliffe KA, Campbell WW. Statin myopathy. Curr Neurol Neurosci Rep 2008; 8:66–72.
- Pasternak RC, Smith SC, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. Circulation 2002; 106:1024–1028.
- Laaksonen R, Jokelainen K, Laakso J, et al. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am J Cardiol 1996; 77:851–854.
- Golomb BA, Dimsdale JE, White HL, Ritchie JB, Criqui MH. Reduction in blood pressure with statins: results from the UCSD Statin Study, a randomized trial. Arch Intern Med 2008; 168:721–727.
- Rosenfeldt FL, Haas SJ, Krum H, et al. Coenzyme Q10 in the treatment of hypertension: a meta-analysis of the clinical trials. J Hum Hypertens 2007; 21:297–306.
- Yamagami T, Shibata N, Folkers K. Bioenergetics in clinical medicine. Studies on coenzyme Q10 and essential hypertension. Res Commun Chem Pathol Pharmacol 1975; 11:273–288.
- Yamagami T, Shibata N, Folkers K. Study of coenzyme Q10. In:Folkers K, Yamamura Y, editors. Biomedical and clinical aspects of coenzyme Q10: proceedings of the International Symposium on Coenzyme Q10, held at Lake Yamanaka, Japan, September 16/17, 1976, a Naito Foundation symposium. Amsterdam: Elsevier Scientific Publishing Company; 1977:231–242.
- Digiesi V, Cantini F, Brodbeck B. Effect of coenzyme Q10 on essential arterial hypertension. Curr Ther Res; 1990; 47:841–845.
- Thibault A, Samid D, Tompkins AC, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res 1996; 2:483–491.
- Kim WS, Kim MM, Choi HJ, et al. Phase II study of high-dose lova-statin in patients with advanced gastric adenocarcinoma. Invest New Drugs 2001; 19:81–83.
- Hidaka T, Fujii K, Funahashi I, Fukutomi N, Hosoe K. Safety assessment of coenzyme Q10 (CoQ10). Biofactors 2008; 32:199–208.
- US Food and Drug Administration. Consumer Information on Dietary Supplements. Overview of Dietary Supplements. http://www.fda.gov/Food/DietarySupplements/ConsumerInformation/. Accessed May 25, 2010.
- US Pharmacopeia. The USP Dietary Supplement Verification Program http://www.usp.org/USPVerified/dietary-Supplements/. Accessed May 25, 2010.
- Serebruany VL, Ordonez JV, Herzog WR, et al. Dietary coenzyme Q10 supplementation alters platelet size and inhibits human vitronectin (CD51/CD61) receptor expression. J Cardiovasc Pharmacol 1997; 29:16–22.
- A close look at coenzyme Q10 and policosanol. Do these supplements live up to their claims for improving heart health? Harv Heart Lett 2002; 13:6.
- Greenberg S, Frishman WH. Co-enzyme Q10: a new drug for cardiovascular disease. J Clin Pharmacol 1990; 30:596–608.
- Singh U, Devaraj S, Jialal I. Coenzyme Q10 supplementation and heart failure. Nutr Rev 2007; 65:286–293.
- Bonakdar RA, Guarneri E. Coenzyme Q10. Am Fam Physician 2005; 72:1065–1070.
- Hodgson JM, Watts GF, Playford DA, Burke V, Croft KD. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr 2002; 56:1137–1142.
- Pepping J. Coenzyme Q10. Am J Health Syst Pharm 1999; 56:519–521.
- Tomono Y, Hasegawa J, Seki T, Motegi K, Morishita N. Pharmacokinetic study of deuterium-labelled coenzyme Q10 in man. Int J Clin Pharmacol Ther Toxicol 1986; 24:536–541.
- Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998; 68:109–113.
- Liu ZX, Artmann C. Relative bioavailability comparison of different coenzyme Q10 formulations with a novel delivery system. Altern Ther Health Med 2009; 15:42–46.
- Bhagavan HN, Chopra RK. Plasma coenzyme Q10 response to oral ingestion of coenzyme Q10 formulations. Mitochondrion 2007; 7(suppl 1):S78–S88.
- The United States Pharmacopeia. Ubidecarenone Capsules Monograph. 32nd Revision. Baltimore: United Book Press, 2009:1080.
KEY POINTS
- In some clinical trials, coenzyme Q10 supplements significantly lowered diastolic and systolic blood pressure.
- Statins may lower coenzyme Q10 serum levels, and some investigators have evaluated the relationship between coenzyme Q10 deficiency and statin-related myalgia, but more evidence is needed to support the use of coenzyme Q10 supplements to prevent or treat myalgia.
- Coenzyme Q10 supplementation appears to be relatively safe. Most clinical trials have not reported significant side effects that necessitated stopping therapy. Gastrointestinal effects include abdominal discomfort, nausea, vomiting, diarrhea, and anorexia. Allergic rash and headache have also been reported.
Update on the management of hirsutism
Hirsutism causes significant anxiety and lack of self-esteem in women. Although it is itself a benign condition, it is often the sign of an underlying and possibly serious endocrine condition.
As we will discuss, the diagnosis begins with a detailed history and physical examination, with laboratory testing and imaging as needed to confirm or rule out underlying causes. Management begins with patient education and support and includes hair removal and drug treatment of any underlying metabolic derangement.
PREVALENCE AND IMPACT
Hirsutism is a common disorder of excess growth of terminal hair in an androgen-dependent male distribution in women, including the chin, upper lip, breasts, upper back, and abdomen.1 It affects 5% to 10% of women of reproductive age.1,2
Hirsutism should be differentiated from hypertrichosis, which can be hereditary or acquired, and which is defined as increased general hair growth in androgen-independent areas.1
Excess hair is cosmetically concerning for women and can significantly affect self-esteem. 3 Normal or acceptable hair growth depends on a woman’s ethnicity and her perception of familial, cultural, and societal norms for the quantity and distribution of hair. Mediterranean women generally have a medium amount of body and facial hair, whereas Asian women have a minimal amount.1,4,5
Hirsutism can be clinically graded according to the Ferriman-Gallwey scale2,6 and is defined as a Ferriman-Gallwey score of 8 or higher.1
HOW CIRCULATING ANDROGENS AFFECT HAIR FOLLICLES
In androgen-dependent areas, circulating androgens influence hair follicle characteristics. Androgens increase the size and diameter of the hair fibers in certain androgen-dependent sites, as seen in puberty with the transformation of vellus hairs (small, nonpigmented hairs) into terminal hairs (large, pigmented hairs) in the pubic and axillary regions in women, as well as the beard area in men.2,7 Interestingly, the same circulating androgens cause miniaturization of the susceptible hair follicles of the central scalp.7 The susceptibility of the hair follicle to the effects of the androgens may be genetically determined.7,8
Hirsutism is a sign of hyperandrogenism and increased action of androgens on hair follicles. In women, about half of circulating testosterone arises from the ovaries and adrenal glands; the rest originates from peripheral conversion of weaker androgens (such as androstenedione produced by the adrenals and ovaries) into testosterone.9 Dehydroepiandrosterone sulfate (DHEAS) originates mainly in the adrenal glands.9,10 Testosterone is converted to the more potent dihydrotestosterone (DHT) by type II 5-alpha reductase in the skin, which can then act on susceptible hair follicles.7,11 Therefore, hirsutism can be a consequence of endogenous androgen over-production from the ovaries or the adrenal glands (or both), of exposure to an exogenous source of androgen such as a drug, or of heightened hair follicle sensitivity and metabolism of normal circulating androgen levels (target end-organ dysfunction).1
‘IDIOPATHIC’ HIRSUTISM: A MISLEADING DIAGNOSIS
Many women with hirsutism are found to have polycystic ovary syndrome as the underlying cause, but hirsutism is also commonly labeled as idiopathic when it occurs without an obvious cause, eg, in women with regular menses and normal androgen levels and without features suspicious for other causes of hirsutism. 1,2,12,13 But while this term is commonly used,1,12 it may be misleading, especially if the diagnosis of idiopathic hirsutism is based on standard laboratory tests, which do not always detect androgen excess.2,13 Minor ovarian or adrenal functional hyperandrogenism,14 increased peripheral activity of 5-alpha reductase in the hair follicle, or abnormalities in the androgen receptor have been implicated in the pathogenesis of so-called idiopathic hirsutism. 2,15
HIRSUTISM AND POLYCYSTIC OVARY SYNDROME
Polycystic ovary syndrome, a metabolic syndrome, presents clinically with menstrual irregularities such as oligomenorrhea or amenorrhea, infertility, and signs of hyperandrogenism such as hirsutism, acne, or androgenetic alopecia.16,17 Metabolic disturbances including insulin resistance, impaired glucose tolerance, hyperlipidemia, and obesity (body mass index > 30 kg/m2) also can occur, thus increasing cardiovascular risk.16–18
The finding of polycystic ovaries is not required to make the diagnosis of polycystic ovary syndrome, and their presence does not prove the diagnosis.16,19 Gonadotropin-dependent functional ovarian hyperandrogenism is believed to cause this syndrome; however, mild adrenocorticotropic-dependent functional adrenal hyperandrogenism also is a feature in many cases. In rare cases, polycystic ovary syndrome presents with an isolated elevation of DHEAS.16,20
OTHER CONDITIONS OF EXCESS ANDROGEN
The syndrome of hyperandrogenism, insulin resistance, and acanthosis nigricans, abbreviated as HAIR-AN, is separate from polycystic ovary syndrome; it characterizes a group of inherited syndromes associated with severe metabolic abnormalities of insulin and glucose metabolism and with marked clinical signs of hyperandrogenism.12
The syndrome of seborrhea, acne, hirsutism, and acanthosis nigricans, abbreviated as SAHA, while not itself a diagnosis, is a clinical spectrum of dermatologic signs and symptoms also associated with hyperandrogenism. These are signs that may present with the HAIR-AN syndrome or with another cause of excess androgens, such as idiopathic, ovarian, adrenal, or hyperprolactinemic hyperandrogenism.21
Thyroid disease, hyperprolactinemia, acromegaly, Cushing syndrome, exogenous factors such as androgenic drugs, and nonclassical congenital adrenal hyperplasia can also produce hirsutism.12 In nonclassical congenital adrenal hyperplasia, which is typically caused by a deficiency of 21-hydroxylase, patients present with premature pubarche, hirsutism in the prepubertal years, and menstrual irregularities including primary amenorrhea.22,23
Important rare causes of hirsutism include benign and malignant androgen-secreting tumors of adrenal or ovarian origin. In such cases, hirsutism can have an acute onset or rapid progression and may be associated with features of virilization, such as deepened voice, increased muscle mass, androgenetic alopecia, clitoromegaly, and increased libido.12
A THOROUGH HISTORY IS CRITICAL TO DIAGNOSIS
A thorough medical history can provide important diagnostic clues in women with hirsutism. The clinician should elicit details about the onset and progression of the hair growth,12,15 previous treatments, and any cutaneous signs of hyperandrogenism, such as acne, seborrhea, acanthosis nigricans, or patterned hair loss.
Also important are the menstrual history and a history of infertility. Primary amenorrhea is defined as failure to menstruate by 16 years of age if secondary sexual characteristics have developed, or by 14 years of age if no secondary sexual characteristics have developed, and it can indicate nonclassical congenital adrenal hyperplasia.
The clinician should also try to determine if the patient has a history of galactorrhea or symptoms of virilization (eg, deepened voice, clitoromegaly, increased muscle mass); a family history of hirsutism, polycystic ovary syndrome, HAIR-AN syndrome, metabolic conditions such as type 2 diabetes mellitus, or cardiovascular disease12,15; or a history of symptoms of any condition known to produce hirsutism, such as Cushing disease, acromegaly, or a thyroid disorder. Also important is a drug history to determine if the patient has taken drugs such as androgens, anabolic steroids, or valproic acid (Depakote).20
THE PHYSICAL EXAMINATION
Another proposed predictor of hirsutism is that terminal hair on the chin or the lower abdomen (Ferriman-Gallwey score ≥ 2) is nearly 100% sensitive and 27% specific at predicting total-body hirsutism.24
As part of the physical examination, the clinician should also look for other cutaneous signs of hyperandrogenism, such as acne, androgenetic alopecia, and seborrhea. Acanthosis nigricans is a sign of insulin resistance. Height and weight should be measured and the body mass index calculated. Blood pressure should be recorded, as high blood pressure may be seen in Cushing syndrome and is an important cardiovascular risk factor. Signs of virilization should be identified. Indicators of Cushing disease such as striae, moon facies, fat redistribution, fragile skin, and proximal myopathy should be noted as well as signs of thyroid disease, such as textural skin changes, goiter, and hair loss. Expressible or spontaneous galactorrhea suggests hyperprolactinemia. Acromegaly is associated with coarse facies and enlarged hands and feet. Many of the endocrinopathies can be caused by a pituitary adenoma, which can manifest as a visual field defect, so visual fields should be examined.25 The examination should also exclude any palpable ovarian or adrenal mass.12
WHEN IS ADDITIONAL TESTING NEEDED?
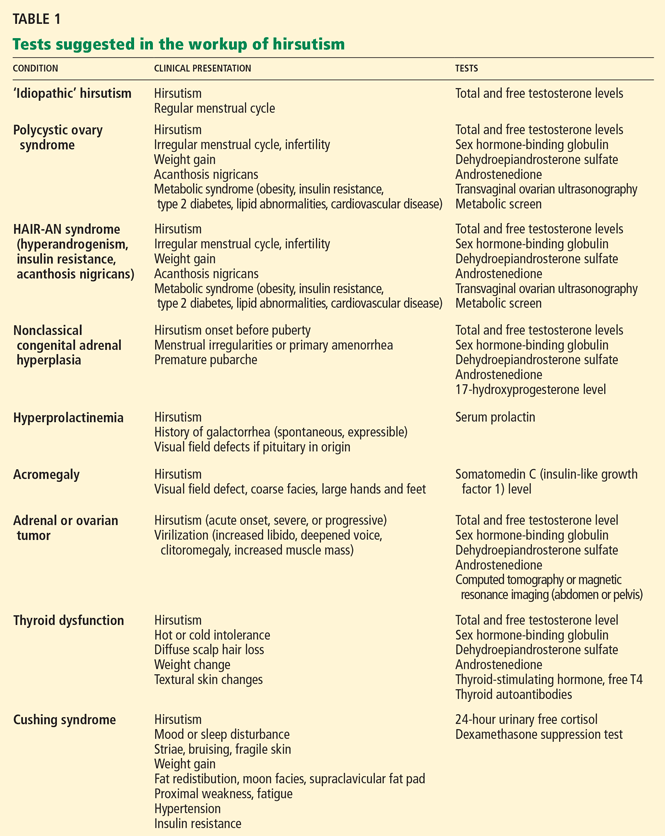
The current Endocrine Society guidelines20 recommend obtaining an early-morning testosterone blood level in the following patients:
- Women with moderate or severe hirsutism
- Women with hirsutism of any degree with sudden onset or rapid progression, or accompanied by signs or symptoms suggesting malignancy or polycystic ovary syndrome: eg, menstrual irregularity, infertility, central obesity, clitoromegaly, or acanthosis nigricans.15,20
Testing androgen levels in mild, isolated hirsutism has not been proven to be useful or to alter management.20
Free testosterone level
An early-morning total or free testosterone level is the initial test in the laboratory evaluation of hirsutism.12,15 Additional specialized laboratory testing may be needed to determine the free testosterone level,15 as the free testosterone test is not available at all laboratories. A normal total testosterone level does not exclude hyperandrogenism but can suggest the diagnosis of idiopathic hirsutism.15
Further testing is needed if the total testosterone level is normal or only slightly elevated, or if there is a strong clinical suspicion of an underlying condition such as endocrinopathy or tumor. It is also useful in patients whose hirsutism responds poorly to medical treatments15 (see discussion below).
If the total testosterone level is elevated, if the hirsutism is moderate to severe, if there are associated symptoms, or if hirsutism is acute or progressive, a further endocrinologic workup is needed,15 possibly including measurement of free testosterone, sex hormone-binding globulin, DHEAS, and androstenedione.15 Free testosterone, unbound to sex hormone-binding globulin, is the biologically active fraction, with the levels of binding globulin increased by drugs such as oral contraceptives15 and decreased by high insulin levels in insulin resistance.25
Test in patients with mild hirsutism?
Although the guidelines suggest that no additional workup is necessary for women with mild hirsutism, we evaluate all patients with hirsutism and those with the SAHA clinical spectrum by measuring free and total testosterone and DHEAS. In our experience, even women with mild hirsutism with subtle symptoms and signs of hyperandrogenism and mild hirsutism often have elevated androgen levels.
Test in women with idiopathic hirsutism?
In women with idiopathic hirsutism, minor forms of functional ovarian and adrenal hyperandrogenism are believed to play a role and are thought to be undetectable with conventional testing.25 The gonadotropin-releasing hormone (GnRH) analogue stimulation test may uncover occult hyperandrogenism in this setting, but it is used as a research tool and does not currently have application in routine clinical practice.14
It is important to remember that some women with apparent idiopathic hirsutism and a history of regular menstrual cycles are actually oligo-ovulatory or anovulatory. In these instances, another diagnosis should be considered,13 and referral to an endocrinologist for further evaluation of ovulatory function is recommended.13
CURRENT USE OF DIAGNOSTIC IMAGING
When malignancy is suspected
A testosterone level above 200 ng/dL suggests an ovarian tumor, and a DHEAS level above 700 μg/dL suggests an adrenal tumor.26 However, not all tumors present with such high androgen levels, and sudden onset of hirsutism, rapid progression of hirsutism, or signs of virilization suggest a tumor.15 In such cases, transvaginal ultrasonography, computed tomography, or magnetic resonance imaging (MRI) of the abdomen can exclude an ovarian or adrenal tumor.
When polycystic ovary syndrome is suspected
The diagnosis of polycystic ovary syndrome is confirmed by two out of three criteria:
- Oligo-ovulation or anovulation
- Clinical or laboratory signs of hyperandrogenism
- Ultrasonographic evidence of polycystic ovaries, with exclusion of other causes of hyperandrogenism.
ADDITIONAL LABORATORY TESTING
Tests for polycystic ovary syndrome
Assessment of polycystic ovary syndrome involves transvaginal ultrasonography, but ultrasonographic evidence of a polycystic ovary is not necessary for the diagnosis.16 A fasting lipid profile and fasting serum glucose are recommended, and if the fasting serum glucose is normal, an oral glucose tolerance test is recommended. 17
Some have reported measuring the ratio of luteinizing hormone to follicle-stimulating hormone in the workup of polycystic ovary syndrome, and a ratio greater than 2 has been considered indicative but not diagnostic.16,25 The individual levels of luteinizing hormone, follicle-stimulating hormone, and estradiol are more important in the evaluation of infertility and ovulatory dysfunction. In patients with elevations of these hormones or with these symptoms, referral for infertility screening with an endocrinologist or gynecologist is recommended. 25
Additional testing and referral for Cushing syndrome, other conditions
Cushing syndrome can be tested for with a 24-hour urine cortisol, overnight low-dose dexamethasone suppression test, and late-night salivary cortisol.27,28 Referral to an endocrinologist for further testing can differentiate between corticotropin-dependent or corticotropin-independent Cushing syndrome.25 Cushing syndrome is often associated with hyperandrogenism, particularly in those cases caused by adrenal tumors.29
The prolactin level and the level of somatomedin C (insulin-like growth factor 1) can be used to rule out hyperprolactinemia and acromegaly, respectively.12 If Cushing syndrome, hyperprolactinemia, or acromegaly is diagnosed by endocrinologic testing, pituitary MRI should be performed.12,25
Referral to specialist centers with experience with these conditions is essential. Nonclassical congenital adrenal hyperplasia can be screened for by a serum 17-hydroxyprogesterone level measured in the follicular phase.12 Measurement of thyroid-stimulating hormone, free thyroxine, and thyroid peroxidase antibodies screens for thyroid disease.12 Hirsutism has been reported with the commencement of L-thyroxine therapy.30
THE PRINCIPLES OF TREATMENT
Patient education regarding the cause of hirsutism and reasonable treatment expectations and emotional support are important in the management of hirsutism. Also important is regular follow-up to measure and document the response to treatment; this can include repeating Ferriman-Gallwey scoring, taking photographs of affected areas, and retesting androgen levels after 3 to 6 months.12
Treatment must be continued for an ongoing effect, and most pharmacologic treatments can take up to 3 to 6 months to produce significant improvement.1
When an underlying condition is diagnosed, treatment of the condition is essential. Androgen-secreting tumors require surgical management.12 Cushing disease, hyperprolactinemia, and acromegaly should be clinically apparent from examination and testing, and appropriate referral and standard management should be instigated. Exogenous sources of androgen such as androgenic progestins or anabolic steroids should be discontinued. Lifestyle management is important, and weight loss in obese patients with polycystic ovary syndrome can improve hirsutism as well as mitigate cardiovascular risk factors.31
In classic congenital adrenal hyperplasia, glucocorticoid therapy manages both ovulation induction and hirsutism.20 However, in nonclassical congenital adrenal hyperplasia, glucocorticoid therapy supports ovulation induction, but hirsutism usually requires both systemic antiandrogen and hair removal.20
CURRENT OPTIONS FOR HAIR REMOVAL
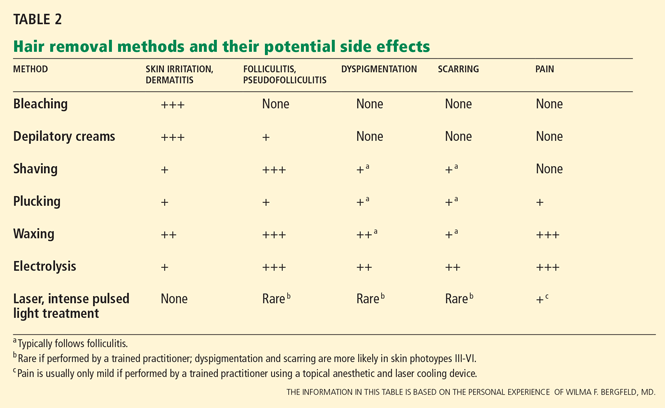
The choice of method depends on patient preference, adverse effects, the degree of hirsutism, the level of distress, previous treatments, and cost.1,15,32
Self-care methods
Self-care methods offer only temporary reduction of excess hairs.
Plucking removes the entire hair, including the root, but it is painful and time-consuming, and it is only practical for areas where few hairs exist, such as on the face.1
Shaving is an easy, inexpensive, and painless choice for hair removal. Although a common belief is that shaving causes faster or thicker hair regrowth, shaving affects neither the diameter nor the rate of growth of the hair.32 Given its masculine association, shaving is not acceptable to most women except perhaps for use on the legs and axillae.1,32 Shaving can cause irritation, folliculitis, pseudofolliculitis, and infection.1
Waxing removes the entire hair. While it is more expensive than plucking, regrowth is slower, occurring over weeks. It is painful and can cause thermal burns, irritation, folliculitis, scarring, and postinflammatory dyspigmentation.1
Chemical depilatories, usually thioglycollic acid preparations, are inexpensive, painless, and easy to use. However, the resulting hair reduction is of short duration because the hair shafts are only removed at the level of the skin surface.1 They can also cause irritant dermatitis. 1
Bleaching with hydrogen peroxide is inexpensive and can camouflage dark facial hair, but it can also cause skin discoloration and irritation. 1
Clinic-based methods
Electrolysis often results in a permanent reduction in hair growth.1,32 A fine needle is placed into the hair follicle and an electrical current is applied. Each follicle is treated individually. 1,32 Best results are seen on darker hairs in patients with lighter skin, but it can be used on all skin types and hair colors.1,32
Electrolysis is operator-dependent, and there are US Food and Drug Administration (FDA) regulations regarding electrolysis techniques. It requires multiple treatments, and it is painful and can cause erythema, folliculitis, pseudofolliculitis, infection, scarring, and postinflammatory dyspigmentation.1,32 Some reports suggest that prior waxing and plucking of hairs damages the hair by twisting the hair shaft, making electrolysis more difficult.32
Laser treatment uses light of certain wavelengths to damage the hair follicles. While laser hair removal does not result in complete or persistent hair removal, it is more effective than shaving, waxing, and electrolysis, producing partial hair reduction for up to 6 months; the effect is enhanced with multiple treatments.33,34 The number of treatments required depends on the laser type and on the nature of the patient’s hair follicles.35
Laser systems for hair removal are of various wavelengths and also include intense pulsed light systems. The choice of system depends on the patient’s skin type and hair color. Women with fair skin and dark hair are ideal candidates; longer-wavelength lasers are preferred for darker or tanned skin types.
Adverse effects of laser hair removal include pain, erythema, burns, dyspigmentation, and scarring. Laser cooling devices can prevent or minimize some of these effects. Laser treatment has also been known to cause a paradoxical increase in hair growth.1,33,34
DRUG THERAPIES FOR HIRSUTISM

The drugs most commonly used for hirsutism are oral contraceptives (off-label use) and antiandrogenic drugs (off-label use). Topical eflornithine cream (Vaniqa) is FDA-approved for hirsutism but is less commonly used. Insulin sensitizers, GnRH analogues, and other drugs are occasionally used (off-label) to treat hirsutism.
Topical eflornithine cream
Topical eflornithine cream treats facial hirsutism by slowing the rate of hair growth; it does this by irreversibly inhibiting ornithine decarboxylase, an enzyme essential for hair growth.39,40 Studies showed that twice-daily application reduced unwanted facial hair in women after 24 weeks of treatment.39,40 Treatment must be continuous, since hair growth rapidly returns to the pretreatment rate by 8 weeks after discontinuing eflornithine.39,40 White women have been shown to respond better than black women.39 Adverse effects include a mild burning sensation, acne, pseudofolliculitis barbae, irritation, and allergic contact dermatitis.39,40 Improved outcomes have been suggested when eflornithine cream is combined with laser hair removal.41
Oral contraceptives
Oral contraceptives are commonly used off-label for the management of hirsutism.20 Oral contraceptives suppress the secretion of luteinizing hormone and, hence, the synthesis of ovarian androgen, thereby increasing levels of sex hormone-binding globulin and decreasing free plasma testosterone.1,20 Adrenal androgen production is also slightly reduced.20
Oral contraceptives usually combine a synthetic estrogen and a progestin. Certain progestins are more androgenic and should be avoided.1
For treating hirsutism, oral contraceptives should be used that contain low-androgenic progestins such as cyproterone acetate (not available in the United States), drosperinone (eg, in Yasmin), norgestimate (eg, in Ortho Tri-Cyclen), or desogestrel (eg, in Mircette).1,20
Side effects of oral contraceptives include breast tenderness, gastrointestinal upset, headache, loss of libido, hypertension, and the potential risk of venous thromboembolism.1,15,32,36
Antiandrogenic drugs
Several antiandrogenic drugs are used off-label to treat hirsutism.
Spironolactone (Aldactone), a competitive inhibitor of the androgen receptor and 5-alpha reductase activity,20 can be effective in the treatment of hirsutism. Monotherapy with spironolactone, without an oral contraceptive or other reliable form of contraception, is not recommended because of the teratogenic potential of all antiandrogens to feminize a developing male fetus.20 Thus, reliable contraception should be used in females of childbearing age when starting antiandrogen therapy.
The dosage of spironolactone for hirsutism is usually 100 mg to 200 mg daily.1,20 Hyperkalemia, polyuria, postural hypotension, irregular menses, and liver abnormalities are among the possible adverse effects (Table 3). Spironolactone was found to be tumorigenic in animal studies, although this has unknown relevance in humans.36
Cyproterone, an antiandrogen not available in the United States,42 competitively inhibits the androgen receptor and 5-alpha-reductase activity.1,20,36 It can be used for only the first 10 days of the menstrual cycle (50-mg or 100-mg dose) with an oral contraceptive pill, or in a low dose in a combined oral contraceptive pill (Diane-35 in Canada and the United Kingdom).1
Side effects are similar to those of oral contraceptives and include fatigue, mood change, risk of venous thromboembolism, and decreased libido.1,15,36 Importantly, in woman of childbearing age, there is the potential risk of feminization of a male fetus, so reliable contraception must be used.15,36
Flutamide, an investigational antiandrogen, has shown promise in the treatment of hirsutism.20 Flutamide is a nonsteroidal competitive inhibitor of androgen receptor binding. It carries a significant risk of hepatotoxicity. 1,15
Finasteride (Propecia) 1 mg is only occasionally used in the treatment of hirsutism (off-label usage). It inhibits type II 5-alphareductase to suppress dihydrotestosterone levels. 32 It carries a risk of gastrointestinal disturbance, decreased libido, hepatotoxicity, and feminization of a male fetus (pregnancy category X), so reliable contraception is required in all females of childbearing age, as with all antiandrogens1 (Table 3).
Dutasteride (Avodart), a type I and II 5-alpha-reductase inhibitor, has not been studied for the treatment of hirsutism (pregnancy category X).
Insulin sensitizers
Metformin (Glucophage) and other insulin sensitizers are less effective than antiandrogens at reducing hirsutism.20,38 However, metformin is effective at inducing ovulation in patients with polycystic ovary syndrome.38 Gastrointestinal upset is a common side effect; lactic acidosis is a serious but rare adverse effect.1
Gonadotropin-releasing hormone analogues
GnRH analogues are an option only if oral contraceptives and antiandrogen drugs are unsuccessful in patients with severe hyperandrogenism. 20 They suppress secretion of luteinizing hormone and the synthesis of ovarian androgen.1,20 These drugs are given as monthly intramuscular injections, usually with some form of estrogen-progestin replacement, since GnRH analogues cause estrogen levels to fall to menopausal levels.1
Side effects include signs and symptoms of menopause including hot flushes, atrophic vaginitis, and osteoporosis.1,15 These drugs completely inhibit ovulation, and some endocrinologists and gynecologists do not suggest further contraception in women of childbearing years for this reason. However, GnRH analogues are not approved as a contraceptive and are pregnancy category X.
Other drugs
Other drugs with antiandrogen activity include cimetidine and ketoconazole.12 Cimetidine (Tagamet) is not effective for the treatment of hirsutism, and ketoconazole (Nizoral) is associated with significant risk for adrenocortical suppression12 and hepatotoxicity in addition to multiple drug interactions, given its effect on the hepatic P450 enzyme system.
Acknowledgment: Many thanks to Rebecca Tung, MD, dermatologic surgeon, Cleveland Clinic, for her advice on lasers.
- Mofid A, Seyyed Alinaghi SA, Zandieh S, Yazdani T. Hirsutism. Int J Clin Pract 2008; 62:433–443.
- Azziz R, Carmina E, Sawaya ME. Idiopathic hirsutism. Endocr Rev 2000; 21:347–362.
- Himelein MJ, Thatcher SS. Polycystic ovary syndrome and mental health: a review. Obstet Gynecol Surv 2006; 61:723–732.
- Williamson K, Gunn AJ, Johnson N, Milsom SR. The impact of ethnicity on the presentation of polycystic ovarian syndrome. Aust N Z J Obstet Gynaecol 2001; 41:202–206.
- Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999; 84:4006–4011.
- Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 1961; 21:1440–1447.
- Messenger AG. The control of hair growth: an overview. J Invest Dermatol 1993; 101(suppl 1):4S–9S.
- Rosenfield RL. Hirsutism and the variable response of the pilosebaceous unit to androgen. J Investig Dermatol Symp Proc 2005; 10:205–208.
- Longcope C. Adrenal and gonadal androgen secretion in normal females. Clin Endocrinol Metab 1986; 15:213–228.
- Braunstein GD. Testis. In:Gardner DG, Shoback D, editors. Greenspan’s Basic & Clinical Endocrinology. 8th ed. New York: McGraw-Hill, 2007.
- Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev 2000; 21:363–392.
- Practice Committee of the American Society for Reproductive Medicine. The evaluation and treatment of androgen excess. Fertil Steril 2006; 86(suppl 5):S241–S247.
- Azziz R, Waggoner WT, Ochoa T, Knochenhauer ES, Boots LR. Idiopathic hirsutism: an uncommon cause of hirsutism in Alabama. Fertil Steril 1998; 70:274–278.
- Rossi R, Tauchmanovà L, Luciano A, et al. Functional hyperandrogenism detected by corticotropin and GnRH-analogue stimulation tests in women affected by apparently idiopathic hirsutism. J Endocrinol Invest 2001; 24:491–498.
- Rosenfield RL. Clinical practice. Hirsutism. N Engl J Med 2005; 353:2578–2588.
- Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 2004; 19:41–47.
- Salley KE, Wickham EP, Cheang KI, Essah PA, Karjane NW, Nestler JE. Glucose intolerance in polycystic ovary syndrome—a position statement of the Androgen Excess Society. J Clin Endocrinol Metab 2007; 92:4546–4556.
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365:1415–1428.
- Azziz R. Diagnostic criteria for polycystic ovary syndrome: a reappraisal. Fertil Steril 2005; 83:1343–1346.
- Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. http://www.endo-society.org/guidelines/final/upload/Hirsutism_Guideline.pdf. Accessed March 30, 2010.
- Orfanos CE, Adler YD, Zouboulis CC. The SAHA syndrome. Horm Res 2000; 54:251–258.
- New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab 2006; 91:4205–4214.
- Kohn B, Levine LS, Pollack MS, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 1982; 55:817–827.
- Knochenhauer ES, Hines G, Conway-Myers BA, Azziz R. Examination of the chin or lower abdomen only for the prediction of hirsutism. Fertil Steril 2000; 74:980–983.
- Somani N, Harrison S, Bergfeld WF. The clinical evaluation of hirsutism. Dermatol Ther 2008; 21:376–391.
- Waggoner W, Boots LR, Azziz R. Total testosterone and DHEAS levels as predictors of androgen-secreting neoplasms: a populational study. Gynecol Endocrinol 1999; 13:394–400.
- Crapo L. Cushing’s syndrome: a review of diagnostic tests. Metabolism 1979; 28:955–977.
- Blethen SL, Chasalow FI. Overnight dexamethasone suppression test: normal responses and the diagnosis of Cushing’s syndrome. Steroids 1989; 54:185–193.
- Bertagna C, Orth DN. Clinical and laboratory findings and results of therapy in 58 patients with adrenocortical tumors admitted to a single medical center (1951 to 1978). Am J Med 1981; 71:855–875.
- Kologlu S, Baskal N, Kologlu LB, Laleli Y, Tuccar E. Hirsutism due to the treatment with L-thyroxine in patients with thyroid pathology. Endocrinologie 1988; 26:179–185.
- Gambineri A, Patton L, Vaccina A, et al. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. J Clin Endocrinol Metab 2006; 91:3970–3980.
- Dawber RP. Guidance for the management of hirsutism. Curr Med Res Opin 2005; 21:1227–1234.
- Haedersdal M, Wulf HC. Evidence based review of hair removal using lasers and light sources. J Eur Acad Dermatol Venereol 2006; 20:9–20.
- Sadighha A, Mohaghegh Zahed G. Meta-analysis of hair removal laser trials. Lasers Med Sci 2009; 24:21–25.
- Casey AS, Goldberg D. Guidelines for laser hair removal. J Cosmet Laser Ther 2008; 10:24–33.
- Wakelin SH, Maibach HI, editors. Handbook of Systemic Drug Treatment in Dermatology. London: Manson Publishing Ltd, 2004.
- Swiglo BA, Cosma M, Flynn DN, et al. Clinical review: antiandrogens for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab 2008; 93:1153–1160.
- Cosma M, Swiglo BA, Flynn DN, et al. Clinical review: insulin sensitizers for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab 2008; 93:1135–1142.
- Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol 2001; 2:197–201.
- Wolf JE, Shander D, Huber F, et al; Eflornithine HCl Study Group. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair. Int J Dematol 2007; 46:94–98.
- Hamzavi I, Tan E, Shapiro J, Lui H. A randomized bilateral vehicle-controlled study of eflornithine cream combined with laser treatment versus laser treatment alone for facial hirsutism in women. J Am Acad Dermatol 2007; 57:54–59.
- Van der Spuy ZM, le Roux PA. Cyproterone acetate for hirsutism. Cochrane Database Syst Rev 2003; 4:CD001125.
Hirsutism causes significant anxiety and lack of self-esteem in women. Although it is itself a benign condition, it is often the sign of an underlying and possibly serious endocrine condition.
As we will discuss, the diagnosis begins with a detailed history and physical examination, with laboratory testing and imaging as needed to confirm or rule out underlying causes. Management begins with patient education and support and includes hair removal and drug treatment of any underlying metabolic derangement.
PREVALENCE AND IMPACT
Hirsutism is a common disorder of excess growth of terminal hair in an androgen-dependent male distribution in women, including the chin, upper lip, breasts, upper back, and abdomen.1 It affects 5% to 10% of women of reproductive age.1,2
Hirsutism should be differentiated from hypertrichosis, which can be hereditary or acquired, and which is defined as increased general hair growth in androgen-independent areas.1
Excess hair is cosmetically concerning for women and can significantly affect self-esteem. 3 Normal or acceptable hair growth depends on a woman’s ethnicity and her perception of familial, cultural, and societal norms for the quantity and distribution of hair. Mediterranean women generally have a medium amount of body and facial hair, whereas Asian women have a minimal amount.1,4,5
Hirsutism can be clinically graded according to the Ferriman-Gallwey scale2,6 and is defined as a Ferriman-Gallwey score of 8 or higher.1
HOW CIRCULATING ANDROGENS AFFECT HAIR FOLLICLES
In androgen-dependent areas, circulating androgens influence hair follicle characteristics. Androgens increase the size and diameter of the hair fibers in certain androgen-dependent sites, as seen in puberty with the transformation of vellus hairs (small, nonpigmented hairs) into terminal hairs (large, pigmented hairs) in the pubic and axillary regions in women, as well as the beard area in men.2,7 Interestingly, the same circulating androgens cause miniaturization of the susceptible hair follicles of the central scalp.7 The susceptibility of the hair follicle to the effects of the androgens may be genetically determined.7,8
Hirsutism is a sign of hyperandrogenism and increased action of androgens on hair follicles. In women, about half of circulating testosterone arises from the ovaries and adrenal glands; the rest originates from peripheral conversion of weaker androgens (such as androstenedione produced by the adrenals and ovaries) into testosterone.9 Dehydroepiandrosterone sulfate (DHEAS) originates mainly in the adrenal glands.9,10 Testosterone is converted to the more potent dihydrotestosterone (DHT) by type II 5-alpha reductase in the skin, which can then act on susceptible hair follicles.7,11 Therefore, hirsutism can be a consequence of endogenous androgen over-production from the ovaries or the adrenal glands (or both), of exposure to an exogenous source of androgen such as a drug, or of heightened hair follicle sensitivity and metabolism of normal circulating androgen levels (target end-organ dysfunction).1
‘IDIOPATHIC’ HIRSUTISM: A MISLEADING DIAGNOSIS
Many women with hirsutism are found to have polycystic ovary syndrome as the underlying cause, but hirsutism is also commonly labeled as idiopathic when it occurs without an obvious cause, eg, in women with regular menses and normal androgen levels and without features suspicious for other causes of hirsutism. 1,2,12,13 But while this term is commonly used,1,12 it may be misleading, especially if the diagnosis of idiopathic hirsutism is based on standard laboratory tests, which do not always detect androgen excess.2,13 Minor ovarian or adrenal functional hyperandrogenism,14 increased peripheral activity of 5-alpha reductase in the hair follicle, or abnormalities in the androgen receptor have been implicated in the pathogenesis of so-called idiopathic hirsutism. 2,15
HIRSUTISM AND POLYCYSTIC OVARY SYNDROME
Polycystic ovary syndrome, a metabolic syndrome, presents clinically with menstrual irregularities such as oligomenorrhea or amenorrhea, infertility, and signs of hyperandrogenism such as hirsutism, acne, or androgenetic alopecia.16,17 Metabolic disturbances including insulin resistance, impaired glucose tolerance, hyperlipidemia, and obesity (body mass index > 30 kg/m2) also can occur, thus increasing cardiovascular risk.16–18
The finding of polycystic ovaries is not required to make the diagnosis of polycystic ovary syndrome, and their presence does not prove the diagnosis.16,19 Gonadotropin-dependent functional ovarian hyperandrogenism is believed to cause this syndrome; however, mild adrenocorticotropic-dependent functional adrenal hyperandrogenism also is a feature in many cases. In rare cases, polycystic ovary syndrome presents with an isolated elevation of DHEAS.16,20
OTHER CONDITIONS OF EXCESS ANDROGEN
The syndrome of hyperandrogenism, insulin resistance, and acanthosis nigricans, abbreviated as HAIR-AN, is separate from polycystic ovary syndrome; it characterizes a group of inherited syndromes associated with severe metabolic abnormalities of insulin and glucose metabolism and with marked clinical signs of hyperandrogenism.12
The syndrome of seborrhea, acne, hirsutism, and acanthosis nigricans, abbreviated as SAHA, while not itself a diagnosis, is a clinical spectrum of dermatologic signs and symptoms also associated with hyperandrogenism. These are signs that may present with the HAIR-AN syndrome or with another cause of excess androgens, such as idiopathic, ovarian, adrenal, or hyperprolactinemic hyperandrogenism.21
Thyroid disease, hyperprolactinemia, acromegaly, Cushing syndrome, exogenous factors such as androgenic drugs, and nonclassical congenital adrenal hyperplasia can also produce hirsutism.12 In nonclassical congenital adrenal hyperplasia, which is typically caused by a deficiency of 21-hydroxylase, patients present with premature pubarche, hirsutism in the prepubertal years, and menstrual irregularities including primary amenorrhea.22,23
Important rare causes of hirsutism include benign and malignant androgen-secreting tumors of adrenal or ovarian origin. In such cases, hirsutism can have an acute onset or rapid progression and may be associated with features of virilization, such as deepened voice, increased muscle mass, androgenetic alopecia, clitoromegaly, and increased libido.12
A THOROUGH HISTORY IS CRITICAL TO DIAGNOSIS
A thorough medical history can provide important diagnostic clues in women with hirsutism. The clinician should elicit details about the onset and progression of the hair growth,12,15 previous treatments, and any cutaneous signs of hyperandrogenism, such as acne, seborrhea, acanthosis nigricans, or patterned hair loss.
Also important are the menstrual history and a history of infertility. Primary amenorrhea is defined as failure to menstruate by 16 years of age if secondary sexual characteristics have developed, or by 14 years of age if no secondary sexual characteristics have developed, and it can indicate nonclassical congenital adrenal hyperplasia.
The clinician should also try to determine if the patient has a history of galactorrhea or symptoms of virilization (eg, deepened voice, clitoromegaly, increased muscle mass); a family history of hirsutism, polycystic ovary syndrome, HAIR-AN syndrome, metabolic conditions such as type 2 diabetes mellitus, or cardiovascular disease12,15; or a history of symptoms of any condition known to produce hirsutism, such as Cushing disease, acromegaly, or a thyroid disorder. Also important is a drug history to determine if the patient has taken drugs such as androgens, anabolic steroids, or valproic acid (Depakote).20
THE PHYSICAL EXAMINATION
Another proposed predictor of hirsutism is that terminal hair on the chin or the lower abdomen (Ferriman-Gallwey score ≥ 2) is nearly 100% sensitive and 27% specific at predicting total-body hirsutism.24
As part of the physical examination, the clinician should also look for other cutaneous signs of hyperandrogenism, such as acne, androgenetic alopecia, and seborrhea. Acanthosis nigricans is a sign of insulin resistance. Height and weight should be measured and the body mass index calculated. Blood pressure should be recorded, as high blood pressure may be seen in Cushing syndrome and is an important cardiovascular risk factor. Signs of virilization should be identified. Indicators of Cushing disease such as striae, moon facies, fat redistribution, fragile skin, and proximal myopathy should be noted as well as signs of thyroid disease, such as textural skin changes, goiter, and hair loss. Expressible or spontaneous galactorrhea suggests hyperprolactinemia. Acromegaly is associated with coarse facies and enlarged hands and feet. Many of the endocrinopathies can be caused by a pituitary adenoma, which can manifest as a visual field defect, so visual fields should be examined.25 The examination should also exclude any palpable ovarian or adrenal mass.12
WHEN IS ADDITIONAL TESTING NEEDED?
The current Endocrine Society guidelines20 recommend obtaining an early-morning testosterone blood level in the following patients:
- Women with moderate or severe hirsutism
- Women with hirsutism of any degree with sudden onset or rapid progression, or accompanied by signs or symptoms suggesting malignancy or polycystic ovary syndrome: eg, menstrual irregularity, infertility, central obesity, clitoromegaly, or acanthosis nigricans.15,20
Testing androgen levels in mild, isolated hirsutism has not been proven to be useful or to alter management.20
Free testosterone level
An early-morning total or free testosterone level is the initial test in the laboratory evaluation of hirsutism.12,15 Additional specialized laboratory testing may be needed to determine the free testosterone level,15 as the free testosterone test is not available at all laboratories. A normal total testosterone level does not exclude hyperandrogenism but can suggest the diagnosis of idiopathic hirsutism.15
Further testing is needed if the total testosterone level is normal or only slightly elevated, or if there is a strong clinical suspicion of an underlying condition such as endocrinopathy or tumor. It is also useful in patients whose hirsutism responds poorly to medical treatments15 (see discussion below).
If the total testosterone level is elevated, if the hirsutism is moderate to severe, if there are associated symptoms, or if hirsutism is acute or progressive, a further endocrinologic workup is needed,15 possibly including measurement of free testosterone, sex hormone-binding globulin, DHEAS, and androstenedione.15 Free testosterone, unbound to sex hormone-binding globulin, is the biologically active fraction, with the levels of binding globulin increased by drugs such as oral contraceptives15 and decreased by high insulin levels in insulin resistance.25
Test in patients with mild hirsutism?
Although the guidelines suggest that no additional workup is necessary for women with mild hirsutism, we evaluate all patients with hirsutism and those with the SAHA clinical spectrum by measuring free and total testosterone and DHEAS. In our experience, even women with mild hirsutism with subtle symptoms and signs of hyperandrogenism and mild hirsutism often have elevated androgen levels.
Test in women with idiopathic hirsutism?
In women with idiopathic hirsutism, minor forms of functional ovarian and adrenal hyperandrogenism are believed to play a role and are thought to be undetectable with conventional testing.25 The gonadotropin-releasing hormone (GnRH) analogue stimulation test may uncover occult hyperandrogenism in this setting, but it is used as a research tool and does not currently have application in routine clinical practice.14
It is important to remember that some women with apparent idiopathic hirsutism and a history of regular menstrual cycles are actually oligo-ovulatory or anovulatory. In these instances, another diagnosis should be considered,13 and referral to an endocrinologist for further evaluation of ovulatory function is recommended.13
CURRENT USE OF DIAGNOSTIC IMAGING
When malignancy is suspected
A testosterone level above 200 ng/dL suggests an ovarian tumor, and a DHEAS level above 700 μg/dL suggests an adrenal tumor.26 However, not all tumors present with such high androgen levels, and sudden onset of hirsutism, rapid progression of hirsutism, or signs of virilization suggest a tumor.15 In such cases, transvaginal ultrasonography, computed tomography, or magnetic resonance imaging (MRI) of the abdomen can exclude an ovarian or adrenal tumor.
When polycystic ovary syndrome is suspected
The diagnosis of polycystic ovary syndrome is confirmed by two out of three criteria:
- Oligo-ovulation or anovulation
- Clinical or laboratory signs of hyperandrogenism
- Ultrasonographic evidence of polycystic ovaries, with exclusion of other causes of hyperandrogenism.
ADDITIONAL LABORATORY TESTING
Tests for polycystic ovary syndrome
Assessment of polycystic ovary syndrome involves transvaginal ultrasonography, but ultrasonographic evidence of a polycystic ovary is not necessary for the diagnosis.16 A fasting lipid profile and fasting serum glucose are recommended, and if the fasting serum glucose is normal, an oral glucose tolerance test is recommended. 17
Some have reported measuring the ratio of luteinizing hormone to follicle-stimulating hormone in the workup of polycystic ovary syndrome, and a ratio greater than 2 has been considered indicative but not diagnostic.16,25 The individual levels of luteinizing hormone, follicle-stimulating hormone, and estradiol are more important in the evaluation of infertility and ovulatory dysfunction. In patients with elevations of these hormones or with these symptoms, referral for infertility screening with an endocrinologist or gynecologist is recommended. 25
Additional testing and referral for Cushing syndrome, other conditions
Cushing syndrome can be tested for with a 24-hour urine cortisol, overnight low-dose dexamethasone suppression test, and late-night salivary cortisol.27,28 Referral to an endocrinologist for further testing can differentiate between corticotropin-dependent or corticotropin-independent Cushing syndrome.25 Cushing syndrome is often associated with hyperandrogenism, particularly in those cases caused by adrenal tumors.29
The prolactin level and the level of somatomedin C (insulin-like growth factor 1) can be used to rule out hyperprolactinemia and acromegaly, respectively.12 If Cushing syndrome, hyperprolactinemia, or acromegaly is diagnosed by endocrinologic testing, pituitary MRI should be performed.12,25
Referral to specialist centers with experience with these conditions is essential. Nonclassical congenital adrenal hyperplasia can be screened for by a serum 17-hydroxyprogesterone level measured in the follicular phase.12 Measurement of thyroid-stimulating hormone, free thyroxine, and thyroid peroxidase antibodies screens for thyroid disease.12 Hirsutism has been reported with the commencement of L-thyroxine therapy.30
THE PRINCIPLES OF TREATMENT
Patient education regarding the cause of hirsutism and reasonable treatment expectations and emotional support are important in the management of hirsutism. Also important is regular follow-up to measure and document the response to treatment; this can include repeating Ferriman-Gallwey scoring, taking photographs of affected areas, and retesting androgen levels after 3 to 6 months.12
Treatment must be continued for an ongoing effect, and most pharmacologic treatments can take up to 3 to 6 months to produce significant improvement.1
When an underlying condition is diagnosed, treatment of the condition is essential. Androgen-secreting tumors require surgical management.12 Cushing disease, hyperprolactinemia, and acromegaly should be clinically apparent from examination and testing, and appropriate referral and standard management should be instigated. Exogenous sources of androgen such as androgenic progestins or anabolic steroids should be discontinued. Lifestyle management is important, and weight loss in obese patients with polycystic ovary syndrome can improve hirsutism as well as mitigate cardiovascular risk factors.31
In classic congenital adrenal hyperplasia, glucocorticoid therapy manages both ovulation induction and hirsutism.20 However, in nonclassical congenital adrenal hyperplasia, glucocorticoid therapy supports ovulation induction, but hirsutism usually requires both systemic antiandrogen and hair removal.20
CURRENT OPTIONS FOR HAIR REMOVAL
The choice of method depends on patient preference, adverse effects, the degree of hirsutism, the level of distress, previous treatments, and cost.1,15,32
Self-care methods
Self-care methods offer only temporary reduction of excess hairs.
Plucking removes the entire hair, including the root, but it is painful and time-consuming, and it is only practical for areas where few hairs exist, such as on the face.1
Shaving is an easy, inexpensive, and painless choice for hair removal. Although a common belief is that shaving causes faster or thicker hair regrowth, shaving affects neither the diameter nor the rate of growth of the hair.32 Given its masculine association, shaving is not acceptable to most women except perhaps for use on the legs and axillae.1,32 Shaving can cause irritation, folliculitis, pseudofolliculitis, and infection.1
Waxing removes the entire hair. While it is more expensive than plucking, regrowth is slower, occurring over weeks. It is painful and can cause thermal burns, irritation, folliculitis, scarring, and postinflammatory dyspigmentation.1
Chemical depilatories, usually thioglycollic acid preparations, are inexpensive, painless, and easy to use. However, the resulting hair reduction is of short duration because the hair shafts are only removed at the level of the skin surface.1 They can also cause irritant dermatitis. 1
Bleaching with hydrogen peroxide is inexpensive and can camouflage dark facial hair, but it can also cause skin discoloration and irritation. 1
Clinic-based methods
Electrolysis often results in a permanent reduction in hair growth.1,32 A fine needle is placed into the hair follicle and an electrical current is applied. Each follicle is treated individually. 1,32 Best results are seen on darker hairs in patients with lighter skin, but it can be used on all skin types and hair colors.1,32
Electrolysis is operator-dependent, and there are US Food and Drug Administration (FDA) regulations regarding electrolysis techniques. It requires multiple treatments, and it is painful and can cause erythema, folliculitis, pseudofolliculitis, infection, scarring, and postinflammatory dyspigmentation.1,32 Some reports suggest that prior waxing and plucking of hairs damages the hair by twisting the hair shaft, making electrolysis more difficult.32
Laser treatment uses light of certain wavelengths to damage the hair follicles. While laser hair removal does not result in complete or persistent hair removal, it is more effective than shaving, waxing, and electrolysis, producing partial hair reduction for up to 6 months; the effect is enhanced with multiple treatments.33,34 The number of treatments required depends on the laser type and on the nature of the patient’s hair follicles.35
Laser systems for hair removal are of various wavelengths and also include intense pulsed light systems. The choice of system depends on the patient’s skin type and hair color. Women with fair skin and dark hair are ideal candidates; longer-wavelength lasers are preferred for darker or tanned skin types.
Adverse effects of laser hair removal include pain, erythema, burns, dyspigmentation, and scarring. Laser cooling devices can prevent or minimize some of these effects. Laser treatment has also been known to cause a paradoxical increase in hair growth.1,33,34
DRUG THERAPIES FOR HIRSUTISM
The drugs most commonly used for hirsutism are oral contraceptives (off-label use) and antiandrogenic drugs (off-label use). Topical eflornithine cream (Vaniqa) is FDA-approved for hirsutism but is less commonly used. Insulin sensitizers, GnRH analogues, and other drugs are occasionally used (off-label) to treat hirsutism.
Topical eflornithine cream
Topical eflornithine cream treats facial hirsutism by slowing the rate of hair growth; it does this by irreversibly inhibiting ornithine decarboxylase, an enzyme essential for hair growth.39,40 Studies showed that twice-daily application reduced unwanted facial hair in women after 24 weeks of treatment.39,40 Treatment must be continuous, since hair growth rapidly returns to the pretreatment rate by 8 weeks after discontinuing eflornithine.39,40 White women have been shown to respond better than black women.39 Adverse effects include a mild burning sensation, acne, pseudofolliculitis barbae, irritation, and allergic contact dermatitis.39,40 Improved outcomes have been suggested when eflornithine cream is combined with laser hair removal.41
Oral contraceptives
Oral contraceptives are commonly used off-label for the management of hirsutism.20 Oral contraceptives suppress the secretion of luteinizing hormone and, hence, the synthesis of ovarian androgen, thereby increasing levels of sex hormone-binding globulin and decreasing free plasma testosterone.1,20 Adrenal androgen production is also slightly reduced.20
Oral contraceptives usually combine a synthetic estrogen and a progestin. Certain progestins are more androgenic and should be avoided.1
For treating hirsutism, oral contraceptives should be used that contain low-androgenic progestins such as cyproterone acetate (not available in the United States), drosperinone (eg, in Yasmin), norgestimate (eg, in Ortho Tri-Cyclen), or desogestrel (eg, in Mircette).1,20
Side effects of oral contraceptives include breast tenderness, gastrointestinal upset, headache, loss of libido, hypertension, and the potential risk of venous thromboembolism.1,15,32,36
Antiandrogenic drugs
Several antiandrogenic drugs are used off-label to treat hirsutism.
Spironolactone (Aldactone), a competitive inhibitor of the androgen receptor and 5-alpha reductase activity,20 can be effective in the treatment of hirsutism. Monotherapy with spironolactone, without an oral contraceptive or other reliable form of contraception, is not recommended because of the teratogenic potential of all antiandrogens to feminize a developing male fetus.20 Thus, reliable contraception should be used in females of childbearing age when starting antiandrogen therapy.
The dosage of spironolactone for hirsutism is usually 100 mg to 200 mg daily.1,20 Hyperkalemia, polyuria, postural hypotension, irregular menses, and liver abnormalities are among the possible adverse effects (Table 3). Spironolactone was found to be tumorigenic in animal studies, although this has unknown relevance in humans.36
Cyproterone, an antiandrogen not available in the United States,42 competitively inhibits the androgen receptor and 5-alpha-reductase activity.1,20,36 It can be used for only the first 10 days of the menstrual cycle (50-mg or 100-mg dose) with an oral contraceptive pill, or in a low dose in a combined oral contraceptive pill (Diane-35 in Canada and the United Kingdom).1
Side effects are similar to those of oral contraceptives and include fatigue, mood change, risk of venous thromboembolism, and decreased libido.1,15,36 Importantly, in woman of childbearing age, there is the potential risk of feminization of a male fetus, so reliable contraception must be used.15,36
Flutamide, an investigational antiandrogen, has shown promise in the treatment of hirsutism.20 Flutamide is a nonsteroidal competitive inhibitor of androgen receptor binding. It carries a significant risk of hepatotoxicity. 1,15
Finasteride (Propecia) 1 mg is only occasionally used in the treatment of hirsutism (off-label usage). It inhibits type II 5-alphareductase to suppress dihydrotestosterone levels. 32 It carries a risk of gastrointestinal disturbance, decreased libido, hepatotoxicity, and feminization of a male fetus (pregnancy category X), so reliable contraception is required in all females of childbearing age, as with all antiandrogens1 (Table 3).
Dutasteride (Avodart), a type I and II 5-alpha-reductase inhibitor, has not been studied for the treatment of hirsutism (pregnancy category X).
Insulin sensitizers
Metformin (Glucophage) and other insulin sensitizers are less effective than antiandrogens at reducing hirsutism.20,38 However, metformin is effective at inducing ovulation in patients with polycystic ovary syndrome.38 Gastrointestinal upset is a common side effect; lactic acidosis is a serious but rare adverse effect.1
Gonadotropin-releasing hormone analogues
GnRH analogues are an option only if oral contraceptives and antiandrogen drugs are unsuccessful in patients with severe hyperandrogenism. 20 They suppress secretion of luteinizing hormone and the synthesis of ovarian androgen.1,20 These drugs are given as monthly intramuscular injections, usually with some form of estrogen-progestin replacement, since GnRH analogues cause estrogen levels to fall to menopausal levels.1
Side effects include signs and symptoms of menopause including hot flushes, atrophic vaginitis, and osteoporosis.1,15 These drugs completely inhibit ovulation, and some endocrinologists and gynecologists do not suggest further contraception in women of childbearing years for this reason. However, GnRH analogues are not approved as a contraceptive and are pregnancy category X.
Other drugs
Other drugs with antiandrogen activity include cimetidine and ketoconazole.12 Cimetidine (Tagamet) is not effective for the treatment of hirsutism, and ketoconazole (Nizoral) is associated with significant risk for adrenocortical suppression12 and hepatotoxicity in addition to multiple drug interactions, given its effect on the hepatic P450 enzyme system.
Acknowledgment: Many thanks to Rebecca Tung, MD, dermatologic surgeon, Cleveland Clinic, for her advice on lasers.
Hirsutism causes significant anxiety and lack of self-esteem in women. Although it is itself a benign condition, it is often the sign of an underlying and possibly serious endocrine condition.
As we will discuss, the diagnosis begins with a detailed history and physical examination, with laboratory testing and imaging as needed to confirm or rule out underlying causes. Management begins with patient education and support and includes hair removal and drug treatment of any underlying metabolic derangement.
PREVALENCE AND IMPACT
Hirsutism is a common disorder of excess growth of terminal hair in an androgen-dependent male distribution in women, including the chin, upper lip, breasts, upper back, and abdomen.1 It affects 5% to 10% of women of reproductive age.1,2
Hirsutism should be differentiated from hypertrichosis, which can be hereditary or acquired, and which is defined as increased general hair growth in androgen-independent areas.1
Excess hair is cosmetically concerning for women and can significantly affect self-esteem. 3 Normal or acceptable hair growth depends on a woman’s ethnicity and her perception of familial, cultural, and societal norms for the quantity and distribution of hair. Mediterranean women generally have a medium amount of body and facial hair, whereas Asian women have a minimal amount.1,4,5
Hirsutism can be clinically graded according to the Ferriman-Gallwey scale2,6 and is defined as a Ferriman-Gallwey score of 8 or higher.1
HOW CIRCULATING ANDROGENS AFFECT HAIR FOLLICLES
In androgen-dependent areas, circulating androgens influence hair follicle characteristics. Androgens increase the size and diameter of the hair fibers in certain androgen-dependent sites, as seen in puberty with the transformation of vellus hairs (small, nonpigmented hairs) into terminal hairs (large, pigmented hairs) in the pubic and axillary regions in women, as well as the beard area in men.2,7 Interestingly, the same circulating androgens cause miniaturization of the susceptible hair follicles of the central scalp.7 The susceptibility of the hair follicle to the effects of the androgens may be genetically determined.7,8
Hirsutism is a sign of hyperandrogenism and increased action of androgens on hair follicles. In women, about half of circulating testosterone arises from the ovaries and adrenal glands; the rest originates from peripheral conversion of weaker androgens (such as androstenedione produced by the adrenals and ovaries) into testosterone.9 Dehydroepiandrosterone sulfate (DHEAS) originates mainly in the adrenal glands.9,10 Testosterone is converted to the more potent dihydrotestosterone (DHT) by type II 5-alpha reductase in the skin, which can then act on susceptible hair follicles.7,11 Therefore, hirsutism can be a consequence of endogenous androgen over-production from the ovaries or the adrenal glands (or both), of exposure to an exogenous source of androgen such as a drug, or of heightened hair follicle sensitivity and metabolism of normal circulating androgen levels (target end-organ dysfunction).1
‘IDIOPATHIC’ HIRSUTISM: A MISLEADING DIAGNOSIS
Many women with hirsutism are found to have polycystic ovary syndrome as the underlying cause, but hirsutism is also commonly labeled as idiopathic when it occurs without an obvious cause, eg, in women with regular menses and normal androgen levels and without features suspicious for other causes of hirsutism. 1,2,12,13 But while this term is commonly used,1,12 it may be misleading, especially if the diagnosis of idiopathic hirsutism is based on standard laboratory tests, which do not always detect androgen excess.2,13 Minor ovarian or adrenal functional hyperandrogenism,14 increased peripheral activity of 5-alpha reductase in the hair follicle, or abnormalities in the androgen receptor have been implicated in the pathogenesis of so-called idiopathic hirsutism. 2,15
HIRSUTISM AND POLYCYSTIC OVARY SYNDROME
Polycystic ovary syndrome, a metabolic syndrome, presents clinically with menstrual irregularities such as oligomenorrhea or amenorrhea, infertility, and signs of hyperandrogenism such as hirsutism, acne, or androgenetic alopecia.16,17 Metabolic disturbances including insulin resistance, impaired glucose tolerance, hyperlipidemia, and obesity (body mass index > 30 kg/m2) also can occur, thus increasing cardiovascular risk.16–18
The finding of polycystic ovaries is not required to make the diagnosis of polycystic ovary syndrome, and their presence does not prove the diagnosis.16,19 Gonadotropin-dependent functional ovarian hyperandrogenism is believed to cause this syndrome; however, mild adrenocorticotropic-dependent functional adrenal hyperandrogenism also is a feature in many cases. In rare cases, polycystic ovary syndrome presents with an isolated elevation of DHEAS.16,20
OTHER CONDITIONS OF EXCESS ANDROGEN
The syndrome of hyperandrogenism, insulin resistance, and acanthosis nigricans, abbreviated as HAIR-AN, is separate from polycystic ovary syndrome; it characterizes a group of inherited syndromes associated with severe metabolic abnormalities of insulin and glucose metabolism and with marked clinical signs of hyperandrogenism.12
The syndrome of seborrhea, acne, hirsutism, and acanthosis nigricans, abbreviated as SAHA, while not itself a diagnosis, is a clinical spectrum of dermatologic signs and symptoms also associated with hyperandrogenism. These are signs that may present with the HAIR-AN syndrome or with another cause of excess androgens, such as idiopathic, ovarian, adrenal, or hyperprolactinemic hyperandrogenism.21
Thyroid disease, hyperprolactinemia, acromegaly, Cushing syndrome, exogenous factors such as androgenic drugs, and nonclassical congenital adrenal hyperplasia can also produce hirsutism.12 In nonclassical congenital adrenal hyperplasia, which is typically caused by a deficiency of 21-hydroxylase, patients present with premature pubarche, hirsutism in the prepubertal years, and menstrual irregularities including primary amenorrhea.22,23
Important rare causes of hirsutism include benign and malignant androgen-secreting tumors of adrenal or ovarian origin. In such cases, hirsutism can have an acute onset or rapid progression and may be associated with features of virilization, such as deepened voice, increased muscle mass, androgenetic alopecia, clitoromegaly, and increased libido.12
A THOROUGH HISTORY IS CRITICAL TO DIAGNOSIS
A thorough medical history can provide important diagnostic clues in women with hirsutism. The clinician should elicit details about the onset and progression of the hair growth,12,15 previous treatments, and any cutaneous signs of hyperandrogenism, such as acne, seborrhea, acanthosis nigricans, or patterned hair loss.
Also important are the menstrual history and a history of infertility. Primary amenorrhea is defined as failure to menstruate by 16 years of age if secondary sexual characteristics have developed, or by 14 years of age if no secondary sexual characteristics have developed, and it can indicate nonclassical congenital adrenal hyperplasia.
The clinician should also try to determine if the patient has a history of galactorrhea or symptoms of virilization (eg, deepened voice, clitoromegaly, increased muscle mass); a family history of hirsutism, polycystic ovary syndrome, HAIR-AN syndrome, metabolic conditions such as type 2 diabetes mellitus, or cardiovascular disease12,15; or a history of symptoms of any condition known to produce hirsutism, such as Cushing disease, acromegaly, or a thyroid disorder. Also important is a drug history to determine if the patient has taken drugs such as androgens, anabolic steroids, or valproic acid (Depakote).20
THE PHYSICAL EXAMINATION
Another proposed predictor of hirsutism is that terminal hair on the chin or the lower abdomen (Ferriman-Gallwey score ≥ 2) is nearly 100% sensitive and 27% specific at predicting total-body hirsutism.24
As part of the physical examination, the clinician should also look for other cutaneous signs of hyperandrogenism, such as acne, androgenetic alopecia, and seborrhea. Acanthosis nigricans is a sign of insulin resistance. Height and weight should be measured and the body mass index calculated. Blood pressure should be recorded, as high blood pressure may be seen in Cushing syndrome and is an important cardiovascular risk factor. Signs of virilization should be identified. Indicators of Cushing disease such as striae, moon facies, fat redistribution, fragile skin, and proximal myopathy should be noted as well as signs of thyroid disease, such as textural skin changes, goiter, and hair loss. Expressible or spontaneous galactorrhea suggests hyperprolactinemia. Acromegaly is associated with coarse facies and enlarged hands and feet. Many of the endocrinopathies can be caused by a pituitary adenoma, which can manifest as a visual field defect, so visual fields should be examined.25 The examination should also exclude any palpable ovarian or adrenal mass.12
WHEN IS ADDITIONAL TESTING NEEDED?
The current Endocrine Society guidelines20 recommend obtaining an early-morning testosterone blood level in the following patients:
- Women with moderate or severe hirsutism
- Women with hirsutism of any degree with sudden onset or rapid progression, or accompanied by signs or symptoms suggesting malignancy or polycystic ovary syndrome: eg, menstrual irregularity, infertility, central obesity, clitoromegaly, or acanthosis nigricans.15,20
Testing androgen levels in mild, isolated hirsutism has not been proven to be useful or to alter management.20
Free testosterone level
An early-morning total or free testosterone level is the initial test in the laboratory evaluation of hirsutism.12,15 Additional specialized laboratory testing may be needed to determine the free testosterone level,15 as the free testosterone test is not available at all laboratories. A normal total testosterone level does not exclude hyperandrogenism but can suggest the diagnosis of idiopathic hirsutism.15
Further testing is needed if the total testosterone level is normal or only slightly elevated, or if there is a strong clinical suspicion of an underlying condition such as endocrinopathy or tumor. It is also useful in patients whose hirsutism responds poorly to medical treatments15 (see discussion below).
If the total testosterone level is elevated, if the hirsutism is moderate to severe, if there are associated symptoms, or if hirsutism is acute or progressive, a further endocrinologic workup is needed,15 possibly including measurement of free testosterone, sex hormone-binding globulin, DHEAS, and androstenedione.15 Free testosterone, unbound to sex hormone-binding globulin, is the biologically active fraction, with the levels of binding globulin increased by drugs such as oral contraceptives15 and decreased by high insulin levels in insulin resistance.25
Test in patients with mild hirsutism?
Although the guidelines suggest that no additional workup is necessary for women with mild hirsutism, we evaluate all patients with hirsutism and those with the SAHA clinical spectrum by measuring free and total testosterone and DHEAS. In our experience, even women with mild hirsutism with subtle symptoms and signs of hyperandrogenism and mild hirsutism often have elevated androgen levels.
Test in women with idiopathic hirsutism?
In women with idiopathic hirsutism, minor forms of functional ovarian and adrenal hyperandrogenism are believed to play a role and are thought to be undetectable with conventional testing.25 The gonadotropin-releasing hormone (GnRH) analogue stimulation test may uncover occult hyperandrogenism in this setting, but it is used as a research tool and does not currently have application in routine clinical practice.14
It is important to remember that some women with apparent idiopathic hirsutism and a history of regular menstrual cycles are actually oligo-ovulatory or anovulatory. In these instances, another diagnosis should be considered,13 and referral to an endocrinologist for further evaluation of ovulatory function is recommended.13
CURRENT USE OF DIAGNOSTIC IMAGING
When malignancy is suspected
A testosterone level above 200 ng/dL suggests an ovarian tumor, and a DHEAS level above 700 μg/dL suggests an adrenal tumor.26 However, not all tumors present with such high androgen levels, and sudden onset of hirsutism, rapid progression of hirsutism, or signs of virilization suggest a tumor.15 In such cases, transvaginal ultrasonography, computed tomography, or magnetic resonance imaging (MRI) of the abdomen can exclude an ovarian or adrenal tumor.
When polycystic ovary syndrome is suspected
The diagnosis of polycystic ovary syndrome is confirmed by two out of three criteria:
- Oligo-ovulation or anovulation
- Clinical or laboratory signs of hyperandrogenism
- Ultrasonographic evidence of polycystic ovaries, with exclusion of other causes of hyperandrogenism.
ADDITIONAL LABORATORY TESTING
Tests for polycystic ovary syndrome
Assessment of polycystic ovary syndrome involves transvaginal ultrasonography, but ultrasonographic evidence of a polycystic ovary is not necessary for the diagnosis.16 A fasting lipid profile and fasting serum glucose are recommended, and if the fasting serum glucose is normal, an oral glucose tolerance test is recommended. 17
Some have reported measuring the ratio of luteinizing hormone to follicle-stimulating hormone in the workup of polycystic ovary syndrome, and a ratio greater than 2 has been considered indicative but not diagnostic.16,25 The individual levels of luteinizing hormone, follicle-stimulating hormone, and estradiol are more important in the evaluation of infertility and ovulatory dysfunction. In patients with elevations of these hormones or with these symptoms, referral for infertility screening with an endocrinologist or gynecologist is recommended. 25
Additional testing and referral for Cushing syndrome, other conditions
Cushing syndrome can be tested for with a 24-hour urine cortisol, overnight low-dose dexamethasone suppression test, and late-night salivary cortisol.27,28 Referral to an endocrinologist for further testing can differentiate between corticotropin-dependent or corticotropin-independent Cushing syndrome.25 Cushing syndrome is often associated with hyperandrogenism, particularly in those cases caused by adrenal tumors.29
The prolactin level and the level of somatomedin C (insulin-like growth factor 1) can be used to rule out hyperprolactinemia and acromegaly, respectively.12 If Cushing syndrome, hyperprolactinemia, or acromegaly is diagnosed by endocrinologic testing, pituitary MRI should be performed.12,25
Referral to specialist centers with experience with these conditions is essential. Nonclassical congenital adrenal hyperplasia can be screened for by a serum 17-hydroxyprogesterone level measured in the follicular phase.12 Measurement of thyroid-stimulating hormone, free thyroxine, and thyroid peroxidase antibodies screens for thyroid disease.12 Hirsutism has been reported with the commencement of L-thyroxine therapy.30
THE PRINCIPLES OF TREATMENT
Patient education regarding the cause of hirsutism and reasonable treatment expectations and emotional support are important in the management of hirsutism. Also important is regular follow-up to measure and document the response to treatment; this can include repeating Ferriman-Gallwey scoring, taking photographs of affected areas, and retesting androgen levels after 3 to 6 months.12
Treatment must be continued for an ongoing effect, and most pharmacologic treatments can take up to 3 to 6 months to produce significant improvement.1
When an underlying condition is diagnosed, treatment of the condition is essential. Androgen-secreting tumors require surgical management.12 Cushing disease, hyperprolactinemia, and acromegaly should be clinically apparent from examination and testing, and appropriate referral and standard management should be instigated. Exogenous sources of androgen such as androgenic progestins or anabolic steroids should be discontinued. Lifestyle management is important, and weight loss in obese patients with polycystic ovary syndrome can improve hirsutism as well as mitigate cardiovascular risk factors.31
In classic congenital adrenal hyperplasia, glucocorticoid therapy manages both ovulation induction and hirsutism.20 However, in nonclassical congenital adrenal hyperplasia, glucocorticoid therapy supports ovulation induction, but hirsutism usually requires both systemic antiandrogen and hair removal.20
CURRENT OPTIONS FOR HAIR REMOVAL
The choice of method depends on patient preference, adverse effects, the degree of hirsutism, the level of distress, previous treatments, and cost.1,15,32
Self-care methods
Self-care methods offer only temporary reduction of excess hairs.
Plucking removes the entire hair, including the root, but it is painful and time-consuming, and it is only practical for areas where few hairs exist, such as on the face.1
Shaving is an easy, inexpensive, and painless choice for hair removal. Although a common belief is that shaving causes faster or thicker hair regrowth, shaving affects neither the diameter nor the rate of growth of the hair.32 Given its masculine association, shaving is not acceptable to most women except perhaps for use on the legs and axillae.1,32 Shaving can cause irritation, folliculitis, pseudofolliculitis, and infection.1
Waxing removes the entire hair. While it is more expensive than plucking, regrowth is slower, occurring over weeks. It is painful and can cause thermal burns, irritation, folliculitis, scarring, and postinflammatory dyspigmentation.1
Chemical depilatories, usually thioglycollic acid preparations, are inexpensive, painless, and easy to use. However, the resulting hair reduction is of short duration because the hair shafts are only removed at the level of the skin surface.1 They can also cause irritant dermatitis. 1
Bleaching with hydrogen peroxide is inexpensive and can camouflage dark facial hair, but it can also cause skin discoloration and irritation. 1
Clinic-based methods
Electrolysis often results in a permanent reduction in hair growth.1,32 A fine needle is placed into the hair follicle and an electrical current is applied. Each follicle is treated individually. 1,32 Best results are seen on darker hairs in patients with lighter skin, but it can be used on all skin types and hair colors.1,32
Electrolysis is operator-dependent, and there are US Food and Drug Administration (FDA) regulations regarding electrolysis techniques. It requires multiple treatments, and it is painful and can cause erythema, folliculitis, pseudofolliculitis, infection, scarring, and postinflammatory dyspigmentation.1,32 Some reports suggest that prior waxing and plucking of hairs damages the hair by twisting the hair shaft, making electrolysis more difficult.32
Laser treatment uses light of certain wavelengths to damage the hair follicles. While laser hair removal does not result in complete or persistent hair removal, it is more effective than shaving, waxing, and electrolysis, producing partial hair reduction for up to 6 months; the effect is enhanced with multiple treatments.33,34 The number of treatments required depends on the laser type and on the nature of the patient’s hair follicles.35
Laser systems for hair removal are of various wavelengths and also include intense pulsed light systems. The choice of system depends on the patient’s skin type and hair color. Women with fair skin and dark hair are ideal candidates; longer-wavelength lasers are preferred for darker or tanned skin types.
Adverse effects of laser hair removal include pain, erythema, burns, dyspigmentation, and scarring. Laser cooling devices can prevent or minimize some of these effects. Laser treatment has also been known to cause a paradoxical increase in hair growth.1,33,34
DRUG THERAPIES FOR HIRSUTISM
The drugs most commonly used for hirsutism are oral contraceptives (off-label use) and antiandrogenic drugs (off-label use). Topical eflornithine cream (Vaniqa) is FDA-approved for hirsutism but is less commonly used. Insulin sensitizers, GnRH analogues, and other drugs are occasionally used (off-label) to treat hirsutism.
Topical eflornithine cream
Topical eflornithine cream treats facial hirsutism by slowing the rate of hair growth; it does this by irreversibly inhibiting ornithine decarboxylase, an enzyme essential for hair growth.39,40 Studies showed that twice-daily application reduced unwanted facial hair in women after 24 weeks of treatment.39,40 Treatment must be continuous, since hair growth rapidly returns to the pretreatment rate by 8 weeks after discontinuing eflornithine.39,40 White women have been shown to respond better than black women.39 Adverse effects include a mild burning sensation, acne, pseudofolliculitis barbae, irritation, and allergic contact dermatitis.39,40 Improved outcomes have been suggested when eflornithine cream is combined with laser hair removal.41
Oral contraceptives
Oral contraceptives are commonly used off-label for the management of hirsutism.20 Oral contraceptives suppress the secretion of luteinizing hormone and, hence, the synthesis of ovarian androgen, thereby increasing levels of sex hormone-binding globulin and decreasing free plasma testosterone.1,20 Adrenal androgen production is also slightly reduced.20
Oral contraceptives usually combine a synthetic estrogen and a progestin. Certain progestins are more androgenic and should be avoided.1
For treating hirsutism, oral contraceptives should be used that contain low-androgenic progestins such as cyproterone acetate (not available in the United States), drosperinone (eg, in Yasmin), norgestimate (eg, in Ortho Tri-Cyclen), or desogestrel (eg, in Mircette).1,20
Side effects of oral contraceptives include breast tenderness, gastrointestinal upset, headache, loss of libido, hypertension, and the potential risk of venous thromboembolism.1,15,32,36
Antiandrogenic drugs
Several antiandrogenic drugs are used off-label to treat hirsutism.
Spironolactone (Aldactone), a competitive inhibitor of the androgen receptor and 5-alpha reductase activity,20 can be effective in the treatment of hirsutism. Monotherapy with spironolactone, without an oral contraceptive or other reliable form of contraception, is not recommended because of the teratogenic potential of all antiandrogens to feminize a developing male fetus.20 Thus, reliable contraception should be used in females of childbearing age when starting antiandrogen therapy.
The dosage of spironolactone for hirsutism is usually 100 mg to 200 mg daily.1,20 Hyperkalemia, polyuria, postural hypotension, irregular menses, and liver abnormalities are among the possible adverse effects (Table 3). Spironolactone was found to be tumorigenic in animal studies, although this has unknown relevance in humans.36
Cyproterone, an antiandrogen not available in the United States,42 competitively inhibits the androgen receptor and 5-alpha-reductase activity.1,20,36 It can be used for only the first 10 days of the menstrual cycle (50-mg or 100-mg dose) with an oral contraceptive pill, or in a low dose in a combined oral contraceptive pill (Diane-35 in Canada and the United Kingdom).1
Side effects are similar to those of oral contraceptives and include fatigue, mood change, risk of venous thromboembolism, and decreased libido.1,15,36 Importantly, in woman of childbearing age, there is the potential risk of feminization of a male fetus, so reliable contraception must be used.15,36
Flutamide, an investigational antiandrogen, has shown promise in the treatment of hirsutism.20 Flutamide is a nonsteroidal competitive inhibitor of androgen receptor binding. It carries a significant risk of hepatotoxicity. 1,15
Finasteride (Propecia) 1 mg is only occasionally used in the treatment of hirsutism (off-label usage). It inhibits type II 5-alphareductase to suppress dihydrotestosterone levels. 32 It carries a risk of gastrointestinal disturbance, decreased libido, hepatotoxicity, and feminization of a male fetus (pregnancy category X), so reliable contraception is required in all females of childbearing age, as with all antiandrogens1 (Table 3).
Dutasteride (Avodart), a type I and II 5-alpha-reductase inhibitor, has not been studied for the treatment of hirsutism (pregnancy category X).
Insulin sensitizers
Metformin (Glucophage) and other insulin sensitizers are less effective than antiandrogens at reducing hirsutism.20,38 However, metformin is effective at inducing ovulation in patients with polycystic ovary syndrome.38 Gastrointestinal upset is a common side effect; lactic acidosis is a serious but rare adverse effect.1
Gonadotropin-releasing hormone analogues
GnRH analogues are an option only if oral contraceptives and antiandrogen drugs are unsuccessful in patients with severe hyperandrogenism. 20 They suppress secretion of luteinizing hormone and the synthesis of ovarian androgen.1,20 These drugs are given as monthly intramuscular injections, usually with some form of estrogen-progestin replacement, since GnRH analogues cause estrogen levels to fall to menopausal levels.1
Side effects include signs and symptoms of menopause including hot flushes, atrophic vaginitis, and osteoporosis.1,15 These drugs completely inhibit ovulation, and some endocrinologists and gynecologists do not suggest further contraception in women of childbearing years for this reason. However, GnRH analogues are not approved as a contraceptive and are pregnancy category X.
Other drugs
Other drugs with antiandrogen activity include cimetidine and ketoconazole.12 Cimetidine (Tagamet) is not effective for the treatment of hirsutism, and ketoconazole (Nizoral) is associated with significant risk for adrenocortical suppression12 and hepatotoxicity in addition to multiple drug interactions, given its effect on the hepatic P450 enzyme system.
Acknowledgment: Many thanks to Rebecca Tung, MD, dermatologic surgeon, Cleveland Clinic, for her advice on lasers.
- Mofid A, Seyyed Alinaghi SA, Zandieh S, Yazdani T. Hirsutism. Int J Clin Pract 2008; 62:433–443.
- Azziz R, Carmina E, Sawaya ME. Idiopathic hirsutism. Endocr Rev 2000; 21:347–362.
- Himelein MJ, Thatcher SS. Polycystic ovary syndrome and mental health: a review. Obstet Gynecol Surv 2006; 61:723–732.
- Williamson K, Gunn AJ, Johnson N, Milsom SR. The impact of ethnicity on the presentation of polycystic ovarian syndrome. Aust N Z J Obstet Gynaecol 2001; 41:202–206.
- Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999; 84:4006–4011.
- Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 1961; 21:1440–1447.
- Messenger AG. The control of hair growth: an overview. J Invest Dermatol 1993; 101(suppl 1):4S–9S.
- Rosenfield RL. Hirsutism and the variable response of the pilosebaceous unit to androgen. J Investig Dermatol Symp Proc 2005; 10:205–208.
- Longcope C. Adrenal and gonadal androgen secretion in normal females. Clin Endocrinol Metab 1986; 15:213–228.
- Braunstein GD. Testis. In:Gardner DG, Shoback D, editors. Greenspan’s Basic & Clinical Endocrinology. 8th ed. New York: McGraw-Hill, 2007.
- Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev 2000; 21:363–392.
- Practice Committee of the American Society for Reproductive Medicine. The evaluation and treatment of androgen excess. Fertil Steril 2006; 86(suppl 5):S241–S247.
- Azziz R, Waggoner WT, Ochoa T, Knochenhauer ES, Boots LR. Idiopathic hirsutism: an uncommon cause of hirsutism in Alabama. Fertil Steril 1998; 70:274–278.
- Rossi R, Tauchmanovà L, Luciano A, et al. Functional hyperandrogenism detected by corticotropin and GnRH-analogue stimulation tests in women affected by apparently idiopathic hirsutism. J Endocrinol Invest 2001; 24:491–498.
- Rosenfield RL. Clinical practice. Hirsutism. N Engl J Med 2005; 353:2578–2588.
- Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 2004; 19:41–47.
- Salley KE, Wickham EP, Cheang KI, Essah PA, Karjane NW, Nestler JE. Glucose intolerance in polycystic ovary syndrome—a position statement of the Androgen Excess Society. J Clin Endocrinol Metab 2007; 92:4546–4556.
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365:1415–1428.
- Azziz R. Diagnostic criteria for polycystic ovary syndrome: a reappraisal. Fertil Steril 2005; 83:1343–1346.
- Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. http://www.endo-society.org/guidelines/final/upload/Hirsutism_Guideline.pdf. Accessed March 30, 2010.
- Orfanos CE, Adler YD, Zouboulis CC. The SAHA syndrome. Horm Res 2000; 54:251–258.
- New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab 2006; 91:4205–4214.
- Kohn B, Levine LS, Pollack MS, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 1982; 55:817–827.
- Knochenhauer ES, Hines G, Conway-Myers BA, Azziz R. Examination of the chin or lower abdomen only for the prediction of hirsutism. Fertil Steril 2000; 74:980–983.
- Somani N, Harrison S, Bergfeld WF. The clinical evaluation of hirsutism. Dermatol Ther 2008; 21:376–391.
- Waggoner W, Boots LR, Azziz R. Total testosterone and DHEAS levels as predictors of androgen-secreting neoplasms: a populational study. Gynecol Endocrinol 1999; 13:394–400.
- Crapo L. Cushing’s syndrome: a review of diagnostic tests. Metabolism 1979; 28:955–977.
- Blethen SL, Chasalow FI. Overnight dexamethasone suppression test: normal responses and the diagnosis of Cushing’s syndrome. Steroids 1989; 54:185–193.
- Bertagna C, Orth DN. Clinical and laboratory findings and results of therapy in 58 patients with adrenocortical tumors admitted to a single medical center (1951 to 1978). Am J Med 1981; 71:855–875.
- Kologlu S, Baskal N, Kologlu LB, Laleli Y, Tuccar E. Hirsutism due to the treatment with L-thyroxine in patients with thyroid pathology. Endocrinologie 1988; 26:179–185.
- Gambineri A, Patton L, Vaccina A, et al. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. J Clin Endocrinol Metab 2006; 91:3970–3980.
- Dawber RP. Guidance for the management of hirsutism. Curr Med Res Opin 2005; 21:1227–1234.
- Haedersdal M, Wulf HC. Evidence based review of hair removal using lasers and light sources. J Eur Acad Dermatol Venereol 2006; 20:9–20.
- Sadighha A, Mohaghegh Zahed G. Meta-analysis of hair removal laser trials. Lasers Med Sci 2009; 24:21–25.
- Casey AS, Goldberg D. Guidelines for laser hair removal. J Cosmet Laser Ther 2008; 10:24–33.
- Wakelin SH, Maibach HI, editors. Handbook of Systemic Drug Treatment in Dermatology. London: Manson Publishing Ltd, 2004.
- Swiglo BA, Cosma M, Flynn DN, et al. Clinical review: antiandrogens for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab 2008; 93:1153–1160.
- Cosma M, Swiglo BA, Flynn DN, et al. Clinical review: insulin sensitizers for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab 2008; 93:1135–1142.
- Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol 2001; 2:197–201.
- Wolf JE, Shander D, Huber F, et al; Eflornithine HCl Study Group. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair. Int J Dematol 2007; 46:94–98.
- Hamzavi I, Tan E, Shapiro J, Lui H. A randomized bilateral vehicle-controlled study of eflornithine cream combined with laser treatment versus laser treatment alone for facial hirsutism in women. J Am Acad Dermatol 2007; 57:54–59.
- Van der Spuy ZM, le Roux PA. Cyproterone acetate for hirsutism. Cochrane Database Syst Rev 2003; 4:CD001125.
- Mofid A, Seyyed Alinaghi SA, Zandieh S, Yazdani T. Hirsutism. Int J Clin Pract 2008; 62:433–443.
- Azziz R, Carmina E, Sawaya ME. Idiopathic hirsutism. Endocr Rev 2000; 21:347–362.
- Himelein MJ, Thatcher SS. Polycystic ovary syndrome and mental health: a review. Obstet Gynecol Surv 2006; 61:723–732.
- Williamson K, Gunn AJ, Johnson N, Milsom SR. The impact of ethnicity on the presentation of polycystic ovarian syndrome. Aust N Z J Obstet Gynaecol 2001; 41:202–206.
- Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999; 84:4006–4011.
- Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 1961; 21:1440–1447.
- Messenger AG. The control of hair growth: an overview. J Invest Dermatol 1993; 101(suppl 1):4S–9S.
- Rosenfield RL. Hirsutism and the variable response of the pilosebaceous unit to androgen. J Investig Dermatol Symp Proc 2005; 10:205–208.
- Longcope C. Adrenal and gonadal androgen secretion in normal females. Clin Endocrinol Metab 1986; 15:213–228.
- Braunstein GD. Testis. In:Gardner DG, Shoback D, editors. Greenspan’s Basic & Clinical Endocrinology. 8th ed. New York: McGraw-Hill, 2007.
- Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev 2000; 21:363–392.
- Practice Committee of the American Society for Reproductive Medicine. The evaluation and treatment of androgen excess. Fertil Steril 2006; 86(suppl 5):S241–S247.
- Azziz R, Waggoner WT, Ochoa T, Knochenhauer ES, Boots LR. Idiopathic hirsutism: an uncommon cause of hirsutism in Alabama. Fertil Steril 1998; 70:274–278.
- Rossi R, Tauchmanovà L, Luciano A, et al. Functional hyperandrogenism detected by corticotropin and GnRH-analogue stimulation tests in women affected by apparently idiopathic hirsutism. J Endocrinol Invest 2001; 24:491–498.
- Rosenfield RL. Clinical practice. Hirsutism. N Engl J Med 2005; 353:2578–2588.
- Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 2004; 19:41–47.
- Salley KE, Wickham EP, Cheang KI, Essah PA, Karjane NW, Nestler JE. Glucose intolerance in polycystic ovary syndrome—a position statement of the Androgen Excess Society. J Clin Endocrinol Metab 2007; 92:4546–4556.
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365:1415–1428.
- Azziz R. Diagnostic criteria for polycystic ovary syndrome: a reappraisal. Fertil Steril 2005; 83:1343–1346.
- Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. http://www.endo-society.org/guidelines/final/upload/Hirsutism_Guideline.pdf. Accessed March 30, 2010.
- Orfanos CE, Adler YD, Zouboulis CC. The SAHA syndrome. Horm Res 2000; 54:251–258.
- New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab 2006; 91:4205–4214.
- Kohn B, Levine LS, Pollack MS, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab 1982; 55:817–827.
- Knochenhauer ES, Hines G, Conway-Myers BA, Azziz R. Examination of the chin or lower abdomen only for the prediction of hirsutism. Fertil Steril 2000; 74:980–983.
- Somani N, Harrison S, Bergfeld WF. The clinical evaluation of hirsutism. Dermatol Ther 2008; 21:376–391.
- Waggoner W, Boots LR, Azziz R. Total testosterone and DHEAS levels as predictors of androgen-secreting neoplasms: a populational study. Gynecol Endocrinol 1999; 13:394–400.
- Crapo L. Cushing’s syndrome: a review of diagnostic tests. Metabolism 1979; 28:955–977.
- Blethen SL, Chasalow FI. Overnight dexamethasone suppression test: normal responses and the diagnosis of Cushing’s syndrome. Steroids 1989; 54:185–193.
- Bertagna C, Orth DN. Clinical and laboratory findings and results of therapy in 58 patients with adrenocortical tumors admitted to a single medical center (1951 to 1978). Am J Med 1981; 71:855–875.
- Kologlu S, Baskal N, Kologlu LB, Laleli Y, Tuccar E. Hirsutism due to the treatment with L-thyroxine in patients with thyroid pathology. Endocrinologie 1988; 26:179–185.
- Gambineri A, Patton L, Vaccina A, et al. Treatment with flutamide, metformin, and their combination added to a hypocaloric diet in overweight-obese women with polycystic ovary syndrome: a randomized, 12-month, placebo-controlled study. J Clin Endocrinol Metab 2006; 91:3970–3980.
- Dawber RP. Guidance for the management of hirsutism. Curr Med Res Opin 2005; 21:1227–1234.
- Haedersdal M, Wulf HC. Evidence based review of hair removal using lasers and light sources. J Eur Acad Dermatol Venereol 2006; 20:9–20.
- Sadighha A, Mohaghegh Zahed G. Meta-analysis of hair removal laser trials. Lasers Med Sci 2009; 24:21–25.
- Casey AS, Goldberg D. Guidelines for laser hair removal. J Cosmet Laser Ther 2008; 10:24–33.
- Wakelin SH, Maibach HI, editors. Handbook of Systemic Drug Treatment in Dermatology. London: Manson Publishing Ltd, 2004.
- Swiglo BA, Cosma M, Flynn DN, et al. Clinical review: antiandrogens for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab 2008; 93:1153–1160.
- Cosma M, Swiglo BA, Flynn DN, et al. Clinical review: insulin sensitizers for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab 2008; 93:1135–1142.
- Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol 2001; 2:197–201.
- Wolf JE, Shander D, Huber F, et al; Eflornithine HCl Study Group. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair. Int J Dematol 2007; 46:94–98.
- Hamzavi I, Tan E, Shapiro J, Lui H. A randomized bilateral vehicle-controlled study of eflornithine cream combined with laser treatment versus laser treatment alone for facial hirsutism in women. J Am Acad Dermatol 2007; 57:54–59.
- Van der Spuy ZM, le Roux PA. Cyproterone acetate for hirsutism. Cochrane Database Syst Rev 2003; 4:CD001125.
KEY POINTS
- The finding of polycystic ovaries is not required for the diagnosis of polycystic ovary syndrome, nor does their presence prove the diagnosis. Gonadotropin-dependent functional ovarian hyperandrogenism is believed to cause this syndrome; however, mild adrenocorticotropic-dependent functional adrenal hyperandrogenism also is a feature in many cases.
- Even women with mild hirsutism with subtle symptoms and signs of hyperandrogenism can have elevated androgen levels, and thus, they deserve a laboratory evaluation.
- Laser treatment does not result in complete, permanent hair reduction, but it is more effective than shaving, waxing, and electrolysis, producing partial hair reduction for up to 6 months.
Incidence, outcomes, and management of bleeding in non-ST-elevation acute coronary syndromes
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?

Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13

BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
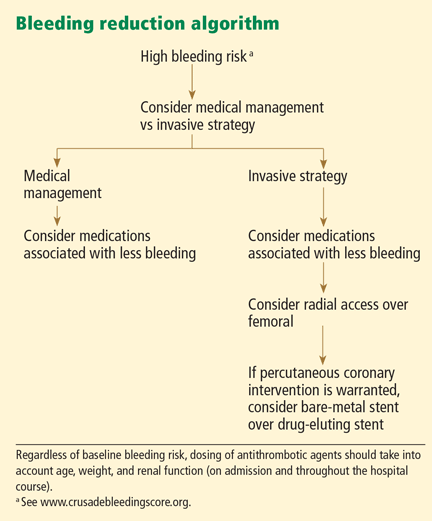
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
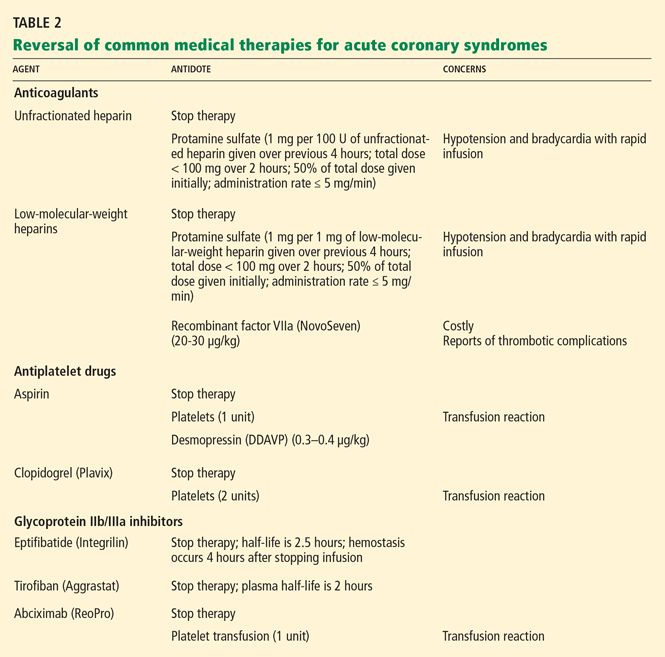
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?
Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13
BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?
Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13
BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
KEY POINTS
- The reported incidence of bleeding after treatment for non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%, depending on a number of factors.
- Bleeding is strongly associated with adverse outcomes, although a causal relationship has not been established.
- Patients should be assessed for risk of bleeding so that the antithrombotic and antiplatelet regimen can be adjusted, safer alternatives can be considered, and percutaneous interventions can be used less aggressively for those at high risk.
- If bleeding develops and the risk of continued bleeding outweighs the risk of recurrent ischemia, antithrombotic and antiplatelet drug therapy can be interrupted and other agents given to reverse the effects of these drugs.
Should patients with mild asthma use inhaled steroids?
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
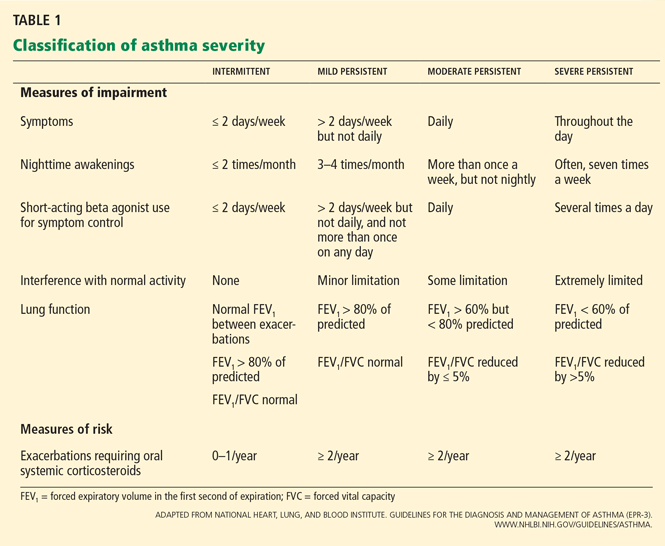
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT

The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
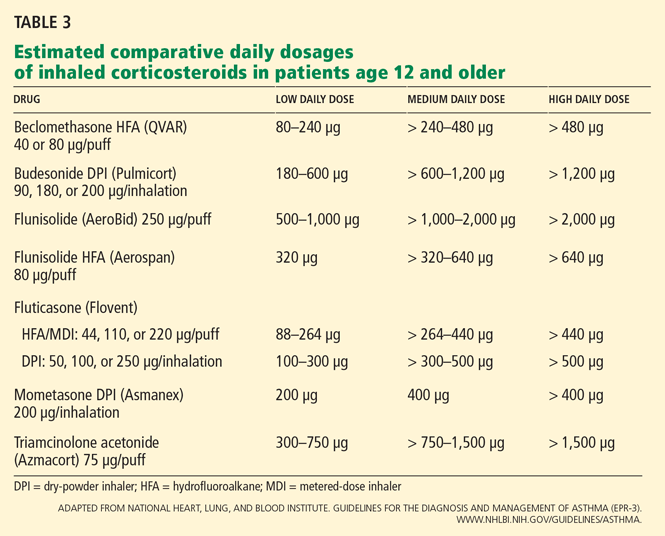
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT
The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
Yes. A number of large randomized controlled trials have shown inhaled corticosteroids to be beneficial in low doses for patients who have mild persistent asthma, and therefore these drugs are strongly recommended in this situation.1
Asthma care providers should, however, consider this “yes” in the context of asthma severity, the goals of therapy, and the benefits and risks associated with inhaled corticosteroids.
CLASSIFICATION OF ASTHMA SEVERITY
Although the studies of asthma prevalence had methodologic limitations and therefore the true prevalence of mild persistent asthma cannot be determined, it is common. Fuhlbrigge et al2 reported that most asthma patients have some form of persistent asthma. In contrast, Dusser et al3 reviewed available studies and concluded that most patients with asthma have either intermittent or mild persistent asthma.
GOALS: REDUCE IMPAIRMENT AND RISK
The goals of asthma management are to:
Reduce impairment by controlling symptoms so that normal activity levels can be maintained, by minimizing the need for short-acting bronchodilator use, and by maintaining normal pulmonary function; and to
Reduce risk by preventing progressive loss of lung function and recurrent exacerbations, and by optimizing pharmacotherapy while minimizing potential adverse effects.1
EVIDENCE OF BENEFIT
The OPTIMA trial4 (Low Dose Inhaled Budesonide and Formoterol in Mild Persistent Asthma) was a double-blind, randomized trial carried out in 198 centers in 17 countries. Compared with those randomized to receive placebo, patients who were randomized to receive an inhaled corticosteroid, ie, budesonide (Pulmicort) 100 μg twice daily, had 60% fewer severe exacerbations (relative risk [RR] 0.4, 95% confidence interval [CI] 0.27–0.59) and 48% fewer days when their asthma was poorly controlled (RR 0.52, 95% CI 0.4–0.67). Adding a long-acting beta-agonist did not change this outcome.
The START study5 (Inhaled Steroid Treatment as Regular Therapy in Early Asthma) showed that, compared with placebo, starting inhaled budesonide within the first 2 years of asthma symptoms in patients with mild persistent asthma was associated with better asthma control and less need for additional asthma medication.
The IMPACT study6 (Improving Asthma Control Trial) showed that inhaled steroids need to be taken daily, on a regular schedule, rather than intermittently as needed. Patients received either inhaled budesonide as needed, budesonide 200 μg twice daily every day, or zafirlukast (Accolate) 20 mg twice daily. Daily budesonide therapy resulted in better asthma control, less bronchial hyperresponsiveness, and less airway inflammation compared with intermittent use, zafirlukast therapy, or placebo. Daily zafirlukast and intermittent steroid treatment produced similar results for all outcomes measured.
Despite this strong evidence supporting regular use of inhaled corticosteroids in patients with mild persistent asthma, many patients choose to take them intermittently.
Suissa et al7 found, in a large observational cohort study, that fewer patients died of asthma if they were receiving low-dose inhaled corticosteroids than if they were not. The rate of death due to asthma was lower in patients who had used more inhaled corticosteroids over the previous year, and the death rate was higher in those who had discontinued inhaled corticosteroids in the previous 3 months than in those who continued using them.
STEROIDS DO NOT SLOW THE LOSS OF LUNG FUNCTION
Compared with people without asthma, asthma patients have substantially lower values of forced expiratory volume in the first second of expiration (FEV1). They also have a faster rate of functional decline: the average decrease in FEV1 in asthma patients is 38 mL per year, compared with 22 mL per year in nonasthmatic people.9
Although inhaled corticosteroids have been shown to increase lung function in asthma patients in the short term, there is little convincing evidence to suggest that they affect the rate of decline in the long term.10 In fact, airway inflammation and bronchial hyperresponsiveness return to baseline within 2 weeks after inhaled corticosteroids are discontinued.10
DO INHALED CORTICOSTEROIDS STUNT CHILDREN’S GROWTH?
The safety of long-term low-dose inhaled corticosteroids is well established in adults. However, two large randomized controlled trials found that children treated with low-dose inhaled steroids (budesonide 200–400 μg per day) grew 1 to 1.5 cm less over 3 to 5 years of treatment than children receiving placebo.11 However, this effect was primarily evident within the first year of therapy, and growth velocity was similar to that with placebo at the end of the treatment period (4 to 6 years).12
Agertoft and Pedersen13 found that taking inhaled corticosteroids long-term is unlikely to have an effect on final height. Children who took inhaled budesonide (up to an average daily dose of 500 μg) into adulthood ended up no shorter than those who did not.
Based on these and other data, inhaled corticosteroids are generally considered safe at recommended doses. However, the decision to prescribe them for long-term therapy should be based on the risks and benefits to the individual patient.1
ALTERNATIVE DRUGS FOR MILD PERSISTENT ASTHMA
Leukotriene-modifying drugs include the leukotriene receptor antagonists montelukast (Singulair) and zafirlukast and the 5-lipoxygenase inhibitor zileuton (Zyflo CR). These drugs have been associated with statistically significant improvement in FEV1 compared with placebo in patients with mild to moderate asthma, reductions in both blood and sputum eosinophils,14 and attenuation of bronchoconstriction with exercise.11
Large randomized trials comparing leukotriene modifier therapy with low-dose inhaled steroids in adults and children with mild persistent asthma have found that although outcomes improve with either therapy, the improvement is statistically superior with inhaled steroids for most asthma-control measures. 6,8 Low-dose inhaled steroid therapy in patients with mild persistent and moderate persistent asthma has been associated with superior clinical outcomes as well as greater improvement in pulmonary function than treatment with antileukotriene drugs (Table 2).8
Asthma is heterogeneous, and properly selected patients with mild persistent asthma may achieve good control with leukotrienemodifier monotherapy.15 Alternatives for patients with mild persistent asthma include the methylxanthine theophylline, but this drug is less desirable due to its narrow therapeutic index. 1 The inhaled cromones nedocromil (Tilade) and cromolyn (Intal) were other options in this patient population, but their short half-lives made them less practical, and US production has been discontinued.
THE BOTTOM LINE
Though leukotriene receptor antagonists can be effective, the daily use of inhaled corticosteroids results in higher asthma control test scores, more symptom-free days, greater pre-bronchodilator FEV1, and decreased percentage of sputum eosinophils6 in patients with mild persistent asthma, and the addition of a long-acting beta agonist does not provide additional benefit.4 Furthermore, daily use of inhaled corticosteroids in these patients has also been associated with a lower rate of asthma-related deaths and with less need for systemic corticosteroid therapy,7,8 even though inhaled corticosteroids have not yet been shown to alter the progressive loss of lung function.10
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
- National Heart, Lung, and Blood Institute. Guidelines for the Diagnosis and Management of Asthma (EPR-3). www.nhlbi.nih.gov/guidelines/asthma/. Accessed March 26, 2010.
- Fuhlbrigge AL, Adams RJ, Guilbert TW, et al. The burden of asthma in the United States: level and distribution are dependent on interpretation of the National Asthma Education and Prevention Program. Am J Respir Crit Care Med 2002; 166:1044–1049.
- Dusser D, Montani D, Chanez P, et al. Mild asthma: an expert review on epidemiology, clinical characteristics and treatment recommendations. Allergy 2007; 62:591–604.
- O’Byrne PM, Barnes PJ, Rodriguez-Roisin R, et al. Low dose inhaled budesonide and formoterol in mild persistent asthma: the OPTIMA randomized trial. Am J Respir Crit Care Med 2001; 164:1392–1397.
- Busse WW, Pedersen S, Pauwels RA, et al; START Investigators Group. The Inhaled Steroid Treatment As Regular Therapy in Early Asthma (START) study 5-year follow-up: effectiveness of early intervention with budesonide in mild persistent asthma. J Allergy Clin Immunol 2008; 121:1167–1174.
- Boushey HA, Sorkness CA, King TS, et al; National Heart, Lung, and Blood Institute’s Asthma Clinical Research Network. Daily versus as-needed corticosteroids for mild persistent asthma. N Engl J Med 2005; 352:1519–1528.
- Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med 2000; 343:332–356.
- Busse W, Wolfe J, Storms W, et al. Fluticasone propionate compared with zafirlukast in controlling persistent asthma: a randomized double-blind, placebo-controlled trial. J Fam Pract 2001; 50:595–602.
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N Engl J Med 1998; 339:1194–1200.
- Fanta CH. Asthma. N Engl J Med 2009; 360:1002–1014.
- O’Byrne PM, Parameswaran K. Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 368:794–803.
- The Childhood Asthma Management Program Research Group. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med 2000; 343:1054–1063.
- Agertoft L, Pedersen S. Effect of long-term treatment with inhaled budesonide on adult height in children with asthma. N Engl J Med 2000; 343:1064–1069.
- Pizzichini E, Leff JA, Reiss TF, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J 1999; 14:12–18.
- Kraft M, Israel E, O’Connor GT. Clinical decisions. Treatment of mild persistent asthma. N Engl J Med 2007; 356:2096–2100.
LVH and hypertension: Is treating the pressure not enough?

In patients with “borderline” hypertension or those in whom the duration of blood pressure elevation is hard to ascertain, the finding of end-organ damage has traditionally been used as an argument to institute aggressive antihypertensive therapy. In this setting, retinal hypertensive disease, an S4 gallop, and left ventricular hypertrophy (LVH) are often specifically sought.
LVH and otherwise unexplained chronic kidney disease in patients with hypertension have generally been believed to be products of the elevated arterial pressure, and primary treatment has targeted pressure control. Bauml and Underwood, in this issue of the Journal, emphasize some published clinical trial data indicating that LVH may be an independent risk factor for poorer cardiovascular outcome. Even more provocative is the suggestion that LVH can be reversed, as can the associated increased risk of cardiovascular morbidity, independently of the hypertension.
Given our current understanding that LVH, under some conditions, can be induced by products of the renin-angiotensin system, this would suggest that pharmacologic blockade of this enzyme system should have extra benefit, above that seen from other antihypertensive agents. Conceivably, this may be true only in patients with LVH, and the time course of benefit may not directly parallel that seen with the control of hypertension. That theoretically may explain the lack of uniform advantage of angiotensin blockade over other effective antihypertensive approaches.
Since electrocardiography is a specific but not very sensitive test for LVH, the authors suggest that patients with hypertension be routinely screened for LVH using echocardiography. I am not sure the weight of the evidence supports this approach at present, particularly in the current frenzy of cost containment. Nonetheless, this concept warrants consideration, and at the least, large patient databases might be screened retrospectively to further validate or refute the concept that hypertension-associated LVH is an independent, reversible risk factor for cardiovascular morbidity.
In patients with “borderline” hypertension or those in whom the duration of blood pressure elevation is hard to ascertain, the finding of end-organ damage has traditionally been used as an argument to institute aggressive antihypertensive therapy. In this setting, retinal hypertensive disease, an S4 gallop, and left ventricular hypertrophy (LVH) are often specifically sought.
LVH and otherwise unexplained chronic kidney disease in patients with hypertension have generally been believed to be products of the elevated arterial pressure, and primary treatment has targeted pressure control. Bauml and Underwood, in this issue of the Journal, emphasize some published clinical trial data indicating that LVH may be an independent risk factor for poorer cardiovascular outcome. Even more provocative is the suggestion that LVH can be reversed, as can the associated increased risk of cardiovascular morbidity, independently of the hypertension.
Given our current understanding that LVH, under some conditions, can be induced by products of the renin-angiotensin system, this would suggest that pharmacologic blockade of this enzyme system should have extra benefit, above that seen from other antihypertensive agents. Conceivably, this may be true only in patients with LVH, and the time course of benefit may not directly parallel that seen with the control of hypertension. That theoretically may explain the lack of uniform advantage of angiotensin blockade over other effective antihypertensive approaches.
Since electrocardiography is a specific but not very sensitive test for LVH, the authors suggest that patients with hypertension be routinely screened for LVH using echocardiography. I am not sure the weight of the evidence supports this approach at present, particularly in the current frenzy of cost containment. Nonetheless, this concept warrants consideration, and at the least, large patient databases might be screened retrospectively to further validate or refute the concept that hypertension-associated LVH is an independent, reversible risk factor for cardiovascular morbidity.
In patients with “borderline” hypertension or those in whom the duration of blood pressure elevation is hard to ascertain, the finding of end-organ damage has traditionally been used as an argument to institute aggressive antihypertensive therapy. In this setting, retinal hypertensive disease, an S4 gallop, and left ventricular hypertrophy (LVH) are often specifically sought.
LVH and otherwise unexplained chronic kidney disease in patients with hypertension have generally been believed to be products of the elevated arterial pressure, and primary treatment has targeted pressure control. Bauml and Underwood, in this issue of the Journal, emphasize some published clinical trial data indicating that LVH may be an independent risk factor for poorer cardiovascular outcome. Even more provocative is the suggestion that LVH can be reversed, as can the associated increased risk of cardiovascular morbidity, independently of the hypertension.
Given our current understanding that LVH, under some conditions, can be induced by products of the renin-angiotensin system, this would suggest that pharmacologic blockade of this enzyme system should have extra benefit, above that seen from other antihypertensive agents. Conceivably, this may be true only in patients with LVH, and the time course of benefit may not directly parallel that seen with the control of hypertension. That theoretically may explain the lack of uniform advantage of angiotensin blockade over other effective antihypertensive approaches.
Since electrocardiography is a specific but not very sensitive test for LVH, the authors suggest that patients with hypertension be routinely screened for LVH using echocardiography. I am not sure the weight of the evidence supports this approach at present, particularly in the current frenzy of cost containment. Nonetheless, this concept warrants consideration, and at the least, large patient databases might be screened retrospectively to further validate or refute the concept that hypertension-associated LVH is an independent, reversible risk factor for cardiovascular morbidity.
Left ventricular hypertrophy: An overlooked cardiovascular risk factor
Left ventricular hypertrophy (LVH) strongly predicts cardiovascular morbidity and overall mortality in hypertensive patients. 1–7 Antihypertensive treatment that causes LVH to regress decreases the rates of adverse cardiovascular events and improves survival, independent of how much the blood pressure is lowered.8–11 It is clinically important to recognize that LVH is a modifiable risk factor and that management is more complex than just blood pressure control.
This paper reviews the definition of LVH, compares the diagnostic tests for it, and discusses the current evidence-based approach to managing this dangerous risk factor.
A CHRONICALLY ELEVATED CARDIAC WORKLOAD CAUSES LVH
LVH is an abnormal increase in the mass of the left ventricular myocardium caused by a chronically increased workload on the heart.12 This most commonly results from the heart pumping against an elevated afterload, as in hypertension and aortic stenosis. Another notable cause is increased filling of the left ventricle (ie, diastolic overload), which is the underlying mechanism for LVH in patients with aortic or mitral regurgitation and dilated cardiomyopathy. Coronary artery disease can also play a role in the pathogenesis of LVH, as the normal myocardium attempts to compensate for the ischemic or infarcted tissue.13
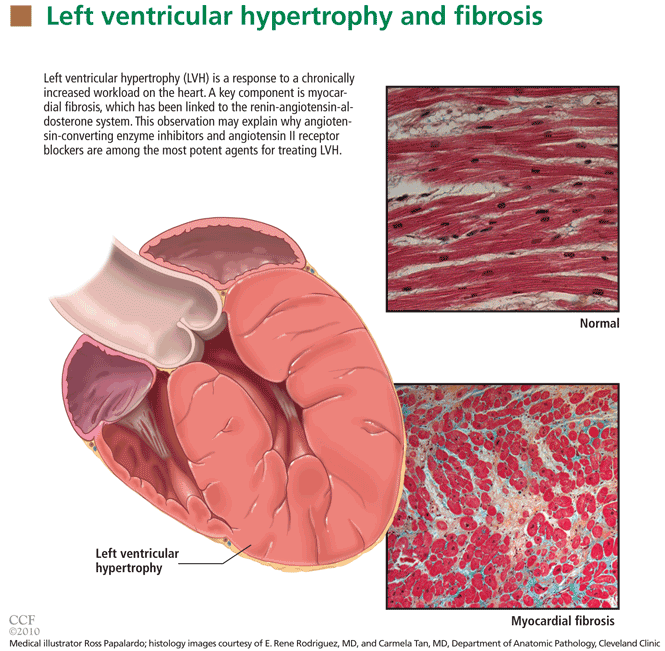
The development of myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system. Specifically, there is evidence that angiotensin II has a profibrotic effect on the myocardium of hypertensive patients.15 This may explain why angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are among the most potent agents for treating LVH, as we will discuss later in this review.
DIAGNOSIS BY ELECTROCARDIOGRAPHY, ECHOCARDIOGRAPHY, OR MRI
Many different criteria for electrocardiographic LVH have been proposed over the years. Most use the voltage in one or more leads, with or without additional factors such as QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. The most well known electrocardiographic criteria are the Cornell voltage,21 the Cornell product,22 the Sokolow-Lyon index,23 and the Romhilt-Estes point score system (Table 1).24

- Cornell voltage—median sensitivity 15%, median specificity 96%
- Cornell product—median sensitivity 19.5%, median specificity 91%
- Sokolow-Lyon voltage—median sensitivity 21%, median specificity 89%
- Romhilt-Estes point score—median sensitivity 17%, median specificity 95%.
Of note, the ranges of the published values were extremely broad. For example, the ranges in sensitivity were:
- Cornell voltage—2% to 41%
- Cornell product—8% to 32%
- Sokolow-Lyon voltage—4% to 51%
- Romhilt-Estes point score—0% to 41%.
While the studies with the extreme values may have had issues of small sample size or poor study quality, the wide range in values may primarily be the result of diverse study populations as well as different validation methods and cutoff values to define LVH. Regardless, the overall message of high specificity and low sensitivity is indisputable.
Electrocardiography is insensitive for diagnosing LVH because it relies on measuring the electrical activity of the heart by electrodes on the surface of the skin to predict the left ventricular mass. The intracardiac electrical activity is problematic to measure externally because the measurements are affected by everything between the myocardium and the electrodes, most notably fat, fluid, and air. Because of this effect, electrocardiography underdiagnoses LVH in patients with obesity, pleural effusions, pericardial effusions, anasarca, or chronic obstructive pulmonary disease. In addition, the diagnosis of LVH by electrocardiography is strongly influenced by age and ethnicity.25–26
While electrocardiography is not sensitive and cannot be used to rule out LVH, it still has a role in its diagnosis and management. In the landmark Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study, regression of LVH (diagnosed electrocardiographically by the Sokolow-Lyon index or the Cornell product criteria) in response to losartan (Cozaar) improved cardiovascular outcomes independent of blood pressure.10 Based on this, it is reasonable that all hypertensive patients and other patients at risk of LVH who undergo electrocardiography be screened with these two criteria.
Echocardiography is the test of choice
Echocardiography, if available, should be the test of choice to assess for LVH. It is much more sensitive than electrocardiography and can also detect other abnormalities such as left ventricular dysfunction and valvular disease.
This test uses transthoracic or transesophageal ultrasonography to measure the left ventricular end-diastolic diameter, posterior wall thickness, and interventricular septum thickness. From these measurements and the patient’s height and weight, the left ventricular mass index can be calculated.27
Several different cutoff values for the left ventricular mass index have been proposed; the LIFE study used values of > 104 g/m2 in women and > 116 g/m2 in men to define LVH.
When using echocardiography to assess for LVH, it is imperative that the left ventricular mass index be used and not just the left ventricular wall thickness, as often happens in clinical practice. This is necessary because diagnosis by wall thickness alone is not a good indicator of LVH, with a concordance between wall thickness and a left ventricular mass index of only 60%.28 In addition, wall thickness tends to underestimate LVH in women and overestimate it in men.
Is echocardiography cost-effective?
Despite its clear advantages, an important consideration about echocardiography as a screening test for all hypertensive patients is its cost.
A suggested way to reduce cost is to measure the left ventricular mass index only.29 A limited echocardiographic examination is much less expensive than a complete two-dimensional echocardiogram ($255 vs $431 per the 2009 Medicare Ambulatory Payment Classification30) and should be the examination performed if the patient has no other clinical indication for echocardiography.
Another way to control cost is to stratify patients by risk and to do echocardiography only in those who would benefit most from it. Based on the prevalence of LVH, one study concluded that echocardiography is most cost-effective in men 50 years or older.31
Further study is necessary to more precisely define the cost-effectiveness of echocardiographic screening for LVH in terms of potentially preventable cardiovascular morbidity and death.
Cardiac MRI: The costly gold standard
Cardiac MRI is the gold standard test for LVH, as it is even more accurate and reproducible than echocardiography.32 It can precisely estimate a patient's left ventricular mass and assess for other structural cardiac abnormalities.
MRI’s use, however, is severely restricted in clinical practice due to its high cost and limited availability. While it may never be used for general screening for LVH, it certainly has a role in clinical research and for assessing cardiac anatomy in special clinical situations.
TREATMENT SHOULD INCLUDE AN ACE INHIBITOR OR ARB
Once LVH has been diagnosed, the next step is to decide on an appropriate treatment plan.
While the choice of therapy will always depend on other comorbidities, a 2003 metaanalysis of antihypertensive medications in the treatment of LVH (controlling for the degree of blood pressure lowering) showed that ARBs were the most efficacious class of agents for reducing the left ventricular mass.33 Specifically, ARBs decreased the mass by 13%, followed by calcium-channel blockers at 11%, ACE inhibitors at 10%, diuretics at 8%, and beta-blockers at 6%. In pairwise comparison, ARBs, calcium-channel blockers, and ACE inhibitors were all significantly more effective in reducing the left ventricular mass than beta-blockers.
As previously discussed, LVH appears to be pathophysiologically linked to myocardial fibrosis and the renin-angiotensin-aldosterone system. For this reason and based on the data presented above regarding the degree of LVH regression, ACE inhibitors or ARBs should be used as the first-line agents for LVH unless they are contraindicated in the individual patient.
The LIFE study
The LIFE study offers the strongest evidence that treating LVH is beneficial. It showed that in hypertensive patients with electrocardiographic LVH by the Cornell product or Sokolow-Lyon criteria, treatment with antihypertensive drugs that resulted in less-severe LVH on electrocardiography was associated with lower rates of cardiovascular morbidity and death, independent of the blood pressure achieved or the drug used.10
The end point in this study was a composite of stroke, myocardial infarction, and cardiovascular death. Regression of electrocardiographic LVH in hypertensive patients has also been shown to decrease the incidence of diabetes mellitus,34 atrial fibrillation,35 and hospitalizations for heart failure.36
The LIFE study also examined the prognostic implications of treating LVH detected by echocardiography. In this prospective cohort substudy, patients who had a lower left ventricular mass index during treatment with antihypertensive drugs had lower rates of cardiovascular morbidity and all-cause mortality, independent of the effects of blood pressure and treatment used.11
These results suggest that there may be a role not only for treating LVH, but also for monitoring for a reduction in the left ventricular mass index as a goal of therapy (similar to the way hemoglobin A1c is used in diabetic patients). If the index is used in this way, one could potentially adjust the dose of current drugs, switch classes, or add an additional drug based on a persistently elevated left ventricular mass index in order to optimize the patient's overall cardiovascular risk. A randomized controlled trial of therapy directed by the mass index vs conventional therapy of LVH would be necessary to assess the clinical utility of this approach.
RECOMMENDATIONS
LVH is a common and potentially modifiable cardiovascular risk factor often overlooked in clinical practice. Ideally, all hypertensive patients should be screened with echocardiography to look for LVH, using the calculated left ventricular mass index rather than wall thickness alone to make the diagnosis. While electrocardiography is specific and also has prognostic implications, it is not sensitive enough to be used alone to screen for LVH.
Once the diagnosis of LVH is made, the initial therapy should be an ARB or an ACE inhibitor. Response to therapy can be assessed by monitoring for a reduction in left ventricular mass index or regression of electrocardiographic LVH.
Treatment-induced regression of LVH decreases adverse cardiovascular events and improves overall survival. When modifying medications in hypertensive patients, it is important to remember that the treatment of LVH is not synonymous with blood pressure control.
- Casale PN, Devereux RB, Milner M, et al. Value of echocardiographic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105:173–178.
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566.
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352.
- Verdecchia P, Carini G, Circo A, et al; MAVI (MAssa Ventricolare sinistra nell’Ipertensione) Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 2001; 38:1829–1835.
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998; 32:1454–1459.
- Verdecchia P, Porcellati C, Reboldi G, et al. Left ventricular hypertrophy as an independent predictor of acute cerebrovascular events in essential hypertension. Circulation 2001; 104:2039–2044.
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35:580–586.
- Verdecchia P, Schillaci G, Borgioni C, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation 1998; 97:48–54.
- Mathew J, Sleight P, Lonn E, et al; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621.
- Okin PM, Devereux RB, Jern S, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004; 292:2343–2349.
- Devereux RB, Wachtell K, Gerdts E, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292:2350–2356.
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470–479.
- Zabalgoitia M, Berning J, Koren MJ, et al; LIFE Study Investigators. Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 2001; 88:646–650.
- Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol 1990; 65:1G–7G.
- González A, López B, Querejeta R, Díez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 2002; 34:1585–1593.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320.
- Liebson PR, Grandits G, Prineas R, et al. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993; 87:476–486.
- Martinez MA, Sancho T, Armada E, et al; Vascular Risk Working Group Grupo Monitorizacíon Ambulatoria de la Presión Arterial (MAPA)-Madrid. Prevalence of left ventricular hypertrophy in patients with mild hypertension in primary care: impact of echocardiography on cardiovascular risk stratification. Am J Hypertens 2003; 16:556–563.
- Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ 2007; 335:711.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990; 81:1144–1146.
- Casale PN, Devereux RB, Kligfield P, et al. Electrocardiographic detection of left ventricular hypertrophy: development and prospective validation of improved criteria. J Am Coll Cardiol 1985; 6:572–580.
- Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol 1992; 20:1180–1186.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949; 37:161–186.
- Romhilt DW, Estes EH. A point-score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968; 75:752–758.
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation 1990; 81:815–820.
- Okin PM, Wright JT, Nieminen MS, et al. Ethnic differences in electrocardiographic criteria for left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint. Am J Hypertens 2002; 15:663–671.
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986; 57:450–458.
- Leibowitz D, Planer D, Ben-Ibgi F, Rott D, Weiss AT, Bursztyn M. Measurement of wall thickness alone does not accurately assess the presence of left ventricular hypertrophy. Clin Exp Hypertens 2007; 29:119–125.
- Black HR, Weltin G, Jaffe CC. The limited echocardiogram: a modification of standard echocardiography for use in the routine evaluation of patients with systemic hypertension. Am J Cardiol 1991; 67:1027–1030.
- American Society of Echocardiography Coding and Reimbursement Newsletter, January 2009. http://www.asecho.org/files/public/CodingnewsJan09.pdf. Accessed May 13, 2010.
- Cuspidi C, Meani S, Valerio C, Fusi V, Sala C, Zanchetti A. Left ventricular hypertrophy and cardiovascular risk stratification: impact and cost-effectiveness of echocardiography in recently diagnosed essential hypertensives. J Hypertens 2006; 24:1671–1677.
- Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, Gottdiener JS. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 1995; 8:221–228.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003; 115:41–46.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. In-treatment resolution or absence of electrocardiographic left ventricular hypertrophy is associated with decreased incidence of new-onset diabetes mellitus in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2007; 50:984–990.
- Okin PM, Wachtell K, Devereux RB, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006; 296:1242–1248.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Ann Intern Med 2007; 147:311–319.
Left ventricular hypertrophy (LVH) strongly predicts cardiovascular morbidity and overall mortality in hypertensive patients. 1–7 Antihypertensive treatment that causes LVH to regress decreases the rates of adverse cardiovascular events and improves survival, independent of how much the blood pressure is lowered.8–11 It is clinically important to recognize that LVH is a modifiable risk factor and that management is more complex than just blood pressure control.
This paper reviews the definition of LVH, compares the diagnostic tests for it, and discusses the current evidence-based approach to managing this dangerous risk factor.
A CHRONICALLY ELEVATED CARDIAC WORKLOAD CAUSES LVH
LVH is an abnormal increase in the mass of the left ventricular myocardium caused by a chronically increased workload on the heart.12 This most commonly results from the heart pumping against an elevated afterload, as in hypertension and aortic stenosis. Another notable cause is increased filling of the left ventricle (ie, diastolic overload), which is the underlying mechanism for LVH in patients with aortic or mitral regurgitation and dilated cardiomyopathy. Coronary artery disease can also play a role in the pathogenesis of LVH, as the normal myocardium attempts to compensate for the ischemic or infarcted tissue.13
The development of myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system. Specifically, there is evidence that angiotensin II has a profibrotic effect on the myocardium of hypertensive patients.15 This may explain why angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are among the most potent agents for treating LVH, as we will discuss later in this review.
DIAGNOSIS BY ELECTROCARDIOGRAPHY, ECHOCARDIOGRAPHY, OR MRI
Many different criteria for electrocardiographic LVH have been proposed over the years. Most use the voltage in one or more leads, with or without additional factors such as QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. The most well known electrocardiographic criteria are the Cornell voltage,21 the Cornell product,22 the Sokolow-Lyon index,23 and the Romhilt-Estes point score system (Table 1).24
- Cornell voltage—median sensitivity 15%, median specificity 96%
- Cornell product—median sensitivity 19.5%, median specificity 91%
- Sokolow-Lyon voltage—median sensitivity 21%, median specificity 89%
- Romhilt-Estes point score—median sensitivity 17%, median specificity 95%.
Of note, the ranges of the published values were extremely broad. For example, the ranges in sensitivity were:
- Cornell voltage—2% to 41%
- Cornell product—8% to 32%
- Sokolow-Lyon voltage—4% to 51%
- Romhilt-Estes point score—0% to 41%.
While the studies with the extreme values may have had issues of small sample size or poor study quality, the wide range in values may primarily be the result of diverse study populations as well as different validation methods and cutoff values to define LVH. Regardless, the overall message of high specificity and low sensitivity is indisputable.
Electrocardiography is insensitive for diagnosing LVH because it relies on measuring the electrical activity of the heart by electrodes on the surface of the skin to predict the left ventricular mass. The intracardiac electrical activity is problematic to measure externally because the measurements are affected by everything between the myocardium and the electrodes, most notably fat, fluid, and air. Because of this effect, electrocardiography underdiagnoses LVH in patients with obesity, pleural effusions, pericardial effusions, anasarca, or chronic obstructive pulmonary disease. In addition, the diagnosis of LVH by electrocardiography is strongly influenced by age and ethnicity.25–26
While electrocardiography is not sensitive and cannot be used to rule out LVH, it still has a role in its diagnosis and management. In the landmark Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study, regression of LVH (diagnosed electrocardiographically by the Sokolow-Lyon index or the Cornell product criteria) in response to losartan (Cozaar) improved cardiovascular outcomes independent of blood pressure.10 Based on this, it is reasonable that all hypertensive patients and other patients at risk of LVH who undergo electrocardiography be screened with these two criteria.
Echocardiography is the test of choice
Echocardiography, if available, should be the test of choice to assess for LVH. It is much more sensitive than electrocardiography and can also detect other abnormalities such as left ventricular dysfunction and valvular disease.
This test uses transthoracic or transesophageal ultrasonography to measure the left ventricular end-diastolic diameter, posterior wall thickness, and interventricular septum thickness. From these measurements and the patient’s height and weight, the left ventricular mass index can be calculated.27
Several different cutoff values for the left ventricular mass index have been proposed; the LIFE study used values of > 104 g/m2 in women and > 116 g/m2 in men to define LVH.
When using echocardiography to assess for LVH, it is imperative that the left ventricular mass index be used and not just the left ventricular wall thickness, as often happens in clinical practice. This is necessary because diagnosis by wall thickness alone is not a good indicator of LVH, with a concordance between wall thickness and a left ventricular mass index of only 60%.28 In addition, wall thickness tends to underestimate LVH in women and overestimate it in men.
Is echocardiography cost-effective?
Despite its clear advantages, an important consideration about echocardiography as a screening test for all hypertensive patients is its cost.
A suggested way to reduce cost is to measure the left ventricular mass index only.29 A limited echocardiographic examination is much less expensive than a complete two-dimensional echocardiogram ($255 vs $431 per the 2009 Medicare Ambulatory Payment Classification30) and should be the examination performed if the patient has no other clinical indication for echocardiography.
Another way to control cost is to stratify patients by risk and to do echocardiography only in those who would benefit most from it. Based on the prevalence of LVH, one study concluded that echocardiography is most cost-effective in men 50 years or older.31
Further study is necessary to more precisely define the cost-effectiveness of echocardiographic screening for LVH in terms of potentially preventable cardiovascular morbidity and death.
Cardiac MRI: The costly gold standard
Cardiac MRI is the gold standard test for LVH, as it is even more accurate and reproducible than echocardiography.32 It can precisely estimate a patient's left ventricular mass and assess for other structural cardiac abnormalities.
MRI’s use, however, is severely restricted in clinical practice due to its high cost and limited availability. While it may never be used for general screening for LVH, it certainly has a role in clinical research and for assessing cardiac anatomy in special clinical situations.
TREATMENT SHOULD INCLUDE AN ACE INHIBITOR OR ARB
Once LVH has been diagnosed, the next step is to decide on an appropriate treatment plan.
While the choice of therapy will always depend on other comorbidities, a 2003 metaanalysis of antihypertensive medications in the treatment of LVH (controlling for the degree of blood pressure lowering) showed that ARBs were the most efficacious class of agents for reducing the left ventricular mass.33 Specifically, ARBs decreased the mass by 13%, followed by calcium-channel blockers at 11%, ACE inhibitors at 10%, diuretics at 8%, and beta-blockers at 6%. In pairwise comparison, ARBs, calcium-channel blockers, and ACE inhibitors were all significantly more effective in reducing the left ventricular mass than beta-blockers.
As previously discussed, LVH appears to be pathophysiologically linked to myocardial fibrosis and the renin-angiotensin-aldosterone system. For this reason and based on the data presented above regarding the degree of LVH regression, ACE inhibitors or ARBs should be used as the first-line agents for LVH unless they are contraindicated in the individual patient.
The LIFE study
The LIFE study offers the strongest evidence that treating LVH is beneficial. It showed that in hypertensive patients with electrocardiographic LVH by the Cornell product or Sokolow-Lyon criteria, treatment with antihypertensive drugs that resulted in less-severe LVH on electrocardiography was associated with lower rates of cardiovascular morbidity and death, independent of the blood pressure achieved or the drug used.10
The end point in this study was a composite of stroke, myocardial infarction, and cardiovascular death. Regression of electrocardiographic LVH in hypertensive patients has also been shown to decrease the incidence of diabetes mellitus,34 atrial fibrillation,35 and hospitalizations for heart failure.36
The LIFE study also examined the prognostic implications of treating LVH detected by echocardiography. In this prospective cohort substudy, patients who had a lower left ventricular mass index during treatment with antihypertensive drugs had lower rates of cardiovascular morbidity and all-cause mortality, independent of the effects of blood pressure and treatment used.11
These results suggest that there may be a role not only for treating LVH, but also for monitoring for a reduction in the left ventricular mass index as a goal of therapy (similar to the way hemoglobin A1c is used in diabetic patients). If the index is used in this way, one could potentially adjust the dose of current drugs, switch classes, or add an additional drug based on a persistently elevated left ventricular mass index in order to optimize the patient's overall cardiovascular risk. A randomized controlled trial of therapy directed by the mass index vs conventional therapy of LVH would be necessary to assess the clinical utility of this approach.
RECOMMENDATIONS
LVH is a common and potentially modifiable cardiovascular risk factor often overlooked in clinical practice. Ideally, all hypertensive patients should be screened with echocardiography to look for LVH, using the calculated left ventricular mass index rather than wall thickness alone to make the diagnosis. While electrocardiography is specific and also has prognostic implications, it is not sensitive enough to be used alone to screen for LVH.
Once the diagnosis of LVH is made, the initial therapy should be an ARB or an ACE inhibitor. Response to therapy can be assessed by monitoring for a reduction in left ventricular mass index or regression of electrocardiographic LVH.
Treatment-induced regression of LVH decreases adverse cardiovascular events and improves overall survival. When modifying medications in hypertensive patients, it is important to remember that the treatment of LVH is not synonymous with blood pressure control.
Left ventricular hypertrophy (LVH) strongly predicts cardiovascular morbidity and overall mortality in hypertensive patients. 1–7 Antihypertensive treatment that causes LVH to regress decreases the rates of adverse cardiovascular events and improves survival, independent of how much the blood pressure is lowered.8–11 It is clinically important to recognize that LVH is a modifiable risk factor and that management is more complex than just blood pressure control.
This paper reviews the definition of LVH, compares the diagnostic tests for it, and discusses the current evidence-based approach to managing this dangerous risk factor.
A CHRONICALLY ELEVATED CARDIAC WORKLOAD CAUSES LVH
LVH is an abnormal increase in the mass of the left ventricular myocardium caused by a chronically increased workload on the heart.12 This most commonly results from the heart pumping against an elevated afterload, as in hypertension and aortic stenosis. Another notable cause is increased filling of the left ventricle (ie, diastolic overload), which is the underlying mechanism for LVH in patients with aortic or mitral regurgitation and dilated cardiomyopathy. Coronary artery disease can also play a role in the pathogenesis of LVH, as the normal myocardium attempts to compensate for the ischemic or infarcted tissue.13
The development of myocardial fibrosis appears to be pathophysiologically linked to the renin-angiotensin-aldosterone system. Specifically, there is evidence that angiotensin II has a profibrotic effect on the myocardium of hypertensive patients.15 This may explain why angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are among the most potent agents for treating LVH, as we will discuss later in this review.
DIAGNOSIS BY ELECTROCARDIOGRAPHY, ECHOCARDIOGRAPHY, OR MRI
Many different criteria for electrocardiographic LVH have been proposed over the years. Most use the voltage in one or more leads, with or without additional factors such as QRS duration, secondary ST-T wave abnormalities, or left atrial abnormalities. The most well known electrocardiographic criteria are the Cornell voltage,21 the Cornell product,22 the Sokolow-Lyon index,23 and the Romhilt-Estes point score system (Table 1).24
- Cornell voltage—median sensitivity 15%, median specificity 96%
- Cornell product—median sensitivity 19.5%, median specificity 91%
- Sokolow-Lyon voltage—median sensitivity 21%, median specificity 89%
- Romhilt-Estes point score—median sensitivity 17%, median specificity 95%.
Of note, the ranges of the published values were extremely broad. For example, the ranges in sensitivity were:
- Cornell voltage—2% to 41%
- Cornell product—8% to 32%
- Sokolow-Lyon voltage—4% to 51%
- Romhilt-Estes point score—0% to 41%.
While the studies with the extreme values may have had issues of small sample size or poor study quality, the wide range in values may primarily be the result of diverse study populations as well as different validation methods and cutoff values to define LVH. Regardless, the overall message of high specificity and low sensitivity is indisputable.
Electrocardiography is insensitive for diagnosing LVH because it relies on measuring the electrical activity of the heart by electrodes on the surface of the skin to predict the left ventricular mass. The intracardiac electrical activity is problematic to measure externally because the measurements are affected by everything between the myocardium and the electrodes, most notably fat, fluid, and air. Because of this effect, electrocardiography underdiagnoses LVH in patients with obesity, pleural effusions, pericardial effusions, anasarca, or chronic obstructive pulmonary disease. In addition, the diagnosis of LVH by electrocardiography is strongly influenced by age and ethnicity.25–26
While electrocardiography is not sensitive and cannot be used to rule out LVH, it still has a role in its diagnosis and management. In the landmark Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study, regression of LVH (diagnosed electrocardiographically by the Sokolow-Lyon index or the Cornell product criteria) in response to losartan (Cozaar) improved cardiovascular outcomes independent of blood pressure.10 Based on this, it is reasonable that all hypertensive patients and other patients at risk of LVH who undergo electrocardiography be screened with these two criteria.
Echocardiography is the test of choice
Echocardiography, if available, should be the test of choice to assess for LVH. It is much more sensitive than electrocardiography and can also detect other abnormalities such as left ventricular dysfunction and valvular disease.
This test uses transthoracic or transesophageal ultrasonography to measure the left ventricular end-diastolic diameter, posterior wall thickness, and interventricular septum thickness. From these measurements and the patient’s height and weight, the left ventricular mass index can be calculated.27
Several different cutoff values for the left ventricular mass index have been proposed; the LIFE study used values of > 104 g/m2 in women and > 116 g/m2 in men to define LVH.
When using echocardiography to assess for LVH, it is imperative that the left ventricular mass index be used and not just the left ventricular wall thickness, as often happens in clinical practice. This is necessary because diagnosis by wall thickness alone is not a good indicator of LVH, with a concordance between wall thickness and a left ventricular mass index of only 60%.28 In addition, wall thickness tends to underestimate LVH in women and overestimate it in men.
Is echocardiography cost-effective?
Despite its clear advantages, an important consideration about echocardiography as a screening test for all hypertensive patients is its cost.
A suggested way to reduce cost is to measure the left ventricular mass index only.29 A limited echocardiographic examination is much less expensive than a complete two-dimensional echocardiogram ($255 vs $431 per the 2009 Medicare Ambulatory Payment Classification30) and should be the examination performed if the patient has no other clinical indication for echocardiography.
Another way to control cost is to stratify patients by risk and to do echocardiography only in those who would benefit most from it. Based on the prevalence of LVH, one study concluded that echocardiography is most cost-effective in men 50 years or older.31
Further study is necessary to more precisely define the cost-effectiveness of echocardiographic screening for LVH in terms of potentially preventable cardiovascular morbidity and death.
Cardiac MRI: The costly gold standard
Cardiac MRI is the gold standard test for LVH, as it is even more accurate and reproducible than echocardiography.32 It can precisely estimate a patient's left ventricular mass and assess for other structural cardiac abnormalities.
MRI’s use, however, is severely restricted in clinical practice due to its high cost and limited availability. While it may never be used for general screening for LVH, it certainly has a role in clinical research and for assessing cardiac anatomy in special clinical situations.
TREATMENT SHOULD INCLUDE AN ACE INHIBITOR OR ARB
Once LVH has been diagnosed, the next step is to decide on an appropriate treatment plan.
While the choice of therapy will always depend on other comorbidities, a 2003 metaanalysis of antihypertensive medications in the treatment of LVH (controlling for the degree of blood pressure lowering) showed that ARBs were the most efficacious class of agents for reducing the left ventricular mass.33 Specifically, ARBs decreased the mass by 13%, followed by calcium-channel blockers at 11%, ACE inhibitors at 10%, diuretics at 8%, and beta-blockers at 6%. In pairwise comparison, ARBs, calcium-channel blockers, and ACE inhibitors were all significantly more effective in reducing the left ventricular mass than beta-blockers.
As previously discussed, LVH appears to be pathophysiologically linked to myocardial fibrosis and the renin-angiotensin-aldosterone system. For this reason and based on the data presented above regarding the degree of LVH regression, ACE inhibitors or ARBs should be used as the first-line agents for LVH unless they are contraindicated in the individual patient.
The LIFE study
The LIFE study offers the strongest evidence that treating LVH is beneficial. It showed that in hypertensive patients with electrocardiographic LVH by the Cornell product or Sokolow-Lyon criteria, treatment with antihypertensive drugs that resulted in less-severe LVH on electrocardiography was associated with lower rates of cardiovascular morbidity and death, independent of the blood pressure achieved or the drug used.10
The end point in this study was a composite of stroke, myocardial infarction, and cardiovascular death. Regression of electrocardiographic LVH in hypertensive patients has also been shown to decrease the incidence of diabetes mellitus,34 atrial fibrillation,35 and hospitalizations for heart failure.36
The LIFE study also examined the prognostic implications of treating LVH detected by echocardiography. In this prospective cohort substudy, patients who had a lower left ventricular mass index during treatment with antihypertensive drugs had lower rates of cardiovascular morbidity and all-cause mortality, independent of the effects of blood pressure and treatment used.11
These results suggest that there may be a role not only for treating LVH, but also for monitoring for a reduction in the left ventricular mass index as a goal of therapy (similar to the way hemoglobin A1c is used in diabetic patients). If the index is used in this way, one could potentially adjust the dose of current drugs, switch classes, or add an additional drug based on a persistently elevated left ventricular mass index in order to optimize the patient's overall cardiovascular risk. A randomized controlled trial of therapy directed by the mass index vs conventional therapy of LVH would be necessary to assess the clinical utility of this approach.
RECOMMENDATIONS
LVH is a common and potentially modifiable cardiovascular risk factor often overlooked in clinical practice. Ideally, all hypertensive patients should be screened with echocardiography to look for LVH, using the calculated left ventricular mass index rather than wall thickness alone to make the diagnosis. While electrocardiography is specific and also has prognostic implications, it is not sensitive enough to be used alone to screen for LVH.
Once the diagnosis of LVH is made, the initial therapy should be an ARB or an ACE inhibitor. Response to therapy can be assessed by monitoring for a reduction in left ventricular mass index or regression of electrocardiographic LVH.
Treatment-induced regression of LVH decreases adverse cardiovascular events and improves overall survival. When modifying medications in hypertensive patients, it is important to remember that the treatment of LVH is not synonymous with blood pressure control.
- Casale PN, Devereux RB, Milner M, et al. Value of echocardiographic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105:173–178.
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566.
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352.
- Verdecchia P, Carini G, Circo A, et al; MAVI (MAssa Ventricolare sinistra nell’Ipertensione) Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 2001; 38:1829–1835.
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998; 32:1454–1459.
- Verdecchia P, Porcellati C, Reboldi G, et al. Left ventricular hypertrophy as an independent predictor of acute cerebrovascular events in essential hypertension. Circulation 2001; 104:2039–2044.
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35:580–586.
- Verdecchia P, Schillaci G, Borgioni C, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation 1998; 97:48–54.
- Mathew J, Sleight P, Lonn E, et al; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621.
- Okin PM, Devereux RB, Jern S, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004; 292:2343–2349.
- Devereux RB, Wachtell K, Gerdts E, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292:2350–2356.
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470–479.
- Zabalgoitia M, Berning J, Koren MJ, et al; LIFE Study Investigators. Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 2001; 88:646–650.
- Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol 1990; 65:1G–7G.
- González A, López B, Querejeta R, Díez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 2002; 34:1585–1593.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320.
- Liebson PR, Grandits G, Prineas R, et al. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993; 87:476–486.
- Martinez MA, Sancho T, Armada E, et al; Vascular Risk Working Group Grupo Monitorizacíon Ambulatoria de la Presión Arterial (MAPA)-Madrid. Prevalence of left ventricular hypertrophy in patients with mild hypertension in primary care: impact of echocardiography on cardiovascular risk stratification. Am J Hypertens 2003; 16:556–563.
- Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ 2007; 335:711.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990; 81:1144–1146.
- Casale PN, Devereux RB, Kligfield P, et al. Electrocardiographic detection of left ventricular hypertrophy: development and prospective validation of improved criteria. J Am Coll Cardiol 1985; 6:572–580.
- Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol 1992; 20:1180–1186.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949; 37:161–186.
- Romhilt DW, Estes EH. A point-score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968; 75:752–758.
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation 1990; 81:815–820.
- Okin PM, Wright JT, Nieminen MS, et al. Ethnic differences in electrocardiographic criteria for left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint. Am J Hypertens 2002; 15:663–671.
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986; 57:450–458.
- Leibowitz D, Planer D, Ben-Ibgi F, Rott D, Weiss AT, Bursztyn M. Measurement of wall thickness alone does not accurately assess the presence of left ventricular hypertrophy. Clin Exp Hypertens 2007; 29:119–125.
- Black HR, Weltin G, Jaffe CC. The limited echocardiogram: a modification of standard echocardiography for use in the routine evaluation of patients with systemic hypertension. Am J Cardiol 1991; 67:1027–1030.
- American Society of Echocardiography Coding and Reimbursement Newsletter, January 2009. http://www.asecho.org/files/public/CodingnewsJan09.pdf. Accessed May 13, 2010.
- Cuspidi C, Meani S, Valerio C, Fusi V, Sala C, Zanchetti A. Left ventricular hypertrophy and cardiovascular risk stratification: impact and cost-effectiveness of echocardiography in recently diagnosed essential hypertensives. J Hypertens 2006; 24:1671–1677.
- Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, Gottdiener JS. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 1995; 8:221–228.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003; 115:41–46.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. In-treatment resolution or absence of electrocardiographic left ventricular hypertrophy is associated with decreased incidence of new-onset diabetes mellitus in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2007; 50:984–990.
- Okin PM, Wachtell K, Devereux RB, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006; 296:1242–1248.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Ann Intern Med 2007; 147:311–319.
- Casale PN, Devereux RB, Milner M, et al. Value of echocardiographic measurement of left ventricular mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med 1986; 105:173–178.
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322:1561–1566.
- Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 1991; 114:345–352.
- Verdecchia P, Carini G, Circo A, et al; MAVI (MAssa Ventricolare sinistra nell’Ipertensione) Study Group. Left ventricular mass and cardiovascular morbidity in essential hypertension: the MAVI study. J Am Coll Cardiol 2001; 38:1829–1835.
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol 1998; 32:1454–1459.
- Verdecchia P, Porcellati C, Reboldi G, et al. Left ventricular hypertrophy as an independent predictor of acute cerebrovascular events in essential hypertension. Circulation 2001; 104:2039–2044.
- Schillaci G, Verdecchia P, Porcellati C, Cuccurullo O, Cosco C, Perticone F. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension 2000; 35:580–586.
- Verdecchia P, Schillaci G, Borgioni C, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation 1998; 97:48–54.
- Mathew J, Sleight P, Lonn E, et al; Heart Outcomes Prevention Evaluation (HOPE) Investigators. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation 2001; 104:1615–1621.
- Okin PM, Devereux RB, Jern S, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 2004; 292:2343–2349.
- Devereux RB, Wachtell K, Gerdts E, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA 2004; 292:2350–2356.
- Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470–479.
- Zabalgoitia M, Berning J, Koren MJ, et al; LIFE Study Investigators. Impact of coronary artery disease on left ventricular systolic function and geometry in hypertensive patients with left ventricular hypertrophy (the LIFE study). Am J Cardiol 2001; 88:646–650.
- Weber KT, Janicki JS, Pick R, Capasso J, Anversa P. Myocardial fibrosis and pathologic hypertrophy in the rat with renovascular hypertension. Am J Cardiol 1990; 65:1G–7G.
- González A, López B, Querejeta R, Díez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol 2002; 34:1585–1593.
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320.
- Liebson PR, Grandits G, Prineas R, et al. Echocardiographic correlates of left ventricular structure among 844 mildly hypertensive men and women in the Treatment of Mild Hypertension Study (TOMHS). Circulation 1993; 87:476–486.
- Martinez MA, Sancho T, Armada E, et al; Vascular Risk Working Group Grupo Monitorizacíon Ambulatoria de la Presión Arterial (MAPA)-Madrid. Prevalence of left ventricular hypertrophy in patients with mild hypertension in primary care: impact of echocardiography on cardiovascular risk stratification. Am J Hypertens 2003; 16:556–563.
- Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ 2007; 335:711.
- Devereux RB. Is the electrocardiogram still useful for detection of left ventricular hypertrophy? Circulation 1990; 81:1144–1146.
- Casale PN, Devereux RB, Kligfield P, et al. Electrocardiographic detection of left ventricular hypertrophy: development and prospective validation of improved criteria. J Am Coll Cardiol 1985; 6:572–580.
- Molloy TJ, Okin PM, Devereux RB, Kligfield P. Electrocardiographic detection of left ventricular hypertrophy by the simple QRS voltage-duration product. J Am Coll Cardiol 1992; 20:1180–1186.
- Sokolow M, Lyon TP. The ventricular complex in left ventricular hypertrophy as obtained by unipolar precordial and limb leads. Am Heart J 1949; 37:161–186.
- Romhilt DW, Estes EH. A point-score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968; 75:752–758.
- Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation 1990; 81:815–820.
- Okin PM, Wright JT, Nieminen MS, et al. Ethnic differences in electrocardiographic criteria for left ventricular hypertrophy: the LIFE study. Losartan Intervention For Endpoint. Am J Hypertens 2002; 15:663–671.
- Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986; 57:450–458.
- Leibowitz D, Planer D, Ben-Ibgi F, Rott D, Weiss AT, Bursztyn M. Measurement of wall thickness alone does not accurately assess the presence of left ventricular hypertrophy. Clin Exp Hypertens 2007; 29:119–125.
- Black HR, Weltin G, Jaffe CC. The limited echocardiogram: a modification of standard echocardiography for use in the routine evaluation of patients with systemic hypertension. Am J Cardiol 1991; 67:1027–1030.
- American Society of Echocardiography Coding and Reimbursement Newsletter, January 2009. http://www.asecho.org/files/public/CodingnewsJan09.pdf. Accessed May 13, 2010.
- Cuspidi C, Meani S, Valerio C, Fusi V, Sala C, Zanchetti A. Left ventricular hypertrophy and cardiovascular risk stratification: impact and cost-effectiveness of echocardiography in recently diagnosed essential hypertensives. J Hypertens 2006; 24:1671–1677.
- Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, Gottdiener JS. Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 1995; 8:221–228.
- Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003; 115:41–46.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. In-treatment resolution or absence of electrocardiographic left ventricular hypertrophy is associated with decreased incidence of new-onset diabetes mellitus in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) Study. Hypertension 2007; 50:984–990.
- Okin PM, Wachtell K, Devereux RB, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006; 296:1242–1248.
- Okin PM, Devereux RB, Harris KE, et al; LIFE Study Investigators. Regression of electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Ann Intern Med 2007; 147:311–319.
KEY POINTS
- LVH is caused by a chronically increased cardiac workload, most commonly from hypertension.
- Ideally, all hypertensive patients should undergo echocardiography to screen for LVH, using the calculated left ventricular mass index.
- Electrocardiography is too insensitive to be used alone to screen for LVH.
- In hypertensive patients, initial therapy of LVH should consist of an angiotensin II receptor blocker or an angiotensin-converting enzyme inhibitor.
- Treatment-induced regression of LVH improves cardiovascular outcomes independent of blood pressure.
- Further study is necessary to examine the utility of following the left ventricular mass index as a treatment goal.
Laryngopharyngeal reflux: More questions than answers
The scenario is common: a patient complains of chronic hoarseness, cough, throat-clearing, sore throat, dysphagia, or a lump in the throat and undergoes laryngoscopy. If this test rules out cancer, the patient is given a diagnosis of laryngopharyngeal reflux (LPR), ie, a form of gastroesophageal reflux disease (GERD) in which the stomach contents get all the way up into the pharynx and down into the larynx. A proton pump inhibitor (PPI) is often prescribed, usually twice daily for 2 months.1–6

In this article, we review the current understanding of the pathophysiology of LPR and evaluate current diagnostic tests and treatment regimens for patients with suspected LPR.
THE PATHOPHYSIOLOGY OF LPR IS POORLY UNDERSTOOD
Transient relaxation of the lower esophageal sphincter
In a study in 10 healthy volunteers, Dent et al7 found that the pressure in the lower esophageal sphincter varies considerably over a 12-hour period. Episodes of reflux were not related to low basal (resting) pressure. Rather, 70% to 100% of reflux episodes occurred during random episodes of transient, complete, and inappropriate relaxation of the sphincter that lasted about 5 to 30 seconds. The mechanism of this relaxation is not known but is thought to be related to activation of the vagus nerve, possibly as a consequence of gastric distention.8
Gastric, not duodenal products seem to cause the damage
In a study in dogs, Adhami et al9 evaluated the possible role of gastric juices (acid and pepsin) vs duodenal juices (bile acids and trypsin) in laryngeal tissue damage. After taking baseline biopsy samples of the larynx, the investigators applied a variety of gastric and duodenal enzymes at varying pH levels (pH 1–7) to the larynxes. After 9 to 12 applications, they took another biopsy and assessed the changes visually and histologically.
At low (ie, acidic) pH levels, pepsin and conjugated bile acids were the most injurious, causing erythema and histologic evidence of inflammation. The authors concluded that gastric and not duodenal substances cause laryngeal injury and that acid-suppressive therapy “should eliminate the injurious potential” of acid reflux.9
The larynx is more sensitive than the esophagus
Monitoring of esophageal pH has shown that healthy people can tolerate as many as 50 episodes a day of acid reflux (pH < 4) in the esophagus. However, Koufman10 found that as few as three episodes of laryngeal reflux per week can cause severe laryngeal inflammation and injury.
Does pepsin deplete buffers, worsening acid damage?
Johnston et al11 took biopsies from a control group of healthy volunteers and from patients diagnosed with LPR. They detected pepsin in the samples from eight of the nine patients with LPR but in none of the controls. Furthermore, the tissue from patients with LPR had low levels of carbonic anhydrase III. The authors hypothesized that pepsin depletes the laryngopharynx of carbonic anhydrase III, and that therefore these tissues cannot produce enough bicarbonate to buffer the gastric acid. Less bicarbonate would mean greater acidity, so the pepsin would remain active and would be more likely to cause cellular damage.11
However, this contention is controversial. What is universally agreed upon is that reflux of gastric or gastroduodenal contents is most likely causing injury, most likely through direct exposure, although indirect effects through vagal mechanisms cannot be ruled out.
CURRENT DIAGNOSTIC TESTS FOR LPR HAVE SHORTCOMINGS
A careful history is important. Many patients report they have sore throat, hoarseness, cough, dysphasia, or chronic throat-clearing.13 Factors that may predispose a patient to esophageal reflux should be discussed, eg:
- Tobacco use
- Diet (eg, soda, spicy foods, fatty foods)
- Alcohol use
- Certain drugs (calcium channel blockers, nitrates, steroids).
Up to 50% of patients presenting with extraesophageal symptoms may not have classic reflux symptoms such as heartburn and regur-gitation.14 However, the existence of “silent reflux” is currently controversial.
Laryngoscopy is nonspecific and subjective
Because the key symptoms of LPR are nonspecific, many patients who present to an otolaryngologist undergo laryngoscopy, mainly to rule out malignancy. Once cancer is ruled out, many patients are given a diagnosis of LPR.
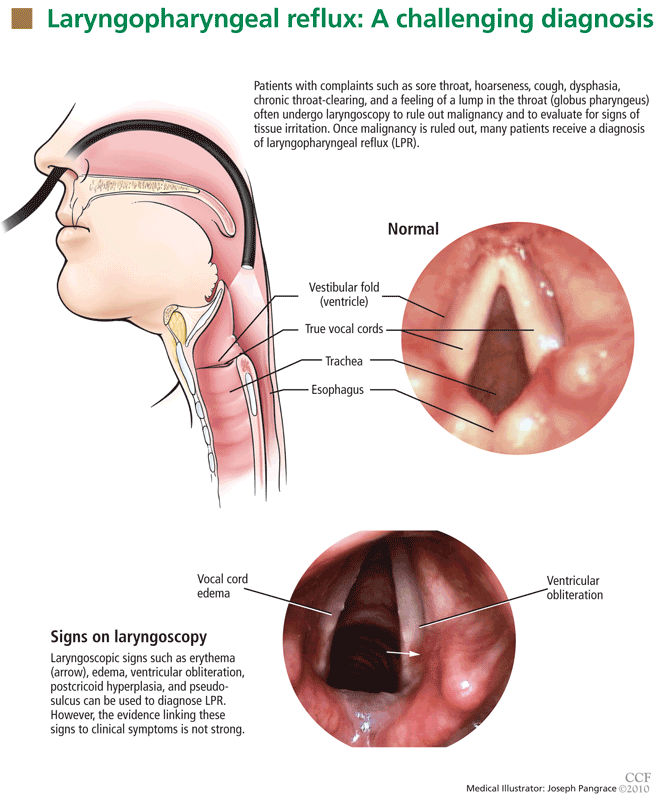
Furthermore, Branski et al17 performed transoral rigid laryngoscopy with videorecording in 100 consecutive patients presenting with a chief complaint of dysphonia. Five board-certified otolaryngologists individually viewed each recording, scored the degree of erythema and edema, and assessed the likelihood that LPR played a role in dysphonia and the severity of the LPR findings. The physicians’ ratings showed considerable interobserver variability. In other words, this study showed that laryngeal findings are often nonspecific and that the laryngoscopic diagnosis of LPR tends to be subjective.17
The Reflux Finding Score. Concerned by the lack of consistency in the diagnosis of LPR, Belafsky et al18 created a scoring system for documenting the physical findings and severity of disease on a standardized scale. Their Reflux Finding Score is based on eight laryngoscopic findings: subglottic edema, ventricular edema, erythema, vocal cord edema, diffuse laryngeal edema, hypertrophy of the posterior commissure, granuloma or granulation tissue, and thick endolaryngeal mucus. The total score can range from 0 (best) to 26 (worst).
In 40 patients with LPR confirmed by pH monitoring, the mean score was 11.5, compared with 5.2 in 40 age-matched controls. The authors calculated they could be 95% certain that a person with a score higher than 7 has LPR.18
However, this diagnostic method has not been validated in a large-scale randomized trial and so has yet to be incorporated into routine otolaryngology practice.
Ambulatory pH monitoring is not so golden for diagnosing LPR
Although pH monitoring was once the gold standard for diagnosing reflux, it has since been shown to be unreliable in patients who have laryngeal symptoms.4
How high or low in the esophagus the probe is placed is clearly critical for useful results. 4 But the test is subject to variability: different physicians place the probe in different locations, and the probe may shift. Another problem is that reflux may occur during untested periods.19
A pH of less than 4 in the esophagus had originally been shown to have high sensitivity and specificity,20 but Reichel and Issing21 suggested using a pH of less than 5 as the cutoff, which would identify more patients as having LPR. Further trials are needed to more precisely determine the pH threshold for the diagnosis of LPR.
Enthusiasm is waning for pharyngeal pH monitoring
In LPR, it was initially thought that pH monitoring in the pharynx was more accurate than in the distal or proximal esophagus.
Shaker et al22 monitored the pH in the pharynx, proximal esophagus, and distal esophagus in four groups: 14 patients who had both laryngeal signs and symptoms, 12 patients who had laryngeal symptoms only, 16 patients who had GERD but no laryngeal symptoms, and 12 healthy volunteers. They found that pharyngeal reflux was more frequent and in greater quantity in patients with laryngeal signs and symptoms than in the other groups. This study suggested that pharyngeal pH monitoring may be useful in diagnosing LPR in patients who have laryngeal signs and symptoms.
However, hypopharyngeal pH monitoring has several problems. One issue is that, even in this trial, 2 of 12 healthy volunteers had episodes of pharyngeal reflux.22 In other studies, the rate of false-positive results ranged from 7% to 17%.23,24 Additionally, in 12 previous studies, only 54% of 1,217 patients with suspected LPR had esophageal acid exposure, regardless of where the pH probe was placed.25
More importantly, another study found that patients with pharyngeal reflux documented by pH monitoring were no more likely to respond to acid-suppressive therapy than patients with no documented reflux.26 These findings dampen the enthusiasm for pharyngeal pH monitoring in LPR.
Impedance monitoring on therapy may be useful in refractory cases
Esophageal impedance monitoring, a newer test, uses a catheter that measures electrical resistance (impedance) between different points along the esophagus. Thus, it can detect the reflux of acid and nonacid liquid or gaseous material.
Pritchett et al27 performed esophageal impedance and pH monitoring in 39 patients who were on twice-daily PPI therapy and then evaluated the same patients with wireless pH monitoring while they were off therapy. The most prevalent complaint in the study group was cough (56%), followed by heartburn (18%) and sore throat (10%).
Of the 39 patients, 25 (64%) had normal results on impedance/pH monitoring while on therapy, ruling out reflux. On pH monitoring off therapy, 28 (72%) of the 39 patients had abnormal results; this group included 13 (93%) of the 14 patients who had abnormal results on impedance/pH monitoring while on therapy. The authors recommended on-therapy testing with impedance monitoring in patients with refractory reflux, since it provides more useful clinical information.27 If the results of impedance/pH monitoring are negative in these patients, a diagnosis other than reflux should be considered.
EMPIRIC PPI TREATMENT HAS SHOWN DISAPPOINTING RESULTS
Because laryngoscopy and pH monitoring are not very sensitive or specific for LPR, experts recommend empiric therapy with a PPI twice daily. However, the results have been disappointing when PPIs were compared with placebo in clinical trials.
In a randomized controlled trial,28 we found that patients who had complaints of chronic throat-clearing, cough, globus, sore throat, and hoarseness had a similar response to twice-daily esomeprazole (Nexium) compared with placebo: their primary symptom had resolved by 16 weeks in 14.7% of the esomeprazole group vs 16.0% of the placebo group (P = .799). Similarly, the final findings on laryngoscopy such as edema, erythema, and surface irregularity were not significantly different between groups.
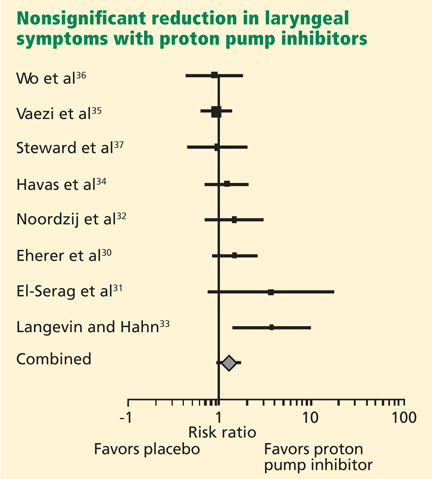
Adding a histamine-2 receptor antagonist is not recommended
Adding a histamine-2 receptor antagonist to PPI therapy has also been considered as a treatment for LPR.
Fackler et al38 studied 16 GERD patients and 18 healthy volunteers to determine if adding ranitidine (Pepcid) to the PPI omeprazole (Prilosec) could improve GERD symptoms. Patients underwent baseline manometry and then gastroesophageal pH monitoring before starting the drugs. They first received omeprazole 20 mg twice daily alone for 2 weeks, and then added ranitidine 300 mg at bedtime. A pH test was done again after the first day of treatment with ranitidine, at the end of 1 week of combination therapy, and after 4 weeks of combination therapy. The combination reduced nocturnal acid breakthrough on day 1; however, due to tolerance to ranitidine, no significant difference in acid suppression was seen after 1 week of therapy. Therefore, this combination is not recommended.
Surgery is not recommended either
Some experts have argued for surgical fundoplication in patients whose symptoms persist despite drug therapy.
Swoger et al39 treated 72 patients who had symptoms consistent with LPR with a PPI for 4 months; 25 patients in this group had less than a 50% improvement despite maximal drug therapy. Ten of these patients underwent surgical fundoplication, and 15 remained on drug therapy alone. At 1 year of follow-up, only one surgical patient (10%) reported improvement in laryngeal symptoms.
In view of this report and prior studies of surgical fundoplication,40 surgery is not recommended for patients whose symptoms do not respond to aggressive PPI therapy.
IF A PPI FAILS, LOOK FOR OTHER CAUSES OF SYMPTOMS
Although gastroesophageal reflux and laryngeal signs and symptoms have been associated with one another, this relation may have been overstated, leading to the overdiagnosis of LPR.
The diagnosis of LPR is difficult, as laryngoscopy has high interrater variability and as the results of pH monitoring do not dependably predict who will respond to treatment.
Because PPI therapy is easy and appears to be safe, patients with extraesophageal symptoms thought to be related to reflux should undergo a trial of twice-daily PPI therapy for at least 2 months. If the patient responds to therapy, then tapering to once-daily therapy initially and then to minimal acid suppression to control symptoms would be prudent.
In patients who show no improvement, other causes of symptoms should be explored. Diseases that can mimic LPR include postnasal drip, allergies, sinus inflammation, and various pulmonary diseases. These patients should also be advised to adopt lifestyle modifications—eg, to stop smoking, lose weight, and decrease activities that cause stress on the voice. Surgery is not likely to provide any benefit in this situation. The patient should be tapered off the PPI to make sure no rebound acid reflux occurs.
- Nebel OT, Fornes MF, Castell DO. Symptomatic gastroesophageal reflux: incidence and precipitating factors. Am J Dig Dis 1976; 21:953–956.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute; Clinical Practice and Quality Management Committee. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Jonaitis L, Pribuisiene R, Kupcinskas L, Uloza V. Laryngeal examination is superior to endoscopy in the diagnosis of the laryngopharyngeal form of gastroesophageal reflux disease. Scand J Gastroenterol 2006; 41:131–137.
- Vaezi MF, Hicks DM, Abelson TI, Richter JE. Laryngeal signs and symptoms and gastroesophageal reflux disease (GERD): a critical assessment of cause and effect association. Clin Gastroenterol Hepatol 2003; 1:333–344.
- Jaspersen D, Kulig M, Labenz J, et al. Prevalence of extra-oesophageal manifestations in gastro-oesophageal reflux disease: an analysis based on the ProGERD study. Aliment Pharmacol Ther 2003; 17:1515–1520.
- Karkos PD, Benton J, Leong SC, et al. Trends in laryngopharyngeal reflux: a British ENT survey. Eur Arch Otorhinolaryngol 2007; 264:513–517.
- Dent J, Dodds WJ, Friedman RH, et al. Mechanism of gastroesophageal reflux in recumbent asymptomatic human subjects. J Clin Invest 1980; 65:256–267.
- Schreiber S, Garten D, Sudhoff H, et al. Pathophysiological mechanisms of extraesophageal reflux in otolaryngeal disorders. Eur Arch Otorhinolaryngol 2009; 266:17–24.
- Adhami T, Goldblum JR, Richter JE, Vaezi MF. The role of gastric and duodenal agents in laryngeal injury: an experimental canine model. Am J Gastroenterol 2004; 99:2098–2106.
- Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24-hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope 1991; 101(4 pt 2 suppl 53):1–78.
- Johnston N, Knight J, Dettmar PW, Lively MO, Koufman J. Pepsin and carbonic anhydrase isoenzyme III as diagnostic markers for laryngopharyngeal reflux disease. Laryngoscope 2004; 114:2129–2134.
- Vaezi MF. Therapy insight: gastroesophageal reflux disease and laryngopharyngeal reflux. Nat Clin Pract Gastroenterol Hepatol 2005; 2:595–603.
- Farrokhi F, Vaezi MF. Extra-esophageal manifestations of gastroesophageal reflux. Oral Dis 2007; 13:349–359.
- Koufman JA. Laryngopharyngeal reflux is different from classic gastroesophageal reflux disease. Ear Nose Throat J 2002; 81( 9 suppl 2):7–9.
- Koufman JA, Amin MR, Panetti M. Prevalence of reflux in 113 consecutive patients with laryngeal and voice disorders. Otolaryngol Head Neck Surg 2000; 123:385–388.
- Milstein CF, Charbel S, Hicks DM, Abelson TI, Richter JE, Vaezi MF. Prevalence of laryngeal irritation signs associated with reflux in asymptomatic volunteers: impact of endoscopic technique (rigid vs flexible scope). Laryngoscope 2005; 115;2256–2261.
- Branski RC, Bhattacharyya N, Shapiro J. The reliability of the assessment of endoscopic laryngeal findings associated with laryngopharyngeal reflux disease. Laryngoscope 2002; 112;1019–1024.
- Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS). Laryngoscope 2001; 111:1313–1317.
- Gupta R, Sataloff RT. Laryngopharyngeal reflux: current concepts and questions. Curr Opin Otolaryngol Head Neck Surg 2009; 17:143–148.
- Jamieson JR, Stein HJ, DeMeester TR, et al. Ambulatory 24-h esophageal pH monitoring: normal values, optimal thresholds, specificity, sensitivity, and reproducibility. Am J Gastroenterol 1992; 87:1102–1111.
- Reichel O, Issing WJ. Impact of different pH thresholds for 24-h dual probe pH monitoring in patients with suspected laryngopharyngeal reflux. J Laryngol Otol 2008; 122:485–489.
- Shaker R, Milbrath M, Ren J, et al. Esophagopharyngeal distribution of refluxed gastric acid in patients with reflux laryngitis. Gastroenterology 1995; 109:1575–1582.
- Jacob P, Kahrilas PJ, Herzon G. Proximal esophageal pH-metry in patients with “reflux laryngitis.” Gastroenterology 1991; 100:305–310.
- Eubanks TR, Omelanczuk PE, Maronian N, Hillel A, Pope CE, Pellegrini CA. Pharyngeal pH monitoring in 222 patients with suspected laryngeal reflux. J Gastrointest Surg 2001; 5:183–190.
- Vaezi MF. Gastroesophageal reflux disease and the larynx. J Clin Gastroenterol 2003; 36:198–203.
- Ulualp SO, Toohill RJ, Shaker R. Outcomes of acid suppressive therapy in patients with posterior laryngitis. Otolaryngol Head Neck Surg 2001; 124:16–22.
- Pritchett JM, Aslam M, Slaughter JC, Ness RM, Garrett CG, Vaezi MF. Efficacy of esophageal impedance/pH monitoring in patients with refractory gastroesophageal reflux disease, on and off therapy. Clin Gastroenterol Hepatol 2009; 7:743–748.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116;254–260.
- Qadeer MA, Phillips CO, Lopez AR, et al. Proton pump inhibitor therapy for suspected GERD-related chronic laryngitis: a meta analysis of randomized controlled trials. Am J Gastroenterol 2006; 101:2646–2654.
- Eherer AJ, Habermann W, Hammer HF, et al. Effect of pantoprazole on the course of reflux-associated laryngitis: a placebo-controlled double-blind crossover study. Scand J Gastroenterol 2003; 38:462–467.
- El-Serag HB, Lee P, Buchner A, et al. Lansoprazole treatment of patients with chronic idiopathic laryngitis: a placebo-controlled trial. Am J Gastroenterol 2001; 96:979–983.
- Noordzij JP, Khidr A, Evans BA, et al. Evaluation of omeprazole in the treatment of reflux laryngitis: a prospective, placebo-controlled, randomized, double-blind study. Laryngoscope 2001; 111:2147–2151.
- Langevin S, Hanh N. GERD-induced ENT symptoms: a prospective placebo controlled study with omeprazole 40 mg a day. Gastroenterology 2001; 120:A-16.
- Havas T, Huang S, Levy M, et al. Posterior pharyngolaryngitis. Double blind randomized placebo-controlled trial of proton pump inhibitor therapy. Aust J Otolaryng 1999; 3:243–246.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Wo JM, Koopman JI, Harrell SP, et al. Double-blind placebo-controlled trial with single-dose pantoprazole for laryngopharyngeal reflux. Am J Gastroenterol 2006; 101:1972–1978.
- Steward DL, Wilson KM, Kelly DH, et al. Proton pump inhibitor therapy for chronic laryngo-pharyngitis: a randomized placebo-control trial. Otolaryngol Head Neck Surg 2004; 131:342–350.
- Fackler WK, Ours TM, Vaezi MF, Richter JE. Long term effect of H2RA therapy on nocturnal gastric acid breakthrough. Gastroenterology 2002; 122:625–632.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- So JB, Zeitels SM, Rattner DW. Outcome of atypical symptoms attributed to gastroesophageal reflux treated by laparoscopic fundoplication. Surgery 1998; 124:28–32.
The scenario is common: a patient complains of chronic hoarseness, cough, throat-clearing, sore throat, dysphagia, or a lump in the throat and undergoes laryngoscopy. If this test rules out cancer, the patient is given a diagnosis of laryngopharyngeal reflux (LPR), ie, a form of gastroesophageal reflux disease (GERD) in which the stomach contents get all the way up into the pharynx and down into the larynx. A proton pump inhibitor (PPI) is often prescribed, usually twice daily for 2 months.1–6
In this article, we review the current understanding of the pathophysiology of LPR and evaluate current diagnostic tests and treatment regimens for patients with suspected LPR.
THE PATHOPHYSIOLOGY OF LPR IS POORLY UNDERSTOOD
Transient relaxation of the lower esophageal sphincter
In a study in 10 healthy volunteers, Dent et al7 found that the pressure in the lower esophageal sphincter varies considerably over a 12-hour period. Episodes of reflux were not related to low basal (resting) pressure. Rather, 70% to 100% of reflux episodes occurred during random episodes of transient, complete, and inappropriate relaxation of the sphincter that lasted about 5 to 30 seconds. The mechanism of this relaxation is not known but is thought to be related to activation of the vagus nerve, possibly as a consequence of gastric distention.8
Gastric, not duodenal products seem to cause the damage
In a study in dogs, Adhami et al9 evaluated the possible role of gastric juices (acid and pepsin) vs duodenal juices (bile acids and trypsin) in laryngeal tissue damage. After taking baseline biopsy samples of the larynx, the investigators applied a variety of gastric and duodenal enzymes at varying pH levels (pH 1–7) to the larynxes. After 9 to 12 applications, they took another biopsy and assessed the changes visually and histologically.
At low (ie, acidic) pH levels, pepsin and conjugated bile acids were the most injurious, causing erythema and histologic evidence of inflammation. The authors concluded that gastric and not duodenal substances cause laryngeal injury and that acid-suppressive therapy “should eliminate the injurious potential” of acid reflux.9
The larynx is more sensitive than the esophagus
Monitoring of esophageal pH has shown that healthy people can tolerate as many as 50 episodes a day of acid reflux (pH < 4) in the esophagus. However, Koufman10 found that as few as three episodes of laryngeal reflux per week can cause severe laryngeal inflammation and injury.
Does pepsin deplete buffers, worsening acid damage?
Johnston et al11 took biopsies from a control group of healthy volunteers and from patients diagnosed with LPR. They detected pepsin in the samples from eight of the nine patients with LPR but in none of the controls. Furthermore, the tissue from patients with LPR had low levels of carbonic anhydrase III. The authors hypothesized that pepsin depletes the laryngopharynx of carbonic anhydrase III, and that therefore these tissues cannot produce enough bicarbonate to buffer the gastric acid. Less bicarbonate would mean greater acidity, so the pepsin would remain active and would be more likely to cause cellular damage.11
However, this contention is controversial. What is universally agreed upon is that reflux of gastric or gastroduodenal contents is most likely causing injury, most likely through direct exposure, although indirect effects through vagal mechanisms cannot be ruled out.
CURRENT DIAGNOSTIC TESTS FOR LPR HAVE SHORTCOMINGS
A careful history is important. Many patients report they have sore throat, hoarseness, cough, dysphasia, or chronic throat-clearing.13 Factors that may predispose a patient to esophageal reflux should be discussed, eg:
- Tobacco use
- Diet (eg, soda, spicy foods, fatty foods)
- Alcohol use
- Certain drugs (calcium channel blockers, nitrates, steroids).
Up to 50% of patients presenting with extraesophageal symptoms may not have classic reflux symptoms such as heartburn and regur-gitation.14 However, the existence of “silent reflux” is currently controversial.
Laryngoscopy is nonspecific and subjective
Because the key symptoms of LPR are nonspecific, many patients who present to an otolaryngologist undergo laryngoscopy, mainly to rule out malignancy. Once cancer is ruled out, many patients are given a diagnosis of LPR.
Furthermore, Branski et al17 performed transoral rigid laryngoscopy with videorecording in 100 consecutive patients presenting with a chief complaint of dysphonia. Five board-certified otolaryngologists individually viewed each recording, scored the degree of erythema and edema, and assessed the likelihood that LPR played a role in dysphonia and the severity of the LPR findings. The physicians’ ratings showed considerable interobserver variability. In other words, this study showed that laryngeal findings are often nonspecific and that the laryngoscopic diagnosis of LPR tends to be subjective.17
The Reflux Finding Score. Concerned by the lack of consistency in the diagnosis of LPR, Belafsky et al18 created a scoring system for documenting the physical findings and severity of disease on a standardized scale. Their Reflux Finding Score is based on eight laryngoscopic findings: subglottic edema, ventricular edema, erythema, vocal cord edema, diffuse laryngeal edema, hypertrophy of the posterior commissure, granuloma or granulation tissue, and thick endolaryngeal mucus. The total score can range from 0 (best) to 26 (worst).
In 40 patients with LPR confirmed by pH monitoring, the mean score was 11.5, compared with 5.2 in 40 age-matched controls. The authors calculated they could be 95% certain that a person with a score higher than 7 has LPR.18
However, this diagnostic method has not been validated in a large-scale randomized trial and so has yet to be incorporated into routine otolaryngology practice.
Ambulatory pH monitoring is not so golden for diagnosing LPR
Although pH monitoring was once the gold standard for diagnosing reflux, it has since been shown to be unreliable in patients who have laryngeal symptoms.4
How high or low in the esophagus the probe is placed is clearly critical for useful results. 4 But the test is subject to variability: different physicians place the probe in different locations, and the probe may shift. Another problem is that reflux may occur during untested periods.19
A pH of less than 4 in the esophagus had originally been shown to have high sensitivity and specificity,20 but Reichel and Issing21 suggested using a pH of less than 5 as the cutoff, which would identify more patients as having LPR. Further trials are needed to more precisely determine the pH threshold for the diagnosis of LPR.
Enthusiasm is waning for pharyngeal pH monitoring
In LPR, it was initially thought that pH monitoring in the pharynx was more accurate than in the distal or proximal esophagus.
Shaker et al22 monitored the pH in the pharynx, proximal esophagus, and distal esophagus in four groups: 14 patients who had both laryngeal signs and symptoms, 12 patients who had laryngeal symptoms only, 16 patients who had GERD but no laryngeal symptoms, and 12 healthy volunteers. They found that pharyngeal reflux was more frequent and in greater quantity in patients with laryngeal signs and symptoms than in the other groups. This study suggested that pharyngeal pH monitoring may be useful in diagnosing LPR in patients who have laryngeal signs and symptoms.
However, hypopharyngeal pH monitoring has several problems. One issue is that, even in this trial, 2 of 12 healthy volunteers had episodes of pharyngeal reflux.22 In other studies, the rate of false-positive results ranged from 7% to 17%.23,24 Additionally, in 12 previous studies, only 54% of 1,217 patients with suspected LPR had esophageal acid exposure, regardless of where the pH probe was placed.25
More importantly, another study found that patients with pharyngeal reflux documented by pH monitoring were no more likely to respond to acid-suppressive therapy than patients with no documented reflux.26 These findings dampen the enthusiasm for pharyngeal pH monitoring in LPR.
Impedance monitoring on therapy may be useful in refractory cases
Esophageal impedance monitoring, a newer test, uses a catheter that measures electrical resistance (impedance) between different points along the esophagus. Thus, it can detect the reflux of acid and nonacid liquid or gaseous material.
Pritchett et al27 performed esophageal impedance and pH monitoring in 39 patients who were on twice-daily PPI therapy and then evaluated the same patients with wireless pH monitoring while they were off therapy. The most prevalent complaint in the study group was cough (56%), followed by heartburn (18%) and sore throat (10%).
Of the 39 patients, 25 (64%) had normal results on impedance/pH monitoring while on therapy, ruling out reflux. On pH monitoring off therapy, 28 (72%) of the 39 patients had abnormal results; this group included 13 (93%) of the 14 patients who had abnormal results on impedance/pH monitoring while on therapy. The authors recommended on-therapy testing with impedance monitoring in patients with refractory reflux, since it provides more useful clinical information.27 If the results of impedance/pH monitoring are negative in these patients, a diagnosis other than reflux should be considered.
EMPIRIC PPI TREATMENT HAS SHOWN DISAPPOINTING RESULTS
Because laryngoscopy and pH monitoring are not very sensitive or specific for LPR, experts recommend empiric therapy with a PPI twice daily. However, the results have been disappointing when PPIs were compared with placebo in clinical trials.
In a randomized controlled trial,28 we found that patients who had complaints of chronic throat-clearing, cough, globus, sore throat, and hoarseness had a similar response to twice-daily esomeprazole (Nexium) compared with placebo: their primary symptom had resolved by 16 weeks in 14.7% of the esomeprazole group vs 16.0% of the placebo group (P = .799). Similarly, the final findings on laryngoscopy such as edema, erythema, and surface irregularity were not significantly different between groups.
Adding a histamine-2 receptor antagonist is not recommended
Adding a histamine-2 receptor antagonist to PPI therapy has also been considered as a treatment for LPR.
Fackler et al38 studied 16 GERD patients and 18 healthy volunteers to determine if adding ranitidine (Pepcid) to the PPI omeprazole (Prilosec) could improve GERD symptoms. Patients underwent baseline manometry and then gastroesophageal pH monitoring before starting the drugs. They first received omeprazole 20 mg twice daily alone for 2 weeks, and then added ranitidine 300 mg at bedtime. A pH test was done again after the first day of treatment with ranitidine, at the end of 1 week of combination therapy, and after 4 weeks of combination therapy. The combination reduced nocturnal acid breakthrough on day 1; however, due to tolerance to ranitidine, no significant difference in acid suppression was seen after 1 week of therapy. Therefore, this combination is not recommended.
Surgery is not recommended either
Some experts have argued for surgical fundoplication in patients whose symptoms persist despite drug therapy.
Swoger et al39 treated 72 patients who had symptoms consistent with LPR with a PPI for 4 months; 25 patients in this group had less than a 50% improvement despite maximal drug therapy. Ten of these patients underwent surgical fundoplication, and 15 remained on drug therapy alone. At 1 year of follow-up, only one surgical patient (10%) reported improvement in laryngeal symptoms.
In view of this report and prior studies of surgical fundoplication,40 surgery is not recommended for patients whose symptoms do not respond to aggressive PPI therapy.
IF A PPI FAILS, LOOK FOR OTHER CAUSES OF SYMPTOMS
Although gastroesophageal reflux and laryngeal signs and symptoms have been associated with one another, this relation may have been overstated, leading to the overdiagnosis of LPR.
The diagnosis of LPR is difficult, as laryngoscopy has high interrater variability and as the results of pH monitoring do not dependably predict who will respond to treatment.
Because PPI therapy is easy and appears to be safe, patients with extraesophageal symptoms thought to be related to reflux should undergo a trial of twice-daily PPI therapy for at least 2 months. If the patient responds to therapy, then tapering to once-daily therapy initially and then to minimal acid suppression to control symptoms would be prudent.
In patients who show no improvement, other causes of symptoms should be explored. Diseases that can mimic LPR include postnasal drip, allergies, sinus inflammation, and various pulmonary diseases. These patients should also be advised to adopt lifestyle modifications—eg, to stop smoking, lose weight, and decrease activities that cause stress on the voice. Surgery is not likely to provide any benefit in this situation. The patient should be tapered off the PPI to make sure no rebound acid reflux occurs.
The scenario is common: a patient complains of chronic hoarseness, cough, throat-clearing, sore throat, dysphagia, or a lump in the throat and undergoes laryngoscopy. If this test rules out cancer, the patient is given a diagnosis of laryngopharyngeal reflux (LPR), ie, a form of gastroesophageal reflux disease (GERD) in which the stomach contents get all the way up into the pharynx and down into the larynx. A proton pump inhibitor (PPI) is often prescribed, usually twice daily for 2 months.1–6
In this article, we review the current understanding of the pathophysiology of LPR and evaluate current diagnostic tests and treatment regimens for patients with suspected LPR.
THE PATHOPHYSIOLOGY OF LPR IS POORLY UNDERSTOOD
Transient relaxation of the lower esophageal sphincter
In a study in 10 healthy volunteers, Dent et al7 found that the pressure in the lower esophageal sphincter varies considerably over a 12-hour period. Episodes of reflux were not related to low basal (resting) pressure. Rather, 70% to 100% of reflux episodes occurred during random episodes of transient, complete, and inappropriate relaxation of the sphincter that lasted about 5 to 30 seconds. The mechanism of this relaxation is not known but is thought to be related to activation of the vagus nerve, possibly as a consequence of gastric distention.8
Gastric, not duodenal products seem to cause the damage
In a study in dogs, Adhami et al9 evaluated the possible role of gastric juices (acid and pepsin) vs duodenal juices (bile acids and trypsin) in laryngeal tissue damage. After taking baseline biopsy samples of the larynx, the investigators applied a variety of gastric and duodenal enzymes at varying pH levels (pH 1–7) to the larynxes. After 9 to 12 applications, they took another biopsy and assessed the changes visually and histologically.
At low (ie, acidic) pH levels, pepsin and conjugated bile acids were the most injurious, causing erythema and histologic evidence of inflammation. The authors concluded that gastric and not duodenal substances cause laryngeal injury and that acid-suppressive therapy “should eliminate the injurious potential” of acid reflux.9
The larynx is more sensitive than the esophagus
Monitoring of esophageal pH has shown that healthy people can tolerate as many as 50 episodes a day of acid reflux (pH < 4) in the esophagus. However, Koufman10 found that as few as three episodes of laryngeal reflux per week can cause severe laryngeal inflammation and injury.
Does pepsin deplete buffers, worsening acid damage?
Johnston et al11 took biopsies from a control group of healthy volunteers and from patients diagnosed with LPR. They detected pepsin in the samples from eight of the nine patients with LPR but in none of the controls. Furthermore, the tissue from patients with LPR had low levels of carbonic anhydrase III. The authors hypothesized that pepsin depletes the laryngopharynx of carbonic anhydrase III, and that therefore these tissues cannot produce enough bicarbonate to buffer the gastric acid. Less bicarbonate would mean greater acidity, so the pepsin would remain active and would be more likely to cause cellular damage.11
However, this contention is controversial. What is universally agreed upon is that reflux of gastric or gastroduodenal contents is most likely causing injury, most likely through direct exposure, although indirect effects through vagal mechanisms cannot be ruled out.
CURRENT DIAGNOSTIC TESTS FOR LPR HAVE SHORTCOMINGS
A careful history is important. Many patients report they have sore throat, hoarseness, cough, dysphasia, or chronic throat-clearing.13 Factors that may predispose a patient to esophageal reflux should be discussed, eg:
- Tobacco use
- Diet (eg, soda, spicy foods, fatty foods)
- Alcohol use
- Certain drugs (calcium channel blockers, nitrates, steroids).
Up to 50% of patients presenting with extraesophageal symptoms may not have classic reflux symptoms such as heartburn and regur-gitation.14 However, the existence of “silent reflux” is currently controversial.
Laryngoscopy is nonspecific and subjective
Because the key symptoms of LPR are nonspecific, many patients who present to an otolaryngologist undergo laryngoscopy, mainly to rule out malignancy. Once cancer is ruled out, many patients are given a diagnosis of LPR.
Furthermore, Branski et al17 performed transoral rigid laryngoscopy with videorecording in 100 consecutive patients presenting with a chief complaint of dysphonia. Five board-certified otolaryngologists individually viewed each recording, scored the degree of erythema and edema, and assessed the likelihood that LPR played a role in dysphonia and the severity of the LPR findings. The physicians’ ratings showed considerable interobserver variability. In other words, this study showed that laryngeal findings are often nonspecific and that the laryngoscopic diagnosis of LPR tends to be subjective.17
The Reflux Finding Score. Concerned by the lack of consistency in the diagnosis of LPR, Belafsky et al18 created a scoring system for documenting the physical findings and severity of disease on a standardized scale. Their Reflux Finding Score is based on eight laryngoscopic findings: subglottic edema, ventricular edema, erythema, vocal cord edema, diffuse laryngeal edema, hypertrophy of the posterior commissure, granuloma or granulation tissue, and thick endolaryngeal mucus. The total score can range from 0 (best) to 26 (worst).
In 40 patients with LPR confirmed by pH monitoring, the mean score was 11.5, compared with 5.2 in 40 age-matched controls. The authors calculated they could be 95% certain that a person with a score higher than 7 has LPR.18
However, this diagnostic method has not been validated in a large-scale randomized trial and so has yet to be incorporated into routine otolaryngology practice.
Ambulatory pH monitoring is not so golden for diagnosing LPR
Although pH monitoring was once the gold standard for diagnosing reflux, it has since been shown to be unreliable in patients who have laryngeal symptoms.4
How high or low in the esophagus the probe is placed is clearly critical for useful results. 4 But the test is subject to variability: different physicians place the probe in different locations, and the probe may shift. Another problem is that reflux may occur during untested periods.19
A pH of less than 4 in the esophagus had originally been shown to have high sensitivity and specificity,20 but Reichel and Issing21 suggested using a pH of less than 5 as the cutoff, which would identify more patients as having LPR. Further trials are needed to more precisely determine the pH threshold for the diagnosis of LPR.
Enthusiasm is waning for pharyngeal pH monitoring
In LPR, it was initially thought that pH monitoring in the pharynx was more accurate than in the distal or proximal esophagus.
Shaker et al22 monitored the pH in the pharynx, proximal esophagus, and distal esophagus in four groups: 14 patients who had both laryngeal signs and symptoms, 12 patients who had laryngeal symptoms only, 16 patients who had GERD but no laryngeal symptoms, and 12 healthy volunteers. They found that pharyngeal reflux was more frequent and in greater quantity in patients with laryngeal signs and symptoms than in the other groups. This study suggested that pharyngeal pH monitoring may be useful in diagnosing LPR in patients who have laryngeal signs and symptoms.
However, hypopharyngeal pH monitoring has several problems. One issue is that, even in this trial, 2 of 12 healthy volunteers had episodes of pharyngeal reflux.22 In other studies, the rate of false-positive results ranged from 7% to 17%.23,24 Additionally, in 12 previous studies, only 54% of 1,217 patients with suspected LPR had esophageal acid exposure, regardless of where the pH probe was placed.25
More importantly, another study found that patients with pharyngeal reflux documented by pH monitoring were no more likely to respond to acid-suppressive therapy than patients with no documented reflux.26 These findings dampen the enthusiasm for pharyngeal pH monitoring in LPR.
Impedance monitoring on therapy may be useful in refractory cases
Esophageal impedance monitoring, a newer test, uses a catheter that measures electrical resistance (impedance) between different points along the esophagus. Thus, it can detect the reflux of acid and nonacid liquid or gaseous material.
Pritchett et al27 performed esophageal impedance and pH monitoring in 39 patients who were on twice-daily PPI therapy and then evaluated the same patients with wireless pH monitoring while they were off therapy. The most prevalent complaint in the study group was cough (56%), followed by heartburn (18%) and sore throat (10%).
Of the 39 patients, 25 (64%) had normal results on impedance/pH monitoring while on therapy, ruling out reflux. On pH monitoring off therapy, 28 (72%) of the 39 patients had abnormal results; this group included 13 (93%) of the 14 patients who had abnormal results on impedance/pH monitoring while on therapy. The authors recommended on-therapy testing with impedance monitoring in patients with refractory reflux, since it provides more useful clinical information.27 If the results of impedance/pH monitoring are negative in these patients, a diagnosis other than reflux should be considered.
EMPIRIC PPI TREATMENT HAS SHOWN DISAPPOINTING RESULTS
Because laryngoscopy and pH monitoring are not very sensitive or specific for LPR, experts recommend empiric therapy with a PPI twice daily. However, the results have been disappointing when PPIs were compared with placebo in clinical trials.
In a randomized controlled trial,28 we found that patients who had complaints of chronic throat-clearing, cough, globus, sore throat, and hoarseness had a similar response to twice-daily esomeprazole (Nexium) compared with placebo: their primary symptom had resolved by 16 weeks in 14.7% of the esomeprazole group vs 16.0% of the placebo group (P = .799). Similarly, the final findings on laryngoscopy such as edema, erythema, and surface irregularity were not significantly different between groups.
Adding a histamine-2 receptor antagonist is not recommended
Adding a histamine-2 receptor antagonist to PPI therapy has also been considered as a treatment for LPR.
Fackler et al38 studied 16 GERD patients and 18 healthy volunteers to determine if adding ranitidine (Pepcid) to the PPI omeprazole (Prilosec) could improve GERD symptoms. Patients underwent baseline manometry and then gastroesophageal pH monitoring before starting the drugs. They first received omeprazole 20 mg twice daily alone for 2 weeks, and then added ranitidine 300 mg at bedtime. A pH test was done again after the first day of treatment with ranitidine, at the end of 1 week of combination therapy, and after 4 weeks of combination therapy. The combination reduced nocturnal acid breakthrough on day 1; however, due to tolerance to ranitidine, no significant difference in acid suppression was seen after 1 week of therapy. Therefore, this combination is not recommended.
Surgery is not recommended either
Some experts have argued for surgical fundoplication in patients whose symptoms persist despite drug therapy.
Swoger et al39 treated 72 patients who had symptoms consistent with LPR with a PPI for 4 months; 25 patients in this group had less than a 50% improvement despite maximal drug therapy. Ten of these patients underwent surgical fundoplication, and 15 remained on drug therapy alone. At 1 year of follow-up, only one surgical patient (10%) reported improvement in laryngeal symptoms.
In view of this report and prior studies of surgical fundoplication,40 surgery is not recommended for patients whose symptoms do not respond to aggressive PPI therapy.
IF A PPI FAILS, LOOK FOR OTHER CAUSES OF SYMPTOMS
Although gastroesophageal reflux and laryngeal signs and symptoms have been associated with one another, this relation may have been overstated, leading to the overdiagnosis of LPR.
The diagnosis of LPR is difficult, as laryngoscopy has high interrater variability and as the results of pH monitoring do not dependably predict who will respond to treatment.
Because PPI therapy is easy and appears to be safe, patients with extraesophageal symptoms thought to be related to reflux should undergo a trial of twice-daily PPI therapy for at least 2 months. If the patient responds to therapy, then tapering to once-daily therapy initially and then to minimal acid suppression to control symptoms would be prudent.
In patients who show no improvement, other causes of symptoms should be explored. Diseases that can mimic LPR include postnasal drip, allergies, sinus inflammation, and various pulmonary diseases. These patients should also be advised to adopt lifestyle modifications—eg, to stop smoking, lose weight, and decrease activities that cause stress on the voice. Surgery is not likely to provide any benefit in this situation. The patient should be tapered off the PPI to make sure no rebound acid reflux occurs.
- Nebel OT, Fornes MF, Castell DO. Symptomatic gastroesophageal reflux: incidence and precipitating factors. Am J Dig Dis 1976; 21:953–956.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute; Clinical Practice and Quality Management Committee. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Jonaitis L, Pribuisiene R, Kupcinskas L, Uloza V. Laryngeal examination is superior to endoscopy in the diagnosis of the laryngopharyngeal form of gastroesophageal reflux disease. Scand J Gastroenterol 2006; 41:131–137.
- Vaezi MF, Hicks DM, Abelson TI, Richter JE. Laryngeal signs and symptoms and gastroesophageal reflux disease (GERD): a critical assessment of cause and effect association. Clin Gastroenterol Hepatol 2003; 1:333–344.
- Jaspersen D, Kulig M, Labenz J, et al. Prevalence of extra-oesophageal manifestations in gastro-oesophageal reflux disease: an analysis based on the ProGERD study. Aliment Pharmacol Ther 2003; 17:1515–1520.
- Karkos PD, Benton J, Leong SC, et al. Trends in laryngopharyngeal reflux: a British ENT survey. Eur Arch Otorhinolaryngol 2007; 264:513–517.
- Dent J, Dodds WJ, Friedman RH, et al. Mechanism of gastroesophageal reflux in recumbent asymptomatic human subjects. J Clin Invest 1980; 65:256–267.
- Schreiber S, Garten D, Sudhoff H, et al. Pathophysiological mechanisms of extraesophageal reflux in otolaryngeal disorders. Eur Arch Otorhinolaryngol 2009; 266:17–24.
- Adhami T, Goldblum JR, Richter JE, Vaezi MF. The role of gastric and duodenal agents in laryngeal injury: an experimental canine model. Am J Gastroenterol 2004; 99:2098–2106.
- Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24-hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope 1991; 101(4 pt 2 suppl 53):1–78.
- Johnston N, Knight J, Dettmar PW, Lively MO, Koufman J. Pepsin and carbonic anhydrase isoenzyme III as diagnostic markers for laryngopharyngeal reflux disease. Laryngoscope 2004; 114:2129–2134.
- Vaezi MF. Therapy insight: gastroesophageal reflux disease and laryngopharyngeal reflux. Nat Clin Pract Gastroenterol Hepatol 2005; 2:595–603.
- Farrokhi F, Vaezi MF. Extra-esophageal manifestations of gastroesophageal reflux. Oral Dis 2007; 13:349–359.
- Koufman JA. Laryngopharyngeal reflux is different from classic gastroesophageal reflux disease. Ear Nose Throat J 2002; 81( 9 suppl 2):7–9.
- Koufman JA, Amin MR, Panetti M. Prevalence of reflux in 113 consecutive patients with laryngeal and voice disorders. Otolaryngol Head Neck Surg 2000; 123:385–388.
- Milstein CF, Charbel S, Hicks DM, Abelson TI, Richter JE, Vaezi MF. Prevalence of laryngeal irritation signs associated with reflux in asymptomatic volunteers: impact of endoscopic technique (rigid vs flexible scope). Laryngoscope 2005; 115;2256–2261.
- Branski RC, Bhattacharyya N, Shapiro J. The reliability of the assessment of endoscopic laryngeal findings associated with laryngopharyngeal reflux disease. Laryngoscope 2002; 112;1019–1024.
- Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS). Laryngoscope 2001; 111:1313–1317.
- Gupta R, Sataloff RT. Laryngopharyngeal reflux: current concepts and questions. Curr Opin Otolaryngol Head Neck Surg 2009; 17:143–148.
- Jamieson JR, Stein HJ, DeMeester TR, et al. Ambulatory 24-h esophageal pH monitoring: normal values, optimal thresholds, specificity, sensitivity, and reproducibility. Am J Gastroenterol 1992; 87:1102–1111.
- Reichel O, Issing WJ. Impact of different pH thresholds for 24-h dual probe pH monitoring in patients with suspected laryngopharyngeal reflux. J Laryngol Otol 2008; 122:485–489.
- Shaker R, Milbrath M, Ren J, et al. Esophagopharyngeal distribution of refluxed gastric acid in patients with reflux laryngitis. Gastroenterology 1995; 109:1575–1582.
- Jacob P, Kahrilas PJ, Herzon G. Proximal esophageal pH-metry in patients with “reflux laryngitis.” Gastroenterology 1991; 100:305–310.
- Eubanks TR, Omelanczuk PE, Maronian N, Hillel A, Pope CE, Pellegrini CA. Pharyngeal pH monitoring in 222 patients with suspected laryngeal reflux. J Gastrointest Surg 2001; 5:183–190.
- Vaezi MF. Gastroesophageal reflux disease and the larynx. J Clin Gastroenterol 2003; 36:198–203.
- Ulualp SO, Toohill RJ, Shaker R. Outcomes of acid suppressive therapy in patients with posterior laryngitis. Otolaryngol Head Neck Surg 2001; 124:16–22.
- Pritchett JM, Aslam M, Slaughter JC, Ness RM, Garrett CG, Vaezi MF. Efficacy of esophageal impedance/pH monitoring in patients with refractory gastroesophageal reflux disease, on and off therapy. Clin Gastroenterol Hepatol 2009; 7:743–748.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116;254–260.
- Qadeer MA, Phillips CO, Lopez AR, et al. Proton pump inhibitor therapy for suspected GERD-related chronic laryngitis: a meta analysis of randomized controlled trials. Am J Gastroenterol 2006; 101:2646–2654.
- Eherer AJ, Habermann W, Hammer HF, et al. Effect of pantoprazole on the course of reflux-associated laryngitis: a placebo-controlled double-blind crossover study. Scand J Gastroenterol 2003; 38:462–467.
- El-Serag HB, Lee P, Buchner A, et al. Lansoprazole treatment of patients with chronic idiopathic laryngitis: a placebo-controlled trial. Am J Gastroenterol 2001; 96:979–983.
- Noordzij JP, Khidr A, Evans BA, et al. Evaluation of omeprazole in the treatment of reflux laryngitis: a prospective, placebo-controlled, randomized, double-blind study. Laryngoscope 2001; 111:2147–2151.
- Langevin S, Hanh N. GERD-induced ENT symptoms: a prospective placebo controlled study with omeprazole 40 mg a day. Gastroenterology 2001; 120:A-16.
- Havas T, Huang S, Levy M, et al. Posterior pharyngolaryngitis. Double blind randomized placebo-controlled trial of proton pump inhibitor therapy. Aust J Otolaryng 1999; 3:243–246.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Wo JM, Koopman JI, Harrell SP, et al. Double-blind placebo-controlled trial with single-dose pantoprazole for laryngopharyngeal reflux. Am J Gastroenterol 2006; 101:1972–1978.
- Steward DL, Wilson KM, Kelly DH, et al. Proton pump inhibitor therapy for chronic laryngo-pharyngitis: a randomized placebo-control trial. Otolaryngol Head Neck Surg 2004; 131:342–350.
- Fackler WK, Ours TM, Vaezi MF, Richter JE. Long term effect of H2RA therapy on nocturnal gastric acid breakthrough. Gastroenterology 2002; 122:625–632.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- So JB, Zeitels SM, Rattner DW. Outcome of atypical symptoms attributed to gastroesophageal reflux treated by laparoscopic fundoplication. Surgery 1998; 124:28–32.
- Nebel OT, Fornes MF, Castell DO. Symptomatic gastroesophageal reflux: incidence and precipitating factors. Am J Dig Dis 1976; 21:953–956.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute; Clinical Practice and Quality Management Committee. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Jonaitis L, Pribuisiene R, Kupcinskas L, Uloza V. Laryngeal examination is superior to endoscopy in the diagnosis of the laryngopharyngeal form of gastroesophageal reflux disease. Scand J Gastroenterol 2006; 41:131–137.
- Vaezi MF, Hicks DM, Abelson TI, Richter JE. Laryngeal signs and symptoms and gastroesophageal reflux disease (GERD): a critical assessment of cause and effect association. Clin Gastroenterol Hepatol 2003; 1:333–344.
- Jaspersen D, Kulig M, Labenz J, et al. Prevalence of extra-oesophageal manifestations in gastro-oesophageal reflux disease: an analysis based on the ProGERD study. Aliment Pharmacol Ther 2003; 17:1515–1520.
- Karkos PD, Benton J, Leong SC, et al. Trends in laryngopharyngeal reflux: a British ENT survey. Eur Arch Otorhinolaryngol 2007; 264:513–517.
- Dent J, Dodds WJ, Friedman RH, et al. Mechanism of gastroesophageal reflux in recumbent asymptomatic human subjects. J Clin Invest 1980; 65:256–267.
- Schreiber S, Garten D, Sudhoff H, et al. Pathophysiological mechanisms of extraesophageal reflux in otolaryngeal disorders. Eur Arch Otorhinolaryngol 2009; 266:17–24.
- Adhami T, Goldblum JR, Richter JE, Vaezi MF. The role of gastric and duodenal agents in laryngeal injury: an experimental canine model. Am J Gastroenterol 2004; 99:2098–2106.
- Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24-hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope 1991; 101(4 pt 2 suppl 53):1–78.
- Johnston N, Knight J, Dettmar PW, Lively MO, Koufman J. Pepsin and carbonic anhydrase isoenzyme III as diagnostic markers for laryngopharyngeal reflux disease. Laryngoscope 2004; 114:2129–2134.
- Vaezi MF. Therapy insight: gastroesophageal reflux disease and laryngopharyngeal reflux. Nat Clin Pract Gastroenterol Hepatol 2005; 2:595–603.
- Farrokhi F, Vaezi MF. Extra-esophageal manifestations of gastroesophageal reflux. Oral Dis 2007; 13:349–359.
- Koufman JA. Laryngopharyngeal reflux is different from classic gastroesophageal reflux disease. Ear Nose Throat J 2002; 81( 9 suppl 2):7–9.
- Koufman JA, Amin MR, Panetti M. Prevalence of reflux in 113 consecutive patients with laryngeal and voice disorders. Otolaryngol Head Neck Surg 2000; 123:385–388.
- Milstein CF, Charbel S, Hicks DM, Abelson TI, Richter JE, Vaezi MF. Prevalence of laryngeal irritation signs associated with reflux in asymptomatic volunteers: impact of endoscopic technique (rigid vs flexible scope). Laryngoscope 2005; 115;2256–2261.
- Branski RC, Bhattacharyya N, Shapiro J. The reliability of the assessment of endoscopic laryngeal findings associated with laryngopharyngeal reflux disease. Laryngoscope 2002; 112;1019–1024.
- Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS). Laryngoscope 2001; 111:1313–1317.
- Gupta R, Sataloff RT. Laryngopharyngeal reflux: current concepts and questions. Curr Opin Otolaryngol Head Neck Surg 2009; 17:143–148.
- Jamieson JR, Stein HJ, DeMeester TR, et al. Ambulatory 24-h esophageal pH monitoring: normal values, optimal thresholds, specificity, sensitivity, and reproducibility. Am J Gastroenterol 1992; 87:1102–1111.
- Reichel O, Issing WJ. Impact of different pH thresholds for 24-h dual probe pH monitoring in patients with suspected laryngopharyngeal reflux. J Laryngol Otol 2008; 122:485–489.
- Shaker R, Milbrath M, Ren J, et al. Esophagopharyngeal distribution of refluxed gastric acid in patients with reflux laryngitis. Gastroenterology 1995; 109:1575–1582.
- Jacob P, Kahrilas PJ, Herzon G. Proximal esophageal pH-metry in patients with “reflux laryngitis.” Gastroenterology 1991; 100:305–310.
- Eubanks TR, Omelanczuk PE, Maronian N, Hillel A, Pope CE, Pellegrini CA. Pharyngeal pH monitoring in 222 patients with suspected laryngeal reflux. J Gastrointest Surg 2001; 5:183–190.
- Vaezi MF. Gastroesophageal reflux disease and the larynx. J Clin Gastroenterol 2003; 36:198–203.
- Ulualp SO, Toohill RJ, Shaker R. Outcomes of acid suppressive therapy in patients with posterior laryngitis. Otolaryngol Head Neck Surg 2001; 124:16–22.
- Pritchett JM, Aslam M, Slaughter JC, Ness RM, Garrett CG, Vaezi MF. Efficacy of esophageal impedance/pH monitoring in patients with refractory gastroesophageal reflux disease, on and off therapy. Clin Gastroenterol Hepatol 2009; 7:743–748.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116;254–260.
- Qadeer MA, Phillips CO, Lopez AR, et al. Proton pump inhibitor therapy for suspected GERD-related chronic laryngitis: a meta analysis of randomized controlled trials. Am J Gastroenterol 2006; 101:2646–2654.
- Eherer AJ, Habermann W, Hammer HF, et al. Effect of pantoprazole on the course of reflux-associated laryngitis: a placebo-controlled double-blind crossover study. Scand J Gastroenterol 2003; 38:462–467.
- El-Serag HB, Lee P, Buchner A, et al. Lansoprazole treatment of patients with chronic idiopathic laryngitis: a placebo-controlled trial. Am J Gastroenterol 2001; 96:979–983.
- Noordzij JP, Khidr A, Evans BA, et al. Evaluation of omeprazole in the treatment of reflux laryngitis: a prospective, placebo-controlled, randomized, double-blind study. Laryngoscope 2001; 111:2147–2151.
- Langevin S, Hanh N. GERD-induced ENT symptoms: a prospective placebo controlled study with omeprazole 40 mg a day. Gastroenterology 2001; 120:A-16.
- Havas T, Huang S, Levy M, et al. Posterior pharyngolaryngitis. Double blind randomized placebo-controlled trial of proton pump inhibitor therapy. Aust J Otolaryng 1999; 3:243–246.
- Vaezi MF, Richter JE, Stasney CR, et al. Treatment of chronic posterior laryngitis with esomeprazole. Laryngoscope 2006; 116:254–260.
- Wo JM, Koopman JI, Harrell SP, et al. Double-blind placebo-controlled trial with single-dose pantoprazole for laryngopharyngeal reflux. Am J Gastroenterol 2006; 101:1972–1978.
- Steward DL, Wilson KM, Kelly DH, et al. Proton pump inhibitor therapy for chronic laryngo-pharyngitis: a randomized placebo-control trial. Otolaryngol Head Neck Surg 2004; 131:342–350.
- Fackler WK, Ours TM, Vaezi MF, Richter JE. Long term effect of H2RA therapy on nocturnal gastric acid breakthrough. Gastroenterology 2002; 122:625–632.
- Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol 2006; 4:433–441.
- So JB, Zeitels SM, Rattner DW. Outcome of atypical symptoms attributed to gastroesophageal reflux treated by laparoscopic fundoplication. Surgery 1998; 124:28–32.
KEY POINTS
- Laryngoscopy has high interrater variability, and results of pH monitoring do not reliably predict who will respond to treatment.
- A proton pump inhibitor twice daily for 2 months is currently recommended for patients with laryngeal signs and symptoms. If the condition responds to therapy, tapering to once-daily therapy and then to minimal acid-suppression to control symptoms is prudent.
- Patients whose symptoms do not respond to a proton pump inhibitor are unlikely to benefit from surgery. Other diagnoses should be entertained, while the drug is tapered to prevent rebound acid reflux.
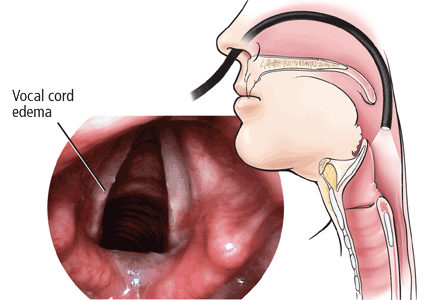
Preventing and treating orthostatic hypotension: As easy as A, B, C
Orthostatic hypotension is a chronic, debilitating illness associated with common neurologic conditions (eg, diabetic neuropathy, Parkinson disease). It is common in the elderly, especially in those who are institutionalized and are using multiple medications.
Treatment can be challenging, especially if the problem is neurogenic. This condition has no cure, symptoms vary in different circumstances, treatment is nonspecific, and aggressive treatment can lead to marked supine hypertension.
This review focuses on the prevention and treatment of neurogenic causes of orthostatic hypotension. We emphasize a simple but effective patient-oriented approach to management, using a combination of nonpharmacologic strategies and drugs clinically proven to be efficacious. The recommendations and their rationale are organized in a practical and easy-to-remember format for both physicians and patients.
WHAT HAPPENS WHEN WE STAND UP?
When we stand up, the blood goes down from the chest to the distensible venous capacitance system below the diaphragm. This fluid shift produces a decrease in venous return, ventricular filling, cardiac output, and blood pressure.1
This gravity-induced drop in blood pressure, detected by arterial baroreceptors in the aortic arch and carotid sinus, triggers a compensatory reflex tachycardia and vasoconstriction that restores normotension in the upright position. This compensatory mechanism is termed a baroreflex; it is mediated by afferent and efferent autonomic peripheral nerves and is integrated in autonomic centers in the brainstem.2
Orthostatic hypotension is the result of baroreflex failure (autonomic failure), end-organ dysfunction, or volume depletion. Injury to any limb of the baroreflex causes neurogenic orthostatic hypotension, although with afferent lesions alone, the hypotension tends to be modest and accompanied by wide fluctuations in blood pressure, including severe hypertension. Drugs can produce orthostatic hypotension by interfering with the autonomic pathways or their target end-organs or by affecting intravascular volume. Brain hypoperfusion, resulting from orthostatic hypotension from any cause, can lead to symptoms of orthostatic intolerance (eg, lightheadedness) and falls, and if the hypotension is severe, to syncope.
A DECREASE OF 20 MM HG SYSTOLIC OR 10 MM HG DIASTOLIC
The consensus definition of orthostatic hypotension is a reduction of systolic blood pressure of at least 20 mm Hg or a reduction of diastolic blood pressure of at least 10 mm Hg within 3 minutes of erect standing.3 A transient drop that occurs with abrupt standing and resolves rapidly suggests a benign condition, such as dehydration, rather than autonomic failure.
In the laboratory, patients are placed on a tilt table in the head-up position at an angle of at least 60 degrees to detect orthostatic changes in blood pressure. In the office, 1 minute of standing probably detects nearly all cases of orthostatic hypotension; however, standing beyond 2 minutes helps establish the severity (a further drop in blood pressure).4 Orthostatic hypotension developing after 3 minutes of standing is uncommon and may represent a reflex presyncope (eg, vasovagal) or a mild or early form of sympathetic adrenergic dysfunction.4, 5
NEUROGENIC AND NONNEUROGENIC CAUSES
Orthostatic hypotension may result from neurogenic and nonneurogenic causes.
Neurogenic orthostatic hypotension can be due to neuropathy (eg, diabetic or autoimmune neuropathies) or to central lesions (eg, Parkinson disease or multiple system atrophy). Its presence, severity, and temporal course can be important clues in diagnosing Parkinson disease and differentiating it from other parkinsonian syndromes with a more ominous prognosis, such as multiple system atrophy and Lewy body dementia.
Nonneurogenic causes include cardiac impairment (eg, from myocardial infarction or aortic stenosis), reduced intravascular volume (eg, from dehydration, adrenal insufficiency), and vasodilation (eg, from fever, systemic mastocytosis).
Common drugs that cause orthostatic hypotension are diuretics, alpha-adrenoceptor blockers for prostatic hypertrophy, antihypertensive drugs, and calcium channel blockers. Insulin, levodopa, and tricyclic antidepressants can also cause vasodilation and orthostatic hypotension in predisposed patients. Poon and Braun,6 in a retrospective study in elderly veterans, identified hydrochlorothiazide, lisinopril (Prinivil, Zestril), trazodone (Desyrel), furosemide (Lasix), and terazosin (Hytrin) as the most common culprits.
ORTHOSTATIC HYPOTENSION IS COMMON IN THE ELDERLY
The prevalence of orthostatic hypotension is high in the elderly and depends on the characteristics of the population studied, such as age, use of medications, and comorbidities known to be associated with this problem. Orthostatic hypotension is more common in institutionalized elderly people (up to 68%)7 than in those living in the community (6%).8 The high prevalence among institutionalized patients likely reflects multiple disease processes, including neurologic and cardiac conditions, as well as medications associated with orthostatic hypotension.
CLINICAL MANIFESTATIONS ARE DUE TO HYPOPERFUSION, OVERCOMPENSATION
Symptoms are related to cerebral hypoperfusion, with resulting lack of cerebral oxygenation (causing lightheadedness, dizziness, weakness, difficulty thinking, headache, syncope, or feeling faint) and a compensatory autonomic overreaction (causing palpitations, tremulousness, nausea, coldness of extremities, chest pain, and syncope).
Lightheadedness is a common symptom, but subtler issues such as difficulty thinking, weakness, and neck discomfort are also common in the elderly. Recurrent or unexplained falls in older adults may be a manifestation of syncope due to orthostatic hypotension.
PROGNOSIS DEPENDS ON CAUSE
Orthostatic hypotension is a syndrome, and its prognosis depends on its specific cause, its severity, and the distribution of its autonomic and nonautonomic involvement. In patients who have extrapyramidal and cerebellar disorders (eg, Parkinson disease, multiple system atrophy), the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis.9,10
In hypertensive patients with diabetes mellitus, the risk of death is higher if they have orthostatic hypotension.11 Diastolic orthostatic hypotension is associated with a higher risk of vascular death in older persons.12
MANAGEMENT: FROM A TO F
The goal of management of orthostatic hypotension is to raise the patient’s standing blood pressure without also raising his or her supine blood pressure, and specifically to reduce orthostatic symptoms, increase the time the patient can stand, and improve his or her ability to perform daily activities. No specific treatment is currently available that achieves all these goals, and drugs alone are never completely adequate.

Because the mainstays of treatment are volume expansion and vasoconstriction, it is difficult to improve the symptoms of orthostatic hypotension without inducing some degree of supine hypertension. Strategies to minimize nocturnal hypertension and to treat orthostatic hypotension in special circumstances are summarized in Table 1.


If hypovolemia is playing a major role, and the patient cannot ingest enough salt or plasma volume fails to increase despite salt supplementation, fludrocortisone (Florinef) should be considered. Untreated hypovolemia will decrease the efficacy of vasoconstrictor drugs.
Pyridostigmine (Mestinon) has a putative vasoconstrictor effect only during standing, but because its effect is modest it should be used in mild orthostatic hypotension that does not improve with nonpharmacologic measures and in moderate cases. Its effect can be enhanced with additional low doses of midodrine (ProAmatine). Midodrine with or without fludrocortisone should be used in severe orthostatic hypotension.

A: Abdominal compression
In conditions in which there is adrenergic denervation of vascular beds, there is an increase in vascular capacitance and peripheral venous pooling. Compression of capacitance beds (ie, the legs and abdomen) improves orthostatic symptoms.14 The improvement is due to a reduction of venous capacitance and an increase in total peripheral resistance.14
On standing, healthy adults experience an orthostatic shift of approximately 500 mL of blood to the lower extremities15 that, when added to an increased vascular capacitance in those with orthostatic hypotension, results in a relative state of hypovolemia.
Compression of the legs alone is not as beneficial as compression of the abdomen because the venous capacitance of the calves and thighs is relatively small compared with that of the splanchnic mesenteric bed, which accounts for 20% to 30% of total blood volume.16 Moreover, compression garments and stockings that are strong enough to produce a measurable effect on orthostatic hypotension are cumbersome to put on and uncomfortable to wear. Because some patients gain significant benefit from abdominal compression alone, this should be considered the first step in reducing venous capacitance.
In a laboratory experiment, Smit et al17 found that an elastic abdominal binder that exerted 15 to 20 mm Hg of pressure on the abdomen raised the standing blood pressure by about 11/6 mmHg, which was comparable to the effect of a gravity suit (such as those worn by fighter pilots to prevent syncope during violent aircraft maneuvers) inflated to 20 mm Hg—an increase of about 17/8 mm Hg. Higher gravity-suit pressures had a greater effect.
In practical terms, the binder should be tight enough to exert gentle pressure. It should be put on before rising from bed in the morning and taken off when lying supine, to avoid supine hypertension. Advantages are that a binder’s effects are immediate, its benefits can be easily assessed, and it can be used on an as-needed basis by patients who need it only during periods of prolonged orthostatic stress. Binders are also easy to fit and are available in most sporting good stores and on the Web (try searching for “abdominal binder”).
When abdominal compression alone is not enough, the addition of compression of the lower extremities can result in further benefits. This can be achieved by using compression garments that ideally extend to the waist or, at the least, to the proximal thigh.
B: Boluses of water
Rapidly drinking two 8-oz (500-mL) glasses of cold water helps expand plasma volume. It also, within a few minutes, elicits a significant pressor effect that is in part norepinephrine-mediated,18,19 increasing the standing systolic blood pressure by more than 20 mm Hg for about 2 hours and improving symptoms and orthostatic endurance.18,20 This easy technique can be used when prolonged standing is expected (eg, shopping).
B (continued): Bed up
The head of the bed of a patient with orthostatic hypotension should be elevated by 10 to 20 degrees or 4 inches (10 cm) to decrease nocturnal hypertension and nocturnal diuresis.21 During the day, adequate orthostatic stress, ie, upright activity, should be maintained. If patients are repeatedly tilted up, their orthostatic hypotension is gradually attenuated, presumably by increasing venomotor tone.22
C: Countermaneuvers
Physical countermaneuvers involve isometrically contracting the muscles below the waist for about 30 seconds at a time, which reduces venous capacitance, increases total peripheral resistance, and augments venous return to the heart.23,24 These countermeasures can help maintain blood pressure during daily activities and should be considered at the first symptoms of orthostatic intolerance and in situations of orthostatic stress (eg, standing for prolonged periods).
Specific techniques include23:
- Toe-raising
- Leg-crossing and contraction
- Thigh muscle co-contraction
- Bending at the waist
- Slow marching in place
- Leg elevation.
D: Drugs
Midodrine, a vasopressor, is effective and safe when used for treating neurogenic orthostatic hypotension.25 It has been shown to increase standing systolic blood pressure, reduce orthostatic lightheadedness, and increase standing and walking time.
A common starting dose is 5 mg three times a day; most patients respond best to 10 mg three times a day. As its duration of action is short (2 to 4 hours),25–27 it should be taken before arising in the morning, before lunch, and in the midafternoon. To avoid nocturnal supine hypertension, doses should not be taken after the midafternoon, and a dose should be omitted if the supine or sitting blood pressure is greater than 180/100 mm Hg.
Midodrine’s main side effects are supine hypertension, scalp paresthesias, and pilomotor reactions (goosebumps). Vasoconstrictors such as midodrine are ineffective when plasma volume is reduced.
Fludrocortisone is a synthetic mineralocorticoid that has a pressor effect as a result of its ability to expand plasma volume and increase vascular alpha-adrenoceptor sensitivity.28–30 This medication is helpful when plasma volume fails to adequately increase with salt supplementation31 and for patients who cannot ingest enough salt or do not respond adequately to midodrine.
The usual dose is 0.1 to 0.2 mg/day, but it may be increased to 0.4 to 0.6 mg/day in patients with refractory orthostatic hypotension.
If the patient gains 3 to 5 pounds (1.2–2.3 kg) and develops mild dependent edema, you can infer that the plasma volume has expanded adequately. However, in view of these effects, fludrocortisone is contraindicated in congestive heart failure and chronic renal failure. The potential risks are severe hypokalemia and excessive supine hypertension. Frequent monitoring of serum potassium, a diet high in potassium, and regular checks of supine blood pressure are advised, especially at higher doses, when added to midodrine, or in elderly patients who tend to poorly tolerate the medication.28,29,32
Pyridostigmine is a cholinesterase inhibitor that improves ganglionic neurotransmission in the sympathetic baroreflex pathway. Because this pathway is activated primarily during standing, this drug improves orthostatic hypotension and total peripheral resistance without aggravating supine hypertension. Because the pressor effect is modest, it is most adequate for patients with mild to moderate orthostatic hypotension.33,34
Dosing is started at 30 mg two to three times a day and is gradually increased to 60 mg three times a day. The drug’s effectiveness can be enhanced by combining each dose of pyridostigmine with 5 mg of midodrine without occurrence of supine hypertension.34 Mestinon Timespan, a 180-mg slow-release pyridostigmine tablet, can be taken once a day and may be a convenient alternative.
The main side effects are cholinergic (abdominal colic, diarrhea).
Review the patient’s medications. If he or she is taking any drug that may cause orthostatic hypotension, consider discontinuing it, substituting another drug, or changing the dosage (Table 2). In the elderly, antiparkinsonian, nitrate, antidepressant, diuretic, prostate, and antihypertensive medications35 may be particularly suspect.
E: Education
Education is probably the single most important factor in the proper control of orthostatic hypotension. A number of issues should be considered.
- Patients should be taught, in simple terms, the mechanisms that maintain postural normotension and how to recognize the onset of orthostatic symptoms.
- They must realize that there is no specific treatment of the underlying cause and that drug treatment alone is not adequate.
- They should be taught nonpharmacologic approaches and be aware that other drugs they start may worsen symptoms.
It is also important that the patient learn the conditions (and their mechanisms) that can lower blood pressure (Table 3). Such conditions include prolonged or motionless standing, alcohol ingestion (causing vasodilation), carbohydrate-heavy meals (causing postprandial orthostatic hypotension related to an increase in the splanchnic-mesenteric venous capacitance), early morning orthostatic hypotension related to nocturnal diuresis and arising from bed, physical activity sufficient to cause muscle vasodilation, heat exposure (eg, hot weather or a hot bath or shower) producing skin vessel vasodilation, sudden postural changes, and prolonged recumbency. Once these stressors are explained, patients have no difficulty recognizing them.
The patient should also be instructed in how to manage situations of increased orthostatic stress and periods of orthostatic decompensation, to minimize nocturnal hypertension, and to modify their activities of daily living. Keeping a log of supine and upright blood pressures (taken with an automated sphygmomanometer) during situations of orthostatic stress can help establish whether worsening symptoms are related to orthostatic hypotension or to another mechanism. Once patients discover that they can actively deal with these situations, they develop a great sense of empowerment.
E (continued): Exercise
Mild physical exercise improves orthostatic tolerance by reducing venous pooling and increasing plasma volume.36 Deconditioning from lack of exercise exacerbates orthostatic hypotension.37 Because upright exercise may increase the orthostatic drop in blood pressure, training in a supine or sitting position (eg, swimming, recumbent biking) is advisable. Isotonic exercise (eg, light weight-lifting) is recommended because the incorrect straining and breath-holding during isometric exercise (eg, holding weights in the same position) may decrease venous return.
F: Fluid and salt (volume expansion)
Maintaining an adequate plasma volume is crucial. Patients should drink five to eight 8-ounce glasses (1.25 to 2.5 L) of water or other fluid per day. Many elderly people do not take in this much. The patient should have at least 1 glass or cup of fluid with meals and at least twice at other times of each day to obtain 1 L/day.
Salt intake should be between 150 and 250 mmol of sodium (10 to 20 g of salt) per day. Sodium helps with retention of ingested fluids and should be maximized if tolerated. However, caution should be exercised in patients who have severe refractory supine hypertension, uncontrolled hypertension, or comorbidities characterized by insterstitial edema (eg, heart failure, liver failure). Some patients are very sensitive to sodium supplementation and can fine-tune their orthostatic control with salt alone. If salting food is not desired, prepared soups, pretzels, potato chips, and 0.5- or 1.0-g salt tablets can be an option.
Patients need to maintain a high-potassium diet, as the high sodium intake combined with fludrocortisone promotes potassium loss. Fruits (especially bananas) and vegetables have high potassium content.
The combination of fludrocortisone and a high-salt diet can also cause sustained supine hypertension, which can be minimized by the interventions noted in Table 2.
Appropriate salt supplementation and fluid intake leading to an adequate volume expansion can be verified by checking the 24-hour urinary sodium content: patients who excrete less than 170 mmol can be treated with 1 to 2 g of supplemental sodium three times a day.38
- Sjostrand T. The regulation of the blood distribution in man. Acta Physiol Scand 1952; 26:312–327.
- Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977; 296:293–297.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996; 46:1470.
- Gehrking JA, Hines SM, Benrud-Larson LM, Opher-Gehrking TL, Low PA. What is the minimum duration of head-up tilt necessary to detect orthostatic hypotension? Clin Auton Res 2005; 15:71–75.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005; 30:173–178.
- Weiss A, Grossman E, Beloosesky Y, Grinblat J. Orthostatic hypotension in acute geriatric ward: is it a consistent finding? Arch Intern Med 2002; 162:2369–2374.
- Mader SL, Josephson KR, Rubenstein LZ. Low prevalence of postural hypotension among community-dwelling elderly. JAMA 1987; 258:1511–1514.
- Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991; 1:147–155.
- Saito Y, Matsuoka Y, Takahashi A, Ohno Y. Survival of patients with multiple system atrophy. Intern Med 1994; 33:321–325.
- Davis BR, Langford HG, Blaufox MD, Curb JD, Polk BF, Shulman NB. The association of postural changes in systolic blood pressure and mortality in persons with hypertension: the Hypertension Detection and Follow-up Program experience. Circulation 1987; 75:340–346.
- Luukinen H, Koski K, Laippala P, Kivelä SL. Prognosis of diastolic and systolic orthostatic hypotension in older persons. Arch Intern Med 1999; 159:273–280.
- Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. N Engl J Med 1993; 329:611–615.
- Denq JC, Opfer-Gehrking TL, Giuliani M, Felten J, Convertino VA, Low PA. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clin Auton Res 1997; 7:321–326.
- Sjostrand T. Volume and distribution of blood and their significance in regulating the circulation. Physiol Rev 1953; 33:202–228.
- Rowell LB, Detry JM, Blackmon JR, Wyss C. Importance of the splanchnic vascular bed in human blood pressure regulation. J Appl Physiol 1972; 32:213–220.
- Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res 2004; 14:167–175.
- Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000; 101:504–509.
- Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002; 112:355–360.
- Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water [letter]. Lancet 1999; 353:723.
- MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia: treatment with the “head-up” bed. JAMA 1940; 115:2162–2167.
- Ector H, Reybrouck T, Heidbüchel H, Gewillig M, Van de Werf F. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol 1998; 21:193–196.
- Bouvette CM, McPhee BR, Opfer-Gehrking TL, Low PA. Role of physical countermaneuvers in the management of orthostatic hypotension: efficacy and biofeedback augmentation. Mayo Clin Proc 1996; 71:847–853.
- Ten Harkel AD, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci (Lond) 1994; 87:553–558.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993; 95:38–48.
- Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med 1995; 99:604–610.
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007; 7:63–70.
- Axelrod FB, Goldberg JD, Rolnitzky L, et al. Fludrocortisone in patients with familial dysautonomia—assessing effect on clinical parameters and gene expression. Clin Auton Res 2005; 15:284–291.
- Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301:68–73.
- van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. Clin Auton Res 2000; 10:35–42.
- Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart 1996; 76:507–509.
- Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry 2003; 74:1294–1298.
- Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63:513–518.
- Lipsitz LA, Pluchino FC, Wei JY, Rowe JW. Syncope in institutionalized elderly: the impact of multiple pathological conditions and situational stress. J Chronic Dis 1986; 39:619–630.
- Mtinangi BL, Hainsworth R. Effects of moderate exercise training on plasma volume, baroreceptor sensitivity and orthostatic tolerance in healthy subjects. Exp Physiol 1999; 84:121–130.
- Bonnin P, Ben Driss A, Benessiano J, Maillet A, Pavy le Traon A, Levy BI. Enhanced flow-dependent vasodilatation after bed rest, a possible mechanism for orthostatic intolerance in humans. Eur J Appl Physiol 2001; 85:420–426.
- El-Sayed H, Hainsworth R. Salt supplementation increases plasma volume and orthostatic tolerance in patients with unexplained syncope. Heart 1996; 75:134–140.
Orthostatic hypotension is a chronic, debilitating illness associated with common neurologic conditions (eg, diabetic neuropathy, Parkinson disease). It is common in the elderly, especially in those who are institutionalized and are using multiple medications.
Treatment can be challenging, especially if the problem is neurogenic. This condition has no cure, symptoms vary in different circumstances, treatment is nonspecific, and aggressive treatment can lead to marked supine hypertension.
This review focuses on the prevention and treatment of neurogenic causes of orthostatic hypotension. We emphasize a simple but effective patient-oriented approach to management, using a combination of nonpharmacologic strategies and drugs clinically proven to be efficacious. The recommendations and their rationale are organized in a practical and easy-to-remember format for both physicians and patients.
WHAT HAPPENS WHEN WE STAND UP?
When we stand up, the blood goes down from the chest to the distensible venous capacitance system below the diaphragm. This fluid shift produces a decrease in venous return, ventricular filling, cardiac output, and blood pressure.1
This gravity-induced drop in blood pressure, detected by arterial baroreceptors in the aortic arch and carotid sinus, triggers a compensatory reflex tachycardia and vasoconstriction that restores normotension in the upright position. This compensatory mechanism is termed a baroreflex; it is mediated by afferent and efferent autonomic peripheral nerves and is integrated in autonomic centers in the brainstem.2
Orthostatic hypotension is the result of baroreflex failure (autonomic failure), end-organ dysfunction, or volume depletion. Injury to any limb of the baroreflex causes neurogenic orthostatic hypotension, although with afferent lesions alone, the hypotension tends to be modest and accompanied by wide fluctuations in blood pressure, including severe hypertension. Drugs can produce orthostatic hypotension by interfering with the autonomic pathways or their target end-organs or by affecting intravascular volume. Brain hypoperfusion, resulting from orthostatic hypotension from any cause, can lead to symptoms of orthostatic intolerance (eg, lightheadedness) and falls, and if the hypotension is severe, to syncope.
A DECREASE OF 20 MM HG SYSTOLIC OR 10 MM HG DIASTOLIC
The consensus definition of orthostatic hypotension is a reduction of systolic blood pressure of at least 20 mm Hg or a reduction of diastolic blood pressure of at least 10 mm Hg within 3 minutes of erect standing.3 A transient drop that occurs with abrupt standing and resolves rapidly suggests a benign condition, such as dehydration, rather than autonomic failure.
In the laboratory, patients are placed on a tilt table in the head-up position at an angle of at least 60 degrees to detect orthostatic changes in blood pressure. In the office, 1 minute of standing probably detects nearly all cases of orthostatic hypotension; however, standing beyond 2 minutes helps establish the severity (a further drop in blood pressure).4 Orthostatic hypotension developing after 3 minutes of standing is uncommon and may represent a reflex presyncope (eg, vasovagal) or a mild or early form of sympathetic adrenergic dysfunction.4, 5
NEUROGENIC AND NONNEUROGENIC CAUSES
Orthostatic hypotension may result from neurogenic and nonneurogenic causes.
Neurogenic orthostatic hypotension can be due to neuropathy (eg, diabetic or autoimmune neuropathies) or to central lesions (eg, Parkinson disease or multiple system atrophy). Its presence, severity, and temporal course can be important clues in diagnosing Parkinson disease and differentiating it from other parkinsonian syndromes with a more ominous prognosis, such as multiple system atrophy and Lewy body dementia.
Nonneurogenic causes include cardiac impairment (eg, from myocardial infarction or aortic stenosis), reduced intravascular volume (eg, from dehydration, adrenal insufficiency), and vasodilation (eg, from fever, systemic mastocytosis).
Common drugs that cause orthostatic hypotension are diuretics, alpha-adrenoceptor blockers for prostatic hypertrophy, antihypertensive drugs, and calcium channel blockers. Insulin, levodopa, and tricyclic antidepressants can also cause vasodilation and orthostatic hypotension in predisposed patients. Poon and Braun,6 in a retrospective study in elderly veterans, identified hydrochlorothiazide, lisinopril (Prinivil, Zestril), trazodone (Desyrel), furosemide (Lasix), and terazosin (Hytrin) as the most common culprits.
ORTHOSTATIC HYPOTENSION IS COMMON IN THE ELDERLY
The prevalence of orthostatic hypotension is high in the elderly and depends on the characteristics of the population studied, such as age, use of medications, and comorbidities known to be associated with this problem. Orthostatic hypotension is more common in institutionalized elderly people (up to 68%)7 than in those living in the community (6%).8 The high prevalence among institutionalized patients likely reflects multiple disease processes, including neurologic and cardiac conditions, as well as medications associated with orthostatic hypotension.
CLINICAL MANIFESTATIONS ARE DUE TO HYPOPERFUSION, OVERCOMPENSATION
Symptoms are related to cerebral hypoperfusion, with resulting lack of cerebral oxygenation (causing lightheadedness, dizziness, weakness, difficulty thinking, headache, syncope, or feeling faint) and a compensatory autonomic overreaction (causing palpitations, tremulousness, nausea, coldness of extremities, chest pain, and syncope).
Lightheadedness is a common symptom, but subtler issues such as difficulty thinking, weakness, and neck discomfort are also common in the elderly. Recurrent or unexplained falls in older adults may be a manifestation of syncope due to orthostatic hypotension.
PROGNOSIS DEPENDS ON CAUSE
Orthostatic hypotension is a syndrome, and its prognosis depends on its specific cause, its severity, and the distribution of its autonomic and nonautonomic involvement. In patients who have extrapyramidal and cerebellar disorders (eg, Parkinson disease, multiple system atrophy), the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis.9,10
In hypertensive patients with diabetes mellitus, the risk of death is higher if they have orthostatic hypotension.11 Diastolic orthostatic hypotension is associated with a higher risk of vascular death in older persons.12
MANAGEMENT: FROM A TO F
The goal of management of orthostatic hypotension is to raise the patient’s standing blood pressure without also raising his or her supine blood pressure, and specifically to reduce orthostatic symptoms, increase the time the patient can stand, and improve his or her ability to perform daily activities. No specific treatment is currently available that achieves all these goals, and drugs alone are never completely adequate.
Because the mainstays of treatment are volume expansion and vasoconstriction, it is difficult to improve the symptoms of orthostatic hypotension without inducing some degree of supine hypertension. Strategies to minimize nocturnal hypertension and to treat orthostatic hypotension in special circumstances are summarized in Table 1.
If hypovolemia is playing a major role, and the patient cannot ingest enough salt or plasma volume fails to increase despite salt supplementation, fludrocortisone (Florinef) should be considered. Untreated hypovolemia will decrease the efficacy of vasoconstrictor drugs.
Pyridostigmine (Mestinon) has a putative vasoconstrictor effect only during standing, but because its effect is modest it should be used in mild orthostatic hypotension that does not improve with nonpharmacologic measures and in moderate cases. Its effect can be enhanced with additional low doses of midodrine (ProAmatine). Midodrine with or without fludrocortisone should be used in severe orthostatic hypotension.
A: Abdominal compression
In conditions in which there is adrenergic denervation of vascular beds, there is an increase in vascular capacitance and peripheral venous pooling. Compression of capacitance beds (ie, the legs and abdomen) improves orthostatic symptoms.14 The improvement is due to a reduction of venous capacitance and an increase in total peripheral resistance.14
On standing, healthy adults experience an orthostatic shift of approximately 500 mL of blood to the lower extremities15 that, when added to an increased vascular capacitance in those with orthostatic hypotension, results in a relative state of hypovolemia.
Compression of the legs alone is not as beneficial as compression of the abdomen because the venous capacitance of the calves and thighs is relatively small compared with that of the splanchnic mesenteric bed, which accounts for 20% to 30% of total blood volume.16 Moreover, compression garments and stockings that are strong enough to produce a measurable effect on orthostatic hypotension are cumbersome to put on and uncomfortable to wear. Because some patients gain significant benefit from abdominal compression alone, this should be considered the first step in reducing venous capacitance.
In a laboratory experiment, Smit et al17 found that an elastic abdominal binder that exerted 15 to 20 mm Hg of pressure on the abdomen raised the standing blood pressure by about 11/6 mmHg, which was comparable to the effect of a gravity suit (such as those worn by fighter pilots to prevent syncope during violent aircraft maneuvers) inflated to 20 mm Hg—an increase of about 17/8 mm Hg. Higher gravity-suit pressures had a greater effect.
In practical terms, the binder should be tight enough to exert gentle pressure. It should be put on before rising from bed in the morning and taken off when lying supine, to avoid supine hypertension. Advantages are that a binder’s effects are immediate, its benefits can be easily assessed, and it can be used on an as-needed basis by patients who need it only during periods of prolonged orthostatic stress. Binders are also easy to fit and are available in most sporting good stores and on the Web (try searching for “abdominal binder”).
When abdominal compression alone is not enough, the addition of compression of the lower extremities can result in further benefits. This can be achieved by using compression garments that ideally extend to the waist or, at the least, to the proximal thigh.
B: Boluses of water
Rapidly drinking two 8-oz (500-mL) glasses of cold water helps expand plasma volume. It also, within a few minutes, elicits a significant pressor effect that is in part norepinephrine-mediated,18,19 increasing the standing systolic blood pressure by more than 20 mm Hg for about 2 hours and improving symptoms and orthostatic endurance.18,20 This easy technique can be used when prolonged standing is expected (eg, shopping).
B (continued): Bed up
The head of the bed of a patient with orthostatic hypotension should be elevated by 10 to 20 degrees or 4 inches (10 cm) to decrease nocturnal hypertension and nocturnal diuresis.21 During the day, adequate orthostatic stress, ie, upright activity, should be maintained. If patients are repeatedly tilted up, their orthostatic hypotension is gradually attenuated, presumably by increasing venomotor tone.22
C: Countermaneuvers
Physical countermaneuvers involve isometrically contracting the muscles below the waist for about 30 seconds at a time, which reduces venous capacitance, increases total peripheral resistance, and augments venous return to the heart.23,24 These countermeasures can help maintain blood pressure during daily activities and should be considered at the first symptoms of orthostatic intolerance and in situations of orthostatic stress (eg, standing for prolonged periods).
Specific techniques include23:
- Toe-raising
- Leg-crossing and contraction
- Thigh muscle co-contraction
- Bending at the waist
- Slow marching in place
- Leg elevation.
D: Drugs
Midodrine, a vasopressor, is effective and safe when used for treating neurogenic orthostatic hypotension.25 It has been shown to increase standing systolic blood pressure, reduce orthostatic lightheadedness, and increase standing and walking time.
A common starting dose is 5 mg three times a day; most patients respond best to 10 mg three times a day. As its duration of action is short (2 to 4 hours),25–27 it should be taken before arising in the morning, before lunch, and in the midafternoon. To avoid nocturnal supine hypertension, doses should not be taken after the midafternoon, and a dose should be omitted if the supine or sitting blood pressure is greater than 180/100 mm Hg.
Midodrine’s main side effects are supine hypertension, scalp paresthesias, and pilomotor reactions (goosebumps). Vasoconstrictors such as midodrine are ineffective when plasma volume is reduced.
Fludrocortisone is a synthetic mineralocorticoid that has a pressor effect as a result of its ability to expand plasma volume and increase vascular alpha-adrenoceptor sensitivity.28–30 This medication is helpful when plasma volume fails to adequately increase with salt supplementation31 and for patients who cannot ingest enough salt or do not respond adequately to midodrine.
The usual dose is 0.1 to 0.2 mg/day, but it may be increased to 0.4 to 0.6 mg/day in patients with refractory orthostatic hypotension.
If the patient gains 3 to 5 pounds (1.2–2.3 kg) and develops mild dependent edema, you can infer that the plasma volume has expanded adequately. However, in view of these effects, fludrocortisone is contraindicated in congestive heart failure and chronic renal failure. The potential risks are severe hypokalemia and excessive supine hypertension. Frequent monitoring of serum potassium, a diet high in potassium, and regular checks of supine blood pressure are advised, especially at higher doses, when added to midodrine, or in elderly patients who tend to poorly tolerate the medication.28,29,32
Pyridostigmine is a cholinesterase inhibitor that improves ganglionic neurotransmission in the sympathetic baroreflex pathway. Because this pathway is activated primarily during standing, this drug improves orthostatic hypotension and total peripheral resistance without aggravating supine hypertension. Because the pressor effect is modest, it is most adequate for patients with mild to moderate orthostatic hypotension.33,34
Dosing is started at 30 mg two to three times a day and is gradually increased to 60 mg three times a day. The drug’s effectiveness can be enhanced by combining each dose of pyridostigmine with 5 mg of midodrine without occurrence of supine hypertension.34 Mestinon Timespan, a 180-mg slow-release pyridostigmine tablet, can be taken once a day and may be a convenient alternative.
The main side effects are cholinergic (abdominal colic, diarrhea).
Review the patient’s medications. If he or she is taking any drug that may cause orthostatic hypotension, consider discontinuing it, substituting another drug, or changing the dosage (Table 2). In the elderly, antiparkinsonian, nitrate, antidepressant, diuretic, prostate, and antihypertensive medications35 may be particularly suspect.
E: Education
Education is probably the single most important factor in the proper control of orthostatic hypotension. A number of issues should be considered.
- Patients should be taught, in simple terms, the mechanisms that maintain postural normotension and how to recognize the onset of orthostatic symptoms.
- They must realize that there is no specific treatment of the underlying cause and that drug treatment alone is not adequate.
- They should be taught nonpharmacologic approaches and be aware that other drugs they start may worsen symptoms.
It is also important that the patient learn the conditions (and their mechanisms) that can lower blood pressure (Table 3). Such conditions include prolonged or motionless standing, alcohol ingestion (causing vasodilation), carbohydrate-heavy meals (causing postprandial orthostatic hypotension related to an increase in the splanchnic-mesenteric venous capacitance), early morning orthostatic hypotension related to nocturnal diuresis and arising from bed, physical activity sufficient to cause muscle vasodilation, heat exposure (eg, hot weather or a hot bath or shower) producing skin vessel vasodilation, sudden postural changes, and prolonged recumbency. Once these stressors are explained, patients have no difficulty recognizing them.
The patient should also be instructed in how to manage situations of increased orthostatic stress and periods of orthostatic decompensation, to minimize nocturnal hypertension, and to modify their activities of daily living. Keeping a log of supine and upright blood pressures (taken with an automated sphygmomanometer) during situations of orthostatic stress can help establish whether worsening symptoms are related to orthostatic hypotension or to another mechanism. Once patients discover that they can actively deal with these situations, they develop a great sense of empowerment.
E (continued): Exercise
Mild physical exercise improves orthostatic tolerance by reducing venous pooling and increasing plasma volume.36 Deconditioning from lack of exercise exacerbates orthostatic hypotension.37 Because upright exercise may increase the orthostatic drop in blood pressure, training in a supine or sitting position (eg, swimming, recumbent biking) is advisable. Isotonic exercise (eg, light weight-lifting) is recommended because the incorrect straining and breath-holding during isometric exercise (eg, holding weights in the same position) may decrease venous return.
F: Fluid and salt (volume expansion)
Maintaining an adequate plasma volume is crucial. Patients should drink five to eight 8-ounce glasses (1.25 to 2.5 L) of water or other fluid per day. Many elderly people do not take in this much. The patient should have at least 1 glass or cup of fluid with meals and at least twice at other times of each day to obtain 1 L/day.
Salt intake should be between 150 and 250 mmol of sodium (10 to 20 g of salt) per day. Sodium helps with retention of ingested fluids and should be maximized if tolerated. However, caution should be exercised in patients who have severe refractory supine hypertension, uncontrolled hypertension, or comorbidities characterized by insterstitial edema (eg, heart failure, liver failure). Some patients are very sensitive to sodium supplementation and can fine-tune their orthostatic control with salt alone. If salting food is not desired, prepared soups, pretzels, potato chips, and 0.5- or 1.0-g salt tablets can be an option.
Patients need to maintain a high-potassium diet, as the high sodium intake combined with fludrocortisone promotes potassium loss. Fruits (especially bananas) and vegetables have high potassium content.
The combination of fludrocortisone and a high-salt diet can also cause sustained supine hypertension, which can be minimized by the interventions noted in Table 2.
Appropriate salt supplementation and fluid intake leading to an adequate volume expansion can be verified by checking the 24-hour urinary sodium content: patients who excrete less than 170 mmol can be treated with 1 to 2 g of supplemental sodium three times a day.38
Orthostatic hypotension is a chronic, debilitating illness associated with common neurologic conditions (eg, diabetic neuropathy, Parkinson disease). It is common in the elderly, especially in those who are institutionalized and are using multiple medications.
Treatment can be challenging, especially if the problem is neurogenic. This condition has no cure, symptoms vary in different circumstances, treatment is nonspecific, and aggressive treatment can lead to marked supine hypertension.
This review focuses on the prevention and treatment of neurogenic causes of orthostatic hypotension. We emphasize a simple but effective patient-oriented approach to management, using a combination of nonpharmacologic strategies and drugs clinically proven to be efficacious. The recommendations and their rationale are organized in a practical and easy-to-remember format for both physicians and patients.
WHAT HAPPENS WHEN WE STAND UP?
When we stand up, the blood goes down from the chest to the distensible venous capacitance system below the diaphragm. This fluid shift produces a decrease in venous return, ventricular filling, cardiac output, and blood pressure.1
This gravity-induced drop in blood pressure, detected by arterial baroreceptors in the aortic arch and carotid sinus, triggers a compensatory reflex tachycardia and vasoconstriction that restores normotension in the upright position. This compensatory mechanism is termed a baroreflex; it is mediated by afferent and efferent autonomic peripheral nerves and is integrated in autonomic centers in the brainstem.2
Orthostatic hypotension is the result of baroreflex failure (autonomic failure), end-organ dysfunction, or volume depletion. Injury to any limb of the baroreflex causes neurogenic orthostatic hypotension, although with afferent lesions alone, the hypotension tends to be modest and accompanied by wide fluctuations in blood pressure, including severe hypertension. Drugs can produce orthostatic hypotension by interfering with the autonomic pathways or their target end-organs or by affecting intravascular volume. Brain hypoperfusion, resulting from orthostatic hypotension from any cause, can lead to symptoms of orthostatic intolerance (eg, lightheadedness) and falls, and if the hypotension is severe, to syncope.
A DECREASE OF 20 MM HG SYSTOLIC OR 10 MM HG DIASTOLIC
The consensus definition of orthostatic hypotension is a reduction of systolic blood pressure of at least 20 mm Hg or a reduction of diastolic blood pressure of at least 10 mm Hg within 3 minutes of erect standing.3 A transient drop that occurs with abrupt standing and resolves rapidly suggests a benign condition, such as dehydration, rather than autonomic failure.
In the laboratory, patients are placed on a tilt table in the head-up position at an angle of at least 60 degrees to detect orthostatic changes in blood pressure. In the office, 1 minute of standing probably detects nearly all cases of orthostatic hypotension; however, standing beyond 2 minutes helps establish the severity (a further drop in blood pressure).4 Orthostatic hypotension developing after 3 minutes of standing is uncommon and may represent a reflex presyncope (eg, vasovagal) or a mild or early form of sympathetic adrenergic dysfunction.4, 5
NEUROGENIC AND NONNEUROGENIC CAUSES
Orthostatic hypotension may result from neurogenic and nonneurogenic causes.
Neurogenic orthostatic hypotension can be due to neuropathy (eg, diabetic or autoimmune neuropathies) or to central lesions (eg, Parkinson disease or multiple system atrophy). Its presence, severity, and temporal course can be important clues in diagnosing Parkinson disease and differentiating it from other parkinsonian syndromes with a more ominous prognosis, such as multiple system atrophy and Lewy body dementia.
Nonneurogenic causes include cardiac impairment (eg, from myocardial infarction or aortic stenosis), reduced intravascular volume (eg, from dehydration, adrenal insufficiency), and vasodilation (eg, from fever, systemic mastocytosis).
Common drugs that cause orthostatic hypotension are diuretics, alpha-adrenoceptor blockers for prostatic hypertrophy, antihypertensive drugs, and calcium channel blockers. Insulin, levodopa, and tricyclic antidepressants can also cause vasodilation and orthostatic hypotension in predisposed patients. Poon and Braun,6 in a retrospective study in elderly veterans, identified hydrochlorothiazide, lisinopril (Prinivil, Zestril), trazodone (Desyrel), furosemide (Lasix), and terazosin (Hytrin) as the most common culprits.
ORTHOSTATIC HYPOTENSION IS COMMON IN THE ELDERLY
The prevalence of orthostatic hypotension is high in the elderly and depends on the characteristics of the population studied, such as age, use of medications, and comorbidities known to be associated with this problem. Orthostatic hypotension is more common in institutionalized elderly people (up to 68%)7 than in those living in the community (6%).8 The high prevalence among institutionalized patients likely reflects multiple disease processes, including neurologic and cardiac conditions, as well as medications associated with orthostatic hypotension.
CLINICAL MANIFESTATIONS ARE DUE TO HYPOPERFUSION, OVERCOMPENSATION
Symptoms are related to cerebral hypoperfusion, with resulting lack of cerebral oxygenation (causing lightheadedness, dizziness, weakness, difficulty thinking, headache, syncope, or feeling faint) and a compensatory autonomic overreaction (causing palpitations, tremulousness, nausea, coldness of extremities, chest pain, and syncope).
Lightheadedness is a common symptom, but subtler issues such as difficulty thinking, weakness, and neck discomfort are also common in the elderly. Recurrent or unexplained falls in older adults may be a manifestation of syncope due to orthostatic hypotension.
PROGNOSIS DEPENDS ON CAUSE
Orthostatic hypotension is a syndrome, and its prognosis depends on its specific cause, its severity, and the distribution of its autonomic and nonautonomic involvement. In patients who have extrapyramidal and cerebellar disorders (eg, Parkinson disease, multiple system atrophy), the earlier and the more severe the involvement of the autonomic nervous system, the poorer the prognosis.9,10
In hypertensive patients with diabetes mellitus, the risk of death is higher if they have orthostatic hypotension.11 Diastolic orthostatic hypotension is associated with a higher risk of vascular death in older persons.12
MANAGEMENT: FROM A TO F
The goal of management of orthostatic hypotension is to raise the patient’s standing blood pressure without also raising his or her supine blood pressure, and specifically to reduce orthostatic symptoms, increase the time the patient can stand, and improve his or her ability to perform daily activities. No specific treatment is currently available that achieves all these goals, and drugs alone are never completely adequate.
Because the mainstays of treatment are volume expansion and vasoconstriction, it is difficult to improve the symptoms of orthostatic hypotension without inducing some degree of supine hypertension. Strategies to minimize nocturnal hypertension and to treat orthostatic hypotension in special circumstances are summarized in Table 1.
If hypovolemia is playing a major role, and the patient cannot ingest enough salt or plasma volume fails to increase despite salt supplementation, fludrocortisone (Florinef) should be considered. Untreated hypovolemia will decrease the efficacy of vasoconstrictor drugs.
Pyridostigmine (Mestinon) has a putative vasoconstrictor effect only during standing, but because its effect is modest it should be used in mild orthostatic hypotension that does not improve with nonpharmacologic measures and in moderate cases. Its effect can be enhanced with additional low doses of midodrine (ProAmatine). Midodrine with or without fludrocortisone should be used in severe orthostatic hypotension.
A: Abdominal compression
In conditions in which there is adrenergic denervation of vascular beds, there is an increase in vascular capacitance and peripheral venous pooling. Compression of capacitance beds (ie, the legs and abdomen) improves orthostatic symptoms.14 The improvement is due to a reduction of venous capacitance and an increase in total peripheral resistance.14
On standing, healthy adults experience an orthostatic shift of approximately 500 mL of blood to the lower extremities15 that, when added to an increased vascular capacitance in those with orthostatic hypotension, results in a relative state of hypovolemia.
Compression of the legs alone is not as beneficial as compression of the abdomen because the venous capacitance of the calves and thighs is relatively small compared with that of the splanchnic mesenteric bed, which accounts for 20% to 30% of total blood volume.16 Moreover, compression garments and stockings that are strong enough to produce a measurable effect on orthostatic hypotension are cumbersome to put on and uncomfortable to wear. Because some patients gain significant benefit from abdominal compression alone, this should be considered the first step in reducing venous capacitance.
In a laboratory experiment, Smit et al17 found that an elastic abdominal binder that exerted 15 to 20 mm Hg of pressure on the abdomen raised the standing blood pressure by about 11/6 mmHg, which was comparable to the effect of a gravity suit (such as those worn by fighter pilots to prevent syncope during violent aircraft maneuvers) inflated to 20 mm Hg—an increase of about 17/8 mm Hg. Higher gravity-suit pressures had a greater effect.
In practical terms, the binder should be tight enough to exert gentle pressure. It should be put on before rising from bed in the morning and taken off when lying supine, to avoid supine hypertension. Advantages are that a binder’s effects are immediate, its benefits can be easily assessed, and it can be used on an as-needed basis by patients who need it only during periods of prolonged orthostatic stress. Binders are also easy to fit and are available in most sporting good stores and on the Web (try searching for “abdominal binder”).
When abdominal compression alone is not enough, the addition of compression of the lower extremities can result in further benefits. This can be achieved by using compression garments that ideally extend to the waist or, at the least, to the proximal thigh.
B: Boluses of water
Rapidly drinking two 8-oz (500-mL) glasses of cold water helps expand plasma volume. It also, within a few minutes, elicits a significant pressor effect that is in part norepinephrine-mediated,18,19 increasing the standing systolic blood pressure by more than 20 mm Hg for about 2 hours and improving symptoms and orthostatic endurance.18,20 This easy technique can be used when prolonged standing is expected (eg, shopping).
B (continued): Bed up
The head of the bed of a patient with orthostatic hypotension should be elevated by 10 to 20 degrees or 4 inches (10 cm) to decrease nocturnal hypertension and nocturnal diuresis.21 During the day, adequate orthostatic stress, ie, upright activity, should be maintained. If patients are repeatedly tilted up, their orthostatic hypotension is gradually attenuated, presumably by increasing venomotor tone.22
C: Countermaneuvers
Physical countermaneuvers involve isometrically contracting the muscles below the waist for about 30 seconds at a time, which reduces venous capacitance, increases total peripheral resistance, and augments venous return to the heart.23,24 These countermeasures can help maintain blood pressure during daily activities and should be considered at the first symptoms of orthostatic intolerance and in situations of orthostatic stress (eg, standing for prolonged periods).
Specific techniques include23:
- Toe-raising
- Leg-crossing and contraction
- Thigh muscle co-contraction
- Bending at the waist
- Slow marching in place
- Leg elevation.
D: Drugs
Midodrine, a vasopressor, is effective and safe when used for treating neurogenic orthostatic hypotension.25 It has been shown to increase standing systolic blood pressure, reduce orthostatic lightheadedness, and increase standing and walking time.
A common starting dose is 5 mg three times a day; most patients respond best to 10 mg three times a day. As its duration of action is short (2 to 4 hours),25–27 it should be taken before arising in the morning, before lunch, and in the midafternoon. To avoid nocturnal supine hypertension, doses should not be taken after the midafternoon, and a dose should be omitted if the supine or sitting blood pressure is greater than 180/100 mm Hg.
Midodrine’s main side effects are supine hypertension, scalp paresthesias, and pilomotor reactions (goosebumps). Vasoconstrictors such as midodrine are ineffective when plasma volume is reduced.
Fludrocortisone is a synthetic mineralocorticoid that has a pressor effect as a result of its ability to expand plasma volume and increase vascular alpha-adrenoceptor sensitivity.28–30 This medication is helpful when plasma volume fails to adequately increase with salt supplementation31 and for patients who cannot ingest enough salt or do not respond adequately to midodrine.
The usual dose is 0.1 to 0.2 mg/day, but it may be increased to 0.4 to 0.6 mg/day in patients with refractory orthostatic hypotension.
If the patient gains 3 to 5 pounds (1.2–2.3 kg) and develops mild dependent edema, you can infer that the plasma volume has expanded adequately. However, in view of these effects, fludrocortisone is contraindicated in congestive heart failure and chronic renal failure. The potential risks are severe hypokalemia and excessive supine hypertension. Frequent monitoring of serum potassium, a diet high in potassium, and regular checks of supine blood pressure are advised, especially at higher doses, when added to midodrine, or in elderly patients who tend to poorly tolerate the medication.28,29,32
Pyridostigmine is a cholinesterase inhibitor that improves ganglionic neurotransmission in the sympathetic baroreflex pathway. Because this pathway is activated primarily during standing, this drug improves orthostatic hypotension and total peripheral resistance without aggravating supine hypertension. Because the pressor effect is modest, it is most adequate for patients with mild to moderate orthostatic hypotension.33,34
Dosing is started at 30 mg two to three times a day and is gradually increased to 60 mg three times a day. The drug’s effectiveness can be enhanced by combining each dose of pyridostigmine with 5 mg of midodrine without occurrence of supine hypertension.34 Mestinon Timespan, a 180-mg slow-release pyridostigmine tablet, can be taken once a day and may be a convenient alternative.
The main side effects are cholinergic (abdominal colic, diarrhea).
Review the patient’s medications. If he or she is taking any drug that may cause orthostatic hypotension, consider discontinuing it, substituting another drug, or changing the dosage (Table 2). In the elderly, antiparkinsonian, nitrate, antidepressant, diuretic, prostate, and antihypertensive medications35 may be particularly suspect.
E: Education
Education is probably the single most important factor in the proper control of orthostatic hypotension. A number of issues should be considered.
- Patients should be taught, in simple terms, the mechanisms that maintain postural normotension and how to recognize the onset of orthostatic symptoms.
- They must realize that there is no specific treatment of the underlying cause and that drug treatment alone is not adequate.
- They should be taught nonpharmacologic approaches and be aware that other drugs they start may worsen symptoms.
It is also important that the patient learn the conditions (and their mechanisms) that can lower blood pressure (Table 3). Such conditions include prolonged or motionless standing, alcohol ingestion (causing vasodilation), carbohydrate-heavy meals (causing postprandial orthostatic hypotension related to an increase in the splanchnic-mesenteric venous capacitance), early morning orthostatic hypotension related to nocturnal diuresis and arising from bed, physical activity sufficient to cause muscle vasodilation, heat exposure (eg, hot weather or a hot bath or shower) producing skin vessel vasodilation, sudden postural changes, and prolonged recumbency. Once these stressors are explained, patients have no difficulty recognizing them.
The patient should also be instructed in how to manage situations of increased orthostatic stress and periods of orthostatic decompensation, to minimize nocturnal hypertension, and to modify their activities of daily living. Keeping a log of supine and upright blood pressures (taken with an automated sphygmomanometer) during situations of orthostatic stress can help establish whether worsening symptoms are related to orthostatic hypotension or to another mechanism. Once patients discover that they can actively deal with these situations, they develop a great sense of empowerment.
E (continued): Exercise
Mild physical exercise improves orthostatic tolerance by reducing venous pooling and increasing plasma volume.36 Deconditioning from lack of exercise exacerbates orthostatic hypotension.37 Because upright exercise may increase the orthostatic drop in blood pressure, training in a supine or sitting position (eg, swimming, recumbent biking) is advisable. Isotonic exercise (eg, light weight-lifting) is recommended because the incorrect straining and breath-holding during isometric exercise (eg, holding weights in the same position) may decrease venous return.
F: Fluid and salt (volume expansion)
Maintaining an adequate plasma volume is crucial. Patients should drink five to eight 8-ounce glasses (1.25 to 2.5 L) of water or other fluid per day. Many elderly people do not take in this much. The patient should have at least 1 glass or cup of fluid with meals and at least twice at other times of each day to obtain 1 L/day.
Salt intake should be between 150 and 250 mmol of sodium (10 to 20 g of salt) per day. Sodium helps with retention of ingested fluids and should be maximized if tolerated. However, caution should be exercised in patients who have severe refractory supine hypertension, uncontrolled hypertension, or comorbidities characterized by insterstitial edema (eg, heart failure, liver failure). Some patients are very sensitive to sodium supplementation and can fine-tune their orthostatic control with salt alone. If salting food is not desired, prepared soups, pretzels, potato chips, and 0.5- or 1.0-g salt tablets can be an option.
Patients need to maintain a high-potassium diet, as the high sodium intake combined with fludrocortisone promotes potassium loss. Fruits (especially bananas) and vegetables have high potassium content.
The combination of fludrocortisone and a high-salt diet can also cause sustained supine hypertension, which can be minimized by the interventions noted in Table 2.
Appropriate salt supplementation and fluid intake leading to an adequate volume expansion can be verified by checking the 24-hour urinary sodium content: patients who excrete less than 170 mmol can be treated with 1 to 2 g of supplemental sodium three times a day.38
- Sjostrand T. The regulation of the blood distribution in man. Acta Physiol Scand 1952; 26:312–327.
- Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977; 296:293–297.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996; 46:1470.
- Gehrking JA, Hines SM, Benrud-Larson LM, Opher-Gehrking TL, Low PA. What is the minimum duration of head-up tilt necessary to detect orthostatic hypotension? Clin Auton Res 2005; 15:71–75.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005; 30:173–178.
- Weiss A, Grossman E, Beloosesky Y, Grinblat J. Orthostatic hypotension in acute geriatric ward: is it a consistent finding? Arch Intern Med 2002; 162:2369–2374.
- Mader SL, Josephson KR, Rubenstein LZ. Low prevalence of postural hypotension among community-dwelling elderly. JAMA 1987; 258:1511–1514.
- Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991; 1:147–155.
- Saito Y, Matsuoka Y, Takahashi A, Ohno Y. Survival of patients with multiple system atrophy. Intern Med 1994; 33:321–325.
- Davis BR, Langford HG, Blaufox MD, Curb JD, Polk BF, Shulman NB. The association of postural changes in systolic blood pressure and mortality in persons with hypertension: the Hypertension Detection and Follow-up Program experience. Circulation 1987; 75:340–346.
- Luukinen H, Koski K, Laippala P, Kivelä SL. Prognosis of diastolic and systolic orthostatic hypotension in older persons. Arch Intern Med 1999; 159:273–280.
- Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. N Engl J Med 1993; 329:611–615.
- Denq JC, Opfer-Gehrking TL, Giuliani M, Felten J, Convertino VA, Low PA. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clin Auton Res 1997; 7:321–326.
- Sjostrand T. Volume and distribution of blood and their significance in regulating the circulation. Physiol Rev 1953; 33:202–228.
- Rowell LB, Detry JM, Blackmon JR, Wyss C. Importance of the splanchnic vascular bed in human blood pressure regulation. J Appl Physiol 1972; 32:213–220.
- Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res 2004; 14:167–175.
- Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000; 101:504–509.
- Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002; 112:355–360.
- Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water [letter]. Lancet 1999; 353:723.
- MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia: treatment with the “head-up” bed. JAMA 1940; 115:2162–2167.
- Ector H, Reybrouck T, Heidbüchel H, Gewillig M, Van de Werf F. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol 1998; 21:193–196.
- Bouvette CM, McPhee BR, Opfer-Gehrking TL, Low PA. Role of physical countermaneuvers in the management of orthostatic hypotension: efficacy and biofeedback augmentation. Mayo Clin Proc 1996; 71:847–853.
- Ten Harkel AD, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci (Lond) 1994; 87:553–558.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993; 95:38–48.
- Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med 1995; 99:604–610.
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007; 7:63–70.
- Axelrod FB, Goldberg JD, Rolnitzky L, et al. Fludrocortisone in patients with familial dysautonomia—assessing effect on clinical parameters and gene expression. Clin Auton Res 2005; 15:284–291.
- Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301:68–73.
- van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. Clin Auton Res 2000; 10:35–42.
- Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart 1996; 76:507–509.
- Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry 2003; 74:1294–1298.
- Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63:513–518.
- Lipsitz LA, Pluchino FC, Wei JY, Rowe JW. Syncope in institutionalized elderly: the impact of multiple pathological conditions and situational stress. J Chronic Dis 1986; 39:619–630.
- Mtinangi BL, Hainsworth R. Effects of moderate exercise training on plasma volume, baroreceptor sensitivity and orthostatic tolerance in healthy subjects. Exp Physiol 1999; 84:121–130.
- Bonnin P, Ben Driss A, Benessiano J, Maillet A, Pavy le Traon A, Levy BI. Enhanced flow-dependent vasodilatation after bed rest, a possible mechanism for orthostatic intolerance in humans. Eur J Appl Physiol 2001; 85:420–426.
- El-Sayed H, Hainsworth R. Salt supplementation increases plasma volume and orthostatic tolerance in patients with unexplained syncope. Heart 1996; 75:134–140.
- Sjostrand T. The regulation of the blood distribution in man. Acta Physiol Scand 1952; 26:312–327.
- Ziegler MG, Lake CR, Kopin IJ. The sympathetic-nervous-system defect in primary orthostatic hypotension. N Engl J Med 1977; 296:293–297.
- The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology 1996; 46:1470.
- Gehrking JA, Hines SM, Benrud-Larson LM, Opher-Gehrking TL, Low PA. What is the minimum duration of head-up tilt necessary to detect orthostatic hypotension? Clin Auton Res 2005; 15:71–75.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Poon IO, Braun U. High prevalence of orthostatic hypotension and its correlation with potentially causative medications among elderly veterans. J Clin Pharm Ther 2005; 30:173–178.
- Weiss A, Grossman E, Beloosesky Y, Grinblat J. Orthostatic hypotension in acute geriatric ward: is it a consistent finding? Arch Intern Med 2002; 162:2369–2374.
- Mader SL, Josephson KR, Rubenstein LZ. Low prevalence of postural hypotension among community-dwelling elderly. JAMA 1987; 258:1511–1514.
- Sandroni P, Ahlskog JE, Fealey RD, Low PA. Autonomic involvement in extrapyramidal and cerebellar disorders. Clin Auton Res 1991; 1:147–155.
- Saito Y, Matsuoka Y, Takahashi A, Ohno Y. Survival of patients with multiple system atrophy. Intern Med 1994; 33:321–325.
- Davis BR, Langford HG, Blaufox MD, Curb JD, Polk BF, Shulman NB. The association of postural changes in systolic blood pressure and mortality in persons with hypertension: the Hypertension Detection and Follow-up Program experience. Circulation 1987; 75:340–346.
- Luukinen H, Koski K, Laippala P, Kivelä SL. Prognosis of diastolic and systolic orthostatic hypotension in older persons. Arch Intern Med 1999; 159:273–280.
- Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. N Engl J Med 1993; 329:611–615.
- Denq JC, Opfer-Gehrking TL, Giuliani M, Felten J, Convertino VA, Low PA. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clin Auton Res 1997; 7:321–326.
- Sjostrand T. Volume and distribution of blood and their significance in regulating the circulation. Physiol Rev 1953; 33:202–228.
- Rowell LB, Detry JM, Blackmon JR, Wyss C. Importance of the splanchnic vascular bed in human blood pressure regulation. J Appl Physiol 1972; 32:213–220.
- Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin Auton Res 2004; 14:167–175.
- Jordan J, Shannon JR, Black BK, et al. The pressor response to water drinking in humans: a sympathetic reflex? Circulation 2000; 101:504–509.
- Shannon JR, Diedrich A, Biaggioni I, et al. Water drinking as a treatment for orthostatic syndromes. Am J Med 2002; 112:355–360.
- Jordan J, Shannon JR, Grogan E, Biaggioni I, Robertson D. A potent pressor response elicited by drinking water [letter]. Lancet 1999; 353:723.
- MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia: treatment with the “head-up” bed. JAMA 1940; 115:2162–2167.
- Ector H, Reybrouck T, Heidbüchel H, Gewillig M, Van de Werf F. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol 1998; 21:193–196.
- Bouvette CM, McPhee BR, Opfer-Gehrking TL, Low PA. Role of physical countermaneuvers in the management of orthostatic hypotension: efficacy and biofeedback augmentation. Mayo Clin Proc 1996; 71:847–853.
- Ten Harkel AD, van Lieshout JJ, Wieling W. Effects of leg muscle pumping and tensing on orthostatic arterial pressure: a study in normal subjects and patients with autonomic failure. Clin Sci (Lond) 1994; 87:553–558.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993; 95:38–48.
- Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med 1995; 99:604–610.
- Maule S, Papotti G, Naso D, Magnino C, Testa E, Veglio F. Orthostatic hypotension: evaluation and treatment. Cardiovasc Hematol Disord Drug Targets 2007; 7:63–70.
- Axelrod FB, Goldberg JD, Rolnitzky L, et al. Fludrocortisone in patients with familial dysautonomia—assessing effect on clinical parameters and gene expression. Clin Auton Res 2005; 15:284–291.
- Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med 1979; 301:68–73.
- van Lieshout JJ, Ten Harkel AD, Wieling W. Fludrocortisone and sleeping in the head-up position limit the postural decrease in cardiac output in autonomic failure. Clin Auton Res 2000; 10:35–42.
- Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart 1996; 76:507–509.
- Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry 2003; 74:1294–1298.
- Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol 2006; 63:513–518.
- Lipsitz LA, Pluchino FC, Wei JY, Rowe JW. Syncope in institutionalized elderly: the impact of multiple pathological conditions and situational stress. J Chronic Dis 1986; 39:619–630.
- Mtinangi BL, Hainsworth R. Effects of moderate exercise training on plasma volume, baroreceptor sensitivity and orthostatic tolerance in healthy subjects. Exp Physiol 1999; 84:121–130.
- Bonnin P, Ben Driss A, Benessiano J, Maillet A, Pavy le Traon A, Levy BI. Enhanced flow-dependent vasodilatation after bed rest, a possible mechanism for orthostatic intolerance in humans. Eur J Appl Physiol 2001; 85:420–426.
- El-Sayed H, Hainsworth R. Salt supplementation increases plasma volume and orthostatic tolerance in patients with unexplained syncope. Heart 1996; 75:134–140.
KEY POINTS
- Treatment is directed at increasing blood volume, decreasing venous pooling, and increasing vasoconstriction while minimizing supine hypertension.
- Patient education and nondrug strategies alone can be effective in mild cases. Examples: consuming extra fluids and salt, wearing an abdominal binder, drinking boluses of water, raising the head of the bed, and performing countermaneuvers and physical activity.
- Moderate and severe cases require additional drug treatment. Pyridostigmine (Mestinon) is helpful in moderate cases. Fludrocortisone (Florinef) and midodrine (ProAmatine) are indicated in more severe cases.
Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival?
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
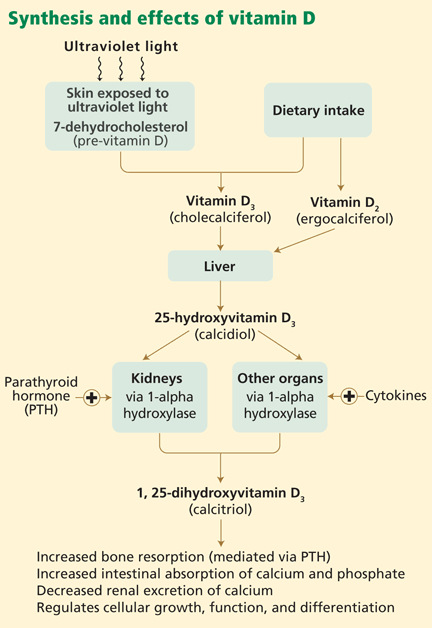
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE

The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.